
Clusterin interacts with Paclitaxel and confer Paclitaxel resistance in ovarian cancer
Upload
independentCategory
view
3download
0
2007;67:1979-1987. Cancer Res George Z. Cheng, Joseph Chan, Qi Wang, et al. Resistance to PaclitaxelCells Leading to Increased Migration, Invasion, and Twist Transcriptionally Up-regulates AKT2 in Breast Cancer
Updated version
http://cancerres.aacrjournals.org/content/67/5/1979
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2007/02/26/67.5.1979.DC1.html
Access the most recent supplemental material at:
Cited Articles
http://cancerres.aacrjournals.org/content/67/5/1979.full.html#ref-list-1
This article cites by 58 articles, 28 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/67/5/1979.full.html#related-urls
This article has been cited by 34 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

Twist Transcriptionally Up-regulates AKT2 in Breast Cancer
Cells Leading to Increased Migration, Invasion, and
Resistance to Paclitaxel
George Z. Cheng,1Joseph Chan,
1Qi Wang,
2Weizhou Zhang,
1Calvin D. Sun,
1and Lu-Hai Wang
1
1Department of Microbiology, Mount Sinai School of Medicine, New York, New York and 2Molecular Oncology Program, H. Lee MoffittCancer Center and Research Institute, University of South Florida College of Medicine, Tampa, Florida
Abstract
Metastasis, the cardinal feature of malignant tumors, is animportant clinical variable in patient prognosis. To under-stand the basis for metastasis, we systematically selected forhighly invasive cells from breast cancer cell lines, MCF7 andMDA-MB-453, with moderate to low invasive ability usingBoyden chamber invasion assay. The four-cycle selectedinvasive lines, named MCF7-I4 and MDA-MB-453-I4, respec-tively, displayed epithelial-mesenchymal transition (EMT)and dramatically enhanced invasive ability. EMT changeswere corroborated with decreased level of E-cadherin andincreased vimentin, fibronectin, and B1 integrin. Twist, a basichelix-loop-helix transcription factor, and AKT2, a knownproto-oncogene, were found to be elevated in the invasivecells compared with the parental. Ectopic expression andknockdown of Twist by short interference RNA resulted insignificant increase and reduction, respectively, of AKT2protein and mRNA expression. Twist bound to E-box elementson AKT2 promoter and enhanced its transcriptional activity.Moreover, silencing AKT2 decreased Twist-promoted migra-tion, invasion, and paclitaxel resistance. Reintroducing AKT2largely rescued the phenotype resulted from knockdown ofTwist in I4 cells, suggesting that AKT2 is a downstream targetand functional mediator of Twist. Finally, we observed a 68.8%correlation of elevated Twist and AKT2 expression in late-stage breast cancers as oppose to 13% in early-stage breastcancers. Our study identifies Twist as a positive transcrip-tional regulator of AKT2 expression, and Twist-AKT2 signalingis involved in promoting invasive ability and survival of breastcancer cells. [Cancer Res 2007;67(5):1979–87]
Introduction
The molecular mechanism of metastasis is still poorly under-stood. Current models propose a sequential program of metastasiswhere cancer cells penetrate the basement membrane, intravasateinto blood/lymphatic vessels, survive the journey in vasculature,extravasate into secondary sites, and adapt to new hostenvironment (1). Metastatic tumors ultimately lead to poor clinicaloutcomes; for breast cancer, a significant reduction in the 5-yearsurvival rate from 90% to 20% is observed when comparinglocalized versus metastasized tumors (2). Metastasis-associated
molecular changes include decreased cell-cell junction proteins,such as E-cadherin (3–5), and increased basement membranedegradation proteins, such as matrix metalloproteinases andcollagenases (6, 7).
The serine/threonine kinase AKT/protein kinase B, a down-stream effector of phosphatidylinositol 3-kinase pathway, has beenshown to play a key role in cell survival and growth (8–10).Activated AKT prevents apoptosis via enhancing glucose uptakeand utilization, activating nuclear factor-nB pathway, promotingMDM2 nuclear translocation, increasing Bcl-2 or Bcl-xL levels,cytoplasmic sequestering of forkhead-related ligand 1, inactivatingBad and Bax, and inhibiting cytochrome c release from mitochon-dria (11–13). Additionally, AKT contributes to neoplastic growthvia antagonizing p21Cip1/Waf1 and p27Kip1, inhibiting glycogen syn-thase kinase-3h, and promoting mammalian target of rapamycinactivity (11). Of the three AKT isoforms, AKT2 has been shownto promote cell motility, invasiveness, and metastasis (14, 15).Moreover, AKT2 is amplified or activated in prostate (9), hepa-tocellular (16), colon (17), follicular thyroid (18, 19), pancreatic(20–22), ovarian (8, 23, 24), and breast carcinomas (10, 25, 26).
Twist, a basic helix-loop-helix (bHLH) transcription factor, ischaracterized by a basic DNA binding domain that targets theconsensus E-box sequence 5¶-CANNTG-3¶ and a helix-loop-helixdomain that mediates heterodimerization or homodimerization(27). The bHLH protein family has well-described functions in cellgrowth and differentiation in both vertebrates and invertebrates(28, 29). Developmentally, Twist inhibits myogenic differentiationvia interfering with myoD and MEF2 activity (30, 31). Moreover,Twist mediates transcriptional repression by inhibiting twodifferent coactivators, p300 and PCAF (32). Recently, Twist hasgained attention as a putative oncogene (33–35), as a contributorto acquired paclitaxel resistance (36), and as a key regulator ofmetastasis (37). Exogenous overexpression of Twist inhibitsapoptosis and promotes colony formation via suppression of theADP ribosylation factor/MDM2/p53 pathway (33). In the processof metastasis, Twist plays a crucial role by down-regulatingE-cadherin and h-catenin, promoting epithelial-mesenchymaltransition (EMT), and mediating cell motility and invasiveness(37, 38). Elevated Twist expression leads to higher vascularendothelial growth factor expression, promotes angiogenesis, andcorrelates with chromosomal instability in breast cancer (39). Inaddition, increased Twist expression is found in rhabdomyosarco-ma (33), melanoma (34), pediatric osteosarcoma (40), T-celllymphoma (41), gastric (42), prostate (38), and breast carcinoma(37, 43). The elevated Twist expression positively correlates withaggressiveness of cancer and poor survival rate (34, 37, 38).
In an effort to study the molecular mechanism underlyingmetastasis, we have established a model system using Boydenchamber invasion assay to select highly invasive cells from a
Note: Supplementary data for this article are available at Cancer Research Online(http://cancerres.aacrjournals.org/).
Requests for reprints: Lu-Hai Wang, Department of Microbiology, Mount SinaiSchool of Medicine, Box 1124, One Gustave L. Levy Place, New York, NY 10029-6574.Phone: 212-241-3759; Fax: 212-534-1684; E-mail: [email protected].
I2007 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-06-1479
www.aacrjournals.org 1979 Cancer Res 2007; 67: (5). March 1, 2007
Research Article
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

population of moderate to low invasive breast cancer lines (MCF7and MDA-MB-453, respectively). We observed increased Twist andAKT2 expression in the selected highly invasive cells andinvestigated the possible functional connection between thesetwo proteins. We show that Twist increased mRNA and proteinlevels of AKT2 in a dosage-dependent manner. In addition, weshow that Twist transactivated AKT2 by binding to E-boxes on theAKT2 promoter and drove its expression. Furthermore, blockingAKT2 largely abolished Twist-mediated paclitaxel resistance andinvasion activity. Ectopic expression of AKT2 rescued thephenotype of knockdown of Twist. Thus, our results indicate thatAKT2 is downstream target of Twist and is a critical player inTwist-promoted metastatic process.
Materials and Methods
Cloning and construction of Twist and related plasmids. Full-lengthcDNA of Twist was generated by reverse transcription using normal human
mammary total RNA as template and followed by nest PCR amplification
with the primers derived from human Twist (nest primers: 5¶-GCTCTTC-
TCCTCTGCCCCGG-3¶ and 5¶-CATCTAGGTCTCCGGCCCTG-3¶). Green fluo-rescent protein (GFP)–fused and Myc-tagged Twist were created by
digestion of the PCR products with BamHI/EcoRI and by subcloning the
PCR products into pEGFP-C2 and pCMV-Tag3B vectors, respectively. The
resulted constructs were confirmed by DNA sequencing. AKT2 promoter–driven luciferase reporter plasmids were described previously (44). Deletion
mutants of AKT2 promoters were created by PCR.
Antibodies and reagents. Antibodies against E-cadherin, a-catenin, h-
catenin, g-catenin, h1 integrin, and fibronectin were from PharMingen/BDBiosciences (San Jose, CA). Anti-AKT2, anti-Myc, anti-actin, and anti-GFP
antibodies were purchased from Cell Signaling (Danvers, MA). Anti-Twist
antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).Paclitaxel and growth factor–reduced Matrigel were from Sigma (St. Louis,
MO) and BD Biosciences, respectively. Two chamber culture apparatus was
obtained from Falcon (BD BioSciences, San Jose, CA).
Cell lines, transfection, and tumor specimens. Cells were cultured inDMEM with 10% FCS and antibiotics. Cultures were maintained in a
humidified incubator at 37jC in the presence of 5% CO2. Transfections were
done with LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) in the presence of
serum according to the manufacturer’s instructions. Primary human breastcancer specimens were procured anonymously from patients who
underwent surgery at H. Lee Moffitt Cancer Center (Tampa, FL), and each
sample contained at least 70% tumor cells, as was confirmed by microscopicexamination. The tissues were snap frozen and stored at �70jC.
Cell invasion assay and selection. Invasion assays were done in a
Boyden chamber with polyethylene terephthalate filter inserts for 24-well
plates containing 0.8-Am pores (Falcon). Filters were coated on ice with100 AL growth factor–reduced Matrigel at 0.5 to 0.8 mg/mL protein. Cells
(1 � 104 to 1 � 105) were plated in 300 AL of 0.1% bovine serum albumin
(BSA)-DMEM into the upper chamber. The lower chamber was filled with
500 AL of 10% FCS-DMEM. After culture for 6 to 36 h, noninvaded cells inthe inserts were removed with cotton swabs. The invaded cells on the
underside were treated with a fixative/staining solution (0.1% crystal violet,
1% formalin, and 20% ethanol) for visualization. The invaded cells atunderside of the membrane were photographed. To select for invasive cells,
larger inserts and companion plates were used (Falcon). Cells (2 � 106) in a
volume of 1.5 mL medium were seeded in the insert and with 2.5 mL of 10%
FCS-DMEM as the chemoattractant in the lower chamber. After 24 h ofincubation, the invaded cells were recovered by trypsinization and
amplified for the next cycle of selection. Statistical analysis was done using
two-sample t test, assuming equal variance, and P value was calculated
based on two-tailed test.Soft agar colony formation assay. Cells (1 � 105) were suspended in
10% FCS-DMEM containing 0.4% agar. The cells were then placed into a
dish containing a hard agar base composed of 10% FCS-DMEM and 0.75%
agar. The cultures were returned to the incubator and fed every 2 days with
500 AL of normal growth medium. Photomicrographs of colonies weretaken 14 to 21 days later.
Cell survival assay. MCF7 cells stably expressing pCMV-Tag3B or pCMV-
Twist were transfected with siAKT2 or siControl. Thirty-six hours after
transfection, the cells were treated with or without paclitaxel in ethanol.
Thirty-six hours later, percentage cell survival was determined by flow
cytometry or trypan blue staining. Briefly, for flow cytometry, 1� 106 to 2 �106 cells were fixed in 70% ethanol and stained with propidium iodide (PI)
solution (50 Ag PI and 200 Ag RNase A). Cells were resuspended in PBS and
analyzed via flow cytometry. For trypan blue staining, cells were diluted 1:2
with 0.1% trypan blue in PBS and then counted under a light microscope.
Live cells were not permeable to trypan blue and remained clear and phase
bright. Percentage of cell death was calculated via counting cells permeable
to trypan blue. Statistical analysis was done using two-sample t test,
assuming equal variance, and P value was calculated based on two-tailed test.Luciferase reporter assay. HEK293T or MCF7 cells were seeded in six-
well plate and transfected with AKT2-Luc reporter plasmid, pRenilla-
luciferase plasmid, and Twist. The amount of DNA in each transfection waskept constant by the addition of empty vector, pCMV-Tag3B. Thirty-six
hours post-transfection, firefly and Renilla luciferase were assayed accord-
ing to the manufacturer’s protocol (Promega, Madison, WI). Luciferase
activity was expressed as relative light units. Each experiment was repeatedthrice in triplicates.
Chromatin immunoprecipiatation assay. Chromatin immunopreci-
piatation (ChIP) assay was done essentially as described previously with
modifications (45). Briefly, soluble chromatin was prepared from a total
of 2 � 107 asynchronously growing HEK293T cells that were transfected
with Myc-Twist. The chromatin solution was diluted 10-fold with ChIP
dilution buffer [1.1% Triton X-100, 1.2 mmol/L EDTA, 167 mmol/L NaCl,
16.7 mmol/L Tris-HCl (pH 8.1), 0.01% SDS, protease inhibitors] and
precleared with protein-A beads blocked with 2 Ag of sheared salmon sperm
DNA and preimmune serum. The precleared chromatin solution was
divided and used in immunoprecipitation assays with either an anti-Myc
antibody or an anti-HA antibody. Following wash, the antibody-protein-
DNA complex was eluted from the beads by resuspending the pellets in
1% SDS and 0.1 mol/L NaHCO3 at room temperature for 20 min. After
reversal cross-link incubation at 67jC, protein and RNA were removed
by incubation with 10 Ag proteinase K and 10 Ag RNase A at 42jC for
3 h. Purified DNA was subjected to PCR with primers specific for four
proximal E-box sites within the AKT2 promoter. The sequences of the
PCR primers used are as follows: 5¶-TGAGATGGAGTCTTGCTCTGTC-3¶and 5¶-ATCACAAGGTCAGGAGGTCG-3¶ (E-box 1); 5¶-ACCTTCTCACCAAG-
GAGGTAG-3¶ and 5¶-TTATTATTTCCTCTGATGGGTGGAG-3¶ (E-box 2);
5¶-TGCTCCAAGCAGACAGATGGG-3¶ and 5¶-TTTTACCTATTCTACCTCCT-
TGGTG-3¶ (E-box 3); and 5¶-TATTTGACATGATTTCTGTGGGTG-3¶ and
5¶-TCTGTCTGCTTGGAGCAGGTGGGG-3¶ (E-box 4).
Northern and Western blot analysis. Northern blot analysis of totalcellular RNA was done according to standard procedures. RNA was extracted
using the RNeasy purification kits (Qiagen, Valencia, CA). Total RNA was
electrophoresed in 1.0% formaldehyde-agarose gels, transferred to Duralon-
UVTM membrane (Stratagene, La Jolla, CA), and then hybridized withrandomly primed [a-32P]dCTP-labeled cDNA probes for AKT2. Membranes
were exposed to autoradiography and the mRNA levels were visualized and
quantified using PhosphorImager analysis (Amersham Biosciences/GE
Healthcare, Piscataway, NJ). Western blot analysis was done as describedpreviously (36). Briefly, the cells were lysed with radioimmunoprecipitation
assay buffer [50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% Triton X-
100, 1% sodium deoxycholate, 0.1% SDS, 1 mmol/L EDTA, 1 mmol/Lphenylmethylsulfonyl fluoride, 5 Ag/mL aprotinin, 5 Ag/mL leupeptin],
separated by SDS-PAGE, and immunoblotted with appropriate antibodies as
indicated in the figure legends.
Short interference RNA, short hairpin RNA, and retroviral construc-tion. The short interference RNA (siRNA) duplexes were constructed with
SilencerTM siRNA Construction kit (Ambion, Austin, TX) following the
manufacturer’s instructions. Two siRNAs for human AKT2 [GCAGAGA-
TTGTCTCGGCTCTT (siAKT2-2 nucleotides 972–992) and GCACAGG-TTCTTCCTCAGCAT (siAKT2-3 nucleotides 1418–1438)] and human Twist
Cancer Research
Cancer Res 2007; 67: (5). March 1, 2007 1980 www.aacrjournals.org
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
[AAGCTGAGCAAGATTCAGACC (siTwist1 nucleotides 505–525) and AGG-TACATCGACTTCCTCTAC (siTwist4 nucleotides 541–561)] were synthe-
sized. Control siRNA oligonucleotide, which does not match any known
human coding cDNA, was designed and used. The siRNA duplexes were
reconstituted to 10 Amol/L in sterile RNase-free water. Transfection of siRNAfor targeting endogenous genes was done using LipofectAMINE 2000 as per
the manufacturer’s instructions. The same siRNA targeting regions with a
hairpin sequence (TTCAAGAGA) was cloned into pSIREN-RetroQ-linker at
BamHI and EcoRI sites (a generous gift from Dr. Domenico Tortorella, MountSinai School of Medicine, New York, NY). Sequences are as shown: 5¶-gatccGCAGAGATTGTCTCGGCTCTTCAAGAGAGAGCCGAGACAATCTCT-
GCTTTTTTACGCGTg-3¶ (shAKT2-2); 5¶-gatccGCACAGGTTCTTCCTCAGCT-
TCAAGAGAGCTGAGGAAGAACCTGTGCTTTTTTACGCGTg-3¶ (shAKT2-3);5¶-gatccAAGCTGAGCAAGATTCAGACCTTCAAGAGAGGTCTGAATCTTG-
CTCAGCTTTTTTTTg-3¶ (shTwist1); and 5¶-gatccAGGTACATCGACTTCCTC-
TACTTCAAGAGAGTAGAGGAAGTCGATGTACCTTTTTTTg-3¶ (shTwist4).Retrovirus containing short hairpin RNA (shRNA) was packaged in HEK293T
cells and transfected with pVSV-G, pPol-GAG, and respective pSIREN-
RetroQ-shRNA. MCF7-I4 was infected with the shRNA virus, selected with
puromycin, and used for migration, invasion, and survival assays.Cell migration and wound healing assay. Migration assay was done
using Boyden chamber without coating of Matrigel. Cells were stained and
counted. ‘‘Wound healing’’ assay was used to detect the alteration of cell
motility. Cells were initially seeded uniformly onto 60-mm culture plateswith an artificial ‘‘wound’’ carefully created at 0 h, using a P-200 pipette tip
to scratch on the confluent cell monolayer. Microphotographs were taken at
0 and 24 h.Immunohistochemistry. Immunohistochemistry was done as described
previously (39). Anti-Twist and anti-AKT2 antibodies described above were
used for the staining. Briefly, paraffin sections of 3-Am thick were cut and
probed. After rehydration, samples were treated with solution containing0.3% hydrogen peroxide for 30 min to block endogenous peroxidase
activity. After antigen retrieval in citrate buffer, the sections were incubated
with the primary antibody (1:100 in PBS/1% BSA, overnight at 4jC). and
then with biotinylated secondary antibody (Vector Laboratories, Burlin-game, CA). The signal was amplified by avidin-biotin complex formation
and developed with diaminobenzidine followed by counterstaining with
hematoxylin, dehydrated in alcohol and xylene, and mounted. Thepercentage of staining and staining intensity was estimated with ImageJ
(NIH, Bethesda, MD).
Results
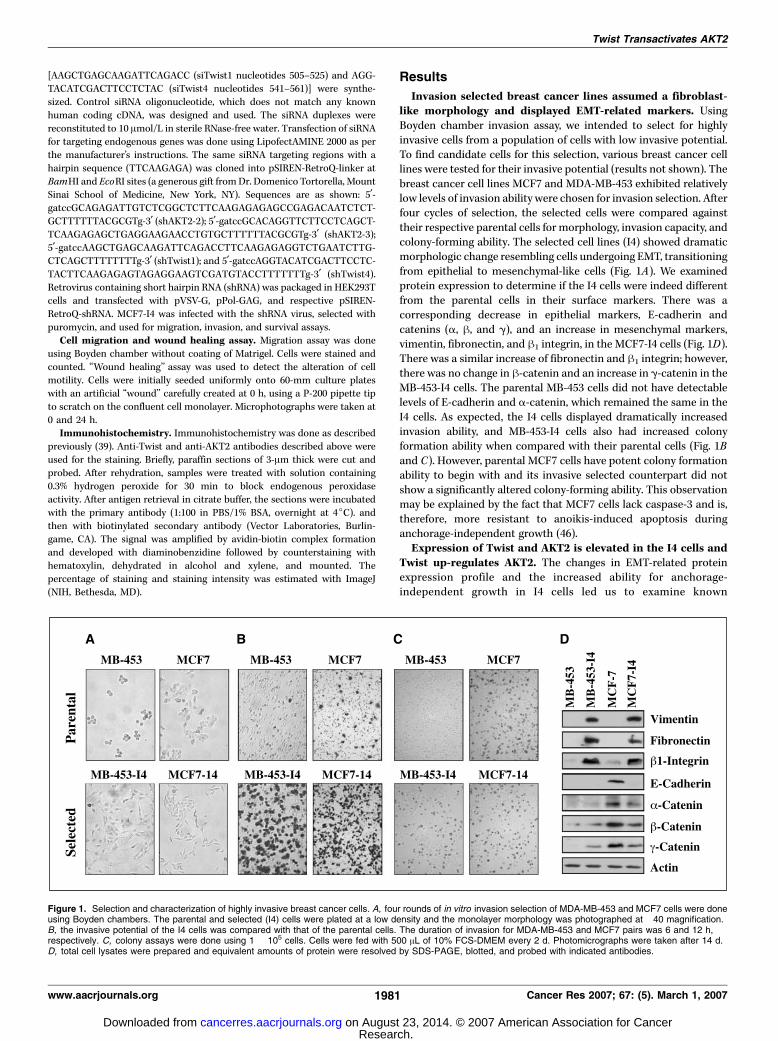
Invasion selected breast cancer lines assumed a fibroblast-like morphology and displayed EMT-related markers. UsingBoyden chamber invasion assay, we intended to select for highlyinvasive cells from a population of cells with low invasive potential.To find candidate cells for this selection, various breast cancer celllines were tested for their invasive potential (results not shown). Thebreast cancer cell lines MCF7 and MDA-MB-453 exhibited relativelylow levels of invasion ability were chosen for invasion selection. Afterfour cycles of selection, the selected cells were compared againsttheir respective parental cells for morphology, invasion capacity, andcolony-forming ability. The selected cell lines (I4) showed dramaticmorphologic change resembling cells undergoing EMT, transitioningfrom epithelial to mesenchymal-like cells (Fig. 1A). We examinedprotein expression to determine if the I4 cells were indeed differentfrom the parental cells in their surface markers. There was acorresponding decrease in epithelial markers, E-cadherin andcatenins (a, h, and g), and an increase in mesenchymal markers,vimentin, fibronectin, and h1 integrin, in the MCF7-I4 cells (Fig. 1D).There was a similar increase of fibronectin and h1 integrin; however,there was no change in h-catenin and an increase in g-catenin in theMB-453-I4 cells. The parental MB-453 cells did not have detectablelevels of E-cadherin and a-catenin, which remained the same in theI4 cells. As expected, the I4 cells displayed dramatically increasedinvasion ability, and MB-453-I4 cells also had increased colonyformation ability when compared with their parental cells (Fig. 1Band C). However, parental MCF7 cells have potent colony formationability to begin with and its invasive selected counterpart did notshow a significantly altered colony-forming ability. This observationmay be explained by the fact that MCF7 cells lack caspase-3 and is,therefore, more resistant to anoikis-induced apoptosis duringanchorage-independent growth (46).Expression of Twist and AKT2 is elevated in the I4 cells and
Twist up-regulates AKT2. The changes in EMT-related proteinexpression profile and the increased ability for anchorage-independent growth in I4 cells led us to examine known
Figure 1. Selection and characterization of highly invasive breast cancer cells. A, four rounds of in vitro invasion selection of MDA-MB-453 and MCF7 cells were doneusing Boyden chambers. The parental and selected (I4) cells were plated at a low density and the monolayer morphology was photographed at �40 magnification.B, the invasive potential of the I4 cells was compared with that of the parental cells. The duration of invasion for MDA-MB-453 and MCF7 pairs was 6 and 12 h,respectively. C, colony assays were done using 1 � 105 cells. Cells were fed with 500 AL of 10% FCS-DMEM every 2 d. Photomicrographs were taken after 14 d.D, total cell lysates were prepared and equivalent amounts of protein were resolved by SDS-PAGE, blotted, and probed with indicated antibodies.
Twist Transactivates AKT2
www.aacrjournals.org 1981 Cancer Res 2007; 67: (5). March 1, 2007
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
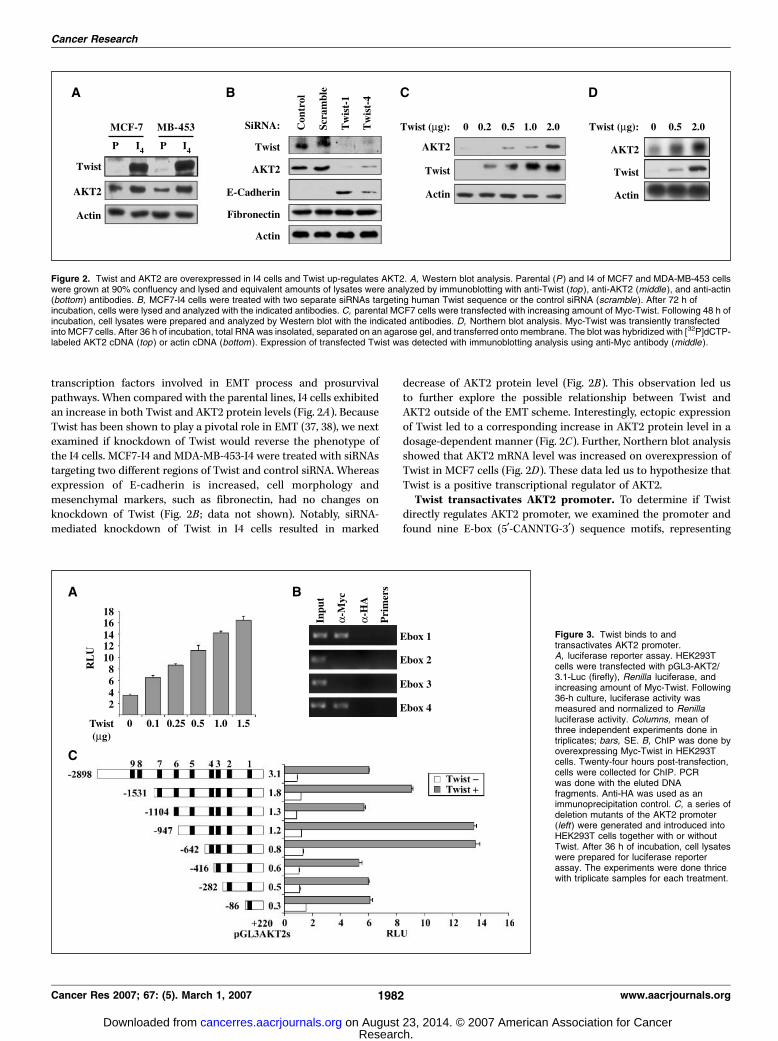
transcription factors involved in EMT process and prosurvivalpathways. When compared with the parental lines, I4 cells exhibitedan increase in both Twist and AKT2 protein levels (Fig. 2A). BecauseTwist has been shown to play a pivotal role in EMT (37, 38), we nextexamined if knockdown of Twist would reverse the phenotype ofthe I4 cells. MCF7-I4 and MDA-MB-453-I4 were treated with siRNAstargeting two different regions of Twist and control siRNA. Whereasexpression of E-cadherin is increased, cell morphology andmesenchymal markers, such as fibronectin, had no changes onknockdown of Twist (Fig. 2B ; data not shown). Notably, siRNA-mediated knockdown of Twist in I4 cells resulted in marked
decrease of AKT2 protein level (Fig. 2B). This observation led usto further explore the possible relationship between Twist andAKT2 outside of the EMT scheme. Interestingly, ectopic expressionof Twist led to a corresponding increase in AKT2 protein level in adosage-dependent manner (Fig. 2C). Further, Northern blot analysisshowed that AKT2 mRNA level was increased on overexpression ofTwist in MCF7 cells (Fig. 2D). These data led us to hypothesize thatTwist is a positive transcriptional regulator of AKT2.Twist transactivates AKT2 promoter. To determine if Twist
directly regulates AKT2 promoter, we examined the promoter andfound nine E-box (5¶-CANNTG-3¶) sequence motifs, representing
Figure 2. Twist and AKT2 are overexpressed in I4 cells and Twist up-regulates AKT2. A, Western blot analysis. Parental (P ) and I4 of MCF7 and MDA-MB-453 cellswere grown at 90% confluency and lysed and equivalent amounts of lysates were analyzed by immunoblotting with anti-Twist (top ), anti-AKT2 (middle ), and anti-actin(bottom ) antibodies. B, MCF7-I4 cells were treated with two separate siRNAs targeting human Twist sequence or the control siRNA (scramble ). After 72 h ofincubation, cells were lysed and analyzed with the indicated antibodies. C, parental MCF7 cells were transfected with increasing amount of Myc-Twist. Following 48 h ofincubation, cell lysates were prepared and analyzed by Western blot with the indicated antibodies. D, Northern blot analysis. Myc-Twist was transiently transfectedinto MCF7 cells. After 36 h of incubation, total RNA was insolated, separated on an agarose gel, and transferred onto membrane. The blot was hybridized with [32P]dCTP-labeled AKT2 cDNA (top ) or actin cDNA (bottom ). Expression of transfected Twist was detected with immunoblotting analysis using anti-Myc antibody (middle ).
Figure 3. Twist binds to andtransactivates AKT2 promoter.A, luciferase reporter assay. HEK293Tcells were transfected with pGL3-AKT2/3.1-Luc (firefly), Renilla luciferase, andincreasing amount of Myc-Twist. Following36-h culture, luciferase activity wasmeasured and normalized to Renillaluciferase activity. Columns, mean ofthree independent experiments done intriplicates; bars, SE. B, ChIP was done byoverexpressing Myc-Twist in HEK293Tcells. Twenty-four hours post-transfection,cells were collected for ChIP. PCRwas done with the eluted DNAfragments. Anti-HA was used as animmunoprecipitation control. C, a series ofdeletion mutants of the AKT2 promoter(left) were generated and introduced intoHEK293T cells together with or withoutTwist. After 36 h of incubation, cell lysateswere prepared for luciferase reporterassay. The experiments were done thricewith triplicate samples for each treatment.
Cancer Research
Cancer Res 2007; 67: (5). March 1, 2007 1982 www.aacrjournals.org
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

possible binding sites for Twist. Luciferase reporter assay was doneto examine if Twist was able to transactivate full-length AKT2promoter. The result indicated that Twist led to a dosage-dependent increase in AKT2 promoter transactivation as measuredby luciferase activity (Fig. 3A).Twist binds to proximal E-box elements on AKT2 promoter.
Serial truncation of AKT2 promoter showed that Twist couldtransactivate AKT2 promoter with a minimal of one E-box, butmaximal transactivation occurred when first four E-boxes werepresent. The addition of E-boxes 2 and 3 contributed minimally tothe transactivation ability of Twist, and the full-length promoterseemed to have repressive elements located between 1.2 and 3.1 kbthat inhibited maximal Twist-mediated transactivation (Fig. 3C).Based on these results, we examined Twist binding to the first fourE-boxes on the endogenous AKT2 promoter using ChIP assay. Theresults showed that Twist bound to endogenous AKT2 promoterE-boxes 1 and 4, but not E-boxes 2 and 3, which corroborated withour observations from the reporter assay (Fig. 3B).Knockdown of AKT2 decreased Twist-mediated paclitaxel
resistance, migration, and invasion. Previous report hadshown that Twist conferred paclitaxel resistance in prostatecancer cells (36). We generated MCF7 cells stably expressingTwist and its control line with pCMV-Tag3B vector. When treated
with increasing concentration of paclitaxel, we found that MCF7-Twist cells had lower amount of poly(ADP-ribose) polymerasecleavage than that of the MCF7-pCMV cells (SupplementaryFig. S1A). In addition, MCF7-Twist cells also had a lower portionof apoptotic cells compared with that of the MCF7-pCMV controlfollowing paclitaxel treatment, suggesting that Twist conferredpaclitaxel resistance in breast cancer cells (SupplementaryFig. S1B). When compared with the control, MCF7-Twist cellsalso had elevated AKT2 level, which was significantly reduced byAKT2 siRNA (Fig. 4A). Knockdown of AKT2 significantly reducedthe prosurvival effect of Twist on treatment of MCF7-Twist cellswith paclitaxel compared with knockdown of AKT2 in MCF7-pCMV cells (Fig. 4B). These data suggest that AKT2 isresponsible for at least in part the Twist-mediated paclitaxelresistance of the cells.
To further show that AKT2 is a downstream target of Twist, weexamined if Twist was able to mediate its known migratory andinvasive functions in the absence of AKT2. Overexpression of Twistin the parental MCF7 cells led to an increase in migration andinvasion ability as measured by Boyden chamber assays (Fig. 4Cand D). However, the observed Twist-mediated increase onmigration and invasion was significantly reverted when AKT2was knocked down by siRNA (Fig. 4C and D). These data suggest
Figure 4. AKT2 mediates the effects ofTwist on migration, invasion, and paclitaxelsensitivity of MCF7 cells. A, Western blot.MCF7-Twist and MCF7-pCMV cells weretreated with siRNA of AKT2 or scramblesiRNA (as siControl). Following 72 h ofincubation, cell lysates were prepared andimmunoblotted with the indicatedantibodies. B, MCF7-Twist andMCF7-pCMV cells were treated withindicated siRNA (box ) for 36 h and thentreated with or without paclitaxel foradditional 36 h. Cell death was assayed bytrypan blue staining. The experimentswere repeated thrice in triplicates samplesfor each. Ectopic expression of Twistinduced paclitaxel resistance (*, P < 0.05),whereas knockdown of AKT2 significantlyabrogated the effects of Twist (P < 0.004or P < 0.005 in MCF7-Twist versusP > 0.05 in MCF7-pCMV cells). C and D,similar siRNA-treated cells were used formigration and invasion assay. Thirty-sixhours post-treatment, 1 � 105 cells eachwere seeded onto the migration/invasionchamber. After 36 h, chambers werestained and counted for migrated/invadedcells. The experiments were repeatedthrice in triplicates samples each. P valuesfor comparisons are indicated.
Twist Transactivates AKT2
www.aacrjournals.org 1983 Cancer Res 2007; 67: (5). March 1, 2007
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

that AKT2 plays an important role in mediating Twist proinvasivefunctions.AKT2 mediates Twist function in MCF7-I4 cells. To further
show the functional link between AKT2 and Twist in controllingcell mobility, invasion, and survival, we asked if AKT2 mediatesTwist functions in the selected MCF7-I4 cells, in which endogenousprotein levels of Twist and AKT2 were elevated (Fig. 2A) andknockdown of Twist decreased AKT2 expression (Fig. 2B). ParentalMCF7 cells, expressing undetectable Twist and low level of AKT2(Figs. 2A and 5A), were used as controls. Using pSIREN-RetroQsystem, AKT2 was stably knocked down by two different shAKT2stargeting different regions of AKT2 in both MCF7 and MCF7-I4cells (Fig. 5A). As expected, Twist expression was not affected byknockdown of AKT2 (Fig. 5A). In MCF7-I4 cells, reduction in AKT2resulted in a significant decrease in cell survival, migration, andinvasion (P < 0.05; Fig. 5B). However, in MCF7 cells, the decrease inAKT2 did not cause a significant decrease in any of the examinedfunctions, although the inhibitory trend was observed (P > 0.05;Fig. 5B). These results further indicate that Twist exerts its cellularfunction in some extent through AKT2.Knockdown of Twist in MCF7-I4 can be rescued by
reexpression of AKT2. To provide further functional link betweenTwist and AKT2, we next examined if AKT2 can rescue thephenotypes resulted from knockdown of Twist in MCF7-I4 cells. Westably knocked down Twist in the MCF7-I4 cells using two differentshRNA targeting different regions of Twist. We then reintroducedHA-AKT2 into these cells. Figure 5C shows that AKT2 level wasdramatically reduced by introducing pSIREN-RetroQ-shTwist,further indicating Twist as a transcriptional regulator of AKT2
expression that becomes deregulated during metastatic progressand confirming our initial observations (Fig. 2B). ReintroducingAKT2 into shTwist-MCF7-I4 cells to a level comparable with that ofthe control significantly rescued the effects of Twist knockdown oncell survival, migration, invasion, and wound healing (Fig. 5D ;Supplementary Fig. S2). These data in combination with thefindings observed from ectopic expression of Twist and knockdownof AKT2 (Fig. 4) establish the functional link between AKT2 andTwist and indicate that AKT2 is a major target of Twist andmediates Twist function.Coexpression of Twist and AKT2 correlates with advanced
breast cancer. Having observed that Twist mediated AKT2increase in cell culture system, we asked if this regulation is seenin vivo . We examined 12 normal breast and 65 primary breasttumor samples for protein expression of Twist and AKT2 (Fig. 6A).Of the 65 breast tumors, 26 had overexpression of AKT2 and 25 hadoverexpression of Twist. Of the 25 tumors with elevated Twist, 17(68%) also had elevated AKT2 levels (P < 0.0001). Immunohisto-chemistry of these tumor samples showed that the coexpression ofTwist and AKT2 are located specifically to the cancer cells and notto the stroma (Fig. 6B). Furthermore, when grouped by the stage oftumors, we found a striking pattern that the Twist-AKT2coexpression increased in the late-stage breast tumors. Of the 16stage III to IV tumors, 11 (69%) tumors had both Twist and AKT2expression. By contrast, of the 45 stage 0 to II tumors, only 6 (13%)had both Twist and AKT2 expression (Fig. 6C). When grouped byhistology, we observed similar elevated trend for coexpression ofTwist and AKT2 in the invasive tumor samples versus thenoninvasive counterpart (Fig. 6D). Although the coexpression of
Figure 5. Effects of AKT on Twist functionin migration, invasion, and paclitaxelsensitivity in MCF7-I4 cells. A and C,Western blot. Parental MCF7 and/orMCF7-I4 cells were infected with retroviruscontaining the indicated shRNAs (A) andsubsequently transfected with HA-AKT2(C ). Western blotting analysis was donewith the indicated antibodies. B and D,functional assays. The indicated cells weretreated with paclitaxel for 36 h. Cell deathwas assayed by trypan blue staining. Cellswere seeded onto the migration/invasionchamber (1 � 104 per chamber). After12 h, chambers were stained andcounted for migrated/invaded cells. Theexperiments were repeated thrice intriplicates samples each. P values forcomparisons are indicated.
Cancer Research
Cancer Res 2007; 67: (5). March 1, 2007 1984 www.aacrjournals.org
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
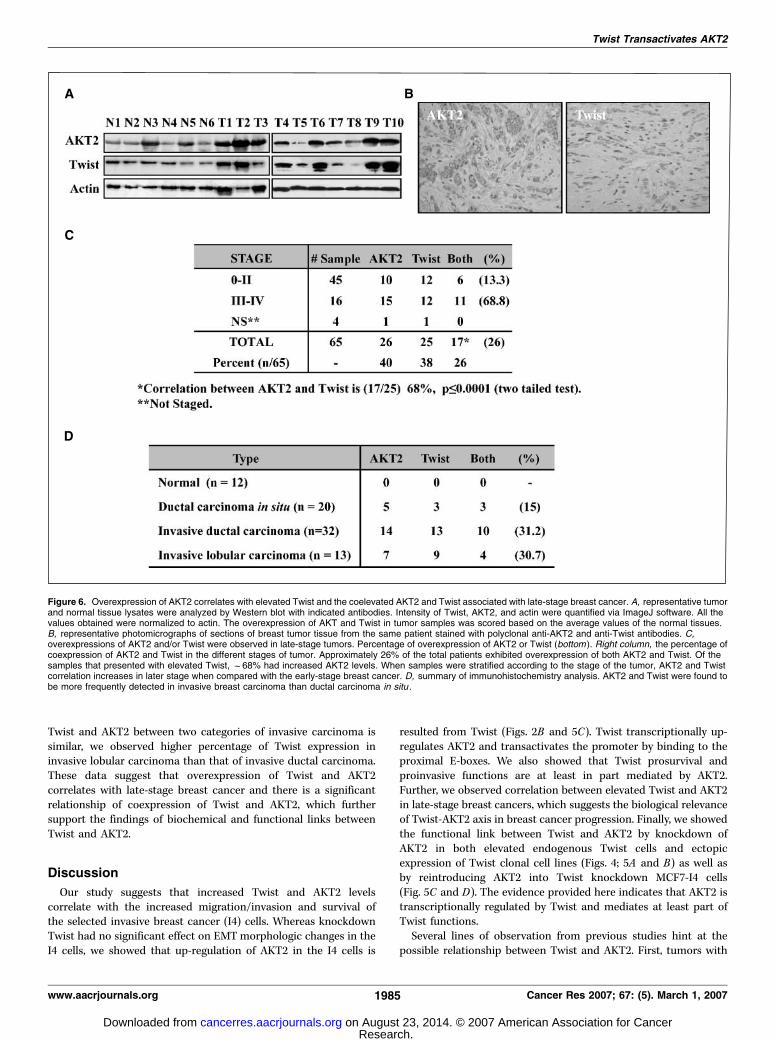
Twist and AKT2 between two categories of invasive carcinoma issimilar, we observed higher percentage of Twist expression ininvasive lobular carcinoma than that of invasive ductal carcinoma.These data suggest that overexpression of Twist and AKT2correlates with late-stage breast cancer and there is a significantrelationship of coexpression of Twist and AKT2, which furthersupport the findings of biochemical and functional links betweenTwist and AKT2.
Discussion
Our study suggests that increased Twist and AKT2 levelscorrelate with the increased migration/invasion and survival ofthe selected invasive breast cancer (I4) cells. Whereas knockdownTwist had no significant effect on EMT morphologic changes in theI4 cells, we showed that up-regulation of AKT2 in the I4 cells is
resulted from Twist (Figs. 2B and 5C). Twist transcriptionally up-regulates AKT2 and transactivates the promoter by binding to theproximal E-boxes. We also showed that Twist prosurvival andproinvasive functions are at least in part mediated by AKT2.Further, we observed correlation between elevated Twist and AKT2in late-stage breast cancers, which suggests the biological relevanceof Twist-AKT2 axis in breast cancer progression. Finally, we showedthe functional link between Twist and AKT2 by knockdown ofAKT2 in both elevated endogenous Twist cells and ectopicexpression of Twist clonal cell lines (Figs. 4; 5A and B) as well asby reintroducing AKT2 into Twist knockdown MCF7-I4 cells(Fig. 5C and D). The evidence provided here indicates that AKT2 istranscriptionally regulated by Twist and mediates at least part ofTwist functions.
Several lines of observation from previous studies hint at thepossible relationship between Twist and AKT2. First, tumors with
Figure 6. Overexpression of AKT2 correlates with elevated Twist and the coelevated AKT2 and Twist associated with late-stage breast cancer. A, representative tumorand normal tissue lysates were analyzed by Western blot with indicated antibodies. Intensity of Twist, AKT2, and actin were quantified via ImageJ software. All thevalues obtained were normalized to actin. The overexpression of AKT and Twist in tumor samples was scored based on the average values of the normal tissues.B, representative photomicrographs of sections of breast tumor tissue from the same patient stained with polyclonal anti-AKT2 and anti-Twist antibodies. C,overexpressions of AKT2 and/or Twist were observed in late-stage tumors. Percentage of overexpression of AKT2 or Twist (bottom ). Right column, the percentage ofcoexpression of AKT2 and Twist in the different stages of tumor. Approximately 26% of the total patients exhibited overexpression of both AKT2 and Twist. Of thesamples that presented with elevated Twist, f68% had increased AKT2 levels. When samples were stratified according to the stage of the tumor, AKT2 and Twistcorrelation increases in later stage when compared with the early-stage breast cancer. D, summary of immunohistochemistry analysis. AKT2 and Twist were found tobe more frequently detected in invasive breast carcinoma than ductal carcinoma in situ .
Twist Transactivates AKT2
www.aacrjournals.org 1985 Cancer Res 2007; 67: (5). March 1, 2007
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

acquired paclitaxel resistance displayed Twist gene amplificationand AKT2 overexpression/activation (36, 47, 48). Second, bothTwist and AKT have been implicated in Wnt signaling pathway(39, 49–51). Wnt pathway inhibitors Frzb/secreted Frizzled-relatedprotein 3 and Wnt inhibitory factor 1 were shown to lead todecreased Twist expression and reduced AKT activation (50, 51).Furthermore, both Twist and AKT promotes angiogenesis, cellgrowth, and survival via altering h-catenin cellular distribution(39). Finally, previous studies have shown that both Twist andAKT2 promote EMT and invasive phenotype of cancer cells (14, 15,34, 39). These observations suggest that Twist and AKT2 may workin similar pathways in tumor development. Our observationsprovide direct evidence that AKT2 is a target gene of Twist.
Previous studies have shown that overexpression of AKT1 andAKT2 is a much more frequent event than their gene amplificationin human malignancies, suggesting transcriptional regulation ofAKT during the tumor development (52). AKT1 has been shownrecently to be regulated by signal transducers and activators oftranscription 3 (STAT3) and mediate STAT3-induced angiogenesisand cell survival (53, 54). In addition, AKT1 is up-regulated byh-catenin/TCF/LEF and frequent elevated expression levels ofAKT1, correlating with enhanced cytoplasmic/nuclear expressionof h-catenin, were detected in colorectal carcinoma (55). WhereasAKT2 is up-regulated by MyoD during the muscle differentiation(44), transcriptional regulation of AKT2 has not been shownpreviously in the cellular processes that are related to carcinogen-esis. We have shown in this report that Twist transcriptionallyregulates AKT2 and that elevated expression levels of Twistcorrelates with overexpression of AKT2 in late-stage breast cancer(Figs. 2, 3, and 6). Knockdown AKT2 reduces the effect of Twist oncell migration, invasion, and survival (Figs. 4 and 5).
Our data show that Twist functions as a transcriptionalactivator of AKT2. Previous studies have shown that Twistfunctions as a transcriptional repressor and that the action ofTwist is regulated by its dimerization with other bHLH-containing
transcriptional factors. Post-translational modifications, suchas phosphorylation, can alter the dimerization preferences ofTwist, either promoting homodimer or heterodimer formation(56). This alteration in Twist dimerization partners ultimatelyaffects the transcription regulatory function of Twist. It has beensuggested that Twist as a heterodimer acts as a transcriptionrepressor, whereas Twist homodimer, whose formation is favoredby elevated expression, acts as a transcription activator. Recentstudies in Drosophila mesoderm development and in humancranial suture patterning suggest that Twist homodimer functionsas a transcriptional activator (57, 58). In addition, recent reportshows Twist as a transcriptional activator of N-cadherin gene inprostate cancer cells (59). The specific mechanism through whichdimerization modulates Twist activity on AKT2 promoter remainsto be elucidated.
In conclusion, recent evidence suggests that Twist is a majorfactor participating in tumor development and progression. Inhuman breast cancer, elevated Twist is found in 70% invasivelobular carcinomas (37). Recently, Twist is found to be increased inmetastatic lesions of prostate cancer (38). As a novel player in themetastatic program, Twist is gaining rapid attention as a regulatorof metastasis. Our finding of the functional link between Twist andAKT2 suggests that targeting Twist and its downstream effectors,such as AKT2, may provide novel therapeutic cocktails for breastcancer intervention.
Acknowledgments
Received 4/25/2006; revised 10/18/2006; accepted 12/29/2006.Grant support: NIH grants CA29339 and Department of Defense grant DAMD 17-
02-0504.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. Domenico Tortorella (Mt. Sinai School of Medicine, New York, NY)for the pSIREN-RetroQ-linker vector and Dr. Jin Q. Cheng (H. Lee Moffitt CancerCenter, Tampa, FL) for his generous gift of AKT2 reporter.
Cancer Research
Cancer Res 2007; 67: (5). March 1, 2007 1986 www.aacrjournals.org
References
1. Fidler IJ. The pathogenesis of cancer metastasis: the’seed and soil’ hypothesis revisited. Nat Rev Cancer 2003;3:453–8.2. Chau NM, Ashcroft M. Akt2: a role in breast cancer
metastasis. Breast Cancer Res 2004;6:55–7.3. Vleminckx K, Vakaet L, Jr., Mareel M, Fiers W, van Roy
F. Genetic manipulation of E-cadherin expression byepithelial tumor cells reveals an invasion suppressorrole. Cell 1991;66:107–19.4. Batlle E, Sancho E, Franci C, et al. The transcription
factor snail is a repressor of E-cadherin gene expressionin epithelial tumour cells. Nat Cell Biol 2000;2:84–9.5. Cano A, Perez-Moreno MA, Rodrigo I, et al. The
transcription factor snail controls epithelial-mesenchy-mal transitions by repressing E-cadherin expression. NatCell Biol 2000;2:76–83.6. Liotta LA, Stetler-Stevenson WG. Metalloproteinases
and cancer invasion. Semin Cancer Biol 1990;1:99–106.7. Maslow DE. Collagenase effects on cancer cell invasive-
ness and motility. Invasion Metastasis 1987;7:297–310.8. Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation
of AKT2 and induction of apoptosis by inhibition ofphosphoinositide-3-OH kinase/Akt pathway in humanovarian cancer. Oncogene 2000;19:2324–30.9. Graff JR, Konicek BW, McNulty AM, et al. Increased
AKT activity contributes to prostate cancer progressionby dramatically accelerating prostate tumor growth anddiminishing p27Kip1 expression. J Biol Chem 2000;275:24500–5.
10. Bacus SS, Altomare DA, Lyass L, et al. AKT2 isfrequently upregulated in HER-2/neu -positive breastcancers and may contribute to tumor aggressiveness byenhancing cell survival. Oncogene 2002;21:3532–40.11. Vivanco I, Sawyers CL. The phosphatidylinositol 3-
kinase AKT pathway in human cancer. Nat Rev Cancer2002;2:489–501.12. Testa JR, Bellacosa A. AKT plays a central role in
tumorigenesis. Proc Natl Acad Sci U S A 2001;98:10983–5.13. Mayo LD, Donner DB. A phosphatidylinositol 3-
kinase/Akt pathway promotes translocation of Mdm2from the cytoplasm to the nucleus. Proc Natl Acad SciU S A 2001;98:11598–603.14. Arboleda MJ, Lyons JF, Kabbinavar FF, et al. Over-
expression of AKT2/protein kinase Bh leads to up-regulation of h1 integrins, increased invasion, andmetastasis of human breast and ovarian cancer cells.Cancer Res 2003;63:196–206.15. Grille SJ, Bellacosa A, Upson J, et al. The protein
kinase Akt induces epithelial mesenchymal transitionand promotes enhanced motility and invasiveness ofsquamous cell carcinoma lines. Cancer Res 2003;63:2172–8.16. Xu X, Sakon M, Nagano H, et al. Akt2 expression
correlates with prognosis of human hepatocellularcarcinoma. Oncol Rep 2004;11:25–32.17. Roy HK, Olusola BF, Clemens DL, et al. AKT proto-
oncogene overexpression is an early event duringsporadic colon carcinogenesis. Carcinogenesis 2002;23:201–5.
18. Ringel MD, Hayre N, Saito J, et al. Overexpression andoveractivation of Akt in thyroid carcinoma. Cancer Res2001;61:6105–11.19. Kim CS, Vasko VV, Kato Y, et al. AKT activation
promotes metastasis in a mouse model of follicularthyroid carcinoma. Endocrinology 2005;146:4456–63.20. Cheng JQ, Ruggeri B, Klein WM, et al. Amplification
of AKT2 in human pancreatic cells and inhibition ofAKT2 expression and tumorigenicity by antisense RNA.Proc Natl Acad Sci U S A 1996;93:3636–41.21. Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR.
Amplification and overexpression of the AKT2 oncogenein a subset of human pancreatic ductal adenocarcino-mas. Mol Carcinog 1998;21:81–6.22. Miwa W, Yasuda J, Murakami Y, et al. Isolation of
DNA sequences amplified at chromosome 19q13.1-q13.2including the AKT2 locus in human pancreatic cancer.Biochem Biophys Res Commun 1996;225:968–74.23. Liu AX, Testa JR, Hamilton TC, Jove R, Nicosia SV,
Cheng JQ. AKT2, a member of the protein kinase Bfamily, is activated by growth factors, v-Ha-ras, and v-srcthrough phosphatidylinositol 3-kinase in human ovarianepithelial cancer cells. Cancer Res 1998;58:2973–7.24. Cheng JQ, Godwin AK, Bellacosa A, et al. AKT2, a
putative oncogene encoding a member of a subfamily ofprotein-serine/threonine kinases, is amplified in humanovarian carcinomas. Proc Natl Acad Sci U S A 1992;89:9267–71.25. Sun M, Paciga JE, Feldman RI, et al. Phosphatidyli-
nositol-3-OH kinase (PI3K)/AKT2, activated in breastcancer, regulates and is induced by estrogen receptor a
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

Twist Transactivates AKT2
www.aacrjournals.org 1987 Cancer Res 2007; 67: (5). March 1, 2007
(ERa) via interaction between ERa and PI3K. CancerRes 2001;61:5985–91.26. Bellacosa A, de Feo D, Godwin AK, et al. Molecular
alterations of the AKT2 oncogene in ovarian and breastcarcinomas. Int J Cancer 1995;64:280–5.27. Castanon I, Baylies MK. A Twist in fate: evolutionary
comparison of Twist structure and function. Gene 2002;287:11–22.28. Desprez PY, Sumida T, Coppe JP. Helix-loop-helix
proteins in mammary gland development and breastcancer. J Mammary Gland Biol Neoplasia 2003;8:225–39.29. Massari ME, Murre C. Helix-loop-helix proteins:
regulators of transcription in eucaryotic organisms.Mol Cell Biol 2000;20:429–40.30. Spicer DB, Rhee J, Cheung WL, Lassar AB. Inhibition
of myogenic bHLH and MEF2 transcription factors bythe bHLH protein Twist. Science 1996;272:1476–80.31. Hamamori Y, Wu HY, Sartorelli V, Kedes L. The basic
domain of myogenic basic helix-loop-helix (bHLH)proteins is the novel target for direct inhibition by anotherbHLH protein, Twist. Mol Cell Biol 1997;17:6563–73.32. Hamamori Y, Sartorelli V, Ogryzko V, et al. Regulation
of histone acetyltransferases p300 and PCAF by thebHLH protein twist and adenoviral oncoprotein E1A.Cell 1999;96:405–13.33. Maestro R, Dei Tos AP, Hamamori Y, et al. Twist is a
potential oncogene that inhibits apoptosis. Genes Dev1999;13:2207–17.34. Hoek K, Rimm DL, Williams KR, et al. Expression
profiling reveals novel pathways in the transformation ofmelanocytes to melanomas. Cancer Res 2004;64:5270–82.35. Valsesia-Wittmann S, Magdeleine M, Dupasquier S,
et al. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. CancerCell 2004;6:625–30.36. Wang X, Ling MT, Guan XY, et al. Identification of a
novel function of TWIST, a bHLH protein, in thedevelopment of acquired Taxol resistance in humancancer cells. Oncogene 2004;23:474–82.37. Yang J, Mani SA, Donaher JL, et al. Twist, a master
regulator of morphogenesis, plays an essential role intumor metastasis. Cell 2004;117:927–39.38. Kwok WK, Ling MT, Lee TW, et al. Up-regulation of
TWIST in prostate cancer and its implication as atherapeutic target. Cancer Res 2005;65:5153–62.39. Mironchik Y, Winnard PT, Jr., Vesuna F, et al. Twist
overexpression induces in vivo angiogenesis and corre-lates with chromosomal instability in breast cancer.Cancer Res 2005;65:10801–9.40. Entz-Werle N, Stoetzel C, Berard-Marec P, et al.
Frequent genomic abnormalities at TWIST in humanpediatric osteosarcomas. Int J Cancer 2005;117:349–55.41. van Doorn R, Dijkman R, Vermeer MH, et al.
Aberrant expression of the tyrosine kinase receptorEphA4 and the transcription factor twist in Sezarysyndrome identified by gene expression analysis. CancerRes 2004;64:5578–86.42. Rosivatz E, Becker I, Specht K, et al. Differential
expression of the epithelial-mesenchymal transitionregulators snail, SIP1, and twist in gastric cancer. Am JPathol 2002;161:1881–91.43. Watanabe O, Imamura H, Shimizu T, et al. Expression
of twist and wnt in human breast cancer. Anticancer Res2004;24:3851–6.44. Kaneko S, Feldman RI, Yu L, et al. Positive feedback
regulation between Akt2 and MyoD during muscledifferentiation. Cloning of Akt2 promoter. J Biol Chem2002;277:23230–5.45. Sierra J, Villagra A, Paredes R, et al. Regulation of the
bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300intrinsic histone acetyltransferase activity. Mol Cell Biol2003;23:3339–51.46. Ofir R, Seidman R, Rabinski T, et al. Taxol-induced
apoptosis in human SKOV3 ovarian and MCF7 breastcarcinoma cells is caspase-3 and caspase-9 independent.Cell Death Differ 2002;9:636–42.47. Page C, Lin HJ, Jin Y, et al. Overexpression of Akt/
AKT can modulate chemotherapy-induced apoptosis.Anticancer Res 2000;20:407–16.48. VanderWeele DJ, Zhou R, Rudin CM. Akt up-
regulation increases resistance to microtubule-directedchemotherapeutic agents through mammalian target ofrapamycin. Mol Cancer Ther 2004;3:1605–13.49. Howe LR, Watanabe O, Leonard J, Brown AM. Twist
is up-regulated in response to Wnt1 and inhibits
mouse mammary cell differentiation. Cancer Res 2003;63:1906–13.50. Ohigashi T, Mizuno R, Nakashima J, Marumo K,
Murai M. Inhibition of Wnt signaling downregulates Aktactivity and induces chemosensitivity in PTEN-mutatedprostate cancer cells. Prostate 2005;62:61–8.51. Zi X, Guo Y, Simoneau AR, et al. Expression of Frzb/
secreted Frizzled-related protein 3, a secreted Wntantagonist, in human androgen-independent prostatecancer PC-3 cells suppresses tumor growth and cellularinvasiveness. Cancer Res 2005;65:9762–70.52. Altomare DA, Testa JR. Perturbations of the AKT
signaling pathway in human cancer. Oncogene 2005;24:7455–64.53. Park S, Kim D, Kaneko S, et al. Molecular cloning and
characterization of the human AKT1 promoter uncoversits up-regulation by the Src/Stat3 pathway. J Biol Chem2005;280:38932–41.54. Xu Q, Briggs J, Park S, et al. Targeting Stat3 blocks
both HIF-1 and VEGF expression induced by multipleoncogenic growth signaling pathways. Oncogene 2005;24:5552–60.55. Dihlmann S, Kloor M, Fallsehr C, von Knebel
Doeberitz M. Regulation of AKT1 expression by h-catenin/Tcf/Lef signaling in colorectal cancer cells.Carcinogenesis 2005;26:1503–12.56. Firulli BA, Krawchuk D, Centonze VE, et al. Altered
Twist1 and Hand2 dimerization is associated withSaethre-Chotzen syndrome and limb abnormalities.Nat Genet 2005;37:373–81.57. Connerney J, Andreeva V, Leshem Y, Muentener C,
Mercado MA, Spicer DB. Twist1 dimer selectionregulates cranial suture patterning and fusion. DevDyn 2006;235:1334–46.58. Castanon I, Von Stetina S, Kass J, Baylies MK.
Dimerization partners determine the activity of theTwist bHLH protein during Drosophila mesodermdevelopment. Development 2001;128:3145–59.59. Alexander NR, Tran NL, Rekapally H, Summers CE,
Glackin C, Heimark RL. N-cadherin gene expression inprostate carcinoma is modulated by integrin-dependentnuclear translocation of Twist1. Cancer Res 2006;66:3365–9.
Research. on August 23, 2014. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
Copyright © 2022 FDOKUMEN




















