
The finiteness of certain sets of attached prime ideals and the length of generalized fractions
Upload
independentCategory
view
1download
0
Tu
SD
a
ARRAA
KAGMc“P
1
fBabrfoathcmslan
t7
0h
Electrochimica Acta 108 (2013) 624– 633
Contents lists available at ScienceDirect
Electrochimica Acta
jou rn al hom ep age: www.elsev ier .com/ locate /e lec tac ta
uning the apparent formal potential of covalently attached ferrocenesing SAM bearing ionizable COOH groups
ohini Mukherjee, Sabyasachi Bandyopadhyay, Abhishek Dey ∗
epartment of Inorganic Chemistry, Indian Association for the Cultivation of Science, Kolkata 700032, India
r t i c l e i n f o
rticle history:eceived 16 March 2013eceived in revised form 8 July 2013ccepted 9 July 2013vailable online 16 July 2013
eywords:pparent formal potential
a b s t r a c t
A method of tuning the apparent formal potential at the interface between the electrode surface cov-ered with carboxylate thiol self assembled monolayer (SAM) and the solution containing electrolytes isreported. Redox active ferrocene moiety has been covalently attached to the terminal azido groups inthe SAM using “click” chemistry. Cyclic voltammetry experiments show that the reduction potential ofthis ferrocene moiety can be tuned easily from 0.345 V to 0.200 V, i.e. by 145 mV by varying the chainlength of the thiol carboxylic acid diluents, changing the pH of the electrolytic solution and by changingthe coverage of the surface. Using the (applying the theory of interfacial potential distribution by Smith
ouy–Chapman–Stern modelixed SAM of azide thiol and thiol
arboxylic acidClick” chemistrylane of electron transfer (PET)
and White and including the Stern layer effect on the potential of the redox species at the interface pro-posed by Fawcett) modified Nernst equation the shift of apparent formal potential of redox species atthe interface has been analyzed when the chain length of the diluents, pH of the solution and coverageof the surface are changed. The data indicate that the phenomenon is governed by the ionizable polarhead groups SAM, i.e. COOH which changes the interfacial microenvironment around the redox activeferrocene centre.
. Introduction
Self assembled monolayers (SAMs) of thiols on Au/Ag haveound applications in different areas of chemistry and biology [1–8].oth aliphatic and aromatic thiols are known to form very stablend organized SAM on Au surfaces. Stable SAM of thiols on Au,earing different solvent exposed terminal functional groups areeported as well [9–18]. Particularly, SAM bearing terminal COOHunctional groups provide a convenient approach for the fabricationf functionalized electrodes for sensing metals and biomolecules,nd catalysis [19–26]. The hydrophilicity and negative charge ofhe carboxylate group, which remains ionized at physiological pHsas enabled successful non covalent immobilization of positivelyharged proteins like cytochrome C as well as the covalent attach-ent of the heme protein like myoglobin on these biocompatible
urfaces [26–28]. Alternatively, NH2 groups of solvent exposedysine residues of horse radish peroxidase have been covalently
ttached to the COOH head group of such carboxylic acid termi-ated SAM by forming amide bonds [29].∗ Corresponding author at: Department of Inorganic Chemistry, Indian Associa-ion for the Cultivation of Science, 2A & 2B Raja S. C. Mullick Road, Jadavpur, Kolkata00032, India. Tel.: +91 33 2473 4971x1366; fax: +91 33 2473 2805.
E-mail addresses: [email protected], [email protected] (A. Dey).
013-4686/$ – see front matter © 2013 Elsevier Ltd. All rights reserved.ttp://dx.doi.org/10.1016/j.electacta.2013.07.065
© 2013 Elsevier Ltd. All rights reserved.
The electrochemical reactions by an electroactive moiety takeplace within the electrical double layer which polarizes speciesand concentrates oppositely charged ions on the electrode sur-face [30]. Gouy–Chapman–Stern model is the most commonly usedtheory to analyze and understand this phenomenon. According tothis model, a linear drop in potential occurs across the Stern layerwhich is the junction between the metal electrode surface and thelocus of the first hydrated counter ions from the aqueous electrolyte(Scheme 1) [31]. Beyond this Stern layer, an exponential decay inpotential occurs out to the diffused bulk electrolyte following thePoisson–Boltzmann equation [32]. This layer is commonly knownas Gouy–Chapman layer (Scheme 1). In the case of SAM modifiedelectrodes, another potential drop occurs between the metal andthe stern layer [33]. Recently, Paddon-Row, Gooding, et al. demon-strated the effect of double layer on the electrochemical reactionsand presented a new method to measure potential drop across thiselectrical double layer using ferrocene terminated norbornylogousbridges [34]. Based on this method the effect of electric field onthe measurement of the formal potential of surface-bound redoxspecies has also been investigated [34]. The effect of chain length ofthe n-alkanethiol diluent and the polar head groups of the diluentmolecule on the change of formal potential of the surface bound
redox species has also been investigated by Creager et al., [35]They showed that only by changing the chain length of the alka-nethiol diluent from C4 to C10, the potential of the redox speciescan be tuned up to 330 mV and by changing the polarity of the
S. Mukherjee et al. / Electrochimica
Scheme 1. Double layer model of mixed SAM containing ferrocene terminated thioland thiol carboxylic acid on Au electrode in the dielectric medium containing aque-ous electrolyte, Na2HPO4 and KPF6. The potential drop of the redox active speciesacross the SAM is shown by brown dotted line. The Stern layer is represented as thethickness enclosed within the black dashed lines. The potential at the metal elec-tbt
hoetBbfiito
pSSptzaatorl
E
watathaaotttm
of the diluent containing COOH polar head group, mole fractionof the linker azidothiol and the pHs of the electrolyte solution on
rode, at the plane of electron transfer (PET) and in the bulk solution are designatedy ϕM, ϕPET and ϕSOL, respectively. (For interpretation of the references to colour inhis figure legend, the reader is referred to the web version of the article.)
ead group that can be varied up to 180 mV. Electric field effectf n-alkanethiol SAM with varying chain length, coverage and theffect of concentration of the electrolyte on the formal potential ofhe surface bound redox species has been widely exploited [36–47].ut the effect of varying the chain length, coverage and pH’s of SAMearing COOH head group on the formal potential of redox activeerrocene species which has been covalently attached to the SAM,s still unrevealed. In particular changing the pH, which controls theonization state of the carboxylate head groups, may be expectedo produce measurable changes in the apparent formal potentialsf surface bound species.
The effect of the electric field on the formal potential was firstostulated in the theory of interfacial potential distribution bymith and White [33] and was further augmented including thetern layer by Fawcett [48]. The Nernst equation assumes that theotential difference between the electrode surface (either the dis-al surface of the SAM or the metal surface) and the bulk solution isero, which is not true in reality. That was first identified by Smithnd White and they mentioned it in their seminal paper. There isn electrical double layer formed to balance the surface charge athe solid–electrolyte interface [34,38]. So, considering the effectf double layer, the potential difference between the plane of theedox active groups and the bulk solution (i.e., across the doubleayer) is expressed as [33]
= E0 + (ϕPET − ϕSOL) + RT
nFln
(�O
�R
)(1)
here R is the ideal gas constant, T is the thermodynamic temper-ture, n is the number of electrons, F is the Faraday constant, E ishe equilibrium potential difference between modified electrodend reference electrode, E0 is the standard reduction potential ofhe cell (including the activity of ions in the reference electrodealf-cell), � O and � R are the equilibrium surface activities (takens the amount coverage (in mol m−2) of surface-bound, redox-ctive species when E0 strictly becomes the formal potential E0′
)f oxidized and reduced species, and (ϕPET − ϕSOL) is the poten-
ial difference between the plane of electron transfer (PET) andhe bulk solution (SOL). The term E0 + (ϕPET − ϕSOL) will be calledhe “apparent formal potential”, as this quantity is what is actuallyeasured in a typical electrochemical experiment. The potential at
Acta 108 (2013) 624– 633 625
which the charge on the surface of the working electrode becomeszero, i.e. at pzc, (ϕPET − ϕSOL) = 0 and Eq. (1) is converted into thetypical Nernstian equation.
The shift of apparent formal potential can be attributed to eitherthe double layer effect or the solvent effect. In the former case it isalready known that it would be due to changes in the spatial dis-tribution of ions in the interfacial region between the surface andthe electrolyte whereas in the latter case, the shift would be due tochanges in the Gibbs energy of solvation in the interfacial region.The difference in formal potentials for a half-cell reaction in twodifferent media is expressed by the difference in Gibbs energies oftransfer of the oxidized and reduced forms of the couple from onemedium to the other [49]. The Gibbs energies of transfer can be esti-mated using Born equation (or modifications thereof) for solvationof charged sphere in dielectric continuum. When the transfer ofoxidized and reduced forms of the redox couple occurs in betweenthe plane of the electron transfer (PET) and the bulk solvent (SOL),the expressions for estimating the difference in formal potentialscan be written as follows:
ϕPET − ϕSOL =(
1nF
)(�Gtransfer,O,PET→SOL
− �Gtransfer,R,PET→SOL) (2)
�Gtransfer,PET→SOL = N(Ze)2
8�ε0r
[1
εPET− 1
εSOL
](3)
where �Gtransfer,O,PET→SOL is the free energy for transfer of the oxi-dized species from the plane of electron transfer to the bulk solutionand �Gtransfer,R,PET→SOL is the free energy for transfer of the reducedspecies from the plane of electron transfer to the bulk solution, zis the charge number, e is the charge of an electron, r is the radiusof the redox group, εPET is the permittivity at the plane of elec-tron transfer, εSOL is the permittivity for the bulk solution, ε0 isthe permittivity in vacuum, n is number of electron transferred,and F is Faradaic constant. Note that, Born equation used to calcu-late Gibbs energies of ion transfer considers only coulombic effectsin ion solvation. It is assumed that other chemical contributionsto ion solvation, like hydrogen bonding will be equal for the oxi-dized and reduced forms of the couple, and hence they will becancelled out in Eq. (2). Thus these two equations ((2) and (3))become useful in analyzing the effect of the charged species onthe apparent formal potential of the redox active species at thePET.
In this paper we have used “click” reaction (Cu(I) catalyzedcycloaddition of alkyl and aryl azides to terminal alkynes) to attachthe redox active ferrocene groups to a mixed SAM containing azidethiol and thiol carboxylic acid (Scheme 1). This method is alreadywell known for functionalizing SAM with electroactive ferrocene,O2 reducing iron and cobalt porphyrin catalysts, heme proteinslike myoglobin, DNA etc. on to the Au electrodes [2,28,50–55]. The“click” reaction is inherently regioselective, high yielding and canbe performed in a wide range of solvents at room temperature[56,60]. Immersing Au surfaces in solutions of alkylthiol contain-ing small mole fractions of azide terminated thiols (azidothiols)result in a surface covered with alkylthiol SAM impregnated withazido thiols [57–59]. These azide groups can be then functional-ized in situ using Cu(I) catalyzed “click” reaction [11,60,61]. In thisreport, we have investigated the effect of varying the chain length
the formal potential of these surface bound ferrocenes. The resultsindicate that significant control of the formal reduction potential ofthis redox active group can be achieved (∼150 mV) by tuning thesefactors.

6 himica Acta 108 (2013) 624– 633
2
2
aeASfaaos∼i
2
siA3a1e
2
SwntotffTo
2
ttEptchdatb
3
3c
Cm
-20
-15
-10
-5
0
5
10
15
20
-0.5-0.3-0.10.10.3
Potential vs (Ag/AgCl) / V
Cu
rren
t / µ µA
C8H17
C2-COO H
C5-COO H
C7-COO H
Fig. 1. Comparison of the background current for different SAM (mole fraction ofdeposition solution = 20%). Green – background current for SAM with 1-octanethioldiluents (HS-C8H17), blue – background current for SAM with HS-C2-COOH diluent,pink – background current for SAM with HS-C5-COOH diluent, yellow – backgroundcurrent for SAM with HS-C7-COOH diluent in pH 7 phosphate buffer at 1 V/s scan
26 S. Mukherjee et al. / Electroc
. Materials and methods
.1. Experimental
3-Mercaptopropionic acid (HS-C2-COOH), 6-mercaptohexanoiccid (HS-C5-COOH), 8-mercaptooctanoic acid (HS-C7-COOH) andthynylferrocene (Fc) are purchased form Sigma–Aldrich. 1-zidoundecane-11-thiol (HS-C11-N3) is synthesized by using-(11-bromoundecyl)thioacetate purchased from Sigma–Aldrich,ollowing the reported procedure [57]. KOH, CuSO4·5H2O andscorbic acid (vitamin C) are purchased form Merck. Au wafersre purchased from Platypus Technologies (1000 A of Au on 50 Af Ti adhesion layer on top of a Si(III) substrate). AFM image ofuch a freshly cut clean Au wafer shows the average roughness of1.2 ± 0.2 nm (SI-1) [62]. All electrochemical experiments are done
n CH Instruments model CHI710D Electrochemical Analyzer.
.2. Mixed self assembled monolayer (SAM) formation
Gold wafers are cleaned both by immersing them into a Piranhaolution (1:3 H2O2/H2SO4) and also electrochemically follow-ng the reported procedures [63,64] (SI-2). This freshly cleanedu surface is then immersed into the deposition solutions of-mercaptopropionic acid (HS-C2-COOH) or 6-mercaptohexanoiccid (HS-C5-COOH) or 8-mercaptooctanoic acid (HS-C7-COOH) and-azidoundecane-11-thiol in the desired ratio (mole fraction) inthanol.
.3. Covalent attachment of ethynylferrocene
It is well established that COOH terminated thiols form goodAM on Au surfaces. The redox active molecule ethynylferroceneas covalently attached to the azide ( N3) groups of azide termi-ated linker of the mixed SAM of 1-azidoundecane-11-thiol andhiol carboxylic acid by using the method of “click” chemistry [65]nto the surface of mixed SAM modified gold electrode (SI-3). Notehat the concentration of copper catalyst (5–10 mM) required tounctionalize the azide groups on a COOH terminated SAM sur-ace is ∼400 times more than the previous results (20 �M) [61,66].his is because of the coordination of copper by COOH head groupsf the SAMs (vide infra) [28].
.4. Cyclic voltammetry
All CV experiments are measured in a CH Instruments bipo-entiostat (model 710D). An aq. Ag/AgCl and a Pt wire is used ashe reference and counter electrode for all of these experiments.xcept for the study of change of apparent formal potential withH, all other CV measurements are done in pH 7 buffered solu-ions (100 mM Na2HPO4) containing 100 mM KPF6. In the study ofhange of apparent formal potential with pH, the buffered solutionsaving a mixture of 100 mM Na2HPO4 and 100 mM KPF6 and withifferent pH are used. For all the CV measurements the Au wafersre sandwiched between the two Teflon® blocks of a plate materialesting cell (ALS, Japan). A Viton® O-ring is used to seal the junctionetween the Au wafer and the Teflon® material.
. Results
.1. Effect of varying the chain length of the diluent thiolarboxylic acid
Diluent thiol carboxylic acid of different chain lengths (HS-C2-OOH, HS-C5-COOH, HS-C7-COOH) is used to create the mixedonolayers. They form a network of H-bonding on the surface
rate using aq. Ag/AgCl as reference electrode and platinum as the counter electrode.(For interpretation of the references to colour in this figure legend, the reader isreferred to the web version of the article.)
as indicated by grazing angle FTIR [67–71]. The background cur-rent obtained using HS-C2-COOH (Fig. 1, blue) is much larger andindicates poorer insulation of the Au surface by a short chain thiol(Fig. 1, green) [72]. The double layer capacitance (Cd,l) of the abovementioned SAM has been determined by using the following equa-tion
Cd,l = ic�A
(4)
where ic is the charging current of the SAM in �A, � is the scanrate in volt (V/s) which is 1 V/s here and A is the area of thesurface which has been scanned. The Cd,l value for HS-C2-COOH,HS-C5-COOH, and HS-C7-COOH are found to be 23.5 ± 0.1 �F/cm2,12.1 ± 0.1 �F/cm2 and 10.4 ± 0.1 �F/cm2, respectively. Whereas thevalue obtained for the mixed SAM of 1-azidoundecane-11-thiol and1-octanethiol (HS-C8H17) is found to be 3.6 ± 0.1 �F/cm2 which ismuch lesser than that for thiol carboxylic acid SAM. Thus, the Cd,lvalues obtained for the thiol carboxylic acid SAM are much higherthan that of octane thiol and it decrease with the increase of thechain length. This is in good agreement with the previous reportsand indicates the increased disorderness with and lower packingdensity with decreased chain length of the diluent [73,74]. More-over it is also reported that the presence of COOH groups increasesthe disorder of the SAM, inducing higher values than those obtainedusing alkylthiol SAM [75]. Note that, no additional peak having acapacitive origin (as reported for homocysteine SAM) is observed(SI-4) for the COOH terminated thiol SAM because of the absenceof any terminal amine group ( NH2) [76].
The equilibrium potential difference (E) between the modi-fied electrode and reference electrode of the ferrocene/ferriceniumredox couple (Fc/Fc+) in pH 7 for the HS-C2-COOH (Fig. 2, blue), HS-C5-COOH (Fig. 2, pink) and HS-C7-COOH (Fig. 2, orange) hydrophilicdiluents are measured to be 342 mV, 340 mV and 313 mV, respec-tively. Note that these small shifts in the formal potential of Fc/Fc+
redox couple with chain length of the hydrophilic thiol carboxylicacid diluent are reproducible and reflects changes in the localmicroenvironment around the Fc moiety (vide infra). With increase
in chain length of the thiol carboxylic acid the thickness of thelayer between the redox centre and the polar head group of thediluent decreases (Scheme 2). Thus the long chain thiol carboxylicacid produces more polar interfacial microenvironment around the
S. Mukherjee et al. / Electrochimica Acta 108 (2013) 624– 633 627
-45
-35
-25
-15
-5
5
15
25
35
45
00.10.20.30.40.5
Potential vs (Ag/AgCl) / V
Cu
rren
t /
µ µA
C8H17C2-COO HC5-COO HC7-COO H
Fig. 2. Shift of E with the variation of chain length of thiol carboxylic acid with thesame linker molecule 1-azidoundecan-11-thiol in the mixed SAM (mole fractionof deposition solution = 20%) (blue: C2; pink: C5; orange: C7) and their comparisonwith that of hydrophobic surface (green: 1-octanethiol diluent) with the same linkermolecule 1-azidoundecan-11-thiol (mole fraction of deposition solution = 10%) at1 V/s scan rate in pH 7 buffer using aq. Ag/AgCl as reference electrode and platinumal
rt
bmucucbcadg
Table 1Full width at half maxima (FWHM) values of both the oxidation and reduction peaksof the CV of FC/Fc+ redox couple for different chain length of the diluent molecule.
Chain length FWHM values
Oxidation peak (mV) Reduction peak (mV)
C8H17 106 108C2-COOH 108 128
�−1 = (5)
Si
s the counter electrode. (For interpretation of the references to colour in this figureegend, the reader is referred to the web version of the article.)
edox centre. Further, as the interfacial microenvironment aroundhe redox centre becomes more polar with negatively chargedCOO− groups at pH 7, the oxidized form of the ferrocene (Fc+)ecomes stabilized and the apparent formal potential shifts toore negative values. The full width half maxima (FWHM) val-
es obtained from the CV of the Fc/Fc+ redox couple for the thiolarboxylic acid diluent with different chain length have been tab-lated in Table 1. These values show that with decrease of thehain length and polarity of the head group, the FWHM value foroth the reduction and the oxidation peak of the Fc/Fc+ redoxouple gradually approaches the ideal value for identical non inter-
cting electroactive groups which is ∼90 mV [58]. This could beue to the prevention of the monolayer disorder by allowing thereater screening of the ferrocenes one from the other by thecheme 2. Schematic representation of the distance between the redox active ferrocene
n chain length of the thiol carboxylic acid. (A) Mixed SAM of ferrocene terminated thiol a
C5-COOH 126 131C7-COOH 140 152
thick layer between the redox centre and the diluent of shorterchain length[57]. However FWHM closer to the ideal value couldbe obtained at lower coverages (vide infra). Previous work hassuccessfully shown that the double layer effect has a significantcontribution towards the rate constant of the redox reaction on theelectrode [34,77,78].
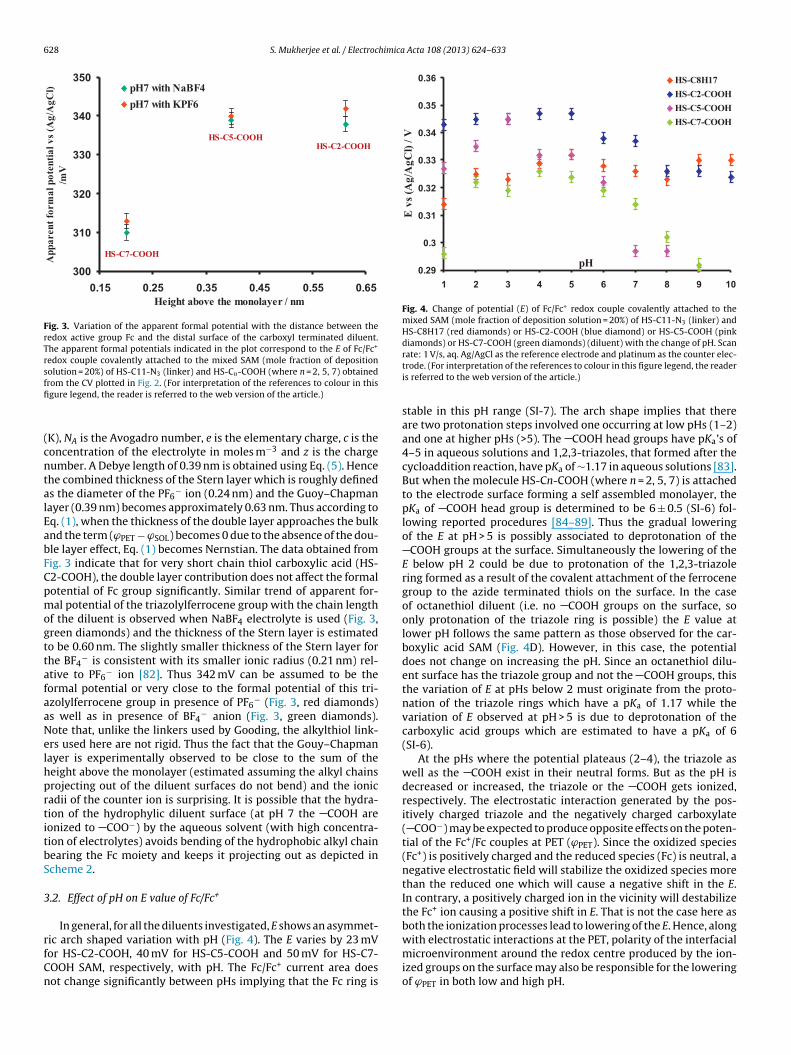
The potential drop across the double layer could be measuredfollowing the method recently reported by Gooding, Paddon-Row,et al. [34]. The plot of distance of the Fc group from the SAM (cal-culated using a 35◦ tilt angle, SI-5) and the measured apparentformal potential shows a saturation behaviour similar to the Good-ing and Paddon-Row report [34,67,79,80]. The electrostatic effectof the deprotonated COOH (pKa = 6 ± 0.5, see SI-6) groups in pH 7phosphate buffer lower the potential as the chain length of the car-boxyl terminated diluent is increased i.e. as the distance betweenthe terminal COO− groups and the redox active Fc group decrease.A steep drop in potential from the distal surface of the monolayerto ∼0.2 nm above it is observed (Fig. 3, red diamonds). This valuecoincides with the diameter of a PF6
− anion, 0.24 nm [81,82]. Thissuggests that the initial drop is created by a layer of PF6
− ion des-ignated as the Stern layer of a double layer model. However, afterthis initial steep drop a very shallow potential drop observed whichsaturates at 0.6 nm. The thickness of the double layer is determinedby adding up the thickness of the Stern layer and the thickness ofthe Gouy–Chapman layer (Debye length, �−1). The Debye length(�−1) is expressed as [34]√
ε0εrkT
2NAe2cz2
where ε0 is the permittivity of the vacuum, εr is the dielectric con-stant, k is the Boltzmann constant, T is the temperature in Kelvin
centre and the polar head group of the thiol carboxylic acid (diluent) with decreasend HS-C7-COOH. (B) Mixed SAM of ferrocene terminated thiol and HS-C2-COOH.
628 S. Mukherjee et al. / Electrochimica Acta 108 (2013) 624– 633
300
310
320
330
340
350
0.15 0.25 0.35 0.45 0.55 0.65
Height a bov e the monolay er / nm
Ap
pa
ren
t fo
rm
al
po
ten
tia
l v
s (A
g/A
gC
l)
/mV
pH7 with NaBF4
pH7 with KPF6
HS-C7-COO H
HS-C2-COOHHS-C5-COO H
Fig. 3. Variation of the apparent formal potential with the distance between theredox active group Fc and the distal surface of the carboxyl terminated diluent.The apparent formal potentials indicated in the plot correspond to the E of Fc/Fc+
redox couple covalently attached to the mixed SAM (mole fraction of depositionsolution = 20%) of HS-C11-N3 (linker) and HS-Cn-COOH (where n = 2, 5, 7) obtainedffi
(cntalEabFCpmogttafaaNelhprtitbS
3
rfCn
0.29
0.3
0.31
0.32
0.33
0.34
0.35
0.36
1 2 3 4 5 6 7 8 9 10
pH
E v
s (A
g/A
gC
l) /
V
HS-C8H17
HS-C2-COO H
HS-C5-COO H
HS-C7-COO H
Fig. 4. Change of potential (E) of Fc/Fc+ redox couple covalently attached to themixed SAM (mole fraction of deposition solution = 20%) of HS-C11-N3 (linker) andHS-C8H17 (red diamonds) or HS-C2-COOH (blue diamond) or HS-C5-COOH (pinkdiamonds) or HS-C7-COOH (green diamonds) (diluent) with the change of pH. Scan
rom the CV plotted in Fig. 2. (For interpretation of the references to colour in thisgure legend, the reader is referred to the web version of the article.)
K), NA is the Avogadro number, e is the elementary charge, c is theoncentration of the electrolyte in moles m−3 and z is the chargeumber. A Debye length of 0.39 nm is obtained using Eq. (5). Hencehe combined thickness of the Stern layer which is roughly defineds the diameter of the PF6
− ion (0.24 nm) and the Guoy–Chapmanayer (0.39 nm) becomes approximately 0.63 nm. Thus according toq. (1), when the thickness of the double layer approaches the bulknd the term (ϕPET − ϕSOL) becomes 0 due to the absence of the dou-le layer effect, Eq. (1) becomes Nernstian. The data obtained fromig. 3 indicate that for very short chain thiol carboxylic acid (HS-2-COOH), the double layer contribution does not affect the formalotential of Fc group significantly. Similar trend of apparent for-al potential of the triazolylferrocene group with the chain length
f the diluent is observed when NaBF4 electrolyte is used (Fig. 3,reen diamonds) and the thickness of the Stern layer is estimatedo be 0.60 nm. The slightly smaller thickness of the Stern layer forhe BF4
− is consistent with its smaller ionic radius (0.21 nm) rel-tive to PF6
− ion [82]. Thus 342 mV can be assumed to be theormal potential or very close to the formal potential of this tri-zolylferrocene group in presence of PF6
− (Fig. 3, red diamonds)s well as in presence of BF4
− anion (Fig. 3, green diamonds).ote that, unlike the linkers used by Gooding, the alkylthiol link-rs used here are not rigid. Thus the fact that the Gouy–Chapmanayer is experimentally observed to be close to the sum of theeight above the monolayer (estimated assuming the alkyl chainsrojecting out of the diluent surfaces do not bend) and the ionicadii of the counter ion is surprising. It is possible that the hydra-ion of the hydrophylic diluent surface (at pH 7 the COOH areonized to COO−) by the aqueous solvent (with high concentra-ion of electrolytes) avoids bending of the hydrophobic alkyl chainearing the Fc moiety and keeps it projecting out as depicted incheme 2.
.2. Effect of pH on E value of Fc/Fc+
In general, for all the diluents investigated, E shows an asymmet-
ic arch shaped variation with pH (Fig. 4). The E varies by 23 mVor HS-C2-COOH, 40 mV for HS-C5-COOH and 50 mV for HS-C7-OOH SAM, respectively, with pH. The Fc/Fc+ current area doesot change significantly between pHs implying that the Fc ring israte: 1 V/s, aq. Ag/AgCl as the reference electrode and platinum as the counter elec-trode. (For interpretation of the references to colour in this figure legend, the readeris referred to the web version of the article.)
stable in this pH range (SI-7). The arch shape implies that thereare two protonation steps involved one occurring at low pHs (1–2)and one at higher pHs (>5). The COOH head groups have pKa’s of4–5 in aqueous solutions and 1,2,3-triazoles, that formed after thecycloaddition reaction, have pKa of ∼1.17 in aqueous solutions [83].But when the molecule HS-Cn-COOH (where n = 2, 5, 7) is attachedto the electrode surface forming a self assembled monolayer, thepKa of COOH head group is determined to be 6 ± 0.5 (SI-6) fol-lowing reported procedures [84–89]. Thus the gradual loweringof the E at pH > 5 is possibly associated to deprotonation of the
COOH groups at the surface. Simultaneously the lowering of theE below pH 2 could be due to protonation of the 1,2,3-triazolering formed as a result of the covalent attachment of the ferrocenegroup to the azide terminated thiols on the surface. In the caseof octanethiol diluent (i.e. no COOH groups on the surface, soonly protonation of the triazole ring is possible) the E value atlower pH follows the same pattern as those observed for the car-boxylic acid SAM (Fig. 4D). However, in this case, the potentialdoes not change on increasing the pH. Since an octanethiol dilu-ent surface has the triazole group and not the COOH groups, thisthe variation of E at pHs below 2 must originate from the proto-nation of the triazole rings which have a pKa of 1.17 while thevariation of E observed at pH > 5 is due to deprotonation of thecarboxylic acid groups which are estimated to have a pKa of 6(SI-6).
At the pHs where the potential plateaus (2–4), the triazole aswell as the COOH exist in their neutral forms. But as the pH isdecreased or increased, the triazole or the COOH gets ionized,respectively. The electrostatic interaction generated by the pos-itively charged triazole and the negatively charged carboxylate( COO−) may be expected to produce opposite effects on the poten-tial of the Fc+/Fc couples at PET (ϕPET). Since the oxidized species(Fc+) is positively charged and the reduced species (Fc) is neutral, anegative electrostatic field will stabilize the oxidized species morethan the reduced one which will cause a negative shift in the E.In contrary, a positively charged ion in the vicinity will destabilizethe Fc+ ion causing a positive shift in E. That is not the case here asboth the ionization processes lead to lowering of the E. Hence, along
with electrostatic interactions at the PET, polarity of the interfacialmicroenvironment around the redox centre produced by the ion-ized groups on the surface may also be responsible for the loweringof ϕPET in both low and high pH.
S. Mukherjee et al. / Electrochimica Acta 108 (2013) 624– 633 629
Table 2Comparison of the actual coverage calculated (SI-1.3) from the experimental datawith the expected coverage for the SAM solution containing measured mole frac-tions of HS-C11-N3.
Mole fraction of thedeposition solution
Expected surfacecoverage
Actual surfacecoverage
0.02 2% 0.5%0.05 5% 3.5%0.1 10% 4.8%0.2 20% 6%
3
ahctSaacctTfmsoalittii
-50
-40
-30
-20
-10
0
10
20
30
40
50
00.10.20.30.40.5
Potential vs (Ag/AgCl) / V
Cu
rren
t / µµ
A
X=0.02
X=0.05
X=0.1
X=0.2
X=0.3
Fig. 5. Change of potential (E) of Fc/Fc+ redox couple covalently attached to themixed SAM of HS-C11-N3 (linker) and HS-C7-COOH (diluent), with the change of
tial of PET (Scheme 3A) [48]. This fact can be further supported by+
Sw
0.3 30% 10.2%
.3. Effect of varying coverage
The Fc/Fc+ current decreases with decreasing coverage (X) ofzide thiol. This is expected as with decreasing coverage of azideead groups on the surface, the ferrocene groups (resulting fromovalent linkage to the azides) decrease as well. Reductive desorp-ion of surfaces fully covered with HS-C7-COOH and HS-C11-N3AM shows that the number of thiol molecules/cm2 are 6.8 × 1014
nd 3.7 × 1014, respectively (Table 4 and Fig. SI-8). These valuesre consistent with the reported values for these SAM [90–93]. Theoverage of the azide, calculated from the integration of the Fc/Fc+
urrent (SI-9), is much lower than the mole fraction of the deposi-ion solution of azidothiol and thiol carboxylic acid used (Table 2).his does not imply that there are free azide groups on the sur-ace as prolonged immersion of these surfaces in the click reaction
ixture does not increase the Fc/Fc+ current. FTIR data of the depo-ition solution indicates that the N3 functionality does not decayver a period of days. Thus this implies that the fractional cover-ge of azide thiol on the surface achieved during SAM formation isess than that of the mole fraction of the deposition solution. Thiss not the case for an alkylthiol SAM diluent where the composi-ion of the deposition solution is very close to the composition of
he resultant surface (Fig. SI-9) [59]. It is likely that the H-bondingnteraction between the diluent HS-C7-COOH molecules disfavoursmpregnation of the monolayer by aliphatic azide thiols [94].cheme 3. Schematic representation of the surfaces with (A) high and (B) low coverageafer.
coverage at 1 V/s scan rate using aq. Ag/AgCl as reference electrode and platinum asthe counter electrode in pH 7 PO4
3− buffer.
Along with the decrease of Fc/Fc+ current with a decrease in cov-erage there is a negative shift in E (Fig. 5). Interestingly, such largenegative shift of thermodynamic reduction potential on loweringcoverage has not been observed in ordinary alkyl thiol SAM or anyother surface for that matter [59]. As the coverage of the surfaceincreases, the redox active Fc moieties may come closer to eachother and thus the repulsive electrostatic interactions betweenthe redox active neighbouring Fc moieties with same charge maybecome operative. As a result of the repulsive interaction, a smearedup charge density may be created on the surface which may beresponsible for the positive shift in the apparent formal poten-
the FWHM values of the reduction peak of the Fc/Fc redox couplefor different surface coverages listed in Table 3. On decreasing thecoverage the FWHM values approach the ideal value (∼90 mV) for
of mixed SAM of ferrocene terminated thiol and thiol carboxylic acid on clean Au

630 S. Mukherjee et al. / Electrochimica Acta 108 (2013) 624– 633
Table 3Full width at half maxima (FWHM) values of both the oxidation and reduction peaksof the CV of FC/Fc+ redox couple at different coverage.
Coverage (%) FWHM values
Oxidationpeak (mV)
Reductionpeak (mV)
0.5 75 963.5 116 115
tlitcn<ob[o6sutttsteihnbw∼w
tfdeiea(eraapn
TNfor
0.2
0.22
0.24
0.26
0.28
0.3
0.32
0.34
0 0.02 0.04 0.06 0.08 0.1
pH1
pH3
pH9
pH7
Mole frac tion
0.2
0.22
0.24
0.26
0.28
0.3
0.32
0.34
0 0.02 0.04 0.06 0.08 0.1
pH1
pH3
pH9
pH7
E v
s(A
g/A
gC
l) /
V
Mole frac tion
Fig. 6. Change of potential of Fc/Fc+ redox couple with change of mole fractionof the mixed SAM solution of HS-C11-N3 (linker) and HS-C7-COOH (diluent), at5 V/s scan rate using aq. Ag/AgCl as reference electrode and platinum as the counter
3−
4.8 116 1246 139 150
10.2 143 153
he identical non-interactive electroactive species. Thus a ∼60 mVarger FWHM relative to the ideal limit, for ∼10.2% coverage isndicative of some electrostatic interaction between the electroac-ive ferrocene sites or it might be due to some inhomogeneityreated at those electroactive sites at higher coverages. Since no sig-ificant broadening of the redox waves are observed for coverages6% (Table 3), possibilities of formation of clusters of redox speciesr the structural disorder associated with the mismatch in stericulk between Fc groups and the diluent molecules are ruled out37,57,95,96]. This trend is also observed by Creager et al., in casef alkyl thiol SAM [35]. On decreasing the surface coverages from% to 0.5% (where FWHM close to the ideal values are observed), amall monotonic shift of apparent formal potential to negative val-es is observed. This reflects the effect of non-polar alkyl group onhe electroactive species at PET. As the population of the electroac-ive Fc species on the surface decreases with decreasing coverage,he charges on the surface become discrete and the redox activepecies goes far apart from each other (Scheme 3B). This enhanceshe electrostatic interaction of the diluent COO− head group withach Fc+ ion leading to greater stabilization of the ferricenium (Fc+)on and thus causes lowering of E (Eq. (1)). Since COOH is polaread group and can be ionized easily, and the alkyl head group ison-polar, the effect produced by it on the redox active species wille more pronounced than that of the non-polar alkyl group. Hence,hile the COOH head group lowers the E of Fc/Fc+ redox couple by101 mV, only a small monotonic shift is observed for alkyl thiolsith decrease of the surface coverage (SI-10).
The extent of electrostatic stabilization depends on the ioniza-ion state of the surface COOH groups. A plot of E vs coverageor HS-C7-COOH SAM at different pHs (Fig. 6) shows a strong pHependence, i.e. the coverage dependence of E is also pH depend-nt. At low pH’s the E for very dilute Fc coverage is 0.26 V and itncreases with increasing coverage saturating at 0.31 V at 5–6% cov-rage (Fig. 6, red and orange points). For higher pH’s the E is 0.21 Vt very dilute coverages and it saturates at 0.31 V at 6% coverageFig. 6, green and blue points). Note that for surfaces having cov-rages <6% the FWHM is close to its ideal value. Thus these effectseflect the tuning of ˚PET by the changes in the microenvironment
round the Fc moiety. At low pH’s the COOH groups are proton-ted and lowers the potential by only 50 mV (Fig. 6, red and orangeoints). At higher pH’s (both 7 and 9) these groups are deproto-ated and induce higher electrostatic field and thereby stabilizingable 4umber of thiol molecules adsorbed per square cm area of the electrode surface to
orm the self assembled monolayer of either the single component or the mixturef two different components, calculated from the area under the peak obtained byeductive desorption.
SAM Area under thepeak, Ah (◦C)
Coverage(molecules/cm2)
HS-C8H17 16 2.22E+14HS-C7-COOH 49 6.80E+14HS-C11-N3 26.6 3.69E+14HS-C7-COOH + HS-C11-N3, 20% 8.83 1.22E+14
electrode in pH 9 (blue), pH 7 (green), pH 3 (orange) and pH 1 (red) PO4 buffers.(For interpretation of the references to colour in this figure legend, the reader isreferred to the web version of the article.)
the oxidized form of Fc (Fc+) which lowers the E by 100 mV (Fig. 6,green and blue points). At very high coverages, both in low andhigh pH’s, the effect of polarity of the microenvironment providedby carboxylic groups around the redox centre becomes ineffectivein stabilizing the Fc+. Hence, at >6–7% coverages (where the FWHMvalue deviate significantly from the ideal limit), the electrostaticinteractions between the ferrocene molecules become dominatingand E vs coverage plot exhibits saturation behaviour at both low andhigh pHs. The value of E at X = 0 is estimated to be 0.2 by extrapo-lation of the curve and it indicates the expected E on the COOHfunctionalized surface at pH 7 at infinite dilution. Note that thisvalue is very close to the value obtained for a ferrocene systemon an alkyl thiol SAM [35]. Thus the E of Fc/Fc+ can be tuned by∼100 mV by controlling the coverage and pH on a SAM having anionizable COOH functionalized diluent.
4. Discussion
Recently it was reported that mixed SAM with thiol azide andthiol carboxylic acids can be functionalized using “click” chemistryby modification of existing procedures [28]. Here we report that theionizable COOH groups present in these surfaces can be utilizedto tune the reduction potential of electroactive species immobi-lized on them. The formal reduction potential (E) of the redox activecentre on the surface deviates from its standard reduction poten-tial (E0) by the contribution of the term (ϕPET − ϕSOL) and the terminvolving the coverage of the oxidized and the reduced species, fol-lowing Eq. (1). As the potential of the bulk solution (ϕSOL) does notchange, the potential at the plane of electron transfer (ϕPET) is onlyresponsible for the change in apparent formal potential. The inter-facial microenvironment around the redox active ferrocene moietyis changed by ionizing the COOH head groups of the SAM at dif-ferent pHs, by varying the chain length of the thiol carboxylic aciddiluent and by varying the surface coverage with an attempt to tune(ϕPET − ϕSOL).
The apparent formal potential changes simultaneously withvariation in pH and chain length (Fig. 7A). At lower pH (pH ∼2–4),on decreasing the chain length of the diluent gradually, fromHS-C7-COOH to HS-C5-COOH to HS-C2-COOH, the (ϕPET − ϕSOL)value first increases and then gradually reaches saturation for
the shortest chain length evolving into a plateau region in theplot (indicated by the deep green shaded region in Fig. 7A). Thishappens because at this range of pH (pH = 2–4) the carboxylic headgroup remains in its neutral form. The polarity of the interfacial
S. Mukherjee et al. / Electrochimica
Fig. 7. (A) 3D graphical presentation of the variation of E with pH and chain length.(B) 3D graphical presentation of the variation of E with pH and mole fraction orcr
mbiadf(vf(bmtpmd(i(mgto
“click” reaction. These results show that by tuning the dilution of
overage. (For interpretation of the references to colour in the text, the reader iseferred to the web version of the article.)
icroenvironment varies with the chain length of the thiol car-oxylic acid diluent. As the chain length increases the polarity of the
nterfacial microenvironment increases. Since, according to Eq. (3),s the polarity around the redox centre increases, �Gtransfer,PET→SOLecreases with increase of εPET. The dielectric constants of the PETor different carboxylic acid thiols can be determined using Eqs.2) and (3), respectively (SI-11). The decrease of �Gtransfer,PET→SOLalue also decreases the value of (ϕPET − ϕSOL) thereby lowering theormal potential of Fc/Fc+ redox couple. Further, at a very low pHpH = 1–2), when both the triazole and the carboxylic head groupsecome protonated, the polarity of the interfacial microenviron-ent around the redox centre increases further. This brings down
he value of the apparent formal potential of the Fc/Fc+ redox cou-le to more negative following the relationships (Eqs. (3) and (2))entioned earlier. The effect is of course maximum for long chain
iluent like HS-C7-COOH (∼30 mV) and minimum for HS-C2-COOH∼5 mV) (SI-12). At higher pHs (pH 10) the Fc/Fc+ formal potentials minimum and is indicated by the deep blue region in the plotFig. 7A). At pH = 10, the carboxylic head group (pKa = 6 ± 0.5) of the
onolayer becomes deprotonated. As these deprotonated head
roups comes closer to the redox active centre with increase ofhe chain length, the potential drops with increase of the polarityf the interfacial microenvironment with negative charges. ThusActa 108 (2013) 624– 633 631
with increase of the polarity of the interfacial microenvironmentaround the redox centre (in this case due to deprotonation),the (ϕPET − ϕSOL) term decreases thereby decreasing the overallapparent formal potential according to Eq. (1). Raising the pHfrom 5 to 10, the drop of apparent formal potential of Fc/Fc+ redoxcouple is maximum (∼50 mV) for the long chain thiol carboxylicacid, i.e. HS-C7-COOH. The drop of formal potential at very high pH,can also be rationalized considering the electrostatic interactionof the oxidized Fc+ with the ionized COO− groups, i.e. the anionicinterfacial microenvironment around the redox active Fc centrestabilizes the oxidized species (Fc+) thereby decreasing the formalpotential of Fc/Fc+ redox couple. The change in the Fc/Fc+ potentialat very low pH (pH 1–2) can not have any contribution from theelectrostatic effect of protonated triazole because this protonatedtriazole destabilizes the Fc+ moiety causing the increase of theformal potential which is not observed here.
The apparent formal potential of the redox couple (here Fc/Fc+)at the plane of electron transfer can also be tuned by changing theextent of dilution of the electroactive species at different pH. Forthe SAM with the diluent of fixed chain length (HS-C7-COOH), theapparent formal potential varies simultaneously with the change ofmole fraction of the electroactive species as well as the pH (Fig. 7).At both high and low pHs the formal reduction potential (E) ofFc/Fc+ redox couple increases with increase of the surface cover-age and finally reaches saturation. That could be either the effect ofthe increased polarity of the interfacial microenvironment or elec-trostatic effect of the deprotonated carboxylate head groups on theformal reduction potential of Fc/Fc+ redox couple at PET. At low pH,when both the triazole rings as well as the carboxylic head groupsare protonated, the polarity of the interfacial microenvironmentaround the redox centre increases. At these pHs, on decreasing thesurface coverage of the Fc from 6% to 0.5%, the E value is lowered by∼50 mV (indicated by light blue triangular region bordered with redsolid line in Fig. 7B). This is due to enhanced polarity around individ-ual Fc species on a dilute surface. Note that for a surface with highercoverage, the charge density of the electroactive species (Fc/Fc+) getsmeared up on the surface due to the repulsive interaction amongthe redox species which in turn increases the E value (Scheme 3and Fig. 5) [48]. But the data used to generate the contour here rep-resents dilute surfaces (maximum 6% coverage where the FWHMis close to its ideal value) and hence charge smearing of Fc+ is pos-sibly not operative in these cases. At high pH, the polarity of theinterfacial microenvironment around the redox centre (due to thedeprotonated COO− head group) and also the electrostatic sta-bilization of the oxidized species Fc+ by the deprotonated COO−
increases. On lowering the coverage at this high pHs the formalreduction potential (E) is lowered by ∼100 mV indicated by thedeep blue shaded region bordered with black in Fig. 7B. At highpH the number of surface ionized COO− groups surrounding eachFc+ moiety increases with decrease of the surface coverage, result-ing greater stabilization of the oxidized Fc+ thereby lowering the Evalue. In fact, at pH 9 for very dilute surfaces (mole fraction < 1%),the E value of the Fc/Fc+ redox couple is minimum and that hasbeen indicated by the deep blue triangular region (Fig. 7B) sep-arated from the other part of the same region by a white solidline.
5. Conclusions
The (ϕPET − ϕSOL) of COOH terminated SAM has been probedby using Fc which is covalently attached on to these surfaces using
the ferrocenated surface, pH of the medium and chain length of thediluent the apparent formal potential of the Fc/Fc+ process can betuned from 0.345 ± 0.005 V to 0.200 ± 0.005 V i.e. over a range of

6 himica
1vlS(pl
A
A
i2
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
32 S. Mukherjee et al. / Electroc
45 mV, on a SAM containing ionizable COOH head groups. The Ealue reaches the maxima at 350 mV for HS-C5-COOH SAM at veryow pH (pH < 3) (Fig. 4B) and minima at 210 mV for HS-C7-COOHAM with very low coverage (<1%) but at very high pH (pH > 8)Fig. 7B). This provides a convenient method for tuning the formalotential (E) of Fc by using thiol carboxylic acid (with different chain
ength, different coverage and in different pH) SAM.
cknowledgement
This research was sponsored by DST grant SR/S1/IC-35/2009.
ppendix A. Supplementary data
Supplementary material related to this article can be found,n the online version, at http://dx.doi.org/10.1016/j.electacta.013.07.065.
eferences
[1] F. Mizutani, Biosensors utilizing monolayers on electrode surfaces, Sensors andActuators B 130 (2008) 14–20.
[2] R.A. Decreau, J.P. Collman, A. Hosseini, Electrochemical applications. Howclick chemistry brought biomimetic models to the next level: electrocatalysisunder controlled rate of electron transfer, Chemical Society Reviews 39 (2010)1291–1301.
[3] J.C. Love, L.A. Estroff, J.K. Kriebel, R.G. Nuzzo, G.M. Whitesides, Self-assembledmonolayers of thiolates on metals as a form of nanotechnology, ChemicalReviews 105 (2005) 1103.
[4] J.H. Fuhrhop, T. Wang, Bolaamphiphiles, Chemical Reviews 104 (2004)2901–2937.
[5] R.K. Iha, K.L. Wooley, A.M. Nyström, D.J. Burke, M.J. Kade, C.J. Hawker, Appli-cations of orthogonal “click” chemistries in the synthesis of functional softmaterials, Chemical Reviews 109 (2009) 5620–5686.
[6] A. Ulman, Formation and structure of self-assembled monolayers, ChemicalReviews 96 (1996) 1533–1554.
[7] C. Léger, P. Bertrand, Direct electrochemistry of redox enzymes as a tool formechanistic studies, Chemical Reviews 108 (2008) 2379–2438.
[8] J. Zhang, A.M. Kuznetsov, I.G. Medvedev, Q. Chi, T. Albrecht, P.S. Jensen, J.Ulstrup, Single-molecule electron transfer in electrochemical environments,Chemical Reviews 108 (2008) 2737–2791.
[9] F. Buckel, F. Effenberger, C. Yan, A. Gölzhäuser, M. Grunze, Influence of aromaticgroups incorporated in long-chain alkanethiol self-assembled monolayers ongold, Advanced Materials 12 (2000) 901–905.
10] R. Haag, M.A. Rampi, R.E. Holmlin, G.M. Whitesides, Electrical breakdown ofaliphatic and aromatic self-assembled monolayers used as nanometer-thickorganic dielectrics, Journal of the American Chemical Society 121 (1999)7895–7906.
11] C. Ramalechume, S. Berchmans, V. Yegnaraman, A.B. Mandal, Electron transferstudies through mixed self-assembled monolayers of thiophenol and thiocticacid, Journal of Electroanalytical Chemistry 580 (2005) 122–127.
12] E.B. Troughton, C.D. Bain, G.M. Whitesides, R.G. Nuzzo, D.L. Allara, M.D. Porter,Monolayer films prepared by the spontaneous self-assembly of symmetricaland unsymmetrical dialkyl sulfides from solution onto gold substrates: struc-ture, properties, and reactivity of constituent functional groups, Langmuir 4(1988) 365–385.
13] E. Cooper, L. Parker, C.A. Scotchford, S. Downes, G.J. Leggett, T.L. Parker, Theeffect of alkyl chain length and terminal group chemistry on the attachmentand growth of murine 3T3 fibroblasts and primary human osteoblasts on self-assembled monolayers of alkanethiols on gold, Journal of Materials Chemistry10 (2000) 133–139.
14] S. Mendez, L.K. Ista, G.P. López, Use of stimuli responsive polymers graftedon mixed self-assembled monolayers to tune transitions in surface energy,Langmuir 19 (2003) 8115–8116.
15] D.F. Siqueira Petri, G. Wenz, P. Schunk, T. Schimmel, An improved method forthe assembly of amino-terminated monolayers on SiO2 and the vapor deposi-tion of gold layers, Langmuir 15 (1999) 4520–4523.
16] S. Chen, L. Li, C.L. Boozer, S. Jiang, Controlled chemical and structural propertiesof mixed self-assembled monolayers of alkanethiols on Au(1 1 1), Langmuir 16(2000) 9287–9293.
17] J.B. Schlenoff, M. Li, H. Ly, Stability and self-exchange in alkanethiol monolayers,Journal of the American Chemical Society 117 (1995) 12528–12536.
18] P. Iqbal, K. Critchley, D. Attwood, D. Tunnicliffe, S.D. Evans, J.A. Preece, Chemical
manipulation by X-rays of functionalized thiolate self-assembled monolayerson Au, Langmuir 24 (2008) 13969–13976.19] A. Myrskog, H. Anderson, T. Aastrup, B. Ingemarsson, B. Liedberg, Esterifica-tion of self-assembled carboxylic-acid-terminated thiol monolayers in acidenvironment: a time-dependent study, Langmuir 26 (2010) 821–829.
[
Acta 108 (2013) 624– 633
20] L.M. Niu, H.Q. Luo, N.B. Li, L. Song, Electrochemical detection of copper(II) ata gold electrode modified with a self-assembled monolayer of penicillamine,Journal of Analytical Chemistry 62 (2007) 470–474.
21] E. Chow, E.L.S. Wong, O. Pascoe, D.B. Hibbert, J.J. Gooding, Extending thedynamic range of electrochemical sensors using multiple modified electrodes,Analytical and Bioanalytical Chemistry 387 (2007) 1489–1498.
22] W. Yang, J.J. Gooding, D.B. Hibbert, Characterisation of gold electrodesmodified with self-assembled monolayers of l-cysteine for the adsorptive strip-ping analysis of copper, Journal of Electroanalytical Chemistry 516 (2001)10–16.
23] R.K. Shervedani, A. Farahbakhsh, M. Bagherzadeh, Functionalization of gold cys-teamine self-assembled monolayer with ethylenediaminetetraacetic acid as anovel nanosensor, Analytica Chimica Acta 587 (2007) 254–262.
24] B. Atmaja, J.N. Cha, C.W. Frank, Adsorbed �-helical diblock copolypeptides:molecular organization, structural properties, and interactions, Langmuir 25(2009) 865–872.
25] K. Niki, W.R. Hardy, M.G. Hill, H. Li, J.R. Sprinkle, E. Margoliash, K. Fujita, R.Tanimura, N. Nakamura, H. Ohno, J.H. Richards, H.B. Gray, Coupling to lysine-13 promotes electron tunneling through carboxylate-terminated alkanethiolself-assembled monolayers to cytochrome c, Journal of Physical Chemistry B107 (2003) 9947–9949.
26] K.L. Davis, B.J. Drews, H. Yue, D.H. Waldeck, Electron-transfer kinetics of cova-lently attached cytochrome c/SAM/Au electrode assemblies, Journal of PhysicalChemistry C 112 (2008) 6571–6576.
27] D.H. Murgida, P.J. Hildebrandt, The heterogeneous electron transfer ofcytochrome c adsorbed on Ag electrodes coated with �-carboxyl alkanethiols.A surface enhanced resonance Raman spectroscopic study, Journal of MolecularStructure 565–566 (2001) 97–100.
28] S. Mukherjee, K. Sengupta, M.R. Das, S.S. Jana, A. Dey, Site-specific covalentattachment of heme proteins on self-assembled monolayers, Journal of Biolog-ical Inorganic Chemistry 17 (2012) 1009–1023.
29] H. Zimmermann, A. Lindgren, W. Schuhmann, L. Gorton, Anisotropic orien-tation of horseradish peroxidase by reconstitution on a thiol-modified goldelectrode, Chemistry – A European Journal 6 (2000) 592–599.
30] J.J. Sumner, S.E. Creager, Redox kinetics in monolayers on electrodes: electrontransfer is sluggish for ferrocene groups buried within the monolayer interior,Journal of Physical Chemistry B 105 (2001) 8739–8745.
31] A.V. Delgado, F. Gonzalez-Caballero, R.J. Hunter, L.K. Koopal, J. Lyklema, Mea-surement and interpretation of electrokinetic phenomena (IUPAC technicalreport), Pure and Applied Chemistry 77 (2005) 1753–1805.
32] F. Fogolari, A. Brigo, H.J. Molinari, The Poisson–Boltzmann equation forbiomolecular electrostatics: a tool for structural biology, Journal of MolecularRecognition 15 (2002) 377–392.
33] C.P. Smith, H.S. White, Theory of the interfacial potential distribution andreversible voltammetric response of electrodes coated with electroactivemolecular films, Analytical Chemistry 64 (1992) 2398–2405.
34] P.K. Eggers, N. Darwish, M.N. Paddon-Row, J.J. Gooding, Surface-bound molec-ular rulers for probing the electrical double layer, Journal of the AmericanChemical Society 134 (2012) 7539–7544.
35] S.E. Creager, G.K. Rowe, Solvent and double-layer effects on redox reactionsin self-assembled monolayers of ferrocenyl-alkanethiolates on gold, Journal ofElectroanalytical Chemistry 420 (1997) 291–299.
36] D. Acevedo, H.D. Abruna, Electron-transfer study and solvent effects on the for-mal potential of a redox-active self-assembling monolayer, Journal of PhysicalChemistry 95 (1991) 9590–9594.
37] C.E.D. Chidsey, T.M. Bertozzi, T.M. Putvinski, A.M. Mujsce, Coadsorption offerrocene-terminated and unsubstituted alkanethiols on gold: electroactiveself-assembled monolayers, Journal of the American Chemical Society 112(1990) 4301–4306.
38] G.K. Rowe, S.E. Creager, Redox and ion-pairing thermodynamics in self-assembled monolayers, Langmuir 7 (1991) 2307–2312.
39] G.K. Rowe, S.E. Creager, Interfacial solvation and double-layer effects on redoxreactions in organized assemblies, Journal of Physical Chemistry 98 (1994)5500–5507.
40] S.E. Creager, G.K. Rowe, Alcohol aggregation at hydrophobic monolayersurfaces and its effect on interfacial redox chemistry, Langmuir 9 (1993)2330–2336.
41] S.E. Creager, G.K. Rowe, Redox properties of ferrocenylalkane thiols coadsorbedwith linear n-alkanethiols on polycrystalline bulk gold electrodes, AnalyticaChimica Acta 9 (1993) 233–239.
42] S.E. Creager, G.K. Rowe, Competitive self-assembly and electrochemistry ofsome ferrocenyl-n-alkanethiol derivatives on gold, Journal of ElectroanalyticalChemistry 370 (1994) 203–211.
43] H.C. Delong, D.A. Buttry, Environmental effects on redox potentials of violo-gen groups embedded in electroactive self-assembled monolayers, Langmuir8 (1992) 2491–2496.
44] H.O. Finklea, D.D. Hanshew, Preparation and reversible behavior of organizedthiol monolayers with attached pentaminepyridineruthenium redox centers,Journal of Electroanalytical Chemistry 347 (1993) 327–340.
45] J. Redepenning, H.M. Tunison, H.O. Finklea, Influence of Donnan potentialson apparent formal potentials measured for organized thiol monolayers with
attached pentaamminepyridineruthenium redox centers, Langmuir 9 (1993)1404–1407.46] D.D. Popenoe, R.S. Deinhammer, M.D. Porter, Infrared spectroelectrochemicalcharacterization of ferrocene-terminated alkanethiolate monolayers at gold,Langmuir 8 (1992) 2521–2530.

himica
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
S. Mukherjee et al. / Electroc
47] P.K. Eggers, D.B. Hibbert, M.N. Paddon-Row, J.J. Gooding, The effect of surfacepolarity on the electrochemical double layer and its influence on the mea-surement of the standard rate constant of electron transfer, Journal of PhysicalChemistry C 113 (2009) 8964–8971.
48] W.R. Fawcett, Discreteness-of-charge effects at an electrode covered with aself-assembled monolayer containing a simple redox couple, Journal of Electro-analytical Chemistry 378 (1994) 117–124.
49] H. Strehlow, in: J.J. Lagowski (Ed.), The Chemistry of Non-Aqueous Solvents,vol. I, Academic Press, New York, 1966, p. 129.
50] H.C. Kolb, M.G. Finn, K.B. Sharpless, Click chemistry: diverse chemical functionfrom a few good reactions, Angewandte Chemie International Edition 40 (2001)2004–2021.
51] D.I. Rozkiewicz, D. Janczewski, W. Verboom, B.J. Ravoo, D.N. Reinhoudt, “Click”chemistry by microcontact printing, Angewandte Chemie International Edition45 (2006) 5292–5296.
52] N.K. Devaraj, G.P. Miller, W. Ebina, B. Kakaradov, J.P. Collman, E.T. Kool, C.E.D.Chidsey, Chemoselective covalent coupling of oligonucleotide probes to self-assembled monolayers, Journal of the American Chemical Society 127 (2005)8600–8601.
53] J.P. Collman, N.K. Devaraj, R.A. Decreau, Y. Yang, Y.L. Yan, W. Ebina, T.A.Eberspacher, C.E.D. Chidsey, A cytochrome c oxidase model catalyzes oxy-gen to water reduction under rate-limiting electron flux, Science 315 (2007)1565–1568.
54] Y. Zhang, S. Luo, Y. Tang, L. Yu, K.Y. Hou, J.P. Cheng, X. Zeng, P.G. Wang,Carbohydrate–protein interactions by “clicked” carbohydrate self-assembledmonolayers, Analytical Chemistry 78 (2006) 2001–2008.
55] R. Zirbs, F. Kienberger, P. Hinterdorfer, W.H. Binder, Directed assembly of Aunanoparticles onto planar surfaces via multiple hydrogen bonds, Langmuir 21(2005) 8414–8421.
56] C.W. Tornoe, C. Christensen, M. Meldal, Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions ofterminal alkynes to azides, Journal of Organic Chemistry 67 (2002) 3057–3064.
57] J.P. Collman, N.K. Devaraj, C.E.D. Chidsey, “Clicking” functionality onto electrodesurfaces, Langmuir 20 (2004) 1051–1053.
58] C.E.D. Chidsey, C.R. Bertozzi, T.M. Putvinski, A.M. Mujsce, Coadsorption offerrocene-terminated and unsubstituted alkanethiols on gold: electroactiveself-assembled monolayers, Journal of the American Chemical Society 112(1990) 4301–4306.
59] J.P. Collman, N.K. Devaraj, T.P.A. Eberspacher, C.E.D. Chidsey, Mixed azide-terminated monolayers: a platform for modifying electrode surfaces, Langmuir22 (2006) 2457–2464.
60] V.V. Rostovtsev, L.G. Green, V.V. Fokin, K.B. Sharpless, A stepwise Huisgencycloaddition process: copper(I)-catalyzed regioselective “ligation” of azidesand terminal alkynes, Angewandte Chemie International Edition 41 (2002)2596–2599.
61] N.K. Devaraj, J.P. Collman, Copper catalyzed azide–alkyne cycloadditions onsolid surfaces: applications and future directions, QSAR and CombinatorialScience 26 (2007) 1253–1260.
62] S.E. Creager, G.K. Rowe, Redox properties of ferrocenylalkane thiols coadsorbedwith linear n-alkanethiols on polycrystalline bulk gold electrodes, AnalyticaChimica Acta 246 (1991) 233–239.
63] L.M. Fischer, M. Tenje, A.R. Heiskanen, N. Masuda, J. Castillo, A. Bentien, J.Émneus, M.H. Jakobsen, A. Boisen, Gold cleaning methods for electrochemicaldetection applications, Microelectronic Engineering 86 (2009) 1282–1285.
64] C. Spégel, A. Heiskanen, J. Acklid, A. Wolff, R. Taboryski, J. Emneus, T. Ruzgas,On-chip determination of dopamine exocytosis using mercaptopropionic acidmodified microelectrodes, Electroanalysis 19 (2007) 263–271.
65] V. Hong, A.K. Udit, R.A. Evans, M.G. Finn, Electrochemically protected copper(I)-catalyzed azide–alkyne cycloaddition, ChemBioChem 9 (2008) 1481–1486.
66] N.K. Devaraj, P.H. Dinolfo, C.E.D. Chidsey, J.P. Collman, Selective functionaliza-tion of independently addressed microelectrodes by electrochemical activationand deactivation of a coupling catalyst, Journal of the American Chemical Soci-ety 128 (2006) 1794–1795.
67] R.G. Nuzzo, L.H. Dubois, D.L. Allara, Fundamental studies of microscopic wettingon organic surfaces. 1. Formation and structural characterization of a self-consistent series of polyfunctional organic monolayers, Journal of the AmericanChemical Society 112 (1990) 558–569.
68] E.L. Smith, C.A. Alves, J.W. Anderegg, M.D. Porter, Deposition of metal overlayersat end-group-functionalized thiolate monolayers adsorbed at gold. 1. Surfaceand interfacial chemical characterization of deposited copper overlayers atcarboxylic acid-terminated structures, Langmuir 8 (1992) 2707–2714.
69] S.S. Cheng, D.A. Scherson, C.N. Sukenik, In situ attenuated total reflectanceFourier transform infrared spectroscopy study of carboxylate-bearing,
siloxane-anchored, self-assembled monolayers: a study of carboxylate reac-tivity and acid–base properties, Langmuir 11 (1995) 1190–1195.70] O. Gershevitz, C.N. Sukenik, In situ FTIR-ATR analysis and titration of carboxylicacid-terminated SAMs, Journal of the American Chemical Society 126 (2004)482–483.
[
Acta 108 (2013) 624– 633 633
71] R. Arnold, W. Azzam, A. Terfort, C. Woll, Preparation, modification, and crys-tallinity of aliphatic and aromatic carboxylic acid terminated self-assembledmonolayers, Langmuir 18 (2002) 3980–3992.
72] H.D. Sikes, J.F. Smalley, S.P. Dudek, A.R. Cook, M.D. Newton, C.E.D. Chidsey, S.W.Feldberg, Rapid electron tunneling through oligophenylenevinylene bridges,Science 291 (2001) 1519–1523.
73] M.D. Porter, T.B. Bright, D.L. Allara, C.E.D. Chidsey, Spontaneously orga-nized molecular assemblies. 4. Structural characterization of n-alkyl thiolmonolayers on gold by optical ellipsometry, infrared spectroscopy, andelectrochemistry, Journal of the American Chemical Society 109 (1987)3559–3568.
74] C.A. Widrig, C. Chung, M.D. Porter, The electrochemical desorption of n-alkanethiol monolayers from polycrystalline Au and Ag electrodes, Journal ofElectroanalytical Chemistry 310 (1991) 335–359.
75] A. Swietlow, M. Skoog, G. Johansson, Double-layer capacitance measure-ments of self-assembled layers on gold electrodes, Electroanalysis 4 (1992)921–928.
76] J. Zhang, A. Demetriou, A.C. Welinder, T. Albrecht, R.J. Nichols, J. Ulstrup,Potential-induced structural transitions of dl-homocysteine monolayers onAu(1 1 1) electrode surfaces, Chemical Physics 319 (2005) 210–221.
77] M. Ohtani, Quasi-reversible voltammetric response of electrodes coated withelectroactive monolayer films, Electrochemistry Communications 1 (1999)488–492.
78] M.J. Honeychurch, G.A. Rechnitz, Voltammetry of adsorbed molecules. Part 2.Irreversible redox systems, Electroanalysis 10 (1998) 453–457.
79] O. Dannenberger, K. Weiss, H.-J. Himmel, B. Jager, M. Buck, Ch. Woll, An orienta-tion analysis of differently endgroup-functionalised alkanethiols adsorbed onAu substrates, Thin Solid Films 307 (1997) 183–191.
80] C. Dubois, F. Stellacci, Self-assembled monolayer of short carboxyl-terminatedmolecules investigated with ex situ scanning tunneling microscopy, Journal ofPhysical Chemistry C 112 (2008) 7431–7435.
81] H. Wang, M. Yoshio, Suppression of PF6− intercalation into graphite by small
amounts of ethylene carbonate in activated carbon/graphite capacitors, Chem-ical Communications 46 (2010) 1544–1546.
82] W. Yu-xin, L. Xin-yong, Proceedings of the 3rd International Conference onFunctional Molecules, 2005.
83] A. Albert, P.J. Taylor, The tautomerism of 1,2,3-triazole in aqueous solution,Journal of the Chemical Society, Perkin Transactions 2 (1989) 1903–1905.
84] J.J. Gooding, P.S. Hale, L.M. Maddox, J.G. Shapter, Surface pKa of self-assembledmonolayers, Journal of Chemical Education 82 (2005) 779.
85] J. Zhao, L. Luo, X. Yang, E. Wang, S. Dong, Determination of surface pKa ofSAM using an electrochemical titration method, Electroanalysis 11 (1999)1108–1111.
86] M.A. Bryant, R.M. Crooks, Determination of surface pKa values of surface con-fined molecules derivatized with pH-sensitive pendant groups, Langmuir 9(1993) 385–387.
87] T. Kakiuchi, M. Iida, S. Imabayashi, K. Niki, Double-layer-capacitance titrationof self-assembled monolayers of �-functionalized alkanethiols on Au(1 1 1)surface, Langmuir 16 (2000) 5397–5401.
88] K. Hu, A.J. Bard, Use of atomic force microscopy for the study of surfaceacid–base properties of carboxylic acid-terminated self-assembled mono-layers, Langmuir 13 (1997) 5114–5119.
89] Q. Cheng, A. Brajter-Toth, Selectivity and sensitivity of self-assembled thiocticacid electrodes, Analytical Chemistry 64 (1992) 1998–2000.
90] K.C. Nguyen, Quantitative analysis of COOH-terminated alkanethiol SAMs ongold nanoparticle surfaces, Advances in Natural Sciences: Nanoscience andNanotechnology 3 (2012) 1–5.
91] S. Imabayashi, M. Iida, D. Hobara, Z.Q. Feng, K. Niki, T. Kakiuchi, Reduc-tive desorption of carboxylic-acid-terminated alkanethiol monolayers fromAu(1 1 1) surfaces, Journal of Electroanalytical Chemistry 428 (1997) 33–38.
92] R. Meunier-Prest, G. Legay, S. Raveau, N. Chiffot, E. Finot, Potential-assisteddeposition of mixed alkanethiol self-assembled monolayers, ElectrochimicaActa 55 (2010) 2712–2720.
93] J. Strutwolf, C.K. O’Sullivan, Microstructures by selective desorption of self-assembled monolayer from polycrystalline gold electrodes, Electroanalysis 19(2007) 1467–1475.
94] N. Winter, J. Vieceli, I. Benjamin, Hydrogen-bond structure and dynam-ics at the interface between water and carboxylic acid-functionalizedself-assembled monolayers, Journal of Physical Chemistry B 112 (2008)227–231.
95] P.A. Brooksby, K.H. Anderson, A.J. Downard, A.D. Abell, Voltammetric and elec-trochemical impedance study of ferrocenyl containing �-peptide monolayerson gold, Journal of Physical Chemistry C 115 (2011) 7516–7526.
96] B.R. Kozub, M.C. Henstridge, C. Batchelor-McAuley, R.G. Compton, Applica-tion of Marcus–Hush–Chidsey theory to two electron systems. The influenceof electrode material on the electro-reduction kinetics of covalently attached2-anthraquinonyl groups: gold vs. carbon, International Journal of Electro-chemical Science 6 (2011) 6047–6062.
Copyright © 2022 FDOKUMEN




![Drug Delivery System Based on Covalently Bonded Poly[N-Isopropylacrylamide-co-2-Hydroxyethylacrylate]-Based Nanoparticle Networks](https://static.fdokumen.com/doc/165x107/6340d5f6e0dac3b265042228/drug-delivery-system-based-on-covalently-bonded-polyn-isopropylacrylamide-co-2-hydroxyethylacrylate-based.jpg)















