
TUMOR NECROSIS FACTOR IS NOT ASSOCIATED WITH INTESTINAL ISCHEMIA/REPERFUSION-INDUCED LUNG...
12
Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited. ARTICLE COVER SHEET LWW—SHK FLA, Editorial Comment, Commentary, Review Article, Rapid Communication, Invited Opinion, Erratum SERVER-BASED Article : SHK21245 (476-09E) Creator : gi51 Date : 2/19/2010 Time : 14:26 Article Title : Number of Pages (including this page) : 11 Template Version : 4.3 06/30/09 Scripts: 1. SC_EXTRACT_XML 2. ScLoadErratum 3. sc_Multifig_Marker 4. Autopagination compliant
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of TUMOR NECROSIS FACTOR IS NOT ASSOCIATED WITH INTESTINAL ISCHEMIA/REPERFUSION-INDUCED LUNG...

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
ARTICLE COVER SHEET
LWW—SHK
FLA, Editorial Comment,
Commentary, Review Article,
Rapid Communication, Invited Opinion,
Erratum
SERVER-BASED
Article : SHK21245 (476-09E)
Creator : gi51
Date : 2/19/2010
Time : 14:26
Article Title :
Number of Pages (including this page) : 11
Template Version : 4.3
06/30/09
Scripts:
1. SC_EXTRACT_XML
2. ScLoadErratum
3. sc_Multifig_Marker
4. Autopagination compliant

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
TUMOR NECROSIS FACTOR IS NOT ASSOCIATED WITHAQ1 INTESTINALISCHEMIA/REPERFUSIONYINDUCED LUNG INFLAMMATION
Alexandre Learth Soares,* Fernando Rodrigues Coelho,* Rodrigo Guabiraba,†
Mamdouh Kamal,† Bernardo Boris Vargaftig,* Lily Li,‡ Jian Li,§
Wothan Tavares de Lima,* and Bernhard Ryffel†
*Institute of Biomedical Sciences, Department of Pharmacology, University of Sao Paulo, Sao Paulo,Brazil; †Laboratory of Molecular Immunology and Embryology, University of Orleans and CNRS UMR
6218, Orleans, France; and ‡Therakos, Department of Immune Cell Therapy, Exton; and §Centocor R&D,Department of Immunobiology, Horsham, Pennsylvania
Received 14 Oct 2009; first review completed 20 Oct 2009; accepted in final form 12 Nov 2009
ABSTRACT—Intestinal ischemia-reperfusion (I/R) injury may cause acute systemic and lung inflammation. Here, werevisited the role of TNF-! in an intestinal I/R model in mice, showing that this cytokine is not required for the local andremote inflammatory response upon intestinal I/R injury using neutralizing TNF-! antibodies and TNF ligandYdeficientmice. We demonstrate increased neutrophil recruitment in the lung as assessed by myeloperoxidase activity and aug-mented IL-6, granulocyte colony-stimulating factor, and KCAQ2 levels, whereas TNF-! levels in serum were not increased andonly minimally elevated in intestine and lung upon intestinal I/R injury. Importantly, TNF-! antibody neutralization neitherdiminished neutrophil recruitment nor any of the cytokines and chemokines evaluated. In addition, the inflammatoryresponse was not abrogated in TNF and TNF receptors 1 and 2Ydeficient mice. However, in view of the damage on theintestinal barrier upon intestinal I/R with systemic bacterial translocation, we asked whether Toll-like receptor (TLR)activation is driving the inflammatory response. In fact, the inflammatory lung response is dramatically reduced in TLR2/4Ydeficient mice, confirming an important role of TLR receptor signaling causing the inflammatory lung response. Inconclusion, endogenous TNF-! is not or minimally elevated and plays no role as a mediator for the inflammatory responseupon ischemic tissue injury. By contrast, TLR2/4 signaling induces an orchestrated cytokine/chemokine response leadingto local and remote pulmonary inflammation, and therefore disruption of TLR signaling may represent an alternativetherapeutic target.
KEYWORDS—Ischemic tissue injury, pulmonary inflammation, cytokines, TNF, TLR
INTRODUCTION
Intestinal ischemia-reperfusion (I/R) injury is a serious and
common clinical entity and can result from different etiologic
factors, including superior mesenteric artery (SMA) occlusion
or hemorrhagic shock (1). This condition may result in severe
local and extensive tissue injury and subsequent distant organ
dysfunction, affecting mainly the lungs. Several studies (2, 3)
point to the important role that intestinal I/R may play in the
triggering of acute respiratory distress syndrome (ARDS), a
syndrome characterized by hypoxemia, reduced lung com-
pliance, fluffy diffuse infiltrates on the chest radiograph, and
the presence of normal pulmonary capillary pressures (4).
Although most patients survive the initial insult that precipi-
tates ARDS, no therapeutic protocol has proven to modify the
course of this condition and mortality remains greater than
40% (5).
Among the mediators that are altered during intestinal I/R,
TNF-! is the most widely studied (6). TNF-! at high levels
leads to the development of an inflammatory response that is
the hallmark of many diseases because the mediator is impli-
cated in acute lung injury and ARDS, and also more chronic
lung diseases such as asthma, chronic bronchitis, and chronic
obstructive pulmonary disease (7, 8).
TNF-! binds to two receptors, the 55-kd and 75-kd recep-
tors, with similar affinity (6). The 55-kd receptor (TNFR1)
mediates cytotoxic and proinflammatory effect, whereas the
75-kd receptor (TNFR2) mediates T-cell proliferation (8). Al-
though there is a well-established link between TNF-! levels
and sepsis, there is still a debate on the role of TNF-! in non-
septic acute inflammatory conditions. In fact, depending on
the model used for the induction of the inflammatory insult,
no significant increase of circulating TNF-! is detected. Kim
et al. (9) showed that the levels of circulating TNF-! did not
change compared with sham controls in a model of hemorrhage
shock, whereas Grotz et al. (10) showed early TNF-! increases
upon intestinal I/R in rats that are primarily caused by the
laparotomy. Chen et al. (11) showed that the Toll-like receptor
(TLR) ligand LPS decreased mesenteric I/R injury through
TNF-! signaling.
The TLR family members recognize pathogen-associated
molecular patterns derived from microbes as well as endoge-
nous ligands from damaged/stressed cells (12). Toll-like recep-
tor 2 and TLR4 are activated not only by LPS and lipoteichoic
1
SHOCK, Vol. 00, No. 00, pp. 00Y00, 2010
Copyeditor: Maria Charissa Paras
Address reprint requests to Bernhard Ryffel, Molecular Immunology and
Embryology, University and CNRS UMR 6218. 45071, Orleans, France. E-mail:
This study was supported by grants from the Fondation de la Recherche Medical
(F.R.M, France); PhenoTec AG, Zug, Switzerland; Immunis SA, Orleans, France;
and Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP), Sao Paulo,
Brazil.
Supplemental digital content is available for this article. Direct URL citation
appears in the printed text and is provided in the HTML and PDF versions of this
article on the Journal’s Website (www.shockjournal.com).
DOI: 10.1097/SHK.0b013e3181cdc585
Copyright � 2010 by the Shock Society

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
acid but also by extracellular matrix components (13) and pos-
sibly by heat shock proteins (14). Toll-like receptor 4 has also
been implicated in the priming effect induced by hemorrhagic
shock for lung inflammation (15).
Here, we tested the hypothesis that TNF-! is not required
for the local and remote inflammatory response upon intes-
tinal I/R injury using neutralizing TNF-! antibodies and TNF
ligandYdeficient mice (KO). We report that after intestinal
I/R in mice, several cytokines are upregulated, in particular of
IL-6, granulocyte colony-stimulating factor (G-CSF), and KC,
but TNF-! levels are very little affected. Importantly, neu-
tralization of TNF-! by antibodies did not affect neutrophil
recruitment to the lung, the cytokine response, and intestinal
and pulmonary injury. Furthermore, the inflammatory re-
sponse in the absence of TNF-! or of TNF receptors was not
significantly different from wild-type controls upon intestinal
I/R injury. Finally, we demonstrate that TLR2/TLR4 receptor
signaling plays a critical role in the inflammatory process
after intestinal I/R in mice.
MATERIALS AND METHODS
AnimalsMale C57Bl/6 mice aged 8 to 10 weeks were purchased from Ace
(Philadelphia, Pa) or obtained from the animal care facility at the TransgenoseInstitute (CNRS, Orleans, France). TNF KO and TNFR1/R2 KO (obtainedfrom Jackson Laboratories, Bar Harbor, Me), and TLR2 and TLR4 KO(obtained from Dr Akira, crossed in-house to obtain TLR2/4 KO mice) (16)
FIG. 1. Effects of intestinal I/R on MPO activity in lung (A) and intestine (B) LDH activity in serum (C) and gut (D). Histological example onhematoxylin-eosin slides. Mice were subjected to 45 min of mesenteric artery occlusion followed by 4 h of intestinal reperfusion. Lung samples from ischemicanimal (F and H). Control group consisted of mice that were sham operated (E and G) (n = 7) (hematoxylin-eosin, original magnification �400). E and F,Representative section of a small branch of pulmonary arteriole. G and H, The alveolar structure, where arrows point to polymorphonuclear cells infiltrated in thetissue. Basal values were obtained from naive mice. Data represent mean T SEM from 2 experiments from 5 to 7 animals. *P G 0.05.
2 SHOCK VOL. 00, NO. 00 SOARES ET AL.
Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
used in this study were bred under specific pathogenYfree conditions at theTransgenose Institute (CNRS). Animals were housed, kept in sterile isolatedventilated cages, and used after an approved, reviewed, and monitoredprotocol from the Committee on Care and Use of Laboratory Animals ofCentocor R&D and from the French Government’s ethical and animalexperiment regulations. All animals had access to water and food ad libitumbefore the experiments.
Animal model of acute lung injuryIntestinal I/R was performed as described elsewhere (17). In brief, lapa-
rotomy was performed under anesthesia with ketamine (100 mg/kg, intra-peritoneally) and xylazine (20 mg/kg, intraperitoneally). In the ischemic groupof mice, a microsurgical clip (Vascu-statt no. 1001-531; Scalan Internat,Minn) was applied to occlude the SMA. After 45 min occlusion, the clip wasremoved and intestinal perfusion was re-established. Animals were kept inanesthesia and killed 4 h after intestinal reperfusion by cervical dislocation.For the 24 h time point, the animals were awake after reperfusion andreceived acetaminophen-supplemented drinking water (3.2 mg/mL) to relievepain. Blood samples were collected from the abdominal aorta of anesthetizedmice. The aliquots were centrifuged (170 g, 10 min), and the sera obtainedwere stored at j80-C until further analyses. Sham-operated mice undergoingidentical surgical intervention except for the occlusion of the SMA were usedas control groups. An additional group of nonmanipulated mice (naive) wasadded to obtain basal values of the parameters studied.
Measurement of lactate dehydrogenaseIntestinal tissue damage caused by intestinal I/R releases lactate dehydro-
genase (LDH) into the systemic circulation and was measured according
to Cavriani et al. (17). Briefly, LDH activity was determined in the serumsamples using a commercially available kit (Labtest Diagnostica, BeloHorizonte, Brazil) and expressed as units per microliter. Absorbance wasread at 340 nm on a microplate reader spectrophotometrically (Bio-TekInstruments).
Lung and intestine myeloperoxidase activityMyeloperoxidase (MPO) activity was measured as an index of the presence
of neutrophils. The lungs were perfused via the pulmonary artery with 3 mLof ice-cold pH 7.2 phosphate-buffered saline (PBS). Intestines were rinsedwith ice-cold PBS, and the small intestine was excised and used in theanalysis. This procedure was also used to obtain the samples for cytokineand histological analyses. Samples were prepared according to Goldblumet al. (18). In brief, to normalize the pulmonary and intestinal MPO activityamong the groups, the whole tissue was homogenized with 3 mL/g of PBScontaining 0.5% of hexadecyl-trimethylammonium bromide and 5 mMEDTA, pH 6.0. The homogenized samples were centrifuged at 37,000g for15 min. Samples of lung and intestine homogenates (10 2L) were thenincubated for 10 min with H2O2 and ortho-dianisidine, and the reaction wasstopped by the addition of 1% NaNO3. The absorbance was determined at450 nm using a microplate reader (Bio-Tek Instruments).
HistologyIntestine and lung were fixed in 10% buffered formalin. Tissues were
dehydrated in ethanol and embedded in paraffin. Sections of 5 2m werestained with hematoxylin and eosin for evaluation of pathological changes.The microscopic slides were reviewed and assessed using a semiquantitative
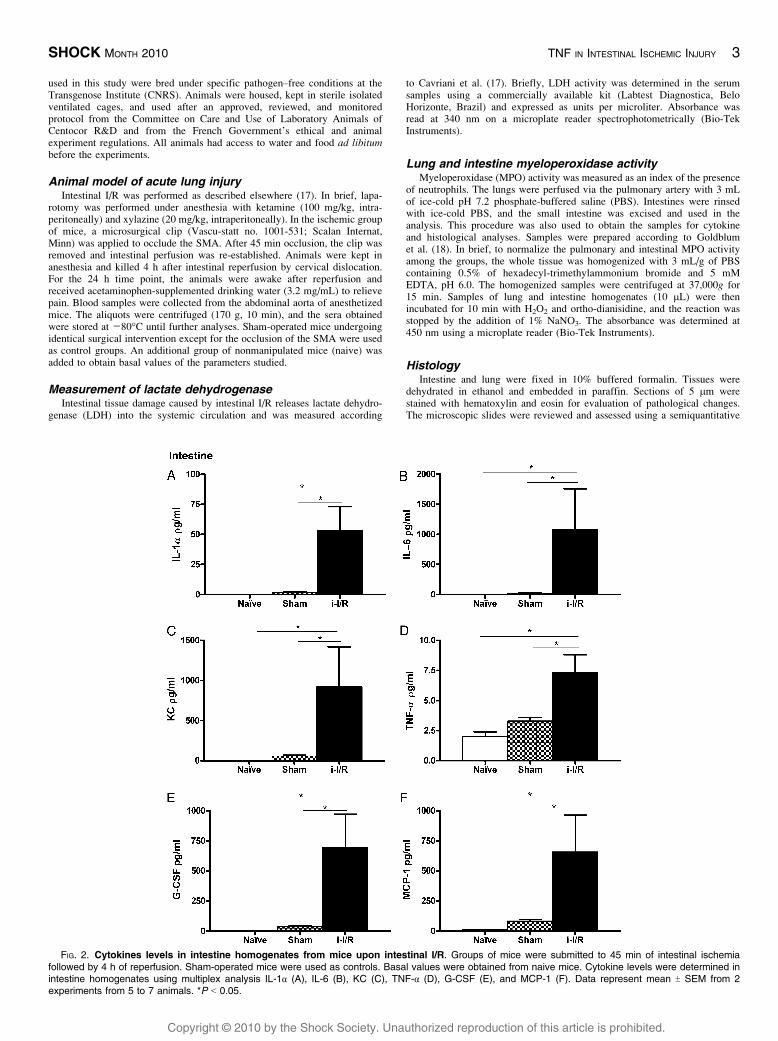
FIG. 2. Cytokines levels in intestine homogenates from mice upon intestinal I/R. Groups of mice were submitted to 45 min of intestinal ischemiafollowed by 4 h of reperfusion. Sham-operated mice were used as controls. Basal values were obtained from naive mice. Cytokine levels were determined inintestine homogenates using multiplex analysis IL-1! (A), IL-6 (B), KC (C), TNF-! (D), G-CSF (E), and MCP-1 (F). Data represent mean T SEM from 2experiments from 5 to 7 animals. *P G 0.05.
SHOCK MONTH 2010 TNF IN INTESTINAL ISCHEMIC INJURY 3

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
score by two investigators (F.R.C., B.R.) who were blinded to the groups andtime points.
Cytokine measurementCytokines levels were evaluated in serum and in lung and gut homogenates
using a 22-cytokine Linco multiplex kit (LINCO Research, Inc, Mo) with aLuminex instrument (Luminex Corporation, Tex).
In vivo neutralization of TNF-!Rat monoclonal antibody (mAb) with neutralizing activity for murine
TNF-! was generated in house (Centocor R&D, Horsham, Pa) using standardhybridoma technology. The neutralizing property of this antibody has beentested in vitro and in vivo in response to TNF (data not shown). Prior (30 min)to intestinal I/R induction, mice were i.v. injected with 10 mL/kg of a solutioncontaining 2.5 mg/mL of TNF-! antibody (final dose, 25 mg/kg of antibody).A group of mice i.v. injected with 10 mL/kg of PBS 30 min before theintestinal I/R induction was used as the control.
Expression of TLR2 and TLR4 mRNA in lung tissueSamples of lungs were excised and frozen in liquid nitrogen after the
experimental procedure. To harvest RNA, samples were lysed using NucleicAcid Purification Lysis solution (Applied Biosystems, Foster City, Calif).RNA was prepared using ABI PRISM 6100 Nucleic Acid PrepStation (DNasestep included). RNA quality was verified with the 2100 BioAnalyzer (AgilentTechnologies, Palo Alto, Calif). Samples that demonstrated high quality (i.e.,the ratio of 28S rRNA to 18S rRNA, 91.7) and had a minimum of 1 2g ofRNA were submitted to real-time polymerase chain reaction (PCR) analysis.
The expression of TLR2 and TLR4 mRNA was evaluated by real-time PCRusing a TaqMan assay kit, following the manufacturer’s recommendations.
Statistical analysisData are given as mean T SEM. Comparisons between the groups were
made by one-way ANOVA followed by unpaired Newman-Keuls post-test. A4.0 version (2005) of the GraphPad Prism software was used for this purpose.Values of P G 0.05 were considered significant.
RESULTS
Model characterization
To study the inflammatory response upon intestinal I/R in-
jury in the local (intestine) and in the remote organ (lung), the
MPO activity was evaluated in tissue homogenates. Figure F11,
A and B, shows that 45 min of ischemia followed by a 4-h
reperfusion of the intestinal tissue caused significant neutro-
phil recruitment both in lung and in intestinal tissues. The
MPO activity reached its peak at 4 h after intestinal reper-
fusion and returned almost to its basal levels after 24 h. Fur-
thermore, intestinal I/R damage induces a significant increase
of LDH activity in serum (Fig. 1C) and a decrease of LDH in
FIG. 3. Cytokines levels in serum from mice upon intestinal I/R. Groups of mice were submitted to 45 min of intestinal ischemia followed by 4 h ofreperfusion. Sham-operated mice were used as controls. Basal values were obtained from naive mice. Cytokine levels were determined in serum usingmultiplex analysis IL-1! (A), IL-6 (B), KC (C), TNF-! (D), G-CSF (E), and MCP-1 (F). Data represent mean T SEM from 2 experiments from 5 to 7 animals.*P G 0.05.
4 SHOCK VOL. 00, NO. 00 SOARES ET AL.

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
the gut homogenates (Fig. 1D) as evidence of tissue injury
caused by intestinal I/R.
The neutrophilic inflammation was confirmed by micro-
scopic investigations of the lungs (Fig. 1F). We observe
neutrophil recruitment in the interstitial space and capillaries
of the lung upon intestinal I/R injury (Fig. 1H). There are also
signs of edema and inflammatory cells in the perivascular
space in the small vessels (Fig. 1F).
Cytokine profile in distinct compartments afterintestinal I/R injury
To identify major changes in the pattern of inflammatory
mediators expressed upon intestinal I/R injury, we evaluated
the expression of 22 cytokines and chemokines using a Linco
Multiplex kit in the homogenates of ileum and lung as well as
in the serum 4 h after intestinal reperfusion.
In the ileum homogenate, the levels of IL-1!, IL-6, KC,G-CSF, monocyte chemotactic protein 1 (MCP-1AQ3 ), and TNF-!are increased (F2 Fig. 2). The levels of the others cytokines and
chemokines are summarized in supplemental Table 1 (see Sup-
plemental Digital Content 1, http://links.lww.com/SHK/A36).
In addition, macrophage inflammatory protein 1! is upregu-
lated upon intestinal I/R injury in the ileum, whereas the other
mediators such as IL-2, IL-4, IL-12, IL-15, IL-17, IFN-+, andregulated upon activation, normal T cell expressed and secreted
were below the detection levels.
In the serum, a similar profile of that in lung homogenates is
observed, where the levels of IL-1!, IL-6, G-CSF, MCP-1, and
KC were increased in the intestinal I/R group as compared with
sham group ( F3Fig. 3 and Table, Supplemental Digital Content 2,
http://links.lww.com/SHK/A37). It is important to note that
there was no significant increase of circulating TNF-! upon
intestinal I/R injury.
In the lung, only IL-1!, IL-6, G-CSF, MCP-1, and KC were
upregulated upon intestinal I/R injury compared with the sham-
operated group, TNF-! concentration was increased less than a
factor of 2 upon intestinal I/R injury ( F4Fig. 4 and Table, Sup-
plemental Digital Content 3, http://links.lww.com/SHK/A38).
Therefore, upon intestinal I/R, the TNF-! levels in intestine
were slightly increased, whereas no increase was observed
in serum. However other cytokines and chemokines must be
important in this model.
FIG. 4. Cytokines levels in lung from mice upon intestinal I/R. Groups of mice were submitted to 45 min of intestinal ischemia followed by 4 h ofreperfusion. Sham-operated mice were used as controls. Basal values were obtained from naive mice. Cytokine levels were determined in lung homogenateusing multiplex analysis: IL-1! (A), IL-6 (B), KC (C), TNF-! (D). Data represent mean T SEM from 2 experiments from 5 to 7 animals. *P G 0.05.
AQ4
SHOCK MONTH 2010 TNF IN INTESTINAL ISCHEMIC INJURY 5

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
Studies of TNF-! involvement in I/R-induced lung andgut inflammation
Although TNF-! levels were not detectable in serum and
lung, we decided to investigate the potential role of this cyto-
kine in the inflammatory process induced by intestinal I/R
injury by the use of neutralizing TNF-! mAb. The admin-
istration of a neutralizing TNF-! mAb 30 min before intes-
tinal ischemia however did not reduce neutrophil recruitment
as assessed by MPO activity in lung and intestinal tissues
( F5Fig. 5, A and B). Moreover, the serum levels of cytokines
and chemokines in mice subjected to intestinal I/R were
not influenced by the administration of the TNF-! mAb
(Fig. 5, CYH, and Table, Supplemental Digital Content 4,
http://links.lww.com/SHK/A39).
To exclude a role of TNF-! in lung neutrophilic inflam-
mation in this model of intestinal I/R, we further used TNF
geneYdeficient mice. However, the MPO activity in the lung
from wild-type and TNF-deficient mice did not differ when
FIG. 5. Effects of the neutralization of soluble TNF-! in lung (A) and intestine (B) MPO activity and in the cytokine profile of mice subjected tointestinal I/R. Mice were treated with 20 mg/kg of antiYTNF-! mAb. Thirty minutes after the treatment, mice were subjected to 45 min of intestinal ischemiafollowed by 4 h of reperfusion. Mice treated with PBS were used as controls. Cytokine levels were determined in serum using multiplex analysis: IL-1! (C), IL-6(D), KC (E), TNF-! (F), G-CSF (G), and MCP-1 (H). Data represent mean T SEM from 2 experiments from 5 to 7 animals. *P G 0.05.
6 SHOCK VOL. 00, NO. 00 SOARES ET AL.

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
submitted to intestinal I/R (F6 Fig. 6A). Furthermore, neutro-
philic inflammation was investigated in mice deficient for
both TNF receptors 1 and 2 (TNFR1/R2 KO). In agreement
with the data from both TNF-! mAb treatment and TNF KO
experiments, MPO activity remains unchanged in the lung
tissues of mice undergoing intestinal I/R injury in the absence
of TNFR1/R2 signaling (Fig. 6B). Therefore, for this model of
intestinal I/R, we can safely conclude that lung inflammation
is TNF-! independent. We also analyzed mice at 8 h after
intestinal I/R injury and did not find any consistent reduction
of the global inflammatory response in TNF or TNFR1/R2
KO mice (data not shown).
TLRs mediate the lung inflammation induced byintestinal I/R
Intestinal ischemia causes substantial damage in the intes-
tinal wall as shown before with leak of intestinal microbes and
microbial products that activate the innate immunity through
TLR ligation. Therefore, we asked whether ablation of the
two major TLRs recognizing intestinal products, TLR2 and
TLR4, would alter the inflammatory response after intestinal
I/R injury. Indeed, pulmonary MPO activity was significantly
decreased in TLR2/4 doubleYdeficient mice when compared
with their wild-type controls (F7 Fig. 7A). We also investigated
the expression of TLR2 and TLR4 in lung tissue in both naive
and intestinal I/R mice and observed that TLR2 mRNA levels
are significantly increased in wild-type mice subjected to
intestinal I/R, whereas no significant upregulation of mRNA
of TLR4 was detected (Fig. 7, B and C), which does not ex-
clude signaling in response to intestinal I/R damage. There-
fore, the data suggest that microbial products augment at least
TLR2 expression and signal through TLR2/4 to induce the
inflammatory response.
DISCUSSION
This study in the murine model of intestinal I/R injury pro-
vides comprehensive data on a wide range of cytokines and
chemokines produced by the intestines and lung tissue and
released in the serum. Despite a pivotal role in many acute
inflammatory responses (19), we confirm our hypothesis that
TNF-! plays no role in remote inflammation induced by in-
testinal I/R injury. Neither the neutralization of soluble TNF-!nor the absence of TNF-! or its receptors prevented lung in-
flammation upon intestinal I/R injury, supporting the view that
TNF-! is not essential for the development of neutrophilic
inflammation nor did it alter the pattern of other proinflamma-
tory cytokines and chemokines in this model of intestinal I/R.
FIG. 6. Lung MPO activity in TNF KO and TNFR1/R2 KO micesubjected to intestinal I/R. Groups of TNF KO (A) and TNFR1/R2 KO(B) mice and wild-type mice were submitted to 45 min of intestinal ischemiafollowed by 4 h of reperfusion. Sham-operated mice were used as controls.Data represent mean T SEM from 2 experiments from 5 to 7 animals.*P G 0.05.
FIG. 7. Lung MPO activity in TLR2/4 KO mice subjected to intestinalI/R and expression of TLR2 and TLR4 mRNA in lung homogenates.Groups of mice C57Bl/6 or TLR2/4 were submitted to 45 min of intestinalischemia followed by 4 h of reperfusion. Basal values were obtained fromnaive mice. Expression of TLR2 (B) and TLR4 (C) mRNA were determinedby real-time PCR. *P G 0.05 relative to naive group.
AQ5
SHOCK MONTH 2010 TNF IN INTESTINAL ISCHEMIC INJURY 7

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
However, we report that TLRs, notably TLR2/4, play a critical
role for the induction of neutrophil recruitment into the lung as
observed by increased lung MPO activity.
The intestinal I/R injury procedure as performed in this
study is a simple and reproducible model of local and sys-
temic inflammation with significant repercussions to lung and
gut homeostasis (17, 20, 21). We used 45 min of intestinal
ischemia followed by 4 h or 24 h of intestinal reperfusion, a
condition considered as a mild model of injury with an in-
flammatory response characterized by neutrophil recruitment
to gut and lung tissues that peaks at 4 h after with no sig-
nificant mortality.
Although experimental data have been reported on the role
of individual cytokines in lung injury after intestinal I/R and
other forms of injury, no report so far exists on a broader panel
of cytokines/chemokines. Here, we investigated a large number
of cytokines and chemokines in ileum, lung, and serum of mice
subjected to intestinal I/R injury using a multiplex platform
(22). Such a comprehensive description of the changes of
cytokines/chemokines in three different compartments might
provide a better understanding of the role of the systemic
inflammation induced by intestinal I/R injury on the multiple
organ disease. We demonstrate here increased levels of IL-1!,IL-6, G-CSF, KC, MCP-1, macrophage inflammatory protein
1!, and TNF-! in the intestines. These factors are clearly
relevant to mediate acute inflammation and intestinal tissue
injury. Therefore, we extend previous data showing upregula-
tion of platelet-activating factorAQ6 (23), IL-1" (24), IL-6, and
TNF-! (25) in the intestine. Furthermore, our data support the
notion that cytokines/chemokines produced upon intestinal
injury drive remote inflammation in the lung. IL-1!, IL-6, G-CSF, and KC were upregulated in lung and in serum after
intestinal I/R injury. The gut, the initial site of ischemic injury
with highest levels of cytokines/chemokines, may spill its in-
flammatory mediators causing remote inflammation as sug-
gested for hemorrhagic shock by Deitch (26) and previously for
intestinal I/R injury (17, 27).
Of great interest was the observation that KC level in lung
homogenates from intestinal I/R mice were greater than
400 pg/mL upon intestinal I/R injury as compared with levels
of 100 pg/mL in the sham-operated group. This elevated level
of KC in lung tissue is even higher than that found in serum,
suggesting that this chemokine is produced locally in the lung
after intestinal I/R. In fact, Wiedermann et al. (28) reportedthat IL-8 (the human equivalent for KC) is increased in the
bronchoalveolar lavage of patients with ARDS, and that this
chemokine may be considered the main factor modulating the
influx of neutrophils in the early stage of the disease.
Unexpectedly, there was no change in TNF-! levels in both
serum and lung homogenates. Serum samples were taken also
1 h after the beginning of reperfusion to exclude a possible
early peak of TNF-!, but no increased serum levels were
observed (8.0 T 1.5 for naive mice, 12.9 T 1.3 for sham mice,
and 10.2 T 2.2 for mice upon intestinal I/R). Therefore, TNF-!seems to be unrelated to induction of lung inflammation in
our model of intestinal I/R. Interestingly, Davidson et al. (29)
did not implicate TNF-! in endothelial cell injury caused
by hemorrhagic shock. Several studies suggested that TNF-!
along with other cytokines may be important mediators of
acute lung injury (30, 31). However, elevated TNF-! levels in
serum and bronchoalveolar lavage fluid have not been con-
sistently correlated with clinical outcomes in patients at risk
of or with established acute lung injury (7).
Another important point in the discussion of the involve-
ment of TNF-! in intestinal I/R is the different roles that
this cytokine may play in the inflammatory process. Chen
et al. (11) showed that the activation of TLR4 is capable of
limiting the murine intestinal injury upon intestinal I/R, and
that this protective effect is mediated by TNF-!. In addition,
Teoh et al. (32) showed that a low dose of TNF-! admin-
istered before liver ischemia may have a protective effect.
The discrepant results from our data may be explained by a
different protocol used for intestinal I/R in the former study.
Noteworthy, these studies did not preclude a pivotal role of
TNF-! in acute inflammation, but suggest that TNF-! could
have a more complex regulatory role. Our experimental data
demonstrated that TNF-! does not participate in the modu-
lation of some of the hallmarks of systemic inflammation
caused by intestinal I/R injury. First, neutralization of TNF-!with antibody treatment did not diminish neutrophilic inflam-
mation and cytokine or chemokine levels in lung and gut upon
intestinal I/R injury. Second, the lack of TNF-! as inves-
tigated in TNF KO AQ7and in TNFR1/R2 double KO mice sub-
mitted to intestinal I/R injury did not abrogate inflammation.
These data represent strong evidence that TNF-! is not asso-
ciated with acute inflammation of the lung.
In contrast to the lack of involvement of TNF-!, TLR2 and
TLR4 showed prominent roles in neutrophil recruitment after
intestinal I/R injury. In fact, when we analyze lung homoge-
nates, there was a significant reduction in MPO activity of
TLR2/4 KO mice submitted to intestinal I/R injury when
compared with their C57Bl/6 controls. Furthermore, quantifi-
cation of mRNA by real-time PCR shows a small but sig-
nificant increase of TLR2 mRNA expression in lung tissue
upon intestinal I/R injury. These data demonstrate that TLR2
and TLR4 are important for neutrophil recruitment and
suggest a possible bacterial translocation as the source of in-
flammation induced by intestinal I/R. However, in a rat model
of intestinal I/R injury inflammation, inflammatory mediators
preceded increased serum endotoxin levels (33). Furthermore,
induction of oral tolerance to LPS or treatment with poly-
myxin B did not alter the inflammation and pulmonary edema
induced by intestinal I/R injury (33). Clearly, more work is
needed to understand the ligand-activating TLR2/4, which
play a critical role in pathophysiology (34). Toll-like receptors
may also recognize endogenous ligands such as heat shock
proteins and extracellular matrix components that could act as
Bdanger signals[ (14, 34, 35).
In conclusion, intestinal I/R injury induced the upregulation
of IL-6, G-CSF, and KC expression in local and remote organs,
which seems to play a role in pulmonary neutrophil recruitment
and inflammation, whereas TNF-! is not important for this
response. Importantly, TLR2/4 signaling caused by microbial
products accessing the body upon intestinal injury is critical to
initiate the inflammatory response and remote inflammation in
the lung.
8 SHOCK VOL. 00, NO. 00 SOARES ET AL.

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
REFERENCES1. Haglund U, Bergqvist D: Intestinal ischemiaVthe basics. Langenbecks Arch
Surg 384:233Y238, 1999.2. Moore FA: The role of the gastrointestinal tract in postinjury multiple organ
failure. Am J Surg 178:449Y453, 1999.3. Thomas S, Karnik S, Balasubramanian KA: Surgical manipulation of the small
intestine and its effect on the lung. J Surg Res 106:145Y156, 2002.4. Bernard GR, Artigas A, Brigham KL, et al.: The American-European Con-
sensus Conference on ARDS. Definitions, mechanisms, relevant outcomes,
and clinical trial coordination. Am J Respir Crit Care Med 149:818Y824, 1994.5. Brun-Buisson C, Minelli C, Bertolini G, et al.: Epidemiology and outcome of
acute lung injury in European intensive care units. Results from the ALIVE
study. Intensive Care Med 30:51Y61, 2004.6. Bazzoni F, Beutler B: The tumor necrosis factor ligand and receptor families.
N Engl J Med 334:1717Y1725, 1996.7. Shimizu M, Hasegawa N, Nishimura T, Endo Y, Shiraishi Y, Yamasawa W,
Koh H, Tasaka S, Shimada H, Nakano Y, et al.: Effects of TNF-alpha-
converting enzyme inhibition on acute lung injury induced by endotoxin in the
rat. Shock 32:535Y540, 2009.8. Ortiz LA, Lasky J, Lungarella G, et al.: Upregulation of the p75 but not the
p55 TNF-alpha receptor mRNA after silica and bleomycin exposure and
protection from lung injury in double receptor knockout mice. Am J RespirCell Mol Biol 20:825Y833, 1999.
9. Kim JY, Park JS, Strassheim D, et al.: HMGB1 contributes to the development
of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol288:L958YL965, 2005.
10. Grotz MR, Ding J, Guo W, et al.: Comparison of plasma cytokine levels in rats
subjected to superior mesenteric artery occlusion or hemorrhagic shock. Shock3:362Y368, 1995.
11. Chen LW, Chang WJ, Chen PH, et al.: TLR ligand decreases mesenteric
ischemia and reperfusion injuryYinduced gut damage through TNF-alpha
signaling. Shock 30:563Y570, 2008.12. Beg AA: Endogenous ligands of Toll-like receptors: implications for regu-
lating inflammatory and immune responses. Trends Immunol 23:509Y512,2002.
13. O’Neill LA: TLRs play good cop, bad cop in the lung. Nat Med 11:1161Y1162,2005.
14. Ohashi K, Burkart V, Flohe S, et al.: Cutting edge: heat shock protein 60 is a
putative endogenous ligand of the toll-like receptor-4 complex. J Immunol164:558Y561, 2000.
15. Fan J, Kapus A, Marsden PA, et al.: Regulation of Toll-like receptor 4
expression in the lung following hemorrhagic shock and lipopolysaccharide.
J Immunol 168:5252Y5259, 2002.16. Takeuchi O, Hoshino K, Kawai T, et al.: Differential roles of TLR2 and TLR4
in recognition of gram-negative and gram-positive bacterial cell wall com-
ponents. Immunity 11:443Y451, 1999.17. Cavriani G, Domingos HV, Soares AL, et al.: Lymphatic system as a path
underlying the spread of lung and gut injury after intestinal ischemia/
reperfusion in rats. Shock 23:330Y336, 2005.18. Goldblum SE, Wu KM, Jay M: Lung myeloperoxidase as a measure of
pulmonary leukostasis in rabbits. J Appl Physiol 59:1978Y1985, 1985.
19. Esposito EME, Muia C, Meli R, Sessa E, Cuzzocrea S: Splanchnic ischemia
and reperfusion injury is reduced by genetic or pharmacological inhibition of
TNF-alpha. J Leukoc Biol 81:1032Y1043, 2007.
20. Kozar RA, Holcomb JB, Hassoun HT, et al.: Superior mesenteric artery occlu-
sion models shock-induced gut ischemia-reperfusion. J Surg Res 116:145Y150,2004.
21. He X, Han B, Mura M, et al.: Anti-human tissue factor antibody ameliorated
intestinal ischemia reperfusionYinduced acute lung injury in human tissue
factor knock-in mice. PLoS ONE 3:e1527, 2008.
22. Khan SS, Smith MS, Reda D, et al.: Multiplex bead array assays for detec-
tion of soluble cytokines: comparisons of sensitivity and quantitative values
among kits from multiple manufacturers. Cytometry B Clin Cytom 61:35Y39,2004.
23. Kim FJ, Moore EE, Moore FA, et al.: Reperfused gut elaborates PAF that
chemoattracts and primes neutrophils. J Surg Res 58:636Y640, 1995.24. Rocourt DV, Mehta VB, Besner GE: Heparin-binding EGF-like growth
factor decreases inflammatory cytokine expression after intestinal ischemia/
reperfusion injury. J Surg Res 139:269Y273, 2007.25. Grotz MR, Deitch EA, Ding J, et al.: Intestinal cytokine response after gut
ischemia: role of gut barrier failure. Ann Surg 229:478Y486, 1999.26. Deitch EA: Role of the gut lymphatic system in multiple organ failure. Curr
Opin Crit Care 7:92Y98, 2001.27. Cavriani G, Domingos HV, Oliveira-Filho RM, et al.: Lymphatic thoracic duct
ligation modulates the serum levels of IL-1beta and IL-10 after intestinal
ischemia/reperfusion in rats with the involvement of tumor necrosis factor
alpha and nitric oxide. Shock 27:209Y213, 2007.28. Wiedermann FJ, Mayr AJ, Kaneider NC, et al.: Alveolar granulocyte colony-
stimulating factor and alpha-chemokines in relation to serum levels, pulmo-
nary neutrophilia, and severity of lung injury in ARDS. Chest 125:212Y219,2004.
29. Davidson MT, Deitch EA, Lu Q, et al.: A study of the biologic activity of
trauma-hemorrhagic shock mesenteric lymph over time and the relative role of
cytokines. Surgery 136:32Y41, 2004.30. Souza DG, Ferreira FL, Fagundes CT, et al.: Effects of PKF242-484 and
PKF241-466, novel dual inhibitors of TNF-alpha converting enzyme and
matrix metalloproteinases, in a model of intestinal reperfusion injury in mice.
Eur J Pharmacol 571:72Y80, 2007.31. Chen LW, Egan L, Li ZW, et al.: The two faces of IKK and NF-kappaB
inhibition: prevention of systemic inflammation but increased local injury
following intestinal ischemia-reperfusion. Nat Med 9:575Y581, 2003.
32. Teoh N, Field J, Sutton J, et al.: Dual role of tumor necrosis factor-alpha in
hepatic ischemia-reperfusion injury: studies in tumor necrosis factor-alpha
gene knockout mice. Hepatology 39:412Y421, 2004.
33. Turnage RH, Guice KS, Oldham KT: Endotoxemia and remote organ injury
following intestinal reperfusion. J Surg Res 56:571Y578, 1994.
34. Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM:
Toll-like receptors in ischemia-reperfusion injury. Shock 32:4Y16, 2009.
35. Chen C, Wang Y, Zhang Z, Wang C, Peng M: Toll-like receptor 4 regulates
heme oxygenase-1 expression after hemorrhagic shock induced acute lung
injury in mice: requirement of P38 mitogen-activated protein kinase activation.
Shock 31:486Y492, 2009.
SHOCK MONTH 2010 TNF IN INTESTINAL ISCHEMIC INJURY 9

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
AUTHOR QUERIES
AUTHOR PLEASE ANSWER ALL QUERIES
AQ1 = WTOW was changed to WWITHW in the article title.
AQ2 = Please provide the expanded form of KC at first mention.
AQ3 = Please check if the expanded forms of MCP-1, MIP-1-alpha, and RANTES are correct.
AQ4 = Please provide significance of part E and F of figure 4 which were not mentioned in thecaption.
AQ5 = Please provide significance of part A of figure 7 which was not mentioned in the caption.
AQ6 = Please check if the expanded form of PAF is correct.
AQ7 = WTNF-W was changed to WTNF KO.W Please check if appropriate.
END OF AUTHOR QUERIES

Copyright @ 2010 by the Shock Society. Unauthorized reproduction of this article is prohibited.
Author(s) Name
Title of Article
*Article # *Publication Mo/Yr ______
*Fields may be left blank if order is placed before article number and publication
month are assigned.
Name
Address Dept/Rm
City State Zip Country
Telephone
Author Reprints
Shock Order
Payment
Ship to
Quantity of Reprints ____ $
Covers (Optional) $
Shipping Cost $
Reprint Color Cost $
Tax $
Total $
REPRINTS ORDERED & PURCHASED UNDER THE AUTHOR REPRINTS PROGRAM MAY NOT BE USED FOR COMMERCIAL PURPOSES
Reprint Pricing 50 copies = $336.00
100 copies = $420.00
200 copies = $494.00
300 copies = $571.00
400 copies = $655.00
500 copies = $732.00
Plain Covers $108.00 for first 100 copies
$18.00 each add’l 100 copies
Reprint Color ($70.00/100 reprints)
ShippingWithin the U.S. - $15.00 up to the first 100 copies and $15.00 for each additional 100 copies
Outside the U.S. - $30.00 up to the first 100 copies and $30.00 for each additional 100 copies
Tax
U.S. and Canadian residents add the appropriate tax or submit a tax exempt form.
• MC • VISA • Discover • American Express
Account # / / Exp. Date
Name
Address Dept/Rm
City State Zip Country
Telephone
Signature
Use this form to order reprints. Publication fees, including color separation charges and page charges will be billed separately, if applicable.
Payment must be received before reprints can be shipped. Payment is accepted in the form of a check or credit card; purchase orders are accepted for orders billed to a U.S. address.
Prices are subject to change without notice.
For quantities over 500 copies contact our Healthcare Dept. For orders shipping in the US and Canada: call 410-528-4396, fax your order to 410-528-4264 or email it to [email protected]. Outside the US: dial 44 1829 772756, fax your order to 44 1829 770330 or email it to [email protected].
MAIL your order to: Lippincott Williams & Wilkins Author Reprints Dept. 351 W. Camden St. Baltimore, MD 21201
FAX: 410.528.4434
For questions regarding reprints or publication fees, E-MAIL: [email protected]
OR PHONE: 1.866.903.6951
For Rapid Ordering go to: www.lww.com/periodicals/author-reprints
For Rapid Ordering go to: www.lww.com/periodicals/author-reprints




















