
Tumor lysis syndrome, case report and review of the literature
6
Annals of Oncology 7: 631-636, 1996. © 1996 Kluwer Academe Publishers. Printed in the Netherlands. Clinical case Tumour lysis syndrome, case report and review of the literature P. C. Lorigan, 1 P. L. Woodings, 1 G. R. Morgenstern 2 & J. H. Scarffe 3 1 YCRC Department of Clinical Oncology, Weston Park Hospital NHS Trust, Sheffield; 2 Department of Haematology, 3 CRC Department of Medical Oncology, Christie Hospital NHS Trust, Withington, Manchester, UK Key words: management, renal failure, tumour lysis syndrome Introduction Tumour lysis syndrome is characterised by the devel- opment of hyperuricaemia, hyperkalaemia, anaemia, hyperphosphataemia and hypocalcaemia as a result of the destruction of a large number of rapidly prolif- erating neoplastic cells. Frequently, acute renal failure occurs. The syndrome has most often been associated with rapidly dividing myeloproliferative and lympho- proliferative disorders, classically Burkitts lymphoma and acute lymphoblastic leukaemia, but has also been documented in chronic leukaemia, breast carcinoma, germ cell rumours, neuroendocrine rumours and small cell lung carcinoma [1-5]. It is usually associated with the initiation of chemotherapy but may occur with radiotherapy, surgery, endocrine therapy, glucocorti- coids, interferon, hyperthermia or spontaneously [6- 13]. With the introduction of increasingly effective drugs and high dose chemotherapy regimes there is potentially increased risk of the development of tu- mour lysis syndrome because of the rapid cell break- down. For this reason, it is also being seen in a wider range of malignant diseases [14,15]. Tumour lysis leads to the release into the systemic circulation of large quantities of potassium from the cytoplasm, urate from purine degradation and phosphate from nucleopro- teins. Clearance of the products of tumour lysis de- pends on renal excretion, hepatic metabolism and phagocytosis by the reticulo-endothelial system. Renal clearance is the primary mechanism for the excretion of uric acid, potassium and phosphate and the metabolic derangements of tumour lysis will be exacerbated by the development of renal failure. Case report A 16-year-old male was transferred to this hospital having been admitted to his local hospital one day pre- viously. He had been unwell for one week with lethargy, bruising, dry cough and swelling of his neck. A diag- nosis of acute leukaemia was made on a blood film. He was started on broad spectrum antibiotics for a pyrexia. On transfer he was pyrexial, had bulky lymph node en- largement in all groups, enlarged tonsils, mild hepato- splenomegaly, petechial haemorrhages on both legs and scattered haemorrhages in the optic fundi. His testes appeared normal and neurological examination revealed no abnormality. His full blood count on admission revealed a haemo- globin (Hb) of 8.9 g/dl, white cell count (WBC) 98 x 10 9 /l and platelet count of 44 x 10 9 /l. The prothrombin time (FT) was prolonged at 24.5 seconds (normal 13-17 sec) but the partial thromboplastin time with kaolin (PTTK), thrombin time, fibrinogen and D-dimer were all normal. Bone marrow aspirate showed a hypercellular marrow completely replaced by lympho- blasts expressing T-cell markers. Cytogenetic studies revealed a t(ll;14)(pl3:qll) translocation. A diagnosis of T-cell ALL, LI, was made. His serum urea, creati- nine and uric acid were normal on admission, but his lactate dehydrogenase (LDH) was greatly elevated at 6275 iu (normal <500). A chest X-ray showed wide- spread adenopathy and a thymic mass. He was com- menced on intravenous fluids and allopurinol 300 mg bd orally. In the 24 hours before chemotherapy, his total fluid intake was 6310 ml and his total urine output was 4120 ml. In the 24 hours after chemotherapy, his total fluid intake was 5190 ml and his total urine output was 4951 ml. His blood pressure was normal. On the second hospital day, he started chemother- apy with daunorubicin 90 mg i.v., vincristine 2 mg i.v. and prednisolone 50 mg/day orally. The WBC before treatment was 73 x 10 9 /l, creatinine 0.15 mmol/1 and phosphate (PO 4 ) 1.91 mmol/1. After 12 hours, he became acutely breathless. Arterial blood gases breath- ing air were PO 2 122 mmHg, PCO 2 31 mmHg, the pH was 7.21, urea 13 mmol/1, creatinine 0.16 mmol/1, potassium 7.4 mmol/1, LDH 13,000 iu, calcium (Ca^) 1.8 mmol/1, PO 4 4.37 mmol/1 (the calcium/phosphate product was 7.9), but the uric acid was normal. Ultra- sound examination of the renal tract was normal. He by guest on February 25, 2013 http://annonc.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Tumor lysis syndrome, case report and review of the literature

Annals of Oncology 7: 631-636, 1996.© 1996 Kluwer Academe Publishers. Printed in the Netherlands.
Clinical case
Tumour lysis syndrome, case report and review of the literature
P. C. Lorigan,1 P. L. Woodings,1 G. R. Morgenstern2 & J. H. Scarffe3
1 YCRC Department of Clinical Oncology, Weston Park Hospital NHS Trust, Sheffield; 2Department of Haematology, 3CRC Department ofMedical Oncology, Christie Hospital NHS Trust, Withington, Manchester, UK
Key words: management, renal failure, tumour lysis syndrome
Introduction
Tumour lysis syndrome is characterised by the devel-opment of hyperuricaemia, hyperkalaemia, anaemia,hyperphosphataemia and hypocalcaemia as a result ofthe destruction of a large number of rapidly prolif-erating neoplastic cells. Frequently, acute renal failureoccurs. The syndrome has most often been associatedwith rapidly dividing myeloproliferative and lympho-proliferative disorders, classically Burkitts lymphomaand acute lymphoblastic leukaemia, but has also beendocumented in chronic leukaemia, breast carcinoma,germ cell rumours, neuroendocrine rumours and smallcell lung carcinoma [1-5]. It is usually associated withthe initiation of chemotherapy but may occur withradiotherapy, surgery, endocrine therapy, glucocorti-coids, interferon, hyperthermia or spontaneously [6-13]. With the introduction of increasingly effectivedrugs and high dose chemotherapy regimes there ispotentially increased risk of the development of tu-mour lysis syndrome because of the rapid cell break-down. For this reason, it is also being seen in a widerrange of malignant diseases [14,15]. Tumour lysis leadsto the release into the systemic circulation of largequantities of potassium from the cytoplasm, urate frompurine degradation and phosphate from nucleopro-teins. Clearance of the products of tumour lysis de-pends on renal excretion, hepatic metabolism andphagocytosis by the reticulo-endothelial system. Renalclearance is the primary mechanism for the excretion ofuric acid, potassium and phosphate and the metabolicderangements of tumour lysis will be exacerbated bythe development of renal failure.
Case report
A 16-year-old male was transferred to this hospitalhaving been admitted to his local hospital one day pre-viously. He had been unwell for one week with lethargy,bruising, dry cough and swelling of his neck. A diag-
nosis of acute leukaemia was made on a blood film. Hewas started on broad spectrum antibiotics for a pyrexia.On transfer he was pyrexial, had bulky lymph node en-largement in all groups, enlarged tonsils, mild hepato-splenomegaly, petechial haemorrhages on both legsand scattered haemorrhages in the optic fundi. Histestes appeared normal and neurological examinationrevealed no abnormality.
His full blood count on admission revealed a haemo-globin (Hb) of 8.9 g/dl, white cell count (WBC) 98 x109/l and platelet count of 44 x 109/l. The prothrombintime (FT) was prolonged at 24.5 seconds (normal13-17 sec) but the partial thromboplastin time withkaolin (PTTK), thrombin time, fibrinogen and D-dimerwere all normal. Bone marrow aspirate showed ahypercellular marrow completely replaced by lympho-blasts expressing T-cell markers. Cytogenetic studiesrevealed a t(ll;14)(pl3:qll) translocation. A diagnosisof T-cell ALL, LI, was made. His serum urea, creati-nine and uric acid were normal on admission, but hislactate dehydrogenase (LDH) was greatly elevated at6275 iu (normal <500). A chest X-ray showed wide-spread adenopathy and a thymic mass. He was com-menced on intravenous fluids and allopurinol 300 mgbd orally. In the 24 hours before chemotherapy, histotal fluid intake was 6310 ml and his total urine outputwas 4120 ml. In the 24 hours after chemotherapy, histotal fluid intake was 5190 ml and his total urine outputwas 4951 ml. His blood pressure was normal.
On the second hospital day, he started chemother-apy with daunorubicin 90 mg i.v., vincristine 2 mg i.v.and prednisolone 50 mg/day orally. The WBC beforetreatment was 73 x 109/l, creatinine 0.15 mmol/1 andphosphate (PO4) 1.91 mmol/1. After 12 hours, hebecame acutely breathless. Arterial blood gases breath-ing air were PO2122 mmHg, PCO2 31 mmHg, the pHwas 7.21, urea 13 mmol/1, creatinine 0.16 mmol/1,potassium 7.4 mmol/1, LDH 13,000 iu, calcium (Ca^)1.8 mmol/1, PO4 4.37 mmol/1 (the calcium/phosphateproduct was 7.9), but the uric acid was normal. Ultra-sound examination of the renal tract was normal. He
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from

632
was treated with intravenous bicarbonate, calcium glu-conate, insulin and dextrose but the potassium correct-ed only slowly.
On the fourth hospital day, he was more unwell witha reduced level of consciousness. He remained pyrex-ial, acidotdc and had progressive oliguria despite diuret-ics. He was markedly hypocalcaemic despite vigoroussupplementation and developed disseminated intravas-cular coagulation (DIC). Urea 44 mmol/1, creatinine0.35 mmol/1, Ca^ 0.86 mmol/1, PT 33 seconds, KCT97 seconds (36-48), thrombin time 18 seconds(n - 13), D-dimer 4 jig/ml (n < 0.5 ng/ml), Hb 5.6 g/dl,WBC 1.2 x 109/l, platelets 9 x 109/l. Cerebrospinalfluid examination was normal. He continued to de-teriorate on the fifth hospital day with oliguria, persis-tent pyrexia, hypocalcaemia and hyperphosphataemia.His serum uric acid was raised at 0.8 mmol/1.
Haemodialysis was started on the sixth day. TheLDH was 40,000 iu, Ca^ 1.3 mmol/1, PO4 6.3 mmol/1.The DIC persisted. CXR showed fluid overload. CTbrain scan showed cerebral oedema His condition wassimilar on the seventh hospital day. Dialysis was repeat-ed on the eighth day. He developed repeated gTand malseizures requiring intravenous phenytoin.
On the ninth day, his temperature was normal, hisconscious level had improved, Ca"1"1" 1.5 mmol/1, PO4
3.5 mmol/1, urea 53 mmol/1, creatinine 0.84 mmol/1,uric acid 1.04 mmol/1, amylase 630 iu. The DIC per-sisted.
On the tenth hospital day, he had a cardiorespiratoryarrest while on dialysis. Resuscitation was prompt andsuccessful, but he made no spontaneous respiratoryeffort. A CT brain scan showed multiple intracerebralhaemorrhages. He subsequently died.
At autopsy, the brain showed marked swelling withgyral flattening and narrowing of the ventricles. Nu-merous small cerebral haemorrhages were seen.Microscopy of the lungs showed marked haemorrhageand oedema in the alveolar spaces. The kidneys ap-peared somewhat enlarged, had a mottled external sur-face and occasional petechial haemorrhages. The renalcortices appeared pale and swollen. Microscopyshowed granular eosinophillic material in Bowman'sspace and features consistent with acute tubular necro-sis. Microscopy of the thymus and lymph nodesshowed scanty lymphoid tissue only, the spleen showedmarkedly congested red pulp with only very scantywhite pulp and the bone marrow was congested andhaemorrhagic with only scanty haemopoietic tissue andno obvious leukaemic infiltrate. Tumour lysis was com-plete.
Metabolic abnormalities
This case illustrates the metabolic consequences in thetumour lysis syndrome resulting from destruction ofmassive numbers of tumour cells following effectivetreatment. The processes leading to these individual
metabolic abnormalities and their effects are discussedbelow.
Uric acid
An association between hyperuricaemia and leukae-mia/lymphoma has been recognised for more than acentury [16]. The hyperuricaemia of malignant diseaseis due to increased cell turnover with breakdown ofnucleic acid purines (Figure 1).
Uric acid is 98% ionised at physiological pH andexists in the blood as its sodium salt, urate. This is whatis measured by most laboratories. As the pH drops,e.g., in the renal tubule, uric acid becomes progressivelyless ionised and in turn becomes less soluble.
Urate filtered at the glomerulus is almost completelyreabsorbed in the proximal tubules, and urinary urate isderived from net tubular secretion, usually 10% of theglomerular filtrate. The pKa of uric acid is 5.6, andeven at normal serum urate levels, the concentration ofuric acid in urine approaches its maximum solubility[17].
In humans, the breakdown of nucleic acid purines touric acid is catalysed by the liver enzyme xanthine oxi-dase [XO]. Allopurinol, a potent inhibitor of xanthineoxidase, has a half life of 39 minutes, being quickly con-verted to its active metabolite oxipurinol, also a potent[XO] inhibitor. Oxipurinol is excreted by the kidneys.Its half life ranges between 14 and 26 hours with nor-mal renal function, and clearance is further prolongedwith renal impairment or concomitant use of thiazides.The main advantage of allopurinol is that the reductionin serum urate levels is not at the expense of an in-crease in urinary urate excretion. Following the admin-istration of allopurinol to patients with rapid tumourlysis a reduction in uric acid excretion would be paral-leled by an increase in total oxipurine (hypoxanthineand xanthine) excretion.
Hypoxanthine
Xanthine Oxidase
Xanthine
Xanthine Oxidase
Uric AcidPH7.3
pH5.5Urate
Figure 1. Metabolic pathway for convention of hypoxanthine andxanthine to uric acid.
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from

633
Tubular xanthine concentration represents 110% ofthat filtered, indicating net secretion, the rate of whichis proportional to the serum level. Xanthine has a pKaof 7.4 and is less soluble in urine than uric acid. Handeet al. measured post chemotherapy purine excretions in11 patients with high grade lymphoma and found anoverall increase in total purine excretion post chemo-therapy [18]. Despite allopurinol, there was an increasein both uric acid and hypoxanthine excretion, but thisdid not exceed their solubility in urine at pH 7. Xan-thine excretion increased by 6.9 fold, and urinary xan-thine concentration exceeded its solubility at pH 7 in 6of 11 patients. In 3 of these, there was a transient in-crease in serum creatinine and fall in creatinine clear-ance, (to a mean of 20 ml/min).
Andreoli et al. studied oxipurine and uric acid excre-tion in 19 children with ALL receiving allopurinol dur-ing tumour lysis [19]. Although the total urinary excre-tion of purine metabolites was higher in those childrenwho developed renal failure, the peak concentrationswere not different, indicating that factors other thantubular precipitation of metabolites may be involved inthe pathogenesis.
Other theories have implicated the precipitation ofpurine derivatives in the vasa recta causing renal failurebut not being reflected in a raised urinary concentra-tion. Micropuncture studies have shown that over-pro-duction of uric acid may result in precipitation of uricacid crystals within both collecting ducts and the deepcortical and medullary vessels, where uric acid concen-tration and acidification is maximal, reducing furtherthe glomerular filtration rate [20].
In addition, factors other than pH are determinantsof purine precipitation and crystal formation. These in-clude ionic strength, salt concentration and otherurinary constituents involved in determining the solu-bility of uric acid and its sodium salt. Precursors of oxi-purines and uric acid include ATP, AMP and adeno-sine. These participate in the regulation of vasculartone, and rapid release may affect renal vascular tone,contributing to the development of renal failure.
A variety of approaches have been adopted in anattempt to limit purine induced renal failure. Firstly,allopurinol increases the total amount of oxypurinesexcreted by the kidney. Each of these, xanthine, hypo-xanthine and uric acid have independent solubilities inurine. Secondly, the solubility of uric acid is increasedby alkalinisation of the urine. As the pKa of xanthine is7.4, the urine pH must be greater than this to make asignificant impact on its solubility. Thirdly, high tubularflow rate may be an important mechanism in protectingagainst acute urate nephropathy [21]. Fourthly, theinduction of tumour breakdown more gradually by lessintensive chemotherapy may reduce the risk of renalfailure. However, this may compromise remission ratesin these curable conditions.
Calcium and phosphate metabolism
There is an inverse relationship between the concentra-tion of calcium and phosphate in plasma. As the phos-phate level rises so the calcium level will fall and viceversa.
The main hormones involved in their regulation areparathyroid hormone (PTH) and vitamin D. In massivetumour lysis, large amounts of phosphate are releasedfrom nucleoproteins into the circulation and renalfunction must be adequate for its excretion. Phosphateis absorbed to a maximum threshold after which excre-tion is dependent on glomerular filtration rate. Therelease of large amounts of phosphate into the circula-tion will have a number of effects.
i) The calcium/phosphate product is close to thesolubility product. Increased levels of phosphatedespite reciprocal fall in calcium may still lead tothe solubility product being exceeded and pre-cipitation of calcium phosphate in the kidneys.This leads to renal interstitial inflammation andlater to tubular atrophy [22]. Worsening renalfailure results, further reducing the amount ofphosphate excreted. Over zealous alkalinisationmay also promote calcium phosphate deposi-tion, and the pH should not be greater than 7.5in the presence of hyperphosphatemia [23,24].
ii) in addition to nephrocalcinosis at a calcium/phosphate product greater than 4.6 there is arisk of extraskeletal calcification, although this isless important in the short term,
iii) in the setting of acute renal failure, hyperphos-phataemia directly depresses the calcium mobil-ising effect of PTH on bone, even in the face ofnormal 1,25-dihydroxycholecalciferal produc-tion [25,26].
iv) acute renal failure may rapidly lead to reducedproduction of 1,25-dihydroxycholecalciferolwhich not only contributes directly to hypocal-caemia but also results in further skeletal resist-ance to PTH [27,28].
Dunlay et al. report on a patient with Burkitts lym-phoma who developed all the stigmata of tumour lysissyndrome, including renal failure [27]. Sustained hypo-calcaemia complicated by obtundation and seizureslasted for almost 2 weeks, even after the plasma phos-phate was returned to normal by dialysis and despite anappropriately elevated PTH level and Ca^ supple-ments. The hypocalcaemia was associated with amarked decrease in plasma 1,25-dihydroxycholecal-ciferol levels, and when replacement therapy was com-menced, the hypocalcaemia rapidly corrected with anappropriate normalisation of PTH levels.
Similarity exists between calcium and phosphatemetabolism in tumour lysis and in rhabdomyolysisinduced acute renal failure. Llach et al. described an as-sociation between acute renal failure, decreased 1,25-dihydroxycalciferol levels and symptomatic hypocal-caemia despite an appropriately elevated PTH, in 6
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from

634
patients with acute rhabdomyolysis [28]. There was asignificant positive correlation between the levels ofserum calcium and 1,25-dihydroxycholecalciferol and asignificant negative correlation between the levels of1,25-dihydroxycholecalciferol and plasma phosphate.
The clinical manifestations of hypocalcaemia in-clude nervous system irritability, confusional states,papilloedema, cardiomyopathy, hypotension, conges-tive cardiac failure and arrhythmias. Rapid onset ofhypocalcaemia in children undergoing treatment foracute leukaemia who develop tumour lysis syndromecauses predominantly CNS symptoms and signs.
Hyperkalaemia
Hyperkalaemia is due to the rapid release of potassiumfrom the cytoplasm of lysed cells. The rise is oftenearlier in onset than other metabolic abnormalities.The normal sodium-potassium gradient across thecell membrane is maintained by sodium/potassiumATPase, which is an energy dependent system. Chemo-therapy may upset this normal cellular control allowingpotassium to leak out of cells, and leading to an earlyrise in blood levels [22]. Hyperkalaemia is then com-pounded by an inability of the kidney to correct thisdue to worsening renal failure.
A rapid rise in potassium is associated with risk ofarrhythmia and sudden death. It should be treated with50 ml of 50% glucose and 15 units of soluble insulininfused over 1 hour, intravenous fluids and diuretics. Inthe longer term oral potassium exchange resins such ascalcium resonium should be used. Levels greater than6.5 mmol/1 should be treated as a medical emergencywith 10-30 ml calcium gluconate (10%) i.v. slowly forcardioprotection, 15 units of soluble insulin and 50 ml50% glucose i.v. followed by insulin and glucose adjust-ed according to the blood glucose. If these measuresdo not control the serum potassium, dialysis may berequired.
Renalfailure
The aetiology of renal failure in tumour lysis syndromeis multifactorial and a number of studies have attempt-ed to define prognostic factors in patients undergoingtreatment for 'high risk' conditions. Two large studies ofBurkitts lymphoma and ALL identified different riskfactors but did not fully agree in their results [7, 29].Important prognostic factors for the development ofpre-treatment renal failure include tumour bulk andhigh admission LDH and uric acid levels. Importantprognostic factors for the development of post-treat-ment renal failure include level of initial LDH, post-treatment serum phosphate, and a reduced urinary out-put before and after cytotoxic chemotherapy.
Renal lymphomatous infiltration is a commonautopsy finding, although renal impairment attributedto this is rare. Richmond et al. reported kidney infiltra-tion in 696 patients at autopsy, of whom only 5 died of
uraemia due to this cause [30]. Kidneys have been mas-sively involved by lymphoma at post mortem withoutsignificant functional impairment during life.
The mechanism of renal failure with diffuse infiltra-tion is not known, but may be partly due to tubularcompression producing intrarenal obstruction, sincehistologically the tubules are compressed, the epithe-lium is flattened but the tubular basement membrane isintact.
In childhood leukaemia, bilateral enlarged kidneysdue to renal infiltrates are found in 3%-5% of cases(and are often confused with polycystic kidneys), buturaemia due primarily to renal infiltration has beenreported in only 1% of patients with acute leukaemia.The kidney may indeed act as a reservoir for leukaemiccells even when the patient is in clinical remission andin an autopsy series of patients who died in 'remission'from causes other than leukaemia, 6 of 10 had micro-scopic evidence of leukaemic involvement of the kid-neys.
Many other factors may affect renal function inpatients with leukaemia or lymphoma. These includeinfection, nephrotoxic antibiotics and antitumouragents, DIC, paraproteinaemia and hydronephrosisdue to lymphadenopathy.
Discussion
The patient reported here had ALL, the treatment ofwhich is attended by significant risk of TLS. Clinicallyand radiologically there was evidence of bulky disease.He had an elevated LDH on admission, which is a poorprognostic sign. His pre-treatment and immediate post-treatment fluid balance showed no signs of oliguria orfluid retention. His blood pressure was normal at allstages. Within 12 hours of treatment, he had developeda marked metabolic acidosis and renal failure. The timeto onset of renal failure suggests the cause was tumourlysis. His phosphate had risen significantly from 1.9mmol/1 before treatment to 4.4 mmol/1 within 12 hoursof treatment. The plasma urate was not measured until3 days post treatment, but was elevated at that stage. Itis interesting to note that his WBC had started to falland the creatinine had started to rise before treatmentbegan, perhaps indicating spontaneous tumour lysis.The renal failure was almost certainly aggravated byunderlying sepsis, DIC and antibiotic therapy.
He developed severe hypocalcaemia within 2 days ofstarting treatment, with marked hyperphosphataemia.His albumin and magnesium were normal at all times.The hypocalcaemia corrected only partially with largeamounts of Ca** supplements, but there was a reduc-tion in PO4 and partial correction in Ca*"1" with theinitiation of dialysis (Figure 2). In retrospect it wouldhave been wise to give activated vitamin D (1,25-di-hydroxycholecalciferol) at the same time. Although theCa x PO4 product exceeded 4.6 for 7 days, duringwhich time large amount of Ca4"1" supplement were
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from
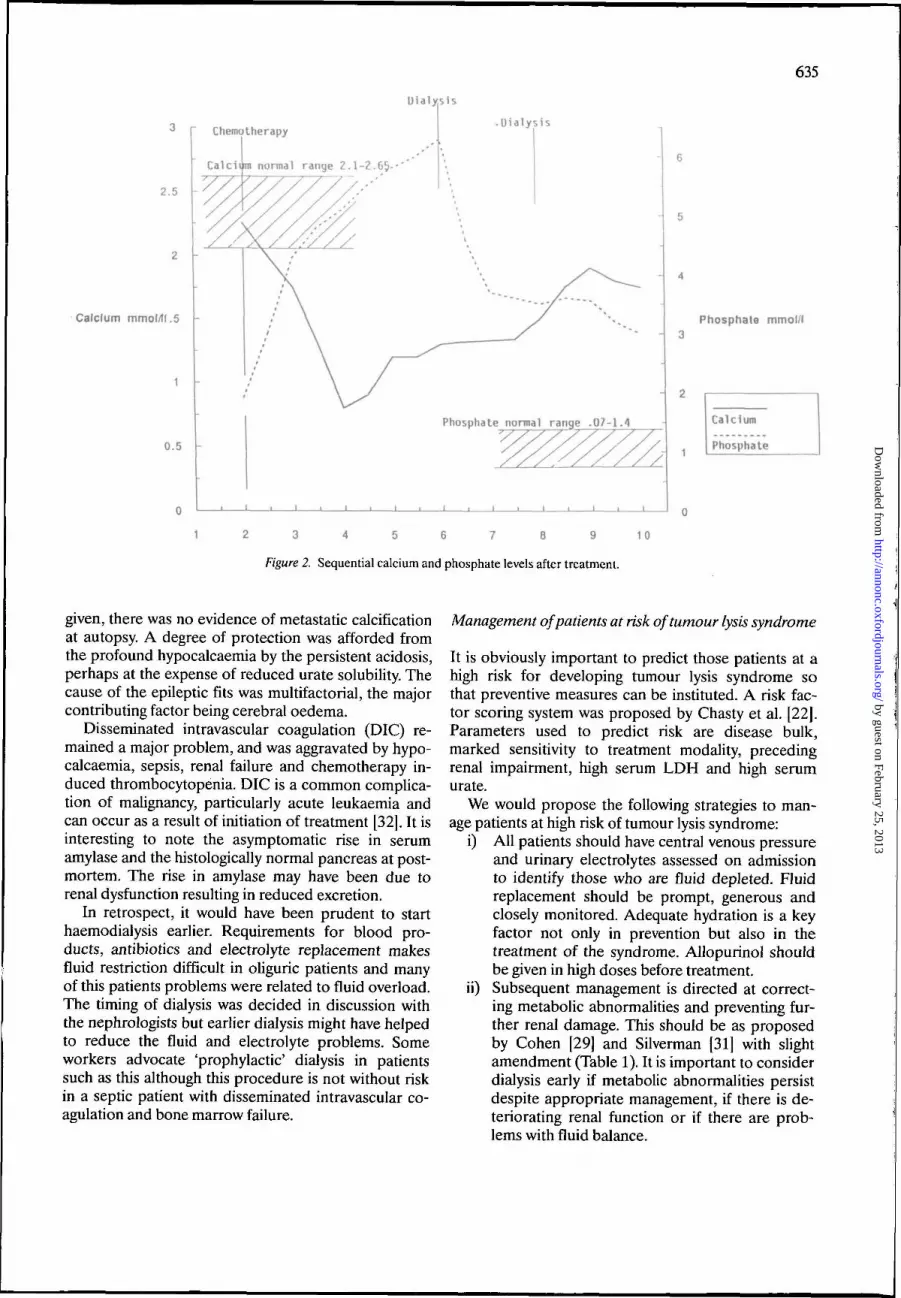
635
Dialysis
2.5
Calcium mmol/tl.5
0.5
Chemotherapy
Calcium normal range Z. 1-2.65-
.Dialysis
Phosphate normal range .07-1.1
i i i i
Phosphate mmol/l
Calcium
Phosphate
1 2 3 4 5 6 7 8 9 10
Figure 2. Sequential calcium and phosphate levels after treatment.
given, there was no evidence of metastatic calcificationat autopsy. A degree of protection was afforded fromthe profound hypocalcaemia by the persistent acidosis,perhaps at the expense of reduced urate solubility. Thecause of the epileptic fits was multifactorial, the majorcontributing factor being cerebral oedema.
Disseminated intravascular coagulation (DIC) re-mained a major problem, and was aggravated by hypo-calcaemia, sepsis, renal failure and chemotherapy in-duced thrombocytopenia. DIC is a common complica-tion of malignancy, particularly acute leukaemia andcan occur as a result of initiation of treatment [32]. It isinteresting to note the asymptomatic rise in serumamylase and the histologically normal pancreas at post-mortem. The rise in amylase may have been due torenal dysfunction resulting in reduced excretion.
In retrospect, it would have been prudent to starthaemodialysis earlier. Requirements for blood pro-ducts, antibiotics and electrolyte replacement makesfluid restriction difficult in oliguric patients and manyof this patients problems were related to fluid overload.The timing of dialysis was decided in discussion withthe nephrologists but earlier dialysis might have helpedto reduce the fluid and electrolyte problems. Someworkers advocate 'prophylactic' dialysis in patientssuch as this although this procedure is not without riskin a septic patient with disseminated intravascular co-agulation and bone marrow failure.
Management of patients at risk of tumour lysis syndrome
It is obviously important to predict those patients at ahigh risk for developing tumour lysis syndrome sothat preventive measures can be instituted. A risk fac-tor scoring system was proposed by Chasty et al. [22].Parameters used to predict risk are disease bulk,marked sensitivity to treatment modality, precedingrenal impairment, high serum LDH and high serumurate.
We would propose the following strategies to man-age patients at high risk of tumour lysis syndrome:
i) All patients should have central venous pressureand urinary electrolytes assessed on admissionto identify those who are fluid depleted. Fluidreplacement should be prompt, generous andclosely monitored. Adequate hydration is a keyfactor not only in prevention but also in thetreatment of the syndrome. Allopurinol shouldbe given in high doses before treatment,
ii) Subsequent management is directed at correct-ing metabolic abnormalities and preventing fur-ther renal damage. This should be as proposedby Cohen [29] and Silverman [31] with slightamendment (Table 1). It is important to considerdialysis early if metabolic abnormalities persistdespite appropriate management, if there is de-teriorating renal function or if there are prob-lems with fluid balance.
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from

636
Table 1. Management of patients at risk of tumour lysis syndrome.
Where no metabolic aberration exists
a) Allopurinol 500 mg/m2/day, reduce to 200 mg/mVday after3 days of chemotherapy.
b) Hydration 3 litre mVday, 1 litre N saline, 2 litre 5% dextrose.c) Chemotherapy initiated within 24—48 hours of admission.d) Monitor urine output, blood pressure and CVP at least 4
hourly.e) Consider prophylactic bicarbonate which can be discontinued
if the serum urate does not rise.f) Monitor electrolytes, urea, creatinine, uric acid, Ca""" and PO4
every 12-24 hours initially.
Where a metabolic aberration exists
a) Allopurinol as above- Reduce dose in renal failure.- Reduce dose if hyperuricaemia controlled.
b) Hydration as above, add a non thiazide diuretic as required.c) Urinary alkalinisation using isotonic sodium bicarbonate
(1.4% w/v) adjust rate to maintain urinary pH > 7. Discon-tinue when serum uric acid normal.
d) Treat hypocalcaemia if symptomatic or ECG abnormality.Use intravenous Ca^ and activated vitamin D.
e) Treat hyperkalaemia appropriately.f) Inform nephrologist at an early stage.
Criteria for dialysis
a) Persistent hyperkalaemia despite conventional treatment.b) Rapidly rising serum phosphate or persistent hyperphos-
phataemia.c) Symptomatic hypocalcaemia despite treatment.d) Fluid overload.e) Hyperuricaemia.
References
1. List AF, Kummett TD, Adams JD, Chan HG. Tumor lysis syn-drome complicating treatment of chronic lymphocytic leukaemiawith fludarabine phosphate. Am J Med 1990; 89(3): 388-90.
2. Stark ME, Dyer MC, Coonley CJ. Fatal tumour lysis syndromewith metastatic breast carcinoma. Cancer 1987; 60: 762-4.
3. Barton, JC. Tumour lysis syndrome in non-haemopoietic neo-plasms. Cancer 1989; 64: 738-40.
4. Hussein AM, Feun LG. Tumour lysis syndrome after inductiontherapy in small-cell lung carcinoma. Am J Clin Oncol 1990;13(1): 10-3.
5. Hain RDW, Rayner L, Weitzman S, Lorenzano A. Acute tumourlysis syndrome complicating treatment of stage IV neuroblas-toma in infants under six months old. Med Paediatr Oncol 1994;23(2): 136-9.
6. Lobe TE, Karkeva MD, Custer MD et al. Fatal refractory hyper-kalaemia due to tumor lysis during primary resection for hepato-blastoma. J Paediatr Surg 1990; 25(2J): 249-50.
7. Stapleton FB, Strother DR, Roy S et al. Acute renal failure at theonset of therapy for advanced stage Burkitts lymphoma and Bcell acute lymphoblastic lymphoma. Paediatrics 1988; 82(6):863-9.
8. Sparano J, Ramirez M, Wiernik PH. Increasing recognition ofcorticosteroid induced tumour lysis syndrome in non-Hodgkin'slymphoma. Cancer 1990,65: 1072-3.
9. Cech P, Block JB, Cone LA, Stone R Tumour lysis syndromeafter tamoxifen flare. NEJM 1986; 315 (4): 263.
10. Fer MF, Bottino EC, Sherwin SA et al. Atypical tumour lysissyndrome in a patient with T-cell lymphoma treated with recom-binant leucocyte interferon. Am J Med 1984; 77: 953-6.
11. Fleming DR, Henslee-Downey PJ, Coffey C. Radiation inducedacute tumour lysis syndrome in the bone marrow transplant set-
ting. Bone Marrow Transplant 1991; 8(3): 235-6.12. Suer RP, Fisher WB, Pearson RW, Triplett DA. Burkitts lym-
phoma: Tumour lysis following malignant hyperthermia. CancerTreat Rep 1980; 64(2-3): 327-30.
13. Tiley C, Grimwade D, Findley M et al. Tumour Lysis followinghydrocortisone prior to a blood product transfusion in T-cellacute lymphoblastic leukaemia Leuk Lymphoma 1992; 81(2):143-6.
14. Montalban C, Liano F, Aguilera A. Tumour Lysis syndromeafter treatment of chronic lymphocytic leukaemia with fludara-bine. Postgrad Med J 1994; 70:651-2.
15. Razis E, Arlin Z, Ahmed T et al. Incidence and treatment ofrumour lysis syndrome in patients with acute leukaemia. ActaHaematol 1994; 91 (4> 171-4.
16. Salkowski F. Beitrage zur Kenntnis der Leuk Omid. VirchowsArch Abt A Pathol Anat 1870; 59:174.
17. Klinenberg JR, Goldfinger S, Seegmiller JE The effectiveness ofthe xanthine oxidase inhibitor allopurinol in the treatment ofgout Ann Intern Med 1965; 62; 638-47.
18. Haude KR, Hixon CV, Chabner BA. Post chemothereapy purineexcretion in lymphoma patients receiving allopurinol. CancerRes 1981; 41: 2273-9.
19. Andreoli SP, Clark JH, McGuire WA, Bergstein JM. Purineexcretions during tumour lysis in children with acute lympho-cytic leukaemia receiving allopurinol: Relationship to acute renalfailure. J Paediatr 1986; 109(2): 292-7.
20. Conger JD, Falk SA, Euggenheim SJ, Burke TJ. A micropunc-ture study of the early phase of acute urate nephropathy. J ClinInvest 1977; 58: 681-9.
21. Conger JD, Falk SA. Intrarenal dynamics in the pathogenesisand prevention of acute urate nephropathy. J Clin Invest 1977;59:786-793.
22. Chasty RC, Liu-Yin JA. Acute tumour lysis syndrome (Review).Br J Hosp Med 1993; 49(7): 488-92.
23. Kanter A, Richet E, Roland J, Chatelet F. Extreme hypelphos-phataemia causing acute anuric nephrocalcinosis in lymphosar-coma.BrMedJ1979;19; 1(6174): 1320-1.
24. Fleming DR, Doukas MA. Acute tumour lysis syndrome inhaematologic malignancies (Review). Lei'1.. Lymphoma 1992;8(4-5): 315-8.
25. Massry SG, Nua S, Garry J, Friedler RM. Role of uraemia in theskeletal resistance to the calcaemic actions of parathyroid hor-mone. Miner Electrolyte Metab 1978; 1:172-80.
26. Somerville PJ, Kaye M. Evidence that resistance to the calcaemicaction of parathyroid hormone in rat, with acute uraemia iscaused by phosphate retention. Kidney Int 1979; 16: 552-60.
27. Dunlay RW, Camp MA, Allon M et al. Calcitriol in prolongedhypocalcaemia due to the tumour lysissyndrome. Ann InternMed 1989; 110(2): 162-4.
28. Llach F, Felsenfeld AJ, Haussler MR. The pathophysiology ofaltered calcium metabolism in rhabdomyolysis - induced acuterenal failure. NEJM 1981; 305(3): 117-23.
29. Cohen LF, Balow JE, Magrath IT et al. Acute tumour lysis syn-drome. A review of 37 patients with Burkitts lymphoma. Am JMed 1980; 68: 486-91.
30. Richmond J, Sherman RS, Diamond HD, Cravor LF. Renallesions associated with malignant lymphomas. Am J Med 1962;32: 184-207.
31. Silverman P, Distelhorst CW. Metabolic emergencies in clinicaloncology. Semin Oncol 1989; 16(6): 504-15.
32. Naitoh M, Andoh K, Sadakata H et al. Prediction of dissemina-ted intravascular coagulation in patients with leukaemia. InternMed 1994; 33(3): 131-5.
Received 15 February 1996; accepted 29 February 1996.
Correspondence to:Dr. P. LoriganYCRC Department of Clinical OncologyWeston Park Hospital NHS TrustWhitham RoadSheffield S10 2SJ.UK
by guest on February 25, 2013http://annonc.oxfordjournals.org/
Dow
nloaded from




















