
Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysm in...
10
ORIGINAL ARTICLES Treatment With Simvastatin Suppresses the Development of Experimental Abdominal Aortic Aneurysms in Normal and Hypercholesterolemic Mice Eric F. Steinmetz, MD,* Celine Buckley, MD,* Murray L. Shames, MD,*† Terri L. Ennis, BS,* Sarah J. Vanvickle-Chavez, BS,* Dongli Mao, MD,* Lee A. Goeddel,* Cherady J. Hawkins,* and Robert W. Thompson, MD*†‡ Objective: To determine if treatment with hydroxymethylglutaryl- coenzyme A reductase inhibitors (statins) can influence the devel- opment of experimental abdominal aortic aneurysms (AAAs). Summary Background Data: AAAs are associated with athero- sclerosis, chronic inflammation, and matrix metalloproteinase (MMP)-mediated connective tissue destruction. Because statins ex- ert antiinflammatory activities independent of their lipid-lowering effects, these agents may help suppress aneurysmal degeneration. Methods: C57Bl/6 wild-type and hypercholesterolemic apoE-defi- cient mice underwent transient perfusion of the aorta with elastase followed by subcutaneous treatment with either 2 mg/kg simvastatin per day or vehicle. Aortic diameter (AD) was measured before and 14 days after elastase perfusion. The extent of aortic dilatation (AD) was determined with AAAs defined as AD 100%. Results: Wild-type mice treated with simvastatin exhibited a 21% reduction in AD and a 33% reduction in AAAs compared with vehicle-treated controls. Suppression of AAAs in simvastatin- treated mice was associated with preservation of medial elastin and vascular smooth muscle cells, as well as a relative reduction in aortic wall expression of MMP-9 and a relative increase in expression of TIMP-1. In hypercholesterolemic apoE-deficient mice, treatment with simvastatin was associated with a 26% reduction in AD and a 30% reduction in AAAs. Treatment with simvastatin had no effect on serum cholesterol levels in either normal or hypercholesterolemic mice. Conclusions: Treatment with simvastatin suppresses the develop- ment of experimental AAAs in both normal and hypercholester- olemic mice. The mechanisms of this effect are independent of lipid-lowering and include preservation of medial elastin and smooth muscle cells, as well as altered aortic wall expression of MMPs and their inhibitors. (Ann Surg 2005;241: 92–101) A bdominal aortic aneurysms (AAAs) are a common and potentially life-threatening disorder associated with ag- ing and atherosclerosis. 1 Despite these associations, there is considerable uncertainty regarding the precise role of athero- sclerosis in the etiology and pathophysiology of AAAs, with opinion ranging from the view that aneurysms arise as a direct consequence of advanced atheromatous disease to speculation that aneurysmal degeneration might be an inde- pendent disease process only coincidentally related to athero- sclerosis. 2–4 This debate has been fueled by the discrepancy between strong evidence linking hypercholesterolemia with atherogenesis and the lack of a compelling relationship be- tween altered lipid metabolism and AAAs. 5–7 It has become evident over the past decade that many of the cellular and molecular mechanisms involved in aneurys- mal degeneration are analogous to those involved in the clinical complications of atherosclerosis such as rupture of atheromatous plaques. 8 –10 The most important of these mech- anisms include: 1) arterial wall accumulation and activation of mononuclear inflammatory cells, including both macro- phages and lymphocytes; 2) increased local expression of proinflammatory cytokines, chemokines, and matrix-degrad- ing proteinases; 3) accelerated degradation of structurally important matrix proteins (ie, elastin and collagen); 4) pro- nounced oxidative and hemodynamic stresses; and 5) deple- tion of medial smooth muscle cells (SMC) through acceler- ated senescence and apoptosis. The occurrence of these pathophysiological processes within both AAAs and vulner- able atherosclerotic plaques suggests that these conditions likely share many potential targets for pharmacologic ther- From the *Departments of *Surgery (Section of Vascular Surgery), †Radi- ology, and ‡Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri. Supported by grants HL56701 and HL64333 from the National Heart, Lung, and Blood Institute. Reprints: Robert W. Thompson, MD, Section of Vascular Surgery, Washington University School of Medicine, 9901 Wohl Hospital, 4960 Children’s Place, St. Louis, MO 63110. E-mail: [email protected]. Copyright © 2004 by Lippincott Williams & Wilkins ISSN: 0003-4932/05/24101-0092 DOI: 10.1097/01.sla.0000150258.36236.e0 Annals of Surgery • Volume 241, Number 1, January 2005 92
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysm in...

ORIGINAL ARTICLES
Treatment With Simvastatin Suppresses the Developmentof Experimental Abdominal Aortic Aneurysms in Normal
and Hypercholesterolemic Mice
Eric F. Steinmetz, MD,* Celine Buckley, MD,* Murray L. Shames, MD,*† Terri L. Ennis, BS,*Sarah J. Vanvickle-Chavez, BS,* Dongli Mao, MD,* Lee A. Goeddel,* Cherady J. Hawkins,* and
Robert W. Thompson, MD*†‡
Objective: To determine if treatment with hydroxymethylglutaryl-coenzyme A reductase inhibitors (statins) can influence the devel-opment of experimental abdominal aortic aneurysms (AAAs).Summary Background Data: AAAs are associated with athero-sclerosis, chronic inflammation, and matrix metalloproteinase(MMP)-mediated connective tissue destruction. Because statins ex-ert antiinflammatory activities independent of their lipid-loweringeffects, these agents may help suppress aneurysmal degeneration.Methods: C57Bl/6 wild-type and hypercholesterolemic apoE-defi-cient mice underwent transient perfusion of the aorta with elastasefollowed by subcutaneous treatment with either 2 mg/kg simvastatinper day or vehicle. Aortic diameter (AD) was measured before and14 days after elastase perfusion. The extent of aortic dilatation(�AD) was determined with AAAs defined as �AD �100%.Results: Wild-type mice treated with simvastatin exhibited a 21%reduction in �AD and a 33% reduction in AAAs compared withvehicle-treated controls. Suppression of AAAs in simvastatin-treated mice was associated with preservation of medial elastin andvascular smooth muscle cells, as well as a relative reduction in aorticwall expression of MMP-9 and a relative increase in expression ofTIMP-1. In hypercholesterolemic apoE-deficient mice, treatmentwith simvastatin was associated with a 26% reduction in �AD anda 30% reduction in AAAs. Treatment with simvastatin had no effecton serum cholesterol levels in either normal or hypercholesterolemicmice.Conclusions: Treatment with simvastatin suppresses the develop-ment of experimental AAAs in both normal and hypercholester-olemic mice. The mechanisms of this effect are independent of
lipid-lowering and include preservation of medial elastin andsmooth muscle cells, as well as altered aortic wall expression ofMMPs and their inhibitors.
(Ann Surg 2005;241: 92–101)
Abdominal aortic aneurysms (AAAs) are a common andpotentially life-threatening disorder associated with ag-
ing and atherosclerosis.1 Despite these associations, there isconsiderable uncertainty regarding the precise role of athero-sclerosis in the etiology and pathophysiology of AAAs, withopinion ranging from the view that aneurysms arise as adirect consequence of advanced atheromatous disease tospeculation that aneurysmal degeneration might be an inde-pendent disease process only coincidentally related to athero-sclerosis.2–4 This debate has been fueled by the discrepancybetween strong evidence linking hypercholesterolemia withatherogenesis and the lack of a compelling relationship be-tween altered lipid metabolism and AAAs.5–7
It has become evident over the past decade that many ofthe cellular and molecular mechanisms involved in aneurys-mal degeneration are analogous to those involved in theclinical complications of atherosclerosis such as rupture ofatheromatous plaques.8–10 The most important of these mech-anisms include: 1) arterial wall accumulation and activationof mononuclear inflammatory cells, including both macro-phages and lymphocytes; 2) increased local expression ofproinflammatory cytokines, chemokines, and matrix-degrad-ing proteinases; 3) accelerated degradation of structurallyimportant matrix proteins (ie, elastin and collagen); 4) pro-nounced oxidative and hemodynamic stresses; and 5) deple-tion of medial smooth muscle cells (SMC) through acceler-ated senescence and apoptosis. The occurrence of thesepathophysiological processes within both AAAs and vulner-able atherosclerotic plaques suggests that these conditionslikely share many potential targets for pharmacologic ther-
From the *Departments of *Surgery (Section of Vascular Surgery), †Radi-ology, and ‡Cell Biology and Physiology, Washington University Schoolof Medicine, St. Louis, Missouri.
Supported by grants HL56701 and HL64333 from the National Heart, Lung,and Blood Institute.
Reprints: Robert W. Thompson, MD, Section of Vascular Surgery, WashingtonUniversity School of Medicine, 9901 Wohl Hospital, 4960 Children’s Place,St. Louis, MO 63110. E-mail: [email protected].
Copyright © 2004 by Lippincott Williams & WilkinsISSN: 0003-4932/05/24101-0092DOI: 10.1097/01.sla.0000150258.36236.e0
Annals of Surgery • Volume 241, Number 1, January 200592

apy, whether they represent distinct diseases or simply diver-gent aspects of the same heterogeneous disease process.
Three-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) are widely used in thetreatment of hypercholesterolemia, atherosclerosis, and cor-onary artery disease. Large clinical trials consistently dem-onstrate significant reductions in cardiac events and mortalityassociated with statin therapy, but discrepancies betweenobserved clinical benefits and the degree of angiographicimprovement has raised the possibility that statins might exertadditional therapeutic effects beyond those attributable solelyto cholesterol-lowering.11–18 In support of this notion, recentlaboratory studies have demonstrated a host of antiinflamma-tory and other “pleiotropic” effects associated with statintherapy.19–21 Because statins influence pathophysiologicalmechanisms in atherosclerosis that appear similar to thoseinvolved in aneurysmal degeneration, we postulated thattreatment with statins might also have an impact on thedevelopment and progression of AAAs.
The primary purpose of this study was to determine iftreatment with an HMG-CoA reductase inhibitor would fa-vorably influence the development of experimental AAAs,and second, to explore whether any effects of statins on AAAdevelopment could be considered independent of changes incirculating cholesterol. To address these questions, we ap-plied a previously characterized mouse model of aortic an-eurysms to normocholesterolemic animals and to mice withgenetically determined hypercholesterolemia, and assessedwhether treatment with simvastatin has an influence on an-eurysmal degeneration under each of these conditions. Weexamined the effects of simvastatin because this drug is oneof the most commonly used agents in clinical practice, andchose apoE-deficient mice as a model for hypercholesterol-emia because these animals do not exhibit reductions inserum cholesterol during treatment with statins.22–27 Thisexperimental strategy thereby allowed us to examine theeffects of statin therapy on aneurysm development in bothnormocholesterolemic and hypercholesterolemic animals in amanner independent of changes in blood cholesterol.
MATERIALS AND METHODS
Animals and Experimental GroupsC57BL/6J wild-type mice and apolipoprotein E (apoE)-
deficient mice on a C57Bl/6 background were purchasedfrom The Jackson Laboratory (Bangor, ME). All experimen-tal procedures were performed in male animals that hadreached maturity (8–10 weeks of age) according to a protocolapproved by the Animal Studies Committee at WashingtonUniversity School of Medicine.
Group I (normocholesterolemia) consisted of 35 wild-type mice that had been maintained on normal laboratorychow beginning at 3 weeks of age (PicoLab Rodent Diet
5053; PMI Nutrition International/El Mel, St. Charles, MO).Group II (hypercholesterolemia) consisted of 53 apoE-defi-cient mice that had been placed on a high-fat Western dietbeginning at 3 weeks of age. The composition of this dietincluded 0.15% (wt/wt) cholesterol, 21% fat, 17% protein,and 48% carbohydrate (Harlan Teklad TD88137; Madison,WI). To verify circulating lipid levels, blood was drawn from6 animals in each experimental group at the time the micewere killed and total serum cholesterol was measured usingthe Infinity Cholesterol Reagent kit from Sigma Diagnostics(St. Louis, MO).
Drug TreatmentSimvastatin tablets (40 mg) were purchased from
Merck, Sharp and Dohme (West Point, PA). Because orallyadministered simvastatin requires metabolism to its pharma-cologically active form by hepatic lactonases, the drug waschemically activated by alkaline hydrolysis before adminis-tration by subcutaneous injection in accord with a previouslyreported protocol.23,28 Preactivated simvastatin was dilutedwith phosphate-buffered saline (PBS) to a final workingconcentration of 0.4 mg/mL. Beginning 7 days before theinduction of experimental AAAs, a subset of animals in eachgroup underwent treatment with 2 mg/kg per day preactivatedsimvastatin by daily subcutaneous injection with the remain-ing animals in each group treated with PBS vehicle. Treat-ment with drug or vehicle was continued for 14 days afterelastase perfusion, at which time all animals were killed byintravenous pentobarbital overdose. The dose regimen forsimvastatin was based on previous studies demonstratingbeneficial effects of the drug in mouse models of atheroscle-rosis and cerebral ischemia, and the recognition that treat-ment with simvastatin does not decrease serum cholesterollevels in wild-type or apoE-deficient mice.23–27
Elastase-Induced Abdominal Aortic Aneurysmsand Aortic Diameter Measurements
Transient elastase perfusion was used to induce thedevelopment of experimental AAAs as previously de-scribed.29–31 Briefly, mice (20–35 g) were anesthetized withintraperitoneal sodium pentobarbital (55–60 mg/kg) and alaparotomy was performed under sterile conditions. The ab-dominal aorta was isolated with the assistance of an operatingstereomicroscope (Leica, Deerfield, IL) and the preperfusionaortic diameter (AD) was measured with a calibrated oculargrid. Temporary ligatures were placed around the proximaland distal aorta, and an aortotomy was created at the bifur-cation using the tip of a 30-gauge needle. A heat-taperedsegment of PE-10 polyethylene tubing was introducedthrough the aortotomy and secured, and the aortic lumen wasperfused for 5 minutes at 100 mm Hg with saline containing0.414 U/mL type I porcine pancreatic elastase (Sigma Chem-ical Co., St. Louis, MO). After removing the perfusion
Annals of Surgery • Volume 241, Number 1, January 2005 Treatment With Simvastatin
© 2004 Lippincott Williams & Wilkins 93

catheter, the aortotomy was repaired without constriction ofthe lumen, and the postperfusion AD was measured at least 5minutes after restoration of flow to the lower extremities.Animals were allowed free access to food and water for 14days, when the aorta was reexposed by laparotomy underanesthesia. Final AD measurements were obtained beforeeuthanasia and tissue procurement. AAAs were defined as aninterval increase in diameter (�AD) greater than 100% be-tween day 0 (preperfusion) and day 14 (final).
Light MicroscopyAbdominal aortic tissues were perfusion-fixed with
10% neutral-buffered formalin (120 mm Hg for 10 minutes).Specimens were embedded in paraffin and 5-�m cross-sec-tions of the aortic wall were stained with hematoxylin andeosin (H&E), Verhoeff-Van Gieson (VVG) for elastin, andpicrosirius red for collagen. Additional sections were stainedwith immunoperoxidase techniques using antialpha-SMC ac-tin monoclonal antibodies (clone 1A4, catalog #A2547;Sigma Chemical Co.), as previously described.32
Quantitative (Real-Time) ReverseTranscriptase–Polymerase Chain Reaction
Aortic wall mRNA expression was measured by real-time reverse transcriptase–polymerase chain reaction (RT-PCR) as previously described.30,31 Briefly, 3 to 4 pooledtissue samples were pulverized under liquid nitrogen and totalRNA was isolated with Trizol reagent (Gibco BRL, GrandIsland, NY). Each sample was normalized to 1 �g of totalRNA and cDNA synthesis was performed by reverse tran-scription on a GeneAmp 2400 thermal cycler system usingreagents provided by the manufacturer (Applied Biosystems,Foster City, CA). The reverse transcription products served asthe template for real-time PCR analysis using reagents andprotocols provided in the SYBR Green PCR kit and theGeneAmp 5700 Sequence Detection System (Applied Bio-systems). The gene-specific primers used were as follows:mouse matrix metalloproteinase-9 (MMP-9) (GenBank RefNM013599), forward primer: 5�-ACA ATC CTT GCA ATGTGG ATG TT-3�, reverse primer: 5�-CGC CCT GGA TCTCAG GAA TA-3�; mouse tissue inhibitor of metalloprotein-ases-1 (TIMP-1) (GenBank Ref NM011593), forward primer:5�-ACG CCT ACA CCC CAG TCA TG-3�, reverse primer:5�-GGG ACT TGT GGG CAT ATC CA-3�; and mouse�-actin (GenBank Ref M12481), forward primer: 5�-CCCTAA GGC CAA CCG TGA A-3�, reverse primer: 5�-GTTGAG GTC TCA AAC ATG ATC TG-3�. PCR reactions (40cycles) were performed in quadruplicate with SYBR GreenPCR Core Reagents (Applied Biosystems), and fluorescencesignals were analyzed using the GeneAmp 5700 SequenceDetection System software (version 1.3; Applied Biosys-tems). Results for each sample were normalized to the con-centration of �-actin mRNA.
StatisticsStatistical analysis was performed with the InStat 3.0a
program (GraphPad Software, Inc., San Diego, CA). Within-group comparisons of AD at different intervals after elastaseperfusion were performed by repeated-measures analysis ofvariance (ANOVA) with the Bonferroni multiple compari-sons test. Between-group comparisons of AD at a giveninterval were performed by nonparametric testing using theMann-Whitney rank-sum U test, as were comparisons of theextent of aortic dilatation (�AD) on day 14. Differencesbetween groups in the incidence of AAAs were evaluatedusing 2 � 2 contingency tables and the 2-tailed Fisher exacttest. Relative differences in gene expression were analyzedusing the paired Student t test. In all cases, a P value �0.05was considered significant.
RESULTS
Effects of Simvastatin in NormocholesterolemicMice
Consistent with our previous studies,29–31 wild-typeC57Bl/6 mice treated with vehicle (n � 15) exhibited asignificant expansion in aortic size after transient elastaseperfusion, with AD increasing from 0.51 � 0.01 mm(mean � standard error of mean �SEM) before perfusionto 1.40 � 0.06 mm on day 14 (P �0.001) (Table 1). Theoverall extent of aortic dilatation in this group was 174 �12%, with 14 of 15 animals (93%) developing AAAs (Fig. 1).
Normocholesterolemic (wild-type) animals treated withsimvastatin (n � 20) also exhibited a significant increase inAD after elastase perfusion, from 0.51 � 0.01 mm beforeperfusion to 1.20 � 0.06 mm on day 14 (P �0.001). Al-though there was no difference between the vehicle-treatedand simvastatin-treated groups with regard to AD measure-ments before or immediately after elastase perfusion, treat-ment with simvastatin was associated with a 17% reduction inmedian AD on day 14 compared with vehicle-treated controls(P �0.05) (Table 1). The overall extent of aortic dilatation insimvastatin-treated animals was 137% � 12%, representing a21% reduction from that observed in the vehicle-treatedcontrol group (P �0.05), and the incidence of AAAs wasreduced by 33% (12 of 20 mice treated with simvastatin�60% vs. 14 of 15 mice treated with vehicle �93%; P �0.05)(Fig. 1). There was no difference in total serum cholesterolbetween wild-type mice treated with vehicle (93 � 5 mg/dL)and those treated with simvastatin (95 � 3 mg/dL).
Morphologic ChangesTo elucidate the structural basis for decreased aneu-
rysm development in simvastatin-treated mice, morphologicchanges in the aortic wall were evaluated by histochemicalstaining for elastin, collagen, and vascular smooth musclecells. As shown in Figure 2, the development of AAAs in
Steinmetz et al Annals of Surgery • Volume 241, Number 1, January 2005
© 2004 Lippincott Williams & Wilkins94

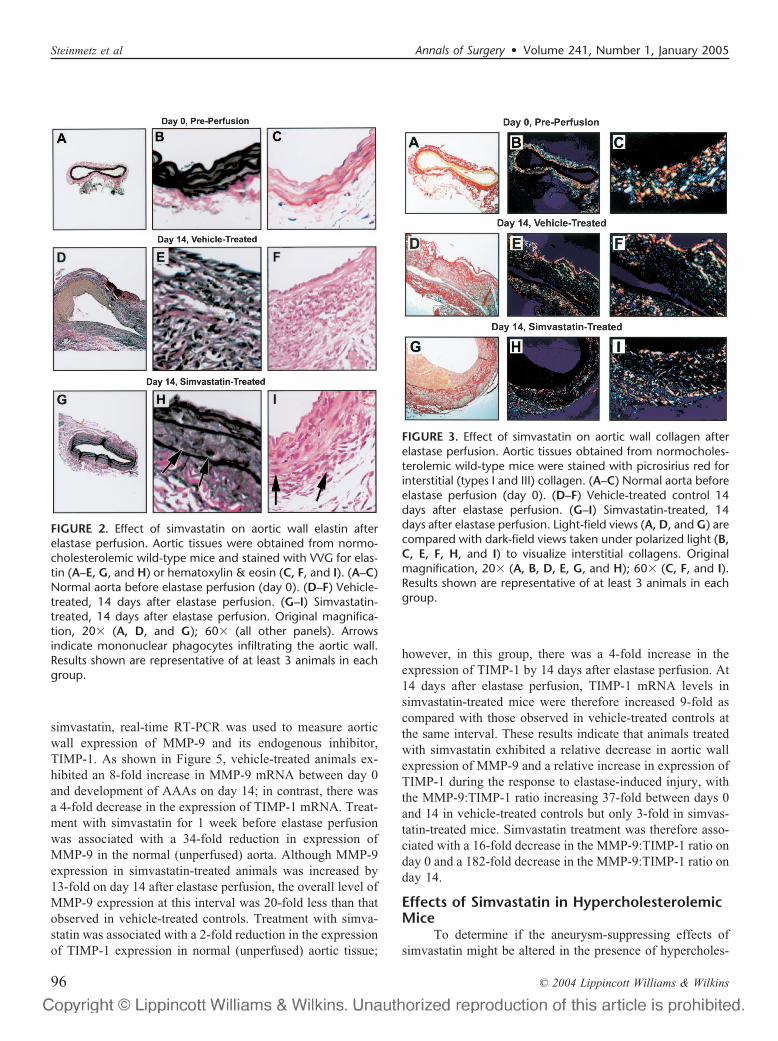
vehicle-treated mice was accompanied by marked thickeningof the aortic wall, a dense transmural inflammatory response,and complete destruction of the elastic media. Althoughsimvastatin-treated animals exhibited inflammatory cell infil-tration similar to vehicle controls (Fig. 2H, I), the mostremarkable and consistent difference was preservation ofintact elastic lamellae within the aortic media. As shown inFigure 3, staining with sirius red for collagen revealed asomewhat more diffuse appearance of interstitial collagenfibers within the media and adventitia of AAAs compared
with normal aorta, but there was no detectable difference inthe overall collagen content between simvastatin-treated miceand vehicle-treated controls. Sections stained for alpha-smooth muscle actin revealed a marked depletion of medialSMC in elastase-induced AAAs, with significant preservationof vascular SMC in simvastatin-treated mice (Fig. 4).
Aortic Wall Gene ExpressionTo examine molecular mechanisms that might underlie
the reduction in aneurysmal dilatation in mice treated with
TABLE 1. Aortic Diameter Measurements in Normocholesterolemic Mice
AD Day 0 (Pre) AD Day 0 (Post) AD Day 14 (Final)
Vehicle-treated (n � 15)Median 0.53 mm 0.95 mm 1.40 mmRange 0.47–0.53 mm 0.84–1.00 mm 1.05–1.68 mmMean � standard error of mean 0.51 � 0.01 mm 0.93 � 0.01 mm* 1.40 � 0.06 mm†‡
Simvastatin-treated (n � 20)Median 0.53 mm 0.95 mm 1.16 mmRange 0.47–0.53 mm 0.84–1.05 mm 0.79–1.79 mmMean � standard error of mean 0.51 � 0.01 mm 0.94 � 0.01 mm* 1.20 � 0.06 mm†‡
P value§ NS NS 0.012
Normocholesterolemic (wild-type) C57Bl/6 mice were treated with either vehicle or simvastatin for 7 d before elastase perfusion and for 14 d after. Aorticdiameter (AD) was measured before (pre) and immediately after (post) elastase perfusion and again at day 14 (final). Within-group comparisons of mean ADat each interval were made with repeated-measures analysis of variance and the Bonferroni multiple comparisons test (*P �0.001 for post vs. pre; †P �0.001for final vs. pre; ‡P �0.001 for final vs. post).
§Between-group comparisons (vehicle vs simvastatin) of median AD were made with the Mann-Whitney rank-sum U test (NS, not significant).
FIGURE 1. Effect of simvastatin on the development of elastase-induced abdominal aortic aneurysms (AAAs) in normocholester-olemic mice. C57Bl/6 wild-type mice were subjected to transient elastase perfusion of the abdominal aorta and treatment witheither vehicle (n � 15) or simvastatin (n � 20) for up to 14 days. (A) The extent of aortic dilatation for each animal was determinedas the percent increase from the normal (preperfusion) aortic diameter. Data shown represent the mean � standard error of mean.Aneurysmal dilatation is defined as an increase �100% (indicated by the horizontal line). *Vehicle versus simvastatin, Mann-Whitney rank-sum U test. (B) Incidence of AAAs. **Vehicle versus Simvastatin, 2-tailed Fisher exact test.
Annals of Surgery • Volume 241, Number 1, January 2005 Treatment With Simvastatin
© 2004 Lippincott Williams & Wilkins 95
simvastatin, real-time RT-PCR was used to measure aorticwall expression of MMP-9 and its endogenous inhibitor,TIMP-1. As shown in Figure 5, vehicle-treated animals ex-hibited an 8-fold increase in MMP-9 mRNA between day 0and development of AAAs on day 14; in contrast, there wasa 4-fold decrease in the expression of TIMP-1 mRNA. Treat-ment with simvastatin for 1 week before elastase perfusionwas associated with a 34-fold reduction in expression ofMMP-9 in the normal (unperfused) aorta. Although MMP-9expression in simvastatin-treated animals was increased by13-fold on day 14 after elastase perfusion, the overall level ofMMP-9 expression at this interval was 20-fold less than thatobserved in vehicle-treated controls. Treatment with simva-statin was associated with a 2-fold reduction in the expressionof TIMP-1 expression in normal (unperfused) aortic tissue;
however, in this group, there was a 4-fold increase in theexpression of TIMP-1 by 14 days after elastase perfusion. At14 days after elastase perfusion, TIMP-1 mRNA levels insimvastatin-treated mice were therefore increased 9-fold ascompared with those observed in vehicle-treated controls atthe same interval. These results indicate that animals treatedwith simvastatin exhibited a relative decrease in aortic wallexpression of MMP-9 and a relative increase in expression ofTIMP-1 during the response to elastase-induced injury, withthe MMP-9:TIMP-1 ratio increasing 37-fold between days 0and 14 in vehicle-treated controls but only 3-fold in simvas-tatin-treated mice. Simvastatin treatment was therefore asso-ciated with a 16-fold decrease in the MMP-9:TIMP-1 ratio onday 0 and a 182-fold decrease in the MMP-9:TIMP-1 ratio onday 14.
Effects of Simvastatin in HypercholesterolemicMice
To determine if the aneurysm-suppressing effects ofsimvastatin might be altered in the presence of hypercholes-
FIGURE 2. Effect of simvastatin on aortic wall elastin afterelastase perfusion. Aortic tissues were obtained from normo-cholesterolemic wild-type mice and stained with VVG for elas-tin (A–E, G, and H) or hematoxylin & eosin (C, F, and I). (A–C)Normal aorta before elastase perfusion (day 0). (D–F) Vehicle-treated, 14 days after elastase perfusion. (G–I) Simvastatin-treated, 14 days after elastase perfusion. Original magnifica-tion, 20� (A, D, and G); 60� (all other panels). Arrowsindicate mononuclear phagocytes infiltrating the aortic wall.Results shown are representative of at least 3 animals in eachgroup.
FIGURE 3. Effect of simvastatin on aortic wall collagen afterelastase perfusion. Aortic tissues obtained from normocholes-terolemic wild-type mice were stained with picrosirius red forinterstitial (types I and III) collagen. (A–C) Normal aorta beforeelastase perfusion (day 0). (D–F) Vehicle-treated control 14days after elastase perfusion. (G–I) Simvastatin-treated, 14days after elastase perfusion. Light-field views (A, D, and G) arecompared with dark-field views taken under polarized light (B,C, E, F, H, and I) to visualize interstitial collagens. Originalmagnification, 20� (A, B, D, E, G, and H); 60� (C, F, and I).Results shown are representative of at least 3 animals in eachgroup.
Steinmetz et al Annals of Surgery • Volume 241, Number 1, January 2005
© 2004 Lippincott Williams & Wilkins96

terolemia, the induction of AAAs was also examined infat-fed apoE-deficient mice that had markedly elevated totalserum cholesterol levels (mean � SEM: 846 � 66 mg/dL).Similar to wild-type mice, hypercholesterolemic apoE-defi-cient mice developed a significant increase in AD aftertransient elastase perfusion, with mean AD increasing from0.50 � 0.01 mm before perfusion to 1.37 � 0.08 mm on day14 (P �0.001) (Table 2). The overall extent of aortic dilata-tion was 176 � 16%, with 20 of 25 animals (80%) develop-ing AAAs (Fig. 6).
In apoE-deficient mice treated with simvastatin, ADincreased significantly from 0.50 � 0.08 mm before perfu-
sion to 1.14 � 0.07 mm on day 14 (P �0.05) (Table 2). Therewas no difference between groups in AD measured before orimmediately after elastase perfusion, yet the median AD onday 14 was 28% lower in simvastatin-treated apoE-deficientanimals compared with vehicle-treated apoE-deficient con-trols (P �0.05). The overall extent of aortic dilatation insimvastatin-treated apoE-deficient animals was 131% �14%, a 26% reduction from that observed in the vehicle-treated apoE-deficient control group (P �0.05), and the
FIGURE 4. Effect of simvastatin on aortic wall smooth musclecells after elastase perfusion. Aortic tissues were obtained fromnormocholesterolemic wild-type mice and stained with immu-noperoxidase techniques for alpha-smooth muscle cell actin(brown reaction product). (A and B) Normal aorta beforeelastase perfusion (day 0). (C and D) Vehicle-treated, 14 daysafter elastase perfusion. (E and F(Simvastatin-treated, 14 daysafter elastase perfusion. Original magnification, 20� (A, C, andE); 60� (B, D, and F). Results shown are representative of atleast 3 animals in each group.
FIGURE 5. Effect of simvastatin on mouse aortic wall geneexpression during elastase-induced aneurysmal degeneration.Aortic wall RNA samples were obtained at the indicated inter-vals from normocholesterolemic C57BL/6 wild-type micetreated with either vehicle or simvastatin, and mRNA transcriptlevels were measured by real-time reverse transcriptase–poly-merase chain reaction. Data shown indicate the mean �standard error of mean transcript levels for MMP-9 (A) andTIMP-1 (B) as normalized to �-actin (n � 3). *P �0.05, day 14versus day 0 for the group indicated. **P �0.05, simvastatinversus vehicle for the interval indicated.
Annals of Surgery • Volume 241, Number 1, January 2005 Treatment With Simvastatin
© 2004 Lippincott Williams & Wilkins 97

incidence of AAAs was 50% in apoE-deficient mice treatedwith simvastatin compared with 80% in apoE-deficient con-trols (P �0.05) (Fig. 6). There was no difference in totalserum cholesterol in apoE-deficient mice treated for 3 weekswith simvastatin (836 � 43 mg/dL) versus vehicle (846 � 66mg/dL).
DISCUSSIONThe most important finding of this study is that treat-
ment with simvastatin reduced the development of elastase-
induced experimental AAAs in both normocholesterolemicand apoE-deficient hypercholesterolemic mice and that thisoccurred in the absence of drug-induced alterations in totalserum cholesterol. Treatment with simvastatin was also as-sociated with structural preservation of aortic wall elastin andmedial smooth muscle cells despite the presence of a chronicinflammatory response, and there was a relative reduction inaortic wall expression of MMP-9 and a relative increase inexpression of TIMP-1. These results provide the first exper-imental evidence that treatment with statins can suppress
TABLE 2. Aortic Diameter Measurements in Hypercholesterolemic Mice
AD Day 0 (Pre) AD Day 0 (Post) AD Day 14 (Final)
Vehicle-treated (n � 25)Median 0.47 mm 0.90 mm 1.42 mmRange 0.42–0.58 mm 0.74–1.16 mm 0.84–2.42 mmMean � standard error of mean 0.50 � 0.01 mm 0.88 � 0.02 mm* 1.37 � 0.08 mm†‡
Simvastatin-treated (n � 28)Median 0.47 mm 0.90 mm 1.03 mmRange 0.42–0.58 mm 0.79–1.00 mm 0.74–1.89 mmMean � standard error of mean 0.50 � 0.01 mm 0.89 � 0.01 mm* 1.14 � 0.07 mm†‡
P value§ NS NS 0.022
Hypercholesterolemic (ApoE-deficient) C57Bl/6 mice were treated with either vehicle or simvastatin for 7 d before elastase perfusion and for 14 d after.Aortic diameter (AD) was measured before (pre) and immediately after (post) elastase perfusion and again at day 14 (final). Within-group comparisons of meanAD at each interval were made with repeated-measures analysis of variance and the Bonferroni multiple comparisons test (*P �0.001 for post vs. pre; †P�0.001 for final vs. pre; ‡P �0.001 for final vs. post).
§Between-group comparisons (vehicle vs simvastatin) of median AD were made with the Mann-Whitney rank-sum U test (NS, not significant).
FIGURE 6. Effect of simvastatin on the development of elastase-induced abdominal aortic aneurysms (AAAs) in hypercholester-olemic mice. C57Bl/6 apoE-deficient mice were subjected to transient elastase perfusion of the abdominal aorta and treatmentwith either vehicle (n � 25) or simvastatin (n � 28) for up to 14 days. (A) The extent of aortic dilatation for each animal wasdetermined as the percent increase from the normal (preperfusion) aortic diameter. Data shown represent the mean � standarderror of mean. Aneurysmal dilatation is defined as an increase �100% as indicated by the horizontal line. *Vehicle versussimvastatin, Mann-Whitney rank-sum U test. (B) Incidence of AAAs. **Vehicle versus simvastatin, 2-tailed Fisher exact test.
Steinmetz et al Annals of Surgery • Volume 241, Number 1, January 2005
© 2004 Lippincott Williams & Wilkins98

aneurysmal degeneration in an animal model of AAAs andthat the underlying mechanism is independent of serumcholesterol levels. These findings have potentially importantclinical implications as a result of the widespread use ofstatins in treatment of patients with atherosclerosis and thefrequency of small asymptomatic AAAs in this population.
In addition to the inhibition of AAAs, perhaps the moststriking observation in this study was the structural preserva-tion of the medial elastic lamellae that occurred in micetreated with simvastatin. Elastin degradation in AAAs isthought to require proteinases elaborated by mononuclearphagocytes, including MMPs, cathepsins, and serine pro-teases. Despite the presence of mononuclear phagocyteswithin the elastase-injured aorta, we found that treatmentwith simvastatin was associated with alterations in the rela-tive balance of aortic wall gene expression that might favordiminished MMP-9 activity. Although the results regardingMMP-9 and TIMP-1 expression need to be further verified atthe protein and enzyme activity levels, these findings indicatethat simvastatin-mediated preservation of elastin may havebeen the result of suppression of elastolytic activities (ie,MMP-9), thereby serving to limit structural damage. Previousreports indicate that suppression of MMP-9 is a consistenteffect of HMG-CoA reductase inhibitors, because statins caninhibit MMP-9 activity produced by monocytes,33,34 macro-phages,35,36 fibroblasts,37 vascular SMC,38 and cultured tu-mor cell lines.39 The functional importance of this mecha-nism is illustrated by studies showing that simvastatin cansuppress migration of THP-1 human monocytic cells inresponse to MCP-1, in part through a reduction in MMP-9.33
The potential clinical relevance of this mechanism was alsohighlighted by a recent study using cultured human AAAtissue explants, in which exposure to cerivastatin suppressedproduction of MMP-9 up to 5-fold in vitro.40 Overproductionof TIMP-1 might also contribute to suppression of MMP-9activity as well as other MMPs involved in the degradation ofmatrix proteins such as interstitial collagenases, therebymaintaining aortic wall tensile strength and diminishing an-eurysmal dilatation. This conclusion is also consistent withobservations on the mechanisms by which HMG-CoA reduc-tase inhibitors favorably influence the behavior of atheroma-tous plaques by nonlipid-lowering activities, including arecent report in which pravastatin affected a number ofmarkers related to plaque stabilization, including TIMP-1expression, in human carotid artery atherosclerosis.41 Finally,although we observed no measurable difference in overallcollagen content in aortas from simvastatin-treated mice andvehicle controls, simvastatin treatment may still be associatedwith functionally important differences in aortic collagensuch as changes in the distribution, alignment, or crosslinkingof collagen fibers that we were unable to assess in this study.
One of the mechanisms suspected to underlie the ac-tivities of HMG-CoA reductase inhibitors in atherosclerosis
is a prominent suppressive effect on inflammatory cell func-tion, which may serve to diminish the propensity of vulner-able plaques to rupture.21,24,42,43 This includes effects onmonocyte/macrophage growth,8,44 MMP expression and se-cretion,45 expression of tissue factor, adhesion molecules, andproinflammatory cytokines,35,46,47 secretion of MCP-1 andinterleukin-8,48 and activation of NF�B.49 Although we ob-served no measurable decrease in the intensity of aortic wallinflammation associated with simvastatin, our finding thatsimvastatin reduced mouse aortic wall expression of MMP-9likely reflects important drug effects on mononuclear phago-cytes because we have previously shown that inflammatorycell production of MMP-9 is critical for AAA formation inthe same mouse model.29 Because inhibition of protein iso-prenylation has emerged as a key cellular mechanism bywhich statins might exert their antiinflammatory activities inatherosclerosis and other conditions, our results suggest thatalterations in protein isoprenylation may also be an importantpathway to be examined in aortic aneurysms.50
A second mechanism potentially explaining the clinicalbenefit of statins is a direct action on vascular SMC. It hasbeen demonstrated that statins can inhibit SMC migration andproliferation,28,51,52 decrease SMC expression of plasmino-gen activator inhibitor-1,53 and decrease SMC free radicalproduction induced by angiotensin II.54 The importance ofthis mechanism is illustrated by the demonstration that sim-vastatin reduces neointima formation in human saphenousvein explants in association with inhibition of SMC prolifer-ation and migration.55 We have previously demonstrated thatmedial SMC depletion is a prominent feature of humanAAAs in association with accelerated replicative senescenceand increased apoptosis.32,56 The preservation of medialSMC we observed in simvastatin-treated animals maythereby reflect the effects of statin treatment on some of thesemolecular mechanisms; moreover, these medial SMC mayhave an important function in the synthesis and secretion ofstructurally important matrix proteins (ie, interstitial collag-ens), thereby contributing to the prevention of aneurysmaldilatation.
Hypercholesterolemia is an important risk factor foratherosclerosis, and the apoE-deficient mouse has providedan important model to investigate the development of com-plex atheromatous lesions.57 Although it is still unclear ifhypercholesterolemia has a specific role in development ofAAAs, we did not observe any deleterious effects of severehypercholesterolemia on elastase-induced aneurysmal degen-eration. Because the apoE-deficient mice used here were 8 to10 weeks of age and exposed to a high-fat diet for only 5weeks at the time of the experimental procedures, the dura-tion of hypercholesterolemia was not sufficient for the devel-opment of the severe and widespread atherosclerotic lesionstypically observed in older apoE-deficient animals. It there-fore remains possible that aneurysm formation might have
Annals of Surgery • Volume 241, Number 1, January 2005 Treatment With Simvastatin
© 2004 Lippincott Williams & Wilkins 99

been more significantly affected by a longer period of hyper-cholesterolemia or in the presence of established atheroma-tous lesions within the abdominal aorta. Another point worthrepeating is that apoE-deficient mice are known to be resis-tant to cholesterol-lowering by treatment with statins asopposed to the effects of these drugs in different mousemodels of hypercholesterolemia or in humans.22–27 It istherefore possible that in other experimental or clinical set-tings in which statins cause a reduction in serum cholesterol,drug treatment might have additional effects on aneurysmaldegeneration beyond those observed in this study.
The results of this investigation suggest that treatmentwith statins might have clinical potential in suppressing thedevelopment and growth of AAAs. Previous studies in ratsand mice have demonstrated effective suppression of elas-tase-induced aneurysm formation by treatment with antiin-flammatory agents,58 hydroxamate- and tetracycline-basedMMP inhibitors,59,60 and angiotensin-converting enzyme an-tagonists,61 and similar results have been observed in micewith genetic deficiencies in either MMP-9 or MMP-2.29,62
Although it remains difficult to compare these different stud-ies, we have consistently observed reductions in aneurysmaldilatation of approximately 60% in MMP-9-deficient mice orafter treatment of wild-type mice with doxycycline, an agentthat suppresses both expression and proteolytic activity ofMMP-9.29,60,63 Because this degree of suppression is some-what greater than that observed with simvastatin, more directcomparisons will be necessary to evaluate the relative effi-cacy of these different pharmacologic strategies. Moreover,because tetracyclines, statins, and other agents act by distinctbut potentially overlapping mechanisms, it remains possiblethat combination therapies might be even more effective thanany single agent alone. Because the cellular and molecularmechanisms underlying aneurysmal degeneration appear toparallel those involved in the vulnerability of atheromatousplaques to rupture, studies with the elastase-induced mousemodel of AAAs can be expected to yield further insights intothe pathogenesis and treatment of these conditions.
REFERENCES1. Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms:
basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:93–232.
2. Zarins CK, Glagov S, Vesselinovitch D, et al. Aneurysm formation inexperimental atherosclerosis: relationship to plaque evolution. J VascSurg. 1990;12:246–256.
3. Tilson MD. Aortic aneurysms and atherosclerosis. Circulation. 1992;85:378–379.
4. Zarins CK, Xu CP, Glagov S. Aneurysmal enlargement of the aortaduring regression of experimental atherosclerosis. J Vasc Surg. 1992;15:90–98.
5. DePalma RG, Sidaway AN, Giordano JM. Associated aetiological andatherosclerotic risk factors in abdominal aneurysms. In: Greenhalgh RM,Mannick JA, Powell JT, eds. The Cause and Management of Aneurysms.London: WB Saunders; 1990:37–46.
6. Lederle FA, Johnson GR, Wilson SE, et al. The aneurysm detection andmanagement study screening program: validation cohort and final re-
sults. Aneurysm Detection and Management Veterans Affairs Cooper-ative Study Investigators. Arch Intern Med. 2000;160:1425–1430.
7. Blanchard JF, Armenian HK, Friesen PP. Risk factors for abdominalaortic aneurysm: results of a case–control study. Am J Epidemiol.2000;151:575–583.
8. Fuster V, Stein B, Ambrose JA, et al. Atherosclerotic plaque rupture andthrombosis: evolving concepts. Circulation 1990;82(suppl II):II47–II59.
9. Gibbons GH, Dzau V. The emerging concept of vascular remodeling.N Engl J Med. 1994;330:1431–1438.
10. Thompson RW. Basic science of abdominal aortic aneurysms: emergingtherapeutic strategies for an unresolved clinical problem. Curr OpinCardiol. 1996;11:504–518.
11. Randomised trial of cholesterol lowering in 4444 patients with coronaryheart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet.1994;344:1383–1389.
12. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heartdisease with pravastatin in men with hypercholesterolemia: West ofScotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307.
13. Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin oncoronary events after myocardial infarction in patients with averagecholesterol levels: Cholesterol and Recurrent Events Trial investigators.N Engl J Med. 1996;335:1001–1009.
14. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acutecoronary events with lovastatin in men and women with average cho-lesterol levels: results of AFCAPS/TexCAPS: Air Force/Texas CoronaryAtherosclerosis Prevention Study. JAMA. 1998;279:1615–1622.
15. Pekkanen J, Linn S, Heiss G, et al. Ten-year mortality from cardiovas-cular disease in relation to cholesterol level among men with and withoutpreexisting cardiovascular disease. N Engl J Med. 1990;322:1700–1707.
16. Brown BG, Zhao XQ, Sacco DE, et al. Lipid lowering and plaqueregression: new insights into prevention of plaque disruption and clinicalevents in coronary disease. Circulation. 1993;87:1781–1791.
17. Brown BG, Hillger L, Zhao XQ, et al. Types of change in coronarystenosis severity and their relative importance in overall progression andregression of coronary disease: observations from the FATS Trial:Familial Atherosclerosis Treatment Study. Ann N Y Acad Sci. 1995;748:407–418.
18. Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatinon early recurrent ischemic events in acute coronary syndromes: theMIRACL study: a randomized controlled trial. JAMA. 2001;285:1711–1718.
19. Bellosta S, Ferri N, Bernini F, et al. Non-lipid-related effects of statins.Ann Med. 2000;32:164–176.
20. Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglu-taryl coenzyme A reductase inhibitors. Arterioscler Thromb Vasc Biol.2001;21:1712–1719.
21. Liao JK. Beyond lipid lowering: the role of statins in vascular protection.Int J Cardiol. 2002;86:5–18.
22. Quarfordt SH, Oswald B, Landis B, et al. In vivo cholesterol kinetics inapolipoprotein E-deficient and control mice. J Lipid Res. 1995;36:1227–1235.
23. Endres M, Laufs U, Huang Z, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothe-lial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–8885.
24. Sparrow CP, Burton CA, Hernandez M, et al. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasmacholesterol lowering. Arterioscler Thromb Vasc Biol. 2001;21:115–121.
25. Wang YX, Martin-McNulty B, Huw LY, et al. Anti-atheroscleroticeffect of simvastatin depends on the presence of apolipoprotein E.Atherosclerosis. 2002;162:23–31.
26. Bea F, Blessing E, Bennett B, et al. Simvastatin promotes atheroscleroticplaque stability in apoE-deficient mice independently of lipid lowering.Arterioscler Thromb Vasc Biol. 2002;22:1832–1837.
27. Bea F, Blessing E, Shelley MI, et al. Simvastatin inhibits expression oftissue factor in advanced atherosclerotic lesions of apolipoprotein Edeficient mice independently of lipid lowering: potential role of simv-astatin-mediated inhibition of Egr-1 expression and activation. Athero-sclerosis. 2003;167:187–194.
Steinmetz et al Annals of Surgery • Volume 241, Number 1, January 2005
© 2004 Lippincott Williams & Wilkins100

28. Laufs U, Marra D, Node K, et al. 3-Hydroxy-3-methylglutaryl-CoAreductase inhibitors attenuate vascular smooth muscle proliferation bypreventing rho GTPase-induced down-regulation of p27(Kip1). J BiolChem. 1999;274:21926–21931.
29. Pyo R, Lee JK, Shipley JM, et al. Targeted gene disruption of matrixmetalloproteinase-9 (gelatinase B) suppresses development of experi-mental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649.
30. Lee JK, Borhani M, Ennis TL, et al. Experimental abdominal aorticaneurysms in mice lacking expression of inducible nitric oxide synthase.Arterioscler Thromb Vasc Biol. 2001;21:1393–1401.
31. Colonnello JS, Hance KA, Shames ML, et al. Transient exposure toelastase induces mouse aortic wall smooth muscle cell production ofMCP-1 and RANTES during development of experimental aortic aneu-rysm. J Vasc Surg. 2003;38:138–146.
32. Lopez-Candales A, Holmes DR, Liao S, et al. Decreased vascularsmooth muscle cell density in medial degeneration of human abdominalaortic aneurysms. Am J Pathol. 1997;150:993–1007.
33. Wong B, Lumma WC, Smith AM, et al. Statins suppress THP-1 cellmigration and secretion of matrix metalloproteinase-9 by inhibitinggeranylgeranylation. J Leuk Biol. 2001;69:959–962.
34. Ganne F, Vasse M, Beaudeux J-L, et al. Cerivastatin, an inhibitor ofHMG-CoA reductase, inhibits urokinase/urokinase receptor expressionand MMP-9 secretion by peripheral blood monocytes. Thromb Haemost.2000;84:680–688.
35. Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductaseinhibitor, cerivastatin, suppresses growth of macrophages expressingmatrix metalloproteinases, and tissue factor in vivo, and in vitro. Cir-culation. 2001;103:276–283.
36. Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitorsreduce MMP-9 secretion by macrophages. Arterioscler Thromb VascBiol. 1998;18:1671–1678.
37. Wang IK, Lin-Shiau SY, Lin JK. Suppression of invasion and MMP-9expression in NIH 3T3 and v-H-Ras 3T3 fibroblasts by lovastatinthrough inhibition of ras isoprenylation. Oncology. 2000;59:245–254.
38. Luan Z, Chase AJ, Newby AC. Statins inhibit secretion of metallopro-teinases-1, -2, -3, and -9 from vascular smooth muscle cells andmacrophages. Arterioscler Thromb Vasc Biol. 2003;23:769–775.
39. Denoyelle C, Vasse M, Korner M, et al. Cerivastatin, an inhibitor ofHMG-CoA reductase, inhibits the signaling pathways involved in theinvasiveness and metastatic properties of highly invasive breast cancercell lines: an in vitro study. Carcinogenesis. 2001;22:1139–1148.
40. Nagashima H, Aoka Y, Sakomura Y, et al. A 3-hydroxy-3-methylglu-taryl coenzyme A reductase inhibitor, cerivastatin, suppresses produc-tion of matrix metalloproteinase-9 in human abdominal aortic aneurysmwall. J Vasc Surg. 2002;36:158–163.
41. Crisby M, Nordin-Fredriksson G, Shah PK, et al. Pravastatin treatmentincreases collagen content and decreases lipid content, inflammation,metalloproteinases, and cell death in human carotid plaques: implica-tions for plaque stabilization. Circulation. 2001;103:926–933.
42. Corsini A, Bellosta S, Baetta R, et al. New insights into the pharmaco-dynamic and pharmacokinetic properties of statins. Pharmacol Ther.1999;84:413–428.
43. Crisby M. Modulation of the inflammatory process by statins. DrugsToday. 2003;39:137–143.
44. Sakai M, Kobori S, Matsumura T, et al. HMG-CoA reductase inhibitorssuppress macrophage growth induced by oxidized low density lipopro-tein. Atherosclerosis. 1997;133:51–59.
45. Fukumoto Y, Libby P, Rabkin E, et al. Statins alter smooth muscle cellaccumulation and collagen content in established atheroma of Watanabeheritable hyperlipidemic rabbits. Circulation. 2001;103:993–999.
46. Niwa S, Totsuka T, Hayashi S. Inhibitory effect of fluvastatin, an
HMG-CoA reductase inhibitor, on the expression of adhesion moleculeson human monocyte cell line. Int J Immunopharmacol. 1996;18:669–675.
47. Pahan K, Sheikh FG, Namboodiri AM, et al. Lovastatin and phenylace-tate inhibit the induction of nitric oxide synthase and cytokines in ratprimary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679.
48. Kothe H, Dalhoff K, Rupp J, et al. Hydroxymethylglutaryl coenzyme Areductase inhibitors modify the inflammatory response of human mac-rophages and endothelial cells infected with Chlamydia pneumoniae.Circulation. 2000;101:1760–1763.
49. Bustos C, Hernandez-Presa MA, Ortego M, et al. HMG-CoA reductaseinhibition by atorvastatin reduces neointimal inflammation in a rabbitmodel of atherosclerosis. J Am Coll Cardiol. 1998;32:2057–2064.
50. Liao JK. Isoprenoids as mediators of the biological effects of statins.J Clin Invest. 2002;110:285–288.
51. Indolfi C, Cioppa A, Stabile E, et al. Effects of hydroxymethylglutarylcoenzyme A reductase inhibitor simvastatin on smooth muscle cellproliferation in vitro and neointimal formation after vascular injury.J Am Coll Cardiol. 1999;35:214–221.
52. Yang Z, Kozai T, van der Loo B, et al. HMG-CoA reductase inhibitionimproves endothelial cell function and inhibits smooth muscle cellproliferation in human saphenous veins. J Am Coll Cardiol. 2000;36:1691–1697.
53. Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasmino-gen activator inhibitor-1 expression by human vascular smooth muscleand endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–562.
54. Wassmann S, Laufs U, Baumer AT, et al. Inhibition of geranylgerany-lation reduces angiotensin II-mediated free radical production in vascu-lar smooth muscle cells: involvement of angiotensin AT1 receptorexpression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–654.
55. Porter KE, Naik J, Turner NA, et al. Simvastatin inhibits humansaphenous vein neointima formation via inhibition of smooth muscle cellproliferation and migration. J Vasc Surg. 2002;36:150–157.
56. Liao S, Curci JA, Kelley B, et al. Accelerated replicative senescence ofmedial smooth muscle cells derived from abdominal aortic aneurysms ascompared to the adjacent inferior mesenteric artery. J Surg Res. 2000;92:85–95.
57. Nakashima Y, Plump AS, Raines EW, et al. ApoE-deficient micedevelop lesions of all phases of atherosclerosis throughout the arterialtree. Arterioscler Thromb. 1994;14:133–140.
58. Holmes DR, Petrinec D, Wester W, et al. Indomethacin prevents elas-tase-induced abdominal aortic aneurysms in the rat. J Surg Res. 1996;63:305–309.
59. Bigatel DA, Elmore JR, Carey DJ, et al. The matrix metalloproteinaseinhibitor BB-94 limits expansion of experimental abdominal aorticaneurysms. J Vasc Surg. 1999;29:130–138.
60. Petrinec D, Liao S, Holmes DR, et al. Doxycycline inhibition ofaneurysmal degeneration in an elastase-induced rat model of abdominalaortic aneurysm: preservation of aortic elastin associated with sup-pressed production of 92 kD gelatinase. J Vasc Surg. 1996;23:336–346.
61. Liao S, Miralles M, Kelley BJ, et al. Suppression of experimentalabdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg. 2001;33:1057–1064.
62. Longo GM, Xiong W, Greiner TC, et al. Matrix metalloproteinases 2and 9 work in concert to produce aortic aneurysms. J Clin Invest.2002;110:625–632.
63. Prall AK, Longo GM, Mayhan WG, et al. Doxycycline in patients withabdominal aortic aneurysms and in mice: comparison of serum levelsand effect on aneurysm growth in mice. J Vasc Surg. 2002;35:923–929.
Annals of Surgery • Volume 241, Number 1, January 2005 Treatment With Simvastatin
© 2004 Lippincott Williams & Wilkins 101




















