
Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular...
10
..................................................................................................................................................................................... ..................................................................................................................................................................................... CLINICAL RESEARCH Coronary heart disease Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: implications for FXIII therapy and antithrombin use in myocardial infarction Matthias Nahrendorf 1 * , Elena Aikawa 1 , Jose-Luiz Figueiredo 1 , Lars Stangenberg 1 , Susanne W. van den Borne 2 , W. Matthijs Blankesteijn 2 , David E. Sosnovik 1,3 , Farouc A. Jaffer 3 , Ching-Hsuan Tung 1 , and Ralph Weissleder 1 1 Center for Molecular Imaging Research, Massachusetts General Hospital and Harvard Medical School, Building 149, 13th Street, Room 5406, Charlestown, MA 02129, USA; 2 Department of Pharmacology and Toxicology, Cardiovascular Research Institute Maastricht, Maastricht University, The Netherlands; 3 Cardiology Division, Department of Medicine, Massachusetts General Hospital, Boston, MA, USA Received 7 September 2007; revised 25 October 2007; accepted 5 November 2007 See page 427 for the editorial comment on this article (doi:10.1093/eurheartj/ehm610) Aims The transglutaminase factor XIII (FXIII) emerges as a key enzyme in healing after myocardial infarction (MI). Here we assess the impact of transglutaminase-modulating therapies on healing and evolution of heart failure using a novel, non-invasive molecular imaging technique. Methods and results Immunoblotting revealed lower FXIII levels in the myocardium of nine patients with infarct rupture when com- pared to MI patients without rupture (P , 0.0045). In a murine model of MI, we assessed healing while modulating local FXIII activity. Infarct tissue activity was monitored with molecular in vivo single photon emission computed tomography-computed tomography (SPECT-CT) imaging, and activity was found to be increased by 80% in FXIII-treated mice (400 IU FXIII/kg iv.), and decreased by 65% in dalteparin (DP)-treated mice (600 IU/kg DP sc., P , 0.05). DP-treated mice exhibited increased mortality due to infarct rupture (64% by day 7, P , 0.018). Serial Magnetic Resonance Imaging (MRI) showed that left ventricular dilation post-MI was attenuated by FXIII treatment when compared to saline control-treated mice with MI (P ¼ 0.04). Quantitative histological and reverse transcription–polymerase chain reaction analyses revealed that FXIII treatment induced a faster resolution of the neutrophil response, enhanced macrophage recruitment, increased collagen content and augmented angio- genesis in the healing infarct (P , 0.05 vs. control-treated mice with MI). Conclusion FXIII tissue levels are decreased in patients with insufficient healing. Therapeutic strategies that modulate FXIII activity impact murine myocardial healing. Molecular imaging of FXIII activity predicts prognosis in mice with experimental MI. ----------------------------------------------------------------------------------------------------------------------------------------------------------- Keywords Imaging † Inflammation † Wound healing † Myocardial infarction † Factor XIII Introduction Adverse left ventricular (LV) remodelling and the subsequent evol- ution of heart failure remain common after myocardial infarction (MI) and result in a poor prognosis. 1 While many factors contrib- ute to remodelling, 2–4 poor infarct healing and infarct expansion alter LV geometry, increase wall tension and lead to progressive remodelling. 4,5 The transglutaminase factor XIII (FXIII) may play an important role in infarct healing, since it is involved in extracellu- lar matrix turnover and regulation of the inflammatory response after ischaemic injury. 6 This plasma protein circulates in its inactive precursor form and is primarily activated by thrombin. 7 * Corresponding author. Tel: þ1 617 726 8226/ þ1 617 726 5788, Fax: þ1 617 726 5708, Email: [email protected] Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2008. For permissions please email: [email protected]. European Heart Journal (2008) 29, 445–454 doi:10.1093/eurheartj/ehm558 at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from at Ernst Mayr Library of the Museum Comp Zoology, Harvard University on September 27, 2013 http://eurheartj.oxfordjournals.org/ Downloaded from
-
Upload
hms-harvard -
Category
Documents
-
view
0 -
download
0
Transcript of Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular...

. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
CLINICAL RESEARCHCoronary heart disease
Transglutaminase activity in acute infarctspredicts healing outcome and left ventricularremodelling: implications for FXIII therapyand antithrombin use in myocardial infarctionMatthias Nahrendorf1*, Elena Aikawa1, Jose-Luiz Figueiredo1, Lars Stangenberg1,Susanne W. van den Borne2, W. Matthijs Blankesteijn2, David E. Sosnovik1,3,Farouc A. Jaffer3, Ching-Hsuan Tung1, and Ralph Weissleder1
1Center for Molecular Imaging Research, Massachusetts General Hospital and Harvard Medical School, Building 149, 13th Street, Room 5406, Charlestown, MA 02129, USA;2Department of Pharmacology and Toxicology, Cardiovascular Research Institute Maastricht, Maastricht University, The Netherlands; 3Cardiology Division, Department ofMedicine, Massachusetts General Hospital, Boston, MA, USA
Received 7 September 2007; revised 25 October 2007; accepted 5 November 2007
See page 427 for the editorial comment on this article (doi:10.1093/eurheartj/ehm610)
Aims The transglutaminase factor XIII (FXIII) emerges as a key enzyme in healing after myocardial infarction (MI). Here weassess the impact of transglutaminase-modulating therapies on healing and evolution of heart failure using a novel,non-invasive molecular imaging technique.
Methodsand results
Immunoblotting revealed lower FXIII levels in the myocardium of nine patients with infarct rupture when com-pared to MI patients without rupture (P , 0.0045). In a murine model of MI, we assessed healing while modulatinglocal FXIII activity. Infarct tissue activity was monitored with molecular in vivo single photon emission computedtomography-computed tomography (SPECT-CT) imaging, and activity was found to be increased by 80% inFXIII-treated mice (400 IU FXIII/kg iv.), and decreased by 65% in dalteparin (DP)-treated mice (600 IU/kg DPsc., P , 0.05). DP-treated mice exhibited increased mortality due to infarct rupture (64% by day 7, P , 0.018).Serial Magnetic Resonance Imaging (MRI) showed that left ventricular dilation post-MI was attenuated by FXIIItreatment when compared to saline control-treated mice with MI (P ¼ 0.04). Quantitative histological andreverse transcription–polymerase chain reaction analyses revealed that FXIII treatment induced a faster resolutionof the neutrophil response, enhanced macrophage recruitment, increased collagen content and augmented angio-genesis in the healing infarct (P , 0.05 vs. control-treated mice with MI).
Conclusion FXIII tissue levels are decreased in patients with insufficient healing. Therapeutic strategies that modulate FXIII activityimpact murine myocardial healing. Molecular imaging of FXIII activity predicts prognosis in mice with experimental MI.
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -Keywords Imaging † Inflammation † Wound healing † Myocardial infarction † Factor XIII
IntroductionAdverse left ventricular (LV) remodelling and the subsequent evol-ution of heart failure remain common after myocardial infarction(MI) and result in a poor prognosis.1 While many factors contrib-ute to remodelling,2– 4 poor infarct healing and infarct expansion
alter LV geometry, increase wall tension and lead to progressiveremodelling.4,5 The transglutaminase factor XIII (FXIII) may playan important role in infarct healing, since it is involved in extracellu-lar matrix turnover and regulation of the inflammatory responseafter ischaemic injury.6 This plasma protein circulates inits inactive precursor form and is primarily activated by thrombin.7
* Corresponding author. Tel: þ1 617 726 8226/þ1 617 726 5788, Fax: þ1 617 726 5708, Email: [email protected]
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2008. For permissions please email: [email protected].
European Heart Journal (2008) 29, 445–454doi:10.1093/eurheartj/ehm558
at Ernst M
ayr Library of the M
useum C
omp Z
oology, Harvard U
niversity on September 27, 2013
http://eurheartj.oxfordjournals.org/D
ownloaded from
at E
rnst Mayr L
ibrary of the Museum
Com
p Zoology, H
arvard University on Septem
ber 27, 2013http://eurheartj.oxfordjournals.org/
Dow
nloaded from
at Ernst M
ayr Library of the M
useum C
omp Z
oology, Harvard U
niversity on September 27, 2013
http://eurheartj.oxfordjournals.org/D
ownloaded from
at E
rnst Mayr L
ibrary of the Museum
Com
p Zoology, H
arvard University on Septem
ber 27, 2013http://eurheartj.oxfordjournals.org/
Dow
nloaded from
at Ernst M
ayr Library of the M
useum C
omp Z
oology, Harvard U
niversity on September 27, 2013
http://eurheartj.oxfordjournals.org/D
ownloaded from
at E
rnst Mayr L
ibrary of the Museum
Com
p Zoology, H
arvard University on Septem
ber 27, 2013http://eurheartj.oxfordjournals.org/
Dow
nloaded from
at Ernst M
ayr Library of the M
useum C
omp Z
oology, Harvard U
niversity on September 27, 2013
http://eurheartj.oxfordjournals.org/D
ownloaded from
at E
rnst Mayr L
ibrary of the Museum
Com
p Zoology, H
arvard University on Septem
ber 27, 2013http://eurheartj.oxfordjournals.org/
Dow
nloaded from
at Ernst M
ayr Library of the M
useum C
omp Z
oology, Harvard U
niversity on September 27, 2013
http://eurheartj.oxfordjournals.org/D
ownloaded from
at E
rnst Mayr L
ibrary of the Museum
Com
p Zoology, H
arvard University on Septem
ber 27, 2013http://eurheartj.oxfordjournals.org/
Dow
nloaded from

Genetically altered mice with 50% reduced FXIII levelshow impaired healing, enhanced ventricular dilation, and infarctrupture.6 Clinically, FXIII gene variant associated with higherplasma activity reduced cardiovascular mortality and the incidenceof heart failure at 1 year follow-up after MI.8 The aim of this studywas to evaluate the clinical significance of reduced FXIII levels inacute MI. This involved the measurement of FXIII levels in myocar-dial tissue specimens from nine patients with fatal infarct ruptures,an examination of the effects of prolonged heparin therapy onmyocardial FXIII levels and infarct healing, and assessing the thera-peutic potential of intravenous administration of FXIII pro-enzyme.
During the first week after MI, an acute phase reduction in FXIIIplasma levels has been described.8,9 In addition, patients who areadmitted with acute MI are often treated with heparin to inhibitclotting. Since all heparin formulations are thrombin inhibitors,they also inhibit FXIII activation and there have been reports thatheparins may interfere with the inflammatory response afterinjury10,11 and delay collagen synthesis during wound healing.12
The current study thus tested the following hypotheses: (i) FXIIItissue levels are lower in patients with acute infarct rupture; (ii)supranormal FXIII pro-enzyme plasma levels lead to higher FXIIIactivity in the MI and improve infarct healing; (iii) treatment withhigh doses of dalteparin (DP) is associated with decreased FXIIIactivity in the MI and with impaired myocardial wound healing.We tested hypotheses 2 and 3 in a murine model of MI using anovel, non-invasive molecular imaging approach to directlyreport FXIII activity in the healing infarct in vivo.
Methods
Myocardial transglutaminase factor XIIIlevels in patientsFXIII levels were studied by western blotting in myocardial samples ofnine patients who died 3–7 days after MI due to infarct rupture.Inclusion criteria were a history of acute MI, haemothorax, and aclearly visible site of rupture at autopsy. In the control group weused samples from nine patients with a cause of death unrelated toinfarct rupture, which was excluded by autopsy. The most commoncauses of death in this group were heart failure and arrhythmia.Both groups were matched for post-infarction survival time, age, andgender. The frozen samples were homogenized, sonicated and centri-fuged. Protein content was measured using the BCA protein Assay(Pierce Biotechnology). Ten microgram of total protein was separatedon a 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresisgel and transferred onto a Hybond C nitrocellulose membrane(Amersham Biosciences). After blocking, membranes were incubatedovernight with primary antibodies directed against FXIIIa 1:1500(Neomarker) and a-tubulin (H-300) 1:500 (Santa Cruz) for loadingcontrol. Collection, storage and use of human heart tissue andpatient data were undertaken in compliance with the ‘Code forProper Secondary Use of Human Tissue in the Netherlands’ (http://www.fmwv.nl), and informed consent was given by the families ofthe patients.
Mouse model and study outlineThis study used 110 C57BL6 mice: validation of 111In-DOTA-FXIII forSPECT-CT imaging of FXIII infarct activity, n ¼ 5; FXIII plasma leveldetermination in control-, DP- and FXIII-treated mice, n ¼ 10;
ex-vivo assessment of FXIII activity in the infarct of control-, DP-and FXIII-treated mice using 111In-DOTA-FXIII, n ¼ 32; longitudinalSPECT-CT and MRI trial for assessment of FXIII and DP effects, n ¼ 18;survival, histology and polymerase chain reaction (PCR) analysesin respective treatment groups, n ¼ 45. In addition, five FXIII2/2 micewere used for in vivo SPECT-CT validation (a gift of Dr Dickneite,ZLB Behring, Marburg, Germany). MI was induced by permanent coron-ary ligation. First, we validated molecular imaging of FXIII activity in theinfarct with SPECT-CT by imaging of FXIII2/2 mice, ex-vivo imaging,autoradiography and scintillation counting of excised hearts. Thereafter,we assessed the natural time course of FXIII activity in murine MI. In thein vivo therapy trial as outlined in Supplementary Figure S1 (see Sup-plementary material online), the following daily treatments werestarted 2 h after MI and continued for 7 days: (i) DP (Pharmacia, Kala-mazoo, MI, USA), 600 IU/kg sc., a dosage comparable to previousstudies investigating non-antithrombotic functions of heparin inmice;11,13–17 (ii) factor XIII (ZLB Behring, Marburg, Germany), 400 IU/kg iv.; (iii) saline control, 100 mL sc. Mice were randomly assigned totreatment prior to infarct surgery. The surgeon was blinded withrespect to treatment groups. We measured the resulting FXIII plasmalevels using a colorimetric assay (Berichrom assay, Behring, Marburg,Germany).18 Three hours after injection, FXIII plasma levels wereunchanged in the DP group (107+5% of control treatment, P ¼ 0.9),and significantly increased in the FXIII injected mice (170+5%,P , 0.01, n ¼ 4 per group). The impact of this treatment on FXIIIactivity in the infarct was then studied ex vivo in a separate group ofmice on day 3 after MI by scintillation counting. Furthermore, healingwas assessed by histopathology and real time reverse transcription–polymerase chain reaction in a group of mice sacrificed on day 7 afterMI. Mice were anesthetized for all procedures by inhalation anaesthesia(isoflurane 1–2% v/vþ2L O2). The institutional subcommittee onresearch animal care at MGH approved all animal studies.
Molecular imaging of in vivo transglutaminasefactor XIII activityTo assess the in vivo enzyme activity of FXIII within the infarct, weemployed an 111Indium labelled affinity peptide (111In-DOTA-FXIII).19
FXIII recognizes the probe as a substrate and cross-links it toextracellular matrix proteins, leading to local entrapment of111In-DOTA-FXIII in the healing infarct. After validation experimentsin five FXIII2/2 mice with MI, the imaging agent (100 mCi/animal iv.)was employed to determine the time course of endogenous FXIIIactivity in the MI. We measured radioactivity of excised hearts insham operated animals and at day 2, 3 and 4 after coronary ligationwith a gamma counter, with subsequent exposure on the phosphorimager.6 These myocardial rings were also stained with triphenyltetra-zolium chloride (TTC),6 to assess the distribution of the signal to theinfarct.
Non-invasive therapy trialWe performed late enhancement and cine MRI volumetry on day 2and day 21 after MI, and SPECT-CT imaging of FXIII activity on day3 after MI to assess the impact of FXIII modulating treatment as out-lined in Supplementary Figure S1. Ten minutes after iv. injection of0.4 mmol/kg Gd-DPTA, a 7 Tesla Pharmascan (Bruker, Billerica, MA,USA) and a gradient echo FLASH-sequence20 were employed with a200 mm�200 mm in-plane resolution and a slice thickness of 1 mm.Images were analysed using OsiriX to determine infarct size(Gd-enhanced myocardial volume as percentage of total LV myocardialvolume), LV volumes and mass, and ejection fraction (EF). FXIII activitywas imaged on an X-SPECT system (Gamma Medica, Northridge, CA,
M. Nahrendorf et al446

USA) 3 days post-MI. Isotropic resolution was 72 mm for CT and2 mm for SPECT. Imaging was performed 1 h after injection of 1mCi111In-DOTA-FXIII. Directly prior CT imaging, 250 mL iodinated CTcontrast agent was injected intravenously. The peak FXIII activity wasdetermined as a target to background ratio by drawing regions ofinterest in the infarct and skeletal muscle in fused SPECT-CT images.Analysis of MRI and SPECT-CT data was performed in a blindedfashion.
Real time polymerase chain reactionIn an additional group of mice, we determined the impact of DP andFXIII treatment on mRNA levels of TSP-1, TSP-2, vascular endothelialgrowth factor (VEGF), MMP-2, MMP-9, and collagen 1-a2 on day 7after MI using TriZol (Invitrogen) to isolate RNA and Oligo(dT)primers to transcribe mRNA into cDNA (StrataScript, Stratagene).Quantitative PCR was performed on an SDS 7000 system (ABI) intriplicates with b-Actin as an internal calibrator. Primer was generatedwith PrimerExpress.
Quantitative histopathological analysisOn day 7, mice of all treatment groups were sacrificed and heartsexcised for histological assessment (see Supplementary materialonline, Figure S1). In addition, neutrophil content was also analysedon day 2 after MI. Serial 6 mm thick sections were produced forimmunoreactive staining of neutrophils, macrophages, myofibroblasts,and endothelial cells forming capillaries with appropriate secondaryantibodies. The collagen content of the scar was visualized with picro-sirius red stain, which was assessed under polarized light.21 We quan-tified neutrophils, macrophages, and myofibroblasts using five highpower fields (�400) per section and per animal with IP Lab Software(Scanlytics, Fairfax, VA, USA).
StatisticsResults are expressed as mean+ SD. The data sets were testedfor normality using the Kolmogorov–Smirnov test with the Dallal–Wilkinson–Lilliefors correction, and for equality of variances usingthe F-test. If normality and equality of variances could not be rejectedat 0.05 significance level, the group means were compared using para-metric tests. Unpaired data were compared using the unpaired two-sided t-test, and for paired data using the paired two-sided t-test.If either normality or equality of variances were rejected, the non-parametric Mann–Whitney test was used. For comparison of multiplegroups, variance was analysed by ANOVA, followed by Bonferroni’smultiple comparison test. Survival was analysed by a Kaplan–Meierplot with a log-rank test for differences between groups. P , 0.05indicates statistical significance. We used Graphpad Prism 4.0c forMacintosh (GraphPad Software, Inc., San Diego, CA, USA) for statisti-cal analysis.
Results
Transglutaminase factor XIII levels arediminished in patients after infarctruptureFXIII protein levels were significantly diminished in the myocar-dium of nine patients who died due to acute infarct rupturewhen compared to patients who also died acutely after MI, butunrelated to infarct rupture (Figure 1, P ¼ 0.0045).
In vivo imaging of transglutaminase factorXIII activity by SPECT-CT is feasible andspecificSPECT-CT fusion imaging detected areas of high FXIIIactivity within the healing infarct (Figure 2). To validate thesource of the signal, we performed ex vivo SPECT-CT on anexplanted mouse heart that had been embedded in agar, confirm-ing the cardiac source of activity (Figure 2A and B). The infarct orthe thoracotomy wound did not enhance in FXIII2/2 mice, demon-strating the specificity of 111In-DOTA-FXIII (Figure 2D). Scintillationcounts yielded a 356% higher % injected dose per gram tissue inexplanted hearts from wild-type than in FXIII2/2 mice (P ¼ 0.0014).
Transglutaminase factor XIII activitypeaks on day 3 after myocardial infarctionWe next investigated the natural time course of FXIII activity byinjecting 111In-DOTA-FXIII on days 0, 2, 3 and 4 after MI usingthree mice per time point. FXIII activity peaked on day 3 after cor-onary ligation (Figure 3). Autoradiography corroborated these find-ings and located the imaging agent to the healing MI.
Dalteparin impairs and exogenoustransglutaminase factor XIII boostsuptake of 111In-DOTA-FXIIITreatment with DP significantly reduced FXIII activity in the healingMI (Figure 4A). Conversely, intravenous FXIII pro-enzyme sup-plementation increased 111In-DOTA-FXIII uptake. These resultswere corroborated by target to background ratios calculated fromautoradiography (control: 2.92+0.88, DP: 1.75+0.89, FXIII:4.18+1.85, P , 0.0001).
Dalteparin reduces survivalDaily subcutaneous injections with DP increased mortality signifi-cantly (Figure 4B). Autopsy established macroscopic rupture ofthe infarct in nine DP-treated mice (Figure 4B) and 1 control-treated mouse and did not show any thoracotomy related bleeding
Figure 1 Immunoblotting of transglutaminase factor XIII (FXIII)in cardiac tissue from patients after myocardial infarction (MI).Myocardial FXIII protein levels were significantly reduced inpatients who died from rupture acutely after MI. Control tissuesamples were retrieved from patients who also died after MI,but did not show signs of infarct rupture
Factor XIII modulation in MI healing 447

as source of death. DP-treated sham-operated mice survived thethoracotomy without increased mortality.
In vivo imaging of infarct healing and leftventricular remodellingLV remodelling was imaged by serial MRI in control and FXIII-treatedmice only, since DP-treated mice died before the second MRI could
be performed (Figure 5A– I). On day 2 we determined infarct size asenhanced % of the LV by late enhancement MRI (control 27+5%,DP 26+5%, FXIII 26+5%, P ¼ 0.95). SPECT-CT showed lowerFXIII activity with DP treatment, and higher activity in FXIII-treatedmice (Figure 5K). Serial MRI on day 2 and 21 post-MI revealed atte-nuated LV dilatation in FXIII treated mice (Figure 5L). LV massincreased due to post-MI hypertrophy, and this change was only sig-nificant in the control group (Table 1). Representative MR andSPECT-CT images are shown in Figure 5, and cine MRI movies areprovided as Supplementary material online.
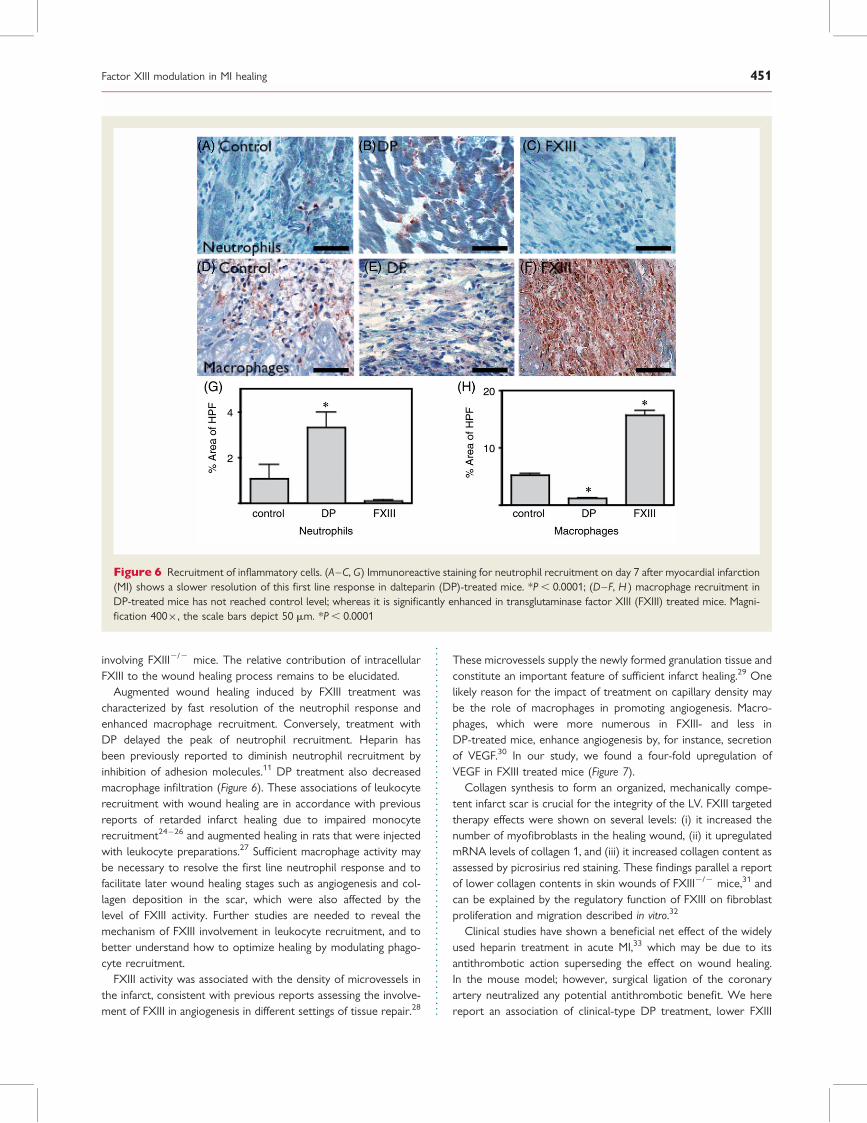
Altered recruitment of inflammatorycellsImmunoreactive staining revealed that treatment with DP signifi-cantly delayed neutrophil recruitment. In a subgroup of mice thatwas sacrificed on day 2 after MI, fewer neutrophils were observed(positive % area of high power field, DP 0.1+0.03%, FXIII 1.3+0.2%, P ¼ 0.0004); however, on day 7 the staining was mostintense in the DP group, thus establishing a delayed time courseof neutrophil influx (Figure 6A–C, G). Macrophages as assessedby MAC-3 staining were less numerous in mice that had receivedDP treatment and showed an enhanced presence in FXIII-treatedmice at day 7 after MI (Figure 6D-F, H).
AngiogenesisSeven days after induction of MI, DP-injected mice exhibited asignificantly lower capillary density in the infarct, whereastreatment with FXIII increased formation of new microvessels(Figure 7A–D). TSP-1, which has been described as downregulatedin FXIII mediated angiogenic activity in a transplant model,22
showed similar expression levels in the treatment groups(control: 0.25+0.08, DP: 0.21+ 0.12, FXIII: 0.22+ 0.03,P ¼ 0.80). TSP-2 was also unchanged by either treatment(control: 1.49+0.71, DP: 2.53+2.52, FXIII 2.35+0.35,P ¼ 0.72). VEGF was upregulated four-fold in FXIII-treated mice,and was unchanged in DP-treated mice (Figure 7E).
Figure 3 Time course of transglutaminase factor XIII (FXIII)activity. Endogenous FXIII activity in infarct healing peaks onday 3 after myocardial infarction (MI) as assessed by scintillationcounting and the target to background ratio of the phosphorimager. Comparison to TTC staining shows that the imagingagent targets the infarct
Figure 2 SPECT-CT imaging of transglutaminase factor XIII (FXIII) activity. (A) CT of an explanted heart with myocardial infarction (MI).Arrows indicate the infarct; (B) fusion of CT with SPECT reveals focal signal in the infarct; (C ) SPECT-CT shows high (FXIII) activity in theMI. Some SPECT signal is observed in the thorax wall, also a site of active wound healing; (D) SPECT-CT of a FXIII2/2 mouse shows nosignal in the infarct
M. Nahrendorf et al448

Dalteparin and transglutaminase factorXIII treatment modulate collagensynthesisThe presence of myofibroblasts, the source of collagen 1-a2,increased in the infarct of FXIII-treated mice, and diminishedby DP treatment (Figure 8A–C, G). In FXIII-treated mice, themRNA level of collagen 1-a2 significantly increased, and decreasedin the DP group (Figure 8H). Collagen staining of the infarctsdemonstrated increased amounts of collagen fibres in FXIII-, butless collagen in DP-treated animals (Figure 8D–F, I ). Levels ofMMP-2 (control: 4.01+0.53, DP: 6.00+2.95, FXIII: 6.74+2.92,P ¼ 0.49) and MMP-9 (control: 0.32+ 0.27, DP: 0.41+0.47,FXIII: 0.27+0.19, P ¼ 0.87) were not affected by treatment.
DiscussionIn patients with acute infarct rupture, FXIII tissue levels were sig-nificantly diminished. These data provide clinical evidence for therole of FXIII in cardiac healing. In a murine model of MI, DP treat-ment is associated with lower FXIII activity and impaired woundhealing. Therefore, heparin therapy in acute infarction might havedifferential effects in the infarct related artery (beneficial) andthe injured myocardium (detrimental). Conversely, supranormalFXIII zymogen plasma levels lead to higher local FXIII activity inthe MI and improve infarct healing. We furthermore demonstratethat molecular imaging of FXIII activity allows direct assessment ofwound healing in vivo and predicts prognosis after MI.
We assessed the time course of endogenous FXIII activity in thehealing murine infarct using an isotope labelled peptide. FXIIIspecifically recognizes this peptide as a substrate19 and cross-linksit to extracellular matrix proteins, leading to local entrapment ofthe radioactive labelled peptide in the healing infarct. The
specificity of this imaging agent has been compared to scrambledcontrol peptides19 and investigated in FXIII2/2 mice (Figure 2).The peak activity occurred on day 3 after MI (Figure 3), coincidingwith the beginning of the proliferative phase of infarct healing.23
This indicates that FXIII participates in the organization of thenew extracellular matrix forming the infarct scar, rather thanserving in its ‘traditional’ clotting-factor role by cross-linkingfibrin monomers. If the latter was true, activity would peakduring haemostasis, which occurs acutely after MI. One limitationof this study is the surgical induction of MI as opposed to throm-botic occlusion in the clinical setting, which is frequently followedby reperfusion. Clinical or large animal studies will have to definehow the present data predicts the situation in patients with MI, alsobecause surgical ligation of the coronary artery neutralizes bothantithrombotic benefits of heparin and potential prothromboticeffects of increased FXIII levels.
Serial MR imaging revealed attenuated LV remodelling in FXIIItreated mice as indicated by reduced LV dilation and less myocar-dial hypertrophy. Also, we found a trend towards preserved EF, andless infarct thinning and expansion (Table 1). It is therefore concei-vable that improved healing in FXIII-treated mice introduced ageometrical advantage with smaller initial LV volumes and lowerwall stress. Since wall stress is a major determinant of LV remodel-ling,4,5 this may have attenuated the evolution of heart failure inFXIII-treated mice. In a previous study, we found acceleratedpost-MI remodelling still occurred in FXIII2/2 mice resubstitutedwith FXIII (200 U/kg bodyweight), suggesting that the resubstitu-tion was only partial.6 In the present study, we treat wild-typemice, with normal basal levels of FXIII, with FXIII injections at400 U/kg in an attempt to create supranormal levels. In addition,wild-type mice do not lack intracellular FXIII as FXIII2/2 mice.These differences may explain why attenuated LV remodellingwas observed in the present study and not in the previous one
Figure 4 Transglutaminase factor XIII (FXIII) modulating therapy and survival. (A) Scintillation counting of explanted hearts showeddiminished FXIII activity in dalteparin (DP)-treated and higher activity in FXIII-treated mice. *P ¼ 0.045 vs. control, **P ¼ 0.0011;(B) Kaplan–Meier survival curve. Autopsy established a blood clot originating from a ruptured myocardial infarction (MI) (arrow)
Factor XIII modulation in MI healing 449

. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 1 Increase in left ventricular mass, and trend towards preserved ejection fraction and less infarct thinning andexpansion
Treatment group(imaging time point)
Ejectionfraction (%)
Myocardial infarctionthickness (mm)
Infarct size(%)
Left ventricularmass (mg)
End-diastolic volume(mL)
Control (day 2) 41+11 690+50 27+5 70.4+3.3 77.5+9.8
Dalteparin (day 2) 40+7 560+20 26+5 81.2+10.2 72.7+4.1
FXIII (day 2) 41+7 680+40 26+5 88.2+4.4 77.7+6.9
Control (day 21) 29+10 380+40* 27+5 103.0+2.7* 153.5+36.6*
FXIII (day 21) 35+5 505+60*,** 23+5 97.0+3.7 110.1+17.9*,**
*P , 0.05 vs. day 2 .**P ¼ 0.05 vs. control treatment.
Figure 5 In vivo molecular imaging of transglutaminase factor XIII (FXIII) activity predicts survival and evolution of heart failure. (A– I ) Longi-tudinal imaging study (MRI day 2, SPECT-CT day 3, second MRI day 21); on day 2 (A, D, G), late enhancement MRI showed similar infarct size inall groups. FXIII-treatment led to higher SPECT signal (H, K). In dalteparin-treated mice, the SPECT signal was lower (E, K ). Serial MRI showedattenuated left ventricular (LV) dilation in FXIII-treated mice (L). Due to reduced survival in dalteparin (DP)-treated mice, the second MRI onday 21 was not acquired (F ). *P , 0.05, **P , 0.001
M. Nahrendorf et al450
involving FXIII2/2 mice. The relative contribution of intracellularFXIII to the wound healing process remains to be elucidated.
Augmented wound healing induced by FXIII treatment wascharacterized by fast resolution of the neutrophil response andenhanced macrophage recruitment. Conversely, treatment withDP delayed the peak of neutrophil recruitment. Heparin hasbeen previously reported to diminish neutrophil recruitment byinhibition of adhesion molecules.11 DP treatment also decreasedmacrophage infiltration (Figure 6). These associations of leukocyterecruitment with wound healing are in accordance with previousreports of retarded infarct healing due to impaired monocyterecruitment24–26 and augmented healing in rats that were injectedwith leukocyte preparations.27 Sufficient macrophage activity maybe necessary to resolve the first line neutrophil response and tofacilitate later wound healing stages such as angiogenesis and col-lagen deposition in the scar, which were also affected by thelevel of FXIII activity. Further studies are needed to reveal themechanism of FXIII involvement in leukocyte recruitment, and tobetter understand how to optimize healing by modulating phago-cyte recruitment.
FXIII activity was associated with the density of microvessels inthe infarct, consistent with previous reports assessing the involve-ment of FXIII in angiogenesis in different settings of tissue repair.28
These microvessels supply the newly formed granulation tissue andconstitute an important feature of sufficient infarct healing.29 Onelikely reason for the impact of treatment on capillary density maybe the role of macrophages in promoting angiogenesis. Macro-phages, which were more numerous in FXIII- and less inDP-treated mice, enhance angiogenesis by, for instance, secretionof VEGF.30 In our study, we found a four-fold upregulation ofVEGF in FXIII treated mice (Figure 7).
Collagen synthesis to form an organized, mechanically compe-tent infarct scar is crucial for the integrity of the LV. FXIII targetedtherapy effects were shown on several levels: (i) it increased thenumber of myofibroblasts in the healing wound, (ii) it upregulatedmRNA levels of collagen 1, and (iii) it increased collagen content asassessed by picrosirius red staining. These findings parallel a reportof lower collagen contents in skin wounds of FXIII2/2 mice,31 andcan be explained by the regulatory function of FXIII on fibroblastproliferation and migration described in vitro.32
Clinical studies have shown a beneficial net effect of the widelyused heparin treatment in acute MI,33 which may be due to itsantithrombotic action superseding the effect on wound healing.In the mouse model; however, surgical ligation of the coronaryartery neutralized any potential antithrombotic benefit. We herereport an association of clinical-type DP treatment, lower FXIII
Figure 6 Recruitment of inflammatory cells. (A–C, G) Immunoreactive staining for neutrophil recruitment on day 7 after myocardial infarction(MI) shows a slower resolution of this first line response in dalteparin (DP)-treated mice. *P , 0.0001; (D–F, H ) macrophage recruitment inDP-treated mice has not reached control level; whereas it is significantly enhanced in transglutaminase factor XIII (FXIII) treated mice. Magni-fication 400�, the scale bars depict 50 mm. *P , 0.0001
Factor XIII modulation in MI healing 451

Figure 8 Collagen synthesis. (A–C, G) Immunoreactive staining for myofibroblasts, the cellular source of collagen, was decreased in dalteparin(DP)-treated and increased in transglutaminase factor XIII (FXIII)-treated mice. Magnification 400�, the scale bars depict 20 nm. *P , 0.0001. Inparallel, mRNA levels of collagen 1-a2 were decreased in DP-treated and increased in FXIII-treated mice (H ) *P ¼ 0.0026. (D–F, I ) Collagencontent in polarized light microscopy of picrosirius red stains revealed less collagen deposition in DP-treated and increased collagen content inFXIII-treated mice. Magnification 200�, the scale bars depict 50 mm. *P , 0.0001
Figure 7 Capillary density and VEGF expression. (A–D) Microvessel density is reduced by dalteparin (DP). Conversely, transglutaminasefactor XIII (FXIII) treatment increased microvessel density. Magnification 400�, the scale bars depict 50 mm. *P , 0.0001; (E) in FXIII-treatedmice, the level of VEGF mRNA was increased four-fold, consistent with the increased vessel density observed in Figure 6C
M. Nahrendorf et al452

activity in the infarct, and increased infarct rupture. Heparin exertsa wide range of effects modulating the inflammatory, proliferativeand fibrotic response, independent of FXIII activation.11,13 –15,34
Given this broad action of heparin, the detrimental effect of DPmay also have been caused or cofounded by mechanisms otherthan the inhibition of FXIII activation. However, the FXIII activitylevels found on day 3 in the infarct of DP-treated mice (65%reduction in scintillation counts) closely resembled the situationin FXIIIþ/2 mice, which have a 50% reduction of FXIII plasmalevels and were also reported to die from infarct rupture.6 Sincethe observed effects may be species-specific and not directly trans-latable, further studies in large animal models and humans areneeded to confirm these data. While heparin therapy has beneficialanticoagulant properties and promotes the patency of the infarctrelated artery, it may retard wound healing in the myocardium.Future strategies should thus aim to maintain the beneficial antic-oagulatory effects of heparin in the infarct related vessel, butprevent its effects on wound healing.
In summary, FXIII levels are significantly decreased in patientswith ruptured infarcts. In the mouse model of coronary ligation,modulation of FXIII activity by therapy impacts myocardialhealing. This finding is of clinical importance, since it potentiallytranslates into novel therapeutic strategies to augment infarcthealing and retard the genesis of heart failure post-MI. Patientswith low FXIII plasma levels, which occur naturally after MI,8,9 maypotentially benefit from transient and local augmentation of FXIIIactivity in the infarct.
Supplementary materialSupplementary material is available at European Heart Journalonline.
AcknowledgementsThe authors would like to acknowledge Gerhard Dickneite, PhD,ZLB Behring (Marburg, Germany) for the generous gift of fibro-gammin and FXIII knock out mice; Peter Whittaker, PhD, Univer-sity of Massachusetts; Georg Ertl, MD, Wurzburg University; andRichard T. Lee, MD, PhD, Brigham and Women’s Hospital,Boston, for fruitful discussions; Jan Grimm, MD, PhD, for helpwith establishing cardiac SPECT-CT imaging; the CMIR PathologyCore (Vincent Lok, BS; Todd Sponholz, MS) for assistance with his-tology; Nan-Hui Ho, PhD and Hanwen Zhang, PhD, for synthesiz-ing the FXIII peptide probe; the CMIR mouse imaging programme(Carlos Rangel, BS; Claire Kaufman, BS; Sheena Hembrador, BS;and Gregory Wojtkiewicz, MS); and the MGH PCR core facility.Support sources: RO1-HL078641 (RW), and UO1-HL080731(RW).
Conflict of interest: none declared.
References1. Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L,
Maggioni AP, Solomon SD, Swedberg K, Van de Werf F,White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S,Sellers MA, Califf RM. Valsartan, captopril, or both in myocardial
infarction complicated by heart failure, left ventricular dysfunction,or both. N Engl J Med 2003;349:1893–1906.
2. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardialinfarction. Experimental observations and clinical implications. Cir-culation 1990;81:1161–1172.
3. Anversa P, Olivetti G, Capasso JM. Cellular basis of ventricularremodeling after myocardial infarction. Am J Cardiol 1991;68:7D–16D.
4. Sutton MG, Sharpe N. Left ventricular remodeling after myocardialinfarction: pathophysiology and therapy. Circulation 2000;101:2981–2988.
5. Nahrendorf M, Wiesmann F, Hiller KH, Hu K, Waller C, Ruff J,Lanz TE, Neubauer S, Haase A, Ertl G, Bauer WR. Serial cine-magnetic resonance imaging of left ventricular remodeling aftermyocardial infarction in rats. J Magn Reson Imaging 2001;14:547–555.
6. Nahrendorf M, Hu K, Frantz S, Jaffer FA, Tung CH, Hiller KH,Voll S, Nordbeck P, Sosnovik D, Gattenlohner S, Novikov M,Dickneite G, Reed GL, Jakob P, Rosenzweig A, Bauer WR,Weissleder R, Ertl G. Factor XIII deficiency causes cardiacrupture, impairs wound healing, and aggravates cardiac remodelingin mice with myocardial infarction. Circulation 2006;113:1196–1202.
7. Muszbek L, Yee VC, Hevessy Z. Blood coagulation factor XIII:structure and function. Thromb Res 1999;94:271–305.
8. Gemmati D, Federici F, Campo G, Tognazzo S, Serino ML, DeMattei M, Valgimigli M, Malagutti P, Guardigli G, Ferraresi P,Bernardi F, Ferrari R, Scapoli GL, Catozzi L. Factor XIIIA-V34Land factor XIIIB-H95R gene variants: effects on survival in myocar-dial infarction patients. Mol Med 2007;13:112–120.
9. Alkjaersig N, Fletcher AP, Lewis M, Ittyerah R. Reduction of coagu-lation factor XIII concentration in patients with myocardial infarc-tion, cerebral infarction, and other thromboembolic disorders.Thromb Haemost 1977;38:863–873.
10. Lucio J, D’Brot J, Guo CB, Abraham WM, Lichtenstein LM,Kagey-Sobotka A, Ahmed T. Immunologic mast cell-mediatedresponses and histamine release are attenuated by heparin.J Appl Physiol 1992;73:1093–1101.
11. Nelson RM, Cecconi O, Roberts WG, Aruffo A, Linhardt RJ,Bevilacqua MP. Heparin oligosaccharides bind L- and P-selectinand inhibit acute inflammation. Blood 1993;82:3253–3258.
12. Thompson RC Jr, Ludewig RM, Wangensteen SL, Rudolf LM.Effects of heparin on wound healing. Surg Gynecol Obstet 1972;134:22–26.
13. Lund EL, Olsen MW, Lipson KE, McMahon G, Howlett AR,Kristjansen PE. Improved effect of an antiangiogenic tyrosinekinase inhibitor (SU5416) by combinations with fractionated radio-therapy or low molecular weight heparin. Neoplasia 2003;5:155–160.
14. Stevenson JL, Choi SH, Varki A. Differential metastasis inhibitionby clinically relevant levels of heparins–correlation with selectininhibition, not antithrombotic activity. Clin Cancer Res 2005;11:7003–7011.
15. Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastaticeffect of tinzaparin, a low-molecular-weight heparin. J ThrombHaemost 2003;1:1972–1976.
16. Shen ZX, Basara N, Xi XD, Caen J, Maffrand JP, Pascal M,Petitou M, Lormeau JC, Han ZC. Fraxiparin, a low-molecular-weight heparin, stimulates megakaryocytopoiesis in vitro and invivo in mice. Br J Haematol 1994;88:608–612.
17. Echtenacher B, Weigl K, Lehn N, Mannel DN. Tumor necrosisfactor-dependent adhesions as a major protective mechanism
Factor XIII modulation in MI healing 453

early in septic peritonitis in mice. Infect Immun 2001;69:3550–3555.
18. Lauer P, Metzner HJ, Zettlmeissl G, Li M, Smith AG, Lathe R,Dickneite G. Targeted inactivation of the mouse locus encodingcoagulation factor XIII-A: hemostatic abnormalities in mutantmice and characterization of the coagulation deficit. ThrombHaemost 2002;88:967–974.
19. Tung CH, Ho NH, Zeng Q, Tang Y, Jaffer FA, Reed GL,Weissleder R. Novel factor XIII probes for blood coagulationimaging. Chembiochem 2003;4:897–899.
20. Yang Z, Berr SS, Gilson WD, Toufektsian MC, French BA. Simul-taneous evaluation of infarct size and cardiac function in intactmice by contrast-enhanced cardiac magnetic resonance imagingreveals contractile dysfunction in noninfarcted regions early aftermyocardial infarction. Circulation 2004;109:1161–1167.
21. Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitativeassessment of myocardial collagen with picrosirius red staining andcircularly polarized light. Basic Res Cardiol 1994;89:397–410.
22. Dardik R, Solomon A, Loscalzo J, Eskaraev R, Bialik A, Goldberg I,Schiby G, Inbal A. Novel proangiogenic effect of factor XIII associ-ated with suppression of thrombospondin 1 expression. Arterios-cler Thromb Vasc Biol 2003;23:1472–1477.
23. Frangogiannis NG. Targeting the inflammatory response in healingmyocardial infarcts. Curr Med Chem 2006;13:1877–1893.
24. Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M.Targeted deletion of CC chemokine receptor 2 attenuates leftventricular remodeling after experimental myocardial infarction.Am J Pathol 2004;165:439–447.
25. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G,Abou-Khamis T, Michael LH, Rollins BJ, Entman ML,Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1regulates inflammatory responses critical to healing myocardialinfarcts. Circ Res 2005;96:881–889.
26. Nahrendorf M, Sosnovik DE, Waterman P, Swirski FK, Pande AN,Aikawa E, Figueiredo JL, Pittet MJ, Weissleder R. Dual channeloptical tomographic imaging of leukocyte recruitment and
protease activity in the healing myocardial infarct. Circ Res 2007;100:1218–1225.
27. Leor J, Rozen L, Zuloff-Shani A, Feinberg MS, Amsalem Y,Barbash IM, Kachel E, Holbova R, Mardor Y, Daniels D,Ocherashvilli A, Orenstein A, Danon D. Ex vivo activated humanmacrophages improve healing, remodeling, and function of theinfarcted heart. Circulation 2006;114(Suppl. 1):I94–I100.
28. Dardik R, Loscalzo J, Inbal A. Factor XIII (FXIII) and angiogenesis.J Thromb Haemost 2006;4:19–25.
29. Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E,Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A,Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM,Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ,Carmeliet P. Inhibition of plasminogen activators or matrix metal-loproteinases prevents cardiac rupture but impairs therapeuticangiogenesis and causes cardiac failure. Nat Med 1999;5:1135–1142.
30. Cianfarani F, Tommasi R, Failla CM, Viviano MT, Annessi G, Papi M,Zambruno G, Odorisio T. Granulocyte/macrophage colony-stimulating factor treatment of human chronic ulcers promotesangiogenesis associated with de novo vascular endothelialgrowth factor transcription in the ulcer bed. Br J Dermatol 2006;154:34–41.
31. Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneitte G,Modis L, Muszbek L, Inbal A. Impaired wound healing in factorXIII deficient mice. Thromb Haemost 2005;94:432–437.
32. Dardik R, Krapp T, Rosenthal E, Loscalzo J, Inbal A. Effect of FXIIIon monocyte and fibroblast function. Cell Physiol Biochem 2007;19:113–120.
33. Yusuf S, Mehta SR, Chrolavicius S, Afzal R, Pogue J, Granger CB,Budaj A, Peters RJ, Bassand JP, Wallentin L, Joyner C, Fox KA.Effects of fondaparinux on mortality and reinfarction in patientswith acute ST-segment elevation myocardial infarction: theOASIS-6 randomized trial. JAMA 2006;295:1519–1530.
34. Dager WE, Andersen J, Nutescu E. Special considerations withfondaparinux therapy: heparin-induced thrombocytopenia andwound healing. Pharmacotherapy 2004;24:88S–94S.
M. Nahrendorf et al454




















