
Towards the understanding of the function and regulation of a ...
226
Towards the understanding of the function and regulation of a membrane protein complex involving SppA and YteJ in Bacillus subtilis. Thèse de doctorat de l'Université Paris-Saclay préparée à l’Université Paris-Sud École doctorale n°577 structure et dynamique des systèmes vivants (SDSV) Spécialité de doctorat: Sciences de la vie et de la santé Thèse présentée et soutenue à Jouy-en-Josas, le 1 Julliet 2019, par Gabriela Fernandes Henriques Composition du Jury: Mme. Anne Galinier Directrice de recherche, CNRS-IMM, Marseille Rapportrice Mme. Nathalie Declerck Directrice de recherche, INRA-CBS, Montpellier Rapportrice Mme. Sylvie Nessler Professeure, Université Paris-Sud XI, Orsay Président du Jury Mme. Nathalie Campo Chargée de recherche, CNRS-LMGM, Toulouse Examinatrice M. Martin Picard Directeur de recherche, CNRS- IBPC, Paris Examinateur M. Jean Luc Pernodet Directeur de Recherche, CNRS-I2BC, Orsay Examinateur M. Matthieu Jules Professeur, AgroParisTech, Micalis, Jouy-en-Josas Directeur de thèse M. Olivier Delumeau Ingénieur de recherche, Micalis, Jouy-en-Josas Co-Directeur de thèse NNT : 2019SACLS191
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Towards the understanding of the function and regulation of a ...

Towards the understanding of the
function and regulation of a membrane
protein complex involving SppA and YteJ
in Bacillus subtilis.
Thèse de doctorat de l'Université Paris-Saclay
préparée à l’Université Paris-Sud
École doctorale n°577 structure et dynamique des systèmes vivants (SDSV)
Spécialité de doctorat: Sciences de la vie et de la santé
Thèse présentée et soutenue à Jouy-en-Josas, le 1 Julliet 2019, par
Gabriela Fernandes Henriques
Composition du Jury:
Mme. Anne Galinier
Directrice de recherche, CNRS-IMM, Marseille Rapportrice
Mme. Nathalie Declerck
Directrice de recherche, INRA-CBS, Montpellier Rapportrice
Mme. Sylvie Nessler
Professeure, Université Paris-Sud XI, Orsay Président du Jury
Mme. Nathalie Campo
Chargée de recherche, CNRS-LMGM, Toulouse Examinatrice
M. Martin Picard
Directeur de recherche, CNRS- IBPC, Paris Examinateur
M. Jean Luc Pernodet
Directeur de Recherche, CNRS-I2BC, Orsay Examinateur
M. Matthieu Jules
Professeur, AgroParisTech, Micalis, Jouy-en-Josas Directeur de thèse
M. Olivier Delumeau
Ingénieur de recherche, Micalis, Jouy-en-Josas Co-Directeur de thèse
NN
T :
20
19
SA
CLS
191


Title : Towards the understanding of the function and regulation of a membrane complex
involving SppA and YteJ in Bacillus subtilis.
Keywords : Bacillus subtilis, Protein secretion, Membrane protein complex, Protease, Lantibiotic resistance,
Quality control
Abstract: Bacillus subtilis is a model organism for Gram-positive bacteria and a workhorse used for large-
scale production of industrial enzymes. Secretory and membrane proteins are mainly translocated through the
SecA-YEG (Sec) pathway. Although this highly conserved pathway has been extensively studied, there is a
lack of knowledge about the protein interactions and complexes involved in this mechanism in B. subtilis.
Therefore, to better understand these membrane protein complexes, we SPA-tagged a set of proteins involved
in the Sec pathway for tandem affinity purification. Through the combination of this technique with mass
spectrometry and Blue Native PAGE, we studied the protein interactions within the (holo)translocon as well
as with the accessory proteins. Our results showed that, under their SPA-tagged forms, SecDF and SpoIIIJ,
could not be retrieved together after immune-capture, as opposed to what was shown in E. coli. This result
indicated that SecDF and SpoIIIJ, unexpectedly, do not form a stable complex in B. subtilis. The other
experiments using SPA-tagged proteins (SecG, BdbB, and BdbC) did not provide evidence for the existence
of stable complexes involving proteins known to be part of the translocon complex, except for one protein, the
so-called signal peptide peptidase, SppA.Using the SPA-tag technique, we have identified a membrane protein
complex, involving SppA and another membrane protein, YteJ. While YteJ function is unknown, SppA is a
known membrane bound serine protease, described as responsible for recycling of the signal peptides of the
translocated proteins left in the membrane and also involved in the resistance to nisin, an antibacterial peptide
Of the lantibiotic family. Interestingly, sppA and yteJ genes are organized in operon, for which we confirmed
the regulation by two sigma factors, σA and σW, using transcriptional fusions. The fluorescent microscopy
experiments revealed a diffused membrane localization for both proteins, and therefore their co-localization
was difficult to prove by this technique. Using deletion mutant strains, we showed that both SppA and YteJ
are involved in the lantibiotic resistance: the sppA- strain was more sensitive while the yteJ- strain showed a
more sensitive phenotype than wild type, but less pronounced than the sppA- strain. The sppA-dependent
resistance was confirmed by the ectopic overexpression of SppA that restored the lantibiotic resistance.
However, the overexpression of SppA also induced the formation of elongated cells. Interestingly, this
phenotype can be suppressed by the simultaneous overexpression of YteJ, indicating an YteJ-dependent
negative control of SppA activity. Furthermore, using the elongated cell phenotype, we tested the
overexpression of SppA along with truncated versions of YteJ. The results pinpointed to the inhibitory role of
the C-terminal domain of YteJ. Finally, we performed in vitro biochemical studies with purified His-tagged
SppA and YteJ which showed that B subtilis SppA and YteJ form a membrane complex when expressed
together in E. coli. That the SppA protease activity of the purified complex His-SppA-YteJ was almost null,
and that, purified from the membrane, the full length SppA protease activity was lower than that of a soluble
truncated version of SppA. This full-length SppA activity was strongly reduced in the presence of YteJ,
supporting the hypothesis of an inhibition by YteJ. Furthermore, we showed that the ability or inability of
SppA to digest fully folded proteins probably depends on the structure of the proteins, suggesting a possible
role in protein quality control. In conclusion, our in vivo and in vitro studies showed that YteJ, via one of its
domain, acts as a negative regulator of the protease activity of SppA in this complex. Although we have
documented a role for this complex in the lantibiotic resistance, a possible role of SppA in protein quality
control might not be excluded

I
Titre : Caractérisation du complexe membranaire impliquant la signal peptide peptidase
SppA et YteJ chez Bacillus subtilis.
Mots clés : Sécretion protéique, complexe protéique membranaire, protéase, résistance aux
lantibiotiques, contrôle qualité
INTRODUCTION :
Les complexes membranaires impliqués dans la sécrétion des protéines chez B. subtilis ont été identifiés
mais ont été peu étudiés biochimiquement contrairement à leurs homologues chez E. coli.
Objectifs de l’étude :Dans le cadre de l’ITN « ProteinFactory », dont le but était d’améliorer les
capacités de production/sécrétion des protéines chez les bactéries, nous avons tenté d’identifier les
complexes membranaires impliqués dans la sécrétion de protéines chez B. subtilis en nous focalisant sur
les protéines de la voie « Sec » (Figure 1). En effet, si les connaissances fondamentales ont été obtenues
chez E. coli, peu de données existent sur les interactions physiques entre ces protéines chez B. subtilis,
ou encore leur dynamique en fonction de la protéine sécrétée ou de la phase de croissance, par exemple.
Grâce à l’emploi de deux techniques biochimiques : le BN-PAGE et l’étiquetage de protéines ciblées
pour réaliser de la purification par affinité en tandem (SPA-tag), nous avons tenté de répondre à ces
questions.
Le modèle d’étude : Bacillus subtilis est une bactérie modèle pour l’étude des bactéries à Gram positif ;
elle est capable de produire des endospores afin de résister aux conditions difficiles et est aussi un
modèle pour l’industrie par sa capacité à sécréter un grand nombre de molécules, et notamment des
protéines (Figure 2).

II
Au cours de cette étude, nous avons identifié un complexe stable formé de deux protéines membranaires,
SppA et YteJ. SppA a été décrite comme une « Signal Peptide Peptidase », une protéase qui dégrade les
peptides signaux qui s’accumulent dans la membrane cytoplasmique après libération des protéines
sécrétées. Une structure de la partie soluble de SppA de B. subtilis par cristallographie existe. Située à
l’extérieur de la cellule et ancrée à la membrane par un seul domaine transmembranaire, SppA forme
un homo-octamère [2, 3]. Son domaine C-terminal est absent dans les structures de la protéine active,
mais est présent dans un mutant catalytique, indiquant sa fonction régulatrice (Figure 3).
Par ailleurs, le gène sppA fait partie du régulon σW, qui est un facteur sigma qui devient actif lors d’un
stress affectant la membrane ou la paroi [4]. Ainsi il a été montré qu’un mutant ∆sppA est plus sensible
à la nisine (peptide antibiotique de la famille des lantibiotiques).
Enfin, le rôle prépondérant de SppA dans le recyclage des peptides signaux de la membrane a été remis
en cause par une étude qui a suggéré que RasP était la SP peptidase majeure chez B. subtilis [5]. Ceci
nous a amené à revisiter le rôle de SppA chez B. subtilis ainsi que celui de YteJ, protéine dont nous
avons découvert qu’elle formait un complexe membranaire avec SppA.
RESULTATS :
1. Identification du complexe SppA/YteJ par BN-PAGE et SPA-Tag :
1.1 Par BN-PAGE :
La technique du Blue-Native PAGE permet de réaliser une électrophorèse des complexes membranaires
dans des conditions où les interactions physiques entre les protéines sont préservées au maximum. Ainsi,
un gel de BN-PAGE d’échantillon de membranes solubilisées avec du DDM, découpé en morceaux le
long de la migration, suivi d’une analyse par spectrométrie de masse montre que les deux protéines se

III
trouvent dans la même portion de gel (Figure 4). Nous en avons conclu que SppA et YteJ co-migrent
dans ce type de gel ce qui suggère que les deux protéines forme potentiellement un complexe protéique
membranaire. YteJ, protéine inconnue, est une protéine de la famille RDD, famille de fonction inconnue,
potentiellement un transporteur.
1.2 Par SPA-tag :
La séquence d’une étiquette permettant une double chromatographie séquentielle par affinité a été
fusionnée en 5’ de la séquence du gène de la protéine d’intérêt par intégration chromosomique du
plasmide pMUTIN-SPA-LIC. Des souches sppA-SPA et yteJ-SPA furent ainsi obtenues mais seule la
souche yteJ-SPA a montré une expression de YteJ-SPA détectable par Western-blot. La purification de
YteJ-SPA à partir des membranes, solubilisées en présence de DDM, a été réalisée et les protéines co-
purifiées ont été analysées par spectrométrie de masse. La protéine la plus abondante ayant été co-
purifiée avec YteJ-SPA était SppA. Nous avons pu estimer d’après ces données de spectrométrie de
masse que le ratio entre SppA et YteJ dans le complexe était de 1:1 ou 2:1.
2. Etude des mutants de délétion de sppA et/ou yteJ :
D’après la littérature, SppA est une signal peptide peptidase, mais est aussi impliquée dans la résistance
à la nisin, un peptide antibiotique de la famille des lantibiotiques, supposément grâce à son activité
peptidase. Afin d’étudier le rôle de YteJ dans le complexe que cette protéine forme avec SppA, des
souches de B. subtilis délétées de sppA et/ou yetJ ont été construites et caractérisées.
2.1 Caractérisation des souches mutantes :
La croissance de ces souches mutantes n’est nullement affectée dans les premières 24 heures (Figure 5
A). Toutefois, la survie à 48 heures (nombre de cellules formant colonie) est particulièrement affectée
pour les souches ∆sppA et ∆sppA-yteJ, alors que la survie de la souche ∆yteJ était moins affectée. La
survie de la souche ∆sppA n’a pas pu être restaurée en co-cultivant celle-ci avec des cellules de la souche
sauvage. Il semblerait donc que l’activité de SppA soit importante sur la survie à long terme de B. subtilis.
En revanche, le phénotype observé pour la souche ∆yteJ est intermédiaire (Figure 5 B).

IV
2.2 La sécrétion des protéines :
Afin de tester le rôle de SppA et de YteJ, potentiellement via son interaction avec SppA, dans la sécrétion
des protéines, nous avons utilisé la protéine modèle AmyM dont le gène est présent sur le plasmide
pCS73. Aucune des souches comportant les délétions de sppA et/ou yteJ, n’ont montré de différence
dans la quantité finale de AmyM sécrétée dans le milieu (Figure 6). Des expériences de « pulse-
chase assays » avec ces mêmes souches mutantes réalisées par des collaborateurs (Pr JM Van Dijl,
Groningen) ont également conclu à l’absence de différence dans la cinétique de sécrétion de AmyQ.
2.3 Résistance à la subtilin
Après avoir testé la nisine, peptide de la famille des lantibiotiques, nous avons testé un autre peptide de
cette famille, la subtiline, que nous avons produite à l’aide d’une souche sécrétrice de B. subtilis
(ATCC6633). Les résultats obtenus avec la nisine et la subtiline ont été identiques, quoique plus
reproductibles avec la subtiline (Figure 7). La présence de subtiline (ou nisine) dans le milieu de culture
LB sur les cellules de la souche sauvage provoque un retard de croissance mesurable par densité optique
et ce retard est accentué chez la souche sppA à partir de 25% de subtiline. La souche yteJ montre un
phénotype intermédiaire à partir de 30%. La résistance aux lantibiotiques liée à SppA décrite dans la
littérature est donc confirmée, et nos expériences montrent que YteJ semble avoir un rôle dans cette
résistance.
A B
Figure 5 : A : Croissance en milieu LB de la souche sauvage (BSB1) et des souches délétées de sppA, yteJ
ou sppA-yteJ. B : survie à long-terme des différentes souches après comptage des cellules viables sur boites
gélosées LB-agar.
Figure 6 : Analyse par SDS-PAGE de la quantité de AmyM sécrétée dans la souche sauvage comparée aux
souches délétées de sppA, yetJ et sppA-yteJ.

V
Afin de vérifier que l’absence de SppA – et celle de YetJ - était bien la raison directe de la sensibilité
accrue à la subtiline chez les souches délétées de ces gènes, nous avons utilisé ces souches et les avons
complémentées en réintroduisant, en ectopique au locus amyE, une copie du gène délété, sppA ou yetJ,
ou encore de l’operon sppA-yteJ, sous un promoteur inductible à l’IPTG. Ces souches furent testées en
présence de 30% de subtiline, concentration retenue car provoquant une différence observable de
résistance à la subtiline entre les souches. Dans ces conditions, des concentrations croissantes d’IPTG
ont été ajouté dans le milieu LB, afin de tester la dose-réponse de l’expression de ces gènes pour la
résistance à la subtiline (Figure 8).
Figure 7 : Croissance de cellules de B. subtilis mesurée par densité optique à 600 nm dans un lecteur de
microplaques en présence de différentes concentrations de subtiline. Chaque courbe représente la moyenne
de 6 réplicats.
Figure 8 : Croissance des cellules de la souche sauvage (BSB1) et des souches délétées sppA et yetJ
complémentées de leurs gènes réciproques sous le contrôle d’un promoteur Phyperspank (Phs) inductible à l’IPTG
(notées Phs-sppA et Phs-yteJ). L’effet de la subtiline (30%) est testé en fonction de concentrations croissantes
d’IPTG (0 à 100 µM).

VI
A partir de 25 µM d’IPTG, les courbes de croissance de la souche Phs-sppA se rapproche de la souche
sauvage et de la souche Phs-yteJ, qui, elle, se comporte comme la souche sauvage. Ceci prouve que la
résistance à la subtiline dépend bien directement de SppA. En revanche, à partir de 100 µM IPTG, nous
avons systématiquement observé que les cellules de la souche Phs-sppA lysaient rapidement après
l’entrée en phase stationnaire. Il semblerait donc que la surexpression de SppA, seule, provoque un effet
délétère sur les cellules. Cet effet n’est en revanche pas observé lorsque les deux protéines SppA et YteJ
sont surexprimées en même temps comme c’est le cas de la souche Phs-sppA-yteJ (Figure 9). Nous
pouvons noter que, alors que sans IPTG cette souche est plus sensible à la subtiline, une meilleure
résistance à la subtiline pour cette souche par rapport à la souche sauvage est observable dès l’ajout de
10 µM d’IPTG dans le milieu de culture.
Ces expériences nous ont permis de confirmer le rôle direct de SppA dans la résistance à la subtiline,
possiblement via son activité protéasique/peptidasique. Cependant, une surexpression de SppA devient
délétère au-delà d’un seuil, ce qui se traduit par une lyse prématurée des cellules. En revanche, la
surexpression simultanée des deux protéines ne provoque aucun problème aux cellules de cette souche
et lui permet même d’être plus résistante que la souche sauvage.
3. Régulation de l’activité protéase de SppA La surexpression de SppA dans la souche Phs-sppA nous avait montré que, pour des concentrations de
100 µM d’IPTG, des effets délétères sur les cellules pouvaient être observés à travers une biomasse plus
faible en phase stationnaire et une chute de la densité optique certainement due à une lyse prématurée
des cellules. Cet effet était totalement annihilé par la surexpression concomitante de YteJ. Nous avons
voulu observé au microscope l’effet sur la morphologie cellulaire.
3.1 Effet de la surexpression de SppA sur la morphologie cellulaire de cellules de B. subtilis Les effets sur la morphologie cellulaire furent observés sur des cellules prises en phase
exponentielle de croissance, en présence de 100 µM d’IPTG, au microscope à contraste de phase ; les
membranes révélées avec une coloration au FM 4-64 et l’ADN chromosomique coloré au DAPI (Figure
10).
Figure 9 : Croissance des cellules de la souche sauvage (BSB1) et de la souche délétée de l’opéron sppA-
yetJ complémentés par cet opéron en ectopique sous le contrôle d’un promoteur Phyperspank (Phs) inductible à
l’IPTG (notées Phs-sppA-yteJ). L’effet de la subtiline (30%) est testé en fonction de concentrations
croissantes d’IPTG (0 à 100 µM).
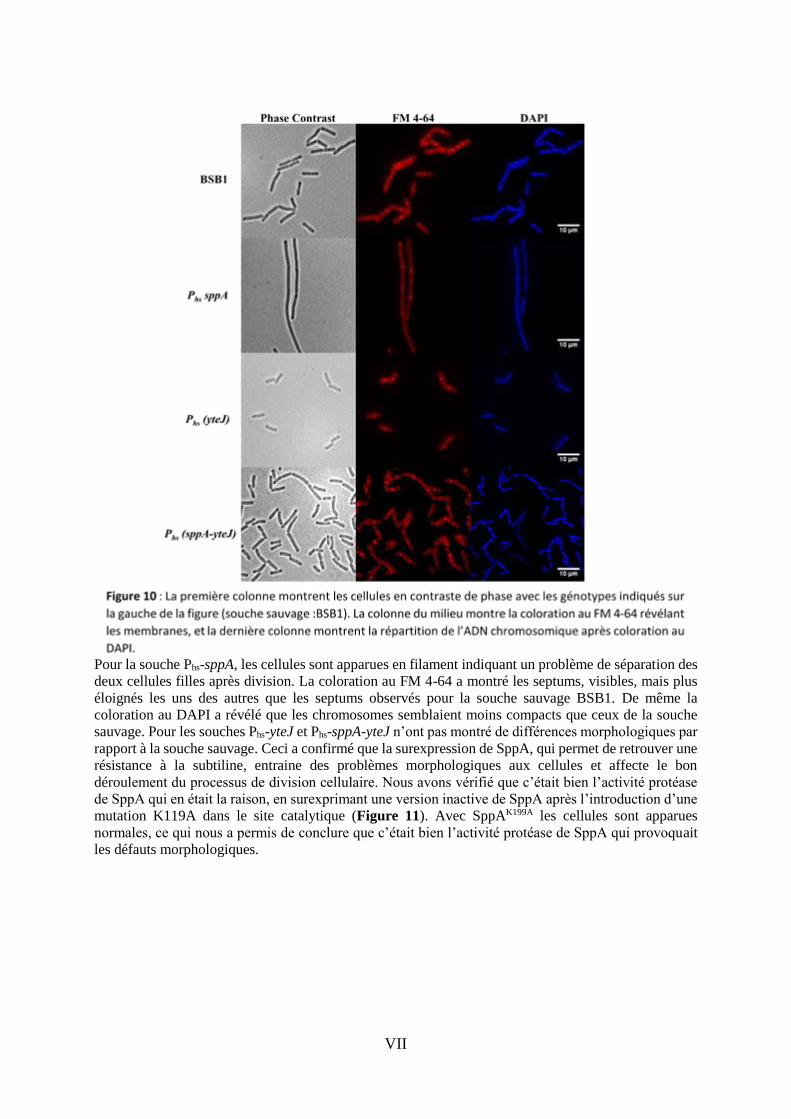
VII
Pour la souche Phs-sppA, les cellules sont apparues en filament indiquant un problème de séparation des
deux cellules filles après division. La coloration au FM 4-64 a montré les septums, visibles, mais plus
éloignés les uns des autres que les septums observés pour la souche sauvage BSB1. De même la
coloration au DAPI a révélé que les chromosomes semblaient moins compacts que ceux de la souche
sauvage. Pour les souches Phs-yteJ et Phs-sppA-yteJ n’ont pas montré de différences morphologiques par
rapport à la souche sauvage. Ceci a confirmé que la surexpression de SppA, qui permet de retrouver une
résistance à la subtiline, entraine des problèmes morphologiques aux cellules et affecte le bon
déroulement du processus de division cellulaire. Nous avons vérifié que c’était bien l’activité protéase
de SppA qui en était la raison, en surexprimant une version inactive de SppA après l’introduction d’une
mutation K119A dans le site catalytique (Figure 11). Avec SppAK199A les cellules sont apparues
normales, ce qui nous a permis de conclure que c’était bien l’activité protéase de SppA qui provoquait
les défauts morphologiques.

VIII
L’effet de l’activité SppA surexprimée sur la morphologie cellulaire est annulé par la surexpression
simultanée de YteJ avec SppA dans la souche Phs-sppA-yteJ (Figure 10). Nous avons donc émis
l’hypothèse que YteJ jouait un rôle régulateur de l’activité de SppA.
3.2 Expression et purification de SppA, SppA∆1-52, du complexe SppA/YteJ et YteJ chez E.
coli.
Afin de répondre à l’hypothèse d’une régulation de l’activité de SppA par YteJ dans le complexe
SppA/YteJ identifié chez B. subtilis, nous avons exprimé chez E. coli les proteines suivantes : SppA,
SppA 1-52, SppA/YteJ (sous forme d’opéron existant nativement chez B. subtilis) et YteJ. Chaque
protéine fut exprimée avec un tag 6xHis fusionné à son extrémité N-terminale (Figure 12).
Après expression chez E. coli, excepté pour SppA∆1-52 qui a été purifiée à partir de la fraction cytosolique,
les autres protéines, étant membranaires, ont été purifiées à partir des membranes de E. coli, isolées par
ultracentrifugation et solubilisées avec 1% DDM. Les échantillons furent chargés sur colonne de Ni-
NTA agarose (Qiagen). Après lavage, les protéines furent éluées avec 200 mM imidazol et concentré
sur Amicon Ultra-15 Centrifugal Filters. L’analyse par SDS-PAGE du résultat des purifications de ces
quatre protéines est montré en Figure 13
Figure 12 : Schéma représentant les différentes expressions chez E. coli des protéines SppA, SppA1-52,
SppA/YteJ et YteJ, dont les formes sont étiquetées His en N-terminal pour une purification par colonne
d’affinité Ni-agarose.
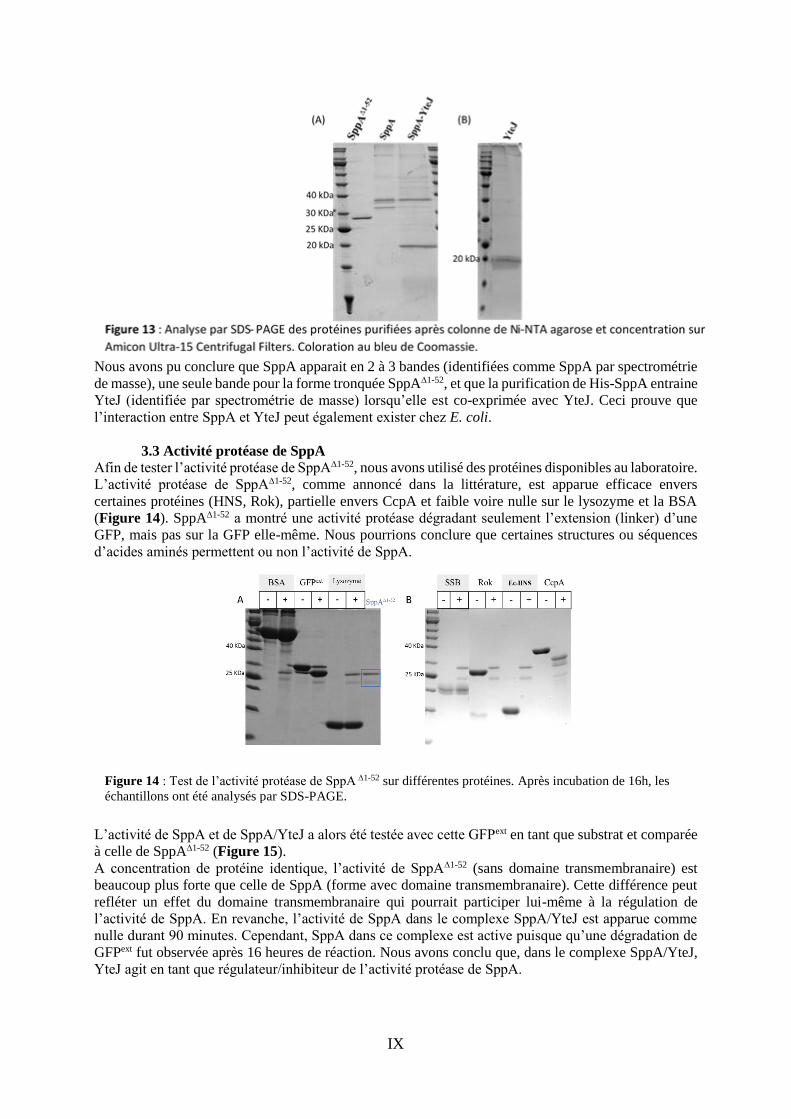
IX
Nous avons pu conclure que SppA apparait en 2 à 3 bandes (identifiées comme SppA par spectrométrie
de masse), une seule bande pour la forme tronquée SppA∆1-52, et que la purification de His-SppA entraine
YteJ (identifiée par spectrométrie de masse) lorsqu’elle est co-exprimée avec YteJ. Ceci prouve que
l’interaction entre SppA et YteJ peut également exister chez E. coli.
3.3 Activité protéase de SppA Afin de tester l’activité protéase de SppA∆1-52, nous avons utilisé des protéines disponibles au laboratoire.
L’activité protéase de SppA∆1-52, comme annoncé dans la littérature, est apparue efficace envers
certaines protéines (HNS, Rok), partielle envers CcpA et faible voire nulle sur le lysozyme et la BSA
(Figure 14). SppA∆1-52 a montré une activité protéase dégradant seulement l’extension (linker) d’une
GFP, mais pas sur la GFP elle-même. Nous pourrions conclure que certaines structures ou séquences
d’acides aminés permettent ou non l’activité de SppA.
L’activité de SppA et de SppA/YteJ a alors été testée avec cette GFPext en tant que substrat et comparée
à celle de SppA∆1-52 (Figure 15).
A concentration de protéine identique, l’activité de SppA∆1-52 (sans domaine transmembranaire) est
beaucoup plus forte que celle de SppA (forme avec domaine transmembranaire). Cette différence peut
refléter un effet du domaine transmembranaire qui pourrait participer lui-même à la régulation de
l’activité de SppA. En revanche, l’activité de SppA dans le complexe SppA/YteJ est apparue comme
nulle durant 90 minutes. Cependant, SppA dans ce complexe est active puisque qu’une dégradation de
GFPext fut observée après 16 heures de réaction. Nous avons conclu que, dans le complexe SppA/YteJ,
YteJ agit en tant que régulateur/inhibiteur de l’activité protéase de SppA.
Figure 14 : Test de l’activité protéase de SppA∆1-52 sur différentes protéines. Après incubation de 16h, les
échantillons ont été analysés par SDS-PAGE.

X
3.4 Le domaine C-terminal de SppA des différentes formes purifiées Le domaine C-terminal de SppA est absent de SppA active mais présent dans un mutant catalytique
(K199A) ce qui suggère que ce domaine participe à la régulation de l’activité de SppA. De façon
intéressante, nous avons remarqué que la purification de SppA conduit à l’obtention de plusieurs bandes
sur gel de SDS-PAGE séparées de quelques kDa. Par spectrométrie de masse, nous avons pu identifier
que toutes ces bandes sont des formes de SppA (Figure 16). Par ailleurs, cette analyse a aussi révélé la
présence du domaine C-terminal de SppA, à travers la détection de deux peptides y appartenant,
uniquement dans SppA purifiée en complexe avec YteJ. SppA dans le complexe SppA/YteJ possède
donc encore son domaine C-terminal, comme pour le mutant catalytique SppAK199A. Ce résultat est en
accord avec le résultat précédent qui montrait que SppA dans le complexe SppA/YteJ est inactive.
3.5 Le domaine C-terminal de YteJ
Afin d’étudier plus précisément comment YteJ régule l’activité de SppA, nous avons construit des
souches qui surexpriment SppA et différentes versions tronquées du domaine C-terminal de YteJ. Des
zones structurales de YteJ ont été ciblées : dernière hélice alpha prédite (12 derniers acides aminés) ou
Figure 16 : A : Analyse par SDS-PAGE et spectrométrie de masse des différentes formes purifiées de SppA
après découpage des bandes sur gel ; B : illustration des peptides (surlignés en vert et jaune) retrouvés par
spectrométrie de masse uniquement dans la bande SppA purifiée en complexe avec YteJ.
Figure 17 : Schéma représentant les insertions de l’opéron modifié sppA-yteJ avec les différentes troncations de
yteJ. Ces constructions ont été insérées dans le chromosome de B. subtilis au locus amyE.

XI
encore le dernier domaine transmembranaire potentiel (35 derniers acides aminés) (Figure 17). Pour
tester l’effet de ces troncations de YteJ sur l’activité SppA, nous nous sommes basés sur le fait que
l’activité SppA est négativement régulée par YteJ et que, si SppA est non-régulée donc active, alors in
vivo les cellules apparaissent allongées au microscope.
Les cellules furent observées au microscope en phase exponentielle de croissance dans un milieu LB
avec 0 ou 100 µM IPTG. Les cellules surexprimant SppA et les troncations de YteJ montrent que
l’activité de SppA est dérégulée dès que YteJ est tronquée de ses 12 derniers acides aminés (cellules
allongées), et encore plus avec la troncation des 35 derniers acides aminés (Figure 18). Nous pouvons
conclure que le domaine C-terminal de YteJ contient des déterminants qui sont importants dans le
contrôle de l’activité de SppA.
CONCLUSION et PERSPECTIVES :
Chez B. subtilis, la protéase SppA forme un complexe membranaire avec YteJ. L’implication de SppA
dans la maturation des protéines sécrétées n’a pas pu être mis en évidence, mais nous avons confirmé
son rôle direct dans la résistance aux peptides antibiotiques de la famille des lantibiotiques. Le rôle de
YteJ a été analysé et nous avons montré, in vivo et in vitro, que YteJ régule négativement l’activité de
SppA. Cette activité de SppA, lorsque surexprimée, apparait délétère pour la cellule. La surexpression
simultanée de YteJ abolit cet effet délétère, renforçant l’idée que YteJ régule l’activité de SppA. Plus
précisément c’est le domaine C-terminal de YteJ qui semble être impliqué dans cette régulation. Afin
de mieux comprendre cette régulation, la topologie du domaine C-terminal de YteJ devrait être étudié
ainsi que les déterminants majeurs de son interaction avec SppA. Une caractérisation du complexe dans
son ensemble par microscopie cryo-microscopie électronique apporterait des réponses sur son
organisation générale dans la membrane. Physiologiquement, il reste encore une question importante :
si YteJ est bien capable d’inhiber SppA, quels sont les signaux extérieurs (comme les lantibiotiques) qui
entraine l’activation de SppA, c’est-à-dire la levée de l’inhibition par YteJ. Et via quel mécanisme
moléculaire cette levée d’inhibition peut-elle se faire au niveau de YteJ et SppA. Une étude récente sur
le gène rdd chez Halobacillus andaensis, potentiellement homologue de yteJ chez B. subtilis, ouvre des
pistes sur une fonction d’antiporteur Na+/H+ si celle-ci a été conservée entre ces deux espèces [6].
REFERENCES:
1. Tjalsma, H., et al., Proteomics of protein secretion by Bacillus subtilis: separating the
"secrets" of the secretome. Microbiol Mol Biol Rev, 2004. 68(2): p. 207-33.
2. Nam, S.E., A.C. Kim, and M. Paetzel, Crystal structure of Bacillus subtilis signal peptide
peptidase A. Journal of Molecular Biology, 2012. 419(5): p. 347-358.
FM 4-64 DAPI Merge
IPTG
Figure 18 : Observation au microscope à contraste de phase et fluorescence de l’état morphologique des cellules
surexprimant SppA et différentes troncations de YteJ, en absence d’IPTG (1ère colonne) et en présence de 100
µM d’IPTG (4 dernières colonnes) dans le milieu de culture.

XII
3. Nam, S.-e. and M. Paetzel, Structure of Signal Peptide Peptidase A with C ‑ Termini Bound
in the Active Sites: Insights into Speci fi city, Self-Processing, and Regulation. Biochemistry,
2013. 52: p. 8811-8822.
4. Kingston, A.W., X. Liao, and J.D. Helmann, Contributions of the σ(W) , σ(M) and σ(X)
regulons to the lantibiotic resistome of Bacillus subtilis. Molecular microbiology, 2013. 90(3):
p. 502-18.
5. Saito, A., et al., Post-liberation cleavage of signal peptides is catalyzed by the site-2
protease (S2P) in bacteria. Proceedings of the National Academy of Sciences of the United
States of America, 2011. 108(33): p. 13740-5.
6. Shao, L., et al., Characterization of a Functionally Unknown Arginine–Aspartate–Aspartate
Family Protein From Halobacillus andaensis and Functional Analysis of Its Conserved
Arginine/Aspartate Residues. Frontiers in Microbiology, 2018. 9: p. 807-807.


Acknowledgments
This study was carry out at the Systems Biology for bacterial Engineering and Redesign
(SyBER) group, part of the Genetic Controls in Bacterial Systems (GCBS) group, at the Microbiologie
de l’Alimentation au service de la Santé (MICALIS) Institute, INRA, Jouy en Josas (France). This PhD
project was part of the European Marie Skłodowska-Curie ITN programme: ProteinFactory -
"Engineering of New-Generation Protein Secretion Systems''. The development of an international PhD
project it is not an easy road but with the support, encouragement and generosity of a large number of
people, including my family, friends, colleagues and collaborators it has been continued towards its
completion. All of them deserve special appreciations.
Je tiens à exprimer mes remerciements aux membres du jury, qui ont accepté d’évaluer mon
travail de thèse: Anne Galinier et Nathalie Declerck pour avoir accepté d’être les
rapporteurs de mon manuscrit. Je remercie également Sylvie Nessler, Nathalie Campo, Martin Picard et
Jean Luc Pernodet d’avoir accepté d’examiner ce travail. Je leur suis reconnaissante pour toutes les
corrections et suggestions qu’ils apporteront.
First of all, I would like to start by thanking to my supervisor Olivier Delumeau for accepting
me as PhD student, welcoming me to the team and for helping me to settle down on my arrival. I greatly
appreciate Olivier for what he taught me during my PhD thesis, not only about the scientific knowledge
he transferred to me, but also the importance of transferable skills as resilience. I would like to express
my profound gratitude to our PI, Matthieu Jules, for his genuine leadership, commitment, support and
encouragement. He believed and motivated me when I most needed, saying the right words at the right
time, as well as pushed me to give my best on the most difficult times. I am grateful for having the
opportunity of working and learning from him. To my bench mate, Stephen McGovern, I will be always
grateful for all what he taught me, helped me and for all our conversations and jokes! I would like to
thank to the rest of our team members for all the nice moments we passed together, great discussions
and shared ideas. Specially to Etinne Dervyn, for all our great conversations and for your advices, to
Claire and Marwa (my Protein factory roommate) for all we have shared and support each other along
the way. Je vous remercie tous de faire partie de ce voyage!
When you live abroad, far from your home town, friends start to be like a family you have there.
I am grateful for the friends I have made these years and for all the great moments we have shared, you
are one of the best things I bring from this experience! Aos melhores vizinhos que podia pedir, obrigado
por tudo Joana, João e Lina! Pela cumplicidade imediata e por toda a amizade que criamos, fica o meu
obrigado do fundo coração Priscilla, Caroline e Camilla, que mesmo transatlântica que se mantenha! À
minha companheira de zumba Transito e à minha italiana favorita Ludovica gracias por toda a amizade,
força e jantares temáticos! To ma coupine and my sweetie Cecile, I have no words to express what I feel

about all the moments we have shared. Thank you my sweetie, we were together in the life ups and
downs, and our craziness!
To the amazing experience that was Protein Factory consortium! It was a privilege work and
learn with this amazing group of talented people. Thank you for all the knowledge shared and for all the
traveling we did together! First I would like to express my gratitude to Jan Maarten van Dijl, for joining
together this consortium, for being an example of true leadership, support and commitment. Thank you,
Jan Maarten van Dijl and Colin Robinson, despite all the tiredness, for stay up late with us after meetings,
for drinks and advices. To Sierd Bron, thank you for helping us and for sharing with us your passion
about science. To all the members of my thesis committee (Jan Maarten van Dijl, Emma Dave, Edda
Klipp, Andreas Otto and Tjeerd van Rij) for all the productive discussions and guidance. I would like to
express my gratitude to David Humphreys, Emma Dave, Neesha Dedi and Gilles Malherbe for
welcoming me to UCB Celltech and for all the help during my secondment. Also to Dörte Becher,
Andreas Otto and Minia Varela for welcoming me to Greifswald and helping me during my secondment.
Os três anos do meu doutoramento foi mais uma prova da quão sortuda sou por ter uma família
e amigos maravilhosos, que independentemente da distância ou quaisquer outros fatores, estiveram
sempre lá para me apoiar! Sou muito grata por isso!
Grata pelas minhas tias e primos, que me ensinam todos os dias a ser o melhor de mim, sendo
exemplos de força e amor, e que estiveram sempre lá para o bom e mau, para me dar colo quando
precisei. Ao meu primo-irmão Alexandre, por tudo o que vivemos juntos, obrigado (e também por me
ensinares as primeiras asneiras em francês deu jeito)! Um enorme obrigado ao meu pai e à Irene pela
ajuda, apoio, pelo carinho e as palavras certas no momento certo, que me deram força quando mais
precisei! Um obrigado aos meus Sogros, ao Hugo e Daniel por também fazerem parte desta jornada,
partilharem comigo bons e maus momentos, e por todo o carinho!
Não tenho palavras para expressar a minha gratidão, pelas minhas companheiras de vida, de
sempre e para sempre, Sofia, Joana e Patrícia! Muito obrigado por estarem sempre presentes! E claro ao
resto da nossa família, claro a que escolhi (sei eu que estou a ser lamechas) João, Filipe e Marisa!
À pessoa que estarei eternamente grata, a minha Mãe, não só porque sempre fez de tudo para
me ajudar a ser a pessoa que sou hoje como é uma guerreira cheia de força. Foi ela que me ensinou o
valor da vida, da família e a ir sempre á luta pelos meus sonhos! Obrigado Mãe!
Como os ingleses dizem, for last and not least, o meu amor para a vida toda, o Diogo! Esta tese
também é tua, sem dúvida que sem ti isto não teria sido possível! Estiveste sempre ao meu lado e muitas
vezes confiaste mais em mim do que eu própria e isso nunca vou saber como te agradecer o suficiente!
Os estudos diziam que a maioria dos relacionamentos não sobrevivem a uma tese de doutoramento, nós

mais uma vez contra estatísticas viemos mostrar que o amor cura, dá força e vence tudo! Obrigado
Diogo!
Queria por último expressar a minha gratidão por o meu caminho se ter cruzado com a lendária
equipa da becoach! A grande turma do Arranque90 que no momento certo veio me ajudar a olhar para
a minha vida de um outro prisma! Com especial agradecimento ao Marco Fonseca e ao Thiago Feder,
pela oportunidade que me deram, pela ajuda e força, para que seja a melhor versão de mim!
“Life doesn’t give you the people you want. It gives you the people you need: to
love you, to help you, to hurt you, to leave you and to make you the person you
were meant to be!”
To all of you that believed in me!


“We must have perseverance and above all confidence in ourselves. We
must believe that we are gifted for something and that this thing must
be attained.”
Marie Curie


LIST OF ABBREVIATIONS
ATP – Adenosine triphosphate
ADP – Adenosine diphosphate
CaM – Calmodulin resin
Cm – Chloramphenicol
DDM – n-Dodecyl β-D-maltoside
EDTA – Ethylenediaminetetraacetic Acid
EGTA – Egtazic Acid
Ery – Erythromycin
FEA – Flagella Export Apparatus
ICE – Integrative and Conjugative Elements
IPTG – Isopropyl β- d-1-thiogalactopyranoside
GFP – Green Fluorescent Protein
GRAS – Generally Recognized as Safe
LCA – Live-Cell Array
LIC – Ligation-independent cloning
LTA – Lipoteichoic Acid
Neo – Neomycin
ORF – Open Reading Frame
PMF – Proton Motor Force
RBS – Ribosomal Binding Site
RNC – Ribosome-Nascent Chain
SP – Signal Peptide
SPA – Sequential Peptide Affinity
SPase – Signal Peptide Peptidase
SppA – Signal Peptide Peptidase A
Spec – Spectinomycin
SRP – Signal Recognition Particle
TCA – Trichloroacetic Acid
T4SS – Type IV Secretion System
WTA – Wall Teichoic Acid


1
TABLE OF CONTENTS

2

3
TABLE OF CONTENT
1 INTRODUCTION ............................................................................................................................. 13
1.1 General introduction ............................................................................................................. 13
1.1.1 Project context .............................................................................................................. 13
1.1.2 Bacillus subtilis, a Gram-positive bacterium ................................................................. 14
1.1.3 B. subtilis: a Gram-positive model ................................................................................. 16
1.1.4 B. subtilis: a bacterium of industrial interest ................................................................ 17
1.2 Protein translocation across cytoplasmic membrane in bacteria ......................................... 19
1.2.1 The general protein secretion pathway: the Sec system .............................................. 20
1.2.2 Substrate recognition .................................................................................................... 22
1.2.3 Intracellular chaperoning and targeting to the Sec translocase ................................... 25
1.2.4 Twin-Arginine Transport (Tat) Pathway ........................................................................ 33
1.2.5 Flagella Export Apparatus (FEA) .................................................................................... 38
1.2.6 Type IV Secretion Systems ............................................................................................. 43
1.2.7 Holins ............................................................................................................................. 47
1.3 A guarantee of a correct protein translocation in B. subtilis ................................................ 49
1.3.1 A bacterial cytoplasmic quality control system: ClpXP protease .................................. 49
1.3.2 Membrane-bound proteases, an important quality control step for membrane and
secretory proteins. ........................................................................................................................ 54
1.3.3 Signal peptide peptidase A (SppA) ................................................................................ 55
1.4 Scope of the Thesis ................................................................................................................ 60
2 Results ........................................................................................................................................... 65
2.1 Identification of membrane protein complexes involved in the B. subtilis Sec pathway ..... 65
2.1.1 SPA-tag constructions .................................................................................................... 65
2.1.2 Purifications of the SPA-tagged proteins ...................................................................... 69
2.1.3 Optimisation of the BN-PAGE technique ....................................................................... 71
2.2 The SppA/YteJ membrane complex ...................................................................................... 74
2.2.1 The sppA yteJ operon is regulated by sigma factors σA and σW ..................................... 74
2.2.2 Deletion of the sppA and yteJ genes ............................................................................. 80
2.2.3 Role of the SppA/YteJ complex in protein secretion ..................................................... 82
2.2.4 The deletion of sppA yteJ affects the long term survival .............................................. 87
2.2.5 The deletion of sppA and yteJ affects the resistance to lantibiotics ............................. 90
2.2.6 The deletions of sppA and yteJ do not affect the resistance to vancomycin,
erythromycin or protect against lysozyme .................................................................................... 94
2.2.7 Overexpression of sppA results in the recovery of the BSB1 phenotype in the presence
of subtilin. ...................................................................................................................................... 97

4
2.2.8 The overexpression of sppA results in a change of the cell morphology. ................... 100
2.2.9 SppA and YteJ cell localization..................................................................................... 103
2.2.10 SppA and YteJ in vitro purification. ............................................................................. 104
2.2.11 SppA can digest fully folded proteins or not ............................................................... 106
2.2.12 YteJ inhibits SppA activity ............................................................................................ 107
2.2.13 The C-terminal domain of YteJ is involved in the regulation of SppA activity ............ 110
2.2.14 SppA digests subtilin .................................................................................................... 112
3 Discussion and perspectives ........................................................................................................ 117
3.1 Protein interactions of the B. subtilis Sec pathway ............................................................. 117
3.2 SppA/YteJ membrane protein complex .............................................................................. 118
3.2.1 Role of SppA and YteJ in protein secretion: ................................................................ 119
3.2.2 Role in the resistance to nisin, subtilin and LP9 .......................................................... 120
3.2.3 Regulation of the sppA yteJ operon ............................................................................ 120
3.2.4 SppA/YteJ, a complex involved in quality control of cell division? ............................. 121
3.2.5 The protease activity of SppA and its regulation by YteJ ............................................ 123
3.2.6 YteJ, another function? ............................................................................................... 125
3.2.7 Model for the interaction of SppA and YteJ ................................................................ 126
3.3 Main results ......................................................................................................................... 127
4 MATERIAL AND METHODS .......................................................................................................... 131
4.1 Techniques for DNA manipulation ...................................................................................... 131
4.1.1 Oligonucleotide ........................................................................................................... 131
4.1.2 Polymerase chain reaction (PCR) ................................................................................ 131
4.1.3 Purification of PCR products ........................................................................................ 131
4.1.4 Electrophoresis of DNA ............................................................................................... 131
4.1.5 Purification of DNA from agarose gel .......................................................................... 131
4.1.6 Purification of plasmid DNA ........................................................................................ 132
4.1.7 Chromosomal DNA extraction ..................................................................................... 132
4.1.8 Digestion of DNA with restriction enzymes ................................................................ 132
4.1.9 Ligation of DNA fragments .......................................................................................... 132
4.1.10 Gibson assembly technology ....................................................................................... 132
4.1.11 Ligation-Independent Cloning (LIC) ............................................................................. 133
4.1.12 DNA sequencing .......................................................................................................... 135
4.1.13 DNA transformation .................................................................................................... 135
4.2 Bacterial strains and growth conditions .............................................................................. 135
4.3 Strain construction .............................................................................................................. 136
4.3.1 SPA tagged strains construction .................................................................................. 136

5
4.3.2 Deletion mutants ......................................................................................................... 138
4.3.3 SppA, YteJ and SppA-YteJ overexpressing strains ....................................................... 139
4.3.4 Construction of the GFP Fusions ................................................................................. 140
4.3.5 His- tag constructions .................................................................................................. 140
4.4 Fluorescence Microscopy .................................................................................................... 141
4.5 Live-cell array (LCA) ............................................................................................................. 141
4.6 Bacteria survival test ........................................................................................................... 141
4.7 Swimming and swarming capacity ...................................................................................... 142
4.8 Biofilm formation ................................................................................................................ 142
4.9 Protein analysis ................................................................................................................... 142
4.9.1 Sodium Dodecyl Sulphate Poly-Acrylamide Gel Electrophoresis (SDS PAGE gels) ...... 142
4.9.2 Blue Native PAGE ......................................................................................................... 143
4.9.3 Western blot analysis .................................................................................................. 143
4.9.4 Secreted proteins analysis ........................................................................................... 144
4.9.5 Sequential Peptide Affinity (SPA) purification ............................................................. 144
4.9.6 His-tag purification ...................................................................................................... 146
4.9.7 Double tag purification ................................................................................................ 147
5 BIBLIOGRAPHY ............................................................................................................................. 151
6 Appendix ...................................................................................................................................... 179
6.1 Contents, sterilization and storage of required solutions ................................................... 179
6.2 UCB Secondment: Purification and characterization of a maltogenic α-amylase from
Bacillus stearothermophilus ............................................................................................................ 198

6

7
LIST OF FIGURES
Figure 1. Schematic representation of the distinct cell fates in Bacillus subtilis biofilms…………....16
Figure 2. Protein export pathways in B. subtilis………………………………...………………….....19
Figure 3. Schematic prediction of the B. subtilis Sec-dependent secretory protein translocase……....21
Figure 4. Components involved in Sec-dependent protein export in B. subtilis………………...…….22
Figure 5. General features of the signal peptides and propeptides of Bacillus secretory proteins…....24
Figure 6. Schematic representation of the different modes of protein translocation across the
membrane of B. subtilis……………………………………………………………………...26
Figure 7. Scheme of SecA-mediated protein translocation………..……………………………….....29
Figure 8. Signal peptide cleavage of precursor proteins by signal peptidases….…….……………….31
Figure 9. Tat pathway components………………………………………………………………...….34
Figure 10. Gram-positive bacterial Tat pathway..…………………………………………………….35
Figure 11. Model of the regulation of TatA pore formation by TatBC and substrate……..………….36
Figure 12. Bacterial flagella structure………………………………………………………………....39
Figure 13. B. subtilis flagellum…..……………………………………………………………….…...40
Figure 14. Model for the assembly process of the Gram-negative flagellar type III export apparatus 41
Figure 15. An entropic chain mechanism for flagellum growth outside the cell………….…….…….42
Figure 16. Schematic representation of type IV secretion architecture and functions in bacteria….....44
Figure 17. T4SS of ICEBs1……………………………………….………………………..………....46
Figure 18. Schematic depiction of the proteins involved in Gram-negative bacterial cell envelope
disruption by holin-type lysis systems………………………………………………………48
Figure 19. Scanning electron micrographs……………………………………………………………50
Figure 20. Cartoon model of substrate recognition and degradation by the ClpXP protease….….…..51
Figure 21. Interaction of ClpX and ClpP……………………………………………………………...51
Figure 22. Representation of the expansion and contraction of the axial pore by a snake-jaws
model………………………………………………………………………………………...52
Figure 23. The B. subtilis SppA protein………………………………………….…………………...56
Figure 24. Comparison between octameric SppABS and tetrameric SppAEC…………….………....57
Figure 25. Cross section representation of the oligomeric dome shape structure of SppAEC and
SppABS………………………………………………………………………………….…..58
Figure 26. Growth analysis of strains harbouring C-terminal SPA-tag fusions………………..……..67
Figure 27. Expression of the SPA tag fused proteins…………………………………..….………….68
Figure 28. Purification of SpoIIIJ-SPA and SecDF-SPA protein with different concentrations of
DDM………………….………………………………………………………….……….….70
Figure 29. Analysis by Western blot of a Blue native PAGE 4-16% Bis-Tris Gel of SecDF-SPA in the
presence of different concentrations of DDM………………………………….……………72

8
Figure 30. Number of peptides identified, by LC-MS/MS for each of the proteins purified in the
presence of 1% DDM………………………………………………………………………..73
Figure 31. Alignment of sppA and yteJ genes with other bacteria of firmicutes phylum……..…..…..74
Figure 32. Transcriptional fusions of the PσAW and PσW, promoters and sfGFP………………...……75
Figure 33. Nisin induced increasing expression of sfGFP, regulated by σA and σW sigma factors…...77
Figure 34. Subtilin induced increasing expression of sfGFP, regulated by σA σW sigma factors……..79
Figure 35. Growth of B.subtilis sppA and yteJ deletion mutant strains……………………………….81
Figure 36. Cell morphology of the sppA yteJ deletion mutant……………………………..…………82
Figure 37. Analysis of AmyM extracellular amount by SDS PAGE………………………..………..84
Figure 38. Analysis of AmyM extracellular amount by Western Blot………………………..………85
Figure 39. SDS PAGE analysis of AmyM extracellular amount produced by B. subtilis WT and
overexpression mutant strains……………………………………………………………….86
Figure 40. Capacity of survival of BSB1 and ∆yteJ, ∆sppA and ∆sppA yteJ deletion mutants...…….88
Figure 41. Long-term survival of BSB1 and ∆sppA yteJ deletion mutant, in LB and M9 liquid
media.......................................................................................................................................89
Figure 42. Complementation assays in LB with BSB1 and ∆sppA yteJ deletion mutant strains……..90
Figure 43. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of Nisin……………………………………………………...…………….…91
Figure 44. Growth of the B. subtilis sppA and yteJ mutant strains in the presence of different
concentrations of subtilin…………………………………………………….……………...92
Figure 45. The growth rates of BSB1, ∆sppA and ∆yteJ in the presence of different concentrations of
subtilin………….……………………………………………………………………………94
Figure 46. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of vancomycin………………………………………………………………95
Figure 47. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of erythromycin……………………………………………………………..96
Figure 48. The B. subtilis sppA and yteJ deletion mutant strains treated with different concentrations
of lysozyme…………………………….................................................……………………97
Figure 49. The B. subtilis sppA and yteJ overexpressing strains……………………………...………99
Figure 50. The B. subtilis sppA yteJ overexpressing strain…………………..……..……………….100
Figure 51. Cell morphology of the sppA, yteJ or sppA yteJ overexpression mutants……….………102
Figure 52. Cell morphology of the sppA and sppAK199A overexpression mutants………..…….…….103
Figure 53. SppA and YteJ cell localization………………………………………………………….104
Figure 54. Scheme representing the different His-tag constructions………………..……………….105
Figure 55. SDS PAGE analysis of SppA∆1-52, SppA, SppA/YteJ and YteJ His-tagged purified
proteins……………………………………………………………………………………..106
Figure 56. SDS PAGE analysis of SppA∆1-52 activity………………………………………………..107

9
Figure 57. SDS PAGE analysis of SppA∆1-52, SppA and SppA/YteJ activity……………………….109
Figure 58. SDS-PAGE and mass spectrometry analysis of SppA and SppA/YteJ complex…...……109
Figure 59. Scheme of SppA/YteJ protein complex in the different YteJ truncated constructions…..110
Figure 60. Cell morphology of the overexpressing strains…………………….……………….……111
Figure 61. Growth of BSB1 and ∆sppA in the presence of different subtilin solutions……….…….113
Figure 62. Cells mutated for minJ are filamentous……………….……………………………….…122
Figure 63. Morphology of B. subtilis cells lacking multiple penicillin-binding proteins (PBP)…….122
Figure 64. Amino acid sequence alignment between H. andaensis RDD and B. subtilis YteJ…..….125
Figure 65. Model for the regulation of SppA by YteJ………………………………..……………...126
Figure 66. LIC integration into B. subtilis…………………………………...………………………136
Figure 67. Sequential peptide affinity purification……..…………………….……………………...146
LIST OF TABLES
Table 1. Set of proteins involved in the Sec pathway chosen to be SPA-tagged for tandem affinity
purification……………………………….…………………..……………………………...65
Table 2. Oligonucleotides used for LIC constructions………………………………………...……..137
LIST OF APPENDIX TABLES
Table S 1. Stock and working solutions of growth supplements…………………..………………...180
Table S 2. Stock and working solutions of antibiotics…………………………...…………………..180
Table S 3. List oligonucleotides………………………………………………………...……………181
Table S 4. List of B. subtilis strains………………….………….…………………………………...188
Table S 5 List of E. coli strains……………………………………………………………...……….190
Table S 6 List of plasmids………………………………………………………..….……………….191
LIST OF APPENDIX FIGURES
Figure S 1 YteJ predicted structure by Protter…………………………………...…………………..193
Figure S 2. Purification of SppA, YteJ, SppA/YteJ and SppAK199A by Gel Filtration…………….....194
Figure S 3 Pulse chase assays for sppA, yteJ and sppA yteJ deletion mutants and complemented
strains……………………………………………………………………………………….195
Figure S 4. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of LP9………………………………………………………………...…….196
Figure S 5. The growth of B. subtilis sppA and yteJ overexpressing mutant strains in the presence of
different concentrations of LP9…………………………………………………………….197
Figure S 6. AmyM supernatant purification…………………………………...…………………….199
Figure S 7. AmyM Size exclusion purification results……………………………………...……….200

10

11
INTRODUCTION

12

Chapter 1. INTRODUCTION
13
1 INTRODUCTION
1.1 General introduction
1.1.1 Project context
Biotechnology has become one of the most important industries in our modern society. This
industry promotes the development of technology and products based on the use of living organisms
and their products for the benefit of mankind in different areas and, at the same time, contributes to a
resource-efficient and sustainable economy (Gavanji 2013; Chu and Robinson 2001). The biotech
industry is hugely important for the EU, as it is a sector that employs around 7.5 million people and it
has an annual turnover over 100 € billion (European Commission 2017). Within this industry, the
production of recombinant enzymes and biopharmaceutical proteins is of major importance. The
global market for such recombinant proteins has grown significantly in the past decade, worth 180 €
millions in 2017 and is forecast to exceed 225 € million by 2021. The constant growth of this market
leads to a continuous need for optimization of the production platforms that can deliver protein
products in greater yields, with higher quality and at lower costs. Therefore there is an overwhelming
need for the development of next-generation super-secreting cell factories (van Dijl and Hecker 2013;
Browning et al. 2017; Matthews et al. 2017).
This thesis was carried out within the frame of the EU-funded innovative training network
(ITN), Protein Factory (2015-2018), of the Horizon 2020 Marie Skłodowska-Curie actions program.
This project brings together 6 academic Institutes, which provide the expertise in synthetic biology
and protein secretion, and 5 non-academic partners which include some of the world's premier
biotechnology companies (Novozymes, DSM, Abera Bioscience, UCB-Celltech and FGen) with the
main objective of creating super-secreting strains, bypassing major production bottlenecks such as
secretion stress. A variety of different host expression systems for the production of recombinant
proteins has been described over the years, and each has its own advantages and disadvantages. The
host organisms should be precisely selected for each specific product, considering the product
development and running cost, yield, and easiness of downstream processing, time to market,
necessity of correct glycosylation, and available infrastructure (Chu and Robinson 2001). The two
most popular industrial workhorses for the production of biopharmaceuticals and industrial enzymes
are Escherichia coli and Bacillus subtilis, respectively (Tu, Hong, and Chen 2009). These bacteria are
the two model organisms used in Protein Factory. Both are fast growing bacteria able to replicate in
cheap media to high cell density, their genomes are far better studied than other microorganisms which
makes available many genetic tools, both are amenable to genetic manipulation and can express and
produce proteins with high yields. Due to E. coli very high level of expression of recombinant
proteins, that riches up to 30% of total cellular protein, this bacterium is preferably used for the

Chapter 1. INTRODUCTION
14
production of biopharmaceuticals, such as the human growth hormone (e.g. Humatrope) or insulin
(e.g. Lispro) (Ramadan et al. 2015). E. coli is a Gram-negative bacterium, characteristically having a
periplasm and an outer membrane bilayer composed of lipopolysaccharides. As such, the secreted
proteins are exported from the cytoplasm to the periplasmic space from which they can be recovered
by the rupture of the outer membrane, which gives advantages in downstream processing (Blount
2015). However, the presence of this outer membrane can be a limitation as it works as a permeability
barrier and obstructs the secreted proteins of being released to the medium. The overexpression of the
recombinant proteins in this host can origin misfolded proteins that accumulate in the cytoplasm as
inclusion bodies (Villaverde and Carrió 2003). To recover these proteins the inclusion bodies need to
be solubilized and the proteins refolded into bioactive molecules, a process that bring major costs to
the production process with a final poor recovery of the recombinant proteins (Sørensen and
Mortensen 2005). At an industrial scale and depending on the proteins to be obtained, in the case of
secreted proteins, using B. subtilis as a host organism might be a better option. As a non-pathogenic
Gram-positive bacteria, B. subtilis have a cytoplasmic membrane surrounded by a permeable cell
envelope that allows the release of secretory proteins directly into the culture medium. Therefore, B.
subtilis was used as a model organism for the development of this thesis project and, will be described
in detail in the next section.
1.1.2 Bacillus subtilis, a Gram-positive bacterium
Bacillus subtilis is a Gram-positive rod-shaped bacterium that belongs to the Firmicute
phylum (Bacillaceae family). The main classes of this phylum are the Clostridia, Bacilli (Bacillus,
Listeria, Enterococcus, Staphylococcus and Streptococcus) and Mollicute (Wolf et al. 2004). B.
subtilis was discovered in 1835 by Christian Gottfried Ehrenberg and originally named “Vibrio
subtilis”, however in 1872 Ferdinand Cohn renamed it “Bacillus subtilis”. B. subtilis is considered as
an obligate aerobe, but can also grow anaerobically by respiration on nitrate or by fermentation on
glucose (Nakano et al. 1997; Hoffmann et al. 1995). The B. subtilis genome exhibits a low % GC
content and, although Gram-positive and Gram-negative bacteria diverged about one billion years ago,
it shares similarities with the E. coli genome sequence, as about one-quarter of the entire B. subtilis
Box1: Project context
This thesis is part of the EU-funded innovative training network (ITN), part of the
Horizon 2020 Marie Skłodowska-Curie actions program, entitled Protein Factory. A project
that intends to overcome the major bottlenecks of recombinant protein production, such as
secretion stress, through the creation of engineered super-secreting strains, capable of
secreting an unprecedented range of target molecules. Bacillus subtilis was the model
organisms used in this project.

Chapter 1. INTRODUCTION
15
genome have clear orthologous counterparts in E. coli (Kunst et al. 1997). Besides the core of the
translation and transcription machineries, other operons involved in major integrated functions, such
as ATP synthesis (atp operon) and electron transfer machinery (cta and qox operons), are well
conserved between the two organisms (Kunst et al. 1997). In contrast, these two bacteria are
structurally different, B. subtilis, as a Gram-positive bacterium, possesses a single cytoplasmic
membrane and a thick cell wall matrix of peptidoglycans and teichoic acids, thus lacking a true
periplasmic space (Matias and Beveridge 2005; Christopher Weidenmaier and Peschel 2008), while E.
coli has a characteristic Gram-negative envelope composed by three principal layers, the inner (IM)
and outer (OM) membrane flanking their cell wall within the periplasmic space (Salton 1953;
Christopher Weidenmaier and Peschel 2008).
B. subtilis natural niche is the rhizosphere soil, it grows associated to the plant roots, a healthy
and mutually beneficial association. By one side, B. subtilis benefits from the components excreted by
plant roots (such as amino acids, organic acids, sugars, phenolic acids and other secondary
metabolites), in turn it contributes to the plant defence against pathogenic bacteria (Gaskins, Albrecht,
and Hubbell 1985; Haney et al. 2015).
B. subtilis is able to survive in or adapt to adverse conditions, to engage various life-styles by
triggering different genetic programs, in response to unfavourable environmental signals (Mielich-
Süss and Lopez 2015; López and Kolter 2010). B. subtilis responds to different environmental changes
or to the lack of an essential nutrient, by differentiating into subpopulations of specialized cells:
I. motile cells, give the capacity to the cells to move through liquid and over surfaces;
II. endospore, a small robust, enduring and metabolically inactive cells are produce in
response to nutritional limitations. Spores can survive years in the absence of
nutrients, and can resist to a wide variety of extreme conditions such as heat, cold,
drying and radiations (Burbulys, Trach, and Hoch 1991; Gonzalez-Pastor 2003).
During the sporulation process, cells secrete extracellular killing factors that induce
the lysis of nonsporulating cells that have not developed immunity to these toxins.
This lysing process releases nutrients into the medium that the surviving sporulating
cells can feed on, and thus this behaviour is referred to cannibalism (González-Pastor
2011; Gonzalez-Pastor 2003);
III. competent cells, a type of cells able to scavenge DNA from environment to use as a
nutrient source or, when possible, integrated into the chromosome by recombination
to gain new functions (Anagnostopoulos and Spizizen 1961);
Connected to the cell fate is their social behaviour, bacteria do not grow as dispersed
individual organisms, they communicate and cooperate with each other, originating bacterial
communities. This social behaviour gives to the cell the ability to perceive environmental changes and
react accordingly (López and Kolter 2010). The biofilms are an example of how bacterial cells can

Chapter 1. INTRODUCTION
16
form multicellular structures able to adhere to surface encased in a self-produced extracellular matrix,
as it is visible on Figure 1 (Vlamakis et al. 2013; Kearns et al. 2005; Aleti et al. 2016).
Undomesticated B. subtilis strains form robust, multicellular biofilms that exhibit complex
architectural features (Lemon et al. 2008). Swarming is another social behaviour through which a
synchronised group of flagellum-driven bacterial cells moves on a surface (Partridge and Harshey
2013; Kearns and Losick 2003; Ke et al. 2015). Bacteria communicate between each other and
respond to fluctuations in external cues as a global bacterial population by a complex sensing system
called quorum sensing. This mechanism allows the cells to produce signal molecules to activate
distinct pathways and trigger changes in gene expression in response to changes in cell density,
leading bacteria to adapt to new scenarios (Abisado et al. 2018; Camilli and Bassler 2006; Ke et al.
2015).
Figure 1. Schematic representation of the distinct cell fates in Bacillus subtilis biofilms. A mature biofilm is
composed of distinct subpopulations of cell types that coexist and exhibit different spatiotemporal distribution
patterns. The different subpopulations interact and cooperate with each other with the objective of overcome
changes of external and environmental factors. Adapted from Mielich-Süss and Lopez 2015.
1.1.3 B. subtilis: a Gram-positive model
In 1958, John Spizizen showed for the first time that the strain B. subtilis 168, a non-
pathogenic and auxotrophic for tryptophan strain, is transformable by exogenous chromosomal DNA
(Spizizen 1958). This discovery opened a door that allowed the scientific community to study and
deeply understand the genetics and molecular biology of B. subtilis, adopting this organism as a
genetic tool, which is one of the most studied microorganisms along with E. coli and Salmonella
typhimurium. The 4215 kb entire genome of B. subtilis was sequenced in 1997 and completely re-

Chapter 1. INTRODUCTION
17
sequenced and annotated in 2009, with 4406 predicted ORFs (Open Reading Frame) (Kunst et al.
1997; Barbe et al. 2009). Studies showed that only around 260 genes were found to be essential for
growth, corresponding to proteins involved in DNA replication, protein synthesis (i.e. ribosomes,
synthesis, translation, secretion, protein quality control, lipid biosynthesis, cell metabolism and cell
division) (Kobayashi et al. 2003; Commichau, Pietack, and Stülke 2013; Koo et al. 2017). The
acquirement of such information, as the genome sequence, has allowed more comprehensive and
global experimental approaches such as transcriptomic, proteomics and interactomics (Mäder et al.
2002; Otto et al. 2010; Marchadier et al. 2011; Nicolas et al. 2012). Along with the creation of
dedicated and reliable B. subtilis databases. These databases are either part of global efforts such as
BsubCyc or specialized to specific scientific problems such as transcriptional regulation in B. subtilis
(DBTBS) or proteome databases (KEGG, UniProt and PDB) (Nicolas et al., 2012; Kanehisa et al.,
2016; Burley et al., 2017). SubtiWiki, a highly detailed database, has been developed to gather
together all information of B. subtilis, from specific databases of genes, messenger RNAs, protein
expression, protein-protein interactions to regulatory and metabolic pathways (Michna et al. 2014;
Zhu and Stülke 2018).
1.1.4 B. subtilis: a bacterium of industrial interest
In addition to its GRAS (Generally Recognized as Safe) status, B. subtilis and close relatives
are a source of products with industrial importance for the production of numerous proteins such as
amylases (used in the bread industry), proteases, cellulases (used in the detergent industry) or
antibiotics such as bacitracin and polymyxin (van Dijl and Hecker 2013; Westers, Westers, and Quax
2004; H. Cao et al. 2017). This capability is mainly due to two intrinsic characteristics of this group of
bacteria:
I. In their natural habitat, the soil, they have the ability to produce a wide range of hydrolytic
enzymes that allows them to breakdown soil-based macromolecules into vital nutrients (Pohl
and Harwood 2010b).
II. They are also able to export and secrete proteins directly into the extracellular medium at
concentrations as high as grams per litre, due in part to the single membrane system of Gram-
positive bacteria, in contrast to the double membrane system of Gram-negative bacteria.
The fact that B. subtilis is easy to grow at high cell densities and its natural ability to release
secretory proteins into the culture medium is of great industrial and commercial interest. It reduces
downstream processing costs, makes purification more straightforward and reduces the likelihood of
contamination with cytoplasmic proteins (van Dijl and Hecker 2013; H. Cao et al. 2017) .

Chapter 1. INTRODUCTION
18
Box2: B. subtilis
Commonly found in the soil, B.subtilis, is a rod shaped ubiquitous bacterium that
secretes numerous enzymes to the culture medium, enabling the bacterium to survive in a
continuously changing environment. It is considered as a GRAS organism, it can be easily
cultivated up to high cell densities and handled in the laboratory. Along with its high genetic
tractability and amenability to adopt different life-styles, B. subtilis has become the Gram-
positive model organism, fundamental for research studies, and a cell factory used for large-
scale production of industrial enzymes.

Chapter 1. INTRODUCTION
19
1.2 Protein translocation across cytoplasmic membrane in
bacteria
Membrane and exported proteins are crucial players for maintenance and survival of bacterial
organisms and their correct compartmentalization is a key step for molecular function (Figure 2).
After being synthesized by ribosomes in the bacteria cytoplasm, these proteins need to be inserted in
the cytoplasmic membrane, anchored to the membrane or released into the extracellular medium
(Green and Mecsas 2015; Schneewind and Missiakas 2012; Costa et al. 2015).
Figure 2. Protein export pathways in B. subtilis. Schematic representation of B. subtilis cell and its export
pathways. After the protein synthesis by the ribosomes, proteins undergo different destinations depending on the
presence (+SP) or absence (-SP) of an N-terminal signal peptide (SP) and its category. Proteins without a signal
peptide remain in the cytoplasm. Proteins that have a typical secretory signal peptide are exported via the Sec
pathway, but if the SP contains a twin-arginine motif (+RR) the protein is exported by the Tat pathway. These
proteins can be then secreted to the medium, retained at the membrane by a transmembrane segment (TM),
anchored to the extra-cytoplasmic side of the membrane by a lipid modification (+lipobox) or anchored to the
cell wall, the mature parts of these proteins contain cell wall-binding repeats (+CWB). Pseudopilins are exported
by the Com system. ABC transporters can export peptides too, as it is the case of pheromones and lantibiotics.
(H Tjalsma et al. 2004)
Exported proteins have important functions as, among others, hydrolysis of extracellular
polymers (proteins, nucleic acids, polysaccharides), synthesis of the cell wall and intercellular matrix,
such as biofilm, modification of the host cell, and sensing of the environmental conditions (Ivankov et
al. 2013). In the present chapter, I will describe all the export routes that membrane and secretory
proteins can undertake in bacteria, with greater emphasis on the Gram-positive B. subtilis.

Chapter 1. INTRODUCTION
20
1.2.1 The general protein secretion pathway: the Sec system
In bacteria, the Sec system is highly conserved and it represents the major route for protein
transportation through the hydrophobic membrane barrier. The Sec system allows the translocation of
proteins in an unfolded conformation, without loss of small molecules such as ions and valuable
metabolites. This system was originally identified in E. coli back in the early 90’s. By the use of
liposomes, a preprotein substrate (proOmpA) mixed with Sec purified proteins, respectively SecB,
SecA and SecY/E proteins, these early studies showed that these components form a binding cascade
from SecB to SecA to SecY/E and that they may be enough for the basic reactions involved in the
targeting and the translocation of a cargo protein (Brundage et al. 1990; Hartl et al. 1990). The Sec
translocase, in its minimal form, consists of a protein-conducting channel formed by the heterotrimeric
membrane protein complex SecYEG, and the essential ATPase SecA that acts as a molecular motor
mediating protein transport in this pathway (Brundage et al. 1990; Akimaru et al. 1991; Beckwith
2013).
In general, the main components of the Sec system are highly conserved, and homologous of
the SecYEG complex (Figure 3) have been found in the cytoplasmic membrane of archaea, the
chloroplast thylakoid membrane and the eukaryotic endoplasmic reticulum (Pohlschröder et al. 1997).
However, recent studies on the Gram-positive Sec system revealed that a large number of Gram-
positive bacteria possess accessory Sec components that are not found in other organisms. A second
SecA paralog, termed SecA2, and a second SecY paralog, termed SecY2, are present and although
being involved in protein translocation, in most cases, they are not essential (Prabudiansyah and
Driessen 2016). This accessory pathway is found in different Gram positive bacterial species as
Mycobacterium, Listeria, Clostridium difficile, Streptococcus, Staphylococcus and Bacillus. Bacillus
subtilis is devoid of accessory SecA2 protein, however this is present in some Bacillus species such as
Bacillus anthracis, Bacillus cereus, Bacillus thuringiensis, Bacillus smithi, and Bacillus methanolicus.
SecA2 promotes the export of a small subset of proteins and plays an important role in virulence
(Kurtz et al. 2006; Sullivan et al. 2012). SecY2 is predicted to form an accessory membrane channel
that is responsible for the export of specific proteins, which cannot be exported by the canonical
SecYEG (Prabudiansyah and Driessen 2016). These accessory SecA2/Y2 pathway appear to mediate
the transport of the surface protein GspB, the superoxide dismutase SodA and the catalase peroxidase
KatG (Bensing and Sullam 2002; Kurtz et al. 2006).

Chapter 1. INTRODUCTION
21
Figure 3. Schematic prediction of the B. subtilis Sec-dependent secretory protein translocase. A preprotein
substrate is pushed, in an unfolded state, through the heterotrimeric pore (SecYEG) by the SecA motor protein.
The SecDF–YrbF complex and the main type I signal peptidases (SipS and SipT) are involved in the substrate
translocation by helping on the translocation mechanism and cleaving the signal peptide, respectively. After
translocation, the preprotein is folded into its correct formation by Sec accessory proteins (folding factors).
Adapted from Harwood and Cranenburgh 2008.
Briefly, the translocation through the Sec pathway can be divided in to three main steps
(Figure 4):
I. Targeting: The secreted proteins are synthesised as precursors (pre-pro-protein) with a N-
terminal signal peptide (SP), which is recognized by cytoplasmic factors (Signal Recognition Particle-
SRP) or chaperones, such as SecB in E. coli and CsaA in B. subtilis (Collinson 2005; H Tjalsma et al.
2000). These factors are responsible for maintaining the substrate in an unfolded secretory state and
for targeting the pre-protein to the translocon formed by the SecYEG complex.
II. Translocation: The secreted or membrane proteins are pushed into the SecYEG channel. This
movement is driven by the translocon motor SecA, an ATPase that interacts with the substrate proteins
and binds to the SecYEG complex (Bauer et al. 2014). The repeated cycles of binding, hydrolysis of
ATP, conformational changes of the dimeric SecA leads to the translocation of the secretory protein
through the SecYEG complex (Robson et al. 2009).
III. Release and maturation: Subsequently, the signal peptide is cleaved resulting in the release of
the protein from the membrane. Finally the protein undergoes a maturation process carried out by
other accessory proteins, such as the foldase PrsA, or the Bdb thiol-disulfide oxido-reductases
(Bolhuis et al. 1999; van Wely et al. 2001; Driessen and Nouwen 2008).

Chapter 1. INTRODUCTION
22
Figure 4. Components involved in Sec-dependent protein export in B. subtilis. Secretory proteins are
ribosomally synthesized as precursor proteins with an N-terminal signal peptide (SP). Cytoplasmic chaperones,
such as SRP/FtsY, keep the precursors in a translocation-competent state and facilitate their targeting to the
translocase in the membrane, consisting of SecA, SecY, SecE, SecG, and SecDF. During or shortly after
translocation, the preprotein is cleaved by one of the type I signal peptidases (SipS-W) or lipid modified by the
diacylglyceryl-transferase (Lgt) and cleaved by the lipoprotein-specific signal peptidase (Lsp). SppA may be
involved in the degradation of cleaved signal peptides, whereas the folding of several secreted proteins depends
on the activities of PrsA, BdbBC, and/or SpoIIIJ/YqjG. HtrA, HtrB, and WprA are involved in the quality
control of secretory proteins. It should be noted that for reasons of simplicity, HtrAB are depicted in the cell
wall, although HtrA is detected in both the membrane and the medium. On passage through the cell wall, the
mature protein is released into the environment. Adapted fom Tjalsma, H., et al. (2004).
1.2.2 Substrate recognition
A crucial early event in the Sec secretion pathway is the identification of substrates that are
destined for secretion by cytoplasmic components. The first evidence of a substrate recognition system
for protein translocation dates back to the 70’s, when Milstein and his colleagues (1972) provided
insights into how secreted immunoglobulins were made in precursor form with an amino-terminal
hydrophobic sequence that was cleaved to the mature form of the protein, raising the signal sequence
hypothesis (Milstein et al. 1972). Few years later, Blobel and Dobberstein provided substantial and
persuasive evidences into how secreted proteins are recognized. They showed, through in vitro studies,

Chapter 1. INTRODUCTION
23
that the product synthesized from the mRNA for the light chain of lgG was larger by 4,000 g/mol than
the actual secreted light chain (Blobel and Dobberstein 1975).
The non-mature preproteins have a hydrophobic N-terminal extension, the signal sequence or
signal peptide (SP), that can be recognised by the targeting system (as SRP, SecB and CsaA)
responsible for directing the preproteins to the cytoplasmic membrane (Ivankov et al. 2013). The
signal sequences are typically between 15 up to 30 amino acids long and they are characterized by
three different domains:
i. N-domain: a 5- to 8- residue-long positively charged amino terminal that interacts with the
translocation machinery and the negatively charged phospholipids of the membrane;
ii. H-domain: a 8- to 12- residues-long hydrophobic core region decisive for the signal
peptide function. The presence of these hydrophobic residues promotes an α-helical
conformation and consequently a formation of a hairpin-like structure that might result in
the insertion of the signal peptide into the membrane;
iii. C-domain: a 5- to 7- residue long sequence containing a cleavage site for specific signal
peptidases (SPase) responsible for removing the signal peptide from the exported protein,
during or shortly after its translocation.
Although, the signal peptides have this structure in common there is a high variability of the
amino acids sequence Figure 5). In general, the signal peptides of Gram-positive bacteria are typically
longer and more hydrophobic than the SP of Gram-negative bacteria (Zanen et al. 2005; Harwood and
Cranenburgh 2008). Responsible for the targeting of membrane proteins and secretory proteins to the
correct secretion machinery. The SP can be classified according to the SPase cleavage sites and the
pathway by which the preproteins are predicted to be exported. The Sec-dependent SPs, which are the
most abundant type of SPs, contain a characteristic AXA motif that allows the targeting to the Sec
translocation machinery.

Chapter 1. INTRODUCTION
24
Figure 5. General features of the signal peptides and propeptides of Bacillus secretory proteins. The signal
peptides tripartite structure, N-terminal (N) containing lysine and/or arginine residues (indicated by +),
hydrophobic (H, indicated by a grey box) and cleavage (C) regions are schematically represented. The export
pathways via which the preproteins are exported and the SPases responsible for their cleavage are indicated. The
break between the boxes represent cleaved sites and the most frequently occurring first amino acid of the mature
protein (+1) is indicated. Tat-dependent signal peptides with twin arginine motif (SRRxFLK) and the Sec-
dependent signal peptides containing an AXA cleavage site, are cleaved by a type I signal peptidase (SPase).
Lipoprotein signal peptide cleaved by the type II SP. The pseudopilin-like signal peptides and bacteriocin and
pheromone signal peptides have specific SPases. The length of the signal peptides and their subdomains is drawn
to the same scale. (H Tjalsma et al. 2004)
The signal peptides present in B. subtilis can be classified into five distinct classes: twin-
arginine (SRRxFLK) signal peptides, secretory (Sec-type) signal peptides, lipoprotein signal peptides,
pseudopilin-like signal peptides, and bacteriocin and pheromone signal peptides (H Tjalsma et al.
2004). The Tat-dependent SPs have a twin arginine motif (RR/KR) specific that directs them to the
Tat secretion machinery (Natale, Bruser, and Driessen 2008). The difference between the SP of the

Chapter 1. INTRODUCTION
25
secretory proteins and lipoproteins is the presence of a conserved lipobox. This sequence contains a N-
terminal invariable cysteine residue that have to be diacylglycerol-modified before their attachment to
the outer surface of the membrane (Harwood and Cranenburgh 2008). In B. subtilis four proteins have
been identified with pseudopilins SPs (ComGC, ComGD, ComGE, and ComGG) which are cleaved
by the SPase ComC (Chung and Dubnau 1998). Finally, the fifth class of SPs refers to the ribosomally
synthesized pheromones and lantibiotics, which contain only a N- and C-domains and completely
devoid of a hydrophobic H-domain (Fig. 4). These types of proteins are exported and cleaved by ABC
transporters (Harold Tjalsma et al. 2000).
Since the SPs are an essential prerequisite for a successful targeting and secretion of secreted
proteins, it is a good starting point for the optimization of secretion of heterologous proteins. In
general, the signal peptides of Gram-positive bacteria are typically longer and more hydrophobic than
the SP of Gram-negative bacteria (Zanen et al. 2005; Harwood and Cranenburgh 2008). Zanen et al,
have showed that the hydrophobicity is critical for early signal peptide recognition. B. subtilis is not
able to secrete preproteins with signal peptides of low hydrophobicity but secretory proteins with
signal peptides with high hydrophobicity would be more efficiently translocated in this bacterium
(Zanen et al. 2005).
As an attempt to optimize the secretion of heterologous proteins, Brockmeier et al. (2006,)
created a library of all natural Sec type SPs from B. subtilis fused to reporter proteins that were then
tested with a screening approach, making use of two lipolytic enzymes- cutinase from Fusarium solani
pisi and a cytoplasmatic esterase of metagenomic origin. They observed that the best SP of one target
protein may not be the best or even work for other secreted proteins. As an example, the fusion of the
SP of AmyE to the cutinase did not inevitably result in an improvement of the secretion. As a
conclusion, the authors showed that the secretion efficiency for a certain target protein in B. subtilis is
determined by complex modifications and interactions between the signal peptide and the mature
protein (Brockmeier et al. 2006).
1.2.3 Intracellular chaperoning and targeting to the Sec translocase
The targeting of the secretory preproteins to the Sec pathway in bacteria is an important step
for a correct protein exportation. During this stage, preproteins interact with cytosolic chaperones to
undertake the correct route, to be maintained in the correct unfolded state and to ensure a driving force
that provides directionality to the secretion, because polypeptides can slide by Brownian motion in
either direction inside the channel (Rapoport, Li, and Park 2017). The protein targeting to the channel
can occur by two distinct routes, by a co-translational or post-translational route (Figure 6).

Chapter 1. INTRODUCTION
26
Figure 6. Schematic representation of the different modes of protein translocation across the membrane of
B. subtilis. (A) Different stages of cotranslational translocation. The polypeptide is moved directly from the
ribosome tunnel into the SecYEG channel. The ribosome is anchored to the membrane by the SRP-receptor
complex. (B) In the model for posttranslational translocation, the preprotein translocation is mediated by the
SecA ATPase.
1.2.3.1 Co-translational translocation route
In the co-translational path, the new emerging protein chain is simultaneously synthesized,
pushed directly from the ribosome exit tunnel through the translocon and exported or laterally inserted
into the lipid bilayer. This co-translational route is performed by the signal recognition particle (SRP),
which is a highly conserved ribonucleoprotein complex, in all three kingdoms of life, and which is
responsible for recognizing the hydrophobic signal sequence of the ribosome-nascent chain (RNC) as
it emerges from the ribosomes and targeting it to the cytosolic membrane (Koch, Moser, and Müller
2003). In general, SRP is composed of a small cytoplasmic RNA (scRNA) and signal sequence
binding subunit (Ménétret et al. 2005; Collinson, Corey, and Allen 2015). First, this protein complex
binds to a membrane receptor, forming a heterodimeric SRP-receptor complex, which helps the
targeting of the emerging nascent pre-protein from the ribosome to the membrane after the binding to
the signal peptide (Saraogi and Shan 2014). E. coli SRP comprises a scRNA domain and a single
essential protein, Ffh. After the recognition of the hydrophobic signal sequence of the emerging
protein, the SRP interacts with the SRP membrane receptor FtsY, which leads to the transfer of the
emerging protein to the Sec translocase for translocation or insertion into the membrane (Angelini,
Deitermann, and Koch 2005; S. Chen et al. 2008). Ffh and FtsY belongs to the SRP-GTPase family,
depending on GTP-binding, and both are essential for SRP-dependent protein secretion and cell
viability (S. Chen et al. 2008; Neher et al. 2008). B. subtilis has a co-translational route too. As the E.
coli SRP, B. subtilis SRP consists of a scRNA and a GTPase Ffh, however it additionally contains two
molecules of the Histone-like protein Hbsu (Nakamura et al. 1999). The SRP can be divided into two
functional domains: the S domain recognizes the N-terminal signal sequences as soon as the pre-
protein emerges from the ribosomal tunnel exit and the Alu domain imposes an elongation arrest by

Chapter 1. INTRODUCTION
27
blocking the elongation factor entry site, retarding translation (Kempf, Wild, and Sinning 2014;
Rodnina and Wintermeyer 2016). Therefore, by stopping the elongation, SRP prevents membrane
proteins from being prematurely released from the ribosome before the ribosome-nascent chain
complex (RNC) has correctly engaged with the translocation channel (Kempf, Wild, and Sinning
2014). In B. subtilis SRP, the HbsU protein binds to the SRP Alu-Like domain, being essential to this
process. In a study using β-lactamase as a reporter protein, the repression of hbs gene and depletion of
Hbsu leads to slow growth and defect in the translocation with a high accumulation of the preprotein
form of the β-lactamase and an induced scRNA upregulation (Yamazaki et al. 1999).
1.2.3.2 Post-translational translocation route
The post-translational translocation route involves the binding of specialized energy-
transducing factors, such as ATPases. Although in eukaryotes the translocation occurs across the
endoplasmic reticulum (ER) membrane and is realised by a larger complex formed by trimeric Sec61
complex associated with additional components, the post-translocational mechanism works as in
bacteria (Osborne, Rapoport, and van den Berg 2005). It needs a chaperone that acts as a molecular
clamp to grab the emerging polypeptide, directing to the pore and preventing its backsliding. This
mechanism occurs by cycles of hydrolyses of ATP, that consequently increase the affinity for
unfolded polypeptide (Collinson, Corey, and Allen 2015). The SecB protein is the secretory chaperone
in E. coli. This protein binds to the mature regions of a subset of preproteins maintaining them in a
translocation-competent state (unfolded and extended state) and to the carboxy terminus of SecA,
which suggests that SecB promotes protein translocation of the associated preproteins by its specific
binding to SecA. Therefore, the SP interacts post-translationally with SecA, the protein motor of the
pore SecYEG, that subsequently undergoes different conformational states and cycles of ATP and
ADP binding states promoting the translocation through the SecYEG pore by a “push and slide”
movement (Bauer et al. 2014). Then the SecD, SecF and YajC, associated membrane proteins,
prevents the slide back of the translocated protein (Corey et al. 2016).
SecB protein is absent from B. subtilis and all other Gram-positive bacteria, and many studies
have focused on identifying a functional homologue of this protein in this group of microorganisms.
To date these studies have failed to identify a convincing homologue. It might be important to note
that SecB is not essential for E. coli viability and it is only needed for the secretion of a specific subset
of proteins, many of which are outer membrane proteins (van Wely et al. 2001). The absence of outer
membrane proteins in Gram-positive bacteria might explain why a homologue of SecB has not yet
been found in this group of organisms. However, according to Fekkes et al. (1997) the B. subtilis
SecA protein contains a highly conserved binding domain that is present in SecB, which suggests that
SecA has a chaperone-like function (Fekkes, Van der Does, & Driessen, 1997). In 2012, Diao et al.
showed a significant improvement on the secretion of a maltose binding protein (MalE11) and an
alkaline phosphatase (PhoA) with the implementation of an artificial posttranslational protein targeting

Chapter 1. INTRODUCTION
28
pathway in B. subtilis by co-expressing SecB from E. coli together with a B. subtilis hybrid SecA
protein, in which the C-terminal 32 amino acids were replaced by the corresponding part of SecA from
E. coli (Diao et al. 2012). This data indicated that, in this route, SecA plays part of the role of SecB.
However, B. subtilis possibly has another homologous protein of SecB that contains other binding
domains that might serve as a docking site for the SecB analogue. The cytosolic CsaA protein is the
possible homologue of SecB in B. subtilis. It was demonstrated, that, a CsaA has a chaperone-like
activity both in vitro and in vivo, with affinity for SecA and preproteins, as well as a role in the protein
secretion, as the suppression of the csaA gene leads to a decrease of the secretion efficiency in B.
subtilis (Müller et al. 2000). Furthermore, it was shown that the intracellular and general chaperones
GroES and GroEL or DnaK, DnaJ and GrpE can work on pre-proteins and maintain them in a
translocation-competent form. By inactivating the gene encoding HrcA, a negative regulator of the
groE and dnaK operons, the authors bserved that the synthesis of all these chaperones. This increases
the yields of the single-chain antibody (SCA) fragment and a reduced inclusion body formation (S. C.
Wu et al. 1998).
1.2.3.3 SecA and the Sec translocase
In B. subtilis, the membrane bound SecYEG complex forms the translocon (De Keyzer, Van
Der Does, and Driessen 2003). As the name implies, this complex includes the SecY, SecE, and SecG
proteins. SecY is the largest component of the translocase and is essential for translocation and
viability. The secY gene is located within a ribosomal operon, the spc operon. SecY is predicted to
span the cytoplasmic membrane 10 times and is found in randomly dispersed foci that dynamically
assemble and disassemble in the membrane (Dajkovic et al. 2016). SecE is a small integral membrane
protein that is essential for translocation and viability (Jeong, Yoshikawa, and Takahashi 1993). In E.
coli, the SecE three transmembrane domains (TM), surround the back of SecY and appear to stabilize
SecY by preventing it from being degraded by FtsH, an ATP-dependent protease present in the
cytoplasmic membrane (Lycklama A Nijeholt et al. 2013). As mentioned before, SecE is part of the B.
subtilis translocon, however experimental evidences do not exist that it has the same function than in
E. coli. It is assumed that this small protein has the same function because of its homology to E. coli
SecE. SecG is the third component of this integral membrane complex. It is the only component that is
not essential for the cell viability and the protein translocation. In B. subtilis, the deletion of this gene
causes cold sensitivity when secretory stress is imposed (van Wely et al. 1999). These observations
suggest that SecG only contributes to the efficiency of the translocation reaction. During the
translocation cycle, the SecYEG translocon assume different conformational stages (Zheng et al.
2016).
SecA is an ATPase located on the cis side of the membrane which is responsible for both the
preprotein targeting and the coupling of the energy required for translocation. It is the motor
component responsible for pushing the preprotein through the SecYEG pore, as illustrated on

Chapter 1. INTRODUCTION
29
Figure 7 (Bauer et al. 2014). Therefore, SecA is considered as part of the core of the
translocation channel as the motor protein of the SecYEG translocon interacts with both proteins
involved in the translocation, the secretory preprotein and the membrane proteins that compose the
translocon (Bauer et al. 2014; Tanaka et al. 2015).
Figure 7. Scheme of SecA-mediated protein translocation. (A) SecYEG closed channel; (B) Formation of the
SecA–SecY complex; (C and D) At this stage the channel is active and the translocation starts; (E) The final
stage where the signal peptide is cleaved and the protein is released. The translocating polypeptide is shown in
green and the signal peptide as a green cylinder. The lateral gate of the channel is shown as a broken line on the
left. (Alguel et al. 2016)
Kakeshita et al. 2010 showed that the SecA C-terminal region is not conserved in eubacteria
and by deleting 61 amino acids of this region led to significant enhanced production of heterologous
proteins in B. subtilis. The membrane translocase complex itself is of great interest for the
optimisation of production strains. In an attempt to solve the problem of jamming of the membrane
caused by the overexpression of secretory proteins through the increase of the number of translocon,
Mulder et al (2013), constructed an artificial SecYEG operon under the control of an inducible
promoter. They demonstrated that the yield of α-amylase could be improved by increasing the
production of the SecYEG proteins (Mulder, Bandola, and Schumann 2013). To investigate the
bottleneck of the Sec pathway in B. subtilis Chen et al. (2015) developed a combinatorial approach to
evaluate the effect of a systematic overexpression of 23 genes or operons involved or related to this
route for the production of AmyL and AmyS. Contrary to Mulder et al. (2013) results, this new
approach showed that the overexpression of the translocase genes did not significantly increase the
production of heterologous proteins, but the overexpression of post-translocation molecular chaperone
(PrsA) leads to an optimization of AmyL and AmyS secretion (J. Chen et al. 2015). Furthermore, in
2016, Dajkovic and colleagues revealed by internal reflection microscopy (TIRFM) that SecA and
SecY form foci throughout the cytoplasmic membrane. These foci were in constant flux with freely
diffusing SecA and SecY molecules, concluding, this way that the secretion system is remarkably
dynamic and is in constant reorganization (Dajkovic et al. 2016).

Chapter 1. INTRODUCTION
30
1.2.3.4 Protein release and maturation
The final step of the Sec translocation consists in the cleavage of the signal peptide (SP) by
Signal peptidases (SPases), resulting in the release of the protein from the membrane to the media or
lipid modified by the diacylglyceryl-transferase and cleaved by the lipoprotein-specific signal
peptidase. SPases are membrane proteins responsible for the cleavage of the signal peptide and they
are divided in Type I, Type II and TypeIV (Figure 8).
In bacteria, SPaseI cleaves the majority of SP of the secretory pro-proteins, mainly proteins
released to the medium or cell wall proteins. Lep is the only type I signal peptidase that E. coli has,
while B. subtilis has five: SipS, SipT, SipU, SipV, and SipW. SipS and SipT are the main signal
peptidases and their deletion leads to cell death (Harold Tjalsma, Van Den Dolder, et al. 1999).
SPasesI are general signal peptidases, catalysing the release of exported proteins through the Sec
pathway and, also, by the Tat pathway, which facilitates the export of fully folded proteins (Lüke et al.
2009). These peptidases, that have a large C-terminal domain characterized by two transmembrane
helixes and an active site extended to the extra-cytoplasmic membrane surface, are unusual serine
protease, as the active site exhibits a Ser-Lys dyad configuration instead of the canonical Ser-His-Asp
triad architecture (Dalbey, Wang, and van Dijl 2012).
The SPaseII, the lipoprotein signal peptidase, has a crucial role in recognizing and processing
the lipid-modified bacterial membrane proteins, which have to be diacylglycerol-modified at an N-
terminal cysteine residue prior to their attachment to the outer surface of the membrane (Pohl and
Harwood 2010). In B. subtilis, Lgt is the diacylglyceryl-transferase responsible for this lipid
modification, prior to the SP cleavage by the SPaseII Lsp, contrary to E. coli this protein is not
essential for cell viability (Harold Tjalsma, Zanen, et al. 1999). This peptidase is an aspartic acid
protease, whose catalytic residues are fixed just beneath the extra-cytoplasmic membrane surface.
The SPaseIV, the prepilin signal peptidase, are responsible for the processing of the precursors
of type IV pilins and related pseudopilins involved in the secretion of proteins across the outer
membranes of Gram-negative bacteria (type II secretion) or in DNA uptake by Gram-positive bacteria.
In contrast to the other types of peptidases, this type has a specific recognition sequence, Gly-Phe-Thr-
Leu-Ile-Glu, where cleavage occurs between the Gly and Phe residues, and is also responsible for N-
methylation of the phenylalanine at position +1 relative to the cleavage site. Similar to SPaseII, these
peptidases are intramembrane-cleaving aspartic acid proteases, characterize by eight transmembrane
helixes with the active site positioned on the cytoplasmic side close to the membrane, within short
cytoplasmic loops (Strom, Bergman, and Lory 1993).

Chapter 1. INTRODUCTION
31
Figure 8. Signal peptide cleavage of precursor proteins by signal peptidases. Signal peptidase I (SPaseI)
employs a Ser-Lys catalytic dyad for signal peptide cleavage from secretory precursor proteins at the
extracytoplasmic surface of the membrane. The Protein Data Bank (PDB) structure of the catalytic domain
(accession number 1T7D) and the program JMol were used to generate the three-dimensional (3D) structure
image of SPI. Signal peptidase II (SPII) is an aspartic acid protease that cleaves signal peptides from bacterial
lipoprotein precursors just beneath the extracytoplasmic membrane surface. Signal peptidase IV (SPIV) is an
aspartic protease that cleaves signal peptides from prepilins and pseudopilins at the cytoplasmic surface of
bacterial membranes. Transmembrane helices of signal peptidases are depicted as blue barrels, and substrate
helices are depicted as red barrels. A zoomed-in view of the active site residues of SPI is shown. The locations of
the N and C termini of the signal peptidases and their substrates are indicated. Dalbey, Wang, and van Dijl 2012.
Beyond the cleavage of the signal peptide and subsequent release of the mature protein, there
can be other limitation step for the optimization of the secretion, which implies an optimization of the
rate of the signal peptides processing by the signal peptidases. Therefore, Tjalsm et al (2000)
described different signal peptidases present in B. subtillis which have different affinities for different
substrates, suggesting that the overexpression of the right combination of signal peptides will lead to
an increase of the protein of interest (Harold Tjalsma et al. 1997; H Tjalsma et al. 2000). The rate of
signal peptide processing by signal peptidases has also been considered as a possible limiting factor
for protein production. Vitikainen et al. (2001) showed that the proportion of mature AmyQ in SipT-
overproducing cells was higher than in the wild type (Vitikainen et al. 2001).
After this cleavage reaction the released SP need to be recycled, otherwise these peptides
could have an inhibitory action to protein translocation. SppA and RasP have been described as
possible signal peptide peptidases responsible for this degradation in B. subtilis (Bolhuis et al. 1999;
Neef et al. 2017).
Gram-positive bacteria are known by their characteristic cell envelope architecture that
facilitates the release of secretory proteins directly into the culture medium. Therefore, it is expected
that this architecture would be compatible with the protein export process. However, a closer look at
the cell wall reveals that it offers a very challenging environment for proteins to fold after emerging
from the translocase. Unfolded proteins that emerge from the translocase must be able to fold into

Chapter 1. INTRODUCTION
32
their native configuration, a crucial step not only for their functionality but also for their stability. The
folding process should be quick without forming intra- or inter-molecular interactions that could block
the translocation machinery and/or block cell wall synthesis, either of which would compromise cell
viability. B. subtilis contains several post-translocational and quality control proteases in the
membrane and cell wall, as well as extracellular proteases that rapidly target heterologous proteins or
misfolded native proteins with exposed protease recognition sites. In B. subtilis, key roles are played
by PrsA, the folding catalyst, various thiol-disulphide oxidoreductases (BdbA, BdbB, BdbC and
BdbD), cleaning proteases such as the membrane-associated HtrA and HtrB proteases and
extracellular proteases as WprA.
PrsA is a peptidyl-prolyl cis-trans isomerase (PPIase), an essential membrane anchored
lipoprotein, bound to the outer surface of the cytoplasmic membrane, that assists proteins with cis-
prolyl residues to fold (Kontinen, Saris, and Sarvas 1991; Kontinen and Sarvas 1993). Its deletion
results in altered cellular morphology and cell death given that this protein is required for the folding
of one or more proteins involved in cell wall synthesis (Kontinen and Sarvas 1993). Moreover, several
studies have revealed that there is a linear relationship between the amount of PrsA and the yield of
certain secretory proteins (notably amylases), making PrsA a popular target when engineering host
strains dedicated to the production of native, as well as heterologous secretory protein (Vitikainen et
al. 2005).
Due to the oxidized environment into which secretory proteins are translocated, wich favours
disulfide bond formation, B. subtilis expresses four thiol-disulphide oxidoreductase/isomerase
enzymes (BdbA, BdbB, BdbC and BdbD) involved in the disulphide bond formation (Sarvas et al.
2004). However, secretory proteins from B. subtilis and other Gram-positive bacteria generally lack
disulphide bonds. Nevertheless, these enzymes have an important role in the production of
heterologous proteins that require such bonds.
HtrA and HtrB are serine proteases, belonging to the quality control protease family,
combating the extracellular folding stress, and preventing potentially fatal obstruction of the secretory
translocase and cell wall synthesis (Darmon et al. 2002; Antelmann et al. 2003). It was recently
suggested that, in addition to their already described roles as quality control proteases, HtrA and HtrB
might also function to release lipoproteins from their membrane anchorage, to degrade membrane
proteins and also to facilitate the folding of native secretory proteins (Krishnappa et al. 2013).
WprA is one of the eight extracellular proteases encoded in B. subtilis (AprE, Bpr, Epr, Mpr,
NprB, NprE, Vpr), which is also localized on the cell wall (Krishnappa et al. 2014). It is a serine
protease involved in the degradation of peptides for recycling and nutrient provision and the
proteolytic processing of other proteins (Babe and Schmidt 1998). Although it was believed that,
correctly folded Sec-secreted proteins would be highly resistant to the extra-cytoplasmic proteases,
Krishnappa et al. (2014) have shown that many of these proteins are in fact degraded. They showed

Chapter 1. INTRODUCTION
33
that the deletion of WprA leads to an increase in the amount of PrsA secreted into the medium,
demonstrating that WprA has a role in the degradation of PrsA (Krishnappa et al. 2014).
1.2.4 Twin-Arginine Transport (Tat) Pathway
In contrast to other transport systems, the twin-arginine translocase (Tat) is an unusual
protein-targeting export pathway, found in all the kingdoms of life, that uniquely facilitates the
translocation of fully folded and cofactor-attached proteins across the cytoplasmic membrane (Sargent
2007; B C Berks 2015). The active translocation is driven only by the transmembrane proton motive
force (PMF) (Anné, Economou, and Bernaerts 2016; Patel, Smith, and Robinson 2014). This system
was identified for the first time in the thylakoids of plant chloroplasts in 1997, it was shown that a
metallo-enzyme was translocated across the thylakoid membrane in a folded conformation through a
∆pH-dependent pathway (Settles and Martienssen 1998). This pathway has been evolutionarily
conserved in plant chloroplasts where it mediates protein import across the thylakoid membrane and is
essential for the formation of a functional photosynthetic apparatus (Alcock et al. 2016). Later in
1998, Weiner and his colleagues proved the existence of this pathway in prokaryotes. They identified,
in E. coli, a Sec-independent pathway which is required for the correct localization of periplasmic
redox proteins and for the targeting of DMSO reductase to the membrane (Weiner et al. 1998).
Consequently, Tat pathway plays a key role in a range of different cellular processes, because it
mediate the transport, in its majority, of proteins that bind cofactors in the cytoplasm and, thus, fold
Box3: Sec Pathway
The Sec pathway is highly conserved in bacteria and represents the main route for
protein transportation. It mediates the translocation of secretory and membrane proteins in
an unfolded conformation. This secretion process can be divided into three steps: targeting,
translocation and release. The secreted proteins are synthesised as precursors (pre-pro-
protein) with a N-terminal signal peptide (SP), which is recognized by cytoplasmic factors
(Signal Recognition Particle- SRP) or chaperones (CsaA) involved in targeting of the pre-
protein to the translocon SecYEG. The translocation of these proteins across the channel is
driven by the translocon motor SecA, an ATPase that interacts with the substrate proteins
and binds to the SecYEG complex. The repeated cycles of binding, hydrolysis of ATP,
conformational changes of a dimeric SecA leads to the translocation of the secretory protein
through the SecYEG complex. Subsequently, the signal peptide is cleaved resulting in the
protein release from the membrane. Finally, the protein undergoes a maturation process
carried out by accessory proteins, such as the foldase PrsA, or the Bdb thiol-disulfide oxido-
reductases (Driessen and Nouwen 2008; Kang et al. 2014; H Tjalsma et al. 2004).

Chapter 1. INTRODUCTION
34
before transport. Such co-factored proteins have essential roles in photosynthetic and respiratory
energy metabolism, iron and phosphate nutrition, resistance to heavy metals and antimicrobial
peptides, cell separation, symbiotic nitrogen fixation and membrane biosynthesis (Ben C. Berks,
Palmer, and Sargent 2003; De Buck, Lammertyn, and Anné 2008).
Similar to the Sec pathway, Tat substrates are targeted to the Tat apparatus by an N-terminal
signal peptide. Although Tat and Sec signal peptides share a similar overall architecture (Figure 9),
Tat signal peptides specifically exhibit a larger hydrophobic region, a general low hydrophobicity and
bear a conserved S/TRR-x-FLK (with x representing a polar aminoacid) eponymous motif that
contains the characteristic pair of adjacent arginine residues (twin arginines)(Alcock et al. 2016;
Goosens et al. 2015). A fact that gives to this pathway its name. The twin arginine residues are critical
for the Tat translocation in chloroplasts, but may not be an absolute prerequisite in bacteria, as the rate
of translocation is affected by mutating a single arginine, whereas mutation of both completely
abolishes the transport.
Figure 9. Tat pathway components. (A) Schematic representation of the Sec signal peptide and Tat signal
peptide. The different regions of the respective signal peptides (N-terminal (N), hydrophobic (H) and cleavage
(C) domains), mature protein parts, and conserved SP recognition sites are indicated. The black arrow indicate
the SPase cleavage site. (B) Illustration of the basic components of the Tat apparatus. The large transmembrane
protein TatC is shown in purple and the TatA-like proteins TatA, TatB and TatE are shown in pink, green and
orange respectively. Adapted from (Dalbey, Wang, and van Dijl 2012; Goosens, Monteferrante, and Van Dijl
2014).
In general, two types of integral membrane proteins compose the Tat translocon
(Monteferrante et al. 2013; Goosens et al. 2015; Alcock et al. 2016):
TatC: a highly hydrophobic and relatively large membrane protein with six transmembrane
helices, whose N- and C-termini faces the cytoplasm;
TatA-like proteins (TatA, TatB or TatE): smaller proteins with a single hydrophobic
transmembrane domain (TMD) at the N-terminus immediately followed by an amphipathic helix

Chapter 1. INTRODUCTION
35
(APH) and a C-terminal tail densely charged. The TatA-like components resulted from gene
duplication and they have evolved towards different functions, namely substrate binding (TatB)
and pore formation (TatA). E. coli possesses a third TatA paralog, TatE, which is functionally
equivalent to TatA but is present at a much lower concentration in the cell and is not essential for
Tat transport.
The Tat transport requires (i) a docking complex composed by TatB and TatC, which together
form a multivalent receptor complex that binds Tat substrates (Fröbel et al. 2012); and (ii) a pore
complex, which results from the assembly of multiple protomers of TatA (Gohlke et al. 2005).
Translocation is initiated when a substrate protein with the correct signal peptide is recognized and
bound to the docking complex located in the membrane (Figure 10). As soon as it binds, the TatC
component of the docking complex inserts the substrate proteins into the membrane (Blümmel et al.
2017). This binding event triggers the proton motor force (PMF) and, consequently, the recruitment of
pore-forming TatA protomers from a pool in the membrane to form the active TatABC-containing
translocation site (Sargent 2007; Alcock et al. 2016; Wojnowska et al. 2018). Upon completion of
transport, the substrate is released from the translocation site and the signal peptide is cleaved by
signal peptidase.
Figure 10. Gram-positive bacterial Tat pathway. Schematic representation of the envelope structure Gram-
positive bacteria and its Tat translocation machinery. Tat substrates can be targeted to the membrane, the cell
wall or the growth medium. Adapted from Goosens 2014.
Crosslinking and fluorescent microscopy experiments, showed the dynamic oligomerization of
TatA. Particularly, it was possible to visualize directly the dynamics in E. coli through the fusion of
TatA with a green fluorescent protein (GFP). When substrates are scarce, TatA is localized in the
membrane surrounding the cells, while it is found in large oligomers when the substrate is present in
high concentrations (Leake et al. 2008; Rose et al. 2013).
The Tat machinery transports folded proteins of widely varying size across the membrane,
these substrates may vary from 20 Å up to 70 Å of diameter comparing to an unfolded polypeptide
chain with 12 A° diameter transported by the Sec pathway (Gohlke et al. 2005; B C Berks 2015). The

Chapter 1. INTRODUCTION
36
variation of the TatA channel diameter according to large or small substrates, ensuring a tight seal
substrate transportation across the membrane without an uncontrolled ion leakage, it is a challenge
faced by the Tat machinery. The membrane-weakening and pulling mechanism hypothesis explains
how the Tat system manage this process. The model suggests that the N-terminal of the TatA
oligomers, that form the Tat translocon site, are capable of destabilize the membrane by weakening or
even rupturing the membrane upon substrate-binding, allowing the TatC component to pull the
substrate through the destabilized membrane (Frobel et al. 2012).
In 2013, Rodriguez and colleagues provided the first experimental evidence supporting this
model, schematically presented on Figure 11. They showed by NMR that E. coli TatA forms a ring-
like complex in the presence of the detergent C12E9, where the transmembrane helices form a ring
inside the membrane from which the APH extends allowing the formation of complexes with variable
subunit numbers. They also showed by molecular dynamics simulation that the periplasm side of the
lipid bilayer surrounding the TatA complex was destabilized because of the unusual short TMD
(Rodriguez et al. 2013).
Later in 2018, Hou and colleagues demonstrated in vivo that in the presence of a Tat substrate
the N-terminus TMD of TatA can destabilize the membrane. However in the absence of substrate, the
full length TatA counterbalances such weakening effects (Hou et al. 2018). Therefore, they proposed a
two-state model: (i) the activated state - in which the binding of the substrate to the TatABC complex
promotes the position switch of the TatA APH, which locally weakens the membrane, allowing the
substrate translocation across the membrane; (ii) the resting state- in the absence of the substrate, the
N-terminal membrane part of the APH compensates for distorting effects of the short TatA membrane
TMD anchor (Hou et al. 2018).
Figure 11. Model of the regulation of TatA pore formation by TatBC and substrate. (A) The inactive
monomeric form of TatA accommodates the depth of the membrane by tilting the short TMH, increasing the
angle between the TMH and the APH, and deeply inserting the APH into the membrane. (B) Interactions with
TatBC/substrate complex force an orientation of the TatA TMH roughly parallel to the membrane normally
favoring TatA polymerization. The N ter- minus is pulled into the membrane thinning and distorting the bilayer
in the pore and sensitizing the membrane to disruption. The TatA APH serves as a platform for the substrate that
is tethered to the TatBC complex by a signal peptide. In the drawing the TatA oligomeric ring is represented by
two opposing subunits. (C) Pulling of substrate protein into the membrane forces TatA subunits apart and
ruptures the membrane (Rodriguez et al. 2013).
In the case of Gram-positive bacteria as the actinomycetes (including the Streptomycetes,
Mycobacterium and Corynebacterium), the Tat system is similar to the Gram-negative bacteria and is

Chapter 1. INTRODUCTION
37
composed by TatA, TatB and TatC components (Anné et al. 2014). In contrast, Staphylococci and
Bacillus have a smaller core Tat pathway, mainly composed by a single pair of TatA and TatC and
devoid of TatB. B. subtilis have genes that encode for two TatC (TatCd and TatCy) and three TatA
(TatAd, TatAy and TatAc). These Tat components can assemble into two distinct Tat translocases that
operate in parallel, working independently of each other and translocating specific substrates. Both
systems appear to be localized at the cell poles and division sites (Monteferrante et al. 2013). The first
of the two systems, TatAy-TatCy (TatAyCy), was expressed constitutively over many tested
conditions and exports several substrates, including YwbN, an iron-dependent DyP-peroxidase, the
Rieske iron-sulfur protein QcrA and the alkaline phosphatase YkuE. In contrast, the second system,
TatAd-TatCd (TatAdCd), whose the only known substrate is the phosphodiesterase (PhoD), is only
expressed in conditions of phosphate limitation (Goosens et al. 2015; Patel, Smith, and Robinson
2014; Jongbloed, van der Ploeg, and van Dijl 2006).
In bacteria the Tat and Sec pathways work in parallel. Even if their systems have specific
components, they share associated proteins, allowing a crosstalk between the cellular systems
(Goosens, Monteferrante, and Van Dijl 2014). This have been documented in E. coli, the production of
heterologous membrane bound Rieske iron–sulfur protein, depends on the secretion of both pathways,
as a portion of this protein was shown to use a Sec–YidC-dependent membrane insertion mechanism
while the remaining portion was translocated in a Tat-dependent manner (Keller et al. 2012). In B.
subtilis, the TatAy–TatCy membrane complex interacts with the cell wall and quality control protease
WprA. Through yeast two-hybrid experiments, it was shown that WprA resulted in a decrease of the
amount of the TatAy–TatCy -translocated EfeB and YkuE proteins, suggesting a functional WprA–
TatAy–TatCy–EfeB/ YkuE interaction (Monteferrante et al. 2013).
Box4: Tat pathway
The twin-arginine translocase (Tat) is a unusual protein-targeting export pathway,
found in all the kingdoms of life, that uniquely facilitates the translocation of fully folded and
cofactor-attached proteins across the cytoplasmic membrane (Sargent 2007; B C Berks 2015).
An event, that remarkably has no requirement for ATP as energy source, the active
translocation is driven only by the transmembrane proton motive force (PMF) (Anné,
Economou, and Bernaerts 2016; Patel, Smith, and Robinson 2014). This pathway have this
name due to its signal peptides distinguish themselves by bearing a conserved S/TRR-x-FLK
(with x representing a polar aminoacid) eponymous motif that contains the characteristic pair
of adjacent arginine residues (twin arginines)(Alcock et al. 2016; Goosens et al. 2015). The
variation of the Tat machinery pore is remarkable, since it is able to transport folded proteins
of widely varying size across the membrane, from 20 Å up to 70 Å of diameter compared to
unfolded polypeptide chains with 12 A° diameter transported by the Sec pathway (Gohlke et
al. 2005; B C Berks 2015).

Chapter 1. INTRODUCTION
38
1.2.5 Flagella Export Apparatus (FEA)
Bacteria are motile microorganisms. To survive they need to swim or swarm depending on
their environment. The bacterial motor that allows this movement is called the flagellum, a rotating
semi-rigid helical filament. The flagellum was observed for the first time in 1681, when Antony van
Leeuwenhoek, the father of protozoology and bacteriology, observed his own faeces (Dobell 1932). In
one of his letters describing this observations, he clearly described a flagellated microscopic parasite
named two centuries later as Giardia intestinalis, and wrote that this “little animal” has “sundry little
paws” capable of strongly stirring the medium, a clear observation of flagella (Dobell 1932).
The bacterial flagellum is a highly efficient molecular nanomachine with approximately 120-
140 nm of diameter and about 10 to 20 µm long, composed by three main structures : the basal body,
the hook and the filament, which are made of about 25 different proteins (Erhardt, Namba, and Hughes
2010). The basal body, a membrane-anchored structure that works as a structural anchor, a reversible
rotary motor and flagellar protein export system (Minamino, Imada, and Namba 2008). The hook is a
curved, thicker and flexible cylinder that joins the basal body to the filament, contributing to the
synchronized rotation of the several flagella on the cell (Depamphilis and Adler 1971). The filament is
a helical propeller composed by thousands of copies of the protein monomer flagelin (F. Wang et al.
2017). All the components of these protein complexes are exported by the flagellum protein export
apparatus, using ATP and PMF as the energy source (Forster and Marquis 2012; Diepold and
Armitage 2015). The flagellum is a great example of how membrane protein super-complexes are of
importance for cell integrity.
Due to cell envelope differences, Gram-positive and Gram-negative flagellum are structurally
different. As the basal body is a transmembrane structure that attaches the filament and, the hook to
the cell, it is the structure that has more differences between the two types of bacteria. While the
Gram-negative bacteria have a longer basal body composed of four rings – the cytoplasmic disk (C-
ring), the membrane disk (M-ring), the periplasm disk (P-ring) and the outer membrane disk (L-ring);
the Gram-positive bacteria have only two basal body rings, a M-ring and a P-ring, as shown on Figure
12 (Erhardt, Namba, and Hughes 2010; Mukherjee and Kearns 2014). As I am reviewing the B.
subtilis secretion systems, I will describe in detail the flagellar protein export system.
(Berg 2003)

Chapter 1. INTRODUCTION
39
Figure 12. Bacterial flagella structure. Schematic representation of the flagellar motor of Gram-negative (A)
and Gram-positive (B) bacteria. Electron micrographic reconstruction of the flagellar basal body side view (C).
All the components that compose the flagellar motor are specified on the Figure 13. Adapted from Berg 2003.
As described above, the flagellar membrane complex, named basal body, works as a rotary
motor and flagellar protein export system, the two key elements for the existence and well-functioning
of the flagellum. The B. subtilis flagellar motor comprises an inner membrane FliF ring that sits on top
of a cytoplasmic gear-like rotor complex composed by FliG, a C-ring protein complex FliM and FliY
responsible for controlling the flagellar direction, and the stator transmembrane complex composed by
MotA and MotB proteins (Depamphilis and Adler 1971; Minamino, Imada, and Namba 2008). The
stator complex allows the flow of protons across the membrane. This flux leads to a conformational
change of MotA, which promotes the electrostatic interaction with the C-terminus of FliG, generating
the torque for flagellar rotation (Blair 2003; Berg 2003). Through FliF, the torque movement is
transferred to the rod complex, a conduction duct that drives the rotation from the basal body to the
hook and filament (Gabel and Berg 2003; Mukherjee and Kearns 2014). B. subtilis encodes five
homologs of rod proteins - FlgB, FlgC, FlhO, FlhP and FliE. The four proteins, FlgB, FlgC, FlhO and
FlhP, were co-purified with the B. subtilis hook–basal body, results that support their role in the rod-
shape structure (Depamphilis and Adler 1971; Kubori et al. 1997). The study of mutants depleted for

Chapter 1. INTRODUCTION
40
FlhO and FlhP showed that these two proteins are part of the upper part of the rod structure, close to
the hook assembly, since their deletion leads to the secretion of the hook subunits, to the media,
abolishing the hook formation (Courtney, Cozy, and Kearns 2012). In contrast, the deletion of fliE
does not abolish the hook formation, but reduces rod and hook proteins secretion. Therefore, FliE is
not only a structural component of the rod, but a regulatory component necessary for full and correct
secretion of the flagellar components (Burrage, Vanderpool, and Kearns 2018).
The flagellar protein secretion system translocates the flagellar components, across the
membrane, in an unfolded conformation, to the channel of the growing flagellum (a structure with 20
Å of diameter) to maintain the proteins unfolded while they move to the flagellum tip up to 10-20 µm
long (Evans et al. 2013). This translocation process requires cellular energy, using ATP and PMF as
the energy source.
Figure 13. B. subtilis flagellum. Cartoon depicting the putative structure of the B. subtilis flagellum based on
empirical data and similarity to S. enterica. Peptidoglycan is indicated in light gray. Membrane is indicated in
dark gray. Flagellar components are colored and labeled. Micrographs are superresolution fluorescence
micrographs of the indicated structures in wild type cells: filament (maleimide-stained HagT209C), hook
(maleimide-strained FlgET123C) and basal body (FliMGFP fusion protein). Adapted from Mukherjee and
Kearns 2014.
Despite their differences, the flagellar secretion apparatus and the type III secretion system
(T3SS) of pathogenic bacteria, the injectisome, are remarkably conserved. These two systems are
structurally similar and their secretion mechanism are analogous, and most part of the membrane and
ATPase complex components share sequence homology (Carpenter, Zuberi, and Ordal 1993; Diepold
and Armitage 2015; Deng et al. 2017).
The secretion machinery comprises a membrane gate complex, inserted into the FliF M-ring
and composed by FlhA, FlhB, FliP, FliQ, and FliR, five higly conserved transmembrane proteins, and
a cytoplasmic ATPase complex formed by FliH, FliI, and FliJ (Erhardt, Namba, and Hughes 2010).
The transmembrane protein FliO, part of this apparatus too, is responsible for an efficient assembly of
the export gate complex. FliP and FliR form the core structure of the gate complex, a starting point for

Chapter 1. INTRODUCTION
41
the assembly of the remaining components (Figure 14) (Fukumura et al. 2017). Recently, by
performing purification experiments, it was shown that salmonella FliP forms a homohexamer
dependent on the FliO complex help, their interactions are required for a FliP correct assembly
(Fukumura et al. 2017).
Figure 14. Model for the assembly process of the Gram-negative flagellar type III export apparatus. The
export apparatus is composed of a transmembrane export gate complex made of FlhA, FlhB, FliP, FliQ, and FliR
and a cytoplasmic ATPase ring complex consisting of FliH, FliI, and FliJ. The FliP dimers form a homohexamer
with the help of the FliO complex, followed by the assembly of FliQ, FliR, and FlhB and finally of FlhA during
MS ring formation in the cytoplasmic membrane. Then, the FliM/FliN complex binds to FliG to form the C ring
on the cytoplasmic face of the MS ring. Finally, the FliH/FliI/FliJ ATPase ring complex is formed at the flagellar
base through interactions of FliH with FlhA and FliN (FliY in B. subtilis), allowing export of substrates though
the central cavity of the FliP6 ring complex (Fukumura et al. 2017).
FlhA and FlhB are responsible for coordinating the flagellar protein export, through their C-
terminal cytoplasmic domains, and form a docking complex platform for the substrates, ATPase
complex (FliH, FliI and FliJ) and chaperones associated to this system (Fukumura et al. 2017). The
Salmonella and E. coli FlhA forms a homononamer. In E. coli, it was shown that this protein plays
also a key role in the energy transduction mechanism, acting as an ion channel to conduct H+ and
Na+. FlhA works along with the cytoplasmic ATPase complex or as a genetic backup that
compensates for the loss or inactivation of the ATPase complex (Minamino et al. 2016).

Chapter 1. INTRODUCTION
42
Figure 15. An entropic chain mechanism for flagellum growth outside the cell. Subunit crystallization
beneath the cap foldase provides a strong anchor (force to break anchor, FA). Sequential subunits are linked
(force to break link, FL) head-to-tail in a chain by juxtaposed terminal helices forming parallel coiled-coils.
Subunits docked at the export ATPase are unfolded and docked by the export machinery. The N-terminal helix
of the docked subunit is then captured (force to break docking, FM) into the subunit chain by the free C-terminal
helix of an existing subunit in the flagellar channel. Adapted from Evans et al. 2013.
During the flagellum growth, the exported substrates remain unfolded while they move
through the flagellum channel, from the bottom to the end, it is other example of the great importance
of protein-protein interactions. Evans et al. (2013) demonstrated that this mechanism uses the entropic
force of each unfolded substrate and that these substrates gather together thus creating a multi-subunit
chain through the flagellum channel by the isolation of the growing filaments (Evans et al. 2013).
Through these experiments, they also revealed that, each subunit is linked head-to-tail to the other via
an interaction between the juxtaposed amino N-terminal and carboxyl C-terminal helices, as it is
possible to see on Figure 15 (Evans et al. 2013). The first subunit on the filament bottom is docked to
the export machinery and its C-terminal is free to be captured by a free unfolded subunit, through
sequential rounds of linkage. Hence, a multi-subunit chain is created until the flagellum tip, where it is
predicted that the N-terminal of each unfolded subunit crystallizes (Evans et al. 2013). Furthermore,
Evans et al. (2013) confirmed, by thermodynamic analysis, that subunit link regions in the chain resist
to the pulling force generated, an energy that is automatically adjusted over the increasing length of
the growing flagellum (Evans et al. 2013).

Chapter 1. INTRODUCTION
43
1.2.6 Type IV Secretion Systems
The type IV Secretion System (T4SS) is a long and thin multi-subunit, membrane-spanning
translocation system that is present on the surface of many bacterial species. T4SSs have evolved from
a self-transmissible, single-stranded DNA conjugation system with VirB4-like AAA + ATPase to
systems with an enormous diversity in their overall structure and the types of substrates secreted.
Therefore, this pathway serves to translocate a highly diverse family of substrates, since it has a
functional astounding collective capacity of (Figure 16 A-E):
recognizing and translocating single-stranded (ss) DNA substrates across the membrane to
bacterial or eukaryotic target cell, whereby direct cell contact is required (conjugation
machines);
delivering effector proteins (effector translocator systems) to the cytosol of eukaryotic target
cells in a contact-dependent mechanism;
exporting/importing molecules from/to the extracellular milieu;
delivering killing toxins to bacterial neighbours;
contributing to biofilm development.
Consequently, T4SSs are involved in a variety of functions including pilus formation, toxin
and other protein secretion, gene transfer, and biofilm formation (Grohmann et al. 2018). This type of
secretion system can be found in Gram-positive as well in Gram-negative bacteria and in some
archaea. Furthermore, many pathogenic bacteria use this secretion system as virulence factor and
several intracellular symbionts use it for supporting their colonization and propagation in the
eukaryotic host (Figure 16 E)(Cascales and Christie 2003).
Box5:Flagella Export Apparatus (FEA)
Bacteria are motile microorganisms, to survive they need to swim or swarm depending
on their environment. The bacterial motor that allows this movement it is called the flagellum,
a rotating semi-rigid helical filament. The bacterial flagellum is a highly efficient molecular
nanomachine with approximately 120-140 nm of diameter and about 10 to 20 µm long,
composed by three main structures : the basal body, the hook and the filament, which are made
of about 25 different proteins (Erhardt, Namba, and Hughes 2010). All the components of
these protein complexes are exported, in an unfolded conformation, by the flagellum protein
export apparatus, using ATP and PMF as the energy source.

Chapter 1. INTRODUCTION
44
Figure 16. Schematic representation of type IV secretion architecture and functions in bacteria. (A) Conjugative T4SSs translocate DNA from the donor bacterium into various recipients, including other bacteria
or eukaryotic cells. (B) DNA release systems facilitate an exchange of DNA with the extracellular space as well
as biofilm formation. (C) DNA uptake from the environment proceeds by the ComB T4SS. (D) The
Xanthomonas citri T4SS can deliver a protein toxin to kill neighboring G-ve bacterial competitors. (E) Various
pathogenic bacteria and symbionts have evolved T4SSs to deliver effector proteins or DNA–protein complexes
into their host (either eukaryotic target cells or protozoan hosts). The T4SSs can either inject their effectors
directly into the host cell or secrete them into the medium, thereby exerting remarkably different effects on host
cell functions during infection. (F) To achieve genetic exchange during bacterial conjugation, two relaxase
monomers collaborate, adopting distinct structural conformations to provide the two necessary enzymatic
activities for processing the DNA. Adapted from Grohmann et al. 2018)

Chapter 1. INTRODUCTION
45
The integrative and conjugative elements (ICEs) are a major form of horizontal gene transfer
mechanism present on bacterial host chromosomal DNA that, when needed, are excised to form a
conjugative DNA circle that can be transferred into other cells (I. Chen and Dubnau 2004; Laverde-
gomez, Sarkar, and Christie 2012). These elements play a key role in bacterial evolution by spreading
genes, which can be involved in cell metabolism, symbiosis, antibiotic resistance, degradation of
pollutants and pathogenesis (Goessweiner-Mohr et al. 2013). ICEBs1 is a ~20.5 kb model ICE for
conjugation in Gram-positive bacteria, found in the B. subtilis genome. It helps to the bacterial
colonization and DNA uptake mechanism (Auchtung et al. 2005; C. A. Lee, Thomas, and Grossman
2012). To the DNA be transfered into the recipient, during the bacterial conjugation, a relaxase
conjugative DNA nucleoprotein complex is delivered by a coupling protein to the DNA translocation
channel. I will now focus and describe in detail the ICEBs1 membrane conjugation machinery required
for this DNA mobilization.
The ICEBs1 T4SS machinery comprises a coupling protein (ConQ), a cell wall hydrolase
(CwlT), an ATPase (ConE) and the membrane channel components (ConB, ConC, ConD, and ConG)
(Leonetti et al. 2015).
The ConQ coupling protein is responsible for the docking of the conjugative DNA to the T4SS
translocation channel and belongs to the FtsK clade of the FtsK-HerA superfamily of ATPases
(Leonetti et al. 2015). ConQ, as the ATPases of its superfamily, forms doughnut-shaped hexamer
with a central channel large enough to hold single or double-stranded DNA (Figure 17), through
its two N-terminal transmembrane helices and a cytoplasmic C-terminal ATPase domain (Iyer et
al. 2004). This protein is essential to the mobilization and conjugation by ICEBs1 (C. A. Lee,
Thomas, and Grossman 2012).
The Gram-positive cell wall is a thick and dense layer. The presence of a cell wall hydrolase is a
key element that allows the correct assembly of the translocation channel, and, allowing the
conjugation. CwlT has two functional domains, a C-terminal domain with DL-endopeptidase
function and an N-terminal domain that works as an N-acetylmuramidase. Mutations in the
catalytic site of CwlT muramidase domain completely abolish the conjugation, while if the
catalytic site of the endopeptidase domain is mutated, it leads to a significant decrease of the
conjugation (Auchtung et al. 2016).
The ICEBs1 membrane channel is a membrane protein complex composed of four integral
membrane proteins ConB, ConC, ConD, and ConG (Figure 17). ConG is the larger component of
this complex, predicted to have seven transmembrane helices, and ConB, as the second larger
component, together may form the bulk of the translocation channel. In contrast, ConC and ConD,
smaller proteins, may work as scaffolding or assembly factors, a role that is still unclear.
ConE is a doughnut-shaped hexamer ATPase, homolog to the Gram-negative canonical VirD4,
which belongs to the VirB4 clade of the FtsK-HerA superfamily, is essential for the DNA
translocation upon the channel (Iyer et al. 2004). The study of its subcellular localization, through

Chapter 1. INTRODUCTION
46
fluorescent microscopy using a ConE tagged with the green fluorescent protein (GFP), revealed,
the localization of the ICEBs1 apparatus. In ICEBs1 donor cells ConE-GFP had a polar
localization, and it is possible to see clear dots in the cell pole in close association with the
membrane, while when observed in cells lacking ICEBs1 ConE-GFP was dispersed within the cell
cytoplasm (Berkmen et al. 2010). Leonetti et al. 2015, demonstrated by two-hybrid assays that
ConE directly interacts with ConB, proving that ConB is essential for the recruitment and
maintenance of ConE (Leonetti et al. 2015).
Figure 17. T4SS of ICEBs1. Working model of the ICEBs1 T4SS. This speculative model relies on data from
other T4SSs. The single-stranded conjugative DNA is shown in red, covalently attached to the NicK relaxase.
The presumed coupling protein ConQ likely delivers NicK and associated T-DNA to the membrane-associated
T4SS. ConB and ConG may make up the bulk of the membrane channel. The ConE ATPase may provide energy
for T4SS assembly and/or DNA transfer. Adapted from Auchtung et al. 2016.

Chapter 1. INTRODUCTION
47
1.2.7 Holins
The term “holin” or hole formation protein originally refers to phage-encoded pore forming
membrane proteins that control access of phage-encoded endolysins to the peptidoglycan layer. Recent
studies showed that these proteins and their homologues may also be important for a variety of other
functions in Gram-positive phage-free bacteria such as (Saier and Reddy 2015):
spore morphogenesis and germination in B. subtilis;
biofilm formation and DNA release for S. aureus;
programmed cell death and acetate metabolism in S. aureus;
biofilm formation and oxidative stress adaptation in Streptococcus mutans.
Holins are small membrane proteins that play two clear roles in the phage infection cycle.
These proteins can be classified in three different membrane topologies- (I) Class I holins that have
three TMDs, beginning with the N-terminal outside and ending with the C-terminal inside the
cytoplasm; (II) Class II have two TMDs, whereas both ends N- and C-terminal are in the cytoplasm;
(III) Class III just have a single highly hydrophilic TMD, with a disposition of the N- terminal in and
C-terminal out.
The detailed pore formation mechanism, its regulation and the pore dynamics remain
unknown. Studies have shown that until a signal triggers their oligomerization, the small-pore-forming
pin-holins accumulate in the cytoplasmic membrane until it dissipates the PMF. As a consequence this
leads to a membrane permeabilization and large-pore formation that allows the release of endolysins,
which are produced fully folded in the cytoplasm (Figure 18) (Young 2002; Saier and Reddy 2015).
When the endolysins reach the cell wall they cleave various bonds in the peptidoglycan polymer,
depending on the type of endolysin. The genes that encode all the elements involved on this holin-
Box6: Type IV Secretion Systems (T4SSs)
The type IV Secretion Systems (T4SSs) are long and thin multi-subunit, membrane-
spanning translocation systems that are present on the surface of many bacterial species. This
pathway serves to translocate a highly diverse family of substrates, it possesses the collective
capacity to (a) recognize and translocate DNA substrates (conjugation machines) to bacterial
recipients, (b) deliver effector proteins to eukaryotic target cells, (c) exchange DNA with the
milieu, (d) contribute to biofilm development and (e) deliver a killing toxin to bacterial
neighbours. The integrative and conjugative elements (ICEs) are a major form of horizontal gene
transfer mechanism present on bacterial host chromosomal DNA, playing a key role in bacterial
evolution by spreading antibiotic resistance and moving genes, which can be involved in cell
metabolism, symbiosis, degradation of pollutants and pathogenesis. The B. subtilis ICEBs1 T4SS
machinery comprises a coupling protein (ConQ), a cell wall hydrolase (CwlT), an ATPase (ConE)
and the membrane channel components (ConB, ConC, ConD, and ConG) (Leonetti et al. 2015).

Chapter 1. INTRODUCTION
48
endolysin system are clustered in a ‘‘lysis cassette’’(I.-N. Wang, Smith, and Young 2000). Therefore,
it is possible to define the holins as “clock” proteins, as they coordinate the timing of the end of the
infection cycle (I.-N. Wang, Smith, and Young 2000).
Real et al. (2005) demonstrated that B. subtilis has a gene, ywcE, which encodes for a holin-
like protein. Performing transcriptional analysis, they prove that this protein is involved in B. subtilis
sporulation (Real et al. 2005). Furthermore, fluorescent microscopy analysis of a strain bearing an
ywcE-gfp translational fusion showed that ywcE localizes at the cell and spore membranes. Its deletion
was also shown to affect the spore morphology. By transmission electron microscopy analysis of the
∆ywcE spores, they indeed showed that the ∆ywcE mutant had a reduced outer coat without the
characteristic striated pattern, and the outer coat failed to attach to the underlying inner coat (Real et
al. 2005).
Figure 18. Schematic depiction of the proteins involved in Gram-negative bacterial cell envelope
disruption by holin-type lysis systems. Pinholins form small heptameric pores that collapse the membrane
potential (the PMF) across the inner membrane, while the more conventional holins form large multisubunit
pores of variable sizes that allow release of fully folded endolysins from the cytoplasm, which in the periplasm
hydrolyze specific bonds in the peptidoglycan cell wall, depending on the lysin type. C+, a cation. Adapted from
Saier and Reddy 2015.

Chapter 1. INTRODUCTION
49
1.3 A guarantee of a correct protein translocation in B.
subtilis
As described in the previous chapter, membrane and secreted proteins carry out a multiplicity
of essential functions that directly affect the cellular protein homeostasis, so that the maintenance of
this proteome is of extreme importance for the cell viability. These proteins, from their synthesis until
reaching their final destiny, pass through different structural and folding states. To avoid cell damage
coming from defective and misfolded proteins, which can be caused by different cellular stresses and
leads to an increase of the cytotoxicity level, the cells use specific machineries to overcome these
problems, called quality control mechanism. One part of the quality control strategies is protein
degradation carried out by protease complexes, with the main goal of removing deleterious species and
recycling of amino acids.
Proteases enzymes, that preform protein catabolism by hydrolysis of peptide bonds, are part of
these defense mechanism to eliminate the damaged and misfolded proteins, regulating this way the
fate, activity, localization and protein-protein interactions of many different proteins (Lo and Bond
2009). Some bacterial proteases act as chaperones too, and have a key role in cellular protein quality
control (e.g. B. subtilis WprA feeding protease). Initially, these enzymes were differentiated into two
classes, based on their interaction with the substrate, the endopeptidases, which target internal peptide
bonds, and exopeptidases (aminopepti-dases and carboxypeptidases), the action of which is directed to
the NH2 and COOH terminal domains. Later studies allowed a more detailed classification. Based on
their mechanism of catalysis, proteases can be divided into six distinct classes, aspartic, glutamic, and
metalloproteases, cysteine, serine, and threonine proteases (Lo and Bond 2009). Some bacterial
proteases do not work as individual catalytic devices to hydrolyze the substrate, they have the ability
to interact with each other forming protease complexes. Some of these complexes are crucial, as the
case of the AAA+ proteases (ATPase associated with cellular activities), to the proper functioning and
maintenance of the bacterial cell, as well as to its response to the stress, since these multimeric
machines are able to recognize and degrade misfolded, damaged or aggregated proteins (Dougan et al.
2002).
1.3.1 A bacterial cytoplasmic quality control system: ClpXP protease
Cytoplasmic protease complexes, as ClpXP, are responsible for the degradation of defective or
denatured membrane or secretory proteins that, due to stress conditions, cannot be translocated into or
out of the membrane and are arrested in the cytoplasm. ClpXP consists of two subunits, the ClpX
subunit, which has ATPase activity and form a hexameric ring, and the ClpP, which is a double-
heptameric serine protease (Figure 20). The interaction between these proteins was observed for the
first time in the early 1990s, Gottesma et al. named and identified E. coli ClpX as the protein that, in
the presence of ClpP, induces the ATP-dependent degradation of λ0 protein. They showed that the

Chapter 1. INTRODUCTION
50
clpX and clpP genes form an operon and are co-transcribed in a single heat-induced mRNA
(Gottesman et al. 1993). As for E. coli ClpP, it has been demonstrated that B. subtilis clpP is also
induced by heat shock stress. Additionally, it was shown that B. subtilis clpP is induced by salt and
ethanol stress, as well as after a puromycin treatment. Indeed after such a stress exposure the amount
of clpP specific mRNA increased; however the highest increase was shown after heat shock stress
(Gerth et al. 1998). Therefore, these experiments suggested that B. subtilis ClpP is involved in the cell
response to puromycin treatment or heat shock stress, through the degradation of abnormal proteins
resulting from the stress. It was also demonstrated that the deletion of clpP or clpX in B. subitilis
grown in stress conditions, induced the production of elongated cells (Figure 19) and an impaired
growth, indicating that B. subtilis ClpXP have substrates involved in cell morphology or cell division
and emphasized the importance of ClpXP as proteases or chaperones in stress tolerance (Gerth et al.
1998).
Figure 19. Scanning electron micrographs of B. subtilis IS58 (A), the clpP (B) and clpX (C) mutant strains
grown exponentially. Adapted from Gerth et al. 1998.
ClpP exhibits a cylindrical structure composed of two associated heptameric rings and, as a
typical serine protease, contains internal (inner surface) proteolytic sites composed of the conserved
catalytic triad (Ser-His-Asp) (B. G. Lee, Kim, and Song 2011). The hexameric ClpX ATPase ring
binds to the heptameric ClpP ring in a symmetry mismatch, aligning the two axial pores that creates a
continuous central channel between them (Figure 20). Allowing the translocation, of the denatured
and linear protein substrate, from the narrow ATPase pore into the concealed proteolytic chamber
(Olivares, Baker, and Sauer 2015). Despite the fact that high-resolution structures of the ClpXP
complex do not exist, it was demonstrated, by different studies and techniques, that the stability of this
complex relies on interactions involving flexible loops near the axial pores of each ring and another set
between peripheral structural elements, as represented on Figure 21 (Baker and Sauer 2012). Joshi
and colleagues demonstrated that the major determinant of interaction within E. coli ClpXP complex,
are surface ClpX loops, which contains a IGF conserved motif. They observed mutations that ClpX
IGF loop lead to a weakening or elimination of the ClpX-ClpP interaction without affecting other
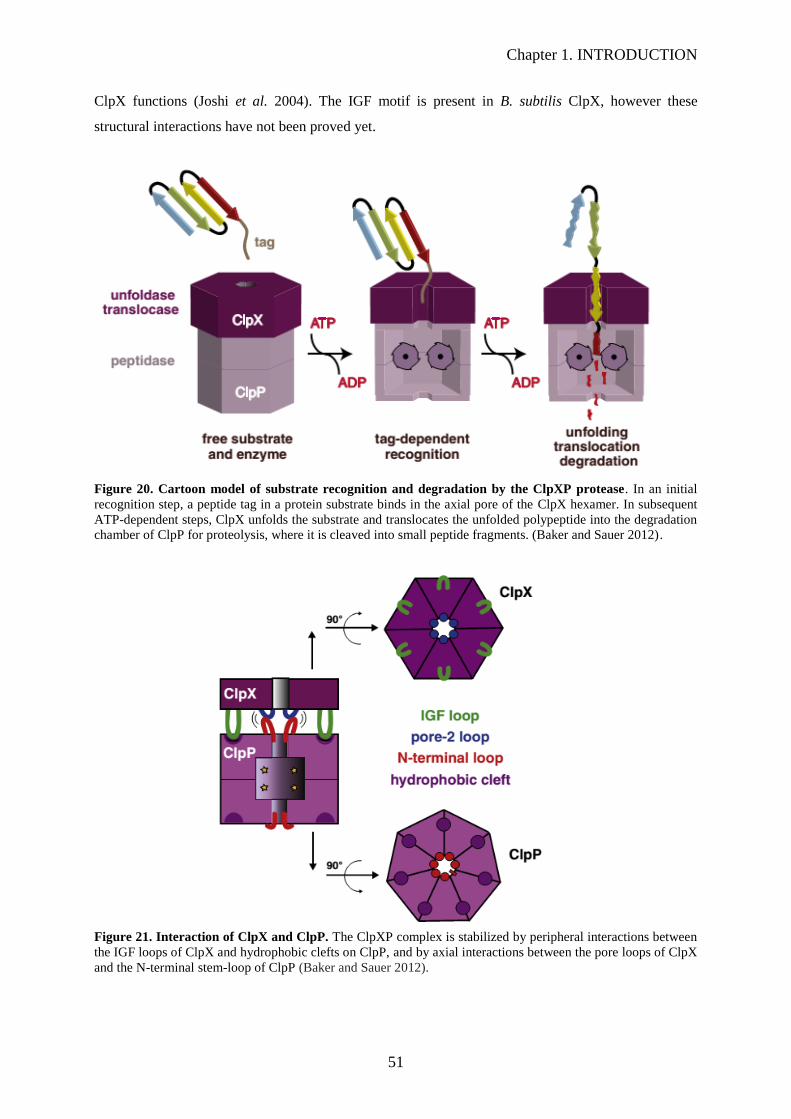
Chapter 1. INTRODUCTION
51
ClpX functions (Joshi et al. 2004). The IGF motif is present in B. subtilis ClpX, however these
structural interactions have not been proved yet.
Figure 20. Cartoon model of substrate recognition and degradation by the ClpXP protease. In an initial
recognition step, a peptide tag in a protein substrate binds in the axial pore of the ClpX hexamer. In subsequent
ATP-dependent steps, ClpX unfolds the substrate and translocates the unfolded polypeptide into the degradation
chamber of ClpP for proteolysis, where it is cleaved into small peptide fragments. (Baker and Sauer 2012).
Figure 21. Interaction of ClpX and ClpP. The ClpXP complex is stabilized by peripheral interactions between
the IGF loops of ClpX and hydrophobic clefts on ClpP, and by axial interactions between the pore loops of ClpX
and the N-terminal stem-loop of ClpP (Baker and Sauer 2012).

Chapter 1. INTRODUCTION
52
The interaction between this two proteins is essential, as the ClpX protein is responsible for
recognition and binding of the substrates, and it allows the degradation of larger protein substrates as
for example disulphide-bonded substrates which require the translocation of three polypeptide chains.
The crystal structure of the ClpX structure revealed a small pore almost completely closed, that will
need to expand and adapt to the different substrate sizes (Baker and Sauer 2012; Gersch et al. 2015).
So, how does the ClpX narrow pore expand according to the substrate size?
Each ClpX protomer has two domains, the large and the small domains, that assemble,
through the binding of the small domain of one protomer to the large domain of the next one, in an
asymmetric hexamer that can bind a maximum of four molecules of ATP and/or ADP (Glynn et al.
2009). Due to its asymmetry and domain-domain orientation the ClpX hexamer can be divided into
subunits of two main classes, the “loadable” or L subunits and the “unloadable” or U subunits (Glynn
et al. 2009; Baker and Sauer 2012). In the L subunit, the large and small AAA+ domains of each
protomer connect in a nucleotide-binding conformation, corresponding to four subunits of the
hexamer. Interestingly, on the two U subunits can occur a rotation of 80° between the bound domains
of the protomers (large and small AAA+ of each protomer), originating movements as large as 30 Å
that destroy the nucleotide-binding site (Glynn et al. 2009; Baker and Sauer 2012). Consequently, the
two U subunits, create two flexible sides of the hexamer that allow the adaptation of the pore to
different substrate sizes, extending the pore to accommodate larger substrates and then refold and
shortening it to contact with smaller substrates (Glynn et al. 2009; Baker and Sauer 2012). This
movement can be compared to the unhinging of the jaws of a snake, which allows it to swallow large
prey, as it is demonstrated on Figure 22.
Figure 22. Representation of the expansion and contraction of the axial pore by a snake-jaws model. The
main contacts between these jaws are formed by the interfaces between the large and small AAA+ domains of
the unloadable (U) ClpX subunits. In which pore size is controlled by the size of the substrates and the
conformation of the hinge region and flanking structure of the unloadable (U) ClpX subunits. Opening of these
interfaces, as shown in the exaggerated right cartoon, provides a potential mechanism for pore expansion to
accommodate large substrates, including those with multiple chains. Each colour represents a ClpX protomer.
(L) loadable subunits. (Baker and Sauer 2012).
This mechanism starts by the recognition of substrates by ClpX, through the binding to short
unstructured peptide sequences called degradation tags or degrons, which are typically located at the

Chapter 1. INTRODUCTION
53
N-terminal or C-terminal end of a target protein, or to adaptor proteins, that modulate and enhance the
substrate recognition by the respective ATPase partner (Dougan et al. 2002; Kirstein et al. 2006). The
adaptor proteins bind to specific substrates and form a delivery complex with the ATPase partner, by
binding to its N-terminal domain (Kirstein et al. 2006; Molière and Turgay 2009; Elsholz, Birk, and
Charpentier 2017). The ClpX adaptor proteins of B. subtilis are YjbH and CmpA (Kirstein et al. 2009;
Elsholz, Birk, and Charpentier 2017).
YjbH is a cytosolic protein involved in sensing various stresses and is an adaptor protein for
the ClpXP catalyzed degradation of Spx factor, being, this way, involved in the resistance against
nitrosating agent. The name of transcription factor Spx was originated from the identification of
genetic mutations on its encoding gene (yjbD), that caused a suppression clpP or clpX (Nakano et al.
2001). High concentrations of Spx were observed in the clpP mutant, higher than in the wild-type
strain, indicating that ClpP is necessary to maintain the Spx concentration low (Nakano et al. 2001).
YjbH exposes the Spx degradation tag, the ClpX recognition site, mediating its degradation. This
recognition site, part of the Spx 28-amino-acid C-terminal segment, is buried by its own structure
buries, a structure that is disrupted by YjbH interaction, thereby promoting Spx degradation by ClpXP
(Garg et al. 2009; Chan, Hahn, and Zuber 2014). This way, under normal conditions, the YjbH-ClpXP
interaction suppress Spx activity through its degradation, while under stress conditions, e.g. heat or
oxidative stress, leads to the YjbH aggregation that promotes the stabilization and accumulation of
Spx (Chan, Hahn, and Zuber 2014; Elsholz, Birk, and Charpentier 2017).
On the other hand, CmpA adaptor protein, together with ClpXP, are involved in the quality-
control pathway that maintain the integrity of the spore envelope, by removing defective sporulating
cells (Tan et al. 2015; Elsholz, Birk, and Charpentier 2017). Under correct sporulation conditions, the
cells produce spores with a proper envelope, ClpXP degrades CmpA and the sporulation continues.
However, in those cells harbouring defects or misassembled spore envelopes, CmpA stabilizes and
delivers the coat morphogenetic protein SpoIVA for degradation by ClpXP, that, consequently,
destabilizes the forespore and leads to cell lysis, thereby eliminating the defective cells and ensuring
the correct spores assembly within the population (Tan et al. 2015; Elsholz, Birk, and Charpentier
2017).
This structural and activity coordination between the six-fold symmetric ClpX ATPase and
seven-fold symmetric ClpP peptidase allows the unfolding of bound substrates in an ATP consuming
reaction and the transportation of the unfolded polypeptide into the ClpP proteolytic chamber, that are
consequently degraded to small peptides that exit the chamber by diffusion, as represented at Figure
20 (Glynn et al. 2009; Baker and Sauer 2012; Elsholz, Birk, and Charpentier 2017).

Chapter 1. INTRODUCTION
54
1.3.2 Membrane-bound proteases, an important quality control step for
membrane and secretory proteins.
The bacterial cell envelope, membrane and cell wall, the external layer of bacterial cells, is very
important for the integrity and viability of bacteria, as it provides physical protection, determines the
cell shape and is the principal stress-bearing element (Scheffers and Pinho 2005; Thomas, Daniel, and
Suzanne 2010). These are sophisticated, dynamic and complex structures that, which in addition to
protecting, allows selective passage of nutrients from the outside, translocate secretory proteins and
waste products from the inside. Gram-negative bacteria cell envelope, such for E. coli, is composed of
two membranes, the cytoplasmic membrane and an outer membrane, and between these structures a
membrane-enclosed periplasm, a dynamic and metabolically highly active environment. The outer
membrane of Gram-negative bacteria contains lipopolysaccharides in its outer leaflet and
phospholipids in the inner leaflet.
In contrast, Gram-positive bacteria have a cytoplasmic membrane and a cell wall. The cell
wall is a complex structure composed of surface proteins, teichoic acids and a thick layer of
peptidoglycan (PG). Peptidoglycan, also called murein, is a heteropolymer composed of long glycan
chains, made up of alternating β-1,4-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid
(MurNAc) subunits, which are cross-linked by peptide bridges to form a strong but flexible structure
(ref). Peptidoglycan is present in almost all bacteria, in B. subtilis these glycan strands are considered
to be long (between 50 and 250 disaccharide units) when compared to, for instance, Staphylococcus
aureus strands (between 3 and 10 disaccharide units), except in Mycoplasma and a few other species
that lack detectable cell walls (Vollmer, Blanot, and De Pedro 2008). To recoup the absence of
periplasm, Gram-positive bacteria are able to lipo-modify homologues of periplasmic proteins and
attach them to the outer leaflet of the cytoplasmic membrane, as well as retain some secretory proteins
in the cell wall by ionic or covalent attachment to the peptidoglycan (Sarvas et al. 2004; Schneewind
and Missiakas 2014). This way, the Gram-positive cell wall can be described as a thicker but dynamic
and flexible structure rich in cell surface proteins, including quality control proteases,
extracytoplasmic chaperones, autolysins, surface layer proteins, and substrate binding proteins (Pohl
and Harwood 2010a; Schneewind and Missiakas 2014). In addition to peptidoglycan, an important
class of cell surface glycopolymers in Gram‐positive bacteria are the phosphate rich teichoic acids
(TAs). These molecules play a role in a large variety of functions, such as in maintaining the
physicochemical properties of the cell surface, cation homeostasis, resistance to antimicrobial peptides
and lytic enzymes, acting as phage receptors, in cell division, biofilm formation and host adhesion.
There are two types of TAs, distinguished by the way they are covalently linked to the surface, the
lipo-teichoic acids (LTAs), which are anchored to the cytoplasmic membrane, extending from the cell
into the peptidoglycan layer, and the wall teichoic acids (WTAs), which are covalently attached to the
peptidoglycan layers and extend beyond them. Together, the LTAs and the WTAs, create a negative

Chapter 1. INTRODUCTION
55
gradient that goes from the bacterial cell surface until the outer most layers of the PG (C Weidenmaier
and Peschel 2008; Pasquina, Santa Maria, and Walker 2013). B. subtilis has two distinct phosphate
rich WTAs, the poly(glycerol phosphate) and poly(glucosyl N-acetylgalactosamine 1-phosphate),
although the last one is present in minor amounts (Freymond et al. 2006). These anionic polymers and
the lipo-teichoic acids create a high density of negative charge in the cell wall, an environment into
which secretory proteins emerge in an unfolded state from the translocase (Hyyrylainen et al. 2000).
As described before in this introduction the Gram-positive architecture is compatible to
protein export, a closer look at the cell envelope reveals that it offers a very challenging environment
for proteins. From the correct insertion of membrane proteins and their maintenance to the unfolded
proteins that emerge from the translocase, which should fold into their native configuration quickly
without forming intra or intermolecular interactions that could block the translocation machinery
and/or block cell wall synthesis sites, either or which would compromise cell viability. This reinforce
the importance of having membrane quality control systems. As is the case of HtrA, HtrB and WprA
for the Sec pathway, as they are serine proteases, belonging to the quality control protease family,
combating the extracellular folding stress, and preventing potentially fatal obstruction of the secretory
translocase. Another serine protease of the Sec pathway is the Signal peptide peptidase A (SppA), that
will be described in the next subchapter.
1.3.3 Signal peptide peptidase A (SppA)
As detailed in the second chapter of this introduction the rapid removal and recycling of the
cleaved signal peptide is crucial for maintaining a proper protein translocation, an action engaged by a
membrane protease named signal peptide peptidase. The E. coli signal peptide peptidase, encoded by
the gene sppA, was purified and characterized in 1987 by Ichihara and his colleagues (Ichihara et al.
1986; Suzuki et al. 1987). They showed that this protein forms a tetramer, in which each protomer is
estimated to have 67 kDa, and it is able to digest, in the presence of the cell envelop, the accumulated
signal peptides and non-specifically other proteins (Ichihara et al. 1986).
Through sequence comparison between E. coli and B. subtilis genomes, a homologue of the E.
coli sppA gene was found in B. subtilis, the yteI. YteI was renamed signal peptide peptidase A (SppA)
of B. subtilis as E. coli SppA (SppAEC), since these two proteins have 49% of sequence similarity
(Bolhuis et al. 1999). In contrast, B. subtilis SppA (SppABS) is a smaller membrane protease with
about 36 kDa that assembles as an octamer, which protomer have a globular and extension part (Nam,
Kim, and Paetzel 2012).
As characteristic of serine proteases, the SppA catalytic domain consists in a dyad of a serine
and a lysine (Nam, Kim, and Paetzel 2012). In order to improve the structure resolution, SppAEC and
SppABS, were purified without the N-terminal transmembrane region and crystalized. The SppAEC
protomer structure showed that the active site residues are in opposite domains, the lysine general base
(K209) was identified in the N-terminal domain while the serine nucleophile (S409) is in the C-

Chapter 1. INTRODUCTION
56
terminal domain, which means that they just interact with the chains are side-positioned (Kim, Oliver,
and Paetzel 2008; P. Wang et al. 2008). Differently, the crystal structure of SppABS revealed that a
serine (S147) residue, located at the globular region of the protomer, and a lysine (K199), located at
the extension region, are quite distant from each other, as shown on Fig.23. Demonstrating that
SppABS on its own is not able to catalyse a substrate, as the active-site catalytic dyad is promoted by
the interaction between the S147 of one promoter and the K199 of the adjacent protomer (Nam, Kim,
and Paetzel 2012).
Figure 23. The B. subtilis SppA protein. (A) A schematic diagram showing the full-length SppA B. subtilis
with its predicted transmembrane segment (pink) and the confirmed amino- terminal thermolysin cleavage site.
The light-green region is what is observed in the electron density. (B) A topology diagram of SppA B. subtilis
showing the full-length protein with β-strand as arrows and α- helices as cylinders. The protein is colored as a
gradient from N- terminus (blue) to C-terminus (red). The regions not seen in the electron density are shown as
bro- ken lines. (C) A cartoon diagram showing the tertiary structure of the SppA B. subtilis protomer. The β-
strands are shown as arrows, and the α- helices are shown as cylinders. The color scheme is the same as in (B).
The nucleophile Ser147 (red) and the general base Lys199Ala (blue) are shown as spheres (Nam, Kim, and
Paetzel 2012).
A
B
C

Chapter 1. INTRODUCTION
57
Figure 24. Comparison between octameric SppABS and tetrameric SppAEC. (A) Schematic representation of
SppAEC tetramer. (B) Schematic representation of SppABS octomer. The catalytic dyads are shown (general base,
blue spheres; nucleophile, red spheres). One protomer in each oligomer is shown in black in (A) and (B). (C) A
cartoon diagrams of an SppA E. coli protomer: the C-terminal domain is colored darker gray, and the region
linking the domains is colored yellow. (D) A cartoon diagrams of an SppA B. subtilis protomer.
Both oligomeric forms of SppAEC and SppABS assemble into a dome-shaped structure (
Figure 24), with a large opening of the bowl, created by the globular region, facing the outer
leaflet of the cytoplamic membrane and a axial hole (small hole), originated by the extension region,
facing the periplasm or extracellular media, respectively (Kim, Oliver, and Paetzel 2008). The interior
of the dome is predominantly hydrophobic and it has an inside midway up a continuous concave
groove that encircles the entire inner bowl, containing the active sites and the substrate binding
pockets (Kim, Oliver, and Paetzel 2008; Nam, Kim, and Paetzel 2012). The substrate binding pockets
of SppABS were described as deeper and wider than the SppAEC pockets, as it is represented on the
cross plan of Figure 25. Although the substrates, E. coli and B. subtilis signal peptides, are predicted
and experimentally shown to have a similar residues content the signal peptides of Gram-negative
bacteria are in general shorter than the Gram-positive ones (Nam, Kim, and Paetzel 2012).
In 2013, Nam and colleagues, revealed an important step of SppABS activity regulation
through its crystal structure. They solved the crystal structure of the SppABS∆2-54
deleted of the first 54
amino acids, corresponding to the N-terminal transmembrane domain, and of an active-site mutant
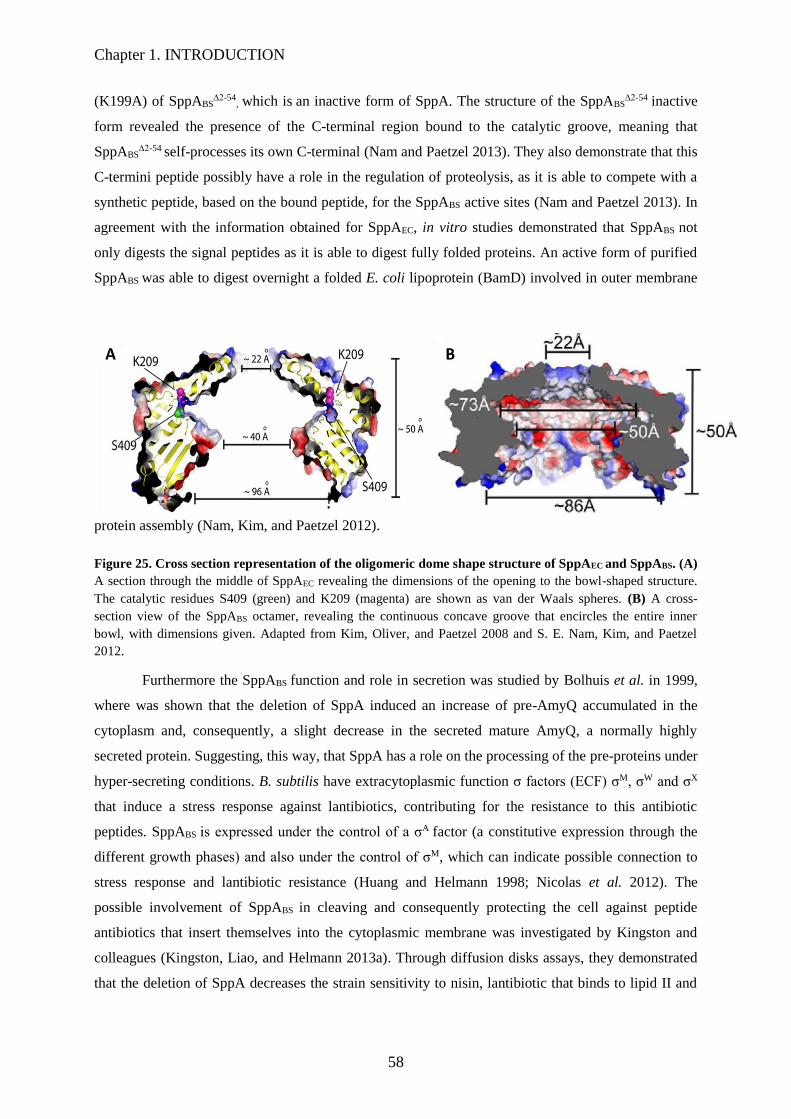
Chapter 1. INTRODUCTION
58
(K199A) of SppABS∆2-54
, which is an inactive form of SppA. The structure of the SppABS∆2-54
inactive
form revealed the presence of the C-terminal region bound to the catalytic groove, meaning that
SppABS∆2-54 self-processes its own C-terminal (Nam and Paetzel 2013). They also demonstrate that this
C-termini peptide possibly have a role in the regulation of proteolysis, as it is able to compete with a
synthetic peptide, based on the bound peptide, for the SppABS active sites (Nam and Paetzel 2013). In
agreement with the information obtained for SppAEC, in vitro studies demonstrated that SppABS not
only digests the signal peptides as it is able to digest fully folded proteins. An active form of purified
SppABS was able to digest overnight a folded E. coli lipoprotein (BamD) involved in outer membrane
protein assembly (Nam, Kim, and Paetzel 2012).
Figure 25. Cross section representation of the oligomeric dome shape structure of SppAEC and SppABS. (A)
A section through the middle of SppAEC revealing the dimensions of the opening to the bowl-shaped structure.
The catalytic residues S409 (green) and K209 (magenta) are shown as van der Waals spheres. (B) A cross-
section view of the SppABS octamer, revealing the continuous concave groove that encircles the entire inner
bowl, with dimensions given. Adapted from Kim, Oliver, and Paetzel 2008 and S. E. Nam, Kim, and Paetzel
2012.
Furthermore the SppABS function and role in secretion was studied by Bolhuis et al. in 1999,
where was shown that the deletion of SppA induced an increase of pre-AmyQ accumulated in the
cytoplasm and, consequently, a slight decrease in the secreted mature AmyQ, a normally highly
secreted protein. Suggesting, this way, that SppA has a role on the processing of the pre-proteins under
hyper-secreting conditions. B. subtilis have extracytoplasmic function σ factors (ECF) σM, σW and σX
that induce a stress response against lantibiotics, contributing for the resistance to this antibiotic
peptides. SppABS is expressed under the control of a σA factor (a constitutive expression through the
different growth phases) and also under the control of σM, which can indicate possible connection to
stress response and lantibiotic resistance (Huang and Helmann 1998; Nicolas et al. 2012). The
possible involvement of SppABS in cleaving and consequently protecting the cell against peptide
antibiotics that insert themselves into the cytoplasmic membrane was investigated by Kingston and
colleagues (Kingston, Liao, and Helmann 2013a). Through diffusion disks assays, they demonstrated
that the deletion of SppA decreases the strain sensitivity to nisin, lantibiotic that binds to lipid II and

Chapter 1. INTRODUCTION
59
inhibiting cell wall synthesis and forming pores in the cytoplasmic membrane, which suggests a
possible role on antibiotic cell protection (Kingston, Liao, and Helmann 2013a).

Chapter 1. INTRODUCTION
60
1.4 Scope of the Thesis
Around 5-10% of the bacterial proteins produced are transported from the cytoplasm to the
extracellular compartment to perform several important functions, such as the provision of nutrients,
cell-to-cell communication, and release of toxins and detoxification of the environment. In B. subtilis,
the major route for the export of these proteins involves mainly the highly conserved Sec pathway
(Harwood, C. R. and R. Cranenburgh 2008; Tjalsma, H., et al. 2004).
The Sec pathway, described in the introduction section 1.2.1 of the introduction, mediates the
translocation across the hydrophobic membrane barrier of secretory and membrane proteins in an
unfolded conformation. Briefly, this secretion process can be divided into three steps: targeting,
translocation and release. First the secreted proteins are synthesised as precursors (pre-pro-protein)
with a N-terminal signal peptide (SP), which is recognized by cytoplasmic factors (Signal Recognition
Particle- SRP) or chaperones (CsaA) involved in targeting the pre-protein to the translocon SecYEG.
The translocation of these proteins across the channel is driven by the translocon motor SecA, an
ATPase that interacts with the substrate proteins and binds to the SecYEG complex. The repeated
cycles of binding, hydrolysis of ATP, conformational changes of a dimeric SecA leads to the
translocation of the secretory protein through the SecYEG complex. Subsequently, the signal peptide
is cleaved resulting in the release of the protein from the membrane. Finally, the protein undergoes a
maturation process carried out by other accessory proteins, such as the foldase PrsA, or the Bdb thiol-
disulfide oxido-reductases (Driessen and Nouwen 2008; Kang et al. 2014; H Tjalsma et al. 2004).
Although these mechanisms involved in the Sec pathway has been extensively studied in B.
subtilis, the knowledge about the protein complexes is limited. For example, it was shown, in E. coli,
that the holotranslocon is a large complex where the YidC and SecYEG are linked by a subcomplex
consisting of YajC and SecDF. SecDF regulates the interaction and activity of SecA with SecYEG.
Physical interactions, organization, activity and bioenergetics of these two sub-complexes SecYEG-
YidC and SecDF-YajC were studied in vivo and in vitro (Duong et al. 1997; Schulze et al. 2014).
Although paralogous proteins have been identified in B. subtilis, such detailed data on these
complexes are not available. This raises questions about the organisation of the holotranslocon in B.
subtilis.. In the first chapter of results, we will show the efforts made to detect physical interactions
involving the B. subtilis (holo)translocon proteins as well as the possible interactions between the
accessory proteins involved in the maturation of secreted proteins. The goal was also to
discoverunknown protein partners interacting with these complexes. In this chapter we showed a
possible protein complex between a known protease, called SppA, and an unknown protein YteJ.
The chapter 2 of the results is dedicated to the study of the membrane complex SppA/YteJ.
The signal peptide peptidase A (SppA), a component of the Sec machinery of B. subtilis, was named
as such because sequence comparison with that of E. colirevealed that these two proteins share 49% of
sequence similarity (Bolhuis et al. 1999). SppA is a membrane bound serine protease that forms a

Chapter 1. INTRODUCTION
61
homo-octameric dome-shaped complex described to be involved in the recycling of the remaining
signal peptides (SPs), cleaved off the translocated proteins, in the membrane. SppA is also involved in
the resistance to nisin, an antibacterial peptide of the lantibiotic family (Bolhuis et al. 1999; M. Cao et
al. 2002; Nam and Paetzel 2013). Its function and role in protein secretion were studied by Bolhuis et
al. (1999), who showed that the deletion of SppA induced a mild decrease in the secretion of mature
secretory proteins, suggesting that SppA has a role on the processing of the pre-proteins under hyper-
secreting conditions (Bolhuis et al. 1999). However, Saito et al. (2011), showed that the disruption of
E. coli sppA gene did not affect in vivo cleavage of the SPs and that the B. subtilis RasP, a S2P
protease, is involved in proteolysis of SPs of some secretory proteins and. Recently in 2017, Neef et
al. gave a proof that the overexpression of RasP in B. subtilis improved the secretion of an
heterologous and highly secreted AmyAc amylase by tenfold, proving that the overexpression of a
bacterium membrane protease can boost the protein production (Neef et al. 2017). These results
changed the view on the SppA function, rising new questions about this topic. In B. subtilis sppA gene
is in an operon with another gene called yteJ. From its sequence, the protein YteJ, of unknown
function, is predicted to be an integral membrane protein belonging to the RDD family, a family of
proteins of unknown functions. To better understand the influence of YteJ on SppA function and to try
to clarify SppA role, we divided this study in two main parts – the in vivo and in vitro study. In vivo,
we tried to uncloak the main role of the SppA/YteJ complex in the cell, starting to study the effect of
the deletion of these two genes, their regulation by two sigma factors (σA and σW), their cellular
localization by fluorescent microscopy, the involvement of both SppA and YteJ in the lantibiotic
resistance.

62

63
RESULTS

64

65
2 RESULTS
2.1 Identification of membrane protein complexes involved
in the B. subtilis Sec pathway
2.1.1 SPA-tag constructions
In order to study the interactions within the B. subtilis (holo)translocon, as well as the possible
interactions between the accessory proteins or unknown protein partners interacting with these protein
complexes, we constructed strains harbouring the 3'-translational SPA sequence fusions in the genetic
background of the BSB1 strain. This strain is tryptophan-prototrophic (trp+) derivative of the 168
trpC2 strain as described in (Nicolas et al. 2012).
A set of proteins of the Sec secretion pathway (see Table 1 below) was chosen to be SPA-
tagged for tandem affinity purification. For that purpose, the 3' ends of the corresponding genes were
fused to the SPA-tag sequence present in the plasmid pMutin-SPA-LIC. The constructed plasmids
were used to transform the BSB1 strain by a Campbell-like integration. Moreover, the pMutin-SPA-
LIC is an integrative plasmid that contains an IPTG inducible / LacI repressible Pspac promoter that,
once integrated in the chromosome, is just upstream of the downsteam genes to ensure their
expression.
The construction of these plasmids had some setbacks: it had been only possible to obtain nine
strains out of ten, as PrsA-SPA strain was not obtained. PrsA is an essential protein for extracellular
folding of some secreted proteins after their translocation. The addition of the SPA-tag might have
impaired its activity, therefore leading to an unviable strain.
Table 1. Set of proteins involved in the Sec pathway chosen to be SPA-tagged for tandem affinity
purification.
Name Protein Function Molecular weight
(kDa)
SecDF
Preprotein
translocase subunit
(ATPase)
Regulates the
interaction and
activity of SecA with
SecYEG
81.47
SecG Sec tanslocase
subunit Protein secretion 7.86
SipT Signal peptidase I
Cleavage of the
signal peptide from
the emerging protein
21.71
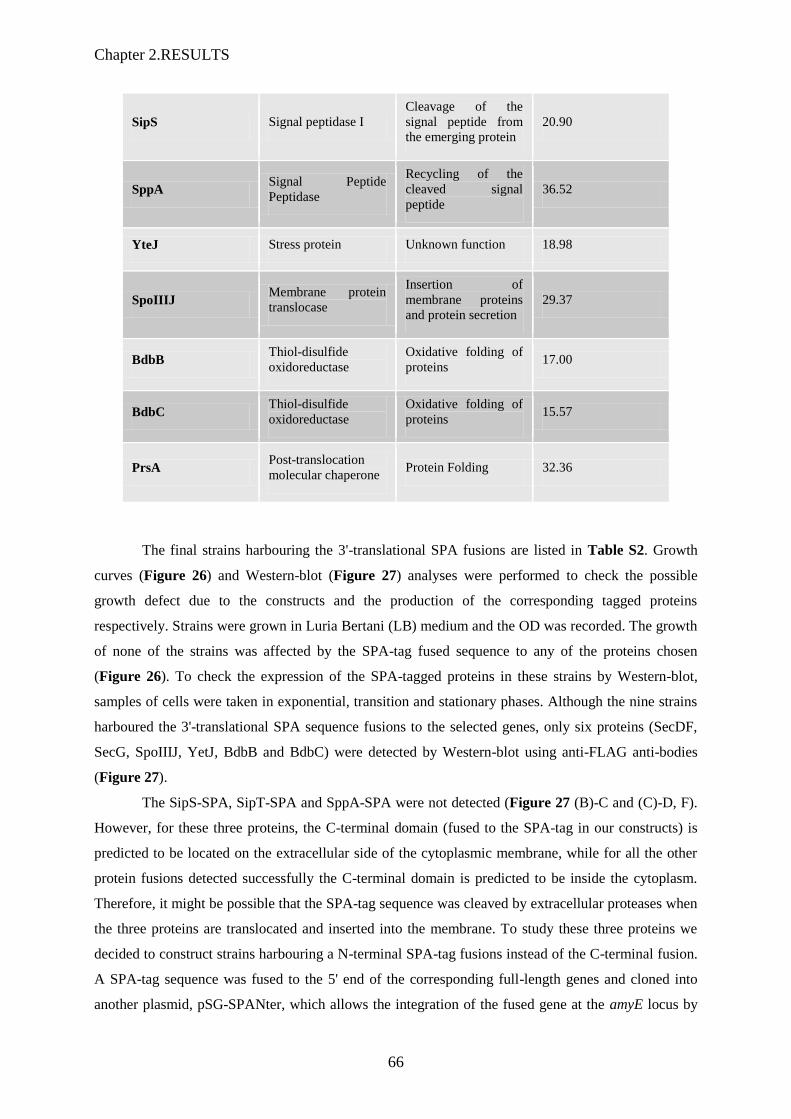
Chapter 2.RESULTS
66
SipS Signal peptidase I
Cleavage of the
signal peptide from
the emerging protein
20.90
SppA Signal Peptide
Peptidase
Recycling of the
cleaved signal
peptide
36.52
YteJ Stress protein Unknown function 18.98
SpoIIIJ Membrane protein
translocase
Insertion of
membrane proteins
and protein secretion
29.37
BdbB Thiol-disulfide
oxidoreductase
Oxidative folding of
proteins 17.00
BdbC Thiol-disulfide
oxidoreductase
Oxidative folding of
proteins 15.57
PrsA Post-translocation
molecular chaperone Protein Folding 32.36
The final strains harbouring the 3'-translational SPA fusions are listed in Table S2. Growth
curves (Figure 26) and Western-blot (Figure 27) analyses were performed to check the possible
growth defect due to the constructs and the production of the corresponding tagged proteins
respectively. Strains were grown in Luria Bertani (LB) medium and the OD was recorded. The growth
of none of the strains was affected by the SPA-tag fused sequence to any of the proteins chosen
(Figure 26). To check the expression of the SPA-tagged proteins in these strains by Western-blot,
samples of cells were taken in exponential, transition and stationary phases. Although the nine strains
harboured the 3'-translational SPA sequence fusions to the selected genes, only six proteins (SecDF,
SecG, SpoIIIJ, YetJ, BdbB and BdbC) were detected by Western-blot using anti-FLAG anti-bodies
(Figure 27).
The SipS-SPA, SipT-SPA and SppA-SPA were not detected (Figure 27 (B)-C and (C)-D, F).
However, for these three proteins, the C-terminal domain (fused to the SPA-tag in our constructs) is
predicted to be located on the extracellular side of the cytoplasmic membrane, while for all the other
protein fusions detected successfully the C-terminal domain is predicted to be inside the cytoplasm.
Therefore, it might be possible that the SPA-tag sequence was cleaved by extracellular proteases when
the three proteins are translocated and inserted into the membrane. To study these three proteins we
decided to construct strains harbouring a N-terminal SPA-tag fusions instead of the C-terminal fusion.
A SPA-tag sequence was fused to the 5' end of the corresponding full-length genes and cloned into
another plasmid, pSG-SPANter, which allows the integration of the fused gene at the amyE locus by

67
homologous recombination. Unfortunately, although several attempts were done, these constructions
could not be obtained.
Figure 26. Growth analysis of strains harbouring C-terminal SPA-tag fusions. The different strain cultures
were grown overnight in LB supplemented with 0.6 µg/ml Erythromycin and 0.5mM IPTG at 37°C, and in the
next day diluted 1:200 into a new medium with the same supplements. Compared to the BSB1 WT strain the
SPA-tag fusions do not affect the growth of the mutant strains. Samples were taken on specific time points.
SecDF-SPA was detected (Figure 27(B)-A) as a main band of the right molecular weight in
the three growth phases, with additional bands corresponding to proteins of lower molecular weight,
probably products of SecDF-SPA degradation.
SecG-SPA was detected (Figure 27(B)-B) just in transition and stationary phase with the
lower apparent expression level compared to that of the other SPA-tagged proteins of our analysis.
BdbC-SPA (Figure 27(C)-E), YteJ-SPA (Figure 27(D)-G), SpoIIIJ-SPA (Figure 27(D)-H)
and BdbC-SPA (Figure 27(D)-H) were also detected in the three growth phases, showing three
intense bands. These results showed the six detectable SPA-tagged proteins (SecDF, SecG, SpoIIIJ,
YetJ, BdbB and BdbC) could then be purified using this tandem affinity purification technique.
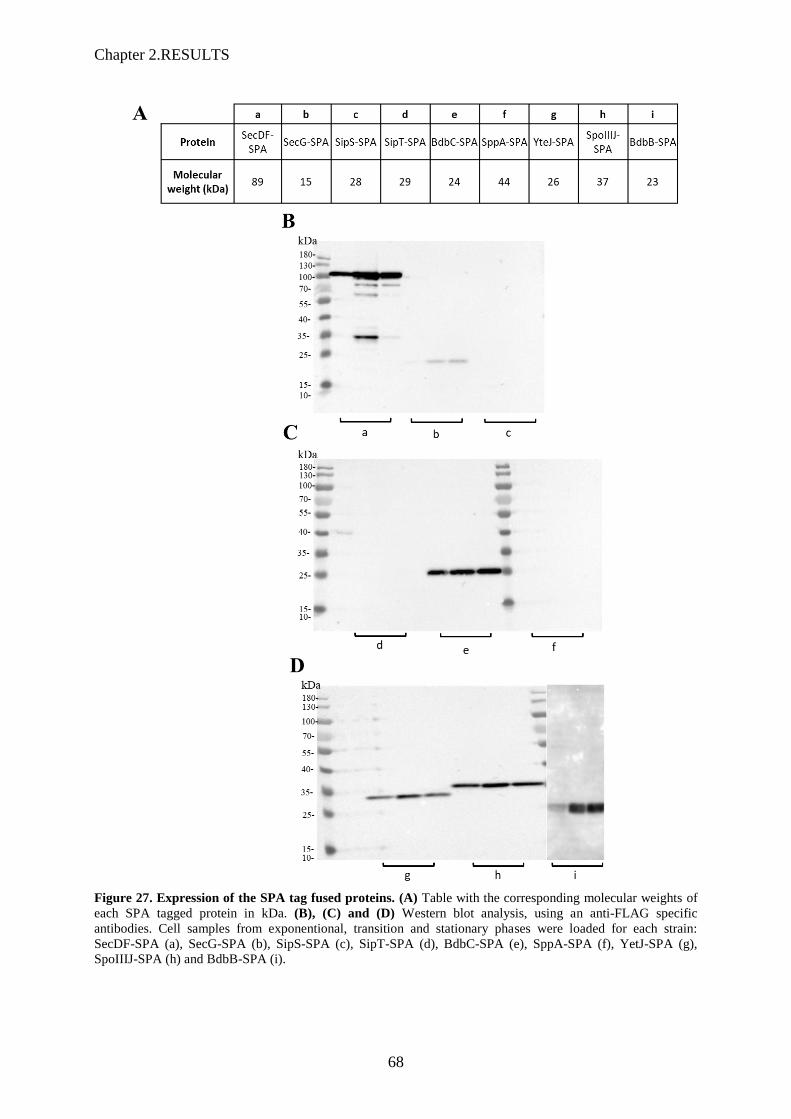
Chapter 2.RESULTS
68
Figure 27. Expression of the SPA tag fused proteins. (A) Table with the corresponding molecular weights of
each SPA tagged protein in kDa. (B), (C) and (D) Western blot analysis, using an anti-FLAG specific
antibodies. Cell samples from exponentional, transition and stationary phases were loaded for each strain:
SecDF-SPA (a), SecG-SPA (b), SipS-SPA (c), SipT-SPA (d), BdbC-SPA (e), SppA-SPA (f), YetJ-SPA (g),
SpoIIIJ-SPA (h) and BdbB-SPA (i).

69
2.1.2 Purifications of the SPA-tagged proteins
The tandem affinity purification approach is based on two sequential affinity purifications as
the SPA-tag is composed of two affinity tags (FLAG and CaM peptide). This double affinity
purification allows the elimination of more contaminants compared to a single affinity purification.
This technique is also use in native conditions to purify protein complexes, i.e. without using cross-
linkers that sometimes induce “false” interactions. The C-terminal SPA-tagged proteins (SecDF,
SecG, SpoIIIJ, YteJ, BdbB and BdbC) that were proved to be well expressed (see western-blot Figure
27) were purified using this procedure. For the identification of the co-purified proteins by liquid
chromatography coupled to tandem mass spectrometry (LC-MS/MS), the purified samples were
loaded on SDS-PAGE for a short migration. Large bands (0.5 cm), which comprises all the co-purified
proteins, were cut out of the gel and were sent for analysis by LC:MS/MS.
According to Schulze et al. (2014), SecDF, an ATPase, and YidC, an membrane protein
translocase, form a subcomplex that interacts with the SecYEG in E. coli, and all together constitute
the holo-translocon (Schulze et al. 2014). Therefore, we expected that SecDF and SpoIIIJ, respective
B. subtilis homologous of E. coli SecDF and YidC, form a sub-complex in B. subtilis as well. As such,
we expected to identify and detect, by LC-MS/MS, these proteins together in the same sample after the
SPA-purification of SecDF-SPA or SpoIIIJ-SPA. However, SecDF was not found in the co-purified
proteins of the SpoIIIJ-SPA purified sample, and conversely, SpoIIIJ was not detected either in the
SecDF-SPA purified sample (data not shown). These results showed that, as opposed to E.coli
(Schulze et al. 2014), SecDF and SpoIIIJ do not co-purify in B. subtilis. Perhaps, in B. subtilis this two
proteins form a weak interaction that could be disrupted by the presence of a SPA-tag. Although
counter-intuitive, the difference could come from the detergent (n-Dodecyl β-D-maltoside- DDM)
concentration used to solubilize the membranes, since in E. coli 2% DDM was used (Schulze et al.
2014) and our experiments were performed with 1% DDM. In B. subtilis, these complexes might be
more susceptible to the DDM concentration and even 1% is too high so that the weak interactions
might be abolished. In order to test this hypothesis, we used four different concentrations 0.5%,
0.75%, 1% and 2% of DDM to solubilize the B. subtilis membranes from the SecDF-SPA and SpoIIIJ-
SPA strains. To maximise the chances to get these two proteins co-purified in at least one condition,
we also used only one affinity chromatography, i.e. the calmoduline resin. After purification in these
different conditions of DDM concentrations, to check the quantity levels of the captured proteins,
SpoIIJ-SPA or SecDF-SPA, we analysed each purified sample by Western-blot (Figure 28 (A)).
These results showed that the purification of both proteins were more or less the same whatever the
different concentrations of DDM (Figure 28 (A)). The LC-MS/MS results corresponding to the
analysis of these samples, that have identified numerous proteins (mainly contaminants), are focused
only on the presence or absence of the other possible partners (SpoIIIJ for SecDF-SPA and SecDF for
SpoIIIJ-SPA) and are showed as two tables in Figure 28 (B). These results showed that the SpoIIIJ

Chapter 2.RESULTS
70
was not detected in the SecDF-SPA purified samples. However, low amounts of SecDF were detected
in all the SpoIIIJ-SPA samples. Altogether, these results showed that SecDF and SpoIIIJ do not form a
stable complex in B. subtilis in the conditions tested. The small amounts of SecDF found in the
SpoIIIJ-SPA purified samples might be an indication of a weak interaction between the two proteins
or a simple contamination as only one affinity column was used. In either cases we could conclude
that these two proteins form a stable complex, at least in our tested conditions.
Figure 28. Purification of SpoIIIJ-SPA and SecDF-SPA protein with different concentrations of DDM. (A)
Western blot analysis, using an anti-FLAG specific antibodies, of SecDF-SPA and SpoIIIJ-SPA in the presence
of a range of n-Dodecyl β-D-maltoside (DDM) concentrations, from 0.5% up to 2%. (B) Identification by LC-
MS/MS of the expected protein partner: poitive (٧) negative (-).

71
The other experiments using SPA-tagged proteins (SecG, BdbB, and BdbC) did not provide
evidence for the existence of any stable complexes involving proteins known to be part of the
translocon complex.
The only exception came with the finding of SppA, the so-called signal peptide peptidase, that
co-purified with YteJ-SPA. This result was expected as preliminary results obtained in our lab from
Blue native PAGE (BN-PAGE) experiments had indicated that these two proteins might form a
complex. Two tandem affinity purifications of the YteJ-SPA protein and LC-MS/MS analysis showed
that the relative quantities of the two proteins could be inferred from the LC-MS/MS data. The
SppA/YteJ ratios found were 2:1 and 1:1. As both genes are in operon, it can be expected that the two
proteins are translated in a 1:1 ratio, which would be in accordance with our experimental results.
2.1.3 Optimisation of the BN-PAGE technique
The BN-PAGE (for Blue Native PAGE) can be used to isolate protein complexes
from the membrane fraction, to determine the molecular weight of native protein complex, to obtain
information about their oligomeric states and to identify physiological protein–protein interactions
(Wittig, Braun, and Schägger 2006). We therefore used the BN-PAGE as a complementary technique
to identify the components of membrane protein complexes involved in the Sec pathway. As this
electrophoretic technique is performed in native conditions, theoritically, the different protein
components of a stable complex are not separated and migrate together in the gel. The n-Dodecyl β-D-
maltoside (DDM) was the detergent chosen to solubilize the membrane samples obtained after
ultracentrifugation of disrupted B. subtilis cells. After solubilisation (1% DDM) the samples are
supplemented with an anionic dye, Coomassie blue G-250, and the protein complexes are separated
according to their sizes by electrophoresis on a gradient 4-16% polyacrylamide gel. High molecular
weight complexes are running high in the gel. If the concentration of detergent is too high, these
complexes dissociate and individual proteins are running lower in the gel.
We used the SecDF-SPA strain to analyse if the BN-PAGE can used for our study as SecDF-
SPA is well expressed and can be easily detected by Western-Blot. Again different concentrations of
DDM were used to solubilize the SecDF-SPA cell membranes. To obtain better results we have loaded
the maximum quantity of protein possible (in this case 17.5µg, all the samples were loaded with same
amount) and to avoid contamination between samples, we have loaded the samples with four empty
wells between. In BN-PAGE, proteins often appear as a smear and not as single bands as in SDS-
PAGE gels (Figure 29). Nonetheless, at low DDM concentrations the smears corresponding to
SecDF-SPA appeared higher in the gel, indicating that SecDF-SPA is part of a higher molecular
weight complex. In contrast, at higher DDM concentrations this complex is dissociated as SecDF-SPA
appeared lower in the gel (Figure 29).

Chapter 2.RESULTS
72
Figure 29. Analysis by Western blot of a Blue native PAGE 4-16% Bis-Tris Gel of SecDF-SPA in the
presence of different concentrations of DDM. The triangle on the top represents the increase of DDM
concentration, from 0.05% up to 2%. The red circles represent the loaded samples, which with the increase of the
DDM concentration decreases its position in the gel.
For the analysis of BN-PAGE by LC-MS/MS each gel lane was divided in ten small pieces,
in-gel trypsin digested to identify proteins present in each part of the lane (Figure 30). The results we
obtained from the LC-MS/MS analysis of these samples indicated that 1% DDM is the best
concentration to solubilize the membrane samples of B. subtilis, as it was the condition with a higher
number of proteins detected. We needed a membrane protein complex that can be used as a control in
our experiment, and, according to preliminary data obtained at our laboratory SppA and YteJ could
co-migrate together in BN-PAGE. In our experiment, in the presence of 1% DDM, the LC-MS/MS
results showed that the presence together of SppA and YteJ in the gel fraction number 4 (Figure 30).
Interestingly, YteJ was identified in the piece of gel in which the highest number of SppA peptides
were detected. At lower or higher DDM concentrations, YteJ was not detected.
Unfortunately, peptides belonging to SpoIIIJ were identified in almost all pieces of gel, with
an equal number of spectra in each fraction, indicating that SpoIIIJ might dissociate easily from a
possible membrane complex. Moreover, for an unknown reason, SecDF was not detected in any piece
of gel, in any DDM conditions (Figure 30). Altogether, these results reinforced the hypothesis that
SppA and YteJ form a stable complex, while, if SpoIIIJ and SecDF form a membrane protein
complex, it is unstable.
In conclusion, using SPA purification combined with BN-PAGE analysis, we were
unsuccessful to reveal any expected complexes of the Sec pathway. We focused on using native
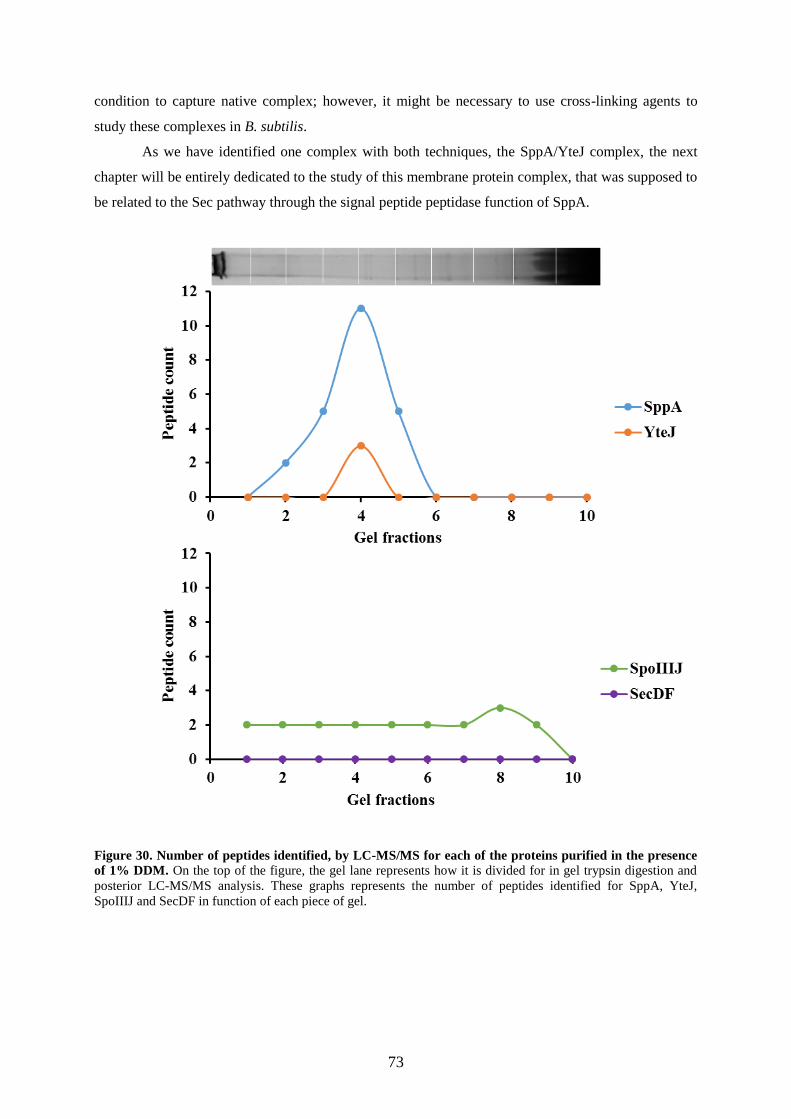
73
condition to capture native complex; however, it might be necessary to use cross-linking agents to
study these complexes in B. subtilis.
As we have identified one complex with both techniques, the SppA/YteJ complex, the next
chapter will be entirely dedicated to the study of this membrane protein complex, that was supposed to
be related to the Sec pathway through the signal peptide peptidase function of SppA.
Figure 30. Number of peptides identified, by LC-MS/MS for each of the proteins purified in the presence
of 1% DDM. On the top of the figure, the gel lane represents how it is divided for in gel trypsin digestion and
posterior LC-MS/MS analysis. These graphs represents the number of peptides identified for SppA, YteJ,
SpoIIIJ and SecDF in function of each piece of gel.

Chapter 2.RESULTS
74
2.2 The SppA/YteJ membrane complex
The function of B. subtilis SppA has been studied, as described at the introduction (Signal
peptide peptidase A (SppA) section). However, yteJ which in an operon with sppA encodes for a
protein of unknown function and unknown structure. As a first step towards the understanding of this
unknown protein, we analysed (i) its genomic context using bioinformatics tools, such as BLAST
(Singh and Raghava 2016) and microbial genomic context viewer (Overmars et al. 2013), and (ii) its
putative structure using structure prediction softwares such as Protter (Figure S 1 of appendix chapter)
(Omasits et al. 2014). Using BLAST-P to align the sequence of the Bacillus subtilis sppA yteJ operon
with different bacterial genomes of different bacterial species, the results showed that yteJ is often
present following sppA. The sppA gene is not only present in B. subtilis but also in most of the bacteria
from the firmicutes phylum, including pathogenic bacteria as listeria monocytogenes (Figure 31).
Figure 31. Alignment of sppA and yteJ genes with other bacteria of firmicutes phylum. The red highlighted
box indicate the sppA yteJ operon. (Overmars et al. 2013).
2.2.1 The sppA yteJ operon is regulated by sigma factors σA and σW
Bacterial sigma factors interact with the RNA polymerase core and enable its binding to
specific promoter sequences, inducing the initiation of transcription (Britton et al. 2002). Sigma-A
(σA) is the major sigma factor required for expression of many essential housekeeping genes and the
majority of the RNAs necessary during bacterial growth (Britton et al. 2002).
The extracytoplasmic function (ECF) subfamily of sigma factors is a distinct group that allows
bacteria to adequately respond to stressful environmental conditions and that are involved in the
control of cell envelope-related functions, such as secretion, synthesis of exopolysaccharides or
macromolecules (Missiakas and Raina 1998). B. subtilis has seven members of this sigma factor
family, identified through bacterial genome sequencing. The sigma-W factor (σW), a member of this
subfamily, is normally induced in response to cell envelope stress caused by antibiotics, alkaline and
salt shocks (Zweers et al. 2012).
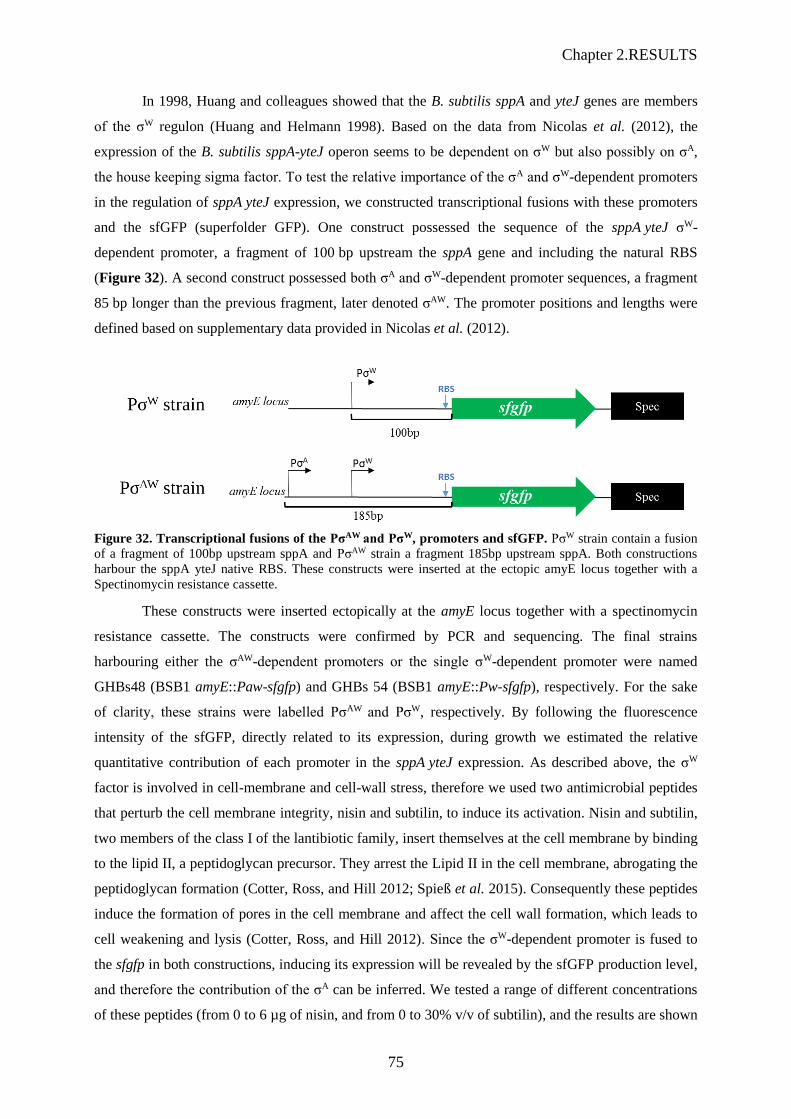
Chapter 2.RESULTS
75
In 1998, Huang and colleagues showed that the B. subtilis sppA and yteJ genes are members
of the σW regulon (Huang and Helmann 1998). Based on the data from Nicolas et al. (2012), the
expression of the B. subtilis sppA-yteJ operon seems to be dependent on σW but also possibly on σA,
the house keeping sigma factor. To test the relative importance of the σA and σW-dependent promoters
in the regulation of sppA yteJ expression, we constructed transcriptional fusions with these promoters
and the sfGFP (superfolder GFP). One construct possessed the sequence of the sppA yteJ σW-
dependent promoter, a fragment of 100 bp upstream the sppA gene and including the natural RBS
(Figure 32). A second construct possessed both σA and σW-dependent promoter sequences, a fragment
85 bp longer than the previous fragment, later denoted σAW. The promoter positions and lengths were
defined based on supplementary data provided in Nicolas et al. (2012).
Figure 32. Transcriptional fusions of the PσAW and PσW, promoters and sfGFP. PσW strain contain a fusion
of a fragment of 100bp upstream sppA and PσAW strain a fragment 185bp upstream sppA. Both constructions
harbour the sppA yteJ native RBS. These constructs were inserted at the ectopic amyE locus together with a
Spectinomycin resistance cassette.
These constructs were inserted ectopically at the amyE locus together with a spectinomycin
resistance cassette. The constructs were confirmed by PCR and sequencing. The final strains
harbouring either the σAW-dependent promoters or the single σW-dependent promoter were named
GHBs48 (BSB1 amyE::Paw-sfgfp) and GHBs 54 (BSB1 amyE::Pw-sfgfp), respectively. For the sake
of clarity, these strains were labelled PσAW and PσW, respectively. By following the fluorescence
intensity of the sfGFP, directly related to its expression, during growth we estimated the relative
quantitative contribution of each promoter in the sppA yteJ expression. As described above, the σW
factor is involved in cell-membrane and cell-wall stress, therefore we used two antimicrobial peptides
that perturb the cell membrane integrity, nisin and subtilin, to induce its activation. Nisin and subtilin,
two members of the class I of the lantibiotic family, insert themselves at the cell membrane by binding
to the lipid II, a peptidoglycan precursor. They arrest the Lipid II in the cell membrane, abrogating the
peptidoglycan formation (Cotter, Ross, and Hill 2012; Spieß et al. 2015). Consequently these peptides
induce the formation of pores in the cell membrane and affect the cell wall formation, which leads to
cell weakening and lysis (Cotter, Ross, and Hill 2012). Since the σW-dependent promoter is fused to
the sfgfp in both constructions, inducing its expression will be revealed by the sfGFP production level,
and therefore the contribution of the σA can be inferred. We tested a range of different concentrations
of these peptides (from 0 to 6 µg of nisin, and from 0 to 30% v/v of subtilin), and the results are shown

Chapter 2.RESULTS
76
in Figure 33 and Figure 34. The strains were grown in LB medium, supplemented with 100 µg.mL-1
spectinomycin, at 37°C with vigorous shaking in flasks. Overnight cultures were diluted 20-fold in
100 µL per well of pre-warmed fresh CH defined medium (pre-culture) into 96-well microtiter plates.
The CH pre-cultures were grown until an OD600nm of 0.3-0.4 and then diluted 400-fold into a pre-
warmed fresh medium. The antimicrobial peptides were added when the cell cultures reached early
exponential phase, around 0.2 OD600nm. OD600nm and fluorescence were recorded at time intervals of 10
minutes. The Figure 33 and Figure 34 display the PσAW and PσW strains growth curves and the
fluorescence/OD600nm ratio of sfGFP after induction (time point 0 min) with nisin or subtilin,
respectively. The background signal and the auto-fluorescence intensity were subtracted from all the
graphs.
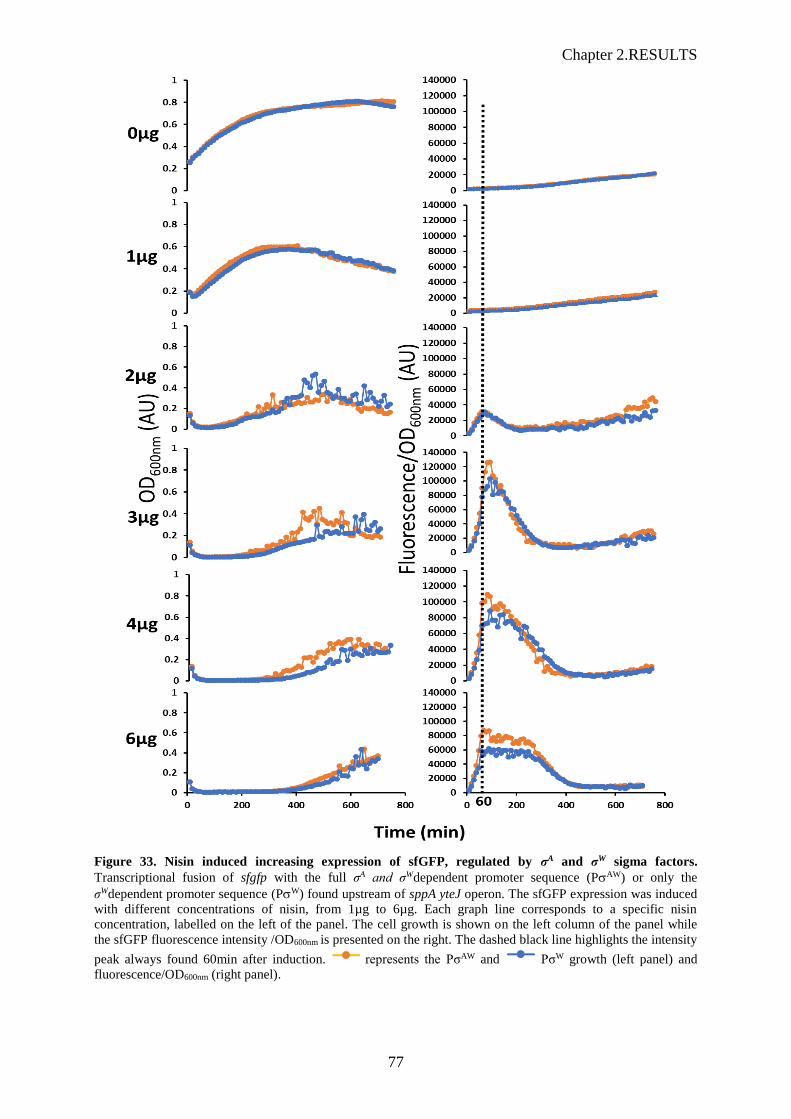
Chapter 2.RESULTS
77
Figure 33. Nisin induced increasing expression of sfGFP, regulated by σA and σW sigma factors.
Transcriptional fusion of sfgfp with the full σA and σWdependent promoter sequence (PAW) or only the
σWdependent promoter sequence (PW) found upstream of sppA yteJ operon. The sfGFP expression was induced
with different concentrations of nisin, from 1µg to 6µg. Each graph line corresponds to a specific nisin
concentration, labelled on the left of the panel. The cell growth is shown on the left column of the panel while
the sfGFP fluorescence intensity /OD600nm is presented on the right. The dashed black line highlights the intensity
peak always found 60min after induction. represents the PσAW and PσW growth (left panel) and
fluorescence/OD600nm (right panel).

Chapter 2.RESULTS
78
At the top of Figure 33, the graphs labelled 0 µg corresponds to the controls, i.e. the PσAW and
PσW strains growing in LB without nisin. Both strains showed identical growth rates, while the
intensity of measured sfGFP fluorescence remained low. With 1 µg of nisin (second line panel of
Figure 33), although the three characteristic phases of the bacterial growth (exponential, transition and
stationary phase) can be observed, when compared with the controls 0 µg, the growths of both strains
are affected. Just after induction, a decrease in the cell growth was observed followed by a rapid
growth recovery. However, these cells do not reach the same optical density as the controls and as in
stationary phase, the OD of the nisin-treated cultures showed a slight decrease due to cell lysis. When
treated with 2 µg of nisin the strain growths were affected, as a drop in the OD600nm was measured just
after induction. Probably a part of the cells died, by cell lysis, and it took around 200 min to the
remaining living cells to produce enough biomass to be measurable by OD. The corresponding ratio of
sfGFP fluorescence intensity/OD600nm showed, 60 min (represented by a dashed line) after the
induction, a clear peak, meaning that the remaining living cells were producing higher amount of
sfGFP. This was interpreted as a boost of activity of the sigma-dependent promoters. For higher
concentrations of nisin, from 3 µg up to 6 µg, a longer lag phase was observed during which the
OD600nm was not measurable. Regardless of nisin concentrations, the levels of the fluorescence/OD
ratio increased during the first 60 min. The highest levels were recorded with 3 µg.ml-1 of nisin, while
the addition of 4 or 6 µg.ml-1 of nisin led to lower levels. For 2 and 3 µg.mL-1 of nisin, a peak of
fluorescence/OD ratio was observed - i.e. same duration for the increase and decrease of the signal,
while for higher concentrations (4 and 6 µg), a longer lag phase and even a plateau than what we
observed for lower nisin concentrations. Whatever the nisin concentration applied to the cells, the
fluorescence/OD600nm ratios followed the same patterns and levels in the two strains, PW and PAW.
Therefore, whether the sfgfp gene was under the control of a single promoter sequence, i.e. in the PW
strain, or the two adjacent promoters sequences found natively in the chromosome, i.e. in the PAW
strain, the results were identical. The exception was a slight increase of this ratio that was observed for
the highest concentrations of nisin (6 µg.ml-1), which indicated a very marginal contribution of A to
the overall control of the operon expression. We concluded that the main promoter responsible for the
genetic regulation of the sppA yteJ operon was the W-dependent promoter. It should be noted that the
fluorescence/OD ratio decreased to reach back the minimal levels after a period of time which
increased proportionally to the nisin concentration. Moreover, whatever the nisin concentration
considered, when the signal ratio was back to the minimum level, the cell growth concomitantly
resumed. As W is activated in response to nisin, one might suggest that the nisin concentrations
decreased below the activation threshold of W, and that these concentration levels were compatible
with cell division.

Chapter 2.RESULTS
79
Figure 34. Subtilin induced increasing expression of sfGFP, regulated by σA σW sigma factors. Transcriptional fusion of sfgfp.with the σA and σWdependent promoter sequences, native of the sppA yteJ operon.
The sfGFP expression was induced with different concentrations of of subtilin, from 5% up to 30%. Each graph
line corresponds to a specific subtilin concentration, labelled on the left of the panel. The cell growth is shown
on the left column of the panel while the sfGFP fluorescence intensity /OD600nm is presented on the right. The
dashed black line highlights the intensity peak always found 60min after induction. represents the PσAW
and PσW growth.

Chapter 2.RESULTS
80
Similar results were obtained when cells were treated with subtilin (Figure 34), an
antimicrobial peptide of the same family. At low concentration of subtilin (5%), we did not observe
any difference between the control and the treated strains (first and second lines of the Figure 34),
neither for the growth nor for the sfGFP production. When we used 10% of subtilin, the sfGFP
expression was induced, displaying a small peak in the fluorescence intensity. When increasing the
treatment to 15% of subtilin we reproduce the results observed with 2 µg of nisin. After 60 min
(represented by the dashed line) of subtilin treatment, a clear peak on the sfGFP fluorescence intensity
ratio was observed. As observed for nisin, treating the strains with high concentrations of subtilin,
20% and 30%, leads to an increase in the lag phase and a W-dependent response 60 min after
induction, with higher and wider peaks of fluorescence intensity. In the presence of 30% of subtilin,
cells did not grow. but a high fluorescence peak with a larger plateau, than the lower concentrations of
subtilin, was observed. In conclusion, we observed that regardless of the nisin or subtilin
concentrations, the major contributor for the regulation of the expression of the sppA yteJ operon was
σW.
2.2.2 Deletion of the sppA and yteJ genes
To better understand the role of the B. subtilis SppA/YteJ complex, we deleted these two
genes either separetly or together to study the effect of their deletion(s). The sppA, yteJ and both genes
were deleted from their native locus and replaced by a chloramphenicol resistance cassette (cat). The
expression of this cassette remained under the control of the sppA yteJ native promoter, as described at
the section 4.3.2 of the Material and Methods chapter. These constructs were confirmed by PCR and
sequencing. The final strains were named: GHBs 023 (BSB1 ∆yteJ::cat), GHBs 025 (BSB1
∆sppA::cat) and GHBs 028 (BSB1 ∆sppA yteJ::cat). For the sake of clarity, strains GHBs 023, 025
and 028 will be named ∆yteJ, ∆sppA and ∆sppA yteJ, respectively. The growth of the strains were
recorded by measuring, at regular intervals, the optical density at 600 nm (OD600nm). The Figure 35
shows the growth curves plotted for each strain. The wild type strain (BSB1) and the deletion mutants
presented the same growth rate and cell density at all growth stages. These results allowed us to
conclude that the deletion of the sppA and/or yteJ genes did not affect the growth of the mutant strains
in LB medium. This is in agreement with the result reported in Bolhuis et al (1999) for the deletion of
sppA.
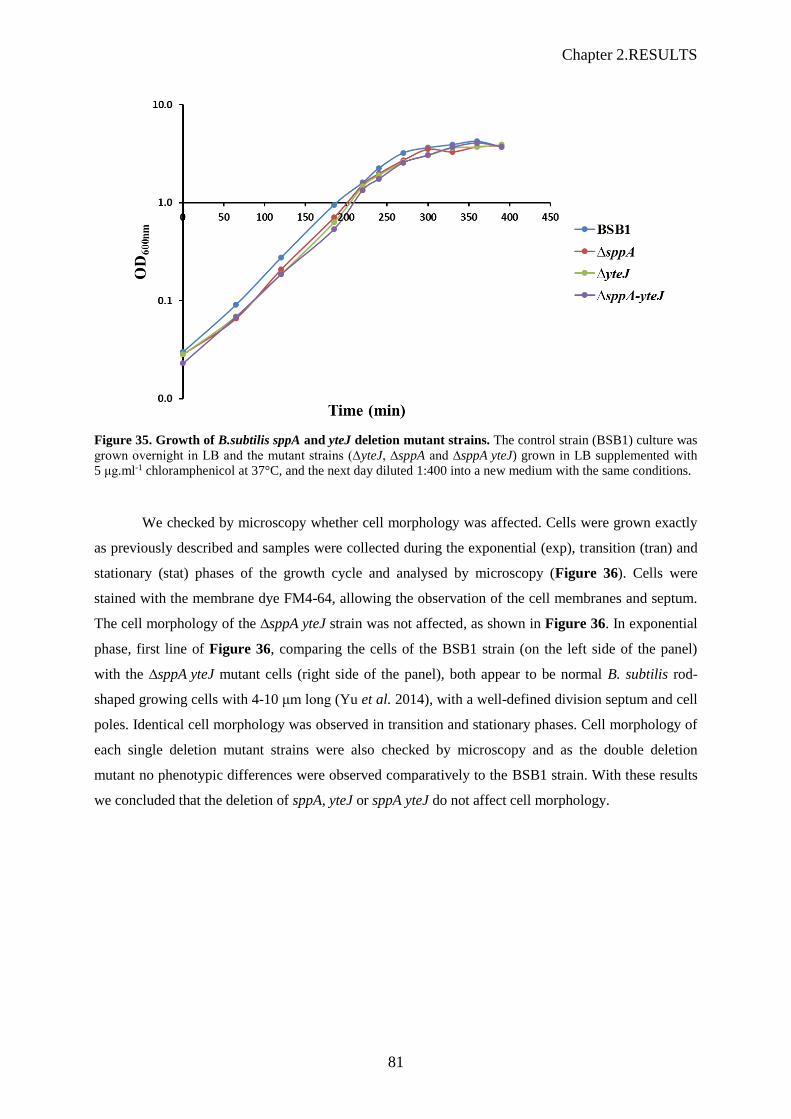
Chapter 2.RESULTS
81
Figure 35. Growth of B.subtilis sppA and yteJ deletion mutant strains. The control strain (BSB1) culture was
grown overnight in LB and the mutant strains (∆yteJ, ∆sppA and ∆sppA yteJ) grown in LB supplemented with
5 μg.ml-1 chloramphenicol at 37°C, and the next day diluted 1:400 into a new medium with the same conditions.
We checked by microscopy whether cell morphology was affected. Cells were grown exactly
as previously described and samples were collected during the exponential (exp), transition (tran) and
stationary (stat) phases of the growth cycle and analysed by microscopy (Figure 36). Cells were
stained with the membrane dye FM4-64, allowing the observation of the cell membranes and septum.
The cell morphology of the ∆sppA yteJ strain was not affected, as shown in Figure 36. In exponential
phase, first line of Figure 36, comparing the cells of the BSB1 strain (on the left side of the panel)
with the ∆sppA yteJ mutant cells (right side of the panel), both appear to be normal B. subtilis rod-
shaped growing cells with 4-10 μm long (Yu et al. 2014), with a well-defined division septum and cell
poles. Identical cell morphology was observed in transition and stationary phases. Cell morphology of
each single deletion mutant strains were also checked by microscopy and as the double deletion
mutant no phenotypic differences were observed comparatively to the BSB1 strain. With these results
we concluded that the deletion of sppA, yteJ or sppA yteJ do not affect cell morphology.

Chapter 2.RESULTS
82
Figure 36. Cell morphology of the sppA yteJ deletion mutant. The strains, control strain (BSB1) and
∆sppA yteJ, were grown overnight in LB at 37°C, supplemented with 5μg/ml chloramphenicol when required,
and in the next day diluted 400-fold into fresh medium with the same conditions. Samples were collected during
the exponential (exp), transition (tran) and stationary (stat) phases for microscopy analysis. Phase contrast
microscopy pictures and corresponding red fluorescence microscopy (515/640nm) are shown. Membranes (false
coloured red) were stained with FM4-64. Scale bar is 10 micron.
2.2.3 Role of the SppA/YteJ complex in protein secretion
Bolhuis et al., (1999) showed that the deletion of sppA affected the maturation of a highly
secreted protein, AmyQ. AmyQ, a secreted protein through the Sec pathway, is maintained in an
unfolded pre-protein form in the cytoplasm and reaches its folded mature form after being translocated
through the cell membrane and released to the extracellular medium. Bolhuis et al., (1999) observed
that the sppA deletion caused a delay in the processing of the AmyQ mature form which led to an
accumulation of the pre-AmyQ, which resulted in a mild decrease in the secretion of AmyQ. These
results suggested that the SppA activity was important in the efficient processing of the pre-secreted
proteins, supposedly by degrading the signal peptides from secreted proteins (Bolhuis et al. 1999).
While the role of SppA in protein secretion was documented, we wondered what was the role of YteJ,
and also what was the role of the SppA/YteJ complex in this cellular process. To address these issues,

Chapter 2.RESULTS
83
we checked the secretion levels of a highly secreted protein, the maltogenic amylase (AmyM). Dr.
Van Rij, from DSM (Delft, The Netherlands), provided us with a multi copy plasmid containing the
gene encoding AmyM (pCS73) and the corresponding empty plasmid (pCS74), all containing a
neomycin (neo) resistance cassette. Each one of these plasmids was transformed into the B. subtilis
sppA, yteJ or sppA yteJ deletion strains and the BSB1 wild type strain, giving rise to eight new
strains (listed in Table S5 in the appendix section) from GHBs029 to GHBs040. AmyM protein has a
molecular weight of 75.2 kDa and can be followed by SDS-PAGE.
To analyse the extracellular fraction, the cells were harvested in exponential phase of growth
and centrifuged. The obtained cell-free supernatant was supplemented with TCA to precipitate the
extracellular secreted proteins that were then analysed by SDS-PAGE. Before running the gel, the
concentration of the protein samples were measured by the Bradford method and equally loaded. The
results are shown in Figure 37. The two first lanes correspond to the control samples, the BSB1 wild-
type strain and BSB1 with pCS74, respectively. These two strains do not contain the amyM gene. The
sample BSB1 containing pCS74 empty plasmid was used as a control to check if the presence of a
multicopy plasmid affects the normal protein content. Compared to the BSB1 sample without plasmid,
first lane, the presence of the empty plasmid do not affect the protein content, as both lanes showed the
same protein pattern. The remaining four lanes on this gel correspond to the extracellular protein
samples of the strains expressing AmyM, thus containing the pCS73 plasmid, including the respective
control BSB1 and deletion mutant strains. All these samples showed a band around 75 kDa that
corresponds to the molecular weight of AmyM.
All three deleted strains showed the same intensity for the AmyM band on the gel. This
indicated that the deletion of sppA, yteJ or the operon sppA yteJ had no impact on the overall amount
of the hyper-secreted AmyM protein.
The delay in the processing of the AmyQ mature form observed by Bolhuis et al., (1999) was
a mild effect and the results were obtained through pulse-chase protein labelling. These facts can
explain why it is not possible to see a difference on the final amount of the mature form in our
experiments. To further study the premature and mature form of the secreted protein and analyse the
differences between the amount of secreted protein in the wild-type and mutant strains, we needed to
use specific antibodies for this protein. The production of AmyM antibodies, from the protein
purification up to rabbit immunization, was part of a secondment project developed in collaboration
with one of our industrial partners of the ProteinFactory ITN, UCB-Celltech company (Slough, UK),
including an internship of six weeks on UCB premises. The details of this work are described in the
Appendix 6.2.

Chapter 2.RESULTS
84
Figure 37. Analysis of AmyM extracellular amount by SDS PAGE. pCS74 empty plasmid; pCS73 plasmid
encoding amyM gene. The BSB1 wild type strain, BSB1 with pCS74 empty plasmid and with the pCS73 AmyM
plasmid were used as controls. The black arrow points for the correct band of AmyM.
With the serum of antibodies against AmyM we analysed the extracellular fraction, obtained
as previously explained, by Western Blot (WB). To do the WB we ran the samples in a 10%SDS-
PAGE gel exactly as described for the previous experiment. In the first two lanes, corresponding to the
controls, we cannot (as expected) observe a band corresponding to AmyM, since these strains do not
produce AmyM. The remaining samples, expressing AmyM, showed a band around 75KDa
corresponding to the molecular weight of AmyM and some lower bands corresponding to nonspecific
antibodies reaction or possible AmyM degradation. However, once more, this technique did not show
significant or measurable differences in the amount of secreted AmyM, between the strains. We also
tried to use a second reporter protein, the highly secreted endo-1,4-beta-xylanase (XynA). The xynA
gene was also provided by Dr. Van Rij, on a multi copy plasmid, and we proceeded to the same
experiment as just described above for AmyM. Unfortunately, it was not possible to detect XynA on
SDS-PAGE , maybe due to low expression and concentration of this secreted protein in the medium.

Chapter 2.RESULTS
85
Figure 38. Analysis of AmyM extracellular amount by Western Blot. This WB was performed with
antibodies against AmyM. pCS74 empty plasmid; pCS73 plasmid encoding amyM gene. The BSB1 wild type
strain, BSB1 with pCS74 empty plasmid and with the pCS73 AmyM plasmid were used as controls. The black
arrow points for the correct band of AmyM.
2.2.3.1 Effect of the sppA, yteJ or sppA yteJ overexpression in the secretion
Since we could not observe a difference in secretion caused by the deletion of the sppA and
yteJ genes, we wondered what would be the effect of their overexpression. Therefore, we constructed
overexpressing strains by re-inserting, in the WT BSB1 background, each gene or the full operon in
the amyE ectopic locus under the control of the hyperspank promoter (Phs), an IPTG inducible
promoter (as described in the material and methods section 4.3.3). These constructs were confirmed by
PCR and sequencing. The final strains are GHBs 059 (BSB1 amyE::Phs yteJ /spec), GHBs 066 (BSB1
amyE::Phs sppA /spec) and GHBs 071 (BSB1 amyE::Phs sppA yteJ /spec). For the sake of clarity, the
strains GHBs 059, 066 and 071 will be named BSB1 PhsyteJ, BSB1 PhssppA and BSB1 PhssppA yteJ,
respectively. To address the effect of the overexpression of SppA, YteJ and of the SppA/YteJ complex
in protein secretion we transformed these strains with either the multi copy plasmids containing the
gene encoding AmyM (pCS73) or an empty plasmid (pCS74) as a control, as we proceeded before
with the deletion mutants. This way, we could followed by SDS-PAGE the levels of AmyM secreted
into the extracellular medium. It gave rise to nine new strains (listed in Table S5 in the appendix
section) from GHBs073 up to GHBs081.
The overexpressing strains, listed above, were grown in LB at 37°C, supplemented with the
correct antibiotics when needed, and the cells were harvested in exponential phase and centrifuged.
The extracellular cell-free medium was TCA precipitated and analysed by 10% SDS-PAGE gels.
Before loading the samples onto the gel, the concentrations in total protein were measured by the
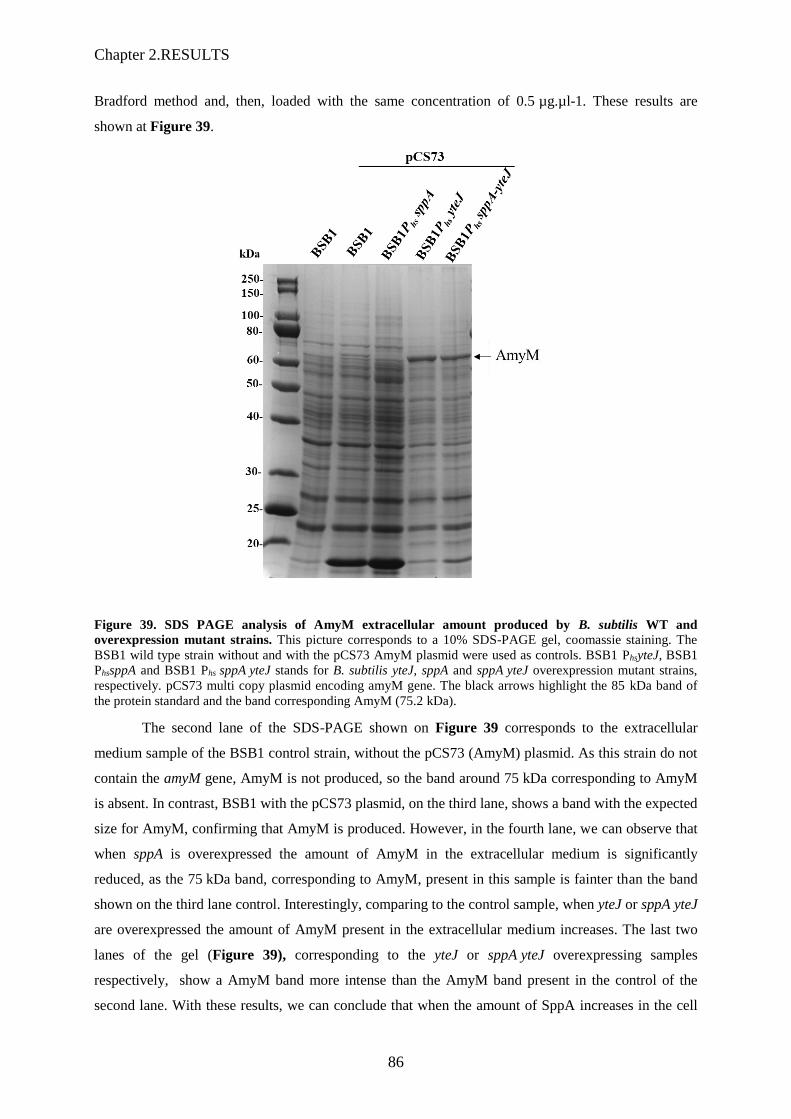
Chapter 2.RESULTS
86
Bradford method and, then, loaded with the same concentration of 0.5 µg.µl-1. These results are
shown at Figure 39.
Figure 39. SDS PAGE analysis of AmyM extracellular amount produced by B. subtilis WT and
overexpression mutant strains. This picture corresponds to a 10% SDS-PAGE gel, coomassie staining. The
BSB1 wild type strain without and with the pCS73 AmyM plasmid were used as controls. BSB1 PhsyteJ, BSB1
PhssppA and BSB1 Phs sppA yteJ stands for B. subtilis yteJ, sppA and sppA yteJ overexpression mutant strains,
respectively. pCS73 multi copy plasmid encoding amyM gene. The black arrows highlight the 85 kDa band of
the protein standard and the band corresponding AmyM (75.2 kDa).
The second lane of the SDS-PAGE shown on Figure 39 corresponds to the extracellular
medium sample of the BSB1 control strain, without the pCS73 (AmyM) plasmid. As this strain do not
contain the amyM gene, AmyM is not produced, so the band around 75 kDa corresponding to AmyM
is absent. In contrast, BSB1 with the pCS73 plasmid, on the third lane, shows a band with the expected
size for AmyM, confirming that AmyM is produced. However, in the fourth lane, we can observe that
when sppA is overexpressed the amount of AmyM in the extracellular medium is significantly
reduced, as the 75 kDa band, corresponding to AmyM, present in this sample is fainter than the band
shown on the third lane control. Interestingly, comparing to the control sample, when yteJ or sppA yteJ
are overexpressed the amount of AmyM present in the extracellular medium increases. The last two
lanes of the gel (Figure 39), corresponding to the yteJ or sppA yteJ overexpressing samples
respectively, show a AmyM band more intense than the AmyM band present in the control of the
second lane. With these results, we can conclude that when the amount of SppA increases in the cell

Chapter 2.RESULTS
87
the amount of secreted AmyM decreases. As SppA is a membrane protease, it might degrade directly
the AmyM produced or it could be an indirect effect. When YteJ is overexpressed, AmyM levels seem
to be higher than those observed with BSB1 strain, as if AmyM was less degraded. Furthermore, when
SppA is co-overexpressed with YteJ, the levels of AmyM are higher than BSB1. If SppA is able to
degrade AmyM directly – not yet proven – one can hypothesize that the SppA protease activity might
be inhibited or regulated by YteJ.
2.2.4 The deletion of sppA yteJ affects the long term survival
The ability to replicate in a specific environment is named bacterial fitness. The fitness of a
bacteria can be tested by analysing the capacity of survival of two strains, one antibiotic sensitive
strain and one antibiotic resistant strain, under the same conditions for a predefined time. Furthermore,
at the end of the predefined time, the number of bacteria is counted and the one which has produced a
higher number is by definition "Fitter".
In order to characterise the ∆yteJ, ∆sppA and ∆sppA yteJ deletion mutant strains we studied
their capacity of survival by performing some fitness tests. First the strains were grown in liquid rich
medium (LB), at 37°C for 48h. At different time points (after 18h, 24h and 48h), samples of the
cultures were plated out onto LB agar plates. The number of colonies were counted after the plates
have been incubated for 18h at 37°C. As shown in Figure 40, after 18h of growth, the number of
colonies counted is just slightly affected by the deletion of sppA, yteJ and the full operon, as compared
with the number of WT colonies. The lowest number of colonies was counted for the ∆sppA strain,
showing around 200 CFU/ml, which is 1.5-times less than the wild type. After 24h of growth in the
same medium, the capacity of survival of all strains was affected, because the number of colonies was
lower than the numbers after 18h of growth, even for the WT control strain. However, the capacity of
survival of the deletion mutants was even more affected, as in the absence of these genes the number
of surviving colonies is lower than the WT. The ∆sppA strain was the more affected, showing the
lowest value of colonies counted, around 150 CFU/ml lower than the wild type control.
After of 48h of growth, as shown on Figure 40, although all the strains showed a lower
number of surviving colonies, the mutant strains have a lower capacity of survival. The ∆sppA yteJ
and ∆sppA strains are both the less fit, as both have the lowest number of colonies counted. The yteJ
strain was the least affected strain compared to the other mutant strains. The results of these tests
showed that in LB medium the survival of the deletion strains is affected comparing to the wild type
strain. We concluded that the deletion of sppA was the most deleterious one for survival, indicating
that SppA function provides an advantage for long term survival. The same conclusion applied to the
deletion of sppA yteJ operon. However, as the deletion of yteJ was the less deleterious, we concluded
that YteJ function was not important for survival.

Chapter 2.RESULTS
88
Figure 40. Capacity of survival of BSB1 and ∆yteJ, ∆sppA and ∆sppA yteJ deletion mutants. The strains
were grown in rich liquid media (LB), at 37°C for 48h. After 18h, 24h and 48h, samples of the cultures were
collected and plated out onto LB agar plates. The number of colonies were counted after the plates have been
incubated for 18h at 37°C.
The survival ability of the ∆sppA yteJ mutant was as much affected as for the ∆sppA mutant
strain. As our main objective was to understand the function of the SppA/YteJ complex we performed
further fitness tests with only BSB1 and ∆sppA yteJ strains. We wondered whether the composition of
the media may affect the long-term survival of the strains. New fitness tests in LB rich media (Figure
41A) and in M9 minimal medium (M9 supplemented with glucose) were realised (Figure 41B). After
24h and 48h, samples were collected and plated on LB agar plates, and colonies were counted after
18h of growth at 37°C.
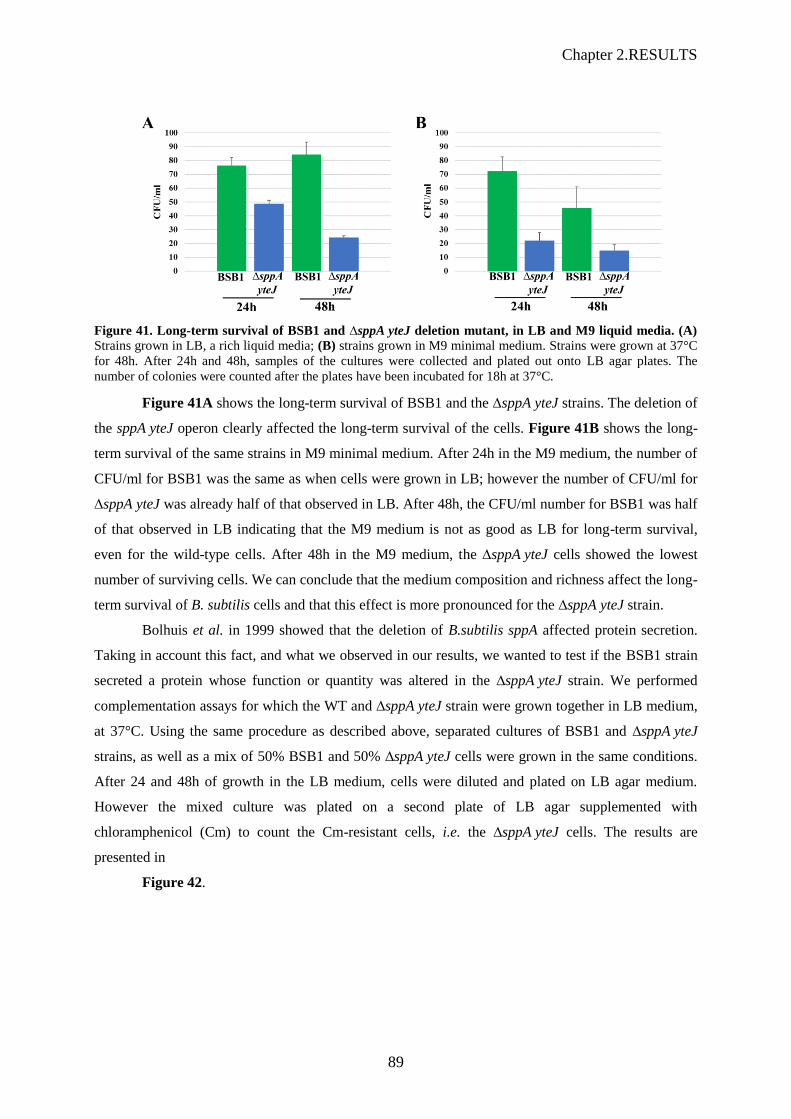
Chapter 2.RESULTS
89
Figure 41. Long-term survival of BSB1 and ∆sppA yteJ deletion mutant, in LB and M9 liquid media. (A)
Strains grown in LB, a rich liquid media; (B) strains grown in M9 minimal medium. Strains were grown at 37°C
for 48h. After 24h and 48h, samples of the cultures were collected and plated out onto LB agar plates. The
number of colonies were counted after the plates have been incubated for 18h at 37°C.
Figure 41A shows the long-term survival of BSB1 and the ∆sppA yteJ strains. The deletion of
the sppA yteJ operon clearly affected the long-term survival of the cells. Figure 41B shows the long-
term survival of the same strains in M9 minimal medium. After 24h in the M9 medium, the number of
CFU/ml for BSB1 was the same as when cells were grown in LB; however the number of CFU/ml for
∆sppA yteJ was already half of that observed in LB. After 48h, the CFU/ml number for BSB1 was half
of that observed in LB indicating that the M9 medium is not as good as LB for long-term survival,
even for the wild-type cells. After 48h in the M9 medium, the ∆sppA yteJ cells showed the lowest
number of surviving cells. We can conclude that the medium composition and richness affect the long-
term survival of B. subtilis cells and that this effect is more pronounced for the ∆sppA yteJ strain.
Bolhuis et al. in 1999 showed that the deletion of B.subtilis sppA affected protein secretion.
Taking in account this fact, and what we observed in our results, we wanted to test if the BSB1 strain
secreted a protein whose function or quantity was altered in the ∆sppA yteJ strain. We performed
complementation assays for which the WT and ∆sppA yteJ strain were grown together in LB medium,
at 37°C. Using the same procedure as described above, separated cultures of BSB1 and ∆sppA yteJ
strains, as well as a mix of 50% BSB1 and 50% ∆sppA yteJ cells were grown in the same conditions.
After 24 and 48h of growth in the LB medium, cells were diluted and plated on LB agar medium.
However the mixed culture was plated on a second plate of LB agar supplemented with
chloramphenicol (Cm) to count the Cm-resistant cells, i.e. the ∆sppA yteJ cells. The results are
presented in
Figure 42.
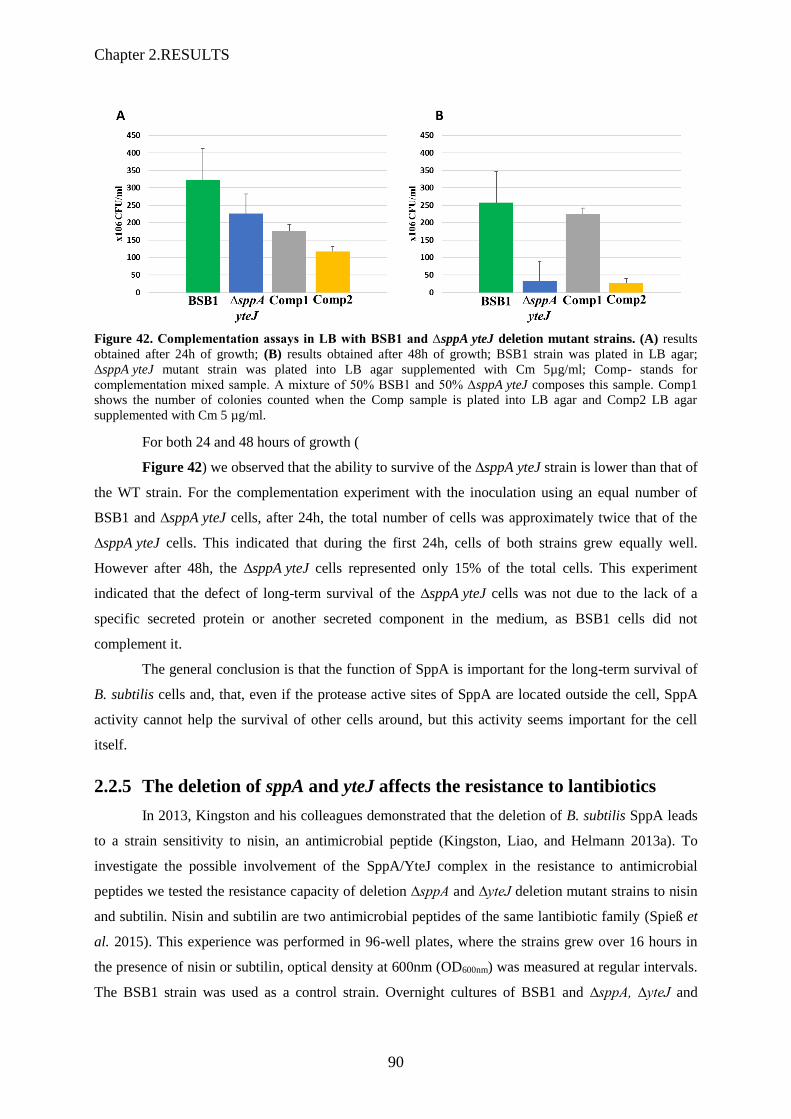
Chapter 2.RESULTS
90
Figure 42. Complementation assays in LB with BSB1 and ∆sppA yteJ deletion mutant strains. (A) results
obtained after 24h of growth; (B) results obtained after 48h of growth; BSB1 strain was plated in LB agar;
∆sppA yteJ mutant strain was plated into LB agar supplemented with Cm 5µg/ml; Comp- stands for
complementation mixed sample. A mixture of 50% BSB1 and 50% ∆sppA yteJ composes this sample. Comp1
shows the number of colonies counted when the Comp sample is plated into LB agar and Comp2 LB agar
supplemented with Cm 5 µg/ml.
For both 24 and 48 hours of growth (
Figure 42) we observed that the ability to survive of the ∆sppA yteJ strain is lower than that of
the WT strain. For the complementation experiment with the inoculation using an equal number of
BSB1 and ∆sppA yteJ cells, after 24h, the total number of cells was approximately twice that of the
∆sppA yteJ cells. This indicated that during the first 24h, cells of both strains grew equally well.
However after 48h, the ∆sppA yteJ cells represented only 15% of the total cells. This experiment
indicated that the defect of long-term survival of the ∆sppA yteJ cells was not due to the lack of a
specific secreted protein or another secreted component in the medium, as BSB1 cells did not
complement it.
The general conclusion is that the function of SppA is important for the long-term survival of
B. subtilis cells and, that, even if the protease active sites of SppA are located outside the cell, SppA
activity cannot help the survival of other cells around, but this activity seems important for the cell
itself.
2.2.5 The deletion of sppA and yteJ affects the resistance to lantibiotics
In 2013, Kingston and his colleagues demonstrated that the deletion of B. subtilis SppA leads
to a strain sensitivity to nisin, an antimicrobial peptide (Kingston, Liao, and Helmann 2013a). To
investigate the possible involvement of the SppA/YteJ complex in the resistance to antimicrobial
peptides we tested the resistance capacity of deletion ∆sppA and ∆yteJ deletion mutant strains to nisin
and subtilin. Nisin and subtilin are two antimicrobial peptides of the same lantibiotic family (Spieß et
al. 2015). This experience was performed in 96-well plates, where the strains grew over 16 hours in
the presence of nisin or subtilin, optical density at 600nm (OD600nm) was measured at regular intervals.
The BSB1 strain was used as a control strain. Overnight cultures of BSB1 and ∆sppA, ∆yteJ and

Chapter 2.RESULTS
91
∆sppA yteJ strains were grown at 37 ᵒC, in LB and LB supplemented with Cm 5 µg/ml respectively,
with aeration and vigorous shaking in flasks. The overnight cultures were diluted 20-fold into 100µl
(into the 96 well plate) of fresh pre-warmed LB medium and grown until beginning of exponential
phase. Then the cells were re-diluted 400-fold into a new plate, containing different concentrations of
nisin or subtilin. The Figure 43 shows the results obtained with nisin.
Figure 43. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of Nisin. The BSB1, ∆sppA and ∆yteJ strains grown in LB in a 96-well plate, where the OD600
was measured at specific time points. The different graphs represent the growth of the strains in the presence of
different concentrations (2 µg, 3 µg, 4 µg, 6 µg and 8 µg/ml) of nisin, labelled on the left of the graph.
In Figure 43, BSB1, ∆sppA and ∆yteJ strains showed identical growth rates when grown in
LB without nisin. With 3µg/ml of nisin (graph middle left), although the three characteristic phases of
the bacterial growth (exponential, transition and stationary phase), the growth of the strains was
affected. The strains took around 100 extra minutes to resume growth showing a longer lag phase than
in the control conditions. We defined the time necessary to resume growth as the time needed for the
strain to reach an OD600 of 0.1. In the presence of 3µg/ml of nisin, the deletion of yteJ led to a slightly
increased resistance to nisin, as it took around 20 minutes less than the BSB1 strain to reach an OD600
of 0.1. On the contrary the deletion of sppA turn the cells more susceptible to nisin, as it took around
20 minutes more than the BSB1 strain to reach an OD600 of 0.1. When the concentration of nisin was
increased to 4 µg/ml in the medium, the same results were observed but with an exacerbate difference

Chapter 2.RESULTS
92
between the strains. When treated with 6µg/ml of nisin the BSB1 strain showed a lag phase of 250
minutes longer than in the control condition without nisin. In this condition, the ∆yteJ strain showed
the same lag phase and growth rate as the BSB1 strain, while the ∆sppA strain took slightly longer to
resume growth. The ∆sppA strain is therefore more susceptible to nisin than the two other strains. In
the presence of 8µg/ml of nisin, the strains did not grow.
To simplify we did not represent the ∆sppA yteJ results, as this mutant strain behaved exactly
as the yteJ mutant strain. With these results we concluded that the deletion of sppA affects de
resistance to nisin. However the deletion of yteJ or the full operon, yteJ sppA, showed non
reproducible results with either an intermediate phenotype or a nisin resistance higher than that of the
BSB1 strain.
Although the trend for the nisin resistance of the BSB1 strain compared to the deletion strains
were reproducible, the effect of a specific nisin concentration could vary from one experiment to
another. The stability of nisin might explain these differences. Therefore we decided to test another
lantibiotic of the same class with a highly similar ring structure, subtilin. (Spieß et al. 2015). The
experiments were performed as explained before for nisin. As shown in Figure 44, the results
obtained with subtilin were similar to that observed with nisin.
Figure 44. Growth of the B. subtilis sppA and yteJ mutant strains in the presence of different
concentrations of subtilin. The BSB1, ∆sppA and ∆yteJ strains were grown in LB in a 96-well plate, and the
OD600 was measured at specific time points. The different graphs represent the growth of the strains in the
presence of different concentrations (20%, 25%, 30%, 34% and 38%) of subtilin, labelled on the left of the
graph.

Chapter 2.RESULTS
93
With 20% of subtilin (graph top right, Figure 44), although we observed the three
characteristic phases of the bacterial growth (exponential, transition and stationary phase), when
compared with the control conditions 0% (graph top left), the growth of the strains was altered. The
cells took slightly longer to resume growth, did not reach the same optical density in stationary phase
as in the control conditions. The growth of the two deletion strains showed a small delay compared to
the BSB1 strain. When treated with 25% of subtilin, the BSB1 and ∆yteJ mutant strains had identical
growth rates, and similar 'lag phase' of around 200 min. The ∆sppA strain showed an even longer lag
phase, around 100 extra minutes to resume growth, indicating a higher susceptibilty to subtilin.
Increasing the subtilin to 30% led to a striking difference between the growths of the three strains.
Comparing BSB1 with ∆yteJ, we can see that the deletion of yteJ led to a decrease in the resistance to
subtilin, as it took around 200 minutes more than the BSB1 strain to reach an OD600 of 0.1. The
deletion of sppA turn the strain even more susceptible, as it took 300 minutes more than the BSB1
strain to reach an OD600 of 0.1. The deletion of yteJ showed an intermediate phenotype, between the
BSB1 and ∆sppA strains. When the concentration of subtilin was increased to 34%, the BSB1 strain
was able to resume growth after 300 min while it took 600 min for the ∆sppA strain and while the
∆yteJ strain showed almost no growth. Altogether these results led us to conclude that the deletion of
either yteJ or sppA turned the cells more susceptible to subtilin. In the presence of 38% subtilin, the
strains did not grow. As stated before for the nisin results, with subtilin we did not show the
∆sppA yteJ results, as this strain had the same behaviour as the yteJ strain. With these results,
comparable to the nisin results, we can conclude that the deletion of sppA affects de resistance to
subtilin. However, depending on the concentration of subtilin, the deletion of yteJ or of the full operon
showed again non reproducible results (as for nisin) with either an intermediate phenotype or a subtilin
resistance higher than that of the BSB1 strain.
Figure 45 illustrates the effect of increasing subtilin concentrations on the growth rate. It
revealed that the growth rates were strongly affected when the concentration of subtilin was higher
than 30%. As the results obtained with subtilin were more reproducible than the experiments done
with nisin, and as a 30% subtilin concentration showed significant differences in the lag phase
duration of the three strains without affecting too much the growth rates (and so allowing to
discriminate strains based on their subtilin resistance capacity), we selected this concentration to setup
the lantibiotic resistance test used later in this work.
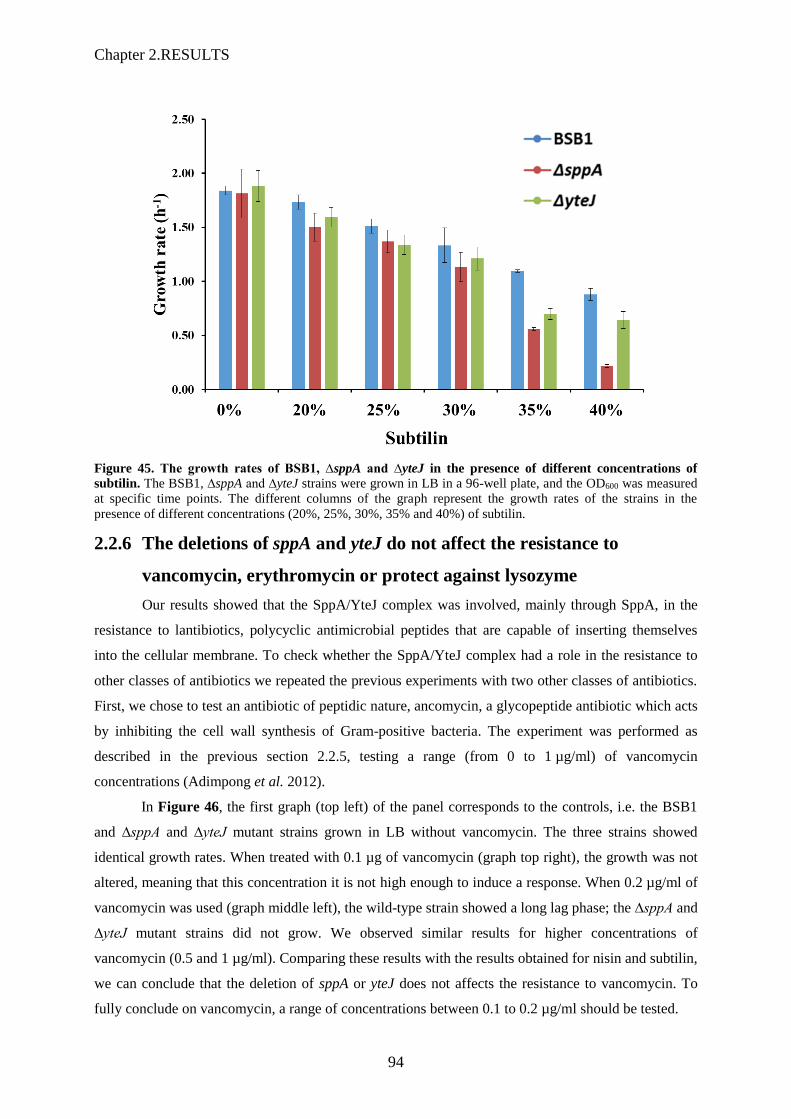
Chapter 2.RESULTS
94
Figure 45. The growth rates of BSB1, ∆sppA and ∆yteJ in the presence of different concentrations of
subtilin. The BSB1, ∆sppA and ∆yteJ strains were grown in LB in a 96-well plate, and the OD600 was measured
at specific time points. The different columns of the graph represent the growth rates of the strains in the
presence of different concentrations (20%, 25%, 30%, 35% and 40%) of subtilin.
2.2.6 The deletions of sppA and yteJ do not affect the resistance to
vancomycin, erythromycin or protect against lysozyme
Our results showed that the SppA/YteJ complex was involved, mainly through SppA, in the
resistance to lantibiotics, polycyclic antimicrobial peptides that are capable of inserting themselves
into the cellular membrane. To check whether the SppA/YteJ complex had a role in the resistance to
other classes of antibiotics we repeated the previous experiments with two other classes of antibiotics.
First, we chose to test an antibiotic of peptidic nature, ancomycin, a glycopeptide antibiotic which acts
by inhibiting the cell wall synthesis of Gram-positive bacteria. The experiment was performed as
described in the previous section 2.2.5, testing a range (from 0 to 1 µg/ml) of vancomycin
concentrations (Adimpong et al. 2012).
In Figure 46, the first graph (top left) of the panel corresponds to the controls, i.e. the BSB1
and ∆sppA and ∆yteJ mutant strains grown in LB without vancomycin. The three strains showed
identical growth rates. When treated with 0.1 µg of vancomycin (graph top right), the growth was not
altered, meaning that this concentration it is not high enough to induce a response. When 0.2 µg/ml of
vancomycin was used (graph middle left), the wild-type strain showed a long lag phase; the ∆sppA and
∆yteJ mutant strains did not grow. We observed similar results for higher concentrations of
vancomycin (0.5 and 1 µg/ml). Comparing these results with the results obtained for nisin and subtilin,
we can conclude that the deletion of sppA or yteJ does not affects the resistance to vancomycin. To
fully conclude on vancomycin, a range of concentrations between 0.1 to 0.2 µg/ml should be tested.

Chapter 2.RESULTS
95
Figure 46. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of vancomycin. The BSB1, ∆sppA and ∆yteJ deletion strains grown in LB in a 96-well plate.
The OD600 was measured at specific time points. The different graphs represent the growth of the strains in the
presence of different concentrations (0µg, 0.1µg, 0.2µg, 0.5µg and 1µg) of vancomycin, labelled on the left of
the graph. Colours, on the right side of the picture, indicate the wild type or mutated strains.
To test a different class of antibiotic not related to cell wall or membrane synthesis or
integrity, we chose erythromycin. Erythromycin is part of the macrolide family of antibiotics, which
decreases the bacterial protein production. The experiment was performed as described earlier, testing
a range (from 0-1 µg/ml) of different concentrations of erythromycin, taking into account the minimal
inhibitory concentration (MIC) of this antibiotic against B. subtilis, 4 µg/ml (Adimpong et al. 2012).
The results obtained with erythromycin (Figure 47) are similar to that observed with vancomycin. In
the absence of erythromycin, the BSB1, ∆sppA and ∆yteJ strains showed identical growth rates.
However, when we started to treat with 0.1 µg of erythromycin (Figure 47, graph top right) strains did
not grow, meaning that at this concentration the antibiotic have a bactericidal effect. All the
concentrations used of erythromycin showed the same results. Furthermore, for all concentrations of
erythromycin used so far, the WT strain started to grow after 600 min, reaching an OD600 of 0.1.
Comparing these results with the results obtained with the lantibiotics, we concluded that the deletion
of sppA or yteJ do not affects de resistance to erythromycin.

Chapter 2.RESULTS
96
Figure 47. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of erythromycin. The BSB1, ∆sppA and ∆yteJ deletion strains grown in LB in a 96-well plate.
The OD600 was measured at specific time points. The different graphs represent the growth of the strains in the
presence of different concentrations (0 µg, 0.1 µg, 0.2 µg, 0.5 µg and 1 µg) of erythromycin, labelled on the left
of the graph. Colours, on the right side of the picture, indicate the wild type or mutated strains.
SppA is a serine protease which is, according to Nam et al., 2012, capable of digesting fully
folded proteins. Therefore, we decided to test if the SppA/YteJ complex could be involved in a
mechanism that protects the cell against the activity of lysozyme, glycoside hydrolase that is used as
an antimicrobial enzyme due to its capacity to digest peptidoglycan, the major component of the
Gram-positive bacterial cell wall. The experiment was performed as previously described, testing
different concentrations of lysozyme, from 0 to 250 µg/ml. The lysozyme results are shown in Figure
48. Whatever the concentrations of lysozyme added in the medium, no marked differences were
observed between the BSB1 and the ∆sppA and ∆yteJ strains. (At 150 µg/ml lysozyme, the ∆yteJ
strain took slightly longer to reach OD600 0.1 compared to the BSB1 and ∆sppA strain. As the results
obtained for vancomycin and erythromycin, it seems that SppA/YteJ complex does not protect the
cells against the lysozyme action.
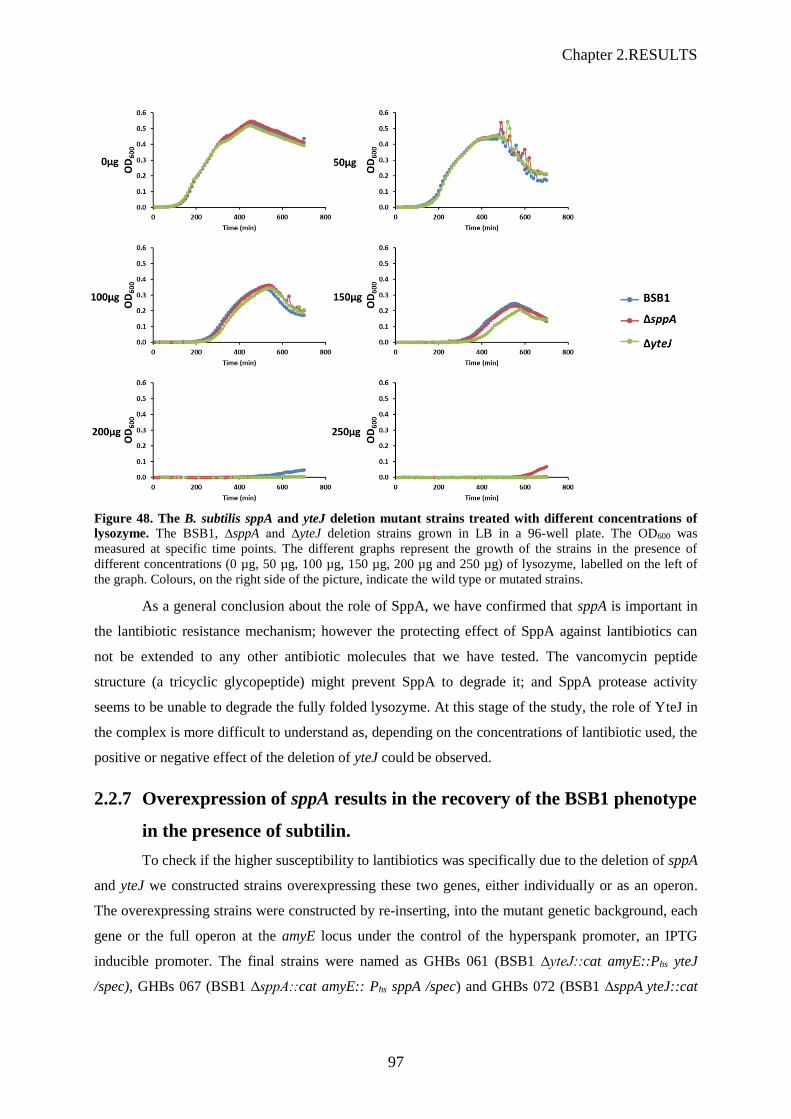
Chapter 2.RESULTS
97
Figure 48. The B. subtilis sppA and yteJ deletion mutant strains treated with different concentrations of
lysozyme. The BSB1, ∆sppA and ∆yteJ deletion strains grown in LB in a 96-well plate. The OD600 was
measured at specific time points. The different graphs represent the growth of the strains in the presence of
different concentrations (0 µg, 50 µg, 100 µg, 150 µg, 200 µg and 250 µg) of lysozyme, labelled on the left of
the graph. Colours, on the right side of the picture, indicate the wild type or mutated strains.
As a general conclusion about the role of SppA, we have confirmed that sppA is important in
the lantibiotic resistance mechanism; however the protecting effect of SppA against lantibiotics can
not be extended to any other antibiotic molecules that we have tested. The vancomycin peptide
structure (a tricyclic glycopeptide) might prevent SppA to degrade it; and SppA protease activity
seems to be unable to degrade the fully folded lysozyme. At this stage of the study, the role of YteJ in
the complex is more difficult to understand as, depending on the concentrations of lantibiotic used, the
positive or negative effect of the deletion of yteJ could be observed.
2.2.7 Overexpression of sppA results in the recovery of the BSB1 phenotype
in the presence of subtilin.
To check if the higher susceptibility to lantibiotics was specifically due to the deletion of sppA
and yteJ we constructed strains overexpressing these two genes, either individually or as an operon.
The overexpressing strains were constructed by re-inserting, into the mutant genetic background, each
gene or the full operon at the amyE locus under the control of the hyperspank promoter, an IPTG
inducible promoter. The final strains were named as GHBs 061 (BSB1 ∆yteJ::cat amyE::Phs yteJ
/spec), GHBs 067 (BSB1 ∆sppA::cat amyE:: Phs sppA /spec) and GHBs 072 (BSB1 ∆sppA yteJ::cat

Chapter 2.RESULTS
98
amyE::Phs sppA yteJ /spec). For the sake of clarity, strains GHBs 061, 067 and 072 will be named
PhsyteJ, PhssppA and PhssppA yteJ, respectively.
Cells were grown in 96 well plates, over 16 hours in the presence of 30% subtilin and different
concentrations of IPTG, and the optical density at 600 nm (OD600nm) was measured at regular intervals.
The BSB1 strain was used as the control strain. Overnight cultures of the BSB1 and PhsyteJ, PhssppA
and Phs sppA yteJ deletion mutant strains were grown at 37ᵒC with aeration and vigorous shaking in
flasks. BSB1 in LB and the overexpressing mutants in LB supplemented with 5 µg/ml Cm and
100 µg/ml Spec. The overnight cultures were diluted 20-fold into 100 µl (in the 96 well plate) of fresh
pre-warmed LB medium and grown until the beginning of the exponential phase. Then the cells were
re-diluted 400-fold into a new plate in the following conditions: LB as control, LB supplemented with
30% subtilin and LB supplemented with 30% subtilin and different concentrations of IPTG.
The Figure 49 shows the results for the overexpression of sppA and yteJ. In LB (0% subtilin
0 µM IPTG), the BSB1, PhsyteJ and PhssppA strains showed identical growth rates. In the presence of
30% subtilin and 0 µM IPTG, the PhssppA (no SppA is produced) showed a longer lag phase, around
100 extra minutes, compared to BSB1 and PhsyteJ. When 10 µM of IPTG was added, inducing the
overexpression of sppA and yteJ, we could observe that the lag phase of strain PhssppA decreased.
Increasing the concentrations of IPTG to 25 and 50 µM induced an even shorter lag phase for PhssppA
strain. The PhssppA resistance to 30 % subtilin was almost recovered at 50µM ITPG. With 50µM of
IPTG, the PhsyteJ showed a very slight increase in the resistance to 30% subtilin, as compared to the
BSB1 strain. However, when induced with 100 µM, the three strains first grew similarly but then the
high overexpression of sppA rapidly led to cell lysis in the stationary phase.
With these results, we can conclude that the deletion of sppA affects the resistance to subtilin
directly through the enzymatic activity of SppA, as the complementation with sppA overexpression
allowed the recovery of the subtilin resistance. However, a new phenomenon appeared with the early
cell lysis observed when 100 µM IPTG were used to induce sppA.
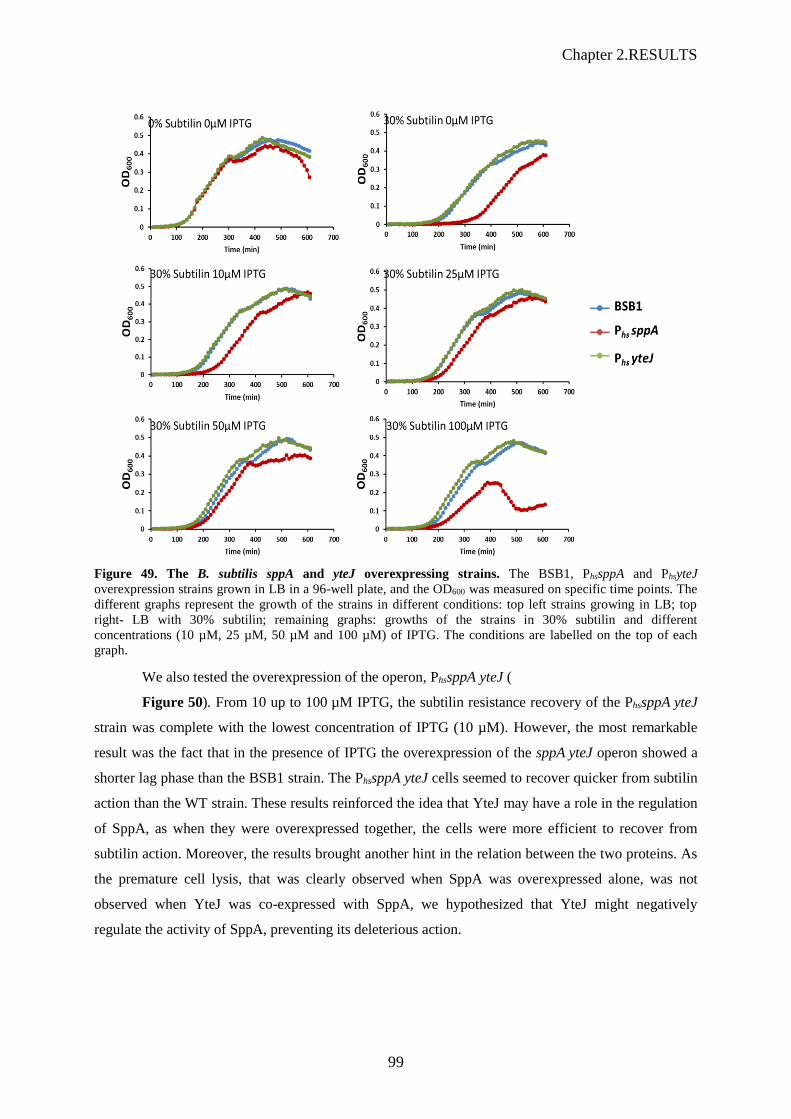
Chapter 2.RESULTS
99
Figure 49. The B. subtilis sppA and yteJ overexpressing strains. The BSB1, PhssppA and PhsyteJ
overexpression strains grown in LB in a 96-well plate, and the OD600 was measured on specific time points. The
different graphs represent the growth of the strains in different conditions: top left strains growing in LB; top
right- LB with 30% subtilin; remaining graphs: growths of the strains in 30% subtilin and different
concentrations (10 µM, 25 µM, 50 µM and 100 µM) of IPTG. The conditions are labelled on the top of each
graph.
We also tested the overexpression of the operon, PhssppA yteJ (
Figure 50). From 10 up to 100 µM IPTG, the subtilin resistance recovery of the PhssppA yteJ
strain was complete with the lowest concentration of IPTG (10 µM). However, the most remarkable
result was the fact that in the presence of IPTG the overexpression of the sppA yteJ operon showed a
shorter lag phase than the BSB1 strain. The PhssppA yteJ cells seemed to recover quicker from subtilin
action than the WT strain. These results reinforced the idea that YteJ may have a role in the regulation
of SppA, as when they were overexpressed together, the cells were more efficient to recover from
subtilin action. Moreover, the results brought another hint in the relation between the two proteins. As
the premature cell lysis, that was clearly observed when SppA was overexpressed alone, was not
observed when YteJ was co-expressed with SppA, we hypothesized that YteJ might negatively
regulate the activity of SppA, preventing its deleterious action.

Chapter 2.RESULTS
100
Figure 50. The B. subtilis sppA yteJ overexpressing strain. The BSB1 and PhssppA yteJ strains were grown in
LB in a 96-well plate, and the OD600 was measured at specific time points. The different graphs represent the
growth of the strains in different conditions: top left in LB; top right- 30% subtilin; remaining graphs: growth of
the strains in 30% subtilin and different concentrations (10 µM, 25 µM, 50 µM and 100 µM) of IPTG. The
conditions are labelled on the top of each graph.
2.2.8 The overexpression of sppA results in a change of the cell
morphology.
The overexpression of sppA led to premature cell lysis. As the effect was drastic, we checked
by microscopy if the overexpression of sppA, yteJ or sppA yteJ would affect the morphology of the
cells. The cells were grown overnight at 37 ᵒC with aeration and vigorous shaking in flasks, BSB1 was
inoculated into fresh LB media and the overexpression mutants were inoculated into LB medium
supplemented with 100 μg/ml Spec and 5 µg/ml Cm. Overnight cultures were diluted 400-fold into
fresh pre-warmed LB media without any supplements. Biomass (OD600) was measured at regular
intervals and samples were collected during the early stationary (stat) phase of the growth cycle and
observed by microscopy. The early stationary phase was chosen as, when induced with 100 µM IPTG,
at this growth stage it is possible to observed a growth defect for PhssppA cells (see Figure 49). Cells
were stained with the membrane dye FM4-64, allowing the observation of the cell membranes and
septum, and with DAPI, allowing the observation of chromosomal segregation. The cells of the BSB1
strain (Figure 51-first line on the panel) appeared to be normal B. subtilis rod-shaped growing cells, 4-
10 μm long (Yu et al. 2014), well-defined division septa, cell poles and well segregated chromosomes.
The overexpression of sppA affected cell morphology (second line Figure 51). The PhssppA cells were

Chapter 2.RESULTS
101
longer than the BSB1 cells, with septal formation problems and an abnormal chromosomal
segregation, as the cells did not divide equally and the chromosomal DNA was not well condensed in
the middle of the cell. On other hand, the PhsyteJ and PhssppA yteJ cells appeared as normal B. subtilis
rod-shaped cells with a well-defined division septa, cell poles and well segregated chromosomes. We
concluded that the presence of a high amount of SppA leads to a morphological and division defect.
However, when overexpressed together with YteJ, it seems that YteJ somehow counteracted the
morphological defect caused by the overexpression of SppA.
The observed phenotype, “elongated/early lysing cells” of the PhssppA strain, could be the
result of an membrane overloading by the overexpressed membrane protein SppA itself or by its
protease activity. To discriminate between these two hypotheses, we constructed an inactive form of
SppA and inserted it at the amyE locus under the control of the hyperspank promoter (IPTG
inducible). The active site of SppA is composed of a catalytic dyad, between a serine (S147) of one
SppA protomer and a lysine (K199) of the adjacent protomer. To obtain an inactive form of SppA, we
mutated the codon for the Lys199 changing it to a codon for alanine (K199A), as described in (Nam,
Kim, and Paetzel 2012). The final strain was named GHBs 088 (BSB1 amyE::PhssppAK199A /spec), and
to simplify Phs sppAK199A.
Figure 52 shows the cell morphology expected for BSB1 without (Figure 52A) and with
(Figure 52B) IPTG: normal B.subtilis rod-shaped cells, while Phs sppA showed damaged long cells.
Interestingly, Phs sppAK199A (Figure 52B) cells did not show a cell growth defect but the normal cell
morphology observed for the BSB1 strain. These results allowed us to conclude that the cell
morphology defect is not a consequence of overexpressing a membrane protein but that the
morphology defect was due to the protease activity of SppA.

Chapter 2.RESULTS
102
Figure 51. Cell morphology of the sppA, yteJ or sppA yteJ overexpression mutants. The strains, BSB1, Phs
sppA, Phs yteJ and Phs sppA yteJ were grown overnight in LB at 37°C, supplemented with 5 μg/ml Cm and 100
µg/ml Spec when required, and diluted 400-fold into fresh LB medium. Then samples were collected during the
early stationary (stat) phase for microscopy analysis. Phase contrast microscopy and fluorescence microscopy
pictures are shown. Membranes (false coloured red) were stained with FM4-64 and DNA (false coloured blue)
were stained with DAPI. Scale bar is 10 micron.
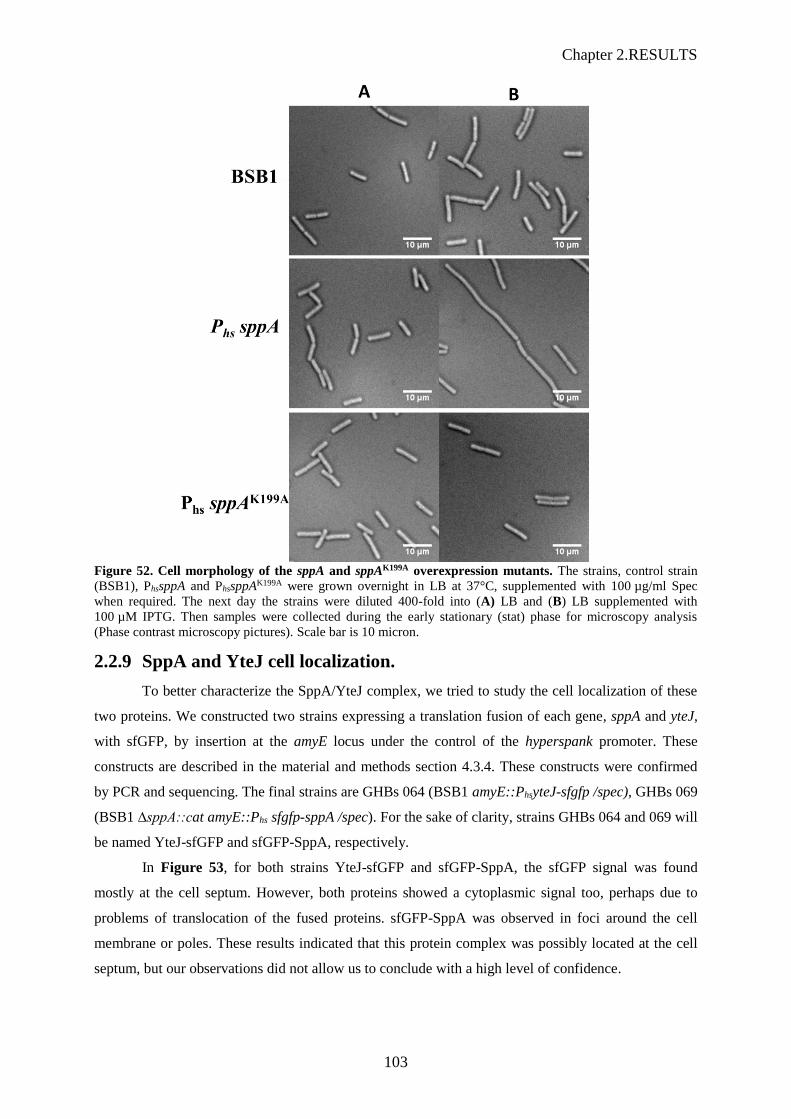
Chapter 2.RESULTS
103
Figure 52. Cell morphology of the sppA and sppAK199A overexpression mutants. The strains, control strain
(BSB1), PhssppA and PhssppAK199A were grown overnight in LB at 37°C, supplemented with 100 µg/ml Spec
when required. The next day the strains were diluted 400-fold into (A) LB and (B) LB supplemented with
100 µM IPTG. Then samples were collected during the early stationary (stat) phase for microscopy analysis
(Phase contrast microscopy pictures). Scale bar is 10 micron.
2.2.9 SppA and YteJ cell localization.
To better characterize the SppA/YteJ complex, we tried to study the cell localization of these
two proteins. We constructed two strains expressing a translation fusion of each gene, sppA and yteJ,
with sfGFP, by insertion at the amyE locus under the control of the hyperspank promoter. These
constructs are described in the material and methods section 4.3.4. These constructs were confirmed
by PCR and sequencing. The final strains are GHBs 064 (BSB1 amyE::PhsyteJ-sfgfp /spec), GHBs 069
(BSB1 ∆sppA::cat amyE::Phs sfgfp-sppA /spec). For the sake of clarity, strains GHBs 064 and 069 will
be named YteJ-sfGFP and sfGFP-SppA, respectively.
In Figure 53, for both strains YteJ-sfGFP and sfGFP-SppA, the sfGFP signal was found
mostly at the cell septum. However, both proteins showed a cytoplasmic signal too, perhaps due to
problems of translocation of the fused proteins. sfGFP-SppA was observed in foci around the cell
membrane or poles. These results indicated that this protein complex was possibly located at the cell
septum, but our observations did not allow us to conclude with a high level of confidence.

Chapter 2.RESULTS
104
Figure 53. SppA and YteJ cell localization. The strains, YteJ-sfGFP and sfGFP-SppA, contain a sfGFP
translational fusion with yteJ and sppA genes, respectively. Strains were grown in LB at 37°C; samples were
collected during the early stationary (stat) phase for microscopy analysis. Phase contrast microscopy and
fluorescence microscopy pictures are shown. sfGFP is false coloured of green. Scale bar is 10 micron.
2.2.10 SppA and YteJ in vitro purification.
To analyse the role of YteJ in the complex and the SppA protease activity in vitro, we
produced and purified B. subtilis SppA, YteJ and SppA/YteJ (from an artificial operon) from E. coli.
For that, from the pJ411 plasmid, we constructed plasmids harbouring a copy of each gene or the full
operon fused to a His-tag sequence, under the control of the IPTG-inducible pT7 promoter, and a
kanamycin resistance cassette. These plasmids were used to transform E. coli. The final genes
sequences on these plasmids were confirmed by PCR and sequencing. The his-tagged proteins were
purified as described on the section 4.9.6 of material and method. To simplify, we will refer to the his-
tagged purified proteins as SppA, YteJ and SppA/YteJ.
Purifying active membrane proteins is a challenging task, as it means extracting proteins from
the membrane maintaining its native environment to allow the proteins to preserve its fully folded
conformation. This process can destabilize the protein structure impairing its activity. A strategy to
avoid this problem and study a membrane protein activity is to purify the protein without its
transmembrane domains, transforming the membrane protein in a soluble protein that can be recovered
from the cell cytoplasm. This strategy was used by Nam et al., 2012, which allowed to obtain the
crystallographic structure of SppA and discover that this protein is able to digest fully folded proteins
(Nam, Kim, and Paetzel 2012; Nam and Paetzel 2013).

Chapter 2.RESULTS
105
Figure 54. Scheme representing the different His-tag constructions. The rectangles represents SppA and YteJ
proteins, orange represents cytosolic domain, green the transmembrane domains and blue extracellular domain.
The numbers represent the length in amino-acids.
As such, we constructed and purified, as described by Nam et al., 2012 , a SppA truncated of
its transmembrane domain, the first 52 amino acids from SppA N-terminus. This protein was
overexpressed and purified from E. coli cytoplasm and the protein was named SppA∆1-52. In this study,
we used SppA∆1-52 as a control for SppA activity. The purified SppA∆1-52 (lane 2, Figure 55A) showed
an expected band at around 28 kDa. In the case of the full length SppA (lane 3), we observed at least
three bands, one at 36 kDa which was expected as the theoretical molecular weigth (MW) is 36.6 kDa
and two lower bands. These bands of lower MW may indicate two different forms of SppA might
result from a partial autoprocessing of the C-terminal domain of SppA. The purified YteJ (lane 2,
Figure 55B) shows a band of the expected size for YteJ-His, around 20 kDa. When SppA is expressed
together with YteJ-His (lane 4, Figure 55A), SppA (highest band ≈36 kDa) was pulled down with
YteJ-His (lowest band ≈20 kDa), indicating that these two proteins formed a membrane protein
complex even when produced in E.coli.. Comparing with the full length SppA sample (lane 3), the
SppA in the SppA/YteJ complex (lane 4) – i.e. pulled down with the YteJ-His, was mainly observed as
a single band. This single band corresponded to the highest band of SppA full length (≈36 kDa). In
addition, all the full length proteins, purified from the E. coli membrane solubilized with DDM
showed additional faint bands that corresponded to contaminants.
SppA has been described to form an octomer (Nam, Kim, and Paetzel 2012), a structure
obtained by crystallography of SppA∆1-52. We wondered whether this octomeric structure could also be
that of the full length membrane SppA. The same question applied toYteJ. To get rid of contaminants
we performed a second setp of purification: a gel filtration using a Superdex 200 HR 10/300 GL
columnthat allows the separation of proteins from 10 up to 600 kDa. The curves and SDS-PAGE gels
showing the gel filtration results can be found Figure S 2. These results showed that SppA possibly
forms an octomer around 300 kDa, as described in the literature, and YteJ may form an octomer
around 160 kDa.
When loaded on the gel filtration column, the complex SppA/YteJ dissociated as the two
proteins were eluted separately from the column, as two separated octomers. This result indicated that,
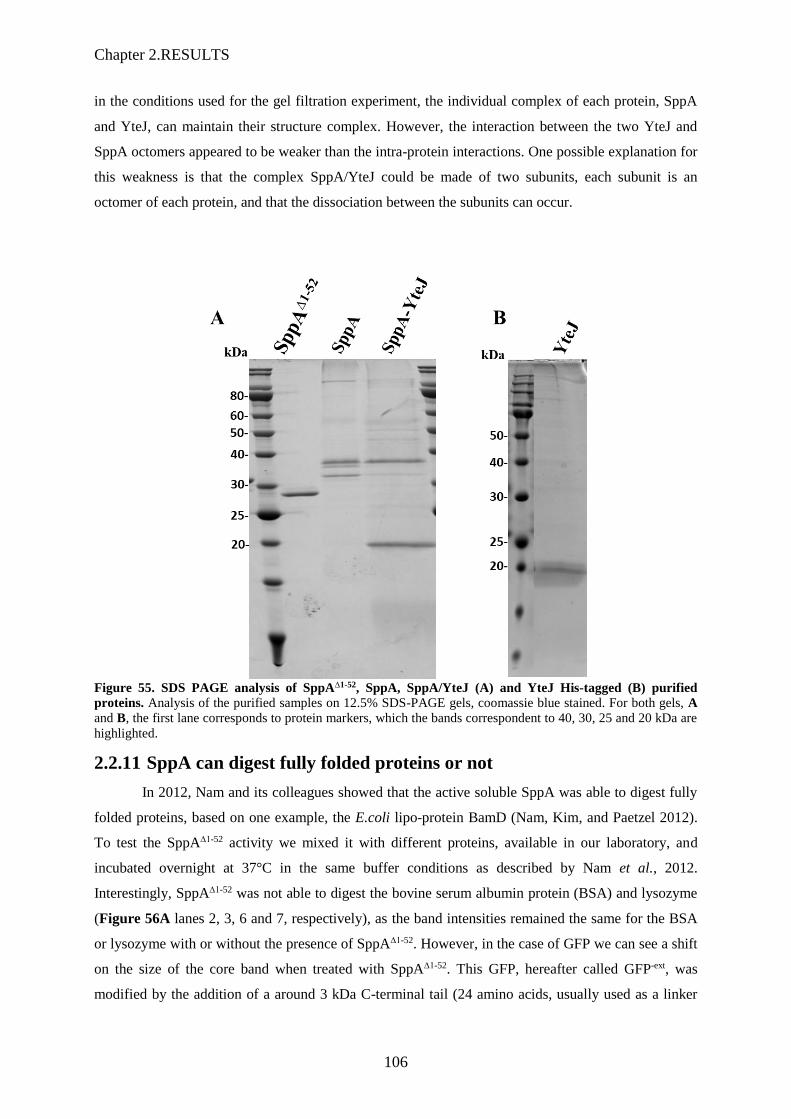
Chapter 2.RESULTS
106
in the conditions used for the gel filtration experiment, the individual complex of each protein, SppA
and YteJ, can maintain their structure complex. However, the interaction between the two YteJ and
SppA octomers appeared to be weaker than the intra-protein interactions. One possible explanation for
this weakness is that the complex SppA/YteJ could be made of two subunits, each subunit is an
octomer of each protein, and that the dissociation between the subunits can occur.
Figure 55. SDS PAGE analysis of SppA∆1-52, SppA, SppA/YteJ (A) and YteJ His-tagged (B) purified
proteins. Analysis of the purified samples on 12.5% SDS-PAGE gels, coomassie blue stained. For both gels, A
and B, the first lane corresponds to protein markers, which the bands correspondent to 40, 30, 25 and 20 kDa are
highlighted.
2.2.11 SppA can digest fully folded proteins or not
In 2012, Nam and its colleagues showed that the active soluble SppA was able to digest fully
folded proteins, based on one example, the E.coli lipo-protein BamD (Nam, Kim, and Paetzel 2012).
To test the SppA∆1-52 activity we mixed it with different proteins, available in our laboratory, and
incubated overnight at 37°C in the same buffer conditions as described by Nam et al., 2012.
Interestingly, SppA∆1-52 was not able to digest the bovine serum albumin protein (BSA) and lysozyme
(Figure 56A lanes 2, 3, 6 and 7, respectively), as the band intensities remained the same for the BSA
or lysozyme with or without the presence of SppA∆1-52. However, in the case of GFP we can see a shift
on the size of the core band when treated with SppA∆1-52. This GFP, hereafter called GFP-ext, was
modified by the addition of a around 3 kDa C-terminal tail (24 amino acids, usually used as a linker

Chapter 2.RESULTS
107
for GFP fusion). The shift on the band size corresponded to the digestion of this 3 kDa tail, the core of
the GFP remaining intact. We then tested different proteins, four are shown on Figure 56B. SppA∆1-52
could not digest the E. coli DNA single strand binding protein (SSB) (lane 3). When treated with
SppA∆1-52, the bands corresponding to the following proteins disappeared: B. subtilis nucleoid
associated protein (Rok) and E. coli DNA-binding protein (Ec-HNS). These two proteins were
completely digested (Figure 56B, lane 5 and 7), therefore we aslo proved that SppA can digest fully
folded proteins. However, the B. subtilis catabolite control protein A (CcpA), when mixed overnight
with SppA, disappeared as a whole protein leaving a lower MW band still present. This is an example
of a partial digestion of a protein by SppA∆1-52 (lane 9). These results allowed us to conclude that, as
described in the literature, SppA∆1-52 can digest fully folded proteins. However, as the digestion was
sometimes not possible for some substrates, occured sometimes only partially for others, it is highly
probable that the protease activity of SppA∆1-52.depends on the structure of the substrate.
Figure 56. SDS PAGE analysis of SppA∆1-52 activity. For both gels, (A) and (B), the first lane corresponds to
protein markers, for which the bands corresponding to 40 and 25 kDa are indicated. SppA∆1-52 alone at the last
right lane of gel A.. Protein substrates: bovine serum albumin (BSA), GFPext, lysozyme, E. coli DNA single
strand binding protein (SSB), B. subtilis nucleoid associated protein (Rok), E. coli DNA-binding protein (Ec-
HNS) and B. subtilis catabolite control protein A (CcpA). (-) protein substrate alone and (+) SppA∆1-52 and the
substrate protein. The samples were incubated 16 h at 37°C.
2.2.12 YteJ inhibits SppA activity
Based on the previous results we decided to use the GFP containing C-terminal unstructured
tail as substrate to study the protease activity of SppA as well as SppA in the SppA/YteJ complex.
Their activity were tested overnight at 37°C (Figure 57A). SppA∆1-52 and SppA were able to digest the
substrate, as a MW downshift of the GFPext was observed. However, when in complex with YteJ and
mixed with the GFPext substrate, SppA did not completely digest the substrate, as two bands
corresponding to both, GFP and GFPext, were visible on the gel. Altogether these results revealed the
negative effect of the presence of YteJ on the activity of SppA.

Chapter 2.RESULTS
108
As it seemed that the activity of SppA was slower when SppA was in complex with YteJ, we
performed more precise kinetic experiments following the protease activity of the different forms of
SppA by taking samples at intervals of one hour at 37°C. As the Bradford assay to determine the
concentration of SppA was not very accurate with membrane proteins, we chose to estimate the
quantities of SppA for our “different” SppAs on SDS-PAGE. The band intensities were used to adjust
the quantity of SppAs mixed with the GFPext substrate.
Figure 57. SDS PAGE analysis of SppA∆1-52, SppA and SppA/YteJ activity. A and B corresponds to 12.5%
SDS-PAGE gels, coomassie blue stained. A. activity after 16h at 37°C; B. kinetics of 60 minutes of activity at
37°C. C. Graph of protease activity kinetics of the different forms of SppA. The intensity of the product bands
were measured by Image Lab (Biorad software).
In Figure 57B and C, after 15 min SppA∆1-52 was able to digest almost 100% of the substrate,
while SppA was only able to digest 50% of the substrate after 60 min. The protease activity of the full
length membrane SppA appeared therefore lower than that of the soluble SppA. The presence of the
transmembrane domain of SppA might be involved in the regulation of SppA activity. For one hour, in
complex with YteJ, SppA was not able to digest the substrate. This result indicated that YteJ may act
as a negative regulator of SppA activity.
An active SppA is only purified without its C-terminal, that the enzyme self-processes as
described in (Nam and Paetzel 2013). The authors also showed that an inactive SppA, SppAK199A, was
purified with its unprocessed C-terminal domain.

Chapter 2.RESULTS
109
On our SDS-PAGE analysis, SppA appeared as three bands (lane 2, Figure 58A), while the
inactive SppAK199A appeared as a single band, whose MW was higher than that of the active SppA
(lane 3). When SppA is expressed together with YteJ, we observed a band for SppA which was higher
than active form of SppA but slightly lower than the SppAK199A inactive form. The hypothesis was
that:
1) when active SppA is expressed and purified alone, it can autocatalitycally remove its own
C-terminal domain and appeared as multiple bands on gel (different stages of self-processing of the C-
terminus)
2) SppA in the SppA/YteJ complex probably still contains its C-terminus and appears as a
higher MW band comparable to that of the inactive SppAK119A.
To confirm this hypothesis we performed a MS/MS analysis of the major bands for SppA and
SppA+YteJ shown on Figure 58A. The results showed that (i) the 3 bands of lane 2 were SppA,
although the peptides from the C-terminal could not be detected (ii) for SppA purified in complex with
YteJ, two peptides belonging to the C-terminal domain of SppA were present (peptides are highlighted
in Figure 58B (highlighted by green and yellow). (iii) finally the second major band on lane 4 was
YteJ. We therefore concluded that the presence of YteJ inhibits the SppA-mediated self-cleavage of its
C-terminal part. The presence of a normally absent C-terminal domain in an active SppA indicated
that SppA was inactive in the complex SppA/YteJ, and it confirmed the results obtained with the
enzymatic experiments shown in Figure 57.
Figure 58. SDS-PAGE and mass spectrometry analysis of SppA and SppA/YteJ complex (A) The first lane
corresponds to protein marker, for which the band corresponding to 40 kDa is highlighted. All the proteins
contain a his-tag. Expected bands for SppAK199A and SppA≈37 kDa and YteJ 19 kDa. (B) SppA protein
sequence. The major bands of SppA and SppA/YteJ were analysed by MS/MS as indicated in the figure with
labels i, ii, iii. The MS/MS analysis showed that the band of SppA, in the SppA/YteJ sample, contains two
peptides belonging to the C-terminal domain of SppA (highlighted in green and yellow).
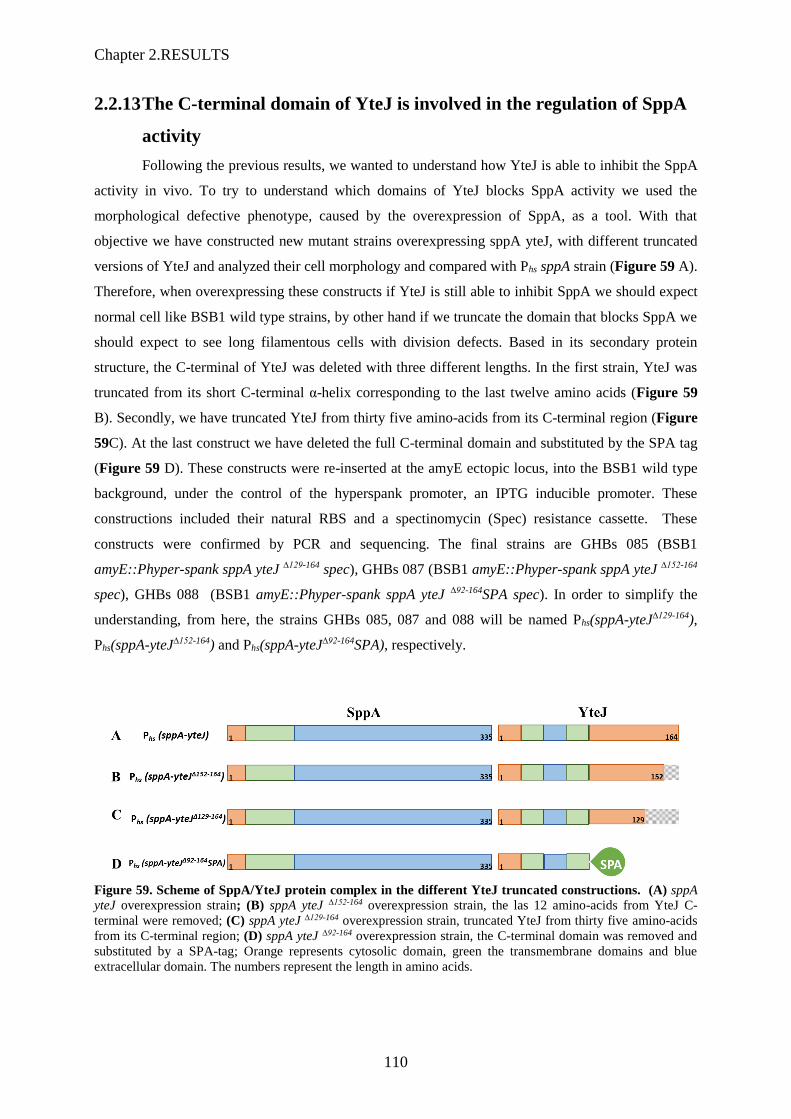
Chapter 2.RESULTS
110
2.2.13 The C-terminal domain of YteJ is involved in the regulation of SppA
activity
Following the previous results, we wanted to understand how YteJ is able to inhibit the SppA
activity in vivo. To try to understand which domains of YteJ blocks SppA activity we used the
morphological defective phenotype, caused by the overexpression of SppA, as a tool. With that
objective we have constructed new mutant strains overexpressing sppA yteJ, with different truncated
versions of YteJ and analyzed their cell morphology and compared with Phs sppA strain (Figure 59 A).
Therefore, when overexpressing these constructs if YteJ is still able to inhibit SppA we should expect
normal cell like BSB1 wild type strains, by other hand if we truncate the domain that blocks SppA we
should expect to see long filamentous cells with division defects. Based in its secondary protein
structure, the C-terminal of YteJ was deleted with three different lengths. In the first strain, YteJ was
truncated from its short C-terminal α-helix corresponding to the last twelve amino acids (Figure 59
B). Secondly, we have truncated YteJ from thirty five amino-acids from its C-terminal region (Figure
59C). At the last construct we have deleted the full C-terminal domain and substituted by the SPA tag
(Figure 59 D). These constructs were re-inserted at the amyE ectopic locus, into the BSB1 wild type
background, under the control of the hyperspank promoter, an IPTG inducible promoter. These
constructions included their natural RBS and a spectinomycin (Spec) resistance cassette. These
constructs were confirmed by PCR and sequencing. The final strains are GHBs 085 (BSB1
amyE::Phyper-spank sppA yteJ ∆129-164 spec), GHBs 087 (BSB1 amyE::Phyper-spank sppA yteJ ∆152-164
spec), GHBs 088 (BSB1 amyE::Phyper-spank sppA yteJ ∆92-164SPA spec). In order to simplify the
understanding, from here, the strains GHBs 085, 087 and 088 will be named Phs(sppA-yteJ∆129-164),
Phs(sppA-yteJ∆152-164) and Phs(sppA-yteJ∆92-164SPA), respectively.
Figure 59. Scheme of SppA/YteJ protein complex in the different YteJ truncated constructions. (A) sppA
yteJ overexpression strain; (B) sppA yteJ ∆152-164 overexpression strain, the las 12 amino-acids from YteJ C-
terminal were removed; (C) sppA yteJ ∆129-164 overexpression strain, truncated YteJ from thirty five amino-acids
from its C-terminal region; (D) sppA yteJ ∆92-164 overexpression strain, the C-terminal domain was removed and
substituted by a SPA-tag; Orange represents cytosolic domain, green the transmembrane domains and blue
extracellular domain. The numbers represent the length in amino acids.
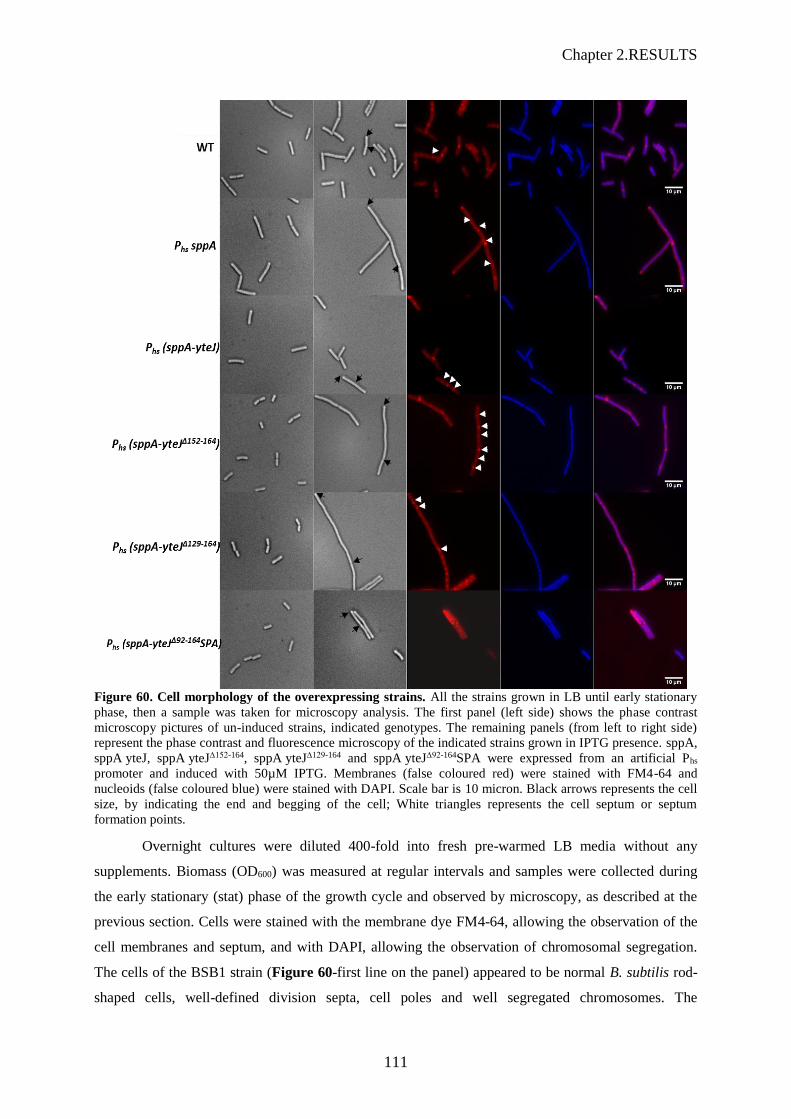
Chapter 2.RESULTS
111
Figure 60. Cell morphology of the overexpressing strains. All the strains grown in LB until early stationary
phase, then a sample was taken for microscopy analysis. The first panel (left side) shows the phase contrast
microscopy pictures of un-induced strains, indicated genotypes. The remaining panels (from left to right side)
represent the phase contrast and fluorescence microscopy of the indicated strains grown in IPTG presence. sppA,
sppA yteJ, sppA yteJ∆152-164, sppA yteJ∆129-164 and sppA yteJ∆92-164SPA were expressed from an artificial Phs
promoter and induced with 50µM IPTG. Membranes (false coloured red) were stained with FM4-64 and
nucleoids (false coloured blue) were stained with DAPI. Scale bar is 10 micron. Black arrows represents the cell
size, by indicating the end and begging of the cell; White triangles represents the cell septum or septum
formation points.
Overnight cultures were diluted 400-fold into fresh pre-warmed LB media without any
supplements. Biomass (OD600) was measured at regular intervals and samples were collected during
the early stationary (stat) phase of the growth cycle and observed by microscopy, as described at the
previous section. Cells were stained with the membrane dye FM4-64, allowing the observation of the
cell membranes and septum, and with DAPI, allowing the observation of chromosomal segregation.
The cells of the BSB1 strain (Figure 60-first line on the panel) appeared to be normal B. subtilis rod-
shaped cells, well-defined division septa, cell poles and well segregated chromosomes. The

Chapter 2.RESULTS
112
overexpression of sppA affected the cell morphology creating elongated cells, as the observed in our
previous results (second line Figure 60). Surprisingly, when we overexpressed sppA-yteJ∆152-164
(fourth line Figure 60), YteJ with the short truncation at its C-terminal, we could observe an effect in
the cell morphology. Even though the cells were not so long as PhssppA cells, the cell were longer
than the BSB1 cells, with an evident septa mal formation and an abnormal chromosomal segregation.
Furthermore, the overexpressed sppA-yteJ∆129-164 (fifth line Figure 60) restored the long phenotype
observed for the sppA overexpression. Contrarily, the strain containing a swap between the C-terminal
region of YteJ by the SPA-tag (sixth line Figure 60), Phs (sppA-yteJ∆92-164SPA), show shorter cells
similar to the BSB1 and PhssppA yteJ cells (first and third lines Figure 60). However, these cells
show cell wall and division problems. These results allowed us to conclude that the C-terminal domain
of YteJ is involved and important for the regulation of SppA activity, as the removal of the last 25
amino acids affected the cell growth, and that removing the last 45 amino acids of YteJ led to a a cell
phenotype similar to what was observed with the overexpression of SppA alone, i.e. a complete
deregulation.of SppA protease activity.
2.2.14 SppA digests subtilin
Our previous results showed that SppA∆1-52 can digest fully folded proteins, depending on the structure
of the substrate. In this chapter of results we have also demonstrated that SppA have an important role
in the lantibiotic resistance mechanism. Assuming that the lantibiotics are peptides, we were
wondering if in this resistance mechanism SppA is directly digesting the lantibiotics. As such we used
the previous purified and active SppA∆1-52 to test our hypothesis. SppA∆1-52 was incubated overnight in
a subtilin-containing reaction buffer (20 mM Tris-HCl pH7.5, 200mM NaCl) at 37°C, as well as the
controls, SppA∆1-52 alone in the reaction buffer, subtilin solution and a positive control with proteinase
K. Proteinase K is a broad range serine protease, the same family that SppA (MORIHARA and
TSUZUK 1975). Then these compounds were tested with the wild type BSB1 and ΔsppA strain. The
experiment was done as described at section. At Figure 61, the first graph top left of the panel
corresponds to the controls, i.e. the BSB1 and ∆sppA mutant strain grown in LB without subtilin. The
two strains showed identical growth rates. When treated with 30% subtilin (graph top right), the
growth of the strains was affected, with a visible difference in the resistance to subtilin between the
BSB1 and the ∆sppA. ∆sppA took longer to recover its growth and reach an OD600 of 0.1. The subtilin
was stable and did not degrade overnight at 37°C, as it induced the same type of growth delay (graph
middle left), than the fresh subtilin used. When the subtilin was treated overnight with the SppA∆1-52,
we can observe that the growth is similar to the control without subtilin (graph top left), without
differences between the strains. The same was obtained for with the subtilin treated with proteinase K.
With these results we can conclude that the subtilin was degraded by both SppA∆1-52 and proteinase K.
Therefore, we can hypothesize that, in vivo, the direct degradation of subtilin by SppA is part of the
mechanism of action against lantibiotics activity.
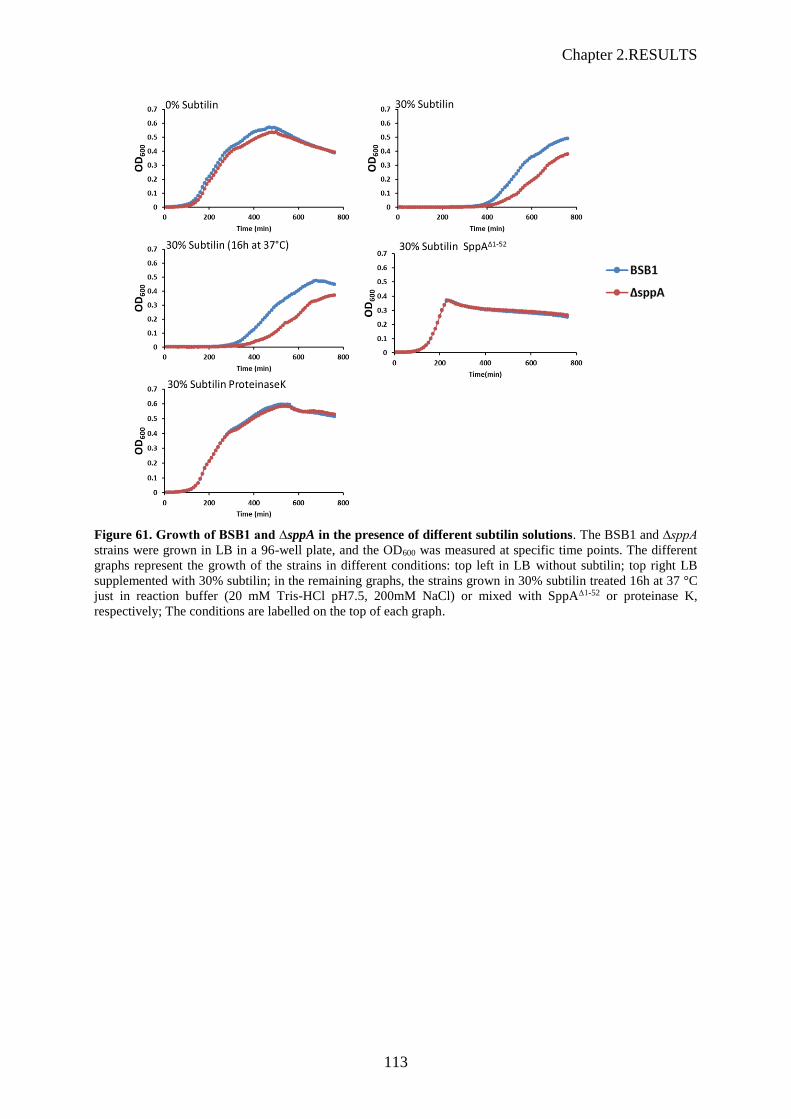
Chapter 2.RESULTS
113
Figure 61. Growth of BSB1 and ∆sppA in the presence of different subtilin solutions. The BSB1 and ∆sppA
strains were grown in LB in a 96-well plate, and the OD600 was measured at specific time points. The different
graphs represent the growth of the strains in different conditions: top left in LB without subtilin; top right LB
supplemented with 30% subtilin; in the remaining graphs, the strains grown in 30% subtilin treated 16h at 37 °C
just in reaction buffer (20 mM Tris-HCl pH7.5, 200mM NaCl) or mixed with SppA∆1-52 or proteinase K,
respectively; The conditions are labelled on the top of each graph.

Chapter 2.RESULTS
114

115
DISCUSSION AND
PERSPECTIVES

116

Chapter 3. DISCUSSION AND PERSPECTIVES
117
3 DISCUSSION AND PERSPECTIVES
The ability to transport newly synthesized proteins across and into the bacterial cytoplasmic
membrane is an essential mechanism to maintain cell integrity. This transportation mechanism
involves universally conserved protein-conducting machineries that can transport a wide variety of
potential substrates. For example, the Sec translocon mediates the translocation of secretory and
membrane proteins in an unfolded conformation. The SecYEG, the core channel of the bacterial Sec
translocon, is the minimal membrane-embedded unit required for protein transport (Collinson 2005).
However, the full translocation process from the recognition of the pre-protein, its translocation and
final maturation involve a dynamic between the protein-channel and associated proteins, which
interact and work together (de Keyzer et al. 2003). Although the mechanisms involved in the Sec
pathway has been extensively studied, the knowledge about B. subtilis protein complexes is limited.
The possible protein-protein interactions within the B. subtilis Sec pathway were studied and will be
discussed in section 3.1. As SppA was initially identified as an accessory protein involved in the Sec
pathway, the section 3.2 will focus on its interaction with YteJ, its role and activity in the cell.
3.1 Protein interactions of the B. subtilis Sec pathway
In E. coli, it has been established that the Sec holo-translocon is a supramolecular complex
composed of the membrane protein complexes, SecYEG (Sec translocon), SecD/SecF, YajC and YidC
(Schulze et al. 2014; Komar et al. 2016; Botte et al. 2016). Based on the multiple interactions found in
E. coli, we intended to study these interactions in B. subtilis.
Our experiments of sequential peptide affinity purifications with the SPA-tagged proteins
SecDF and SpoIIIJ in B. subtilis showed that the B. subtilis SecDF, homologous to a fusion of the E.
coli SecD and SecF, and B. subtilis SpoIIIJ, homologous to E. coli YidC, did not co-purify. According
to these results, it seems that SecDF and SpoIIIJ do not form a stable complex in B. subtilis, unlike
what was shown in E. coli. However, the absence of co-purification of B. subtilis SecDF and SpoIIIJ
does not completely rule out the possibility that these two proteins interact. Their interaction can be
weaker than that in E. coli, and might have been abolished by the use of a too high concentration of
detergent, the n-dodecyl β-D-maltoside (DDM). In our procedure, we classically used a DDM
concentration of 1% which might affect protein interactions, although 2% DDM concentration was
used in E. coli to solubilize the membranes. We therefore tested a range of different DDM
concentrations, from 0.5% to 2%, and also reduced the purification steps to only one affinity column
to avoid losing some protein partners. Analysed by LC-MS/MS, the results of these experiments
showed that the SpoIIIJ did not co-purified with the SecDF-SPA protein. However, in the reciprocal
experiment, after affinity purification of SpoIIIJ-SPA, low amounts of SecDF were detected regardless

Chapter 3. DISCUSSION AND PERSPECTIVES
118
of the DDM concentrations. This suggested that B. subtilis SecDF and SpoIIIJ may eventually interact.
If so, this interaction might be rather weak as the addition of a second affinity column had previously
abolished it. Moreover, as this result was only obtained when the SPA tag was added to SpoIIIJ, it also
suggests that the SPA tag fused to SecDF might have precluded the interaction. However, in
conclusion, if B. subtilis SecDF and SpoIIIJ interact, these two proteins do not form a stable complex
like in E. coli.
Unfortunately, in our attempt to study the translocon SecYEG, B. subtilis strains expressing
SecY-SPA and SecE-SPA could not be obtained. Therefore we analysed only B. subtilis SecG-SPA by
tandem affinity purification. It turned out that SecG-SPA did not co-purify with the expected SecY or
SecE, nor SecDF, unlike what had been observed in E. coli. Again, we cannot rule out the possibility
that the SPA tag fused to SecG have induced a dissociation of the other proteins, SecY and SecE, or
weakened their interactions. However, it is highly unlikely as the translocon, SecYEG, is essential to
the cell and as any defect in the formation of this complex should harm the cell. Our result rather
reflects the fact that the translocon, SecYEG, or the holotranslocon, SecYEG-SecDF-SpoIIIJ, might
not be a very stable protein complex in B. subtilis.
Combined together, these results suggest that B. subtilis do not possess a stable holo-
translocon as the E. coli SecYEG-SecDF-YajC-YidC supracomplex. In E. coli the existence of this
holo-translocon as a supracomplex was shown by different methods including BN-PAGE and the
direct interactions within the proteins forming the holotranslocon were further refined by crosslinking
experiments (Schulze et al. 2014; Komar et al. 2016). To study the “holotranslocon” in B. subtilis,
cross-linkers should be used and added to the cells before purification of any of the translocon
components.
After translocation, the secretory proteins are released from the membrane and undergo a
maturation process carried out by other accessory proteins to the Sec machinery, as for example the
thiol-disulfide oxido-reductases (Bdb proteins), which during protein folding, are responsible for
rearranging incorrect disulfide bonds (Tjalsma et al. 2004; Driessen & Nouwen 2008). Structural
studies showed that the E. coli thiol-disulfide oxido-reductases DsbC and DsbD form a complex.
DsbD binds to the central cleft of the V-shaped DsbC dimer causing a shift into a close conformation
and it activates DsbC (Fabianek et al. 2000; Haebel et al. 2002). We also investigated this connection
between the B. subtilis BdbB and BdbD, two thiol-disulfide oxido-reductases homologous to E. coli
DsbC and DsbD. We did not find evidences that these two proteins form a complex either, as our
results showed that, when SPA-tagged, BdbB or BdbD did not co-purify with each other.
3.2 SppA/YteJ membrane protein complex
As shown in Chapter II of the results, we have found evidences of a stable complex between
the B. subtilis SppA, the signal peptide peptidase, and the unknown membrane protein YteJ.

119
The first evidence for the existence of this complex was obtained by BN-PAGE experiments,
which showed that these two proteins co-migrated together, suggesting that they might interact. The
tandem affinity purification of YteJ-SPA showed that SppA was co-purified with YteJ-SPA,
indicating again that these two proteins interact and form a stable complex. For the in vitro study,
when the his6-sppA yteJ operon was expressed in E. coli, the purification of the his-tagged SppA
clearly showed the presence of YteJ, reinforcing the idea that a membrane protein complex made of B.
subtilis SppA and YteJ can be formed in E. coli.
The existence of this complex in the cytoplasmic membrane of B. subtilis prompted us to
study whether YteJ could play a role in this complex. I will discuss our results in the following order:
1. the role of SppA in protein secretion ?
2. the role of SppA (and YteJ) in the resistance to nisin, subtilin and LP9
3. the protease activity of SppA is regulated by YteJ
4. SppA/YteJ, a complex involved in cell division ?
5. YteJ, another function?
3.2.1 Role of SppA and YteJ in protein secretion:
In 1999, Bolhuis et al. observed that the deletion of sppA induced a slight decrease in the
secretion of mature secretory proteins, and more importantly a delay in the maturation process of the
hypersecreted protein AmyQ. As SppA was believed to be involved in the degradation of the signal
peptides of the secreted proteins, this result was interpreted as a consequence of the presence of
remnant signal peptides in the membrane, that would cause a slow-down of the processing of the pre-
proteins under hyper-secreting conditions. Here, in this study, we have shown that the deletion of sppA
and yteJ do not affect the secretion of a highly secreted protein, AmyM. However, Bolhuis et al.
(1999) used pulse chase experiments to show the maturation delay of the hypersecreted AmyQ.
Therefore, our deletion strains were also tested by pulse chase experiments (see Figure S 3), in Prof.
van Dijl’s laboratory (Groningen, The Neterlands). Their results showed no major effect of any
deletion (ΔsppA, ΔyteJ or Δsppa yteJ) on the secretion of the model protein AmyM. Taking into
account these results, our conclusions are that neither SppA nor YteJ are directly involved in protein
secretion. It does not rule out any indirect effect that could explain what was previously observed.
The results obtained with the SppA, or YteJ overexpression strains, overexpressing, showed a
negative effect on the AmyM secretion level when SppA is overexpressed, while the overexpression
of YteJ, or SppA and YteJ together, did not lead to any difference. This indicated that, SppA, able to
degrade secreted proteins, could be detrimental when overexpressed for the production of secreted
proteins. Our result is in complete disagreement with previous results in Bacillus licheniformis for
which the overexpression of SppA proved to be beneficial for the secretion levels of nattokinase and

Chapter 3. DISCUSSION AND PERSPECTIVES
120
AmyL (Cai et al. 2017). In this study, sppA was under the control of the constitutive promoter P43, a
promoter which is thought to be active during the lag and exponential phase of growth but not during
the stationary phase. In our study, we used the promoter Phs induced by the addition of IPTG, therefore
SppA was expressed, regardless of the growth phase. This might explain the difference of results
between the two studies, as in both cases, the extracellular secreted protein contents were assessed in
late stationary phase. In conclusion, the role of SppA in protein secretion appeared to be quite limited
as worst negative effects of 20 % or best positive effects of 60% in the protein secretion levels were
reported (Bolhuis et al. 1999)(Cai et al. 2017), as opposed to the positive effects of 250-1000 %
obtained after the overexpression of RasP (Neef et al. 2017).
3.2.2 Role in the resistance to nisin, subtilin and LP9
The role of SppA was more obvious in the resistance to nisin, subtilin and LP9 as the deletion
of sppA induced a higher sensitivity of the cells to these antibiotic peptides. These results were in
agreement with previous observations that showed that sppA, a member of the σW regulon, was
involved in the lantibiotic resistance (Kingston, Liao, and Helmann 2013b).
Our experiments with the sppA complementation strain confirmed the direct role of SppA in
this resistance as the complemented cells showed a lantiobic or LP9 resistance identical to the wild-
type cells. We also tested the direct digestion of subtilin by SppA by incubating the purified SppA in a
subtilin-containing LB medium for 12 hours at 37°C, and we observed that the ΔsppA cells grew as
well as the wild-type cells in this SppA-treated medium (Figure 61).
The role of YteJ appeared more complicated to understand with the antibiotic peptide
experiments. The observed phenotypes of the ΔyteJ cells showed either a slightly higher sensitivity to
the antibiotic peptides than the wild-type cells, or a greater resistance. It seemed to be dependent on
the concentrations of the peptide used. If the resistance to the lantibiotic peptides or LP9 is only due to
the protease activity of SppA, the ΔyteJ phenotype might be explained by the SppA regulatory role of
YteJ. We showed that YteJ negatively regulates the activity of SppA; however, YteJ has to release its
inhibitory action on SppA, presumably by responding to a signal, induced by the antibiotic peptides. In
the absence of YteJ, the activity of SppA might not be timely regulated, hence the lower antibiotic
resistance. This hypothesis could not account for the higher antibiotic resistance observed in ΔyteJ
cells in some cases.
3.2.3 Regulation of the sppA yteJ operon
In parallel with the antibiotic resistance study, we also studied the transcription regulation of
the operon sppA yteJ. In Nicolas et al (2012), a second promoter, σA-dependent, was detected
upstream of the σW-dependent promoter. Our experiments using sGFP fusions with one or potentially
two promoters showed that the major promoter was the σW-dependent promoter. As such, we
confirmed that sppA and yteJ are part of the σW regulon, a regulon that is expressed when cells are

121
subjected to an envelope or cell wall stress. Interestingly, the operon structure of these two genes,
sppA and yteJ, is well conserved across the bacilli class, which includes Bacillus subtilis but also
Listeria monocytogenes.
3.2.4 SppA/YteJ, a complex involved in quality control of cell division?
As shown in the chapter II of the results, we observed that high levels of expression of SppA
create a severe cell growth defect, giving rise to a filamentous phenotype (see Figure 51). This
phenotype could be due to the overexpression of a membrane protein. The overexpression of
membrane proteins and proteases normally result in the accumulation of cytoplasmic aggregates
containing the overexpressed proteins, accumulation of chaperones (e.g. DnaK), soluble proteases (e.g.
ClpXP) or precursors of secreted and membrane proteins (Wagner et al., 2007). To discard this
hypothesis, we constructed a strain that overexpressed an inactive form of SppA, the SppAK199A. The
analysis of the morphology of the cells showed a normal cell morphology (see Figure 52). These
results allowed us to conclude that the activity of SppA is responsible for the cell growth defect. In
Figure 51 the affected long cells of the PhssppA strain are not just only filamentous but they seem to
show a defect on cell division, malformation of the septum and impaired chromosomal segregation.
This morphological phenotype defect is reminiscent of the cell defect phenotype described in deletion
mutants involving protein regulators of the divisome. The B. subtilis cell division is well coordinated
with the elongation, as it requires a switch in the orientation of the cell wall synthesis, and the
formation of the mid-cell Z ring for cell constriction and division (Adams and Errington, 2009; Leaver
et al., 2009; Errington and Wu, 2017). The Min system prevents the polar division of the cells. It was
shown that the deletion of B. subtilis noc combined with the depletion of MinD lead to a defect in the
division, absence of division septa formation and elongated filaments phenotype, similar to what we
observed on our results obtained for the overexpression of SppA (Figure Figure 51 and Figure 60) (L.
J. Wu and Errington 2004). It was shown too that the B. subtilis minJ deletion mutant, comparing with
the wild type strain, grew as long filaments of cells with sporadic septa (Figure 62), as we observed in
our mutants (Patrick and Kearns 2008). However, in both cases the segregation of the chromosomal
DNA was not affected, as it is possible to observe a well divided nucleoids on Figure 62, the contrary
of our results, that showed long filamentous cells with a unique long nucleoid (Figure 51).

Chapter 3. DISCUSSION AND PERSPECTIVES
122
Figure 62. Cells mutated for minJ are filamentous. Fluorescence microscopy of wild type (A), ∆minJ (B)
were grown to mid-exponential phase and stained with FM 4-64 (membrane, false coloured red) and DAPI
(chromosome, false coloured blue). Scale bar is 4 mm. (Patrick and Kearns 2008).
Other types of proteins could be the targets of an overexpressed, active and unregulated SppA,
as for example the penicillin binding proteins (Pbp). Mutants of some Pbps show similar phenotype of
elongated cells as well (Figure 63).
Figure 63. Morphology of B. subtilis cells lacking multiple penicillin-binding proteins (Pbp). (A) wild-type
strain; (B) cells lacking Pbp1; (C) cells lacking Pbp1 and 4; (D) cells lacking (Adapted from Popham and Setlow
1996)).
In conclusion, the clear cell morphology phenotype defect observed in a strain overexpressing
an active SppA points out that proteins involved in the division process or cell wall synthesis are
affected. However, no specific substrate for SppA can be inferred from this observed phenotype, as
too many proteins can be involved.

123
3.2.5 The protease activity of SppA and its regulation by YteJ
3.2.5.1 The protease activity itself:
Nam et al. (2012) performed in vitro studies with a soluble form, truncated B. subtilis SppAΔ2-
52, deleted from its transmembrane domain (amino acids from position 2 to 52). These authors showed
that SppA∆2-54 could digest signal peptides as well as fully folded proteins. Their conclusion was based
on their assay of a successful overnight digestion of a fully folded E. coli lipoprotein (BamD) (Nam,
Kim, and Paetzel 2012). In our study, we have shown that B. subtilis SppA∆2-54 is not able to digest all
fully folded proteins, as we tested SppA activity on seven different proteins available in the lab as
shown in Figure 53. Our study of the activity of SppA∆2-54 with more proteins indicates a new feature
about the enzymatic properties of SppA. As some proteins showed a SppA proteolytically-resistant
domain, our hypothesis is that some protein structures or specific sequences of amino-acids might
have the ability to resist to the proteolytic activity of SppA. To be demonstrated this hypothesis would
require a specific experimental setting involving, for example, a synthetic peptide that includes a
variety of structures or amino acid sequences.
3.2.5.2 The complex SppA/YteJ and its activity:
In vitro, a soluble SppA, truncated from its N-terminal transmembrane domain, SppA∆2-54, has
been crystallized as an octomer (Nam, Kim, and Paetzel 2012). In our hands, the full-length SppA,
expressed and purified from the E. coli membrane fraction, is also an octomer. Its partner, B. subtilis
YteJ contains two to three transmembrane spanning domains, depending on the prediction structural
programs. YteJ was purified as an apparent molecular weight corresponding to an octomer as well.
The results from the two SPA purification experiments showed that the ratio was more probably a 1:1
between SppA and YteJ, which would be in accordance with the fact that the two genes are in operon.
It would imply that the SppA/YteJ complex would be made of two octomers of each protein. Although
the two proteins were purified as a complex from the sequential affinity purification, in which each
protein was fused to a different tag, the subsequent gel filtration experiment showed a complete
dissociation of the two proteins during the chromatography, suggesting a relative weakly interaction
between the two octomers. This interaction between multimers of different proteins is reminiscent of
the membrane protein complex formed by the protease FtsH and HflK and HflC (Ito and Akiyama
2005). FtsH is a hexamer with most of its protease domain facing the cytoplasm. HflK and HflC, two
membrane proteins that associate first as heterodimers, interact with E. coli FtsH, to form a huge
complex of 1 MDa, the FtsH holoenzyme. It was shown that HflKC inhibits the protease activity of
FtsH for one of its substrate, SecY (Kihara, Akiyama, and Ito 1996).
We also showed that, in vitro, in the SppA/YteJ complex, the SppA protease activity was
strongly diminished when compared to that of SppA alone. In vivo, the overexpression of SppA
created morphological defect to the cells, whereas the cell morphology was unchanged compared to
the wild-type cells, when YteJ was co-expressed with SppA. Our conclusion is that YteJ is a regulator

Chapter 3. DISCUSSION AND PERSPECTIVES
124
of the protease activity of SppA. In vivo, we further showed that the last 25 amino acids of the C-
terminal domain of YteJ were involved in this inhibition mechanism, and that removing the last 45
amino acids of YteJ led to a completely unregulated SppA. Nam and Paetzel (2013) had proposed that
an active SppA processes its own C-terminus, and therefore that SppA C-terminus would act as a
negative regulator domain, presumably preventing SppA to become active too early during
translocation. Our findings provide another level of regulation for SppA through its interaction with
YteJ. Our MS data showed that the C-terminal domain of SppA was detectable by mass spectrometry
only when SppA was purified as a complex with YteJ. The two peptides belonging to the C-terminal
domain of SppA that were detected are highlighted in the Figure 58 are 309SEIDFLNMR317 and
318EILSQSGSPR327.
Therefore one could conclude that SppA in the SppA/YteJ complex is maintained in an
inactive form by the presence of YteJ, and SppA in the SppA/YteJ complex is unable to self-process
its C-terminus as an active SppA would do. However, as the SppA last seven amino acids, i.e. the
peptide 328MMYLYAK335, have not been found in our MS analysis, we cannot be certain that SppA,
even in the complex SppA/YteJ, has not self-processed at least part of its C-terminus. Noticeably,
when SppA was purified, it invariably showed multiple bands on SDS-PAGE, at least 3 visible bands,
separated by an apparent molecular weight difference of 3 to 4 kDa, the highest molecular weight band
corresponding exactly to the band of SppA in the SppA/YteJ complex (See Figure 58). This suggested
that SppA, purified alone from the E. coli membrane fraction, was able to self-process its C-terminus
partially, and that this process was slow. On the opposite, Nam and Paetzel (2013) noticed that, for the
N-terminal transmembrane domain truncated SppA, SppA∆2-54, the self-processing of the C-terminus
of SppA was probably very fast and complete in E. coli even before the enzyme was purified. It
suggests that, in our work, the N-terminal transmembrane domain of the full length SppA may also
slow down the self-processing of the C-terminus. Accordingly, the protease activity of the full-length
SppA was slower than that of the soluble form, SppA∆1-52. E. coli FtsH has been shown to self-process
the last seven amino acids of its own C-terminal domain in vivo, that this self-processing was growth
phase-dependent, and that the deletion of hflKC slowed down the C-terminus self-processing of FtsH
(Akiyama 1999). However, for E. coli FtsH, both full length and processed proteins are active. In the
case of B. subtilis SppA,YteJ inhibits SppA by preventing its C-terminus self-processing. By
interacting with SppA, YteJ is able to maintain SppA in its inactive form presumably until a
physiological harm caused by antibiotic peptides requires the activation of SppA. In this view, SppA
activity, that could be detrimental to the cell if not regulated – see next paragraph -, would act only
when necessary.
Finally, this raises the question of the nature of the signal that triggers the activation of SppA
and how this happens. For example, the PDZ domain of E. coli DegS acts as an inhibitory domain of
DegS until it binds hydrophobic peptides of the C-terminal of misfolded proteins leading to the release

125
of the inhibition (Walsh et al. 2003). This model of activation could be applied to YteJ and SppA, the
C-terminal domain of YteJ acting as the inhibitory/activating domain for SppA.
3.2.6 YteJ, another function?
YteJ, by analogy to the PDZ domain for DegS, could be viewed as a sensor of a signal
induced when the cell is subjected to a cell wall stress due to antibiotic peptides. The overall YteJ
amino-acid sequence analysis shows that the protein belongs to the RDD superfamily, which is
characterized by the presence of three highly conserved residues, arginine-aspartate-aspartate. This
domain is found in three other proteins in the B. subtilis genome (Yczc, YckC, YxaI), and the Pfam
database indicates that this RDD domain is present in many transmembrane proteins, mainly in
bacteria, but also with some occurrences in Archeae and Eukaryota. It could be the main domain of the
protein or associated with other domains of various functions. Until recently the molecular function of
this RDD domain remained completely elusive. However, looking for novel Na+/H+ antiporters, Shao
et al (2018) identified a protein named RDD in the moderate halophile Halobacillus andaensis. These
authors demonstrated that the RDD protein functions as an Na+ (Li+, K+)/H+ antiporter, but also that
four out of six conserved arginine or aspartate residues were indispensable to the antiporter activity
(Shao et al. 2018). Although B. subtilis YteJ did not appear in the sequence alignment showed by Li
Shao et al (2018), it is clearly related to H. andaensis RDD (Figure 64). Moreover the corresponding
rdd gene was identified adjacent to a putative sppA gene. B. subtilis YteJ shares all six conserved
arginine and aspartate residues identified in the homologs of H. andaensis RDD (Shao et al. 2018),
and overall the two protein sequences show 39% identity.
Figure 64. Amino acid sequence alignment between H. andaensis RDD and B. subtilis YteJ. Conserved
amino acids are highlighted in red.
Therefore one can hypothesize that YteJ might act as a cation antiporter in B. subtilis as in H.
andaensis. In case of salt stress and/or leakage of cations induced by lantibiotic peptides, thought to
create holes in the membrane, YteJ would become more active inducing a conformation change in its
own structure that, in turn, would induce the release of the YteJ C-terminus inhibitory effect on SppA.
The complex SppA/YteJ would be a two-protein system dedicated to fight the lantibiotic effects. In
vivo experimental evidences that B. subtilis YteJ acts as a cation antiporter are needed. Within the B.
subtilis genome, YteJ and its closest related proteins (YxaI, YckC and YczC sharing over 30%
identity) share a comparable predicted structure made of three transmembrane domains and these
proteins belong to the RDD family; however, none of their genes seem to be in operon with another
gene. Therefore, SppA and YteJ might be a unique example of a two-protein system involving a

Chapter 3. DISCUSSION AND PERSPECTIVES
126
possible cation transporter regulating the activity of a protease. In vitro detailed enzyme kinetic studies
combined with structural studies involving mutations in SppA as well as in the C-terminus of YteJ
could reveal how this activating mechanism takes place.
3.2.7 Model for the interaction of SppA and YteJ
We propose the following model in which SppA and YteJ formed a membrane protein
complex in which the two proteins are arranged as two homoactomers bound to each other (Figure
65). Based on our experimental data, SppA in the SppA/YteJ complex would be inactive because YteJ
might interfere with the active site of SppA. In this conformation, SppA could not self-process its own
C-terminal domain, a domain that has been shown to block its active site. Upon a signal, whose nature
remains to be determined, YteJ, probably undergoing a conformational change, would release its
inhibitory effect on SppA that in turn would become active. As lantibiotic peptides are able to create
holes in the cytoplasmic membrane and that YteJ amino acid sequence shows homology with a cation
antiporter of a distant relative of B. subtilis, the nature of the signal that could be sensed by YteJ could
be related to ion homeostasis.
Figure 65. Model for the regulation of SppA by YteJ. (A) shows the SppA² inactive state, where YteJ is
structurally blocking SppA active site; A cell stress (represented by the black icon) induces a cell response that
leads to complex conformational change (represented by a black arrow). This structural modification liberated
the SppA active site, passing to its active state (B).

127
3.3 Main results
All of these in vitro and in vivo observations suggest that SppA and YteJ form a strong
membrane protein complex. This study established that YteJ plays a key regulatory role in B.
subtilis SppA activity, highlighting a possible new mechanism of a protein regulating a
protease. The main conclusions of this work are listed below.
BN-PAGE experiments showed us that SppA and YteJ co-migrate together in a native
gradient SDS-PAGE gel, the first evidence of the protein-protein interactions of these
two proteins.
SppA was co-purified with YteJ-SPA.
In B.subtilis, the deletion of sppA affects the resistance to antimicrobial peptides,
while deleting yteJ reveals an intermediate phenotype. The overexpression of SppA
restores the resistance to subtilin.
The high levels of expression of SppA create a cell growth defect and the presence of
YteJ counteracts this effect.
SppA activity is responsible for the cell morphology defect, as when we
overexpressed an inactive version of SppA the cells have a normal phenotype.
In vitro experiments showed that SppA is able to digest fully folded proteins,
depending on their structure.
YteJ inhibit SppA activity, possibly through its C-terminal region.

Chapter 3. DISCUSSION AND PERSPECTIVES
128

129
MATERIAL AND
METHODS

130

Chapter 4. MATERIAL AND METHODS
131
4 MATERIAL AND METHODS
In this chapter, the molecular biology, microbiology, protein purification and microscopy methods
used throughout this study are described.
4.1 Techniques for DNA manipulation
4.1.1 Oligonucleotide
Oligonucleotides or primers were designed using the Geneious software (Gateway® cloning).
All oligonucleotides, upon arrival, were solubilized in sterilised deionized water and stored at -20°C as
100 pmol/µl stock solution. A detailed list of all primers used in this study is given in Table S1
Appendix.
4.1.2 Polymerase chain reaction (PCR)
The amplification of DNA fragments was done in vitro in by Polymerase Chain Reaction
(PCR). Different DNA polymerases were used according to specific requirements: the High-Fidelity
DNA polymerase Q5TMDNA polymerase (NEB) was used for gene amplification; the
InvitrogenTMPlatinumTMSuperFiTM polymerase was used to amplify long fragments for building
genetic constructs requiring a very accurate replication; and for colony-screening purposes the Thermo
ScientificTMDreamTaq polymerase was used. All the enzymes used on this study were handled
according to the supplier recommendations.
4.1.3 Purification of PCR products
PCR products were purified using the QIAquick PCR Purification Kit (QIAGEN), according
to the manufacturer’s instructions.
4.1.4 Electrophoresis of DNA
The DNA was analysed by electrophoresis of DNA. This technique was performed according
to standard methods using 1% agarose gels and Midori Green Advance (NIPPON Genetics) as
fluorescent dye for detecting DNA.
4.1.5 Purification of DNA from agarose gel
For cloning purposes, after DNA electrophoresis, the fragments of interest were purified from
the agarose gel slices using the QIAquick Gel Extraction Kit (QIAGEN) according to the
manufacturer’s instructions.

Chapter 4. MATERIAL AND METHODS
132
4.1.6 Purification of plasmid DNA
Plasmid DNA was extracted and purified from E. coli or B. subtilis using the QIAprep Spin
Miniprep Kit (QIAGEN), according to manufacturer’s instructions. For the isolation of plasmid DNA
from B. subtilis, lysozyme from chicken egg white (Sigma-Aldrich) was included in the resuspension
buffer of the kit, at a final concentration of 2 mg/ml and the cell suspensions incubated at 37 °C for 15
min. All plasmid DNA was eluted with sterile deionized H2O.
4.1.7 Chromosomal DNA extraction
The genomic DNA of B. subtilis was extracted using the GenElute Bacterial Genomic DNA
(SigmaAldrich) kit, according to supplier’s instructions.
4.1.8 Digestion of DNA with restriction enzymes
The DNA strands were digested by restriction endonucleases (NEB). DNA was digested
from 30 min up to 16 hours and inactivated for 20 min, in the conditions recommended by the
manufacturer and using the buffers provided with the enzymes. The digestion products were either
analysed by DNA electrophoresis, purified from the remaining components of the restriction digestion
reaction, as explained in section 1.3, or directly added to the ligation reaction mix.
4.1.9 Ligation of DNA fragments
Digested DNA fragments were ligated by the T4 DNA ligase (INVITROGEN) in the conditions
recommended by the manufacturer. The ligation reaction volume was typically 20µL and contained
the vector and insert DNA in an approximate molar ratio of 1:3. The ligation mixtures were incubated
at room temperature (RT), from 1h up to an overnight, and then used to transform E. coli.
4.1.10 Gibson assembly technology
This method, developed by Dr. Daniel Gibson and his colleagues, allows assembly of multiple
DNA fragments with overlapping regions in a single-tube isothermal-reaction (Gibson et al. 2009).
Typically, the adjacent fragments were designed to contain a homology region of 20 to 40 base pairs at
their extremities. The amplified fragments were added to a assembly master mix (NEBuilder R HiFi
DNA Assembly Master Mix), which possesses three different enzymatic activities - exonuclease,
DNA polymerase, and DNA ligase activities that work in a single buffer. The exonuclease component
creates single-stranded 3´overhangs that facilitate the annealing of fragments that share
complementarity at one end (overlap region). The DNA polymerase incorporates nucleotides to fill in
any gaps within each annealed fragment. The DNA ligase seals the nicks and covalently bounds the
DNA adjacent segments. In more detail, 10µl of assembly master mix were mixed with 0.02–0.5pmols
of DNA fragments, when up to three fragments are being assembled into a vector, and 0.2–1.0pmoles
of DNA fragments when from four to six fragments are being assembled, as recommended by the

Chapter 4. MATERIAL AND METHODS
133
supplier’s. The reaction mixture was incubated at 50◦C for 15 min, when two or three fragments were
being assembled, or 60 min, when more fragments were assembled. The total volume of the resulting
product was then transformed into competent E. coli MC1061 strains.
4.1.11 Ligation-Independent Cloning (LIC)
Ligation-independent cloning (LIC) is a method that allows the ligation of a PCR fragment to
a plasmid without using ligase or an restriction enzyme (Botella et al. 2010). First the The pMUTIN-
SPALIC vector was linearized by digestion with the restriction enzyme AscI and treated with T4 DNA
polymerase. T4 polymerase has a exonuclease activity 3’→5’ that creates the cohesive extremities in
the presence of dATP for the vector, creating a linearized vector with ssDNA extremities.
Oligonucleotides used to amplify the insert (400bp of the end of the gene of interest) were constructed
in order to contain specific extensions, LIC-P1 and LIC-P2 (Fig.57-A) respectively, which are
homologous with the pMUTIN-SPALIC extremities. The insert weas treated with T4 polymerase too,
creating the ssDNA extremities. The two products were then annealed at room temperature with each
other to form a new pMUTIN-SPA plasmid with the PCR fragment inserted upstream of the SPA
sequence. The obtained plasmid is sufficiently stable for direct transformation into competent E. coli
MC1061 strains. The extracted product plasmid was then transformed into B. subtilis and inserted into
the chromosome by single cross-over (Fig. 57-B). pMUTIN-SPA has a Pspac inducible promotor to
assure that after its integration into the chromosome the downstream genes are expressed. In this
study, we used the LIC methodology to generate C-terminal SPA tag (sequential peptide affinity)
translational fusions with genes of interest, through a pMUTIN-SPALIC integrative plasmid
containing a LIC site generated synthetically.
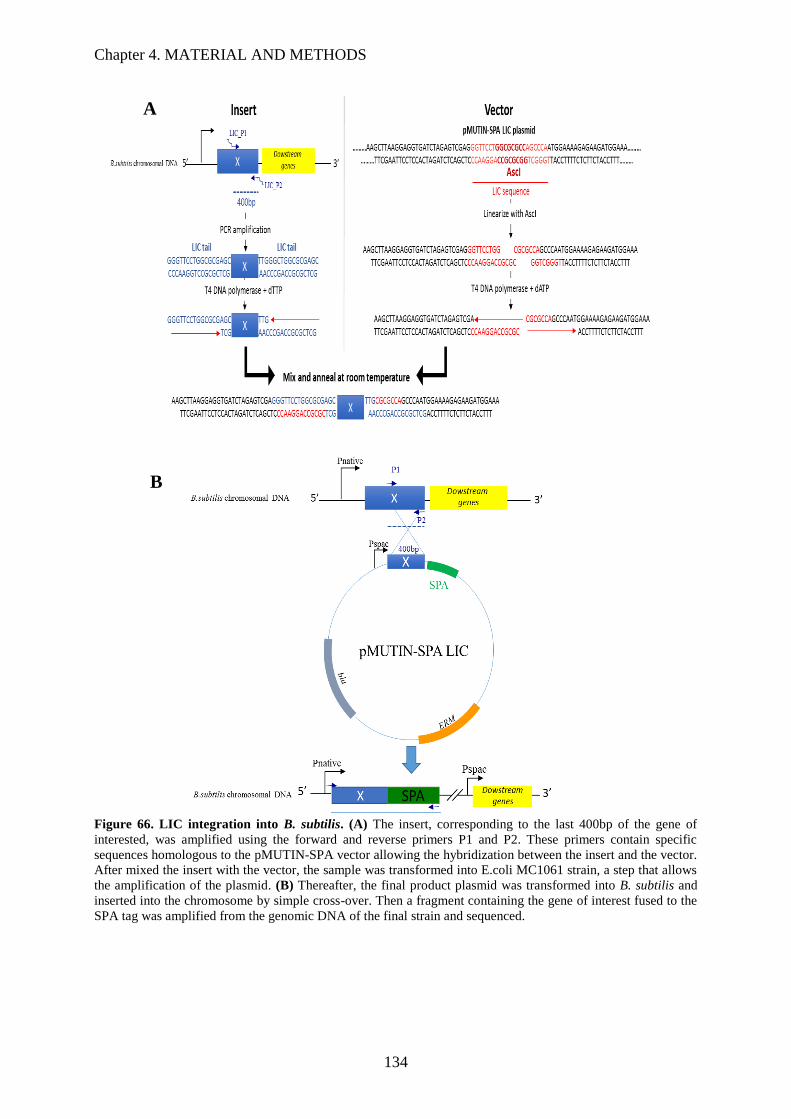
Chapter 4. MATERIAL AND METHODS
134
Figure 66. LIC integration into B. subtilis. (A) The insert, corresponding to the last 400bp of the gene of
interested, was amplified using the forward and reverse primers P1 and P2. These primers contain specific
sequences homologous to the pMUTIN-SPA vector allowing the hybridization between the insert and the vector.
After mixed the insert with the vector, the sample was transformed into E.coli MC1061 strain, a step that allows
the amplification of the plasmid. (B) Thereafter, the final product plasmid was transformed into B. subtilis and
inserted into the chromosome by simple cross-over. Then a fragment containing the gene of interest fused to the
SPA tag was amplified from the genomic DNA of the final strain and sequenced.
A
B

Chapter 4. MATERIAL AND METHODS
135
4.1.12 DNA sequencing
Confirmation of DNA sequences was outsourced to GATC Biothec. The sequencing
chromatograms were analysed and the sequences were confirmed using the Geneious software
alignment features, using as background the BSB1 complete genome.
4.1.13 DNA transformation
4.1.13.1 Transformation of Escherichia coli (Heat shock)
The transformation of DNA into E. coli MC1061, BL21 or ER2566 chimiocompetent strains
were performed by heat shock. 20-50µl of the frozen competent stocked cells were mixed with the
LIC sample, ligation mix or 1-5µl of 10pg to 100 ng/µl of plasmid DNA and incubated on ice during
20 minutes. The mixture was heat shocked for 45s at 42˚C and then put on ice again for 2 min. 500 µL
of fresh LB were added to the cells and incubated at 37˚C for 45 min. Finally, the cells were harvested
by centrifugation (1 min at 12000 xg), resuspended in 100 µL of fresh LB and plated on selective agar
medium.
4.1.13.2 Transformation of Bacillus subtilis using natural competence
Transformation of B. subtilis cells was performed using a quick procedure protocol which is
based on the natural competence of vegetative cells. A fresh plate was prepared by spreading the stock
culture on plate, supplemented with the appropriate selection markers, and incubated overnight at
37˚C. After overnight incubation, a single colony was used to inoculate 2 ml of MC complemented
medium (see appendix,section 6.1) and the culture was grown for 5h, at 37°C in a shaking incubator
(200 rpm). Then 400µl of culture were mixed with 10µl of plasmid DNA or 1µl of chromosomal DNA
and grew for additional 2 hours at 37°C with agitation (200rpm). The culture was plated on LB plates
supplemented with the appropriate antibiotic and incubated overnight at 37°C.
4.2 Bacterial strains and growth conditions
E. coli and B. subtilis strains used in this work are listed in Table S2 and Table S3
respectively. B. subtilis strains used were derived from the BSB1 reference strain, which is a
tryptophan-prototrophic (trp+) derivative of B. subtilis 168-trpC2 strain (Nicolas et al. 2012).
Nutrient Luria-Bertani medium (LB) was used for general-purpose growth of E. coli and B.
subtilis. For B. subtilis DNA transformation, MC media was used (described at the transformation of
B. subtilis section). For specific experiments we used CH defined medium, M9 or Belitsky minimal
media. In general, the bacterial growth was at 37 °C with orbital shaking at 200 rpm, unless otherwise
stated. Growth supplements (Table S 1) and antibiotics (Table S 2) were added to the media when
appropriate. Growth was monitored by measuring optical density at 600 nm (OD600nm) in a
spectrophotometer. For long-term storage of bacterial strains, LB broth cultures, after overnight

Chapter 4. MATERIAL AND METHODS
136
growth, were supplemented with 30 % (v/v) glycerol and stored at -80 °C in cryovials. Growth media
and buffers are described below in the Appendix.
4.3 Strain construction
4.3.1 SPA tagged strains construction
The utilization of a SPA tag allows the purification of proteins in their native conditions. In
order to study the possible protein-protein interactions in between the proteins involved in the Sec
machinery, we have constructed translational fusions of a SPA-tag DNA sequence to the to the 3’ end
of the gene of interest. The cloning of a selected group of proteins involved in the Sec machinery
(secDF, secG, spoIIIJ, bdbB, bdbC, sipT, sipS, sppA, yteJ and prsA), into the BSB1 background, were
carried out according the LIC system described in section 4.1.11. The fragments contained ≥400bp of
the end of the gene of interest, excluding the stop codon, and were amplified from chromosomal DNA
using the appropriate primers listed in Table 2. The primers had a designed LIC tail sequence
homologous to the LIC sequence present on the pMUTIN-SPA LIC plasmid. The vector and the insert
were processed as described on the section 4.1.11 and Figure 66. After the annealing, of the vector
and insert, at room temperature, the mixture was transformed into E.coli MC1061 and the
transformants were selected in 100 µg.ml-1 ampicillin. The final product plasmids were extracted
from E. coli and used to transform BSB1 strain. B. subtilis transformants were selected on LB agar
plates supplemented with 0.6 µg.ml-1 Erythromycin. The 400 bp homology with the targeted gene
allows the plasmid to insert into the chromosome by a single crossover in B.subtilis. To select the
positive clones, the colonies that grew in the presence of 0.6 µg.ml-1 Erythromycin were re-streaked
into 30 µg.ml-1 and grow at 37°C for overnight. The final strains harbouring a C-terminal SPA fusion
under the control of the gene native promotor are listed at Table S 4.
To obtain N-terminal SPA tagged strains, the whole sequences of ORF corresponding to the
genes of interest were amplified by PCR from the BSB1 chromosomal DNA, including the start codon
and stop codon. For this, we used a specific pair of primers for each gene, carrying NotI and XhoI
restriction sites at their 5’ and 3’ ends, respectively. The resulting PCR product was digested with NotI
and XhoI restriction enzymes and ligated into pSPANter plasmid, previously digested with the same
enzymes. E. coli MC1061 cells were transformed with the ligation mixtures and, then, single colonies
were selected on LB-agar plates supplemented with 100 µg.ml-1 ampicillin. The plasmids harbouring
the N-terminal SPA construction were transformed into BSB1 and integrated into the amyE locus by
double crossing over. This integration gives origin to the SPA-tagged protein of interest expressing
strains, under the control of the xylose inducible promoter Pxyl. The positive clones, were selected in
100 µg.ml-1 spectinomycin, confirmed by PCR from the amyE flanking region, tested by the inability
of the strain to produce amylase (starch-iodine test) and sequenced. All the strains constructed are

Chapter 4. MATERIAL AND METHODS
137
listed in Table S2 and S3 of appendix. The final strains harboring a SPA fusion were confirmed by
PCR and sequenced.
The starch-iodine test is used to check the presence of starch, when present the starch reacts
with the iodine crystals and turns into an intense blue/dark color (Dhar 1923). As the amyE gene
encodes for an amylase, this test allows a quick selection of the positive clones. The insertion of the
gene of interest disrupts the amyE gene, consequently the starch is cannot be not degraded, so when
the clones are streaked into a plate containing starch the positive ones turn dark in the presence of
iodine.
Table 2 Oligonucleotides used for LIC constructions
(Sequences required for LIC are underlined)
Sequence
amplified
Primer name Primer sequence
secDF secDF_P1 GGGTTCCTGGCGCGAGCTTGCTATAGCTTCTATTGG
secDF_P2 TTGGGCTGGCGCGAGC
TTGCGCCGAATCTTTTTTCAG
secDF_seq ACGGATCTGATCCAAATGTCAGC
secG secG_P1 GGGTTCCTGGCGCGAGCCACGCAGTTTTGATTACC
secG_P2 TTGGGCTGGCGCGAGC TAGGATATAAGCAAGCGC
secG_seq TCTGATACAGCGTGACATGC
spoIIIJ SpoIIIJ_P1 GGGTTCCTGGCGCGAGC
AGCTTCAACAGGAAACAATGGC
SpoIIIJ_P2 TTGGGCTGGCGCGAGC CTTTTTCTTTCCTCCGGC
SpoIIIJ_seq AGTTCGAAAGCGATGCAGGC
bdbB BdbB_P1 GGGTTCCTGGCGCGAGC
TGGCACAATGGCTAGTTTATTC
BdbB_P2 TTGGGCTGGCGCGAGC ATTATATACATGTTGATTTTG
BdbB_seq TCTTGCCAACTCACTCCAC
bdbC BdbC_P1 GGGTTCCTGGCGCGAGCTATGCTTCCTGGGTTGTG

Chapter 4. MATERIAL AND METHODS
138
BdbC_P2 TTGGGCTGGCGCGAGCTTCAGATTTTTCGCCTTTCAG
C
BdbC_seq TGCGGATTATGATGAAATC
sipS SipS_P1 GGGTTCCTGGCGCGAGC
TGACTCTATGTATCCTACAC
SipS_P2 TTGGGCTGGCGCGAGC ATTTGTTTTGCGCATTTCG
SipS_seq AGCAATTGTGATTGCTGTCG
sipT SipT_P1 GGGTTCCTGGCGCGAGC
CACATTACATGACGGAGAAAGG
SipT_P2 TTGGGCTGGCGCGAGC TTTTGTTTGACGCATTTCG
SipT_seq TCATCGCTGTTCTGCTGGC
sppA SppA_P1 GGGTTCCTGGCGCGAGCCCATAAGGACATTATGTCTC
SppA_P2 TTGGGCTGGCGCGAGCCTTCGCATAGAGATACATC
SppA_seq TCGGCGTCATTATGGAAAGC
yteJ YteJ_P1 GGGTTCCTGGCGCGAGC TTCCTGCTTGATTGGCTC
YteJ_P2 TTGGGCTGGCGCGAGC
TTTGCGGTATAATTTTTCATGC
YteJ_seq AGAGTTAGAGCGCAATGACATC
prsA PrsA_P1 GGGTTCCTGGCGCGAGC
AGGCGAGAAGTTTGAAGACC
PrsA_P2 TTGGGCTGGCGCGAGC TTTAGAATTGCTTGAAGATG
PrsA_seq AAGGCAAAATCCGTGCAAGC
4.3.2 Deletion mutants
The deletion of sppA, yteJ and the sppA yteJ operon at their native locus in the BSB1 strain
was performed by sequence replacement with a chloramphenicol resistance cassette (CmR) expressed
under the control of the native promoter. Therefore, PCR fragments were obtained by amplifying the

Chapter 4. MATERIAL AND METHODS
139
upstream and downstream regions, approximately 1 Kb each, of the gene sequence, from the BSB1
genomic DNA in two sequential PCR steps using the appropriate pair of primers, KO-P1 plus KO-P2
and KO-P5 plus KO-P6 respectively, listed in Table S1. Then a resistance cassette of chloramphenicol
(CmR), approximately 1 Kb, was amplified from pUC19::chloramph plasmid using the primers KO-P3
and KO-P4. The primers P2, P3, P4 and P5, of each construction, have an extension containing an
overlap sequence between the upstream, chloramphenicol and downstream fragments. These three
amplified products were then purified and joined by an overlap PCR reaction. The final PCR product
was purified from agarose gel, directly transformed into BSB1 and plated on LB agar supplemented
with 5 µg.ml-1 chloramphenicol. The deletion of the genes of interest from the native locus was
confirmed by PCR and the final constructs were sequenced, resulting in GHBs023, GHBs025 and
GHBs028 strains. Primers sequences and strains are listed inTable S1 and Table S2 of appendix.
4.3.3 SppA, YteJ and SppA-YteJ overexpressing strains
With the goal of re-insert sppA and yteJ into the deletion mutants, under the control of
inducible promotor, to observe if we could recover the effect on lantibiotics resistance caused by their
deletion we have constructed strains overexpressing sppA and yteJ. To obtain strains overexpressing
SppA or YteJ, or both, we inserted the gene sequences fused to the Phyperspank promoter in the BSB1
chromosome at the amyE locus. For this, the entire gene sequences of sppA and yteJ and the sppA-yteJ
operon, including the RBS sequence, were amplified by PCR from BSB1 genomic DNA , and fused to
the Phyperspank promoter sequence. This promoter is Isopropyl-β-D-thiogalactopyranoside (IPTG)
inducible and lacI-repressible promoter (Overkamp et al. 2013),. For this, the RBS+genes sequences
were amplified by PCR using a specific pair of primers, the Hspank_P1 and Hspank_P2 of each gene,
carrying the HindIII and SalI restriction sites. The resulting PCR products were digested with HindIII
and SalI fast restriction enzymes and ligated into pDR111 plasmid, previously digested with the same
enzymes. HindIII cuts into the sppA sequence, consequently the constructions involving this gene
were done with a blunt 5’ end and a sticky 3’ end created by SalI digestion. The vector and the
fragments were ligated and transformed into E. coli MC1061 competent cells. After the selection of
transformants on 100 µg.ml-1 ampicillin LB-agar plates, the plasmids were extracted, purified and
transformed into BSB1. The positive clones, harboring an integration of the Phyperspank+gene of
interest at the amyE locus, were selected on 100 µg.ml-1 spectinomycin LB-agar plates, confirmed by
PCR using primers in the amyE flanking regions, tested for the inability of the strains to produce
amylase (starch-iodine reaction) and sequenced. With the objective of complementing the deletion
mutants by reintroducing the deleted gene at the ectopic amyE locus, under the control of the inducible
Phyperspank promoter, the final plasmids were also transformed into deletion mutant strains (GHBs023,
GHBs025 and GHBs028). Table S2 of appendix.

Chapter 4. MATERIAL AND METHODS
140
4.3.4 Construction of the GFP Fusions
Two types of superfolder green fluorescent protein (sfGFP) (Pédelacq et al. 2006) fusions
were constructed during this study: transcriptional fusions with the native σW and σA-dependent
promoters, of sppA yteJ operon and translation fusions of SppA and YteJ.
The expression of the sppA-yteJ operon is under the control of two adjacent promoters,
dependent of σW and σA. To study the sppa-yteJ native promoters activity we constructed two strains
harboring a fusion of one and the two promoters with the DNA sequence of the sfgfp introduced at the
ectopic locus amyE.. First, a fragment of 100bp upstream of sppA, containing the natural RBS and the
σW promoter sequence, and a 185bp fragment, including the natural RBS, σW and σA promoter
sequences (Nicolas et al. 2012), were amplified from BSB1 genomic DNA by PCR, and then ligated
upstream of the pSG13 plasmid-borne sequence of the fluorescent protein (Guiziou et al. 2016) by
Gibson assembly. The final mix was transformed into E. coli MC1061 competent cells and plated onto
100 µg.ml-1 ampicillin LB-agar. The plasmid was extracted from positive E. coli clones purified and,
after transformation of B. subtilis BSB1 competent cells, was integrated into the amyE locus. The
positive clones were selected onto 100 µg.ml-1 spectinomycin LB-agar plates, the presence of the
promoter-sfGFP sequence fusions were confirmed by PCR, the clones were also tested by starch-
iodine reaction and, finally, sequenced. The final strains were tested by LCA, as described on section
4.5.
To study SppA and YteJ localization, N-terminal tagged sfGFP-SppA and C-terminal tagged
YteJ-GFP proteins were conditionally expressed from the IPTG inducible Phyper-spank promoter, at the
ectopic amyE locus of B. subtilis chromosome. The appropriate primers, one carrying a HindIII or SalI
restriction site and the second carrying an extension with a linker, were used to PCR amplify the sppA
and yteJ genes from BSB1 chromosomal DNA and the sfgfp gene from the pSG13 plasmid. These
fragments were joined by PCR, digested by HindIII or SalI and ligated into the pDR111 plasmid,
previously digested with the same enzymes. E. coli MC1061 cells were transformed by the ligation
mixtures and transformants were selected onto 100 µg.ml-1 ampicillin LB-agar. The plasmid was
extracted from positive E. coli clones and purified. The sfgfp-sppA and yteJ-sfgfp constructs were then
integrated at the amyE locus of BSB1 competent cells by transformation, and transformant clones
were selected using 100 µg.ml-1 spectinomycin LB-agar plates. The presence of the integrated sfGFP
fusions were verified by PCR amplification from the amyE flanking regions and by the inability of the
strain to produce amylase. The final constructs were sequenced.
4.3.5 His- tag constructions
To obtain E. coli strains overexpressing N-terminal 6xHis tagged proteins construct, the genes
of interest were amplified by PCR from the BSB1 chromosomal DNA, excluding the start codon. For
this, we used a specific pair of primers for each gene, carrying NdeI and XhoI restriction sites (

Chapter 4. MATERIAL AND METHODS
141
Table S 3). The resulting PCR product was digested with NotI and XhoI fast restriction
enzymes and ligated into pJ411_6His, previously digested with the same enzymes, a plasmid
harboring the 6XHis-tag sequence under the control of the PT7 promoter. E. coli MC1061 cells were
transformed by the ligation mixtures and transformants were selected onto 30 µg.ml-1 kanamycin LB-
agar plates. The plasmids were extracted by minipprep and the presence of the right inserts were
confirmed by PCR and sequenced. Thereafter, the final plasmids were used to transform into E.coli
2566 overexpressing strains. The final strains obtained, from GHEc 054 up to GHEc 058, is listed on
Table S 4.
4.4 Fluorescence Microscopy
B. subtilis strains were grown overnight in LB at 37 ºC with the appropriate antibiotic and, the
next day, were diluted (1:200) in 20 ml of fresh LB supplemented with 0.05 mM IPTG and grown
until OD600nm 0.3-0.5 - exponential growth phase - and until OD600nm 1-2 - early stationary growth
phase. Thereafter at regulated intervals samples were taken and cells were observed by fluorescence
microscopy. For that, 2 µl of cell culture were placed onto a thin film of 1.2% agarose. The cell
membranes were stained with FM4-64 (Molecular probes) and nucleoids were stained with DAPI.
Fluorescence microscopy was performed using a Leica DMRA2, controlled by Metamorph software
(Molecular devices). Processing of fluorescence images was performed using Metamorph and ImageJ
software.
4.5 Live-cell array (LCA)
The Live Cell Array is a method (Botella et al. 2010), which allows the high throughput
acquisition of time-course profiles of bacterial cellular growth and gene expression. It measures both
the optical density for recording cell growth and the fluorescence emission of the fluorescent reporter
protein, usually the GFP, expressed in specifically modified strains of interest. The B. subtilis strains
were grown overnight in 5ml of LB medium at 37°C and then diluted 20-fold in 100µL/well of fresh
LB medium (pre-culture) into 96-well microtiter plate. The LB pre-cultures were grown until an
OD600nm of 0.3-0.4 and then diluted 400-fold into a fresh medium, LB or CH defined medium. OD600nm
and fluorescence (excitation 485, emission 528) were measured at time intervals of 10 minutes. Only
the 60 central wells of the 96-well microtiter plates were inoculated since the wells around the edges
exhibited evaporation higher than 5 % over 20 hours; therefore these peripheral wells were only filled
in with sterile medium.
4.6 Bacteria survival test
The BSB1 and deletion mutant strains, described in section 2.1.2, were grown overnight in LB
medium, supplemented with the appropriate antibiotic, at 37°C. On the following day, the overnight

Chapter 4. MATERIAL AND METHODS
142
cultures were diluted 200-fold into fresh LB or M9 medium and then the cultures grew for 48h at 37
°C while shaking. Samples of 100µl were taken after 18h, 24h and 48h. Serial dilutionswere done and
the last two dilutions, 10-6 and 10-7 fold, were plated onto LB agar supplemented with the appropriate
antibiotic. The number of colonies, corresponding to the number of cells that survived, were counted
after overnight incubation at 37°C.
To test if the WT cells could complement or help the growth of the mutants, the WT cells and
deletion mutants were mixed and grew for 48h at 37 °C while shaking. The overnight cultures were
diluted 200-fold into fresh LB and grown at 37°C until reach the exponential phase, then the cultures
were diluted 1000-fold and mixed 1:1 (50% WT culture+50% Deletion mutant). The mixed cultures
and the controls grew for 48h at 37 °C while shaking. From this point the remaining steps were done
as described above.
4.7 Swimming and swarming capacity
The surfaces of agar concentrations above 1% prohibit swarming and swimming of many
bacterial species, consequently, the humidity of the media is crucial for conducting these experiments.
Therefore, it is necessary to define a rigorous protocol in order to control the water content of the
medium, we tested the ability of the bacteria swim at 0.3% of agar and the capacity of swarm at 0.7%
(Kearns 2010). All the plates were consistently prepared, 25 ml of the LB media were poured when
the agar is approximately 50°C, let to solidify and to dry open for 15 minutes - in a laminar flow
chamber. The cells were harvested (100µl) from overnight cultures or at mid exponential phase
(OD600nm ~0.5), centrifuged at 2000rpm to avoid breaking the flagella, resuspended in PBS buffer and
spotted (5µl) in the middle of the plate. Thereafter, the halo created by the cell movement were
measured at time intervals of 1h during 6h.
4.8 Biofilm formation
B. subtilis strains in study were grown in LB shaking at 37°C, supplemented with the
appropriate antibiotic, to an OD600nm of 1,0. To observe the architectural differences between biofilm
from distinct strains, 10 µL cultures were then spotted on a dried MSgg plate (1.5% agar) and
incubated for 70 hours at 30 °C. Colonies were measured and photographed.
4.9 Protein analysis
4.9.1 Sodium Dodecyl Sulphate Poly-Acrylamide Gel Electrophoresis (SDS
PAGE gels)
All the proteins purified in this project were analysed by 10% or 12.5% SDS-PAGE gels.
Samples were mixed with protein loading buffer (0.5 M Tris-HCl pH = 6.8, 1 M β-mercaptoethanol,
20 % (v/v) SDS, 0.2% (v/v) bromophenol blue, 12.5 % (v/v) glycerol), usually incubated 5 min at 95

Chapter 4. MATERIAL AND METHODS
143
°C and electrophoresis was performed in Laemlli 1X buffer (25 mM Tris, 192 mM glycine, 0.1%
SDS) at 200 V. After staining with Coomassie blue staining solution (0.1% Coomassie Brilliant Blue
R-250, 50% ethanol and 10% glacial acetic acid) protein sizes were estimated relative to a protein
marker ladder.
4.9.2 Blue Native PAGE
For Blue-Native electrophoresis, the strains of interest were pre-cultured overnight in LB,
supplemented with antibiotics if needed, at 37°C. The following day the culture was diluted 1000-fold
(to an OD600nm of 0.001) into 1-2 liters of LB medium, supplemented if needed. When the OD600nm
reached 0.5 - 1, cells were harvested by centrifugation at 6000 rpm for 10 min at 4°C. The pellet was
washed with 40 ml of cold resuspension buffer (buffer R: 10 mM Tris-Cl pH 7.5, 150 mM NaCl),
centrifuged at 4500 rpm for 10 min at 4°C and instantly frozen in liquid nitrogen. The cells were
resuspended in 20 ml of buffer R supplemented with 1 mg/ml of lysozyme, 1 mM EDTA and 5 µl of
Benzonase (Invitrogen), and incubated in a water bath at 37 °C for 15-30 min. To remove the cell
debris the samples were centrifuged at 4500rpm for 30min at 4°C and the pellet was discarded.
Membrane and cytosolic fractions were then separated by ultracentrifugation at 100,000 g for 1 h at
4°C. The membrane protein samples were solubilized into 1% n-Dodecyl β-D-maltoside (DDM), were
mixed with NativePAGE sample buffer and with NativePAGE 5% G-250 Sample Additive
(Invitrogen), then incubated for 1h on ice. The samples were separated by electrophoresis on
NativePAGE 3%–12% Bis-Tris gels (Invitrogen) with NativePAGE running buffer and NativePAGE
Dark Blue Cathode Buffer (Invitrogen) for 20 to 30 min at 150 V, substituted with NativePAGE Light
Blue Cathode Buffer (Invitrogen) for 60 min at 150V. The electrophoresis tank was maintained on ice
throughout the procedure. The gel was then used for Western blot analysis or stained by Blue Safe
Protein Stain. This staining allows the analysis by nanoscale liquid chromatography coupled to tandem
mass spectrometry (nanoLC-MS/MS, Plateforme PAPPSO, INRA centre de Jouy en Josas, or Dr
Becher’s lab, Greifswald University).
4.9.3 Western blot analysis
To analyze if the tagged proteins were expressed, Western blots were performed using the
appropriate primary and secondary antibodies for each specific tag. In the case of FLAG-tag the
Monoclonal ANTI-FLAG M2 antibody (F3165 Sigma) as a primary antibody and Anti-Mouse IgG
(A2304 Sigma) as a secondary antibody. For the AmyM the primary antibody, we used, was raised
during my UCB secondments, this antibody was raised in rabbits, as such we used Anti-Rabbit IgG
(A0545 Sigma) (Appendix section 6.2) Equal amounts of protein, from each sample, were heated to
100 ºC for 5 minutes, loaded onto a 10% SDS-PAGE gel and separated at 200V. Proteins were then
transferred to a Hybond-P Polyvinylidene fluoride (PVDF) membrane (GE Healthcare) using a
semidry transfer cell (Bio-Rad) according to standard Western blotting techniques(Burnette 1982).

Chapter 4. MATERIAL AND METHODS
144
The detection was performed, after a last wash step, using Clarity™ Western ECL Blotting detection
substrates (Biorad), according to the manufacturers guidelines, through the ChemiDoc™ XRS+
System and Image lab imaging software.
4.9.4 Secreted proteins analysis
To check if a protein complex is involved in protein secretion we analyzed the if extracellular
amount of a highly secreted reporter protein, as the case of AmyM, is affected. For that purpose, the
B.subtilis strains of interest were naturally transformed with a multi copy plasmid containing the gene
encoding AmyM (pCS73) and an empty plasmid (pCS74) to use as a control, all carrying a neomycin
(Neo) resistance cassette. Dr. Van Rij provided these plasmids from DSM company, our collaborator
from the Protein Factory project. The final strains, carrying the pCS73 and pCS74, were pre-cultured
overnight at 37°C in LB medium supplemented with 10µg/ml Neo and other antibiotic markers
correspondent to the background strains, if needed. The following day the culture was diluted 1000-
fold (to an OD600nm of 0.001) into 50ml of LB medium with the same supplements. Grown at 37°C
until the OD600nm reached 0.5-1 and the cells were harvested by centrifugation at 4500 rpm for 15 min
at 4°C. The cell pellet was discarded and the culture media was filtered using a 0.2µm filter, removing
possible remaining cells. The clear culture media was precipitated overnight at 4°C by 10%
trichloroacetic acid (TCA). After centrifugation at 15000 rpm for 30 min, the supernatant was
discarded and the protein pellet was washed two times with 200 µl cold acetone, repeating the
centrifuge steps each time. The protein pellets were then dissolved by the addition of 3µl of 10 mM
Tris-HCl pH 8, 8 M urea.
4.9.5 Sequential Peptide Affinity (SPA) purification
The sequential peptide affinity (SPA) technique has been developed, from the TAP-tag
technique, to study protein-protein interactions (Rigaut et al. 1999; Zeghouf et al. 2004). The
translational fusion of the SPA-tag, composed of three FLAG sequences (3×FLAG) and the
calmodulin binding peptide (CBP) separated by a TEV protease cleavage site, to a protein of interest
allows its purification together with the protein(s) to which it interacts under native conditions, i.e.
without adding any cross-linking agents ( Delumeau et al. 2011).
The strains expressing SPA-tagged proteins were pre-cultured overnight in the presence of
erythromycin at 37°C. The following day the culture was diluted 1000-fold (to an OD600nm of 0.001)
into 2 liters of LB medium supplemented with 0.6 µg/ml erythromycin and 0.5 mM IPTG. When the
OD600nm reached 0.5 - 1, cells were harvested by centrifugation at 6000 rpm for 10 min at 4°C. The
pellet was washed with 40 ml of cold resuspension buffer (buffer R: 10 mM Tris-Cl pH 7.5, 150 mM
NaCl), centrifuged at 4500 rpm for 10 min at 4°C and instantly frozen in liquid nitrogen. The cells
were resuspended in 20 ml of buffer R supplemented with 1 mg/ml of lysozyme, 1 mM EDTA and 5

Chapter 4. MATERIAL AND METHODS
145
µl of Benzonase (Invitrogen), and incubated in a water bath at 37 °C for 15-30 min. To remove the cell
debris the samples were centrifuged at 4500rpm for 30min at 4°C and the pellet was discarded.
Membrane and cytosolic fractions were then separated by ultracentrifugation at 100,000 g for 1 h at
4°C. The membrane pellet was solubilized using buffer R supplemented with 1% DDM. Identical
protein amounts of the membrane and soluble protein fractions were loaded on 10% SDS-
polyacrylamide gels for Coomassie staining and Western blotting using anti-FLAG antibodies.
In addition, protein complexes from the membrane fraction were pulled down using a SPA
purification method (Charbonnier et al. 2017). The solubilized membrane fraction, 2 mM CaCl22 and
1%DDM, was mixed with 200 µl of pre-equilibrated calmodulin resin (CaM) in 2 ml tubes, which
were placed on a rotating wheel for a minimum of 2 h at 4°C. The resin was transferred in a column,
the eluate was discarded and the CaM resin was washed with 2 ml of buffer R supplemented with 1%
DDM and 0.1 mM CaCl2. The proteins remaining bound to the CaM resin were eluted with a total
volume of 0.75 ml of buffer R supplemented with 1% DDM and 3 mM EGTA. The eluate was
collected and mixed with 200 µl of anti-FLAG resin. After its incubation under gentle agitation on the
rotating wheel for, at least two hours, the eluate from the anti-FLAG resin was discarded, the resin was
washed with 2 ml of buffer R supplemented with 1% DDM. The proteins remaining bound to the anti-
FLAG resin were eluted with a total volume of 1ml of 0.1M glycine pH 3.
Proteins from the eluate were precipitated overnight at 4°C by 10% trichloroacetic acid
(TCA). After centrifugation at 15000 rpm for 30 min, the supernatant was discarded and the protein
pellet was washed two times with 200 µl cold acetone, repeating the centrifuge steps each time. The
protein pellets were then dissolved by the addition of 3µl of 10 mM Tris-HCl pH 8, 8 M urea. The
volume was then extended up to 25µl with 10 mM Tris-HCl pH 8, decreasing the urea concentration to
1 M. Then to analyze if the SPA tagged protein was captured, with possible interacting partners, the
protein concentrations of the samples were measured by the Bradford’s method and the same amount
of protein was loaded and the samples were separated by native polyacrylamide gel electrophoresis
(PAGE), which was coomassie stained after. These samples were analyzed by a second technique,
nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC-MS/MS). For this
the samples were equal loaded into SDS-Page gel, as before, but the samples just run 1 cm into the
resolving part of the gel. The gel was stained by Bio-Safe Coomassie G-250 stain solution and the
small pieces of the gel containing the samples were cut and sent to be treated and analyzed by mass
spectrometry.

Chapter 4. MATERIAL AND METHODS
146
Figure 67. Sequential peptide affinity purification. (A) The SPA-tag is composed of three FLAG sequences
(3×FLAG) and the calmodulin binding peptide (CBP) separated by a TEV protease cleavage site. (B) Schematic
representation of the double step SPA purification. This technique allows the purification of protein complexes
under native conditions.
4.9.6 His-tag purification
The E. coli strains harboring a translational 6XHis-tag fusion were pre-cultured overnight in
the presence of 30 µg.ml-1 kanamycin at 37°C. The following day the culture was diluted 1000-fold (to
an OD600nm of 0.001) into 2 liters of LB medium supplemented with 30 µg.ml-1 kanamycin and grown
until an OD600nm of 0.8 at 37°C. Thereafter the expression was induced by the addition of 250 µM
IPTG for overnight at 16°C. Cells were harvested by centrifugation 4500 rpm for 10 min, and the
pellet was resuspended in 40 ml of 20mM Tris-HCl pH7,5 and 250mM NaCl (BufferR2) and stored at
-20°C. Cells were lysed by sonication and centrifuged at 4500rpm for 30 min to remove the cell
debris. The membrane and cytosolic fractions were separated by ultracentrifugation at 100,000 g for 1
h at 4°C. The membrane fraction was solubilized using buffer R2 supplemented with 1% n-Dodecyl β-
D-maltoside (DDM) for overnight at 4°C. The sample was then loaded onto a Ni2+ affinity column
(Ni-NTA agarose, Qiagen) pre-equilibrated in the same buffer. The Ni-NTA columns were washed
with 50ml of 20mM Tris-HCl pH7,5, 250mM NaCl and 0.01% DDM (Buffer R3) supplemented with
5mM imidazole, and then with 20 ml Buffer R3 suplemented with 20mM imidazole. After washing,
the proteins bound to the Ni-NTA resin were eluted, sequentially, with 1ml of Buffer R3 supplemented
with 50 mM imidazole and then 4ml of the same buffer with 200mM imidazole. All the eluted samples
A
A
A
A
A
A
A
B
B
B
B
Fig
ure
1|
Seq
ue
nti
al
pe

Chapter 4. MATERIAL AND METHODS
147
were analyzed by SDS-PAGE. The samples containing the highest concentration of the His-taged
protein were joined and concentrated by Amicon Ultra-15 Centrifugal Filters, mixed with 50%
glycerol and stored at -20°C.
4.9.7 Double tag purification
With the objective of increasing the purity of the membrane protein complex, a E. coli stain
harboring SppA with a N-terminal His tag and YteJ with a C-terminal Flag ta ( 6xHis-SppA/YteJ-Tev-
3xFlag), a double tagged SppA-YteJ complex, was constructed as described above on section 4.3.5.
The membrane fraction was obtained and solubilized as described above. The sample was mixed with
200 µl of anti-FLAG resin and incubated under gentle agitation on the rotating wheel. The eluate from
the anti-FLAG resin was discarded and the resin was washed with 2 ml of buffer R supplemented with
1% DDM. The resin was resuspended in 200 ml of TeV buffer (50 mM Tris-HCl pH 8, 150 mM NaCl,
0.5mM EDTA, 1 mM DTT) with 50units of Ac-TeV protease. After incubation at 4°C for overnight,
the eluate was collected, diluted up to 2ml (4-fold dilution) and mixed with 200µl Ni-NTA pre-
equilibrated agarose resin. The Ni-NTA columns were washed with 50ml 20mM Tris-HCl pH7,5,
250mM NaCl and 0.01% DDM (Buffer R3) supplemented with 5mM imidazole ,and then with 20 ml
Buffer R3 supplemented with 20mM imidazole. After washing the proteins bounded to the Ni-NTA
resin were eluted, sequentially, with 1ml of Buffer R3 supplemented with 50 mM imidazole and then
4ml of the same buffer with 200mM imidazole. The eluted proteins were concentrated by Amicon
Ultra-15 Centrifugal Filters, mixed with 50% glycerol and storage at -20°C.

Chapter 4. MATERIAL AND METHODS
148

149
BIBLIOGRAPHY

150

Chapter 5. BIBLIOGRAPHY
151
5 BIBLIOGRAPHY
Abisado, Rhe G., Saida Benomar, Jennifer R. Klaus, Ajai A. Dandekar, and Josephine R. Chandler.
2018. “Bacterial Quorum Sensing and Microbial Community Interactions.” MBio 9 (3): 1–13.
https://doi.org/10.1128/mBio.02331-17.
Adimpong, David B., Kim I. Sørensen, Line Thorsen, Birgitte Stuer-Lauridsen, Warda S. Abdelgadir,
Dennis S. Nielsen, Patrick M. F. Derkx, and Lene Jespersen. 2012. “Antimicrobial susceptibility
of Bacillus strains isolated from primary starters for african traditional bread production and
characterization of the bacitracin operon and bacitracin biosynthesis.” Applied and
Environmental Microbiology 78 (22): 7903–14. https://doi.org/10.1128/aem.00730-12.
Akimaru, J, S Matsuyama, H Tokuda, and S Mizushima. 1991. “Reconstitution of a Protein
Translocation System Containing Purified SecY, SecE, and SecA from Escherichia coli.”
Proceedings of the National Academy of Sciences of the United States of America 88 (15): 6545–
49. https://doi.org/10.1073/pnas.88.15.6545.
Akiyama, Yoshinori. 1999. “Self-Processing of FtsH and Its Implication for the Cleavage Specificity
of This Protease.” Biochemistry 38 (36): 11693–99. https://doi.org/10.1021/bi991177c.
Alcock, Felicity, Phillip J. Stansfeld, Hajra Basit, Johann Habersetzer, Matthew Ab Baker, Tracy
Palmer, Mark I. Wallace, and Ben C. Berks. 2016. “Assembling the Tat Protein Translocase.”
ELife 5: 1–28. https://doi.org/10.7554/eLife.20718.
Aleti, Gajender, Sylvia Lehner, Markus Bacher, Stéphane Compant, Branislav Nikolic, Maja Plesko,
Rainer Schuhmacher, Angela Sessitsch, and Günter Brader. 2016. “Surfactin variants mediate
species-specific biofilm formation and root colonization in Bacillus.” Environmental
Microbiology 18 (8): 2634–45. https://doi.org/10.1111/1462-2920.13405.
Alguel, Yilmaz, Sotiris Amillis, James Leung, George Lambrinidis, Stefano Capaldi, Nicola J. Scull,
Gregory Craven, et al. 2016. “Structure of eukaryotic purine/H+ symporter UapA suggests a
Role for homodimerization in transport activity.” Nature Communications 7: 11336.
https://doi.org/10.1038/ncomms11336.
Anagnostopoulos, C, and J Spizizen. 1961. “Requirements for transformation in Bacillus subtilis.”
Journal of Bacteriology 81 (5): 741–46. http://www.ncbi.nlm.nih.gov/pubmed/16561900.
Angelini, Sandra, Sandra Deitermann, and Hans-Georg Koch. 2005. “FtsY, the bacterial signal-
recognition particle receptor, interacts functionally and physically with the SecYEG translocon.”
EMBO Reports 6 (5): 476–81. https://doi.org/10.1038/sj.embor.7400385.
Anné, Jozef, Anastassios Economou, and Kristel Bernaerts. 2016. “Protein secretion in gram-positive

Chapter 5. BIBLIOGRAPHY
152
bacteria: from multiple pathways to biotechnology.” In Life Science Journal, 6:267–308.
https://doi.org/10.1007/82_2016_49.
Anné, Jozef, Kristof Vrancken, Lieve Van Mellaert, Jan Van Impe, and Kristel Bernaerts. 2014.
“Protein secretion biotechnology in gram-positive bacteria with special emphasis on
Streptomyces lividans.” Biochimica et Biophysica Acta - Molecular Cell Research 1843 (8):
1750–61. https://doi.org/10.1016/j.bbamcr.2013.12.023.
Antelmann, Haike, Elise Darmon, David Noone, Jan Willem Veening, Helga Westers, Sierd Bron,
Oscar P. Kuipers, Kevin M. Devine, Michael Hecker, and Jan Maarten Van Dijl. 2003. “The
extracellular proteome of Bacillus Subtilis under Secretion Stress Conditions.” Molecular
Microbiology 49 (1): 143–56. https://doi.org/10.1046/j.1365-2958.2003.03565.x.
Auchtung, Jennifer M., Naira Aleksanyan, Artemisa Bulku, and Melanie B. Berkmen. 2016. “Biology
of ICEBs1, an integrative and conjugative element in Bacillus subtilis.” Plasmid 86: 14–25.
https://doi.org/10.1016/j.plasmid.2016.07.001.
Auchtung, Jennifer M, Catherine A Lee, Rita E Monson, Alisa P Lehman, and Alan D Grossman.
2005. “Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the
global dna damage response.” Proceedings of the National Academy of Sciences of the United
States of America 102 (35): 12554–59. https://doi.org/10.1073/pnas.0505835102.
Babe, L M, and B Schmidt. 1998. “Purification and biochemical analysis of Wpra, a 52-Kda serine
protease secreted by B. subtilis as an active complex with its 23 kDa propeptide.” Biochimica et
Biophysica Acta - Protein Structure & Molecular Enzymology 1386 (1): 211–19.
https://doi.org/10.1016/S0167-4838(98)00110-1.
Baker, Tania A, and Robert T Sauer. 2012. “ClpXP, an ATP-powered unfolding and protein-
degradation machine.” Biochimica et Biophysica Acta 1823 (1): 15–28.
https://doi.org/10.1016/j.bbamcr.2011.06.007.
Barbe, Valérie, Stéphane Cruveiller, Frank Kunst, Patricia Lenoble, Guillaume Meurice, Agnieszka
Sekowska, David Vallenet, et al. 2009. “From a consortium sequence to a unified Sequence: The
Bacillus subtilis 168 reference genome a decade later.” Microbiology 155 (6): 1758–75.
https://doi.org/10.1099/mic.0.027839-0.
Bauer, Benedikt W., Tom Shemesh, Yu Chen, and Tom A. Rapoport. 2014. “A ‘Push and Slide’
mechanism allows sequence-insensitive translocation of secretory proteins by the SecA
ATPase.” Cell 157 (6): 1416–29. https://doi.org/10.1016/j.cell.2014.03.063.
Beauchemin, Diane. 2009. “Inductively coupled plasma mass spectrometry.” Trends in Analytical
Chemistry 80 (12): 4455–86. https://doi.org/10.1017/CBO9781107415324.004.

Chapter 5. BIBLIOGRAPHY
153
Beckwith, Jon. 2013. “The Sec-Dependent Pathway.” Research in Microbiology 164 (6): 497–504.
https://doi.org/10.1016/j.resmic.2013.03.007.
Bensing, Barbara A., and Paul M. Sullam. 2002. “An accessory Sec Locus of Streptococcus gordonii
is required for export of the surface protein GspB and for normal levels of binding to human
platelets.” Molecular Microbiology 44 (4): 1081–94. https://doi.org/10.1046/j.1365-
2958.2002.02949.x.
Berg, Howard C. 2003. “The rotary motor of bacterial flagella.” Annual Review of Biochemistry 72
(1): 19–54. https://doi.org/10.1146/annurev.biochem.72.121801.161737.
Berkmen, Melanie B., Catherine A. Lee, Emma Kate Loveday, and Alan D. Grossman. 2010. “Polar
positioning of a conjugation protein from the integrative and conjugative element ICEBs1 of
Bacillus subtilis.” Journal of Bacteriology 192 (1): 38–45. https://doi.org/10.1128/JB.00860-09.
Berks, B C. 2015. “The Twin-Arginine protein translocation pathway.” Annu Rev Biochem 84: 843–
64. https://doi.org/10.1146/annurev-biochem-060614-034251.
Berks, Ben C., T. Palmer, and F. Sargent. 2003. “The Tat protein translocation pathway and its role in
microbial physiology.” Advances in Microbial Physiology 47 (0): 187–254.
Blair, David F. 2003. “Flagellar movement driven by proton translocation.” FEBS Letters 545 (1): 86–
95. https://doi.org/10.1016/S0014-5793(03)00397-1.
Blobel, G, and B Dobberstein. 1975. “Transfer of proteins across membranes. I. Presence of
proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-
bound ribosomes of murine myeloma.” The Journal of Cell Biology 67 (3): 835–51.
https://doi.org/10.1083/jcb.67.3.835.
Blount, Zachary D. 2015. “The unexhausted potential of E. Coli.” ELife 4: 1–12.
https://doi.org/10.7554/eLife.05826.
Blümmel, Anne Sophie, Friedel Drepper, Bettina Knapp, Ekaterina Eimer, Bettina Warscheid,
Matthias Müller, and Julia Fröbel. 2017. “Structural features of the tatc membrane protein that
determine docking and insertion of a twin-arginine signal peptide.” Journal of Biological
Chemistry 292 (52): 21320–29. https://doi.org/10.1074/jbc.M117.812560.
Bolhuis, Albert, Arne Matzen, Hanne Leena Hyyryläinen, Vesa P. Kontinen, Rob Meima, Jerome
Chapuis, Gerard Venema, Sierd Bron, Roland Freudl, and Jan Maarten Van Dijl. 1999. “Signal
peptide peptidase- and ClpP-like proteins of Bacillus subtilis required for efficient translocation
and processing of secretory proteins.” Journal of Biological Chemistry 274 (35): 24585–92.
https://doi.org/10.1074/jbc.274.35.24585.

Chapter 5. BIBLIOGRAPHY
154
Botella, Eric, Mark Fogg, Matthieu Jules, Sjouke Piersma, Geoff Doherty, Annette Hansen, Emma L.
Denham, et al. 2010. “PBaSysBioII: an integrative plasmid generating Gfp transcriptional
fusions for high-throughput analysis of gene expression in Bacillus subtilis.” Microbiology 156
(6): 1600–1608. https://doi.org/10.1099/mic.0.035758-0.
Britton, Robert A, Patrick Eichenberger, Jose Eduardo Gonzalez-pastor, Paul Fawcett, Rita Monson,
Richard Losick, Alan D Grossman, and J B Acteriol. 2002. “Genome-Wide analysis of the
stationary-phase sigma factor ( sigma-h ) regulon of Bacillus subtilis” 184 (17): 4881–90.
https://doi.org/10.1128/JB.184.17.4881.
Brockmeier, Ulf, Michael Caspers, Roland Freudl, Alexander Jockwer, Thomas Noll, and Thorsten
Eggert. 2006. “Systematic screening of all signal peptides from bacillus subtilis: a powerful
strategy in optimizing heterologous protein secretion in gram-positive bacteria.” Journal of
Molecular Biology 362 (3): 393–402. https://doi.org/10.1016/j.jmb.2006.07.034.
Browning, Douglas F., Kirsty L. Richards, Amber R. Peswani, Jo Roobol, Stephen J.W. Busby, and
Colin Robinson. 2017. “Escherichia Coli ‘TatExpress’ strains super-secrete human growth
hormone into the bacterial periplasm by the Tat pathway.” Biotechnology and Bioengineering
114 (12): 2828–36. https://doi.org/10.1002/bit.26434.
Brundage, Lorna, Joseph P. Hendrick, Elmar Schiebel, Arnold J.M. Driessen, and William Wickner.
1990. “The purified E. coli integral membrane protein SecYE is sufficient for reconstitution of
seca-dependent precursor protein translocation.” Cell 62 (4): 649–57.
https://doi.org/10.1016/0092-8674(90)90111-Q.
Buck, Emmy De, Elke Lammertyn, and Jozef Anné. 2008. “The importance of the twin-arginine
translocation pathway for bacterial virulence.” Trends in Microbiology 16 (9): 442–53.
https://doi.org/10.1016/j.tim.2008.06.004.
Burbulys, David, Kathleen A. Trach, and James A. Hoch. 1991. “Initiation of sporulation in B. subtilis
is controlled by a multicomponent phosphorelay.” Cell 64 (3): 545–52.
https://doi.org/10.1016/0092-8674(91)90238-T.
Burnette, S. 1982. “‘Western Blotting’: electrophoresis of protein from sodium dodecyl sulphate
polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and
radioiodinated Protein A.” Anal. Biochem 112: 195–203.
https://doi.org/10.1177/104063870601800613.
Burrage, Andrew M., Eric Vanderpool, and Daniel B. Kearns. 2018. “The assembly order of flagellar
rod subunits in Bacillus subtilis.” Journal of Bacteriology, no. September: JB.00425-18.
https://doi.org/10.1128/JB.00425-18.

Chapter 5. BIBLIOGRAPHY
155
Cai, Dongbo, Hao Wang, Penghui He, Chengjun Zhu, Qin Wang, Xuetuan Wei, Christopher T.
Nomura, and Shouwen Chen. 2017. “A novel strategy to improve protein secretion via
overexpression of the sppa signal peptide peptidase in Bacillus Licheniformis.” Microbial Cell
Factories 16 (1): 1–10. https://doi.org/10.1186/s12934-017-0688-7.
Camilli, Andrew, and Bonnie Bassler. 2006. “Bacterial small-molecule signalling pathways.” Science
311 (February): 1113–16. https://doi.org/doi:10.1126/science.1121357.
Cao, Haojie, Auke J. Heel, Hifza Ahmed, Maarten Mols, and Oscar P. Kuipers. 2017. “Cell surface
engineering of bacillus subtilis improves production yields of heterologously expressed α-
amylases.” Microbial Cell Factories 16 (1): 1–9. https://doi.org/10.1186/s12934-017-0674-0.
Cao, Min, Tao Wang, Rick Ye, and John D Helmann. 2002. “Antibiotics that inhibit cell wall
biosynthesis induce expression of the Bacillus subtilis sigma(W) and sigma(M) regulons.”
Molecular Microbiology 45 (5): 1267–76. https://doi.org/papers2://publication/uuid/DFAB4E11-
2A39-4580-922D-28C991BAB0E1.
Carpenter, Phillip B, Aamir R Zuberi, and George W Ordal. 1993. “Bacillus subtilis flagellar proteins
Flip , FliQ , FliR and FlhB are related to Shigella flexneri virulence factors” 137: 243–45.
Cascales, Eric, and Peter J. Christie. 2003. “The versatile bacterial Type IV secretion systems.” Nature
Reviews Microbiology 1 (2): 137–49. https://doi.org/10.1038/nrmicro753.
Cha, Hyun Ju, Hyun Geun Yoon, Young Wan Kim, Hee Seob Lee, Jung Wan Kim, Ki Sung Kweon,
Byung Ha Oh, and Kwan Hwa Park. 1998. “molecular and enzymatic characterization of a
maltogenic amylase that hydrolyzes and transglycosylates acarbose.” European Journal of
Biochemistry 253 (1): 251–62. https://doi.org/10.1046/j.1432-1327.1998.2530251.x.
Chan, Chio Mui, Erik Hahn, and Peter Zuber. 2014. “adaptor bypass mutations of Bacillus subtilis spx
suggest a mechanism for YjbH-enhanced proteolysis of the regulator Spx by ClpXP.” Molecular
Microbiology 93 (3): 426–38. https://doi.org/10.1111/mmi.12671.
Charbonnier, Teddy, Dominique Le Coq, Stephen McGovern, Magali Calabre, Olivier Delumeau,
Stéphane Aymerich, and Matthieu Jules. 2017. “ molecular and physiological logics of the
pyruvate-induced response of a novel transporter in Bacillus subtilis .” MBio 8 (5): 1–18.
https://doi.org/10.1128/mbio.00976-17.
Chen, Inês, and David Dubnau. 2004. “DNA uptake during bacterial transformation.” Nature Reviews
Microbiology 2 (3): 241–49. https://doi.org/10.1038/nrmicro844.
Chen, J, G Fu, Y Gai, P Zheng, D Zhang, and J Wen. 2015. “Combinatorial Sec pathway analysis for
improved heterologous protein secretion in Bacillus subtilis: identification of bottlenecks by
systematic gene overexpression.” Microb Cell Fact 14: 92. https://doi.org/10.1186/s12934-015-

Chapter 5. BIBLIOGRAPHY
156
0282-9.
Chen, Shifei, Yi Fan, Xueling Shen, Peng Sun, Guosong Jiang, Yi Shen, Wenfei Xue, Yongquan Li,
and Xin Chen. 2008. “A molecular modeling study of the interaction between SRP-Receptor
complex and peptide translocon.” Biochemical and Biophysical Research Communications 377
(2): 346–50. https://doi.org/10.1016/j.bbrc.2008.09.119.
Chu, Lily, and DK David K. Robinson. 2001. “Industrial choises for protein production by large scale
cell culture.” Current Opinion in Biotechnology 12 (2): 180–87. https://doi.org/10.1016/S0958-
1669(00)00197-X.
Chung, YS, and D Dubnau. 1998. “All seven ComG Open reading frames are required for DNA
binding during transformation of competent Bacillus subtilis.” Journal of Bacteriology 180 (1):
41–45. http://jb.asm.org/content/180/1/41.short.
Collinson, I. 2005. “The structure of the bacterial protein translocation complex SecYEG,” 1225–30.
Collinson, I, R A Corey, and W J Allen. 2015. “Channel Crossing: How are proteins shipped across
the bacterial plasma membrane?” Philos Trans R Soc Lond B Biol Sci 370.
https://doi.org/10.1098/rstb.2015.0025.
Commichau, Fabian M., Nico Pietack, and Jörg Stülke. 2013. “Essential genes in Bacillus subtilis: a
re-evaluation after ten years.” Molecular BioSystems 9 (6): 1068–75.
https://doi.org/10.1039/c3mb25595f.
Corey, Robin A, William J Allen, Joanna Komar, Simonas Masiulis, Sam Menzies, and Alice Robson.
2016. “Unlocking the bacterial SecY translocon article unlocking the bacterial secy translocon
structure.” Structure/Folding and Design 24 (4): 518–27.
https://doi.org/10.1016/j.str.2016.02.001.
Costa, Tiago R D, Catarina Felisberto-rodrigues, Amit Meir, Marie S Prevost, Adam Redzej, Martina
Trokter, and Gabriel Waksman. 2015. “Secretion systems in gram-negative insights.” Nature
Publishing Group 13 (6): 343–59. https://doi.org/10.1038/nrmicro3456.
Cotter, Paul D., R. Paul Ross, and Colin Hill. 2012. “Bacteriocins — a viable alternative to
antibiotics?” Nature Reviews Microbiology 11 (2): 95–105. https://doi.org/10.1038/nrmicro2937.
Courtney, Colleen R., Loralyn M. Cozy, and Daniel B. Kearns. 2012. “Molecular characterization of
the flagellar hook in Bacillus subtilis.” Journal of Bacteriology 194 (17): 4619–29.
https://doi.org/10.1128/JB.00444-12.
Dajkovic, Alex, Elizabeth Hinde, Calum MacKichan, and Rut Carballido-Lopez. 2016. “Dynamic
organization of SecA and SecY secretion complexes in the B. subtilis membrane.” PLoS ONE 11

Chapter 5. BIBLIOGRAPHY
157
(6): 1–17. https://doi.org/10.1371/journal.pone.0157899.
Dalbey, R E, P Wang, and J M van Dijl. 2012. “Membrane proteases in the bacterial protein secretion
and quality control pathway.” Microbiol Mol Biol Rev 76 (2): 311–30.
https://doi.org/10.1128/MMBR.05019-11.
Darmon, Elise, David Noone, Anne Masson, Sierd Bron, Oscar P. Kuipers, Kevin M. Devine, and Jan
Maarten Van Dijl. 2002. “A novel class of heat and secretion stress-responsive genes is
controlled by the autoregulated CssRS two-component system of Bacillus subtilis.” Journal of
Bacteriology 184 (20): 5661–71. https://doi.org/10.1128/JB.184.20.5661-5671.2002.
Dauter, Zbigniew, Miroslawa Dauter, A. Marek Brzozowski, Søren Christensen, Torben V. Borchert,
Lars Beier, Keith S. Wilson, and Gideon J. Davies. 1999. “X-Ray structure of Novamyl, the five-
domain ‘maltogenic’ α-Amylase from Bacillus Stearothermophilus: Maltose and Acarbose
complexes at 1.7 Å resolution.” Biochemistry 38 (26): 8385–92.
https://doi.org/10.1021/bi990256l.
Delumeau, Olivier, François Lecointe, Jan Muntel, Alain Guillot, Eric Guédon, Véronique Monnet,
Michael Hecker, Dörte Becher, Patrice Polard, and Philippe Noirot. 2011. “The dynamic protein
partnership of rna polymerase in Bacillus subtilis.” Proteomics 11 (15): 2992–3001.
https://doi.org/10.1002/pmic.201000790.
Deng, Wanyin, Natalie C. Marshall, Jennifer L. Rowland, James M. McCoy, Liam J. Worrall, Andrew
S. Santos, Natalie C.J. Strynadka, and B. Brett Finlay. 2017. “Assembly, structure, function and
regulation of Type III secretion systems.” Nature Reviews Microbiology 15 (6): 323–37.
https://doi.org/10.1038/nrmicro.2017.20.
Depamphilis, M L, and Julius Adler. 1971. “Fine structure and isolation of the hook-basal body
complex of flagella from Escherichia coli and Bacillus subtilis” 105: 384–95.
Dhar, N. R. 1923. “The Starch-Iodine reaction.” The Journal of Physical Chemistry 28 (2): 125–30.
https://doi.org/10.1021/j150236a002.
Diao, Liuyang, Qilei Dong, Zhaohui Xu, Sheng Yang, Jiahai Zhou, and Roland Freudl. 2012.
“Functional implementation of the posttranslational SecB-SecA protein-targeting pathway in
Bacillus subtilis.” Applied and Environmental Microbiology 78 (3): 651–59.
https://doi.org/10.1128/AEM.07209-11.
Diepold, A, and J P Armitage. 2015. “Type III Secretion Systems: The bacterial flagellum and the
injectisome.” Philos Trans R Soc Lond B Biol Sci 370 (1679).
https://doi.org/10.1098/rstb.2015.0020.
Dijl, Jan Maarten van, and Michael Hecker. 2013. “Bacillus subtilis: from soil bacterium to super-

Chapter 5. BIBLIOGRAPHY
158
secreting cell factory.” Microbial Cell Factories 12 (1): 3. https://doi.org/10.1186/1475-2859-12-
3.
Dobell, Clifford. 1932. “Antony van Leeuwenhoek and his ‘Little Animals,’” 520.
https://doi.org/10.1007/s13398-014-0173-7.2.
Dougan, David A, Axel Mogk, Kornelius Zeth, and Bernd Bukau. 2002. “AAA + proteins and
substrate recognition , it all depends on their partner in crime” 529: 6–10.
Driessen, A J, and N Nouwen. 2008. “Protein translocation across the bacterial cytoplasmic
membrane.” Annu Rev Biochem 77: 643–67.
https://doi.org/10.1146/annurev.biochem.77.061606.160747.
Duong, Franck, Jerry Eichler, Albert Price, Marilyn Rice Leonard, and William Wickner. 1997.
“Biogenesis of the gram-negative bacterial envelope.” Cell 91 (5): 567–73.
https://doi.org/10.1016/S0092-8674(00)80444-4.
Elsholz, Alexander K W, Marlene S Birk, and Emmanuelle Charpentier. 2017. “Functional diversity
of AAA + protease complexes in Bacillus subtilis” 4 (July): 1–15.
https://doi.org/10.3389/fmolb.2017.00044.
Erhardt, Marc, Keiichi Namba, and Kelly T. Hughes. 2010. “Bacterial Nanomachines: The Flagellum
and Type III Injectisome.” Cold Spring Harbor Perspectives in Biology 2 (11): 1–23.
https://doi.org/10.1101/cshperspect.a000299.
Evans, Lewis D B, Simon Poulter, Eugene M. Terentjev, Colin Hughes, and Gillian M. Fraser. 2013.
“A chain mechanism for flagellum growth.” Nature 504 (7479): 287–90.
https://doi.org/10.1038/nature12682.
Forster, Brian M., and Hélène Marquis. 2012. “Protein transport across the cell wall of monoderm
gram-positive bacteria.” Molecular Microbiology 84 (3): 405–13. https://doi.org/10.1111/j.1365-
2958.2012.08040.x.
Freymond, Pierre Philippe, Vladimir Lazarevic, Blazenka Soldo, and Dimitri Karamata. 2006.
“Poly(Glucosyl-N-Acetylgalactosamine 1-Phosphate), a wall teichoic acid of Bacillus subtilis
168: its biosynthetic pathway and mode of attachment to peptidoglycan.” Microbiology 152 (6):
1709–18. https://doi.org/10.1099/mic.0.28814-0.
Frobel, J, P Rose, F Lausberg, A S Blummel, R Freudl, and M Muller. 2012. “Transmembrane
insertion of twin-arginine signal peptides is driven by TatC and regulated by TatB.” Nat
Commun 3: 1311. https://doi.org/10.1038/ncomms2308.
Fröbel, Julia, Patrick Rose, Frank Lausberg, Anne-Sophie Blümmel, Roland Freudl, and Matthias

Chapter 5. BIBLIOGRAPHY
159
Müller. 2012. “Transmembrane insertion of twin-arginine signal peptides is driven by TatC and
regulated by TatB.” Nature Communications 3: 1311. https://doi.org/10.1038/ncomms2308.
Fukumura, Takuma, Fumiaki Makino, Tobias Dietsche, Miki Kinoshita, Takayuki Kato, Samuel
Wagner, Keiichi Namba, Katsumi Imada, and Tohru Minamino. 2017. “Assembly and
stoichiometry of the core structure of the bacterial flagellar Type III export gate complex.” PLoS
Biology 15 (8): e2002281. https://doi.org/10.1371/journal.pbio.2002281.
Gabel, Christopher V, and Howard C Berg. 2003. “The speed of the flagellar rotary motor of
Escherichia coli varies linearly with protonmotive force.” Proceedings of the National Academy
of Sciences of the United States of America 100: 8748–51.
https://doi.org/10.1073/pnas.1533395100.
Garg, Saurabh K., Sushma Kommineni, Luke Henslee, Ying Zhang, and Peter Zuber. 2009. “The
YjbH protein of Bacillus Subtilis enhances ClpXP-catalyzed proteolysis of Spx.” Journal of
Bacteriology 191 (4): 1268–77. https://doi.org/10.1128/JB.01289-08.
Gaskins, M. H., S. L. Albrecht, and D. H. Hubbell. 1985. “Rhizosphere bacteria and their use to
increase plant productivity: a review.” Agriculture, Ecosystems and Environment 12 (2): 99–116.
https://doi.org/10.1016/0167-8809(85)90071-4.
Gavanji, Shahin. 2013. “Application of recombinant DNA technology- A Review.” Applied Science
Reports 2 (2): 29–31.
Gersch, Malte, Kirsten Famulla, Maria Dahmen, Christoph Göbl, Imran Malik, Klaus Richter, Vadim
S. Korotkov, et al. 2015. “AAA+ chaperones and acyldepsipeptides activate the ClpP Protease
via conformational control.” Nature Communications 6. https://doi.org/10.1038/ncomms7320.
Gerth, Ulf, Elke Krüger, Isabelle Derré, Tarek Msadek, and Michael Hecker. 1998. “Stress induction
of the Bacillus subtilis ClpP gene encoding a homologue of the proteolytic component of the Clp
protease and the involvement of ClpP and ClpX in stress tolerance.” Molecular Microbiology 28
(4): 787–802. https://doi.org/10.1046/j.1365-2958.1998.00840.x.
Gibson, Daniel G., Lei Young, Ray Yuan Chuang, J. Craig Venter, Clyde A. Hutchison, and Hamilton
O. Smith. 2009. “Enzymatic assembly of DNA molecules up to several hundred kilobases.”
Nature Methods 6 (5): 343–45. https://doi.org/10.1038/nmeth.1318.
Glynn, Steven E., Andreas Martin, Andrew R. Nager, Tania A. Baker, and Robert T. Sauer. 2009.
“Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+
protein-unfolding machine.” Cell 139 (4): 744–56. https://doi.org/10.1016/j.cell.2009.09.034.
Goessweiner-Mohr, Nikolaus, Karsten Arends, Walter Keller, and Elisabeth Grohmann. 2013.
“Conjugative Type IV secretion systems in gram-positive bacteria.” Plasmid 70 (3): 289–302.

Chapter 5. BIBLIOGRAPHY
160
https://doi.org/10.1016/j.plasmid.2013.09.005.
Gohlke, Ulrich, Lee Pullan, Christopher A Mcdevitt, Ida Porcelli, Erik De Leeuw, Tracy Palmer,
Helen R Saibil, and Ben C Berks. 2005. “The TatA component of the twin-arginine protein
transport system forms channel complexes of variable diameter.”
Gonzalez-Pastor, J. E. 2003. “Cannibalism by sporulating bacteria.” Science 301 (5632): 510–13.
https://doi.org/10.1126/science.1086462.
González-Pastor, José Eduardo. 2011. “Cannibalism: a social behavior in sporulating Bacillus
subtilis.” FEMS Microbiology Reviews 35 (3): 415–24. https://doi.org/10.1111/j.1574-
6976.2010.00253.x.
Goosens, Vivianne J., Alba De-San-Eustaquio-Campillo, Rut Carballido-López, and Jan Maarten van
Dijl. 2015. “A Tat Ménage à Trois - the role of Bacillus subtilis TatAc in twin-arginine protein
translocation.” Biochimica et Biophysica Acta - Molecular Cell Research 1853 (10): 2745–53.
https://doi.org/10.1016/j.bbamcr.2015.07.022.
Goosens, Vivianne J., Carmine G. Monteferrante, and Jan Maarten Van Dijl. 2014. “The Tat system of
gram-positive bacteria.” Biochimica et Biophysica Acta - Molecular Cell Research 1843 (8):
1698–1706. https://doi.org/10.1016/j.bbamcr.2013.10.008.
Gottesman, S., W. P. Clark, V. De Crecy-Lagard, and M. R. Maurizi. 1993. “ClpX, an alternative
subunit for the ATP-Dependent Clp protease of Escherichia coli. sequence and in vivo
activities.” Journal of Biological Chemistry 268 (30): 22618–26.
Green, Erin R, and Joan Mecsas. 2015. “Bacterial secretion systems – an overview.” American Society
for Microbiology 4 (1): 1–32. https://doi.org/10.1128/microbiolspec.VMBF-0012-2015.Bacterial.
Grohmann, Elisabeth, Peter J Christie, Gabriel Waksman, and Steffen Backert. 2018. “Micro Review
Type IV secretion in gram-negative and gram-positive bacteria” 107 (January): 455–71.
https://doi.org/10.1111/mmi.13896.
Guiziou, Sarah, Vincent Sauveplane, Hung-ju Chang, Caroline Clerté, Nathalie Declerck, Matthieu
Jules, and Jerome Bonnet. 2016. “A part toolbox to tune genetic expression in Bacillus subtilis.”
Nucleic Acids Research 44 (15): gkw624. https://doi.org/10.1093/nar/gkw624.
Haney, Cara H., Buck S. Samuel, Jenifer Bush, and Frederick M. Ausubel. 2015. “Associations with
Rhizosphere bacteria can confer an adaptive advantage to plants.” Nature Plants 1 (6): 15051.
https://doi.org/10.1038/nplants.2015.51.
Hartl, F.-U., S. Lecker, E. Schiebel, J.P. Hendrick, and W. Wickner. 1990. “The binding cascade of
SecB to SecA to SecY/E mediates preprotin targeting to the E. coli plasma membrane.” Cell 63:

Chapter 5. BIBLIOGRAPHY
161
269–79.
Harwood, C R, and R Cranenburgh. 2008. “Bacillus protein secretion: an unfolding story.” Trends
Microbiol 16 (2): 73–79. https://doi.org/10.1016/j.tim.2007.12.001.
Hoffmann, Tamara, Barbara Troup, Alexandra Szabo, Christoph Hungerer, and Dieter Jahn. 1995.
“The anaerobic life of Bacillus subtilis : cloning of the genes encoding the respiratory nitrate
reductase system” 131 (95): 219–25.
Hou, Bo, Eyleen S Heidrich, Denise Mehner-Breitfeld, and Thomas Brüser. 2018. “The TatA
component of the twin-arginine translocation system locally weakens the cytoplasmic membrane
of Escherichia Coli upon protein substrate binding.” Journal of Biological Chemistry, no. 17:
jbc.RA118.002205. https://doi.org/10.1074/jbc.RA118.002205.
Huang, Xuejun, and J D Helmann. 1998. “Identification of target promoters for the Bacillus subtilis
SigmaX factor using a consensus-directed search.” J Mol Biol 279 (1): 165–73.
https://doi.org/10.1006/jmbi.1998.1765.
Hyyrylainen, H.-L., Hanne Leena Hyyryläinen, Marika Vitikainen, Joanne Thwaite, Hongyan Wu,
Matti Sarvas, Colin R. Harwood, Vesa P. Kontinen, and Keith Stephenson. 2000. “D-Alanine
substitution of teichoic acids as a modulator of protein folding and stability at the cytoplasmic
membrane/cell wall interface of Bacillus Subtilis.” Journal of Biological Chemistry 275 (35):
26696–703. https://doi.org/10.1074/jbc.M003804200.
Ichihara, S., T. Suzuki, M. Suzuki, and S. Mizushima. 1986. “Molecular cloning and sequencing of the
SppA gene and characterization of the encoded protease IV, a Signal Peptide Peptidase, of
Escherichia coli.” Journal of Biological Chemistry 261 (20): 9405–11.
Ito, Koreaki, and Yoshinori Akiyama. 2005. “Cellular functions , mechanism of action , and regulation
of FtsH protease.” https://doi.org/10.1146/annurev.micro.59.030804.121316.
Ivankov, Dmitry N, Samuel H Payne, Michael Y Galperin, Stefano Bonissone, Pavel A Pevzner, and
Dmitrij Frishman. 2013. “How many signal peptides are there in bacteria?” Environmental
Microbiology 15 (4): 983–90. https://doi.org/10.1111/1462-2920.12105.
Iyer, Lakshiminarayan M., Kira S. Makarova, Eugene V. Koonin, and L. Aravind. 2004.
“Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: implications for
the origins of chromosome segregation, cell division and viral capsid packaging.” Nucleic Acids
Research 32 (17): 5260–79. https://doi.org/10.1093/nar/gkh828.
Jeong, Sang Min, Hirofumi Yoshikawa, and Hideo Takahashi. 1993. “Isolation and characterization of
the SecE homologue gene of Bacillus subtilis.” Molecular Microbiology 10 (1): 133–42.
https://doi.org/10.1111/j.1365-2958.1993.tb00910.x.

Chapter 5. BIBLIOGRAPHY
162
Jongbloed, J D, R van der Ploeg, and J M van Dijl. 2006. “Bifunctional TatA subunits in minimal Tat
protein translocases.” Trends Microbiol 14 (1): 2–4. https://doi.org/10.1016/j.tim.2005.11.001.
Joshi, Shilpa A, Greg L Hersch, Tania A Baker, Robert T Sauer. 2004. “Communication between
ClpX and ClpP during substrate processing and degradation” 11 (5): 404–11.
https://doi.org/10.1038/nsmb752.
Kang, Zhen, Sen Yang, Guocheng Du, and Jian Chen. 2014. “Molecular engineering of secretory
machinery components for high-level secretion of proteins in Bacillus species.” Journal of
Industrial Microbiology and Biotechnology 41 (11): 1599–1607. https://doi.org/10.1007/s10295-
014-1506-4.
Ke, Wan Ju, Yi Huang Hsueh, Yu Chieh Cheng, Chih Ching Wu, and Shih Tung Liu. 2015. “Water
surface tension modulates the swarming mechanics of Bacillus subtilis.” Frontiers in
Microbiology 6 (SEP): 1–12. https://doi.org/10.3389/fmicb.2015.01017.
Kearns, Daniel B., Frances Chu, Steven S. Branda, Roberto Kolter, and Richard Losick. 2005. “A
master regulator for biofilm formation by Bacillus Subtilis.” Molecular Microbiology 55 (3):
739–49. https://doi.org/10.1111/j.1365-2958.2004.04440.x.
Kearns, Daniel B., and Richard Losick. 2003. “Swarming motility in undomesticated Bacillus
subtilis.” Molecular Microbiology 49 (3): 581–90. https://doi.org/10.1046/j.1365-
2958.2003.03584.x.
Kearns, Daniel B. 2010. “A field guide to bacterial swarming motility.” Nature Reviews. Microbiology
8 (9): 634–44. https://doi.org/10.1038/nrmicro2405.
Keller, Rebecca, Jeanine de Keyzer, Arnold J.M. Driessen, and Tracy Palmer. 2012. “Co-Operation
between different targeting pathways during integration of a membrane protein.” Journal of Cell
Biology 199 (2): 303–15. https://doi.org/10.1083/jcb.201204149.
Kempf, Georg, Klemens Wild, and Irmgard Sinning. 2014. “Structure of the complete bacterial SRP
Alu domain” 42 (19): 12284–94. https://doi.org/10.1093/nar/gku883.
Kernphysik, Institut, T U Darmstadt, and Karl Wien. 1999. “100 Years of Ion Beams : Willy Wien ’ s
Canal Rays Introduction : How Wien Came to Physics with Canal Rays Wien ’ s Experiments
with Cathode and Canal Rays in” 29 (3).
Keyzer, J. De, C. Van Der Does, and A. J M Driessen. 2003. “The Bacterial Translocase: A dynamic
protein channel complex.” Cellular and Molecular Life Sciences 60 (10): 2034–52.
https://doi.org/10.1007/s00018-003-3006-y.
Kihara, A, Y Akiyama, and K Ito. 1996. “A protease complex in the Escherichia coli plasma

Chapter 5. BIBLIOGRAPHY
163
membrane: HflKC (HflA) forms a complex with FtsH (HflB), regulating its proteolytic activity
against SecY.” The EMBO Journal 15 (22): 6122–31.
Kim, Apollos C., David C. Oliver, and Mark Paetzel. 2008. “Crystal structure of a bacterial Signal
Peptide Peptidase.” Journal of Molecular Biology 376 (2): 352–66.
https://doi.org/10.1016/j.jmb.2007.11.080.
Kingston, Anthony W, Xiaojie Liao, and John D Helmann. 2013b. “Contributions of the σ(W) , σ(M)
and σ(X) regulons to the lantibiotic resistome of Bacillus subtilis.” Molecular Microbiology 90
(3): 502–18. https://doi.org/10.1111/mmi.12380.
Kirstein, Janine, Noël Molière, David A. Dougan, and Kürşad Turgay. 2009. “Adapting the machine:
adaptor proteins for Hsp100/Clp and AAA+ Proteases.” Nature Reviews Microbiology 7 (8):
589–99. https://doi.org/10.1038/nrmicro2185.
Kirstein, Janine, Tilman Schlothauer, David A. Dougan, Hauke Lilie, Gilbert Tischendorf, Axel
Mogk, Bernd Bukau, and Kürşad Turgay. 2006. “Adaptor protein controlled oligomerization
activates the AAA + protein ClpC.” EMBO Journal 25 (7): 1481–91.
https://doi.org/10.1038/sj.emboj.7601042.
Kobayashi, K., S. D. Ehrlich, A. Albertini, G. Amati, K. K. Andersen, M. Arnaud, K. Asai, et al.
2003. “Essential Bacillus subtilis genes.” Proceedings of the National Academy of Sciences 100
(8): 4678–83. https://doi.org/10.1073/pnas.0730515100.
Koch, H-G, M Moser, and M Müller. 2003. “Signal Recognition Particle- dependent protein targeting,
universal to all kingdoms of life.” Reviews of Physiology, Biochemistry and Pharmacology 146:
55–94. https://doi.org/10.1007/s10254-002-0002-9.
Kontinen, V P, P Saris, and M Sarvas. 1991. “A gene (PrsA) of Bacillus subtilis involved in a novel,
late stage of protein export.” Molecular Microbiology 5 (5): 1273–83.
Kontinen, V P, and M Sarvas. 1993. “The PrsA lipoprotein is essential for protein secretion in Bacillus
subtilis and sets a limit for high-level secretion.” Molecular Microbiology 8 (4): 727–37.
https://doi.org/10.1111/j.1365-2958.1993.tb01616.x.
Koo, Byoung Mo, George Kritikos, Jeremiah D. Farelli, Horia Todor, Kenneth Tong, Harvey Kimsey,
Ilan Wapinski, et al. 2017. “Construction and analysis of two genome-scale deletion libraries for
Bacillus subtilis.” Cell Systems 4 (3): 291-305.e7. https://doi.org/10.1016/j.cels.2016.12.013.
Krishnappa, Laxmi, Carmine G. Monteferrante, Jolanda Neef, Annette Dreisbach, and Jan Maarten
Van Dijl. 2014. “Degradation of extracytoplasmic catalysts for protein folding in Bacillus
subtilis.” Applied and Environmental Microbiology 80 (4): 1463–68.
https://doi.org/10.1128/AEM.02799-13.

Chapter 5. BIBLIOGRAPHY
164
Kubori, Tomoko, Mitsumasa Okumura, Dai Nakamura, and Masahiro Iwakura. 1997. “Purification
and characterization of the flagellar hook – basal body complex of Bacillus subtilis” 24: 399–
410.
Kunst, F., N. Ogasawara, I. Moszer, A.M. Albertini, G. Alloni, V. Azevedo, M.G. Bertero, et al. 1997.
“The complete genome sequence of the gram-positive Bacterium Bacillus subtilis.” Nature 390
(6657): 249–56. https://doi.org/10.1038/36786.
Kurtz, Sherry, Karen P. McKinnon, Marschall S. Runge, Jenny P Y Ting, and Miriam Braunstein.
2006. “The SecA2 secretion factor of mycobacterium tuberculosis promotes growth in
macrophages and inhibits the host immune response.” Infection and Immunity 74 (12): 6855–64.
https://doi.org/10.1128/IAI.01022-06.
Laverde-gomez, Jenny A, Mayukh Sarkar, and Peter J Christie. 2012. “Regulation of bacterial Type
IV secretion.” Regulation of Bacterial Virulence, 335–62.
https://doi.org/10.1128/9781555818524.ch18.
Leake, M. C., N. P. Greene, R. M. Godun, T. Granjon, G. Buchanan, S. Chen, R. M. Berry, T. Palmer,
and B. C. Berks. 2008. “Variable stoichiometry of the TatA component of the twin-arginine
protein transport system observed by in vivo single-molecule imaging.” Proceedings of the
National Academy of Sciences 105 (40): 15376–81. https://doi.org/10.1073/pnas.0806338105.
Lee, Byung Gil, Min Kyung Kim, and Hyun Kyu Song. 2011. “Structural insights into the
conformational diversity of ClpP from Bacillus subtilis.” Molecules and Cells 32 (6): 589–95.
https://doi.org/10.1007/s10059-011-0197-1.
Lee, Catherine A., Jacob Thomas, and Alan D. Grossman. 2012. “The Bacillus subtilis conjugative
transposon ICEBs1 mobilizes plasmids lacking dedicated mobilization functions.” Journal of
Bacteriology 194 (12): 3165–72. https://doi.org/10.1128/JB.00301-12.
Lemon, K P, A M Earl, H C Vlamakis, C Aguilar, and R Kolter. 2008. “Biofilm development with an
emphasis on Bacillus subtilis.” Current Topics in Microbiology and Immunology 322 (8): 1–16.
https://doi.org/10.1021/nn300902w.Release.
Leonetti, Cori T., Matt A. Hamada, Stephanie J. Laurer, Matthew P. Broulidakis, Kyle J. Swerdlow,
Catherine A. Lee, Alan D. Grossman, and Melanie B. Berkmen. 2015. “Critical components of
the conjugation machinery of the integrative and conjugative element ICEBs1 of Bacillus
subtilis.” Journal of Bacteriology 197 (15): 2558–67. https://doi.org/10.1128/JB.00142-15.
Lo, Carlos, and Judith S Bond. 2009. “Proteases : multifunctional enzymes in life and disease” 283
(45): 30433–37. https://doi.org/10.1074/jbc.R800035200.
López, Daniel, and Roberto Kolter. 2010. “Extracellular signals that define distinct and coexisting cell

Chapter 5. BIBLIOGRAPHY
165
fates in Bacillus subtilis.” FEMS Microbiology Reviews 34 (2): 134–49.
https://doi.org/10.1111/j.1574-6976.2009.00199.x.
Lüke, Iris, Jennifer I. Handford, Tracy Palmer, and Frank Sargent. 2009. “Proteolytic processing of
Escherichia coli twin-arginine signal peptides by LepB.” Archives of Microbiology 191 (12):
919–25. https://doi.org/10.1007/s00203-009-0516-5.
Mäder, Ulrike, Georg Homuth, Christian Scharf, Rüdiger Bode, Michael Hecker, Ulrike Ma, and Knut
Bu. 2002. “Transcriptome and proteome analysis of Bacillus subtilis gene expression modulated
by amino acid availability transcriptome and proteome analysis of Bacillus subtilis Gene
expression modulated by amino acid availability.” Journal of Bacteriology 184 (15): 4288–95.
https://doi.org/10.1128/JB.184.15.4288.
Marchadier, Elodie, Rut Carballido-López, Sophie Brinster, Céline Fabret, Peggy Mervelet, Philippe
Bessières, Marie Françoise Noirot-Gros, Vincent Fromion, and Philippe Noirot. 2011. “An
expanded protein-protein interaction network in Bacillus subtilis reveals a group of hubs:
exploration by an integrative approach.” Proteomics 11 (15): 2981–91.
https://doi.org/10.1002/pmic.201000791.
Matias, V. R F, and Terry J. Beveridge. 2005. “Cryo-Electron microscopy reveals native polymeric
cell wall structure in bacillus subtilis 168 and the existence of a periplasmic space.” Molecular
Microbiology 56 (1): 240–51. https://doi.org/10.1111/j.1365-2958.2005.04535.x.
Matthews, Catherine B., Chapman Wright, Angel Kuo, Noelle Colant, Matthew Westoby, and J.
Christopher Love. 2017. “Reexamining opportunities for therapeutic protein production in
eukaryotic microorganisms.” Biotechnology and Bioengineering 114 (11): 2432–44.
https://doi.org/10.1002/bit.26378.
Ménétret, J. F., Ramanujan S. Hegde, Sven U. Heinrich, Preethi Chandramouli, Steven J. Ludtke, Tom
A. Rapoport, and Christopher W. Akey. 2005. “Architecture of the ribosome-channel complex
derived from native membranes.” Journal of Molecular Biology 348 (2): 445–57.
https://doi.org/10.1016/j.jmb.2005.02.053.
Michna, Raphael H., Fabian M. Commichau, Dominik Tödter, Christopher P. Zschiedrich, and jörg
stülke. 2014. “subtiwiki- a database for the model organism bacillus subtilis that links pathway,
interaction and expression information.” Nucleic Acids Research 42 (D1): 692–98.
https://doi.org/10.1093/nar/gkt1002.
Mielich-Süss, Benjamin, and Daniel Lopez. 2015. “Molecular mechanisms involved in Bacillus
subtilis biofilm formation.” Environmental Microbiology 17 (3): 555–65.
https://doi.org/10.1111/1462-2920.12527.

Chapter 5. BIBLIOGRAPHY
166
Milstein, C, G G Brownlee, T M Harrison, and M B Mathews. 1972. “A possible precursor of
immunoglobulin light chains.” Nature: New Biology 239 (91): 117–20.
http://www.ncbi.nlm.nih.gov/pubmed/4507519.
Minamino, Tohru, Katsumi Imada, and Keiichi Namba. 2008. “Molecular motors of the bacterial
flagella.” Current Opinion in Structural Biology 18 (6): 693–701.
https://doi.org/10.1016/j.sbi.2008.09.006.
Minamino, Tohru, Yusuke V. Morimoto, Noritaka Hara, Phillip D. Aldridge, and Keiichi Namba.
2016. “The bacterial flagellar Type III Export gate complex is a dual fuel engine that can use
both H+ and Na+ for flagellar protein export.” PLoS Pathogens 12 (3): 1–18.
https://doi.org/10.1371/journal.ppat.1005495.
Missiakas, Dominique, and Satish Raina. 1998. “MicroReview the extracytoplasmic function sigma
factors : role and regulation” 28: 1059–66.
Molière, Noël, and Kürşad Turgay. 2009. “Chaperone-Protease systems in regulation and protein
quality control in Bacillus subtilis.” Research in Microbiology 160 (9): 637–44.
https://doi.org/10.1016/j.resmic.2009.08.020.
Monteferrante, C G, C MacKichan, E Marchadier, M V Prejean, R Carballido-Lopez, and J M van
Dijl. 2013. “Mapping the twin-arginine protein translocation network of Bacillus subtilis.”
Proteomics 13 (5): 800–811. https://doi.org/10.1002/pmic.201200416.
Mukherjee, Sampriti, and Daniel B Kearns. 2014. “The structure and regulation of flagella in Bacillus
subtilis.” Annual Review of Genetics 48 (1): 319–40. https://doi.org/10.1146/annurev-genet-
120213-092406.
Mulder, Kelly C L, Joanna Bandola, and Wolfgang Schumann. 2013. “Construction of an artificial
SecYEG operon allowing high level secretion of α-amylase.” Protein Expression and
Purification 89 (1): 92–96. https://doi.org/https://doi.org/10.1038/newbio239117a0.
Müller, J P, J Ozegowski, S Vettermann, J Swaving, K H Van Wely, and A J Driessen. 2000.
“Interaction of Bacillus subtilis CsaA with SecA and precursor proteins.” The Biochemical
Journal 348 Pt 2 (2): 367–73. https://doi.org/10.1042/0264-6021:3480367.
Nakamura, Kouji, Shou-ichi Yahagi, Takao Yamazaki, and Kunio Yamane. 1999. “Bacillus subtilis
histone-like protein , HBsu , is an integral component of a SRP-like particle that can bind the Alu
domain of small cytoplasmic RNA” 274 (19): 13569–76.
Nakano, Michiko M., Yves P. Dailly, Peter Zuber, and David P. Clark. 1997. “Characterization of
anaerobic fermentative growth of Bacillus Subtilis: identification of fermentation end products
and genes required for growth.” Journal of Bacteriology 179 (21): 6749–55.

Chapter 5. BIBLIOGRAPHY
167
https://doi.org/10.1128/jb.179.21.6749-6755.1997.
Nakano, Michiko M, Farin Hajarizadeh, Yi Zhu, and Peter Zuber. 2001. “loss-of-function mutations in
YjbD Result in ClpX- and ClpP-independent competence development of Bacillus Subtilis” 42:
383–94.
Nam, Sung-eun, Apollos C. Kim, and Mark Paetzel. 2012. “Crystal structure of Bacillus subtilis signal
peptide peptidase A.” Journal of Molecular Biology 419 (5): 347–58.
https://doi.org/10.1016/j.jmb.2012.03.020.
Nam, Sung-eun, and Mark Paetzel. 2013. “Structure of Signal Peptide Peptidase A with C ‐ Termini
bound in the active sites: insights into specificity, self-processing, and regulation.”
Natale, P, T Bruser, and A J Driessen. 2008. “Sec- and Tat-Mediated protein secretion across the
bacterial cytoplasmic membrane distinct translocases and mechanisms.” Biochim Biophys Acta
1778 (9): 1735–56. https://doi.org/10.1016/j.bbamem.2007.07.015.
Neef, Jolanda, Cristina Bongiorni, Vivianne J. Goosens, Brian Schmidt, and Jan Maarten van Dijl.
2017. “Intramembrane protease RasP boosts protein production in Bacillus.” Microbial Cell
Factories 16 (1): 57. https://doi.org/10.1186/s12934-017-0673-1.
Neher, Saskia B, Niels Bradshaw, Stephen N Floor, John D Gross, and Peter Walter. 2008. “SRP RNA
controls a conformational switch regulating the SRP-SRP receptor interaction.” Nature
Structural & Molecular Biology 15 (9): 916–23. https://doi.org/10.1021/nn300902w.Release.
Nicolas, Pierre, U. Mader, Etienne Dervyn, Tatiana Rochat, Aurélie Leduc, Nathalie Pigeonneau,
Elena Bidnenko, et al. 2012. “Condition-dependent transcriptome reveals high-level regulatory
architecture in Bacillus subtilis.” Science 335 (6072): 1103–6.
https://doi.org/10.1126/science.1206848.
Olivares, Adrian O., Tania A. Baker, and Robert T. Sauer. 2015. “Mechanistic insights into bacterial
AAA+ proteases and protein-remodelling machines.” Nature Reviews Microbiology 14 (1): 33–
44. https://doi.org/10.1038/nrmicro.2015.4.
Omasits, Ulrich, Christian H. Ahrens, Sebastian Muller, and Bernd Wollscheid. 2014. “Protter:
interactive protein feature visualization and integration with experimental proteomic data.”
Bioinformatics 30 (6): 884–86. https://doi.org/10.1093/bioinformatics/btt607.
Osborne, Andrew R., Tom A. Rapoport, and Bert van den Berg. 2005. “Protein translocation by the
Sec61/Secy channel.” Annual Review of Cell and Developmental Biology 21 (1): 529–50.
https://doi.org/10.1146/annurev.cellbio.21.012704.133214.
Otto, Andreas, Jörg Bernhardt, Hanna Meyer, Marc Schaffer, Florian-A Herbst, Juliane Siebourg,

Chapter 5. BIBLIOGRAPHY
168
Ulrike Mäder, Michael Lalk, Michael Hecker, and Dörte Becher. 2010. “Systems-wide temporal
proteomic profiling in glucose-starved Bacillus subtilis.” Nature Communications 1: 137.
https://doi.org/10.1038/ncomms1137.
Outtrup, H. 1984. “Properties and Application,” 405–11.
Overkamp, Wout, Katrin Beilharz, Ruud Detert, Oude Weme, Ana Solopova, Harma Karsens, Jan
Kok, Oscar P Kuipers, and Jan-willem Veening. 2013. “Benchmarking various green fluorescent
protein variants in Bacillus subtilis , Streptococcus pneumoniae , and Lactococcus lactis for live
cell” 79 (20): 6481–90. https://doi.org/10.1128/AEM.02033-13.
Overmars, Lex, Robert Kerkhoven, Roland J. Siezen, and Christof Francke. 2013. “MGcV: The
microbial genomic context viewer for comparative genome analysis.” BMC Genomics 14 (1).
https://doi.org/10.1186/1471-2164-14-209.
Park, Jong-Tae, Antonius Suwanto, Irawan Tan, Tommy Nuryanto, Rudy Lukman, Kan Wang, and
Jay-lin Jane. 2013. “Molecular cloning and characterization of a thermostable α-amylase
exhibiting an unusually high activity.” Food Science and Biotechnology 23 (1): 125–32.
https://doi.org/10.1007/s10068-014-0017-4.
Partridge, Jonathan D., and Rasika M. Harshey. 2013. “Swarming: flexible roaming plans.” Journal of
Bacteriology 195 (5): 909–18. https://doi.org/10.1128/JB.02063-12.
Pasquina, Lincoln W, John P Santa Maria, and Suzanne Walker. 2013. “Teichoic acid biosynthesis as
an antibiotic target.” Current Opinion in Microbiology 16 (5): 531–37.
https://doi.org/10.1016/j.mib.2013.06.014.
Patel, Roshani, Sarah M. Smith, and Colin Robinson. 2014. “Protein transport by the bacterial Tat
pathway.” Biochimica et Biophysica Acta - Molecular Cell Research 1843 (8): 1620–28.
https://doi.org/10.1016/j.bbamcr.2014.02.013.
Patrick, Joyce E., and Daniel B. Kearns. 2008. “MinJ (YvjD) is a topological determinant of cell
division in Bacillus subtilis.” Molecular Microbiology 70 (5): 1166–79.
https://doi.org/10.1111/j.1365-2958.2008.06469.x.
Pédelacq, Jean Denis, Stéphanie Cabantous, Timothy Tran, Thomas C. Terwilliger, and Geoffrey S.
Waldo. 2006. “Engineering and characterization of a superfolder green fluorescent protein.”
Nature Biotechnology 24 (1): 79–88. https://doi.org/10.1038/nbt1172.
Pohl, Susanne, and Colin R. Harwood. 2010a. “Heterologous Protein Secretion by Bacillus Species:
from the cradle to the grave.” Advances in Applied Microbiology 73: 1–25.
https://doi.org/10.1016/S0065-2164(10)73001-X.

Chapter 5. BIBLIOGRAPHY
169
Pohlschröder, M, W a Prinz, E Hartmann, and J Beckwith. 1997. “Protein translocation in the three
domains of life: variations on a theme.” Cell 91: 563–66. https://doi.org/10.1016/S0092-
8674(00)80443-2.
Popham, David L., and Peter Setlow. 1996. “Phenotypes of Bacillus subtilis mutants lacking multiple
class a high-molecular-weight penicillin-binding proteins.” Journal of Bacteriology 178 (7):
2079–85. https://doi.org/10.1128/jb.178.7.2079-2085.1996.
Prabudiansyah, Irfan, and Arnold J M Driessen. 2016. “The Canonical and accessory sec system of
gram-positive bacteria.” In , 45–67. https://doi.org/10.1007/82_2016_9.
Ramadan, Hassan A I, Kulvinder S Saini, Nabih A Baeshen, and Elrashdy M Redwan. 2015.
“Production of Biopharmaceuticals in E. Coli : Current Scenario and Future Perspectives” 25 (7):
953–62.
Rapoport, Tom A, Long Li, and Eunyong Park. 2017. “Structural and mechanistic insights into protein
translocation.”
Real, Gonçalo, Sérgio M Pinto, Ghislain Schyns, Adriano O Henriques, Charles P Moran Jr, M Pinto,
Teresa Costa, and Charles P Moran. 2005. “A gene encoding a holin-like protein involved in
spore morphogenesis and spore germination in Bacillus subtilis” 187 (18): 6443–53.
https://doi.org/10.1128/JB.187.18.6443.
Rigaut, Guillaume, Anna Shevchenko, Berthold Rutz, Matthias Wilm, Matthias Mann, and Bertrand
Séraphin. 1999. “A generic protein purification method for protein complex characterization and
proteome exploration.” Nature Biotechnology 17 (10): 1030–32. https://doi.org/10.1038/13732.
Robson, Alice, Vicki a M Gold, Skye Hodson, Anthony R Clarke, and Ian Collinson. 2009. “Energy
transduction in protein transport and the ATP hydrolytic cycle of SecA.” Proceedings of the
National Academy of Sciences of the United States of America 106 (13): 5111–16.
https://doi.org/10.1073/pnas.0809592106.
Rodnina, Marina V., and Wolfgang Wintermeyer. 2016. “Protein elongation, co-translational folding
and targeting.” Journal of Molecular Biology. https://doi.org/10.1016/j.jmb.2016.03.022.
Rodriguez, F., S. L. Rouse, C. E. Tait, J. Harmer, A. De Riso, C. R. Timmel, M. S. P. Sansom, B. C.
Berks, and J. R. Schnell. 2013. “Structural model for the protein-translocating element of the
twin-arginine transport system.” Proceedings of the National Academy of Sciences 110 (12):
E1092–1101. https://doi.org/10.1073/pnas.1219486110.
Rose, Patrick, Julia Fröbel, Peter L. Graumann, and Matthias Müller. 2013. “Substrate-dependent
assembly of the tat translocase as observed in live Escherichia coli cells.” PLoS ONE 8 (8): 1–
17. https://doi.org/10.1371/journal.pone.0069488.

Chapter 5. BIBLIOGRAPHY
170
Saier, Milton H., and Bhaskara L. Reddy. 2015. “Holins in bacteria, eukaryotes, and archaea:
multifunctional xenologues with potential biotechnological and biomedical applications.”
Journal of Bacteriology 197 (1): 7–17. https://doi.org/10.1128/JB.02046-14.
Saito, Akira, Yohei Hizukuri, Ei-ichi Matsuo, Shinobu Chiba, Hiroyuki Mori, Osamu Nishimura, and
Koreaki Ito. 2011. “Post-Liberation cleavage of signal peptides is catalyzed by the site-2 protease
(S2P) in Bacteria.” https://doi.org/10.1073/pnas.1108376108/-
/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1108376108.
Salton, M R J. 1953. “Studies of the bacterial cell wall: IV. The composition of the cell walls of some
gram-positive and gram-negative bacteria.” Biochimica et Biophysica Acta 10 (21522): 512–23.
Saraogi, Ishu, and Shu ou Shan. 2014. “Co-translational protein targeting to the bacterial membrane.”
Biochimica et Biophysica Acta - Molecular Cell Research 1843 (8): 1433–41.
https://doi.org/10.1016/j.bbamcr.2013.10.013.
Sargent, Frank. 2007. “The Twin-Arginine transport system : moving folded proteins across
membranes.” Biochemical Society Transactions 35 (5): 835–47.
https://doi.org/10.1042/BST0350835.
Sarvas, Matti, Colin R. Harwood, Sierd Bron, and Jan Maarten Van Dijl. 2004. “Post-translocational
folding of secretory proteins in gram-positive bacteria.” Biochimica et Biophysica Acta -
Molecular Cell Research 1694 (1-3 SPEC.ISS.): 311–27.
https://doi.org/10.1016/j.bbamcr.2004.04.009.
Scheffers, D.-J., and M. G. Pinho. 2005. “Bacterial cell wall synthesis: new insights from localization
studies.” Microbiology and Molecular Biology Reviews 69 (4): 585–607.
https://doi.org/10.1128/MMBR.69.4.585-607.2005.
Schneewind, Olaf, and Dominique Missiakas. 2014. “Sec-secretion and sortase-mediated anchoring of
proteins in gram-positive bacteria.” Biochimica et Biophysica Acta - Molecular Cell Research
1843 (8): 1687–97. https://doi.org/10.1016/j.bbamcr.2013.11.009.
Schneewind, Olaf, and Dominique M Missiakas. 2012. “Protein secretion and surface display in gram-
positive bacteria,” 1123–39. https://doi.org/10.1098/rstb.2011.0210.
Schulze, Ryan J, Joanna Komar, Mathieu Botte, William J Allen, Sarah Whitehouse, Vicki A M Gold,
Jelger A Lycklama A Nijeholt, et al. 2014. “Membrane protein insertion and proton-motive-
force-dependent secretion through the bacterial holo-translocon SecYEG-SecDF-YajC-YidC.”
Proceedings of the National Academy of Sciences of the United States of America 111 (13):
4844–49. https://doi.org/10.1073/pnas.1315901111.
Settles, A M, and R Martienssen. 1998. “Old and New pathways of protein export in chloroplasts and

Chapter 5. BIBLIOGRAPHY
171
bacteria.” Trends Cell Biol 8 (12): 494–501.
Shao, Li, Heba Abdel-Motaal, Jin Chen, Huiwen Chen, Tong Xu, Lin Meng, Zhenglai Zhang, Fankui
Meng, and Juquan Jiang. 2018. “Characterization of a functionally unknown arginine-aspartate-
aspartate family protein from halobacillus andaensis and functional analysis of its conserved
arginine/aspartate residues.” Frontiers in Microbiology 9 (APR): 1–17.
https://doi.org/10.3389/fmicb.2018.00807.
Sigaud, L., V. L. B. de Jesus, Natalia Ferreira, and E. C. Montenegro. 2016. “A Novel double-focusing
time-of-flight mass spectrometer for absolute recoil ion cross sections measurements.” Review of
Scientific Instruments 87 (8): 083112. https://doi.org/10.1063/1.4961664.
Singh, Harinder, and Gajendra P.S. Raghava. 2016. “BLAST-based structural annotation of protein
residues using protein data bank.” Biology Direct 11 (1): 1–13. https://doi.org/10.1186/s13062-
016-0106-9.
Sørensen, Hans Peter, and Kim Kusk Mortensen. 2005. “Soluble expression of recombinant proteins
in the cytoplasm of Escherichia coli.” Microbial Cell Factories 4 (Figure 1): 1–8.
https://doi.org/10.1186/1475-2859-4-1.
Spieß, Tobias, Sophie Marianne Korn, Peter Kötter, and Karl Dieter Entian. 2015. “Autoinduction
specificities of the lantibiotics subtilin and nisin.” Applied and Environmental Microbiology 81
(22): 7914–23. https://doi.org/10.1128/AEM.02392-15.
Spizizen, John. 1958. “Transformation of biochemically deficient strains of Bacillus subtilis by
deoxyribonucleate.” Proceedings of the National Academy of Sciences 44 (10): 1072–78.
https://doi.org/10.1073/pnas.44.10.1072.
Strom, M. S., P. Bergman, and S. Lory. 1993. “Identification of active-site cysteines in the conserved
domain of PilD, the bifunctional Type IV Pilin leader peptidase/n-methyltransferase of
Pseudomonas aeruginosa.” Journal of Biological Chemistry 268 (21): 15788–94.
Sullivan, Jonathan Tabb, Ellen F. Young, Jessica R. Mccann, and Miriam Braunstein. 2012. “The
Mycobacterium Tuberculosis SecA2 system subverts phagosome maturation to promote growth
in macrophages.” Infection and Immunity 80 (3): 996–1006. https://doi.org/10.1128/IAI.05987-
11.
Suzuki, T., A. Itoh, S. Ichihara, and S. Mizushima. 1987. “Characterization of the SppA gene coding
for Protease IV, a Signal Peptide Peptidase of Escherichia coli.” Journal of Bacteriology 169 (6):
2523–28.
Svensson, H. 1961. “Isoelectric fractionation, analysis, and characterization of ampholytes in natural
ph gradients. I. The differential equation of solute concentrations at a steady state and its solution

Chapter 5. BIBLIOGRAPHY
172
for simple cases.” Acta Chemica Scandinavica.
Tan, Irene S., Cordelia A. Weiss, David L. Popham, and Kumaran S. Ramamurthi. 2015. “A quality-
control mechanism removes unfit cells from a population of sporulating bacteria.”
Developmental Cell 34 (6): 682–93. https://doi.org/10.1016/j.devcel.2015.08.009.
Tanaka, Yoshiki, Yasunori Sugano, Mizuki Takemoto, Takaharu Mori, Arata Furukawa, Tsukasa
Kusakizako, Kaoru Kumazaki, et al. 2015. “Crystal structures of SecYEG in lipidic cubic phase
elucidate a precise resting and a peptide-bound state.” Cell Reports 13 (8): 1561–68.
https://doi.org/10.1016/j.celrep.2015.10.025.
Thomas, J. Silhavy, Kahne Daniel, and Walker Suzanne. 2010. “The bacterial cell envelope.” Cold
Spring Harbor Perspectives in Biology 2 (5): 1–16. https://doi.org/10.1101/cshperspect.a000414.
Tjalsma, H, H Antelmann, J D Jongbloed, P G Braun, E Darmon, R Dorenbos, J Y Dubois, et al.
2004. “Proteomics of protein secretion by Bacillus subtilis: separating the ‘secrets’ of the
secretome.” Microbiol Mol Biol Rev 68 (2): 207–33. https://doi.org/10.1128/MMBR.68.2.207-
233.2004.
Tjalsma, H, A Bolhuis, J D Jongbloed, S Bron, and J M van Dijl. 2000. “Signal Peptide-dependent
protein transport in Bacillus subtilis: a genome-based survey of the secretome.” Microbiology
and Molecular Biology Reviews : MMBR 64 (3): 515–47.
https://doi.org/10.1128/MMBR.64.3.515-547.2000.
Tjalsma, Harold, Albert Bolhuis, Jan D H Jongbloed, Sierd Bron, and J M van Dijl. 2000. “Signal
peptide-dependent protein transport in Bacillus subtilis: A Genome-Based Survey of the
Secretome.” Microbiol Mol Biol Rev 64 (3): 515–47. https://doi.org/PMC99003.
Tjalsma, Harold, Juliëtte Van Den Dolder, Wilfried J J Meijer, Gerard Venema, Sierd Bron, and Jan
Maarten Van Dijl. 1999. “The plasmid-encoded Signal Peptidase SipP can functionally replace
the major signal peptidases SipS and SipT of Bacillus subtilis.” Journal of Bacteriology 181 (8):
2448–54.
Tjalsma, Harold, Michiel a Noback, Gerard Venema, Kunio Yamane, Sierd Bron, and Jan Maarten
Van Dijl. 1997. “Bacillus subtilis contains four closely related type i signal peptidases with
overlapping substrate specificities” 272 (41): 25983–92.
https://doi.org/10.1074/jbc.272.41.25983.
Tjalsma, Harold, Geeske Zanen, Gerard Venema, Sierd Bron, and Jan Maarten Van Dijl. 1999. “The
potential active site of the lipoprotein-specific (Type II) signal peptidase of Bacillus subtilis.”
Journal of Biological Chemistry 274 (40): 28191–97. https://doi.org/10.1074/jbc.274.40.28191.
Tu, Yuan Kun, You Yao Hong, and Yung Chuan Chen. 2009. “Finite element modeling of kirschner

Chapter 5. BIBLIOGRAPHY
173
pin and bone thermal contact during drilling.” Life Science Journal 6 (4): 23–27.
https://doi.org/10.1007/82.
Villaverde, Antonio, and M. Mar Carrió. 2003. “Protein aggregation in recombinant bacteria:
biological role of inclusion bodies.” Biotechnology Letters 25 (17): 1385–95.
https://doi.org/10.1023/A:1025024104862.
Vitikainen, M., H. L. Hyyryläinen, A. Kivimäki, V. P. Kontinen, and Matti Sarvas. 2005. “Secretion
of heterologous proteins in Bacillus subtilis can be improved by engineering cell components
affecting post-translocational protein folding and degradation.” Journal of Applied Microbiology
99 (2): 363–75. https://doi.org/10.1111/j.1365-2672.2005.02572.x.
Vitikainen, M., T. Pummi, U. Airaksinen, E. Wahlström, H. Wu, M. Sarvas, and V. P. Kontinen. 2001.
“Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and
secretion of alpha-amylase in Bacillus subtilis.” Journal of Bacteriology 183 (6): 1881–90.
https://doi.org/10.1128/JB.183.6.1881-1890.2001.
Vlamakis, Hera, Yunrong Chai, Pascale Beauregard, Richard Losick, and Roberto Kolter. 2013.
“Sticking Together: building a biofilm the Bacillus subtilis way.” Nature Reviews Microbiology
11 (3): 157–68. https://doi.org/10.1038/nrmicro2960.
Vollmer, Waldemar, Didier Blanot, and Miguel A. De Pedro. 2008. “Peptidoglycan structure and
architecture.” FEMS Microbiology Reviews. https://doi.org/10.1111/j.1574-6976.2007.00094.x.
Walsh, Nathan P, Benjamin M Alba, Baundauna Bose, Carol A Gross, and Robert T Sauer. 2003.
“OMP peptide signals initiate the envelope-stress response by activating degs protease via relief
of inhibition mediated by its PDZ domain.” Cell 113 (1): 61–71. https://doi.org/10.1016/S0092-
8674(03)00203-4.
Wang, Fengbin, Andrew M Burrage, Sandra Postel, Reece E Clark, Albina Orlova, Eric J Sundberg,
Daniel B Kearns, and Edward H Egelman. 2017. “A structural model of flagellar filament
switching across multiple bacterial species.” Nature Communications 8 (1): 960.
https://doi.org/10.1038/s41467-017-01075-5.
Wang, Ing-Nang, David L. Smith, and Ry Young. 2000. “Holins: the protein clocks of bacteriophage
infections.” Annual Review of Microbiology 54 (1): 799–825.
https://doi.org/10.1146/annurev.micro.54.1.799.
Wang, Peng, Eunjung Shim, Benjamin Cravatt, Richard Jacobsen, Joe Schoeniger, Apollos C. Kim,
Mark Paetzel, and Ross E. Dalbey. 2008. “Escherichia coli Signal Peptide Peptidase A is a
serine-lysine protease with a lysine recruited to the nonconserved amino-terminal domain in the
S49 protease family.” Biochemistry 47 (24): 6361–69. https://doi.org/10.1021/bi800657p.

Chapter 5. BIBLIOGRAPHY
174
Weidenmaier, C, and A Peschel. 2008. “Teichoic Acids and related cell-wall glycopolymers in gram-
positive physiology and host interactions.” Nature Reviews Microbiology 6 (1740–1534): 276–
87. https://doi.org/10.1038/nrmicro1861.
Weidenmaier, Christopher, and Andreas Peschel. 2008. “Teichoic acids and related cell-wall
glycopolymers in gram-positive physiology and host interactions.” Nature Reviews Microbiology
6 (4): 276–87. https://doi.org/10.1038/nrmicro1861.
Weiner, Joel H., Peter T. Bilous, Gillian M. Shaw, Shannon P. Lubitz, Laura Frost, Gavin H. Thomas,
Jeff A. Cole, and Raymond J. Turner. 1998. “A novel and ubiquitous system for membrane
targeting and secretion of cofactor-containing proteins.” Cell 93 (1): 93–101.
https://doi.org/10.1016/S0092-8674(00)81149-6.
Wely, K H van, J Swaving, R Freudl, and a J Driessen. 2001. “Translocation of proteins across the cell
envelope of gram-positive bacteria.” FEMS Microbiology Reviews 25 (4): 437–54.
https://doi.org/10.1111/j.1574-6976.2001.tb00586.x.
Westers, Lidia, Helga Westers, and Wim J. Quax. 2004. “Bacillus subtilis as cell factory for
pharmaceutical proteins: a biotechnological approach to optimize the host organism.” Biochimica
et Biophysica Acta - Molecular Cell Research 1694 (1-3 SPEC.ISS.): 299–310.
https://doi.org/10.1016/j.bbamcr.2004.02.011.
Wittig, Ilka, Hans-Peter Braun, and Hermann Schägger. 2006. “Blue Native PAGE.” Nature Protocols
1 (1): 418–28. https://doi.org/10.1038/nprot.2006.62.
Wojnowska, Marta, Joseph Gault, Shee Chien Yong, Carol V. Robinson, and Ben C. Berks. 2018.
“Precursor-receptor interactions in the twin arginine protein transport pathway probed with a new
receptor complex preparation.” Biochemistry 57 (10): 1663–71.
https://doi.org/10.1021/acs.biochem.8b00026.
Wolf, Matthias, Tobias Müller, Thomas Dandekar, and J. Dennis Pollack. 2004. “Phylogeny of
firmicutes with special reference to mycoplasma (mollicutes) as inferred from phosphoglycerate
kinase amino acid sequence data.” International Journal of Systematic and Evolutionary
Microbiology 54 (3): 871–75. https://doi.org/10.1099/ijs.0.02868-0.
Wu, Ling Juan, and Jeff Errington. 2004. “Coordination of cell division and chromosome segregation
by a nucleoid occlusion protein in bacillus subtilis.” Cell 117 (7): 915–25.
https://doi.org/10.1016/j.cell.2004.06.002.
Wu, S C, R Ye, X C Wu, S C Ng, and S L Wong. 1998. “Enhanced secretory production of a single-
chain antibody fragment from Bacillus Subtilis by coproduction of molecular chaperones.”
Journal of Bacteriology 180 (11): 2830–35.

Chapter 5. BIBLIOGRAPHY
175
Yamazaki, T, S Yahagi, K Nakamura, and K Yamane. 1999. “Depletion of Bacillus subtilis Histone-
like protein, HBsu, causes defective protein translocation and induces upregulation of small
cytoplasmic rna.” Biochemical and Biophysical Research Communications 258 (1): 211–14.
https://doi.org/S0006291X99906150 [pii].
Young, Ry. 2002. “Bacteriophage Holins: deadly diversity.” Journal of Molecular Microbiology and
Biotechnology 4 (1): 21–36.
Yu, Allen C.S., Jacky F.C. Loo, Samuel Yu, S. K. Kong, and Ting Fung Chan. 2014. “Monitoring
bacterial growth using tunable resistive pulse sensing with a pore-based technique.” Applied
Microbiology and Biotechnology 98 (2): 855–62. https://doi.org/10.1007/s00253-013-5377-9.
Zanen, Geeske, Edith N G Houben, Rob Meima, Harold Tjalsma, Jan D H Jongbloed, Helga Westers,
Bauke Oudega, Joen Luirink, Jan Maarten Van Dijl, and Wim J. Quax. 2005. “Signal peptide
hydrophobicity is critical for early stages in protein export by Bacillus subtilis.” FEBS Journal
272 (18): 4617–30. https://doi.org/10.1111/j.1742-4658.2005.04777.x.
Zeghouf, Mahel, Joyce Li, Gareth Butland, Anna Borkowska, Veronica Canadien, Dawn Richards,
Bryan Beattie, Andrew Emili, and Jack F. Greenblatt. 2004. “Sequential Peptide Affinity (SPA)
system for the identification of mammalian and bacterial protein complexes.” Journal of
Proteome Research 3 (3): 463–68. https://doi.org/10.1021/pr034084x.
Zheng, Zeliang, Amy Blum, Tithi Banerjee, Qianyu Wang, Virginia Dantis, and Donald Oliver. 2016.
“determination of the oligomeric state of SecYEG protein secretion channel complex using in
vivo photo- and disulfide cross-linking.” Journal of Biological Chemistry 291 (11): 5997–6010.
https://doi.org/10.1074/jbc.M115.694844.
Zhu, Bingyao, and Jörg Stülke. 2018. “SubtiWiki in 2018: from genes and proteins to functional
network annotation of the model organism Bacillus subtilis.” Nucleic Acids Research 46 (D1):
D743–48. https://doi.org/10.1093/nar/gkx908.
Zweers, Jessica C, Pierre Nicolas, Thomas Wiegert, Jan Maarten Van Dijl, and Emma L Denham.
2012. “Definition of the W regulon of Bacillus subtilis in the absence of stress” 7 (11).
https://doi.org/10.1371/journal.pone.0048471.

176

177
APPENDIX

178

Chapter 6. APPENDIX
179
6 APPENDIX
6.1 Contents, sterilization and storage of required
solutions
Luria Bertani (LB) medium- For 1L of media join 10 g Bacto-Tryptone (Becton Dickinson
and Co); 10 g NaCl (Merck); 5 g Yeast extract (Becton Dickinson and Co) and nanopure H2O up to
1L. Medium can be autoclaved at 121°C for 20 minutes and stored at RT.
Luria Bertani (LB) agar medium- For 1L of media join 10 g Bacto-Tryptone (Becton
Dickinson and Co); 10 g NaCl (Merck); 5 g Yeast extract (Becton Dickinson and Co); 15 g Agar
(Invitogen) and nanopure H2O up to 1L. Medium can be autoclaved at 121°C for 20 minutes and
stored at RT.
M9 minimal medium- For 1L of media add the following solutions (that were previously
sterilized separately): 1 ml 100 mM CaCl2; 10 ml trace saltsA
100x stock; 1 ml 1 M MgSO4; 1 ml 50
mM FeCl3/100 mM C6H8O7; 6 ml of glucose 50% (w/v) stock solution and 200 ml M9B
5x stock.
Trace salts 100x stock : 100 mg MnCl2.4H2O (Merck), 170 mg ZnCl2 (Sigma); 43 mg
CuCl2.2H2O (Merck); 60 mg CoCl2.6H2O (Merck); 60 mg Na2MoO4.2H2O (Aldrich), per liter.
Contrary to what is described by Harwood and Cutting (1) MgSO4, CaCl2 are added as separate
solutions to prevent precipitation problems. FeCl3 is also added as a separate solution as this
compound is unstable. Filter-sterilize the solution and store in the dark at RT.
M9 5x stock: 42.5 g Na2HPO4.2H2O(Merck); 15 g KH2PO4 (Merck); 5.0 g NH4Cl (Merck);
2.5 g NaCl (Merck). Adjust to pH 7.0 using 4M NaOH. Solution is autoclavable at 121°C for 20
minutes and stored at RT.
Belitsky-Minimal Medium- For 1L of media join 2 g (NH4)2SO4; 2 g MgSO4 * 7H2O; 2g
KCl, 2g Na3-Citrate * H2O and 50 ml 1M Tris-HCl pH 7.5. Add nanopure H2O up to 1L and adjust pH
to 7.5. Then this media needs to be supplemented with 5 ml 0.2M KH2PO4; 9 ml 0,5 M Glutamic
Acid; 10 ml 20% Glucose , 2 ml 1M CaCl2; 2 ml 0,0005M FeSO4 and 0,4 ml 0,025M MnSO4.
Completed Competence Medium (MC completed)- For 100ml of solution: 14g
K2HPO4*3H2O, 5.2g KH2PO4, 20g glucose, 8.8g C6H5Na3O7, 2.2g C6H5O7FeNH4, 1g casein
hydrolysate, 2g C5H8KNO4*H2O and 3.35mM MgSO4.
MSgg medium- it is composed of 5mM K3PO4 (pH7),100mM MOPS (pH7), 2mM MgCl2,
700μM CaCl2, 50μM MnCl2, 5μM FeCl3, 1μM ZnCl2, 2μM thiamine, 0.5% glycerol, 0.5% glutamate,
50 μg ml–1 tryptophan and 50 μg ml–1 phenylalanine.

Chapter 6. APPENDIX
180
Subtilin preparation- The subtilin is produced by the B.subtilis ATCC6633 strain, which was
supplied by Jan Maarten van Dijl group. To extract it, this strain was grown overnight in LB medium
at 37°C. The supernatant was collected by centrifugation at 1400 rpm for 10 min, the pellet was
discarded. Aliquots of 25 ml of supernatant were heated at 80°C for 10 min and filtered. These
samples were freeze with liquid nitrogen and then freeze dry overnight. Initial experiments were done
with 30% m/v subtilin. However, to avoid the effect of volume in the 96 well plate culture we
concentrated 10X times the stock solution. The sample powder was weighed and concentrated 10
times by resuspensing in water, by other words the stock powder corresponding to 25 ml of liquid
sample was ressuspended into 2.5 ml. This 10X stock solution was then used to induce the strains at
30% v/v or other concentrations when needed for testing. The powder sample was kept at -20°C.
Table S 1. Stock and working solutions of growth supplements
Supplement Solute Stock concentration Usual working
concentration
IPTG dH2O 1 M 0.5 mM
Xylose dH2O 20% 1%
Table S 2. Stock and working solutions of antibiotics.
Antibiotic Solute Stock concentration
(mg.ml-1)
Working
concentration
(µg.ml-1) E.coli
Working
concentration
(µg.ml-1) B.subtilis
Ampicillin (Amp) dH2O 50 100 -
Spectinomycin
(Spec) dH2O 50 - 100
Neomycin (Neo) dH2O 50 - 10
Erythromycin
(Erm) 100% ethanol (v/v) 6 - 0.6 or 30
Chloramphenicol
(Cm) 50% ethanol (v/v) 10 - 5
Kanamycin (Kan) dH2O 50 30 -
Chapter 6. APPENDIX
181
Table S 3 . List oligonucleotides
Construction Primer name Sequence (5’-> 3’)
C-terminal SPA tag
fusion
SecDF_P1 GGGTTCCTGGCGCGAGCTTGCTATAGCTTCTATT
GG
SecDF_P2 TTGGGCTGGCGCGAGC
TTGCGCCGAATCTTTTTTCAG
SecDF_seq ACGGATCTGATCCAAATGTCAGC
SecG_P1 GGGTTCCTGGCGCGAGCCACGCAGTTTTGATTA
CC
SecG_P2 TTGGGCTGGCGCGAGC
TAGGATATAAGCAAGCGC
SecG_seq TCTGATACAGCGTGACATGC
BdbB_P1 GGGTTCCTGGCGCGAGC
TGGCACAATGGCTAGTTTATTC
BdbB_P2 TTGGGCTGGCGCGAGC
ATTATATACATGTTGATTTTG
BdbB_seq TCTTGCCAACTCACTCCAC
BdbC_P1 GGGTTCCTGGCGCGAGCTATGCTTCCTGGGTTGT
G
BdbC_P2 TTGGGCTGGCGCGAGCTTCAGATTTTTCGCCTTTCA
GC
BdbC_seq TGCGGATTATGATGAAATC
SppA_P1 GGGTTCCTGGCGCGAGCCCATAAGGACATTATG
TCTC
SppA_P2 TTGGGCTGGCGCGAGCCTTCGCATAGAGATACA
TC
SppA_seq TCGGCGTCATTATGGAAAGC
YteJ_P1 GGGTTCCTGGCGCGAGC
Chapter 6. APPENDIX
182
TTCCTGCTTGATTGGCTC
YteJ_P2 TTGGGCTGGCGCGAGC
TTTGCGGTATAATTTTTCATGC
YteJ_seq AGAGTTAGAGCGCAATGACATC
SipS_P1 GGGTTCCTGGCGCGAGC
TGACTCTATGTATCCTACAC
SipS_P2 TTGGGCTGGCGCGAGC
ATTTGTTTTGCGCATTTCG
SipS_seq AGCAATTGTGATTGCTGTCG
SipT_P1 GGGTTCCTGGCGCGAGC
CACATTACATGACGGAGAAAGG
SipT_P2 TTGGGCTGGCGCGAGC
TTTTGTTTGACGCATTTCG
SipT_seq TCATCGCTGTTCTGCTGGC
PrsA_P1 GGGTTCCTGGCGCGAGC
AGGCGAGAAGTTTGAAGACC
PrsA_P2 TTGGGCTGGCGCGAGC
TTTAGAATTGCTTGAAGATG
PrsA_seq AAGGCAAAATCCGTGCAAGC
SpoIIIJ_P1 GGGTTCCTGGCGCGAGC
AGCTTCAACAGGAAACAATGGC
SpoIIIJ_P2 TTGGGCTGGCGCGAGC CTTTTTCTTTCCTCCGGC
SpoIIIJ_seq AGTTCGAAAGCGATGCAGGC
N-terminal SPA tag
fusion
SipS_1N cagCTCGAGATTGGAGGAAATCAGTTG
SipS_2N cagGCGGCCGCGCTGCTTGATCCTAATTTG
SipT_1N catCTCGAGCTAGGAGGAAACACTTTGACC
SipT_2N ataGCGGCCGCAAACGCCTGCTGTCCTAGG

Chapter 6. APPENDIX
183
SppA_1N gtgCTCGAGGGAGAAAAAATGAATGC
SppA_2N attGCGGCCGCTTGTTCCCTCCTACTTCGC
pSPANter_P1 GGAGATTCCTAGGATGGACTAC
pSPANter_P2 ACTGGTCTGATCGATGGGATGTC
Deletion mutants
Ko_ytej_P1 GGATTGCATTAGTGATTGCTCTGG
Ko_yteJ_P2 ACTATTGCCGATGATAAGCTGTATTGTTCCCTCC
TACTTCGCATAG
Ko_yteJ_P3 TGCGAAGTAGGAGGGAACAATACAGCTTATCAT
CGGCAATAGTTAC
KO_yteJ_P4 ACGGAAGTCCGGGTTATCGGGGCGATATAGGCG
CCAGCAAC
Ko_yteJ_P5 TTGCTGGCGCCTATATCGCCCCGATAACCCGGA
CTTCCGTC
Ko_yteJ_P6 TCTGATGCCTGTCGATCCGAC
ko_YteJ_P7 AGCTTCGGATTAGGCTCACTG
Ko_yteJ_P8 AGATACTCCAGCGCGACATG
KO_sppA_P1 TCATAAAAGGACAGGCAGCAGC
Ko_sppA_P2 AGGGTAACTATTGCCGATGATAAGCTGCCCAGA
GCAATCACTAATGC
Ko_sppA_P3 GGATTGCATTAGTGATTGCTCTGGGCAGCTTATC
ATCGGCAATAG
ko_sppA_P4 TGTCGCGTCCATTATTGTTCCCTCGGCGATATAG
GCGCCAGCAACC
Ko_sppA_P5 TTGCTGGCGCCTATATCGCCGAGGGAACAATAA
TGGACGCGAC
Ko_sppA_P6 GTTAACACACCTGTCACTGC
Ko_sppA_P7 TCTGCCATCCAGTATTTGTG
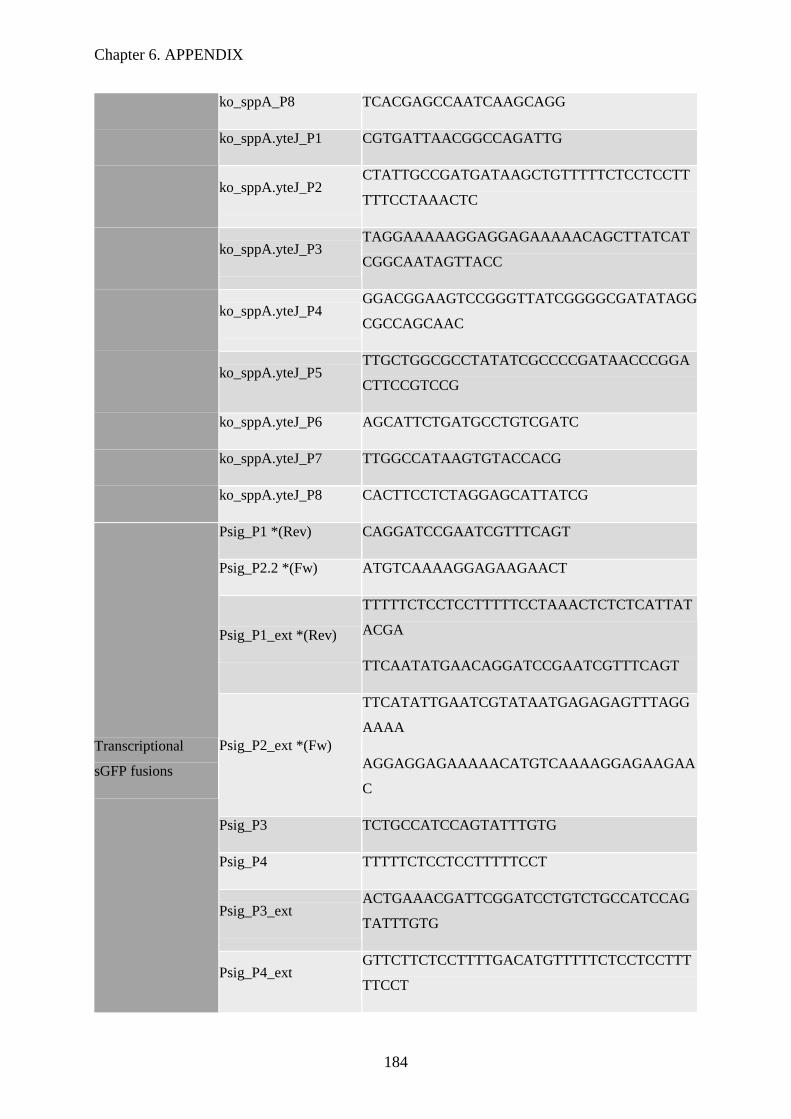
Chapter 6. APPENDIX
184
ko_sppA_P8 TCACGAGCCAATCAAGCAGG
ko_sppA.yteJ_P1 CGTGATTAACGGCCAGATTG
ko_sppA.yteJ_P2 CTATTGCCGATGATAAGCTGTTTTTCTCCTCCTT
TTTCCTAAACTC
ko_sppA.yteJ_P3 TAGGAAAAAGGAGGAGAAAAACAGCTTATCAT
CGGCAATAGTTACC
ko_sppA.yteJ_P4 GGACGGAAGTCCGGGTTATCGGGGCGATATAGG
CGCCAGCAAC
ko_sppA.yteJ_P5 TTGCTGGCGCCTATATCGCCCCGATAACCCGGA
CTTCCGTCCG
ko_sppA.yteJ_P6 AGCATTCTGATGCCTGTCGATC
ko_sppA.yteJ_P7 TTGGCCATAAGTGTACCACG
ko_sppA.yteJ_P8 CACTTCCTCTAGGAGCATTATCG
Transcriptional
sGFP fusions
Psig_P1 *(Rev) CAGGATCCGAATCGTTTCAGT
Psig_P2.2 *(Fw) ATGTCAAAAGGAGAAGAACT
Psig_P1_ext *(Rev)
TTTTTCTCCTCCTTTTTCCTAAACTCTCTCATTAT
ACGA
TTCAATATGAACAGGATCCGAATCGTTTCAGT
Psig_P2_ext *(Fw)
TTCATATTGAATCGTATAATGAGAGAGTTTAGG
AAAA
AGGAGGAGAAAAACATGTCAAAAGGAGAAGAA
C
Psig_P3 TCTGCCATCCAGTATTTGTG
Psig_P4 TTTTTCTCCTCCTTTTTCCT
Psig_P3_ext ACTGAAACGATTCGGATCCTGTCTGCCATCCAG
TATTTGTG
Psig_P4_ext GTTCTTCTCCTTTTGACATGTTTTTCTCCTCCTTT
TTCCT

Chapter 6. APPENDIX
185
PsigAW_P3_ext ACTGAAACGATTCGGATCCTGATCCCATTTCCCC
TTTGCTG
PsigAW_P4_ext AGTTCTTCTCCTTTTGACATTTTTTCTCCTCCTTT
TTCCT
PsigW_P3 ACTGAAACGATTCGGATCCTGGTTTGCCGGCAG
CCCGTTAAG
Translational sGFP
fusions
SG13_P1 TGGTCTGATCGCTCGGATACC
SG13_P2 ACATCACCATCCAATTCAACC
YteJ-GFP_P1 AACTAATAAGGAGGACAAACATGGACGCGACAT
ATGAAGA
YteJ-GFP_P2 TGAGCCGCCGCCGCCTGAGCCGCCGCCGCCTTT
GCGGTATAATTTTTCAT
SG13_YteJ-GFP_P3 GGCGGCGGCGGCTCAGGCGGCGGCGGCTCAAT
GTCAAAAGGAGAAGAACT
SG13_YteJ-GFP_P4 TCTTCATATGTCGCGTCCATGTTTGTCCTCCTTATT
AGTT
GFP-SppA_P1 GGCGGCGGCGGCTCAGGCGGCGGCGGCTCAAT
GAATGCAAAAAGATGGAT
GFP-SppA_P2 TGACTATTCTCAGATAAAGTCTACTTCGCATAG
AGATACATC
SG13_GFP-SppA_P3 GATGTATCTCTATGCGAAGTAGACTTTATCTGA
GAATAGTCA
SG13_GFP-SppA_P4 TGAGCCGCCGCCGCCTGAGCCGCCGCCGCCTTT
ATAAAGTTCGTCCATAC
GFPseq_P1 TGCTTCCAGATAACCACTACC
GFP seq_P2 ACCGTTAGTTGCATCACCTTC
sGFP_RBS_P1 AGCGATTAACTAATAAGGAG
sGFPSppA_P2.2 CAGGTCGACCTACTTCGCATAGAGATACATC
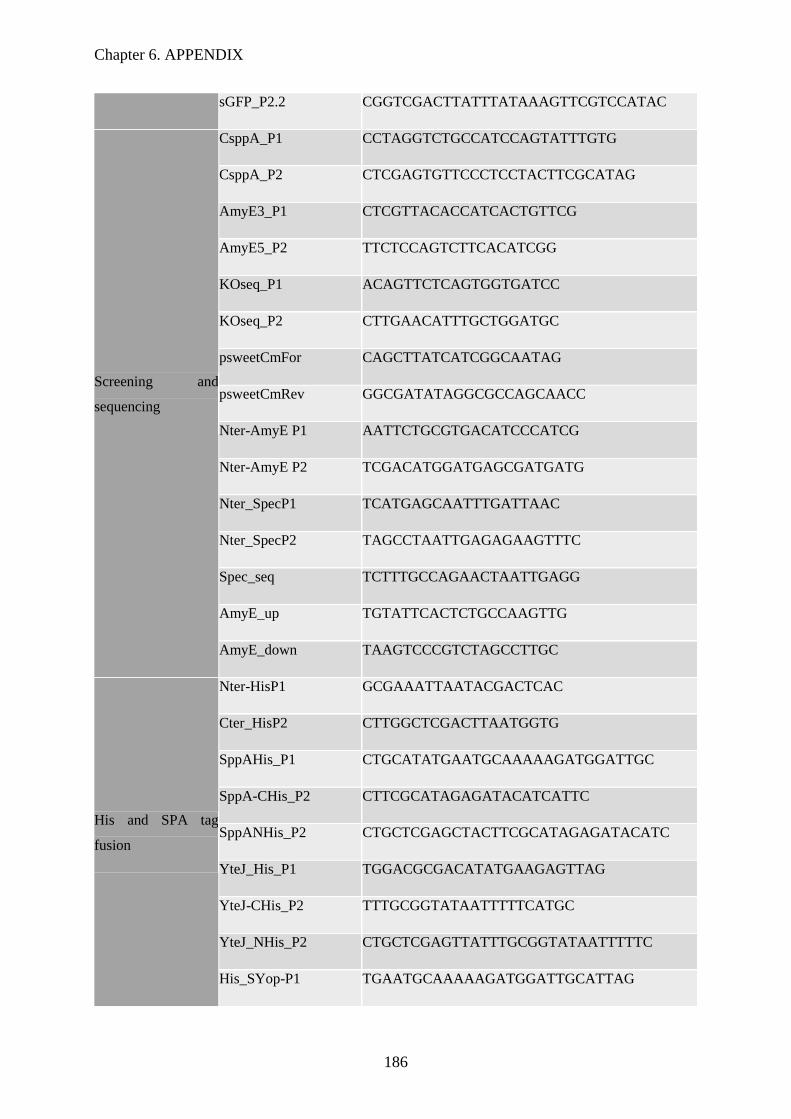
Chapter 6. APPENDIX
186
sGFP_P2.2 CGGTCGACTTATTTATAAAGTTCGTCCATAC
Screening and
sequencing
CsppA_P1 CCTAGGTCTGCCATCCAGTATTTGTG
CsppA_P2 CTCGAGTGTTCCCTCCTACTTCGCATAG
AmyE3_P1 CTCGTTACACCATCACTGTTCG
AmyE5_P2 TTCTCCAGTCTTCACATCGG
KOseq_P1 ACAGTTCTCAGTGGTGATCC
KOseq_P2 CTTGAACATTTGCTGGATGC
psweetCmFor CAGCTTATCATCGGCAATAG
psweetCmRev GGCGATATAGGCGCCAGCAACC
Nter-AmyE P1 AATTCTGCGTGACATCCCATCG
Nter-AmyE P2 TCGACATGGATGAGCGATGATG
Nter_SpecP1 TCATGAGCAATTTGATTAAC
Nter_SpecP2 TAGCCTAATTGAGAGAAGTTTC
Spec_seq TCTTTGCCAGAACTAATTGAGG
AmyE_up TGTATTCACTCTGCCAAGTTG
AmyE_down TAAGTCCCGTCTAGCCTTGC
His and SPA tag
fusion
Nter-HisP1 GCGAAATTAATACGACTCAC
Cter_HisP2 CTTGGCTCGACTTAATGGTG
SppAHis_P1 CTGCATATGAATGCAAAAAGATGGATTGC
SppA-CHis_P2 CTTCGCATAGAGATACATCATTC
SppANHis_P2 CTGCTCGAGCTACTTCGCATAGAGATACATC
YteJ_His_P1 TGGACGCGACATATGAAGAGTTAG
YteJ-CHis_P2 TTTGCGGTATAATTTTTCATGC
YteJ_NHis_P2 CTGCTCGAGTTATTTGCGGTATAATTTTTC
His_SYop-P1 TGAATGCAAAAAGATGGATTGCATTAG

Chapter 6. APPENDIX
187
SSppAHis_P1 ACGCATATGAACGGATCTCACATCACTGACG
His-SSppA_P1.35 CAGCATATGCAAACGGATCTCACATCACTGACG
His-SSppA_P1.53 CGCATATGGGCAGTCCCTCAAGTAAAATTGCC
FHis-SppA
TAGGAGGTAAAAAAAATGCACCATCACCAT
CACCATATGAATGCAAAAAGATGGATTGC
SPA_Rev CTGGTCGACCTACTTGTCATCGTCATCCTTG
YteJSPA-P2 GCCGCTAGCTTTGCGGTATAATTTTTCATGC
Overexpression
strains
pDR111-P1 TTGCTCGAGGGTAAATGTGAGC
Hspank_SppA_P2 CAGGTCGACCTACTTCGCATAGAGATAC
Hspank_SppA_P1 AGGAGGAGAAAAAATGAATGC
Hspank_YteJ_P1 CAGAAGCTTAGGAGGGAACAATAATGGACG
Hspank_YteJ_P2 CAGGTCGACTTATTTGCGGTATAATTTTTC
FsppA-K199A AGCGGGGCCCATGCGGACATTATGTCT
BsppA-K119A AGACATAATGTCCGCATGGGCCCCGCT
YteJ_LacA-P1 TGCGGATCCAGGAGGTAAAAAAAATGGACGCG
ACATATGAAGAG
YteJ_LacA-P2 CTGCCGCGGTTATTTGCGGTATAATTTTTC
XylR_seq TACTTGAGAGGAGACAGTAGAC
LacA_up TGCCGTCATCTTTATTATGCTGCA
LacA_down CCTGTTGAACTACATGCACTCCAC
RYteJ_93 CTGGTCGACTTACTGCCTGAAGTATTTTGTCATG
AG
RYteJ_129 CTGGTCGACTTAATCAATATACCGTCCAACCAC
RYteJ_152 CTGGTCGACTTACGCGATGTAATCATGAATCCC
RYteJ_55 CTGGTCGACTTAAAAGAGGGGAGAAACGAGCA
A

Chapter 6. APPENDIX
188
SppA-SeqP2 TCTGTGGTTATATCCGTCTGCACC
Table S 4. List of B. subtilis strains
Strain Genotype Source
GHBs 001 BSB1 (Trp+) strain Laboratory
stock
GHBs 006 BSB1 sipT::sipT-SPA lacImC;EryR This study
GHBs 008 BSB1 sipS::sipS-SPA lacImC;EryR This study
GHBs 010 BSB1 secG::secG-SPA lacImC;EryR This study
GHBs 012 BSB1 sppA::sppA-SPA lacImC;EryR This study
GHBs 014 BSB1 bdbC::bdbC-SPA lacImC;EryR This study
GHBs 017 BSB1 secDF::secDF-SPA lacImC;EryR This study
GHBs 018 BSB1 yteJ::yteJ-SPA lacImC;EryR This study
GHBs 020 BSB1 spoIIIJ::spoIIIJ-SPAlacImC;EryR This study
GHBs 023 BSB1 ∆yteJ::cm This study
GHBs 025 BSB1 ∆sppA::cm This study
GHBs 028 BSB1 ∆sppA-yteJ::cm This study
GHBs 029 BSB1 pCS74 This study
GHBs 030 BSB1 pCS73 This study
GHBs 031 BSB1 pCS58 This study
GHBs 032 BSB1 ∆yteJ::cm pCS74 This study
GHBs 033 BSB1 ∆yteJ::cm pCS73 This study
GHBs 034 BSB1 ∆yteJ::cm pCS58 This study
GHBs 035 BSB1 ∆sppA::cm pCS74 This study
GHBs 036 BSB1 ∆sppA::cm pCS73 This study
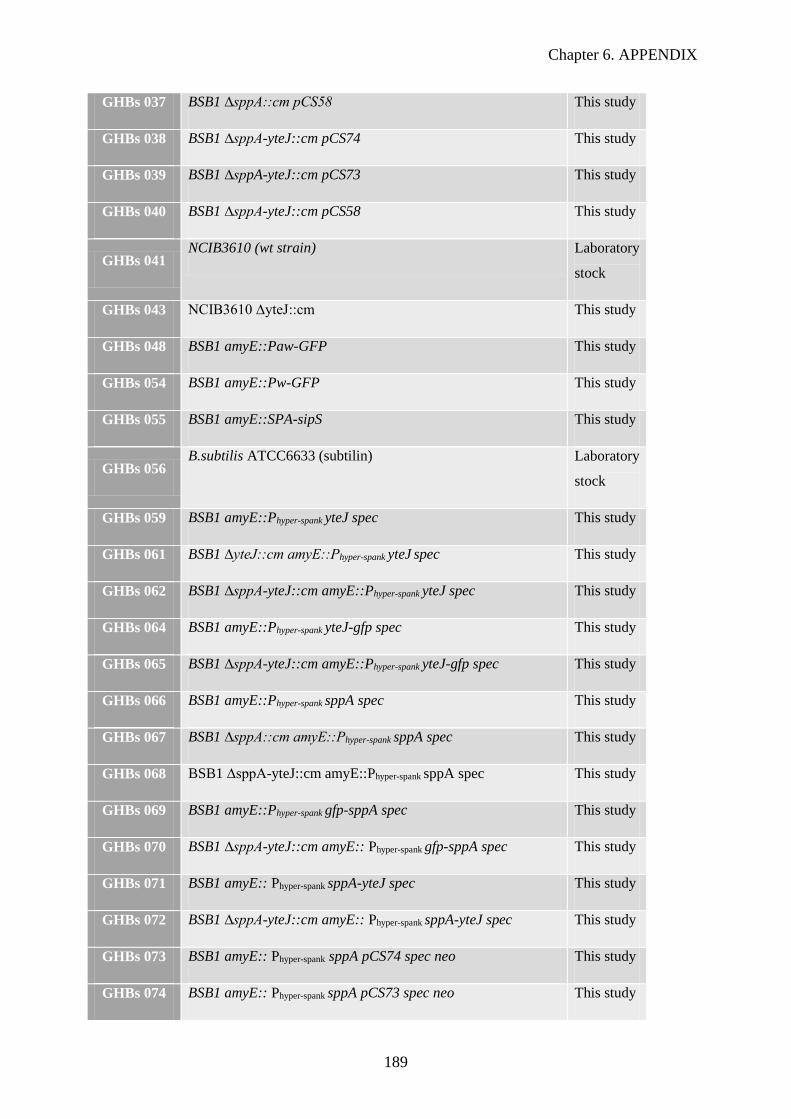
Chapter 6. APPENDIX
189
GHBs 037 BSB1 ∆sppA::cm pCS58 This study
GHBs 038 BSB1 ∆sppA-yteJ::cm pCS74 This study
GHBs 039 BSB1 ∆sppA-yteJ::cm pCS73 This study
GHBs 040 BSB1 ∆sppA-yteJ::cm pCS58 This study
GHBs 041 NCIB3610 (wt strain) Laboratory
stock
GHBs 043 NCIB3610 ∆yteJ::cm This study
GHBs 048 BSB1 amyE::Paw-GFP This study
GHBs 054 BSB1 amyE::Pw-GFP This study
GHBs 055 BSB1 amyE::SPA-sipS This study
GHBs 056 B.subtilis ATCC6633 (subtilin) Laboratory
stock
GHBs 059 BSB1 amyE::Phyper-spank yteJ spec This study
GHBs 061 BSB1 ∆yteJ::cm amyE::Phyper-spank yteJ spec This study
GHBs 062 BSB1 ∆sppA-yteJ::cm amyE::Phyper-spank yteJ spec This study
GHBs 064 BSB1 amyE::Phyper-spank yteJ-gfp spec This study
GHBs 065 BSB1 ∆sppA-yteJ::cm amyE::Phyper-spank yteJ-gfp spec This study
GHBs 066 BSB1 amyE::Phyper-spank sppA spec This study
GHBs 067 BSB1 ∆sppA::cm amyE::Phyper-spank sppA spec This study
GHBs 068 BSB1 ∆sppA-yteJ::cm amyE::Phyper-spank sppA spec This study
GHBs 069 BSB1 amyE::Phyper-spank gfp-sppA spec This study
GHBs 070 BSB1 ∆sppA-yteJ::cm amyE:: Phyper-spank gfp-sppA spec This study
GHBs 071 BSB1 amyE:: Phyper-spank sppA-yteJ spec This study
GHBs 072 BSB1 ∆sppA-yteJ::cm amyE:: Phyper-spank sppA-yteJ spec This study
GHBs 073 BSB1 amyE:: Phyper-spank sppA pCS74 spec neo This study
GHBs 074 BSB1 amyE:: Phyper-spank sppA pCS73 spec neo This study
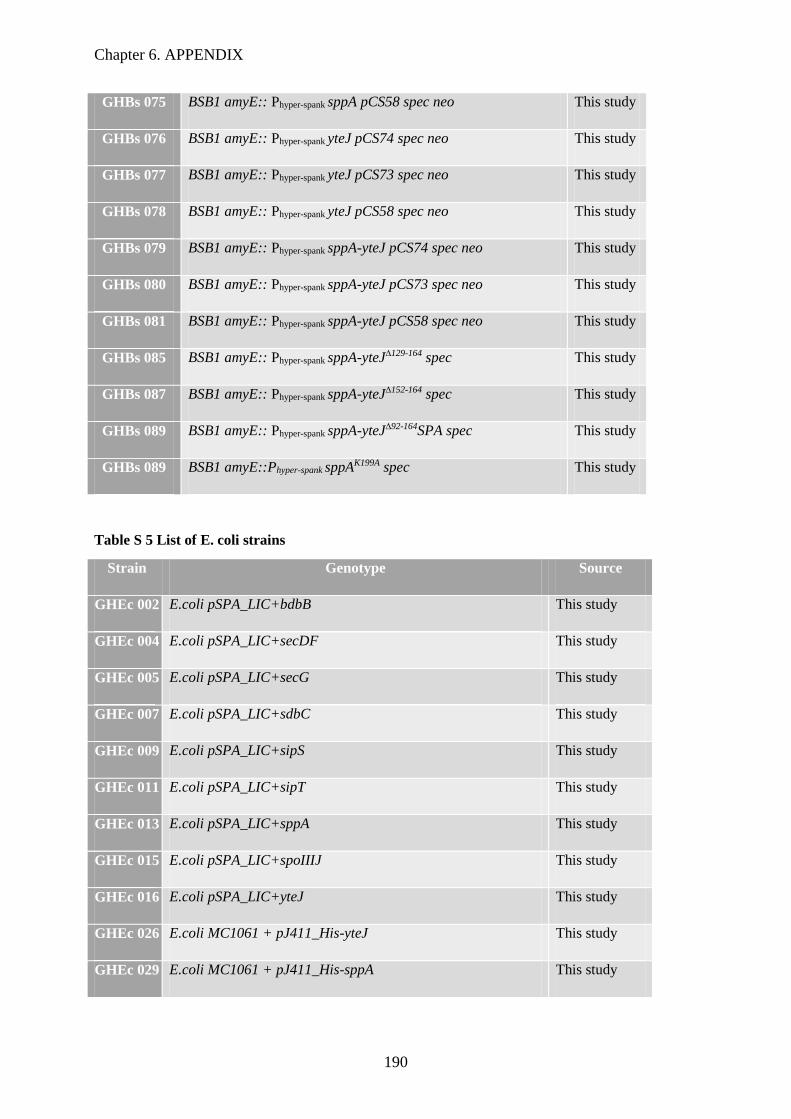
Chapter 6. APPENDIX
190
GHBs 075 BSB1 amyE:: Phyper-spank sppA pCS58 spec neo This study
GHBs 076 BSB1 amyE:: Phyper-spank yteJ pCS74 spec neo This study
GHBs 077 BSB1 amyE:: Phyper-spank yteJ pCS73 spec neo This study
GHBs 078 BSB1 amyE:: Phyper-spank yteJ pCS58 spec neo This study
GHBs 079 BSB1 amyE:: Phyper-spank sppA-yteJ pCS74 spec neo This study
GHBs 080 BSB1 amyE:: Phyper-spank sppA-yteJ pCS73 spec neo This study
GHBs 081 BSB1 amyE:: Phyper-spank sppA-yteJ pCS58 spec neo This study
GHBs 085 BSB1 amyE:: Phyper-spank sppA-yteJ∆129-164 spec This study
GHBs 087 BSB1 amyE:: Phyper-spank sppA-yteJ∆152-164 spec This study
GHBs 089 BSB1 amyE:: Phyper-spank sppA-yteJ∆92-164SPA spec This study
GHBs 089 BSB1 amyE::Phyper-spank sppAK199A spec This study
Table S 5 List of E. coli strains
Strain Genotype Source
GHEc 002 E.coli pSPA_LIC+bdbB This study
GHEc 004 E.coli pSPA_LIC+secDF This study
GHEc 005 E.coli pSPA_LIC+secG This study
GHEc 007 E.coli pSPA_LIC+sdbC This study
GHEc 009 E.coli pSPA_LIC+sipS This study
GHEc 011 E.coli pSPA_LIC+sipT This study
GHEc 013 E.coli pSPA_LIC+sppA This study
GHEc 015 E.coli pSPA_LIC+spoIIIJ This study
GHEc 016 E.coli pSPA_LIC+yteJ This study
GHEc 026 E.coli MC1061 + pJ411_His-yteJ This study
GHEc 029 E.coli MC1061 + pJ411_His-sppA This study
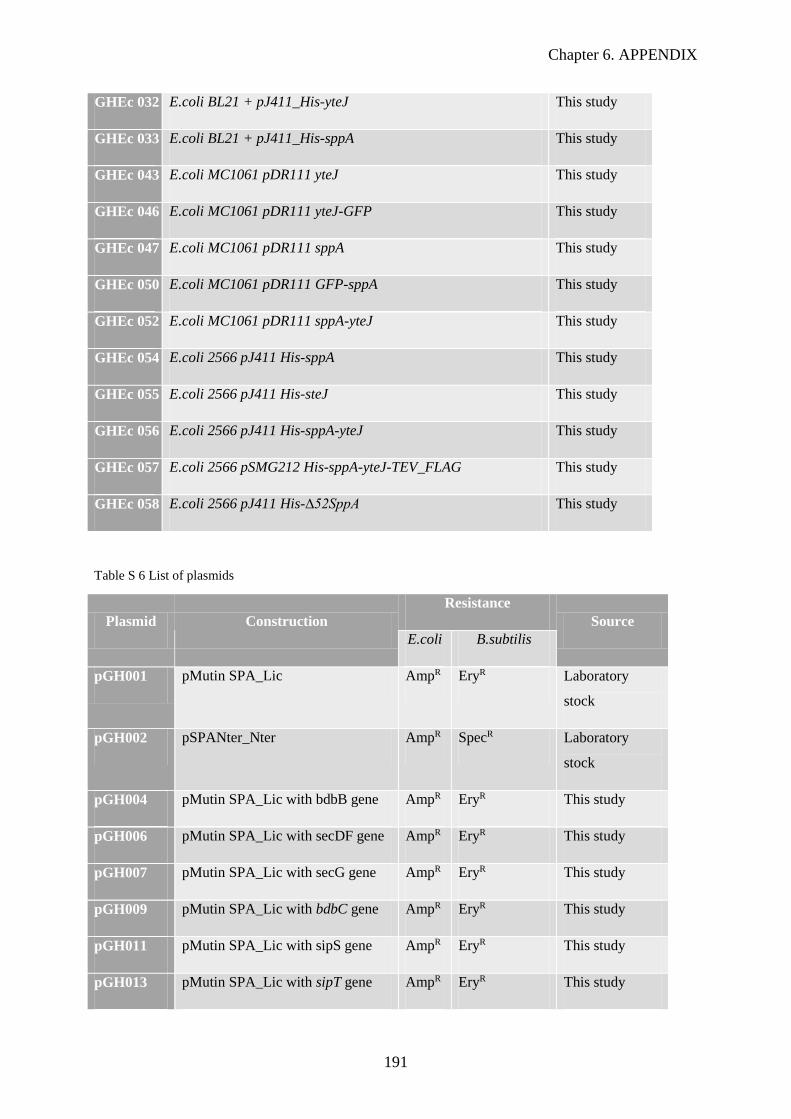
Chapter 6. APPENDIX
191
GHEc 032 E.coli BL21 + pJ411_His-yteJ This study
GHEc 033 E.coli BL21 + pJ411_His-sppA This study
GHEc 043 E.coli MC1061 pDR111 yteJ This study
GHEc 046 E.coli MC1061 pDR111 yteJ-GFP This study
GHEc 047 E.coli MC1061 pDR111 sppA This study
GHEc 050 E.coli MC1061 pDR111 GFP-sppA This study
GHEc 052 E.coli MC1061 pDR111 sppA-yteJ This study
GHEc 054 E.coli 2566 pJ411 His-sppA This study
GHEc 055 E.coli 2566 pJ411 His-steJ This study
GHEc 056 E.coli 2566 pJ411 His-sppA-yteJ This study
GHEc 057 E.coli 2566 pSMG212 His-sppA-yteJ-TEV_FLAG This study
GHEc 058 E.coli 2566 pJ411 His-∆52SppA This study
Table S 6 List of plasmids
Plasmid Construction
Resistance
Source
E.coli B.subtilis
pGH001 pMutin SPA_Lic AmpR EryR Laboratory
stock
pGH002 pSPANter_Nter AmpR SpecR Laboratory
stock
pGH004 pMutin SPA_Lic with bdbB gene AmpR EryR This study
pGH006 pMutin SPA_Lic with secDF gene AmpR EryR This study
pGH007 pMutin SPA_Lic with secG gene AmpR EryR This study
pGH009 pMutin SPA_Lic with bdbC gene AmpR EryR This study
pGH011 pMutin SPA_Lic with sipS gene AmpR EryR This study
pGH013 pMutin SPA_Lic with sipT gene AmpR EryR This study

Chapter 6. APPENDIX
192
pGH015 pMutin SPA_Lic with sppA gene AmpR EryR This study
pGH017 pMutin SPA_Lic with spoIIIJ gene AmpR EryR This study
pGH018 pMutin SPA_Lic with yteJ gene AmpR EryR This study
pGH021 pMutin SPA_Lic with prsA gene AmpR EryR This study
pGH022 pCS74 NeoR From DSM
pGH023 pCS73 NeoR From DSM
pGH024 pCS58 NeoR From DSM
pGH025 pSG13 AmpR SpecR Laboratory
stock
pGH026 pSG13_PAW-GFP AmpR SpecR This study
pGH029 pSG13_PW-GFP AmpR SpecR This study
pJ411_6His KanR Laboratory
stock
pGH032 pJ114_His-YteJ KanR This study
pGH035 pJ114_His-SppA KanR This study
pGH037 pSPANter_Nter SPA-Sips AmpR SpecR This study
pGH032 pJ114_His-SSppA KanR This study
pJ114_His-operon KanR This study
pGH042 pDR111 AmpR SpecR Laboratory
stock
pGH043 pDR111 yteJ AmpR SpecR This study
pGH046 pDR111 yteJ-GFP AmpR SpecR This study
pGH047 pDR111 SppA AmpR SpecR This study
pGH050 pDR111 GFP-SppA AmpR SpecR This study
pGH052 pDR111 operon AmpR SpecR This study
pGH053 pJ114 His-SppA-YteJ KanR This study

Chapter 6. APPENDIX
193
pSMG212 KanR Laboratory
stock
pGH054 pSMG212 His-SppA-YteJ-
TEV_FLAG
KanR This study
Figure S 1 YteJ predicted structure by Protter. Extra- extracellular compartment; Intra- cytosolic
compartment; The structure shows iYteJ amino acid composition, the amino acids are represented by the unique
letter symbol (Omasits et al. 2014).

Chapter 6. APPENDIX
194
Figure S 2. Purification of SppA, YteJ, SppA/YteJ and SppAK199A by Gel Filtration. (A)
Chromatograph obtained with the gel filtration of SppA, YteJ, SppA/YteJ and SppAK199A samples ;
(B) The protein samples were injected in a Superdex 200 10/300 GL gel filtration and all the fractions
from 4-23 were analysed by SDS-PAGE. The two bands 40kDa and 25KDa are highlighted in the
protein ladder.

Chapter 6. APPENDIX
195
Figure S 3 Pulse chase assays for sppA, yteJ and sppA yteJ deletion mutants and complemented strains. All the strains were transformed with pHTK10 (AmyQ secretion),as such were grown at MBU supplemented
with Kanamycin. (A) pulse chase gel results. P-pre-protein. M-mature protein; (B,C) Graphic representation of
the AmyQ processing results.

Chapter 6. APPENDIX
196
Figure S 4. The growth of B. subtilis sppA and yteJ deletion mutant strains in the presence of different
concentrations of LP9. The BSB1, ∆sppA and ∆yteJ deletion strains grown in LB in a 96-well plate, where the
OD600 was measured on specific time points. The different graphs represent the growth of the strains in the
presence of different concentrations (0 µg, 50 µg, 75 µg, 100 µg, 150 µg and 200µg) LP9, labelled on the Top of
the graph. Colours, on the picture, indicate the wild type or mutated strains; Black arrow represents the time
point of induction with LP9. Data from our collaborators at the Prof. Jan Maarten van Dijl.

Chapter 6. APPENDIX
197
Figure S 5. The growth of B. subtilis sppA and yteJ overexpressing mutant strains in the presence of
different concentrations of LP9. The BSB1, ∆sppA and ∆yteJ deletion strains grown in LB in a 96-well plate,
where the OD600 was measured on specific time points. The different graphs represent the growth of the strains
in the presence of different concentrations (0 µg, 50 µg, 75 µg, 100 µg, 150 µg and 200µg) LP9, labelled on the
Top of the graph. Colours, on the picture, indicate the wild type or mutated strains; Black arrow represents the
time point of induction with LP9. Data from our collaborators at the Prof. Jan Maarten van Dijl.

Chapter 6. APPENDIX
198
6.2 UCB Secondment: Purification and characterization
of a maltogenic α-amylase from Bacillus
stearothermophilus
A maltogenic amylase from B. stearothermophilus was overexpressed in B. subtilis at the
DSM company. Dr. Van Rij, one of our collaborators from the Protein Factory consortium, sent us a
crude medium supernatant sample from B. subtilis AmyM-secreting cells to UCB Company, at
Slough. For my secondment in this company, I started a collaboration with the group of Dr. David
Humphreys at UCB, which project objective it is the purification of the AmyM protein. This work has
been developed under the supervision of Dr. Emma Dave and Dr. Neesha Dedi.
This AmyM protein has a molecular weight of 75.2KDa, but in SDS-Page gels run below the
expected, around 66KDa as is possible to see on the Figure S 6. Its theoretical isoelectric point is 5.38.
This calculation is based on the protein sequence. According, to this information and to the
purification process published by Cha et al. in 1998, we begun a small scale purification with a
volume of 1ml of the supernatant sample (20mg/ml). The sample was diluted up to 2ml, applied to a
Q-Sepharose resin (a strong anion exchanger) equilibrated in 20mM Tris HCl pH7,5 and 5mM
CaCl2 and eluted with a linear gradient of NaCl from 100mM up to 1M. Consequently, as is
possible to see on the Figure 1 below, the protein was collected from the flow-through. The protein
did not bind to the resin beads, which may be due to a high salt concentration of the supernatant
buffer, making impossible the exchange of ions.

Chapter 6. APPENDIX
199
Figure S 6. AmyM supernatant purification. The AmyM sample was purified by anionic exchange
(Q-Sepharose resin). The different eluted samples and fractions were analysed by 10% SDS-PAGE gel. The
description for each lane shown at the right side of the picture.
This experiment was repeated but the result obtained was the same. The expiry date can may
be the reason why the AmyM sample is not binding to the Q-rresin. To test this hypothesis, the
concentrated enzyme sample was applied to a new Q-resin charged with two columns volumes of 20
mM Tris HCl and 1 M NaCl, washed with and equilibrated with 20 mM Tris HCl pH 7.5 and 5 mM
CaCl2. Like the previous experiments the protein was collected from the run-trough. This fraction was
reapplied to a Size exclusion Hitrap 16/60 Superdex 200 column, that allows the separation of the
protein and contaminants based on their size. The protein was eluted as a single peak with an
apparent molecular weight of 75 KDa, according with the molecular weight predicted. Although, the
protein has been eluted in a single peak the analysis by SDS-PAGE gel of each fractions
correspondent to the protein elution showed that some contaminants were eluted at the same peak
(Figure S 7).
The observation of some contaminants in the protein sample after the gel filtration raised the
question, is the pH buffers used for the anionic exchange chromatography correct?
Therefore, to test this hypothesis the protein sample was subjected to a new anion exchange
chromatography at 4 °C in a 1 ml Hitrap HP-Q column equilibrated in 20 mM Tris HCl pH 8.5 and 5
mM CaCl2. The protein was eluted with a linear gradient of NaCl from 0 mM up to 1 M. Through
the analysis of the UV value measured at 280 nm it was possible to see that the protein did not bind
to the resin and was eluted during the flow-through. The flow-through sample was concentrated
(with an Amicon Ultra-15 column) and analysed by SDS-PAGE gel, were we could confirm the
presence of AmyM protein at the flow-through.

Chapter 6. APPENDIX
200
Figure S 7. AmyM Size exclusion purification results. The protein sample was size-fractionated at 4 °C using
a Hitrap 16/60 Superdex 200 column equilibrated with 20 mM Tris HCl pH 7.5, 5 mM CaCl2 and 100 mM NaCl
buffer and eluted at a flow rate of 2 ml/min. The legend of the loaded samples is described at the right side of the
figure.
Consequently, it was performed a cationic exchange chromatography at 4 °C in a 1 ml Hitrap
HP-S column equilibrated in 20 mM Tris HCl pH 7.5 and 5 mM CaCl2. The protein was eluted with a
linear gradient of NaCl from 0 mM up to 1 M. The measurements of the UV values at 280 nm
showed that the protein had not bound to the resin and, once more, the protein was eluted with the
flow-through.
Then the AmyM sample was reapplied to a Hitrap 26/60 Superdex 200 column equilibrated
with TBS and 5 mM CaCl2 and eluted at a flow rate of 2 ml/min for 5 hours. This column allows a
better separation of the different fractions of the sample since have a bigger size. The protein was
eluted as a single peak with an apparent molecular weight of 75 KDa. However, the sample fractions
correspondent to the protein elution showed the contaminants were kept.
Although the theoretically pI be 5.38 (Dauter et al. 1999; Cha et al. 1998; Park et al. 2013)
it is possible that this is not the real value, what could explain the why the protein is not binding to
the anionic or cationic resin of the exchange chromatography process. In 1984, H. Outtrup and B. E.
Norman studied the B. stearothermophilus maltogenic amylase and determined by thin layer gel-
electrofocusing that this protein has a pI of 8.5. To confirm the correct pI value we measured the pI
of our sample by iCE3 Capillary IEF System. This is a HPLC-like method that performs free solution
Isoelectric Focusing (IEF) in a capillary column, in which a pH gradient is created to separate and
focus the proteins according to their pI. The all process is monitored in real time by a whole-column
UV detector. Before run, the samples are premixed with carrier ampholytes, additives and pI markers.
The pI measured for our sample was 8.35. To complement the pI measurement a molecular weight
analysis was performed by time-of-flight mass spectrometry (LC- QTOF). The used technique allows
the obtainment of the protein molecular weight thought the determination of an ion mass-to-charge
ratio that is measured according to the time the ion takes to reach a detector at a known distance
(Sigaud et al. 2016; Kernphysik, Darmstadt, and Wien 1999; Beauchemin 2009). The measure

Chapter 6. APPENDIX
201
molecular weight was 75157 Da. This value is in good agreement with the information received from
DSM with the details of the supernatant sample but is slightly different from those reported previously
(Cha et al. 1998; Outtrup 1984).
The real pI value (8.35) of the AmyM sample can explain the problems that we had with the
binding of the protein to the cationic and anionic resins, because the range of pH tested was from 7.5
up to 8.5 and to be possible the exchange of ions the pH value used should be two values above or
below of the protein pI value (Svensson 1961) .
Furthermore, to avoid high pH values in which the protein is denatured we tested a cationic
exchange chromatography at pH 6.5. The buffer of a 2 ml supernatant sample was exchange to 50 mM
MES pH 6.5 and 50 mM NaCl with the PD-10 Desalting columns GE Health Care. The sample was
then diluted up 20 ml and applied to a cationic a 1 ml Hitrap HP-S column equilibrated in 50 mM
MES pH 6.5 and 50 mM NaCl. The protein was eluted with a linear gradient of NaCl from 50 mM up
to 1 M. During the collection of the flow-through fraction it was possible to observe an increase of
the UV values at 280 nm what normally indicates that the protein is not binding to the column resin,
but because it is not a high value as the values previous measured it can be some contaminants that
did not bound to the resin. Therefore, a 50 mM MES pH 6.5 buffer with 2 M NaCl was loaded into
the purification system to create a gradient of NaCl up to 2 M. This additional step has the objective of
elute the sample in the case that it binds tightly and it is necessary to have higher concentrations of salt
to elute the protein. However, it was not the case and the protein was collected in the flow-through
fraction once again.Since the previous purifications were not successful we procedure to a new anionic
exchange chromatography with an increase of the pH buffer up to 9,5 (Dauter et al. 1999). A 2 ml
protein sample from the previous experiment Flow-through fractions sample was buffer exchanged to
20 mM Tris-HCl pH 9.5 with the PD-10 Desalting columns. The sample applied to an anionic 1 ml
Hitrap HP-Q column equilibrated in 20 mM Tris-HCl pH 9.5. The protein was eluted with a linear
gradient of NaCl from 50 mM up to 2 M. Following the UV measured values during the
chromatography process it is possible to see that the protein bound to the resin and it was eluted as a
single peack. However, after SDS-PAGE analysis the protein sample had yet other bands besides the
band corresponding to the AmyM molecular weight as shown at Figure S 7. These bands could
represent contaminants or cleaved protein, because by either processes, by ions exchange process or
by molecular exclusion size this, it seems impossible to rid of the contaminant bands. By the
comparison of the abundance of the AmyM and the other specimens presents in the sample is possible
to know that the AmyM sample is clean enough for the rabbit Immunization. This purification step
was applied to all the remaining protein sample. After, all the sample passed through a endotoxin free
removal protocol and were send to the rabbit Immunization department.

Université Paris-Saclay
Espace Technologique / Immeuble Discovery
Route de l’Orme aux Merisiers RD 128 / 91190 Saint-Aubin, France
Titre : Caractérisation du complexe membranaire impliquant la signal peptide peptidase
SppA et YteJ chez Bacillus subtilis.
Mots clés : Sécretion protéique, complexe protéique membranaire, protéase, résistance aux
lantibiotiques, contrôle qualité
Résumé : Chez Bacillus subtilis nous avons identifié un complexe protéique membranaire impliquant
une protéine inconnue, YteJ, et une autre protéine membranaire, SppA, une signal peptide peptidase
également impliquée dans la résistance aux peptides antibactériens de la famille des lantibiotiques.
Après délétion des gènes correspondant, nous avons montré que les deux protéines sont impliquées dans
cette résistance. Dans la souche ∆sppA, la surexpression ectopique de SppA a non seulement restauré la
résistance, mais elle a également induit la formation de cellules allongées, un phénotype supprimé par
la surexpression simultanée de YteJ. L'expression de versions tronquées de YteJ a mis en évidence le
rôle inhibiteur d'un domaine spécifique de YteJ. Enfin, des études biochimiques in vitro ont confirmé
que l'activité de la protéase SppA était fortement réduite par la présence de YteJ, confirmant l'hypothèse
d'une inhibition par YteJ. Nos études in vivo et in vitro ont montré que YteJ, via l'un de ses domaines,
agit comme régulateur négatif de l'activité protéase de SppA dans ce complexe. En conclusion, nous
avons montré que le complexe SppA/YteJ est impliqué dans la résistance aux lantibiotiques à travers
l’activité protéase de SppA, elle-même régulée par YteJ.
Title : Towards the understanding of the function and regulation of a membrane complex
involving SppA and YteJ in Bacillus subtilis.
Keywords : Bacillus subtilis, Protein secretion, Membrane protein complex, Protease, Lantibiotic
resistance, Quality control
Abstract: We have identified a membrane protein complex of Bacillus subtilis involving an
unknown protein, YteJ, and SppA, a membrane protein first described as a signal peptide
peptidase and later shown to be also involved in the resistance to antibacterial peptides of the
lantibiotic family. Using deletion mutant strains, we showed that both proteins are involved in
this resistance. In the ∆sppA strain, the ectopic overexpression of SppA not only restored the
resistance, it also induced the formation of elongated cells, a phenotype suppressed by the
simultaneous overexpression of YteJ. Furthermore, the expression of truncated versions of YteJ
pinpointed the inhibitory role of a specific domain of YteJ. Finally, in vitro biochemical studies
showed that SppA protease activity was strongly reduced by the presence of YteJ, supporting
the hypothesis of an inhibition by YteJ. Our in vivo and in vitro studies showed that YteJ, via
one of its domain, acts as a negative regulator of the protease activity of SppA in this complex.
In conclusion, we have shown that SppA/YteJ complex is involved in lantibiotic resistance
through the protease activity of SppA, which is regulated by YteJ.




















