
DISSIPATIVE QUANTUM-SYSTEMS WITH A POTENTIAL BARRIER - GENERAL-THEORY AND THE PARABOLIC BARRIER
Upload
northwesternCategory
view
3download
0
iRHOM2-dependent regulation of ADAM17 incutaneous disease and epidermal barrier function
Matthew A. Brooke1, Sarah L. Etheridge1, Nihal Kaplan2, Charlotte Simpson1, Edel A. O’Toole1,
Akemi Ishida-Yamamoto3, Olivier Marches1, Spiro Getsios2 and David P. Kelsell1,∗
1Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK,2Department of Dermatology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA and 3Department
of Dermatology, Asahikawa Medical University, Asahikawa, Japan
Received October 24, 2013; Revised and Accepted March 13, 2014
iRHOM2 is a highly conserved, catalytically inactive member of the Rhomboid family, which has recently beenshown to regulate the maturation of the multi-substrate ectodomain sheddase enzyme ADAM17 (TACE) inmacrophages. Dominant iRHOM2 mutations are the cause of the inherited cutaneous and oesophagealcancer-susceptibility syndrome tylosis with oesophageal cancer (TOC), suggesting a role for this protein inepithelial cells. Here, using tissues derived from TOC patients, we demonstrate that TOC-associated mutationsin iRHOM2 cause an increase in the maturation and activity of ADAM17 in epidermal keratinocytes, resultingin significantly upregulated shedding of ADAM17 substrates, including EGF-family growth factors and pro-inflammatory cytokines. This activity is accompanied by increased EGFR activity, increased desmosomeprocessing and the presence of immature epidermal desmosomes, upregulated epidermal transglutaminaseactivity and heightened resistance to Staphylococcal infection in TOC keratinocytes. Many of these featuresare consistent with the presence of a constitutive wound-healing-like phenotype in TOC epidermis, whichmay shed light on a novel pathway in skin repair, regeneration and inflammation.
INTRODUCTION
iRHOM2 is a highly conserved, catalytically inactive proteinbelonging to the Rhomboid family of intramembrane serine pro-teases (1,2). Inaddition to its role in the regulation of EGFprocess-ing(3), iRHOM2was recently identifiedas a novel regulator of themulti-substrate ectodomain sheddase enzyme ADAM17 (TNFa-converting enzyme; TACE) (4,5). ADAM17 is an enzymerequired for the proteolytic cleavage and release of a wide spec-trum of substrates from the cell surface (6,7), including TNFa,multiple members of the EGF family of growth factors (8–10)and components of the desmosome (11,12). In some cell types,iRHOM2 controls ADAM17 maturation, by regulating thetransit of Pro-ADAM17 from the endoplasmic reticulum (ER)to the Golgi apparatus (4), where ADAM17 is activated byremoval of its inhibitory pro-domain by pro-protein convertaseenzymes such as Furin (13). Although iRhom22/2 mice areviable and show no obvious defects, they fail to control the repli-cation of the pathogenic bacterium Listeria monocytogenes, asa result of impaired shedding of the ADAM17 substrate
TNFa (5). Similarly, iRhom22/2 mice were recently shown tobe protected from inflammatory arthritis to the same extent as
mice lacking either ADAM17 or TNFa (14). In contrast,Adam172/2 mice die perinatally (9), recapitulating the pheno-
types of mice lacking various EGFR ligands shed under the
control of ADAM17, such as severe failures of epithelial (9) andcardiac (15,16) morphogenesis and maturation, which result
from knockouts of TGFa and HB-EGF, respectively. The appar-
ent paradox of the requirement for iRHOM2 in ADAM17 matur-
ation, the lethality of Adam17 knockouts in mice and the seeminglack of effect of iRhom2 knockout may in part be explained by
recent evidence showing that the closely related iRHOM1 may
support ADAM17 maturation in iRHOM2-deficient cells (14).Indeed, both iRHOM1 and 2 have been shown to support the traf-
ficking and maturation of ADAM17 in numerous murine cell
types (17), with a high degree of redundancy observed betweenthe two iRHOMs in tissues where both are highly expressed (in-
cluding the skin). Murine knockouts of iRhom1 result in lethality
after between 9 days and 6 weeks (dependent on mouse genetic
∗To whom correspondence should be addressed at: Centre for Cutaneous Research, Blizard Institute, Barts and The London School of Medicine andDentistry, Queen Mary University of London, London E1 2AT, UK. Tel: +44 2078827167; Fax: +44 2078827172; Email: [email protected]
# The Author 2014. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2014 1–13doi:10.1093/hmg/ddu120
HMG Advance Access published March 28, 2014 at G
alter Health Sciences L
ibrary on June 22, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
background) and a phenotype much more severe than knockout ofiRhom2, the phenotype of which appears to be immune cell-specific in mice (17).
Recently, we described the first example of ADAM17-deficiency in humans; unlike in the mouse model, this was com-patible with life with one affected individual surviving intoadulthood (18). This recessive syndrome was characterizedby a severe inflammatory skin phenotype, greatly increasedsusceptibility to cutaneous and paronychial infection, bowelinflammation and a moderate cardiac phenotype. Tissue-specificknockouts of Adam17 in mice have shed light on this phenotype,with keratinocyte-restricted knockouts producing a skin pheno-type very similar to that seen in humans, resulting from a defect-ive epidermal barrier and impaired transglutaminase activity(19). Additionally, hypomorphic Adam17 mice show substan-tially increased susceptibility to inflammatory colitis (20).
Tylosis with oesophageal cancer (TOC; OMIM: 148500) is adominantly inherited syndrome of palmoplantar keratoderma,oral and oesophageal leukoplakia, follicular keratosis and astriking susceptibility to oesophageal squamous cell carcinoma(OSCC). We recently reported the association of TOC in threelarge families (from the UK, USA and Germany) with heterozy-gous point mutations in RHBDF2, the gene encoding iRHOM2(21). This finding was subsequently confirmed in a study on aseparate Finnish family (22). Interestingly, although the muta-tions in each family arose independently, they display a remark-able clustering, with all four families’ mutations found within thesame four residues (p.Ile186Thr in the UK and US, p.Asp188Ansin the Finnish and p.Pro189Leu in the German families) in ahighly conserved N-terminal cytoplasmic tail domain uniqueto the iRHOMs. Intriguingly, these residues are also conservedin iRHOM1, which has been shown to share the ability ofiRHOM2 to support ADAM17 maturation. This suggests thatthe affected domain may play a role in this process. Prior tothe reports of a direct iRHOM2/ADAM17 interaction, wedescribed abnormal, plasma-membranous iRHOM2 localiza-tion and dysregulated EGFR signalling in TOC keratinocytes(21), making the manner in which ADAM17 is affected byTOC-associated iRHOM2 mutations of particular interest.Here, we describe investigations of the iRHOM2/ADAM17axis in the epidermis, keratinocytes and peripheral blood mono-nuclear cells (PBMCs) from TOC-affected individuals and un-affected controls. We demonstrate that ADAM17 maturationand activity is increased in TOC. These data support a key rolefor this axis in EGFR ligand shedding in the epithelium, kerati-nocyte barrier function, migration and bacterial infection.
RESULTS
iRHOM2 has a role in the regulation of ADAM17in keratinocytes
To investigate the effect of TOC-associated iRHOM2 mutationson ADAM17, studies were carried out in immortalized keratino-cyte cell lines derived from two TOC patients (TYLK1 andTYLK2—a male and a female) carrying the UK iRHOM2 muta-tion.Thesekeratinocyteshave beenpreviouslyused in the studyofthe pathophysiology of TOC(21) andwere immortalized by trans-fection with human papilloma virus (HPV)-16 open readingframes E6 and E7 (23,24). Throughout, these cell lines were
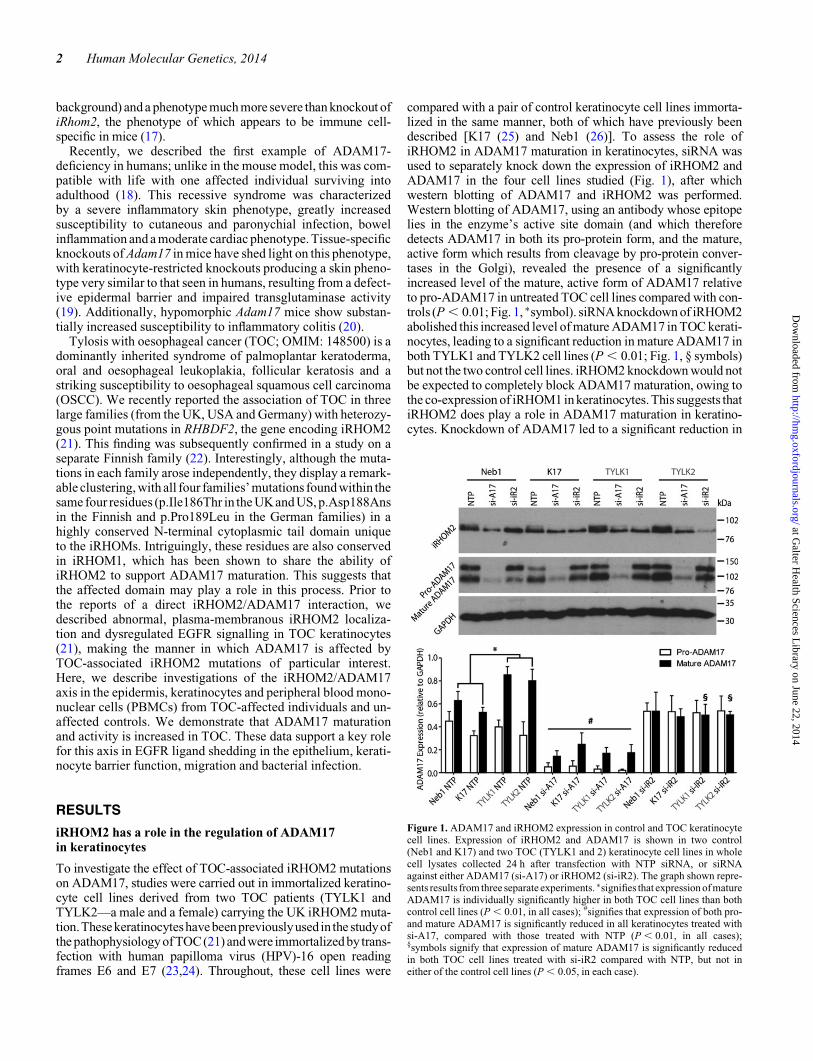
compared with a pair of control keratinocyte cell lines immorta-lized in the same manner, both of which have previously beendescribed [K17 (25) and Neb1 (26)]. To assess the role ofiRHOM2 in ADAM17 maturation in keratinocytes, siRNA wasused to separately knock down the expression of iRHOM2 andADAM17 in the four cell lines studied (Fig. 1), after whichwestern blotting of ADAM17 and iRHOM2 was performed.Western blotting of ADAM17, using an antibody whose epitopelies in the enzyme’s active site domain (and which thereforedetects ADAM17 in both its pro-protein form, and the mature,active form which results from cleavage by pro-protein conver-tases in the Golgi), revealed the presence of a significantlyincreased level of the mature, active form of ADAM17 relativeto pro-ADAM17 in untreated TOC cell lines compared with con-trols (P , 0.01; Fig. 1, ∗symbol). siRNA knockdown of iRHOM2abolished this increased level of mature ADAM17 in TOC kerati-nocytes, leading to a significant reduction in mature ADAM17 inboth TYLK1 and TYLK2 cell lines (P , 0.01; Fig. 1, § symbols)but not the two control cell lines. iRHOM2 knockdown would notbe expected to completely block ADAM17 maturation, owing tothe co-expression of iRHOM1 in keratinocytes. This suggests thatiRHOM2 does play a role in ADAM17 maturation in keratino-cytes. Knockdown of ADAM17 led to a significant reduction in
Figure 1. ADAM17 and iRHOM2 expression in control and TOC keratinocytecell lines. Expression of iRHOM2 and ADAM17 is shown in two control(Neb1 and K17) and two TOC (TYLK1 and 2) keratinocyte cell lines in wholecell lysates collected 24 h after transfection with NTP siRNA, or siRNAagainst either ADAM17 (si-A17) or iRHOM2 (si-iR2). The graph shown repre-sents results from three separate experiments. ∗signifies that expression of matureADAM17 is individually significantly higher in both TOC cell lines than bothcontrol cell lines (P , 0.01, in all cases); #signifies that expression of both pro-and mature ADAM17 is significantly reduced in all keratinocytes treated withsi-A17, compared with those treated with NTP (P , 0.01, in all cases);§symbols signify that expression of mature ADAM17 is significantly reducedin both TOC cell lines treated with si-iR2 compared with NTP, but not ineither of the control cell lines (P , 0.05, in each case).
2 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

both pro- and mature ADAM17 in all four cell lines (P , 0.05, inall cases; Fig. 1, # symbol) but also interestingly caused a corre-sponding reduction in expression of iRHOM2 in all four celllines, further illustrating the close relationship between the twoproteins in keratinocytes. iRHOM2 expression in NTP-treatedkeratinocytes did appear slightly higher in TOC keratinocytes,but when this was quantified over three separate experiments,this difference was not found to be significant. iRHOM2 reductionwas also observed in the epidermis from a human ADAM17knockout individual (Supplementary Material, Fig. S1) and, inmurine iRHOM1 and 2 knockouts, a reduction in the overalllevel of ADAM17 expression has also been observed (17).
ADAM17 maturation and localization is enhancedin TOC keratinocytes
To further investigate the observation of increased ADAM17maturation in TOC keratinocytes, immunocytochemicalco-staining of ADAM17 alongside markers of the ER, Golgi ap-paratus and plasma membrane was performed in the four celllines previously described. ADAM17 displayed clear stainingat the plasma membrane and also within the Golgi apparatus(Fig. 2A). However, when comparing these cell lines, it wasnoticeable that the two TOC cell lines displayed greater levelsof ADAM17 at the plasma membrane—its major site of activity(Fig. 2A). Western blotting using an antibody directed againstthe ADAM17 pro-domain revealed a greater presence of the55-kDa band representing the pro-domain—produced byADAM17 cleavage by pro-protein convertase enzymes—inTOC keratinocytes compared with controls (Fig. 2B). Next,organotypic three-dimensional cultures of these TOC cell linesand a control (K17) were performed. These 3D culturesyielded fully differentiated skin equivalent models for eachcell line, which expressed a range of differentiation markers,including involucrin and loricrin displayed robust transglutami-nase 1 activity in their upper layers (Figs 2C and 5, Supplemen-tary Material, Fig. S2). The two TOC cell lines yielded stratifiedepithelial tissues with distinct morphologies; however, all linesgenerate epidermal equivalents that express differentiation pro-ducts similar to controls. Also importantly, the effects onADAM17 and the EGFR observed were consistent betweenTYLK1 and TYLK2. Western blotting of lysates from these3D cultures revealed similar results to those seen in monolayer,with increased levels of mature ADAM17 and the free ADAM17pro-domain present (Fig. 2D). Significantly greater levels ofADAM17 were again observed at the plasma membrane ofboth the TOC organotypic cultures compared with the control(Fig. 2E and F). These results in monolayer and organotypicculture suggested the possibility of increased levels ofADAM17 maturation and activity in TOC cells compared withcontrols and led us to investigate the effect of the apparentlyincreased ADAM17 activity in both systems.
TOC keratinocytes display increased sheddingof ADAM17 substrates
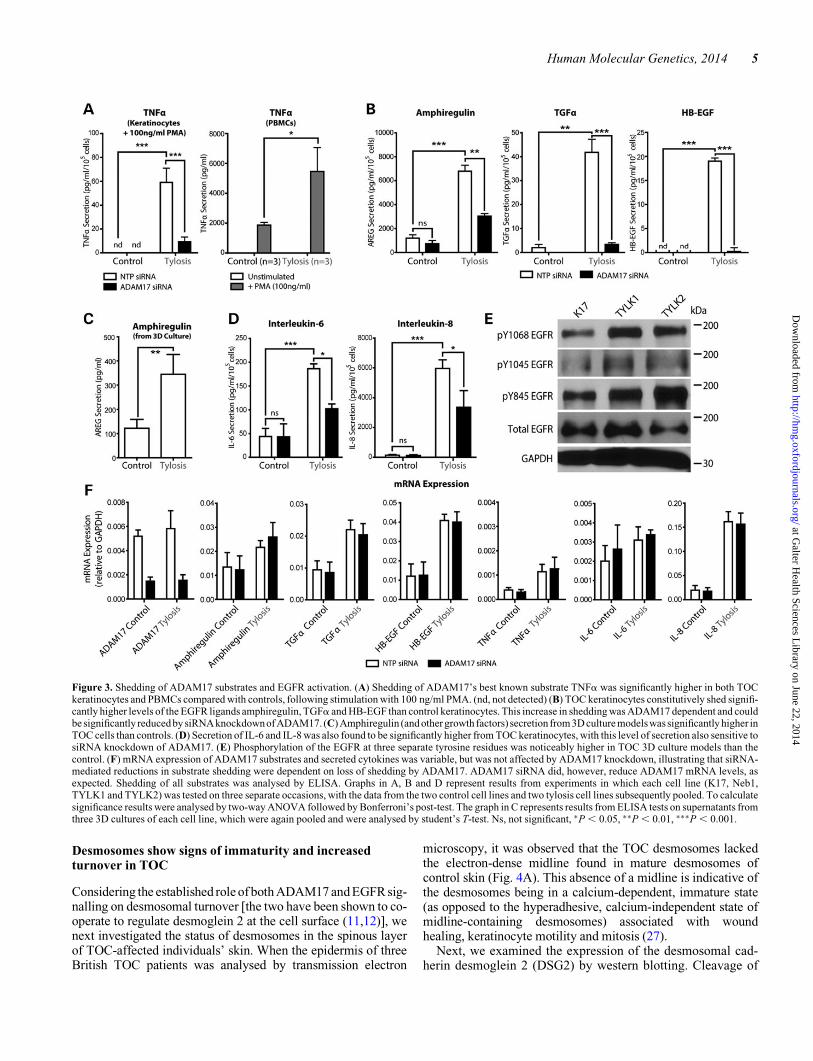
To investigate ADAM17 activity, we examined the shedding of anumber of ADAM17 substrates from TOC keratinocytes andTOC PBMCs compared with controls. ADAM17’s best known
substrate—TNFa—was shed at a significantly higher level byTOC keratinocytes than controls (in which levels of TNFashed were below the limit of detection; Fig. 3A) when thesecells were stimulated with the irreversible Protein Kinase C ac-tivator, Phorbol myristate acetate (PMA; 100 ng/ml). Important-ly, this excess TNFa shedding was found to be dependent onADAM17, as siRNA-mediated knockdown of ADAM17(Figs 1 and 3F) was sufficient to inhibit the excess sheddingobserved. Similarly, secretion of TNFa from PBMCs isolatedfrom three TOC patients was also significantly higher thanmatched controls following PMA stimulation (Fig. 3A). Nosignificant difference in TNFa secretion was observed constitu-tively, or when these cells were stimulated with varying concen-trations of lipopolysaccharide (LPS). These findings show thatTOC keratinocytes harbouring mutant iRHOM2 protein arenot only capable of shedding TNFa [in contrast to macrophagesof iRHOM2-knockout mice, which cannot (4)] but do so in amore efficient manner than wild-type cells.
In a previous study, TOC keratinocytes proliferated andmigrated significantly more than controls in a scratch assayand did so independently of exogenous EGF (21). Consequently,the shedding of ADAM17’s substrates in the EGF growth factorfamily, namely amphiregulin, TGFa and HB-EGF, was exam-ined when cells were cultured in the absence of exogenousEGF. Shedding of all three growth factors was shown to be sig-nificantly higher in TOC keratinocytes than controls, withamphiregulin shedding around two orders of magnitude higherthan that of TGFa and HB-EGF. In each of these cases, the ele-vated shedding observed could be significantly reduced bysiRNA-mediated knockdown of ADAM17 in TOC keratino-cytes (Fig. 3B), illustrating the ADAM17-dependent nature ofthe observed increase. Importantly, there was no change at themRNA transcript level of each protein when ADAM17 siRNAwas applied (Fig. 3F), illustrating the ADAM17-dependentnature of the shedding observed. mRNA transcript levels ofEGFR ligands were in general marginally higher in TOC kerati-nocytes, although these differences would not appear to explainthe large degree to which shedding levels were elevated in TOCcells.
Secretion of EGFR ligands from organotypic culture modelsof TOC was also examined. Amphiregulin secretion was againobserved to be significantly higher in TOC cells than controls(Fig. 3C), whereas TGFa and HB-EGF shedding levels werebelow the limit of detection (not shown). As expected, theenhanced EGFR ligand shedding was associated with a corre-sponding increase in the phosphorylation of the EGFR at threeseparate tyrosine residues (Y845, Y1045 and Y1068) in TOC3D cultures (Fig. 3E), suggesting that EGFR signalling is upre-gulated in these models.
We also examined the secretion of a number of pro-inflamm-atory cytokines that, although not directly shed by ADAM17,are important mediators of inflammation, epidermal woundhealing and keratinocyte migration. These included interleukins6 and 8, both of which were found to be secreted at significantlyincreased levels in TOC keratinocytes, with secretion of bothsensitive to siRNA knockdown of ADAM17 (Fig. 3D). Owingto ADAM17’s inability to directly shed these cytokines, this ispresumed to be an indirect effect of the increased shedding of,and signalling by, other ADAM17 substrates.
Human Molecular Genetics, 2014 3
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
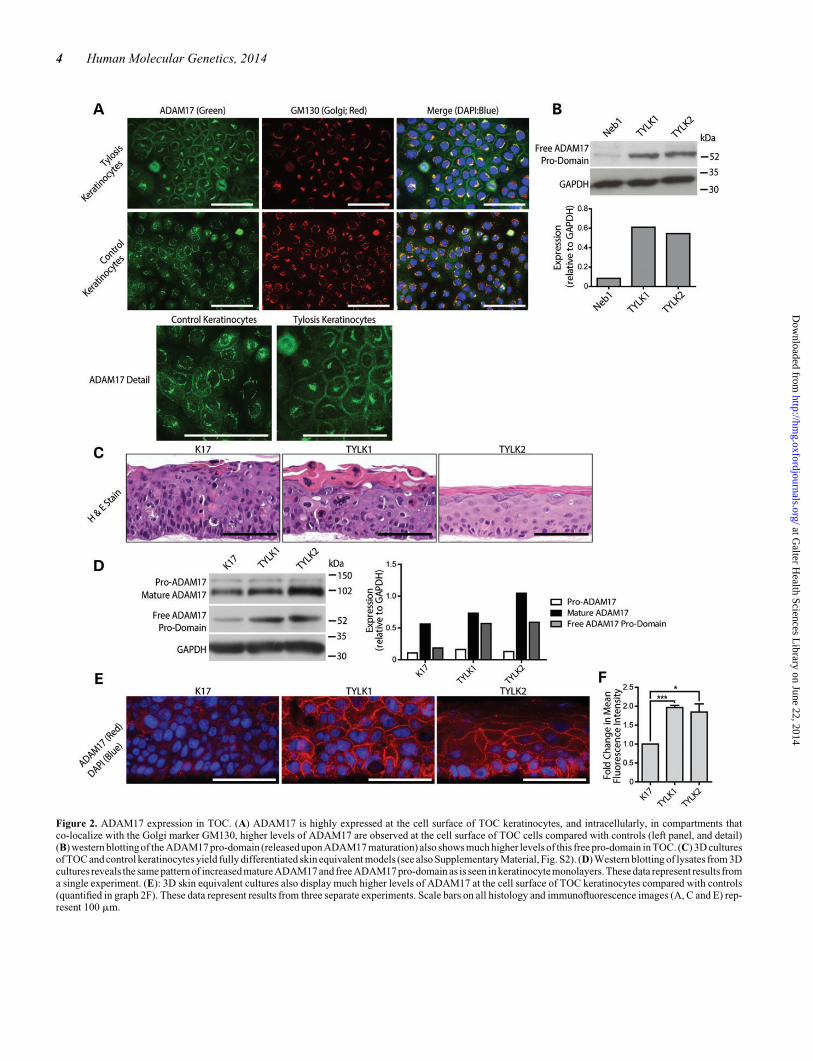
Figure 2. ADAM17 expression in TOC. (A) ADAM17 is highly expressed at the cell surface of TOC keratinocytes, and intracellularly, in compartments thatco-localize with the Golgi marker GM130, higher levels of ADAM17 are observed at the cell surface of TOC cells compared with controls (left panel, and detail)(B) western blotting of the ADAM17 pro-domain (released upon ADAM17 maturation) also shows much higher levels of this free pro-domain in TOC. (C) 3D culturesof TOC and control keratinocytes yield fully differentiated skin equivalent models (see also Supplementary Material, Fig. S2). (D) Western blotting of lysates from 3Dcultures reveals the same pattern of increased matureADAM17 and free ADAM17 pro-domain as is seen in keratinocytemonolayers. These data represent results froma single experiment. (E): 3D skin equivalent cultures also display much higher levels of ADAM17 at the cell surface of TOC keratinocytes compared with controls(quantified in graph 2F). These data represent results from three separate experiments. Scale bars on all histology and immunofluorescence images (A, C and E) rep-resent 100 mm.
4 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Desmosomes show signs of immaturity and increasedturnover in TOC
Considering the established role of both ADAM17 and EGFR sig-nalling on desmosomal turnover [the two have been shown to co-operate to regulate desmoglein 2 at the cell surface (11,12)], wenext investigated the status of desmosomes in the spinous layerof TOC-affected individuals’ skin. When the epidermis of threeBritish TOC patients was analysed by transmission electron
microscopy, it was observed that the TOC desmosomes lackedthe electron-dense midline found in mature desmosomes ofcontrol skin (Fig. 4A). This absence of a midline is indicative ofthe desmosomes being in a calcium-dependent, immature state(as opposed to the hyperadhesive, calcium-independent state ofmidline-containing desmosomes) associated with woundhealing, keratinocyte motility and mitosis (27).
Next, we examined the expression of the desmosomal cad-herin desmoglein 2 (DSG2) by western blotting. Cleavage of
Figure 3. Shedding of ADAM17 substrates and EGFR activation. (A) Shedding of ADAM17’s best known substrate TNFa was significantly higher in both TOCkeratinocytes and PBMCs compared with controls, following stimulation with 100 ng/ml PMA. (nd, not detected) (B) TOC keratinocytes constitutively shed signifi-cantly higher levels of the EGFR ligands amphiregulin, TGFa and HB-EGF than control keratinocytes. This increase in shedding was ADAM17 dependent and couldbe significantly reduced by siRNA knockdownof ADAM17. (C) Amphiregulin (and other growth factors) secretion from 3D culture modelswas significantly higher inTOC cells than controls. (D) Secretion of IL-6 and IL-8 was also found to be significantly higher from TOC keratinocytes, with this level of secretion also sensitive tosiRNA knockdown of ADAM17. (E) Phosphorylation of the EGFR at three separate tyrosine residues was noticeably higher in TOC 3D culture models than thecontrol. (F) mRNA expression of ADAM17 substrates and secreted cytokines was variable, but was not affected by ADAM17 knockdown, illustrating that siRNA-mediated reductions in substrate shedding were dependent on loss of shedding by ADAM17. ADAM17 siRNA did, however, reduce ADAM17 mRNA levels, asexpected. Shedding of all substrates was analysed by ELISA. Graphs in A, B and D represent results from experiments in which each cell line (K17, Neb1,TYLK1 and TYLK2) was tested on three separate occasions, with the data from the two control cell lines and two tylosis cell lines subsequently pooled. To calculatesignificance results were analysed by two-way ANOVA followed by Bonferroni’s post-test. The graph in C represents results from ELISA tests on supernatants fromthree 3D cultures of each cell line, which were again pooled and were analysed by student’s T-test. Ns, not significant, ∗P , 0.05, ∗∗P , 0.01, ∗∗∗P , 0.001.
Human Molecular Genetics, 2014 5
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
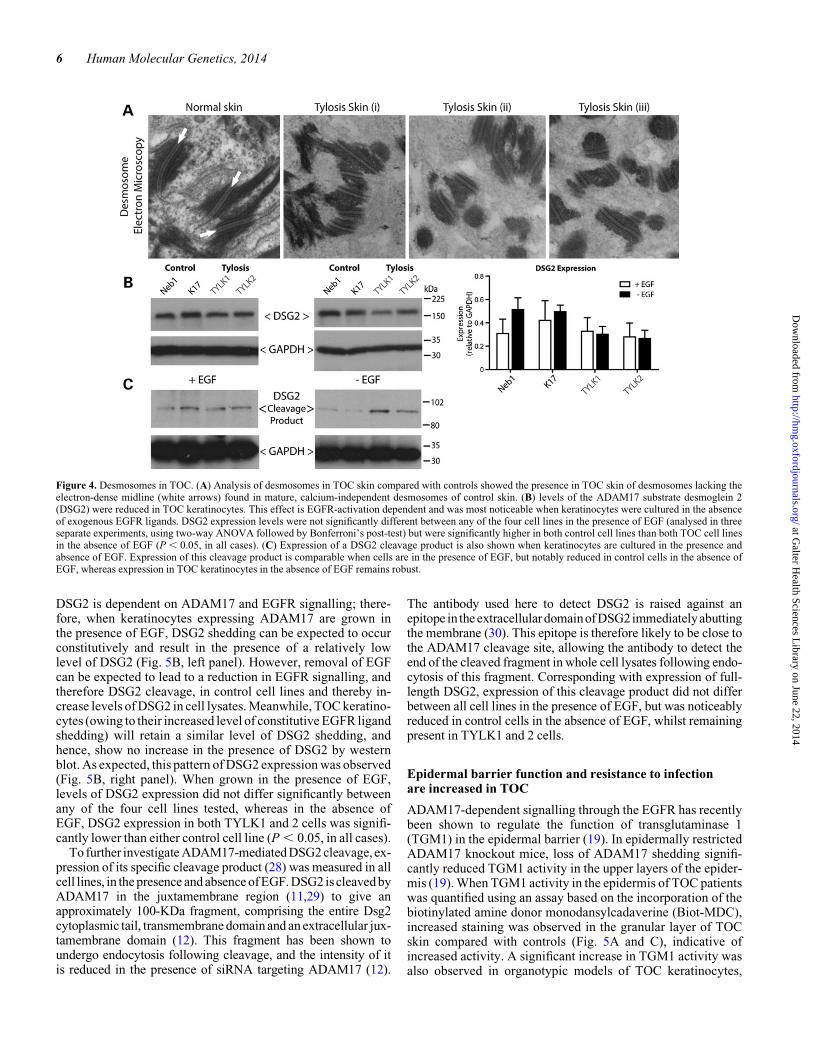
DSG2 is dependent on ADAM17 and EGFR signalling; there-fore, when keratinocytes expressing ADAM17 are grown inthe presence of EGF, DSG2 shedding can be expected to occurconstitutively and result in the presence of a relatively lowlevel of DSG2 (Fig. 5B, left panel). However, removal of EGFcan be expected to lead to a reduction in EGFR signalling, andtherefore DSG2 cleavage, in control cell lines and thereby in-crease levels of DSG2 in cell lysates. Meanwhile, TOC keratino-cytes (owing to their increased level of constitutive EGFR ligandshedding) will retain a similar level of DSG2 shedding, andhence, show no increase in the presence of DSG2 by westernblot. As expected, this pattern of DSG2 expression was observed(Fig. 5B, right panel). When grown in the presence of EGF,levels of DSG2 expression did not differ significantly betweenany of the four cell lines tested, whereas in the absence ofEGF, DSG2 expression in both TYLK1 and 2 cells was signifi-cantly lower than either control cell line (P , 0.05, in all cases).
To further investigate ADAM17-mediated DSG2 cleavage, ex-pression of its specific cleavage product (28) was measured in allcell lines, in the presence and absence of EGF. DSG2 is cleaved byADAM17 in the juxtamembrane region (11,29) to give anapproximately 100-KDa fragment, comprising the entire Dsg2cytoplasmic tail, transmembrane domain and an extracellular jux-tamembrane domain (12). This fragment has been shown toundergo endocytosis following cleavage, and the intensity of itis reduced in the presence of siRNA targeting ADAM17 (12).
The antibody used here to detect DSG2 is raised against anepitope in the extracellulardomainof DSG2immediatelyabuttingthe membrane (30). This epitope is therefore likely to be close tothe ADAM17 cleavage site, allowing the antibody to detect theend of the cleaved fragment in whole cell lysates following endo-cytosis of this fragment. Corresponding with expression of full-length DSG2, expression of this cleavage product did not differbetween all cell lines in the presence of EGF, but was noticeablyreduced in control cells in the absence of EGF, whilst remainingpresent in TYLK1 and 2 cells.
Epidermal barrier function and resistance to infectionare increased in TOC
ADAM17-dependent signalling through the EGFR has recentlybeen shown to regulate the function of transglutaminase 1(TGM1) in the epidermal barrier (19). In epidermally restrictedADAM17 knockout mice, loss of ADAM17 shedding signifi-cantly reduced TGM1 activity in the upper layers of the epider-mis (19). When TGM1 activity in the epidermis of TOC patientswas quantified using an assay based on the incorporation of thebiotinylated amine donor monodansylcadaverine (Biot-MDC),increased staining was observed in the granular layer of TOCskin compared with controls (Fig. 5A and C), indicative ofincreased activity. A significant increase in TGM1 activity wasalso observed in organotypic models of TOC keratinocytes,
Figure 4. Desmosomes in TOC. (A) Analysis of desmosomes in TOC skin compared with controls showed the presence in TOC skin of desmosomes lacking theelectron-dense midline (white arrows) found in mature, calcium-independent desmosomes of control skin. (B) levels of the ADAM17 substrate desmoglein 2(DSG2) were reduced in TOC keratinocytes. This effect is EGFR-activation dependent and was most noticeable when keratinocytes were cultured in the absenceof exogenous EGFR ligands. DSG2 expression levels were not significantly different between any of the four cell lines in the presence of EGF (analysed in threeseparate experiments, using two-way ANOVA followed by Bonferroni’s post-test) but were significantly higher in both control cell lines than both TOC cell linesin the absence of EGF (P , 0.05, in all cases). (C) Expression of a DSG2 cleavage product is also shown when keratinocytes are cultured in the presence andabsence of EGF. Expression of this cleavage product is comparable when cells are in the presence of EGF, but notably reduced in control cells in the absence ofEGF, whereas expression in TOC keratinocytes in the absence of EGF remains robust.
6 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
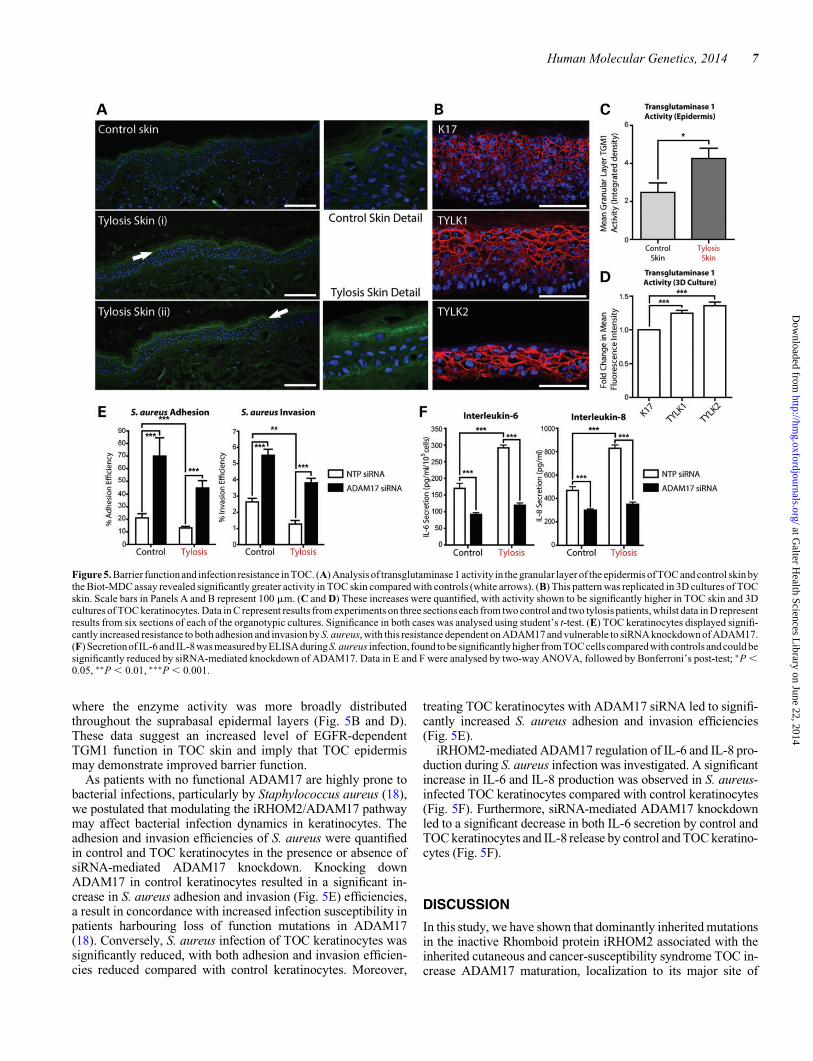
where the enzyme activity was more broadly distributedthroughout the suprabasal epidermal layers (Fig. 5B and D).These data suggest an increased level of EGFR-dependentTGM1 function in TOC skin and imply that TOC epidermismay demonstrate improved barrier function.
As patients with no functional ADAM17 are highly prone tobacterial infections, particularly by Staphylococcus aureus (18),we postulated that modulating the iRHOM2/ADAM17 pathwaymay affect bacterial infection dynamics in keratinocytes. Theadhesion and invasion efficiencies of S. aureus were quantifiedin control and TOC keratinocytes in the presence or absence ofsiRNA-mediated ADAM17 knockdown. Knocking downADAM17 in control keratinocytes resulted in a significant in-crease in S. aureus adhesion and invasion (Fig. 5E) efficiencies,a result in concordance with increased infection susceptibility inpatients harbouring loss of function mutations in ADAM17(18). Conversely, S. aureus infection of TOC keratinocytes wassignificantly reduced, with both adhesion and invasion efficien-cies reduced compared with control keratinocytes. Moreover,
treating TOC keratinocytes with ADAM17 siRNA led to signifi-cantly increased S. aureus adhesion and invasion efficiencies(Fig. 5E).
iRHOM2-mediated ADAM17 regulation of IL-6 and IL-8 pro-duction during S. aureus infection was investigated. A significantincrease in IL-6 and IL-8 production was observed in S. aureus-infected TOC keratinocytes compared with control keratinocytes(Fig. 5F). Furthermore, siRNA-mediated ADAM17 knockdownled to a significant decrease in both IL-6 secretion by control andTOC keratinocytes and IL-8 release by control and TOC keratino-cytes (Fig. 5F).
DISCUSSION
In this study, we have shown that dominantly inherited mutationsin the inactive Rhomboid protein iRHOM2 associated with theinherited cutaneous and cancer-susceptibility syndrome TOC in-crease ADAM17 maturation, localization to its major site of
Figure 5. Barrier functionand infection resistance in TOC. (A) Analysis of transglutaminase 1 activity in the granular layer of the epidermis of TOC and control skin bythe Biot-MDC assay revealed significantly greater activity in TOC skin compared with controls (white arrows). (B) This pattern was replicated in 3D cultures of TOCskin. Scale bars in Panels A and B represent 100 mm. (C and D) These increases were quantified, with activity shown to be significantly higher in TOC skin and 3Dcultures of TOC keratinocytes. Data in C represent results from experiments on three sections each from two control and two tylosis patients, whilst data in D representresults from six sections of each of the organotypic cultures. Significance in both cases was analysed using student’s t-test. (E) TOC keratinocytes displayed signifi-cantly increased resistance to both adhesion and invasion by S. aureus, with this resistance dependent on ADAM17 and vulnerable to siRNA knockdown of ADAM17.(F) Secretionof IL-6 and IL-8 was measured by ELISAduring S. aureus infection, found to be significantly higher from TOC cells compared with controls and could besignificantly reduced by siRNA-mediated knockdown of ADAM17. Data in E and F were analysed by two-way ANOVA, followed by Bonferroni’s post-test; ∗P ,0.05, ∗∗P , 0.01, ∗∗∗P , 0.001.
Human Molecular Genetics, 2014 7
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

activity at the plasma membrane and shedding of many of its sub-strates. During the genetic studies of TOC, the remarkable cluster-ing of TOC-associated mutations was noted: in the four large,unrelated families with RHBDF2 mutations so far identified, thethree distinct mutations observed cluster within a motif of justfour codons. All three mutated residues are very highly conservedin vertebrates and found within a long N-terminal loop domainthat is unique to the iRHOM proteins iRHOM1 and iRHOM2.This mutation clustering therefore strongly implies that a particu-lar site-specific function of iRHOM2 is affected in TOC. Interest-ingly, iRHOM1 has recently been shown to share the ability ofiRHOM2 to regulate the maturation of ADAM17 (14), withhigh levels of functional redundancy between the two proteinsobserved in multiple cell types (17). The effect of theseiRHOM2 mutations on ADAM17, and the confinement of theaffected domain to proteins that regulate ADAM17 maturation,suggests that the mutated domain is intimately involved in thisADAM17-regulatory function, perhaps being involved in thedirect association of the two proteins, or the transit of iRHOM2between the ER and Golgi compartments. Recently, knockoutof iRhom2 in mouse embryonic fibroblasts (mEFs) was shownto strongly reduce the ability of these cells to shed amphiregulin,HB-EGF and epiregulin (but not TGFa), as well as the non-EGFRligands (and ADAM17 substrates) EphB4, KitL2 and Tie2 (31).Furthermore, the abrogation of the shedding of KitL2 (used as amodel substrate) in iRhom22/2 mEFs could be rescued by over-expression of wild-type iRhom2, but not by an iRhom2 mutantlacking the N-terminal cytoplasmic domain, in which TOC-associated mutations are located (31).
ADAM17 acts as the ectodomain sheddase for five members ofthe EGF family of growth factors: TGFa, HB-EGF, amphiregulin,betacellulin and epigen [whilst the closely related ADAM10 isresponsible for the shedding of EGF and epiregulin (8,9,32)].In culture, keratinocytes express amphiregulin, with muchlower levels of TGFa andHB-EGF(33). Itwas thereforeespecial-ly interesting to note that shedding of all three of these growthfactors was upregulated in TOC keratinocytes, even in unstimu-lated conditions. Amphiregulin—the growth factor known toprovide by far the strongest degree of autocrine stimulation of ker-atinocyte growth (34)—was shed at a particularly high level.Upregulation of EGFR ligands in vivo is characteristic of numer-ous benign and malignant hyperproliferative conditions of theskin. For example, hyperproliferative psoriatic lesions overex-press TGFa (35), HB-EGF (36) and amphiregulin (37) as wellas theEGFR(38).Amphiregulin isalsoknowntobestronglyupre-gulated in actinic keratoses, verrucas and squamous cell carcin-omas (37). Meanwhile, transgenic overexpression of TGFa inmurine epidermis leads to cutaneous hyperplasia and hyperkera-tosis, accompanied by the spontaneous formation of squamouspapillomas at sites of wounding (39). EGFR ligand signallingalso underlies the hyperproliferative effects of retinoids on kerati-nocytes, with retinoid-induced hyperplasia mediated by themarked induction of HB-EGF and amphiregulin (40,41). The sig-nificant increase in shedding of these factors by keratinocytesfrom TOC-affected areas could therefore reasonably be expectedto play a role in the epidermal hyperproliferation that charac-terizes TOC. Signalling through the EGFR is also known toplay an important role in cutaneous wound healing—particularlyin the early phase response to wounding—by increasing keratino-cyte proliferation and migration (42). The importance of this
response is illustrated in EGFR knockout mice, whose skinlesions heal at a slower rate compared with controls (43). ADAM-mediated shedding of EGFR ligands is required for keratinocytemigration during wound healing and is immediately inducedupon wounding (44), with factors upregulated in wounded skinincluding amphiregulin, TGFa and HB-EGF (but not EGF orbetacellulin) (42). In particular, TGFa is responsible for �80%of keratinocyte migration activity during wound healing (45)and also promotes proliferation (46), whilst HB-EGF promotesre-epithelialization (47) and accelerates cutaneous woundhealing when applied topically (48,49).
Excessive secretion of EGFR ligands may also offer clues asto the striking oesophageal cancer susceptibility observed inTOC patients [TOC remains the only known highly penetrantsyndrome of genetic predisposition to OSCC (21,50)]. In sporad-ic OSSC, overexpression of the EGFR has been observed in45.6–72.1% of primary tumours (51,52), 88% of OSCC lymphnode metastases and 90% of oesophageal squamous dysplasias(53)—suggesting EGFR signalling may play a role in tumour ini-tiation. Direct EGFR gene amplification is seen in 12–28% ofOSCC (53–55) and is associated with significantly reduced cu-mulative survival (54), whilst EGFR overexpression has beenshown to correlate with poor prognosis (56). Increased levelsof EGFR ligand secretion have been associated with the develop-ment and progression of squamous cell carcinomas in both theoesophagus—where upregulation of TGFa is associated withpoor OSCC prognosis (57)—and numerous other tissues, includ-ing the epidermis (58,59), lung (60), mouth (61) and head andneck tissues (62). The observed presence of excessive EGFRligand secretion may therefore play an important role in oe-sophageal cancer development. Independently of the EGFR, ex-pression of ADAM17 itself has also been recently shown tocorrelate with progression of OSCC (63).
Furthermore, ADAM17-dependent signalling through theEGFR has recently been described to regulate the function ofthe epidermal barrier, via its effects on transglutaminase 1(TGM1) in the upper epidermal layers. Epidermal-restrictedknockout of murine Adam17 results in reduced TGM1 activity,reduced barrier function and an inflammatory dermatitis (19)(that could be rescued by the topical application of TGFa)similar to that seen in human individuals with loss-of-functionmutations in ADAM17 (18). Conversely, both epidermal sec-tions from TOC patients and TOC keratinocytes in 3D cultureshowed increased levels of TGM1 activity. This is presumedto reflect the increased level of EGFR ligand shedding observedin TOC keratinocytes and suggests that TOC patients may dem-onstrate increased epidermal barrier function.
The phenotype observed in TOC tissues shows several hall-marks that may be indicative of the presence of a constitutivewound-healing-like state in the palmoplantar epidermis of TOCpatients. In addition to the upregulation of EGFR ligand sheddingand signalling, electron microscopy data demonstrate the absenceof electron-dense midlines in the desmosomes of TOC skin—astateassociated witha migratory,wound-healingstate inkeratino-cytes (27)—and a reduction in presence of desmoglein 2 in TOCkeratinocytes. Shedding of TNFa and secretion of IL-6 are alsoupregulated in wound-healing epidermis (64), making constitu-tive expression of these cytokines also suggestive of a wound-healing state. IL-6 in particular is increased in wounding, persistsin chronic wounds (42,65,66) and acts in a mitogenic (67) and
8 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

pro-proliferative (68) manner on keratinocytes. Meanwhile, IL-8plays important roles in wound-healing epidermis (69,70) as wellas its better-known roles in local inflammation. The sensitivity ofIL-6 and IL-8 production to ADAM17 inhibition may appearsomewhat surprising; however, secretion of both IL-6 and IL-8has been shown to be stimulated by EGFR activation in anumber of cell types (71,72), meaning this cytokine responsemay reflect reduced EGFR ligand signalling followingADAM17 knockdown. The tissue-restricted pattern of epidermalhyperproliferation observed in TOC may therefore be representa-tiveofanupregulatedwound-healingstatebeingpresent inepithe-lia that undergo particularly high levels of stress, such as thepalmoplantar epidermis and the oral mucosa. These data demon-strate that the iRHOM2/ADAM17 axis plays a key role in skinbarrier maintenance, inflammation and migration. Thus, activa-tion of this pathway may be an attractive target for improvedchronic and acute cutaneous wound repair.
MATERIALS AND METHODS
Keratinocyte cell culture
Control and TOC keratinocyte cell lines [as previously described(21)] were cultured in Dulbecco’s Modified Eagle’s Medium(DMEM), supplemented with 10% foetal calf serum, penicillin,streptomycin, L-Glutamine and the keratinocyte growth supple-ment RM+/2 (RM+: containing EGF, RM2: with all EGFRligands absent), at 378C and 5% CO2.
3D culture
The 3D organotypic raft models of human epidermis usingcontrol and TOC keratinocytes were established as previouslydescribed (73,74). Briefly, J2-3T3 fibroblasts maintained inDMEM containing 10% heat-inactivated foetal calf serumwere trypsinized and resuspended (2.5 × 105/ml) in anice-cold, pH-neutralized DMEM-rat tail collagen (4 mg/ml;BD Biosciences, San Jose, CA, USA) solution. The collagen–fibroblast slurry was polymerized in a 12-well plate (2 ml/well) at 378C for 30 min and maintained in J2-3T3 culturemedium for 24 h in a humidified tissue culture incubator at378C and 5% CO2. Control or TOC keratinocyte lines were tryp-sinized and resuspended in DMEM RM containing 5 ng/mlEGF; 5 × 105/ml keratinocytes were seeded onto a collagen–fibroblast plug established in duplicate for each experiment.Keratinocyte–collagen plugs were maintained as submergedcultures for 48 h to ensure an intact, confluent epidermal sheetof keratinocytes covered the entire surface of the dermal equiva-lent prior to air exposure. A sterilized metal spatula was used totransfer the keratinocyte–collagen plug to the top of a stainlesssteel meshed wire grid raised above the surface of a 60-mm Petridish. FAD RM2 was added to the bottom chamber of the wiremesh in contact with the base of the collagen plug, thereby ex-posing the keratinocytes to an air-liquid interface. The culturemedium was replaced every other day from organotypic culturesmaintained in a humidified tissue culture incubator with 5% CO2
at 378C for 12 days. Duplicate cultures were established fromeach control and TOC keratinocyte line, and the raft cultureexperiment was repeated on three separate occasions.
Western blotting
For western blotting studies, isolates of keratinocytes or PBMCswere lysed directly by the addition of lysis buffer containing0.125 M Tris–HCl pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol,0.001% (w/v) Bromophenol Blue and 1.44 Mb-mercaptoethanol.Cell lysates were separated by SDS–PAGE, transferred ontonitrocellulose membranes and immunoblotted following standardprocedures. Membranes were incubated overnight at 48C, withprimary antibodies against full-length ADAM17 (AbcamAb2051, Cambridge, UK), the ADAM17 Pro-Domain (AbcamAb39161), desmoglein 2 (a gift from My Mahoney; Jefferson In-stitute, Philadelphia, PA, USA), three different phospho-EGFRs(Cell signalling Technology 3777, 2237 and 6963, Danvers,MA, USA) or glyceraldehyde-3-phosphate dehydrogenase(GAPDH; Abcam ab9485) and were developed using ECL pluschemoluminescence agent (GE Healthcare). Densitometric quan-tification of western blots was performed using ImageJ image ana-lysis software [National Institutes of Health (NIH), Bethesda, MD,USA], with levels of each protein normalized to the loadingcontrol (GAPDH) for each individual sample.
Immunocytochemistry
For immunocytochemical studies, keratinocytes were culturedon 16-mm glass cover slips and fixed using a solution of 4%paraformaldehyde in PBS. Immunocytochemistry was thenperformed according to standard procedures with primary anti-bodies against ADAM17 (Abcam Ab2051) and the Golgimarker GM130 (BD 610822, Oxford, UK) followed byincubation with (green-fluorescently tagged) anti-rabbit and(red-fluorescently tagged) anti-mouse secondary antibodies.Mounting medium containing DAPI was used as a nuclear stain.
Histology and immunohistochemistry
For histology of 3D organotypic cultures, formalin-fixed,paraffin-embedded tissue sections were stained with haematoxy-lin and eosin. For IHC, tissue sections were deparaffinized andthen heated for 45 min at 968C in a water bath and immunos-tained as previously described (75) using ADAM17 antibody(Abcam Ab2051). IHC imaging of 3D organotypic cultureswas performed using an epifluorescence Zeiss Axiovision Z1microscope containing an Apotome slide module and a high-resolution AxioCam MRm digital camera. Image analysis wasperformed with the Zeiss AxioVision software (Thornwood,NY, USA). The ImageJ software (NIH) was used to calculatethe pixel intensity of ADAM17 at cell-cell borders; thesevalues were normalized for total area to calculate the mean fluor-escence intensity.
ELISA
To assess the production of ADAM17 substrates (and othercytokines), keratinocytes were incubated for 24 h in mediumcontaining no exogenous EGFR ligands, which was then col-lected. For assessment of TNFa production, this medium wassupplemented by the addition of 100 ng/ml PMA. PBMCswere isolated from fresh blood specimens using Ficoll PaquePremium isolation medium (GE Healthcare Life sciences,
Human Molecular Genetics, 2014 9
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Buckinghamshire, UK) and were cultured for 24 h in the pres-ence and absence of PMA, at 378C in RPMI medium. Collectedmedium specimens were analysed for the proteins of interestusing relevant DuoSet ELISA kits (R&D Systems, Abingdon,UK). In all cases, the data are presented as mean+SEM. Collec-tion of supernatants was performed in triplicate for each cell line,and results from the two control and two TOC keratinocyte celllines were pooled for presentation. Results were analysed by Stu-dent’s t-test using GraphPad Prism (Graphpad, San Diego, CA,USA).
siRNA knockdown of ADAM17
siRNA knockdown was performed on cells in culture usingOnTarget SMARTPool siRNA specific to ADAM17 (ThermoFisher Dharmacon, Lafayette, Colorado, USA). In brief, keratino-cytes were incubated for 24 h with penicillin- and streptomycin-free medium, containing 100 nM ADAM17 or non-targetingpool (NTP) siRNA, alongside the transfection reagent Dharma-FECT 1 (Thermo Fisher Dharmacon). ADAM17 expressionwas assessed by western blot or quantitative real-time PCR.
Quantitative real-time PCR
RNA was extracted from cultured cells using the Qiagen RNeasymini kit (Qiagen, Crawley, UK) according to the manufacturer’sinstructions. RNA was converted to cDNA using InvitrogenSuperScript II reverse transcriptase (Invitrogen, Grand Island,NY, USA) as the active enzyme. Quantitative real-time PCRusing TaqMan chemistry on a Rotorgene Q thermocycler(Qiagen) was performed using primers against ADAM17,AREG, TGFA, HBEGF, TNFA, IL6, IL8 and GAPDH designedin-house. Primers were designed to bind across exon–exonboundaries, ensuring only cDNA was amplified. Specimenswere assayed simultaneously for GAPDH and the gene of inter-est via the use of probers labelled with different fluorophores.Expression of all genes was quantified relative to GAPDHusing the DCt method (76).
Electron microscopy
For electron microscopy, TOC and control epidermis specimenswere fixed in phosphate buffered 4% glutaraldehyde, post-fixedin 1% osmium tetroxide and dehydrated through a gradedethanol series. They were then cleared in propylene oxide andinfiltrated with Araldite. The cells were embedded by inverting‘BEEM’ capsules filled with partly cured Araldite over the mono-layer and incubating them at 608C for 48 h. After cutting away thesilicone membrane, semi-thin sections (0.5 mm) for light micros-copy were cut and stained with Toluidine Blue. Ultrathin sections(60–80 nm) were cut, mounted on copper grids and stained withuranyl acetate and lead citrate. They were examined in a J.E.O.L.JEM 1230 electron microscope and images collected with anOlympus ‘Morada’ 2 × 2 K digital camera.
Transglutaminase activity
For in situ detection of epidermal transglutaminase activity,we used the biotinylated amine donor substrate monodansylcada-verine (biot-MDC) assay, as previously described (77). In brief,
frozen epidermal sections from normal controls and tylosispatients were preincubated with 1% BSA in 0.1 M Tris–HCl, atpH 7.4 for 30 min. The sections were then incubated for 2 hwith 100-mM biot-MDC (or 10 mM EDTA as a negativecontrol) and 5 mM CaCl2 in 0.1 M Tris–HCl at pH 7.4 (to assesstransglutaminase 1 activity). The reaction was halted by the add-ition of 10 mM EDTA solution and washing with PBS, followingwhich the sections were stained using Alexa Fluor 488 (Invitro-gen) conjugated to Streptavidin and mounted in medium contain-ing DAPI. To quantify TGM1 activity in the granular layer, theaverage fluorescence intensity of the epidermis of negativecontrol skin was calculated using ImageJ image analysis software(as previous), then subtracted from Biot-MDC-positive specimenimages. The fluorescence intensity in the granular layer of thesenormalized images was then measured.
Staphylococcus aureus invasion and adhesion
Staphylococcus aureus (NCTC 6571) was grown in lysogenybroth (LB) (Sigma Aldrich, Dorset, UK) in an orbital shaker in5% CO2 atmosphere overnight. In duplicate, keratinocyteswere cultured and treated with siRNA as previously described.After 48 h, cells were processed for S. aureus infection: themedia on cells was replaced with 1 ml of serum and antibiotic-free media. S. aureus was added at a multiplicity of infectionof 100 bacteria per cell. The plate was centrifuged at 1000RPM for 10 min to initiate infection and incubated at 378C/10% CO2 for 3 h. The media on the cells was collected as super-natants for ELISA experiments. One well that was used to deter-mine the cell-associated bacteria was lysed immediatelypost-infection. Media containing 75mg/ml of gentamycin wasadded to the other well, which was used to determine the interna-lized bacteria. The plate was incubated at 378C for 1 h andwashed three times with 1× PBS. Cells were trypsinized, col-lected in a tube and lysed with 0.5% Triton X-100 in PBS.Lysates were serially diluted in PBS and were plated onto LBagar. Bacteria were grown overnight at 378C, and the numberof colonies was counted. The number of adhering bacteria wascalculated by subtracting the internalized bacteria from thecell-associated bacteria. The percentage adhesion and invasionefficiencies were calculated relative to the inoculum dose. Foranalysis adhesion and invasion, efficiencies for two controland two TOC keratinocyte cell lines were pooled and comparedacross three independent experiments to give an n value of6. Statistical analysis was carried out using a two-tailed unpairedt-test.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We are grateful to Claus-Werner Franzke (University ofFreiburg, Germany) for the gift of the Biotinylated MDC usedin the transglutaminase activity assays, to MyMahoney (JeffersonInstitute, Philadelphia, PA, USA) for the gift of the anti-DSG2monoclonal antibody used for western blotting studies and toGraham McPhail (Department of Cellular Pathology, Barts and
10 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

The London NHS Trust, London) for the embedding of tissue forelectron microscopy.
Conflict of Interest statement. None declared.
FUNDING
This study was supported by a British Skin Foundation PhD stu-dentship (2008s) and a Medical Research Council/Queen MaryUniversity of London PhD studentship (all awarded to D.P.K).Additional funding came in part from a Barts and The LondonCharity project grant (458/1493; awarded to D.P.K).
REFERENCES
1. Etheridge, S.L., Brooke, M.A., Kelsell, D.P. and Blaydon, D.C. (2013)Rhomboid proteins: a role in keratinocyte proliferation and cancer. CellTissue Res., 351, 301–307.
2. Freeman, M. (2008) Rhomboid proteases and their biological functions.Ann. Rev. Genet., 42, 191–210.
3. Zettl, M., Adrain, C., Strisovsky, K., Lastun, V. and Freeman, M. (2011)Rhomboid family pseudoproteases use the ER quality control machinery toregulate intercellular signaling. Cell, 145, 79–91.
4. Adrain, C., Zettl, M., Christova, Y., Taylor, N. and Freeman, M. (2012)Tumor necrosis factor signaling requires iRhom2 to promote trafficking andactivation of TACE. Science, 335, 225–228.
5. McIlwain, D.R., Lang, P.A., Maretzky, T., Hamada, K., Ohishi, K., Maney,S.K., Berger, T., Murthy, A., Duncan, G., Xu, H.C. et al. (2012) iRhom2regulation of TACE controls TNF-mediated protection against Listeria andresponses to LPS. Science, 335, 229–232.
6. Black, R.A., Rauch, C.T., Kozlosky,C.J., Peschon, J.J., Slack, J.L., Wolfson,M.F., Castner, B.J., Stocking, K.L., Reddy, P., Srinivasan, S. et al. (1997) Ametalloproteinase disintegrin that releases tumour-necrosis factor-alphafrom cells. Nature, 385, 729–733.
7. Moss, M.L., Jin, S.L., Milla, M.E., Bickett, D.M., Burkhart, W., Carter, H.L.,Chen, W.J., Clay, W.C., Didsbury, J.R., Hassler, D. et al. (1997) Cloning of adisintegrin metalloproteinase that processes precursor tumour-necrosisfactor-alpha. Nature, 385, 733–736.
8. Sahin, U., Weskamp, G., Kelly, K., Zhou, H.M., Higashiyama, S., Peschon,J., Hartmann, D., Saftig, P. and Blobel, C.P. (2004) Distinct roles forADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J.Cell Biol., 164, 769–779.
9. Peschon, J.J., Slack, J.L., Reddy, P., Stocking, K.L., Sunnarborg, S.W., Lee,D.C., Russell, W.E., Castner, B.J., Johnson, R.S., Fitzner, J.N. et al. (1998)An essential role for ectodomain shedding in mammalian development.Science, 282, 1281–1284.
10. Gschwind, A., Hart, S., Fischer, O.M. and Ullrich, A. (2003) TACE cleavageof proamphiregulin regulates GPCR-induced proliferation and motility ofcancer cells. EMBO J., 22, 2411–2421.
11. Bech-Serra, J.J., Santiago-Josefat, B., Esselens, C., Saftig, P., Baselga, J.,Arribas, J. and Canals, F. (2006) Proteomic identification of desmoglein 2and activated leukocyte cell adhesion molecule as substrates of ADAM17and ADAM10 by difference gel electrophoresis. Mol. Cell Biol., 26,5086–5095.
12. Klessner, J.L., Desai, B.V., Amargo, E.V., Getsios, S. and Green, K.J. (2009)EGFR and ADAMs cooperate to regulate shedding and endocytic traffickingof the desmosomal cadherin desmoglein 2. Mol. Biol. Cell, 20, 328–337.
13. Schlondorff, J., Becherer, J.D. and Blobel, C.P. (2000) Intracellularmaturation and localization of the tumour necrosis factor alpha convertase(TACE). Biochem J., 347, 131–138.
14. Issuree, P.D., Maretzky, T., McIlwain, D.R., Monette, S., Qing, X., Lang,P.A., Swendeman, S.L., Park-Min, K.H., Binder, N., Kalliolias, G.D. et al.(2013) iRHOM2 is a critical pathogenic mediator of inflammatory arthritis.J. Clin. Invest., 123, 928–932.
15. Jackson, L.F., Qiu, T.H., Sunnarborg, S.W., Chang, A., Zhang, C., Patterson,C. and Lee, D.C. (2003) Defective valvulogenesis in HB-EGF andTACE-null mice is associated with aberrant BMP signaling. EMBO J., 22,2704–2716.
16. Shi, W., Chen, H., Sun, J., Buckley, S., Zhao, J., Anderson, K.D., Williams,R.G. and Warburton, D. (2003) TACE is required for fetal murine cardiacdevelopment and modeling. Dev. Biol., 261, 371–380.
17. Christova, Y., Adrain, C., Bambrough, P., Ibrahim, A. and Freeman, M.(2013) Mammalian iRhoms have distinct physiological functions includingan essential role in TACE regulation. EMBO Rep., 14, 884–890.
18. Blaydon, D.C., Biancheri, P., Di, W.L., Plagnol, V., Cabral, R.M., Brooke,M.A., van Heel, D.A., Ruschendorf, F., Toynbee, M., Walne, A. et al. (2011)Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl.
J. Med., 365, 1502–1508.19. Franzke, C.W., Cobzaru, C., Triantafyllopoulou, A., Loffek, S., Horiuchi,
K., Threadgill, D.W., Kurz, T., van Rooijen, N., Bruckner-Tuderman, L. andBlobel, C.P. (2012) Epidermal ADAM17 maintains the skin barrier byregulating EGFR ligand-dependent terminal keratinocyte differentiation.J. Exp. Med., 209, 1105–1119.
20. Chalaris, A., Adam, N., Sina, C., Rosenstiel, P., Lehmann-Koch, J.,Schirmacher, P., Hartmann, D., Cichy, J., Gavrilova, O., Schreiber, S. et al.
(2010) Critical role of the disintegrin metalloprotease ADAM17 forintestinal inflammation and regeneration in mice. J. Exp. Med., 207,1617–1624.
21. Blaydon, D.C., Etheridge, S.L., Risk, J.M., Hennies, H.C., Gay, L.J., Carroll,R., Plagnol, V., McRonald, F.E., Stevens, H.P., Spurr, N.K. et al. (2012)RHBDF2 mutations are associated with tylosis, a familial esophageal cancersyndrome. Am. J. Hum. Genet., 90, 340–346.
22. Saarinen, S., Vahteristo, P., Lehtonen, R., Aittomaki, K., Launonen, V.,Kiviluoto, T. and Aaltonen, L.A. (2012) Analysis of a Finnish familyconfirms RHBDF2 mutations as the underlying factor in tylosis withesophageal cancer. Fam. Cancer. 11, 525–528.
23. Munger, K., Phelps, W.C., Bubb, V., Howley, P.M. and Schlegel, R. (1989)The E6 and E7 genes of the human papillomavirus type 16 together arenecessary and sufficient for transformation of primary human keratinocytes.J. Virol, 63, 4417–4421.
24. Hawley-Nelson, P., Vousden, K.H., Hubbert, N.L., Lowy, D.R. and Schiller,J.T. (1989) HPV16 E6 and E7 proteins cooperate to immortalize humanforeskin keratinocytes. EMBO J., 8, 3905–3910.
25. Morley, S.M., Dundas, S.R., James, J.L., Gupta, T., Brown, R.A., Sexton,C.J., Navsaria, H.A., Leigh, I.M. and Lane, E.B. (1995) Temperaturesensitivity of the keratin cytoskeleton and delayed spreading of keratinocytelines derived from EBS patients. J. Cell Sci., 108, 3463–3471.
26. Morley, S.M., D’Alessandro, M., Sexton, C., Rugg, E.L., Navsaria, H.,Shemanko, C.S., Huber, M., Hohl, D., Heagerty, A.I., Leigh, I.M. et al.
(2003) Generation and characterization of epidermolysis bullosa simplexcell lines: scratch assays show faster migration with disruptive keratinmutations. Br. J. Dermatol., 149, 46–58.
27. Brooke, M.A., Nitoiu, D. and Kelsell, D.P. (2012) Cell-cell connectivity:desmosomes and disease. J. Pathol., 226, 158–171.
28. Nava, P., Laukoetter, M.G., Hopkins, A.M., Laur, O., Gerner-Smidt, K.,Green, K.J., Parkos, C.A. and Nusrat, A. (2007) Desmoglein-2: a novelregulator of apoptosis in the intestinal epithelium. Mol. Biol. Cell, 18,4565–4578.
29. Lorch, J.H., Klessner, J., Park, J.K., Getsios, S., Wu, Y.L., Stack, M.S. andGreen, K.J. (2004) Epidermal growth factor receptor inhibition promotesdesmosome assembly and strengthens intercellular adhesion in squamouscell carcinoma cells. J. Biol. Chem., 279, 37191–37200.
30. Brennan, D. and Mahoney, M.G. (2009) Increased expression of Dsg2 inmalignant skin carcinomas: a tissue-microarray based study. Cell Adhes.
Migrat., 3, 148–154.31. Maretzky, T., McIlwain, D.R., Issuree, P.D., Li, X., Malapeira, J., Amin, S.,
Lang, P.A., Mak, T.W. and Blobel, C.P. (2013) iRhom2 controls thesubstrate selectivity of stimulated ADAM17-dependent ectodomainshedding. Proc. Natl. Acad. Sci. USA, 110, 11433–11438.
32. Sahin, U. and Blobel, C.P. (2007) Ectodomain shedding of the EGF-receptorligand epigen is mediated by ADAM17. FEBS Lett., 581, 41–44.
33. Pastore, S., Mascia, F., Mariani, V. and Girolomoni, G. (2008) Theepidermal growth factor receptor system in skin repair and inflammation. J.
Invest. Dermatol., 128, 1365–1374.34. Piepkorn, M., Lo, C. and Plowman, G. (1994) Amphiregulin-dependent
proliferation of cultured human keratinocytes: autocrine growth, the effectsof exogenous recombinant cytokine, and apparent requirement forheparin-like glycosaminoglycans. J. Cell. Physiol., 159, 114–120.
35. Elder, J.T., Fisher, G.J., Lindquist, P.B., Bennett, G.L., Pittelkow, M.R.,Coffey, R.J. Jr, Ellingsworth, L., Derynck, R. and Voorhees, J.J. (1989)
Human Molecular Genetics, 2014 11
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Overexpression of transforming growth factor alpha in psoriatic epidermis.Science, 243, 811–814.
36. Zheng, Y., Peng, Z., Wang, Y., Tan, S., Xi, Y. and Wang, G. (2003)Alteration and significance of heparin-bindingepidermal-growth-factor-like growth factor in psoriatic epidermis.Dermatology, 207, 22–27.
37. Piepkorn, M. (1996) Overexpression of amphiregulin, a major autocrinegrowth factor for cultured human keratinocytes, in hyperproliferative skindiseases. Am. J. Dermatopathol., 18, 165–171.
38. Nanney, L.B., Stoscheck, C.M., Magid, M. and King, L.E. Jr. (1986) Altered[125I]epidermal growth factor binding and receptor distribution in psoriasis.J. Invest. Dermatol., 86, 260–265.
39. Dominey, A.M., Wang, X.J., King, L.E. Jr, Nanney, L.B., Gagne, T.A.,Sellheyer, K., Bundman, D.S., Longley, M.A., Rothnagel, J.A., Greenhalgh,D.A. et al. (1993) Targeted overexpression of transforming growth factoralpha in the epidermis of transgenic mice elicits hyperplasia, hyperkeratosis,and spontaneous, squamous papillomas. Cell Growth Diff., 4, 1071–1082.
40. Rittie, L., Varani, J., Kang, S., Voorhees, J.J. and Fisher, G.J. (2006)Retinoid-induced epidermal hyperplasia is mediated by epidermal growthfactor receptor activation via specific induction of its ligandsheparin-binding EGF and amphiregulin in human skin in vivo. J. Invest.
Dermatol., 126, 732–739.41. Stoll, S.W. and Elder, J.T. (1998) Retinoid regulation of heparin-binding
EGF-like growth factor gene expression in human keratinocytes and skin.Experim. Dermatol., 7, 391–397.
42. Barrientos, S., Stojadinovic, O., Golinko, M.S., Brem, H. and Tomic-Canic,M. (2008) Growth factors and cytokines in wound healing. Wound Repair
Regen., 16, 585–601.43. Repertinger, S.K., Campagnaro, E., Fuhrman, J., El-Abaseri, T., Yuspa, S.H.
and Hansen, L.A. (2004) EGFR enhances early healing after cutaneousincisional wounding. J. Invest. Dermatol., 123, 982–989.
44. Tokumaru, S., Higashiyama, S., Endo, T., Nakagawa, T., Miyagawa, J.I.,Yamamori, K., Hanakawa, Y., Ohmoto, H., Yoshino, K., Shirakata, Y. et al.
(2000) Ectodomain shedding of epidermal growth factor receptor ligands isrequired for keratinocyte migration in cutaneous wound healing. J. Cell
Biol., 151, 209–220.45. Li, Y., Fan, J., Chen, M., Li, W. and Woodley, D.T. (2006) Transforming
growth factor-alpha: a major human serum factor that promotes humankeratinocyte migration. J. Invest. Dermatol., 126, 2096–2105.
46. Cha, D., O’Brien, P., O’Toole, E.A., Woodley, D.T. and Hudson, L.G.(1996) Enhanced modulation of keratinocyte motility by transforminggrowth factor-alpha (TGF-alpha) relative to epidermal growth factor (EGF).J. Invest. Dermatol., 106, 590–597.
47. Hashimoto, K., Higashiyama, S., Asada, H., Hashimura, E., Kobayashi, T.,Sudo, K., Nakagawa, T., Damm, D., Yoshikawa, K. and Taniguchi, N.(1994) Heparin-binding epidermal growth factor-like growth factor is anautocrine growth factor for human keratinocytes. J. Biol. Chem., 269,20060–20066.
48. Tolino, M.A., Block, E.R. and Klarlund, J.K. (2011) Brief treatment withheparin-binding EGF-like growth factor, but not with EGF, is sufficient toaccelerate epithelial wound healing. Biochim. Biophys. Acta., 1810,875–878.
49. Shirakata, Y., Kimura, R., Nanba, D., Iwamoto, R., Tokumaru, S.,Morimoto, C., Yokota, K., Nakamura, M., Sayama, K., Mekada, E. et al.
(2005) Heparin-binding EGF-like growth factor accelerates keratinocytemigration and skin wound healing. J. Cell Sci., 118, 2363–2370.
50. Enzinger, P.C. and Mayer, R.J. (2003) Esophageal cancer. N. Engl. J. Med.,349, 2241–2252.
51. Gibault, L., Metges, J.P., Conan-Charlet, V., Lozac’h, P., Robaszkiewicz,M., Bessaguet, C., Lagarde, N. and Volant, A. (2005) Diffuse EGFR stainingis associated with reduced overall survival in locally advanced oesophagealsquamous cell cancer. Br. J. Cancer, 93, 107–115.
52. Abedi-Ardekani, B., Dar, N.A., Mir, M.M., Zargar, S.A., Lone, M.M.,Martel-Planche, G., Villar, S., Mounawar, M., Saidi, F., Malekzadeh, R.et al. (2012) Epidermal growth factor receptor (EGFR) mutations andexpression in squamous cell carcinoma of the esophagus in central Asia.BMC Cancer, 12, 602.
53. Itakura, Y., Sasano, H., Shiga, C., Furukawa, Y., Shiga, K., Mori, S. andNagura, H. (1994) Epidermal growth factor receptor overexpression inesophageal carcinoma. An immunohistochemical study correlated withclinicopathologic findings and DNA amplification. Cancer, 74, 795–804.
54. Kitagawa, Y., Ueda, M., Ando, N., Ozawa, S., Shimizu, N. and Kitajima, M.(1996) Further evidence for prognostic significance of epidermal growthfactor receptor gene amplification in patients with esophageal squamous cellcarcinoma. Clin. Cancer Res., 2, 909–914.
55. Hanawa, M., Suzuki, S., Dobashi, Y., Yamane, T., Kono, K., Enomoto, N.and Ooi, A. (2006) EGFR protein overexpression and gene amplification insquamous cell carcinomas of the esophagus. Int. J. Cancer, 118, 1173–1180.
56. Okines, A., Cunningham, D. and Chau, I. (2011) Targeting the human EGFRfamily in esophagogastric cancer. Nat. Revi. Clin. Oncol., 8, 492–503.
57. Yamabuki, T., Daigo, Y., Kato, T., Hayama, S., Tsunoda, T., Miyamoto, M.,Ito, T., Fujita, M., Hosokawa, M., Kondo, S. et al. (2006) Genome-wide geneexpression profile analysis of esophageal squamous cell carcinomas. Int. J.Oncol., 28, 1375–1384.
58. Rittie,L., Kansra, S.,Stoll,S.W.,Li, Y.,Gudjonsson, J.E., Shao, Y.,Michael,L.E., Fisher, G.J., Johnson, T.M. and Elder, J.T. (2007) Differential ErbB1signaling in squamous cell versus basal cell carcinoma of the skin. Am. J.Pathol., 170, 2089–2099.
59. Cataisson, C., Salcedo, R., Hakim, S., Moffitt, B.A., Wright, L., Yi, M.,Stephens, R., Dai, R.M., Lyakh, L., Schenten, D. et al. (2012) IL-1R-MyD88signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med.,209, 1689–1702.
60. Fujimoto, N., Wislez, M., Zhang, J., Iwanaga, K., Dackor, J., Hanna, A.E.,Kalyankrishna, S., Cody, D.D., Price, R.E., Sato, M. et al. (2005) Highexpression of ErbB family members and their ligands in lungadenocarcinomas that are sensitive to inhibition of epidermal growth factorreceptor. Cancer Res., 65, 11478–11485.
61. Tsai, S.T., Yang, K.Y., Jin, Y.T., Lin, Y.C., Chang, M.T. and Wu, L.W.(2006) Amphiregulin as a tumor promoter for oral squamous cell carcinoma:involvement of cyclooxygenase 2. Oral Oncol., 42, 381–390.
62. Rubin Grandis, J., Melhem, M.F., Gooding, W.E., Day, R., Holst, V.A.,Wagener, M.M., Drenning, S.D. and Tweardy, D.J. (1998) Levels ofTGF-alpha and EGFR protein in head and neck squamous cell carcinoma andpatient survival. J. Natl. Cancer Inst., 90, 824–832.
63. Liu, H.B., Yang, Q.C., Shen, Y., Zhu, Y., Zhang, X.J. and Chen, H. (2013)Clinicopathological and prognostic significance of the expression ofADAM17 mRNA and protein in esophageal squamous cell carcinoma.Zhonghua Zhong Liu Za Zhi, 35, 361–365.
64. Singer, A.J. and Clark, R.A. (1999) Cutaneous wound healing. N. Engl. J.Med., 341, 738–746.
65. Finnerty, C.C., Herndon, D.N., Przkora, R., Pereira, C.T., Oliveira, H.M.,Queiroz, D.M., Rocha, A.M. and Jeschke, M.G. (2006) Cytokine expressionprofile over time in severely burned pediatric patients. Shock, 26, 13–19.
66. Grellner, W., Georg, T. and Wilske, J. (2000) Quantitative analysis ofproinflammatory cytokines (IL-1beta, IL-6, TNF-alpha) in human skinwounds. Forensic Sci. Int., 113, 251–264.
67. Gallucci, R.M., Sloan, D.K., Heck, J.M., Murray, A.R. and O’Dell, S.J.(2004) Interleukin 6 indirectly induces keratinocyte migration. J. Invest.Dermatol., 122, 764–772.
68. Sato, M., Sawamura, D., Ina, S., Yaguchi, T., Hanada, K. and Hashimoto, I.(1999) In vivo introduction of the interleukin 6 gene into humankeratinocytes: induction of epidermal proliferation by the fully spliced formof interleukin 6, but not by the alternatively spliced form. Arch. Dermatol.Res., 291, 400–404.
69. Jiang, W.G., Sanders, A.J., Ruge, F. and Harding, K.G. (2012) Influence ofinterleukin-8 (IL-8) and IL-8 receptors on the migration of humankeratinocytes, the role of PLC-gamma and potential clinical implications.Experim. Therapeut. Med., 3, 231–236.
70. Rennekampff, H.O., Hansbrough, J.F., Kiessig, V., Dore, C., Sticherling, M.and Schroder, J.M. (2000) Bioactive interleukin-8 is expressed in woundsand enhances wound healing. J. Surg. Res., 93, 41–54.
71. Hwang, Y.S., Lee, S.K., Park, K.K. and Chung, W.Y. (2012) Secretion ofIL-6 and IL-8 from lysophosphatidic acid-stimulated oral squamous cellcarcinoma promotes osteoclastogenesis and bone resorption. Oral Oncol.,48, 40–48.
72. Alberti, C., Pinciroli, P., Valeri, B., Ferri, R., Ditto, A., Umezawa, K., Sensi,M., Canevari, S. and Tomassetti, A. (2012) Ligand-dependent EGFRactivation induces the co-expression of IL-6 and PAI-1via the NFkB pathwayin advanced-stage epithelial ovarian cancer. Oncogene, 31, 4139–4149.
73. Getsios, S., Simpson, C.L., Kojima, S., Harmon, R., Sheu, L.J., Dusek, R.L.,Cornwell, M. and Green, K.J. (2009) Desmoglein 1-dependent suppressionof EGFR signaling promotes epidermal differentiation and morphogenesis.J. Cell Biol., 185, 1243–1258.
12 Human Molecular Genetics, 2014
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

74. Simpson, C.L., Kojima, S. and Getsios, S. (2010) RNA interference inkeratinocytes and an organotypic model of human epidermis. Meth. Mol.Biol., 585, 127–146.
75. Kaplan, N., Fatima, A., Peng, H., Bryar, P.J., Lavker, R.M. and Getsios, S.(2012) EphA2/Ephrin-A1 signaling complexes restrict corneal epithelialcell migration. Invest. Ophthalmol. Vis. Sci., 53, 936–945.
76. Livak, K.J. and Schmittgen, T.D. (2001) Analysisof relative gene expressiondata using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.Methods, 25, 402–408.
77. Raghunath, M., Hennies, H.C., Velten, F., Wiebe, V., Steinert, P.M., Reis, A.and Traupe, H. (1998) A novel in situ method for the detection of deficienttransglutaminase activity in the skin. Arch. Dermatol. Res., 290, 621–627.
Human Molecular Genetics, 2014 13
at Galter H
ealth Sciences Library on June 22, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















