
Towards a Phylogeny and Definition of Species at the Molecular Level within the Genus Mycobacterium
8
INTERNATIONAL JOURNAL OF SYSTEMATIC BACTERIOLOGY, OCt. 1990, p. 323-330 0020-7713/90/040323-08$02.00/0 Copyright 0 1990, International Union of Microbiological Societies Vol. 40, No. 4 Towards a Phylogeny and Definition of Species at the Molecular Level within the Genus Mycobacterium TILL ROGALL,l JORN WOLTERS,* THOMAS FLOHR,' AND ERIK C. BOTTGER1* Institut fur Medizinische Mikrobiologie, Medizinische Hochschule Hannover, Konstanty-Gutschow-Strasse 8, 3000 Hannover 61 ,l and Institut fur Allgemeine Mikrobiologie, Christian-Albrechts- Universitat, 2300 Kiel,2 Federal Republic of Germany 16s rRNA sequences from Mycobacterium tuberculosis, M. avium, M. gastri, M. kansasii, M. murinum, M. chelonue, M. smegmatis, M. terrae, M. gordonae, M. scrofuluceum, M. szulgai, M. intracellulare, M. nonchromogenicum, M. xenopi, M. malmoense, M. simiae, M. flavescens, M. fortuitum, and M. paratuberculosis were determined and compared. The sequence data were used to infer a phylogenetic tree, which provided the basis for a systematic phylogenetic analysis of the genus Mycobacterium. The groups of slow- and fast-growing mycobacteria could be differentiated as distinct entities. We found that M. simiae occupies phylogenetically an intermediate position between these two groups. The phylogenetic relatedness within the slow-growing species did not reflect the Runyon classification of photochromogenic, scotochromogenic, and nonchromogenic mycobacteria. In general, the phylogenetic units identified by using rRNA sequences confirmed the validity of phenotypically defined species; an exception was M. gastri, which was indistinguishable from M. kunsasii when this kind of analysis was used. Mycobacteria are aerobic, nonmobile bacteria that are characteristically acid fast. The property of acid fastness, which is due to waxy materials in cell walls, is particularly important for recognizing mycobacteria. Members of the genus Mycobacterium are widespread in nature and range from soil-dwelling saprophytes to pathogens of humans and animals (22, 34). A major descriptive division of mycobac- teria is related to growth rate and pigmentation. On the basis of these criteria, the genus Mycobacterium has been divided into four groups. Group I consists of the photochromogenic (pigmented) species of slow growers; members of group I1 are scotochromogenic slow growers; group I11 contains the nonchromogenic slow growers; and group IV consists of rapid growers (defined as maturing in less than 1 week) (22, 34). Taxonomic analysis of the genus Mycobacterium is com- plicated by the fact that a variety of specialized and complex tests must be used. Numerical taxonomic analysis, which requires that some dozens of characters be tested (e.g., enzymatic activity, growth, morphology, and drug suscepti- bility), is now being applied to circumscription of clusters and description of strains. Early on, the problems and difficulties of traditional taxonomy with respect to mycobac- teria were recognized, prompting a number of investigators in the field to organize themselves into the International Working Group on Mycobacterial Taxonomy and to under- take a number of cooperative taxonomic studies (18, 29, Attempts to subdivide mycobacterial species by using immunological approaches, DNA composition, and similar characteristics (1-3,9,10) proved to be taxonomically useful but gave little phylogenetic information. The use of macro- molecular comparisons to infer phylogenetic relationships is generally accepted and well established. Of the macromol- ecules used for phylogenetic analysis, the rRNAs, in partic- ular 16s rRNA, have proven to be the most useful for establishing phylogenetic relationships because of their high information content, conservative nature, and universal dis- 31-33). * Corresponding author. tribution (7,35). We recently developed a general procedure for the isolation and direct complete nucleotide determina- tion of entire genes coding for 16s rRNA in which the polymerase chain reaction is used (4, 5). In contrast to sequence determinations of 16s rRNAs in which reverse transcriptase is used (15), a procedure which is frequently prone to sequencing anomalies resulting in a level of inac- curacy of the sequence information of roughly 5% (ll), our method allows contiguous and highly accurate sequence determinations. The inaccuracy of the 16s rRNA sequence data obtained when sequences are determined by using reverse transcriptase makes the definition of phylogenetic relationships difficult for species which are related at a rRNA sequence similarity level greater than 95%. To prove the feasibility, accuracy, and usefulness of the sequencing strat- egy based on the polymerase chain reaction, we applied this procedure to the genus Mycobacterium. In this study, which is based on the almost complete 16s rRNA sequences of 19 species of mycobacteria, we demon- strate high levels of 16s rRNA sequence similarity within the genus Mycobacterium (greater than 94.3%); our data provide the basis for a phylogenetically valid taxonomy of this genus. MATERIALS AND METHODS Bacterial strains. Table 1 shows the strains whose 16s rRNA sequences were determined for this study. Determination of sequences. DNA was extracted by using standard procedures (16). Amplification of gene fragments coding for 16s rRNA and direct sequencing of the amplified DNA fragments were performed as described previously (4, 5). Polymerase chain reaction-mediated synthesis was per- formed by using oligonucleotide AGA GTT TGA TCC TGG CTC AG (positions 8 to 28) in combination with CCC TCA ATT CCT TTG AGT TT (positions 928 to 908) and oligonu- cleotide CAG CAG CCG CGG TAA TAC (positions 518 to 536) in combination with AAG GAG GTG ATC CAG CCG CA (positions 1542 to 1522), resulting in two overlapping DNA fragments that covered the 16s rRNA gene. The nucleotide positions indicated above are the target sites of the synthetic oligonucleotides in procaryotic 16s rRNAs, as represented by Escherichia coli. The oligonucleotide prim- 323
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Towards a Phylogeny and Definition of Species at the Molecular Level within the Genus Mycobacterium

INTERNATIONAL JOURNAL OF SYSTEMATIC BACTERIOLOGY, OCt. 1990, p. 323-330 0020-7713/90/040323-08$02.00/0 Copyright 0 1990, International Union of Microbiological Societies
Vol. 40, No. 4
Towards a Phylogeny and Definition of Species at the Molecular Level within the Genus Mycobacterium
TILL ROGALL,l JORN WOLTERS,* THOMAS FLOHR,' AND ERIK C. BOTTGER1* Institut fur Medizinische Mikrobiologie, Medizinische Hochschule Hannover, Konstanty-Gutschow-Strasse 8,
3000 Hannover 61 , l and Institut fur Allgemeine Mikrobiologie, Christian-Albrechts- Universitat, 2300 Kiel,2 Federal Republic of Germany
16s rRNA sequences from Mycobacterium tuberculosis, M. avium, M. gastri, M. kansasii, M. murinum, M. chelonue, M . smegmatis, M. terrae, M. gordonae, M. scrofuluceum, M . szulgai, M . intracellulare, M. nonchromogenicum, M. xenopi, M. malmoense, M . simiae, M. flavescens, M . fortuitum, and M. paratuberculosis were determined and compared. The sequence data were used to infer a phylogenetic tree, which provided the basis for a systematic phylogenetic analysis of the genus Mycobacterium. The groups of slow- and fast-growing mycobacteria could be differentiated as distinct entities. We found that M. simiae occupies phylogenetically an intermediate position between these two groups. The phylogenetic relatedness within the slow-growing species did not reflect the Runyon classification of photochromogenic, scotochromogenic, and nonchromogenic mycobacteria. In general, the phylogenetic units identified by using rRNA sequences confirmed the validity of phenotypically defined species; an exception was M. gastri, which was indistinguishable from M. kunsasii when this kind of analysis was used.
Mycobacteria are aerobic, nonmobile bacteria that are characteristically acid fast. The property of acid fastness, which is due to waxy materials in cell walls, is particularly important for recognizing mycobacteria. Members of the genus Mycobacterium are widespread in nature and range from soil-dwelling saprophytes to pathogens of humans and animals (22, 34). A major descriptive division of mycobac- teria is related to growth rate and pigmentation. On the basis of these criteria, the genus Mycobacterium has been divided into four groups. Group I consists of the photochromogenic (pigmented) species of slow growers; members of group I1 are scotochromogenic slow growers; group I11 contains the nonchromogenic slow growers; and group IV consists of rapid growers (defined as maturing in less than 1 week) (22, 34).
Taxonomic analysis of the genus Mycobacterium is com- plicated by the fact that a variety of specialized and complex tests must be used. Numerical taxonomic analysis, which requires that some dozens of characters be tested (e.g., enzymatic activity, growth, morphology, and drug suscepti- bility), is now being applied to circumscription of clusters and description of strains. Early on, the problems and difficulties of traditional taxonomy with respect to mycobac- teria were recognized, prompting a number of investigators in the field to organize themselves into the International Working Group on Mycobacterial Taxonomy and to under- take a number of cooperative taxonomic studies (18, 29,
Attempts to subdivide mycobacterial species by using immunological approaches, DNA composition, and similar characteristics (1-3,9,10) proved to be taxonomically useful but gave little phylogenetic information. The use of macro- molecular comparisons to infer phylogenetic relationships is generally accepted and well established. Of the macromol- ecules used for phylogenetic analysis, the rRNAs, in partic- ular 16s rRNA, have proven to be the most useful for establishing phylogenetic relationships because of their high information content, conservative nature, and universal dis-
31-33).
* Corresponding author.
tribution (7,35). We recently developed a general procedure for the isolation and direct complete nucleotide determina- tion of entire genes coding for 16s rRNA in which the polymerase chain reaction is used (4, 5). In contrast to sequence determinations of 16s rRNAs in which reverse transcriptase is used (15), a procedure which is frequently prone to sequencing anomalies resulting in a level of inac- curacy of the sequence information of roughly 5% (ll) , our method allows contiguous and highly accurate sequence determinations. The inaccuracy of the 16s rRNA sequence data obtained when sequences are determined by using reverse transcriptase makes the definition of phylogenetic relationships difficult for species which are related at a rRNA sequence similarity level greater than 95%. To prove the feasibility, accuracy, and usefulness of the sequencing strat- egy based on the polymerase chain reaction, we applied this procedure to the genus Mycobacterium.
In this study, which is based on the almost complete 16s rRNA sequences of 19 species of mycobacteria, we demon- strate high levels of 16s rRNA sequence similarity within the genus Mycobacterium (greater than 94.3%); our data provide the basis for a phylogenetically valid taxonomy of this genus.
MATERIALS AND METHODS Bacterial strains. Table 1 shows the strains whose 16s
rRNA sequences were determined for this study. Determination of sequences. DNA was extracted by using
standard procedures (16). Amplification of gene fragments coding for 16s rRNA and direct sequencing of the amplified DNA fragments were performed as described previously (4, 5) . Polymerase chain reaction-mediated synthesis was per- formed by using oligonucleotide AGA GTT TGA TCC TGG CTC AG (positions 8 to 28) in combination with CCC TCA ATT CCT TTG AGT TT (positions 928 to 908) and oligonu- cleotide CAG CAG CCG CGG TAA TAC (positions 518 to 536) in combination with AAG GAG GTG ATC CAG CCG CA (positions 1542 to 1522), resulting in two overlapping DNA fragments that covered the 16s rRNA gene. The nucleotide positions indicated above are the target sites of the synthetic oligonucleotides in procaryotic 16s rRNAs, as represented by Escherichia coli. The oligonucleotide prim-
323

INT. J . SYST. BACTERIOL. 324 ROGALL ET AL.
TABLE 1. Bacteria used in this study
Species or strain Run yon group
Sourcea
M . tuberculosis M . bovis M . bovis BCG M . tuberculosis H37 M . marinum M . kansasii DSM 43224 M . simiae ATCC 2527STb M . scrofulaceum ATCC 19981T M . szulgai ATCC 25799= M . gordonae ATCC 14470T M . xenopi ATCC 19250T M . jlavescens ATCC 14474T M . avium DSM 43216 M . intracellulare ATCC 15985 M . paratuberculosis ATCC 19698 M . gastri ATCC 15754T M . malmoense ATCC 29571T M . nonchromogenicum ATCC 19530T M . terrae ATCC 15755= M . chelonae ATCC 14472 M . smegmatis ATCC 14468 M . fortuitum ATCC 6841T N . asteroides ATCC 3306
IMM
Schroder DSM ATCC ATCC ATCC ATCC ATCC ATCC DSM DSM Jorgensen ATCC ATCC ATCC DSM DSM DSM ATCC DSM
I I I I1 I1 I1 I1 I1 I11 I11 I11 I11 I11 I11 I11 IV IV IV
a ATCC, American Type Culture Collection, Rockville, Md.; DSM, Deut- sche Stammsammlung fur Mikroorganismen, Braunschweig, Federal Repub- lic of Germany; IMM, Institut fur Medizinische Mikrobiologie, Hannover, Federal Republic of Germany; Schroder, K. H. Schroder, Forschungsinstitut, Borstel, Federal Republic of Germany; Jorgensen, J. Jorgensen, National Veterinary Laboratory, Copenhagen, Denmark.
T = type strain.
ers used to sequence the amplified DNA fragments have been described previously (5). The sequencing strategy which we used resulted in almost complete nucleotide deter- mination of the 16s rRNA gene, in which approximately 40% of the sequence was determined for both strands. The complete sequences are available from the European Molec- ular Biology Laboratory data library (accession numbers X 52917 through X 52934) or from one of us (E.C.B.).
Data analysis. The sequences of 19 mycobacteria and Norcardia asteriodes were aligned by using the multise- quence alignment algorithm of Kriiger and Osterburg (12). This algorithm was manually adjusted to account for com- mon secondary structure. For the phylogenetic analysis regions of alignment uncertainty were omitted, reducing the number of positions from 1,481 to 1,431. Pairwise distances were calculated by weighting nucleotide differences and insertions-deletions equally (Hamming distance). The phy- logenetic tree was constructed by using the neighborliness method (6). This method clusters sequences according to their neighborliness in all possible quadruples. In simulation studies this method has been shown to be superior to simple
clustering methods, such as unweighted pair group matrix analysis, and even superior to the distance Wagner, modified Farries, Fitch-Margoliash, and maximum parsimony meth- ods for recovering a given tree topology from distance data when there is a varying rate of nucleotide substitution (20, 23). The necessary algorithms are implemented in the pro- gram package SAGE (technoma GmbH, Heidelberg, Federal Republic of Germany) designed for IBM XTIATIPS’L com- puters and compatibles.
RESULTS AND DISCUSSION
The sequencing strategy used in this study generated nearly complete 16s rRNA sequences (1,481 positions, covering 95.4% of the 16s rRNA molecule). The 16s rRNAs of the mycobacterial species which we investigated are shown aligned in Fig. 1. Similarity values were calculated, and values greater than 94.3% were obtained (Table 2). The same level of similarity was recently reported for other mycobacterial species by Stahl and Urbance (25). To detect varying rates of evolution, a phylogenetic analysis was carried out by using the neighborliness method (6). A phy- logenetic tree displaying the natural relationships among mycobacteria (Fig. 2) was constructed by using equally weighted (Hamming) distances between sequence pairs for 19 mycobacteria by using N . asteroides as an outgroup (Table 3).
In this scheme (Fig. 2) Mycobacterium fortuitum, M . chelonae, M . smegmatis, and M . flavescens formed a tight cluster that was separated from all of the other mycobacte- rial species investigated. This finding correlated with previ- ously described growth characteristics; M . fortuitum, M. chelonae, and M . smegmatis are defined as rapidly growing mycobacteria (34), whereas M . flavescens has a growth rate which is intermediate between the growth rates of the rapidly growing and slowly growing acid-fast bacteria (13). The segregation of M . flavescens into this cluster confirmed previous phenotypic observations, as well as the results of previous DNA-DNA hybridization studies, which indicated that this species is very unlike other scotochromogens (9,13, 14).
Within the genus Mycobacteriurn all of the slow-growing species were highly related (similarity values greater 94.8%), and these species formed a shallow, heterogenous group that was separated from the tight cluster defined by the fast- growing species (Fig. 2). Interestingly, M. simiae, a slow- growing species, occupied an intermediate position in our phylogenetic scheme (Fig. 2); this organism exhibited rRNA sequence elements that were characteristic of rapidly grow- ing species, as well as elements that were characteristic of slowly growing species (see below).
The phylogenetic relatedness between M. terrae and M . nonchromogenicum at the small-subunit rRNA level con- firmed the International Working Group on Mycobacterial
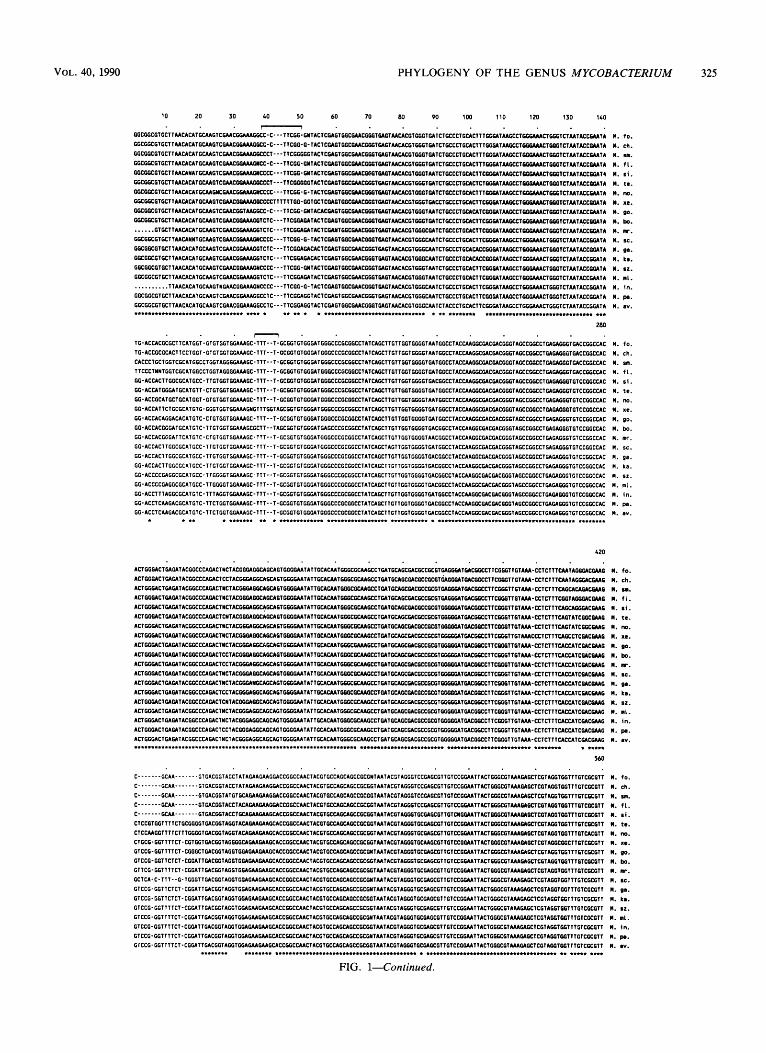
FIG. 1. Alignment of 16s rRNA sequences (total length, 1,481 positions) from M . fortuitum (M.fo.), M . chelonae (M.ch.), M . smegmatis (M.sm.), M . flavescens (M.fl.), M . simiae (M.si.), M . terrae (M.te.), M . nonchromogenicum (M.no.), M . xenopi (M.xe), M . gordonae (M.go.), M . bovis (M.bo.), M . marinum (M.mr.) M . scrofulaceum (M.sc.), M . gastri (M.ga.), M.kansasii (M.ka.), M . szulgai (M.sz.), M . malmoense ( M d . ) , M . intracellulare (M.in.), M . paratuberculosis (M.pa.), and M . avium (M.av.). The sequence of the noncoding (RNA-like) strand is shown. For uniformity, uridine residues were changed to thymidine. A dot or N indicates an undetermined nucleotide, dashes indicate deletions, and nucleotides common to all mycobacterial species are indicated by asteriks. The sequences of M. tuberculosis, M . bovis BCG, and M . tuberculosis H37 are identical to the sequence of M . bovis and therefore are not shown. Compared with the previously published sequence of M . bovis (27), our sequence has an additional cytosine at position 1439; the lack of this residue in the previously published sequence (27) was probably due to a sequencing or cloning artifact. The first and last nucleotides correspond to E. coli 16s rRNA positions 38 and 1510, respectively. Only the relevant portions of the sequences are shown, and the positions of alignment uncertainty that were omitted for the phylogenetic analysis are indicated by brackets.
VOL. 40, 1990 PHYLOGENY OF THE GENUS MYCOBACTERIUM 325
M. fo. M. ch. M. M.
M. f l . M. c i . M. te. M. no. M. xe. M. go. M. bo. M. mr. M. sc. M. ga. M. ke. M. cz. M. nl. M. in. M. pa. M. av.
M. fo. M. ch. M. sm. 11. 11. M. s i . M. te. M. no. M. xe. M. go. M. bo. 16. mr. M. sc. M. ga. M. ka.
M. 92 .
M. ml. M. in. M. pa. M. av.
M. fo. M. ch. M. u. M. f l . M. s i . M. re. k. no. M. xe. M. go.
M. bo. M. M. M. sc. M. 0..
M. to . M. 82.
M. nl. M. in. M. pa. M. av.
M. fo. M. ch. M. M.
M. f l . M. s i . M. te. M. no. M. me. M. go. M. bo. M. mr. M. sc. M. ga. 11. Ira. M. 82.
M. nl. M. in . M. pa. M. ev.

326 ROGALL ET AL. INT. J. SYST. BACTERIOL.
M. fo. M. ch. M. sm. M. f l . M. rl . M. te. M. no. M. X..
M. go. M. bo. M. IK.
M. ac.
M. ka. M. az. M. InI. M. in. (1. p.. M. w.
M. 98.
M. fo. M. ch. M. m. M. 11. M. ri. M. te. M. m. M. xe. M. go. M. bo. M. w. M. sc. M. ga. ti. ka.
M. m l . M. in. M. pa. M. av.
M. 61.
FIG. 1-Continued.

VOL. 40, 1990 PHYLOGENY OF THE GENUS MYCOBACTERKJM 327
TABLE 2. Homology values derived from 16s rRNA sequences"
% Homology with:
M. paratuberculosis M. intracellulare M. malmoense M. smlgai M. kansasii M. gastn' M. scrofulaceum M. marinum M. bovis M. gordonae M. xenopi M. nonchromogenicum M. terrae M. simiae M. fravescens M. smegmatis M. chelonae M. fortuitum N. asteroides
99.9 99.4 99.5 99.1 99.2 99.1 99.1 99.2 99.2 99.9 99.0 99.0 99.0 99.2 99.3 99.0 99.0 99.0 99.2 99.3 100 98.5 98.5 98.7 98.7 98.7 98.8 98.8 98.6 98.7 98.5 98.6 98.7 98.7 98.7 98.2 98.6 98.7 98.5 98.7 98.9 98.8 98.8 98.0 99.4 98.0 98.0 97.9 98.2 98.2 98.1 98.1 97.6 97.9 97.8 95.6 95.7 95.8 95.8 95.9 95.7 95.7 95.4 96.1 96.1 95.6 96.3 96.4 96.2 96.3 96.3 96.1 96.1 96.0 96.7 96.6 96.5 95.8 96.6 96.7 96.6 96.6 96.8 96.7 96.7 96.2 97.5 97.5 97.3 96.3 98.4 96.3 96.4 96.4 96.8 96.8 96.8 96.8 97.0 96.0 96.1 96.7 94.8 95.5 96.0 94.5 94.6 94.6 94.9 94.9 94.7 94.7 94.9 94.9 95.1 95.2 94.5 96.1 96.3 96.9 95.0 95.0 95.0 95.2 95.2 95.1 95.1 95.4 95.4 95.5 95.5 94.7 96.0 96.4 96.8 99.2 95.0 95.0 94.9 94.9 94.9 95.0 95.0 95.3 95.6 95.4 95.3 94.3 96.2 96.1 96.6 97.6 97.4 95.0 95.0 94.9 95.2 95.2 95.1 95.1 95.4 95.9 95.7 95.4 94.8 94.4 96.7 96.8 98.2 97.9 99.0 92.2 92.3 92.2 92.2 92.3 92.2 92.2 92.4 92.3 92.2 92.9 91.7 92.5 92.9 94.3 94.3 93.9 95.0 94.8
a Values were based on the data shown in Fig. 1; 1,431 nucleotides were used.
Taxonomy finding that these two species resemble each other but are distinguishable (18). M. terrae and M. nonchro- mogenicum formed a tight subgroup with individual lines of descent and a common root. M. xenopi emerged phyloge- netically as a clearly distinct species, as indicated by its isolated position among the slow-growing species.
The remaining slow-growing mycobacteria (M. gordonae, M. bovis, M. marinum, M. scrofulaceum, M. gastri, M. kansasii, M. szulgai, M. malmoense, M. intracellulare, M. paratuberculosis, M. avium) were grouped together and had a common line of descent.
M. scrofulaceum could not be clearly separated from the M. avium group by numerical classification; thus, the M. avium-M. intracellulare-M. scrofulaceum complex was formed (28). Phylogenetically, M. scrofulaceum is clearly separated from M. avium and M. intracellulare.
M. malmoense has been described as a species that is different from other species (10, 30, 33). Phylogenetically, M. malmoense, a nonchromogen, is closely related to M. szulgai, a scotochromogen, as demonstrated by a level of 16s rRNA similarity of 99.9%. Consequently, these two species form a tight subgroup.
I M. foRuitum M. chelonae
RAPID GROWERS
M. simiae SLOW GROWERS -------- ns - - - - - - - - - - - //
I M. terrae a - M. nonchromogenicum
a M. xenopi SLOW GROWTH - M. Qordonae
LONG M. bovis HELIX M. marinum 1
M. scrofuiaceum M. Qastri / M. kansasii
(Pas. 390-W)
M. lntracelluiare M. paratuberculosis -
FIG. 2. Phylogenetic tree showing the relationships of species belonging to the genus Mycobacterium. This tree was constructed by using the neighborliness method (6). The tree was rooted by using N. asteriodes as an outgroup. Bar = 10 nucleotide differences. pos., Positions.

328 ROGALL ET AL. INT. J. SYST. BACTERIOL.
TABLE 3. Hamming distances derived from 16s rRNA sequencesa
Hamming distances with:
Species
N. asteroides M. fortuitum M. chelonae M. smegmatis M. flavescens M. simiae M. terrae M. nonchromogenicum M. xenopi M. gordonae M. bovis M. marinum M. scrofulaceum M. gastri M. kansasii M. szulgai M. malmoense M. intracellulare M. paratuberculosis
75 72 88 82 81 102 108 119 15 30 26 46 47 51 75
37 34 49 56 54 81 12 45 51 57 76
44 52 56 79 57 64 74
23 52 60
102 66 67 64 69 47 38 50 63
111 110 61 58 66 63 64 66 70 72 55 57 36 36 48 47 55 56 31 30
8
109 112 112 110 112 66 70 70 68 68 67 71 71 68 68 66 70 70 69 69 72 76 76 73 73 42 46 46 45 45 54 47 47 46 48 57 56 56 52 52 65 61 61 58 60 34 27 27 26 26 28 17 17 16 18 26 19 19 18 20
17 17 19 19 0 10 12
10 12 2
111 110 112 73 71 72 73 71 72 72 71 72 77 77 78 51 51 52 48 47 48 54 51 52 60 62 63 30 28 29 22 19 20 22 19 20 19 21 22 14 14 15 14 14 15 12 12 13 13 12 13
7 8 1
Values were based on the data shown in Fig. 1; 1,431 nucleotides were used.
Numerical taxonomic analysis frequently differentiates M. gastri and M, kansasii as distinct species. M. gastri is classified as a nonchromogenic slow grower, and M. kansasii is classified as a photochromogenic slow-growing species, although nonpigmented M. kansasii strains have been de- scribed (18). At the 16s rRNA sequence level M. gastri is identical to M. kansasii. The close phylogenetic relationship between these two taxa is reflected by the results of an immunodiffusion analysis of cell extracts which identified four antigens that M. kansasii shares with no other species but M. gastri (26), as well as by the results of studies which indicated that the T-catalase of M. gastri is closely related to the T-catalase of M. kansasii (30).
M. intracellulare, M. avium, and M. paratuberculosis form a discrete cluster. In many characteristics, M. paratu- berculosis resembles M. avium (19), and a close genetic relationship between M. paratuberculosis and M. avium has been suspected previously (8). The phenotypic diversity of organisms in the M. avium complex was illustrated by the results of recent DNA studies which indicated that M. paratuberculosis may be synonymous with M. avium (17, 21). Our sequence analysis confirmed and reinforced this notion by finding a level of 16s rRNA similarity of 99.9%.
The sequence divergence among 16s rRNAs is not ran- dom but is confined to certain areas, as comparative analysis of 16s rRNAs revealed regions of highly conserved primary sequences and other regions with a considerable amount of variability (35). The variable portions of rRNA characterize taxa at a genus, group, or species level (35), and natural relationships are also reflected in common sequence patterns or structures among members of a common line of descent
Stackebrandt and Smida found that there is a substantial difference among fast- and slow-growing mycobacteria (24)
(7) -
between positions 370 and 450 (in the numbering system shown in Fig. 1). These authors found that while fast growers consistently form a 21-base-long loop, all slow growers exhibit a longer version in which a 15-base-long loop is interrupted by a 7-base-pair-long stem and a 4-base-long loop. This observation was recently confirmed by Stahl and Urbance (25). However, the data show that there is no exact correlation between the length of the helix comprising posi- tions 390 to 430 and the growth rate since M. simiae, which is characterized as a slow-growing species (33), has the short 21-base-long loop version in this region. These seemingly inconsistent attributes of M. simiae are no contradiction in phylogenetic terms. From a phylogenetic standpoint only groups defined by synapomorphic (shared derived) charac- ters are monophyletic (natural) groups. As Fig. 2 shows, species with a long helix, including M. terrae, M. nonchro- mogenicurn, M. xenopi, M. gordonae, M. bovis, M. mari- num, M. scrofulaceum, M. gastri, M. kunsasii, M. szulgai, M. malmoense, M. intracellulare, M. paratuberculosis, and M. avium, form one monophyletic group. With respect to growth as a different character, fast growth is the plesiomor- phic (primitive) condition, and slow-growing mycobacteria, including M. simiae and the members of the group described above, represent another monophyletic group. Inspection of the collection of mycobacterial small-subunit rRNA se- quences shown in Fig. 1 for sequences that differentiate between fast- and slow-growing species did not reveal sub- stantial differences; instead, both groups could be distin- guished by the composition of a certain few nucleotides (Table 4).
A broad group of slow-growing mycobacteria (including M. gordonae, M. bovis, M. marinum, M. scrofulaceum, M. gastri, M, kansasii, M. szulgai, M. rnalrnoense, M. intra- cellulare, M. paratuberculosis, and M. avium) can be de-

VOL. 40, 1990 PHYLOGENY OF THE GENUS MYCOBACTERIUM 329
TABLE 4. Signature nucleotides
Fast growersb Position“ Slow
growersb Exceptions
141 272
373 412
413
449
450
462
505
1237 1238 1251
T or C A
A G
G
C
C
G
C
G G C
G T
G T
C
G
G
C
G
T A, T
T, G, or deletion
M . phlei, M .
M . phlei (G) M . smegmatis (C),
M . simiae (G) M . smegmatis (A),
M . simiae (G) M . smegmatis (T),
M . simiae (C) M . smegmatis (G),
M . simiae (C) M . phlei, M .
sphagni (C) M . phlei, M.
sphagni (G)
sphagni (T)
M . komossense (deletion)
a Numbering as in Fig. 1. The organisms which we studied are shown in Table 1. In addition, the
relevant portions of the 16s rRNA sequences from M. phlei, M . komossense, “ M . cookii,” and M . sphagni (unpublished data) were included for this analysis.
fined in terms of shared characters (synapomorphy), CCATC at positions 409 to 413, GGTGG at positions 449 to 453, CACG at positions 574 to 577, and GA at positions 588 and 589. Signature nucleotides characteristic for certain slow-growing species ( M . bovis, M . rnarinurn, M . scrofula- ceum, M . gastri, M . kansasii, M . szulgai, M . malrnoense, M . intracellulare, M . paratuberculosis, M . aviurn) can be defined at positions 973 to 977 (CGTCT) and positions 984 to 988 (AGGCG).
Techniques for measuring evolutionary divergence in the structure of semantides (i.e., large information-bearing mol- ecules, such as rRNA, nucleic acids, and proteins) and numerical taxonomy are complementary methods. Numeri- cal taxonomy analyses have proven to be useful for identi- fying previously unrecognized strain clusters, as well as providing information on frequency distribution of features that can be used for strain identification. Numerical taxon- omy may set the basis for a determination system, the basic purpose of which is to identify species, but classification based on phenotypes does not correlate well with natural (i.e., evolutionary) relationships, as defined by macromolec- ular sequence comparisons (35). M. gastri and M . kansasii, as well as M . rnalmoense and M . szulgai, are examples of organisms that have been assigned to different groups ac- cording to the Runyon classification system (22), yet phylo- genetically these pairs of species are closely related, as shown by their 16s rRNA sequences.
Despite the high degree of similarity among mycobacterial 16s rRNAs, the collection of mycobacterial 16s rRNA sequences provided in this paper was used to establish a phylogenetically valid taxonomy of the genus Mycobacte- riurn and should allow rapid and definitive classification of strains of mycobacteria that do not belong to well-estab- lished or thoroughly characterized species, as well as clas- sification of new species which are assigned to this genus.
ACKNOWLEDGMENTS We thank K. Schroder and J. Jorgensen for providing strains, H.
Blocker for providing oligonucleotides, S. Maibom for typing the
manuscript, E. Stackebrandt for critical review of the manuscript, and D. Bitter-Suermann for continuous encouragement.
J.W. was supported by a research grant from the Deutsche Forschungsgemeinsc haft.
1.
2.
3.
4.
5 .
6.
7.
8.
9.
10.
11.
12 <
13.
14.
15.
16.
17.
18.
19.
LITERATURE CITED Baess, I. 1979. Deoxyribonucleic acid relatedness among spe- cies of slowly growing mycobacteria. Acta Pathol. Microbiol. Immunol. Scand. Sect. B 87:221-226. Baess, I. 1982. Deoxyribonucleic acid relatedness among spe- cies of rapidly growing mycobacteria. Acta Pathol. Microbiol. Immunol. Scand. Sect. B 90:371-375. Baess, I. 1983. Deoxyribonucleic acid relationships between different serovars of Mycobacterium avium, Mycobacterium intracellulare and Mycobacterium scrofulaceum. Acta Pathol. Microbiol. Immunol. Scmd. Sect. B 91:201-203. Bottger, E. C. 1989. Rapid determination of bacterial ribosomal RNA sequences by direct sequencing of enzymatically amplified DNA. FEMS Microbiol. Lett. 65171-176. Edwards, U., T. Rogall, H. Blocker, M. Emde, and E. C. Bottger. 1989. Isolation and direct sequencing of entire genes. Characterization of a gene coding for 16s ribosomal RNA. Nucleic Acids Res. 17:7843-7853. Fitch, W. M. 1981. A non-sequential method for constructing trees and hierarchical classifications. J. Mol. Evol. 18:30-37. Fox, G. E., E. Stackebrandt, R. B. Hespell, J. Gibson, J. Maniloff, T. A. Dyer, R. S. Wolfe, W. E. Balch, R. S. Tanner, L. J. Magrum, L. B. Zablen, R. Blakemore, R. Gupta, L. Bonen, B. J. Lewis, D. A. Stahl, K. R. Luehrsen, K. N. Chen, and C. R. Woese. 1980. The phylogeny of prokaryotes. Science 209: 457-463. Grange, J. M. 1984. Mycobacterium avium. Eur. J. Respir. Dis. 65399-401. Gross, W. M., and L. G. Wayne. 1970. Nucleic acid homology in the genus Mycobacterium. J. Bacteriol. 104:630-634. Imaeda, T., G. Broslawski, and S. Imaeda. 1988. Genomic relatedness among mycobacterial species by nonisotopic blot hybridization. Int. J. Syst. Bacteriol. 38:151-156. Johnson, A. M., and P. R. Bavenstock. 1989. Rapid ribosomal RNA sequencing and the phylogenetic analysis of protists. Parasitol. Today 5102-105. Kriiger, M., and G. Osterburg. 1983. On the alignment of two or more molecular sequences. Comput. Programs Biomed. 16:
Kubica, G. P., I. Baess, R. E. Gordon, P. A. Jenkins, J. B. G. Kwapinski, C. McDurmont, S. R. Pattyn, H. Saito, V. Silcox, J. L. Stanford, K. Takeya, and M. Tsukamura. 1972. A cooper- ative numerical analysis of rapidly growing mycobacteria. J. Gen. Microbiol. 7355-70. Kubica, G. P., V. A. Silcox, and E. Hall. 1973. Numerical taxonomy of selected slowly growing mycobacteria. J. Gen. Microbiol. 74:159-167. Lane, D. J., B. Pace, G. J. Ohlsen, D. A. Stahl, M. L. Sogin, and N. R. Pace. 1985. Rapid determination of 16s ribosomal RNA sequences for phylogenetic analysis. Proc. Natl. Acad. Sci. USA 82:6955-6959. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. McFadden, J. J., P. D. Butcher, J. Thompson, R. Chiodini, and J. Hermon-Taylor. 1987. The use of DNA probes identifying restriction-fragment-length polymorphisms to examine the My- cobacterium avium complex. Mol. Microbiol. 1:283-291. Meissner, G., K. H. Schroder, G. E. Amadio, W. Anz, S. Chaparas, H. W. B. Engel, P. A. Jenkins, W. Kappler, H. H. Kleeberg, E. Kubala, M. Kubin, D. Lauterbach, A. Lind, M. Magnusson, Z. Mikova, S. R. Pattyn, W. B. Schaeffer, J. L. Stanford, M. Tsukamura, L. G. Wayne, I. Willers, and E. Wolinsky. 1974. A cooperative numerical analysis of nonscoto- and nonphotochromogenic slowly growing mycobacteria. J. Gen. Microbiol. 83:207-235. Merkal, R. S. 1984. Paratuberculosis, p. 1237-1249. In G. P. Kubica and L. G. Wayne (ed.), The mycobacteria. A source-
61-70.

330 ROGALL ET AL. INT. J. SYST. BACTERIOL.
book, part B. Marcel Dekker, Inc., New York. 20. Saitou, N., and T. Imanishi. 1989. Relative efficiencies of the
Fitch-Margoliash, maximum-parsimony, maximum-likelihood, minimum-evolution, and neighbor-joining methods of phyloge- netic tree construction in obtaining the correct tree. Mol. Biol. Evol. 6514-525.
21. Saxegaard, F., and I. Baess. 1988. Relationship between Myco- bacterium avium, Mycobacterium paratuberculosis and “wood pigeon mycobacteria.” Acta Pathol. Microbiol. Immunol. Scand. Sect. B 96:37-42.
22. Sommers, H. M., and R. C. Good. 1985. Mycobacterium, p. 216-248. In E. H. Lennette, A. Balows, W. J. Hausler, Jr., and H. J. Shadomy (ed.), Manual of clinical microbiology, 4th ed. American Society for Microbiology, Washington, D.C.
23. Sourdis, J., and M. Nei. 1988. Relative efficiencies of the maximum parsimony and distance-matrix methods in obtaining the correct phylogenetic tree. Mol. Biol. Evol. 5298-311.
24. Stackebrandt, E., and J. Smida. 1988. The phylogeny of the genus Mycobacterium as determined by 16s rRNA sequences, and development of DNA probes, p. 244-250. In Biology of actinomycetes. Japan Scientific Societies Press, Tokyo.
25. Stahl, D. A., and J. W. Urbance. 1990. The division between fast- and slow-growing species corresponds to natural relation- ships among the mycobacteria. J. Bacteriol. 172:116-124.
26. Stanford, J. L., and J. M. Grange. 1974. The meaning and structure of species as applied to mycobacteria. Tubercle 55:
27. Suzuki, Y., A. Nagata, Y. Ono, and T. Yamada. 1988. Complete nucleotide sequence of the 16s rRNA gene of Mycobacterium bovis BCG. J. Bacteriol. 170:2886-2889.
28. Tsukamura, M. 1976. Numerical classification of slowly grow- ing mycobacteria. Int. J. Syst. Bacteriol. 26:409420.
29. Wayne, L. G., L. Andrade, S. Froman, W. Kappler, E. Kubala, G. Meissner, and M. Tsukamura. 1978. A cooperative numerical analysis of Mycobacterium gastri, Mycobacterium kansasii and Mycobacterium marinum. J. Gen. Microbiol. 109:319-327.
30. Wayne, L. G., and G. A. Diaz. 1985. Identification of mycobac-
143-152.
teria by specific precipitation of catalase with absorbed sera. J. Clin. Microbiol. 21:721-725.
31. Wayne, L. G., R. C. Good, M. I. Krichevsky, R. E. Beam, Z. Blacklock, S. D. Chaparas, D. Dawson, S. Froman, W. Gross, J. Hawkins, P. A. Jenkins, I. Juhlin, W. Kapper, H. H. Kleeberg, I. Krasnow, M. J. Lefford, E. Mankiewicz, C. McDurmont, G. Meissner, P. Morgan, E. E. Nel, S. R. Pattyn, F. Portaels, P. A. Richards, S. Rusch, K. H. Schroder, V. A. Silcox, I. Szabo, M. Tsukamura, and B. Vergmann. 1981. First report of the coop- erative, open-ended study of slowly growing mycobacteria by the International Working Group on Mycobacterial Taxonomy. Int. J. Syst. Bacteriol. 31:l-20.
32. Wayne, L. G., R. C. Good, M. I. Krichevsky, R. E. Beam, Z. Blacklock, H. L. David, D. Dawson, W. Gross, J. Hawkins, P. A. Jenkins, I. Juhlin, W. Kappler, H. H. Kleeberg, I. Krasnow, M. J. Lefford, E. Mankiewicz, C. McDurmont, E. E. Nel, F. Portaels, P. A. Richards, S. Riisch, K. H. Schroder, V. A. Silcox, I. Szabo, M. Tsukamura, L. Van den Breen, and B. Vergmann. 1983. Second report of the cooperative, open-ended study of slowly growing mycobacteria by the International Working Group on Mycobacterial Taxonomy. Int. J . Syst. Bacteriol.
33. Wayne, L. G., R. C. Good, M. J. Krichevsky, Z. Blacklock, H. L. David, D. Dawson, W. Gross, J. Hawkins, P. A. Jenkins, I. Juhlin, W. Kiippler, H. H. Kleeberg, V. Levy-Frebault, C. McDurmont, E. E. Nel, F. Portaels, S. Rusch-Gerdes, K. H. Schroder, V. A. Silcox, I. Szabo, M. Tsukamura, L. Van den Breen, B. Vergmann, and M. A. Yakrus. 1989. Third report of the cooperative open-ended study of slowly growing mycobac- teria by the International Working Group on Mycobacterial Taxonomy. Int. J. Syst. Bacteriol. 39:267-278.
34. Wayne, L. G., and G. P. Kubica. 1986. The mycobacteria, p. 1435-1457. In P. H. A. Sneath, N. S. Mair, M. E. Sharpe, and J. G. Holt (ed.), Bergey’s manual of systematic bacteriology, vol. 2. The Williams & Wilkins Co., Baltimore.
35. Woese, C. R. 1987. Bacterial evolution. Microbial. Rev. 51:
33:265-274.
221-271.




















