
Seven Gene Phylogeny of Heterokonts
14
Protist, Vol. ], ]]]—]]], ]] ]]]] http://www.elsevier.de/protis Published online date 9 February 2009 ORIGINAL PAPER Seven Gene Phylogeny of Heterokonts Ingvild Riisberg a,d,1 , Russell J.S. Orr b,d,1 , Ragnhild Kluge b,c,2 , Kamran Shalchian-Tabrizi d , Holly A. Bowers e , Vishwanath Patil b,c , Bente Edvardsen a,d , and Kjetill S. Jakobsen b,d,3 a Marine Biology, Department of Biology, University of Oslo, P.O. Box 1066, Blindern, NO-0316 Oslo, Norway b Centre for Ecological and Evolutionary Synthesis (CEES),Department of Biology, University of Oslo, P.O. Box 1066, Blindern, NO-0316 Oslo, Norway c Department of Plant and Environmental Sciences, P.O. Box 5003, The Norwegian University of Life Sciences, N-1432, A ˚ s, Norway d Microbial Evolution Research Group (MERG), Department of Biology, University of Oslo, P.O. Box 1066, Blindern, NO-0316, Oslo, Norway e Center of Marine Biotechnology, 701 East Pratt Street, Baltimore, MD 21202, USA Submitted May 23, 2008; Accepted November 15, 2008 Monitoring Editor: Mitchell L. Sogin Nucleotide ssu and lsu rDNA sequences of all major lineages of autotrophic (Ochrophyta) and heterotrophic (Bigyra and Pseudofungi) heterokonts were combined with amino acid sequences from four protein-coding genes (actin, b-tubulin, cox1 and hsp90) in a multigene approach for resolving the relationship between heterokont lineages. Applying these multigene data in Bayesian and maximum likelihood analyses improved the heterokont tree compared to previous rDNA analyses by placing all plastid-lacking heterotrophic heterokonts sister to Ochrophyta with robust support, and divided the heterotrophic heterokonts into the previously recognized phyla, Bigyra and Pseudofungi. Our trees identified the heterotrophic heterokonts Bicosoecida, Blastocystis and Labyrinthulida (Bigyra) as the earliest diverging lineages. A separate analysis of the phototrophic lineages, by adding the rbcL gene, further resolved the Ochrophyta lineages by increased support for several important nodes. Except for the positioning of Chrysophyceae, Eustigmatophyceae, Raphidophyceae and Pinguiophyceae, all main branches of Ochrophyta were resolved. Our results support the transfer of classes Dictyochophyceae and Pelagophyceae from subphylum Phaeista to Khakista. Based on all our trees, in combination with current knowledge about ultrastructure of heterokonts we suggest that a more advanced flagellar apparatus originated at one occasion in the ancestor of Phaeista whereas, Khakista independently reduced their flagellar apparatus and gained chlorophyll c 3 . & 2008 Elsevier GmbH. All rights reserved. Key words: heterokont phylogeny; covarion; Bayesian; maximum likelihood; multigene; stramenopiles. Introduction Heterokonta was established as a phylum by Cavalier-Smith (1986), comprising all eukaryotic motile biflagellate cells having typically a forward directed flagellum (cilium) with tripartite rigid tubular flagellar hairs (mastigonemes) and a trailing hairless (smooth) flagellum, plus all their descendants having secondarily lost one or both ARTICLE IN PRESS e-mail [email protected] (K.S. Jakobsen). 1 These authors contributed equally to this work. 2 Present address: Norwegian Pollution Control Authority, Box 8100 Dep, 0032 Oslo, Norway. 3 Corresponding author; fax +47 22854001. & 2008 Elsevier GmbH. All rights reserved. doi:10.1016/j.protis.2008.11.004 Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004
Transcript of Seven Gene Phylogeny of Heterokonts

ARTICLE IN PRESS
http://www.elsevier.de/protisPublished online date 9 February 2009
e-mail k.s.jako1These author2Present addr8100 Dep, 003Correspondin
& 2008 Elsevdoi:10.1016/j
Please cite th
], ]]]—]]], ]] ]]]]
Protist, Vol.ORIGINAL PAPER
Seven Gene Phylogeny of Heterokonts
Ingvild Riisberga,d,1, Russell J.S. Orrb,d,1, Ragnhild Klugeb,c,2, Kamran Shalchian-Tabrizid,Holly A. Bowerse, Vishwanath Patilb,c, Bente Edvardsena,d, and Kjetill S. Jakobsenb,d,3
aMarine Biology, Department of Biology, University of Oslo, P.O. Box 1066, Blindern, NO-0316 Oslo, NorwaybCentre for Ecological and Evolutionary Synthesis (CEES),Department of Biology, University of Oslo, P.O. Box1066, Blindern, NO-0316 Oslo, Norway
cDepartment of Plant and Environmental Sciences, P.O. Box 5003, The Norwegian University of LifeSciences, N-1432, As, Norway
dMicrobial Evolution Research Group (MERG), Department of Biology, University of Oslo, P.O. Box 1066,Blindern, NO-0316, Oslo, Norway
eCenter of Marine Biotechnology, 701 East Pratt Street, Baltimore, MD 21202, USA
Submitted May 23, 2008; Accepted November 15, 2008Monitoring Editor: Mitchell L. Sogin
Nucleotide ssu and lsu rDNA sequences of all major lineages of autotrophic (Ochrophyta) andheterotrophic (Bigyra and Pseudofungi) heterokonts were combined with amino acid sequences fromfour protein-coding genes (actin, b-tubulin, cox1 and hsp90) in a multigene approach for resolving therelationship between heterokont lineages. Applying these multigene data in Bayesian and maximumlikelihood analyses improved the heterokont tree compared to previous rDNA analyses by placing allplastid-lacking heterotrophic heterokonts sister to Ochrophyta with robust support, and divided theheterotrophic heterokonts into the previously recognized phyla, Bigyra and Pseudofungi. Our treesidentified the heterotrophic heterokonts Bicosoecida, Blastocystis and Labyrinthulida (Bigyra) as theearliest diverging lineages. A separate analysis of the phototrophic lineages, by adding the rbcL gene,further resolved the Ochrophyta lineages by increased support for several important nodes. Exceptfor the positioning of Chrysophyceae, Eustigmatophyceae, Raphidophyceae and Pinguiophyceae, allmain branches of Ochrophyta were resolved. Our results support the transfer of classesDictyochophyceae and Pelagophyceae from subphylum Phaeista to Khakista. Based on all our trees,in combination with current knowledge about ultrastructure of heterokonts we suggest that a moreadvanced flagellar apparatus originated at one occasion in the ancestor of Phaeista whereas,Khakista independently reduced their flagellar apparatus and gained chlorophyll c3.& 2008 Elsevier GmbH. All rights reserved.
Key words: heterokont phylogeny; covarion; Bayesian; maximum likelihood; multigene; stramenopiles.
[email protected] (K.S. Jakobsen).s contributed equally to this work.ess: Norwegian Pollution Control Authority, Box32 Oslo, Norway.g author; fax +47 22854001.
ier GmbH. All rights reserved..protis.2008.11.004
is article as: Riisberg I, et al. Seven Gene Phylogeny
Introduction
Heterokonta was established as a phylum byCavalier-Smith (1986), comprising all eukaryoticmotile biflagellate cells having typically a forwarddirected flagellum (cilium) with tripartite rigidtubular flagellar hairs (mastigonemes) and atrailing hairless (smooth) flagellum, plus all theirdescendants having secondarily lost one or both
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
2 I. Riisberg et al.
flagella. The diversity among heterokonts is strik-ing; they range from large multicellular seaweedsto tiny unicellular species, they are present infreshwater, marine and terrestrial habitats andembrace many ecologically important algal (e.g.diatoms, brown algae, chrysophytes), and hetero-trophic (e.g. oomycetes) groups. Due to thediversity in Heterokonta it was later raised toinfrakingdom (Cavalier-Smith 1997) with two maingroups; Ochrophyta (Cavalier-Smith 1986) con-sisting mainly of autotrophic heterokonts, and apurely heterotrophic group, which was againfurther subdivided in two phyla: Bigyra andPseudofungi (Cavalier-Smith and Chao 2006).
Due to their diversity and ecological signifi-cance, a number of molecular phylogeneticstudies have been performed on separate hetero-konts groups; predominantly single gene infer-ences (Andersen et al. 1999; Guillou et al.1999; Moriya et al. 2000; Negrisolo et al. 2004;Potter et al. 1997). Investigations on the globalphylogeny of heterokonts are limited to relativelyfew studies of nuclear-encoded ribosomal RNAs(rDNA) or chloroplast-encoded rbcL (ribulose-1,5-bisphosphate carboxylase/oxygenase) (Ben Aliet al. 2001; Daugbjerg and Guillou 2001; Edvard-sen et al. 2007). Recently, the most species-richphylogeny of all three heterokont phyla (Ochro-phyta, Bigyra, Pseudofungi) employing a compre-hensive ssu rDNA dataset was performed (Cava-lier-Smith and Chao 2006). The ssu rDNA studyclarified some sister taxa relationships, and dis-played the heterotrophic groups as deep branch-ing heterokonts (Cavalier-Smith and Chao 2006).The phylogenetic inferences of heterokonts havebeen congruent in placing the plastid-free hetero-trophic species basal to the plastid-containingforms (Cavalier-Smith and Chao 2006), but thestatistical support for this phylogeny has beenonly moderate. According to the chromalveolatehypothesis heterokonts, haptophytes, crypto-phytes and alveolates acquired their red-algalderived plastids in a single secondary endosym-biotic event (Cavalier-Smith 1999). However,recurrent plastid loss and gains have beensuggested for several chromalveolate groups(Bachvaroff et al. 2006; Bodyl 2005; Sanchez-Puerta and Delwiche 2008; Shalchian-Tabrizi et al.2006b). A robust phylogeny for Ochrophyta andthe entirely heterotrophic heterokont groups mayimprove our understanding of the type of pro-cesses forming the current plastid distribution.
Multigene approaches for resolving phyloge-netic relationships using a moderate number ofprotein encoding genes have during the last few
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
years been successfully applied to several protistgroups (Fast et al. 2002; Kim et al. 2006; Nosenkoand Bhattacharya 2007; Shalchian-Tabrizi et al.2006a; Simpson et al. 2006). For heterokonts,two-gene analyses (lsu+ssu rDNA) have beenperformed — but, with the main emphasis onOchrophyta (Ben Ali et al. 2002; Edvardsen et al.2007). In the present study, we have performed atwo-gene lsu+ssu rDNA analysis of all threeheterokont phyla (Ochrophyta, Pseudofungi andBigyra). In order to further enhance the phyloge-netic resolution we have combined the rDNA datawith amino acids from five protein encoding genes(actin, beta-tubulin [b-tubulin], cytochrome oxi-dase subunit I (cox1), heat-shock protein 90(hsp90) and rbcL). Our largest concatenatedalignment consisted of 5643 characters (3886nucleotides and 1757 amino acids) thereby repre-senting the most gene- and character-rich hetero-kont alignment published to date.
Results
Thirty-six new heterokont nucleotide sequencesincluding lsu (rRNA), ssu (rRNA), actin, b-tubulin,cox1 and hsp90 were PCR-amplified for thepurpose of this study and have been madepublicly available with accession numbers fromFJ030880 to FJ030915. In addition, we haveextracted all available accessions from GenBankfor the various heterokont groups including theabove-mentioned genes plus rbcL (Table 1).
Global Phylogeny of Ochrophyta andHeterotrophic Heterokonts
Our dataset included 35 taxa representing tenheterokont classes (Bacillariophyceae, Chryso-phyceae, Dictyochophyceae, Eustigmatophy-ceae, Pelagophyceae, Phaeophyceae, Phaeo-thamniophyceae, Pinguiophyceae, Raphidophy-ceae, Xanthophyceae) as well as the parasitictaxa Blastocystis hominis and Developayella ele-gans. Thus, all major lineages of Ochrophyta,Bigyra and Pseudofungi (heterotrophic hetero-konts) except Bolidiophyceae, Chrysomerophy-ceae, Placididea and Opalinidea (excluded due tolack of available sequence data or lack ofavailable cultures or DNA) were represented.
For all our analyses the Bayesian covarionsequence evolution model (Huelsenbeck 2002)fitted the data better than competing models withthe highest marginal likelihoods. Our phylogeneticanalysis based on lsu+ssu rDNA sequence data
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
Table 1. Taxa, genes and accession numbers of sequences used in the phylogenetic inferences (Figs 1—3)
Taxon lsu ssu actin b-tubulin cox1 hsp90 rbcL Missing%
Aconoidasida AF0134191* AF0134181 ABO696292 XP_0013473692 AAC633902 BM1624043 Heterotroph 0.4CX0201683
Aureococcusanophagereffens
AF289042 U40257 EH117431 AAP42295 — EH117437 AAD39107 14.1
Bacillariophycidae EF5534584 EF1406224 AAV328315 AAW580834 BAA866106 AAX109454 YP_8744184 0.2CT8998704
Blastocystis hominis CT899870 AB023499 EC649064 EC650343 — — Heterotroph 23.2EC649592 EC650685EC651237EC649837EC649960EC645290
Caecitellus parvulus FJ030883 AY642126 ABC85753 — FJ030895 — Heterotroph 17.4Chattonella sp. FJ0308917 AB2176267 — — AAC949767 — AAC029208 16.9Chlorarachniophyceae AF2890369 AF0548329 AAK923639 AAC685069 BAA9635510 DR0415479 Heterotroph 0.4
DR0402089
DR0388999
Cryptomonas sp. AY75299011 AF50827112 AAG3147413 ABR2255111 BAA9635813 — CAJ1966013 11.2Developayella elegans FJ030882 U37107 — — — — Heterotroph 25.3Dictyocha speculum AF289046 U14385 — — AM850252 — AAK85735 19.5Ectocarpales AF33115514 AY30739815 — P3015616 AAC9498017 — AAD2259518 11.2Eimeriorina AF04425219 AF00924419 AAC1376620 XP_62780321 ABL9629022 XP_62692421 Heterotroph 4.8Fucales AF33115123 AB01142324 Q3975823 DV67006325 YP_44862323 ACF0618426 CAC6751827 9.7Glossomastixchrysoplasta
AF409128 AF438324 FJ030905 — FJ030899 FJ030915 AAM45877 9.8
Guillardia theta AF289037 X57162 AAG31473 AAD02570 — AAX10949 NP_050705 6.3Gymnodiniaceae AY83141228 AY83141229 ABI1438729 ABV2220029 ABK5796530 CAJ7575129 Excluded 0.2Heterosigma akashiwo AF409124 DQ191681 AAW56948 AAW58080 — AAX10940 BAD151144 6.3Hyphochytriumcatenoides
X80345 AF163294 — — — — Heterotroph 25.1
Isochrysis galbana DQ202389 AJ246266 AAW56949 AAW58081 BAA24905 AAX10942 BAB20783 6.5EC143993EC146828EC144527EC142332EC137274
Japonochytrium sp. FJ03088731 AB02210431 ABC8574332 ABC6149332 — — Heterotroph 33.6Laminariaceae AF33115333 AB02281834 ABB0244535 CN46811233 NP_65927433 CN46820433 AAR2445033 10.0Mallomonas sp. FJ03088536 FJ03089337 AAW5695036 — — FJ03091336 ABN4695638 15.8
EF16514737
Mischococcales FJ03088139 FJ03089239 — — BAA2496740 FJ03090739 CAE4570039 11.8Nannochloropsisgaditana
FJ030880 AF067957 FJ030901 — — FJ030909 BAC10559 13.7
Nannochloropsis salina Y07975 AF045048 — — — — BAC10567 23.1Nerada mexicana FJ030884 AY520453 FJ030902 — — FJ030912 Heterotroph 15.7Ochromonadaceae Y0797741 EF16513642 AAR2754943 AAW5808743 NP_06645041 AAR2754043 ABN4693444 2.3Pavlova sp. DQ20238845 AF10237346 EC17666247 AAW5808247 AAD1206847 AAX1094447 BAA0828148 12.9
EC17494845 EC17713147
EC17448445
EC17428845
EC17717945
Pedinellales AF28904549 U1438750 — — FJ03089650 FJ03091050 AAC0281750 13.6Pelagococcussubviridis
FJ030889 U14386 — — — — AAC02919 23.4
Pelagomonascalceolata
AF289047 U14389 ABX25970 ABX25960 — — AAC02816 14.3
Peridiniales AY11274651 AF07705551 AAO4934052 AAO4934352 AAM9769451 AAX1094152 Excluded 6.6Phaeothamniales FJ03089053 AF04484654 — — — — AAC6246254 34.4Phytophtora sp. X7563155 AY74276156 P2213156 ABV0008157 AAA3202355 AAX1094658 Heterotroph 0.4Pinguiococcuspyrenoidosa
AF409130 AF438325 FJ030906 — FJ030900 FJ030908 AAM45878 9.9
Prymnesiaceae AF28903859 AB18361260 AAW5695461 ABJ8099762 — AAX1095161 BAB2078861 6.8Pseudochattonellafarcimen
FJ030886 AM075624 FJ030903 FJ030894 FJ030897 — AM850235 27.9
Pythium sp. AY59864763 EF41892564 AAW5695565 AAY4045766 AAM9873867 AAX1094865 Heterotroph 0.3EL77723063
EL77716463
EL774542
3Heterokont Phylogeny
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004
ARTICLE IN PRESS
Table 1. (continued )
Taxon lsu ssu actin b-tubulin cox1 hsp90 rbcL Missing%
Rhizosolenia setigera AF289048 M87329 — — BAA86611 — AAC02907 16.9Saprolegniaceae AF23594368 M3270569 P2618269 P2080270 YP_05291571 AAM9067472 Heterotroph 20.2
AF32058768
Synura sp. DQ98047573 U7322173 ABJ8092573 ABJ8100473 — FJ03091174 ABN4695275 9.5Thalassiosirales Y1151276 AJ63220976 ABC5473877 AAF8190678 YP_31658679 FC50395079 YP_87449879 0.4
FC49718279
FC53609579
Thaumatomonas sp. DQ98047780 AF41125981 ABJ8092780 ABJ8100780 — AAP7216280 Heterotroph 7.5Thraustochytrium sp. FJ03088882 AB02211082 FJ03090482 ABC6149283 FJ03089882 FJ03091482 Heterotroph 18.8Tribonema sp. Y0797984 AF08339785 — — BAA2497584 — AAD0284384 16.9Vacuolaria virescens AF409125 U41651 — — — — AAC02921 24.9
Character endposition
2473 3886 4134 4521 4873 5185 5643
Alignmentaccession
ALIGN_001291 ALIGN_001292 ALIGN_001293 ALIGN_001294 ALIGN_001295 ALIGN_001296 ALIGN_001297
Calculated percentage of total missing characters for each taxon in the multigene alignment is presented.Note, percentages of missing characters for species not included in the phylogenetic inference representedin Fig. 3 are calculated without rbcl (labelled; Heterotroph). The dinoflagellate (Dinophyta) taxa,Gymnodiniaceae and Peridiniales were ‘‘Excluded’’ from the rbcL inference, as this gene is highly derived.All seven single-gene alignments have been made available under the provided accession numbers from theEMBL-Align database: ftp://ftp.ebi.ac.uk/pub/databases/embl/align/*Composite (in silico chimeric) sequences constructed from closely related species indicated as follows:1Theileria parva, 2Plasmodium falciparum, 3Plasmodium sp., 4Phaeodactylum tricornutum, 5Nitzschiathermalis, 6Nitzschia frustulum, 7Chattonella antiqua, 8Chattonella subsalsa, 9Bigelowiella natans, 10Chlor-arachnion reptans, 11Cryptomonas paramecium, 12Cryptomonas platyuris, 13Cryptomonas ovata, 14Stre-blonema maculans, 15Ectocarpus siliculosus, 16Ectocarpus variabilis, 17Ectocarpus sp., 18Myriotrichiaclavaeformis, 19Frenkelia microti, 20Toxoplasma gondii, 21Cryptosporidium parvum, 22Eimeria acervulina,23Fucus vesiculosus, 24Fucus distichus, 25Sargassum binderi, 26Fucus serratus, 27Sargassum muticum,28Akashiwo sanguinea, 29Karlodinium micrum, 30Akashiwo sp., 31Japonochytrium sp., 32Japonochytriummarinum, 33Laminaria digitata, 34Laminaria angustata, 35Saccharina japonica, 36Mallomonas tonsurata,37Mallomonas sp., 38Mallomonas rasilis, 39Chlorellidium tetrabotrys, 40Botrydiopsis alpina, 41Ochromonasdanica, 42Ochromonas distigma, 43Spumella uniguttata, 44Ochromonas sp., 45Pavlova sp., 46Pavlova pinguis,47Pavlova lutheri, 48Pavlova salina, 49Apedinella radians, 50Pseudopedinella elastica, 51Pfiesteria piscicida,52Heterocapsa triquetra, 53Phaeobotrys solitaria, 54Phaeothamnion confervicola, 55Phytophthora mega-sperma, 56Phytophthora infestans, 57Phytophthora glovera, 58Phytophthora palmivora, 59Prymnesiumpatelliferum, 60Prymnesium sp., 61Prymnesium parvum, 62Chrysochromulina sp., 63Pythium sp., 64Pythiumhelicoids, 65Pythium graminicola, 66Pythium macrosporum, 67Pythium aphanidermatum, 68Achlya sp.,69Achlya bisexualis, 70Achlya klebsiana, 71Saprolegnia ferax, 72Achlya ambisexualis, 73Synura sphagnicola,74Synura curtispina, 75Synura petersenii, 76Skeletonema pseudocostatum, 77Skeletonema costatum,78Thalassiosira weissflogii, 79Thalassiosira pseudonana, 80Thaumatomonas sp., 81Thaumatomonas seravini,82Thraustochytrium aureum, 83Thraustochytrium kinnei, 84Tribonema aequale, 85Tribonema intermixtum.
4 I. Riisberg et al.
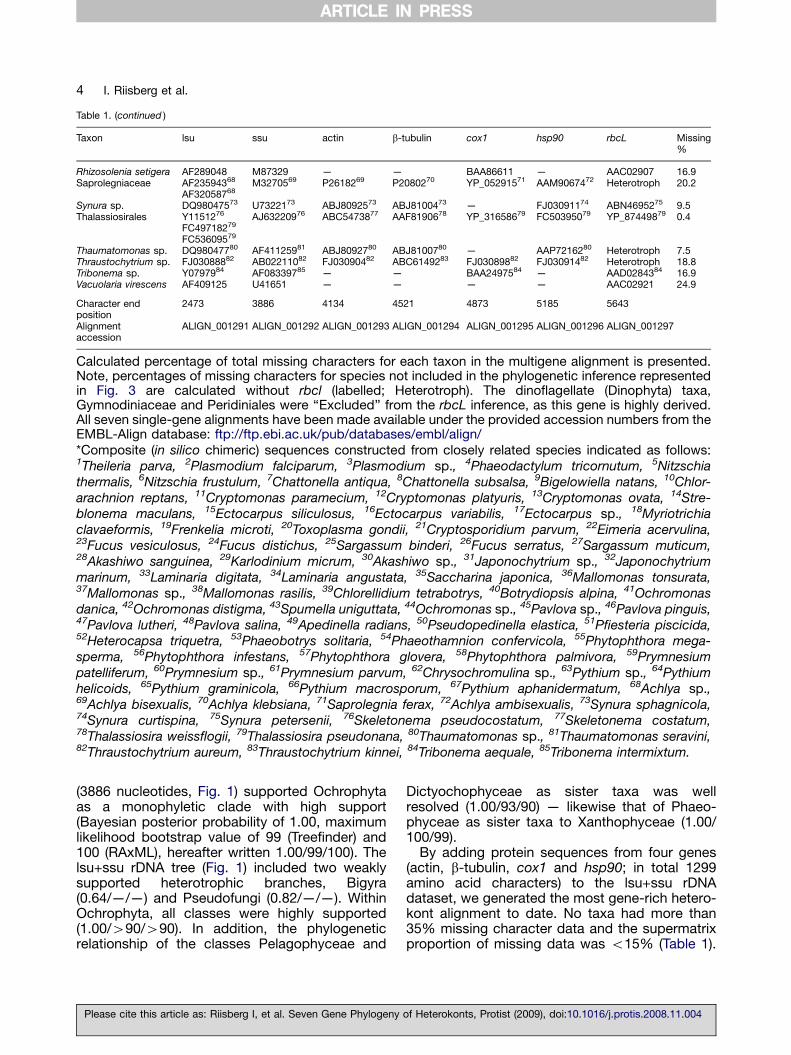
(3886 nucleotides, Fig. 1) supported Ochrophytaas a monophyletic clade with high support(Bayesian posterior probability of 1.00, maximumlikelihood bootstrap value of 99 (Treefinder) and100 (RAxML), hereafter written 1.00/99/100). Thelsu+ssu rDNA tree (Fig. 1) included two weaklysupported heterotrophic branches, Bigyra(0.64/—/—) and Pseudofungi (0.82/—/—). WithinOchrophyta, all classes were highly supported(1.00/490/490). In addition, the phylogeneticrelationship of the classes Pelagophyceae and
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
Dictyochophyceae as sister taxa was wellresolved (1.00/93/90) — likewise that of Phaeo-phyceae as sister taxa to Xanthophyceae (1.00/100/99).
By adding protein sequences from four genes(actin, b-tubulin, cox1 and hsp90; in total 1299amino acid characters) to the lsu+ssu rDNAdataset, we generated the most gene-rich hetero-kont alignment to date. No taxa had more than35% missing character data and the supermatrixproportion of missing data was o15% (Table 1).
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004
ARTICLE IN PRESS
Figure 1. rDNA phylogeny of heterokonts. Combined lsu and ssu phylogeny of Ochrophyta, Bigyra andPseudofungi (3886 characters). The tree is reconstructed with Bayesian inference (MrBayes). Numbers on theinternal nodes represent posterior probability and bootstrap values (450%) for Treefinder and RAxML(ordered; MrBayes/Treefinder/RAxML). Black circles indicate support values of 1.00 posterior probability andbootstrap over 90%. See Table 1 for inferred composite (in silico chimeric) sequences.
5Heterokont Phylogeny
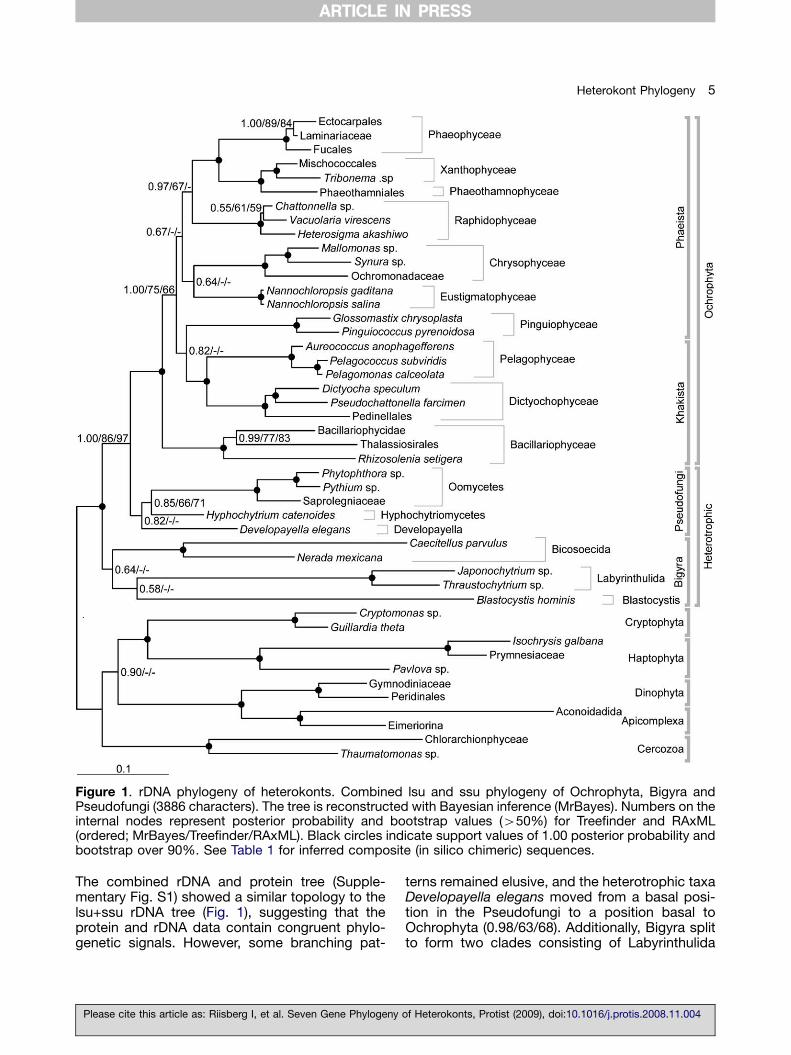
The combined rDNA and protein tree (Supple-mentary Fig. S1) showed a similar topology to thelsu+ssu rDNA tree (Fig. 1), suggesting that theprotein and rDNA data contain congruent phylo-genetic signals. However, some branching pat-
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny
terns remained elusive, and the heterotrophic taxaDevelopayella elegans moved from a basal posi-tion in the Pseudofungi to a position basal toOchrophyta (0.98/63/68). Additionally, Bigyra splitto form two clades consisting of Labyrinthulida
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004
ARTICLE IN PRESS
6 I. Riisberg et al.
and Blastocystis/Bicosoecida (0.96/—/51). Toinvestigate the effect of the fastest evolvingheterotrophic taxa on the topology, PAML wasused to remove fast-evolving classes of sites fromthe alignment (site class 8) (Yang 1997, 2007;
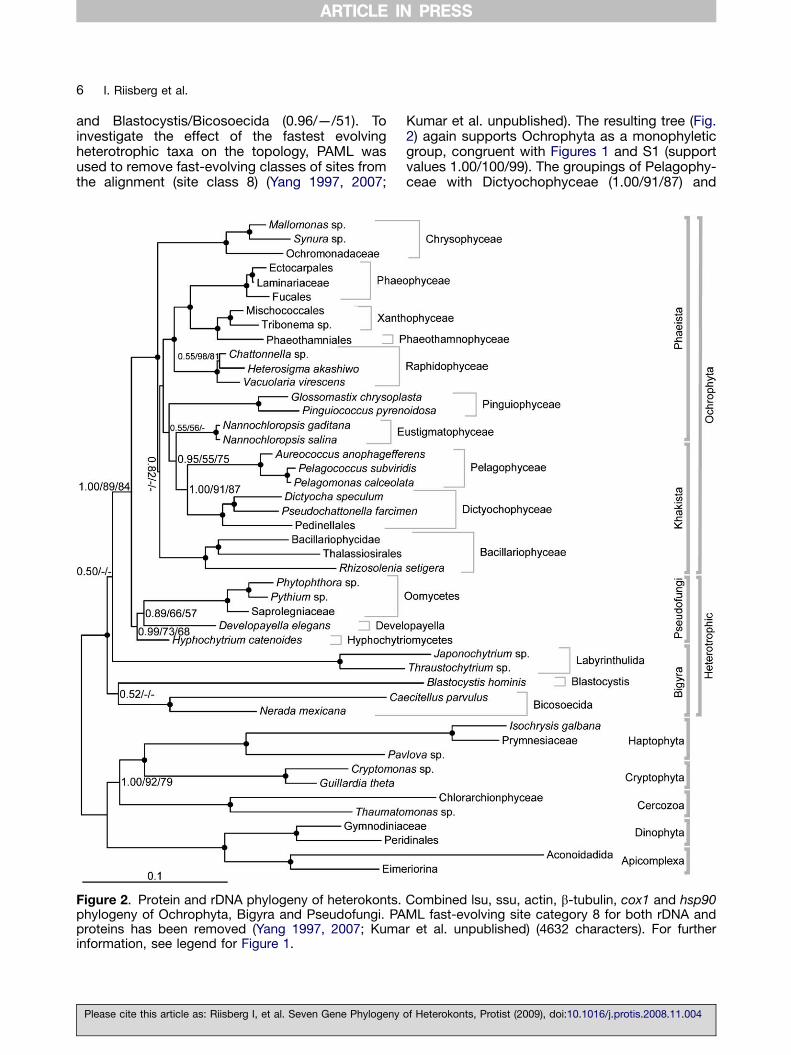
Figure 2. Protein and rDNA phylogeny of heterokonts.phylogeny of Ochrophyta, Bigyra and Pseudofungi. PAproteins has been removed (Yang 1997, 2007; Kumainformation, see legend for Figure 1.
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
Kumar et al. unpublished). The resulting tree (Fig.2) again supports Ochrophyta as a monophyleticgroup, congruent with Figures 1 and S1 (supportvalues 1.00/100/99). The groupings of Pelagophy-ceae with Dictyochophyceae (1.00/91/87) and
Combined lsu, ssu, actin, b-tubulin, cox1 and hsp90ML fast-evolving site category 8 for both rDNA andr et al. unpublished) (4632 characters). For further
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
7Heterokont Phylogeny
Phaeophyceae as sister taxa to Xanthophyceaewere still highly supported (1.00/100/99). Further,Bacillariophyceae clustered at a deep Ochrophytanode, and the placement of Raphidophyceae wasconsistent with Figure 1. The combined rDNA andprotein tree did not resolve the position ofPinguiophyceae, Chrysophyceae and Bacillario-phyceae; all positions received weak support. Incontrast to the tree including fast evolving sites(Fig. S1) and in congruence with Figure 1,Developayella elegans clustered monophyleticallywith the Pseudofungi to form a moderatelysupported clade (0.99/73/68). The branchingpattern for Bigyra remained unresolved. Asbranching pattern for the heterotrophic taxaremained an issue, the approximately unbiased(AU) test (Shimodaira 2002) was applied tocompare several user-defined trees as well asthe optimal topologies obtained from the Bayesiananalyses. The AU test rejected a single plastid loss(heterotrophic monophyly) hypothesis (p-value ¼ 0.00). However, it could not reject aheterotrophic heterokont evolution resulting in 2,3 or 4 separate clades. See Supplementary data;AU tests.
Protein Tree is Congruent with the rDNA andCombined rDNA and Protein Trees
A separate four-gene protein analysis (actin,b-tubulin, cox1, hsp90) of taxa having less than50% missing character data was performed (25taxa, 1299 characters, Fig. S2). As this datasetcontained fewer taxa than Figures 1 and 2, thetree does not reflect the relationship between allclasses, but shows an overall similarity by dividingthe Ochrophyta and the heterotrophic groups(1.00/99/92). The deviations from the other topol-ogies in Figures 1 and 2 were not significantlysupported. Thus, it seems that there are nosignificant conflicts in the phylogenetic signal inthe rDNA and protein datasets. Additionally, thesingle protein gene trees for actin, b-tubulin, cox1and hsp90 did not reveal any supported topolo-gical conflicts with the protein concatenated trees(see Methods and Figs S4—S9).
Addition of rbcL Sequence Data forInference of Ochrophyta Phylogeny
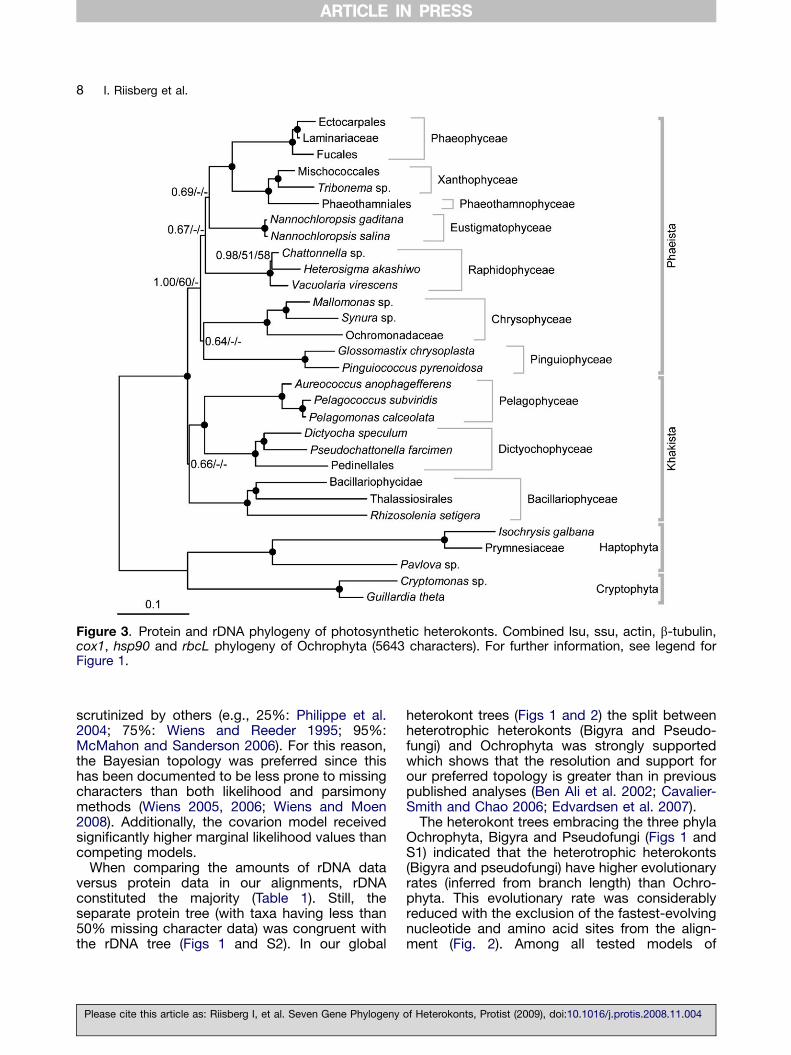
To further improve the resolution of Ochrophyta,we added the rbcL gene to the protein and rDNAsequences (458 amino acids, in total the align-ment constituted 1757 amino acids) and per-
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny
formed a separate analysis of these lineages only(in total 30 taxa, 5643 characters). The hetero-trophic lineages (Bigyra and Pseudofungi) notpossessing the autotrophic rbcL gene wereexcluded and the phylogeny was inferred bothwith and without an outgroup (Figs 3 and S3). Theresulting tree with outgroup (Fig. 3) showed manysimilarities with Figures 1 and 2, but a notablechange was the placement of Pelagophyceae/Dictyochophyceae together with Bacillariophy-ceae (Khakista excluded from Phaeista with1.00/60/—). The exclusion of Khakista fromPhaeista was further highlighted when the rela-tionship was inferred without an outgroup (1.00/80/58) (Fig. S3). The branching pattern withinPhaestia was unresolved with Eustigmatophy-ceae, Raphidophyceae, Chrysophyceae and Pin-guiophyceae receiving no topological support.The single gene tree for rbcL did not reveal anysupported topological conflicts with the concate-nated trees (see Methods and Fig. S10).
Discussion
Multigene Phylogeny and CovarionAnalyses Strongly Divide Ochrophyta andHeterotrophs
In previous studies, usually only a few of theaffiliated classes have clustered together withstatistical support, and many of the deeplydiverging nodes have remained unresolved (BenAli et al. 2001, 2002; Daugbjerg and Andersen1997; Edvardsen et al. 2007). Here we present aconsiderable number of sequences from all themajor classes of the heterokonts, and increase thesupport for class-level phylogenies. Compared toprevious combined lsu+ssu rDNA analyses (BenAli et al. 2002; Edvardsen et al. 2007) weincreased the number of taxa, and instead ofusing only partial sequences including the D1 andD2 domains (Edvardsen et al. 2007) wesequenced and compared the full length of thelsu sequences (Fig. 1).
In all our analyses no taxa had more than 35%missing character data in the inferred alignments(for further details, see Table 1). According toWiens, taxa that are missing up to 50% characterdata can subdivide long branches and improveaccuracy as well as those with no missingcharacter data (Wiens 2005, 2006). In addition,the resulting supermatrices proportion of missingdata never exceeded 15% (14% missing data inFig. 3), a figure significantly lower than that
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004
ARTICLE IN PRESS
Figure 3. Protein and rDNA phylogeny of photosynthetic heterokonts. Combined lsu, ssu, actin, b-tubulin,cox1, hsp90 and rbcL phylogeny of Ochrophyta (5643 characters). For further information, see legend forFigure 1.
8 I. Riisberg et al.
scrutinized by others (e.g., 25%: Philippe et al.2004; 75%: Wiens and Reeder 1995; 95%:McMahon and Sanderson 2006). For this reason,the Bayesian topology was preferred since thishas been documented to be less prone to missingcharacters than both likelihood and parsimonymethods (Wiens 2005, 2006; Wiens and Moen2008). Additionally, the covarion model receivedsignificantly higher marginal likelihood values thancompeting models.
When comparing the amounts of rDNA dataversus protein data in our alignments, rDNAconstituted the majority (Table 1). Still, theseparate protein tree (with taxa having less than50% missing character data) was congruent withthe rDNA tree (Figs 1 and S2). In our global
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
heterokont trees (Figs 1 and 2) the split betweenheterotrophic heterokonts (Bigyra and Pseudo-fungi) and Ochrophyta was strongly supportedwhich shows that the resolution and support forour preferred topology is greater than in previouspublished analyses (Ben Ali et al. 2002; Cavalier-Smith and Chao 2006; Edvardsen et al. 2007).
The heterokont trees embracing the three phylaOchrophyta, Bigyra and Pseudofungi (Figs 1 andS1) indicated that the heterotrophic heterokonts(Bigyra and pseudofungi) have higher evolutionaryrates (inferred from branch length) than Ochro-phyta. This evolutionary rate was considerablyreduced with the exclusion of the fastest-evolvingnucleotide and amino acid sites from the align-ment (Fig. 2). Among all tested models of
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
9Heterokont Phylogeny
sequence evolution the covarion model (Huelsen-beck 2002) fitted the data with the highest loglikelihood. The covarion model is most appro-priate for this dataset due to changes in site-ratepattern, in which sites switch between invariableand variable states across the heterokont tree —similarly to the uneven rates observed among thelineages of other protist nuclear and plastidgenome sequences (Shalchian-Tabrizi et al.2006 a,b,c). Application of the covarion model onthe multigene data resulted in a tree with bettersupport for the basal branches of heterokontsthan earlier shown in studies of global heterokontphylogeny (Ben Ali et al. 2002; Cavalier-Smith andChao 2006; Edvardsen et al. 2007).
In conclusion, some nodes in Ochrophyta werewell resolved (Figs 1 and 2), but despite theamount of data (up to 5185 characters), concate-nating six genes in a multigene analysis, we werenot able to resolve the phylogeny of heterotrophicheterokonts. Our four gene protein tree (Fig. S2) aswell as the combined rDNA+protein tree (Fig. 2)were in congruence with the rDNA tree (Fig. 1). Inall our global heterokont trees, Ochrophyta wasplaced as a monophyletic group clearly dividedfrom the heterotrophic phyla (Bigyra and Pseudo-fungi).
Proposed Relationships of Ochrophyta
In spite of several attempts to resolve theevolutionary relationships among heterokonts,the phylogeny among classes in Ochrophyta hasbeen unclear. Previous analyses (Ben Ali et al.2002; Cavalier-Smith and Chao 2006; Daugbjergand Andersen 1997; Edvardsen et al. 2007)clarified some interclass relationships in Ochro-phyta such as Dictyochophyceae clustering withPelagophyceae (Ben Ali et al. 2001, 2002;Edvardsen et al. 2007) and likewise Phaeophy-ceae as sister taxa to Xanthophyceae (Ben Aliet al. 2002). These sister taxa relationships arestrongly supported in our analyses (Figs 1 and 2)by receiving maximum support values. Whenadding rbcL the separate analysis of Ochrophytadisplayed the three heterokont classes, Bacillar-iophyceae, Pelagophyceae and Dictyochophy-ceae (Khakista) as being excluded from thePhaeista lineages with moderately high support(Fig. 3). This topology is contradictory to Figures 1and 2 as well as Cavalier-Smith and Chao (2006),where the Bacillariophyceae (together with Boli-dophyceae) formed a basal group within Ochro-phyta. Figure 3 shows Phaeothamniophyceaecluster strongly with Xanthophyceae. These two
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny
clades may belong to a single class as earliersuggested (Goertzen and Theriot 2003; Negrisoloet al. 2004). However, more data from deeplydiverging xanthophytes are needed to test thisscenario.
The position of Pinguiophyceae, Chrysophy-ceae, Eustigmatophyceae and Raphidophyceaewithin Phaeista varied in almost all trees, andthese seem to be the clades with weakestaffiliation among all ochrophytes.
Proposed Relationships betweenHeterotrophic Heterokonts (Bigyra andPseudofungi)
The basal heterotrophic branch (Figs 1 and 2)consisting of Bigyra (Bicoecida, Blastocystis andLabyrinthulida) is consistent with previous studies(Cavalier-Smith and Chao 2006) but only weaklysupported. The branch consisting of oomycetesand Hyphochytrium catenoides (forming Pseudo-fungi) was strongly supported, and the mono-phyletic inclusion, with moderate support, ofDevelopayella elegans within the pseudofungiwas achieved when the class of fastest-evolvingsites was excluded from the analysis. Both Bigyraand Pseudofungi were excluded from the Ochro-phyta congruent with earlier studies (Cavalier-Smith and Chao 2006; Leipe et al. 1994, 1996), buthere shown with high support in Bayesian andmaximum likelihood analyses.
Molecular Phylogeny and Ultrastructure
Taken all our trees, plus known ultrastructuraltraits into consideration, we propose that Bacillar-iophyceae, Dictyochophyceae and Pelagophycaeare related and placed within the subphylumKhakista, and separated from subphylum Phaeista(Cavalier-Smith and Chao 2006). This is consistentwith the morphological features as the lineagesbelonging to Khakista have reduced flagellarapparatus (Cavalier-Smith and Chao 2006; Leipeet al. 1994) with no or vestigial flagellar roots(Guillou et al. 1999) and usually no distal transi-tional helix (members of the dictyochophyte orderFlorenciellales is an exception; Edvardsen et al.(2007), but may have a proximal transitional helix(found in some dictyochophytes and pelago-phytes) in the forwardly directed flagellum (Guillouet al. 1999). Furthermore, they lack the pigmentviolaxanthin and possess chlorophyll c3 (Andersen2004; Bjørnland and Liaaen-Jensen 1989; Edvard-sen et al. 2007). The basal bodies are placed in a
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
10 I. Riisberg et al.
depression in the nucleus in Dictyochophyceaeand Pelagophyceae (Edvardsen et al. 2007;Honda et al. 1995) and in Bacillariophyceae, onlyone basal body may be present in the flagellatestage (Preisig 1989). In the remaining taxa, thebasal bodies are placed anterior to the nucleus.Contrary to the original definition of Khakista(Cavalier-Smith and Chao 2006), both the mole-cular and ultrastructural data indicate that Pela-gophyceae and Dictyochophyceae should bemoved from subphylum Phaeista to Khakista.The existence of a monophyletic group consistingof Bacillariophyceae, Pelagophyceae and Dictyo-chophyceae has previously been suggested fromultrastructural, biochemical and ssu rDNA data(Potter et al. 1997; Saunders et al. 1995, Saunderset al. 1997).
This study indicated at least two heterotrophicheterokont clades (Fig. 1) — and possibly more(Figs 2 and S1). Since the number of heterotrophicheterokont taxa is limited in this study, more datafrom other groups will probably further resolve theheterotrophic heterokont tree. Assuming twoheterotrophic clades, as our data may suggest,two independent plastid losses seem to be themost likely scenario given a common photosyn-thetic ancestor of heterokonts (Cavalier-Smith1999). Alternatively, assuming a non-photosyn-thetic origin of heterokonts (see Sanchez-Puertaand Delwiche 2008), only a single acquisition of ared algal (or red algal-derived) plastid in theancestral Ochrophyta needs to be invoked. Con-versely, the evolution of plastids in heterokontsmay be more complex, for example, invokedindependent plastid losses in some Ochrophytalineages (e.g. Oikomonas with the Chrysophyceaeclade; see Cavalier-Smith and Chao 2006).
This study has further resolved the phylogeny ofheterokonts. In particular, the topology withinOchrophyta was better resolved and their separa-tion from heterotrophic heterokonts was robustlydemonstrated. However, despite the substantialincrease in sequence data in this work, there aresome ambiguities among the heterotrophic phyla,Bigyra and Pseudofungi. As most of the characterinformation in our data came from the rDNA geneswe believe it is worth generating an even largeralignment with more genes and increased numberof taxa preferably from the classes Bolidiophy-ceae, Chrysomerophyceae, Placididea and sev-eral of the heterotrophic classes including Opali-nidea as well as the recently identified pico-sizedMAST clades (Kolodziej and Stoeck 2007; Mas-sana et al. 2006). Extending our analyses withimproved taxon and gene sampling combined
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
with ultrastructural characters will enable us tobetter understand the evolution of this diversegroup of organisms.
Methods
Algal cultures: In total 19 different algal strains (Supplemen-tary Table 1) were grown in media and temperature accordingto the specific recommendations from culture collections. Thedictyochophyte Pseudochattonella farcimen strain UIO110was grown in IMR1/2 medium (Eppley et al. 1967) added10 nM selenite and with salinity 25, at the temperature14—15 1C and irradiance of about 100 mmol photons m�2 s�1.
DNA isolation and PCR amplification: Algal cells wereharvested directly from the culture (1 ml) by centrifugation(Eppendorf 5415R, Eppendorf, Hamburg, Germany) at 3220gfor 5 min. Organisms grown on solid surfaces (bottom of Petridishes or agar surfaces) were scraped off and dissolved inmolecular biology grade water prior to centrifugation. DNAfrom the algal pellets was isolated using standard procedureof Dynabeads DNA direct Universal kit (Invitrogen, CA, USA)according to manufactures recommendation.
The primers used for amplifications by PCR are shown inSupplementary Table 2. For degenerate primers the finalconcentration was doubled (20mM). When nested PCRreactions were required, the initial PCR products were diluted1:20 in molecular biology grade water. Amplifications of PCRproducts were optimized and carried out in a Master Cycler(Eppendorf). Two different DNA polymerases HotMaster(Eppendorf) and Phusion (Finnzymes OY, Espoo, Finland)were used (Supplementary Table 2).
Molecular cloning and sequencing: All PCR productswere examined for correct length on a 1.0% agarose gelstained with ethidium bromide. PCR products were purifiedfrom agarose gels, with WizardSV Gel and PCR Clean-UpSystem (Promega, WI, USA). Depending on polymerase usedin PCR we cloned PCR products into TOPO cloning pCR 2.1vector (Invitrogen, CA, USA) or PJET vector (Fermentas, St.Leon-Rot, Germany) and transformed into TOP10 competentbacterial cells (Invitrogen). Plasmids were purified by use ofWizard plus SV miniprep (Promega) and bidirectionallysequenced using the standard M13 sequencing primers orPJET sequencing primers (Fermentas). Internal sequencingprimers (Supplementary Table 2) were also used. One to fiveclones from each PCR product were sequenced andcompared.
Single and multigene alignments: Additional non-ampli-fied sequences used during this study were obtained from thepublicly available GenBank databases (http://www.ncbi.nlm.-nih.gov/). Sequences were compared and manually edited inVector NTI (Invitrogen). Identification of different paralogousgene copies was performed using Pfam gene family seedalignments (Finn et al. 2006) and non-orthologous gene copieswere subsequently removed prior to concatenation.Sequences were aligned to the Pfam alignments usingClustalW (Thompson et al. 1994) and subsequently editedmanually in BioEdit v.7.0.5 (Tippmann 2004) and MacClade(Vazquez 2004). The single gene alignments were then inferredusing RAxML version 7.0.3 (Stamatakis 2006) with 100bootstrap replicates, random starting trees and either theGTRMIX (rDNA) or PROTMIXrtREV(F) (protein) substitutionmodel. This determined if the genes had phylogenticallycompatible histories (no strongly supported conflictingclades). In all cases, no paralogous evidence and no strongly
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
11Heterokont Phylogeny
supported (i.e. bootstrap support 455%) topological conflictswere observed (Figs S4—S10). The only exceptions to thisrule were the placement of Ochromonas danica (Ochromona-daceae) as sister to Caecitelius parvulus in the cox1 tree (Fig.S8) and the placement of Nerada mexicana within theoutgroup in the hsp90 tree (Fig. S9). Both these topologiesand high bootstrap support values are most likely due toartefacts caused by biased taxon sampling; for cox1,Ochromonas danica and Caecitelius are the singular repre-sentatives of Chrysophyceae and Bigyra, whilst for hsp90,Nerada mexicana is adversely affected as being the onlyrepresentative from the basal Bigyra taxa (Bicosoecida andBlastocystis). Removing Ochromonas danica cox1 and Ner-ada mexicana hsp90 sequences for the multigene inferencesdid not change the topology nor maximum likelihood boot-strap values significantly from that presented in Figures 1 and2 (results not shown).
The choice of outgroup has been a contentious issue inresolving heterokont evolutionary relationships (Cavalier-Smith and Chao 2006). An outgroup distant to the ingroupcan result in long branch attraction (LBA) affecting the ingrouprelationships (Hillis 1998; Rannala et al. 1998). However,recent articles that have inferred global eukaryotic phyloge-nies based upon 123+ genes (29908+ amino acid characters)show a clear relationship between the heterokonts, Rhizariaand the alveolates (ciliates, Apicomplexa and dinoflagellates),which form the SAR clade (Burki et al. 2007, 2008). It wastherefore important to include taxa from these lineages inaddition to the chromist divisions of Cryptophyta andHaptophyta, which make up the chromalveolates with theheterokonts and alveolates, for the enhanced resolution ofheterokont evolution.
Two genes (b-tubulin and rbcL) from the dictyochophytePseudochattonella farcimen strain UIO110 originated from anEST library of this organism (Riisberg et al. unpublished;accessions FJ030894 and AM850235, respectively).
Taxa were chosen to prevent missing character data for themultiple gene alignments. However, as missing character datawas extensive for some species, composite (in silico chimeric)sequences were constructed from closely related species. Alltaxa had less than 35% missing character data in the resultingalignments (for further details, see Table 1) and in the resultingsupermatrices the proportion of missing data never exceeded15% (14.37% missing data in Fig. 3).
Phylogenetic analyses and AU test: The evolutionarymodels were chosen on the basis of BIC and AIC criterionsimplemented in ProtTest version 1.3, Modeltest and Treefinder(Abascal et al. 2005; Jobb et al. 2004; Posada and Crandall1998). The best fitting evolutionary models for the proteinpartitions were estimated to be the rtREV+F (frequency)model. For the rDNA partition the GTR evolutionary modelwas preferred. For all analyses we accounted for invariablesites and among-site rate variation by assuming a gammadistribution of site rates approximated with four rate cate-gories. In analyses of concatenated data was divided intorDNA and protein data partitions and the best fitting models ofevolution for each partition applied.
Bayesian analyses were carried out with MrBayes MPIversion 3.1.2 (Huelsenbeck et al. 2001; Ronquist andHuelsenbeck 2003) with the best fitting BIC model estimatedin Prottest, Modeltest and Treefinder. Bayesian covarion treeswere generated from two runs with one heated and three coldchains in the Metropolis-coupled Monte Carlo Markov Chain(MCMC) with 4,000,000 generations implemented. Bothcovarion and non-covarion phylogenies were inferred, how-ever the covarion model, which allows for different between-
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny
site evolutionary rates was preferred in all cases withsignificantly higher marginal likelihoods for the runs. Burn-intrees were set based on the assessment of likelihood plotsand convergence diagnostics implemented in MrBayes. Treesampling was done every 100 generations, and after burn-inthe consensus of the sampled trees used to calculate theposterior probability of the trees topology.
Maximum likelihood (ML) analyses were performed withTreefinder (Jobb et al. 2004) and RAxML version 7.0.3(Stamatakis 2006). Bootstrap analyses were done by 100pseudoreplicates with the same evolutionary model as theinitial search. Trees were inferred from RAxML underGTR(PROT)MIX, with an initial inference under theGTR(PROT)CAT approximation (optimization of individualper-site substitution rates and classification of those individualrates under 25 specified rate categories), before theGTR(PROT)GAMMA model with 4 discrete GAMMA ratecategories was used to evaluate the final tree topology, suchthat it yielded stable likelihood values (Stamatakis 2006).
To investigate the impact of the fast-evolving heterotrophictaxa on the phylogenetic inference, fast-evolving sites wereidentified using PAML (Yang 1997, 2007). The rDNA (lsu andssu) dataset was assessed by estimation with baseML, whilstthe protein dataset (actin, b-tubulin, cox1 and hsp90) wasassessed by estimation with codonML. For both estimations,sites were classified under 8-rate categories, and subsequentsite removal script applied to the alignment (Kumar et al.unpublished). Alignments were constructed with sites 8 andsites 7 and 8 removed; however, subsequent analysis showedthat removal of more than site 8 resulted in a highly reducedalignment and therefore phylogenetic resolution (results notshown) (Kumar et al. unpublished).
The paired-site approximately unbiased (AU) test(Shimodaira 2002) was used to compare incongruent hetero-trophic topologies and implemented in Treefinder (Jobb et al.2004). The heterotrophic topologies to be tested werereoptimized (multifurcations resolved) in Treefinder beforeAU testing. The alignment used was based on Figure S1 (lsu,ssu, actin, b-tubulin, cox1 and hsp90), where no fast evolvingwere sites excluded. Taxon choice for the autotrophicheterokonts was also reduced, with the exclusion of Phaeistataxa from the dataset. The AU test was performed with1,000,000 bootstrap replicates with the same evolutionarymodels and partitions as described earlier (GTR and rtREV+F).The tested topologies and results are shown in Supplemen-tary data.
All model estimation and phylogenetic analyses were doneon the freely available Bioportal at the University of Oslo(http://www.bioportal.uio.no/).
Note added in proof
Tsui et al. (2008) recently published a globalheterokont phylogeny using three protein-codinggenes focusing mainly on the relationshipbetween Labyrinthulida and Bicosoecida (Tsui etal. 2008). In congruence with our topologies theyobtained statistical support for the separation ofBigyra from Pseudofungi and separation betweenthe heterotrophs and Ochrophyta. However in Tsuiet al., Labyrinthulida was moderately supported assister to Bicosoecida. In our trees, based on seven
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
12 I. Riisberg et al.
gene sequences and different taxon sampling, wefound the latter two as weakly supported para-phyletic groups.
Acknowledgements
We would like to thank Thomas Cavalier-Smith fordiscussions and Ema Chao for DNA from Neradamexicana, Cedric Berney for lsu and ssu align-ments of available heterokont sequences, and AveTooming-Klunderud for PCR amplifying and clon-ing of the actin sequence for the LabyrinthulidaThraustochytrium aureum. We thank the Bioportalat the University of Oslo for computer resources.The study was financially supported by grantsfrom the Norwegian Research Council (Projectsno. 166555 and 172572) to KSJ, PhD and by theUniversity of Oslo and the Norwegian University ofLife Sciences for strategic funding (Trippelallian-sen). The seven single-gene alignments used inthis study have been made freely available forpublic access at the EMBL-Align database: ftp://ftp.ebi.ac.uk/pub/databases/embl/align/ underthe accession numbers ALIGN_001291—ALIGN_001297.
Appendix A. Supplementary material
Supplementary data associated with this articlecan be found in the online version at doi:10.1016/j.protis.2008.11.004.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection ofbest-fit models of protein evolution. Bioinformatics 21:2104—2105
Andersen RA, Van de Peer Y, Potter D, Sexton JP, KawachiM, LaJeunesse T (1999) Phylogenetic analysis of the SSUrRNA from members of the Chrysophyceae. Protist 150:71—84
Andersen RA (2004) Biology and systematics of heterokontand haptophyte algae. Am J Bot 91: 1508—1522
Bachvaroff TR, Sanchez-Puerta MV, Delwiche CF (2006)Rate variation as a function of gene origin in plastid-derivedgenes of peridinin-containing dinoflagellates. J Mol Evol 6242-U27
Ben Ali A, De Baere R, Van der Auwera G, De Wachter R,Van de Peer Y (2001) Phylogenetic relationships among algaebased on complete large-subunit rRNA sequences. Int J SystEvol Microbiol 51: 737—749
Ben Ali A, De Baere R, De Wachter R, Van de Peer Y (2002)Evolutionary relationships among heterokont algae (the auto-
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
trophic stramenopiles) based on combined analyses of smalland large subunit ribosomal RNA. Protist 153: 123—132
Bjørnland T, Liaaen-Jensen S (1989) Distribution Patterns ofCarotenoids in Relation to Chromophyte Phylogeny andSystematics. In Green JC, Leadbeater BSC, Diver WL (eds)The Chromophyte Algae: Problems and Perspectives. Sys-temat Assoc, Clarendon Press, Oxford, pp 37—60
Bodyl A (2005) Do plastid-related characters support thechromalveolate hypothesis? J Phycol 41: 712—719
Burki F, Shalchian-Tabrizi K, Minge M, Skjaeveland A,Nikolaev SI, Jakobsen KS, Pawlowski J (2007) Phylogenomicsreshuffles the eukaryotic supergroups. PLoS ONE 2: e790
Burki F, Shalchian-Tabrizi K, Pawlowski J (2008) Phyloge-nomics reveals a new ‘megagroup’ including most photosyn-thetic eukaryotes. Biol Lett 4: 366—369
Cavalier-Smith T (1986) The Kingdom Chromista: Origin andSystematics. In Round FE, Chapman DJ (eds) Progress inPhycological Research, Vol. 4. Biopress Ltd., Bristol,pp 309—347
Cavalier-Smith T (1997) Sagenista and Bigyra, two phyla ofheterotrophic heterokont chromists. Arch Protistenkd 148:253—267
Cavalier-Smith T (1999) Principles of protein and lipidtargeting in secondary symbiogenesis: euglenoid, dinoflagel-late, and sporozoan plastid origins and the eukaryote familytree. J Eukaryot Microbiol 46: 347—366
Cavalier-Smith T, Chao EEY (2006) Phylogeny and mega-systematics of phagotrophic heterokonts (kingdom Chro-mista). J Mol Evol 62: 388—420
Daugbjerg N, Guillou L (2001) Phylogenetic analyses ofBolidophyceae (Heterokontophyta) using rbcL genesequences support their sister group relationship to diatoms.Phycologia 40: 153—161
Daugbjerg N, Andersen RA (1997) A molecular phylogeny ofthe heterokont algae based on analyses of chloroplast-encoded rbcL sequence data. J Phycol 33: 1031—1041
Edvardsen B, Eikrem W, Shalchian-Tabrizi K, Riisberg I,Johnsen G, Naustvoll L, Throndsen J (2007) Verrucophorafarcimen gen. et sp. nov. (Dictyochophyceae, Heterokonta) —a bloom-forming ichthyotoxic flagellate from the Skagerrak,Norway. J Phycol 43: 1054—1070
Eppley R, Holmes RW, E P (1967) Periodicity in cell divisionand physiological behaviour of Ditylum brightwellii, a marineplanktonic diatom during growth in light—dark cycles. ArchMikrobiol 56: 305—323
Fast NM, Xue LR, Bingham S, Keeling PJ (2002) Re-examining alveolate evolution using multiple protein molecularphylogenies. J Eukaryot Microbiol 49: 30—37
Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S,Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A,Durbin R, Eddy SR, Sonnhammer ELL, Bateman A (2006)Pfam: clans, web tools and services. Nucleic Acids Res 34:D247—D251
Goertzen LR, Theriot EC (2003) Effect of taxon sampling,character weighting, and combined data on the interpretationof relationships among the heterokont algae. J Phycol 39:423—439
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
13Heterokont Phylogeny
Guillou L, Chretiennot-Dinet MJ, Medlin LK, Claustre H,Loiseaux-de Goer S, Vaulot D (1999) Bolidomonas: a newgenus with two species belonging to a new algal class, theBolidophyceae (Heterokonta). J Phycol 35: 368—381
Hillis DM (1998) Taxonomic sampling, phylogenetic accuracy,and investigator bias. Syst Biol 47: 3—8
Honda D, Kawachi M, Inouye I (1995) Sulcochrysis biplastidagen. et sp. nov.: cell structure and absolute configuration ofthe flagellar apparatus of an enigmatic chromophyte alga.Phycol Res 43: 1—16
Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP (2001)Bayesian inference of phylogeny and its impact on evolu-tionary biology. Science 294: 2310—2314
Huelsenbeck JP (2002) Testing a covariotide model of DNAsubstitution. Mol Biol Evol 19: 698—707
Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: apowerful graphical analysis environment for molecular phylo-genetics. BMC Evol Biol 4: 18
Kim E, Simpson AGB, Graham LE (2006) Evolutionaryrelationships of apusomonads inferred from taxon-rich ana-lyses of 6 nuclear encoded genes. Mol Biol Evol 23:2455—2466
Kolodziej K, Stoeck T (2007) Cellular identification of a noveluncultured marine stramenopile (MAST-12 clade) small-sub-unit rRNA gene sequence from a Norwegian estuary by use offluorescence in-situ hybridization-scanning electron micro-scopy. Appl Environ Microbiol 73: 2718—2726
Leipe DD, Wainright PO, Gunderson JH, Porter D,Patterson DJ, Valois F, Himmerich S, Sogin ML (1994)The Stramenopiles from a molecular perspective — 16S-likeribosomal-RNA sequences from Labyrinthuloides minuta andCafeteria roenbergensis. Phycologia 33: 369—377
Leipe DD, Tong SM, Goggin CL, Slemenda SB, PieniazekNJ, Sogin ML (1996) 16S-like rDNA sequences from Devel-opayella elegans, Labyrinthuloides haliotidis, and Proteromo-nas lacertae confirm that the stramenopiles are a primarilyheterotrophic group. Eur J Protistol 32: 449—458
Massana R, Terrado R, Forn I, Lovejoy C, Pedros-Alio C(2006) Distribution and abundance of uncultured heterotrophicflagellates in the world oceans. Environ Microbiol 8:1515—1522
McMahon MM, Sanderson MJ (2006) Phylogenetic super-matrix analysis of GenBank sequences from 2228 papilionoidlegumes. Syst Biol 55: 818—836
Moriya M, Nakayama T, Inouye I (2000) Ultrastructure and18S rDNA sequence analysis of Wobblia lunata gen. et an.nov., a new heterotrophic flagellate (Stramenopiles, Incertaesedis). Protist 151: 41—55
Negrisolo E, Maistro S, Incarbone M, Moro I, Dalla Valle L,Broady PA, Andreoli C (2004) Morphological convergencecharacterizes the evolution of Xanthophyceae (Heterokonto-phyta): evidence from nuclear SSU rDNA and plastidial rbcLgenes. Mol Phylogenet Evol 33: 156—170
Nosenko T, Bhattacharya D (2007) Horizontal gene transferin chromalveolates. BMC Evol Biol 7: 173
Philippe H, Snell EA, Bapteste E, Lopez P, Holland PWH,Casane D (2004) Phylogenomics of eukaryotes: impact of
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny
missing data on large alignments. Mol Biol Evol 21:1740—1752
Posada D, Crandall KA (1998) MODELTEST: testing themodel of DNA substitution. Bioinformatics 14: 817—818
Potter D, Saunders GW, Andersen RA (1997) Phylogeneticrelationships of the Raphidophyceae and Xanthophyceae asinferred from nucleotide sequences of the 18S ribosomal RNAgene. Am J Bot 84: 966—972
Preisig HR (1989) The Flagellar Base Ultrastructure andPhylogeny of Chromophytes. In Green JC, Leadbeater BSC,Diver WL (eds) The Chromophyte Algae: Problems andPerspectives. Clarendon Press, Oxford, pp 167—187
Rannala B, Huelsenbeck JP, Yang ZH, Nielsen R (1998)Taxon sampling and the accuracy of large phylogenies. SystBiol 47: 702—710
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesianphylogenetic inference under mixed models. Bioinformatics19: 1572—1574
Sanchez-Puerta MV, Delwiche CF (2008) A hypothesis forplastid evolution in chromalveolates. J Phycol 44: 1097—1107
Saunders GW, Potter D, Paskind MP, Andersen RA (1995)Cladistic analyses of combined traditional and molecular-datasets reveal an algal lineage. Proc Natl Acad Sci USA 92:244—248
Saunders GW, Potter D, Andersen RA (1997) Phylogeneticaffinities of the Sarcinochrysidales and Chrysomeridales(Heterokonta) based on analyses of molecular and combineddata. J Phycol 33: 310—318
Shalchian-Tabrizi K, Eikrem W, Klaveness D, Vaulot D,Minge MA, Le Gall F, Romari K, Throndsen J, Botnen A,Massana R, Thomsen HA, Jakobsen KS (2006a) Telonemia,a new protist phylum with affinity to chromist lineages. Proc RSoc Biol Sci B 273: 1833—1842
Shalchian-Tabrizi K, Minge MA, Cavalier-Smith T, Nedrek-lepp JM, Klaveness D, Jakobsen KS (2006b) Combined heatshock protein 90 and ribosomal RNA sequence phylogenysupports multiple replacements of dinoflagellate plastids. JEukaryot Microbiol 53: 217—224
Shalchian-Tabrizi K, Skanseng M, Ronquist F, KlavenessD, Bachvaroff TR, Delwiche CF, Botnen A, Tengs T,Jakobsen KS (2006c) Heterotachy processes in rhodo-phyte-derived secondhand plastid genes: Implications foraddressing the origin and evolution of dinoflagellate plastids.Mol Biol Evol 23: 1504—1515
Shimodaira H (2002) An approximately unbiased test ofphylogenetic tree selection. Syst Biol 51: 492—508
Simpson AGB, Inagaki Y, Roger AJ (2006) Comprehensivemultigene phylogenies of excavate protists reveal the evolu-tionary positions of ‘‘primitive’’ eukaryotes. Mol Biol Evol 23:615—625
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa andmixed models. Bioinformatics 22: 2688—2690
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal-W —improving the sensitivity of progressive multiple sequencealignment through sequence weighting, position-specific gap
of Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004

ARTICLE IN PRESS
14 I. Riisberg et al.
penalties and weight matrix choice. Nucleic Acids Res 22:4673—4680
Tippmann HF (2004) Analysis for free: comparing programsfor sequence analysis. Brief Bioinform 5: 82—87
Tsui CK, Marshall W, Yokoyama R, Honda D, Lippmeier JC,Craven KD, Peterson PD, Berbee ML (2008) Labyrinthulo-mycetes phylogeny and its implications for the evolutionaryloss of chloroplasts and gain of ectoplasmic gliding. MolPhylogenet Evol 50: 129—140
Vazquez J (2004) Phylogenetics — MacClade 4: analysis ofphylogeny and character evolution, version 4.06. Am BiolTeacher 66: 511—512
Wiens JJ (2005) Can incomplete taxa rescue phylogeneticanalyses from long-branch attraction? Syst Biol 54: 731—742
Please cite this article as: Riisberg I, et al. Seven Gene Phylogeny o
Wiens JJ (2006) Missing data and the design of phylogenetic
analyses. J Biomed Inform 39: 34—42
Wiens JJ, Moen DS (2008) Missing data and the accuracy of
Bayesian phylogenetics. J Syst Evol 46: 307—314
Wiens JJ, Reeder TW (1995) Combining data sets with
different numbers of taxa for phylogenetic analysis. Syst Biol
44: 548—558
Yang Z (1997) PAML: a program package for phylogenetic
analysis by maximum likelihood. Comput Appl Biosci 13:
555—556
Yang Z (2007) PAML 4: phylogenetic analysis by maximum
likelihood. Mol Biol Evol 24: 1586—1591
f Heterokonts, Protist (2009), doi:10.1016/j.protis.2008.11.004




















