
Theoretical prediction and experimental verification of temperature distribution in FGM cylindrical...
7
Chemical Physics Letters 623 (2015) 101–107 Contents lists available at ScienceDirect Chemical Physics Letters jou rn al hom epage: www.elsevier.com/locate/cplett Theoretical prediction and experimental verification on enantioselectivity of haloacid dehalogenase l-DEX YL with chloropropionate Hirotaka Kondo a,∗ , Kazuhiro J. Fujimoto b,∗ , Shigenori Tanaka b,∗ , Hiroyuki Deki c , Takashi Nakamura c,∗ a Graduate School of Human Development and Environment, Kobe University, 3-11 Tsurukabuto, Nada, Kobe 657-8501, Japan b Graduate School of System Informatics, Kobe University, 1-1 Rokkodai, Nada, Kobe 657-8501, Japan c Nagahama Institute of Bio-Science and Technology, 1266 Tamura, Nagahama, Shiga 526-0829, Japan a r t i c l e i n f o Article history: Received 1 December 2014 In final form 29 January 2015 Available online 7 February 2015 a b s t r a c t l-2-Haloacid dehalogenase (l-DEX YL) is a member of a family of enzymes that decontaminate a variety of environmental pollutants such as l-2-chloropropionate (l-2-CPA). This enzyme specifically catalyzes the hydrolytic dehalogenation of l-2-haloacid to produce d-2-hydroxy acid, and does not catalyze that of d-2-haloacid. Here, using the quantum-mechanical/molecular-mechanical and the fragment molecular orbital calculations, the enzymatic reaction of l-DEX YL to d-2-CPA was compared with that to l-2-CPA. As a result, Tyr12, Leu45 and Phe60 were predicted to affect the enantioselectivity. We then performed the site-directed-mutagenesis experiments and the activity measurement of these mutants, thus finding that the F60Y mutant had the enzymatic activity with d-2-CPA. © 2015 Elsevier B.V. All rights reserved. 1. Introduction Many organohalogen compounds are known to be produced biologically or by natural abiogenic process [1]. Produced in large quantities by the chemical industry due to their usefulness as solvents, pharmaceuticals and agrochemicals, these chemicals also cause serious environmental pollution and health problems. Dehalogenases detoxify various halogen compounds, catalyzing the removal of a halogen atom from their substrates, and are use- ful in environmental technology to decontaminate environments polluted with harmful organohalogen compounds. In this con- text, 2-haloacid dehalogenases have been extensively studied and reviewed [2,3]. l-2-Haloacid dehalogenase (l-DEX) is a representative enzyme of the haloacid dehalogenase superfamily, which catalyzes the hydrolytic dehalogenation of l-2-haloalkanoic acids to produce the corresponding d-2-hydroxyalkanoic acids. l-DEX has been isolated and purified from several microbial strains [4]. l-DEX YL was iso- lated from Pseudomonas sp. strain YL cell culture in a medium ∗ Corresponding authors. E-mail addresses: [email protected] (H. Kondo), [email protected] (K.J. Fujimoto), [email protected] (S. Tanaka), t [email protected] (T. Nakamura). containing 2-chloropropionate (2-CPA) as the sole carbon source [5]. The reaction mechanism of l-DEX YL has been analyzed by site-directed mutagenesis [6], X-ray crystallography [7–9], mass spectrometry [10–12], 18 O-incorporation experiments [13], and computer simulations with molecular dynamics (MD), fragment molecular orbital (FMO) [14] and quantum-mechanical/molecular- mechanical (QM/MM) methods [15]. Site-directed mutagenesis experiments have revealed that some amino acid residues are essential for the catalytic function of l-DEX YL [6]. 18 O- incorporation experiments have revealed that Asp10 plays a pivotal role in a nucleophilic attack on a substrate -carbon to form an ester intermediate [13]. These studies suggest the reaction mech- anism shown in Figure 1. Mass spectrometry analysis has shown the involvement of Arg41 and Asp180 in the ester intermediate formation step [12]. X-ray crystallographic analysis of l-DEX YL has provided information on the functions of active-site amino acid residues [7,8]. The contribution of each amino acid residue in l-DEX YL was examined through computer simulations with MD and FMO methods [14]. Recently, QM/MM simulations determined the transition state (TS) structure of the enzyme–substrate com- plex and elucidated its catalytic mechanism in the reaction of ester intermediate formation at the molecular level [15]. The issue on enantioselectivity in dehalogenase has attracted much attention. Previous experiments [4] showed that wild- type of l-DEX YL had the catalytic activity with l-2-CPA but no http://dx.doi.org/10.1016/j.cplett.2015.01.053 0009-2614/© 2015 Elsevier B.V. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Theoretical prediction and experimental verification of temperature distribution in FGM cylindrical...

Tec
HTa
b
c
a
ARIA
1
blaaDtfptr
ohcal
ft
h0
Chemical Physics Letters 623 (2015) 101–107
Contents lists available at ScienceDirect
Chemical Physics Letters
jou rn al hom epage: www.elsev ier .com/ locate /cp le t t
heoretical prediction and experimental verification onnantioselectivity of haloacid dehalogenase l-DEX YL withhloropropionate
irotaka Kondoa,∗, Kazuhiro J. Fujimotob,∗, Shigenori Tanakab,∗, Hiroyuki Dekic,akashi Nakamurac,∗
Graduate School of Human Development and Environment, Kobe University, 3-11 Tsurukabuto, Nada, Kobe 657-8501, JapanGraduate School of System Informatics, Kobe University, 1-1 Rokkodai, Nada, Kobe 657-8501, JapanNagahama Institute of Bio-Science and Technology, 1266 Tamura, Nagahama, Shiga 526-0829, Japan
r t i c l e i n f o
rticle history:eceived 1 December 2014
n final form 29 January 2015vailable online 7 February 2015
a b s t r a c t
l-2-Haloacid dehalogenase (l-DEX YL) is a member of a family of enzymes that decontaminate a varietyof environmental pollutants such as l-2-chloropropionate (l-2-CPA). This enzyme specifically catalyzesthe hydrolytic dehalogenation of l-2-haloacid to produce d-2-hydroxy acid, and does not catalyze that of
d-2-haloacid. Here, using the quantum-mechanical/molecular-mechanical and the fragment molecularorbital calculations, the enzymatic reaction of l-DEX YL to d-2-CPA was compared with that to l-2-CPA.As a result, Tyr12, Leu45 and Phe60 were predicted to affect the enantioselectivity. We then performedthe site-directed-mutagenesis experiments and the activity measurement of these mutants, thus findingthat the F60Y mutant had the enzymatic activity with d-2-CPA.© 2015 Elsevier B.V. All rights reserved.
. Introduction
Many organohalogen compounds are known to be producediologically or by natural abiogenic process [1]. Produced in
arge quantities by the chemical industry due to their usefulnesss solvents, pharmaceuticals and agrochemicals, these chemicalslso cause serious environmental pollution and health problems.ehalogenases detoxify various halogen compounds, catalyzing
he removal of a halogen atom from their substrates, and are use-ul in environmental technology to decontaminate environmentsolluted with harmful organohalogen compounds. In this con-ext, 2-haloacid dehalogenases have been extensively studied andeviewed [2,3].l-2-Haloacid dehalogenase (l-DEX) is a representative enzyme
f the haloacid dehalogenase superfamily, which catalyzes theydrolytic dehalogenation of l-2-haloalkanoic acids to produce the
orresponding d-2-hydroxyalkanoic acids. l-DEX has been isolatednd purified from several microbial strains [4]. l-DEX YL was iso-ated from Pseudomonas sp. strain YL cell culture in a medium∗ Corresponding authors.E-mail addresses: [email protected] (H. Kondo),
[email protected] (K.J. Fujimoto), [email protected] (S. Tanaka), [email protected] (T. Nakamura).
ttp://dx.doi.org/10.1016/j.cplett.2015.01.053009-2614/© 2015 Elsevier B.V. All rights reserved.
containing 2-chloropropionate (2-CPA) as the sole carbon source[5]. The reaction mechanism of l-DEX YL has been analyzed bysite-directed mutagenesis [6], X-ray crystallography [7–9], massspectrometry [10–12], 18O-incorporation experiments [13], andcomputer simulations with molecular dynamics (MD), fragmentmolecular orbital (FMO) [14] and quantum-mechanical/molecular-mechanical (QM/MM) methods [15]. Site-directed mutagenesisexperiments have revealed that some amino acid residuesare essential for the catalytic function of l-DEX YL [6]. 18O-incorporation experiments have revealed that Asp10 plays a pivotalrole in a nucleophilic attack on a substrate �-carbon to form anester intermediate [13]. These studies suggest the reaction mech-anism shown in Figure 1. Mass spectrometry analysis has shownthe involvement of Arg41 and Asp180 in the ester intermediateformation step [12]. X-ray crystallographic analysis of l-DEX YLhas provided information on the functions of active-site aminoacid residues [7,8]. The contribution of each amino acid residuein l-DEX YL was examined through computer simulations with MDand FMO methods [14]. Recently, QM/MM simulations determinedthe transition state (TS) structure of the enzyme–substrate com-plex and elucidated its catalytic mechanism in the reaction of ester
intermediate formation at the molecular level [15].The issue on enantioselectivity in dehalogenase has attractedmuch attention. Previous experiments [4] showed that wild-type of l-DEX YL had the catalytic activity with l-2-CPA but no

102 H. Kondo et al. / Chemical Physics Letters 623 (2015) 101–107
Figure 1. The reaction mechanism of l-DEX YL. l-2-haloacid dehalogenases catalyze hydrolytic dehalogenation of l-2-haloalkanoic acids to produce the correspondingd kyl gro
atppincabohbQgtiitl
oi2plmaew
erc
2
2
aeesgtatiaobgbg
-2-hydroxyalkanoic acids with release of halide ion. X and R mean halogen and al
ctivity with d-2-CPA. These experimental results indicate thathe catalytic reaction was prevented at least in one of the tworocesses, substrate binding process and dehalogenation reactionrocess. In addition to haloacid dehalogenase, the TS structures
n haloalkane dehalogenase [16,17] and in fluoroacetate dehaloge-ase [18] have been computationally determined to elucidate theiratalytic mechanisms. A couple of theoretical studies on the mech-nism of the specific substrate recognition in dehalogenases haveeen reported [19–22]. For example, the recognition mechanismf the fluoroacetate dehalogenase to substrates that have variousalogen atoms (fluorine, chlorine and bromine) was accounted fory X-ray crystallographic analysis, fluorescence spectroscopy andM calculations [19]. An enantioselectivity of haloalkane dehalo-enase DbjA was analyzed by X-ray crystallographic analysis,hermodynamic analysis and MD simulations [20,21]. The signif-cant amino acid residues to recognize the enantiomeric substratesn l-DEX were examined through computer simulations based onhe crystal structure [22]. However, the mechanism of enantiose-ectivity of l-DEX YL has not been clarified yet.
In the present study, we focus on the reason why the wild-typef l-DEX YL does not catalyze the dehalogenation of d-2-CPA. Tonvestigate the difference in the reactions of l-DEX YL between l--CPA and d-2-CPA, QM/MM (ONIOM) and FMO calculations wereerformed, then identifying the important residues for enantiose-
ectivity. Subsequently, we have confirmed the specific activities ofutant enzymes with d-2-CPA by site-directed mutagenesis and
ctivity measurement. Consequently, we have obtained a mutantnzyme (F60Y) which catalyzes the dehalogenation of d-2-CPA asell as the l-2-CPA.
In Section 2, the computational and experimental methodsmployed in the present study are addressed in detail. The mainesults and associated discussions are given in Section 3. The con-lusions with summary are described in Section 4.
. Methods
.1. Computational details
A structure of the enzyme–substrate complex of l-DEX YLnd l-2-CPA in our previous study [15] was used as a refer-nce structure designated as WT/L for comparison. Structures ofnzyme–substrate complex of l-DEX YL and d-2-CPA were con-tructed by using GaussView5 [23]. The positions of functionalroups of l-2-CPA in the structure of WT/L were replaced withhose of other functional groups except the position of chlorine. As
result, three candidate structures were obtained as initial struc-ures for geometry optimization. One complex was called WT/D1,n which the positions of a methyl group and a carboxyl groupt the asymmetric carbon atom in l-2-CPA were exchanged eachther. Another complex called WT/D2 was similarly determined
y exchanging the positions of a hydrogen atom and a methylroup. The other complex called WT/D3 was similarly determinedy exchanging the positions of a hydrogen atom and a carboxylroup. These structures were optimized with the ONIOM methodup including hydrogen, respectively.
[24,25] using Gaussian09 program [26]. The d-2-CPA molecule, acatalytic water, and the side chains of twelve amino acid residueswithin 5 A distance around any substrate atom were describedquantum mechanically, thus including Asp10, Leu11, Tyr12, Arg41,Leu45, Phe60, Ser118, Asn119, Lys151, Asn177, Trp179, and Asp180(155 atoms in total). The whole of the complex was included inMM region (3504 atoms in total). The ONIOM calculations wereperformed with the PM3 [27] method for the QM region and theAmber 96 force field [28] for the MM region (PM3:Amber) using theGaussian09. It is noted that the use of PM3 method in the presentstudy was validated in the earlier work [15] through comparisonwith the density-functional-theory calculations. One hundred fifty-five atoms including the side chains of all residues of the QM regionwere then structurally optimized, while the other atoms were fixedduring the optimization. In the WT/D1 and WT/D2 cases, the car-boxylate of CPA attracted Lys151. In the case of WT/D3, however,the carboxylate attracted Arg41, so that the hydrogen atom linkedto the asymmetric carbon atom in CPA was faced on Asp10. Sincethe hydrogen atom of CPA was too closely placed near Asp10, thepotential energy increased, destabilizing the WT/D3 structure. Thusthe optimization for WT/D3 was not converged. We studied onlyWT/D1 and WT/D2 cases in the following.
Potential energy curves of the first half reaction (i.e., ester inter-mediate formation) for each initial structure were investigated withthe ONIOM method (PM3:Amber) using Gaussian09. The distancebetween nucleophilic oxygen atom closer to the d-2-CPA of theaspartic acid (Asp10) and the asymmetric carbon atom of the d-2-CPA was selected as an internal reaction coordinate. The structurein the QM region was fully optimized with the exception of the driv-ing coordinate, where �-carbons of the twelve residues in the QMregion were fixed [15]. Then a TS structure was obtained from themaximum-point of the potential energy curve, and a reactant struc-ture (enzyme–substrate complex) and a product structure (esterintermediate) were determined with the intrinsic reaction coordi-nate method [29].
To compare the specific roles of amino acid residues amongWT/L, WT/D1 and WT/D2 in the reaction of ester intermediate for-mation, twelve kinds of particular cluster models were extractedfrom the series of whole enzyme structures at the individual pointsof the potential energy curve. One cluster was called ‘control’ modelwhich contained d-2-CPA, catalytic water and the side chain ofAsp10, and it was used as a reference model. The other modelscontained all atoms of ‘control’ model and the side chains of elevenkinds of one additional residue of the QM region. These twelve mod-els were employed for single-point calculations with PM3 method.
To investigate the amino acid residues recognizing the enan-tiomeric substrates in enzyme–substrate complex (i.e., reactant)structure, the single-point FMO calculations were also performedfor WT/L, WT/D1 and WT/D2 at the MP2/6-31G* level [30] usingthe abinit-mp program [31]. Each of amino acid residue, CPA and
catalytic water were set to one fragment in the calculations. Inter-fragment interaction energies (IFIEs) [31] for CPA in their structureswere then calculated. Total IFIE for CPA was also used for analyzingthe affinity between enzyme and substrate, so that total IFIEs of
ysics
Wgio
2
C((eTGwwP[pJ
pTCtripa6HYdacpfYaAt
wpsiGC5G(Cwt
iowttT4(5(
H. Kondo et al. / Chemical Ph
T/L, WT/D1 and WT/D2 were compared. Furthermore, an analo-ous FMO calculation was carried out for l-DEX YL mutant as well,n which an amino acid residue was computationally mutated andptimized with ONIOM method.
.2. Experimental details
Both enantiomers of 2-CPA were purchased from Tokyohemical Industry Co., Ltd. (Tokyo, Japan); BromothymolblueBTB) and N-2-hydroxyethyl-piperazine-N′-2-ethanesulfonic acidHEPES) from Nacalai Tesque Inc. (Kyoto, Japan). Restrictionnzymes and kits for genetic manipulation were purchased fromakara Bio Inc. (Shiga, Japan), Toyobo Co., Ltd. (Osaka, Japan), andE Healthcare Japan (Tokyo, Japan). The synthetic primers usedere from Eurofins Genomics K.K. (Tokyo, Japan). All other reagentsere of analytical grade and obtained from Nacalai and Wako
ure Chemical Industries, Ltd. (Osaka, Japan). The plasmid pBA5016,32], which contains l-DEX YL gene (l-dex YL) on the pUC19-basedlasmid, was a kind gift from T. Kurihara (Kyoto University, Kyoto,
apan).The l-dex YL was amplified by PCR with pBA501 as a tem-
late, KOD plus (Toyobo) and the following primer pairs, 5′-TTGGATCCATGGACTACATCAAGGGTATTGCC-3′ and 5′-TTTAAG-TTTCAGCCCTTTTCTGCCTTCC-3′. Underlined sequences indicatehe restriction endonuclease cleavage site, BamHI and HindIII,espectively. The composition of PCR reaction solution was accord-ng to the procedure recommended by the manufacturer. PCR waserformed by using these primers for 30 cycles of denaturationt 94 ◦C for 15 s, annealing at 62 ◦C for 30 s, and extension at8 ◦C for 1 min. The PCR product was digested with BamHI andindIII, and the 0.7-kb fragment containing the full-length l-dexL sequence was ligated into pQE30Xa (QIAGEN, Hilden, Germany)igested with the same restriction enzymes. The pQE30Xa hasn N-terminal histidine tag sequence, followed by a Factor Xaleavage sequence. The resultant plasmid was designated asQE30/His-LDEX. The Escherichia coli JM109 (Toyobo) was trans-ormed with the pQE30/His-LDEX. The sequence of cloned l-dexL was confirmed by the dideoxy-chain termination method withn automated DNA sequencer (Applied Biosystems 3130xl Geneticnalyzer, Life Technologies Japan, Tokyo, Japan), compared with
hat of the known l-dex YL (accession no. S74078).The site-directed mutants of l-DEX YL (Y12A, L45A, F60A, F60Y)
ere generated using the KOD-plus-mutagenesis kit (Toyobo).QE30/His-LDEX was used as the template, and the primers forite-directed mutagenesis were as follows (underlined codonsndicate the substitutions encoding the mutation): Y12A (5′-CCGGTACGCTGTTCGACGTC-3′ and 5′-AAGGTCGAAGGCAATA-CCT-3′), L45A (5′-GCCGAGTACACATGGCTGCGAAGC-3′ and′-CTGCTTTTGCCGCCACAGC-3′), F60A (5′-GCCCAACAGGCGACC-AAGAC-3′ and 5′-GTTAACGTAGCGATTCATCAGGC-3′), and F60Y
5′-TACCAACAGGCGACCGAAGACG-3′ and 5′-GTTAACGTAGCGATT-ATCAGGC-3′). All of the nucleotide sequences of the mutantsere confirmed by the dideoxy-chain termination method with
he automated DNA sequencer as described above.The glycerol stocks of recombinant E. coli JM109 cells contain-
ng pQE30/His-LDEX or the mutant gene were first grown in 3 mlf Lysogeny Broth (LB) medium containing 100 �g/ml carbenicillinith shaking at 37 ◦C and 140 strokes/min during overnight and
hen the preculture was inoculated into 100 ml of LB medium con-aining 100 �g/ml carbenicillin and grown in the same manner.he cells were collected by centrifugation at 5500 × g for 5 min at
◦C. The collected cells were resuspended in binding/wash buffer50 mM potassium phosphate (KPi) at pH 7.5, 0.3 M NaCl, and mM imidazole) and sonicated with ultrasonic generator US-150TNIHONSEIKI KAISHA LTD., Tokyo, Japan).
Letters 623 (2015) 101–107 103
The expressed l-DEX YL was purified from the culturesupernatant by Ni2+ attachment to immobilized metal affinitychromatography (IMAC) resins (Bio-Rad, CA) according to thebatch-mode procedure recommended by the manufacturer. Theresins were washed by the binding/wash buffer and l-DEX YL or themutants were eluted by elution buffer (50 mM KPi, 0.3 M NaCl, and0.5 M imidazole, pH 7.5). Each fraction was verified by SDS-PAGE.The l-DEX YL fractions were desalted by PD-10 columns (GE health-care) and the buffer was changed into 50 mM KPi buffer (pH 7.5).The solution was concentrated by Amicon Ultra-15 (10 000 MW;Millipore, Billerica, MA, USA) and stored at −80 ◦C at a final con-centration of more than 10 mg/ml. The amount of protein wasdetermined using Coomassie Brilliant Blue Solution for ProteinAssay (Nacalai) with bovine serum albumin (Thermo Fisher Sci-entific Inc., IL) as a standard.
Dehalogenation activity was monitored through the protonreleased in a weakly buffered system, which is coupled to the colorchange of a pH indicator for detection [33,34]. The reaction wasperformed in a volume of 0.8 ml. The final concentrations of theassay components were 5 mM l- or d-2-CPA, which was neutral-ized at around pH 8 by KOH, 1 mM HEPES at pH 8.0, 20 �g/ml BTB,and various amount of l-DEX YL to maintain the conversion within10%. The amounts of enzymes were as follows when l-2-CPA wasused as a substrate: 20 nM for the wild-type; 130 nM for the Y12Amutant; 570 nM for the L45A mutant; 130 nM for the F60A mutantand 67 nM for the F60Y mutant. Similarly, when d-2-CPA was usedas a substrate, the amounts of enzymes were as follows: 6700 nMfor the wild-type; 130 nM for the Y12A mutant; 57 nM for theL45A mutant; 130 nM for the F60A mutant and 67 nM for the F60Ymutant. The enzymatic reaction was monitored for 3 min per 5 s at30 ◦C using a JASCO Model V-630 BIO spectrophotometer (JASCOCorporation, Tokyo, Japan) and the net change of the absorbance at616 nm (A616) was recorded. The specific activity was determinedfrom the calibration curve of the proton concentration to the dif-ference in A616 between the standard samples of various knownconcentration of HCl from 0.025 mM to 0.8 mM and the blank sam-ple without HCl. One unit of enzyme activity was defined as theamount of enzyme required to catalyze the formation of 1 �molof proton per 1 min. Each experiment was repeated 3 times, andthe average and standard deviation of the specific activity werecalculated.
3. Results and discussion
We first constructed the structure of d-2-CPA because the crys-tallographic structure of the complex was unavailable. Based onthe structure of WT/L complex, two d-2-CPA structures were pre-pared (WT/D1 and WT/D2). Both structures were then optimizedwith the ONIOM (PM3:Amber) method. The optimized structuresof WT/D1 and WT/D2 are illustrated in Figure 2, where each struc-ture is superimposed on WT/L. The structural differences from WT/Lwere observed particularly in Tyr12, Arg41, Phe60, Asn119, Lys151,and Asp180, which were caused by the replacement of l-2-CPA byd-2-CPA.
Next, we analyzed the binding energies between l-DEX YL andl-/d-CPA. To this end, the FMO calculations at the MP2/6-31G* levelwere performed to the optimized structures of WT/L, WT/D1, andWT/D2. The FMO calculation can provide the IFIEs among frag-ments. As a result of the calculations, FMO gave the total IFIE of−155.50 kcal/mol, −113.16 kcal/mol and −97.74 kcal/mol for WT/L,WT/D1 and WT/D2, respectively. The total IFIEs calculated were
large negative values, indicating that these substrates bound tothe enzyme l-DEX YL. From these results, we conclude that theenzyme l-DEX YL should bind not only to l-2-CPA but also to d-2-CPA.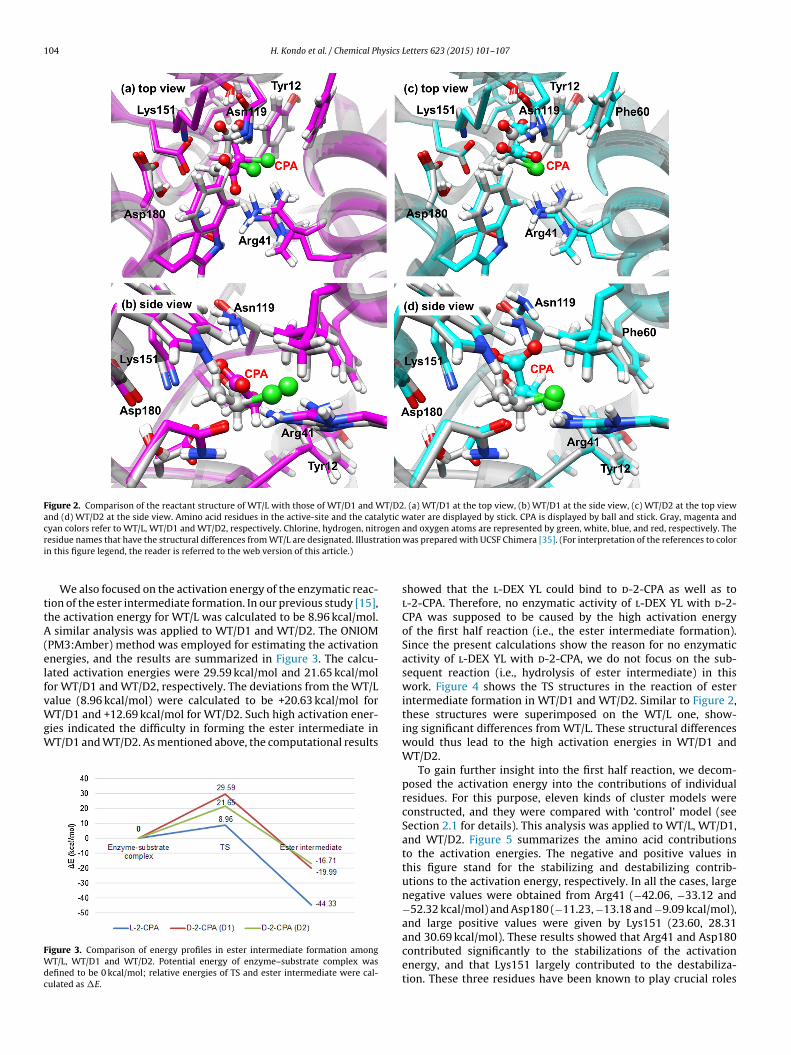
104 H. Kondo et al. / Chemical Physics Letters 623 (2015) 101–107
Figure 2. Comparison of the reactant structure of WT/L with those of WT/D1 and WT/D2. (a) WT/D1 at the top view, (b) WT/D1 at the side view, (c) WT/D2 at the top viewand (d) WT/D2 at the side view. Amino acid residues in the active-site and the catalytic water are displayed by stick. CPA is displayed by ball and stick. Gray, magenta andcyan colors refer to WT/L, WT/D1 and WT/D2, respectively. Chlorine, hydrogen, nitrogen and oxygen atoms are represented by green, white, blue, and red, respectively. Ther ation
i
ttA(elfvWgW
FWdc
esidue names that have the structural differences from WT/L are designated. Illustrn this figure legend, the reader is referred to the web version of this article.)
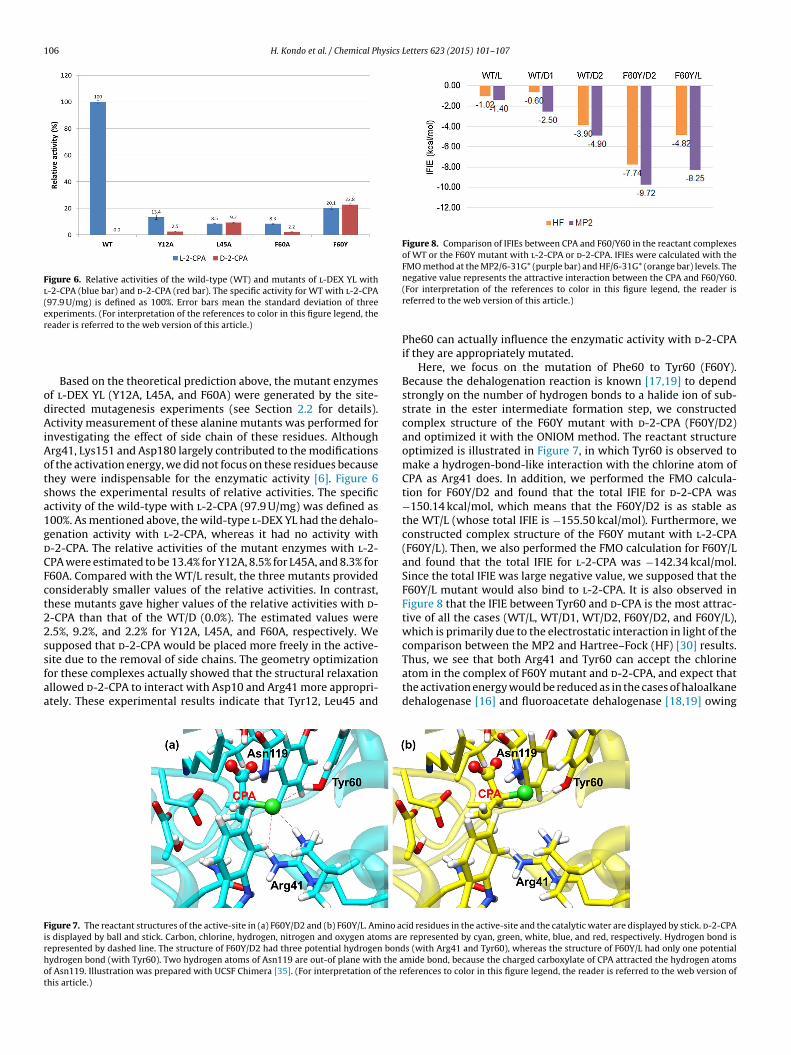
We also focused on the activation energy of the enzymatic reac-ion of the ester intermediate formation. In our previous study [15],he activation energy for WT/L was calculated to be 8.96 kcal/mol.
similar analysis was applied to WT/D1 and WT/D2. The ONIOMPM3:Amber) method was employed for estimating the activationnergies, and the results are summarized in Figure 3. The calcu-ated activation energies were 29.59 kcal/mol and 21.65 kcal/molor WT/D1 and WT/D2, respectively. The deviations from the WT/L
alue (8.96 kcal/mol) were calculated to be +20.63 kcal/mol forT/D1 and +12.69 kcal/mol for WT/D2. Such high activation ener-ies indicated the difficulty in forming the ester intermediate inT/D1 and WT/D2. As mentioned above, the computational results
igure 3. Comparison of energy profiles in ester intermediate formation amongT/L, WT/D1 and WT/D2. Potential energy of enzyme–substrate complex was
efined to be 0 kcal/mol; relative energies of TS and ester intermediate were cal-ulated as �E.
was prepared with UCSF Chimera [35]. (For interpretation of the references to color
showed that the l-DEX YL could bind to d-2-CPA as well as tol-2-CPA. Therefore, no enzymatic activity of l-DEX YL with d-2-CPA was supposed to be caused by the high activation energyof the first half reaction (i.e., the ester intermediate formation).Since the present calculations show the reason for no enzymaticactivity of l-DEX YL with d-2-CPA, we do not focus on the sub-sequent reaction (i.e., hydrolysis of ester intermediate) in thiswork. Figure 4 shows the TS structures in the reaction of esterintermediate formation in WT/D1 and WT/D2. Similar to Figure 2,these structures were superimposed on the WT/L one, show-ing significant differences from WT/L. These structural differenceswould thus lead to the high activation energies in WT/D1 andWT/D2.
To gain further insight into the first half reaction, we decom-posed the activation energy into the contributions of individualresidues. For this purpose, eleven kinds of cluster models wereconstructed, and they were compared with ‘control’ model (seeSection 2.1 for details). This analysis was applied to WT/L, WT/D1,and WT/D2. Figure 5 summarizes the amino acid contributionsto the activation energies. The negative and positive values inthis figure stand for the stabilizing and destabilizing contrib-utions to the activation energy, respectively. In all the cases, largenegative values were obtained from Arg41 (−42.06, −33.12 and−52.32 kcal/mol) and Asp180 (−11.23, −13.18 and −9.09 kcal/mol),and large positive values were given by Lys151 (23.60, 28.31
and 30.69 kcal/mol). These results showed that Arg41 and Asp180contributed significantly to the stabilizations of the activationenergy, and that Lys151 largely contributed to the destabiliza-tion. These three residues have been known to play crucial roles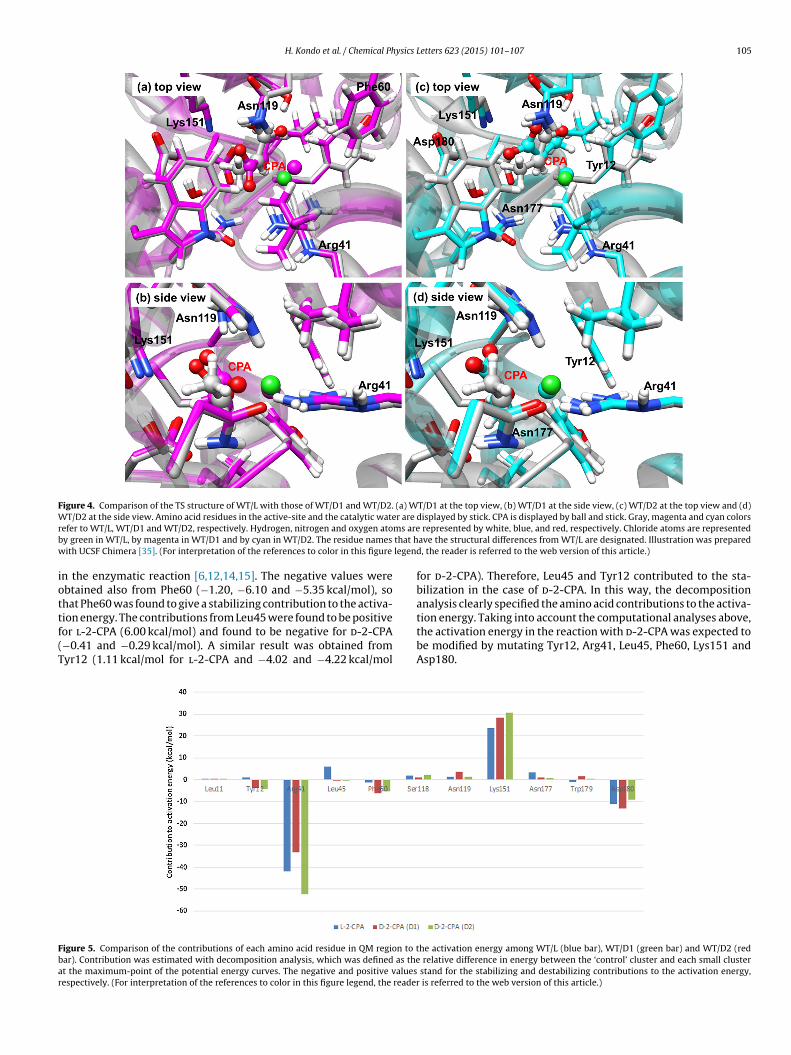
H. Kondo et al. / Chemical Physics Letters 623 (2015) 101–107 105
Figure 4. Comparison of the TS structure of WT/L with those of WT/D1 and WT/D2. (a) WT/D1 at the top view, (b) WT/D1 at the side view, (c) WT/D2 at the top view and (d)W r are
r ms areb that hw legend
iottf(T
Fbar
T/D2 at the side view. Amino acid residues in the active-site and the catalytic wateefer to WT/L, WT/D1 and WT/D2, respectively. Hydrogen, nitrogen and oxygen atoy green in WT/L, by magenta in WT/D1 and by cyan in WT/D2. The residue names
ith UCSF Chimera [35]. (For interpretation of the references to color in this figure
n the enzymatic reaction [6,12,14,15]. The negative values werebtained also from Phe60 (−1.20, −6.10 and −5.35 kcal/mol), sohat Phe60 was found to give a stabilizing contribution to the activa-ion energy. The contributions from Leu45 were found to be positive
or l-2-CPA (6.00 kcal/mol) and found to be negative for d-2-CPA−0.41 and −0.29 kcal/mol). A similar result was obtained fromyr12 (1.11 kcal/mol for l-2-CPA and −4.02 and −4.22 kcal/moligure 5. Comparison of the contributions of each amino acid residue in QM region to tar). Contribution was estimated with decomposition analysis, which was defined as thet the maximum-point of the potential energy curves. The negative and positive valuesespectively. (For interpretation of the references to color in this figure legend, the reader
displayed by stick. CPA is displayed by ball and stick. Gray, magenta and cyan colors represented by white, blue, and red, respectively. Chloride atoms are representedave the structural differences from WT/L are designated. Illustration was prepared, the reader is referred to the web version of this article.)
for d-2-CPA). Therefore, Leu45 and Tyr12 contributed to the sta-bilization in the case of d-2-CPA. In this way, the decompositionanalysis clearly specified the amino acid contributions to the activa-tion energy. Taking into account the computational analyses above,
the activation energy in the reaction with d-2-CPA was expected tobe modified by mutating Tyr12, Arg41, Leu45, Phe60, Lys151 andAsp180.he activation energy among WT/L (blue bar), WT/D1 (green bar) and WT/D2 (red relative difference in energy between the ‘control’ cluster and each small cluster
stand for the stabilizing and destabilizing contributions to the activation energy, is referred to the web version of this article.)
106 H. Kondo et al. / Chemical Physics Letters 623 (2015) 101–107
Figure 6. Relative activities of the wild-type (WT) and mutants of l-DEX YL withl-2-CPA (blue bar) and d-2-CPA (red bar). The specific activity for WT with l-2-CPA(97.9 U/mg) is defined as 100%. Error bars mean the standard deviation of threeexperiments. (For interpretation of the references to color in this figure legend, ther
odAiAotsa1gdCFct22ssfaa
Figure 8. Comparison of IFIEs between CPA and F60/Y60 in the reactant complexesof WT or the F60Y mutant with l-2-CPA or d-2-CPA. IFIEs were calculated with theFMO method at the MP2/6-31G* (purple bar) and HF/6-31G* (orange bar) levels. Thenegative value represents the attractive interaction between the CPA and F60/Y60.
atom in the complex of F60Y mutant and d-2-CPA, and expect that
Firhot
eader is referred to the web version of this article.)
Based on the theoretical prediction above, the mutant enzymesf l-DEX YL (Y12A, L45A, and F60A) were generated by the site-irected mutagenesis experiments (see Section 2.2 for details).ctivity measurement of these alanine mutants was performed for
nvestigating the effect of side chain of these residues. Althoughrg41, Lys151 and Asp180 largely contributed to the modificationsf the activation energy, we did not focus on these residues becausehey were indispensable for the enzymatic activity [6]. Figure 6hows the experimental results of relative activities. The specificctivity of the wild-type with l-2-CPA (97.9 U/mg) was defined as00%. As mentioned above, the wild-type l-DEX YL had the dehalo-enation activity with l-2-CPA, whereas it had no activity with-2-CPA. The relative activities of the mutant enzymes with l-2-PA were estimated to be 13.4% for Y12A, 8.5% for L45A, and 8.3% for60A. Compared with the WT/L result, the three mutants providedonsiderably smaller values of the relative activities. In contrast,hese mutants gave higher values of the relative activities with d--CPA than that of the WT/D (0.0%). The estimated values were.5%, 9.2%, and 2.2% for Y12A, L45A, and F60A, respectively. Weupposed that d-2-CPA would be placed more freely in the active-ite due to the removal of side chains. The geometry optimizationor these complexes actually showed that the structural relaxation
llowed d-2-CPA to interact with Asp10 and Arg41 more appropri-tely. These experimental results indicate that Tyr12, Leu45 andigure 7. The reactant structures of the active-site in (a) F60Y/D2 and (b) F60Y/L. Amino acs displayed by ball and stick. Carbon, chlorine, hydrogen, nitrogen and oxygen atoms arepresented by dashed line. The structure of F60Y/D2 had three potential hydrogen bondydrogen bond (with Tyr60). Two hydrogen atoms of Asn119 are out-of plane with the af Asn119. Illustration was prepared with UCSF Chimera [35]. (For interpretation of the rhis article.)
(For interpretation of the references to color in this figure legend, the reader isreferred to the web version of this article.)
Phe60 can actually influence the enzymatic activity with d-2-CPAif they are appropriately mutated.
Here, we focus on the mutation of Phe60 to Tyr60 (F60Y).Because the dehalogenation reaction is known [17,19] to dependstrongly on the number of hydrogen bonds to a halide ion of sub-strate in the ester intermediate formation step, we constructedcomplex structure of the F60Y mutant with d-2-CPA (F60Y/D2)and optimized it with the ONIOM method. The reactant structureoptimized is illustrated in Figure 7, in which Tyr60 is observed tomake a hydrogen-bond-like interaction with the chlorine atom ofCPA as Arg41 does. In addition, we performed the FMO calcula-tion for F60Y/D2 and found that the total IFIE for d-2-CPA was−150.14 kcal/mol, which means that the F60Y/D2 is as stable asthe WT/L (whose total IFIE is −155.50 kcal/mol). Furthermore, weconstructed complex structure of the F60Y mutant with l-2-CPA(F60Y/L). Then, we also performed the FMO calculation for F60Y/Land found that the total IFIE for l-2-CPA was −142.34 kcal/mol.Since the total IFIE was large negative value, we supposed that theF60Y/L mutant would also bind to l-2-CPA. It is also observed inFigure 8 that the IFIE between Tyr60 and d-CPA is the most attrac-tive of all the cases (WT/L, WT/D1, WT/D2, F60Y/D2, and F60Y/L),which is primarily due to the electrostatic interaction in light of thecomparison between the MP2 and Hartree–Fock (HF) [30] results.Thus, we see that both Arg41 and Tyr60 can accept the chlorine
the activation energy would be reduced as in the cases of haloalkanedehalogenase [16] and fluoroacetate dehalogenase [18,19] owing
id residues in the active-site and the catalytic water are displayed by stick. d-2-CPAe represented by cyan, green, white, blue, and red, respectively. Hydrogen bond iss (with Arg41 and Tyr60), whereas the structure of F60Y/L had only one potentialmide bond, because the charged carboxylate of CPA attracted the hydrogen atomseferences to color in this figure legend, the reader is referred to the web version of

ysics
thlTwtto
taplttata
4
iatwo
daOavoCwb
pesmwwaToCth
Pcamr
[
[
[
[[[[
[
[
[[[[
[
[
[
[[[[[
[
[
H. Kondo et al. / Chemical Ph
o the increase in the number of halogen acceptors. On the otherand, the chlorine atom of CPA turned to Tyr60 in F60Y/L, which
ed to only one potential hydrogen bond between CPA and Tyr60.his result indicated that the halogen acceptor residue in F60Y/Las changed from positively charged Arg41 to neutral Tyr60. We
hus expect that the activation energy would be increased and thathe activity of F60Y mutant with l-2-CPA would be lower than thatf the wild-type.
We experimentally generated the F60Y mutant and measuredhe relative activities with d-2-CPA. The relative activity was 22.8%s shown in Figure 6. This is the highest activity with d-2-CPA in ourresent study and higher than the corresponding value (20.1%) for-2-CPA. In this way, we succeeded in generating a novel l-DEX YLhat had the highest activity ever with d-2-CPA. These experimen-al results clearly showed that the theoretical analysis based on thectivation energy decomposition and the binding energy estima-ion successfully predicted the essential residues for the enzymaticctivity with d-2-CPA.
. Conclusions
In this Letter, we analyzed the mechanism of enantioselectiv-ty of the haloacid dehalogenase l-DEX YL with CPA. Theoreticalnalysis predicted that some amino acid residues contributed tohe recognition of enantiomeric substrates. Then, these predictionsere verified experimentally by measuring the enzymatic activity
f the mutants.We focused on the reason why l-DEX YL could not perform the
ehalogenation of d-2-CPA. The FMO calculations gave large neg-tive values of the total IFIEs for WT/L, WT/D1 and WT/D2. TheNIOM calculations estimated the activation energies of WT/D1nd WT/D2. The deviations of the activation energy from the WT/Lalue were found to be large. Thus, the high activation energiesf the WT/D1 and WT/D2 caused no enzymatic activity with d-2-PA in the dehalogenation process, while the wild-type l-DEX YLould bind not only to l-2-CPA but also to d-2-CPA in the substrate
inding process.Moreover, the decomposition analysis on the activation energy
redicted three residues (i.e., Tyr12, Leu45 and Phe60) which werexpected to modify the activation energy. Then, we performed theite-directed mutagenesis experiments, and measured the enzy-atic activity for Y12A, L45A and F60A mutants. These mutationsere confirmed to increase the activity with d-2-CPA. Furthermore,e focused on the F60Y mutant in which the number of halogen
cceptors would be increased in the ester intermediate formation.he FMO calculations were also applied to the F60Y mutant. Webserved that the Tyr60 could also accept the chlorine atom on d-2-PA. We experimentally generated the F60Y mutant, and measuredhe relative activity with d-2-CPA. As a result, F60Y mutant had theighest activity with d-2-CPA in our present study.
Consequently, we confirmed that Tyr12, Leu45 and especiallyhe60 played significant roles in enantioselectivity. Hence, we suc-
eeded in generating a novel l-DEX YL (F60Y) with the enzymaticctivity with d-2-CPA. These theoretical predictions and experi-ental verifications are expected to be useful for investigating theeaction mechanism of l-DEX YL with other substrates as well.
[[
[
Letters 623 (2015) 101–107 107
Further investigations based on the present approach will be per-formed in our future study.
Acknowledgments
We thank Prof. Tatsuo Kurihara (Kyoto University) for kindlyproviding the plasmid DNA pBA501 that contains l-DEX YL gene.We appreciate fruitful discussions with members of Tanaka Lab-oratory and Ebina Laboratory at Kobe University. We used thecomputer resources at the Research Center for Computational Sci-ence, Okazaki Research Facilities, National Institutes of NaturalSciences.
References
[1] G.W. Gribble, Chemosphere 52 (2003) 289.[2] T. Kurihara, Biosci. Biotechnol. Biochem. 75 (2011) 189.[3] D. O’Hagan, J.W. Schmidberger, Nat. Prod. Rep. 27 (2010) 900.[4] J.Q. Liu, T. Kurihara, A.K.M.Q. Hasan, V. Nardi-Dei, H. Koshikawa, N. Esaki, K.
Soda, Appl. Environ. Microbiol. 60 (1994) 2389.[5] A.K.K.Q. Hasan, H. Takada, H. Koshikawa, J.Q. Liu, T. Kurihara, N. Esaki, K. Soda,
Biosci. Biotechnol. Biochem. 58 (1994) 1599.[6] T. Kurihara, J.Q. Liu, V. Nardi-Dei, H. Koshikawa, N. Esaki, K. Soda, J. Biochem.
117 (1995) 1317.[7] T. Hisano, Y. Hata, T. Fujii, J.Q. Liu, T. Kurihara, N. Esaki, K. Soda, J. Biol. Chem.
271 (1996) 20322.[8] Y.F. Li, Y. Hata, T. Fujii, T. Hisano, M. Nishihara, T. Kurihara, N. Esaki, J. Biol.
Chem. 273 (1998) 15035.[9] Y.F. Li, T. Fujii, T. Kurihara, N. Esaki, J. Biochem. 124 (1998) 20.10] J.Q. Liu, T. Kurihara, M. Miyagi, S. Tsunakawa, M. Nishihara, N. Esaki, K. Soda, J.
Biol. Chem. 272 (1997) 3363.11] S. Ichiyama, T. Kurihara, Y.F. Li, Y. Kogure, S. Tsunasawa, N. Esaki, J. Biol. Chem.
275 (2000) 40804.12] Y.F. Li, T. Kurihara, S. Ichiyama, M. Miyagi, S. Tsunasawa, N. Esaki, J. Mol. Catal.
B: Enzym. 23 (2003) 337.13] J.Q. Liu, T. Kurihara, M. Miyagi, N. Esaki, K. Soda, J. Biol. Chem. 270 (1995) 18309.14] T. Nakamura, et al., J. Comput. Chem. 30 (2009) 2625.15] H. Kondo, T. Nakamura, S. Tanaka, J. Mol. Catal. B: Enzym. 110 (2014) 23.16] J. Damborsky, M. Kuty, M. Nemec, J. Koca, J. Chem. Inf. Comput. Sci. 37 (1997)
562.17] M. Kuty, J. Damborsky, M. Prokop, J. Koca, J. Chem. Inf. Comput. Sci. 38 (1998)
736.18] T. Kamachi, T. Nakayama, O. Shitamachi, K. Jitsumori, T. Kurihara, N. Esaki, K.
Yoshizawa, Chem. Eur. J. 15 (2009) 7394.19] T. Nakayama, et al., Chem. Eur. J. 18 (2012) 8392.20] Z. Prokop, et al., Angew. Chem. Int. Ed. 49 (2010) 6111.21] J. Sykora, et al., Nat. Chem. Biol. 10 (2014) 428.22] J.W. Schmidberger, J.A. Wilce, J.S.H. Tsang, M.C.J. Wilce, J. Mol. Biol. 368 (2007)
706.23] R. Dennington, T. Keith, J. Millam, GaussView, Version 5, Semichem Inc.,
Shawnee Mission, KS, 2009.24] S. Dapprich, I. Komáromi, K.S. Byun, K. Morokuma, M.J. Frisch, J. Mol. Struct.
462 (1999) 1.25] T. Vreven, K. Morokuma, Ö. Farkas, H.B. Schlegel, M.J. Frisch, J. Comput. Chem.
24 (2003) 760.26] M.J. Frisch, et al., Gaussian09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009.27] J.J.P. Stewart, J. Comput. Chem. 10 (1989) 209.28] W.D. Cornell, et al., J. Am. Chem. Soc. 117 (1995) 5179.29] K. Fukui, Acc. Chem. Res. 14 (1981) 363.30] T. Helgaker, P. Jørgensen, J. Olsen, Molecular Electronic-Structure Theory,
Wiley, Chichester, 2000.31] S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama, K. Fukuzawa, Phys. Chem.
Chem. Phys. 16 (2014) 10310.32] V. Nardi-Dei, et al., Appl. Environ. Microbiol. 60 (1994) 3375.
33] P. Holloway, J.T. Trevors, H.J. Lee, Microbiol. Methods 32 (1998) 31.34] P.W.Y. Chan, A.F. Yakunin, E.A. Edwards, E.F.J. Pai, J. Am. Chem. Soc. 133 (2011)7461.35] E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng,
T.E. Ferrin, J. Comput. Chem. 13 (2004) 1605.




















