
TheHelicobacter pylori's protein VacA has direct effects on the regulation of cell cycle and...
6
The Helicobacter pylori’s Protein VacA has Direct Effects on the Regulation of Cell Cycle and Apoptosis in Gastric Epithelial Cells L. MANENTE, 1 A. PERNA, 2 E. BUOMMINO, 1 L. ALTUCCI, 3 A. LUCARIELLO, 2 G. CITRO, 4 A. BALDI, 5 G. IAQUINTO, 6 M.A. TUFANO, 1 AND A. DE LUCA 2 * 1 Department of Experimental Medicine, Second University of Naples, Naples, Italy 2 Department of Medicine and Public Health, Second University of Naples, Naples, Italy 3 Department of General Pathology and Oncology, ‘‘Centro Sperimentale S. Andrea delle Dame,’’ Second University of Naples, Naples, Italy 4 S.A.F.U. Regina Elena Cancer Institute, Rome, Italy 5 Department of Biochemistry and Biophysics ‘‘F. Cedrangolo,’’ Section of Anatomical Pathology, Second University of Naples, Naples, Italy 6 Division of Gastroenterology, San G. Moscati Hospital, Avellino, Italy In this study, we have evaluated the effects on cell cycle regulation of VacA alone and in combination with other two Helicobacter pylori proteins, cytotoxin-associated protein (CagA) and HspB, using the human gastric epithelial cells (AGS). Our results indicate that VacA alone was able to inhibit the G1 to S progression of the cell cycle. The VacA capacity of inhibiting cell progression from G1 to S phase was also observed when cells were co-transfected with CagA or HspB. Moreover, VacA over-expression caused apoptosis in AGS cells through activation of caspase 8 and even more of caspase 9, thus indicating an involvement of both the receptor-mediated and the mitochondrial pathways of apoptosis. Indeed, the two pathways probably can co-operate to execute cell death with a prevalence of the mitochondrial pathways. Our data taken together provide additional information to further enhance our understanding of the molecular mechanism by which H. pylori proteins alter the growth status of human gastric epithelial cells. J. Cell. Physiol. 214: 582–587, 2008. ß 2007 Wiley-Liss, Inc. One of the most important breakthroughs in gastroenterology in the latter part of this century was the finding that a bacterial infection causes ulcer diseases (Graham, 1994). At the end of 1983, several groups almost simultaneously reported the presence of spiral bacteria in patients with chronic gastritis and peptic ulceration. Now it is recognized and accepted that Helicobacter pylori infection is the most frequent cause of gastritis around the world. Its infection affects millions of people in the world, with peaks of 90% of the population infected in countries with poor sanitary conditions and a low socio-economical level (Montecucco et al., 1999; De Luca et al., 2004). The association between H. pylori infection and peptic ulcer disease has been well established, and although most infected individuals only develop a chronic inflammation of the stomach, some patients progress to chronic gastritis, duodenal ulceration, or gastric cancer (Nomura et al., 1991; Blaser, 1993; Gerhard et al., 2002; De Luca et al., 2004). The mechanism by which H. pylori causes disease in humans can be described as a multi-step process in which the organism first has to pass the gastric acid barrier and enter the mucous layer (colonization) and then adapt and multiply under the environmental conditions of the gastric mucus (persistence). All these events are due to the ability of the bacterium to produce a number of protein products including urease, cytotoxin, flagellin, VacA, cytotoxin-associated protein (CagA), heat shock proteins, and adherence factors that contribute to its ability to colonize, avoid host defenses, and inflict damage to the host (Rieder et al., 2005). However, it has been demonstrated that only some H. pylori strains causes neoplasia, and enhanced cancer risk may be considered as the summation of the polymorphic nature of the bacterial population in the host, the host genotype, and environmental exposures (Peek and Blaser, 2002; De Luca et al., 2004). It has been demonstrated that approximately 50% of H. pylori strains express the vacuolating cytotoxin Contract grant sponsor: Second University of Naples. Contract grant sponsor: AIRC. Contract grant sponsor: Ministero della Salute. Contract grant sponsor: FUTURA-onlus. Contract grant sponsor: Dottorato in Microbiologia ambientale ed ecosistema cutaneo. *Correspondence to: A. De Luca, Department of Medicine and Public Health, Second University of Naples, Via L. Armanni 5, 80138 Naples, Italy. E-mail: [email protected] Received 3 May 2007; Accepted 6 July 2007 DOI: 10.1002/jcp.21242 ORIGINAL ARTICLE 582 Journal of Journal of Cellular Physiology Cellular Physiology ß 2007 WILEY-LISS, INC.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of TheHelicobacter pylori's protein VacA has direct effects on the regulation of cell cycle and...
ORIGINAL ARTICLE 582J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
The Helicobacter pylori’sProtein VacA has DirectEffects on the Regulation ofCell Cycle and Apoptosis inGastric Epithelial Cells
L. MANENTE,1 A. PERNA,2 E. BUOMMINO,1 L. ALTUCCI,3 A. LUCARIELLO,2 G. CITRO,4A. BALDI,5 G. IAQUINTO,6 M.A. TUFANO,1 AND A. DE LUCA2*1Department of Experimental Medicine, Second University of Naples, Naples, Italy2Department of Medicine and Public Health, Second University of Naples, Naples, Italy3Department of General Pathology and Oncology, ‘‘Centro Sperimentale S. Andrea delle Dame,’’
Second University of Naples, Naples, Italy4S.A.F.U. Regina Elena Cancer Institute, Rome, Italy5Department of Biochemistry and Biophysics ‘‘F. Cedrangolo,’’ Section of Anatomical Pathology,
Second University of Naples, Naples, Italy6Division of Gastroenterology, San G. Moscati Hospital, Avellino, Italy
In this study, we have evaluated the effects on cell cycle regulation of VacA alone and in combination with other two Helicobacter pyloriproteins, cytotoxin-associated protein (CagA) and HspB, using the human gastric epithelial cells (AGS). Our results indicate that VacAalone was able to inhibit the G1 to S progression of the cell cycle. The VacA capacity of inhibiting cell progression from G1 to S phase wasalso observed when cells were co-transfected with CagA or HspB. Moreover, VacA over-expression caused apoptosis in AGS cellsthrough activation of caspase 8 and even more of caspase 9, thus indicating an involvement of both the receptor-mediated and themitochondrial pathways of apoptosis. Indeed, the two pathways probably can co-operate to execute cell death with a prevalence of themitochondrial pathways. Our data taken together provide additional information to further enhance our understanding of the molecularmechanism by which H. pylori proteins alter the growth status of human gastric epithelial cells.
J. Cell. Physiol. 214: 582–587, 2008. � 2007 Wiley-Liss, Inc.
Contract grant sponsor: Second University of Naples.Contract grant sponsor: AIRC.Contract grant sponsor: Ministero della Salute.Contract grant sponsor: FUTURA-onlus.Contract grant sponsor: Dottorato in Microbiologia ambientale edecosistema cutaneo.
*Correspondence to: A. De Luca, Department of Medicine andPublic Health, Second University of Naples, Via L. Armanni 5, 80138Naples, Italy. E-mail: [email protected]
Received 3 May 2007; Accepted 6 July 2007
DOI: 10.1002/jcp.21242
One of the most important breakthroughs in gastroenterologyin the latter part of this century was the finding that a bacterialinfection causes ulcer diseases (Graham, 1994).
At the end of 1983, several groups almost simultaneouslyreported the presence of spiral bacteria in patients with chronicgastritis and peptic ulceration. Now it is recognized andaccepted that Helicobacter pylori infection is the most frequentcause of gastritis around the world. Its infection affects millionsof people in the world, with peaks of 90% of the populationinfected in countries with poor sanitary conditions and a lowsocio-economical level (Montecucco et al., 1999; De Luca et al.,2004).
The association between H. pylori infection and peptic ulcerdisease has been well established, and although most infectedindividuals only develop a chronic inflammation of the stomach,some patients progress to chronic gastritis, duodenalulceration, or gastric cancer (Nomura et al., 1991; Blaser, 1993;Gerhard et al., 2002; De Luca et al., 2004).
The mechanism by which H. pylori causes disease in humanscan be described as a multi-step process in which the organismfirst has to pass the gastric acid barrier and enter the mucouslayer (colonization) and then adapt and multiply under theenvironmental conditions of the gastric mucus (persistence).All these events are due to the ability of the bacterium toproduce a number of protein products including urease,cytotoxin, flagellin, VacA, cytotoxin-associated protein (CagA),heat shock proteins, and adherence factors that contribute to
� 2 0 0 7 W I L E Y - L I S S , I N C .
its ability to colonize, avoid host defenses, and inflict damage tothe host (Rieder et al., 2005).
However, it has been demonstrated that only some H. pyloristrains causes neoplasia, and enhanced cancer risk may beconsidered as the summation of the polymorphic nature of thebacterial population in the host, the host genotype, andenvironmental exposures (Peek and Blaser, 2002; De Lucaet al., 2004). It has been demonstrated that approximately50% of H. pylori strains express the vacuolating cytotoxin

R O L E O F V a c A I N H U M A N G A S T R I C C E L L S 583
protein VacA (Montecucco et al., 2001), a secretedexotoxin encoded by the vacA gene (De Luca and Iaquinto,2004; De Luca et al., 2004). This toxin inserts itself into theepithelial cell membrane allowing bicarbonate and organicanions release and facilitating the formation oftransmembrane pores which permeabilize the gastricepithelium to urea (Szabo et al., 1999; Tombola et al., 1999a;Jungblut et al., 2000) and probably providing the bacteriumwith nutrients (De Luca et al., 2004). The vacA gene was clonedby several groups using degenerate oligonucleotides that weredesigned based on partial amino acid sequence data. DNAsequence analyses identified a single open reading frame thatcorresponds to a protein of 140 kDa in size, which isprocessed to yield a mature 87 kDa protein. The structure hassimilarity with several secreted bacterial serine proteasesuggesting that VacA can have in the cells a proteolytic activity.The active VacA form is able to interact with several networksof family proteins, such as G proteins, receptor-like proteintyrosine phosphatase (Yahiro et al., 2004), and fibronectin(Hennig et al., 2005). H. pylori VacA-secreting strains are morecommon among patients with distal gastric cancer than amongpatients with gastritis alone (Miehlke et al., 2000; De Luca et al.,2004).
H. pylori strains can be divided into two classes, type I and typeII, based on the presence or the absence of the cag pathogenicityisland (PAI), a 40-kb region that encodes over 40 putativebacterial proteins (Censini et al., 1996; Alm and Trust, 1999;Tombola et al., 1999b). The terminal gene in the island is thecagA encoding CagA. Several studies have demonstrated thatinfection with cagA-positive H. pylori strains significantlyincreases the risk of developing severe gastritis, atrophicgastritis, peptic ulcer disease, and distal gastric cancer (Coveret al., 1991; Blaser, 1995; Kuipers et al., 1995; Peek et al., 1995;Parsonnet et al., 1997; De Luca et al., 2003, 2004). Theexplanation is that the genes present in the pathogenicity islandrequire CagA to be able to induce the expression of cytokines,especially IL-8, by epithelial gastric cells (Yuan et al., 2004). Inaddition, it has also been shown that H. pylori strains expressingCagA protein induce a proto-oncogene activation that mayrepresent an important step in the mechanism of H. pylori-induced neoplasia (Higashi et al., 2002). Moreover, it has beenrecently demonstrated that CagA protein when translocatesfrom the bacteria to the gastric cells is able to form a physicalcomplex with SHP-2 tyrosine phosphatase, inducing a growthfactor-like response in gastric epithelial cells (Higashi et al.,2002).
A few years ago, another protein HspB (Dunn et al.,1992; Macchia et al., 1993) was correlated to a major risk todevelop a gastric carcinoma (Iaquinto et al., 2000; De Lucaet al., 2003). Several strains of H. pylori produce this 58 kDaprotein that has been cloned, characterized, and named uponits homology with the family of the heat shock proteins:HspB (Macchia et al., 1993; Dunn et al., 1997). This protein issecreted by the bacterium and it has been shown on the mucosasurface and within epithelial cells (Dunn et al., 1997;Engstrand et al., 1997; Cao et al., 1998; Kamiya et al., 1998).HspB is able to stimulate a strong immune response inpatients with gastritis and to increase the risk of gastriccarcinoma in infected patients (Perez-Perez et al., 1996;Iaquinto et al., 2000).
Drawing from this background, to better investigate themechanisms and relationships between H. pylori’s proteins andtheir effects on gastric epithelial cells, we examined theinfluence of three H. pylori’s proteins (VacA, CagA, and HspB)on cell kinetics of human gastric epithelial (AGS) cell lines.Taken together, our data provide additional information tofurther enhance our understanding on the molecularmechanisms by which H. pylori proteins are able to alter thegrowth status of the host cell.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
Materials and MethodsCell culture and transfection
AGS human gastric epithelial cells (American Type CultureCollection, Manassas, VA) were grown in Ham’s F-12 mediumsupplemented with 10% FBS and 50 mg/ml penicillin-streptomycinin an atmosphere of 5% CO2 at 378C. Plasmid expressionconstruct for VacA was prepared as previously described in ourarticle (De Luca et al., 2003). AGS cells were transiently transfectedwith mammalian expression vector for CagA, HspB, and VacA usingLipofectamine (Invitrogen, Carlsbad, CA) as previously describedin our article (De Luca et al., 2003) and according to themanufacturer’s instruction. Briefly, the cells were plated in 60 mmdishes (1� 106 cells/dish) 24 h before transfection and the sameamount of total DNA was used. We typically transfected between50 and 70% of cells as judged using a control pcDNA3 vectorexpressing EGFP protein. In the experiments, we usedsynchronized cells by withdrawing the serum 24 h before thetransfection and tested with flow cytometry to prove thesynchronization. Cells were collected at 48 and 72 h aftertransfections and were prepared for flow cytometry analysis orprepared for protein extracts.
Flow cytometry for detection of cell cycle and apoptosis
Unsynchronized and synchronized cells after 48 and 72 h fromthe transfection were collected by mild trypsination, washed inPBS, and fixed in 70% ethanol at 48C. Cells were collected andre-suspended in 500 ml of a hypotonic buffer (0.1%TritonX-100,0.1% sodium citrate, 50 mg/ml propidium iodide, and 100 mg/mlRNase). Then the cells were analyzed using a Becton DickinsonFACSCalibur flow cytometer and the percentages of G1, S, G2/M,and sub-G1 (apoptotic cells) populations were calculated,respectively. All of the experiments were performed three times.Cells used for caspase activity assay (Altucci et al., 2005) werecollected 72 h after transfection and the pellets were lysed in 50 ulof ice-cold lysis buffer. After incubation for 10 min in ice, sampleswere centrifuged for 1 min at 10,000g. After measuring proteinlevels (Bio-Rad, Hercules, CA), the activities of the apical caspases8 and 9 and effector caspase 3 were determined using colorimetricassays according to the supplier’s instructions (R&D; Alexis, SanDiego, CA). All the experiments were performed three times.
Immunoblotting
Plates of 70–80% confluent AGS cells were transiently transfectedwith the plasmids by the Lipofectamine protocol. After 72 h, thecells were collected. Briefly, the cells were lysed in 100 ul lysisbuffer (50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 250 mM NaCl,50 mM NaF, 0.1% Triton X-100, 0.1 mM Na3VO4, 1 mMphenylmethylsulfonyl fluoride, and 10 ug/ml leupeptin) for 30 minin ice. Lysates were centrifuged at 14,000g for 10 min at 48C. Totalcell protein extracts were normalized for concentration by theBradford assay (Bio-Rad) and 30 ug of proteins were separated bySDS–PAGE and transferred to polyvinylidene difluoride membrane(Millipore’s Corporate, Billerica, MA). Membranes were incubatedwith rabbit polyclonal cleaved caspase 3 (Cell Signaling Technology,Danvers, MA) and with rabbit polyclonal antibodies againstRb/XZ55 able to recognize the p68/Rb fragment (Ping Dou andBing, 1998). Primary antibodies were detected using anti-rabbithorseradish peroxidase-conjugated secondary antibody(Amersham Biosciences, Inc., Piscataway, NJ) and visualized by theECL detection system (Amersham Biosciences, Inc.) according tothe manufacturer’s instructions. Each membrane was probed withthe Hsp70 antibody (heat shock proteins) (Santa-Cruz,Biotechnology, Santa Cruz, CA) to estimate equal protein loading.The expression levels of the protein transfected were detectedusing the monoclonal antibody against TAG (data not shown)(Novagen, Darmstadt, Germany) as previously described (De Lucaet al., 2003).
584 M A N E N T E E T A L .
ResultsH. pylori protein VacA induced growth arrest bothalone and in combination with CagA and HspB
In a previous work, we have demonstrated that efficientco-expression of CagA and HspB is able to influence humangastric epithelial (AGS) cell growth by inducing cell proliferationthrough an increase in the S-G2-M phase of the cell cycle(De Luca et al., 2003). In order to better investigate theinterplay among the three pathogenic proteins of H. pylori, wehave performed a co-transfection experiments using VacA inseveral combination with CagA and HspB in AGS cells. Vectoralone was used as negative control. Equal amount of exogenousproteins were transfected, as determined by immunoblottingfor TAG (data not shown).
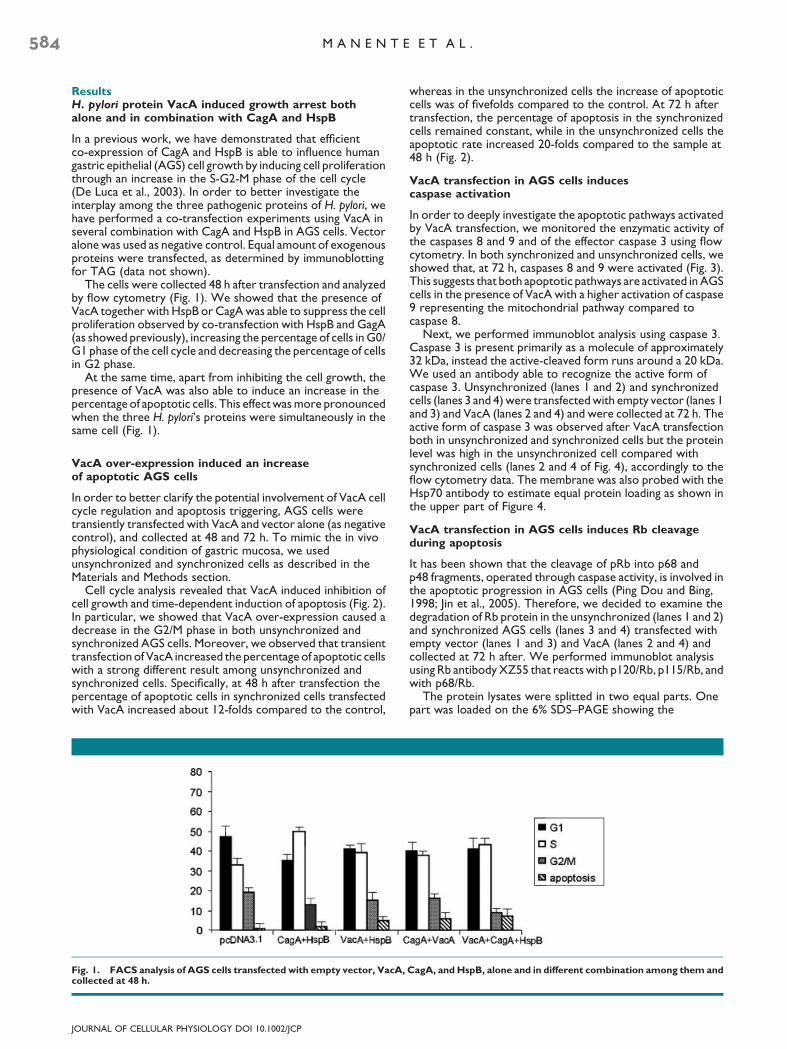
The cells were collected 48 h after transfection and analyzedby flow cytometry (Fig. 1). We showed that the presence ofVacA together with HspB or CagA was able to suppress the cellproliferation observed by co-transfection with HspB and GagA(as showed previously), increasing the percentage of cells in G0/G1 phase of the cell cycle and decreasing the percentage of cellsin G2 phase.
At the same time, apart from inhibiting the cell growth, thepresence of VacA was also able to induce an increase in thepercentage of apoptotic cells. This effect was more pronouncedwhen the three H. pylori’s proteins were simultaneously in thesame cell (Fig. 1).
VacA over-expression induced an increaseof apoptotic AGS cells
In order to better clarify the potential involvement of VacA cellcycle regulation and apoptosis triggering, AGS cells weretransiently transfected with VacA and vector alone (as negativecontrol), and collected at 48 and 72 h. To mimic the in vivophysiological condition of gastric mucosa, we usedunsynchronized and synchronized cells as described in theMaterials and Methods section.
Cell cycle analysis revealed that VacA induced inhibition ofcell growth and time-dependent induction of apoptosis (Fig. 2).In particular, we showed that VacA over-expression caused adecrease in the G2/M phase in both unsynchronized andsynchronized AGS cells. Moreover, we observed that transienttransfection of VacA increased the percentage of apoptotic cellswith a strong different result among unsynchronized andsynchronized cells. Specifically, at 48 h after transfection thepercentage of apoptotic cells in synchronized cells transfectedwith VacA increased about 12-folds compared to the control,
Fig. 1. FACS analysis of AGS cells transfected with empty vector, VacA,collected at 48 h.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
whereas in the unsynchronized cells the increase of apoptoticcells was of fivefolds compared to the control. At 72 h aftertransfection, the percentage of apoptosis in the synchronizedcells remained constant, while in the unsynchronized cells theapoptotic rate increased 20-folds compared to the sample at48 h (Fig. 2).
VacA transfection in AGS cells inducescaspase activation
In order to deeply investigate the apoptotic pathways activatedby VacA transfection, we monitored the enzymatic activity ofthe caspases 8 and 9 and of the effector caspase 3 using flowcytometry. In both synchronized and unsynchronized cells, weshowed that, at 72 h, caspases 8 and 9 were activated (Fig. 3).This suggests that both apoptotic pathways are activated in AGScells in the presence of VacA with a higher activation of caspase9 representing the mitochondrial pathway compared tocaspase 8.
Next, we performed immunoblot analysis using caspase 3.Caspase 3 is present primarily as a molecule of approximately32 kDa, instead the active-cleaved form runs around a 20 kDa.We used an antibody able to recognize the active form ofcaspase 3. Unsynchronized (lanes 1 and 2) and synchronizedcells (lanes 3 and 4) were transfected with empty vector (lanes 1and 3) and VacA (lanes 2 and 4) and were collected at 72 h. Theactive form of caspase 3 was observed after VacA transfectionboth in unsynchronized and synchronized cells but the proteinlevel was high in the unsynchronized cell compared withsynchronized cells (lanes 2 and 4 of Fig. 4), accordingly to theflow cytometry data. The membrane was also probed with theHsp70 antibody to estimate equal protein loading as shown inthe upper part of Figure 4.
VacA transfection in AGS cells induces Rb cleavageduring apoptosis
It has been shown that the cleavage of pRb into p68 andp48 fragments, operated through caspase activity, is involved inthe apoptotic progression in AGS cells (Ping Dou and Bing,1998; Jin et al., 2005). Therefore, we decided to examine thedegradation of Rb protein in the unsynchronized (lanes 1 and 2)and synchronized AGS cells (lanes 3 and 4) transfected withempty vector (lanes 1 and 3) and VacA (lanes 2 and 4) andcollected at 72 h after. We performed immunoblot analysisusing Rb antibody XZ55 that reacts with p120/Rb, p115/Rb, andwith p68/Rb.
The protein lysates were splitted in two equal parts. Onepart was loaded on the 6% SDS–PAGE showing the
CagA, andHspB, alone and in different combination among them and
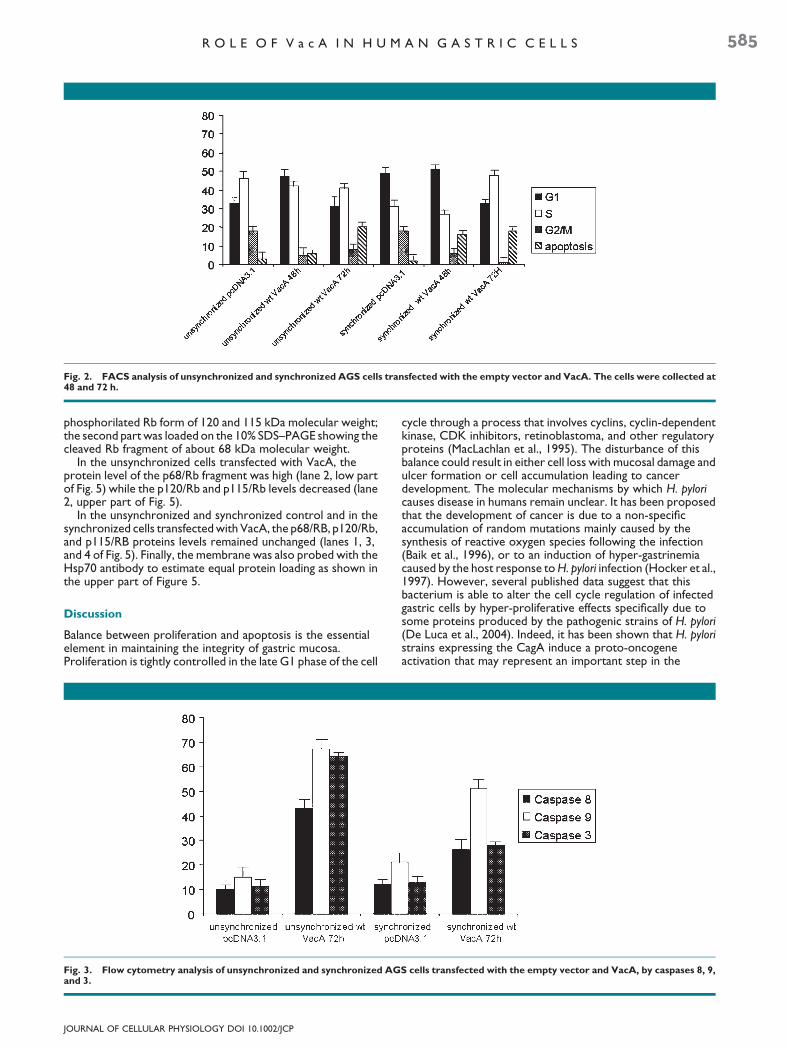
Fig. 2. FACS analysis of unsynchronized and synchronized AGS cells transfected with the empty vector and VacA. The cells were collected at48 and 72 h.
R O L E O F V a c A I N H U M A N G A S T R I C C E L L S 585
phosphorilated Rb form of 120 and 115 kDa molecular weight;the second part was loaded on the 10% SDS–PAGE showing thecleaved Rb fragment of about 68 kDa molecular weight.
In the unsynchronized cells transfected with VacA, theprotein level of the p68/Rb fragment was high (lane 2, low partof Fig. 5) while the p120/Rb and p115/Rb levels decreased (lane2, upper part of Fig. 5).
In the unsynchronized and synchronized control and in thesynchronized cells transfected with VacA, the p68/RB, p120/Rb,and p115/RB proteins levels remained unchanged (lanes 1, 3,and 4 of Fig. 5). Finally, the membrane was also probed with theHsp70 antibody to estimate equal protein loading as shown inthe upper part of Figure 5.
Discussion
Balance between proliferation and apoptosis is the essentialelement in maintaining the integrity of gastric mucosa.Proliferation is tightly controlled in the late G1 phase of the cell
Fig. 3. Flow cytometry analysis of unsynchronized and synchronized AGand 3.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
cycle through a process that involves cyclins, cyclin-dependentkinase, CDK inhibitors, retinoblastoma, and other regulatoryproteins (MacLachlan et al., 1995). The disturbance of thisbalance could result in either cell loss with mucosal damage andulcer formation or cell accumulation leading to cancerdevelopment. The molecular mechanisms by which H. pyloricauses disease in humans remain unclear. It has been proposedthat the development of cancer is due to a non-specificaccumulation of random mutations mainly caused by thesynthesis of reactive oxygen species following the infection(Baik et al., 1996), or to an induction of hyper-gastrinemiacaused by the host response toH. pylori infection (Hocker et al.,1997). However, several published data suggest that thisbacterium is able to alter the cell cycle regulation of infectedgastric cells by hyper-proliferative effects specifically due tosome proteins produced by the pathogenic strains of H. pylori(De Luca et al., 2004). Indeed, it has been shown that H. pyloristrains expressing the CagA induce a proto-oncogeneactivation that may represent an important step in the
S cells transfected with the empty vector and VacA, by caspases 8, 9,

Fig. 4. Immunoblotting analysis of unsynchronized andsynchronized AGS cells transfected with the empty vector and withVacA. The cells were collected at 72 h. Western blotting wasperformed using anti-cleaved caspase 3 antibody. Lanes 1 and 2:unsynchronized cells transfected with empty vector and VacA,respectively; lanes 3 and 4: synchronized cells transfected with emptyvector and VacA, respectively. Upper part showed immunoblottingassay with Hsp70 antibody to estimate equal protein loading.
586 M A N E N T E E T A L .
mechanism of H. pylori-induced neoplasia (Meyer-ter-Vehnet al., 2000). Moreover, it has recently been demonstrated thatCagA protein secreted in the gastric cells is able to form aphysical complex with SHP-2 tyrosine phosphatase, inducing agrowth factor-like response in gastric epithelial cells (Higashiet al., 2002). Our research group has recently found thatefficient co-expression of CagA and HspB is able to influenceAGS cell growth by inducing cell cycle proliferation through anincrease in the S/G2-M phase of the cell cycle. The effects ofH. pylori protein products do not seem less important onapoptosis. It has been demonstrated that H. pylori-associatedchronic gastritis involves apoptotic cell death, and in fact amarkedly elevated number of apoptotic cells is identified insurface epithelium, antral pyloric glands, and lamina propria in83% of biopsies from patients with H. pylori-associated gastritis(Moss et al., 1996). Furthermore, it has been proposed thatH. pylori may induce hyper-proliferation through increasingapoptosis (Peek et al., 1997). Drawing from this background, wedecided to look at the effects on cell cycle regulation andapoptosis of the over-expression of VacA alone and incombination with the other two H. pylori proteins, CagA andHspB proteins in AGS cells, independently from anypathogenetic mechanism induced by the infection.
First of all, we have shown that VacA over-expression wasable, independently by the presence of the other two H. pyloriproteins CagA and HspB to induce a stop in G1 of the cell cyclein AGS cells and an increase in the percentage of apoptotic cells.
Fig. 5. Immunoblotting analysis of unsynchronized andsynchronized AGS cells transfected with the empty vector and withVacA. The cells were collected at 72 h. Western blotting wasperformed using anti-Rb XZ55 antibody. Upper part shows, in a 6%gel, the phosphorilated Rb form and low part shows, in a 10% gel, thecleaved Rb fragment. Lanes 1 and 2: unsynchronized cells transfectedwith empty vector andVacA, respectively; lanes 3 and4: synchronizedcells transfected with empty vector and VacA, respectively. Upperpart showed immunoblotting assay with Hsp70 antibody to estimateequal protein loading.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
In order to mimic more closely the physiological conditions ofthe gastric mucosa, we set up two different conditions of celltreatments, synchronized and unsynchronized cells. In fact, thegastric mucosa is formed by cells (human gastric epithelial cellsthat are proliferating) that have a mitotic turnover, localized inthe isthmus and neck of the gastric glands (Penta et al., 2005).These epithelial cells first proliferating and then migratingupward from these regions, replace superficial cells (humangastric epithelial cells that are not proliferating) that areexfoliated into the lumen after a superficial injury. Our resultssuggest that the epithelial cells of the glands localized in thebottom of the stomach were more sensitive to the damagefrom H. pylori than the cells localized in the outer layers ofgastric mucosa. Specifically, we showed that the percentage ofapoptotic cells was different in synchronized andunsynchronized cells, with the percentage of apoptotic cellshigher in the unsynchronized cells. Interestingly, this differencewas time dependent. Indeed, serum starvation enhanced thesensitivity of gastric epithelial cells so increasing the ability ofVacA to induce apoptosis only for a limited time. Initially, thesynchronized cells were more sensitive to apoptosis induced byVacA but then they acquired resistance. Intriguingly, on theunsynchronized cells, the VacA effect was slower.
A central mechanism in the apoptotic cell death process isthe activation of caspases, a family of cysteine proteases. Thereare two apoptotic pathways: the receptor-mediated pathway,formed by Fas ligand (FasL), Fas and caspase 8, themitochondrial pathway, mediated by the Bcl-2 family, Apaf-1and caspase 9. Activation of caspase 8 occurs as a consequenceof surface receptor binding to its ligand (Ashkenazi and Dixit,1998). Our results on synchronized and unsynchronized cellsshow that caspase 8 was indeed activated but the majorpathway was independent of caspase 8 activation and that VacAis capable of inducing apoptosis mainly through the activation ofcaspase 9, that is, through the mitochondrial pathway.
However, we also have to mention that outer membranevesicles are constantly shed by the bacteria and can provide anadditional mechanism for pathogenicity by releasing non-secretable factors which can then interact with epithelial cells.Ayala et al. showed that external membrane vesicles are able toinduce apoptosis not mediated by mitochondrial pathway ingastric (AGS) epithelial cells, as demonstrated by the lack ofcytochrome c release with an activation of caspases 8 and 3.Apoptosis induced by these vesicles does not require a classicVacAþ phenotype, as a negative strain with a truncated andtherefore non-secretable form of this protein can also inducecell death (Ayala et al., 2006).
Finally, we have to remember that the deregulation of cellcycle progression is essential for the initiation of apoptosis.Retinoblastoma protein, Rb, is an important tumor suppressorand a cell cycle regulator. Different studies suggest that Rb alsoplays a regulatory role in the process of apoptosis; thehyper-phosphorylated form of Rb (p120Rb) is converted to ahypo-phosphorylated form (p115Rb) and this new form isimmediately cleaved by a protease that has properties of thecaspase family (Ping Dou and Bing, 1998). Hypophosphorylatedform is cleaved into p68 and p48 fragments and the interiorcleavage of Rb is tightly associated with the initiation ofapoptotic execution. Indeed, our experimental resultsdemonstrate that there was an increase in the processing ofpRB in the wt-VacA unsynchronized cells with respect to thecontrol, thus suggesting an involvement of pRb cleavage bycaspase in the regulation of the apoptotic process.
In conclusion, we report here that the deregulation ofepithelial proliferation and apoptosis observed in chronicH. pylori infection can be specifically stimulated in AGS cells bysome of the bacterial protein products. Our experimentalstrategy based on the transfection of these proteins, indeed,eliminated all the insults to the cells eventually caused by the

R O L E O F V a c A I N H U M A N G A S T R I C C E L L S 587
infection of the culture with H. pylori. To the best of ourknowledge, this is the first report showing a specific apoptoticeffect on gastric epithelial cells caused by the action of the VacAbacterial protein, independently from any effect due to themechanism of infection.
Finally, some limitations of this study should beacknowledged. First of all, this experimental system looks onlyat the effects of over-expression of some bacterial proteins inthe gastric cells. Therefore, it ignores possible effects of VacA,CagA, or HspB on the other elements present in the stomach,such as the fibroblasts of the gastric lamina propria, that mightalso be targets of their action. Secondly, the possibility cannotbe excluded that with this transfection model, intracellularlevels of bacterial proteins are higher than those actually seen inH. pylori infection. Then, the functional effects analyzed can onlybe the consequence of an artificial situation that is not present inthe gastric cells of the stomach infected with H. pylori. Finally,AGS cells are derived from a tumor and this might partiallyaffect the results, since these cells are already transformed.Moreover, another limitation is the Caucasian origin of the AGScells, which may explain the molecular mechanisms by whichH. pylori proteins are implicated in this tumor only in theWestern world. Further experiment should be performedusing also cell lines KATO-III, YCC from Asia where gastriccancer is more frequent.
Nevertheless, the major contribution of this work, that is,the demonstration of a direct effect of theH. pylori protein VacAon the cell cycle regulation and apoptosis of a gastric carcinomacell line, is not weakened by these limitations.
In conclusion, this study further contributes to theelucidation of the molecular mechanisms involved in the effectsof H. pylori on cell cycle control and provides insights into theroles played by the organism’s proteins in gastriccarcinogenesis. Nevertheless, it indicates possible moleculartargets of diagnosis and therapy for this kind of neoplasm.
Acknowledgments
We thank Dr. Pia Furno for editing the article. This work wassupported in part by grants from Second University of Naplesand AIRC (A.D.L.), by Ministero della Salute (G.C.), and byFUTURA-onlus (A.B.). L.M is supported by ‘‘Dottorato inMicrobiologia ambientale ed ecosistema cutaneo.’’ We thankthe I.S.S.C.O. (president H.E. Kaiser) for the continuoussupport.
Literature Cited
Alm RA, Trust TJ. 1999. Analysis of the genetic diversity ofHelicobacter pylori: The tale of twogenomes. J Mol Med 77:834–846.
Altucci L, Rossin A, Hirsch O, Nebbioso A, Vitoux D, Wilhelm E, Guidez F, De Simone M,Schiavone EM, Grimwade D, Zelent A, de The H, Gronemeyer H. 2005. Rexinoid-triggered differentiation and tumor-selective apoptosis of acute myeloid leukemia byprotein kinase A-mediated desubordination of retinoid X receptor. Cancer Res 65:8754–8765.
Ashkenazi A, Dixit VM. 1998. Death receptor: Signaling and modulation. Science 281:1305–1308.
Ayala G, Torres L, Espinosa M, Fierros-Zarate G, Maldonado V, Melendez-Zajgla J. 2006.External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelialcells. FEMS Microbiol Lett 260:178–185.
Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH.1996. Increased oxidative DNA damage in Helicobacter pylori-infected human gastricmucosa. Cancer Res 56:1279–1282.
Blaser MJ. Intrastrain differences in Helicobacter pylori: A key question in mucosal damage?Ann Med 1995. 27:559–563.
Blaser MJ. 1993. Helicobacter pylori: Microbiology of a ‘‘slow’’ bacterial infection. TrendsMicrobiol 1:225–260.
Cao P, McClain MS, Forsyth MH, Cover TL. 1998. Extracellular release of antigenic proteinsby Helicobacter pylori. Infect Immun 66:2984–2986.
Censini S, Lange C, Xiang Z, Crabtree JE, Chiara P, Borodovky M, Rappuoli R, Covacci A.1996. Cag, a pathogenecity island ofHelicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA 93:14648–14653.
Cover TL, Puryear W, Perez-Perez GI, Blaser MJ. 1991. Effect of urease on HeLa cellvacuolation induced by Helicobacter pylori cytotoxin. Infect Immun 59:1264–1270.
De Luca A, Baldi A, Russo P, Todisco A, Altucci L, Giardullo N, Pasquale L, Iaquinto S,D’Onofrio V, Parodi MC, Paggi MG, Iaquinto G. 2003. Co-expression of Helicobacterpylori’s proteins CagA and HspB induces cell proliferation in AGS gastric epithelial cells,independently from the bacterial infection. Cancer Res 63:6350–6356.
JOURNAL OF CELLULAR PHYSIOLOGY DOI 10.1002/JCP
De Luca A, De Falco M, Iaquinto S, Iaquinto G. 2004. Effects ofHelicobacter pylori infection oncell cycle progression and the expression of cell cycle regulatory proteins. J Cell Physiol200:334–342.
De Luca A, Iaquinto G. 2004.Helicobacter pylori and gastric diseases: A dangerous association.Cancer Lett 213:1–10.
Dunn BE, Roop RM, Sung CC, Sharma SA, Perez-Perez GI, Blaser MJ. 1992. Identificationand purification of a cpn60 heat shock protein homolog from Helicobacter pylori. InfectImmun 60:1946–1951.
Dunn BE, Vakil NB, Schneider BG, Miller MM, Zitzer JB, Peutz T, Phadnis SH. 1997. InfectImmun 65:1181–1188.
Engstrand L, Graham D, Scheynius A, Genta RM, El-Zaatari F. 1997. Is the sanctuary whereHelicobacter pylori avoids antibacterial treatment intracellular? Am J Clin Pathol 108:504–509.
Gerhard M, Rad R, Prinz C, Naumann M. 2002. Pathogenesis of Helicobacter pylori.Helicobacter 1:17–23.
Graham DY. 1994. Evolution of concepts regarding Helicobacter pylori: From a cause ofgastritis to a public health problem. Am J Gastroenterol 89:469–472.
Hennig EE, Godlewski MM, Butruk E, Ostrowski J. 2005. Helicobacter pylori VacA cytotoxininteracts with fibronectin and alters Hela cell adhesion and cytoskeletal organization invitro. FEMS Immunol Med Microbiol 44:143–150.
Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. 2002. SHP-2 tyrosine phosphatase as an intracellular target ofHelicobacter pyloriCagA protein. Science295:683–686.
Hocker M, Henihan RJ, Rosewicz S, Riecken EO, Zhang Z, Koh TJ, Wang TC. 1997. Gastrinand phorbol 12-myristate13-acetate regulate the human histidine decarboxylasepromoter through Raf-dependent activation of extracellular signal-regulated kinase-related signaling pathways in gastric cancer cells. J Biol Chem 272:27015–27024.
Iaquinto G, Todisco A, Giardullo N, D’Onofrio V, Pasquale L, De Luca A, Andriulli A, PerriF, Rega C, De Chiara G, Landi M, Taccone W, Figura N. 2000. Antibody response toHelicobacter pylori CagA and heat-shock proteins in determining the risk of gastric cancerdevelopment. Dig Liver Dis 32:378–383.
Jin Y, Leung WK, Sung JJ, Wu JR. 2005. p53-indipendent pRB degradation contributes to adrug-induced apoptosis in AGS cells. Cell Res 15:695–703.
Jungblut PR, Bumann D, Haas G, Zimny-Arndt U, Holland P, Lamer S, Siejak F, Aebischer A,Meyer TF. 2000. Comparative proteome analysis of Helicobacter pylori. Mol Microbiol36:710–725.
Kamiya S, Yamaguchi H, Osaki T, Taguchi H. 1998. A virulence factor of Helicobacter pylori:Role of heat shock protein in mucosal inflammation after H. pylori infection. J ClinGastroenterol 27S:S35–S39.
Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. 1995.Helicobacter pylori and atrophicgastritis: Importance of the cagA status. J Natl Cancer Inst 87:1777–1780.
Macchia G, Massone A, Burroni D, Covacci A, Censini S, Rappuoli R. 1993. The Hsp60protein of Helicobacter pylori: Structure and immune response in patients withgastroduodenal diseases. Mol Microbiol 9:645–652.
MacLachlan TK, Sang N, Giordano A. 1995. Cyclins, cyclin-dependent kinases and cdkinhibitors: Implications in cell cycle control and cancer. Crit Rev Eukaryot Gene Expr5:127–156.
Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL. 2000. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos andc-jun. J Biol Chem 275:16064–16072.
Miehlke S, Kirsch C, Agha-Amiri K, Gunther T, Lehn N, Malfertheiner P, Stolte M, EhningerG, Bayerdorffer E. 2000. The Helicobacter pylori vacA s1, m1 genotype and cagA isassociated with gastric carcinoma in Germany. Int J Cancer 87:322–327.
Montecucco C, Papini E, de Bernard M, Zoratti M. 1999. Molecular and cellular activities ofHelicobacter pylori pathogenic factors. FEBS Lett 452:16–21.
Montecucco C, De Bernard M, Papini E, Zoratti M. 2001. Helicobacter pylori vacuolatingcytotoxin: Cell intoxication and anion-specific channel activity. Curr Top MicrobiolImmunol 257:113–129.
Moss SF, Calam J, Agarwal B, Wang S, Holt PR. 1996. Induction of gastric epithelial apoptosisby Helicobacter pylori. Gut 38:498–501.
Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. 1991.Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii.New Engl J Med 325:1132–1136.
Parsonnet J, Replogle M, Yang S, Hiat R. 1997. Seroprevalence of CagA-positive strainsamong Helicobacter pylori-infected, healthy young adults. J Infect Dis 175:1240–1242.
Peek RM Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas.Nat Rev Cancer 2:28–37.
Peek RM Jr, Miller GG, Tham KT, Perez-Perez GI, Zhao X, Atherton JC, Blaser MJ. 1995.Heightened inflammatory response and cytokine expression in vivo to cagAþHelicobacterpylori strains. Lab Invest 73:760–770.
Peek RM, Moss SF, Tham KT. 1997. Helicobacter pylori cagAþ strains and dissociation ofgastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst 89:863–868.
Penta R, De Falco M, Iaquinto G, De Luca A. 2005. Helicobacter pylori and gastric epithelialcells: From gastritis to cancer. J Exp Clin Cancer Res 24:337–345.
Perez-Perez GI, Thiberge JM, Labigne A, Blaser MJ. 1996. Relationship of immune responseto heat-shock protein A and characteristics of Helicobacter pylori-infected patients. J InfectDis 174:1046–1050.
Ping Dou Q, Bing An. 1998. Rb and apoptotic cell death. Front Biosci 419–430.Rieder G, Fischer W, Haas R. 2005. Interaction ofHelicobacter pyloriwith host cells: Function
of secreted and translocated molecules. Curr Opin Microbiol 8:67–73.Szabo I, Brutsche S, Tombola F, Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B,
Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. 1999. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori isrequired for its biological activity. EMBO J 18:5517–5527.
Tombola F, Carlesso C, Szabo I, de Bernard M, Reyrat JM, Telford JL, Rappuoli R,Montecucco C, Papini E, Zoratti M. 1999a. Helicobacter pylori vacuolating toxin formsanion-selective channels in planar lipid bilayers: Possible implications for the mechanism ofcellular vacuolation. Biophys J 76:1401–1409.
Tombola F, Oregna F, Brutsche S, Szabo I, Del Giudice G, Rappuoli R, Montecucco C,Papini E, Zoratti M. 1999b. Inhibition of the vacuolating and anion channel activities of theVacA toxin of Helicobacter pylori. FEBS Lett 460:221–225.
Yahiro K, Wada A, Yamasaki E, Nakayama M, Nishi Y, Hisatsune J, Morinaga N, Sap J, NodaM, Moss J, Hirayama T. 2004. Essential domain of receptor tyrosine phosphatase beta(RPTPbeta) for interaction with Helicobacter pylori vacuolating cytotoxin. J Biol Chem279:51013–51021.
Yuan JP, Li T, Chen HB, Li ZH, Yang GZ, Hu BY, Shi XD, Tong SQ, Li YX, Guo XK. 2004.Analysis of gene expression profile in gastric cancer cells stimulated withHelicobacter pyloriisogenic strains. J Med Microbiol 53:965–974.




















