
Organotins pollutions in seawater and sediment around a shipyard

Geochimica Q Cosmochimica Acts Vol. 57, pp. 1705-1718 Copyright Q 1993 Pergamon Press Ltd. Printed in U.S.A.
@II&7037/93/$6.00 + .CHl
The products from the oxidation of H2S in seawater
(Received June 10, 1992; accepted in revised form October 1, 1992 )
Abstract-The oxidation of sulfide in seawater is an overall second-order reaction, first-order with respect to both sulfide and oxygen. The major products formed from the oxidation of H2S with 02 have been measured in water and seawater as a function of pH (4-lo), temperature ( IO-45’C), and salinity (O- 36). The major products formed from the oxidation of H2S were SO:-, S20:-, and SO:-. The pH dependence of the product distribution has been attributed to the effect of pH on the rate of the individual reaction steps. A kinetic model was developed to account for the distribution of the reactants and products. The model is based on the following overall reactions:
H2S + O2 2 Products (S09),
H2S03 + O2 2 Products ( SOa), and
H2S + H2S03 + O2 2 Products ( S203).
Rate constants were determined for the oxidation of HS- and formation of SO:- (k,), for the oxidation of SO:- and production of SO:- (kz) and for the formation of SzO:- from HS- and SO:- (k,). These values of k, , kz, and k3 as a function of pH, temperature, and salinity can predict the concentrations of the measured reactants and products within experimental error. The effect of metals ( Fe3+, Fe*+, Mn2+ , cu2+, and Pb*+) and oxides (ru-FeOOH and MnOz) on the product distribution has also been examined. The field measurements from the Framvaren Fjord (Norway) and Cariaco Trench (Venezuela) are in reasonable agreement with the laboratory results at the same concentration of Fe2+ as in the anoxic basins.
INTRODUCI’ION
IN ANOXIC ENVIRONMENTS, hydrogen sulfide can be produced by bacteria1 anaerobic respiration of organic matter using sulfate as an electron acceptor. This formation of hydrogen sulfide occurs in a variety of natural waters, such as the pore- waters of sediments and stagnant basins (seas, lakes, rivers, and fjords). The production of hydrogen sulfide occurs also in hydrothermal systems. When water containing HIS mixes with oxygenated water at the oxic and anoxic interface, the hydrogen sulfide can be oxidized by 02. This oxidation is frequently coupled to changes in the redox state of metals (JACOBS and EMERSON, 1982; BOULEGUE et al., 1982) and nonmetals ( ZHANG and WHITFIELD, 1986). Dissolved ox- ygen, however, is the most important and abundant oxidant for the oxidation of hydrogen sulfide in natural waters. In recent years, we have studied the oxidation of H2S ( MILLERO, 1986a; MILLERO et al., 1987a) and H2SO3 ( ZHANG and MIL- LERO, 199 1) with O2 in water and seawater in the laboratory ( MILLERO et al., 1987a; ZHANG and MILLERO, 199 1) and in the field ( MILLERO, 199 la-d; ZHANG and MILLERO, 1993). We have attempted to characterize how the rates of oxidation were affected by trace metals in the environment (VAZQUEZ et al., 1989).
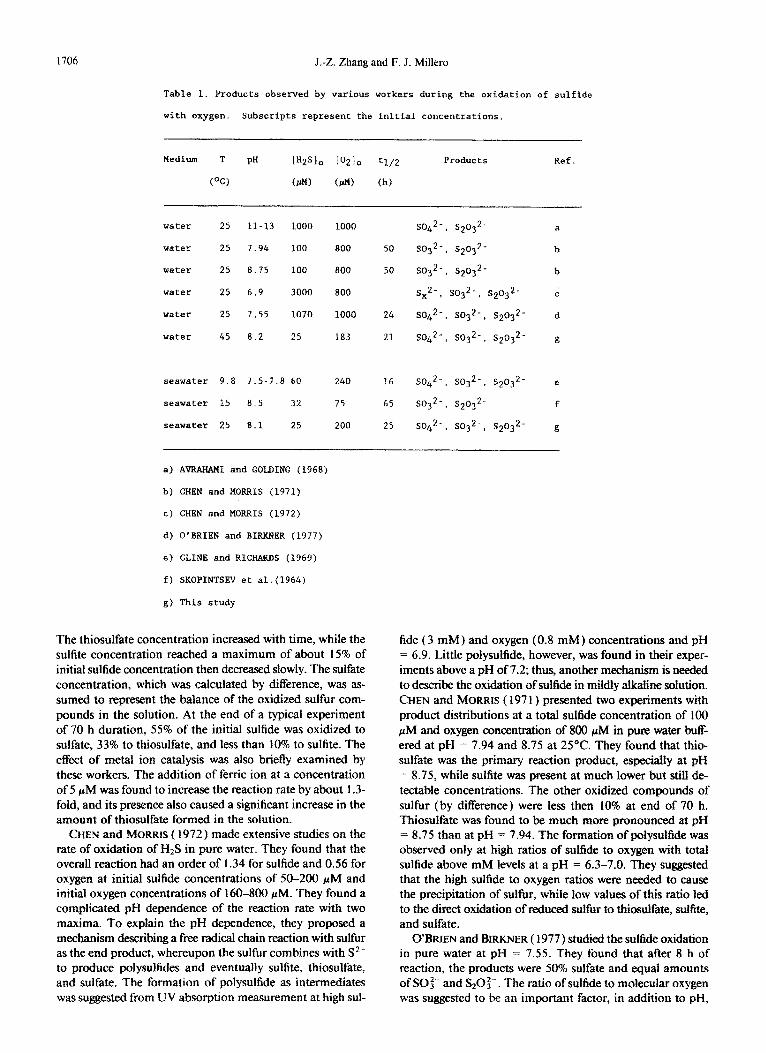
The oxidation of hydrogen sulfide involves a complex mechanism resulting in the formation of several reduced sul- fur species, such as thiosulfate, sulfite, elemental sulfur,.and polysulfide, as well as sulfate (AVRAHAMI and GOLDING, 1968; CHEN and MORRIS, 1972; CLINE and RICHARDS, 1969; O’BRIEN and BIRKNER, 1977). The oxidation products re- ported in previous studies and the conditions under which
they were found are summarized in Table 1. The large range of products found by various workers is due to the different oxidation conditions of the studies. The concentration of reactants and pH of media appear to be the most important parameters that control the product formation.
SKOPINTSEV et al. ( 1964) presented evidence for the for- mation of sulfite and thiosulfate as intermediates in seawater. The sum of sulfite and thiosulfate was estimated from the precipitation of the cadmium salt. The distinction between sulfite and thiosulfate was impossible due to the techniques used. AVRAHAMI and GOLDING ( 1968) studied the oxidation of sulfide in water at pH = 1 I - 13. The disappearance of the sulfide was reported to follow first-order kinetics. Sulfate and thiosulfate were found to be the principal products. They suggested that sulfide is initially oxidized to sulfite and that this initial oxidation is the rate-determining step. This reaction is followed by the rapid removal of sulfite as sulfate and thio- sulfate. They did not, however, measure the formation of the intermediate, sulfite, during the oxidation to support the mechanism. They were unable to quantify the thiosulfate due to the limitations of the analytical techniques used (ab- sorption spectrometry ). By increasing the initial total sulfide concentration to mM levels, they were only able to detect thiosulfate as a shoulder on the HS absorption peak because the wavelengths of the maximum absorption of HS- (230 nm) and S20:- (2 17 nm) were so close.
CLINE and RICHARDS ( 1969) conducted studies of the ox- idation of sulfide in seawater at 9.8”C and pH = 7.5-7.8. They reported first-order kinetics for the disappearance of both oxygen and sulfide. They also followed the distribution of sulfite and thiosulfate during the course of the reaction.
1705
1706 J.-Z. Zhang and F. J. Miller0
Table 1. Products observed by various workers during the oxidation of sulfide
with oxygen. Subscripts represent the initial concentrations.
Medium T pH [N2Slo [02lo t1/2 Products Ref.
(oC) (FM) (Itn) (h)
water 25 11-13 1000
water 25 7.94 100
water 25 8.75 100
water 25 6.9 3000
water 25 7.55 1070
water 45 8.2 25
seawater 9.8 7.5-7.8 60 240 16
seawater 15 8.5 32 75 65
seawater 25 8.1 25 200 25
800 50
800 50
800
1000 24
183 21
so42-, sag-, s2032-
so+, s2032-
so42-, sag- , s2032-
a) AVRAHAMI and GOLDING (1968)
b) CHEN and MORRIS (1971)
C) CHEN and MORRIS (1972)
d) O'BRIEN and BXRKNER (1977)
ef CLINE and RICHARDS (1969)
f) SKOPINTSEV et a1.(1964)
g) This study
The thiosulfate concentration increased with time, while the sulfite concentration reached a maximum of about 15% of initial sulfide concentration then decmased slowly. The sulfate concentration, which was calculated by difference, was as- sumed to represent the balance of the oxidized sulfur com- pounds in the solution. At the end of a typical experiment of 70 h duration, 55% of the initial sulfide was oxidized to sulfate, 33% to thiosulfate, and less than 10% to sulfite. The effect of metal ion catalysis was also briefly examined by these workers. The addition of ferric ion at a concentration of 5 PM was found to increase the reaction rate by about 1.3- fold, and its presence also caused a significant increase in the amount of thiosulfate formed in the solution.
CHEN and MORRIS ( 1972) made extensive studies on the rate of oxidation of H2S in pure water. They found that the overall reaction had an order of 1.34 for sulfide and 0.56 for oxygen at initial sulfide concentrations of 50-200 FM and initial oxygen concentrations of 160-800 PM. They found a compli~t~ pH dependence of the reaction rate with two maxima. To explain the pH dependence, they proposed a mechanism describing a free radical chain reaction with sulfur as the end product, whereupon the sulfur combines with S*- to produce polysulfides and eventually sulfite, thiosulfate, and sulfate. The formation of ~lysul~de as intestates was suggested from UV absorption measurement at high sul-
fide (3 mM) and oxygen (0.8 mM) concentrations and pH = 6.9. Little polysulfide, however, was found in their exper- iments above a pH of 7.2; thus, another mechanism is needed to describe the oxidation of sulfide in mildly alkaline solution. CHEN and MORRIS ( 197 1) presented two experiments with product distributions at a total sulfide concentration of 100 PM and oxygen concentration of 800 PM in pure water buff- ered at pH = 7.94 and 8.75 at 25’C. They found that thio- sulfate was the primary reaction product, especially at pH = 8.75, while sulftte was present at much lower but still de- tectable concentrations. The other oxidized compounds of sulfur (by difference) were less then 10% at end of 70 h. Thiosulfate was found to be much more pronounced at pH = 8.75 than at pH = 7.94. The formation of ~lysul~de was observed only at high mtios of sulfide to oxygen with total sulfide above mM levels at a pH = 6.3-7.0. They suggested that the high sulfide to oxygen ratios were needed to cause the precipitation of sulfur, while low values of this ratio led to the direct oxidation of reduced sulfur to G&sulfate, sulfite, and sulfate.
O'BRIEN and BIRKNER ( 1977) studied the sulfide oxidation in pure water at pH = 7.55. They found that after 8 h of reaction, the products were 50% sulfate and equal amounts of SO{-- and F&O:-. The ratio of sulfide to molecular oxygen was suggested to be an important factor, in addition to pH,

HzS oxidation in seawater 1707
in controlling the type and extent of formation of different sulfur species. For example, a low initial ratio of sulfide to oxygen favors the production of sulfite, thiosulfate, and sulfate over the pH range of 7.5-l 1. At a high ratio of sulfide to oxygen along with a total sulfide concentration greater than 1 mM, the oxidation led to the formation of elemental sulfur and probably polysulfides. They developed a kinetic model with parallel reactions between sulfide and oxygen to form sulfite, thiosulfate, and sulfate and used it to predict the product distribution.
A recent study by KOTRONAROU and HOFFMANN ( 199 1) also provided the evidence for production of elemental sulfur and polysulfide at high concentration of sulfide ( 1 mM ) , pH = 6.7 with 30 PM of Ni2+ in water.
In our laboratory, we have made measurements on the oxidation of Hz!3 ( MILLERO, 1986a; MILLERO et al., 1987a) and H2S03 ( ZHANG and MILLERO, 199 1) in water, NaCl, and seawater solutions as a function of pH, temperature, ionic strength, and composition. Rate equations have been derived for the oxidation of both H2S and HzS09 as a function of these variables. The overall rate equations for the oxidation
kH+ H$ + 02-Products, (1)
and k&=5
HzSO3 + 02- Products (2)
were given by
and
-d[HzfW~t = ki,s[HzSl,[Oz], (3)
-d[H2S0317-/d~ = kH~so,[H2S031~[02]0’5, (4)
where the subscript T refers to the total concentrations. Values
of kHzs and kHm3 have been given as a function of temper- ature, ionic strength, and pH ( MILLERO et al., 1987a; ZHANG and MILLERO, 1991). The effects that metals have on kHzs and kHzw3 have also been examined ( VAZQUEZ et al., 1989; ZHANG and MILLERO, 199 1). Field measurements ( MIL- LERO, 199 la,b,c,d; ZHANG and MILLERO, 1993) have dem- onstrated the importance that iron and manganese have on controlling the initial rates of oxidation of H2S in natural waters.
Only a few of the studies mentioned above have been con- ducted on the distribution of the products of the oxidation of sulfide in aqueous solutions over a wide range of experi- mental conditions and reaction media. By studying the in- termediate products formed and their relation to the exper- imental conditions, it may be possible to elucidate the mech- anism of hydrogen sulfide oxidation in natural waters. In the present study, we present the results of the products from the oxidation of H2S in water and seawater as a function of pH (4-lo), temperature (IO-45’C), and salinity (O-36). We also examine the effect trace metals have on the products formed. These results have been used to develop a chemical model that can be used to predict the products of oxidation of H2S by O2 in natural waters.
EXPERIMENTAL
The water used in all the experiments was ion-exchanged M&pore Super Q ( 18 Ma). All the chemicals used were of reagent grade. The
HzS stock solutions (0.05 M) were prepared with degassed water and NarS .9HzO crystals, which were washed with ionexchanged water to remove any oxidized surface layer. The NaHS stock solution was standardized by potentiometric titration using a sulfide ion-selective electrode and stored under a N2 atmosphere. The NasSQr and NazSzOJ stock solutions were prepared by weight with degassed water. Metal solutions were prepared by diluting standard metal solutions (Sigma). The ~Y-F~QQH was prepared by the addition of 200 mL of 2.5 N KOH to 50 g of Fe( NO,h - 9Hz0 in 825 mL of water to give a final pH near 12, followed by aging in oven at 60°C for 24 h (ATKINSON et al., 1967). The manganese dioxide was prepared by the oxidation of manganous by permanganate in basic solution (MURRAY, 1974). The stoichiometry ofthe manganese dioxide was found to be Mn0i.97. The surface sites of manganese dioxide (p& = 4.6) were determined from potentiometric titrations in 0.7 M NaCl solutions with 2 M HCI and 1 M NaOH and were in agreement with the value obtained by MURRAY ( 1974).
The seawater used was Gulf Stream water collected 10 miles off the coast of Miami and filtered through a 0.45 pm Millipore filter. The salinity was determined with a Guildline Autosal conductance bridge using the Practical Salinity scale. For the measurements at pH from 3-7, 0.02 M acetate buffers made from reagent grade acetic acid, HCI, and NaOH were used. At pH from 7- 11,0.02 M carbonate- bicarbonate buffers made from NaHCOs and NaOH were used. Val- ues of the pH were determined by using Orion 9 I-O I glass electrode and Ag, AgCl double junction reference electrode and a Metrohm pH meter. The electrode system was calibrated with tris (hydroxy- methyl) aminomethane(tris) buffer. The pH of the solutions was measured on the free proton scale ( MILLERO, 1986b).
Most of the kinetic measurements were made in solutions saturated with air at the reaction temperature by bubbling for a half hour. The molal concentrations of O2 in water and seawater at a given tem- perature and salinity were calculated from the equations of BENSON and KRAUSE ( 1984). Independent dissolved oxygen determinations by the Winkler method (CARPENTER, 1965) agreed with the calculated values to 0.5%. In studies of the oxygen dependence of the reaction, mixtures of oxygen and nitrogen at different mixing ratios were used to obtain different concentrations of oxygen. The maximum oxygen concentration was obtained by saturating the solution with pure ox- ygen. The dissolved oxygen concentration in these solutions was de- termined by Winkler titrations.
The oxidation studies were made in a 1000 cm3 water jacket glass cell. The temperature was controlled to +O.O5”C with a Forma tem- perature bath. The vessel was cleaned with 2 M HNO, between runs to minimize the catalytic effect of trace metals on the reaction. The solution in the glass reaction vessel was stirred during the reaction with a magnetic stirrer. Samples were taken from the reaction vessel using a glass piston to displace sample aliquots into a calibrated 5 cm3 pipette for sulfide and sulfate analysis and 1 cm3 pipette for sulfite and thiosulfate analysis.
The total sulfide concentration was determined spectrophotomet- rically by the methylene blue method of CLINE ( 1969 ) . The absor- bance was measured in a I cm cell at 666 nm with a Hewlett Packard 8452A diode array spectrophotometer. The sulfite and thiosulfate concentrations were determined by high-performance liquid chro- matography ( HPLC) after derivatization with 2.2-dithiobis I5-nitro- pyridine), (DTNP, .VAIRAVAMURTHY and MOPPER, 1990). The stronger eluent was a mixture of 80% acetonitrile and 20% pure water. The weaker eluent was a mixture of 45% acetonitrile and 55% pure water containing 0.05 M sodium acetate, 10 mM tetrabutyl am- monium acetate (TBAA). A microbore column containing hypersil was used to obtain the HPLC separations. A microbore guard column packed with spherisorb preceded the analytical column. The HPLC instrument consisted of a gradient system with a piston pump, a sample injector, and a Varian UV detector with a 2 pL flow cell. The complexes were detected at 325 nm. An IBM computer was used to control the system and integrate the area under the peaks. A 1 .O mM DTNP solution in acetonitrile was used for the derivatization. The DTNP ( 100 uL) was added to 1.0 cm’ of the samnle to form the derivative. The reaction was complete within 5 min. The HPLC sep arations were performed by elution at a flow rate of 700 PL per min. The system was calibrated using standard solutions of NazSOp and Na2S203. The precision was +O. I PM at levels of 5 to 10 PM.
The concentration of sulfate in the pure water runs was determined

I708 J.-Z. Zhang and F. J. Miller0
using an ion chromatographic technique. The ion chromatographic instrument consisted of a metal-free column (Dionex MFC- I ), fol- lowed by guard (Dionex AG4A) and analytical columns (Dionex AS4A) with an Eldex metering pump, a sample injector, and a con- ductivity detector. The samples were introduced into the system using a Valco 6 port manual injection valve with a 50 pL sample loop. The eluent solution contained 1.8 mM NaHCOS and 1.7 mM Na2COJ. The eluent solution was pumped through the series columns to separate different anions at a flow rate of 2.8 cm3 per min. A Dionex membrane suppressor ( AMMS) was used to lower the back- ground conductivity and increase the sensitivity. A 12.5 mM HzS04 solution was used as regenerant and was fed through the suppressor using positive air pressure at a flow rate of 4-5 cm3 per min. The anions were detected using a Dionex conductivity detector and quantified using a Spectra-physics integrator ( Model SP4270). The system was calibrated using standard Na$O., solutions prepared by weight. The detection limit was I pM for SO:-.
Polysuhide can be detected qualitatively by the appearance of a yellow-green color in the solutions. Quantitative analyses of poly- sulfide are complicated by the high reactivity of these compounds with oxygen and equilibria between the various polysulfide and monosulfides in solution. CHEN and MORRIS ( 1972) have used ab- sorbance at 290 nm as an indication of the presence of polysulfide in their reaction solution. The molar absorptivities for S$- and S:- at 290 nm are 3900 and 5300 1 cm-’ mol-‘, respectively (CHEN and MORRIS, 1972 ) In this study, solution absorbances at 290 nm were used as a qualitative indication of the presence or absence of poly- sulfide. The detection limit for polysulfides by this method is I PM. Elemental sulfur in the reaction solution at high concentrations can be detected visually since its solubility is about 5 PM. Its appearance was never observed in any experiments reported here.
RESULTS AND DISCUSSION
The overall rate equation for the oxidation of sulfide can be represented by the following:
-dH$3]/dt = k[HzS]“[OJb, (5)
where k is the overall rate constant, a and b are the order of reaction, and the brackets represent concentrations. Mea- surements were made to determine the order of the reaction with respect to H2S and Oz. This was done by measuring the rates of oxidation at different initial concentrations of H2S and Oz. The initial concentration of sulfide was varied from IO-30 PM, and the dissolved oxygen concentration was varied from 2 12-800 PM, depending on the partial pressure of ox- ygen, temperature, and salinity (BENSON and KRAUSE, 1984 ) . Under these experimental conditions, with oxygen in excess, the rate equation can be reduced to
-d[H,S]/dt = k'[ H2Sla. (6)
The order with respect to sulfide was determined from mea- surements made at initial oxygen concentrations [ 0210 = 2 12 PM and initial concentrations of total hydrogen sulfide [ HZS]o = 10 to 30 PM. Plots of In [HIS] vs. time were found to give straight lines with the same slope (Fig. 1) and indicated that a is equal to 1, or the reaction is first-order with respect to the sulfide concentration in agreement with earlier studies ( CLINE and RICHARDS, 1969; MILLERO et al., 1987a). The rate constants, k’ (min-‘), can be derived from the slope of the lines and yield the values of log k’ = -3.33 ? 0.02, -3.32 t 0.02, and -3.32 + 0.02 for initial sulfide concentrations of 30, 17, and 10 PM, respectively. These experiments dem- onstrate that the rates are independent of the initial concen- tration of HIS. All of the kinetic measurements in seawater
‘;; 2.6
N Ed
r
1.8
1 .O 0 6 12 1.9 24 30 36
TIME (HOUR)
FIG. 1. Plot of In [ H2S] (PM ) vs. time for the oxidation of sulfide in seawater at initial concentrations of ]O&, = 212 pM and [H$]o = IO to 30 PM (S = 35.0, pH = 8.20, and t = 25°C).
gave a first-order dependence with respect to sulfide, as was found in our earlier work (MILLERO et al., 1987a) and with previous measurements in buffered solutions at mM levels of sulfide (O’BRIEN and BIRKNER, 1977) and in seawater at
PM levels of sulfide ( CLINE and RICHARDS, 1969).
The value of k' is related to k by
k’ = k[O,lb.
The order of the reaction with respect to the oxygen concen- tration, b, was determined from measurements in seawater at initial concentration of [ H,Slo = 25 PM and [ 0210 = 2 12- 800 PM (Table 2; Fig. 2). The resulting values of log k’ are plotted vs. log [O,] in Fig. 2 and give a slope of 0.97 + 0.02. We accept 1.0 as the order with respect to oxygen. Our results are close to the value (0.8) obtained from initial rate method at mM level of initial sulfide concentrations of O’BRIEN and BIRKNER ( 1977) in NaCI, who accepted an order of unity for the oxidation with respect to oxygen in their modelling work. Our results are higher, however, than the value of OSS- 0.58 found by CHEN and MORRIS ( 1972) in pure water and KOTRONAROU and HOFFMANN ( 199 1) in wastewaters with an added metal catalyst. The differences of order with respect to O2 found in this study and the work of O’BRIEN and BIRK- NER ( 1977) with the work of CHEN and MORRIS ( 1972) and KOTRONAROU and HOFFMANN ( I99 1) may be related to the high levels of H2S used in the latter studies or the effect of trace metals. The measurement made by KOTRONAROU and HOFFMANN ( I99 I ) for waters without added metals give an order of 0.9 +- 0.3, which is in reasonable agreement with our work. In summary, we feel that our results demonstrate that the order of oxidation of H2S with respect to the con- centrations of H2S and O2 in seawater is second order in agreement with Eqn. 1.
The final product of oxidation of sulfide is sulfate, which is the sulfur compound having the highest oxidation state and the most stable sulfur compound in oxic waters. However, various intermediate products, such as sulfite and thiosulfate. can be formed during the course of the reaction. A series of kinetic experiments were made to examine the products dis-

H2S oxidation in seawater I709
Table 2. The pseudo-first order rate constant for
the oxidation of sulfide in seawater at different
concentrations of dissolved oxygen. S - 35,
PH - 8.2 and t - 25OC.
[021 (NM) log k’(min-l)
212.0 -3.36
390.4 -3.09
521.4 -2.97
800.2 -2.80
tribution pattern and its relationship to the reaction condi- tions (ZHANG, 1991). These experiments were carried out at temperatures from 10-45”C in water and seawater. These results are summarized in Tables 3 and 4 and shown in Figs. 3-9. Under these experimental conditions, the oxidation of sulfide by oxygen is a second-order overall reaction, lirst- order with respect to both total sulfide and oxygen.
The first experiment was made in pure water buffered at a pH of 8.2 with 0.002 M bicarbonate at 45°C. In this me- dium, sulfate formed from the oxidation could be measured by an ion ~hromat~raphi~ technique. The major products formed were found to be SO$-, SO:-, and S,O$- (Fig. 3). Elemental sulfur or polysulfides were not found by spectro- scopic techniques during the course of reaction (CHEN and MORRIS, 1972). The total equivalent sulfur of the three products and the remaining sulfide was in good agreement with initial added sulfide, indicating that SO:-, SO:-, and S,O:- are the main products. The initial product of the ox- idation is SO$-, which decreases with time. Thiosulfate and sulfate are the major products for the oxidation of H2S in water under these conditions. The concentrations of thio- sulfate are greater than sulfate and indicate that the oxidation does not go to completion over the 60 h of the experiment. Thiosulfate is known to be stable in neutral and alkaline solutions containing oxygen ( AVRAHAMI and GOLDING, 1968).
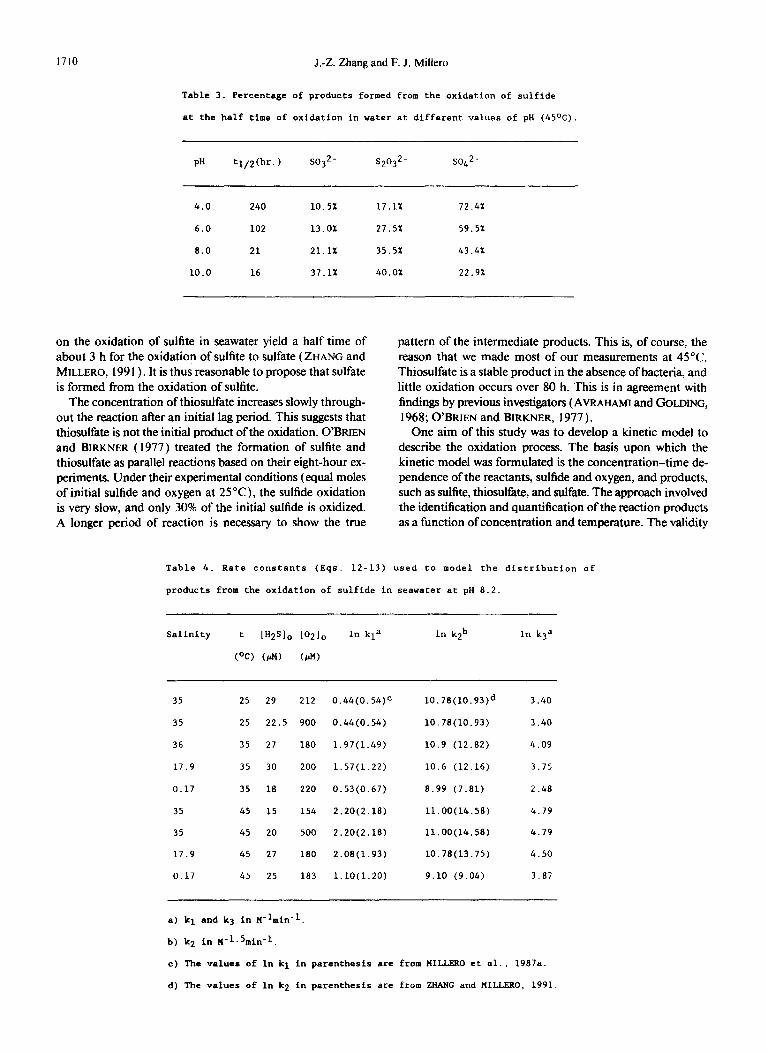
The effect of pH on the distribution of products from the oxidation of HS was studied in water at 45°C (Fig. 4). At
a pH of 4.0, the major product was SO:-. As the pH of the solution is increased from 4 to 10, the percentage of SO:- increases from 10.5% at pH = 4.0 to 37.1% at pH = 10.0. This increase can be attributed to the effect of pH on the oxidation rate of sulfite ( ZHANG and MILLERO, 199 1). Our previous studies have shown that the maximum rate of ox- idation of sulfite occurs at a pH of 6.5. As the pH increases above 7, the rate decreases and sulfite becomes more stable in basic solutions, The percentage of thiosulfate is 17.1% at pH = 4.0 but increases with pH to 40% at pH = 10.0. Thio- sulfate is stable in neutral and alkaline solutions containing oxygen but is not stable in acidic solutions.
The percentage of sulfate decreases from 72.4% at pH = 4.0 to 22.9% at pH = 10.0. Sulfate, the final product of sulfide oxidation, is the main product in acidic solutions but becomes less abundant in basic solutions. The oxidation rate of sulfide increases with an increase of the pH due to the increased concentration of HS-, which is more reactive than H2S ( MILLERO, 1986b). In acidic solutions, the oxidation of sui- fide to sulfite is a slow process, and the oxidation of sulfite to sulfate is relatively faster than the rate of production of sulfite. Under these conditions, sulfite maintains a low con- centration level and sulfate become the primary product. At a high pH, the oxidation of sulfide to intermediate products is faster than the oxidation of intermediate products to sulfate. Thus, suifite and thiosulfate can build up to significant con- centrations in the solutions. In summary, the effect of pH on the product distribution is attributed to the relative magnitude of the pH dependence of individual oxidation steps.
The products formed from the oxidation of H_LS in seawater have been studied at ~fferent tem~ratures and reactant concentrations f ZHANG, 1991). The effect of changes in temperature and reactant concentrations are shown in Figs. 5-8. AVRAHAMI and GOLDING (1968) have proposed that sulfite is the initial product from the oxidation of sulfide with oxygen in alkaline solutions. Unfortunately, they were not able to measure the sulfite in their experiments due to the limitation of their experimental methods. Our measurements in water (Fig. 3) and seawater (Figs. 5-8) at pH = 8.2 support their contention that sulfite is the initial oxidation product. The concentration of sulfite increases rapidly at the beginning of the reaction and decreases slowly when the production is slower then the rate of oxidative removal. The major product is SO:- in all of the seawater runs. The concentration of sulfate is higher at the higher concentrations of oxygen (Figs. 6 and 8) as one might expect. Since we determined SOi- by difference, some of the assigned values could be elemental sulfur or polysulfides present at levels below the detection limit of the spectroscopic method used ( 1 FM ) . O’BRIEN and BIRKNER ( f977) proposed parailel reactions for oxidation of sulfide to sulfite, thiosulfate, and sulfate. Our recent studies
-2.7 t I I 1
-3.5 ! -3.6
I I -3.6
I -3.$
I -3.2 -3.0
m fo2l FIG. 2. Values of log k’ ( min -’ ) vs. log [ 0, ] for the oxidation of
sulfide in seawater at initial concentrations of [ H2Slo = 25 pM and [O& = 212400 NM (S = 35.0, pH = 8.20, and t = 25°C).
1710 J.-Z. Zhang and F. J. Millero
Table 3. Percentage of products formed from the oxidation of sulfide
at the half time of oxidation in water at different values of pH (45OC).
PR tl/2(hr.) so32 - s2032- so42 -
4.0 240 10.5% 17.1% 72.4%
6.0 102 13.0% 27.5% 59.5%
8.0 21 21.1% 35.5% 43.4%
10.0 16 37.1% 40.0% 22.9%
on the oxidation of sulfite in seawater yield a half time of about 3 h for the oxidation of sulfite to sulfate ( ZHANG and MILLERO, 199 1). It is thus reasonable to propose that sulfate is formed from the oxidation of sulfite.
The concentration of thiosulfate increases slowly through- out the reaction after an initial lag period. This suggests that thiosulfate is not the initial product of the oxidation. O’BRIEN
and BIRKNER ( 1977) treated the formation of sulfite and thiosulfate as parallel reactions based on their eight-hour ex- periments. Under their experimental conditions (equal moles of initial sulfide and oxygen at 25’C), the sulfide oxidation is very slow, and only 30% of the initial sulfide is oxidized. A longer period of reaction is necessary to show the true
pattern of the intermediate products. This is, of course, the reason that we made most of our measurements at 45°C. Thiosulfate is a stable product in the absence of bacteria, and little oxidation occurs over 80 h. This is in agreement with findings by previous investigators ( AVRAHAMI and GOLDING, 1968; O’BRIEN and BIRKNER, 1977 ).
One aim of this study was to develop a kinetic model to describe the oxidation process. The basis upon which the kinetic model was formulated is the concentration-time de- pendence of the reactants, sulfide and oxygen, and products, such as sulfite, tbiosulfate, and sulfate. The approach involved the identification and quantification of the reaction products as a function of concentration and temperature. The validity
Table 4. Rate constants (Eqs. 12-13) used to model the distribution of
products from the oxidation of sulfide in seawater at pH 8.2.
Salinity t [H2Sl, l“2l.z In kla ln k2b In k3a
(W (/N (tin)
35 25 29 212 0.44(0.54)C
35 25 22.5 900 0.44(0.54)
36 35 27 180 1.97(1.49)
17.9 35 30 200 1.57(1.22)
0.17 35 18 220 0.53(0.67)
35 45 15 154 2.20(2.16)
35 45 20 500 2.20(2.18)
17.9 45 27 180 2.08(1.93)
0.17 45 25 183 l.lO(1.20)
10.78(10.93)d
10.78(10.93)
10.9 (12.82)
10.6 (12.16)
8.99 (7.81)
ll.OO(14.58)
ll.OO(14.58)
10.78(13.75)
9.10 (9.04)
3.40
3.40
4.09
3.75
2.48
4.79
4.79
4.50
3.87
a) kl and k3 in M-lmin-1.
b) k2 in H-1.5min-1.
c) The values of In kl in parenthesis are from MILLER0 et al., 1987a.
d) The values of In k2 in parenthesis are from ZING and MILLERO, 1991

H2S oxidation in seawater 1711
total S
mtE (HO~JR)
FIG. 3. The sulfur balance during the oxidation of sulfide in water at 4YC, pH = 8.2, [O& = 183 PM, and [HzS]o = 25 FM.
of the model was evaluated by comparison of the model prei dictions with the experimental measurements of reactants and products. AVRAHAMI and GOLDING ( 1968) have sug-
gested that at low concentmtions, the overall oxidation of HS is given by the following:
HS- + 1.50, --+ HSO;, (8)
so:- + 0.502 --, so:-, (9)
SO;- f HS- + 0.502 --* SzO:- + OH-, (10)
and
szo$- + 0.50, --* so:- + s. (11)
With these overall reactions in mind, one can attribute the formation of SO:- to the oxidation of HS-, and the formation of SO:- to the oxidation of SO:-. The formation of SOf from the oxidation of S20:- can be neglected (AVFWHAMI
and GOLDING, 1968 ) for solutions devoid of bacteria (Mft LERO, 199 I c) . The formation of SzO$- can be attributed to the overall reaction of SO:- and HS- with 02. This leads to the following overall rates of oxidation:
R tn
100 u-al sop Hb S$ 2-
rZ- BO-
0-0 so4
60-
40-
20-
,
PH mE (HOUR)
FIG. 4. The etfect of pH on the distribution of products (after one FtG .6. The distribution of products from the oxidation of sulfide half time) fram the oxidation of suhide in water at t = 4YC, [O& in seawater at [O&J = 900 pM, [ HzSb = 22.5 FM, S = 35.0, and t = 183 PM, and fH,Sfo = 25 PM. = 25°C. The smooth curves are cakuiated from the model.
TIME (HOUR)
FIG. 5. The distribution of products from the oxidation of sulfide in seawater at [O& = 212 pM, [H2S]o = 29 pM, S = 35.0, and t = 25°C. The smooth curves are calculated from the model.
and
H*S + 02 2 Products (SO,); (12)
lizSO3 + 02 2 Products (SO& (13)
I&S + &,SOs + 02 9 Products (S203). (14)
The overall rate equations for the formation and production of H$, SO:-, S20$-, and SO:- are given by the following:
d[H,Slldf =-~,WIIS~[W - M-WW:-l[o,l; (15)
d[so:-l/dt = k,[W,S][O21 - k2[s0:-12[021”*
- k,[~2W~O:-l[~21; (16)
dfS,O$-]/dt = ~~[H2S][SO~-][O2]; and (17)
f&SOP]/& = k*[so2,-~2[02]“2, (1st
where [i] are the total concentrations of i. It should

1712 J.-Z. Zhang and F. J. Miller0
20
15
s 10
E
5
0 0 10 20 30 40 50 60
TIME (HOUR)
FIG. 7. The distribution of products from the oxidation of sulfide in seawater at [ O,b = 154 pM, [ HzSk, = 15 PM, S = 35.0, and 1 = 45OC. The smooth curves are calculated from the model.
represent the mechanism of the reactions. The order of the rates of oxidation of H$ (Eqn. 12) and HzS03 (Eqn. 13) are assumed to be equal to the order (Eqns. 3 and 4) found in our earlier studies (MILLERO et al., 1987a; ZHANG and MILLERO, 199 1). The order of the rate of formation of SzO:- has been taken from the work of AVRAHAMI and GOLDING ( 1968 ).
These ordinary differential rate equations can be integrated simultaneously to predict concentrations of all the reactants and products as a function of reaction time as long as the initial concentrations of all species and rate constants are known. We have accomplished this with the fourth-order program of the Runge-Kutta method (0.01 set time steps) on a personal computer (PRESS et al., 1987 ). By simulating the model to fit the experimental time dependence of the reactants and products, one can evaluate the values of k, , k2, and k3. The values of k, , k2, and k, determined from our studies of the rates of oxidation of HIS and the formation of SO:-, SzO:-, and SO:- at various temperatures, salinity, pH in water, and seawater are tabulated in Tables 4 and 5. Com-
BME (HOUR)
FIG. 8. The distribution of products from the oxidation of sulfide in seawater at [ O2 Jo = 500 PM, [ H2S10 = 20 PM, S = 35.0, and f = 45°C. The smooth curves are calculated from the model.
-41 I , . # I , . , 2 4 6 8 10
PH
FIG. 9. The dependence of rate constants (k, , k2, and k,; Eqns. 12-14) on pH for the oxidation of sulfide in water at t = 45°C.
parisons of the predicted concentrations with measured quantities are presented in Figs. 3 and 5-8 (smooth lines). The experimental data are in good agreement with the model predictions up to reaction times of 80 h. The values of k2 in seawater needed to fit the data were slightly smaller than the values determined in our previous study ( ZHANG and MIL- LERO, 199 1)) especially at higher temperatures. This is prob- ably due to the inhibition of the oxidation of sulfite in the presence of sulfide ( CHEN and MORRIS, 1972). This finding is supported by the observations of CLINE and RICHARDS ( 1969) that sulfite in the presence of H$ is more stable in seawater than predicted by its rate of oxidation, Because the oxygen is in excess, the competition for oxidant is unlikely to cause this difference. The complexation of HS with trace metals which can catalyze the oxidation of sulfite in seawater may be a more likely cause. Trace metals have higher ten- dency to complex with HS- than SO:- ( ZHANG, 199 1).
We found that the formation of thiosulfate could be best predicted with a rate equation which is zero-order with respect to oxygen, a finding in agreement with O’BRIEN and BIRKNER ( 1977). The effect of changes in the ratio of H2S to Or on the product distribution over the range of our measurements can be attributed to the order in the rate equations, which is independent of the rate constants.
The values of k, increased by a factor of 27, and k3 increased by a factor of 4 from 25 to 45°C. The rate constant for ox- idation of sulfite, kZ, increased by only 1.25 times for the same temperature change due to the presence of sulfide in the reaction media as mentioned above. The values of k,, kz, and k3 increased by factors of 14,6.7, and 2.5, respectively, when the salinity is increased from O-35. The oxidation of sulfide to sulfite is the rate process most affected by changes in temperature and salinity.
The values of k, , k2, and k3 from the experiments in pure water as a function of pH are given in Table 5 and shown in Fig. 9. The values of k, and k2 increase with an increase in pH, as expected from our earlier studies (MILLERO et al., 1987a; ZHANG and MILLERO, 1991). The values of k3 also increase with an increase in pH. These rate constants in water have been fitted to the second-degree functions of pH (t = 45”C), as follows:

H2S oxidation in seawater 1713
Table 5. R&e censtante (Eqs. 12 -14) at 45% used to model the dfrtributictn
of producte from ths oxidation of sulfide in water as a function of pH.
PH
4.0
6.0
E.0
10.0
In kla
-1.43
-0.5X
1.10
1.44
In kpb
8.19
9.39
9.10
B.?O
In ks8
0.18
2.36
3.87
3.18
a) kl and k3 in M-bin-L.
b) k2 in ~~~%&n-l.
In k, = -4.71 + 0.914 pH - 0.0289 pH2, (19)
In kz = 3.87 -t ISt pH - O.fO3 pH2, (201
and
In k3 = -9.09 + 3.01 pH - 0.177 pH2. (21)
These equations should give reasonable estimates for the effect of pH on the rate constants.
The values of k, , k2, and k3 as a function of salinity (S) and temperature ( Tin K) have been fitted to the equations (PM = 8.2), as follows:
and
In k, =1 26.90 + 0.03228 - 8123.21 IT;
in kz = 14.91 f 0.0524s - 1764.68/T,
(22)
(23)
In k3 = 28.92 + 0.03698 - 8032.68/T. (24)
These equations should be valid for estuarine and seawater over wide ranges of salinity and temperature. The energies of activations (El = RT’[a In kjfilT)] in seawater determined from this study are El = 72, EZ = 17, and Es = 55 kj M-‘. The value for El is in good agreement with the value found in our earlier work (66 + 5 kj M-l; MILLERO et al., 1987a). The value of .& is lower than value found in our earlier work ( 140 kj M-I ; ZWANG and MILLERO, 199 1)) apparently due to the difference in the rates of oxidation of sulfite in the presence of sulfide. Equations 22-24 should yield predicted product distributions for the oxidation of sulfide in natural waters with low levels of metals.
The presence of iron, rnan~n~, and other trace metal in natural waters has been shown to have a sign&ant effect on the rate of oxidation of sulfide in seawater ( VAZQIJEZ et al., 1989). These metals aiso have an effect on the oxidation of the intermediate, sulfite, in seawater (ZHANG and MIL-
LERO, 199 1). We have examined the effect of metals on the oxidation of H$ and the products formed from this oxidation ( ZHANG, 199 1) . Some environmentally important metals, such as Fe, Mn, Cu, and Pb, have been chosen for this study. These metals have been shown to not only increase the rate of oxidation of sulfide but also to change the distribution of products formed during the oxidation f ZHANG, I99 1) .
The effect of metals on the formation of products during the oxidation of I&S can be characterized by the changes of the magnitude of the rate constants, kl , kz, and kj, in the presence of metals at a given concentration. The distribution of the products formed with added metals are given elsewhere @HANG, 199 1). The kinetic model has been used to fit the distribution of products formed in seawater in the presence of metats. The resutting rate constants, k, , k2, and IQ, are summarized in Table 6 ( ZHANG, 199 1). The rate constants as a function of the concentration of metals added to seawater are shown in Figs. IO, 11, and 12.
The effect of Fe’” has been studied at levels of 200 and 1000 nM. Of the metais studied, Fe’” is found to be the most effective catalyst in the oxidation of H2S, which agrees with our previous study (VAZQUEZ et al., 1989). The rate con- stants, k, , k,, and k,, increase with increasing concentration of F$’ but show a similar pattern (ZHANG, 199 1) for the f~ation of products as in the studies devoid of metals (Figs. S-8). The increase in the rate cous~nts by Fe*” is truly a catalytic effect. As discussed elsewhere ( ZHANG and MILLEFIO, 1991), this is presumably caused by the rapid oxidation of Fe2+ with oxygen ~M~LLERO et al., 1987b), as follows:
and
Fe2+ f O2 = Fe3+ + O- 2,
0, + H+ = HQ,
(251
(26)
HO2 + HO2 = H& + 02. (271
The oxidation of Fez’ produces Fe3+ that can oxidice sulfide and sulfite rapidly. The peroxide generated by the oxidation of Fez’ can also increase the rates since it has a higher rate of oxidation with sulfide and sulfite than oxygen ( MILLERO et al., 1989; MADER, 1958). The reduction ofdissolved Fe” by HS- can regenerate Fe2+ to complete the catalytic cycle.
The addition of Fe3” has a smaller effect than Fe” on the rate of oxidation sulfide in seawater. This behavior of Fe3’, when added to seawater, has been shown in our previous study ( ZHANG and MILLERO, 199 1). The addition of Fe3+ only increases the initial rate of the oxidation of sulfite. After 30 min, the rates become constant, similar to the rate in

1714 J.-Z. Zhang and F. J. Mihero
Table 6. Rate constants (Eqs. 12-14) used to model the distribution of
products from the oxidation of sulfide in seawater as a function of the
concentration of metals.
Metal Cont.
W)
In kla In k2b In ksa
Seawater
Pb2+
Pb2+
Pb2+
Fez+
Fe2+
cu2+
Fe3+
un2+
oFeOOH
IQ-lop
1
2
5
0.2
1
5
5
25
12
12
0.54 10.84 3.40
3.40 10.88 5.89
4.29 11.00 7.09
5.37 11.70 7.50
4.09 11.08 4.56
5.19 11.41 5.89
4.28 11.70 6.17
4.62 11.70 7.09
2.40 11.00 4.79
1.57 11.00 4.09
2.83 12.10 4.79
a) kl and k3 in Wb.n~l.
b) kp in K-1.5,in-1.
ordinary seawater without the addition of metals. This is due to the low solubility of Fe3+ in seawater at a pH of 8.2. The Fe3” added precipitates as iron oxides within 30 min after the addition (ZHANG and MILLERO, 1991). Only the dis- solved, not the colloidal, iron was shown to be an effective catalyst for the oxidation of sulfite (MARTIN and HILL, 1989). The addition of Fe’+ has a catalytic effect only before its complete hydrolysis to colloidal iron. This wilf cause the rate of oxidation of sulfite to be greater in the first 30 min and
2.5
Fit. 10. The effect of metal ions and metal oxides on the rate FIG. I I. The effect of metal ions and metal oxides on the rate constant k, (Eqn. 12) in seawater (the superscript zero is used to constant k2 (&I-I. 13) in seawater (the superscript zero is used to denote the values without added metats). denote the values without added metals).
much slower after 30 min. After 8 h of reaction, 56% of the product is thiosulfate, 34% is sulfate, and only 10% is sulfite. This indicates that Fe3+ catalyzed the formation of thiosulfate, which becomes a primary product.
The increase in the rate constants with the addition of Cu*+ is the same magnitude as found for Fe3+. The sulfite decreases more rapidly in the presence of Cu’+, since Cu2+ has been shown to be an effective catalyst for the oxidation of sulfite ( HOFFMANN and JACOB, 1984). The effect of Pb2+

H2S oxidation in seawater 1715
on k, is between the effects found for Fe*’ and Fe3+. The effect of Pb2+ on k2 is similar to the effect of Fe3+ and Cu’+. The effect of Pb2+ on the formation of thiosulfate, however, is as large as found for Fe”. The addition of Pb2+ increases the rate of oxidation of sulfide, causing an initial buildup of sulfite (see Fig. 13). This sulfite is further oxidized to sulfate, causing a maximum. After the maximum, the concentration of sulfite decreases rapidly. The rate of disappearance of sulfite in the presence of Pb’+ was the fastest found among the metals studied. As sulfite decreases, the thiosulfate increases and be- comes almost constant after the sulfite disappears. The sulfur balance indicates that other products besides sulfate might be found in the early stages of the reaction. The most likely intermediate is elemental sulfur or polysulfide, which can react rapidly with sulfite to form thiosulfate, as follows:
s -I- so:- -W szo:- (28)
Sf + XSO:- + H+ + XS,O:- + HS-. (29)
These reactions provide another pa~way to produce thio- sulfate besides Fqn. 10. Sensitive analytical techniques are needed to be able to detect elemental sulfur or polysulfide at these low concentrations to verify this mechanism.
The iron hydroxide and manganese dioxide are thought to be important inorganic particles in natural waters. Iron hydroxides and manganese dioxides can react directly with sulfide and also provide solid surfaces that can act as a catalyst for the oxidation of sulfide. The effect of cu-FeOOH on the rate constants was the same as Mn’+ . The addition of 12 PM of (Y-FeOOH increases the initial rate of oxidation of sulfide by a factor of 4.8, yielding a half time of 7.9 h. The initial rate of oxidation of sulfide decreased after 4 h of reaction to
Mn*+ has the smallest effect on the rate of oxidation among the metals studied. Our previous results (VAZQUEZ et al., 1989; ZHANG and MILLERO, 199 1) indicated that the addition of Mn*+ increased the rate of the oxidation of sulfide and sulfite only at a ratio of f Mn’+]: [ S] larger than 1 .O. The higher oxidation rate at the ratio above 1 .O has been attributed to the formation of the more labile MnHS+ and MnS03.
The primary products are sulfate ( 60% ) and thiosulfate ( 30% ) at the end of 54 h.
FIG. 12. The effect of metal ions and metal oxides on the rate constant Q (Eqn. 14) in seawater (the superscript zero is used to denote the values without added metals).
w H2S
30 a-a so32- z F;‘-
- 3 20
53 IO
0 0 1 2 3 4 5 6 7 8
nta (HOUR)
FIG. 13. The distribution of products from the oxidation of sulfide in seawater with 2 pM of Pba+ at [O& = 212 pM, [H$l, = 27 PM, salinity of 35.0, and 25°C.
a rate in seawater with a haIf time of 22 h, as shown in Fig. 14. The larger initial rate could be attributed to the reactive sites on the surface of a-FeOOH, which increased the rate of oxidation. After 4 h of reaction, all the reactive sites were used up, and the rate decreased to the ordinary rate in sea- water. The primary products are sulfate (57%), thiosulfate (28%), and sulfite ( 15%) at the end of 55 h.
The previous studies indicated that both oxides can react with sulfide. The measurements of PYZIK and SOMMER ( 198 1) show that amorphous iron oxides can oxidiie HzS and produce elemental sulfur and thiosulfate. The measure-
MnO2 is slightly more effective then cw-FeOOH on the ox- idation of sulfide and the formation of ~ios~fate and much more effective on the oxidation of sulfite. The addition of 12
ments of BURDIGE and NEALSON ( 1986) showed that the
PM of Mn02 increased the rate of oxidation of sulfide by a factor of 16.8. The primary products are sulfate (67%), thio-
oxidation of HIS by Mn02 produced elemental sulfur as the
sulfate ( 19%), and sulfite (14%) at end of 7.5 h.
predominant product. The sediment studies of ALLER and RUDE ( 1988) have demonstrated that MnOl can oxidize sul- fide to sulfate, as follows:
3.07. * . * ~....f .,.., .,..,...,, . . ’ , 1
2.5
‘si 2.0 cu
k c 1.5
1.0
os~....,....,.,..,....,....,....~ 0 10 20 30 40 so 60
ntdC: (HOUR)
FIG. 14. The rate of oxidation of sulfide in the presence of 12 pM ofa-FeOOH in seawater at fO& = 212 FM, [H&l, = 16 pM, salinity of 35.0, and 2YC.

1716 J.-Z. Zhang and F. J. Miller0
4Mn02 + HS- + 7H+ --+ 4Mn*+ + SO:- + 4H20. (30)
The oxidation of H2S with iron oxides may also yield sulfate, as follows:
8FeOOH + HS- + 15H + + 8Fe*+ + SO:- + 12H20.
(31)
These studies were made under anoxic conditions. In the presence of oxygen, different reaction pathways may be ex- pected, giving the different product distribution we have found. The manganese oxides react faster than the iron oxides with H2S and may be responsible for the oxidation of HIS in anoxic waters (LUTHER et al., 199 1) ,
We have recently examined the effect of various transition metals on the rates of oxidation of sulfide in the laboratory ( VAZQUEZ et al., 1989) and at sea ( MILLERO, 199 la,c; ZHANG and MILLERO, 1993). The laboratory studies indicate that below concentrations of 300 nM, only Fe2+, Cu*+ , and Pb2+ affect the rates of oxidation of 25 PM H2S in oxygenated seawater. At concentrations below 20 nM, the addition of trace metals has no effect on the rates of oxidation. At higher metal concentrations, the rates of oxidation increase for all the metals studied, except for Zn *+. The field measurements were made in the Black Sea, Framvaren Fjord, and Cariaco Trench to study the oxidation of sulfide in various anoxic environments ( MILLERO, 199 1 a,c; ZHANG and MILLERO, 1993). The measured rates of oxidation in the field were in good agreement with the rates estimated from the laboratory studies at the same concentration of Fe2+ in the waters. These studies indicate that Fe*+ is at sufficiently high concentrations in these anoxic environments to cause the observed increases in the rates of oxidation of H2S.
In our studies in the Framvaren Fjord ( MILLERO, 199 I a), and in the Cariaco Trench ( ZHANG and MILLERO, 1993 ), we have measured the distribution of the products formed during the oxidation of H2S. The kinetic model described in this paper has been used to estimate the values of k, , k2, and k3 in these anoxic waters from these distributions ( MILLERO, 199 la; ZHANG and MILLERO, 1993). The values of k, , k2, and k3 estimated from these field measurements were found to be in reasonable agreement with the values predicted from the results of this study when the comparisons are made at the same concentration level of Fe’+ (see Figs. 15 17 ). These comparisons point out the use of our simple model in de-
scribing the oxidation of H2S in natural waters.
The values of k, estimated for the Framvaren Fjord and Cariaco Trench are slightly higher than the predicted values. This could be due to errors in our estimation of the concen- tration and form of iron in these water. Direct measurements of iron and manganese in the anoxic waters should be made in all future kinetic studies to avoid this problem. The esti- mates based upon only the concentration of iron may also lead to some errors. The concentrations of Mn2+ below the oxic-anoxic interface reach levels of 0.5 and 15 PM in the Cariaco Trench and Framvaren Fjord, respectively. This Mn2+ comes from reduction of Mn02 sinking from above the oxic-anoxic interface. Reoxidation of Mn *+ to MnOz by bacteria occurs when Mn*+ diffuses up to the oxic layer. This cycling of manganese between Mn2+ and Mn02 is an im- portant feature in the oxic-anoxic interface and probably af- fects the distribution of products in the field.
3.0-
2.5- Fromwren /I’
Carioco A+==-
2.0- / I
y’ A’
$ 1.5- 11’ I
1 .o-
0.5-
0.0 I . , , , , , , , , , , . , , . , -7.5
, -7.0 -6.5 -6.0 ‘.5
log [Fez+]
FIG. 15. Comparison of k, (Eqn. 12) from the Cariaco Trench and Framvaren Fjord with the values predicted from concentration of Fe’+. The dash lines are the rate constants predicted from the concentration of Fe*+ based on the this study.
The agreement between the model and the observed dis- tribution of reaction products does not provide conclusive proof that the reaction pathways of the overall model actually describe the series of elemental reactions that occur. The de- tailed mechanisms might involve many elemental reaction steps. At present, there are two detailed mechanisms for the oxidation of H2S with O2 in aqueous solutions (CHEN and MORRIS, 1972; HOFFMANN and LIM, 1979)) the polar mech- anism, and the free radical chain mechanism. The polar mechanism for the formation of H2.S is given in the following (HOFFMANN and LIM, 1979):
HS- + O2 --f HSO;, (32)
HSO; t* H+ + SO:-, (33)
sol- + 02 --t so; + o;, (34)
soi + 02 -+ so* + o;, (35)
SOz+H20++HSOj+H.‘. (36)
The overall reaction is given by the following:
HS- + 30? + Hz0 -+ HSO; + 2HO>. (37)
The superoxide ion formed can react with itself to form hydrogen peroxide, which can also react with HS- ( MILLERO et al., 1989), as follows:
02 + H+ *--) HO*, (38)
HOz + HO2 -+ Hz02 + 02. (39)
The intermediate HSO; or SO:- can react with HS- and form thiosulfate:
HSO; t- HS -+ S202- + H20, (40)
s*02 + 02 + SO-. (41)
The initial reaction can also result in the formation of ele- mental sulfur:
HS + O2 +S+HOy. (42)

HsS oxidation in seawater 1717
Fmmvoren I
5.0- Cariaco -_/+----
AC
YN -----L_/-A
C
H ._/---
4s
4.0, * . * . , * . . . , . . . . * . . , . -7.5 -7.0 -6.5 -6.0 -5.5
log [Fe*+]
RG. 16. Comparison of k2 (Eqn. 13) from the Cariaco Trench and Framvaren Fjord with the values predicted from concentration of Fe’+. The dashed lines are the rate constants predicted from the concentration of Fe*+ based on the this study.
which can react with sulfite to give thiosulfate,
s f so:- -+ sro:-, (43)
or hydrogen sulfide to give polysulfides,
nS + HS- + HS;. (44)
The formation of sulfate comes from the oxidation of sulfite with oxygen,
2so:- + 02 * 2so:-, (45)
or hydrogen peroxide,
HSO, + Hz02 -f HSO; + HrO, (46)
which involves the formation of the intermediate -O&)OH ( HOFFMANN and EDWARDS, 1975; MCARDLE and HOFF- MANN, 1983).
In the presence of trace metals in seawater, a free radical mechanism is a more likely pathway. In the free radical mechanism, oxidation is initiated by an outer-sphere electron transfer from HS- to oxygen to form the HS + radical, as follows:
HS- + O2 + HS= + 03. (47)
In the presence of trace metals in seawater, electron transfer from HS- to the transition metal ions to form HS’ radical is more favorable, as follows:
HS- f lW’+ -+ HS. + M(“-‘)+. (48)
The HS’ radical is further oxidized to sulfite by a free radical chain sequence involving oxygen, as follows:
HS. +O,*HSO.;, (49)
HSOs + H+ + SO;, (50)
soi f 02 + sq + 05, (51)
SO2 f HitO = HSO; + H+, (52)
HSO; = SO:- + H+. (53)
The sulfite formed can react with HS - to produce thiosulfate:
HS. + SO:- + HSzO:- . , (54)
H&O:- - + O2 -+ H&03 + 05, (55)
HSzOi + = SzO:- + H . (56)
The sulfite formed can be further oxidized to sulfate through the free radical mechanism ( BACKSTROM, 1934; ZWANG and MILLERO, 199 1) , as follows:
M”+ + SO;- + I@“-‘)+ + SO;, (57)
so; + o* + so:, (58)
so; + so$- “-+ so:- + so;, (59)
so:- + SOS- * 2so:-. (60)
Different rate equations can be obtained, depending upon the assumptions made for the nature of the rate-determining proration and te~ination steps ( HO~~ANN and JACOB, 1984; KOTRONAROU and HO~ANN, 199 1). Although we cannot propose a unique mechanism for the oxidation of HrS and formation of the resultant products in seawater, it is useful to make some comparison of our findings with the proposed mechanisms given above. The first-order depen- dence of the oxidation of H&l with respect to H&l and O2 can be attributed to the slow steps in the oxidation being Eqn. 32 in polar m~h~ism or Eqn. 49 in free radical mech- anism. The slow step in the formation of thiosulfate can be attributed to Eqn. 54, which will give the correct orders with respect to sulfide and sulfite. As discussed elsewhere ( ZHANG and MILLERO, 199 1 ), the termination steps in the oxidation of sulfite, ( HOFFMANN and JACOB, 1984), such as
so; + so; + !&ok + 02, (61)
2H02 --) Hz02 + 02, (62)
would give the appropriate oxygen dependence for the oxi- dation of sulfite. Further experimentation is needed to prove these speculations. Beside the stable intermediate determined
3.0
Fromvamn / /’
/
2.5- Carioco
/
I /
3 /
B
2.0- /
/ /
1.53 /- /
/
1.0~ . . . . , , . , , , . . , . ( , , , , -7.5 -7.0 -6.5 -6.0 I.5
log [Fe*+]
RG. 17. Comparison of k3 (Eqn. 14) from the Cariaco Trench and Framvaren Fjord with the values predicted from concentration of Fe’+. The dashed lines are the rate constants predicted from the concentration of Fe2+ based on the this study.

1718 J.-Z. Zhang and F. J. Miller0
in this study, identification of those extremely reactive, short- lived intermediates, such as free radical or transition state complexes, would provide evidence for the possible mecha- nism.
In this study, we have derived equations for the rates of product formation from the oxidation of H2S. The model we used, although somewhat simplistic, can be used to estimate the intermediates formed from the oxidation of HIS with O2 in natural waters. Studies are presently underway to examine the oxidation of HIS with manganese and iron oxides and the products formed from this oxidation as a function of temperature, pH, and salinity. When these studies are com- pleted, we should be able to model the abiodic oxidation of HZS and examine the importance of the biological oxidation of H2S, SO:-, and F&O:- in the field.
Acknowledgments-The authors gratefully acknowledge the support of the Ocean Sciences Division of the National Science Foundation (OCE89-22580) and the Office of Naval Research (NOOOl4-90-J- 1225).
Editorial handling: R. A. Schmitt
REFERENCES
ALLER R. C. and RUDE P. D. ( 1988) Complete oxidation of solid phase sulfides by manganese and bacteria in anoxic marine sedi- ments. Geochim. Cosmochim. Acta 52,75 l-765.
ATKINXIN R. J., P~SNER A. M., and QUIRK J. P. ( 1967) Adsorption of potential-determining ions at the ferric oxide-aqueous electrolyte interface. J. Phys. Chem. 71, 550-558.
AVRAHAMI M. and GOLIXNG R. M. ( 1968) The oxidation of the sulfide ion at very low concentrations in aqueous solutions. J. Chem. Sot. A, 647-65 1.
BAC~STROM H. J. ( 1934) The chain mechanism in the autoxidation of sodium sulfite solutions. Z. fhys. Chem. B25,99- 12 1.
BENSON B. B. and KRAUSE D., JR. ( 1984) The concentration and isotopic fractionation of oxygen dissolved in fresh water and sea- water in equilibrium with the atmosphere. Limnol. Oceanogr. 29, 620-632.
BOULEGUE J., LORD C. J., III, and CHURCH T. M. ( 1982) Sulfur speciation and associated trace metals (Fe, Cu) in porewaters of Great Marsh, DeIaware. Geochim. Cosmochim. Acta 46,45 3-464.
BURDIGE D. J. and NEAL~ON K. H. ( 1986) Chemical and micro- biological studies of sulfide-mediated manganese reduction. Geomicrobiol. J. 4, 36 I-387.
CARPENTER J. H. ( 1965) The Chesapeake Bay Institute Technique for the Winkler dissolved oxygen method. Limnol. Oceanogr. 10, 141-143.
CHEN K. Y. and MORRIS J. C. ( 197 I ) Oxidation of aqueous sulfide by O,:l. General characteristics and catalytic influences. In Ad- vances in Water Pollution Research, Vol. 2. (ed. S. H. JENKINS), pp. III-32/l-111-32/17. Pergamon.
CHEN K. Y. and MORRIS J. C. ( 1972) Kinetics of oxidation of aqueous sulfide by 02. Environ. Sci. Tech. 6, 529-537.
CLINE J. D. ( 1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14,454-458.
CLINE J. D. and RICHARDS F. A. ( 1969) Oxygenation of hydrogen sulfide in seawater of constant salinity, temperature, and pH. En- viron. Sci. Tech. 3, 838-843.
HOFFMANN M. R. and EDWARDS J. 0. ( 1975) Kinetics of the oxi- dation of sulfite by hydrogen peroxide in acidic solution. J. Phys. Chem. 79,2096-2098.
HOFFMANN M. R. and JACOB D. J. ( 1984) Kinetics and mechanisms of the catalytic oxidation of dissolved sulfur dioxide in aqueous solution: An application to night time fog water chemistry. In SO,, NO. and NO, Oxidation Mechanisms: Atmospheric Considerations fed. J. G. CALVERT), pp. 101-172. Butterworths.
HOFFMANN M. R. and LIM B. C. ( 1979) Kinetics and mechanism of the oxidation of sulfide by oxygen: Catalysis by homogeneous metal-phthalocyanine complexes. Environ. Sci. Tech. 13, 1406- 1414.
JACOES L. and EMERSON S. ( 1982) Trace metal solubility in an anoxic Fjord. Earth Planet. Sci. Lett. 60,237-252.
KOTRONAROU A. and HOFFMANN M. R. ( I99 I ) Catalytic autoxi- dation of hydrogen sulfide in wastewater. Environ. Sci. Tech. 25, 1153-l 160.
LUTHER G. W., III, CHURCH T. M., and POWELL D. ( I99 1). Sulfur speciation and sulfide oxidation in the water column of the Black Sea. Deep-Sea Res. 38, S 112 1-S 1137.
MADER P. M. ( 1958) Kinetics of the hydrogen Peroxide-sulfite re- action in alkaline solution. J. Amer. Chem. Sot. 80,2634-2639.
MARTIN L. R. and HILL M. R. ( 1989) Sulfur oxidation in aerosols; iron catalysis at high pH. 198th Amer. Chem. Sot. Mtg., Miami Beach. FL 29(Z), 183-l 85 (abstr.).
MCARDLE J. V. and HOFFMANN M. R. (1983) Kinetic and mech- anism of the oxidation of aquated sulfur dioxide by hydrogen per- oxide at low pH. J. Phys. Chem. 87,5425-5429.
MILLERO F. J. ( 1986a) The thermodynamics and kinetics of the hydrogen sulfide system in natural waters. Mar. Chem. 18, 121- 147.
MILLERO F. J. ( 1986b) The pH of estuarine waters. Limnol. Oceanogr. 31,839-847.
MILLERO F. J. (1991a) The oxidation of HIS in Framvaren Fjord. Limnol. Oceanogr. 36, 1007- 10 14.
MILLERO F. J. ( 199 1 b) The oxidation of H2S in the Chesapeake Bay. Estuarine Coasta/ She!fSci. 33, 52 l-527.
MILLERO F. J. ( 199 Ic) The oxidation of H$ in Black Sea Waters. Deep-Sea Res. 38, S I 139-S I 150.
MILLERO F. J. ( 1991d) The oxidation of H2S with O2 in the Black Sea. In Blark Sea Oceanography (eds. E. IZDAR and J. W. MUR- RAY ), pp. 205-227. Kluwer Academic.
MILLERO F. J., HUBINGER S., FERNANDEZ M., and GARNET S. ( I987a) Oxidation of H2S in seawater as a function of temperature, pH, and ionic strength. Environ. Sci. Tech. 21, 439-443.
MILLERO F. J., SOTOLONGO S., and IZAGUIRRE M. ( 1987b) The oxidation kinetics of Fe(II) in seawater. Geochim. Cosmochim. Acta 51, 793-80 1.
MILLERO F. J., LAFERRIERE A. L., FERNANDEZ M., HUBINGER S., and HERSHEY J. P. ( 1989) Oxidation of H2S with Hz02 in natural waters. Environ. Sci. Tech. 23, 209-213.
MURRAY J. W. ( 1974) The surface chemistry of hydrous manganese dioxide. J. Colloid Inte$ce Sci. 46, 357-37 I.
O’BRIEN D. J. and BIRKNER F. G. ( 1977) Kinetics of oxygenation of reduced sulfur species in aqueous solution. Environ. Sci. Tech. II, 1114-1120.
PRESS, W. H., FLANNERY B. P., TEUKOLSKY S. A., and VETTERLING W. T. ( 1987) Numerical Recipes. Cambridge Univ. Press.
PYZIK A. J. and SOMMER S. E. ( 198 1). Sedimentary iron monosul- fides; kinetics and mechanism of formation. Geochim. Cosmochim. Acta 45, 687-698.
SKOPINTSEV B. A., KARPOV A. V., and VERSHININA 0. A. (1964) Study of the dynamics of some sulfur compounds in the Black Sea under experimental conditions. Sov. Oceanogr. 4, 55-73.
VAIRAVAMURTHY A. and MOPPER K. ( 1990) Determination of sulfite and thiosulfate in aqueous samples including anoxic seawater by liquid chromatography after derivation with 2,2’-dithiobis(%ni- tropyridine). Environ. Sci. Tech. 24, 333-337.
VAZQUEZ G. F., ZHANC J-Z., and MILLERO F. J. (1989) Effect of transition metals on the rate of the oxidation of HzS in seawater. Geophys. Res. Lett. 16, I363- 1366.
ZHANG J-Z. ( 199 1) The rates of oxidation of reduced sulfur com- pounds in seawater. Ph.D. diss, Univ. Miami, FL.
ZHANG J-Z. and MILLERO F. J. ( 199 1) The rate of sulfite oxidation in seawater. Geochim. Cosmochim. Acta 55, 677-685.
ZHANG J-Z. and MILLERO F. J. ( 1993) The chemistry of the anoxic waters in the Cariaco Trench. Deep-Sea Res. (in press).
ZHANG J-Z. and WHITFIELD M. ( 1986) Kinetics of inorganic redox reaction in seawater. I. The reduction of iodate by bisulphide. Mar. Chem. 19, 121-137.
Copyright © 2022 FDOKUMEN




















