
Ozone oxidation of polyethoxylated alcohols
10
Was. lles. Vol. 27, No. 8, pp. 1313-1322,1993 0043-1354/93 $6.00+0.00 Printed in GreatBritain.All rights reserved Copyright© 1993Pergamon Pr-,~s Ltd OZONE OXIDATION OF POLYETHOXYLATED ALCOHOLS A ~ A M. BRAMBILLA I, LUCIACALVOSA 2, AMELIA MONTEVERDI I, STEFANO POLESELLO 3 and BRUNO~ * @ IDipartimentodi Chimica Organica • Industriale, Universita' di Milano, via Venezian 21, 20133 Milano, 2Enichem, Via lannozzi, l, San Donato Milanese and 3Dipartimento di Chimica Inorganica, Metallorganica e Analitica, Universita' di Milano, Via Venezian 21, 20133 Milano, Italy (First received July 1992; accepted in revised form January 1993) Almract--The ozonation of a polyethoxylated alcohol (typical of one type of detergent) mixture was performed in aqueous solution under different conditions of pH, reaction time and hydrogen peroxide addition. Surfactant degradation was monitored by TOC measurements. The optimal degradation conditions were found to be at pH ffi7.5 with hydrogen peroxide addition. Inspection of the reaction products was carriedout by GC-MS and GC-FTIR analysis of the silylated mixtures. The main oxidation products of ethoxylated alcohols are dicarboxylic acids. Key words--aqueous ozonation, non-ionic surfactants, polyethoxylated alcohols, GC-MS, GC-FTIR, trimethylsilylderivatives,hydrogen peroxide, dicarboxylic acids INTRODUCTION The ability of ozone to reduce the total organic carbon (TOC), chemical oxygen demand (COD) and biochemical oxygen demand (BOD) of waste- waters has already been demonstrated (Narlds and Schaeidor-Rotel, 1980). Some problems appear in drinking water ozonation since formaldehyde and other carbonyl compounds concentration increased after ozone treatment (Yamada and Somiya, 1991; Glaze et al., 1989; Gilli et al., 1990). Many studies are intended to reveal the by- products of the ozonation of several polluting organic compounds. These might be even more dangerous than the substrates themselves (Legube et al., 1989; Reynolds et al., 1989). The identification of ozona- tion products is also important to investigate the reaction mechanisms that lead to their formation (Caprio et al., 1989; Andreozzi et al., 1990; Takanashi and Katsnki, 1990; Takanasbi, 1990a). Previous work (Niki et al., 1983) had investigated the ozonation in buffered solutions of small aliphatic alcohols, ketones and acids such as 1- and 2-butanol, 2-butanone, 3-pentanone, acetic, propionic, butyric, isobutyric and pivalic acids and small bifunctional molecules containing carboxyl groups, such as methylglyoxal, glycolic, lactic and pyruvic acids. These studies showed that alcohols were oxidized to the corresponding carbonyl compounds and that aliphatic chains were functionalized by introduction of hydroxyl and keto groups. Fragmentation was also observed, with formation of carboxylic acids such as formic, acetic, propionic, oxalic and pyruvic acids. Acetaldehyde and methylglyoxal were also found as *Author to whom all correspondence should be addressed. ozonation products of 2-butanone. In all cases, the rate of ozonation increased with increasing the pH of the medium. At low pH, small saturated acids as acetic, propionic, oxalic and isobutyric acids were stable toward ozone at 30°C, but were attacked at appreciable rate at higher pH. Formic acid was oxidized to carbon dioxide in all conditions. The presence of organic peroxides and hydrogen peroxide was also revealed in the reaction medium. A 1,3 dipolar attack of ozone to C-H bonds and hydrotri- oxide intermediates were postulated. When larger saturated molecules as fatty acids were treated with ozone in water at low pH, little or no consumption of the substrate was observed (Reynolds et al., 1989a), while unsaturated fatty acids were readily modified in the same conditions. In fact, ozone reacts very rapidly with unsaturated compounds, but slowly with saturated compounds. Therefore, it is generally impossible to decompose them completely to CO2 and H20 by ozone itself. Many methods increase the oxidative ability of ozone by simultaneously using other physico- chemical methods (Takanashi and Katsuki, 1990). Ultraviolet irradiation-ozonation (Peyton et al., 1982; Prengle, 1983), ultrasound-ozonation (Dahi, 1976; Sierka and Amy, 1985), gamma radiation- ozonation (Arai et ai., 1986), hydrogen peroxide catalyzed ozonation (Nakayama et al., 1979; Paiilard et al., 1988), electrolysis-ozonation (Takanashi and Katsuki, 1990) have been carded out, and are based on ozone decomposition to more reactive species. One of these (Paillard et al., 1988) showed that the presence of buffers decreased the efficiency of ozonation. Also this study found that ozonation rates were higher at higher pH, because of the presence of the very highly reactive but poorly selective hydroxyl 1313
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Ozone oxidation of polyethoxylated alcohols

Was. lles. Vol. 27, No. 8, pp. 1313-1322, 1993 0043-1354/93 $6.00+0.00 Printed in Great Britain. All rights reserved Copyright © 1993 Pergamon Pr-,~s Ltd
OZONE OXIDATION OF POLYETHOXYLATED ALCOHOLS
A ~ A M. BRAMBILLA I , LUCIA CALVOSA 2, AMELIA MONTEVERDI I, STEFANO POLESELLO 3 and BRUNO ~ * @
IDipartimento di Chimica Organica • Industriale, Universita' di Milano, via Venezian 21, 20133 Milano, 2Enichem, Via lannozzi, l, San Donato Milanese and 3Dipartimento di Chimica Inorganica,
Metallorganica e Analitica, Universita' di Milano, Via Venezian 21, 20133 Milano, Italy
(First received July 1992; accepted in revised form January 1993)
Almract--The ozonation of a polyethoxylated alcohol (typical of one type of detergent) mixture was performed in aqueous solution under different conditions of pH, reaction time and hydrogen peroxide addition. Surfactant degradation was monitored by TOC measurements. The optimal degradation conditions were found to be at pH ffi 7.5 with hydrogen peroxide addition. Inspection of the reaction products was carried out by GC-MS and GC-FTIR analysis of the silylated mixtures. The main oxidation products of ethoxylated alcohols are dicarboxylic acids.
Key words--aqueous ozonation, non-ionic surfactants, polyethoxylated alcohols, GC-MS, GC-FTIR, trimethylsilylderivatives, hydrogen peroxide, dicarboxylic acids
INTRODUCTION
The ability of ozone to reduce the total organic carbon (TOC), chemical oxygen demand (COD) and biochemical oxygen demand (BOD) of waste- waters has already been demonstrated (Narlds and Schaeidor-Rotel, 1980). Some problems appear in drinking water ozonation since formaldehyde and other carbonyl compounds concentration increased after ozone treatment (Yamada and Somiya, 1991; Glaze et al., 1989; Gilli et al., 1990).
Many studies are intended to reveal the by- products of the ozonation of several polluting organic compounds. These might be even more dangerous than the substrates themselves (Legube et al., 1989; Reynolds et al., 1989). The identification of ozona- tion products is also important to investigate the reaction mechanisms that lead to their formation (Caprio et al., 1989; Andreozzi et al., 1990; Takanashi and Katsnki, 1990; Takanasbi, 1990a).
Previous work (Niki et al., 1983) had investigated the ozonation in buffered solutions of small aliphatic alcohols, ketones and acids such as 1- and 2-butanol, 2-butanone, 3-pentanone, acetic, propionic, butyric, isobutyric and pivalic acids and small bifunctional molecules containing carboxyl groups, such as methylglyoxal, glycolic, lactic and pyruvic acids. These studies showed that alcohols were oxidized to the corresponding carbonyl compounds and that aliphatic chains were functionalized by introduction of hydroxyl and keto groups. Fragmentation was also observed, with formation of carboxylic acids such as formic, acetic, propionic, oxalic and pyruvic acids. Acetaldehyde and methylglyoxal were also found as
*Author to whom all correspondence should be addressed.
ozonation products of 2-butanone. In all cases, the rate of ozonation increased with increasing the pH of the medium. At low pH, small saturated acids as acetic, propionic, oxalic and isobutyric acids were stable toward ozone at 30°C, but were attacked at appreciable rate at higher pH. Formic acid was oxidized to carbon dioxide in all conditions. The presence of organic peroxides and hydrogen peroxide was also revealed in the reaction medium. A 1,3 dipolar attack of ozone to C-H bonds and hydrotri- oxide intermediates were postulated.
When larger saturated molecules as fatty acids were treated with ozone in water at low pH, little or no consumption of the substrate was observed (Reynolds et al., 1989a), while unsaturated fatty acids were readily modified in the same conditions. In fact, ozone reacts very rapidly with unsaturated compounds, but slowly with saturated compounds. Therefore, it is generally impossible to decompose them completely to CO2 and H20 by ozone itself.
Many methods increase the oxidative ability of ozone by simultaneously using other physico- chemical methods (Takanashi and Katsuki, 1990). Ultraviolet irradiation-ozonation (Peyton et al., 1982; Prengle, 1983), ultrasound-ozonation (Dahi, 1976; Sierka and Amy, 1985), gamma radiation- ozonation (Arai et ai., 1986), hydrogen peroxide catalyzed ozonation (Nakayama et al., 1979; Paiilard et al., 1988), electrolysis-ozonation (Takanashi and Katsuki, 1990) have been carded out, and are based on ozone decomposition to more reactive species.
One of these (Paillard et al., 1988) showed that the presence of buffers decreased the efficiency of ozonation. Also this study found that ozonation rates were higher at higher pH, because of the presence of the very highly reactive but poorly selective hydroxyl
1313

1314 ANNA M. BRPd~ILLA et al.
radicals, according to the SBH model (Hoign6 and Bader, 1979; Staehelin and Hoign~, 1982; Staehelin et ai., 1984). In this model ozone is decomposed by hydroxide ions to yield hydroxyl radicals. With a normal reduction potential of 2.8 V against 2.07 V for ozone, the hydroxyl radical shows a higher oxidizing power than ozone. In order to increase the amount of this powerful oxidant in aqueous phase, hydrogen peroxide was added to ozonation solutions. The system ozone/hydrogen peroxide is a very efficient tool to obtain high removal of ozone refractory compounds (Duguet et al., 1990; Duguet et al., 1990a). Oxalic acid was used as a substrate to test the efficiency of this system; optimal conditions were found to be at pH 7.5. According to these authors, hydroxyl radicals are decomposed by hydroxide ions at high pH, so their concentration is maximum at neutral pH.
A recent study (Cheikowska et al., 1990) compared the old SBH model for aqueous ozone decomposition with the TFG model (Tomiyasu et al., 1985). This model suggests that the main chain carrier in the decomposition reaction is the ozonide radical ion (O3-) and that the hydroxyl radical is a far less important transient species than it was assumed before. Therefore, also in reactions with organic compounds other radical species than hydroxyl radical, such as the hydroporoxide radical (HO2), the superoxide radical ion (02-) and the ozonide radical ion (03-) might play an important role in their oxidation.
In this paper, we will adopt the SBH model for ozone decomposition, assuming that the hydroxyl radical is the main reactive species in alkaline con- ditions and in the presence of hydrogen peroxide.
In the treatment of non-ionic surfactants ozonation causes alterations in the molecular structure, resulting in changes in characteristics such as foaming ability, complexation with bismuth or cobaltothiocyanate and increased water solubility. These surfactants are not completely broken down into CO2 and H20, as reflected in the high TOC and COD residual concen- trations (Narkis et al., 1987). As a consequence, although surface activity is destroyed by ozonation a large organic load remains and creates potential environmental pollution (Glaze, 1987). Thus ozone treatment alone, despite its many advantages, is inadequate for the total removal of non-ionic suffactants. However ozonation can be used to increase the biodegradability of the biologically resistant compounds: small doses of ozone can be sufficient to change the structure of the non-ionic surfactant, making it more amenable to bacterio- logical breakdown and enhancing the biodegradable properties of the material.
In order to better understand the ability of ozone to degrade non-ionic surfactants in aqueous solutions we studied the behaviour of a mixture of ethoxylated phenols toward ozone (Calvosa et al., 1991). A normal phase HPLC analysis showed decreased
overall polarity of the mixture after ozonation as a result of the partial or total loss of the oxyethyl chain of these compounds.
We report here the results of the ozonation of an ethoxylated alcohol mixture (AEO).
EXPERIMENTAL SECTION
Reagents and materials The non-ionic surfactants investigated were supplied by
Ditta F.11i Lamberti, Albizzate--VA, Italy. The surfactant was obtained by reaction of ethylene oxide
with Dobanol 91 under basic conditions. Dobanoi 91 is a mixture of linear aliphatic alcohols supplied by Shell chemi- cals. The composition of this mixture was:
l-nonanol: 18 ~ 5% l-decanol: 42 :]: 6% l-undecanol: 38 :[: 5% alcohols < C9: < 1% alcohols > Clt: < 2 % carbonyl compounds: < 80 ppm
Average ethoxylation number of this mixture is 3.3. Sodium acetate, acetic acid, sodium carbonate, sodium
bicarbonate and anhydrous sodium sulphate were all analytical grade from Merck (Bracco S.p.A., Milan, Italy). Hexamethyldisilazane (HMSD) and trimethylsilylchloride (TMSC) were reagent grade from Aldrich (BH-Schilling, Milan, Italy). Ethyl acetate was purified by distillation. Benzene was distilled and dried over sodium in nitrogen atmosphere. The AEO samples were derivatized via reaction with HMDS and TMSC (Cross, 1987); diluted with anhydrous benzene and analysed by High Resolution Gas Chromatography (HRGC).
Sample ozonation, extraction and preparation Ozone was produced in a Fisher 501 generator fed with
oxygen. The ozonation was carried out on 1 litre solutions containing 50 mg/l of surfactant in water adjusted at the required pH. Before ozonation the solution of the subetrate was saturated with oxygen for 5 rain. A 70 l/h stream of oxygen was subjected to a 200 mA electrical current and then passed through the solution. The amount of ozone produced in these conditions was 3.3 g/h. After the given time nitrogen was bubbled into the solution for 5 rain.
Acidic conditions (pH = 4.0) were obtained by addition of a dilute solution of sulphuric acid and testing the pH with a Toptronic digital pH-meter: in these conditions the pH remained constant all over the reaction time.
Alkaline conditions (pH -- 9.5) were obtained by addition of a dilute solution of potassium hydroxide. In these conditions the pH moved rapidly to 4-5, and was held constant by addition of a 0.0713 N solution of potassium hydroxide.
Neutral conditions (pH=7.5) were obtained and maintained by addition of the same solution of potassium hydroxide.
The reaction solutions were acidified to pH = 3-4 and extracted three times with 750 ml of ethyl acetate. The collected extracts were dried over anhydrous sodium sulphate for one night, evaporated under reduced pre~ure, and weighed. The AEO samples were diluted with anhydrous benzene, derivatized via reaction with HMDS and TMSC (Cross, 1987) and analy.qed by High Resolution Gas Chromatography (HRGC), HRGC-Positive Ion Electron Impact Mass Spectrometry (PlEIMS) and HRGC-Fourier Transform Infrared Spectroscopy (FTIR).
High Resolution Gas Chromatography (HRGC~ High Resolution Gas Chromatography ~Mass Spectrometry (HRGC/MS) and High Resolution Gas Chromatography/ Fourier Transform Infrared Spectroscopy (GC/FTIR)
d I I
r e , a 9
I I
Ozone oxidation of polyethoxylated alcohols 1315
n = O
I I
m = l O I I I m = l l
I
I I I
I I
I
n = l
I I I
r,-i", m = l O I
I i m = l h
I I I j
I i m = 9
i I
n = 2 I n = 3 I i n = 4 I I ] CmH2m~. 1 - ( O C H 2 C H 2 ) n - 0 - S i M e 3 I",, /IX
/ / I \ , / I ,, ] I" m = l O I / I ~ I I I Ira=X0' , / ,~, , . - 5
: . : ' I , I, ' m lo , m = l l ' L I I n = 6
I ', , m , l l I I I
m : 9 m , L " ~ I I I , / I " ' I i
n 101 I m = 9
I m i t / / l \ 9 f I ",
I I I m = l O I
I I I I
I
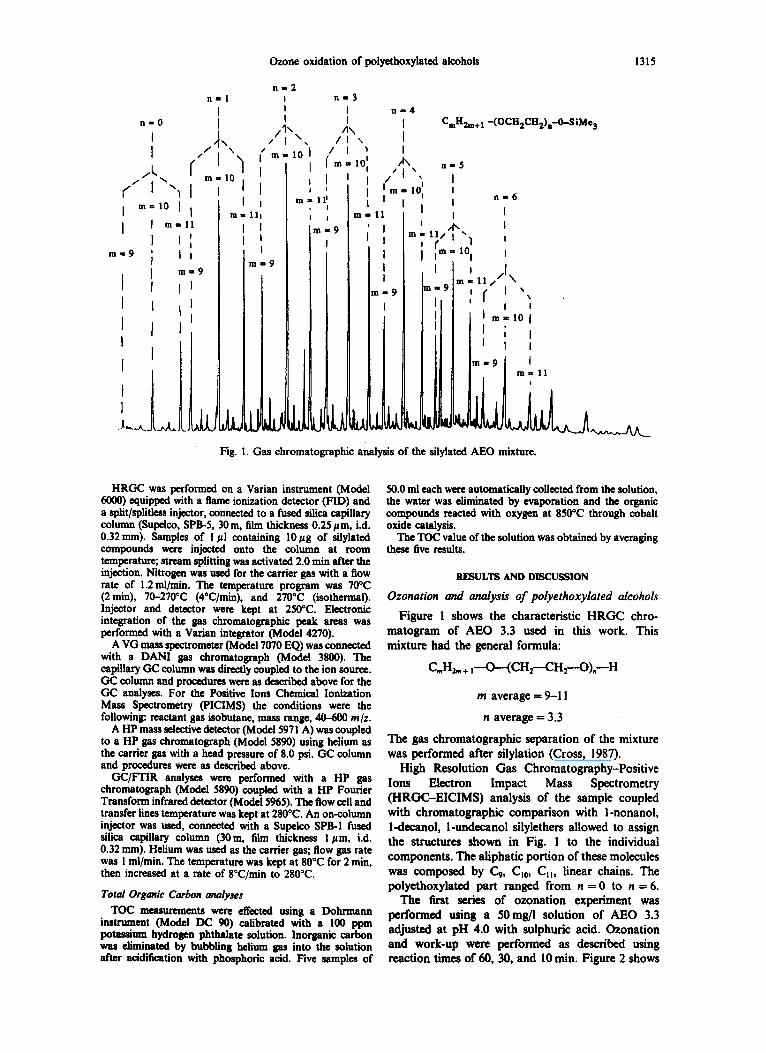
Fig. 1. Gas chromatographic analysis of the silylated AEO mixture.
HRGC was performed on a Varian instrument (Model 6000) equipped with a flame ionization detector (FID) and a split/splitless injector, connected to a fused silica capillary column (Supelco, SPB-5, 30m, film thickness 0.25/tin, i.d. 0.32ram). Samples of 1/~1 containing 10/~g of silylated compounds were injected onto the column at room temperature; stream spfitting was activated 2.0 rain after the injection. Nitrogen was used for the carrier gas with a flow rate of 1.2 mi/min. The temperature program was 70°C (2min), 70-270°C (4°C/rain), and 270°C (isothermal). Injector and detector were kept at 250°C. Electronic integration of the gas chromatographic peak areas was performed with a Varian integrator (Model 4270).
A VG mass spectrometer (Model 7070 EQ) was connected with a DANI gas chromatograph (Model 3800). The capillary GC column was directly coupled to the ion source. GC column and procedures were as described above for the GC analyses. For the Positive Ions Chemical Ionization Mass Spectrometry (PlCIMS) the conditions were the following: reactant gas isobutane, mass range, 40-600 m/z.
A HP mass selective detector (Model 5971 A) was coupled to a HP gas chromatograph (Model 5890) using helium as the carder gas with a head pressure of 8.0 psi. GC column and procedures were as described above.
GC/FTIR analyses were performed with a HP gas chromatograph (Model 5890) coupled with a HP Fourier Transform infrared detector (Model 5965). The flow cell and transfer lines temperature was kept at 280°C. An on-column injector was rated, connected with a Supelco SPB-I fused silica capillary column (30m, film thickness l/~m, i.d. 0.32 ram). Helium was used as the carder gas; flow gas rate was I ml/min. The temperature was kept at 80°C for 2 min, then increased at a rate of 8°C/min to 280°C.
Total Organic Carbon analyses TOC measurements were effected using a Dohrmaun
instrument (Model DC 90) calibrated with a 100 ppm potauium hydrogen phthalate solution. Inorganic carbon was eliminated by bubbling helium gas into the solution after acidification with phosphoric acid. Five samples of
50.0 ml each were automatically collected from the solution, the water was eliminated by evaporation and the organic compounds reacted with oxygen at 850°C through cobalt oxide catalysis.
The TOC value of the solution was obtained by averaging these five results.
RESULTS AND D I S C U S S I O N
Ozonation and analysis of polyethoxylated alcohols
Figure 1 shows the characteristic HRGC chro- matogram of AEO 3.3 used in this work. This mixture had the general formula:
C , H ~ + I - -O-- (CH2--CH2--O) , - -H
m a v e r a g e = 9-11
n average -- 3,3
The gas chromatographic separation of the mixture was performed after silylation (Cross, 1987),
High Resolution Gas Chromatography-Positive Ions Electron Impact Mass Spectrometry (HRGC-EICIMS) analysis of the sample coupled with chromatographic comparison with l-nonanol, l-decanol, l -unde~nol silylethers allowed to assign the structures shown in Fig. l to the individual components. The aliphatic portion of these molecules was composed by C9, Cm0, Ca, linear chains. The polyethoxylated part ranged from n ffi 0 to n = 6.
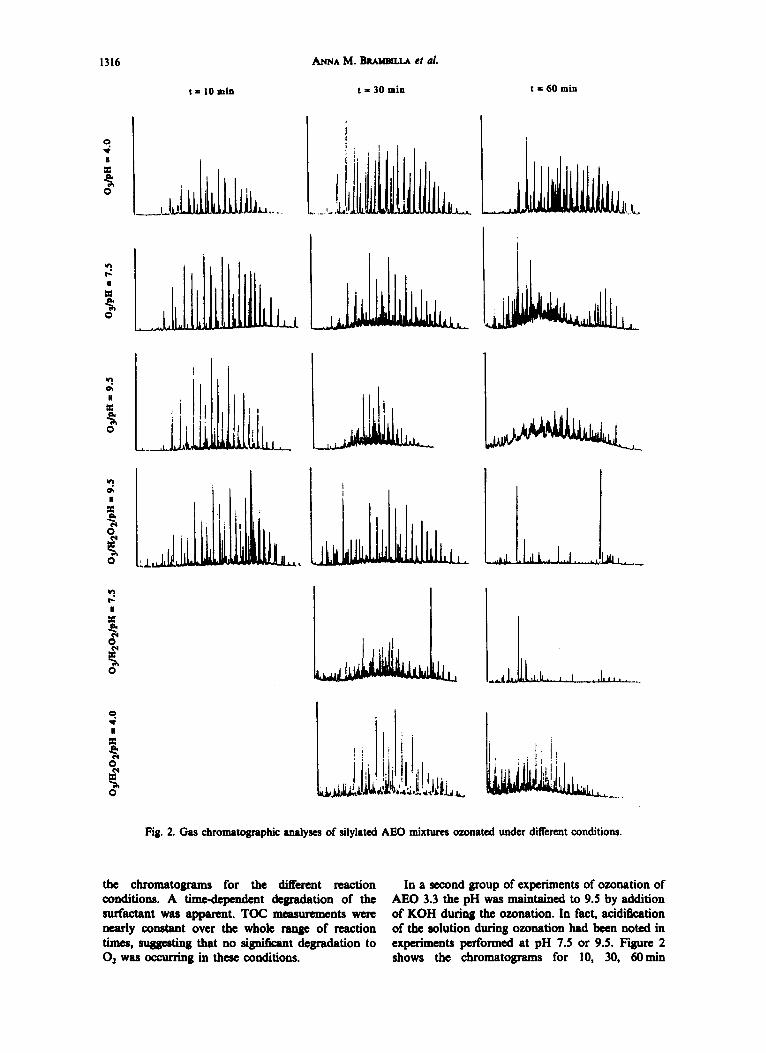
The first series of ozonation experiment was performed using a 50mg/I solution of AEO 3.3 adjusted at pH 4.0 with sulphuric acid. Ozonation and work-up were performed as described using reaction times of 60, 30, and i0 min, Figure 2 shows
1316
0
#
o
t = 10 ra in
A ~ A M. ~ et aL
t = 30 rain
b
t -- 60 rain
i
g
o
t n e~ g
o e t
o
p- g
e o ¢q
o
O
n
o OI
o
I
I,
I , . I , , , ,
I t , i ~ i ~
Fig. 2. Gas chromatographic analyses of silylated AEO mixtures ozonated under different conditions.
the chromatograms for the different reaction conditions. A time-dependent degradation of the surfactant was apparent. TOC measurements were nearly constant over the whole range of reaction times, suggesting that no significant degr~_dR_tion to 02 was occurring in these conditions.
In a second group of experiments of ozonation of AEO 3.3 the pH was maintained to 9.5 by addition of KOH during the ozonation. In fact, acidification of the solution during omnat ion had been noted in experiments performed at pH 7.5 or 9.5. Figure 2 shows the chromatograms for 10, 30, 60min

A
o g
|
Ozone oxidation of polyethoxylated alcohols
o cq ~ , q .
osuodso~
O
"7
0
9
]
qD
e~
0
e~
8
,el
0
m
¢S
ae
ed
1317

1318 ANNA M. lkud~na~ et al.
ozonation time. These data suggested a degradation of the surfactant more pronounced than in acidic conditions. TOC measurements showed a 12% decrease in the first 30 min reaction. TOC remained constant after that time.
A GC/FTIR analysis of the mixture reacted for 30rain showed that many compounds having a carbonyl or carboxyl group had been formed. Figure 3 shows the comparison between the total response chromatogram and the chromatogram related to compounds absorbing in the spectral region between 1700 and 1800 cm -I, e.g. the carbonyl region: it appears that these compounds are relatively more abundant at low retention times than in the total response trace. This indicates that low molecu- lar weight carbonyi compounds might have been formed, thus implying that some fragmentation of the surfactant molecule has occurred. Also when pH 7.5 was maintained by addition of KOH to the solution; inspection of the chromatograms and TOC measurements showed that degradation was even less significant than at pH 4.
An improvement of the ozonation was performed by adding excess hydrogen peroxide (hydrogen peroxide addition ffi HPA) to the solutions of the surfactant. Again, the pH was held constant during the ozonation at pH 7.5 and 9.5 by addition of KOH. Usual work-up allowed to obtain gas chromato- graphic analyses for 10, 30, 60 rain ozonation time in all cases. These show an extensive demolition after 60 rain reaction. In fact, peaks at low retention time appear. Again, GC/FTIR analyses showed that most of these peaks contained a carbonyl or carboxyl group.
In the case of pH 4.0, after 60 rain reaction the surfactant mixture appears to be more modified than at the same pH without HPA, but less modified than at pH 9.5 and 7.5 with HPA. TOC measurements showed a 50% decrease in the ozonation experiment at pH 9.5.
A control experiment was performed by reacting the AEO mixture with the same amount of hydrogen peroxide at pH 9.5 under a stream of dioxygen or dinitrogen. Some oxidation of the surfactant was noticed in the experiment under dioxygen, whereas no change was occurring under nitrogen. However no decrease of TO(] was noticed in both cases. Hence, alkafine and neutral pH and hydrogen peroxide addition gave the most efficient degradation of the surfactant.
Inspection of the ozonation products was per- formed by analyzing the silylated reaction mixtures by HRGC-MS and, in some cases, by chromato- graphic comparison with authentic samples. Samples reacted for 60 rain were chosen as more significant for product identification. At pH 9.5 with no HPA the chromatogram appeared too complex to be analyzed, though relevant mixture modification was apparent. Hence, the mixture ozonated for 30 min at the same pH was used in this case.
HRGC/MS was performed also for mixtures ozonated for 30 rain at pH 7.5 and 9.5 with HPA, in order to determinate the degree of demolition.
The results of the HRGC-PIEIMS analyses are shown in Scheme !.
No quantitative measurements were made, as the amount of highly polar compounds might be heavily influenced by their partition coefficient between water and ethyl acetate used for extraction.
Ethoxylated alcohols silylethers were present in all the samples except at pH 9.5 and 7.5, for a reaction time of 60 rain and with HPA.
Linear carboxylic acids silylesters:
Cm_ iH~_ i---COOSiMe3
with m ffi 9-11 were found in all the samples except at pH 7.5, 60 rain reaction and HPA. The acids might be generated both by oxidation of the corresponding alcohols, which are present in small amounts in the starting mixture, and by the removal of the oxyethyl chain from the ethoxylated alcohols. These acids are the main chromatographic peaks in the mixture ozonated at pH ffi 4 with no HPA.
Acids of the same family but with rn < 9 were found as silylesters in small amounts in all the samples except at pH ~-4 with no HPA. They arise from afiphatic chain fragmentation.
Other products deriving from carbon chain fragmentation were also present as their trimethyl- silylderivatives:
(a) hydroxy and dihydroxy acids, such as 3-hy- droxypropionic acid and 3,4-dihydroxybutanoic acid;
(b) ketoacids as 4-ketopentanoic acid and 5-keto- bexanoic acid;
(c) dicarboxylic aliphatic acids as butanedioic, pentanedioic, hexanedioic, heptanedioic and 4-keto- heptanedioic acids.
These three classes of compounds were found in all the chromatograms except for pH 4 with no HPA. Butanedioic and pentanedioic acids silyldiesters correspond to the main gas chromatographic peaks in samples ozonated for 60 min at pH 9.5 and 7.5 with HPA.
Other silylated products can be attributed to oxyethyi chain modification:
(a) giycolic acid trimethyisilyl derivative was found in small amounts in all the samples except at pH 7.5 and 9.5 for 60 min reaction time with HPA. Glycolic acid might derive from acetic acid oxidation, but can more easily be considered as the oxidative depolimerization product of the oxyethyl chains;
(b) di-, tri- and tetraethyleneglycol silylethers were present in almost all cases. They were absent only at pH 7.5 after 60rain ozonation and with HPA;
(c) the mono- and diacid corresponding to di- and triethyleneglycol were present as trimethylsilyl- derivatives mainly in samples ozonated with HPA.
Hexadecanoic and octadecanoic acids silyl- esters were found in small amounts in many

Ozone oxidation of polyethoxylated alcohols 1319
iq A
-- m
0
"If
~i=,i
= ~ = ~ - ~
ggg ~NgN
NNN ~NNXN
NXN N N N N X
NN X X N N
X NNN
NNN
xlX
xIixI
x~
.~ ,~ .-~ ~.~
--I ~ - ............. "X
~ _ ~ ~ - ~ - - ' - -
X~N~
XX I~
xxXX
Xx X
xx
I.-
II!l
xxxx~
XXXX~ X
X ~X
XXXXX~
XXXX X
XXXX
xx~
XX
XX
XXl
XXl
1i
'Ii
!
~q2~

1320 A~A M. BIt.AM~ILLA et ol.
chromatograms: probably some fatty acids or alco- hols were present as impurities in the starting mix- ture.
Discussion o f results
The primary reactions initiated by ozone in water are strongly pH-depcndent (Hoign~, 1982). Ozone reacts with organic solutes at low pH as trioxygen, but at high pH it decomposes before reacting with the solutes. Ozone decomposition is catalyzed by hydroxide ions, and proceeds more rapidly with increasing pH. It is further accelerated by a radical- type chain reaction in which free radicals, such as OH" and HO 2 produced from decomposed ozone, can act as chain carriers (equations 1 and 2).
03 + OH----,O~- + HO~ (1)
03 + HO~--, 202 + HO" (2)
Organic compounds react with hydroxyl radicals (OH')or hydroperoxyi radicals (HO~ and form sec- ondary radials (R') (equations 3 and 4) that also act as ozone-decomposing catalysts.
OH" + R--H---, R" + H20 (3)
HO2 + R--H R" + H202 (4)
ThUS secondary oxidants such as hydroxyl radical, hydroperoxyl radical and hydrogen peroxide are produced by decomposition of ozone at high pH.
The hydroxyl radical is a very strong oxidant and exhibits little substrate selectivity.
Radicals R" react in ozonation conditions with excess dioxygen and form peroxyl radicals (equation 5):
R" + 02---, RO0" (5)
which behave as radical chain carriers to give hy- droperoxides (equation 6):
ROO" + R'H--* ROOH + R'" (6)
A new radical (R") is formed which again reacts with dioxygen leading to autooxidation.
This radical chain causes more decomposition of ozone and increased formation of hydroxyl radicals (Staehelin and Hoign~, 1983, 1985).
The rearrangement of hydroperoxyl radicals gives carbonylic and carboxylic compounds which are responsible for chain breakdown.
The partial or total removal of the ethoxylated side chain in AEO 3.3 suggests that the reaction occurs through a hydrogen abstraction at one of the ethoxy- lated units or at the carbon ct to the oxygen of the aliphatic chain. The hydrogen abstraction process occurs both at low pH, where ozone itself is the reagent, and at high pH, where the hydroxyl radical is the active species. The relevant presence of the linear ~-~j~ adds and of oligoethyleneglycols at pH 4.0 suggests that attack of ozone to the ~ carbon of the aliphatic moiety is preferred. In fact, other by-
products are negligible in these conditions, indicating that other oxidation pathways are acting in a minor extent. According to Reynolds et al., saturated fatty acids are not modified by ozone at low pH (Reynolds et al., 1989), thus justifying the overall absence of aliphatic reaction products other than nonanoic, decanoic, undecanoic acids.
In the presence of OH" or other radical species (high pH ozonation, hydrogen peroxide addition) the products mentioned above are also found, therefore the same oxidative pathway might be present. Here, many more reaction products are found. This indicates that hydrogen abstraction occurs also in the middle of the aliphatic and oxyethyl chains.
Our results are in good accordance with the findings of Niki et ad., who showed that carbonyl, hydroxyl and carboxyl groups formation occurs during ozonation of small aliphatic compounds (Niki et al., 1983). In our case, some dicarboxylic acids such as butanedioic and pentanedioic seem to be the most relevant products of the degradation of aliphatic chains in the presence of hydrogen peroxide. Keto-acids such as 4-ketopentanoic and 4-keto- heptanedioic might be the precursors of butanedioic acid, while 5-ketohexanoic might be the precursor of pentanedioic acid.
High pH ozonation with no HPA shows deep modifications of the products mixture, but only a few products were identified. Inspection of the chro- matogram of the mixture ozonated for 60 rain at pH--9.5 shows that a high number of by-products have been formed, but the average retention time (which is related to the molecular weights) suggests a small decrease with respect to the AEO mixture, if compared with mixtures ozonated in the presence of hydrogen peroxide.
The three minutes ozonated mixtures with HPA show the presence of about the same by-products. At pH 4.0 the starting ethoxylated alcohols are still the main components. TOC measurements and the ab- sence of the starting compounds suggest that alkaline and neutral pH coupled with hydrogen peroxide addition are the best conditions for ethoxylated alco- hols breakdown: in fact, according to literature data, (Staehelin and Hoign6, 1982) in these conditions the concentration of radical species is maximum due to the presence of the very active ozone decomposing species HO~-.
The selection of the proper ozonation conditions requires some observations:
the primary products nonanoic, decanoic and un. decanoic acids and the non-oxidized tri- and te- traethyleneglycols are absent at pH 7.5 after 60 rain reaction, but are still present in the same conditions after 30 rain reaction. This suggests that these com- pounds have been consumed during the course of the reaction. In fact, oligoethylene glycols are replaced by their corresponding acids, that probably are further reaction products. The same primary products are

Ozone oxidation of polyethoxylated alcohols 1321
present in small amounts at pH ffi 9.5 with HPA after 60rain reaction. These observations suggest that pH ffi 7.5 and HPA are slightly better than pH ffi 9.5 and HPA, in accordance with the findings of Paillard et al. (Paillard et ol., 1988).
These conclusions, which are drawn on the basis of product analysis, are supported by examination of potassium hydroxide consumption to hold the pH constant in alkaline and neutral pH experiments. In fact, nearly I meq of KOH per meq of AEO 3.3 was necessary to hold the pH to 9.5 in the run effected for 60 rain at pH 9.5, where a 12% decrease in TOC had been obtained. On the contrary, a consumption of nearly 3 meq of KOH per meq of AEO 3.3 was necessary to hold the pH at 9.5 in the run effected for 60' at pH 9.5 with additions of hydrogen peroxide. To hold the pH at 7.5 with HPA about 0.5 meq of base per meq of AEO were required. This difference is due to the fact that at pH 7.5 carboxylic acid dissociation is not complete.
Control experiments were effected in absence of the substrate to measure potassium hydroxide consump- tion due exclusively to ozone and hydrogen peroxide decomposition. At pH 9.5 with no hydrogen peroxide less than 10% of added base is required to decompose ozone, while a high percentage of base must be added to dissociate the carboxyfic acids which are formed and to hold the pH at 9.5.
At pH 9.5 with HPA about 38% of base added in the presence of the substrate is required also in its absence, that is, to react with hydrogen peroxide and its by-products.
The consumption of potassium hydroxide to hold the pH at 7.5 with HPA and no substrate was 12% with respect of the reaction in the presence of the substrate. This supports the hypothesis (Paillard et al., 1988) that at pH > 7.5 a certain amount of O H - is required to decompose hydroxyl radicals. Their concentration shows a maximum at pH = 7.5 and decreases with increasing pH beyond 7.5.
CONCLUSIONS
Ozonation of AEO 3.3 at low pH gives no TOC decrease and causes only separation between the aliphatic and oxyethyl part of the molecules.
Some improvements are obtained with high pH ozonation.
pH = 7.5 and hydrogen peroxide addition seems to be the best method to degrade ethoxylated alcohols. In these conditions, some dicarboxylic acids seem to be the main refractory products responsible for residual TOC in AEO 3.3 ozonated solutions.
Acknowledgonents-.We wish to thank Mr G. D'Introno for his collaboration, Mr A. Cremonai for the mare spectra, Dr P. Donag0o for his collaboration in the GC/FTIR analym and Dr To~ani for kindly ~ l y i n g information.
work was supported by a CNR grant.
REFERENCES
Andreozzi R., Caprio V., D'Amore M. G. and Insola A. (1990) Quinoxaline ozonation in aqueous solution. Ozone Sci. Eng. 12, 217.
Arai H., Arai M. and Sakumoto A. (1986) Exhaustive degradation of humic acid in water by simultaneous application of radiation and ozone. War. Res. ~ 885.
Calvosa L., Monteverdi A., Rindone B. and giva G. (1991) Ozone oxidation of compounds resistant to biological degradation. War. Res. 25, 985.
Caprio V., Insola A. and Silvestre A. M. (1989) Glyoxal ozouation process in aqueous solution. Ozone Sci. Eng. 11, 271.
Chelkowska K., Grusso D., Fabian I. and Gordon G. (1990) Mechanistic comparison of residual ozone decompo- sition. Proc. of the International Ozone Association, Shreveport, Louisiana, 427.
Cross J. (1987) Gas-Liquid Chromatography of non-ionic surfactants and their derivatives. Non-ionic Surfactams, Vol. 19, Chemical Analysis (Edited by Cross J.), Suffactant Science Series, pp. 169-224. Marcel Dekker, New York.
Dahi E. (1976) Physico-chemical aspects of disinfection of water by means of ultrasound and ozone. War. Res. 10, 677.
Duguet J. P., Bernazeau F. and MaUevialle J. (1990) Research note. Removal of atrazine by ozone and hydro- gen peroxide combinations in surface water. Ozone Sci. Eng. 12, 195.
Duguet J. P., Anselme C., Mazounie P. and Mallevialle J. (1990a) Application of combined ozone--hydrogen per- oxide for the removal of aromatic compounds from a groundwater. Ozone Sci. Eng. 12, 281.
Gilli G., Scursatone E., Palin L., Bona R., Carraro E. and Meucci L. (1990) Water disinfection: a relationship between ozone and aldehyde production. Ozone Sci. Eng. 12, 231.
Glaze W. H. (1987) Drinking-water treatment with ozone. Envir. $ci. TechnoL 21, 224.
Glaze W. H., Koga M. and Cancilla D. (1989) Ozonation by-products. 2. Formation of formaldehyde and other carbonyl compounds by ozonation of drinking water. Envir. $ci: Technol. 23, 838.
Hoign6 J. (1982) Mechanisms, rates and selectivities of oxidations of o ,r~_nic compounds initiated by ozonation of water. Ozone Technology and its Practical Applications (Edited by Rice R. G. and Net~r A.), Vol. 1, pp. 341-379. Ann Arbor Science, Ann Arbor.
Hoign~ J. and Bader H. (1979) Ozonation of water: selectivity and rate of oxidation of solutes. Ozone Sci. Eng. 1, 73.
Legube B., Crou¢~ J. P., De Laat J. and Dor~ M. (1989) Ozonation of an extracted aquatic fulvic acid: theoretical and practical aspects. Ozone $ci. Eng. 11, 69.
Nakayama S., Esaki K., Namtm K., Taniguchi Y. and Nabata N. (1979) Improved ozonation in aqueous systems. Ozone $ci. Eng. 1, 119.
Narkis N. and Schneidor-Rotei M. (1980) Ozone induced biodegradability of a non-ionic suffactant. War. Res. 14, 1225.
Narkis N., Ben-David B. and Schneidor-Rotel M. (1987) Non-ionic suffactants interactions with ozone. Tens/de Surf. Det. 24, 200.
Niki E., Yamsmoto E., Salto T., Nagano K., Yokoi S. and K~uiya Y. (1983) Ozoni~tion of organic compounda. VII. Carboxyiic acids, alcohols and carbonyl compounds. Ball. Chem. Sac. Jpn ~ , 22.V-228.
Paillard H., Brunet R. and Dot6 M. (1988) Optimal conditions for applying an ozone-hydrogen peroxide oxidizing system. War. Res. 22, 91.
Peyton G. It., Huang F. J., Burleson J. L. and Glaze W. H. (1982) Destruction of pollutants in water with ozone in

1322 ANNA M. Bg~mILLA et ol.
combination with ultraviolet radiation. !. General principles and oxidation of tetracldoro ethylene. Envir. Sci. Technol. 16, 448.
Prengle Jr. H. W. 0983) Experimental rate constants and reactor consideration for the destruction of micro- pollutants and triludomethane precursors by ozone with ultraviolet radiation. F~w/r. $ci. Tecimol. 17, 743.
Reynolds G., Graham N., Perry R. and Rice R. G. (1989) Aqueous ozonation of pesticides: a review. Ozone Sci. Eng. 11, 339.
Reynolds G., Corle, C., Graham N., Perry R., Gibson T. M. and Haley J. (198%) Aqueous ozonation of fatty acids. Ozone Sci, Eng. 11, 143.
Sierka R. A. and Amy G. L. (1985) Catalytic effects of ultraviolet light and/or ultrasound on the ozone oxidation of humic acid and trihalomethane precursors. Ozone Sci. Eng. 7, 47.
Staehelin J. and Hoign~ J. (1982) Decomposition of ozone in water, rate of initiation by hydroxide ions and hydrogen peroxide. Envir. Sci. Technoi. 16, 676.
Staehelin J. and Hoigm~ J. (1983) Mechanism and kinetics
of decomposition of ozone in water in presence of organic solutes. Vom Wass. 61, 337.
Staehefin J. and Hoign~ J. (1985) Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions. Eneir. Sci. Technol. 19, 1206.
Staehelin J., Buhler R. E. and Hoign~ J. (1984) Ozone decomposition in water studied by pulse radiolysis. 2. OH and HO4 as chain intermediates. J. Phys. Chem. N , 5999.
Takanashi N. and Katsuki O. (1990) Decomposition of ethylene glycol by the combined use of ozone oxidation and electrolytic methods. Ozone Sci. Eng. 12, 115.
Takanashi N. (1990a) Ozonation of several organic compounds having low molecular weight under ultra- violet irradiation. Ozone Sci. Eng. 12, 1.
Tomiyasu H., Fukutomi H. and Gordon G. (1985) Kinetics and mechanism of ozone decomposition in basic aqueous solution, lnorg. Chem. 24, 2962.
Yamada H. and Somiya I. (1991) Formation characteristic of formaldehyde during the ozonation of surface water. Proc. of the 10th Ozone World Congress, Monaco 1, 179.




















