
The Homogentisate Pathway: a Central Catabolic Pathway Involved in the Degradation of...
16
JOURNAL OF BACTERIOLOGY, Aug. 2004, p. 5062–5077 Vol. 186, No. 15 0021-9193/04/$08.000 DOI: 10.1128/JB.186.15.5062–5077.2004 Copyright © 2004, American Society for Microbiology. All Rights Reserved. The Homogentisate Pathway: a Central Catabolic Pathway Involved in the Degradation of L-Phenylalanine, L-Tyrosine, and 3-Hydroxyphenylacetate in Pseudomonas putida Elsa Arias-Barrau, 1 Elı ´as R. Olivera, 1 Jose ´ M. Luengo, 1 Cristina Ferna ´ndez, 2 Beatriz Gala ´n, 2 Jose ´ L. Garcı ´a, 2 Eduardo Dı ´az, 2 and Baltasar Min ˜ambres 2 * Departamento de Bioquı ´mica y Biologı ´a Molecular, Facultad de Veterinaria, Universidad de Leo ´n, 24007 Leo ´n, 1 and Departamento de Microbiologı ´a Molecular, Centro de Investigaciones Biolo ´gicas, Consejo Superior de Investigaciones Cientı ´ficas, Madrid, 2 Spain Received 28 February 2004/Accepted 3 May 2004 Pseudomonas putida metabolizes Phe and Tyr through a peripheral pathway involving hydroxylation of Phe to Tyr (PhhAB), conversion of Tyr into 4-hydroxyphenylpyruvate (TyrB), and formation of homogentisate (Hpd) as the central intermediate. Homogentisate is then catabolized by a central catabolic pathway that involves three enzymes, homogentisate dioxygenase (HmgA), fumarylacetoacetate hydrolase (HmgB), and maleylacetoacetate isomerase (HmgC), finally yielding fumarate and acetoacetate. Whereas the phh, tyr, and hpd genes are not linked in the P. putida genome, the hmgABC genes appear to form a single transcriptional unit. Gel retardation assays and lacZ translational fusion experiments have shown that hmgR encodes a specific repressor that controls the inducible expression of the divergently transcribed hmgABC catabolic genes, and homogentisate is the inducer molecule. Footprinting analysis revealed that HmgR protects a region in the Phmg promoter that spans a 17-bp palindromic motif and an external direct repetition from position 16 to position 29 with respect to the transcription start site. The HmgR protein is thus the first IclR-type regulator that acts as a repressor of an aromatic catabolic pathway. We engineered a broad-host-range mobilizable catabolic cassette harboring the hmgABC, hpd, and tyrB genes that allows heterologous bacteria to use Tyr as a unique carbon and energy source. Remarkably, we show here that the catabolism of 3-hydroxyphenylacetate in P. putida U funnels also into the homogentisate central pathway, revealing that the hmg cluster is a key catabolic trait for biodegradation of a small number of aromatic compounds. Eukaryotic organisms catabolize Phe and Tyr by a common peripheral pathway which leads to homogentisate (2,5-OH- PhAc) as a central intermediate (6, 9, 20, 23, 31). The genetic and biochemical interest in this pathway comes from the fact that many severe human diseases (e.g., phenylketonuria, alcap- tonuria, tyrosinemia, tyrosinosis, Richner-Hanhart syndrome, and hawkinsinuria) are associated with enzyme deficiencies in the catabolism of Phe and Tyr (16, 19, 24, 32, 52, 73). First, Phe is transformed into Tyr by a pterin-dependent phenylalanine hydroxylase (PhhA), and later, a tyrosine aminotransferase (TyrB) catalyzes the conversion of Tyr into 4-hydroxyphe- nylpyruvate (4-OH-PhPyr), which is further transformed into 2,5-OH-PhAc by a 4-OH-PhPyr dioxygenase (Hpd) (Fig. 1B). The homogentisate central pathway involves a homogentisate dioxygenase (HmgA) that opens the aromatic ring of 2,5-OH- PhAc, producing maleylacetoacetate, which is isomerized to fumarylacetoacetate by the HmgC isomerase. Finally, fumary- lacetoacetate is hydrolyzed by a specific hydrolase (HmgB) to form fumarate and acetoacetate, which are two compounds of the central metabolism (Fig. 1B). In plants and photosynthetic bacteria the catabolism of Tyr is also crucial because homogen- tisate is a precursor for the biosynthesis of photosynthetic pigments (66). Although the catabolism of Phe and Tyr in eukaryotic or- ganisms has been well established, limited information has been obtained about the degradation of these amino acids in prokaryotes (58, 68, 81). The inability of Escherichia coli, the model prokaryotic organism, to mineralize Phe and Tyr might have contributed to the reduction in interest in this pathway in bacterial systems. However, some studies have shown that the catabolism of Phe and Tyr in bacteria is also carried out by a peripheral pathway similar to that of eukaryotes, with forma- tion of homogentisate as a central intermediate (1, 36, 43, 44, 53, 61, 66, 70). Nevertheless, the genes encoding the catabolic enzymes of the homogentisate central pathway and the regu- latory elements that control the expression of such genes have only been partially identified and characterized (27, 43, 44, 69, 76). This work was aimed at identifying and characterizing for the first time the complete set of genes responsible for degra- dation of Phe and Tyr in bacteria. To this end, we studied the catabolism of Phe and Tyr in Pseudomonas putida,a -pro- teobacterium with great metabolic versatility that can use these amino acids as carbon and energy sources and that is consid- ered a model system for environmental studies (35, 49, 50, 75). By using two strains of P. putida that show different catabolic abilities with some natural aromatic compounds, P. putida U, a strain that is able to grow with 4-hydroxyphenylacetate (4-OH- PhAc) or 3-hydroxyphenylacetate (3-OH-PhAc) as the sole carbon source (48), and P. putida KT2440, a strain whose complete genome is known (47) and that is unable to use * Corresponding author. Present address: Estacio ´n Agrı ´cola Exper- imental, Consejo Superior de Investigaciones Cientı ´ficas, Finca Mar- zanas, 24346 Grulleros, Leo ´n, Spain. Phone: 34-987317156. Fax: 34- 987317161. E-mail: [email protected]. 5062 on February 20, 2016 by guest http://jb.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of The Homogentisate Pathway: a Central Catabolic Pathway Involved in the Degradation of...

JOURNAL OF BACTERIOLOGY, Aug. 2004, p. 5062–5077 Vol. 186, No. 150021-9193/04/$08.00�0 DOI: 10.1128/JB.186.15.5062–5077.2004Copyright © 2004, American Society for Microbiology. All Rights Reserved.
The Homogentisate Pathway: a Central Catabolic Pathway Involved inthe Degradation of L-Phenylalanine, L-Tyrosine, and
3-Hydroxyphenylacetate in Pseudomonas putidaElsa Arias-Barrau,1 Elıas R. Olivera,1 Jose M. Luengo,1 Cristina Fernandez,2 Beatriz Galan,2
Jose L. Garcıa,2 Eduardo Dıaz,2 and Baltasar Minambres2*Departamento de Bioquımica y Biologıa Molecular, Facultad de Veterinaria, Universidad de Leon, 24007 Leon,1 andDepartamento de Microbiologıa Molecular, Centro de Investigaciones Biologicas, Consejo Superior de Investigaciones
Cientıficas, Madrid,2 Spain
Received 28 February 2004/Accepted 3 May 2004
Pseudomonas putida metabolizes Phe and Tyr through a peripheral pathway involving hydroxylation of Pheto Tyr (PhhAB), conversion of Tyr into 4-hydroxyphenylpyruvate (TyrB), and formation of homogentisate(Hpd) as the central intermediate. Homogentisate is then catabolized by a central catabolic pathway thatinvolves three enzymes, homogentisate dioxygenase (HmgA), fumarylacetoacetate hydrolase (HmgB), andmaleylacetoacetate isomerase (HmgC), finally yielding fumarate and acetoacetate. Whereas the phh, tyr, andhpd genes are not linked in the P. putida genome, the hmgABC genes appear to form a single transcriptionalunit. Gel retardation assays and lacZ translational fusion experiments have shown that hmgR encodes a specificrepressor that controls the inducible expression of the divergently transcribed hmgABC catabolic genes, andhomogentisate is the inducer molecule. Footprinting analysis revealed that HmgR protects a region in thePhmg promoter that spans a 17-bp palindromic motif and an external direct repetition from position �16 toposition 29 with respect to the transcription start site. The HmgR protein is thus the first IclR-type regulatorthat acts as a repressor of an aromatic catabolic pathway. We engineered a broad-host-range mobilizablecatabolic cassette harboring the hmgABC, hpd, and tyrB genes that allows heterologous bacteria to use Tyr asa unique carbon and energy source. Remarkably, we show here that the catabolism of 3-hydroxyphenylacetatein P. putida U funnels also into the homogentisate central pathway, revealing that the hmg cluster is a keycatabolic trait for biodegradation of a small number of aromatic compounds.
Eukaryotic organisms catabolize Phe and Tyr by a commonperipheral pathway which leads to homogentisate (2,5-OH-PhAc) as a central intermediate (6, 9, 20, 23, 31). The geneticand biochemical interest in this pathway comes from the factthat many severe human diseases (e.g., phenylketonuria, alcap-tonuria, tyrosinemia, tyrosinosis, Richner-Hanhart syndrome,and hawkinsinuria) are associated with enzyme deficiencies inthe catabolism of Phe and Tyr (16, 19, 24, 32, 52, 73). First, Pheis transformed into Tyr by a pterin-dependent phenylalaninehydroxylase (PhhA), and later, a tyrosine aminotransferase(TyrB) catalyzes the conversion of Tyr into 4-hydroxyphe-nylpyruvate (4-OH-PhPyr), which is further transformed into2,5-OH-PhAc by a 4-OH-PhPyr dioxygenase (Hpd) (Fig. 1B).The homogentisate central pathway involves a homogentisatedioxygenase (HmgA) that opens the aromatic ring of 2,5-OH-PhAc, producing maleylacetoacetate, which is isomerized tofumarylacetoacetate by the HmgC isomerase. Finally, fumary-lacetoacetate is hydrolyzed by a specific hydrolase (HmgB) toform fumarate and acetoacetate, which are two compounds ofthe central metabolism (Fig. 1B). In plants and photosyntheticbacteria the catabolism of Tyr is also crucial because homogen-tisate is a precursor for the biosynthesis of photosyntheticpigments (66).
Although the catabolism of Phe and Tyr in eukaryotic or-ganisms has been well established, limited information hasbeen obtained about the degradation of these amino acids inprokaryotes (58, 68, 81). The inability of Escherichia coli, themodel prokaryotic organism, to mineralize Phe and Tyr mighthave contributed to the reduction in interest in this pathway inbacterial systems. However, some studies have shown that thecatabolism of Phe and Tyr in bacteria is also carried out by aperipheral pathway similar to that of eukaryotes, with forma-tion of homogentisate as a central intermediate (1, 36, 43, 44,53, 61, 66, 70). Nevertheless, the genes encoding the catabolicenzymes of the homogentisate central pathway and the regu-latory elements that control the expression of such genes haveonly been partially identified and characterized (27, 43, 44, 69,76).
This work was aimed at identifying and characterizing forthe first time the complete set of genes responsible for degra-dation of Phe and Tyr in bacteria. To this end, we studied thecatabolism of Phe and Tyr in Pseudomonas putida, a �-pro-teobacterium with great metabolic versatility that can use theseamino acids as carbon and energy sources and that is consid-ered a model system for environmental studies (35, 49, 50, 75).By using two strains of P. putida that show different catabolicabilities with some natural aromatic compounds, P. putida U, astrain that is able to grow with 4-hydroxyphenylacetate (4-OH-PhAc) or 3-hydroxyphenylacetate (3-OH-PhAc) as the solecarbon source (48), and P. putida KT2440, a strain whosecomplete genome is known (47) and that is unable to use
* Corresponding author. Present address: Estacion Agrıcola Exper-imental, Consejo Superior de Investigaciones Cientıficas, Finca Mar-zanas, 24346 Grulleros, Leon, Spain. Phone: 34-987317156. Fax: 34-987317161. E-mail: [email protected].
5062
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
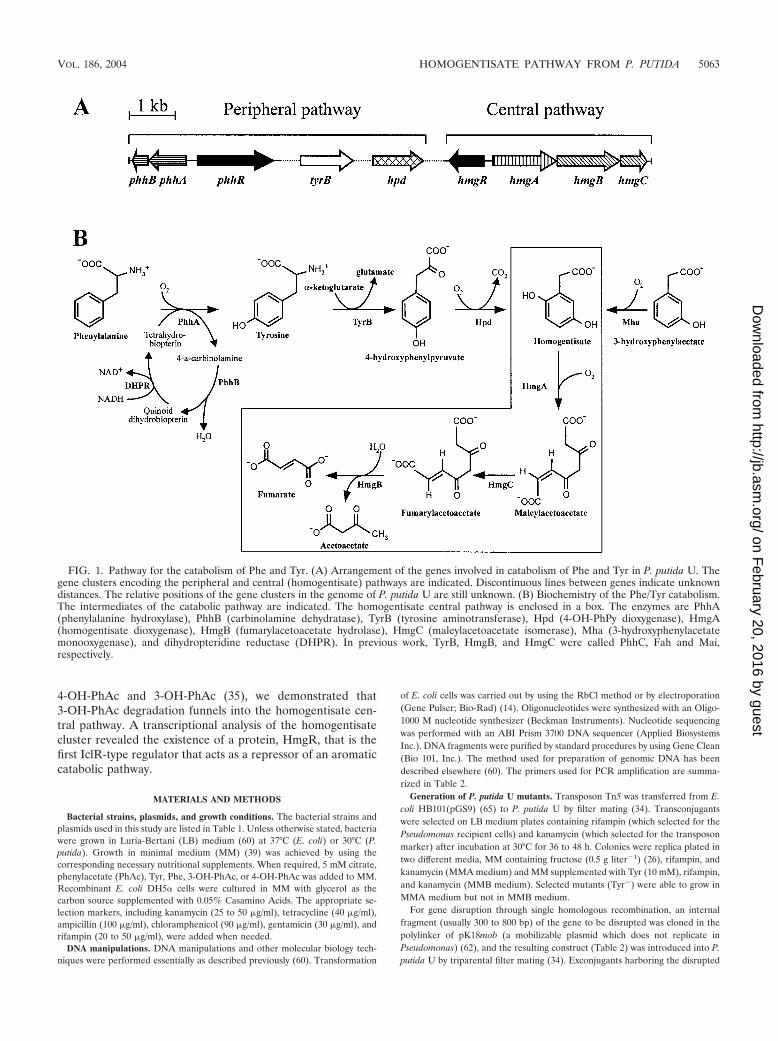
4-OH-PhAc and 3-OH-PhAc (35), we demonstrated that3-OH-PhAc degradation funnels into the homogentisate cen-tral pathway. A transcriptional analysis of the homogentisatecluster revealed the existence of a protein, HmgR, that is thefirst IclR-type regulator that acts as a repressor of an aromaticcatabolic pathway.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions. The bacterial strains andplasmids used in this study are listed in Table 1. Unless otherwise stated, bacteriawere grown in Luria-Bertani (LB) medium (60) at 37°C (E. coli) or 30°C (P.putida). Growth in minimal medium (MM) (39) was achieved by using thecorresponding necessary nutritional supplements. When required, 5 mM citrate,phenylacetate (PhAc), Tyr, Phe, 3-OH-PhAc, or 4-OH-PhAc was added to MM.Recombinant E. coli DH5� cells were cultured in MM with glycerol as thecarbon source supplemented with 0.05% Casamino Acids. The appropriate se-lection markers, including kanamycin (25 to 50 �g/ml), tetracycline (40 �g/ml),ampicillin (100 �g/ml), chloramphenicol (90 �g/ml), gentamicin (30 �g/ml), andrifampin (20 to 50 �g/ml), were added when needed.
DNA manipulations. DNA manipulations and other molecular biology tech-niques were performed essentially as described previously (60). Transformation
of E. coli cells was carried out by using the RbCl method or by electroporation(Gene Pulser; Bio-Rad) (14). Oligonucleotides were synthesized with an Oligo-1000 M nucleotide synthesizer (Beckman Instruments). Nucleotide sequencingwas performed with an ABI Prism 3700 DNA sequencer (Applied BiosystemsInc.). DNA fragments were purified by standard procedures by using Gene Clean(Bio 101, Inc.). The method used for preparation of genomic DNA has beendescribed elsewhere (60). The primers used for PCR amplification are summa-rized in Table 2.
Generation of P. putida U mutants. Transposon Tn5 was transferred from E.coli HB101(pGS9) (65) to P. putida U by filter mating (34). Transconjugantswere selected on LB medium plates containing rifampin (which selected for thePseudomonas recipient cells) and kanamycin (which selected for the transposonmarker) after incubation at 30°C for 36 to 48 h. Colonies were replica plated intwo different media, MM containing fructose (0.5 g liter�1) (26), rifampin, andkanamycin (MMA medium) and MM supplemented with Tyr (10 mM), rifampin,and kanamycin (MMB medium). Selected mutants (Tyr�) were able to grow inMMA medium but not in MMB medium.
For gene disruption through single homologous recombination, an internalfragment (usually 300 to 800 bp) of the gene to be disrupted was cloned in thepolylinker of pK18mob (a mobilizable plasmid which does not replicate inPseudomonas) (62), and the resulting construct (Table 2) was introduced into P.putida U by triparental filter mating (34). Exconjugants harboring the disrupted
FIG. 1. Pathway for the catabolism of Phe and Tyr. (A) Arrangement of the genes involved in catabolism of Phe and Tyr in P. putida U. Thegene clusters encoding the peripheral and central (homogentisate) pathways are indicated. Discontinuous lines between genes indicate unknowndistances. The relative positions of the gene clusters in the genome of P. putida U are still unknown. (B) Biochemistry of the Phe/Tyr catabolism.The intermediates of the catabolic pathway are indicated. The homogentisate central pathway is enclosed in a box. The enzymes are PhhA(phenylalanine hydroxylase), PhhB (carbinolamine dehydratase), TyrB (tyrosine aminotransferase), Hpd (4-OH-PhPy dioxygenase), HmgA(homogentisate dioxygenase), HmgB (fumarylacetoacetate hydrolase), HmgC (maleylacetoacetate isomerase), Mha (3-hydroxyphenylacetatemonooxygenase), and dihydropteridine reductase (DHPR). In previous work, TyrB, HmgB, and HmgC were called PhhC, Fah and Mai,respectively.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5063
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

gene were isolated on LB medium containing rifampin and kanamycin after 2days of incubation at 30°C.
Deletion of the hmg and hpd genes in P. putida U�hmg and P. putida U�hpd(Table 1) was accomplished by using plasmids pJQhmgBX and pJQhpdBX (Ta-ble 2), respectively, through a double-recombination event selected by expressionof a lethal sacB gene (13, 56).
All mutants were analyzed by PCR as previously described (46, 49, 59) to
define the insertion position of the disrupting element (Tn5 or pK18mob deriv-ative) or to confirm the extent of the deletion.
Construction of a DNA cassette for the catabolism of Tyr. For construction ofa DNA cassette containing the genes responsible for catabolism of Tyr in P.putida, the hmgABC, hpd, and tyrB1 genes were PCR isolated and cloned intoplasmid pUC18 to produce plasmids pU-HMG, pU-hpd, and pU-tyrB1, respec-tively (Table 2). The hmgABC and hpd genes were combined as a SacI-KpnI
TABLE 1. Bacterial strains and plasmids used in this study
Strain or plasmid Genotype and/or description Reference or source
E. coli K-12 strainsDH5� F��lacU169 �80 dlacZ�M15 hsdR17 recA1 endA1 gyrA96 thi-1 relA1
supE4477
JM109 F� traD36 proA� proB� lacIq lacZ�MI5/recA1 endA1 gyrA96 (Nalr) thihsdR17 supE44 relA1 �(lac-proAB)
79
HB101 F� �(gpt-proA) 62 leuB6 supE44 ara-14 galK2 lacY1 �(mcrC-mrr) rpsL20(Smr) xyl-5 mtl-1 recA13
3
CC118 pir �(ara-leu) araD �lacX74 galE galK phoA20 thi-I rps-1 rpoB argE(Amp) recAthi pro hsdRM� RP4-2-Tc
34
S17-1 pir pir recA thi pro hsdR M�, RP4:2-Tc::Mu::Km Tn7Tpr Smr 67E. coli W strains
AF141 E. coli W�paa lacZ Rif� derivative 25BMR AF141 with chromosomal insertion of mini-Tn5Km2 Phmg-lacZ This study
P. putida strainsU Wild type 8U-7 P. putida U hmgA due to a Tn5 insertion This studyU-95 P. putida U hmgA due to a Tn5 insertion This studyU-SG6 P. putida U hmgA due to a Tn5 insertion This studyU-111 P. putida U hpd due to a Tn5 insertion This studyU-215 P. putida U phhA due to a Tn5 insertion This studyU-A2 P. putida U hpaB due to a Tn5 insertion Unpublished dataU-dhmgA P. putida U hmgA::pK18mob This studyU-dhmgB P. putida U hmgB::pK18mob This studyU-dhmgC P. putida U hmgC::pK18mob This studyU-dhpd P. putida U hpd::pK18mob This studyU-�hmg P. putida U with hmgABC operon deleted from the genome This studyU-�hpd P. putida U lacking the hpd gene This studyU-dphhA P. putida U phhA::pK18mob This studyU-A2dhpd P. putida U hpaB due to a Tn5 insertion and disrupted in the hpd gene with
the pJQhpd constructionThis study
KT2440 hsdMR 2KT2440Rif� Spontaneous KT2440 rifampin-resistant mutant This studyKT2440-dhmgA KT2440Rif� hmgA::pK18mob This studyKT2440-dhpd KT2440Rif� hpd::pK18mob This studyKT2440-dphhA KT2440Rif� phhA::pK18mob This studyKT2440-dhmgR KT2440Rif� hmgR::pK18mob This study
Plasmidsa
pUC18 Apr oriColE1 lacZ�� lac promoter 79pGEM-T easy Apr oriColE1 lacZ�� SP6 T7 lac promoters, direct cloning of PCR products PromegapK18mob Kmr oriColE1 Mob� lacZ��, used for directed insertional disruption 62pRK600 Cmr oriColE1 oriV Mob�, helper plasmid in triparental matings 37pBBR1MCS-2 Kmr oripBBR1 Mob� lac promoter lacZ��, broad-host-range cloning and
expression vector38
pBBR1MCS-3 Tcr oripBBR1 Mob� lac promoter lacZ��, broad-host-range cloning andexpression vector
38
pJQ200KS Gmr orip15A Mob� lacZ�� sacB, used to generate deletions by double-recombination events
56
pQE32 Apr oriColE1 T5 promoterllac operator, lambda t0/E. coli rrnB T1terminators, N-terminal 6-His
QIAGEN
pSJ3 Apr oriColE1r, lacZ promoter-probe vector 25pUTminiTn5Km2 Apr Kmr oriR6K Mob�, mini-Tn5Km2 transposon delivery plasmid 11pUT-Phmg-lacZ pUTminiTn5Km2 derivative carrying the Phmg-lacZ translational fusion This studypU-HH pUC18 derivative expressing the hmgABC and hpd genes This studypU-HHP pUC18 derivative containing the Tyr catabolic cassette (hmgABC hpd tyrB1) This studypM2-HHP pBBR1MCS-2 derivative harboring the SacI/HindIII Tyr catabolic cassette
from pU-HHPThis study
pGS9 Conjugative plasmid containing the Tn5 transposon 65
a For plasmids obtained by cloning of a PCR-generated fragment see Table 2.
5064 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
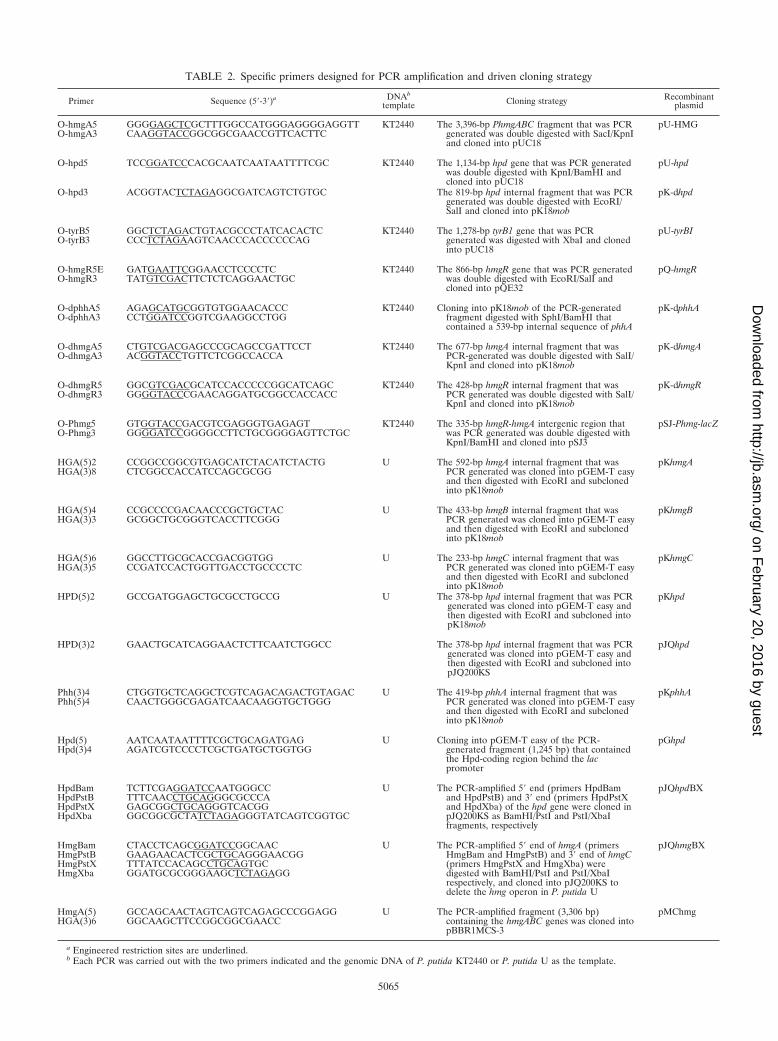
TABLE 2. Specific primers designed for PCR amplification and driven cloning strategy
Primer Sequence (5�-3�)a DNAb
template Cloning strategy Recombinantplasmid
O-hmgA5O-hmgA3
GGGGAGCTCGCTTTGGCCATGGGAGGGGAGGTTCAAGGTACCGGCGGCGAACCGTTCACTTC
KT2440 The 3,396-bp PhmgABC fragment that was PCRgenerated was double digested with SacI/KpnIand cloned into pUC18
pU-HMG
O-hpd5 TCCGGATCCCACGCAATCAATAATTTTCGC KT2440 The 1,134-bp hpd gene that was PCR generatedwas double digested with KpnI/BamHI andcloned into pUC18
pU-hpd
O-hpd3 ACGGTACTCTAGAGGCGATCAGTCTGTGC The 819-bp hpd internal fragment that was PCRgenerated was double digested with EcoRI/SalI and cloned into pK18mob
pK-dhpd
O-tyrB5O-tyrB3
GGCTCTAGACTGTACGCCCTATCACACTCCCCTCTAGAAGTCAACCCACCCCCCAG
KT2440 The 1,278-bp tyrB1 gene that was PCRgenerated was digested with XbaI and clonedinto pUC18
pU-tyrBI
O-hmgR5EO-hmgR3
GATGAATTCGGAACCTCCCCTCTATGTCGACTTCTCTCAGGAACTGC
KT2440 The 866-bp hmgR gene that was PCR generatedwas double digested with EcoRI/SalI andcloned into pQE32
pQ-hmgR
O-dphhA5O-dphhA3
AGAGCATGCGGTGTGGAACACCCCCTGGATCCGGTCGAAGGCCTGG
KT2440 Cloning into pK18mob of the PCR-generatedfragment digested with SphI/BamHI thatcontained a 539-bp internal sequence of phhA
pK-dphhA
O-dhmgA5O-dhmgA3
CTGTCGACGAGCCCGCAGCCGATTCCTACGGTACCTGTTCTCGGCCACCA
KT2440 The 677-bp hmgA internal fragment that wasPCR-generated was double digested with SalI/KpnI and cloned into pK18mob
pK-dhmgA
O-dhmgR5O-dhmgR3
GGCGTCGACGCATCCACCCCCGGCATCAGCGGGGTACCCGAACAGGATGCGGCCACCACC
KT2440 The 428-bp hmgR internal fragment that wasPCR generated was double digested with SalI/KpnI and cloned into pK18mob
pK-dhmgR
O-Phmg5O-Phmg3
GTGGTACCGACGTCGAGGGTGAGAGTGGGGATCCGGGGCCTTCTGCGGGGAGTTCTGC
KT2440 The 335-bp hmgR-hmgA intergenic region thatwas PCR generated was double digested withKpnI/BamHI and cloned into pSJ3
pSJ-Phmg-lacZ
HGA(5)2HGA(3)8
CCGGCCGGCGTGAGCATCTACATCTACTGCTCGGCCACCATCCAGCGCGG
U The 592-bp hmgA internal fragment that wasPCR generated was cloned into pGEM-T easyand then digested with EcoRI and subclonedinto pK18mob
pKhmgA
HGA(5)4HGA(3)3
CCGCCCCGACAACCCGCTGCTACGCGGCTGCGGGTCACCTTCGGG
U The 433-bp hmgB internal fragment that wasPCR generated was cloned into pGEM-T easyand then digested with EcoRI and subclonedinto pK18mob
pKhmgB
HGA(5)6HGA(3)5
GGCCTTGCGCACCGACGGTGGCCGATCCACTGGTTGACCTGCCCCTC
U The 233-bp hmgC internal fragment that wasPCR generated was cloned into pGEM-T easyand then digested with EcoRI and subclonedinto pK18mob
pKhmgC
HPD(5)2 GCCGATGGAGCTGCGCCTGCCG U The 378-bp hpd internal fragment that was PCRgenerated was cloned into pGEM-T easy andthen digested with EcoRI and subcloned intopK18mob
pKhpd
HPD(3)2 GAACTGCATCAGGAACTCTTCAATCTGGCC The 378-bp hpd internal fragment that was PCRgenerated was cloned into pGEM-T easy andthen digested with EcoRI and subcloned intopJQ200KS
pJQhpd
Phh(3)4Phh(5)4
CTGGTGCTCAGGCTCGTCAGACAGACTGTAGACCAACTGGGCGAGATCAACAAGGTGCTGGG
U The 419-bp phhA internal fragment that wasPCR generated was cloned into pGEM-T easyand then digested with EcoRI and subclonedinto pK18mob
pKphhA
Hpd(5)Hpd(3)4
AATCAATAATTTTCGCTGCAGATGAGAGATCGTCCCCTCGCTGATGCTGGTGG
U Cloning into pGEM-T easy of the PCR-generated fragment (1,245 bp) that containedthe Hpd-coding region behind the lacpromoter
pGhpd
HpdBamHpdPstBHpdPstXHpdXba
TCTTCGAGGATCCAATGGGCCTTTCAACCTGCAGGGCGCCCAGAGCGGCTGCAGGGTCACGGGGCGGCGCTATCTAGAGGGTATCAGTCGGTGC
U The PCR-amplified 5� end (primers HpdBamand HpdPstB) and 3� end (primers HpdPstXand HpdXba) of the hpd gene were cloned inpJQ200KS as BamHI/PstI and PstI/XbaIfragments, respectively
pJQhpdBX
HmgBamHmgPstBHmgPstXHmgXba
CTACCTCAGCGGATCCGGCAACGAAGAACACTCGCTGCAGGGAACGGTTTATCCACAGCCTGCAGTGCGGATGCGCGGGAAGCTCTAGAGG
U The PCR-amplified 5� end of hmgA (primersHmgBam and HmgPstB) and 3� end of hmgC(primers HmgPstX and HmgXba) weredigested with BamHI/PstI and PstI/XbaIrespectively, and cloned into pJQ200KS todelete the hmg operon in P. putida U
pJQhmgBX
HmgA(5)HGA(3)6
GCCAGCAACTAGTCAGTCAGAGCCCGGAGGGGCAAGCTTCCGGCGGCGAACC
U The PCR-amplified fragment (3,306 bp)containing the hmgABC genes was cloned intopBBR1MCS-3
pMChmg
a Engineered restriction sites are underlined.b Each PCR was carried out with the two primers indicated and the genomic DNA of P. putida KT2440 or P. putida U as the template.
5065
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

cassette, producing plasmid pU-HH (Table 1). The tyrB1 gene was subcloned asan XbaI fragment in plasmid pU-HH, giving rise to plasmid pU-HHP (Table 1),a pUC18 derivative that contained the 5.8-kb SacI-HindIII hmg-hpd-tyrB1 DNAcassette (Tyr cassette) expressed under control of the tandem Plac and Phmgpromoters. The SacI-HindIII Tyr cassette was then subcloned into a mobilizablebroad-host-range vector, pBBR1MCS-2, producing the recombinant pM2-HHPplasmid (Table 1).
Data analysis. The nucleotide sequence of the P. putida KT2440 genome(accession number AE015451) was analyzed at http://www.tigr.org. Deducedamino acid sequences were analyzed with the Protein Analysis Tool at the WorldWide Web Molecular Biology server of the Geneva University Hospital and theUniversity of Geneva. Protein sequence similarity searches were done with theBLAST program by using the National Center for Biotechnology Informationserver.
Enzyme assays. -Galactosidase activities were measured with permeabilizedcells as described by Miller (45). The 2,5-OH-PhAc dioxygenase activity wasspectrophotometrically determined by measuring the formation of maleylaceto-acetate at 330 nm as described elsewhere (15). Exponentially growing cultureswere centrifuged (7,000 � g, 5 min, 4°C), and cells were resuspended andconcentrated 100-fold in 100 mM potassium phosphate buffer (pH 7.0) contain-ing 20% glycerol. Cell lysis was performed in the same buffer by sonication. Cellextracts were clarified by centrifugation (10,000 � g, 15 min, 4°C) and used in theenzyme assays. The enzyme assay mixtures (final volume, 0.5 ml) contained 100mM potassium phosphate buffer (pH 7.0), 2 mM ascorbate, 50 �M FeSO4, 300�M 2,5-OH-PhAc (unless indicated otherwise), and 5 �l of extract (50 to 100 �gof protein). The reactions were carried out at 37°C with a Beckman DU 520spectrophotometer. One milliunit corresponded to transformation of 1 nmol of2,5-OH-PhAc to maleylacetoacetate per min at 37°C under the conditions de-scribed above. The molar extinction coefficient of maleylacetoacetate is 13,500M�1 cm�1 (64).
Isolation of products that accumulated in the culture broth. The extracellularproducts accumulated by the wild type or by the mutant strains affected in thehomogentisate pathway were identified by high-performance liquid chromatog-raphy (HPLC), nuclear magnetic resonance (NMR), and mass spectrometricanalyses (7, 10, 28).
P. putida U and the mutant strains were cultured in MM containing Phe, Tyr,or 3-OH-PhAc (5 mM) as a source of intermediates and 4-OH-PhAc (5 mM) forsupport of bacterial growth. Moreover, 4-OH-PhAc, which requires a specificcatabolic route, is not degraded through the homogentisate pathway (48), and itis not a substrate of 4-OH-PhPyr dioxygenase. When required, the cultures werecentrifuged (5,000 � g) and filtered through Millipore filters (pore size, 0.45 �m)to eliminate bacteria. The supernatant (culture broth) was acidified with 6 MHCl to pH 1.35 and extracted with n-butanol. The organic phase was washedtwice with Milli-Q water, dried with anhydrous Na2SO4, and lyophilized.
HPLC analyses. Cultures were grown in 500-ml Erlenmeyer flasks containing100 ml of medium and incubated in a rotary shaker (250 rpm) at 30°C. Sampleswere taken at different times, centrifuged (16,000 � g, 20 min) to eliminatebacteria, and filtered through a Millipore filter (pore size, 0.45 �m). Aliquots (50�l) were removed and analyzed by using a high-performance liquid chromato-graph (Spectra Physics SP8800) equipped with a variable-wavelength UV/VISdetector (SP8450), a computing integrator (SP4290), and a microparticulated(particle size, 10 �m; pore size, 100 A) reversed-phase column (Nucleosil C18;length, 250 mm; inside diameter, 4.6 mm; Phenomenex Laboratories, Torrance,Calif.). The mobile phase was 0.05 M K2HPO4 (pH 4)—CH3CN (99:1, vol/vol).The flow rate was 2.5 ml/min, and the eluate was monitored at 254 nm. Underthese conditions the retention times for Tyr, 2,5-OH-PhAc, 3,4-dihydroxypheny-lacetate (3,4-OH-PhAc), 4-OH-PhAc, 3-OH-PhAc, 2-hydroxyphenylacetate (2-OH-PhAc), and PhAc were 3, 7, 11, 20, 23, 29, and 45 min, respectively.
NMR analyses. NMR spectral analyses were recorded at 20°C with a Varian400 Mercury VNMRX spectrometer at 400 MHz (1H) and 100 MHz (13C) byusing tetramethylsilane as the internal standard. Spectra were measured inCD3OD.
Liquid chromatography-MS studies. Mass spectrum analyses were carried outwith a Waters ZMD system by using the following parameters: detection range,200 to 300 nm; capillary power, 3.5 kV; cone power, 25 to 40 V; scan 1, 70 V; scan2, interphase ES�.
Construction of an E. coli strain harboring chromosomal insertions of thePhmg-lacZ translational fusions. Plasmid pUT-Phmg-lacZ (Table 1; see Fig. 7A),which contained a mini-Tn5Km2 hybrid transposon expressing the Phmg-lacZfusion, was transferred from E. coli S17-1pir into the rifampin-resistant E. coliAF141 strain through biparental filter mating as described previously (11).Transconjugants containing the lacZ translational fusions stably inserted into thechromosome were selected for the transposon marker, kanamycin, on rifampin-
containing LB medium. One of these transconjugants, E. coli BMR (Table 1),was used in lacZ gene expression experiments.
Production of the HmgR repressor. The hmgR gene was expressed from thestrong phage T5 promoter (under lac operator control) in the high-copy-numberpQ-hmgR plasmid (Table 1; see Fig. 7B). To prepare crude extract containing theHmgR protein, E. coli JM109(pQ-hmgR) cells were grown in ampicillin-contain-ing LB medium to an A540 of about 0.5, and then 1 volume of LB mediumcontaining 0.5 mM isopropyl--D-thiogalactopyranoside (IPTG) was added andthe culture was incubated until an A540 of about 1.0 was reached. Cells were thencollected by centrifugation (5,000 � g, 10 min, 4°C), washed, and resuspended in0.02 volume of 20 mM Tris-HCl buffer (pH 7.5) containing 10% glycerol, 2 mM-mercaptoethanol, and 50 mM KCl prior to disruption by sonication. The celldebris was removed by centrifugation at 26,000 � g for 30 min at 4°C. The clearsupernatant fluid was decanted and used as the crude cell extract. The controlextract (HmgR�), obtained from E. coli JM109(pQE32) cells, was prepared inexactly the same way. The protein concentration was determined by the methodof Bradford (4) by using bovine serum albumin as the standard.
Mapping the transcription start site by primer extension analysis. E. coliAF141(pSJ-Phmg-lacZ) cells were grown in MM containing 0.5% glycerol untilthe culture reached an A540 of about 1.0. Total RNA was isolated by using anRNA/DNA Midi kit (QIAGEN) according to the instructions of the supplier.Primer extension reactions were carried out with the avian myeloblastosis virusreverse transcriptase (Promega) by using primer O-Phmg3 (which hybridizedwith the coding strand between nucleotides 103 and 124 downstream of the hmgAtranslational start codon [Table 2]). To determine the length of the primerextension product, sequencing reactions with pSJ-Phmg-lacZ were carried outwith the same primer (O-Phmg3) by using a T7 sequencing kit and [�-32P]dCTP(Amershan Pharmacia Biotech) as indicated by the supplier. Products wereanalyzed on 6% polyacrylamide–urea gels. The gels were dried onto Whatman3MM paper and exposed to Hyperfilm MP (Amersham Pharmacia Biotech).
Synthesis of the Phmg probe. The Phmg fragment (335 bp) utilized as a probewas generated by PCR by using plasmid pSJ-Phmg-lacZ (see Fig. 7A) as thetemplate and oligonucleotides O-Phmg5 (which hybridized with the codingstrand between nucleotides 89 and 115 downstream of the hmgR translationalstart codon) and O-Phmg3 as the primers (Table 2). The O-Phmg3 primer waspreviously labeled (50 pmol) at its 5� end with phage T4 polynucleotide kinaseand [�-32P]ATP (3,000 Ci/mmol; Amersham Pharmacia Biotech). To performthe PCR, 10 ng of DNA template (pSJ-Phmg-lacZ), 5 pmol of labeled primerO-Phmg3, and 7.5 pmol of unlabeled primer O-Phmg5 were used; in this way asingly 5�-end-labeled probe at the noncoding strand with respect to the hmgAgene was obtained. The PCR-labeled product was purified with a High Pure PCRproduct purification kit from Roche Molecular Biochemicals.
Gel retardation assays. For gel retardation assays the reaction mixtures (finalvolume, 20 �l) contained in a glutamate buffer solution (20 mM HEPES [pH8.0], 5 mM magnesium chloride, 2 mM dithiothreitol, 50 mM potassium gluta-mate) 0.1 nM DNA probe, 500 �g of bovine serum albumin per ml, 100 �g ofsalmon sperm DNA (competitor) per ml, and cell extract from JM109(pQ-hmgR) or JM109(pQE32) cells. After incubation for 20 min at 20°C, the mixtureswere fractionated by electrophoresis in 4% polyacrylamide gels buffered with0.5� TBE (45 mM Tris-borate, 1 mM EDTA). The gels were dried onto What-man 3MM paper and exposed to Hyperfilm MP (Amersham Pharmacia Bio-tech).
DNase I footprinting assays. The DNase I footprinting assay was carried outin 25 �l (final volume) of a glutamate buffer solution containing 1 nM labeledPhmg probe, 500 �g of bovine serum albumin per ml, and cell extract. Thismixture was incubated for 20 min at 30°C, after which 0.15 U of DNase I(Amersham Pharmacia Biotech) (prepared in a solution containing 10 mMCaCl2, 50 mM MgCl2, 125 mM KCl, and 10 mM Tris-HCl [pH 7.5]) was added,and incubation was continued at 37°C for 20 s. The reaction was stopped byaddition of 180 �l of a solution containing 0.4 M sodium acetate, 2.5 mM EDTA,50 �g of tRNA per ml, and 5 �g of salmon DNA per ml. After phenol-chloro-form extraction, DNA fragments were precipitated with absolute ethanol,washed with 70% ethanol, dried, and directly resuspended in 5 �l of 90%(vol/vol) formamide–loading gel buffer (10 mM Tris-HCl [pH 8.0], 20 mMEDTA [pH 8.0], 0.05% [wt/vol] bromophenol blue, 0.05% [wt/vol] xylene cya-nol). Samples were then denatured at 95°C for 2 min and fractionated in a 6%polyacrylamide–urea gel. A�G Maxam-Gilbert reactions (40) were carried outwith the same fragments, and the mixtures were loaded in the gels along with thefootprinting samples. The gels were dried onto Whatman 3MM paper andexposed to Hyperfilm MP.
Nucleotide sequence accession numbers. The nucleotide sequences reportedin this paper have been submitted to the GenBank/EBI Data Bank; the accessionnumbers are AY168852, AY168853, AY168854, and AY168855.
5066 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

RESULTS AND DISCUSSION
Identification of the genes involved in the central pathwayfor the catabolism of phenylalanine and tyrosine: the ho-mogentisate cluster. During the course of a research programdesigned to characterize the genes involved in the catabolismof aromatic compounds in P. putida U, by using a library ofmutants constructed by Tn5 transposon mutagenesis, we iden-tified three strains, designated P. putida U-7, U-95, and U-SG6(Table 1), that were unable to grow on Phe and Tyr as solecarbon and energy sources. When growing on LB medium,these mutants accumulated a black pigment. The same pheno-type was observed when these strains were cultured in MMcontaining Tyr as the source of colored intermediates and4-OH-PhAc, PhAc, or citrate as the carbon source for support-ing bacterial growth (Fig. 2). HPLC and NMR analyses of theculture broth of these mutants revealed the presence of 2,5-OH-PhAc (see Materials and Methods), suggesting that inser-tion of the Tn5 transposon into the chromosome of the P.putida U mutant strains had disrupted the 2,5-OH-PhAc di-oxygenase activity (see below). The NMR data for the 2,5-OH-PhAc (Fig. 3) are as follows. 1H-NMR (CD3OD): � 3.60(2H, bs, CH2), 6.9 (1H, d, J 8.8 Hz, H-3), 6.7 (1H, dd, J 8.8, J 2.6 Hz, H-4) and 6.8 (1H, d, J 2.6 Hz, H-6). 13C-NMR (CD3OD): � 175.6 (COOH), 32.6 (CH2), 124.4 (C-1),154.1 (C-2), 110.5 (C-3), 114.5 (C-4), 147.8 (C-5�), and 111.7(C-6). The 2,5-OH-PhAc that accumulates in culture brothbecomes oxidized to a quinoid derivative, which by spontane-ous polymerization generates melanic compounds that conferthe characteristic black or brown color to the medium (61).
The accumulation of 2,5-OH-PhAc in the culture broth of theP. putida mutant strains growing in the presence of Tyr sug-gests that this amino acid is metabolized in this microorganismvia the homogentisate pathway. Moreover, sequence analysisof the chromosomal region flanking the transposon insertionsite in the three mutant strains allowed us to identify an openreading frame whose product showed a high level of similarity(56% amino acid sequence identity) with the 2,5-OH-PhAcdioxygenase from Sinorhizobium meliloti (43). Additional ge-netic engineering approaches facilitated cloning and sequenc-ing of a DNA fragment containing a gene cluster (hmg) madeup of four open reading frames (Fig. 1A). Three of these openreading frames, hmgA, hmgB, and hmgC, appear to form asingle transcriptional unit, and they are likely to encode the2,5-OH-PhAc dioxygenase (HmgA), fumarylacetoacetate hy-drolase (HmgB), and maleylacetoacetate isomerase (HmgC)that convert 2,5-OH-PhAc into fumarate and acetoacetate(Fig. 1B). A putative regulatory gene, hmgR, is divergentlytranscribed from the hmgABC catabolic genes and encodes aprotein that shows similarity with members of the IclR familyof transcriptional regulators (72). A homologous cluster(�98% identity) was identified between positions 5241 and5245 of the P. putida KT2440 genome (Fig. 4). The HmgA,HmgB, and HmgC proteins showed significant amino acid se-quence identity with the homogentisate dioxygenase (51%),fumarylacetoacetate hydrolase (44%), and maleylacetoacetateisomerase (39%) involved in degradation of 2,5-OH-PhAc inEmericella nidulans (22). The G�C content of the hmg genesaveraged 64.6%, a value close to the mean G�C content(61%) of the P. putida genome (47), which suggests that thehmg cluster either was imprisoned within the chromosome ofthis bacterium over a long period of evolution or came from adifferent bacterium having a similar G�C content. A genomicsearch in microbial databases revealed the existence of similarhmgRABC clusters in other Pseudomonas species whose ge-nomes are totally or partially known, such as Pseudomonasaeruginosa (71) and Pseudomonas fluorescens; the arrangementof the hmgC gene in Pseudomonas syringae is different (5) (Fig.4). A putative transport gene (hmgT) is located downstream ofhmgC in P. fluorescens and P. aeruginosa. Equivalent hmg clus-ters outside the genus Pseudomonas have been also detected,and they show gene organizations that are different in differentbacteria. Whereas in S. meliloti the hmg genes are organized ina way similar to the way in which they are organized in P.putida, in Ralstonia solanacearum, Bordetella bronchiseptica,Bradyrhizobium japonicum, and Silicibacter pomeroyi, the hmgCgene is located outside the hmgRAB cluster, as has been ob-served in P. syringae (Fig. 4). In Azotobacter vinelandii andXanthomonas axonopodis, hmgA is not associated with thehmgBC genes. It is worth noting that the hmgB gene is dupli-cated in Caulobacter crescentus and Mesorhizobium loti; i.e.,one copy (hmgB1) is clustered with the hmgRA genes, and theother (hmgB2) is linked to hmgC (Fig. 4).
To confirm that the hmg cluster encoded the central ho-mogentisate pathway for the catabolism of Phe and Tyr, as wellas to eliminate the possibility that other undetected mutationswere responsible for the growth deficiencies observed in the P.putida U-7, U-95, and U-SG6 mutant strains, we constructedinsertion mutants with mutations in the hmgA gene (P. putidaU-dhmgA), the hmgB gene (P. putida U-dhmgB), and the hmgC
FIG. 2. Pigment production (browning) by wild-type and mutant P.putida U strains: growth of wild-type P. putida U (a) and the P. putidaU-95 mutant strain (b) on MM containing 5 mM Tyr and 4-OH-PhAc(plate 1) or 5 mM 3-OH-PhAc and 4-OH-PhAc (plate 2).
FIG. 3. Structure of 2,5-OH-PhAc.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5067
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 4. Gene organization of the clusters encoding the homogentisate central pathway and the Phe/Tyr peripheral pathway in P. putida KT2440,and comparisons with equivalent gene clusters from other bacteria. Genes are represented by arrows as follows: black, regulatory genes; stippled,transport genes; vertically striped, genes encoding the homogentisate dioxygenase; hatched, genes encoding the hydrolase and isomerase of thehomogentisate pathway; cross-hatched, genes encoding the 4-OH-PhPyr dioxygenase; white, genes encoding the tyrosine aminotransferase;horizontally striped, genes encoding the phenylalanine hydroxylase and carbinolamine dehydratase. The numbers beneath the arrows indicate thelevels of amino acid sequence identity (expressed as percentages) between the encoded gene products and the equivalent products from P. putida.Identity values are not shown for the hmgR gene products that do not belong to the IclR family of transcriptional regulators. The genomes of P.fluorescens, A. vinelandii, and S. pomeroyi are not completely assembled.
5068 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
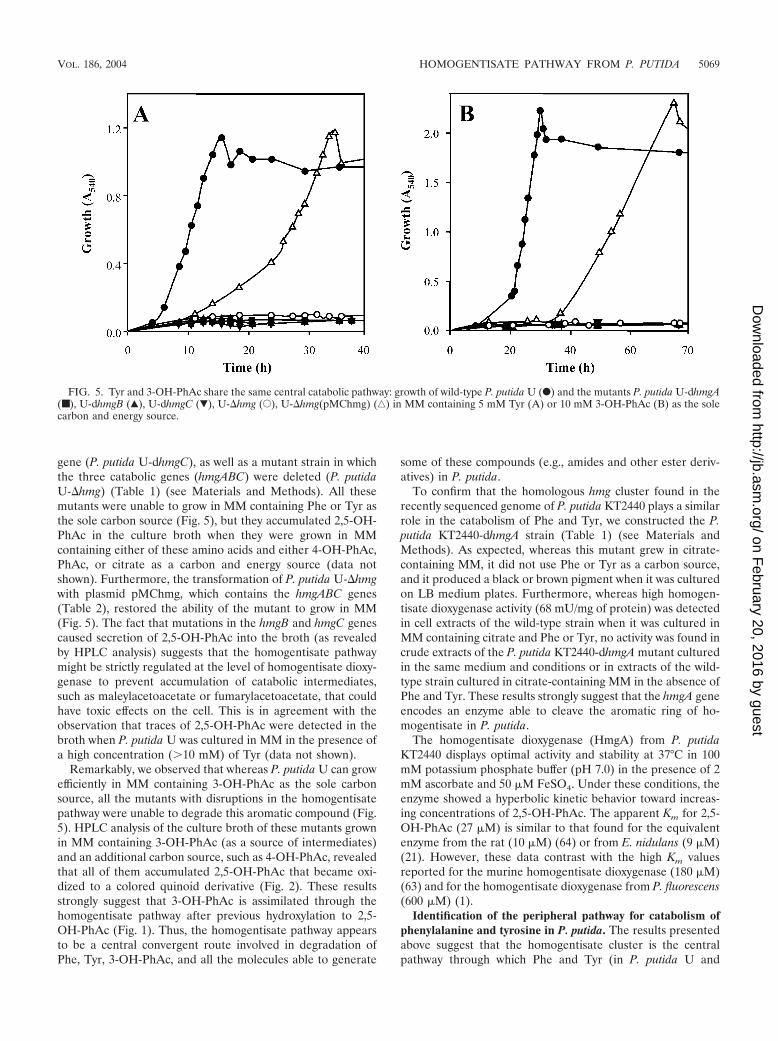
gene (P. putida U-dhmgC), as well as a mutant strain in whichthe three catabolic genes (hmgABC) were deleted (P. putidaU-�hmg) (Table 1) (see Materials and Methods). All thesemutants were unable to grow in MM containing Phe or Tyr asthe sole carbon source (Fig. 5), but they accumulated 2,5-OH-PhAc in the culture broth when they were grown in MMcontaining either of these amino acids and either 4-OH-PhAc,PhAc, or citrate as a carbon and energy source (data notshown). Furthermore, the transformation of P. putida U-�hmgwith plasmid pMChmg, which contains the hmgABC genes(Table 2), restored the ability of the mutant to grow in MM(Fig. 5). The fact that mutations in the hmgB and hmgC genescaused secretion of 2,5-OH-PhAc into the broth (as revealedby HPLC analysis) suggests that the homogentisate pathwaymight be strictly regulated at the level of homogentisate dioxy-genase to prevent accumulation of catabolic intermediates,such as maleylacetoacetate or fumarylacetoacetate, that couldhave toxic effects on the cell. This is in agreement with theobservation that traces of 2,5-OH-PhAc were detected in thebroth when P. putida U was cultured in MM in the presence ofa high concentration (�10 mM) of Tyr (data not shown).
Remarkably, we observed that whereas P. putida U can growefficiently in MM containing 3-OH-PhAc as the sole carbonsource, all the mutants with disruptions in the homogentisatepathway were unable to degrade this aromatic compound (Fig.5). HPLC analysis of the culture broth of these mutants grownin MM containing 3-OH-PhAc (as a source of intermediates)and an additional carbon source, such as 4-OH-PhAc, revealedthat all of them accumulated 2,5-OH-PhAc that became oxi-dized to a colored quinoid derivative (Fig. 2). These resultsstrongly suggest that 3-OH-PhAc is assimilated through thehomogentisate pathway after previous hydroxylation to 2,5-OH-PhAc (Fig. 1). Thus, the homogentisate pathway appearsto be a central convergent route involved in degradation ofPhe, Tyr, 3-OH-PhAc, and all the molecules able to generate
some of these compounds (e.g., amides and other ester deriv-atives) in P. putida.
To confirm that the homologous hmg cluster found in therecently sequenced genome of P. putida KT2440 plays a similarrole in the catabolism of Phe and Tyr, we constructed the P.putida KT2440-dhmgA strain (Table 1) (see Materials andMethods). As expected, whereas this mutant grew in citrate-containing MM, it did not use Phe or Tyr as a carbon source,and it produced a black or brown pigment when it was culturedon LB medium plates. Furthermore, whereas high homogen-tisate dioxygenase activity (68 mU/mg of protein) was detectedin cell extracts of the wild-type strain when it was cultured inMM containing citrate and Phe or Tyr, no activity was found incrude extracts of the P. putida KT2440-dhmgA mutant culturedin the same medium and conditions or in extracts of the wild-type strain cultured in citrate-containing MM in the absence ofPhe and Tyr. These results strongly suggest that the hmgA geneencodes an enzyme able to cleave the aromatic ring of ho-mogentisate in P. putida.
The homogentisate dioxygenase (HmgA) from P. putidaKT2440 displays optimal activity and stability at 37°C in 100mM potassium phosphate buffer (pH 7.0) in the presence of 2mM ascorbate and 50 �M FeSO4. Under these conditions, theenzyme showed a hyperbolic kinetic behavior toward increas-ing concentrations of 2,5-OH-PhAc. The apparent Km for 2,5-OH-PhAc (27 �M) is similar to that found for the equivalentenzyme from the rat (10 �M) (64) or from E. nidulans (9 �M)(21). However, these data contrast with the high Km valuesreported for the murine homogentisate dioxygenase (180 �M)(63) and for the homogentisate dioxygenase from P. fluorescens(600 �M) (1).
Identification of the peripheral pathway for catabolism ofphenylalanine and tyrosine in P. putida. The results presentedabove suggest that the homogentisate cluster is the centralpathway through which Phe and Tyr (in P. putida U and
FIG. 5. Tyr and 3-OH-PhAc share the same central catabolic pathway: growth of wild-type P. putida U (F) and the mutants P. putida U-dhmgA(■), U-dhmgB (Œ), U-dhmgC (�), U-�hmg (E), U-�hmg(pMChmg) (‚) in MM containing 5 mM Tyr (A) or 10 mM 3-OH-PhAc (B) as the solecarbon and energy source.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5069
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

KT2440) and 3-OH-PhAc (in P. putida U) are catabolized. Toidentify the genes involved in the transformation of Phe andTyr into 2,5-OH-PhAc, we isolated by Tn5 transposon mu-tagenesis different P. putida U mutants (strains U-111 andU-215 [Table 1]) which were unable to catabolize Phe and Tyrbut which were able to grow in MM containing 3-OH-PhAcand expressed, therefore, a functional homogentisate pathway.Sequence analysis of the DNA fragment flanking the transpo-son insertion site revealed that whereas P. putida U-111 con-tains the Tn5 transposon within an open reading frame thatencodes a putative 4-OH-PhPyr dioxygenase (Hpd), P. putidaU-215 harbors the Tn5 insertion within a gene cluster encodinga putative pterin-dependent phenylalanine hydroxylase (phhA)that converts Phe into Tyr (81), a putative carbinolamine de-hydratase (phhB) involved in regeneration of the pterin cofac-tor (69), and the putative �54-dependent transcriptional acti-vator (phhR) of the phh operon (68) (Fig. 1). The phhRAB andhpd genes are homologous to the genes previously character-ized in P. aeruginosa (69) and P. fluorescens (66), respectively(Fig. 4).
When we searched for the homologous genes in the genomeof P. putida KT2440, we identified, between positions 5100 and5111 kb of the chromosome, the phhRABT cluster (the phhT
gene encodes a putative transport protein) close to a gene(aroP2) encoding a general aromatic amino acid permease(Fig. 4). In P. aeruginosa the phhC gene encodes a tyrosineaminotransferase that transforms tyrosine into 4-OH-PhPyrand is essential for the catabolism of both Phe and Tyr (33).Although there is no phhC homolog in the phh cluster of P.putida, two genes at positions 2233 kb (tyrB1) and 4080 kb(tyrB2) of the KT2440 genome could encode this tyrosine ami-notransferase function. It is worth noting that while in P.aeruginosa and P. fluorescens the phh genes form a cluster, thetyrB genes are not linked to the phhRAB operon in P. putidaand P. syringae (Fig. 4) (35). This organization is similar to thatfound in other bacteria, like A. vinelandii, X. axonopodis, andR. solanacearum. The hpd gene located at position 3890 kb ofthe P. putida KT2240 genome may encode the 4-OH-PhPyrdioxygenase that converts 4-OH-PhPyr into 2,5-OH-PhAc(Fig. 1). In P. putida, P. fluorescens, A. vinelandii, R. solanacea-rum, and S. pomeroyi the hpd gene is not linked to other genesinvolved in Phe and Tyr degradation, while this gene is asso-ciated with the phh cluster in P. aeruginosa and with the hmggenes in P. syringae, X. axonopodis, C. crescentus, B. japonicum,M. loti, and S. meliloti (Fig. 4).
To confirm that the phh and hpd genes were involved in the
FIG. 6. Catabolic cassette for the catabolism of Tyr. (A) Schematic representation of the construction and expression of a Tyr catabolic cassette.The primer pairs used for PCR amplification are shown in Table 2. Genes are indicated by arrows. The Plac and Phmg promoters are shown. Apr
and Kmr, genes that confer ampicillin and kanamycin resistance, respectively. The pBBR1 and pUC origins of replication (oripBBR1 and oripUC)are indicated. oriTRP4, RP4-mediated mobilization (Mob�) functions. (B) Growth of recombinant strain E. coli(pU-HHP) (F) and control strainE. coli(pUC18) (Œ) in MM supplemented with 0.05% Casamino Acids and 5 mM Tyr as the sole carbon and energy source.
5070 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

catabolism of Phe and Tyr in P. putida, we disrupted some ofthese genes in strain KT2440 and strain U, and then we mon-itored the growth of the resulting mutants in MM containingPhe or Tyr as a carbon source. Insertional inactivation of thephhA gene in P. putida U and in P. putida KT2440 (see Mate-rials and Methods) generated the P. putida U-dphhA and P.putida KT2440-dphhA strains, respectively (Table 1). Thesestrains were unable to grow in MM containing Phe as the solecarbon source, but they grew on Tyr (both mutants) and 3-OH-PhAc (P. putida U-dphhA), producing normal levels of theHmgA enzyme (65 mU/mg of protein). Therefore, these re-sults suggest that the phhA gene is involved in the transforma-tion of Phe into Tyr in P. putida. Since these mutants were notauxotrophs for Tyr, a pathway other than hydroxylation of Pheshould be functional in these bacteria for the biosynthesis ofTyr.
P. putida mutants in which the hpd gene was disrupted (P.putida U-dhpd and P. putida KT2440-dhpd) or deleted (P.putida U-�hpd) (Table 1) were unable to grow in MM con-taining either Phe or Tyr as the sole carbon and energy source,although they grew in MM containing citrate (or 3-OH-PhAcfor P. putida U derivatives). The hpd genes from P. putidaKT2440 and P. putida U were cloned and expressed undercontrol of the Plac promoter in plasmids pU-hpd and pG-hpd,respectively (Table 2). When the recombinant E. coli DH5�cells containing pU-hpd or pG-hpd were grown in LB mediumor in MM containing glycerol (10 mM) and Tyr or 4-OH-PhPyr(1 mM), secretion and accumulation of 2,5-OH-PhAc in theculture broth were observed (data not shown). These dataconfirm that the hpd gene encodes a 4-OH-PhPyr dioxygenase,and they are in agreement with previous observations revealingthat E. coli, as well as other bacteria that do not use Tyr as acarbon source, can synthesize at least one transaminase that isable to convert Tyr into 4-OH-PhPyr (29, 41).
It is worth noting that the hpd mutant of P. putida U (but notthe P. putida KT2440-dhpd mutant) showed a brown pigmen-tation (browning) when it was cultured in MM containing Tyrand 4-OH-PhAc as carbon sources. However, browning wasnot observed when this mutant was cultured in MM containingTyr (as a source of intermediates) and citrate, octanoate, orPhAc as carbon sources. HPLC analysis of the compoundsreleased into the culture medium before the appearance of thebrown pigment revealed the presence of 3,4-dihydroxyphe-nylpyruvate. Thus, these results suggest that the brown com-pound is a derivative of the 3,4-dihydroxyphenylpyruvate pro-duced through hydroxylation of 4-OH-PhPyr by some enzymesof the 4-OH-PhAc catabolic pathway. Since the 4-OH-PhAcmonooxygenase uses a wide range of substrates and is presentin P. putida U (48, 55) but not in P. putida KT2440 (35), itcould be the enzyme responsible for the hydroxylation of4-OH-PhPyr. To confirm this hypothesis, we constructed the P.putida U-A2dhpd mutant strain that harbors a disruption ofboth the hpd and hpaB genes (hpaB encodes the large subunitof the 4-OH-PhAc monooxygenase) (Table 1). The P. putidaU-A2dhpd strain did not show the brown phenotype when itwas grown in citrate-containing MM supplemented with Tyrand 4-OH-PhAc, and the mutant did not produce 3,4-dihy-droxyphenylpyruvate. These data indicate that the productionof 3,4-dihydroxyphenylpyruvate and the browning are specifi-cally related to the presence of Tyr (as a source of 4-OH-
FIG. 7. Scheme for subcloning of the hmg regulatory elements:schematic representation of construction of a Phmg::lacZ translationalfusion cassette (A) and of plasmid pQ-hmgR harboring the regulatoryhmgR gene (B). DNA fragments were PCR amplified by using primersdescribed in Table 2. The Phmg fragment was cloned into the promot-er-probe pSJ3 plasmid. The hmgR fragment was cloned into thepQE32 gene expression vector. �hmgA indicates a truncated hmgAgene (the number of amino acid [aa] residues fused to the LacZprotein is shown in parentheses). T7, to, and rrnB, transcriptionalterminators from the T7 and lambda phages and T1 transcriptionalterminator from the E. coli rrnB operon, respectively; I and O, terminiof the mini-Tn5 transposons; Apr and Kmr, genes that confer ampicil-lin and kanamycin resistance, respectively; tnp*, gene devoid of NotIsites encoding the Tn5 transposase; PT5/lacO, hybrid promoter-oper-ator region composed of the PT5 promoter of phage T5 and the lacOoperator from the E. coli lac cluster; oriTRP4, RP4-mediated mobili-zation functions. The R6K and ColE1 origin of replication (oriR6Kand oriColE1) are indicated. Restriction sites: B, BamHI; E, EcoRI; H,HindIII; K, KpnI; N, NotI; S, SalI.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5071
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

PhPyr) and to the existence of an active 4-OH-PhAc monoox-ygenase whose expression is inducible by 4-OH-PhAc.
Engineering a mobile catabolic cassette for the catabolismof tyrosine. As described above, we characterized a set of genesinvolved in the catabolism of Tyr in P. putida. To demonstratethat these genes encoded all the functions necessary to degradeTyr, we constructed plasmids pU-HHP and pM2-HHP thatcontained the hmgABC, hpd, and tyrB1 genes from P. putidaKT2440 engineered as a 5.8-kb DNA cassette (Tyr cassette)(see Materials and Methods). Whereas the Tyr cassette in
plasmid pU-HHP was expressed under control of the tandemPlac and Phmg promoters, the mobilizable and broad-host-range plasmid pM2-HHP expressed the Tyr cassette only un-der control of the Phmg promoter (Fig. 6A). This cataboliccassette was shown to be functional in heterologous hosts sinceboth plasmids conferred to E. coli the capacity to grow in MMcontaining Tyr as the sole carbon source (Fig. 6B). To ourknowledge, this is the first report of an E. coli strain able tomineralize Tyr. These data confirm, therefore, that the hmg,hpd, and tyr gene products are the only gene products required
FIG. 8. Analysis of the hmgR-hmgA intergenic region. (A) Identification of the transcription start site in Phmg. Primer extension experimentswere carried out by using total RNA isolated from E. coli AF141 cells bearing the lacZ translational fusion plasmid pSJ-Phmg-lacZ (lane 2) andthe control plasmid pSJ3 (lane 1). The size of the extended product was determined by comparison with the DNA sequencing ladder of the Phmgpromoter region (lanes T, C, G, and A). Primer extension and sequencing reactions were performed with primer O-Phmg3 as described inMaterials and Methods. The nucleotide sequence surrounding the transcription initiation site (enclosed in a box) in the coding strand is shown.(B) Schematic representation of regulation of the hmg cluster and nucleotide sequence of the hmgR-hmgA intergenic region. The hmgR regulatorygene is indicated by a thick grey arrow. The hmgABC catabolic genes are indicated by a thick open arrow. The minus sign indicates transcriptionalrepression by the HmgR protein. The plus sign indicates transcriptional activation (induction) promoted by homogentisate. Homogentisate istransformed into fumarate and acetoacetate by the HmgABC proteins. The nucleotide sequence of the Phmg probe (335 bp) is indicated. Thetranslation initiation codon for the hmgA and hmgR genes is indicated by boldface lowercase letters; the bent arrows indicate the direction oftranscription. The transcription start site (position �1) and the inferred �10 and �35 boxes of the Phmg promoter are indicated. The HmgRbinding region is indicated by brackets. The repeated motifs are indicated by thin grey arrows. RBS, ribosome binding site.
TABLE 3. Expression from the Phmg promoter is controlled by HmgRa
E. colistrain Plasmid Relevant
background
-Galactosidase activity (Miller units)
Uninduced Phe Tyr 4-OH-PhPyr 2,5-OH-PhAc 2,5-OH-benzoate
AF141 None lacZ NDb ND ND ND ND NDBMR None Phmg-lacZ 2,520 2,462 2,347 2,269 2,017 2,303BMR pQE32 Phmg-lacZ 1,890 1,895 1,912 1,425 1,343 1,593BMR pQ-hmgR Phmg-lacZ hmgR 9 15 14 27 1,047 198
a E. coli strains were grown in glycerol-containing MM in the absence (uninduced) or in the presence of 5 mM Phe, Tyr, 4-OH-PhPyr, 2,5-OH-PhAc (homogentisate),or 2,5-OH-benzoate (gentisate). -Galactosidase activities were measured with permeabilized cells as described in Materials and Methods.
b ND, not detected.
5072 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

for complete catabolism of Tyr in bacteria. It is worth men-tioning that although the tyrB1 gene in the DNA cassetteensures efficient expression of the tyrosine aminotransferase,E. coli and some other bacteria are equipped with their ownaminotransferases able to transform Tyr into 4-OH-PhPyr (seeabove) (42).
The Tyr cassette, reported for the first time in this work, isa useful tool for metabolic engineering. Thus, this cassette canbe used both to expand the catabolic potential of many mi-crobes lacking a Tyr catabolic pathway and to improve the Tyrdegradation rate in those bacteria that are already able tomineralize this aromatic compound.
In vivo transcriptional analysis of the hmg catabolic genes.Taking into account the fact that the intergenic regions be-tween the hmgA and hmgB genes and between the hmgB andhmgC genes span 3 and 12 bp, respectively, the hmgABC genesmight constitute a catabolic operon. As shown above, the ho-mogentisate pathway is induced when P. putida cells are grownon Phe or Tyr. To investigate in vivo the role of the putativeHgmR regulatory protein in the induction of the hmgABCgenes, we constructed a P. putida strain with the divergentlytranscribed hmgR gene disrupted (Fig. 4). The resulting P.putida KT2440-dhmgR mutant (Table 1) was able to use Pheand Tyr as sole carbon and energy sources, but, in contrast withthe wild-type strain, this mutant constitutively produced a nor-mal amount of HmgA enzyme (72 mU/mg of protein). There-fore, these results suggest that the hmgR gene product is arepressor protein which regulates the inducible hmg catabolicoperon.
To further study the regulatory elements of the hmgABCoperon, a DNA fragment containing the potential promoter(Phmg) was PCR isolated and ligated to the lacZ gene of thepromoter-probe vector pSJ3 (Table 1). The resulting transla-tional fusion plasmid, pSJ-Phmg-lacZ (Fig. 7A), conferred tothe host strain (E. coli CC118) the ability to produce bluecolonies on media containing the -galactosidase indicator5-bromo-4-chloro-3-indolyl--D-galactopyranoside (X-Gal),indicating the presence of a functional promoter in the clonedfragment. To further analyze this regulatory system, we engi-neered the reporter Phmg-lacZ fusion within a mini-Tn5 vec-tor. The resulting construct, pUT-Phmg-lacZ (Fig. 7A), wasused to deliver by transposition the corresponding transla-tional fusion into the chromosome of E. coli AF141 (lacZ),giving rise to the reporter strain E. coli BMR (Table 1). Tocheck the influence of the HmgR protein on the expression ofthe reporter fusion, hmgR was cloned in plasmid pQE32, pro-ducing plasmid pQ-hmgR (Fig. 7B). -Galactosidase assays ofpermeabilized E. coli BMR cells harboring the control plasmidpQE32 showed that there was constitutive expression of thereporter fusion (Table 3). However, when the hmgR gene wasexpressed in trans in E. coli BMR(pQ-hmgR) cells growing inglycerol-containing MM, we observed a drastic decrease (more
FIG. 9. Gel retardation analyses of HmgR binding to the hmgR-hmgA intergenic region. Cell extracts were prepared and gel retarda-tion analyses were performed as described in Materials and Methods.The probe DNA used, Phmg, was PCR amplified from plasmid pSJ-Phmg-lacZ as described in Materials and Methods. (A) Lanes 1 to 7,retardation assay mixtures containing 0, 0.5, 0.7, 1.0, 1.5, 2.0, and 3.0�g of total protein, respectively, of HmgR� extracts obtained fromcells bearing plasmid pQ-hmgR; lane 8, assay mixture containing 3.0 �gof total protein of HmgR� extracts obtained from cells bearing thecontrol plasmid pQE32. (B) Lanes 2 to 8, retardation assay mixturescontaining 3.0 �g of total protein of HmgR� extracts in the absence of2,5-OH-PhAc (lane 2) or in the presence of increasing concentrationsof 2,5-OH-PhAc, as follows: 1 �M (lane 3), 2.5 �M (lane 4), 5.0 �M(lane 5), 10.0 �M (lane 6), 25.0 �M (lane 7), and 50.0 �M (lane 8).Lane 1 shows migration of the Phmg probe without protein extract.(C) Gel retardation assays with 3.0 �g of total protein of HmgR�
extracts and the following different ligands at a concentration of 1 mM:
2,5-OH-PhAc (lane 3), 2,5-OH-benzoate (2,5-OH-Benz) (lane 4),2-OH-PhAc (lane 5), 3-OH-PhAc (lane 6), 3,4-OH-PhAc (lane 7),4-OH-PhPyr (lane 8), PhAc (lane 9), Phe (lane 10), and Tyr (lane 11).Lanes 1 and 2 contained assay mixtures lacking HmgR� extract andligand, respectively. The positions of DNA probes and the DNA-HmgR complexes are indicated by arrows.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5073
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
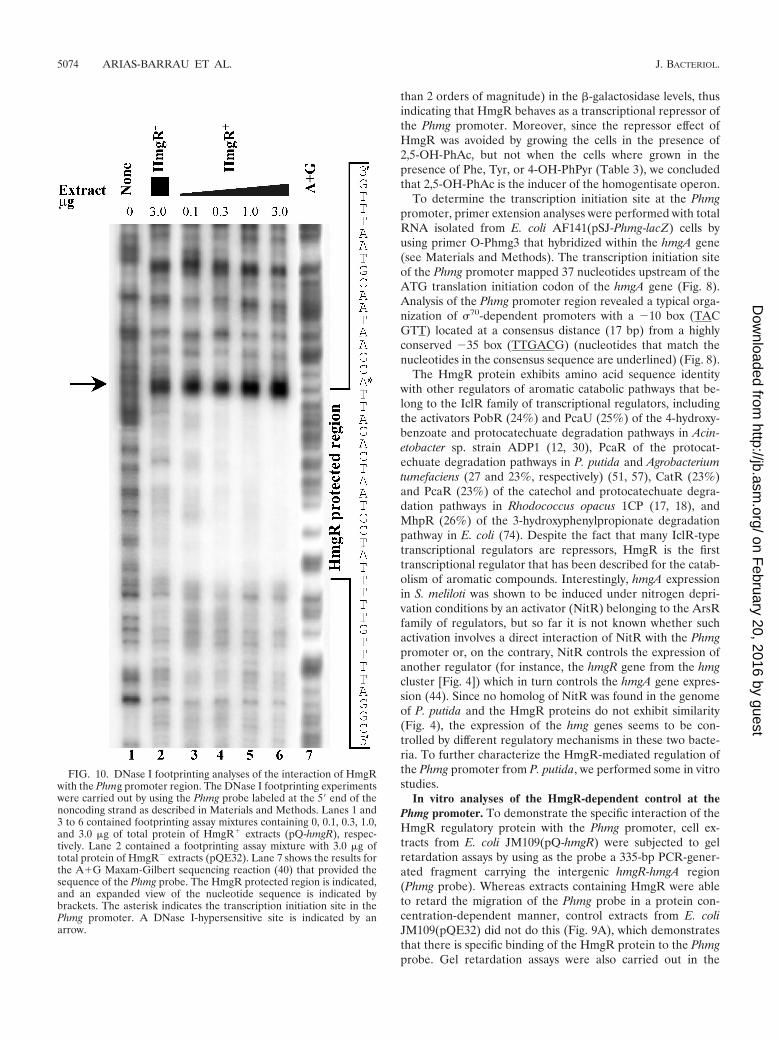
than 2 orders of magnitude) in the -galactosidase levels, thusindicating that HmgR behaves as a transcriptional repressor ofthe Phmg promoter. Moreover, since the repressor effect ofHmgR was avoided by growing the cells in the presence of2,5-OH-PhAc, but not when the cells where grown in thepresence of Phe, Tyr, or 4-OH-PhPyr (Table 3), we concludedthat 2,5-OH-PhAc is the inducer of the homogentisate operon.
To determine the transcription initiation site at the Phmgpromoter, primer extension analyses were performed with totalRNA isolated from E. coli AF141(pSJ-Phmg-lacZ) cells byusing primer O-Phmg3 that hybridized within the hmgA gene(see Materials and Methods). The transcription initiation siteof the Phmg promoter mapped 37 nucleotides upstream of theATG translation initiation codon of the hmgA gene (Fig. 8).Analysis of the Phmg promoter region revealed a typical orga-nization of �70-dependent promoters with a �10 box (TACGTT) located at a consensus distance (17 bp) from a highlyconserved �35 box (TTGACG) (nucleotides that match thenucleotides in the consensus sequence are underlined) (Fig. 8).
The HmgR protein exhibits amino acid sequence identitywith other regulators of aromatic catabolic pathways that be-long to the IclR family of transcriptional regulators, includingthe activators PobR (24%) and PcaU (25%) of the 4-hydroxy-benzoate and protocatechuate degradation pathways in Acin-etobacter sp. strain ADP1 (12, 30), PcaR of the protocat-echuate degradation pathways in P. putida and Agrobacteriumtumefaciens (27 and 23%, respectively) (51, 57), CatR (23%)and PcaR (23%) of the catechol and protocatechuate degra-dation pathways in Rhodococcus opacus 1CP (17, 18), andMhpR (26%) of the 3-hydroxyphenylpropionate degradationpathway in E. coli (74). Despite the fact that many IclR-typetranscriptional regulators are repressors, HmgR is the firsttranscriptional regulator that has been described for the catab-olism of aromatic compounds. Interestingly, hmgA expressionin S. meliloti was shown to be induced under nitrogen depri-vation conditions by an activator (NitR) belonging to the ArsRfamily of regulators, but so far it is not known whether suchactivation involves a direct interaction of NitR with the Phmgpromoter or, on the contrary, NitR controls the expression ofanother regulator (for instance, the hmgR gene from the hmgcluster [Fig. 4]) which in turn controls the hmgA gene expres-sion (44). Since no homolog of NitR was found in the genomeof P. putida and the HmgR proteins do not exhibit similarity(Fig. 4), the expression of the hmg genes seems to be con-trolled by different regulatory mechanisms in these two bacte-ria. To further characterize the HmgR-mediated regulation ofthe Phmg promoter from P. putida, we performed some in vitrostudies.
In vitro analyses of the HmgR-dependent control at thePhmg promoter. To demonstrate the specific interaction of theHmgR regulatory protein with the Phmg promoter, cell ex-tracts from E. coli JM109(pQ-hmgR) were subjected to gelretardation assays by using as the probe a 335-bp PCR-gener-ated fragment carrying the intergenic hmgR-hmgA region(Phmg probe). Whereas extracts containing HmgR were ableto retard the migration of the Phmg probe in a protein con-centration-dependent manner, control extracts from E. coliJM109(pQE32) did not do this (Fig. 9A), which demonstratesthat there is specific binding of the HmgR protein to the Phmgprobe. Gel retardation assays were also carried out in the
FIG. 10. DNase I footprinting analyses of the interaction of HmgRwith the Phmg promoter region. The DNase I footprinting experimentswere carried out by using the Phmg probe labeled at the 5� end of thenoncoding strand as described in Materials and Methods. Lanes 1 and3 to 6 contained footprinting assay mixtures containing 0, 0.1, 0.3, 1.0,and 3.0 �g of total protein of HmgR� extracts (pQ-hmgR), respec-tively. Lane 2 contained a footprinting assay mixture with 3.0 �g oftotal protein of HmgR� extracts (pQE32). Lane 7 shows the results forthe A�G Maxam-Gilbert sequencing reaction (40) that provided thesequence of the Phmg probe. The HmgR protected region is indicated,and an expanded view of the nucleotide sequence is indicated bybrackets. The asterisk indicates the transcription initiation site in thePhmg promoter. A DNase I-hypersensitive site is indicated by anarrow.
5074 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

presence of different concentrations of homogentisate at aconcentration of HmgR that retards completely migration ofthe Phmg probe. As shown in Fig. 9B, increasing concentra-tions of 2,5-OH-PhAc decreased retardation of the DNAprobe, and the interaction of HmgR with Phmg was completelyabolished at 50 �M 2,5-OH-PhAc. These data are in agree-ment with the conclusions provided by lacZ-reporter fusionexperiments reported above, and they confirm that homogen-tisate is the inducer of the hmg catabolic genes.
To check the ligand profile for HmgR, gel retardation ex-periments were performed by using the Phmg probe and dif-ferent structural analogs of 2,5-OH-PhAc, such as 2,5-OH-benzoate (gentisate), 2-OH-PhAc, 3-OH-PhAc, 3,4-OH-PhAc,PhAc, and related compounds of the Phe/Tyr catabolic path-way (Phe, Tyr, and 4-OH-PhPyr). Interestingly, only 2,5-OH-PhAc was able to efficiently inhibit binding of HmgR to thePhmg promoter (Fig. 9C). Gentisate, a structural analog of2,5-OH-PhAc with a side chain that is one carbon shorter, wasalso able to disturb the HmgR-Phmg interaction, but this effectwas more than 2 orders of magnitude less efficient than thatcaused by homogentisate (Fig. 9C). lacZ-reporter fusion ex-periments confirmed that 2,5-OH-benzoate (gentisate) wasable to induce expression from the Phmg promoter in vivo, andthe induction achieved was fivefold lower than that achievedwith 2,5-OH-PhAc (Table 3). These results show that the pres-ence of two hydroxy groups at the para position in the benzenering of the aromatic acid are indispensable for a productiveinteraction of the inducer molecule with the HmgR repressor,and an aromatic acid with a two-carbon side chain (2,5-OH-PhAc) is the best inducer. Such high specificity for the inducermolecule suggests that the regulatory elements were recruiteda long time ago and that they evolved together with the cata-bolic genes during the evolutionary history of the hmg cluster.
To characterize the HmgR binding site within the Phmgpromoter, DNase I footprinting experiments were performedby using the Phmg probe as the target DNA (Fig. 10). Aprotected 45-bp region that spans from position �16 to posi-tion 29 with respect to the Phmg transcription start site wasobserved (Fig. 8B). The 3� end of the HmgR binding regionpartially overlaps the ribosome binding site of the hmgA gene(Fig. 8B), and, as already reported for the IclR regulator (78),a site hypersensitive to DNase I was detected at the 5� end ofthe region (Fig. 10). Analysis of the HmgR binding regionrevealed a 17-bp perfect palindromic motif (TCGTAATCTGATTACGA) with its pseudodyad axis through a central T
residue (located at position 5 with respect to the transcriptioninitiation site) that defines two 8-bp half-sites (Fig. 8B). Otherregulators of aromatic catabolic pathways that belong to theIclR family, such as PobR and PcaU from Acinetobacter sp.strain ADP1, PcaR from P. putida, and MhpR from E. coli,also recognize 17-bp palindromic operator regions with thepseudodyad axis through a central base, but the consensussequence (TGTTCGATAATCGCACA) (30, 74) does not re-semble that of the HmgR binding site. Interestingly, the HmgRbinding region contains a third 6-bp motif (ATTACG), whichis located 4 bp upstream of the palindromic motif, partiallyoverlaps the �10 box, and is arranged as a direct sequencerepetition of the right-half site of the palindrome (Fig. 8B).Analyses of the putative Phmg promoters from other Pseudo-monas species, such as P. fluorescens, P. aeruginosa, and P.syringae, revealed a very similar organization of the presumedoperator regions with a 17-bp imperfect palindromic motifseparated by 4 bp of a highly conserved direct repeat identicalto that of P. putida (Fig. 11). Based on this observation and onthe high amino acid sequence identity among the equivalentHmgR proteins from the four Pseudomonas species (Fig. 4), acommon regulatory mechanism can be suggested for the ho-mogentisate cluster in Pseudomonas.
A similarly structured protein binding region with a palin-dromic motif and an external direct repetition has been de-scribed for promoters controlled by other IclR-type regulators,such as PcaU, PobR, and PcaR activators (54), and for thepromoter controlled by the DeoR repressor from Bacillus sub-tilis (80). However, the architectures of these regulatory re-gions show relevant differences for each individual regulator,which might reflect major differences both in the mechanismby which the regulators interact with the RNA polymerase andin the fundamental method of transcriptional regulation (i.e.,activation or repression) of the cognate promoters. Althoughunraveling the mechanism leading to repression of Phmg byHmgR and to induction by homogentisate requires furtherresearch, the location of the HmgR binding site overlappingboth the �10 box and the transcription initiation site stronglysuggests that HmgR physically competes with the RNA poly-merase in promoter binding and that the presence of the in-ducer must change the nature of the HmgR-Phmg interactionin a way that allows transcription initiation by the RNA poly-merase.
FIG. 11. Comparison of the Phmg promoter regions in several Pseudomonas species. The hmgR-hmgA intergenic regions from P. putida, P.fluorescens, P. aeruginosa, and P. syringae were aligned from position �50 to position 40 with respect to the transcription initiation site in P. putida.The asterisks indicate the conserved nucleotides. The �35 and �10 boxes, the �1 transcription initiation site, the ribosome binding site (RBS),and the ATG translation initiation codon from hmgA in P. putida are indicated by a black background. The HmgR binding region is indicated bybrackets. Repeated motifs are indicated by arrows.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5075
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

ACKNOWLEDGMENTS
This work was supported by EU contract QLRT-2001-02884 and bygrants BMC2000-0125-C04-01/02, BIO2003-05309-C04-01/02, andGEN2001-4698-C05-02 from the Comision Interministerial de Cienciay Tecnologıa. B.M. holds a Contrato Ramon y Cajal from MCYT.E.A.-B. and C.F. have predoctoral fellowships from the Plan Nacionalde Formacion de Personal Investigador, Ministerio de Ciencia y Tec-nologıa.
We thank E. Aporta for help with oligonucleotide synthesis and A.Dıaz, G. Porras, S. Carbajo, and M. Cayuela for help with sequencing.The technical assistance of E. Cano, M. Carrasco, F. Morante, and E.Calvo is gratefully acknowledged.
REFERENCES
1. Adachi, K., Y. Iwayama, H. Tanioka, and Y. Takeda. 1966. Purification andproperties of homogentisate oxygenase from Pseudomonas fluorescens. Bio-chim. Biophys. Acta 118:88–97.
2. Bayley, S. A., C. J. Duggleby, M. J. Worsey, P. A. Williams, K. G. Hardy, andP. Broda. 1977. Two modes of loss of the Tol function from Pseudomonasputida mt-2. Mol. Gen. Genet. 154:203–204.
3. Boyer, H. W., and D. Roulland-Dussoix. 1969. A complementary analysis ofthe restriction and modification of DNA in Escherichia coli. J. Mol. Biol.41:459–472.
4. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72:248–254.
5. Buell, C. R., V. Joardar, M. Lindeberg, J. Selengut, I. T. Paulsen, M. L.Gwinn, R. J. Dodson, R. T. Deboy, A. S. Durkin, J. F. Kolonay, R. Madupu,S. Daugherty, L. Brinkac, M. J. Beanan, D. H. Haft, W. C. Nelson, T.Davidsen, N. Zafar, L. Zhou, J. Liu, Q. Yuan, H. Khouri, N. Fedorova, B.Tran, D. Russell, K. Berry, T. Utterback, S. E. Van Aken, T. V. Feldblyum,M. D’Ascenzo, W. L. Deng, A. R. Ramos, J. R. Alfano, S. Cartinhour, A. K.Chatterjee, T. P. Delaney, S. G. Lazarowitz, G. B. Martín, D. J. Schneider,X. Tang, C. L. Bender, O. White, C. M. Fraser, and A. Collmer. 2003. Thecomplete genome sequence of the Arabidopsis and tomato pathogen Pseudo-monas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. USA 100:10181–10186.
6. Citron, B. A., M. D. Davis, S. Milstien, J. Gutierrez, D. B. Mendel, G. R.Crabtree, and S. Kaufman. 1992. Identity of 4�-carbinolamine dehydratase,a component of the phenylalanine hydroxylation system, and DCoH, a tran-sregulator of homeodomain proteins. Proc. Natl. Acad. Sci. USA 89:11891–11894.
7. Claridge, T. D. W. 1999. High-resolution NMR techniques in organic chem-istry. Elsevier, Amsterdam, The Netherlands.
8. Cooper, R. A., and M. A. Skinner. 1980. Catabolism of 3- and 4-hydroxy-phenylacetate by the 3,4-dihydroxyphenylacetate pathway in Escherichia coli.J. Bacteriol. 143:302–306.
9. Craig, S. P., V. J. Buckle, A. Lamouroux, J. Mallet, and I. Craig. 1986.Localization of the human tyrosine hydroxylase gene to 11p15: gene dupli-cation and evolution of metabolic pathways. Cytogenet. Cell Genet. 42:29–32.
10. Davis, R., and M. Frearson. 1987. Mass spectrometry: analytical chemistry byopen learning. John Wiley and Sons, Chichester, England.
11. de Lorenzo, V., and K. N. Timmis. 1994. Analysis and construction of stablephenotypes in Gram-negative bacteria with Tn5- and Tn10-derived mini-transposons. Methods Enzymol. 235:386–405.
12. DiMarco, A. A., B. Averhoff, and L. N. Ornston. 1993. Regulation of p-hydroxybenzoate hydroxylase synthesis by PobR bound to an operator inAcinetobacter calcoaceticus. J. Bacteriol. 175:4499–4506.
13. Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletionmutant of enteropathogenic Escherichia coli by using a positive-selectionsuicide vector. Infect. Immun. 59:4310–4317.
14. Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency trans-formation of E. coli by high voltage electroporation. Nucleic Acids Res.16:6127–6145.
15. Edwards, S. W., and W. E. Knox. 1955. Enzymes involved in conversion oftyrosine to acetoacetate. Methods Enzymol. 2:287–300.
16. Elpeleg, O. N., A. Shaag, E. Holme, G. Zughayar, S. Ronen, D. Fisher, andH. Hurvitz. 2002. Mutation analysis of the FAH gene in Israeli patients withtyrosinemia type I. Hum. Mutat. 19:80–81.
17. Eulberg, D., and M. Schlomann. 1998. The putative regulator of catecholcatabolism in Rhodococcus opacus 1CP—an IclR-type, not a LysR-type tran-scriptional regulator. Antonie Leeuwenhoek 74:71–82.
18. Eulberg, D., S. Lakner, L. A. Golovleva, and M. Schlomann. 1998. Charac-terization of a protocatechuate catabolic gene cluster from Rhodococcusopacus 1CP: evidence for a merged enzyme with 4-carboxymuconolactone-decarboxylating and 3-oxoadipate enol-lactone-hydrolyzing activity. J. Bac-teriol. 180:1072–1081.
19. Fernandez-Canon, J. M., and M. A. Penalva. 1995. Fungal metabolic model
for human type I hereditary tyrosinaemia. Proc. Natl. Acad. Sci. USA 92:9132–9136.
20. Fernandez-Canon, J. M., and M. A. Penalva. 1995. Molecular characteriza-tion of a gene encoding a homogentisate dioxygenase from Aspergillus nidu-lans and identification of its human and plant homologues. J. Biol. Chem.270:21199–21205.
21. Fernandez-Canon, J. M., and M. A. Penalva. 1997. Spectrophotometricdetermination of homogentisate using Aspergillus nidulans homogentisatedioxygenase. Anal. Biochem. 245:218–221.
22. Fernandez-Canon, J. M., and M. A. Penalva. 1998. Characterization of afungal maleylacetoacetate isomerase gene and identification of its humanhomologue. J. Biol. Chem. 273:329–337.
23. Fernandez-Canon, J. M., M. W. Baetscher, M. Finegold, T. Burlingame,K. M. Gibson, and M. Grompe. 2002. Maleylacetoacetate isomerase (MAAI/GSTZ)-deficient mice reveal aglutathione-dependent nonenzymatic bypassin tyrosine catabolism. Mol. Cell. Biol. 22:4943–4951.
24. Fernandez-Canon, J. M., B. Granadino, D. Beltran-Valero de Bernabe, M.Renedo, E. Fernandez-Ruiz, M. A. Penalva, and S. Rodrıguez de Cordoba.1996. The molecular basis of alkaptonuria. Nat. Genet. 14:19–24.
25. Ferrandez, A., B. Minambres, B. Garcıa, E. R. Olivera, J. M. Luengo, J. L.Garcıa, and E. Dıaz. 1998. Catabolism of phenylacetic acid in Escherichiacoli. J. Biol. Chem. 273:25974–25986.
26. Franklin, F. C., P. R. Lehrbach, R. Lurz, B. Rueckert, M. Bagdasarian, andK. N. Timmis. 1983. Localization and functional analysis of transposonmutations in regulatory genes of the TOL catabolic pathway. J. Bacteriol.156:676–685.
27. Fuqua, W. C., V. E. Coyne, D. C. Stein, C. M. Lin, and R. M. Weiner. 1991.Characterization of melA: a gene encoding melanin biosynthesis from themarine bacterium Shewanella colwelliana. Gene 109:131–136.
28. Garcıa, B., E. R. Olivera, B. Minambres, M. Fernandez-Valverde, L. M.Canedo, M. A. Prieto, J. L. Garcıa, M. Martınez, and J. M. Luengo. 1999.Novel biodegradable aromatic plastics from a bacterial source. Genetic andbiochemical studies on a route of the phenylacetyl-CoA catabolon. J. Biol.Chem. 274:29228–29241.
29. Garcıa, I., M. Rodgers, C. Lenne, A. Roland, A. Saillant, and M. Matringe.1997. Subcellular localization and purification of a p-hydroxyphenylpyruvatedioxygenase from cultured carrot cells and characterization of the corre-sponding cDNA. Biochem. J. 325:761–769.
30. Gerischer, U., A. Segura, and L. N. Ornston. 1998. PcaU, transcriptionalactivator of genes for protocatechuate utilization in Acinetobacter. J. Bacte-riol. 180:1512–1524.
31. Granadino, B., D. Beltran-Valero de Bernabe, J. M. Fernandez-Canon, M. A.Penalva, and S. Rodrıguez de Cordoba. 1997. The human homogentisate1,2-dioxygenase (HGO) gene. Genomics 43:115–122.
32. Grompe, M. 2001. The pathophysiology and treatment of hereditary ty-rosinemia type I. Semin. Liver Dis. 21:563–571.
33. Gu, W., J. Song, C. A. Bonner, G. Xie, and R. A. Jensen. 1998. PhhC, anessential amino transferase for aromatic amino acid catabolism in Pseudo-monas aeruginosa. Microbiology 144:3127–3134.
34. Herrero, M. V., V. de Lorenzo, and K. N. Timmis. 1990. Transposon vectorscontaining non-antibiotic resistance selection markers for cloning and stablechromosomal insertion of foreign genes in gram-negative bacteria. J. Bacte-riol. 172:6557–6567.
35. Jimenez, J. I., B. Minambres, J. L. Garcıa, and E. Dıaz. 2002. Genomicanalysis of the aromatic catabolic pathways from Pseudomonas putidaKT2440. Environ. Microbiol. 4:824–841.
36. Johnson-Winters, K., V. M. Purpero, M. Kavana, T. Nelson, and G. R.Moran. 2003. (4-Hydroxyphenyl)pyruvate dioxygenase from Streptomycesavermitilis: the basis for ordered substrate addition. Biochemistry 42:2072–2080.
37. Kessler, B., V. de Lorenzo, and K. N. Timmis. 1992. A general system tointegrate lacZ fusions into the chromosomes of gram negative eubacteria:regulation of the Pm promoter of the TOL plasmid studied with all control-ling elements in monocopy. Mol. Gen. Genet. 233:293–301.
38. Kovach, M. E., P. H. Elzer, D. S. Hill, G. T. Robertson, M. A. Farris, R. M.Roop, and K. M. Peterson. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistancecassettes. Gene 166:175–176.
39. Martınez-Blanco, H., A. Reglero, L. B. Rodrıguez-Aparicio, and J. M. Lu-engo. 1990. Purification and biochemical characterization of phenylacetyl-CoA ligase from Pseudomonas putida. A specific enzyme for the catabolismof phenylacetic acid. J. Biol. Chem. 265:7084–7090.
40. Maxam, A., and W. Gilbert. 1980. Sequencing end-labeled DNA with basespecific chemical cleavages. Methods Enzymol. 65:499–560.
41. McFall, E., and E. B. Newman. 1996. Amino acids as carbon sources, p.358–379. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B.Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E.Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecularbiology, 2nd ed. ASM Press, Washington, D.C.
42. Mehta, P. K., T. I. Hale, and P. Christen. 1989. Evolutionary relationshipsamong aminotransferases. Tyrosine aminotransferase, histidinol-phosphate
5076 ARIAS-BARRAU ET AL. J. BACTERIOL.
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

aminotransferase, and aspartate aminotransferase are homologous proteins.Eur. J. Biochem. 186:249–253.
43. Milcamps, A., and de F. J. Bruijn. 1999. Identification of a novel nutrient-deprivation-induced Sinorhizobium meliloti gene (hmgA) involved in the deg-radation of tyrosine. Microbiology 145:935–947.
44. Milcamps, A., P. Struffi, and F. J. de Bruijn. 2001. The Sinorhizobiummeliloti nutrient-deprivation-induced tyrosine degradation gene hmgA iscontrolled by a novel member of the arsR family of regulatory genes. Appl.Environ. Microbiol. 67:2641–2648.
45. Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
46. Mullis, K. B., and F. A. Faloona. 1987. Specific synthesis of DNA in vitro viaa polymerase-catalyzed chain reaction. Methods Enzymol. 155:335–350.
47. Nelson, K. E., C. Weinel, I. T. Paulsen, R. J. Dodson, H. Hilbert, V. A.Martins dos Santos, D. E. Fouts, S. R. Gill, M. Pop, M. Holmes, L. Brinkac,M. Beanan, R. T. DeBoy, S. Daugherty, J. Kolonay, R. Madupu, W. Nelson,O. White, J. Peterson, H. Khouri, I. Hance, P. C. Lee, E. Holtzapple, D.Scanlan, K. Tran, A. Moazzez, T. Utterback, M. Rizzo, K. Lee, D. Kosack, D.Moestl, H. Wedler, J. Lauber, D. Stjepandic, J. Hoheisel, M. Straetz, S.Heim, C. Kiewitz, J. Eisen, K. N. Timmis, A. Dusterhoft, B. Tummler, andC. M. Fraser. 2002. Complete genome sequence and comparative analysis ofthe metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol.4:799–808.
48. Olivera, E. R., A. Reglero, H. Martınez-Blanco, A. Fernandez-Medarde,M. A. Moreno, and J. M. Luengo. 1994. Catabolism of aromatics in Pseudo-monas putida U. Formal evidence that phenylacetic acid and 4-hydroxyphe-nylacetic acid are catabolized by two unrelated pathways. Eur. J. Biochem.221:375–381.
49. Olivera, E. R., B. Minambres, B. Garcıa, C. Muniz, M. A. Moreno, A.Ferrandez, E. Dıaz, J. L. Garcıa, and J. M. Luengo. 1998. Molecular char-acterization of the phenylacetic acid catabolic pathway in Pseudomonasputida U: the phenylacetyl-CoA catabolon. Proc. Natl. Acad. Sci. USA 95:6419–6424.
50. Olivera, E. R., D. Carnicero, R. Jodra, B. Minambres, B. Garcıa, G. A.Abraham, A. Gallardo, J. S. Roman, J. L. Garcıa, G. Naharro, and J. M.Luengo. 2001. Genetically engineered Pseudomonas: a factory of new bio-plastics with broad applications. Environ. Microbiol. 3:612–618.
51. Parke, D. 1997. Acquisition, reorganization, and merger of genes—novelmanagement of the -ketoadipate pathway in Agrobacterium tumefaciens.FEMS Microbiol. Lett. 146:3–12.
52. Penalva, M. A. 2001. A fungal perspective on human inborn errors of me-tabolism: alkaptonuria and beyond. Fungal Genet. Biol. 34:1–10.
53. Pometto, A. L., and D. L. Crawford. 1985. L-Phenylalanine and L-tyrosinecatabolism by selected Streptomyces species. Appl. Environ. Microbiol. 49:727–729.
54. Popp, R., T. Kohl, P. Patz, G. Trautwein, and U. Gerischer. 2002. Differen-tial DNA binding of transcriptional regulator PcaU from Acinetobacter sp.strain ADP1. J. Bacteriol. 184:1988–1997.
55. Prieto, M. A., A. Perez-Aranda, and J. L. Garcıa. 1993. Characterization ofan Escherichia coli aromatic hydroxylase with a broad substrate range. J.Bacteriol. 175:2162–2167.
56. Quandt, J., and M. F. Hynes. 1993. Versatile suicide vectors which allowdirect selection for gene replacement in Gram-negative bacteria. Gene 127:15–21.
57. Romero-Steiner, S., R. E. Parales, C. S. Harwood, and J. E. Houghton. 1994.Characterization of the pcaR regulatory gene from Pseudomonas putida,which is required for the complete degradation of p-hydroxybenzoate. J.Bacteriol. 176:5771–5779.
58. Ruetschi, U., B. Odelhog, S. Lindstedt, J. Barros-Soderling, B. Persson, andH. Jornvall. 1992. Characterization of 4-hydroxyphenylpyruvate dioxygen-ase. Primary structure of the Pseudomonas enzyme. J. Biochem. 205:459–466.
59. Saiki, R., S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Ehrlich, andN. Arnheim. 1985. Enzymatic amplification of beta-globin genomic se-quences and restriction site analysis for diagnosis of sickle cell anemia.Science 230:1350–1354.
60. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
61. Sanchez-Amat, A., C. Ruzafa, and F. Solano. 1998. Comparative tyrosinedegradation in Vibrio cholerae strains. The strain ATCC 14035 as a prokary-
otic melanogenic model of homogentisate-releasing cell. Comp. Biochem.Physiol. Part B Biochem. Mol. Biol. 119:557–562.
62. Schafer, A., A. Tauch, W. Jager, J. Kalinowski, G. Thierbach, and A. Puhler.1994. Small mobilizable multi-purpose cloning vectors derived from theEscherichia coli plasmids pK18 and pK19: selection of defined deletions inthe chromosome of Corynebacterium glutamicum. Gene 145:69–73.
63. Schmidt, S. R., C. R. Muller, and W. Kress. 1995. Murine liver homogenti-sate 1,2-dioxygenase. Purification to homogeneity and novel biochemicalproperties. Eur. J. Biochem. 228:425–430.
64. Seegmiller, J. E., V. G. Zannoni, L. Laster, and B. N. La Du. 1961. Anenzymatic spectrophotometric method for the determination of homogen-tisic in plasma and urine. J. Biol. Chem. 236:774–777.
65. Selvaraj, G., and V. N. Iyer. 1983. Suicide plasmid vehicles for insertionmutagenesis in Rhizobium meliloti and related bacteria. J. Bacteriol. 156:1292–1300.
66. Serre, L., A. Sailland, D. Sy, P. Boudec, A. Rolland, E. Pebay-Peyroula, andC. Cohen-Addad. 1999. Crystal structure of Pseudomonas fluorescens 4-hy-droxyphenylpyruvate dioxygenase: an enzyme involved in the tyrosine deg-radation pathway. Struct. Fold Des. 7:977–988.
67. Simon, R., U. Prifer, and A. Puhler. 1983. A broad host range mobilizationsystem for in vivo genetic engineering: transposon mutagenesis in Gram-positive bacteria. Bio/Technology 1:784–791.
68. Song, J., and R. A. Jensen. 1996. PhhR, a divergently transcribed activator ofthe phenylalanine hydroxylase gene cluster of Pseudomonas aeruginosa. Mol.Microbiol. 22:497–507.
69. Song, J., T. Xia, and R. A. Jensen. 1999. PhhB, a Pseudomonas aeruginosahomolog of mammalian pterin 4�-carbinolamine dehydratase/DCoH, doesnot regulate expression of phenylalanine hydroxylase at the transcriptionallevel. J. Bacteriol. 181:2789–2796.
70. Sparnins, V. L., D. G. Burbee, and S. Dagley. 1979. Catabolism of L-tyrosinein Trichosporon cutaneum. J. Bacteriol. 138:425–430.
71. Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J.Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L.Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L.Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D.Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E.Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence ofPseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959–964.
72. Sunnarborg, A., D. Klumpp, T. Chung, and D. C. LaPorte. 1990. Regulationof the glyoxylate bypass operon: cloning and characterization of IclR. J.Bacteriol. 172:2642–2649.
73. Tomoeda, K., H. Awata, T. Matsuura, I. Matsuda, E. Ploechl, T. Milovac, A.Boneh, C. R. Scott, D. M. Danks, and F. Endo. 2000. Mutations in the4-OH-PhPyr dioxygenase gene are responsible for tyrosinemia type III andhawkinsinuria. Mol. Genet. Metab. 71:506–510.
74. Torres, B., G. Porras, J. L. Garcıa, and E. Dıaz. 2003. Regulation of the mhpcluster responsible for 3-(3-hydroxyphenyl)propionic acid degradation inEscherichia coli. J. Biol. Chem. 278:27575–27585.
75. Wackett, L. P. 2003. Pseudomonas putida—a versatile biocatalyst. Nat. Bio-technol. 21:136–138.
76. Wintermeyer, E., M. Flugel, M. Ott, M. Steinert, U. Rdest, K. H. Mann, andJ. Hacker. 1994. Sequence determination and mutational analysis of the llylocus of Legionella pneumophila. Infect. Immun. 62:1109–1117.
77. Woodcock, D. M., D. M. Crowther, J. Doherty, S. Jefferson, E. DeCruz, M.Noyer-Weidner, S. S. Smith, M. Z. Michael, and M. W. Graham. 1989.Quantitative evaluation of Escherichia coli host strains for tolerance to cy-tosine methylation in plasmid and phage recombinants. Nucleic Acids Res.17:3469–3478.
78. Yamamoto, K., and A. Ishihama. 2003. Two different modes of transcriptionrepression of the Escherichia coli acetate operon by IclR. Mol. Microbiol.47:183–194.
79. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phagecloning vectors and host strains: nucleotide sequences of the M13mp18 andpUC vectors. Gene 33:103–119.
80. Zeng, X., H. H. Saxild, and R. L. Switzer. 2000. Purification and character-ization of the DeoR repressor of Bacillus subtilis. J. Bacteriol. 182:1916–1922.
81. Zhao, G., T. Xia, J. Song, and R. A. Jensen. 1994. Pseudomonas aeruginosapossesses homologues of mammalian phenylalanine hydroxylase and 4�-carbinolamine dehydratase/DCoH as part of a three-component gene clus-ter. Proc. Natl. Acad. Sci. USA 91:1366–1370.
VOL. 186, 2004 HOMOGENTISATE PATHWAY FROM P. PUTIDA 5077
on February 20, 2016 by guest
http://jb.asm.org/
Dow
nloaded from




















