
The He + H 2 + reaction: a dynamical test on potential energy surfaces for a system exhibiting a...
10
3 March 2000 Ž . Chemical Physics Letters 318 2000 619–628 www.elsevier.nlrlocatercplett The He q H q reaction: a dynamical test on potential energy 2 surfaces for a system exhibiting a pronounced resonance pattern V. Aquilanti a, ) , G. Capecchi a , S. Cavalli a , D. De Fazio a , P. Palmieri b , C. Puzzarini b , A. Aguilar c , X. Gimenez c , J.M. Lucas c ´ a Dipartimento di Chimica, UniÕersita di Perugia, 06123 Perugia, Italy ` b Dipartimento di Chimica Fisica ed Inorganica, UniÕersita di Bologna, 40126 Bologna, Italy ` c Departament de Quimica Fisica, UniÕersitat de Barcelona, 08028 Barcelona, Spain Received 21 November 1999; in final form 27 December 1999 Abstract Quantum mechanical calculations on three potential energy surfaces for the prototype ion-molecule reaction He q H q HeH q q H have been performed in order to test the influence of their accuracies on reaction probabilities and cross 2 Ž . sections. The ab initio points of McLaughlin and Thompson 1979 fitted by two different functional forms, and a fit of a new set of ab initio points have been used. Dynamical results, in particular the rich resonance pattern, illustrate the dependence both on the nature of the potential energy surface, and on the type of functional form used to fit the same ab initio data. q 2000 Elsevier Science B.V. All rights reserved. 1. Introduction In recent times, there has been a considerable progress in the accuracy of potential energy surface calculations and quantum reaction dynamics studies, so that the comparison between theory and experi- ment has to be attempted, in order to establish benchmark examples. This is a challenge requiring both advanced quantum chemistry codes to deal with multielectronic problems and progress on quantum dynamics leading to converged reactive scattering calculations, at least for simple tri- and tetra-atomic systems. ) Corresponding author. Fax: 75 585 5 606; e-mail: [email protected] In this Letter, a dynamical test is performed on Ž . different potential energy surfaces PES in order to provide a contribution to the present state of the concept of ‘chemical accuracy’. Dynamical features may be very sensitive to even small variations in certain critical regions of the PES. wx Very recently, Kuppermann et al. 1 have shown that even for the ‘simple’ H q H system, a reeval- 2 uation of the whole set of the PES ab initio points proves necessary in order to reach a sufficiently accurate convergence towards results of experimen- tal relevance. The reasons for claiming such an accuracy focus mainly on the resonance features of the reaction probability profile. Therefore, it is ex- pected that in systems showing a prominent reso- nance pattern, the level of accuracy of the PES will 0009-2614r00r$ - see front matter q 2000 Elsevier Science B.V. All rights reserved. Ž . PII: S0009-2614 00 00067-1
Transcript of The He + H 2 + reaction: a dynamical test on potential energy surfaces for a system exhibiting a...

3 March 2000
Ž .Chemical Physics Letters 318 2000 619–628www.elsevier.nlrlocatercplett
The He q Hq reaction: a dynamical test on potential energy2
surfaces for a system exhibiting a pronounced resonance pattern
V. Aquilanti a,), G. Capecchi a, S. Cavalli a, D. De Fazio a, P. Palmieri b,C. Puzzarini b, A. Aguilar c, X. Gimenez c, J.M. Lucas c´
a Dipartimento di Chimica, UniÕersita di Perugia, 06123 Perugia, Italy`b Dipartimento di Chimica Fisica ed Inorganica, UniÕersita di Bologna, 40126 Bologna, Italy`
c Departament de Quimica Fisica, UniÕersitat de Barcelona, 08028 Barcelona, Spain
Received 21 November 1999; in final form 27 December 1999
Abstract
Quantum mechanical calculations on three potential energy surfaces for the prototype ion-molecule reaction He qHq™ HeHq q H have been performed in order to test the influence of their accuracies on reaction probabilities and cross2
Ž .sections. The ab initio points of McLaughlin and Thompson 1979 fitted by two different functional forms, and a fit of anew set of ab initio points have been used. Dynamical results, in particular the rich resonance pattern, illustrate thedependence both on the nature of the potential energy surface, and on the type of functional form used to fit the same abinitio data. q 2000 Elsevier Science B.V. All rights reserved.
1. Introduction
In recent times, there has been a considerableprogress in the accuracy of potential energy surfacecalculations and quantum reaction dynamics studies,so that the comparison between theory and experi-ment has to be attempted, in order to establishbenchmark examples. This is a challenge requiringboth advanced quantum chemistry codes to deal withmultielectronic problems and progress on quantumdynamics leading to converged reactive scatteringcalculations, at least for simple tri- and tetra-atomicsystems.
) Corresponding author. Fax: 75 585 5 606; e-mail:[email protected]
In this Letter, a dynamical test is performed onŽ .different potential energy surfaces PES in order to
provide a contribution to the present state of theconcept of ‘chemical accuracy’.
Dynamical features may be very sensitive to evensmall variations in certain critical regions of the PES.
w xVery recently, Kuppermann et al. 1 have shownthat even for the ‘simple’ H q H system, a reeval-2
uation of the whole set of the PES ab initio pointsproves necessary in order to reach a sufficientlyaccurate convergence towards results of experimen-tal relevance. The reasons for claiming such anaccuracy focus mainly on the resonance features ofthe reaction probability profile. Therefore, it is ex-pected that in systems showing a prominent reso-nance pattern, the level of accuracy of the PES will
0009-2614r00r$ - see front matter q 2000 Elsevier Science B.V. All rights reserved.Ž .PII: S0009-2614 00 00067-1

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628620
have a major decisive role in establishing a definitereactivity profile.
For this purpose, the prototype ion-moleculethree-electron system He q Hq has been considered2
Ž q.as a case study. It belongs to the same X q H 2
family of Ne q Hq system, characterized by a2
resonance pattern which has been recently investi-gated by means of exact quantum dynamical calcula-
w xtions. See Refs. 2,3 and references therein.This system, isoelectronic to H q H , has been2
well studied from both experimental and theoreticalpoints of view in the past decades. In particular,reactive integral cross sections as a function of colli-sion energy and of reactant vibrational excitationw x4–7 as well as state selected differential cross
w xsections 8,9 have been measured.Accurate ab initio calculations were performed for
this system in 1979 by McLaughlin and Thompsonw x10 , who computed a total of 596 ab initio points.These data were subsequently represented by twosuitable functional forms: the first developed by
w x Ž .Joseph and Sathyamurthy 11 in 1987 JS PES andw xthe second by Aguado and Paniagua 12 in 1992
Ž .AP PES . The availability of these PESs allowed thecorresponding dynamical tests, which were per-formed by means of classical methodologies, see
w xRef. 13 and references therein. However, full exactquantum mechanical calculations have been reported
w xonly for the JS PES by Kress et al. 14 and Lepetitw xand Launay 15 .
We have reproduced these results for the JS PES,and present a comparison with the results obtainedfor the AP PES, which is considered to be a moreflexible and accurate fit to the ab initio points. Usingthe Aguado–Paniagua parameterization, we haveproduced a new PES as a fit to our set of ab initiodata and performed quantum calculations in order togive a global account of the sensitivity of the differ-ent PES for the resonance pattern and other signifi-cant features of the reaction. Therefore, for the titlereaction, quantum reactive scattering calculations onthree different surfaces have been performed by us-
w xing the hyperquantization algorithm 16–18 , and, aswill be seen in the following sections, insight will beobtained on the issue of the accuracy required toyield reliable information on cross sections and rates.
The Letter is organized as follows: in the nextsection the potential energy surface fitted to a new
set of ab initio points is presented and comparedwith the previous two surfaces; the third section isfocused on the dynamical results, i.e. the reactionprobabilities for Js0 and the cross sections ob-tained within the adiabatic rotational approximationŽ .ARA approach; further remarks and conclusionsfollow in Section 4.
2. Three potential energy surfaces
First of all, we note the main features of thissystem, which involves a barrierless reaction path,with a 0.76 eV endoergicity. It proceeds through astable intermediate with a minimum of about 0.3 eV,corresponding to a linear configuration He–H–Hq,with both He–H and H–H internuclear distances ofabout 2 a .o
As previously mentioned, two sets of ab initiopoints have been employed as starting data, the first
w xone 10 had been fitted using two different func-w xtional forms 11,12 . Thus, globally, three different
PESs have been used in the present study. The firsttwo surfaces correspond to fits of the McLaughlin
w xand Thompson 10 ab initio points using two differ-ent interpolating functions, the Sorbie–Murrell ex-
Žpansion employed by Joseph and Sathyamurthy JS. w xPES 11 , and a more elaborate many-body expan-
Ž . w xsion by Aguado and Paniagua AP PES 12 . TheŽthird surface studied in this work Modified AP PES,
.available from the authors on request has beenproduced by computing a new grid of ab initio pointsŽ .see below and subsequently fitting them to theAguado–Paniagua functional form, which has beenfound to give a better description of the PES. In fact,the JS PES presents spurious wells, which are due tothe damping functions chosen and seem to be notentirely eliminable. We have established that thesedamping factors introduce spurious extrema andmove the repulsive wall towards larger internucleardistances, possibly producing unphysical effects on
Ž .the dynamics see Fig. 1 .Ž .The regions of the PES investigated see Table 1
for the subsequent dynamical study are the follow-ing: the region around the linear triatomic complexes
q ŽHeHH , which is the stable intermediate of the. qreaction , and HHeH ; the collinear dissociative
asymptotes of HeHHq, which correspond to the
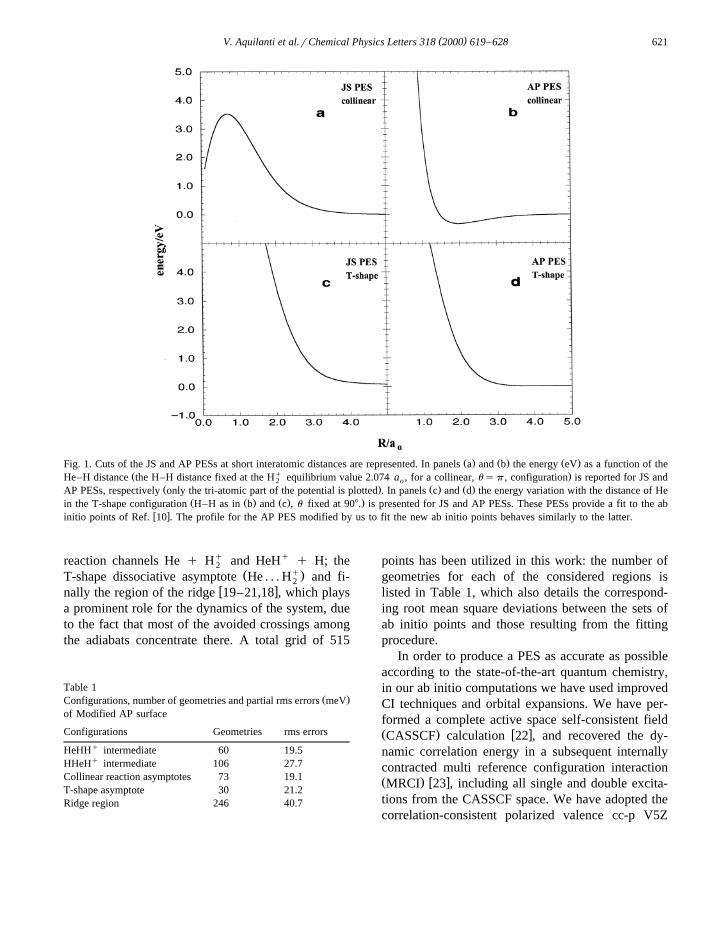
( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628 621
Ž . Ž . Ž .Fig. 1. Cuts of the JS and AP PESs at short interatomic distances are represented. In panels a and b the energy eV as a function of theŽ q .He–H distance the H–H distance fixed at the H equilibrium value 2.074 a , for a collinear, usp , configuration is reported for JS and2 o
Ž . Ž . Ž .AP PESs, respectively only the tri-atomic part of the potential is plotted . In panels c and d the energy variation with the distance of HeŽ Ž . Ž . .in the T-shape configuration H–H as in b and c , u fixed at 908. is presented for JS and AP PESs. These PESs provide a fit to the ab
w xinitio points of Ref. 10 . The profile for the AP PES modified by us to fit the new ab initio points behaves similarly to the latter.
reaction channels He q Hq and HeHq q H; the2Ž q.T-shape dissociative asymptote He . . . H and fi-2
w xnally the region of the ridge 19–21,18 , which playsa prominent role for the dynamics of the system, dueto the fact that most of the avoided crossings amongthe adiabats concentrate there. A total grid of 515
Table 1Ž .Configurations, number of geometries and partial rms errors meV
of Modified AP surface
Configurations Geometries rms errorsqHeHH intermediate 60 19.5qHHeH intermediate 106 27.7
Collinear reaction asymptotes 73 19.1T-shape asymptote 30 21.2Ridge region 246 40.7
points has been utilized in this work: the number ofgeometries for each of the considered regions islisted in Table 1, which also details the correspond-ing root mean square deviations between the sets ofab initio points and those resulting from the fittingprocedure.
In order to produce a PES as accurate as possibleaccording to the state-of-the-art quantum chemistry,in our ab initio computations we have used improvedCI techniques and orbital expansions. We have per-formed a complete active space self-consistent fieldŽ . w xCASSCF calculation 22 , and recovered the dy-namic correlation energy in a subsequent internallycontracted multi reference configuration interactionŽ . w xMRCI 23 , including all single and double excita-tions from the CASSCF space. We have adopted thecorrelation-consistent polarized valence cc-p V5Z

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628622
Fig. 2. Plots of the potential energy surface obtained with the new set of ab initio points. In the four panels, equipotential cuts are shown fordifferent values of the angle between the He–H and H–H distances. The solid curves are contours of the PES corresponding to y6, y3, 0,
w x6, 12, 18, 30, 42 and 54 kcalrmol. This figure is the counterpart of Fig. 2 in Ref. 12 . To facilitate this comparison, energies here are givenŽ .in kcalrmole 1 kcalrmole s 0.0433477 eV and distances in bohr.
w x Ž .basis set of Dunning 24 : 8s4p3d2f1g contracted toŽ .5s4p3d2f1g . Throughout the present work all abinitio calculations have been carried out using theMOLPRO suite of programs 1. Our energies for reac-tants, linear intermediate and products, y3.505776,y3.518181, y3.478190 hartree, respectively, are tobe compared with the energies y3.501484,
w xy3.512920, y3.473703 hartree of Ref. 10 .As mentioned, we have decided to interpolate our
515 energy values using the Aguado–Paniagua pa-Ž w x.rameterization for a detailed account, see Ref. 12 ,
Ž .employing the same restriction (6 on their param-eter M and the same two-body terms. Some contourplots of the surface obtained are shown in Fig. 2, to
w xbe compared with the analogous Fig. 2 of Ref. 12 .Ž .The root mean square deviations rmsd and the
1 MOLPRO is a package of ab initio programs written by H.-J.Werner and P.J. Knowles, with contributions of J. Almlof, R.D.¨Amos, M.J.O. Deegan, S.T. Ebert, C. Hampel, W. Meyer, K.Peterson, R.M. Pitzer, A.J. Stone, P.R. Taylor
Ž .maximum error D of the Modified AP surfacemax
with respect to the new ab initio data and those forw xJS and AP PESs with respect to the data of Ref. 10
are reported in Table 2. We can see that the originaland the Modified AP PESs have comparable rmsds.This means that we are going to compare PESs ofvery similar quality concerning the accuracy in thefitting of two sets of ab initio points. In this work weare considering new ab initio data in the region ofthe ridge separating the reaction channels, whereeffects on the dynamics are expected to be amplified.For the comparison, the original AP parameterization
Table 2Ž .Total rms errors and maximum error D of JS, AP andmax
Modified AP PESs. All data are in meV
rms errors Dmax
JS PES 69.8 ) 150.0AP PES 30.81 184.2Modified AP PES 32.5 98.8

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628 623
has been kept. The original JP and AP PES show ofcourse a higher rmsd with respect to the new ab
Ž .initio data 108.9 and 52.6 meV, respectively .As a measure of the absolute error of our ener-
gies, we compare the asymptotic MRCI energy for Hq Hq q He, y3.40316 hartree, to the exact adia-
w xbatic potential energy of y3.40372 hartree 25 ,Ž .which is lower by 0.00056 hartree 15.2 meV . The
next most important correction, the diagonal nonadiabatic energy term, is of the same order of magni-
Ž . w xtude 0.0003 hartree 26 ; by comparison, the rela-tivistic energy terms are one order of magnitude
Ž . w xsmaller 0.00006 hartree 27 . Since, as discussed inw xdetail in Ref. 26 , these corrections are expected to
modify only slightly relative energies, the accuracyof the MRCI potential energy values is considerablyhigher: for instance, the diatomic dissociation ener-gies are estimated 2.042 and 2.790 eV for HeHq andHq against the experimental values of 2.044 and2
w x2.794 eV 28 , respectively; the vibrational frequen-cies of the diatoms, n s 2913.3 cmy1 for HeHq
and v s 2323.6 cmy1 for Hq, are in very goode 2
agreement with the corresponding experimental val-y1 w x y1 w xues of 2910.96 cm 29 and 2321.7 cm 28 ,
respectively.A full characterization of the new surface, based
on many more ab initio points and more accuratefitting procedures, including an assessment of accu-racies at its critical points, will be presented in thefuture along with with dynamical calculations aimedat realistically simulating experimental results.
3. Quantum dynamics: calculations and results
qŽ .The reactivity for the He q H Õ, js0 ™2qŽ X X.HeH Õ , j q H process, on the three PESs, has
been characterized as follows. Reaction probabilitiesfor total angular momentum Js0, within the 0.95–1.15 eV total energy range, have been computed bymeans of a quantum reactive scattering code basedon the hyperquantization algorithm, previously de-
w xveloped by some of the authors 16–18 . In order totest the reliability of our code, the state-to-statereaction probabilities obtained for the JS surface,have been compared with the results published in
w xRef. 15 . This comparison of the dynamics on thethree surfaces suggests that their differences are ex-
pected to be relevant for differential cross sections.In order to check the sensitivity of the integral crosssections, we use the so called ARA approximationw x32,31 being less demanding in CPU time than exactones and, nevertheless, accurate enough for thesemore averaged quantities.
The hyperquantization algorithm is based on ahyperspherical harmonics expansion of the solutionof the Schrodinger equation at fixed values of the¨hyperradius, the natural variable for the reactivescattering directly related to the total inertia of thetriatomic system. A finite orthonormal basis set ofhyperspherical harmonics in symmetric parameteri-zation of the hypersphere has been used to getconvergent eigenstates parametrically in the hyperra-dial variable. Hahn polynomials, corresponding to a
w xgeneralization of vector coupling coefficients 30 ,are used as discrete analogs of the harmonics so thatan efficient representation of the Hamiltonian matrixis obtained, diagonal in the potential and universaland sparse in the kinetic energy. Two different se-
Žquences of diagonalization schemes compare withw x.the diagonalization truncation in Ref. 18 are em-
ployed to get hyperspherical eigenstates very effi-ciently.
The hyperquantization algorithm leads to a set ofeigenvalues and eigenvectors for the ‘hyperangular’
Ž .problem. The eigenvalues adiabats plotted againstthe hyperradius give a first picture of the mainfeatures characterizing the reactive process. This is asignificant step forward with respect to the fixed-an-gle contour plots in Fig. 2. In particular, it is espe-cially interesting to the purpose of the present workthe comparison between the adiabats obtained foreach of the three PESs. These adiabatic curves areshown in Fig. 3; these plots give a representation ofthe differences among the surfaces, since they con-tain dynamical information concerning the full di-mensionality of the problem. In addition, the compu-tation of the rovibrational manifold incorporates intothese graphs some dynamical information. As a con-sequence of the differences between the PESs, theglobal manifold of avoided crossings between adia-bats changes from one surface to the other. In addi-
Žtion, the adiabatic curves relative to the JS PES Fig..3a tend to be much more ‘long-ranged’ than the
others. Very few differences can be noticed betweenFig. 3b and Fig. 3c both relative to the same

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628624
Fig. 3. Hyperspherical adiabatic curves for the three cases, showing also ridge and valley-bottom lines. The continuous curves correspond toŽ . Ž . Ž .the first 30 eigenvalues calculated at fixed r in the range 2.5–11.0 a . a : JS PES; b : AP PES; c Modified AP PES. The valley bottomo
curves for the entrance and exit channels are represented as dashed curves, while the ridge, which marks the separation between them, isshown as a dotted curve. The zero of the energy scale is taken at the asymptotic minimum of the reactants’ channel.
Aguado–Paniagua fitting on different ab initio points.Finally, to get the radial solution for each rovibra-tional channel, the propagation of the interactionmatrix has been performed by means of the Johnson-Manolopoulos technique. The number of the chan-nels needed in the propagation step, the number ofthe sectors, the maximum and minimum values of
w xthe hyperradius used are the same as in Ref. 15 .Reaction probabilities have been calculated for
specific entrance and exit channels. For the JS sur-face, state-to-state results agree with those of Ref.w x15 , thus validating the numerical consistency of thedynamical calculations.
The total reaction probabilities for js0 entrancechannel summed over all the open exit channels areshown in Fig. 4 for all three surfaces. It can beclearly seen that the reaction probability profiles as afunction of total energy are very different whenconsidering both the two previously proposed fits ofthe same set ab initio data, and the same fittingprocedure applied to two alternative sets of points. Inparticular, differences show up mainly as large varia-tions in position and shape of the resonance peaks, as
can be seen by a simple eye inspection. These differ-ences can produce pronounced variations in the dif-ferential cross section dependence on the collisionenergy.
Ž .As can be seen, reaction probabilities Fig. 4 areextremely sensitive to small variations of the PES,
Ževen when cuts of the surface see our Fig. 2 andw x. Ž .Fig. 2 in Ref. 12 and adiabatic curves Fig. 3 are
very alike. Therefore, reaction probabilities for Js0appear to represent a significant test for the accuracyof PESs. In particular, from Fig. 4 we can concludethat the accuracy required for a PES for a presentreaction to get convergent results for the resonantstructure has to be of the order of the widths of the
Ž .resonances ;10 meV . This is very demanding forpresent day quantum chemistry. However, the exper-imental detection of such resonance patterns, throughhigh angular and energy resolution differential crosssections, appears to be within reach of future devel-opments.
Fig. 5 shows the reaction cross sections whichhave been computed for this reaction using the ARA
w xapproach 31 , as implemented by De Fazio and

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628 625
Ž . qŽ . q Ž X . qŽ .Fig. 4. Reaction probabilities Js0 for the reaction He q H js0 ™ HeH Õ s0 q H summed over all open initial H Õ and2 2qŽ X .all open final HeH j states. From top to bottom: Modified AP PES, AP PES, and JS PES. Total energies as in Fig. 3.
w xCastillo 32 . In short, this approximation, neglectingthe Coriolis coupling in the hyperspherical formula-tion of the scattering problem and treating adiabati-cally the asymmetric top terms of the Hamiltonian,decouples the problem for each projection of thetotal angular momentum on the minimum inertiaprincipal axis. In this way, we obtain an importantCPU and memory saving when we compute thewhole set of total angular momentum partial waves,necessary for cross section convergence. The approx-imation allows a much finer grid of points than the
w xprevious exact calculations 15,33 . The comparisonw xwith published results 33 shows agreement in the
over-all behavior, the largest deviations occurring forthe Õs3 entrance channel but contained within;10%. The resonance pattern is qualitatively simi-lar: although their positions do not match, averagewidths are comparable. In particular amplitudes are
larger in the ARA approach as also found for otherw xreactive systems 32 . By comparing the three inves-
tigated PESs, the overall size of cross sections andtheir dependence on energy and on the vibrationalquantum number of Hq, show differences which are,2
of course, less evident than for the reaction probabili-ties of Fig. 4, but perhaps of easier experimentaldetectability. The requirement is again an energyresolution of ;10 meV. On the other hand, if this isachieved, great insight will be obtained on the de-tailed features of the interaction potentials from anexperimental assessment of the resonance pattern.
Probably, differences among results for the threePESs are in general not large enough to yield signifi-cantly different rate constants, which can be calcu-lated by Maxwell–Boltzmann averaging over thecurves of the type shown in Fig. 5. However, atthermal energy, kT is much larger than the ‘accuracy

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628626
qŽ . qŽ X .Fig. 5. Cross sections in the ARA approach for the reaction He q H Õ, js0 ™ HeH Õ s0 q H summed over all open final2qŽ X .HeH j states. Continuous curves: JS PES. Dotted curves: AP PES. Dashed curves: Modified AP PES. Total energies as in Fig. 3.
limit’ 10 meV, dictated by the requirements thatresonances do not overlap. This is no longer true inastrophysical applications, where this reaction may
play a role and where involved temperatures of theinterstellar medium are in the range 10–50 K and kTvalues are of the order of a few meV.

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628 627
4. Conclusions and perspective remarks
In this work we have studied the sensitivity ofquantum dynamical calculations to small variationsof the potential energy surface for a system exhibit-ing a pronounced resonance pattern. As a benchmarksystem the He q Hq
™ HeHq q H reaction has2
been considered. For the PES of this reaction twodifferent fittings of the same ab initio data withsimilar accuracy were available. We have providednew ab initio data and a fitting of comparable accu-
Ž .racy Table 1 . Fully convergent quantum mechani-cal calculations had been reported previously for oneof these PESs. We have carried out a comparison forthe quantum dynamics on the three surfaces.
The new ab initio points have been calculatedwith improved CI techniques and orbital expansions.The grid has been chosen particularly dense in thecritical points of the reaction and of the intermediatecomplex. The energies obtained have been fittedwith the same functional form of the AP surface at alevel of accuracy similar to the original one.
To obtain Js0 probabilities we have used thehyperquantization algorithm, which appears to be acompetitive technique for exact quantum calculationson few body problems. The calculation of integralreaction cross sections has been performed exploit-ing the ARA approximation in order to reduce thecomputational effort. Check of the accuracy of ourdynamical calculations have been carried out repro-
w xducing the results of Ref. 15 on the JS surface.From these comparisons we can see that the rich
resonance pattern resulting from the Js0 calcula-tions is very sensitive to both the accuracy of the abinitio points and of the quality of the numericalfitting procedure. This suggests that for this type ofsystems the surfaces and their fittings must ensureaccuracies of the order of ;10 meV to produce areliable resonance pattern. Integral cross sections andrate constants are less sensitive to the uncertaintiesof the PES. However, the resonance pattern in theenergy dependence of cross sections can be impor-tant under astrophysical conditions.
To produce state-of-the-art PES and quantum dy-namics, more ab initio points are being generatedand the problem of the full fitting will be faced inthe light of the accuracy requirements emerging frompresent results. An adequately accurate surface should
incorporate also the correct long-range behavior, forwhich the multipole polarization expansion is avail-able. In this direction, a new surface was calculatedand fitted paying special care to the inelastic channel
q w x w xHe q H 34 . Hutson et al. 35 also studied the2
dynamics for inelastic processes, emphasizing theneed of properly matching the surfaces to the long-range behavior in the entrance channel for the reac-tion. The sensitivity of the reaction dynamics to thisregion of the potential will have to be scrutinized infuture work, where the new surface will be used forextensive dynamical studies.
In conclusion, we expect that accurate differentialcross sections or very low energy measurements,sensitive to the resonance pattern with sufficientresolution, would be a stringent test of the accuracyof the potential energy surfaces for a system likethis, for which our work demonstrates that exactquantum dynamics can be carried out very effi-ciently.
In this work, focused on the comparison amongsurfaces and on the role of accuracy for dynamics, alimited energy range was considered and we onlyincluded the lowest rotational initial states for Hq.2
Extensive investigations leading to simulation of theexperiments will require extensions of the energyrange, inclusion of the proper rotational distributionof the reactant and the full treatment of non-zeroangular momentum collisions.
Acknowledgements
The support of the following is acknowledged: theŽ .Italian National Research Council CNR , the Minis-
tero dell’Universita e della Ricerca Scientifica e`Ž .Tecnologica MURST , the European Union within
the TMR Network ‘Potential Energy Surfaces forŽMolecular Spectroscopy and Dynamics’ Contract
.No. ERB-FMRX-CT96-0088 , and the DGICYTProject PB97-0919 of the MEC of Spain. The Centre
Ž .de Supercomputacio de Catalunya CESCA is ac-´knowledged for supplying the CPU time used in thedynamical calculations. D.D.F. is grateful to E.U.
Žand CESCA for the award of a TMR grant Contract.No. ERB-FMGE-CT95-0062 . J.M. Launay is also
acknowledged for supplying some computer codes.

( )V. Aquilanti et al.rChemical Physics Letters 318 2000 619–628628
References
w x1 Y.S.M. Wu, A. Kuppermann, J.B. Anderson, Phys. Chem.Ž .Chem. Phys. 1 1999 929.
w x2 J.D. Kress, R.B. Walker, E.F. Hayes, P. Pendergast, J. Chem.Ž .Phys. 100 1994 2728.
w x3 F. Huarte-Larranaga, X. Gimenez, J.M. Lucas, A. Aguilar,˜ ´Ž .J.M. Launay, Phys. Chem. Chem. Phys. 1 1999 1125.
w x Ž .4 W.A. Chupka, M.E. Russel, J. Chem. Phys. 49 1968 5426.w x Ž .5 T. Turner, O. Dutuit, Y.T. Lee, J. Chem. Phys. 81 1984
3475.w x6 M. Baer, S. Suzuki, K. Tanaka, H. Nakamura, Z. Herman,
Ž .D.J. Kouri, Phys. Rev. A 34 1986 1748.w x Ž .7 T.R. Govers, P.M. Guyon, Chem. Phys. 113 1987 425.w x8 Z. Herman, I. Koyano, J. Chem. Soc. Faraday Trans. 83
Ž .1987 127.w x9 J.E. Pollard, L.K. Johnson, R.B. Cohen, J. Chem. Phys. 95
Ž .1991 4894.w x Ž .10 D.R. McLaughlin, D.L. Thompson, J. Chem. Phys. 70 1979
2748.w x Ž .11 T. Joseph, N. Sathyamurthy, J. Chem. Phys. 86 1987 704.w x Ž .12 A. Aguado, M. Paniagua, J. Chem. Phys. 96 1992 1265.w x13 S. Kumar, H. Kapoor, N. Sathyamurthy, Chem. Phys. Lett.
Ž .289 1998 361.w x14 J.D. Kress, R.B. Walker, E.F. Hayes, J. Chem. Phys. 93
Ž .1990 8085.w x Ž .15 B. Lepetit, J.M. Launay, J. Chem. Phys. 95 1991 5159.w x16 V. Aquilanti, S. Cavalli, D. De Fazio, J. Chem. Phys. 109
Ž .1998 3792.
w x17 V. Aquilanti, S. Cavalli, D. De Fazio, A. Volpi, A. Aguilar,Ž .X. Gimenez, J.M. Lucas, J. Chem. Phys. 109 1998 3805.´
w x18 A. Aquilanti, S. Cavalli, D. De Fazio, A. Volpi, A. Aguilar,Ž .X. Gimenez, J.M. Lucas, Phys. Chem. Chem. Phys. 1 1999´
1091.w x19 V. Aquilanti, S. Cavalli, G. Grossi, Chem. Phys. Lett. 110
Ž .1984 43.w x Ž .20 V. Aquilanti, S. Cavalli, Chem. Phys. Lett. 141 1987 309.w x21 V. Aquilanti, S. Cavalli, G. Grossi, V. Pellizzari, M. Rosi, A.
Ž .Sgamellotti, F. Tarantelli, Chem. Phys. Lett. 162 1989 179.w x Ž .22 H.-J. Werner, P.J. Knowles, J. Chem. Phys. 82 1985 5053.w x Ž .23 H.-J. Werner, P.J. Knowles, J. Chem. Phys. 89 1988 5803.w x Ž .24 T.H. Dunning Jr., J. Chem. Phys. 90 1989 1007.w x Ž . Ž .25 C.L. Pekeris, Phys. Rev. 112 1958 1649; 115 1959 1216.w x26 A. Mitrushenkov, P. Palmieri, C. Puzzarini, R. Tarroni, Mol.
Phys, in press, 2000.w x27 L.C. Allen, E. Clementi, H.M. Gladney, Rev. Mod. Phys. 35
Ž .1963 465.w x28 K.P. Huber, G. Herzberg, Constants of Diatomic Molecules,
van Nostrand-Reinhold, New York, 1979.w x Ž .29 Z. Liu, P.B. Davies, J. Chem. Phys. 107 1997 337.w x30 V. Aquilanti, S. Cavalli, D. De Fazio, J. Phys. Chem. 99
Ž .1995 15964.w x Ž .31 J.M. Bowman, Chem. Phys. Lett. 217 1994 36.w x Ž .32 D. De Fazio, J. Castillo, Phys. Chem. Chem. Phys. 1 1999
1165.w x33 J.M. Launay, M. Le Dourneuf, ICPEAC XVII, Brisbane,
July 1991, Section 15, p. 549.w x Ž .34 M.F. Falcetta, P.E. Siska, Mol. Phys. 97 1999 117.w x Ž .35 M. Meuwly, J.M. Hutson, J. Chem. Phys. 110 1999 3418.







![A C-shaped p-sulfonatocalix[4]arene-based supermolecule exhibiting mutual-inclusion and bilayer insertion of dipicolinate](https://static.fdokumen.com/doc/165x107/632401634d8439cb620d2dad/a-c-shaped-p-sulfonatocalix4arene-based-supermolecule-exhibiting-mutual-inclusion-1677848454.jpg)


![Pyrido[1,2- a ]pyrimidin-4-one Derivatives as a Novel Class of Selective Aldose Reductase Inhibitors Exhibiting Antioxidant Activity](https://static.fdokumen.com/doc/165x107/633af889e2bc852e6509f4ed/pyrido12-a-pyrimidin-4-one-derivatives-as-a-novel-class-of-selective-aldose.jpg)









