
Analysis of the genetic basis of porcine meat quality and coat ...
Upload
independentCategory
view
0download
0
2386 East Heritage Way, Suite B, Salt Lake City, Utah 84109 USA Phone +1-877-628-7300 • Email—[email protected]
www.PC-Project.org
15 March 2005
Use of Articles in the Pachyonychia Congenita Bibliography
The articles in the PC Bibliography may be restricted by copyright laws. These have been made available to you by PC Project for the exclusive use in teaching, scholar-ship or research regarding Pachyonychia Congenita. To the best of our understanding, in supplying this material to you we have followed the guidelines of Sec 107 regarding fair use of copyright materials. That section reads as follows:
Sec. 107. - Limitations on exclusive rights: Fair use Notwithstanding the provisions of sections 106 and 106A, the fair use of a copyrighted work, including such use by reproduction in copies or phonorecords or by any other means specified by that section, for purposes such as criticism, comment, news reporting, teaching (including multiple copies for classroom use), scholarship, or research, is not an infringement of copyright. In determining whether the use made of a work in any particular case is a fair use the factors to be considered shall include - (1) the purpose and character of the use, including whether such use is of a commercial nature or is for nonprofit educational purposes; (2) the nature of the copyrighted work; (3) the amount and substantiality of the portion used in relation to the copyrighted work as a whole; and (4) the effect of the use upon the potential market for or value of the copyrighted work. The fact that a work is unpublished shall not itself bar a finding of fair use if such finding is made upon consideration of all the above factors.
We hope that making available the relevant information on Pachyonychia Congenita will be a means of furthering research to find effective therapies and a cure for PC.

The Genetic Basis of Pachyonychia Congenita
Frances J. D. Smith,� Haihui Liao,� Andrew J. Cassidy,� Arlene Stewart,� Kevin J. Hamill,� Pamela Wood,�
Iris Joval,� Maurice A. M. van Steensel,w Erik Bjorck,z Faith Callif-Daley,y Gerald Pals,z Paul Collins,#Sancy A. Leachman,�� Colin S. Munro,ww and W. H. Irwin McLean��Epithelial Genetics Group, Human Genetics Unit, Ninewells Hospital and Medical School, University of Dundee, Dundee DD1 9SY, UK; wDepartment ofDermatology, University Hospital Maastricht, Maastricht, The Netherlands; zDepartment of Molecular Medicine, Clinical Genetics Unit, Karolinska Hospital,Stockholm, Sweden; yDepartment of Medical Genetics, One Children’s Plaza, Dayton, Ohio, USA; zDepartment of Clinical and Human Genetics, UniversityHospital, Vrije Universiteit, Amsterdam, The Netherlands; #Department of Dermatology, St. Vincent’s University Hospital, Dublin, Ireland; ��Department ofDermatology, University of Utah, Salt Lake City, Utah, USA; wwDepartment of Dermatology, Southern General Hospital, Glasgow, UK
In 1994, the molecular basis of pachyonychia congenita (PC) was elucidated. Four keratin genes are associated
with the major subtypes of PC: K6a or K16 defects cause PC-1; and mutations in K6b or K17 cause PC-2. Mutations
in keratins, the epithelial-specific intermediate filament proteins, result in aberrant cytoskeletal networks which
present clinically as a variety of epithelial fragility phenotypes. To date, mutations in 20 keratin genes are asso-
ciated with human disorders. Here, we review the genetic basis of PC and report 30 new PC mutations. Of these, 25
mutations were found in PC-1 families and five mutations were identified in PC-2 kindreds. All mutations identified
were heterozygous amino acid substitutions or small in-frame deletion mutations with the exception of an unusual
mutation in a sporadic case of PC-1. The latter carried a 117 bp duplication resulting in a 39 amino acid insertion in
the 2B domain of K6a. Also of note was mutation L388P in K17, which is the first genetic defect identified in the
helix termination motif of this protein. Understanding the genetic basis of these disorders allows better counseling
for patients and paves the way for therapy development.
Key words: genodermatosis/keratin intermediate filaments/keratoderma/mutation/nail dystrophyJ Investig Dermatol Symp Proc 10:21 –30, 2005
Keratins Form a Stress-BearingCytoskeleton in Epithelia
Within all eukaryotic cells, there are three cytoskeletal net-works consisting of microfilaments, microtubules, and in-termediate filaments, each of which performs specializedfunctions (Alberts et al, 1989; Morley and Lane, 1994). Mi-crofilaments and microtubules are involved in a variety offunctions including cell division, motility, contraction, differ-entiation, and cell death. Intermediate filament proteins areencoded by a large family of at least 65 genes that can besubdivided into six main classes, of which keratins make uptypes I and II (Quinlan et al, 1994; Fuchs and Cleveland,1998; Hesse et al, 2001, 2004). Before the early 1990s, thefunction of intermediate filaments was largely unknown.Then a number of independent studies focusing on keratinfunction by a variety of methods including in vitro studies,transgenic animals, and human molecular genetics con-cluded that keratins were predominantly structural proteinsgiving cells structural resilience to resist physical damage(McLean and Lane, 1995; Fuchs and Cleveland, 1998; Irvineand McLean, 1999; Porter and Lane, 2003; Omary et al,2004). More recent studies support this but also show thatthese networks are in fact highly dynamic structures(Werner et al, 2003; Windoffer et al, 2004). Keratin genesare clustered to two chromosomal loci; the type I keratins
(K9–K20) on chromosome 17q12–q21 and type II keratins(K1–K8) are located in a gene cluster on chromosome12q11q–14. K18 is the exception as it maps to the type IIgene cluster. In terms of nomenclature, the official genesymbols for keratins are KRT1 for the gene encoding keratinK1 etc, however, in the keratin literature, these are com-monly referred to as the K1 gene etc. With analysis ofrecent data from the human genome project it is now pre-dicted that humans possess at least 54 functional keratingenes (Hesse et al, 2004). In light of the genome data, arevised gene/protein nomenclature system has been sug-gested (Hesse et al, 2004) but this has not yet been adoptedby the field. Importantly, keratins are expressed in pairsconsisting of a type I and type II keratin in a tissue- anddifferentiation-specific manner (Table I). In other words,each of the many different epithelial cells and tissues in thehuman body express particular combinations of keratins(Lane, 1993).
All keratins share the same basic protein structure con-sisting of a central a-helical rod domain of 310 amino acidsflanked by non-helical head (V1) and tail (V2) domains(Quinlan et al, 1994; Smith et al, 2002), as shown in Fig 1a.The rod domain is divided into four a-helical domains, the1A, 1B, 2A, and 2B, which are made up of heptad repeatunits that mediate dimer formation via interaction of recur-rent hydrophobic residues. The helical segments are con-nected together by three non-helical linkers, L1, L12, andL2, which are predicted to provide flexibility to the rod do-main. Type II keratins have two additional subdomains, H1Abbreviation: PC, pachyonychia congenita
Copyright r 2005 by The Society for Investigative Dermatology, Inc.
21
and H2 located between the rod and tail domains. The H1subdomain, specific to type II keratins, is essential for po-lymerization. A ‘‘stutter’’ sequence exists in the centre of the2B domain, where the helix polarity is reversed. Two shortsequences present at either end of the rod domain, the helixinitiation motif and the helix termination motif, are highlyconserved throughout all intermediate filaments. These he-lix-boundary motifs are thought to be critical in mediatingend-to-end overlap interactions during filament assembly(Steinert et al, 1993b).
For each type of keratin, the genomic organization andposition of introns within the coding sequence is the same(Quinlan et al, 1994). Type I keratins have eight exons andtype II keratins have nine exons. Multiple pseudogenes existfor many keratins. Many of these are processed, intron-lesspseuodogenes, notably those related to the simple epithe-lial keratin genes K8 and K18, for which there are nearly 150pseudogenes in the human genome (Hesse et al, 2001).Other keratins have conventional intron-containing pseu-dogenes, in particular the keratins involved in PC, namelyK6, K16, and K17 (Troyanovsky et al, 1992; Takahashi et al,1995; Smith et al, 1999b). The presence of pseudogeneswhose sequences are highly homologous to the functional
gene, makes genetic analysis difficult using genomic DNA,as discussed below.
The keratin cytoskeleton forms a network of filamentsextending from the periphery of the nucleus to the plasmamembrane in all epithelial cells (Fig 1b). The assembly ofthis network involves several steps and the precise spatialarrangement of the protein subunits at some stages is stillcontroversial but should become clearer now that the crys-tal structure for some parts of certain intermediate filamentproteins are emerging (Herrmann et al, 2000; Strelkov et al,2001; Herrmann and Foisner, 2003). The first step in keratinassembly is the formation of obligate heterodimers by par-allel alignment of a type I and type II keratin. Dimers align toform tetramers that then polymerize laterally and longitudi-nally to form 2–3 nm protofilaments. The precise way thatthe dimers align is still uncertain; in vitro cross-linking ex-periments suggest that there could be three possible ways(Steinert et al, 1993a). During the next stage of assemblytwo protofilaments align to form a 4.5 nm protofibril. Theseprotofibrils inter-twine to form a complete 10 nm keratinfilament consisting of 32 monomer chains in cross section.Finally, keratin filaments assemble into structurally resilientcytoskeletal networks.
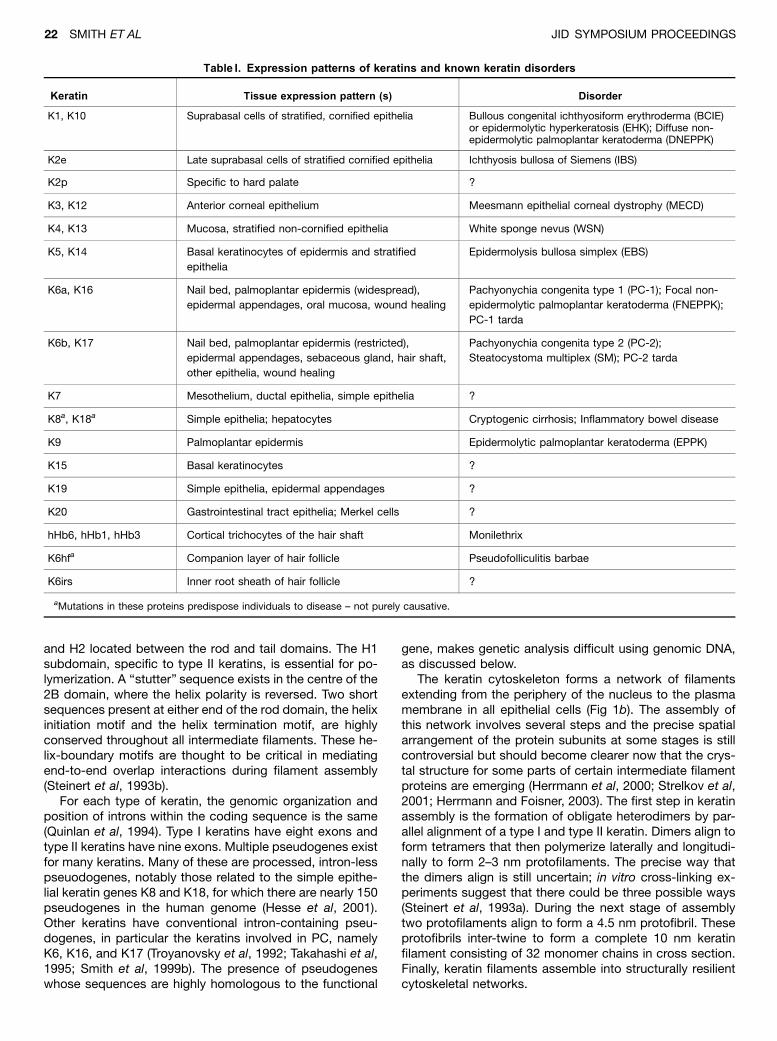
Table I. Expression patterns of keratins and known keratin disorders
Keratin Tissue expression pattern (s) Disorder
K1, K10 Suprabasal cells of stratified, cornified epithelia Bullous congenital ichthyosiform erythroderma (BCIE)or epidermolytic hyperkeratosis (EHK); Diffuse non-epidermolytic palmoplantar keratoderma (DNEPPK)
K2e Late suprabasal cells of stratified cornified epithelia Ichthyosis bullosa of Siemens (IBS)
K2p Specific to hard palate ?
K3, K12 Anterior corneal epithelium Meesmann epithelial corneal dystrophy (MECD)
K4, K13 Mucosa, stratified non-cornified epithelia White sponge nevus (WSN)
K5, K14 Basal keratinocytes of epidermis and stratified
epithelia
Epidermolysis bullosa simplex (EBS)
K6a, K16 Nail bed, palmoplantar epidermis (widespread),
epidermal appendages, oral mucosa, wound healing
Pachyonychia congenita type 1 (PC-1); Focal non-
epidermolytic palmoplantar keratoderma (FNEPPK);
PC-1 tarda
K6b, K17 Nail bed, palmoplantar epidermis (restricted),
epidermal appendages, sebaceous gland, hair shaft,
other epithelia, wound healing
Pachyonychia congenita type 2 (PC-2);
Steatocystoma multiplex (SM); PC-2 tarda
K7 Mesothelium, ductal epithelia, simple epithelia ?
K8a, K18a Simple epithelia; hepatocytes Cryptogenic cirrhosis; Inflammatory bowel disease
K9 Palmoplantar epidermis Epidermolytic palmoplantar keratoderma (EPPK)
K15 Basal keratinocytes ?
K19 Simple epithelia, epidermal appendages ?
K20 Gastrointestinal tract epithelia; Merkel cells ?
hHb6, hHb1, hHb3 Cortical trichocytes of the hair shaft Monilethrix
K6hfa Companion layer of hair follicle Pseudofolliculitis barbae
K6irs Inner root sheath of hair follicle ?
aMutations in these proteins predispose individuals to disease – not purely causative.
22 SMITH ET AL JID SYMPOSIUM PROCEEDINGS

Pachyonychia congenita (PC) is caused by mutations inone of four keratin genes The discovery of PC as a keratindisorder was an extension of earlier studies on epidermo-lysis bullosa simplex (EBS), the first human keratin disorderto be identified (discussed in McLean et al, this issue). Manyother keratin disorders have been identified resulting, todate, in 20 human keratin genes associated with singlegene disorders or predisposing factors in complex traits(Table I). The majority of dominant keratin mutations areclustered to the helix boundary motifs of these proteins.
PC is a group of inherited disorders of the epidermis andits appendages which can be divided into two main clinicalsubtypes, PC-1 (OMIM 167200) and PC-2 (OMIM 167210;Munro, 2001), with other rare variants, including PC-tarda(Paller et al, 1991). The clinical details of these disorders aredescribed in detail by Leachman et al (this issue). The firstclue that PC was a keratin disorder came from geneticlinkage analysis. A study of a large Scottish pedigree pre-senting with a typical PC-2 phenotype showed co-segre-gation of the disease with polymorphic markers within thetype I keratin gene cluster at 17q12–q21, resulting in sta-tistically significant genetic linkage (Munro et al, 1994). Mu-tation analysis in this PC-2 family revealed a heterozygousmissense mutation in the K17 gene, which was detected inall affected members and was absent from all unaffectedfamily members and 50 unrelated controls (McLean et al,1995). Simultaneously, a missense mutation was detectedin a sporadic case of PC-1, this time in the K16 gene(McLean et al, 1995). Another group identified mutations inthe K6a gene in PC-1 families (Bowden et al, 1995; Lin et al,
1999). Later, a mutation was detected in the K6b gene in anextended family with the PC-2 phenotype, which revealedthat this keratin is the expression partner of K17 (Smith et al,1998). Thus, it was established that mutations in K6aor in K16 cause PC-1, whereas mutations in K6b or K17cause PC-2. Subsequently, further genetic testing has re-sulted in the identification of mutations in these keratingenes involving 450 PC families worldwide (http://www.interfil.org).
Methods for diagnosis and problems in PC molecularanalysis By clinical examination, it is usually possible todetermine the type of PC. Subsequently, molecular analysiscan be performed to identify the exact gene defect (Smith,2003). This involves DNA sequencing of the two appropriatecandidate keratin genes; K6a and K16 for PC-1 and K6band K17 for PC-2. Analysis is normally performed on gen-omic DNA extracted from blood samples as this is less in-vasive than obtaining biopsy material. In large families withseveral affected members it may be useful to perform link-age analysis before DNA sequencing (Smith et al, 2004).Using polymorphic genetic markers within the two keratinclusters on chromosome 12q (type II) and 17q (type I) it isoften possible to exclude one locus and therefore halve theamount of DNA sequencing. Even in small families wherestatistically significant linkage cannot be obtained it may bepossible to exclude one keratin locus (Smith et al, 2004).Linkage analysis can also be useful in families where thereare several affected members but clinical distinction be-tween PC-1 and PC-2 is unclear. Mutation screening nor-mally focuses first on the two mutation ‘‘hot spots,’’ thehighly conserved helix-boundary motifs at either end of therod domain, and then if no mutation is found, analysis canbe extended to other regions.
When performing mutation analysis for PC using gen-omic DNA, one needs to be aware of the pseudogenes thatare present for all four keratin genes involved. There are atleast two pseudogenes for each of K16 and K17 and anumber of copies of the K6 gene, some of which are ex-pressed. The early PC mutation studies were carried out onmRNA derived from skin biopsies to overcome the problemof pseudogenes (McLean et al, 1995). The sequences ofthese pseudogenes, however, are now known making itpossible to design PCR primers to specifically amplify onlythe functional genes and avoid amplification of the pseu-dogenes. Mutation detection strategies based on PCR am-plification of genomic DNA have been developed for all fourkeratin genes involved in PC and primers have been de-signed to specifically amplify full length K6a, K6b, K16, andK17 (see Methods). Direct sequencing of the purified PCRproducts is carried out using internal primers. Several mu-tations identified using these methods have also been con-firmed at the mRNA level confirming specificity of the PCRprimers and the conditions. In the UK, genetic diagnosis iscarried out by National Health Service laboratories (all ker-atin disorders are analyzed by the Molecular Genetics Lab-oratory, Human Genetics Unit, Ninewells Hospital, Dundee,UK) and in the USA by local genetics laboratories or byprivate companies such as GeneDx (Gaithersburg, Mary-land). Genetic testing of patients enrolled in the InternationalPC Research Registry (IPCRR) is performed by the Dundee
Figure 1Protein structure of keratins and immunofluorescent staining ofPLK2 cells. (a) Schematic diagram showing the basic protein structureof a keratin filament. The a-helical rod domain is divided into four do-mains, the 1A, 1B, 2A and 2B connected together by linkers L1, L12,and L2. Flanking the rod domain are the highly conserved helix initiationmotif and the helix termination motif, shown in red. Type II keratins haveH1 and H2 subdomains. The ‘‘stutter’’ sequence is marked by S. (b)Antibody staining of keratin filaments (green) in PtK2 epithelial cells inculture, nuclei (blue) stained with 40,6-diamidino-2-phenylindole (DAPI).
PACHYONYCHIA GENES 2310 : 1 OCTOBER 2005

laboratory and the results are independently checked byGeneDx. Useful contacts for PC diagnosis and genetictesting are listed in Table II.
Many laboratories performing mutation analysis of ker-atin disorders have a small percentage of patients where nomutation in the candidate keratin gene(s) is found. This isalso true for PC and based on our experience, about 10% orso of PC patients fall into this category (Smith and McLean,unpublished observations). One explanation is that it can bedifficult to fully exclude a gene; the mutation may be in aregion not normally screened. For example, there may becryptic splicing elements within large introns, mutations af-fecting the promoter, or there could be genomic rearrange-ments because of meiotic mismatch within clusters of highlyhomologous isogenes/pseudogenes, as is the case in theglobin gene clusters (Cooper and Krawczak, 1993). Anotherpossibility is misdiagnosis—in some cases, patients canpresent with very similar clinical symptoms but at the mo-lecular level have mutations in different genes. One exampleis PC and Clouston syndrome (OMIM 129500) where thereis some overlap in clinical symptoms. Recently several pa-tients presenting with variants of PC were screened in ourlaboratory for mutations in the candidate genes, K6a, K6b,K16, and K17. But, no mutations were found and after clin-ical re-examination, alternative diagnoses were considered.Screening of the GJB6 gene encoding the gap junctionprotein connexin 30 led to identification of mutations andreclassification of these patients as Clouston syndrome (vanSteensel et al, 2003). In some cases, PC patients haveblisters at birth that resemble EBS and may be initially di-
agnosed with EBS until more characteristic symptoms ofPC develop (Smith et al, 2001).
Results
Clinical details of PC-1 families The PC families analyzedhere were obtained from IPCRR. This is a research registryresource approved by an Institutional Review Board (IRB)that complies with all principles of the Helsinki Accord(Western IRB #1057496). Participant recruitment is primarilyby self-referral to the web-based registry, although afew families were direct physician referrals to S.A.L. orW.H.I.M.
Seventeen of the cases showed a strong family history ofPC, consistent with autosomal dominant inheritance (TableIII). As an example, the pedigree of Family 5 is shown in Fig2a, which demonstrates typical autosomal dominant inher-itance with male-to-male transmission as seen in both ma-jor forms of PC. There are affected individuals in at least fivegenerations, each of whom presented with hypertrophic naildystrophy (including hyperkeratosis of the nail bed, withthickening and deformation of the nail plate, see Fig 2b) andin a number of cases, oral leukokeratosis. It is notable thatan affected member of this family died at 2 y of age becauseof respiratory infection. Although details of this case are notavailable, potentially fatal laryngeal involvement in child-hood PC-1 has been reported on several occasions (Stieg-litz and Centerwall, 1983; Wudy et al, 1995). The other 13PC cases analyzed here were sporadic.
The clinical phenotypes of the other PC-1 and PC-2families studied here were largely unremarkable and entirelytypical of the classic phenotypic spectrum described inLeachman et al (this issue). In two kindreds (Families 21 and23), the nail dystrophy phenotype was very limited. Specif-ically, the fingernails showed no thickening or hyperkerato-sis whatsoever, although on close examination somesplinter hemorrhages were evident (Fig 2c). There washypertrophic dystrophy affecting the distal portions of thegreat toenails and some but not all of the minor toenails (notshown). Focal keratoderma was moderate-severe and quitetypical of that seen in other PC-1 families (not shown). Theclinical appearance was consistent in the affected membersof Family 21 and the sporadic proband in Family 23. Overall,the phenotype was very similar to that seen in previouslyreported FNEPPK families carrying K16 mutations (Shams-her et al, 1995).
A range of keratin mutations in PC families Here we re-port keratin mutations in 30 new cases of PC from Canada,France, Ireland, the Netherlands, Sweden, Finland, UK, andUSA. In PC-1 patients, eight novel mutations were found inK6a (I167N, L170F, N171S, S176P, I462N, L468P, L469P,E470ins39) and a further three new mutations in K16 (L124P,L124H, N125D). In addition, nine recurrent mutations weredetected in K6a (N171del � 6, N171K, F174S � 2) and afurther five previously reported defects in K16 (N125S � 2,R127C, L132P � 2). In PC-2 patients, we identified onenovel mutation (L388P) and two previously reported muta-tions (N92S � 2) in K17, as well as two substitutions in K6b(E472K � 2). All novel mutations were excluded from 50
Table II. Useful websites for research and diagnosis of
pachyonychia congenita
Website URL
On-line Mendelian Inheritancein Man (OMIM): Clinical andmolecular data on all geneticdisorders
http://www3.ncbi.nlm.nih.gov/Omim/
Intermediate filament mutationdatabase: Clinical andmutational data on keratindisorders and otherintermediate filament diseases
http://www.interfil.org/
GeneDx: US-based companyoffering genetic testing forkeratin disorders, genetic skindiseases and other disorders.IPCRR testing lab
http://www.genedx.com/
Human Genetics Unit, Dundee:UK center for moleculardiagnosis of keratin disorders.IPCRR testing lab
http://www.humangenetics.org.uk/
The Pachyonychia CongenitaProject: Charitable organizationoffering support for PC patientsand sponsoring PC research
http://www.pc-project.org
TransDerm: US-basedcompany set up in partnershipwith the PachyonychiaCongenita Project to developtherapeutics for pachyonychiacongenita
http://www.transderm.org
24 SMITH ET AL JID SYMPOSIUM PROCEEDINGS
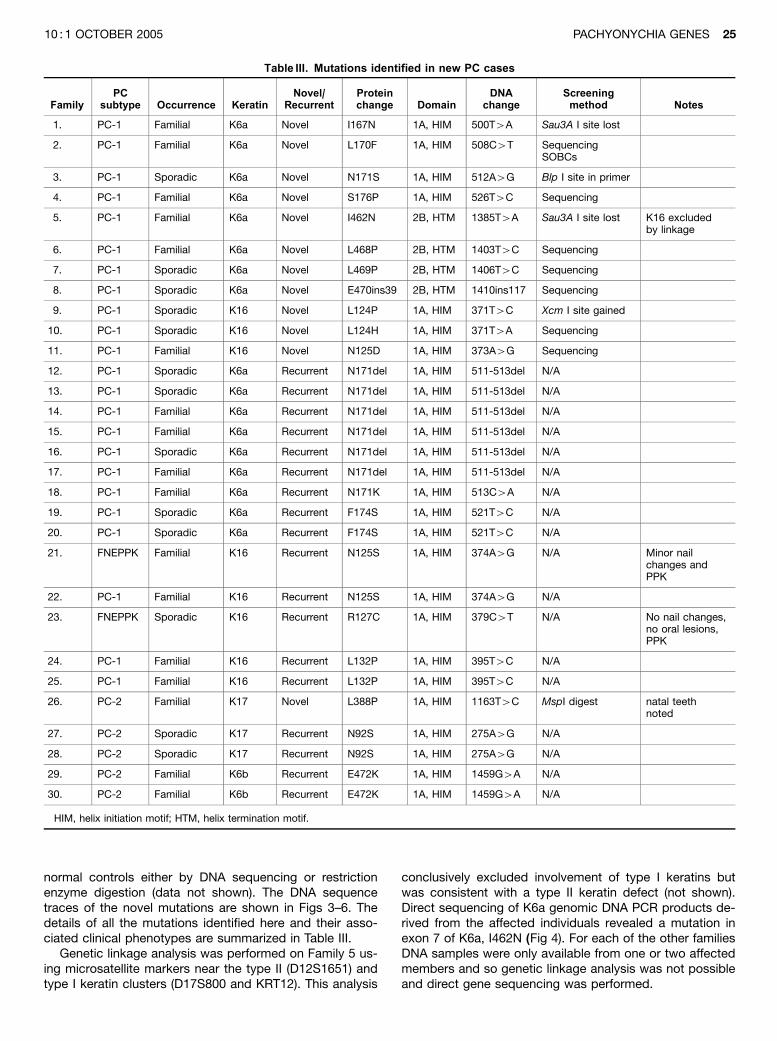
normal controls either by DNA sequencing or restrictionenzyme digestion (data not shown). The DNA sequencetraces of the novel mutations are shown in Figs 3–6. Thedetails of all the mutations identified here and their asso-ciated clinical phenotypes are summarized in Table III.
Genetic linkage analysis was performed on Family 5 us-ing microsatellite markers near the type II (D12S1651) andtype I keratin clusters (D17S800 and KRT12). This analysis
conclusively excluded involvement of type I keratins butwas consistent with a type II keratin defect (not shown).Direct sequencing of K6a genomic DNA PCR products de-rived from the affected individuals revealed a mutation inexon 7 of K6a, I462N (Fig 4). For each of the other familiesDNA samples were only available from one or two affectedmembers and so genetic linkage analysis was not possibleand direct gene sequencing was performed.
Table III. Mutations identified in new PC cases
FamilyPC
subtype Occurrence KeratinNovel/
RecurrentProteinchange Domain
DNAchange
Screeningmethod Notes
1. PC-1 Familial K6a Novel I167N 1A, HIM 500T4A Sau3A I site lost
2. PC-1 Familial K6a Novel L170F 1A, HIM 508C4T SequencingSOBCs
3. PC-1 Sporadic K6a Novel N171S 1A, HIM 512A4G Blp I site in primer
4. PC-1 Familial K6a Novel S176P 1A, HIM 526T4C Sequencing
5. PC-1 Familial K6a Novel I462N 2B, HTM 1385T4A Sau3A I site lost K16 excludedby linkage
6. PC-1 Familial K6a Novel L468P 2B, HTM 1403T4C Sequencing
7. PC-1 Sporadic K6a Novel L469P 2B, HTM 1406T4C Sequencing
8. PC-1 Sporadic K6a Novel E470ins39 2B, HTM 1410ins117 Sequencing
9. PC-1 Sporadic K16 Novel L124P 1A, HIM 371T4C Xcm I site gained
10. PC-1 Sporadic K16 Novel L124H 1A, HIM 371T4A Sequencing
11. PC-1 Familial K16 Novel N125D 1A, HIM 373A4G Sequencing
12. PC-1 Sporadic K6a Recurrent N171del 1A, HIM 511-513del N/A
13. PC-1 Sporadic K6a Recurrent N171del 1A, HIM 511-513del N/A
14. PC-1 Familial K6a Recurrent N171del 1A, HIM 511-513del N/A
15. PC-1 Familial K6a Recurrent N171del 1A, HIM 511-513del N/A
16. PC-1 Sporadic K6a Recurrent N171del 1A, HIM 511-513del N/A
17. PC-1 Familial K6a Recurrent N171del 1A, HIM 511-513del N/A
18. PC-1 Familial K6a Recurrent N171K 1A, HIM 513C4A N/A
19. PC-1 Sporadic K6a Recurrent F174S 1A, HIM 521T4C N/A
20. PC-1 Sporadic K6a Recurrent F174S 1A, HIM 521T4C N/A
21. FNEPPK Familial K16 Recurrent N125S 1A, HIM 374A4G N/A Minor nailchanges andPPK
22. PC-1 Familial K16 Recurrent N125S 1A, HIM 374A4G N/A
23. FNEPPK Sporadic K16 Recurrent R127C 1A, HIM 379C4T N/A No nail changes,no oral lesions,PPK
24. PC-1 Familial K16 Recurrent L132P 1A, HIM 395T4C N/A
25. PC-1 Familial K16 Recurrent L132P 1A, HIM 395T4C N/A
26. PC-2 Familial K17 Novel L388P 1A, HIM 1163T4C MspI digest natal teethnoted
27. PC-2 Sporadic K17 Recurrent N92S 1A, HIM 275A4G N/A
28. PC-2 Sporadic K17 Recurrent N92S 1A, HIM 275A4G N/A
29. PC-2 Familial K6b Recurrent E472K 1A, HIM 1459G4A N/A
30. PC-2 Familial K6b Recurrent E472K 1A, HIM 1459G4A N/A
HIM, helix initiation motif; HTM, helix termination motif.
PACHYONYCHIA GENES 2510 : 1 OCTOBER 2005

Most mutations were missense or small in-frame deletionmutations in the known keratin hotspot regions, as sum-marized in Table III. A small number of mutations, however,were unusual. In Family 8, the proband, who was a sporadic
case, had a very unusual mutation: a 117 bp duplication,1410ins117, in exon 7 of the K6a gene (Fig 4). This resulted ina 39 amino acid insertion in the 2B domain of the K6a po-lypeptide (E470ins39), which duplicates the latter part of the2B domain including a portion of the helix termination motif.The mutation was confirmed by cloning the PCR product (Fig4). Despite the unusual nature of this genetic lesion, the PC-1phenotype of this patient was unremarkable.
In Family 26, the affected persons carried a missensemutation in K17, L388P which is the first reported mutation inthe helix termination motif of this protein (Fig 6). In this case,the phenotype was well within the normal spectrum associ-ated with PC-2 (see Leachman et al, this issue). Specifically,affected persons in this kindred had typical nail dystrophy,pilosebaceous cysts, plantar keratoderma and more thanone affected member of the family had natal teeth.
Interestingly, the two kindreds who presented with amilder, site-restricted PC-1 phenotype of FNEPPK with mildnail changes (Families 21 and 23), carried the same muta-tions as previously reported in similar unrelated FNEPPKfamilies, N125S and R127C, respectively, in K16 (Shamsheret al, 1995). The milder nail changes consisted mainly ofsplinter hemorrhages of the fingernails and mild nail platethickening affecting only one or two toenails. The fingernailsof an affected member of Family 21 are shown in Fig 2. Incontrast, the affected persons in Family 22 were also foundto carry the N125S mutation in K16, but here, the presen-tation was more typical of PC-1, with 17 out of 20 nails inthe proband showing typical hypertrophic nail dystrophy(not shown). Thus, we see here a good example of inter-familial phenotypic variation. Similarly, the affected mem-bers of Family 11, who carried a different mutation in thesame residue of K16, N125D, had a more typical PC-1
Figure2Pedigree of family 5 and clinicalfeatures of families 5 and 4. (a)Part of the pedigree of Family 5showing typical autosomal domi-nant inheritance of PC-1. Asterisksdenote individuals from whom DNAwas obtained. (b) Striking hypertro-phic dystrophy of the fingernails ina patient from Family 5, carrying aheterozygous missense mutationI462N in K6a. (c) The fingernails ofthe proband in Family 21 showingcomplete lack of thickening andonly minor splinter hemorrhages.This patient carries a heterozygousmissense mutation N125S in K16.
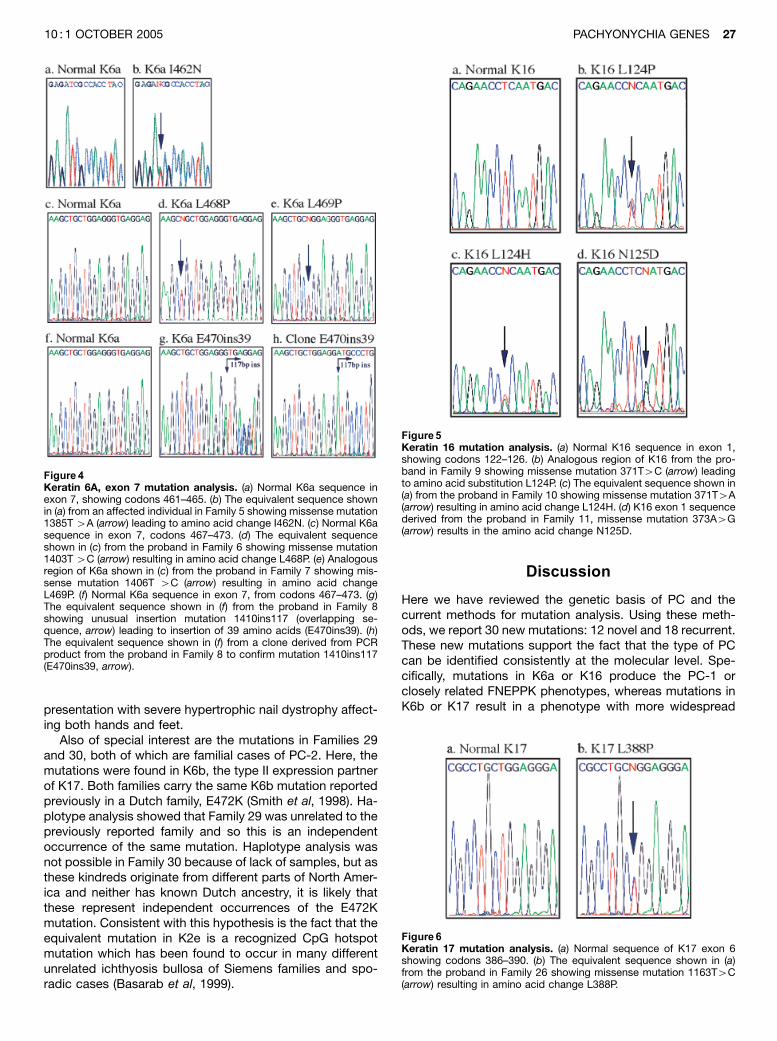
Figure 3Keratin 6A, exon 1 mutation analysis. (a) Normal K6a sequence inexon 1, showing codons 166-170. (b) The equivalent sequence shown in(a) from the proband in Family 1, missense mutation 500T4A (arrow)results in the amino acid substitution I167N. (c) Normal K6a sequence inexon 1, codons 174–178. (d) The equivalent sequence shown in (c) fromthe proband in Family 4 showing missense mutation 526T4C (arrow)resulting in amino acid substitution S176P. (e) Normal K6a sequence inexon 1, from codons 169–173. (f) Analogous region of K6a shown in (e)from the proband in Family 2 showing missense mutation 508C4T (ar-row) resulting in amino acid change L170F. (g) The equivalent sequenceshown in (e) from the proband in Family 3 showing missense mutation512A 4G (arrow) resulting in amino acid change N171S.
26 SMITH ET AL JID SYMPOSIUM PROCEEDINGS
presentation with severe hypertrophic nail dystrophy affect-ing both hands and feet.
Also of special interest are the mutations in Families 29and 30, both of which are familial cases of PC-2. Here, themutations were found in K6b, the type II expression partnerof K17. Both families carry the same K6b mutation reportedpreviously in a Dutch family, E472K (Smith et al, 1998). Ha-plotype analysis showed that Family 29 was unrelated to thepreviously reported family and so this is an independentoccurrence of the same mutation. Haplotype analysis wasnot possible in Family 30 because of lack of samples, but asthese kindreds originate from different parts of North Amer-ica and neither has known Dutch ancestry, it is likely thatthese represent independent occurrences of the E472Kmutation. Consistent with this hypothesis is the fact that theequivalent mutation in K2e is a recognized CpG hotspotmutation which has been found to occur in many differentunrelated ichthyosis bullosa of Siemens families and spo-radic cases (Basarab et al, 1999).
Discussion
Here we have reviewed the genetic basis of PC and thecurrent methods for mutation analysis. Using these meth-ods, we report 30 new mutations: 12 novel and 18 recurrent.These new mutations support the fact that the type of PCcan be identified consistently at the molecular level. Spe-cifically, mutations in K6a or K16 produce the PC-1 orclosely related FNEPPK phenotypes, whereas mutations inK6b or K17 result in a phenotype with more widespread
Figure 4Keratin 6A, exon 7 mutation analysis. (a) Normal K6a sequence inexon 7, showing codons 461–465. (b) The equivalent sequence shownin (a) from an affected individual in Family 5 showing missense mutation1385T 4A (arrow) leading to amino acid change I462N. (c) Normal K6asequence in exon 7, codons 467–473. (d) The equivalent sequenceshown in (c) from the proband in Family 6 showing missense mutation1403T 4C (arrow) resulting in amino acid change L468P. (e) Analogousregion of K6a shown in (c) from the proband in Family 7 showing mis-sense mutation 1406T 4C (arrow) resulting in amino acid changeL469P. (f) Normal K6a sequence in exon 7, from codons 467–473. (g)The equivalent sequence shown in (f) from the proband in Family 8showing unusual insertion mutation 1410ins117 (overlapping se-quence, arrow) leading to insertion of 39 amino acids (E470ins39). (h)The equivalent sequence shown in (f) from a clone derived from PCRproduct from the proband in Family 8 to confirm mutation 1410ins117(E470ins39, arrow).
Figure5Keratin 16 mutation analysis. (a) Normal K16 sequence in exon 1,showing codons 122–126. (b) Analogous region of K16 from the pro-band in Family 9 showing missense mutation 371T4C (arrow) leadingto amino acid substitution L124P. (c) The equivalent sequence shown in(a) from the proband in Family 10 showing missense mutation 371T4A(arrow) resulting in amino acid change L124H. (d) K16 exon 1 sequencederived from the proband in Family 11, missense mutation 373A4G(arrow) results in the amino acid change N125D.
Figure6Keratin 17 mutation analysis. (a) Normal sequence of K17 exon 6showing codons 386–390. (b) The equivalent sequence shown in (a)from the proband in Family 26 showing missense mutation 1163T4C(arrow) resulting in amino acid change L388P.
PACHYONYCHIA GENES 2710 : 1 OCTOBER 2005

pilosebaceous cysts either with hypertrophic nail dystrophy(PC-2) or without it (steatocystoma multiplex). There is sofar only one notable exception to this rule, in a case wherethere was considerable clinical overlap between PC-1 andPC-2 (Ward et al, 2003). This patient reportedly had multiplesoft, yellow papules and nodules that were confirmed his-tologically as steatocysts and here, a mutation was found atthe ‘‘hot spot’’ codon of K6a (N171). As pilosebaceous cystsdo not normally appear until puberty in PC-2 and steatocy-stoma multiplex patients, there is an obvious role for an-drogens in their pathogenesis. It is therefore possible thatinter-individual variation in androgen levels may modulatethe formation of pilosebaceous cysts in PC. It should alsobe noted that PC-1 patients do appear to get more cystsarising from the pilosebaceous unit than was previouslynoted, however, these are usually fewer in number and aremore site-restricted than the cysts seen in PC-2 (seeLeachman et al, this issue).
All the novel PC-1 mutations reported here were in thehelix boundary motifs of either K6a or K16, consistent withprevious findings (Table III). The N171 codon of K6a isemerging as a mutation hotspot in PC-1. Here, we foundone novel mutation N171S and seven recurrent ones(N171K and N171del � 6) further highlighting this codon.Including these mutations, 16 out of 52 reported PC-1 mu-tations (30.8%) occur within this codon (this paper and mu-tation database, http://www.interfil.org). Presently this is themost common codon for mutation in PC-1 although thereare several other codons both in K6a and K16 where re-current mutations have been found in unrelated families(K6a F174; K16 L132). Interestingly, in Families 21 and 23,who presented with FNEPPK and milder nail dystrophyconfined to a few toenails, we found the same K16 muta-tions N125S and R127C as previously reported in FNEPPKfamilies (Shamsher et al, 1995). In Family 22 studied here,however, the same N125S mutation resulted in more typicalPC-1, with most nails being affected. Similarly, Family 11had a different amino acid substitution at the same codon(N125D) and again had the more severe nail phenotypetypical of PC-1. Although it is tempting to speculate thatN125S in K16 is more often associated with more restrictedFNEPPK-like phenotypes, when one compares the pheno-types in affected members of Families 21 and 22, all ofwhom carry this specific defect, interfamilial phenotypicvariation is clearly at play. Thus, it is important not to makefirm conclusions about phenotypes associated with partic-ular genotypes until large numbers of cases are available.
In PC-2 the majority of the mutations are in the helixinitiation motif of K17 with mutations at two codons, N92and R94 accounting for more than 50% of cases (http://www.interfil.org). The two PC-2 cases reported here wereagain at codon N92. Similar to K16, mutations at the samecodon can produce different phenotypes: N92S and N92Dcause PC-2 and in one case N92H resulted in steatocysto-ma multiplex. More surprising is that the same K17 muta-tion, R94C, can lead to PC-2 or the milder steatocystomamultiplex phenotype with no nail changes (Smith et al, 1997;Covello et al, 1998; Wang et al, 2001). Therefore genotype–phenotype correlation in keratin disorders has a level ofcomplexity that is not fully understood at the present time. Itis likely that in addition to the mutation, a combination of
other factors, both genetic and environmental, are involvedin determining the overall clinical phenotype. As more fam-ilies are analyzed it may be possible to draw better geno-type–phenotype correlations in the future (as discussed byMcLean et al, this issue).
In the small number of PC-2 families studied here, weidentified two further instances of the E472K mutation inK6b, confirming that mutations in this gene phenocopymutations in K17 (Smith et al, 1998). This confirms the co-expression of K6b and K17 as a keratin pair in humans atleast, as previously shown (Smith et al, 1998). The first oc-currence of a mutation affecting the K17 helix terminationmotif was also identified here, producing a fairly typical PC-2 presentation in Family 26, including some cases of natalteeth, a hallmark of PC-2. All previous K17 mutations(n¼26) have been located in the 1A domain (see Leachmanet al, this issue and McLean et al, this issue).
Conclusions
In conclusion, PC is caused by mutations in four differen-tiation-specific keratin genes. Methods are now available torapidly diagnose PC at the molecular level despite the dif-ficulties arising from these being multiple copy genes.Knowledge of the exact gene defect allows for better ge-netic counseling and in cases of severe PC, if requested,the option of prenatal diagnosis. When the familial mutationis known, this can be performed at an early stage of preg-nancy by chorionic villus biopsy, as we have previouslydemonstrated (Smith et al, 1999b). In addition, knowing themolecular basis of PC will undoubtedly help in the rationaldesign of novel gene-based or drug-based therapies forthese conditions, which remain incurable at this time.
Methods
Genetic linkage analysis Genomic DNA was extracted from pe-ripheral blood lymphocytes by a standard procedure, with in-formed consent and appropriate ethical approval in all cases.Genetic linkage analysis was performed on Family 5 using fluo-rescently labeled polymorphic markers, as described previously(van Steensel et al, 1999).
Mutation detection and confirmation: general considera-tions The full length K6a, K6b, K16, and K17 genes were ampli-fied by long-range PCR, using primers specific to the respectivefunctional genes to avoid amplification of additional K6 genes orK16/K17 pseudogenes, as previously described (Smith et al, 1998,1999a, b; Terrinoni et al, 2001). As it can be difficult to performlong-range PCR using low quality DNA samples, we have alsodeveloped two shorter PCR reactions covering all exons of the K6agene, described below. PCR reactions were optimized on a Hybaid0.5 mL Multiblock PCR machine but may need to be re-calibratedfor other thermocyclers. The primer pairs and conditions currentlyin use in our laboratory are given below.
K6aP1-P2 fragment (�5.5 kb, encompassing all exons) For-ward primer K6AP1 (50 CCA GCC TCT CAC ACT CTC CTC 30) andreverse primer K6AP2 (50 GAC CGA GAG CTA GCA GAC GC 30)were used in High Fidelity PCR buffer (Roche, Lewes, East Sussex,UK) containing 1.5 mM MgCl2 and 4% dimethylsulphoxide (DMSO).A ‘‘hot start’’ was performed with 1U High Fidelity thermostableDNA polymerase mix (Roche) and the following conditions were
28 SMITH ET AL JID SYMPOSIUM PROCEEDINGS

used: (941C for 5 min) � 1; (941C 30 sec, 611C 1.5 min, 721C 2.5min) � 35; and (721C 5 min) � 1.
K6aSP1L2-R2 fragment (3.8 kb fragment, from promoter to in-tron 6) Forward primer K6aSP1L2 (50 CCG GGC TTA TTG TGTTAG GA 30) and reverse primer K6aSP1R2 (50 TCC AGG GAA GCCAAA ATA CT 30) were used in High Fidelity PCR buffer (Roche)containing 1.0 mM MgCl2. A ‘‘hot start’’ was performed with 1 UHigh Fidelity thermostable DNA polymerase mix (Roche) and thefollowing conditions were used: (941C for 5 min) � 1; (941C 30sec, 551C 1.5 min, 721C 2.5 min) � 35; and (721C 5 min) � 1.
K6aSP2L2-R2 fragment (2.0 kb fragment, from intron 6 todownstream of exon 9) Forward primer K6aSP2L2 (50 ACT CCAAAC AAA CCA GCA GG 30) and reverse primer K6aSPR2 (50 TAACTT GCT CTA GTT GGC CG 30) were used in High Fidelity PCRbuffer (Roche) containing 1.0 mM MgCl2. A ‘‘hot start’’ was per-formed with 1 U High Fidelity thermostable DNA polymerase mix(Roche) and the following conditions were used: (941C for 5 min)� 1; (941C 30 sec, 551C 1.5 min, 721C 2.5 min) � 35; and (721C5 min) � 1.
K16SP2-SP3 fragment (�2.5 kb fragment covering all ex-ons) Primers, K16SP2 (50 AGG GCT CCT GCG GCA TCG GA 30
forward) and K16SP3 (50 GGA TTG GCC AGA TGC TTG CT 30
reverse) were used in High Fidelity PCR buffer (Roche) containing1.5 mM MgCl2 and 4% DMSO. A ‘‘hot start’’ was performed with1 U Taq polymerase (Promega, Southampton, UK). The followingPCR conditions were used: (941C 5 min) � 1; (941C 30 sec, 591C45 sec, 721C 2 min) � 35; and (721C 5 min) � 1.
K6bP1-P2 fragment (�5.5 kb fragment covering all exons) For-ward primer K6bP1 (50 CTC CAG CCT CTC ACA CTC TCC TA 3)and reverse primer K6bP2 (50 CTT CTC AGA ATT ATG GCA GACTCA G 30) were used in High Fidelity PCR buffer (Roche) containing1.5 mM MgCl2 and 4% DMSO. A ‘‘hot start’’ was performed with1 U High Fidelity thermostable DNA polymerase mix (Roche). Re-actions were amplified using the following extended PCR program:(941C 5 min � 1); (941C 20 sec, 561C 1 min 30 sec, 681C 3 min)� 10; (941C 20 sec, 561C 1 min 30 sec, 681C 3 min plus 20 sec
increment/cycle) � 20; and (681C 10 min) � 1.
K17P8-P10 fragment (�1 kb covering exon 1) Primers K17P8 (50
GCC TAT AAA GGA AGC GGG C 30) and reverse primer K17P10 (50
CTC CTT TCT GCC TCC TCC 30) were used with Qiagen PCRbuffer containing 1.0 mM MgCl2 and 1U HotStar Taq DNA polym-erase (Qiagen, Crawley, UK). Reactions were amplified using thefollowing program: (941C for 15 min) � 1; (941C 1 min, 581C1 min, 721C 1 min) � 30; and (721C 5 min) � 1.
K17e6L-E6R (400 bp fragment covering exon 6) PrimersK17e6L (50 CAG AAG AGG GCA TTG ACC AT 30) and reverseprimer K17e6R (50 CCC CAG GGC CGA AGC CAC GC 30) wereused in Qiagen PCR buffer containing 1.0 mM MgCl2 and 1U Hot-Star Taq DNA polymerase (Qiagen). Reactions were amplified us-ing the following program: (941C for 15 min) � 1; (941C 1 min,581C 1 min, 721C 1 min) � 35; and (721C 5 min) � 1.
PCR products were purified for sequencing using QIA quickPCR purification kit (Qiagen) and sequenced using internal primers.Sequencing ladders were analyzed on an ABI 377 or 3100 auto-mated DNA sequencers, according to manufacturer’s instructions.
We thank the many patients who have participated in our research intopachyonychia congenita and the many clinicians who have helped us,including the following who sent DNA from families with recurrent mu-tations reported in this study: Irene Leigh, John McGrath, HowardStevens, Martine Blayau, Elsa Reich, Gail Graham, and Mary Ander-son. Thanks to the Molecular Genetics Laboratory, Ninewells Hospital,Dundee for genomic DNA extraction and to the Molecular GeneticsAnalysis Facility, Division of Pathology and Neuroscience, NinewellsMedical School, Dundee, for DNA sequencing. Erik Bjorck’s work is
supported by the Swedish Medical Research Council. Frances Smithand Irwin McLean are currently funded by a Wellcome Trust SeniorResearch Fellowship (W.H.I.M.). Research in the Epithelial GeneticsGroup is also supported by The Dystrophic Epidermolysis Bullosa Re-search Association UK (http://www.debra.org.uk) and The Pachyony-chia Congenita Project (http://www.pc-project.org).
DOI: 10.1111/j.1087-0024.2005.10204.x
Manuscript received June 9, 2005; revised June 27, 2005; accepted forpublication June 28, 2005
Address correspondence to: Dr Frances J. D. Smith, Human GeneticsUnit, Ninewells Medical School, Dundee DD1 9SY, UK. Emails:[email protected]
References
Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD: Molecular Biology of
the Cell, 2nd edn. New York: Garland Publishing Inc., 1989
Basarab T, Smith FJD, Jolliffe VML, McLean WHI, Neill S, Rustin MHA, Eady RAJ:
Ichthyosis bullosa of Siemens: Report of a family with evidence of a
keratin 2e mutation, and a review of the literature. Br J Dermatol 140:
689–695, 1999
Bowden PE, Haley JL, Kansky A, Rothnagel JA, Jones DO, Turner RJ: Mutation
of a type II keratin gene (K6a) in pachyonychia congenita. Nat Genet
10:363–365, 1995
Cooper DN, Krawczak M: Human Gene Mutation. Oxford: BIOS Scientific Pub-
lishers Ltd, 1993
Covello SP, Smith FJD, Sillevis Smitt JH, et al: Keratin 17 mutations cause either
steatocystoma multiplex or pachyonychia congenita type 2. Br J De-
rmatol 139:475–480, 1998
Fuchs E, Cleveland DW: A structural scaffolding of intermediate filaments in
health and disease. Science 279:514–519, 1998
Herrmann H, Foisner R: Intermediate filaments: Novel assembly models and ex-
citing new functions for nuclear lamins. Cell Mol Life Sci 60:1607–1612,
2003
Herrmann H, Strelkov SV, Feja B, et al: The intermediate filament protein con-
sensus motif of helix 2B: Its atomic structure and contribution to assem-
bly. J Mol Biol 298:817–832, 2000
Hesse M, Magin TM, Weber K: Genes for intermediate filament proteins and the
draft sequence of the human genome: Novel keratin genes and a sur-
prisingly high number of pseudogenes related to keratin genes 8 and 18.
J Cell Sci 114:2569–2575, 2001
Hesse M, Zimek A, Weber K, Magin TM: Comprehensive analysis of keratin
gene clusters in humans and rodents. Eur J Cell Biol 83:19–26,
2004
Irvine AD, McLean WHI: Human keratin diseases: The increasing spectrum of
disease and subtlety of the phenotype–genotype correlation. Br J De-
rmatol 140:815–828, 1999
Lane EB: Keratins. In: Royce PM, Steinmann B (eds). Connective Tissue and its
Heritable Disorders Molecular. New York: Genetic and Medical Aspects
Wiley-Liss Inc., 1993; p 237–247
Leachman SA, Kaspar RL, Fleckman P, et al: Clinical and pathological features of
pachyonychia congenita. J Invest Dermatol 10:3–17, 2005
Lin MT, Levy ML, Bowden PE, Magro C, Baden L, Baden HP, Roop DR: Iden-
tification of sporadic mutations in the helix initiation motif of keratin 6 in
two pachyonychia congenita patients: Further evidence for a mutational
hot spot. Exp Dermatol 8:115–119, 1999
McLean WHI, Lane EB: Intermediate filaments in disease. Curr Opin Cell Biol
7:118–125, 1995
McLean WHI, Rugg EL, Lunny DP, et al: Keratin 16 and keratin 17 mutations
cause pachyonychia congenita. Nat Genet 9:273–278, 1995
McLean WHI, Smith FJD, Cassidy AJ. Insights into genotype-phenotype
correlation in pachyonychia congenita from the human Intermediate fil-
ament mutation database. J Investig Dermatol 10:31–36, 2005
Morley SM, Lane EB: The keratinocyte cytoskeleton. In: Leigh IM, Lane EB, Watt
FM (eds). The Keratinocyte Handbook. Cambridge: Cambridge University
Press, 1994; p 293–321
Munro CS: Pachyonychia congenita: Mutations and clinical presentations. Br J
Dermatol 144:929–930, 2001
Munro CS, Carter S, Bryce S, et al: A gene for pachyonychia congenita is closely
linked to the keratin gene cluster on 17q12–q21. J Med Genet 31:
675–678, 1994
Omary MB, Coulombe PA, McLean WHI: Intermediate filament proteins and their
associated diseases. N Engl J Med 351:2087–2100, 2004
PACHYONYCHIA GENES 2910 : 1 OCTOBER 2005

Paller AS, Moore JA, Scher R: Pachyonychia congenita tarda. A late-onset
form of pachyonychia congenita. Arch Dermatol 127:701–703,
1991
Porter RM, Lane EB: Phenotypes, genotypes and their contribution to under-
standing keratin function. Trends Genet 19:278–285, 2003
Quinlan RA, Hutchison CJ, Lane EB: Intermediate Filaments. London: Academic
Press, 1994
Shamsher MK, Navsaria HA, Stevens HP, et al: Novel mutations in keratin 16 gene
underly focal non-epidermolytic palmoplantar keratoderma (NEPPK) in 2
families. Hum Mol Genet 4:1875–1881, 1995
Smith F: The molecular genetics of keratin disorders. Am J Clin Dermatol 4:
347–364, 2003
Smith FJD, Coleman CM, Bayoumy NM, Tenconi R, Nelson J, David A, McLean
WHI: Novel keratin 17 mutations in pachyonychia congenita type 2.
J Invest Dermatol 116:806–808, 2001
Smith FJD, Corden LD, Rugg EL, Ratnavel R, et al: Missense mutations in keratin
17 cause either pachyonychia congenita type 2 or a phenotype resem-
bling steatocystoma multiplex. J Invest Dermatol 108:220–223, 1997
Smith FJD, Jonkman MF, van Goor H, Coleman C, Covello SP, Uitto J, McLean
WHI: A mutation in human keratin K6b produces a phenocopy of the K17
disorder pachyonychia congenita type 2. Hum Mol Genet 7:1143–1148,
1998
Smith FJD, McKenna KE, Irvine AD, Bingham EA, Coleman CM, Uitto J, McLean
WHI: A mutation detection strategy for the human K6A gene and novel
mutations in two cases of pachyonychia congenita type 1. Exp Dermatol
8:109–114, 1999a
Smith FJD, McKusick VA, Nielsen K, Pfendner E, Uitto J, McLean WHI: Cloning of
multiple keratin 16 genes facilitates prenatal diagnosis of pachyonychia
congenita type 1. Prenatal Diagn 19:941–946, 1999b
Smith FJD, Sandilands A, McLean WHI: Molecular genetics methods for human
intermediate filament diseases. Methods Cell Biol 78:131–161, 2004
Smith TA, Strelkov SV, Burkhard P, Aebi U, Parry DA: Sequence comparisons of
intermediate filament chains: Evidence of a unique functional/structural
role for coiled-coil segment 1A and linker L1. J Struct Biol 137:128–145,
2002
Steinert PM, Marekov LN, Fraser RDB, Parry DAD: Keratin intermediate filament
structure: Crosslinking studies yield quantitative information on molecular
dimensions and mechanism of assembly. J Mol Biol 230:436–452,
1993a
Steinert PM, Yang JM, Bale SJ, Compton JG: Concurrence between the molec-
ular overlap regions in keratin intermediate filaments and the locations of
keratin mutations in genodermatoses. Biochem Biophys Res Commun
197:840–848, 1993b
Stieglitz JB, Centerwall WR: Pachyonychia congenita (Jadassohn–Lewandowsky
syndrome): A seventeen-member, four-generation pedigree with unusual
respiratory and dental involvement. Am J Med Genet 14:21–28,
1983
Strelkov SV, Herrmann H, Geisler N, et al: Divide-and-conquer crystallographic
approach towards an atomic structure of intermediate filaments. J Mol
Biol 306:773–781, 2001
Takahashi K, Paladini RD, Coulombe PA: Cloning and characterization of multiple
human genes and cDNAs encoding highly related type II keratin 6 iso-
forms. J Biol Chem 270:18581–18592, 1995
Terrinoni A, Smith FJD, Didona B, et al: Novel and recurrent mutations in the
genes encoding keratins K6a, K16 and K17 in 13 cases of pachyonychia
congenita. J Invest Dermatol 117:1391–1396, 2001
Troyanovsky SM, Leube RE, Franke WW: Characterization of the human gene
encoding cytokeratin 17 and its expression pattern. Eur J Cell Biol 59:
127–137, 1992
van Steensel MAM, Jonkman MF, van Geel M, Steijlen PM, McLean WHI, Smith
FJD: Clouston syndrome can mimic pachyonychia congenita. J Invest
Dermatol 121:1035–1038, 2003
van Steensel MAM, Smith FJD, Steijlen PM, et al: The gene for hypotrichosis of
Marie Unna maps between D8S258 and D8S298: Exclusion of the hr
gene by cDNA and genomic sequencing. Am J Hum Genet 65:413–419,
1999
Wang X, Shi Y, Ye Y, et al: Keratin 17 gene mutation in patients with steatocy-
stoma multiplex. Zhonghua Yi Xue Za Zhi 81:540–543, 2001
Ward KM, Cook-Bolden FE, Christiano AM, Celebi JT: Identification of a recurrent
mutation in keratin 6a in a patient with overlapping clinical features of
pachyonychia congenita types 1 and 2. Clin Exp Dermatol 28:434–436,
2003
Werner NS, Windoffer R, Strnad P, Grund C, Leube RE, Magin TM: Epidermolysis
bullosa simplex-type mutations alter the dynamics of the keratin cyto-
skeleton and reveal a contribution of actin to the transport of keratin
subunits. Mol Biol Cell 15:990–1002, 2003
Windoffer R, Woll S, Strnad P, Leube RE: Identification of novel principles
of keratin filament network turnover in living cells. Mol Biol Cell 15:
2436–2448, 2004
Wudy SA, Lenders H, Pirsig W, Mohr W, Teller WM: Diagnosis and management
of laryngeal obstruction in childhood pachyonychia congenita. Int J
Pediatr Otorhinolaryngol 31:109–115, 1995
30 SMITH ET AL JID SYMPOSIUM PROCEEDINGS
Copyright © 2022 FDOKUMEN




















