The genetic basis of DOORS syndrome: an exome-sequencing study
64
Page 1 of 39 The genetic basis of DOORS syndrome: an exome sequencing study 1 Philippe M Campeau 1 *, MD, Dalia Kasperaviciute 2 *, PhD, James T Lu 3,4 , PhD, Lindsay C 2 Burrage 1 , PhD, Choel Kim 5 , PhD, Mutsuki Hori 6 , MD, Berkley R Powell 7 , MD, Fiona Stewart 8 , 3 MBBS, Têmis Maria Félix 9 , PhD, Jenneke van den Ende 10 , MD, Marzena Wisniewska 11 , PhD, 4 Hülya Kayserili 12 , MD, Patrick Rump 13 , PhD, Sheela Nampoothiri 14 , MSc, Salim Aftimos 15 , MD, 5 Antje Mey 16 , MD, Lal DV Nair 17 , MD, Michael L Begleiter 18 , MSc, Isabelle De Bie 19 , PhD, Girish 6 Meenakshi 20 , MBBS, Mitzi L. Murray 21 , MD, Gabriela M Repetto 22 , MD, Mahin Golabi 23 , MD, 7 Edward Blair 24 , MD, Alison Male 25 , MD, Fabienne Giuliano 26 , MD, Ariana Kariminejad 27 , MD, 8 William G Newman 28,29 , PhD, Sanjeev S Bhaskar 28,29 , MSc, Jonathan E Dickerson 28,29 , PhD, 9 Bronwyn Kerr 28,29 , MD, Siddharth Banka 28,29 , PhD, Jacques C Giltay 30 , PhD, Dagmar 10 Wieczorek 31 , MD, Anna Tostevin 2 , MSc, Joanna Wiszniewska 1 , MD, Sau Wai Cheung 1 , PhD, 11 Raoul C. Hennekam 32 , PhD, Richard A Gibbs 1,3 , PhD, Brendan H Lee 1,33 , PhD, Sanjay M 12 Sisodiya 2,34 , PhD. 13 1. Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, 14 TX, 77030, USA. 15 2. Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology, London, 16 WC1N 3BG, UK. 17 3. Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, 77030, 18 USA. 19 4. Department of Structural and Computational Biology & Molecular Biophysics, Baylor 20 College of Medicine, Houston, TX, 77030, USA. 21 5. Department of Pharmacology, Baylor College of Medicine, Houston, TX, 77030, USA. 22 6. Department of Pediatrics, Toyohashi Municipal Hospital, Toyohashi, Aichi, 441-8570, 23 Japan. 24 7. Children's Hospital Central California, Madera, California, 93636, USA. 25 8. Genetics Service, Belfast City Hospital, Belfast, BT9 7AB, Ireland. 26 9. Medical Genetics Service, Clinical Hospital of Porto Alegre, Porto Alegre, 90035-903, 27 Brazil. 28 10. Department of Medical Genetics, University Hospital Antwerp, 2650 Antwerp, Belgium. 29
Transcript of The genetic basis of DOORS syndrome: an exome-sequencing study

Page 1 of 39
The genetic basis of DOORS syndrome: an exome sequencing study 1
Philippe M Campeau1*, MD, Dalia Kasperaviciute2*, PhD, James T Lu3,4, PhD, Lindsay C 2
Burrage1, PhD, Choel Kim5, PhD, Mutsuki Hori6, MD, Berkley R Powell7, MD, Fiona Stewart8, 3
MBBS, Têmis Maria Félix9, PhD, Jenneke van den Ende10, MD, Marzena Wisniewska11, PhD, 4
Hülya Kayserili12, MD, Patrick Rump13, PhD, Sheela Nampoothiri14, MSc, Salim Aftimos15, MD, 5
Antje Mey16, MD, Lal DV Nair17, MD, Michael L Begleiter18, MSc, Isabelle De Bie19, PhD, Girish 6
Meenakshi20, MBBS, Mitzi L. Murray21, MD, Gabriela M Repetto22, MD, Mahin Golabi23, MD, 7
Edward Blair24, MD, Alison Male25, MD, Fabienne Giuliano26, MD, Ariana Kariminejad27, MD, 8
William G Newman28,29, PhD, Sanjeev S Bhaskar28,29, MSc, Jonathan E Dickerson28,29, PhD, 9
Bronwyn Kerr28,29, MD, Siddharth Banka28,29, PhD, Jacques C Giltay30, PhD, Dagmar 10
Wieczorek31, MD, Anna Tostevin2, MSc, Joanna Wiszniewska1, MD, Sau Wai Cheung1, PhD, 11
Raoul C. Hennekam32, PhD, Richard A Gibbs1,3, PhD, Brendan H Lee1,33, PhD, Sanjay M 12
Sisodiya2,34, PhD. 13
1. Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, 14
TX, 77030, USA. 15
2. Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology, London, 16
WC1N 3BG, UK. 17
3. Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, 77030, 18
USA. 19
4. Department of Structural and Computational Biology & Molecular Biophysics, Baylor 20
College of Medicine, Houston, TX, 77030, USA. 21
5. Department of Pharmacology, Baylor College of Medicine, Houston, TX, 77030, USA. 22
6. Department of Pediatrics, Toyohashi Municipal Hospital, Toyohashi, Aichi, 441-8570, 23
Japan. 24
7. Children's Hospital Central California, Madera, California, 93636, USA. 25
8. Genetics Service, Belfast City Hospital, Belfast, BT9 7AB, Ireland. 26
9. Medical Genetics Service, Clinical Hospital of Porto Alegre, Porto Alegre, 90035-903, 27
Brazil. 28
10. Department of Medical Genetics, University Hospital Antwerp, 2650 Antwerp, Belgium. 29

Page 2 of 39
11. Department of Medical Genetics, Poznañ University of Medical Sciences, Poznañ, 61-1
701, Poland. 2
12. Medical Genetics Department, Istanbul Medical Faculty, Istanbul University, 34093 3
Turkey. 4
13. Department of Genetics, University of Groningen, Groningen, 9712 CP, The 5
Netherlands. 6
14. Department of Pediatric Genetics, Amrita Institute of Medical Sciences & Research 7
Center, Kerala, 682041, India. 8
15. Genetic Health Service New Zealand – Northern Hub, Auckland City Hospital, 9
Auckland, 92-024, New Zealand. 10
16. Pediatric neurology, Braunschweig Hospital, Braunschweig, 38118, Germany. 11
17. Department of Pediatrics, Saveetha Medical College & Hospital, Saveetha University, 12
Chennai, Tamilnadu, 600077, India. 13
18. Division of Genetics, Children’s Mercy Hospitals and Clinics and the University of 14
Missouri-Kansas City School of Medicine, Kansas City, MO, 64108, USA. 15
19. Department of Medical Genetics, Montreal Children’s Hospital, McGill University Health 16
Center, Quebec, H3H 1P3, Canada. 17
20. Department of Pediatrics, NKP Salve Institute of Medical Sciences & Lata Mangeshkar 18
Hospital, Maharashtra, 400001, India. 19
21. University of Washington Medical Center, Seattle, 98195, WA, USA. 20
22. Center for Human Genetics, Facultad de Medicina, Clínica Alemana-Universidad del 21
Desarrollo, Santiago, 7710162, Chile. 22
23. Department of Pediatrics, University of California San Francisco, San Francisco, CA, 23
94143, USA. 24
24. Department of Clinical Genetics, Churchill Hospital, Oxford, OX3 7LJ, UK. 25
25. Clinical Genetics Department, Great Ormond Street Hospital for Children NHS 26
Foundation Trust, London, WC1N 3JH, UK. 27
26. Centre Référence Anomalie Développement et Syndromes Malformatifs, Centre 28
Hospitalier Universitaire de Nice, 06200, France. 29
27. Kariminejad-Najmabadi Pathology & Genetics Center, 14656 Tehran, Iran. 30
28. Manchester Centre for Genomic Medicine, Institute of Human Development, Faculty of 31
Medical and Human Sciences, University of Manchester, Manchester Academic Health 32
Science Centre (MAHSC), Manchester, M13 9WL, UK 33
29. Manchester Centre for Genomic Medicine, Central Manchester University Hospitals 34
NHS Foundation Trust, MAHSC, Manchester, M13 9WL, UK 35
30. Department of Medical Genetics, University Medical Center Utrecht, 3508 AB Utrecht, 36
The Netherlands. 37
31. Institut für Humangenetik, University of Duisburg-Essen, University Hospital Essen, 38
45147 Essen, Germany. 39

Page 3 of 39
32. Department of Pediatrics and Translational Genetics, Academic Medical Center, 1
University of Amsterdam, Amsterdam, 1017 DS, The Netherlands. 2
33. Howard Hughes Medical Institutes, Houston, TX, 77030, USA. 3
34. Epilepsy Society, Bucks, SL9 0RJ, UK 4
*These authors contributed equally. 5
6
7
Corresponding authors: 8
9
Brendan H Lee 10
Investigator, Howard Hughes Medical Institute 11
Professor, Department of Molecular and Human Genetics, Baylor College of Medicine 12
One Baylor Plaza, R814 - MS225, Houston, TX 77030 [email protected] 13
14
Sanjay M Sisodiya 15
Department of Clinical and Experimental Epilepsy, 16
UCL Institute of Neurology, Queen Square, London, WC1N 3BG, UK. 17
Tel.: + 44 20 3448 8612; fax: + 44 20 3448 8615. [email protected] 18

Page 4 of 39
Abstract 1
2
Background: DOORS syndrome is a recessive condition characterized by Deafness 3
(sensorineural), Onycho/Osteodystrophy (nail and digital anomalies), intellectual disability 4
previously known as mental Retardation, and Seizures. We hypothesized that sequencing 5
most coding exons in affected individuals would lead to the identification of a critical gene. 6
Methods: In this cohort study leveraging an international collaboration, we identified thirty 7
families with individuals presenting at least three of the five features of the DOORS acronym. 8
Four families were excluded based either on clinical features, availability of clinical samples, or 9
ability to contact the family for consent. Clinical information and DNA was collected. We 10
conducted whole exome sequencing in affected individuals as they were enrolled, until we 11
identified a candidate gene. In three families with consanguinity or multiple affected siblings, 12
we concurrently conducted whole genome genotyping by SNP array to identify homozygous 13
regions or shared haplotypes. Mutations were confirmed by Sanger sequencing. Expression 14
studies were performed in human fibroblasts from one individual by real-time PCR and 15
Western blot and in mouse tissues by immunohistochemistry and real-time PCR. 16
Findings: Exome sequencing was performed in the first 17 enrolled families, and TBC1D24 17
was screened by Sanger sequencing in subsequent ones. We identified TBC1D24 mutations 18
in nine families (by exome sequencing in seven, and Sanger in two). All individuals with 19
TBC1D24 mutations in our cohort had the five main features of DOORS syndrome, and they 20
represent half of the 18 families with five main features of DOORS, out of the 26 families 21
enrolled. The seizure types in individuals with TBC1D24 mutations included generalized tonic-22

Page 5 of 39
clonic, complex partial, focal clonic, and infantile spasms. In the individuals without TBC1D24 1
mutations, eight did not have seizures and three did not have deafness. We show that some 2
mutations abrogate TBC1D24 mRNA stability. We further demonstrate expression in mouse 3
phalangeal chondrocytes and calvaria, suggesting a role in skeletogenesis. 4
Interpretation: Mutations in TBC1D24 are an important cause of DOORS syndrome. The 5
breadth of phenotypes now known to be caused by TBC1D24 mutations further illustrates the 6
phenomenon of pleiotropy due to mutations in a single gene, especially amongst the 7
epilepsies. Further studies are needed to dissect the cellular roles of TBC1D24 and identify the 8
genes responsible for the phenotype in other individuals. 9
Funding: This work was funded in part by the NIH (USA), the CIHR (Canada), the NIHR (UK), 10
the Wellcome Trust, the Henry Smith Charity and Action Medical Research. 11
12

Page 6 of 39
Introduction 1
2
DOORS or DOOR syndrome [MIM 220500] is a rare autosomal recessive condition of 3
unknown cause. Cantwell first used the acronym DOOR syndrome in 19751 (referring to the 4
Deafness, Onychodystrophy, Osteodystrophy, and mental Retardation in affected individuals), 5
noting that a few affected individuals had been reported prior to that date. Qazi et al. 6
suggested changing the name to DOORS syndrome to account for the presence of seizures in 7
most individuals2: we use this term. Thirty-two affected individuals were reviewed by James et 8
al. in 20073 and five others have been published since that report4-8. Seizures, present in most 9
patients with DOORS syndrome, usually start in the first year of life. They occasionally occur 10
with increasing frequency or severity and are sometimes refractory to antiepileptic medication. 11
The seizures are more often generalized tonic-clonic, but myoclonic, partial and absence 12
seizures also occur3. Neurological involvement, apart from the epilepsy, intellectual deficiency 13
and profound sensorineural hearing loss, includes occasional optic neuropathy, visual 14
impairment, peripheral neuropathy and abnormalities on brain magnetic resonance imaging. 15
The onycho-osteodystrophy affects the hands and feet equally. Small or absent nails and 16
hypoplastic terminal phalanges are noted in most individuals. A triphalangeal thumb is present 17
in one third of affected individuals. A large base of the nose and a bulbous nose are the most 18
common facial dysmorphisms. Cranial anomalies include microcephaly in a third of individuals 19
and a narrow bifrontal diameter in two thirds. The rarity of DOORS syndrome, the absence of 20
any single pathognomonic feature, the significant clinical variability (including malformations of 21
the brain, eyes, heart, kidneys, skeletal system, adrenals and genitalia3), and features shared 22
with other syndromes, make its clinical diagnosis challenging. In turn, this hinders 23

Page 7 of 39
understanding of its true prevalence and prognosis, limiting accurate counseling. Better 1
diagnostic tools are therefore needed. 2
3
For rare diseases in general, a productive approach to improved understanding and more 4
specific diagnosis has been to identify underlying genetic causation. The genetic cause of 5
DOORS syndrome is unknown9. Strategies to identify disease-causing genes in inherited 6
diseases have changed with the development and availability of new technologies. For 7
example, in epilepsy, channelopathies were mostly identified in the 1990s using positional 8
cloning and candidate gene sequencing10. Next-generation sequencing, such as whole exome 9
sequencing, has accelerated the pace of the identification of epilepsy-associated genes. For 10
example, PPRT2 mutations were identified initially in paroxysmal kinesigenic dyskinesia, and 11
are also seen in infantile seizures and febrile seizures11,12. More recently, DEPDC5 mutations 12
were identified in a variety of dominantly-inherited familial focal epilepsies13,14. The precise role 13
of each protein in the central nervous system remains to be determined. In a cohort of patients 14
with a clinical diagnosis of DOORS syndrome, we sought to identify the genetic basis by whole 15
exome sequencing, aiming thus to provide improved diagnostic resolution and, eventually, 16
better understanding. 17
18

Page 8 of 39
Methods 1
Participants 2
Individuals with DOORS syndrome enrolled in our study were evaluated by clinical geneticists 3
and have at least three of the five features of the disease making its acronym, namely 4
deafness, abnormal nails or digits in the hands and/or feet, developmental delay or intellectual 5
disability (previously known as mental retardation), and seizures. We contacted physicians 6
who had published cases previously and previous collaborators. Recruitment of the families 7
described in this report spanned from December 2010 to March 2013. Families were excluded 8
if the parents could not be contacted for consent, or if DNA could not be obtained from the 9
affected individual. Further exclusion criteria were autosomal dominant inheritance and the 10
absence of both intellectual disability and seizures, the same criteria used by James et al. in 11
their literature review3. Thirty families were thus identified; of which four families were excluded 12
from the study. These include two families which could not be re-contacted for informed 13
consent; in another family, the affected child had died and DNA was not available, and in 14
another family, the phenotype corresponded to dominant deafness-onychodystrophy (DDOD, 15
MIM 124480). In all cases, a parent provided informed written consent on behalf of the affected 16
individual. A clinical questionnaire was completed by the collaborating physician. DNA was 17
collected from the affected individual and both parents when both were available. Our clinical 18
study was conducted with the approval of the institutional review boards of the Baylor College 19
of Medicine, of University College London, of the University of Amsterdam and of the 20
University of Manchester. 21
22

Page 9 of 39
Procedures 1
Exome sequencing and analysis: A detailed description of the method for exome sequencing 2
and data analysis is provided in the Supplement (Supplementary methods and Supplementary 3
Table 1). In summary, genomic DNA is fragmented, enriched for coding regions, and 4
sequenced on an Illumina HiSeq 2000 instrument (Illumina, San Diego, CA). Reads are 5
aligned to the reference human genome, and analyzed for variations from the reference. The 6
variants are filtered to keep only rare or novel variants, and these variants are annotated for 7
conservation data, predicted impact of the variant, variant frequency in various databases, 8
gene expression pattern, function of the gene, and phenotypes in mice and humans. Our 9
variant detection and filtering method is deliberately sensitive but not specific, meaning that 10
there are multiple false-positive variants called. Further filtering and visualization of the 11
variants is required. False-positive variant calls can be due to: artifacts introduced by the next-12
generation technology used15, poor coverage in GC-rich regions, bases missed by next-13
generation sequencing because of homopolymers or dinucleotide repeats, and/or mapping 14
difficulties because of gene homologues or paralogues. The most efficient method to remove 15
these false-positive variants is to visualize alignment files (binary alignment map or BAM files, 16
using Broad Institute’s IGV viewer, www.broadinstitute.org/igv) from affected individuals and 17
compare them to variant files from unaffected individuals or individuals with completely 18
separate conditions (exomes published in our previous papers16-20), which we performed after 19
restricting the candidate gene list by mode of inheritance as described in the results section. 20
21

Page 10 of 39
Single Nucleotide Polymorphism array (SNP array) and analysis: Analysis was performed on 1
the Illumina Infinium HD assay platform using HumanOmni1-Quad BeadChip (Illumina Inc., 2
San Diego, CA) according to the manufacturer’s instructions, and the data were analyzed 3
using Illumina’s GenomeStudio to look for insertions, deletions, and regions of homozygosity 4
as well as haplotypes shared by affected siblings. 5
6
Sanger sequencing: Primers used for sequencing the genomic DNA encoding the complete 7
coding sequence of TBC1D24 variant 1 (NCBI CCDS ID#55980.1) were generated by 8
ExonPrimer (http://ihg.gsf.de/ihg/ExonPrimer.html, see Supplementary Table 2 for primers). 9
Amplicons were generated with 10 ng of genomic DNA using TaqMan polymerase (ABI, Life 10
Technologies, Carlsbad, CA) using the manufacturer’s protocol with an annealing temperature 11
of 55°C and an amplification of 1 minute. Products were sequenced by the Sanger method 12
using the same primers, at Beckman Coulter Genomics (Danvers, MA). Resulting 13
chromatograms were analyzed by comparing the GenBank file for the RefSeq sequence of 14
TBC1D24 using Sequencher version 4.8 (Gene Codes Corporation, Ann Arbor, MI). 15
16
Real time PCR: Fibroblasts were grown in Dulbecco's Modified Eagle's Medium with 10% fetal 17
bovine serum, 1.5 mM glutamine, 100 IU/ml penicillin, and 50ng/ml streptomycin (Invitrogen, 18
Grand Island, NY). Mouse tissues were from C57BL/6J wildtype mice. RNA was extracted 19
from fibroblasts or mouse tissues using TRIzol and GlycoBlue, then cDNA was synthesized 20
using SuperScript III First-Strand Synthesis Kit according to the manufacturer’s instructions (all 21
from Invitrogen, Grand Island, NY). Quantitative real-time PCR was performed using the 22

Page 11 of 39
primers in Supplementary Table 2, the FastStart DNA Master SYBR Green reagent and a 1
LightCycler instrument using an annealing temperature of 65°C according to the 2
manufacturer’s protocol (both from Roche NimbleGen, Madison, WI). 3
4
Immunohistochemistry and western blotting methods are detailed in the Supplement. 5
6
Structural model of human TBC1D24: The homology model was generated by Phyre221 7
(www.sbg.bio.ic.ac.uk/phyre2/) based on the crystal structures of the TBC domain of TBC1D1 8
and TBC1D4 and the TLDc domain of the zebrafish Oxr2 protein22. The figure of the model 9
was generated using Pymol (http://pymol.org/). 10
11
Statistical analyses: Statistical analyses were performed using SigmaPlot software v11.0 12
(Systat Software, San Jose, CA). Differences in mRNA expression in fibroblasts were 13
calculated using Student’s t-test. For the expression in different mouse tissues, a Kruskal-14
Wallis One Way Analysis of Variance (ANOVA) on ranks was performed, as well as a multiple 15
comparison procedure using Dunnett's Method. 16
17
18
Role of the funding source 19

Page 12 of 39
The sponsors of the study had no role in study design, data collection, data analysis, data 1
interpretation, or writing of the report. All authors had full access to all the data in the study and 2
had final responsibility for the decision to submit for publication. 3
4

Page 13 of 39
Results 1
2
We performed exome sequencing in the first 17 enrolled families, and SNP-arrays in three 3
families with consanguinity or multiple affected children. Since sequencing and data 4
preparation took months using our procedures, we analyzed exomes and SNP-arrays after we 5
sequencing was completed in 15 families (see flow diagram in Figure 1). Focusing on genes 6
with rare or novel protein-impacting variants (compared to the frequency in the control group, 7
i.e. the 6,503 exomes in the Exome Variant Server), there were 6,645 genes with at least one 8
rare/novel variant in at least one of fifteen samples. As described in the methods and 9
Supplementary Table 3, to restrict the list of candidate genes, we applied filtering based 10
initially on an autosomal recessive inheritance, considered genes mutated in several samples, 11
and removed the false-positive variants. We visually compared the DOORS syndrome exome 12
alignments to other exomes previously acquired for unrelated conditions16-20, focusing initially 13
on genes following a recessive inheritance pattern (one homozygous mutation or two 14
heterozygous mutations) and variants identified in the highest number of affected individuals. 15
Most variants in genes identified in most exomes by our annotation pipeline proved to be false-16
positive variants upon visualization and comparison with “control” exomes (Supplementary 17
Table 3). 18
19
Using the strategy outlined above, we identified either homozygous or compound 20
heterozygous mutations in TBC1D24 (RefSeq# NM_001199107.1) in affected individuals in six 21
of the initial fifteen families in which we performed exome sequencing (see Table 1). All 22

Page 14 of 39
mutations are novel except for the frameshift mutation, seen in the heterozygous carrier state 1
in two of 6,118 individuals sequenced for that region in the June 2013 release of the Exome 2
Variant Server. This server provides variant frequency data from multiple exome studies 3
performed mostly in adults with various heart and lung diseases and controls, and individual 4
phenotypes are not available. Given that mutations in this gene have been identified as the 5
cause of some epilepsies23-27, that seizures are frequent in DOORS syndrome, and that all 6
mutations we identified were novel or very rare and in some cases truncating, this gene was 7
the best candidate in our cohort. Sanger sequencing of TBC1D24 was conducted in all other 8
enrolled families (primers in Supplementary Table 2) or exome sequencing was completed in 9
two individuals for whom exome sequencing was already started at the time. We identified 10
three more families with TBC1D24 mutations, one through an additional exome and two by 11
Sanger sequencing only, totaling nine families with confirmed mutations in a cohort of 26 12
families affected by DOORS syndrome (see Supplementary Table 1 for the analyses 13
performed on each family and Table 1 for TBC1D24 mutations identified). Annotations for all 14
TBC1D24 variants (from DOORS syndrome, the other associated epileptic conditions, and the 15
Exome Variant Server), including population frequency, conservation scores and PolyPhen2 16
scores can be found in Supplementary Table 4. Collectively, other coding variants in TBC1D24 17
are seen in the heterozygous state in 5% of the population. 18
19
A wide variety of seizure types are noted in this cohort of individuals with TBC1D24 mutations, 20
including generalized tonic-clonic, complex partial, focal clonic seizures, and infantile spasms. 21
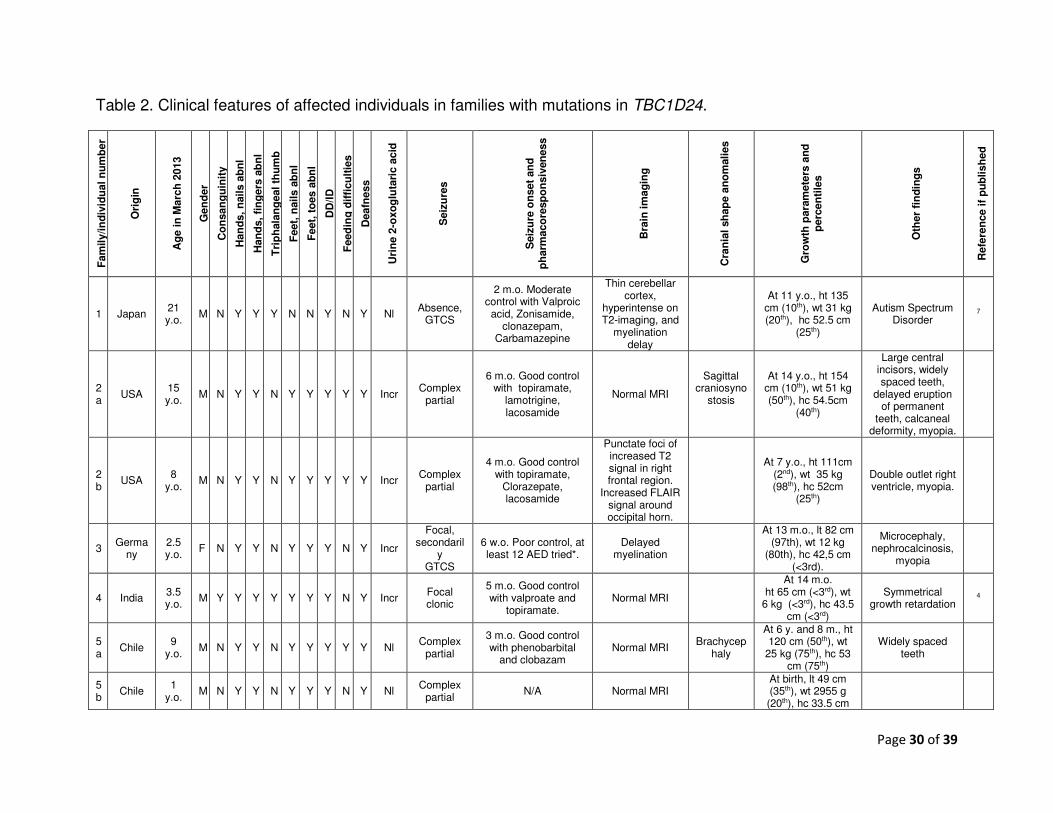
Table 2 lists the clinical features of the individuals with and without TBC1D24 mutations. 22
Photographs of the face, hands, feet and radiographs are shown in Figure 2. The mutations 23

Page 15 of 39
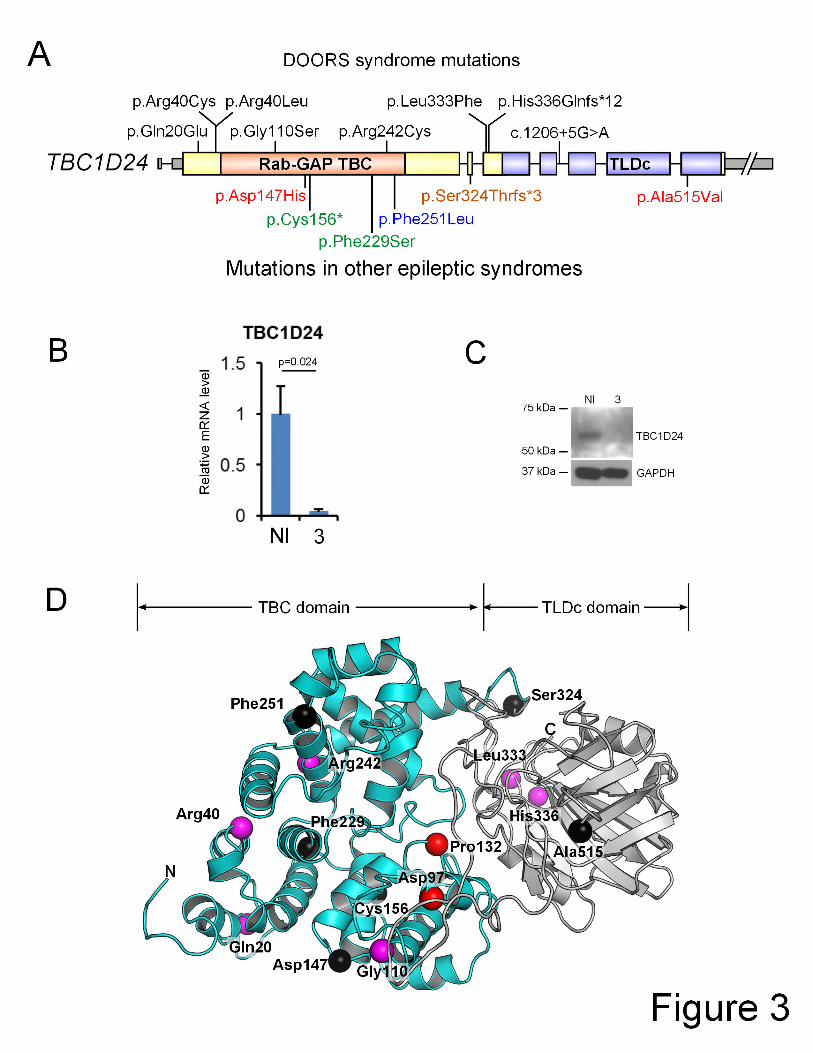
are listed in Table 1 and represented in Figure 3A. The conservation of the affected residues 1
and nucleotides is given in Supplementary Figure 1. In the families with TBC1D24 mutations 2
and consanguinity, the mutations were homozygous and TBC1D24 was in a region of 3
homozygosity as identified by SNP-array or homozygosity mapping from exome data. The 4
regions of homozygosity in these families and other families are given in Supplementary Table 5
5. 6
7
Because of the recessive nature of the disease and the presence of mutations leading to 8
premature protein termination, we proposed that the mutations cause a loss of TBC1D24 9
function. We assessed this hypothesis in fibroblasts from a biopsy previously performed for 10
clinical reasons in individual 3, who was compound heterozygote for a splice site mutation and 11
a frameshift mutation in trans. This frameshift mutation, since it is not in the last exon, is 12
predicted to lead to nonsense-mediated decay (NMD) of the mRNA28. The splice donor 13
mutation after exon five, since it is located five nucleotides away from a splice site 14
(c.1206+5G>A), may or may not affect splicing efficiency. If it affects splicing efficiency, it 15
would lead to an aberrant protein-encoding mRNA, thus engaging NMD. To test for NMD, we 16
performed real-time PCR (Figure 3B). TBC1D24 mRNA levels in the fibroblasts from individual 17
3 were 5% (±1%, standard error) of the levels from three unaffected controls (Student’s t-test: 18
p=0.024), confirming that the mRNA from both alleles undergoes NMD and thus showing a 19
loss of TBC1D24 function. TBC1D24 protein from these fibroblasts could not be detected by 20
Western blot (Figure 3C). 21
22

Page 16 of 39
We next assessed TBC1D24 tissue expression. TBC1D24 is known to be widely expressed, 1
most highly in the brain (especially in pyramidal neurons), kidneys and salivary and lacrimal 2
glands (which have high amounts of secretory vesicles)29,30. In the brain, the regions with the 3
highest expression levels are the hippocampus and the somatomotor areas of the isocortex 4
(see Supplementary Table 6 and Supplementary Figure 2 for a review of available data). Using 5
an antibody against TBC1D24, we studied expression in skeletogenesis. We assessed 6
expression in digital chondrocytes, given the distal phalangeal hypoplasia in DOORS 7
syndrome and the high expression seen in chondrocytes in the Human Protein Atlas. 8
Expression was high in the chondrocytes of the digits (see Supplementary Figure 3). 9
Moreover, we assessed expression by real-time PCR in various newborn mouse tissues and 10
noticed high expression in the calvarium (Supplementary Figure 3F). The high expression in 11
the calvaria correlates well with the cranial shape phenotype and occasional cranial synostosis 12
seen in affected individuals (Table 2). These results, combined with the phenotype in humans, 13
suggest that TBC1D24 might be important in regulating skeletogenesis. 14
15
TBC1D24 contains a TLDc domain and regulates Rab proteins. In our cohort, no mutations 16
were identified in the other TLDc domain-containing genes by exome sequencing (see 17
Supplementary Tables 7-9). By homology modeling, the substitutions we identified do not 18
seem to affect portions of the protein predicted to interact with the TLDc domain, nor are they 19
in the region typically interacting with GTP in other TBC proteins (Figure 3D). Because 20
TBC1D24 regulates Rab proteins and because of clinical overlap with Martsolf syndrome, we 21
also assessed RAB3GAP2. No RAB3GAP2 mutations were identified in families without 22
TBC1D24 mutations (see Supplementary Table 7-9). Moreover, genetic locus mapping by 23

Page 17 of 39
SNP arrays, or homozygosity mapping from exome data in some families without TBC1D24 1
mutations, did not identify a mutated gene in common in these families (details on the regions 2
identified are given in Supplementary Table 5). Analyses are ongoing to identify candidate 3
genes with mutations in three or fewer families following either a de novo dominant or a 4
recessive inheritance pattern. Details on analysis in the other ten exomes without TBC1D24 5
mutations are given in Supplementary Tables 7 to 9. 6
7
Some individuals without TBC1D24 mutations had typical features of DOORS syndrome. 8
including the more specific signs of triphalangeal thumbs and 2-oxoglutaric aciduria, and are 9
clinically indistinguishable from those with TBC1D24 mutation, suggesting genetic 10
heterogeneity in DOORS syndrome, while others lacked some of the typical features such as 11
deafness and seizures and had additional malformations, which suggests that our cohort might 12
include some individuals with different but overlapping conditions (see Table 2 for details). All 13
individuals with TBC1D24 mutations had all five features making the DOORS acronym. If we 14
stratify based on this strict definition of DOORS syndrome, TBC1D24 mutations are found in 15
half of eighteen families. 16
17

Page 18 of 39
Discussion 1
2
We have identified mutations in TBC1D24 as a cause of DOORS syndrome. Previously 3
reported, but different, mutations in TBC1D24 cause various epileptic syndromes. A 4
homozygous p.Phe251Leu substitution in four siblings of an Arab-Israeli family causes focal 5
epilepsy, dysarthria, mild to moderate intellectual disability and cortical thickening with 6
cerebellar atrophy and high T2 and FLAIR signal in the ansiform lobule of the cerebellum on 7
MRI24. In an Italian family, compound heterozygous substitutions (p.Asp147His and 8
p.Ala515Val) cause Familial Infantile Myoclonic Epilepsy (FIME [MIM 605021]) in seven 9
individuals with normal intellect and normal MRI, except for periventricular nodular heterotopia 10
in one individual25. A homozygous truncating mutation (c.969_970delGT, p.Ser324Thrfs*3) in 11
exon 3 was identified in five affected Turkish individuals with a recessive form of myoclonic 12
epilepsy with episodic dystonia, hemiparesis, autonomic signs and lethargy27. This phenotype 13
evolved to chronic dystonia, progressive diffuse cerebral atrophy and early death. Exon 3 is 14
spliced out in isoform 2, which is expressed predominantly in non-neural tissues. Finally, two 15
compound heterozygous substitutions (p.Phe229Ser and p.Cys156*) were identified in two 16
siblings with familial malignant migrating partial seizures of infancy, with progressive diffuse 17
cerebral atrophy of the gray matter sparing the posterior fossa, and early death26. The diversity 18
of seizure types seen in TBC1D24-associated epileptic syndromes and in DOORS syndrome 19
is striking (see Table 2), and may point to a more general epileptogenic mechanism. None of 20
the patients in previous reports of TBC1D24 mutations had digital anomalies or deafness, and 21
none of the above mutations were identified in our DOORS syndrome cohort, demonstrating a 22
clear genotype-phenotype correlation. The reason why certain mutations cause DOORS 23

Page 19 of 39
syndrome while others cause only epilepsy might lie partly in the way the mutations affect 1
interaction patterns with protein partners, an idea which will need to be explored in the future. 2
Some genes implicated in epilepsy seem capable of causing a wide variety of types of 3
epilepsy, with greater or fewer additional neuropsychiatric features, and others have been 4
associated with brain malformations or complex dysmorphic syndromes31. However, 5
phenotypic pleiotropy has rarely been reported to span the spectrum from seizures alone (e.g. 6
previous reports on TBC1D24 mutations) to multi-systemic syndromic disorders (e.g. DOORS 7
syndrome). These emerging phenomena of both genotype-phenotype complexity and genetic 8
pleiotropy support the view that, with accumulating mutational data, the fuller coverage 9
afforded by exome (or genome) sequencing may be more useful than targeted panels in 10
clinical practice. 11
12
In DOORS syndrome, five families have recurrent substitutions affecting the arginine at 13
position 242, and two have substitutions affecting the arginine at position 40, suggesting these 14
are critical in TBC1D24 function. Both are in a CpG island and affect CG dinucleotides. CpG 15
nucleotides are more mutation-prone, which may explain mutation recurrence in individuals of 16
different ethnic background. Two patients also share another recurrent mutation 17
(p.His336Glnfs*12) unique to DOORS. 18
19
TBC1D24 is a member of the Tre2–Bub2–Cdc16 (TBC) domain-containing RAB-specific 20
GTPase-activating proteins which coordinate Rab proteins and other GTPases for the proper 21
transport of intracellular vesicles. It is the only TBC/RabGAP with a TLDc domain (TBC, LysM, 22

Page 20 of 39
Domain catalytic) which is thought to be involved in oxidative stress resistance and to have 1
catalytic activity for unknown substrates32,33. The TLDc domain is also found in the proteins 2
encoded by the human genes OXR1, NCOA7, KIAA1609 and C20orf118 genes, which do not 3
share known functions. Mice lacking Oxr1 exhibit oxidative stress-induced 4
neurodegeneration34, which suggests a possible link with the neurodegeneration seen in some 5
individuals with TBC1D24 mutations27. Mutations were not found in other TLDc domain 6
encoding genes. Several diseases have been associated with other aberrant Rab proteins and 7
Rab-associated proteins. Notably, some clinical overlap exists between DOORS syndrome 8
and Martsolf syndrome (RAB3GAP2 gene, [MIM 212720]; shared aspects include seizures, 9
intellectual deficiency, abnormal toenails and short phalanges), and other syndromes. 10
Mutations were also not identified in RAB3GAP2. 11
12
Further research is needed not only to determine the precise role of TBC1D24 in the nervous 13
system, but also in the skeletal system and other systems affected in DOORS syndrome. In C. 14
elegans, C31H2.1 (a TBC1D24 orthologue) was implicated in synaptic function by an RNAi 15
screen35. In Drosophila, the orthologue Skywalker (Sky) facilitates endosomal trafficking in 16
synaptic vesicles by facilitating GTP hydrolysis by Rab35, thus controlling synaptic vesicle 17
rejuvenation and neurotransmitter release36. Human TBC1D24 has a relatively low similarity to 18
the Drosophila protein36 and neither possesses the arginine and glutamine residues (the so-19
called RQ fingers) which are critical to catalyze GTP hydrolysis by Rab proteins37. Whether 20
human TBC1D24 is able to also facilitate Rab protein-mediated GTP hydrolysis remains to be 21
determined. 22

Page 21 of 39
1
The findings implicate defective vesicular trafficking as the possible basis of the complex 2
phenotype in individuals with DOORS syndrome given previous studies in other TBC proteins. 3
The seizure phenotype present in all individuals with TBC1D24 mutations, and the studies in 4
Drosophila, suggest a role for aberrant neurotransmitter release in the neurological 5
manifestations of the disease. However, further studies in model organisms will be needed to 6
study this in detail. We are currently generating transgenic mice which will also help us to 7
assess these possibilities. 8
9
Exome sequencing, although a very powerful method to identify new genes linked to 10
Mendelian disorders, does have limitations. In a medical genetics clinical setting, exome 11
sequencing has a diagnostic yield of 20-35%38,39. Some limitations are linked to the strategy of 12
sequencing only exons; promoter mutations or deep intronic mutations affecting splicing will be 13
missed. Others limitation are inherent to the analysis strategy; oligogenic inheritance 14
(combination of one mutated allele in each of two or more genes) is not analyzed in our 15
strategy, nor are synonymous exonic variants which could be potentially deleterious. Finally, 16
some limitations are technical: for example, GC-rich regions are difficult to sequence and map, 17
coverage can vary from sample to sample, and reads in genes which have paralogues or 18
homologues can also be difficult to map to the reference genome. Future strategies to identify 19
the causative genes in individuals with DOORS syndrome but without TBC1D24 mutations 20
could include the following: consider oligogenic inheritance models, analyze output for multiple 21
different genes in the same pathway, increase enrollment and number of exomes and/or depth 22

Page 22 of 39
of exome sequencing, perform whole-genome sequencing, and analyze exome data for copy 1
number variants. 2
3
The phenotypic similarity between patients with and without TBC1D24 mutations is highly 4
suggestive of genetic heterogeneity in DOORS syndrome. Individuals with TBC1D24 5
mutations all have the five features making the DOORS acronym. However, other features 6
were greatly variable, in terms of seizure types, pharmacoresponsiveness, brain imaging 7
abnormalities, cranial shape, 2-oxoglutaric aciduria, a triphalangeal thumb, and growth 8
parameters. A similar phenomenon is seen in the individuals without TBC1D24 mutations: nine 9
had the five features making the DOORS acronym, several had 2-oxoglutaric aciduria or a 10
triphalangeal thumb. In the individuals without TBC1D24 mutations, eight did not have seizures 11
and three did not have deafness (including one without seizures or deafness). Further 12
discussion on 2-oxoglutaric aciduria is provided in the supplementary material. 13
14
In conclusion, through a combination of careful clinical phenotyping and exome sequencing, 15
we have identified the molecular basis of DOORS syndrome in approximately a third of 16
individuals included in our cohort, or in half of the eighteen families where affected individuals 17
had the five features making the DOORS acronym. We suggest that individuals without 18
deafness and seizures but with the other features should still be screened for TBC1D24 19
mutations when encountered in a clinical setting since we are only beginning to understand the 20
genetic causation of DOORS syndrome: the discovery of a TBC1D24 mutation in a patient with 21
an appropriate phenotype confirms the diagnosis of DOORS syndrome, but more cases need 22

Page 23 of 39
to be studied to determine the full clinical utility of TBC1D24 mutation testing. As has occurred 1
with other rare diseases that have been genetically solved, we anticipate that this gene 2
discovery will be an important step in galvanizing clinical and scientific progress in 3
understanding and treating DOORS syndrome. 4
5

Page 24 of 39
Research in context 1
Systematic review 2
We searched PubMed and the references of included papers for articles published from 3
January 1970 to March 2013. We included in our search terms: DOOR syndrome, DOORS 4
syndrome, deafness and onychodystrophy, TBC1D24, 2-oxoglutaric aciduria, 2-oxoglutarate, 5
epilepsy and exome sequencing. We collected all previous reports on individuals with DOORS 6
syndrome and assessed the key clinical features. Features present in the majority of patients 7
include sensorineural deafness, nail hypoplasia, terminal phalangeal hypoplasia, triphalangeal 8
thumb,developmental delay, intellectual disability, seizures, craniofacial anomalies, 2-9
oxoglutaric aciduria, and MRI anomalies. Features present in ≥25% include consanguinity or 10
affected siblings, and optic atrophy. Features present in <25% include congenital heart 11
defects, urinary tract anomalies, and peripheral neuropathy. 12
Interpretation 13
This study identifies mutations in TBC1D24 as the genetic cause in a proportion of individuals 14
with DOORS syndrome, and implies that testing for TBC1D24 mutation should be considered 15
when the diagnosis of DOORS syndrome is contemplated. The study confirms the role of this 16
gene in a wide variety of epilepsy syndromes, a phenomenon seen also with other epilepsy-17
related genes such as SCN1A, KCNQ2 and PRRT2. Moreover, it expands the growing 18
concept of pleiotropy in epilepsy genetics because whilst some mutations in TBC1D24 can 19
cause mild epilepsy without significant associated features, other mutations cause epilepsy as 20
part of a multiorgan syndrome, with features beyond the nervous system alone. The resulting 21
important practical consequences of such clinical and genetic heterogeneity for daily 22

Page 25 of 39
neurological practice, with its wealth of rare diseases, are that robust identification of genetic 1
mutations is a valuable adjunct to diagnosis, and that whole exome (or genome) sequencing 2
meeting clinical standards is likely to prove to be the best tool to provide the comprehensive 3
genomic coverage required. 4
5

Page 26 of 39
Supplemental Data 1
Supplemental Data can be found with this article online. 2
3

Page 27 of 39
Contributors 1
PMC designed the study, performed experiments, analyzed the data and wrote the manuscript. 2
DK performed experiments and analyzed the data. JTL, SSB, and JED analyzed the exome 3
data. AT performed experiments. LCB helped with Sanger sequencing. JW and SWC 4
conducted SNP array data analysis. RCH collected clinical data. CK performed 3D model 5
representations. RAG supervised exome sequencing. MH, BRP, FS, TMF, JvdE, MW, HK, PR, 6
SN, SA, AM, LDVN, MLB, IDB, GM, MLM, GMR, MG, EB, AM, FG, AK, WGN, BK, SB, JCG, 7
and DW contributed patient samples and collected clinical data. BHL and SMS jointly 8
supervised the research and revised the manuscript. 9
10
Conflicts of interest 11
All authors have no conflict of interest to declare. 12
13
Acknowledgements 14
We thank David Liu for technical assistance, Alyssa Tran and Dale Kerr for clinical research 15
support, Shalini N. Jhangiani for exome sequencing coordination, Sabrina Hong for the 16
representation of the structural model of TBC1D24, and Costin Leu for assistance with exome 17
sequencing statistics. Funding was provided by NIH grants PO1 HD22657 (BHL), U54 18
HG006542 (RAG) and U54 HG003273-09 (RAG), by The Rolanette and Berdon Lawrence 19
Bone Disease Program of Texas (BL), by the Wellcome Trust (SMS), the Henry Smith Charity 20
(SMS) and Action Medical Research (SMS). This work was supported by the BCM Intellectual 21

Page 28 of 39
and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy 1
Shriver National Institute Of Child Health & Human Development. This work was partly 2
undertaken at UCLH/UCL, which received a proportion of funding from the Department of 3
Health’s NIHR Biomedical Research Centres funding scheme. PMC is supported by a CIHR 4
clinician-scientist training award and the O'Malley Foundation. JTL is supported by Ruth L. 5
Kirschstein National Research Service Award F30 MH098571-01. LCB was supported by the 6
Medical Genetics Research Fellowship Program NIH/NIGMS NIH T32 GM07526. 7
Page 29 of 39
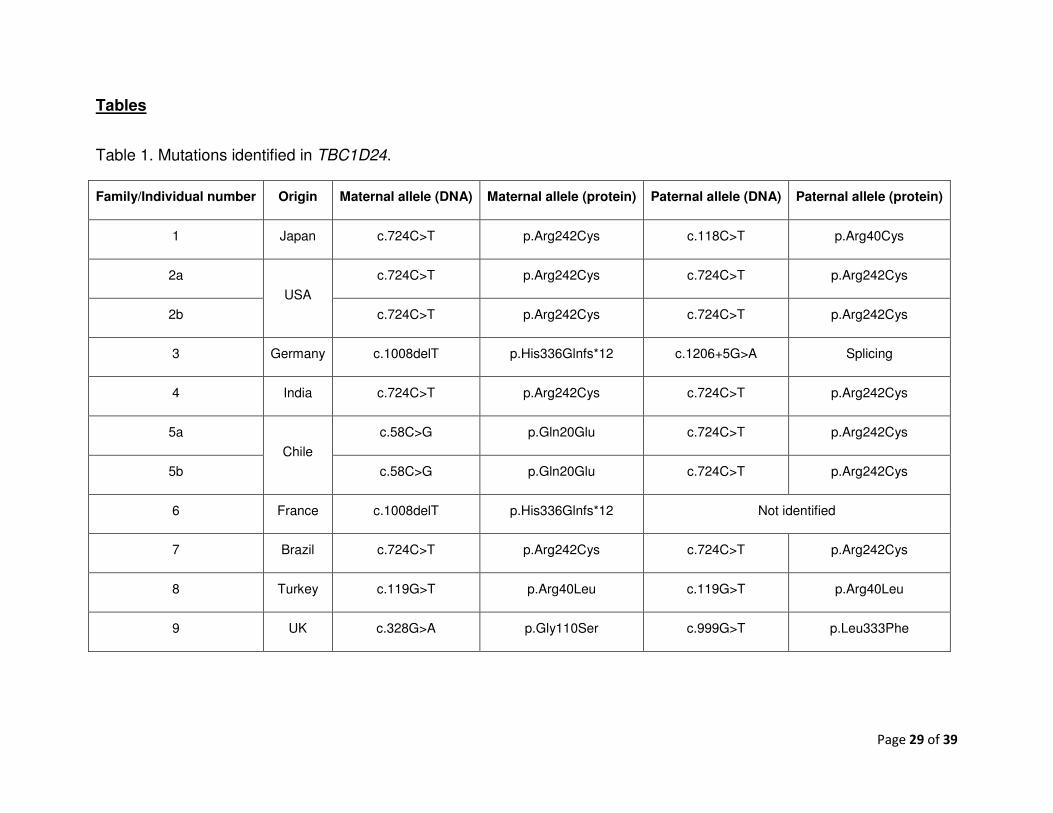
Tables
Table 1. Mutations identified in TBC1D24.
Family/Individual number Origin Maternal allele (DNA) Maternal allele (protein) Paternal allele (DNA) Paternal allele (protein)
1 Japan c.724C>T p.Arg242Cys c.118C>T p.Arg40Cys
2a
USA
c.724C>T p.Arg242Cys c.724C>T p.Arg242Cys
2b c.724C>T p.Arg242Cys c.724C>T p.Arg242Cys
3 Germany c.1008delT p.His336Glnfs*12 c.1206+5G>A Splicing
4 India c.724C>T p.Arg242Cys c.724C>T p.Arg242Cys
5a
Chile
c.58C>G p.Gln20Glu c.724C>T p.Arg242Cys
5b c.58C>G p.Gln20Glu c.724C>T p.Arg242Cys
6 France c.1008delT p.His336Glnfs*12 Not identified
7 Brazil c.724C>T p.Arg242Cys c.724C>T p.Arg242Cys
8 Turkey c.119G>T p.Arg40Leu c.119G>T p.Arg40Leu
9 UK c.328G>A p.Gly110Ser c.999G>T p.Leu333Phe
Page 30 of 39
Table 2. Clinical features of affected individuals in families with mutations in TBC1D24. F
am
ily/in
div
idu
al n
um
ber
Ori
gin
Ag
e i
n M
arc
h 2
013
Gen
der
Co
nsan
gu
init
y
Han
ds,
nail
s a
bn
l
Han
ds,
fin
gers
ab
nl
Tri
ph
ala
ng
eal
thu
mb
Feet,
nail
s a
bn
l
Feet,
to
es a
bn
l
DD
/ID
Feed
ing
dif
ficu
ltie
s
Deafn
ess
Uri
ne 2
-oxo
glu
tari
c a
cid
Seiz
ure
s
Seiz
ure
on
set
an
d
ph
arm
aco
resp
on
siv
en
ess
Bra
in im
ag
ing
Cra
nia
l sh
ap
e a
no
mali
es
Gro
wth
para
mete
rs a
nd
p
erc
en
tile
s
Oth
er
fin
din
gs
Refe
ren
ce i
f p
ub
lish
ed
1 Japan 21 y.o.
M N Y Y Y N N Y N Y Nl Absence,
GTCS
2 m.o. Moderate control with Valproic acid, Zonisamide,
clonazepam, Carbamazepine
Thin cerebellar cortex,
hyperintense on T2-imaging, and
myelination delay
At 11 y.o., ht 135 cm (10th), wt 31 kg (20th), hc 52.5 cm
(25th)
Autism Spectrum Disorder
7
2a
USA 15 y.o.
M N Y Y N Y Y Y Y Y Incr Complex
partial
6 m.o. Good control with topiramate,
lamotrigine, lacosamide
Normal MRI
Sagittal craniosyno
stosis
At 14 y.o., ht 154 cm (10th), wt 51 kg (50th), hc 54.5cm
(40th)
Large central incisors, widely spaced teeth,
delayed eruption of permanent
teeth, calcaneal deformity, myopia.
2b
USA 8
y.o. M N Y Y N Y Y Y Y Y Incr
Complex partial
4 m.o. Good control with topiramate,
Clorazepate, lacosamide
Punctate foci of increased T2 signal in right frontal region.
Increased FLAIR signal around occipital horn.
At 7 y.o., ht 111cm (2nd), wt 35 kg (98th), hc 52cm
(25th)
Double outlet right ventricle, myopia.
3 Germa
ny 2.5 y.o.
F N Y Y N Y Y Y N Y Incr
Focal, secondaril
y GTCS
6 w.o. Poor control, at least 12 AED tried*.
Delayed myelination
At 13 m.o., lt 82 cm (97th), wt 12 kg
(80th), hc 42,5 cm (<3rd).
Microcephaly, nephrocalcinosis,
myopia
4 India 3.5 y.o.
M Y Y Y Y Y Y Y N Y Incr Focal clonic
5 m.o. Good control with valproate and
topiramate. Normal MRI
At 14 m.o. ht 65 cm (<3rd), wt
6 kg (<3rd), hc 43.5 cm (<3rd)
Symmetrical growth retardation
4
5a
Chile 9
y.o. M N Y Y N Y Y Y Y Y Nl
Complex partial
3 m.o. Good control with phenobarbital
and clobazam Normal MRI
Brachycephaly
At 6 y. and 8 m., ht 120 cm (50th), wt
25 kg (75th), hc 53 cm (75th)
Widely spaced teeth
5b
Chile 1
y.o. M N Y Y N Y Y Y N Y Nl
Complex partial
N/A Normal MRI At birth, lt 49 cm (35th), wt 2955 g (20th), hc 33.5 cm
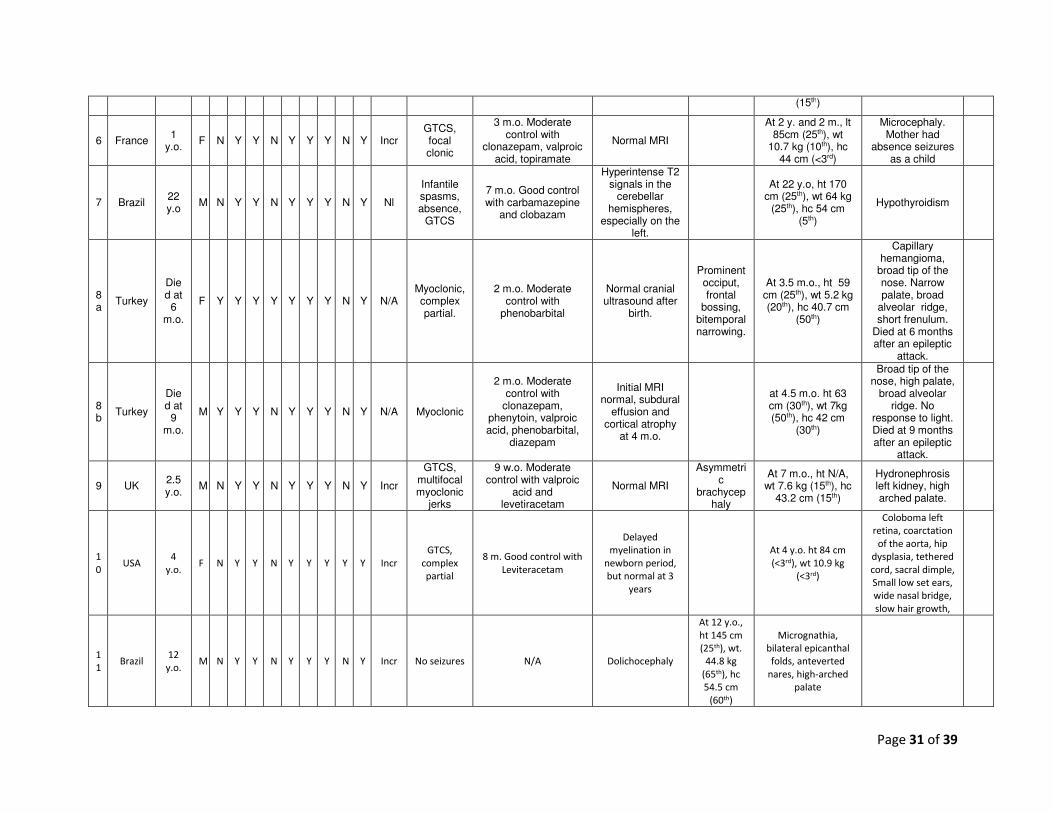
Page 31 of 39
(15th)
6 France 1
y.o. F N Y Y N Y Y Y N Y Incr
GTCS, focal clonic
3 m.o. Moderate control with
clonazepam, valproic acid, topiramate
Normal MRI
At 2 y. and 2 m., lt 85cm (25th), wt
10.7 kg (10th), hc 44 cm (<3rd)
Microcephaly. Mother had
absence seizures as a child
7 Brazil 22 y.o
M N Y Y N Y Y Y N Y Nl
Infantile spasms, absence,
GTCS
7 m.o. Good control with carbamazepine
and clobazam
Hyperintense T2 signals in the
cerebellar hemispheres,
especially on the left.
At 22 y.o, ht 170 cm (25th), wt 64 kg
(25th), hc 54 cm (5th)
Hypothyroidism
8a
Turkey
Died at
6 m.o.
F Y Y Y Y Y Y Y N Y N/A Myoclonic, complex partial.
2 m.o. Moderate control with
phenobarbital
Normal cranial ultrasound after
birth.
Prominent occiput, frontal
bossing, bitemporal narrowing.
At 3.5 m.o., ht 59 cm (25th), wt 5.2 kg (20th), hc 40.7 cm
(50th)
Capillary hemangioma,
broad tip of the nose. Narrow palate, broad
alveolar ridge, short frenulum.
Died at 6 months after an epileptic
attack.
8b
Turkey
Died at
9 m.o.
M Y Y Y N Y Y Y N Y N/A Myoclonic
2 m.o. Moderate control with
clonazepam, phenytoin, valproic acid, phenobarbital,
diazepam
Initial MRI normal, subdural
effusion and cortical atrophy
at 4 m.o.
at 4.5 m.o. ht 63 cm (30th), wt 7kg (50th), hc 42 cm
(30th)
Broad tip of the nose, high palate,
broad alveolar ridge. No
response to light. Died at 9 months after an epileptic
attack.
9 UK 2.5 y.o.
M N Y Y N Y Y Y N Y Incr
GTCS, multifocal myoclonic
jerks
9 w.o. Moderate control with valproic
acid and levetiracetam
Normal MRI
Asymmetric
brachycephaly
At 7 m.o., ht N/A, wt 7.6 kg (15th), hc
43.2 cm (15th)
Hydronephrosis left kidney, high arched palate.
1
0 USA
4
y.o. F N Y Y N Y Y Y Y Y Incr
GTCS,
complex
partial
8 m. Good control with
Leviteracetam
Delayed
myelination in
newborn period,
but normal at 3
years
At 4 y.o. ht 84 cm
(<3rd), wt 10.9 kg
(<3rd)
Coloboma left
retina, coarctation
of the aorta, hip
dysplasia, tethered
cord, sacral dimple,
Small low set ears,
wide nasal bridge,
slow hair growth,
1
1 Brazil
12
y.o. M N Y Y N Y Y Y N Y Incr No seizures N/A Dolichocephaly
At 12 y.o.,
ht 145 cm
(25th), wt.
44.8 kg
(65th), hc
54.5 cm
(60th)
Micrognathia,
bilateral epicanthal
folds, anteverted
nares, high-arched
palate
Page 32 of 39
1
2
a
Belgiu
m
Died
at
3.8
y.o.
F N Y Y Y Y Y Y Y Y Incr GTCS 3 m. Resistant to
therapy.
Hyperintensities in
the pons
Metopic
ridge,
biparietal
narrowing,
At 3 m.o., Ht 3rd, Wt
3rd, hc 10-25th
Metopic ridge,
biparietal
narrowing,
unilateral hearing
loss. Short fixation,
abnormal eye
movements, and
intermittent
strabismus,
Aspiration
pneumonias,
dysplastic ears.
9
1
2
b
Belgiu
m
5
y.o. M N Y Y Y Y Y Y Y Y Incr Myoclonic 3 w. Poor control.
Thin corpus
callosum
At birth, normal
growth parameters
At 2 months, VEP
showed a weak
response, and ERG
was normal. Patent
ductus arteriosus,
dysplastic ears.
9
1
3 India
Died
at 3
m.o.
F Y Y Y Y Y Y Y N N N/A No seizures N/A Broad forehead
At birth,
2200 g
(<3rd), at
2.5 m.o., hc
33 cm
(<3rd)
Microcephaly,
progeroid
appearance, bilateral
low set ears,
dysplastic pinna, high
arched palate,
macrostomia,
submucous clefting
of palate, protruding
tongue, anteverted
nares, long smooth
philtrum,
hypertrichosis, broad
forehead
1
4
Netherl
ands
2.5
y.o. F N Y Y N N N Y Y Y Nl No seizures N/A
At 10 m.o.
ht 69cm
(15th), wt
6.6 kg
(<3rd), hc
42,5cm
(5th)
VSD, long eyelashes,
cleft palate, cup-
shaped ears
1
5 India
8
y.o. F N Y Y N Y Y Y N Y N/A
Focal
motor 4. y.o. Good control. Normal MRI
High
forehead
At 4.5 y.o., ht<3rd,
Wt<3rd
hc<3rd
Microcephaly,
horizontal
nystagmus
1
6
UK of
African
origin
6
y.o. M N Y Y N Y Y Y Y Y N/A GTCS
2y9m. Good control
with valproic acid.
Partial corpus
callosum agenesis.
Small lesion in
right putamen
suggestive of a
At 3y3m, wt 15.6 kg
(65th),
ht 93.1 cm (15th),
hc 48.1 cm (5th)
Severe gastro-
oesophageal reflux,
ASD requiring
surgical repair,
coarse facial
Page 33 of 39
developmental
venous anomaly
features, low
frontal hairline,
webbed neck,
micrognathia,
midline groove of
lower lip, thick lips,
thickened gingiva,
long tongue,
anteverted
abnormally formed
ears. Severe
kyphoscoliosis
(congenital) and
calcaneovalgus.
Tracheomalacia,
tracheostomy,
multiple keloid scar
formation.
1
7
Lebano
n
1
y.o. F N Y Y N Y Y Y Y Y N/A No seizures Aplasia of falx cerebri
At 11w, lt
57 cm (8th),
wt 4.5 kg
(3rd), hc 36
cm (<3rd)
High frontal and
temporal hairline,
hypertelorism,
upslanting palpebral
fissures, broad nose,
low-set and
posteriorly rotated
ears. Hypoplastic
patellae. Uterus
bicornis,
hypercalcemia
1
8 Iran
14
y.o. M Y Y Y N N N Y N Y N/A GTCS
1 y.o. Good control with
phenobarbital N/A
Brachyceph
aly
At 14y.o., hc 50.5
(<3rd)
Multicystic left
kidney,
hypertrophic right
kidney, cleft palate,
bilat inguinal
hernia, short PF,
blepharophimosis,
microcornea,
telecanthus,
prominent nose,
bilateral narrowing
of ear canals, small
mouth,
malocclusion of
teeth, hallux valgus.
Affected sibling
passed away at 1.5
y.o.
Page 34 of 39
1
9 USA
Died
at 3
w.o.
M N Y Y N Y Y Y N Y Nl No seizures
Dandy-Walker
malformation, agenesis
of the corpus callosum
At birth, lt
44 cm
(<3rd), wt
2.26 kg
(<3rd), hc 33
cm (17th)
Blindness, widely-
spaced nipples low-
set ears,
2
0 Canada
2
y.o. F N Y Y N Y Y Y Y Y Nl No seizures
Dandy-Walker
malformation, small
focal area of restricted
diffusion in the right
posterior basal ganglia
area
Large anterior
fontanelle,
microcephaly,
narrow bifrontal
diameter,
At 1 y.o., ht
<3rd, wt
<3rd, hc <3rd
Sparse fine hair,
morning glory
anomaly right optic
nerve, persistent left
superior vena cava
draining into the
coronary sinus, atrial
spetal defect, mild
tricuspid
regurgitation,
laryngomalacia, short
sternum, right
thoracic scoliosis,
low conus
medullaris, rocker
bottom feet, widely-
spaced nipples, high
arched palate, short
neck
2
1 Ireland
Died
at
10
y.o.
M N Y N Y Y N Y Y Y Nl
GTCS,
absence,
myoclonic
6 m.o. Normal MRI Dolichocep
haly
At 3y6m, ht 110 cm
(>97th), wt 19 kg
(95th), hc 58 cm
(>97th)
Double outlet right
ventricle, recurrent
chest infections,
supernumary
nipple, broad
alveolar ridge,
blind,
hydronephrosis.
2
2
UK of
Indian
origin
29
y.o. F N Y N N Y N Y Y N Nl
GTCS,
Complex
partial,
episodes of
status
epilepticus
3 m.o. Moderate
control with valproate,
levetiracetam,
pregabalin, phenytoin,
topiramate
Diffuse atrophy
cerebrum and
cerebellum,
asymmetric lateral
ventricles
At 29 y.o., wt 50 kg
(15th)
Long thin face, high
arched palate.
2
3 India
7
y.o. M Y Y Y Y Y N Y N N Nl GTCS
11m. Good control with
phenytoin and
phenobarbital
Normal MRI
6 y.o., ht 110 cm
(15th), wt 18 kg(15th),
hc 46.5 cm (<3rd)
Delayed permanent
dentition, small
teeth, cavities,
anteverted nares,
low set pinna, open
mouth, Autism
Spectrum Disorder.
Page 35 of 39
Sibling with similar
phenotype died at
1y9m.
2
4 Poland
22
y.o. M Y Y Y Y Y Y Y N Y Nl No seizures N/A
At birth lt
57 cm
(>97th), ht
3.9 kg
(75th), hc 32
cm (4th)
Delayed teething,
haemangioma,
epicanthus of the left
eye, ptosis,
strabismus,
asymmetric face,
high arched palate.
8
2
5
New
Zealan
d
12
y.o. M N Y Y Y Y Y Y Y Y Nl
Infantile
spasms
1 y.o. Good control with
vigabatrin N/A
At 7 y.o., ht 188 cm
(>97th), wt 24 kg
(60th)
Broad nose, dental
misalignment and
irregularities,
Autism Spectrum
Disorder.
2
6 UK
15
y.o. M N Y Y Y Y Y Y N Y Nl No seizures N/A
At 5m, lt
64.5cm
(25th), wt
6.3 kg (5th),
hc 44.5 cm
(75th)
Premature birth
Abbreviations: Incr, Increased; N/A, not available; Nl, normal, Y, yes; N, No; abnl, abnormal; M, male; F, female; DD/ID,
developmental delay or intellectual disability, lt, length; ht, height; wt, weight; hc, head circumference; y.o., year old; m.o.,
month old; w.o., week old. GTCS, generalized tonic-clonic seizures. Individuals with letters a and b after the number are
siblings. Recent cognitive or developmental evaluations were not available for most individuals, but the global
development was estimated for individual 8a as that of 6-week-old when she was 12 weeks old, for individual 5a as that of
a 1 year old when he was 3 years old, and individual 1 had a developmental quotient of 55 with autistic spectrum disorder
when he was 4 years old. Formal audiometric test results were not available for most individuals, but the hearing loss was
qualified as profound sensorineural hearing loss in individuals 1, 2b, 3, 4, and 6. Cochlear implants have been beneficial
in individual 3. Antiepileptic drugs (AEDs) used or tried with limited success in individual 3 include oxcarbazepine,
clobazam, levetiracetam, valproic acid, lamotrigine, sulthiame, diazepam, clonazepam, midazolam, zonisamide, and
cortisone pulses. In family 8, DNA was not available from individual 8b, only for 8a and parents. Sensorineural hearing
loss was unilateral in individual 12a; in individual 15, it was severe on the right and moderate on the left. Recent cognitive
or developmental evaluations were not available for most individuals, but the intellectual disability or developmental delay
was categorised as mild for individual 24, moderate for individual 14, and severe for individuals 10, 11, 12a, 12b, 16, 21,
22, 23 and 25.

Page 36 of 39
References
1. Cantwell RJ. Congenital sensori-neural deafness associated with onycho-osteo dystrophy and mental retardation
(D.O.O.R. syndrome). Humangenetik 1975; 26(3): 261-5.
2. Qazi QH, Nangia BS. Abnormal distal phalanges and nails, deafness, mental retardation, and seizure disorder: A
new familial syndrome. The Journal of Pediatrics 1984; 104(3): 391-4.
3. James AW, Miranda SG, Culver K, Hall BD, Golabi M. DOOR syndrome: clinical report, literature review and
discussion of natural history. Am J Med Genet A 2007; 143A(23): 2821-31.
4. Girish M, Mujawar N, Salodkar A. DOOR syndrome. Indian Pediatr 2011; 48(6): 479-81.
5. Michalek P, Donaldson W, Abraham A. Anaesthetic management of an adult patient with DOOR syndrome: a
case report. Cases J 2009; 2: 7593.
6. Mihci E, Guney K, Velipasaoglu S. DOOR (deafness, onychodystrophy, osteodystrophy, mental retardation)
syndrome in one of the twins after conception with intracytoplasmic sperm injection. Am J Med Genet A 2008; 146A(11):
1483-5.
7. Nomura T, Koyama N, Yokoyama M, Awaya A, Yokochi K. DOOR syndrome concomitant with non-convulsive
status epilepticus and hyperintense cerebellar cortex on T2-weighted imaging. Brain Dev 2009; 31(1): 75-8.
8. Wisniewska M, Siwinska Z, Felczak M, Wielkoszynski T, Krawczynski M, Latos-Bielenska A. A new case of DOOR
syndrome. J Appl Genet 2008; 49(1): 101-3.
9. van Bever Y, Balemans W, Duval EL, et al. Exclusion of OGDH and BMP4 as candidate genes in two siblings with
autosomal recessive DOOR syndrome. Am J Med Genet A 2007; 143(7): 763-7.
10. Helbig I, Lowenstein DH. Genetics of the epilepsies: where are we and where are we going? Curr Opin Neurol
2013; 26(2): 179-85.
11. Marini C, Conti V, Mei D, et al. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and
hemiplegic migraine. Neurology 2012; 79(21): 2109-14.
12. Scheffer IE, Grinton BE, Heron SE, et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile
seizures. Neurology 2012; 79(21): 2104-8.
13. Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet
2013; 45(5): 552-5.
14. Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci.
Nat Genet 2013; 45(5): 546-51.
15. Nothnagel M, Herrmann A, Wolf A, et al. Technology-specific error signatures in the 1000 Genomes Project data.
Hum Genet 2011; 130(4): 505-16.
16. Burrage LC, Lu JT, Liu DS, et al. Early childhood presentation of Czech dysplasia. Clin Dysmorphol 2013; 22(2): 76-
80.
17. Shapiro JR, Lietman C, Grover M, et al. Phenotypic variability of osteogenesis imperfecta type V caused by an
IFITM5 mutation. J Bone Miner Res 2013; 28(7): 1523-30.
18. Campeau PM, Lu JT, Sule G, et al. Whole-exome sequencing identifies mutations in the nucleoside transporter
gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum Mol Genet 2012; 21(22): 4904-9.
19. Campeau PM, Kim JC, Lu JT, et al. Mutations in KAT6B, encoding a histone acetyltransferase, cause
Genitopatellar syndrome. Am J Hum Genet 2012; 90(2): 282-9.
20. Sule G, Campeau PM, Zhang VW, et al. Next-generation sequencing for disorders of low and high bone mineral
density. Osteoporos Int 2013; 24(8): 2253-9.
21. Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat
Protoc 2009; 4(3): 363-71.
22. Blaise M, Alsarraf HM, Wong JE, et al. Crystal structure of the TLDc domain of oxidation resistance protein 2
from zebrafish. Proteins 2012; 80(6): 1694-8.
23. Afawi Z, Mandelstam S, Korczyn AD, et al. TBC1D24 mutation associated with focal epilepsy, cognitive
impairment and a distinctive cerebro-cerebellar malformation. Epilepsy Res 2013; 105(1-2): 240-4.
24. Corbett MA, Bahlo M, Jolly L, et al. A focal epilepsy and intellectual disability syndrome is due to a mutation in
TBC1D24. Am J Hum Genet 2010; 87(3): 371-5.

Page 37 of 39
25. Falace A, Filipello F, La Padula V, et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile
myoclonic epilepsy. Am J Hum Genet 2010; 87(3): 365-70.
26. Milh M, Falace A, Villeneuve N, et al. Novel Compound Heterozygous Mutations in TBC1D24 Cause Familial
Malignant Migrating Partial Seizures of Infancy. Hum Mutat 2013.
27. Guven A, Tolun A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J Med Genet 2013;
50(3): 199-202.
28. Huang L, Wilkinson MF. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip Rev RNA 2012; 3(6):
807-28.
29. Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res
2013; 41(D1): D561-5.
30. Lattin JE, Schroder K, Su AI, et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages.
Immunome Res 2008; 4: 5.
31. Tavyev Asher YJ, Scaglia F. Molecular bases and clinical spectrum of early infantile epileptic encephalopathies.
Eur J Med Genet 2012; 55(5): 299-306.
32. Elliott NA, Volkert MR. Stress induction and mitochondrial localization of Oxr1 proteins in yeast and humans.
Mol Cell Biol 2004; 24(8): 3180-7.
33. Murphy KC, Volkert MR. Structural/functional analysis of the human OXR1 protein: identification of exon 8 as
the anti-oxidant encoding function. BMC Mol Biol 2012; 13: 26.
34. Oliver PL, Finelli MJ, Edwards B, et al. Oxr1 is essential for protection against oxidative stress-induced
neurodegeneration. PLoS Genet 2011; 7(10): e1002338.
35. Sieburth D, Ch'ng Q, Dybbs M, et al. Systematic analysis of genes required for synapse structure and function.
Nature 2005; 436(7050): 510-7.
36. Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic
endosomes as sorting stations for synaptic vesicle proteins. Cell 2011; 145(1): 117-32.
37. Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a
dual-finger mechanism. Nature 2006; 442(7100): 303-6.
38. Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N
Engl J Med 2013; 369(16): 1502-11.
39. Yu Y, Wu BL, Wu J, Shen Y. Exome and whole-genome sequencing as clinical tests: a transformative practice in
molecular diagnostics. Clin Chem 2012; 58(11): 1507-9.
40. Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM. Illuminating the functional and structural repertoire of
human TBC/RABGAPs. Nat Rev Mol Cell Biol 2012; 13(2): 67-73.

Page 38 of 39
Figures Legends
Figure 1. Flow diagram illustrating the enrollment process, genetic analyses and gene identification.
Figure 2. A-F) Photographs of the face in individuals with TBC1D24 mutations. Note the wide base of
the nose and bulbous end of the nose in some individuals (image-individual correspondence: a-3, b-
6, c-5a, d-2b, e-2a, f-7). G-L) Photographs of the hands in individuals with TBC1D24 mutations. Note
the triphalangeal thumbs, the brachydactyly, the short terminal phalanges and the hypoplasia or
aplasia of the nails (image-individual correspondence: g-3, h-6, i-5a, j-2b, k-2a, l-7). M-O) Photograph
of the feet in an individual with TBC1D24 mutations. Note the short terminal phalanges and the
hypoplasia or aplasia of the nails (image-individual correspondence: m-3, n-6, o-5a). P-S)
Radiographs of the hands in individuals with TBC1D24 mutations. Note the triphalangeal thumbs and
the short terminal phalanges (image-individual correspondence: p-4, q-5a, r-6, s-1). T-U)
Radiographs of the feet in individuals with TBC1D24 mutations. Note the short terminal phalanges
(image-individual correspondence: t-6, u-5a).
Figure 3. A) Location of the mutations identified in DOORS syndrome and in other epilepsy
syndromes. p.Phe251Leu (blue – homozygous mutation, affecting four siblings): focal epilepsy,
dysarthria, intellectual disability, cortical thickening, cerebellar atrophy23,24. p.Asp147His and
p.Ala515Val (red – compound heterozygous mutation, affecting seven individuals in one family):
Familial Infantile Myoclonic Epilepsy25. p.Ser324Thrfs*3 (brown – homozygous mutation, affecting five
individuals in one family): myoclonic epilepsy, dystonia, hemiparesis, autonomic signs, lethargy,

Page 39 of 39
progressive diffuse cerebral atrophy27. p.Phe229Ser and p.Cys156* (green – compound
heterozygous mutation affecting two siblings): familial malignant migrating partial seizures of infancy,
progressive diffuse cerebral atrophy26. The diagram also illustrates the exonic structure of TBC1D24,
with the introns not drawn to scale.
B) Real-time PCR of TBC1D24 in fibroblasts from the individual with a frameshift deletion and a
splicing mutation (Nl stands for Normal control fibroblasts, and 3 refers to individual 3) showing
significant reduction of TBC1D24 mRNA in affected fibroblasts. C) Western blot analysis of the cells
used for panel B, showing TBC1D24 protein is undetectable by this method in affected fibroblasts. D)
Structural model of TBC1D24 with the TBC domain colored in cyan and the TLDc domain colored in
gray. The N- and C-termini are labeled, and the red spheres show the alpha carbon atoms of the
residues aligning with the arginine and glutamine fingers interacting with the GTP of Rab proteins in
other TBC proteins40, based on the structure of Gyp1p in complex with Rab3337. The carbon atoms of
residues substituted in DOOR/S syndrome are shown with magenta spheres and those substituted in
other epilepsy syndromes are shown with black spheres.
Page 1 of 22
Supplementary Data for:
The genetic basis of DOORS syndrome: an exome sequencing study.
Philippe M Campeau, Dalia Kasperaviciute, James T Lu, Lindsay C Burrage, Choel Kim, Mutsuki Hori, Berkley R Powell, Fiona Stewart, Têmis Maria
Félix, Jenneke van den Ende, Marzena Wisniewska, Hülya Kayserili, Patrick Rump, Sheela Nampoothiri, Salim Aftimos, Antje Mey, Lal D.V. Nair, Michael
L Begleiter, Isabelle De Bie, Girish Meenakshi, Mitzi L. Murray, Gabriela M Repetto, Mahin Golabi, Edward Blair, Alison Male, Fabienne Giuliano, Ariana
Kariminejad, William G Newman, Sanjeev S Bhaskar, Jonathan E Dickerson, Bronwyn Kerr, Siddharth Banka, Jacques C Giltay, Dagmar Wieczorek, Anna
Tostevin, Joanna Wiszniewska, Sau Wai Cheung, Raoul C. Hennekam, Richard A Gibbs, Brendan H Lee, Sanjay M Sisodiya.
Contents
Supplementary Methods. Exome sequencing and analysis, Immunohistochemistry and Western blotting. .............................................................................. 2
Supplementary Table 1. Genetic analyses performed for each affected individual. ................................................................................................................... 4
Supplementary Table 2. Primers used for Sanger sequencing and real-time PCR. ................................................................................................................... 6
Supplementary Table 3. Table illustrating the process by which we identified recurrent mutations in TBC1D24 in the cohort of individuals with DOOR/S
syndrome. .................................................................................................................................................................................................................................... 7
Supplementary Table 4. Annotations for all known variants in TBC1D24................................................................................................................................... 8
Supplementary Table 5. Candidate regions from SNP array or homozygosity mapping of exome data. ................................................................................. 11
Supplementary Table 6. Review of the expression data on TBC1D24 in human and mouse tissues, detected at the level of mRNA or protein, from
publications or public databases. .............................................................................................................................................................................................. 13
Supplementary Table 7. Coverage data for candidate genes. .................................................................................................................................................. 14
Supplementary Table 8. Candidate gene analysis from exome data in 10 individuals without TBC1D24 mutations. .............................................................. 15
Supplementary Table 9. Gene names for recessive model from exome data in 10 individuals without TBC1D24 mutations. ................................................ 16
Supplementary Figure 1. Conservation across species of the residues affected by missense substitutions. .......................................................................... 17
Supplementary Figure 2. Additional Tbc1d24 expression data from public databases. ........................................................................................................... 18
Supplementary Figure 3. Tbc1d24 expression profiling. ........................................................................................................................................................... 19
Supplementary Discussion. 2-Oxoglutaric aciduria and DOORS syndrome types. .................................................................................................................. 20
References ................................................................................................................................................................................................................................ 21

Page 2 of 22
Supplementary Methods. Exome sequencing and analysis, Immunohistochemistry and Western blotting.
Exome sequencing and analysis: Whole exome sequencing was performed by first fragmenting DNA and then creating libraries that were enriching for
exon-coding regions using various capture reagents. The capture reagent varied depending on where and when the capture was performed as exome
sequencing was completed at the Baylor College of Medicine, the University College London and the University of Manchester (details on each sample are
given in Supplementary Table 1). The capture reagent was either Illumina's TruSeq capture reagent (Illumina Inc., San Diego, CA), Agilent's SureSelect
capture reagent (Agilent Technologies, Santa Clara, CA), or Roche Nimblegen's Baylor VCRome capture reagent (Roche NimbleGen, Madison, WI).
Capture was performed according to the manufacturer’s protocol. Next-generation sequencing was performed on Illumina HiSeq 2000 (Illumina, San
Diego, CA) for all samples. Sequence reads were aligned to the hg19 iteration of the reference human genome using BWA (v 0.5.9)1. Base score
recalibration and local realignment for indel (insertion or deletion) detection and duplicate removal2,3 were performed with GATK. SNVs were called using
Samtools mpileup (version 0.1.17)4 and short indels (insertions and deletion) were called using Samtools5, Atlas-INDEL6, and GATK3 (indels included in
our variant list had to be detected by all three programs, to decrease false-positive rates). Variants were annotated with ANNOVAR7. Protein-impacting
variants that are rare (minor allele frequency <1%) or novel were preferentially explored. Candidate genes and variants were then assessed using
databases such as dbNSFP which annotates the functional impact and the conservation of the mutated residues8, Uniprot for the function of the proteins9,
NeXtProt10 for the expression pattern, Mouse Genome Informatics11 and NCBI’s OMIM12 for the phenotypes in mice and humans, and finally
Genedistiller213 for a combination of some of the above databases. Homozygosity mapping from exome data using VCF variant files was achieved using
HomozygosityMapper14 and regions of homozygosity were analyzed for known disease-causing genes using the Genomic Oligoarray and SNP array
evaluation tool v1.015. We define variants as rare when they have a minor allele frequency (MAF) below 1% in the Exome Variant Server16, as novel when
absent from this database, and as protein-impacting when variants change the coding sequence of the protein (substituting an amino acid, causing an in-
frame or out-of-frame deletion or insertion, a premature stop codon or altering a stop codon) or potentially affect splicing (nucleotide change within five
bases of splice-donor or splice-acceptor sites).
Immunohistochemistry: C57BL/6 whole embryos (E16.5) or limbs (P2) were fixed in 4% PFA at 4°C overnight, washed with PBS, dehydrated using ethanol
then embedded in paraffin for sectioning. Sectioned tissues were deparaffinized with xylene then rehydrated. Sections were rinsed with PBS, blocked with

Page 3 of 22
3% Donkey Serum in 0.1% BSA and 0.1% Triton. Slides were then incubated overnight with a mouse monoclonal IgG1 antibody against an epitope
mapping between amino acids 437-559 at the C-terminus of TBC1D24 of human origin (clone G-6, catalog number sc-390237, Santa Cruz Biotechnology,
Santa Cruz, CA). The antibody is provided at 200 µg/ml and was diluted at 1:200 for the immunohistochemistry (a control slide without primary antibody
was also included). After rinsing, the slides were incubated for 1h in Alexa Fluor® 594 goat anti-rabbit IgG (Invitrogen product number A-11012, 1:600
final dilution in blocking buffer). After further rinsing, slides were mounted with ProLong® Gold antifade reagent with DAPI for imaging. Slides were viewed
with a Zeiss Axioplan 2 fluorescence microscope, and images were taken using the same exposure parameters consistently for all images.
Western blotting: Proteins were extracted in 50 mM Tris-HCl pH7.5, 150 mM NaCl, 1% Triton X100, 1X Complete Protease Inhibitor Cocktail (Roche
NimbleGen, Madison, WI), resolved in 4-15% gradient SDS-PAGE gels (Bio-Rad, Hercules, CA) and transferred to PVDF membranes for western blot
analyses. For TBC1D24, the same antibody was used as for immunohistochemistry, at a dilution of 1:1000 (for the mouse brain lysate) or 1:100 (for the
human fibroblast lysate), then an HRP-conjugated rabbit anti-mouse IgG antibody was used for detection (1:10,000 dilution, BioRad, Hercules, CA), and
for control, a mouse monoclonal antibody to GAPDH directly conjugated to HRP (1:20,000 dilution, Clone GAPDH-71.1, Sigma-Aldrich Co, St-Louis, MO)
was used.
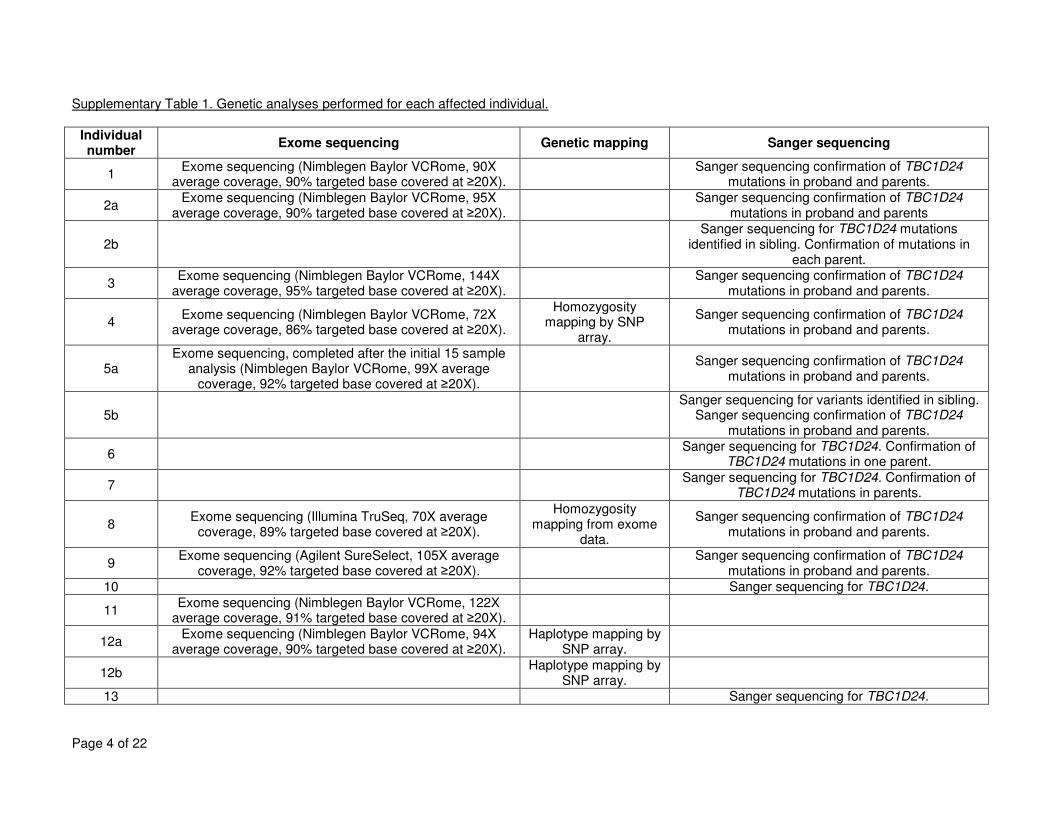
Page 4 of 22
Supplementary Table 1. Genetic analyses performed for each affected individual.
Individual number
Exome sequencing Genetic mapping Sanger sequencing
1 Exome sequencing (Nimblegen Baylor VCRome, 90X
average coverage, 90% targeted base covered at ≥20X).
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
2a Exome sequencing (Nimblegen Baylor VCRome, 95X
average coverage, 90% targeted base covered at ≥20X).
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents
2b Sanger sequencing for TBC1D24 mutations
identified in sibling. Confirmation of mutations in each parent.
3 Exome sequencing (Nimblegen Baylor VCRome, 144X
average coverage, 95% targeted base covered at ≥20X).
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
4 Exome sequencing (Nimblegen Baylor VCRome, 72X
average coverage, 86% targeted base covered at ≥20X).
Homozygosity mapping by SNP
array.
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
5a Exome sequencing, completed after the initial 15 sample
analysis (Nimblegen Baylor VCRome, 99X average coverage, 92% targeted base covered at ≥20X).
Sanger sequencing confirmation of TBC1D24
mutations in proband and parents.
5b Sanger sequencing for variants identified in sibling.
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
6 Sanger sequencing for TBC1D24. Confirmation of
TBC1D24 mutations in one parent.
7 Sanger sequencing for TBC1D24. Confirmation of
TBC1D24 mutations in parents.
8 Exome sequencing (Illumina TruSeq, 70X average
coverage, 89% targeted base covered at ≥20X).
Homozygosity mapping from exome
data.
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
9 Exome sequencing (Agilent SureSelect, 105X average
coverage, 92% targeted base covered at ≥20X).
Sanger sequencing confirmation of TBC1D24 mutations in proband and parents.
10 Sanger sequencing for TBC1D24.
11 Exome sequencing (Nimblegen Baylor VCRome, 122X
average coverage, 91% targeted base covered at ≥20X).
12a Exome sequencing (Nimblegen Baylor VCRome, 94X
average coverage, 90% targeted base covered at ≥20X). Haplotype mapping by
SNP array.
12b Haplotype mapping by
SNP array.
13 Sanger sequencing for TBC1D24.
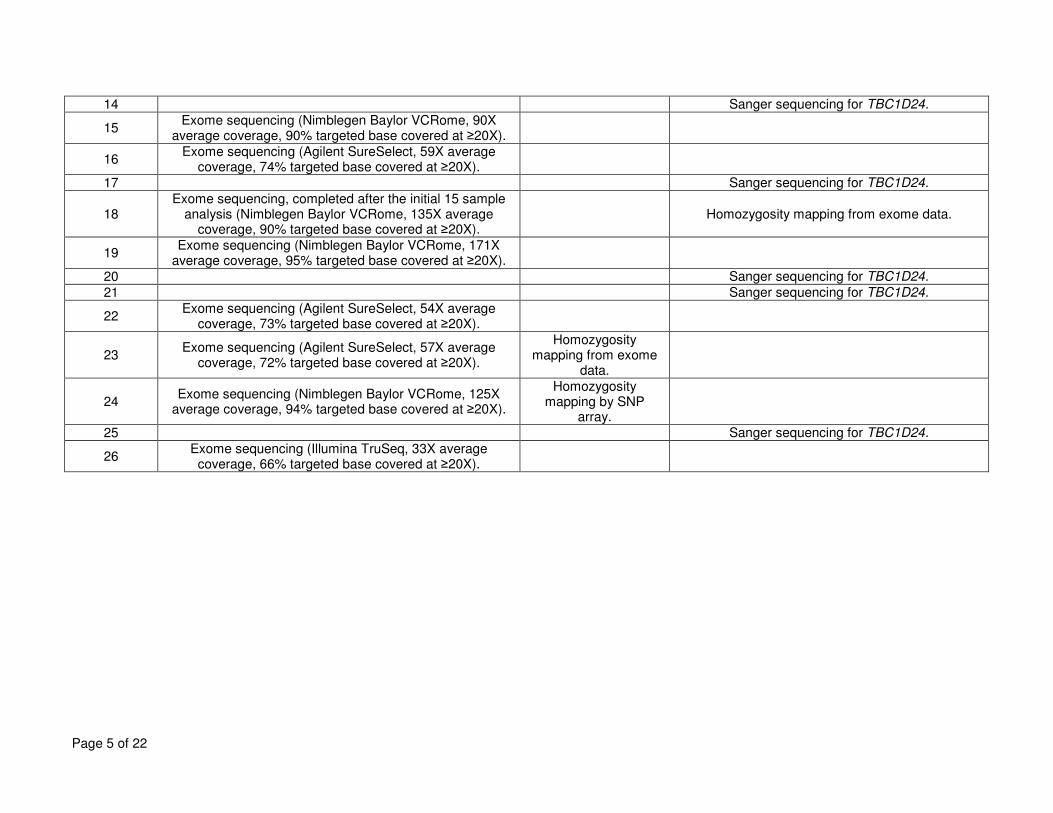
Page 5 of 22
14 Sanger sequencing for TBC1D24.
15 Exome sequencing (Nimblegen Baylor VCRome, 90X
average coverage, 90% targeted base covered at ≥20X).
16 Exome sequencing (Agilent SureSelect, 59X average
coverage, 74% targeted base covered at ≥20X).
17 Sanger sequencing for TBC1D24.
18 Exome sequencing, completed after the initial 15 sample
analysis (Nimblegen Baylor VCRome, 135X average coverage, 90% targeted base covered at ≥20X).
Homozygosity mapping from exome data.
19 Exome sequencing (Nimblegen Baylor VCRome, 171X
average coverage, 95% targeted base covered at ≥20X).
20 Sanger sequencing for TBC1D24.
21 Sanger sequencing for TBC1D24.
22 Exome sequencing (Agilent SureSelect, 54X average
coverage, 73% targeted base covered at ≥20X).
23 Exome sequencing (Agilent SureSelect, 57X average
coverage, 72% targeted base covered at ≥20X).
Homozygosity mapping from exome
data.
24 Exome sequencing (Nimblegen Baylor VCRome, 125X
average coverage, 94% targeted base covered at ≥20X).
Homozygosity mapping by SNP
array.
25 Sanger sequencing for TBC1D24.
26 Exome sequencing (Illumina TruSeq, 33X average
coverage, 66% targeted base covered at ≥20X).
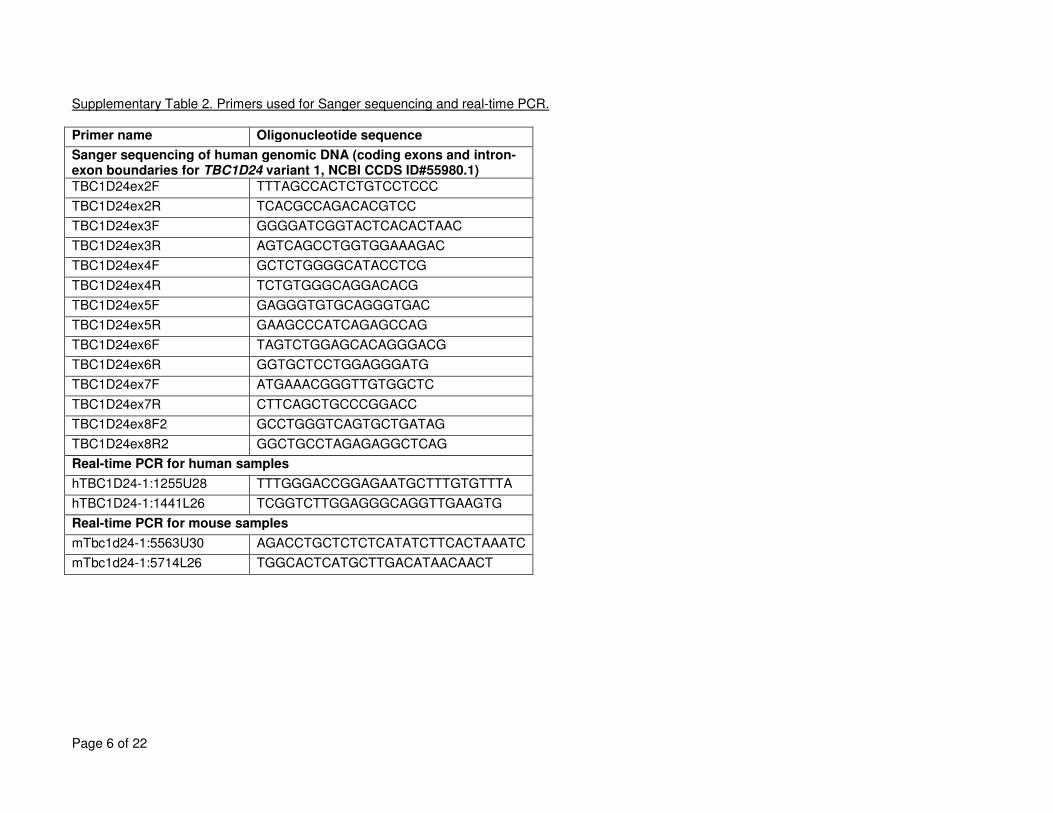
Page 6 of 22
Supplementary Table 2. Primers used for Sanger sequencing and real-time PCR.
Primer name Oligonucleotide sequence
Sanger sequencing of human genomic DNA (coding exons and intron-exon boundaries for TBC1D24 variant 1, NCBI CCDS ID#55980.1)
TBC1D24ex2F TTTAGCCACTCTGTCCTCCC
TBC1D24ex2R TCACGCCAGACACGTCC
TBC1D24ex3F GGGGATCGGTACTCACACTAAC
TBC1D24ex3R AGTCAGCCTGGTGGAAAGAC
TBC1D24ex4F GCTCTGGGGCATACCTCG
TBC1D24ex4R TCTGTGGGCAGGACACG
TBC1D24ex5F GAGGGTGTGCAGGGTGAC
TBC1D24ex5R GAAGCCCATCAGAGCCAG
TBC1D24ex6F TAGTCTGGAGCACAGGGACG
TBC1D24ex6R GGTGCTCCTGGAGGGATG
TBC1D24ex7F ATGAAACGGGTTGTGGCTC
TBC1D24ex7R CTTCAGCTGCCCGGACC
TBC1D24ex8F2 GCCTGGGTCAGTGCTGATAG
TBC1D24ex8R2 GGCTGCCTAGAGAGGCTCAG
Real-time PCR for human samples
hTBC1D24-1:1255U28 TTTGGGACCGGAGAATGCTTTGTGTTTA
hTBC1D24-1:1441L26 TCGGTCTTGGAGGGCAGGTTGAAGTG
Real-time PCR for mouse samples
mTbc1d24-1:5563U30 AGACCTGCTCTCTCATATCTTCACTAAATC
mTbc1d24-1:5714L26 TGGCACTCATGCTTGACATAACAACT
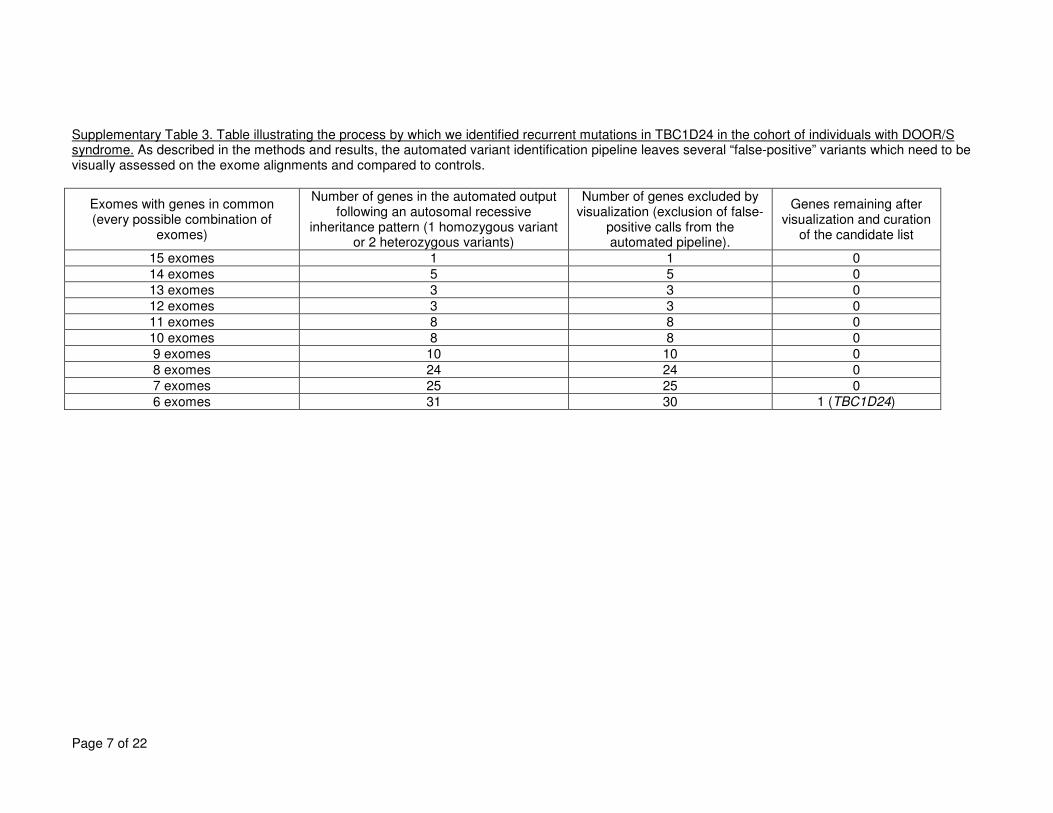
Page 7 of 22
Supplementary Table 3. Table illustrating the process by which we identified recurrent mutations in TBC1D24 in the cohort of individuals with DOOR/S syndrome. As described in the methods and results, the automated variant identification pipeline leaves several “false-positive” variants which need to be visually assessed on the exome alignments and compared to controls.
Exomes with genes in common (every possible combination of
exomes)
Number of genes in the automated output following an autosomal recessive
inheritance pattern (1 homozygous variant or 2 heterozygous variants)
Number of genes excluded by visualization (exclusion of false-
positive calls from the automated pipeline).
Genes remaining after visualization and curation
of the candidate list
15 exomes 1 1 0
14 exomes 5 5 0
13 exomes 3 3 0
12 exomes 3 3 0
11 exomes 8 8 0
10 exomes 8 8 0
9 exomes 10 10 0
8 exomes 24 24 0
7 exomes 25 25 0
6 exomes 31 30 1 (TBC1D24)
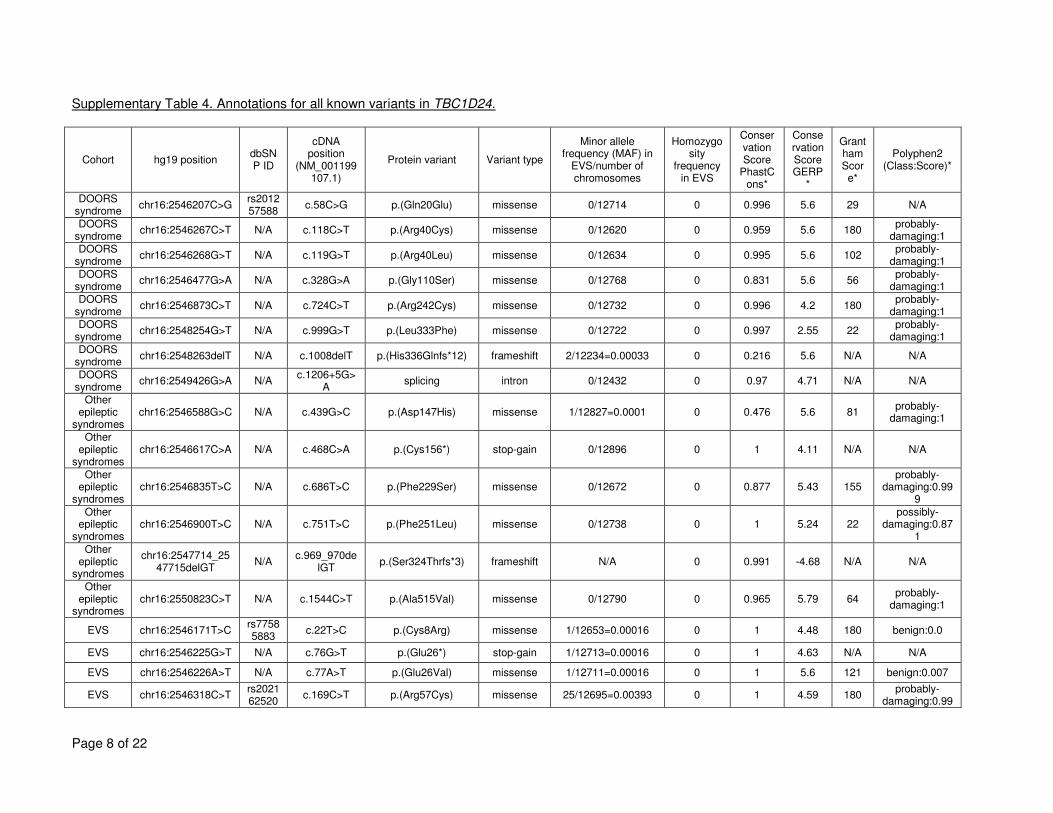
Page 8 of 22
Supplementary Table 4. Annotations for all known variants in TBC1D24.
Cohort hg19 position dbSNP ID
cDNA position
(NM_001199107.1)
Protein variant Variant type
Minor allele frequency (MAF) in
EVS/number of chromosomes
Homozygosity
frequency in EVS
Conservation Score
PhastCons*
Conservation Score GERP
*
Grantham Score*
Polyphen2 (Class:Score)*
DOORS syndrome
chr16:2546207C>G rs201257588
c.58C>G p.(Gln20Glu) missense 0/12714 0 0.996 5.6 29 N/A
DOORS syndrome
chr16:2546267C>T N/A c.118C>T p.(Arg40Cys) missense 0/12620 0 0.959 5.6 180 probably-
damaging:1 DOORS
syndrome chr16:2546268G>T N/A c.119G>T p.(Arg40Leu) missense 0/12634 0 0.995 5.6 102
probably-damaging:1
DOORS syndrome
chr16:2546477G>A N/A c.328G>A p.(Gly110Ser) missense 0/12768 0 0.831 5.6 56 probably-
damaging:1 DOORS
syndrome chr16:2546873C>T N/A c.724C>T p.(Arg242Cys) missense 0/12732 0 0.996 4.2 180
probably-damaging:1
DOORS syndrome
chr16:2548254G>T N/A c.999G>T p.(Leu333Phe) missense 0/12722 0 0.997 2.55 22 probably-
damaging:1 DOORS
syndrome chr16:2548263delT N/A c.1008delT p.(His336Glnfs*12) frameshift 2/12234=0.00033 0 0.216 5.6 N/A N/A
DOORS syndrome
chr16:2549426G>A N/A c.1206+5G>
A splicing intron 0/12432 0 0.97 4.71 N/A N/A
Other epileptic
syndromes chr16:2546588G>C N/A c.439G>C p.(Asp147His) missense 1/12827=0.0001 0 0.476 5.6 81
probably-damaging:1
Other epileptic
syndromes chr16:2546617C>A N/A c.468C>A p.(Cys156*) stop-gain 0/12896 0 1 4.11 N/A N/A
Other epileptic
syndromes chr16:2546835T>C N/A c.686T>C p.(Phe229Ser) missense 0/12672 0 0.877 5.43 155
probably-damaging:0.99
9
Other epileptic
syndromes chr16:2546900T>C N/A c.751T>C p.(Phe251Leu) missense 0/12738 0 1 5.24 22
possibly-damaging:0.87
1
Other epileptic
syndromes
chr16:2547714_2547715delGT
N/A c.969_970de
lGT p.(Ser324Thrfs*3) frameshift N/A 0 0.991 -4.68 N/A N/A
Other epileptic
syndromes chr16:2550823C>T N/A c.1544C>T p.(Ala515Val) missense 0/12790 0 0.965 5.79 64
probably-damaging:1
EVS chr16:2546171T>C rs77585883
c.22T>C p.(Cys8Arg) missense 1/12653=0.00016 0 1 4.48 180 benign:0.0
EVS chr16:2546225G>T N/A c.76G>T p.(Glu26*) stop-gain 1/12713=0.00016 0 1 4.63 N/A N/A
EVS chr16:2546226A>T N/A c.77A>T p.(Glu26Val) missense 1/12711=0.00016 0 1 5.6 121 benign:0.007
EVS chr16:2546318C>T rs202162520
c.169C>T p.(Arg57Cys) missense 25/12695=0.00393 0 1 4.59 180 probably-
damaging:0.99
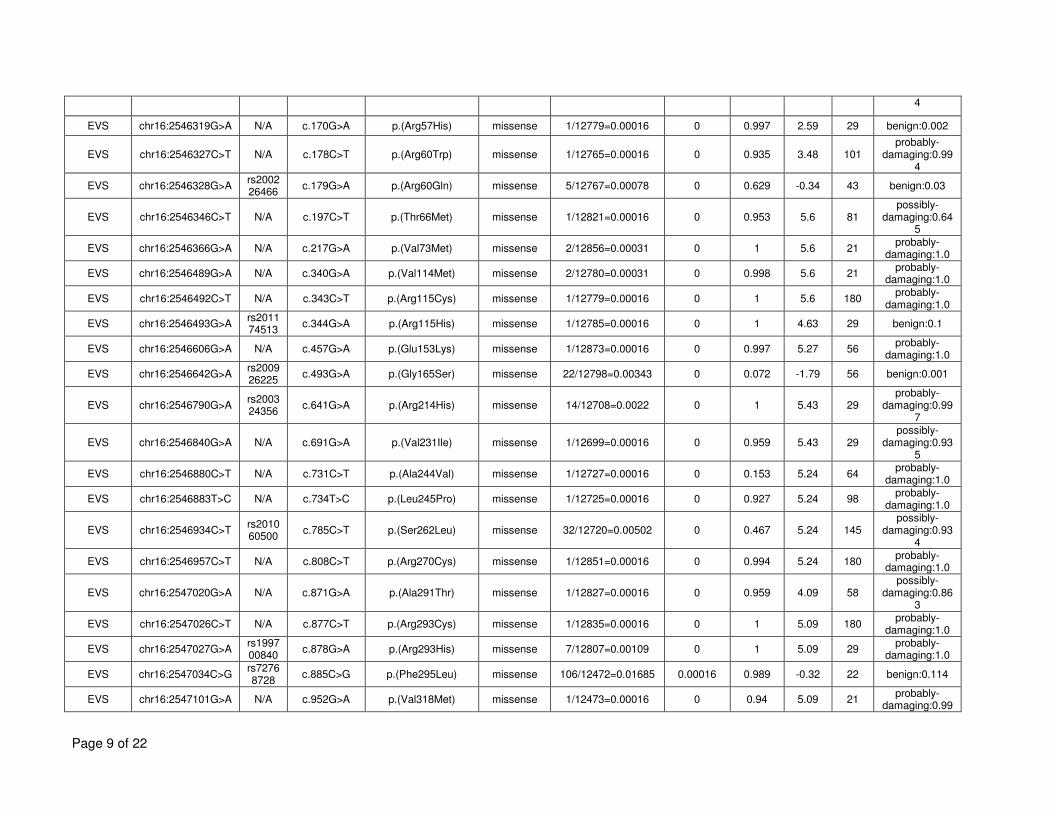
Page 9 of 22
4
EVS chr16:2546319G>A N/A c.170G>A p.(Arg57His) missense 1/12779=0.00016 0 0.997 2.59 29 benign:0.002
EVS chr16:2546327C>T N/A c.178C>T p.(Arg60Trp) missense 1/12765=0.00016 0 0.935 3.48 101 probably-
damaging:0.994
EVS chr16:2546328G>A rs200226466
c.179G>A p.(Arg60Gln) missense 5/12767=0.00078 0 0.629 -0.34 43 benign:0.03
EVS chr16:2546346C>T N/A c.197C>T p.(Thr66Met) missense 1/12821=0.00016 0 0.953 5.6 81 possibly-
damaging:0.645
EVS chr16:2546366G>A N/A c.217G>A p.(Val73Met) missense 2/12856=0.00031 0 1 5.6 21 probably-
damaging:1.0
EVS chr16:2546489G>A N/A c.340G>A p.(Val114Met) missense 2/12780=0.00031 0 0.998 5.6 21 probably-
damaging:1.0
EVS chr16:2546492C>T N/A c.343C>T p.(Arg115Cys) missense 1/12779=0.00016 0 1 5.6 180 probably-
damaging:1.0
EVS chr16:2546493G>A rs201174513
c.344G>A p.(Arg115His) missense 1/12785=0.00016 0 1 4.63 29 benign:0.1
EVS chr16:2546606G>A N/A c.457G>A p.(Glu153Lys) missense 1/12873=0.00016 0 0.997 5.27 56 probably-
damaging:1.0
EVS chr16:2546642G>A rs200926225
c.493G>A p.(Gly165Ser) missense 22/12798=0.00343 0 0.072 -1.79 56 benign:0.001
EVS chr16:2546790G>A rs200324356
c.641G>A p.(Arg214His) missense 14/12708=0.0022 0 1 5.43 29 probably-
damaging:0.997
EVS chr16:2546840G>A N/A c.691G>A p.(Val231Ile) missense 1/12699=0.00016 0 0.959 5.43 29 possibly-
damaging:0.935
EVS chr16:2546880C>T N/A c.731C>T p.(Ala244Val) missense 1/12727=0.00016 0 0.153 5.24 64 probably-
damaging:1.0
EVS chr16:2546883T>C N/A c.734T>C p.(Leu245Pro) missense 1/12725=0.00016 0 0.927 5.24 98 probably-
damaging:1.0
EVS chr16:2546934C>T rs201060500
c.785C>T p.(Ser262Leu) missense 32/12720=0.00502 0 0.467 5.24 145 possibly-
damaging:0.934
EVS chr16:2546957C>T N/A c.808C>T p.(Arg270Cys) missense 1/12851=0.00016 0 0.994 5.24 180 probably-
damaging:1.0
EVS chr16:2547020G>A N/A c.871G>A p.(Ala291Thr) missense 1/12827=0.00016 0 0.959 4.09 58 possibly-
damaging:0.863
EVS chr16:2547026C>T N/A c.877C>T p.(Arg293Cys) missense 1/12835=0.00016 0 1 5.09 180 probably-
damaging:1.0
EVS chr16:2547027G>A rs199700840
c.878G>A p.(Arg293His) missense 7/12807=0.00109 0 1 5.09 29 probably-
damaging:1.0
EVS chr16:2547034C>G rs72768728
c.885C>G p.(Phe295Leu) missense 106/12472=0.01685 0.00016 0.989 -0.32 22 benign:0.114
EVS chr16:2547101G>A N/A c.952G>A p.(Val318Met) missense 1/12473=0.00016 0 0.94 5.09 21 probably-
damaging:0.99
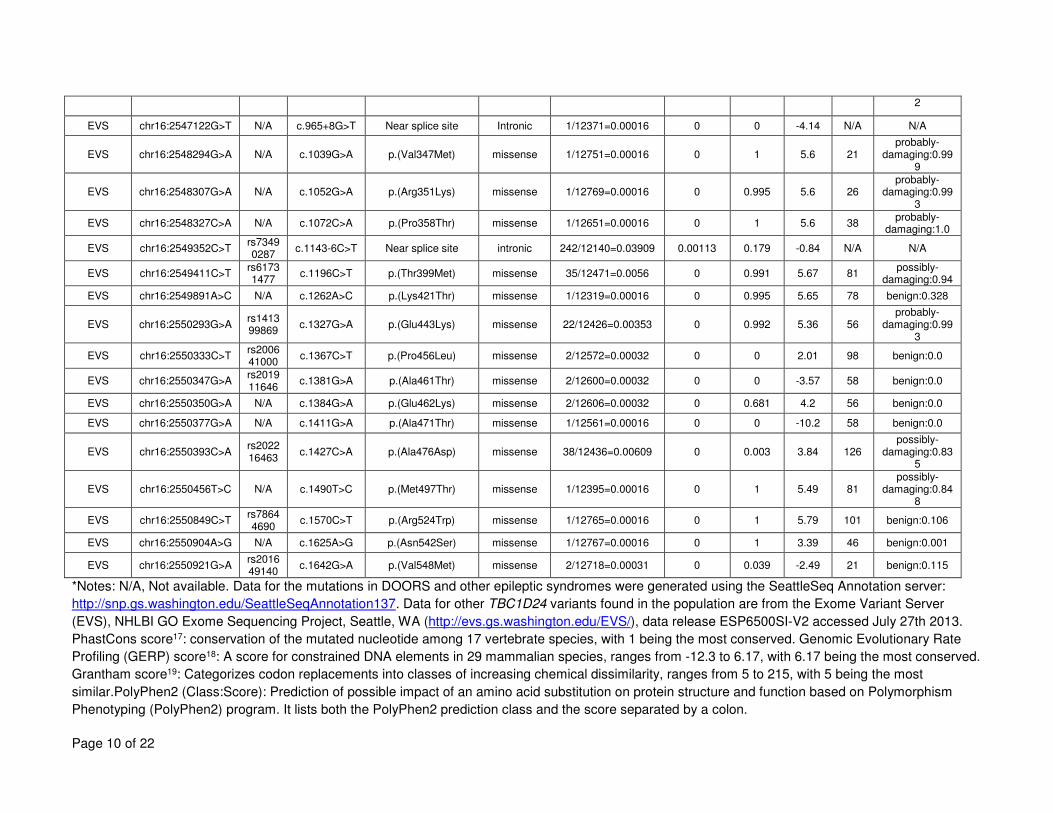
Page 10 of 22
2
EVS chr16:2547122G>T N/A c.965+8G>T Near splice site Intronic 1/12371=0.00016 0 0 -4.14 N/A N/A
EVS chr16:2548294G>A N/A c.1039G>A p.(Val347Met) missense 1/12751=0.00016 0 1 5.6 21 probably-
damaging:0.999
EVS chr16:2548307G>A N/A c.1052G>A p.(Arg351Lys) missense 1/12769=0.00016 0 0.995 5.6 26 probably-
damaging:0.993
EVS chr16:2548327C>A N/A c.1072C>A p.(Pro358Thr) missense 1/12651=0.00016 0 1 5.6 38 probably-
damaging:1.0
EVS chr16:2549352C>T rs73490287
c.1143-6C>T Near splice site intronic 242/12140=0.03909 0.00113 0.179 -0.84 N/A N/A
EVS chr16:2549411C>T rs61731477
c.1196C>T p.(Thr399Met) missense 35/12471=0.0056 0 0.991 5.67 81 possibly-
damaging:0.94
EVS chr16:2549891A>C N/A c.1262A>C p.(Lys421Thr) missense 1/12319=0.00016 0 0.995 5.65 78 benign:0.328
EVS chr16:2550293G>A rs141399869
c.1327G>A p.(Glu443Lys) missense 22/12426=0.00353 0 0.992 5.36 56 probably-
damaging:0.993
EVS chr16:2550333C>T rs200641000
c.1367C>T p.(Pro456Leu) missense 2/12572=0.00032 0 0 2.01 98 benign:0.0
EVS chr16:2550347G>A rs201911646
c.1381G>A p.(Ala461Thr) missense 2/12600=0.00032 0 0 -3.57 58 benign:0.0
EVS chr16:2550350G>A N/A c.1384G>A p.(Glu462Lys) missense 2/12606=0.00032 0 0.681 4.2 56 benign:0.0
EVS chr16:2550377G>A N/A c.1411G>A p.(Ala471Thr) missense 1/12561=0.00016 0 0 -10.2 58 benign:0.0
EVS chr16:2550393C>A rs202216463
c.1427C>A p.(Ala476Asp) missense 38/12436=0.00609 0 0.003 3.84 126 possibly-
damaging:0.835
EVS chr16:2550456T>C N/A c.1490T>C p.(Met497Thr) missense 1/12395=0.00016 0 1 5.49 81 possibly-
damaging:0.848
EVS chr16:2550849C>T rs78644690
c.1570C>T p.(Arg524Trp) missense 1/12765=0.00016 0 1 5.79 101 benign:0.106
EVS chr16:2550904A>G N/A c.1625A>G p.(Asn542Ser) missense 1/12767=0.00016 0 1 3.39 46 benign:0.001
EVS chr16:2550921G>A rs201649140
c.1642G>A p.(Val548Met) missense 2/12718=0.00031 0 0.039 -2.49 21 benign:0.115
*Notes: N/A, Not available. Data for the mutations in DOORS and other epileptic syndromes were generated using the SeattleSeq Annotation server:
http://snp.gs.washington.edu/SeattleSeqAnnotation137. Data for other TBC1D24 variants found in the population are from the Exome Variant Server
(EVS), NHLBI GO Exome Sequencing Project, Seattle, WA (http://evs.gs.washington.edu/EVS/), data release ESP6500SI-V2 accessed July 27th 2013.
PhastCons score17: conservation of the mutated nucleotide among 17 vertebrate species, with 1 being the most conserved. Genomic Evolutionary Rate
Profiling (GERP) score18: A score for constrained DNA elements in 29 mammalian species, ranges from -12.3 to 6.17, with 6.17 being the most conserved.
Grantham score19: Categorizes codon replacements into classes of increasing chemical dissimilarity, ranges from 5 to 215, with 5 being the most
similar.PolyPhen2 (Class:Score): Prediction of possible impact of an amino acid substitution on protein structure and function based on Polymorphism
Phenotyping (PolyPhen2) program. It lists both the PolyPhen2 prediction class and the score separated by a colon.
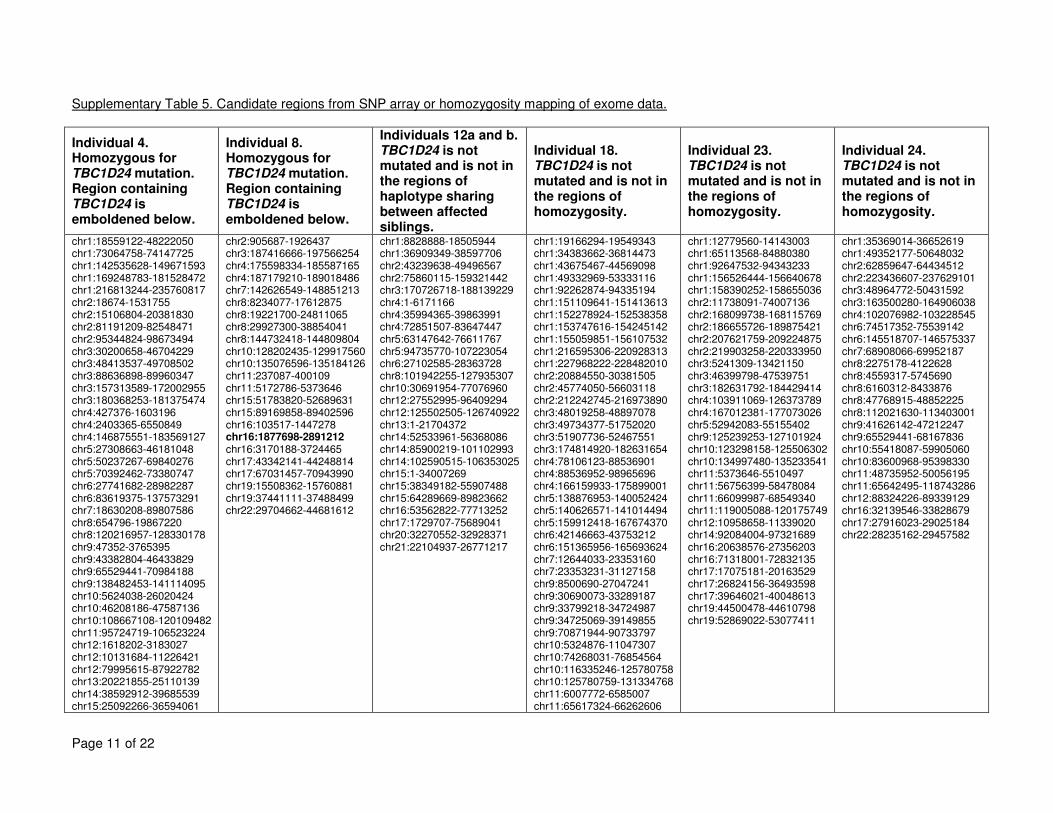
Page 11 of 22
Supplementary Table 5. Candidate regions from SNP array or homozygosity mapping of exome data.
Individual 4. Homozygous for TBC1D24 mutation. Region containing TBC1D24 is emboldened below.
Individual 8. Homozygous for TBC1D24 mutation. Region containing TBC1D24 is emboldened below.
Individuals 12a and b. TBC1D24 is not mutated and is not in the regions of haplotype sharing between affected siblings.
Individual 18. TBC1D24 is not mutated and is not in the regions of homozygosity.
Individual 23. TBC1D24 is not mutated and is not in the regions of homozygosity.
Individual 24. TBC1D24 is not mutated and is not in the regions of homozygosity.
chr1:18559122-48222050 chr1:73064758-74147725 chr1:142535628-149671593 chr1:169248783-181528472 chr1:216813244-235760817 chr2:18674-1531755 chr2:15106804-20381830 chr2:81191209-82548471 chr2:95344824-98673494 chr3:30200658-46704229 chr3:48413537-49708502 chr3:88636898-89960347 chr3:157313589-172002955 chr3:180368253-181375474 chr4:427376-1603196 chr4:2403365-6550849 chr4:146875551-183569127 chr5:27308663-46181048 chr5:50237267-69840276 chr5:70392462-73380747 chr6:27741682-28982287 chr6:83619375-137573291 chr7:18630208-89807586 chr8:654796-19867220 chr8:120216957-128330178 chr9:47352-3765395 chr9:43382804-46433829 chr9:65529441-70984188 chr9:138482453-141114095 chr10:5624038-26020424 chr10:46208186-47587136 chr10:108667108-120109482 chr11:95724719-106523224 chr12:1618202-3183027 chr12:10131684-11226421 chr12:79995615-87922782 chr13:20221855-25110139 chr14:38592912-39685539 chr15:25092266-36594061
chr2:905687-1926437 chr3:187416666-197566254 chr4:175598334-185587165 chr4:187179210-189018486 chr7:142626549-148851213 chr8:8234077-17612875 chr8:19221700-24811065 chr8:29927300-38854041 chr8:144732418-144809804 chr10:128202435-129917560 chr10:135076596-135184126 chr11:237087-400109 chr11:5172786-5373646 chr15:51783820-52689631 chr15:89169858-89402596 chr16:103517-1447278 chr16:1877698-2891212 chr16:3170188-3724465 chr17:43342141-44248814 chr17:67031457-70943990 chr19:15508362-15760881 chr19:37441111-37488499 chr22:29704662-44681612
chr1:8828888-18505944 chr1:36909349-38597706 chr2:43239638-49496567 chr2:75860115-159321442 chr3:170726718-188139229 chr4:1-6171166 chr4:35994365-39863991 chr4:72851507-83647447 chr5:63147642-76611767 chr5:94735770-107223054 chr6:27102585-28363728 chr8:101942255-127935307 chr10:30691954-77076960 chr12:27552995-96409294 chr12:125502505-126740922 chr13:1-21704372 chr14:52533961-56368086 chr14:85900219-101102993 chr14:102590515-106353025 chr15:1-34007269 chr15:38349182-55907488 chr15:64289669-89823662 chr16:53562822-77713252 chr17:1729707-75689041 chr20:32270552-32928371 chr21:22104937-26771217
chr1:19166294-19549343 chr1:34383662-36814473 chr1:43675467-44569098 chr1:49332969-53333116 chr1:92262874-94335194 chr1:151109641-151413613 chr1:152278924-152538358 chr1:153747616-154245142 chr1:155059851-156107532 chr1:216595306-220928313 chr1:227968222-228482010 chr2:20884550-30381505 chr2:45774050-56603118 chr2:212242745-216973890 chr3:48019258-48897078 chr3:49734377-51752020 chr3:51907736-52467551 chr3:174814920-182631654 chr4:78106123-88536901 chr4:88536952-98965696 chr4:166159933-175899001 chr5:138876953-140052424 chr5:140626571-141014494 chr5:159912418-167674370 chr6:42146663-43753212 chr6:151365956-165693624 chr7:12644033-23353160 chr7:23353231-31127158 chr9:8500690-27047241 chr9:30690073-33289187 chr9:33799218-34724987 chr9:34725069-39149855 chr9:70871944-90733797 chr10:5324876-11047307 chr10:74268031-76854564 chr10:116335246-125780758 chr10:125780759-131334768 chr11:6007772-6585007 chr11:65617324-66262606
chr1:12779560-14143003 chr1:65113568-84880380 chr1:92647532-94343233 chr1:156526444-156640678 chr1:158390252-158655036 chr2:11738091-74007136 chr2:168099738-168115769 chr2:186655726-189875421 chr2:207621759-209224875 chr2:219903258-220333950 chr3:5241309-13421150 chr3:46399798-47539751 chr3:182631792-184429414 chr4:103911069-126373789 chr4:167012381-177073026 chr5:52942083-55155402 chr9:125239253-127101924 chr10:123298158-125506302 chr10:134997480-135233541 chr11:5373646-5510497 chr11:56756399-58478084 chr11:66099987-68549340 chr11:119005088-120175749 chr12:10958658-11339020 chr14:92084004-97321689 chr16:20638576-27356203 chr16:71318001-72832135 chr17:17075181-20163529 chr17:26824156-36493598 chr17:39646021-40048613 chr19:44500478-44610798 chr19:52869022-53077411
chr1:35369014-36652619 chr1:49352177-50648032 chr2:62859647-64434512 chr2:223436607-237629101 chr3:48964772-50431592 chr3:163500280-164906038 chr4:102076982-103228545 chr6:74517352-75539142 chr6:145518707-146575337 chr7:68908066-69952187 chr8:2275178-4122628 chr8:4559317-5745690 chr8:6160312-8433876 chr8:47768915-48852225 chr8:112021630-113403001 chr9:41626142-47212247 chr9:65529441-68167836 chr10:55418087-59905060 chr10:83600968-95398330 chr11:48735952-50056195 chr11:65642495-118743286 chr12:88324226-89339129 chr16:32139546-33828679 chr17:27916023-29025184 chr22:28235162-29457582
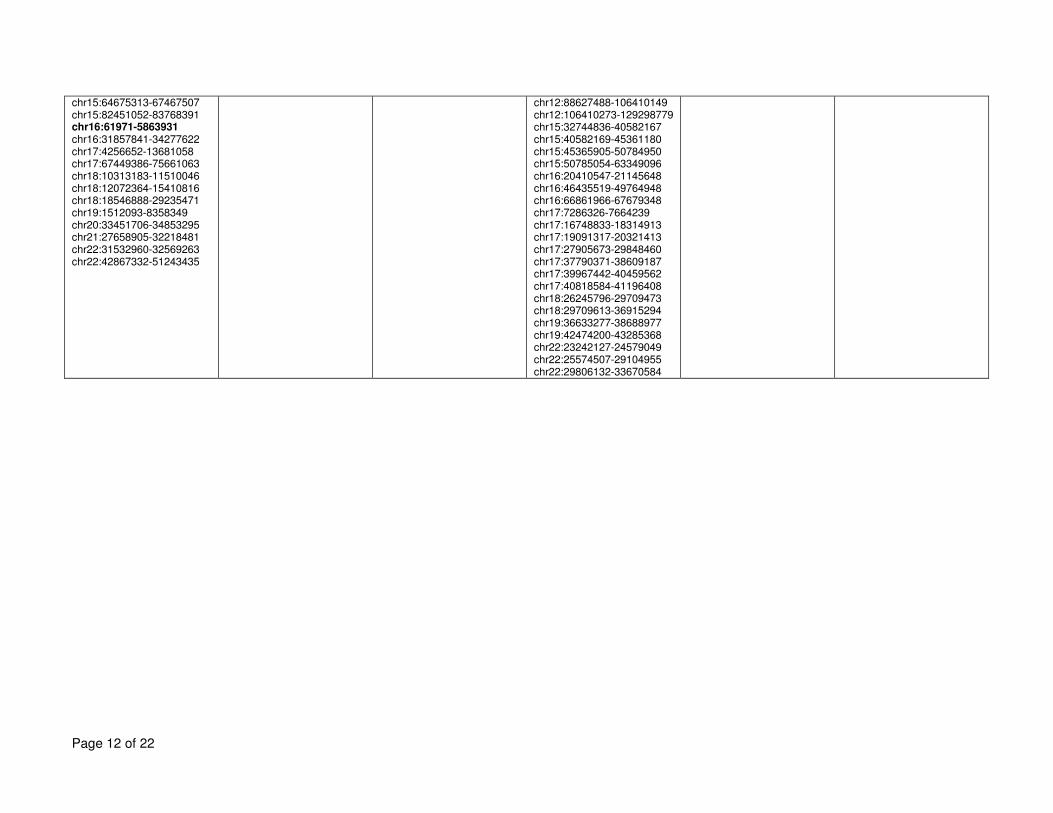
Page 12 of 22
chr15:64675313-67467507 chr15:82451052-83768391 chr16:61971-5863931 chr16:31857841-34277622 chr17:4256652-13681058 chr17:67449386-75661063 chr18:10313183-11510046 chr18:12072364-15410816 chr18:18546888-29235471 chr19:1512093-8358349 chr20:33451706-34853295 chr21:27658905-32218481 chr22:31532960-32569263 chr22:42867332-51243435
chr12:88627488-106410149 chr12:106410273-129298779 chr15:32744836-40582167 chr15:40582169-45361180 chr15:45365905-50784950 chr15:50785054-63349096 chr16:20410547-21145648 chr16:46435519-49764948 chr16:66861966-67679348 chr17:7286326-7664239 chr17:16748833-18314913 chr17:19091317-20321413 chr17:27905673-29848460 chr17:37790371-38609187 chr17:39967442-40459562 chr17:40818584-41196408 chr18:26245796-29709473 chr18:29709613-36915294 chr19:36633277-38688977 chr19:42474200-43285368 chr22:23242127-24579049 chr22:25574507-29104955 chr22:29806132-33670584
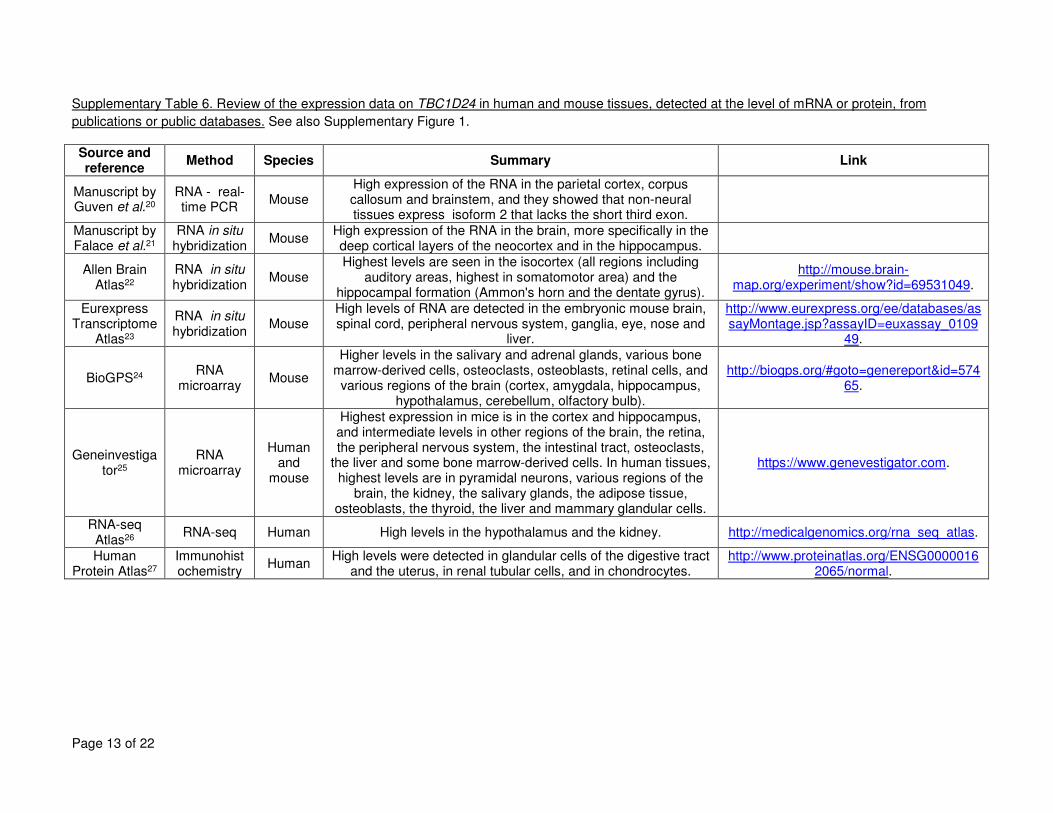
Page 13 of 22
Supplementary Table 6. Review of the expression data on TBC1D24 in human and mouse tissues, detected at the level of mRNA or protein, from
publications or public databases. See also Supplementary Figure 1.
Source and reference
Method Species Summary Link
Manuscript by Guven et al.20
RNA - real-time PCR
Mouse High expression of the RNA in the parietal cortex, corpus callosum and brainstem, and they showed that non-neural tissues express isoform 2 that lacks the short third exon.
Manuscript by Falace et al.21
RNA in situ hybridization
Mouse High expression of the RNA in the brain, more specifically in the deep cortical layers of the neocortex and in the hippocampus.
Allen Brain Atlas22
RNA in situ hybridization
Mouse Highest levels are seen in the isocortex (all regions including
auditory areas, highest in somatomotor area) and the hippocampal formation (Ammon's horn and the dentate gyrus).
http://mouse.brain-map.org/experiment/show?id=69531049.
Eurexpress Transcriptome
Atlas23
RNA in situ hybridization
Mouse High levels of RNA are detected in the embryonic mouse brain, spinal cord, peripheral nervous system, ganglia, eye, nose and
liver.
http://www.eurexpress.org/ee/databases/assayMontage.jsp?assayID=euxassay_0109
49.
BioGPS24 RNA
microarray Mouse
Higher levels in the salivary and adrenal glands, various bone marrow-derived cells, osteoclasts, osteoblasts, retinal cells, and various regions of the brain (cortex, amygdala, hippocampus,
hypothalamus, cerebellum, olfactory bulb).
http://biogps.org/#goto=genereport&id=57465.
Geneinvestigator25
RNA microarray
Human and
mouse
Highest expression in mice is in the cortex and hippocampus, and intermediate levels in other regions of the brain, the retina, the peripheral nervous system, the intestinal tract, osteoclasts,
the liver and some bone marrow-derived cells. In human tissues, highest levels are in pyramidal neurons, various regions of the
brain, the kidney, the salivary glands, the adipose tissue, osteoblasts, the thyroid, the liver and mammary glandular cells.
https://www.genevestigator.com.
RNA-seq Atlas26
RNA-seq Human High levels in the hypothalamus and the kidney. http://medicalgenomics.org/rna_seq_atlas.
Human Protein Atlas27
Immunohistochemistry
Human High levels were detected in glandular cells of the digestive tract
and the uterus, in renal tubular cells, and in chondrocytes. http://www.proteinatlas.org/ENSG0000016
2065/normal.
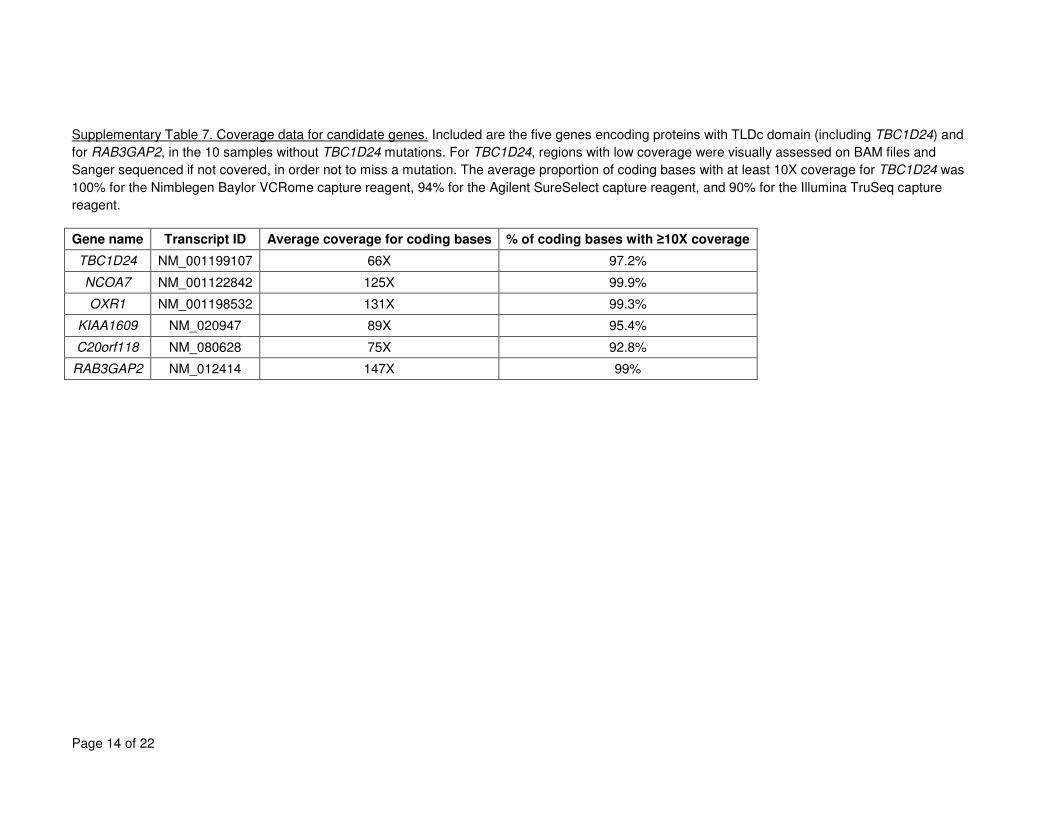
Page 14 of 22
Supplementary Table 7. Coverage data for candidate genes. Included are the five genes encoding proteins with TLDc domain (including TBC1D24) and
for RAB3GAP2, in the 10 samples without TBC1D24 mutations. For TBC1D24, regions with low coverage were visually assessed on BAM files and
Sanger sequenced if not covered, in order not to miss a mutation. The average proportion of coding bases with at least 10X coverage for TBC1D24 was
100% for the Nimblegen Baylor VCRome capture reagent, 94% for the Agilent SureSelect capture reagent, and 90% for the Illumina TruSeq capture
reagent.
Gene name Transcript ID Average coverage for coding bases % of coding bases with ≥10X coverage
TBC1D24 NM_001199107 66X 97.2%
NCOA7 NM_001122842 125X 99.9%
OXR1 NM_001198532 131X 99.3%
KIAA1609 NM_020947 89X 95.4%
C20orf118 NM_080628 75X 92.8%
RAB3GAP2 NM_012414 147X 99%
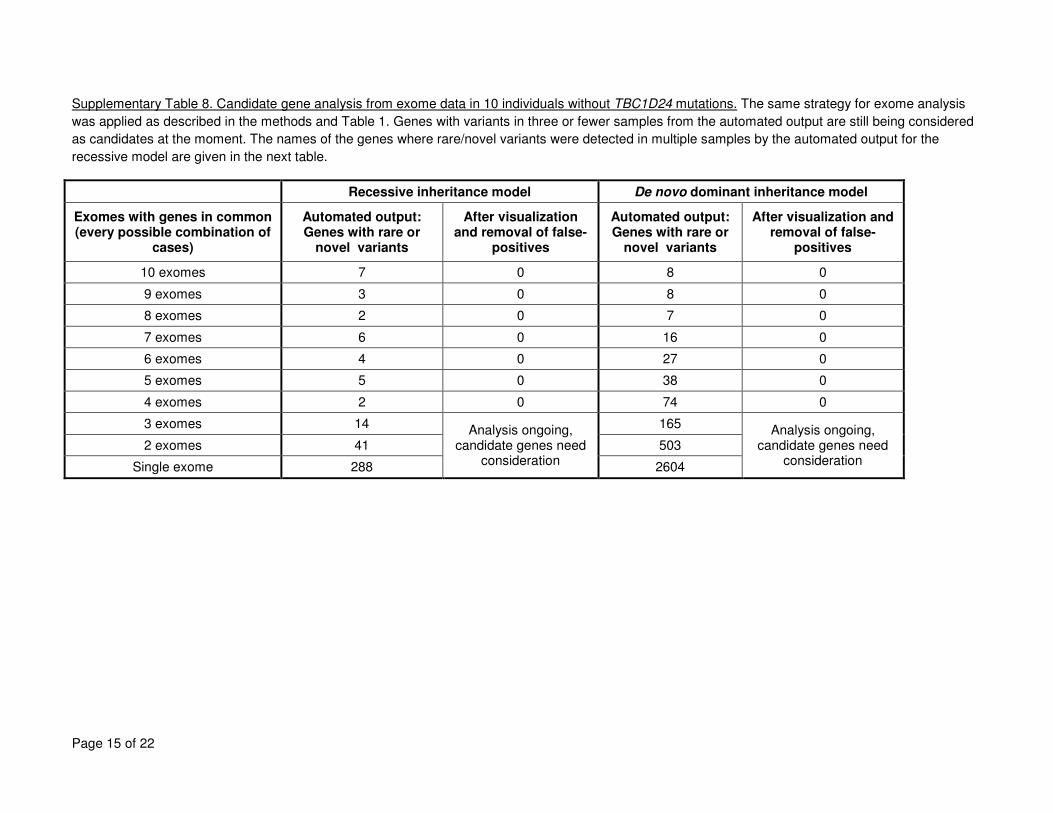
Page 15 of 22
Supplementary Table 8. Candidate gene analysis from exome data in 10 individuals without TBC1D24 mutations. The same strategy for exome analysis
was applied as described in the methods and Table 1. Genes with variants in three or fewer samples from the automated output are still being considered
as candidates at the moment. The names of the genes where rare/novel variants were detected in multiple samples by the automated output for the
recessive model are given in the next table.
Recessive inheritance model De novo dominant inheritance model
Exomes with genes in common (every possible combination of
cases)
Automated output: Genes with rare or
novel variants
After visualization and removal of false-
positives
Automated output: Genes with rare or
novel variants
After visualization and removal of false-
positives
10 exomes 7 0 8 0
9 exomes 3 0 8 0
8 exomes 2 0 7 0
7 exomes 6 0 16 0
6 exomes 4 0 27 0
5 exomes 5 0 38 0
4 exomes 2 0 74 0
3 exomes 14 Analysis ongoing,
candidate genes need consideration
165 Analysis ongoing,
candidate genes need consideration
2 exomes 41 503
Single exome 288 2604
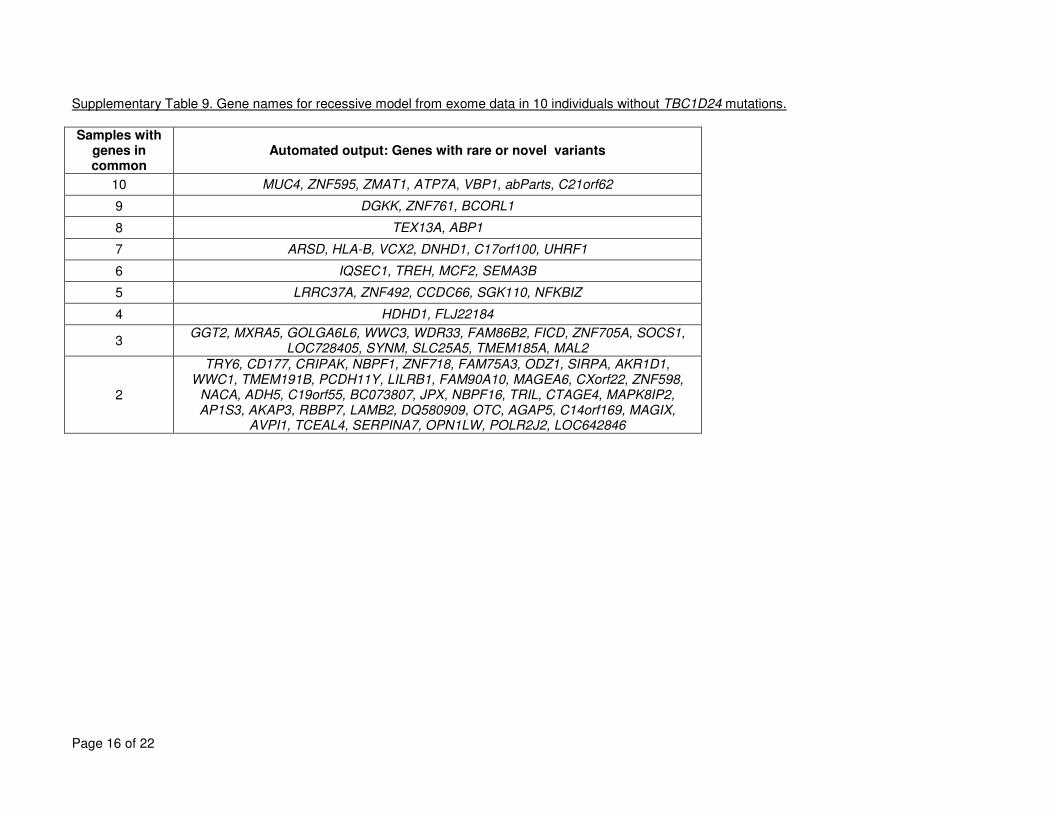
Page 16 of 22
Supplementary Table 9. Gene names for recessive model from exome data in 10 individuals without TBC1D24 mutations.
Samples with genes in common
Automated output: Genes with rare or novel variants
10 MUC4, ZNF595, ZMAT1, ATP7A, VBP1, abParts, C21orf62
9 DGKK, ZNF761, BCORL1
8 TEX13A, ABP1
7 ARSD, HLA-B, VCX2, DNHD1, C17orf100, UHRF1
6 IQSEC1, TREH, MCF2, SEMA3B
5 LRRC37A, ZNF492, CCDC66, SGK110, NFKBIZ
4 HDHD1, FLJ22184
3 GGT2, MXRA5, GOLGA6L6, WWC3, WDR33, FAM86B2, FICD, ZNF705A, SOCS1,
LOC728405, SYNM, SLC25A5, TMEM185A, MAL2
2
TRY6, CD177, CRIPAK, NBPF1, ZNF718, FAM75A3, ODZ1, SIRPA, AKR1D1, WWC1, TMEM191B, PCDH11Y, LILRB1, FAM90A10, MAGEA6, CXorf22, ZNF598,
NACA, ADH5, C19orf55, BC073807, JPX, NBPF16, TRIL, CTAGE4, MAPK8IP2, AP1S3, AKAP3, RBBP7, LAMB2, DQ580909, OTC, AGAP5, C14orf169, MAGIX,
AVPI1, TCEAL4, SERPINA7, OPN1LW, POLR2J2, LOC642846
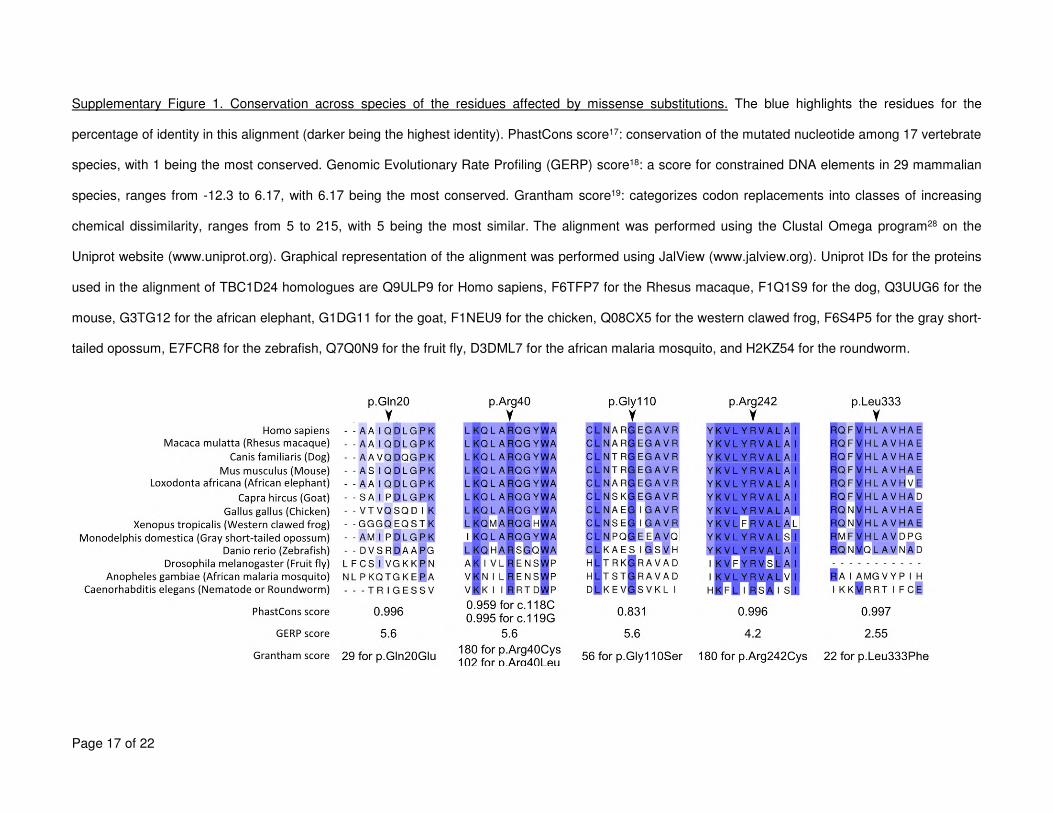
Page 17 of 22
Supplementary Figure 1. Conservation across species of the residues affected by missense substitutions. The blue highlights the residues for the
percentage of identity in this alignment (darker being the highest identity). PhastCons score17: conservation of the mutated nucleotide among 17 vertebrate
species, with 1 being the most conserved. Genomic Evolutionary Rate Profiling (GERP) score18: a score for constrained DNA elements in 29 mammalian
species, ranges from -12.3 to 6.17, with 6.17 being the most conserved. Grantham score19: categorizes codon replacements into classes of increasing
chemical dissimilarity, ranges from 5 to 215, with 5 being the most similar. The alignment was performed using the Clustal Omega program28 on the
Uniprot website (www.uniprot.org). Graphical representation of the alignment was performed using JalView (www.jalview.org). Uniprot IDs for the proteins
used in the alignment of TBC1D24 homologues are Q9ULP9 for Homo sapiens, F6TFP7 for the Rhesus macaque, F1Q1S9 for the dog, Q3UUG6 for the
mouse, G3TG12 for the african elephant, G1DG11 for the goat, F1NEU9 for the chicken, Q08CX5 for the western clawed frog, F6S4P5 for the gray short-
tailed opossum, E7FCR8 for the zebrafish, Q7Q0N9 for the fruit fly, D3DML7 for the african malaria mosquito, and H2KZ54 for the roundworm.
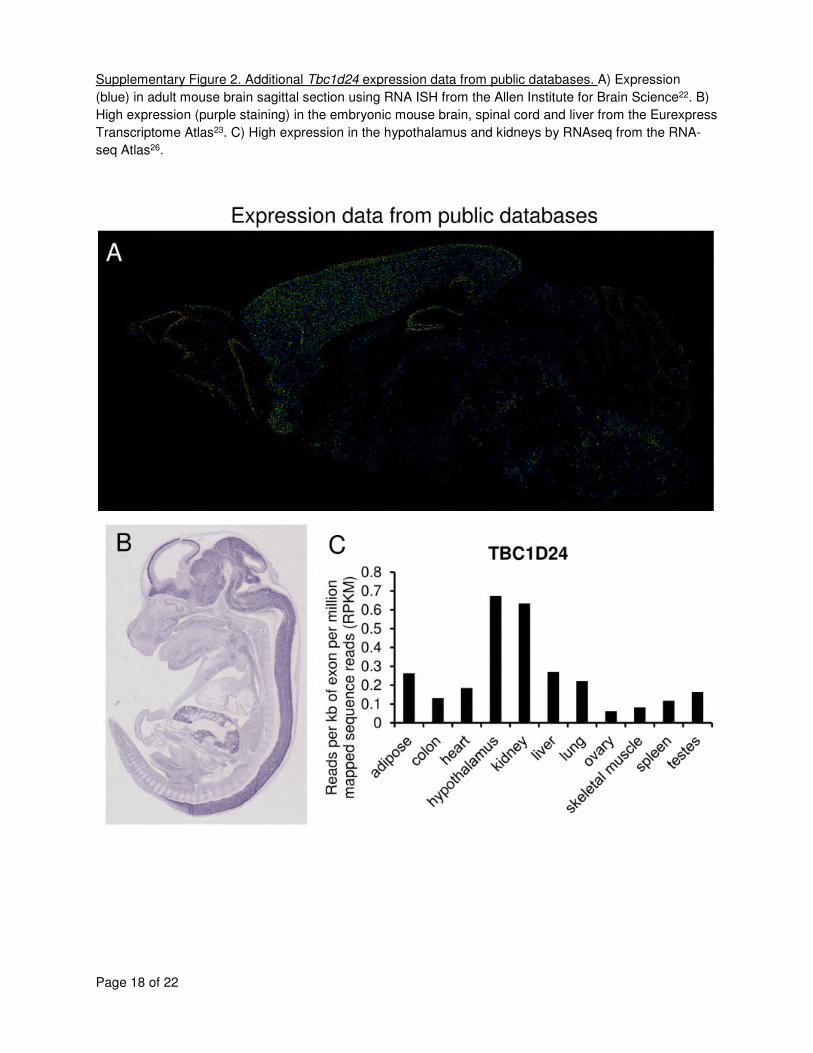
Page 18 of 22
Supplementary Figure 2. Additional Tbc1d24 expression data from public databases. A) Expression
(blue) in adult mouse brain sagittal section using RNA ISH from the Allen Institute for Brain Science22. B)
High expression (purple staining) in the embryonic mouse brain, spinal cord and liver from the Eurexpress
Transcriptome Atlas23. C) High expression in the hypothalamus and kidneys by RNAseq from the RNA-
seq Atlas26.
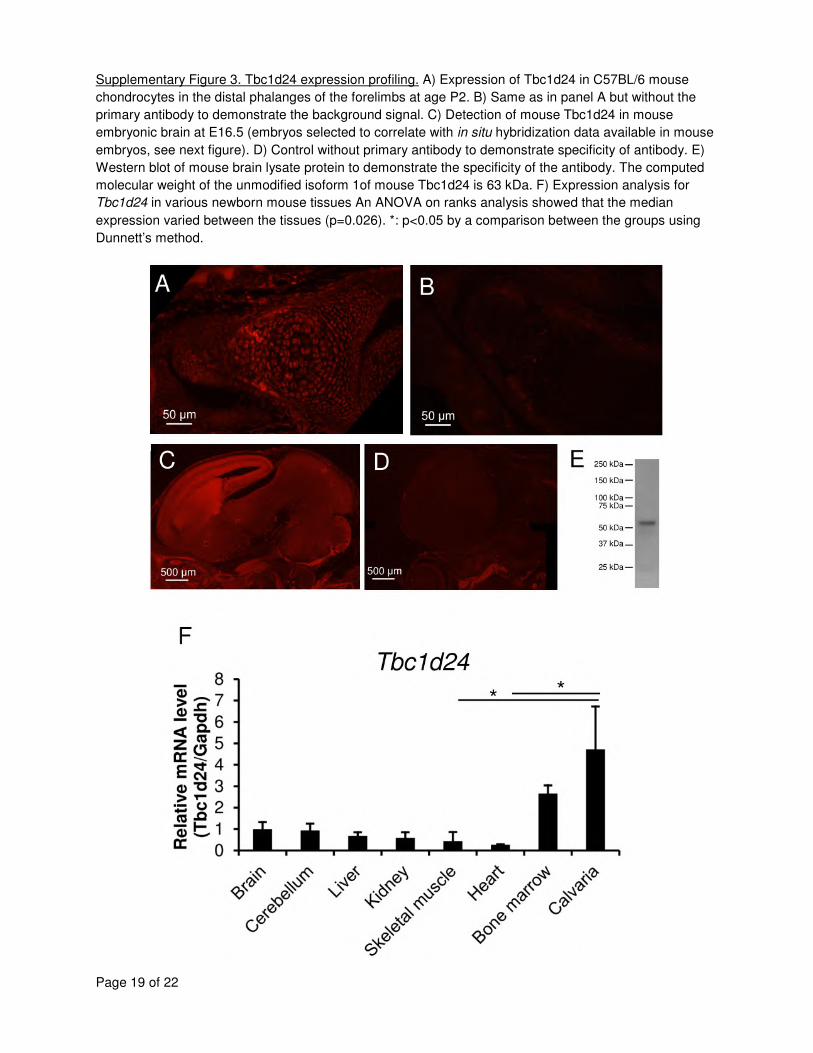
Page 19 of 22
Supplementary Figure 3. Tbc1d24 expression profiling. A) Expression of Tbc1d24 in C57BL/6 mouse
chondrocytes in the distal phalanges of the forelimbs at age P2. B) Same as in panel A but without the
primary antibody to demonstrate the background signal. C) Detection of mouse Tbc1d24 in mouse
embryonic brain at E16.5 (embryos selected to correlate with in situ hybridization data available in mouse
embryos, see next figure). D) Control without primary antibody to demonstrate specificity of antibody. E)
Western blot of mouse brain lysate protein to demonstrate the specificity of the antibody. The computed
molecular weight of the unmodified isoform 1of mouse Tbc1d24 is 63 kDa. F) Expression analysis for
Tbc1d24 in various newborn mouse tissues An ANOVA on ranks analysis showed that the median
expression varied between the tissues (p=0.026). *: p<0.05 by a comparison between the groups using
Dunnett’s method.

Page 20 of 22
Supplementary Discussion. 2-Oxoglutaric aciduria and DOORS syndrome types.
Increased 2-oxoglutaric acid is often found in the blood and urine in DOORS syndrome, but is not
pathognomonic. Levels can fluctuate between normal values to very high values over time in the same
patient29,30 and are not elevated in all affected individuals. As such, its absence cannot be used to
exclude a diagnosis of DOORS syndrome. 2-oxoglutaric aciduria is found in several metabolic disorders,
but rarely in association with dysmorphisms such as in DOORS syndrome31. Patton et al. noted that α-
hydroxyglutarate, a metabolite of 2-oxoglutarate, is also elevated in the urine, thus suggesting that the
activity of the 2-oxoglutarate dehydrogenase complex (made of the E1, E2 and E3 components) was
intact29. Surrendran et al. found decreased E1 activity in patient fibroblasts and white blood cells32, while it
was normal in the patient described by James et al.33 . Patton et al. suggested separating DOORS
syndrome from autosomal dominant deafness and onychodystrophy29, now called DDOD syndrome [MIM
124480], and James et al. also excluded from their review cases with either dominant inheritance or
without intellectual disability or seizures33. Rajab et al. suggested dividing DOORS syndrome into type I,
with more severe neurological involvement (in terms of intellectual disability and seizures) and type II with
a milder neurological disease and course34. They noted that 2-oxoglutaric aciduria was present in the
more severe cases and in none of the milder cases. However, as Felix et al. demonstrated, several
individuals with type I (or more severe) DOORS syndrome do not have 2-oxoglutaric aciduria35. James et
al. also concluded that the division between type I and type II should not be used, in part because of
clinical heterogeneity even within families33.
We suggest three hypotheses for the source of 2-oxoglutaric aciduria found in DOORS in light of our
findings. 2-oxoglutarate might originate from increased glutamate release from neurons (suggested by
increased neurotransmitter release in the Skywalker Drosophila studies36) with subsequent metabolism to
2-oxoglutarate in astrocytes. Another hypothesis is that vesicle-bound aspartate aminotransferase,
converting 2-oxoglutarate and aspartate to glutamate for vesicular transport37, could be secondarily
affected by abnormal vesicular transport. We could partly test these hypotheses in the future using
cerebrospinal fluid or magnetic resonance spectroscopy of the affected individuals. Finally, 2-oxoglutarate
could originate from defective activity of the TLDc domain, the substrates and catalytic activity of which
are unknown, but could involve 2-oxoglutarate. This is less likely given that the mutations are often far
from the TLDc domain (see Figure 3D).

Page 21 of 22
References
1. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform.
Bioinformatics 2009; 25: 1754-60.
2. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using
next-generation DNA sequencing data. Nat Genet 2011; 43(5): 491-8.
3. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res 2010; 20(9): 1297-303.
4. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools.
Bioinformatics 2009; 25: 2078-9.
5. Akyuz M, Cabuk H. Meteorological variations of PM2.5/PM10 concentrations and particle-
associated polycyclic aromatic hydrocarbons in the atmospheric environment of Zonguldak, Turkey. J
Hazard Mater 2009; 170(1): 13-21.
6. Havlak P, Chen R, Durbin KJ, et al. The Atlas genome assembly system. Genome Res 2004; 14(4):
721-32.
7. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-
throughput sequencing data. Nucleic Acids Res 2010; 38(16): e164.
8. Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: A Database of Human Non-synonymous SNVs and Their
Functional Predictions and Annotations. Hum Mutat 2013.
9. Uniprot_Consortium. Reorganizing the protein space at the Universal Protein Resource
(UniProt). Nucleic Acids Res 2012; 40(Database issue): D71-5.
10. Gaudet P, Argoud-Puy G, Cusin I, et al. neXtProt: organizing protein knowledge in the context of
human proteome projects. J Proteome Res 2013; 12(1): 293-8.
11. Shaw DR. Searching the Mouse Genome Informatics (MGI) resources for information on mouse
biology from genotype to phenotype. Curr Protoc Bioinformatics 2009; Chapter 1: Unit1 7.
12. Baxevanis AD. Searching Online Mendelian Inheritance in Man (OMIM) for information on
genetic loci involved in human disease. Curr Protoc Hum Genet 2012; Chapter 9: Unit 9 13 1-0.
13. Seelow D, Schwarz JM, Schuelke M. GeneDistiller--distilling candidate genes from linkage
intervals. PLoS One 2008; 3(12): e3874.
14. Seelow D, Schuelke M. HomozygosityMapper2012--bridging the gap between homozygosity
mapping and deep sequencing. Nucleic Acids Res 2012; 40(Web Server issue): W516-20.
15. Wierenga KJ, Jiang Z, Yang AC, Mulvihill JJ, Tsinoremas NF. A clinical evaluation tool for SNP
arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genet Med
2013; 15(5): 354-60.
16. EVS. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. 2012.
http://evs.gs.washington.edu/EVS/.
17. Felsenstein J, Churchill GA. A Hidden Markov Model approach to variation among sites in rate of
evolution. Mol Biol Evol 1996; 13(1): 93-104.
18. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of
constraint in mammalian genomic sequence. Genome Res 2005; 15(7): 901-13.
19. Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974;
185(4154): 862-4.
20. Guven A, Tolun A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J Med
Genet 2013; 50(3): 199-202.
21. Falace A, Filipello F, La Padula V, et al. TBC1D24, an ARF6-interacting protein, is mutated in
familial infantile myoclonic epilepsy. Am J Hum Genet 2010; 87(3): 365-70.
22. Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse
brain. Nature 2007; 445(7124): 168-76.
23. Diez-Roux G, Banfi S, Sultan M, et al. A high-resolution anatomical atlas of the transcriptome in
the mouse embryo. PLoS Biol 2011; 9(1): e1000582.

Page 22 of 22
24. Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information.
Nucleic Acids Res 2013; 41(D1): D561-5.
25. Hruz T, Laule O, Szabo G, et al. Genevestigator v3: a reference expression database for the meta-
analysis of transcriptomes. Adv Bioinformatics 2008; 2008: 420747.
26. Krupp M, Marquardt JU, Sahin U, Galle PR, Castle J, Teufel A. RNA-Seq Atlas--a reference
database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics
2012; 28(8): 1184-5.
27. Uhlen M, Oksvold P, Fagerberg L, et al. Towards a knowledge-based Human Protein Atlas. Nat
Biotechnol 2010; 28(12): 1248-50.
28. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple
sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7: 539.
29. Patton MA, Krywawych S, Winter RM, Brenton DP, Baraitser M. DOOR syndrome (deafness,
onycho-osteodystrophy, and mental retardation): elevated plasma and urinary 2-oxoglutarate in three
unrelated patients. Am J Med Genet 1987; 26(1): 207-15.
30. van Bever Y, Balemans W, Duval EL, et al. Exclusion of OGDH and BMP4 as candidate genes in
two siblings with autosomal recessive DOOR syndrome. Am J Med Genet A 2007; 143(7): 763-7.
31. Kelley RI, Robinson D, Puffenberger EG, Strauss KA, Morton DH. Amish lethal microcephaly: a
new metabolic disorder with severe congenital microcephaly and 2-ketoglutaric aciduria. Am J Med
Genet 2002; 112(4): 318-26.
32. Surendran S, Michals-Matalon K, Krywawych S, et al. DOOR syndrome: deficiency of E1
component of the 2-oxoglutarate dehydrogenase complex. Am J Med Genet 2002; 113(4): 371-4.
33. James AW, Miranda SG, Culver K, Hall BD, Golabi M. DOOR syndrome: clinical report, literature
review and discussion of natural history. Am J Med Genet A 2007; 143A(23): 2821-31.
34. Rajab A, Riaz A, Paul G, Al-Khusaibi S, Chalmers R, Patton MA. Further delineation of the DOOR
syndrome. Clin Dysmorphol 2000; 9(4): 247-51.
35. Felix TM, de Menezes Karam S, Della Rosa VA, Moraes AM. DOOR syndrome: report of three
additional cases. Clin Dysmorphol 2002; 11(2): 133-8.
36. Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals
synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 2011; 145(1): 117-32.
37. Takeda K, Ishida A, Takahashi K, Ueda T. Synaptic vesicles are capable of synthesizing the VGLUT
substrate glutamate from alpha-ketoglutarate for vesicular loading. J Neurochem 2012; 121(2): 184-96.
Figure 1
17 families without TBC1D24 mutations (including 9 with 5/5 main features
of DOORS syndrome). Analysis of the remaining exomes did not lead to the
identification of another gene mutated in most samples.
TBC1D24 mutations identified
in 2 additional families
9 families with TBC1D24 mutations
(out of 18 with 5/5 main features of
DOORS syndrome)
Sanger sequencing for TBC1D24 was performed in the other 9 families
Data analysis (performed while 2 exomes pending):
TBC1D24 mutations identified in 6 families (7 after all exomes completed)
21 families with 3/5 main features of DOORS syndrome assessed for eligibility
4 ineligible
2 families could not be contacted for consent
1 had DDOD syndrome
1 for which sample was not available on affected individual
9 additional eligible
families enrolled
17 remaining families had exome sequencing in an affected individual
3 families with consanguinity or affected siblings also had SNP array
7 without mutations
10 without mutations
FiguresClick here to download Figure: Figure R4.pdf
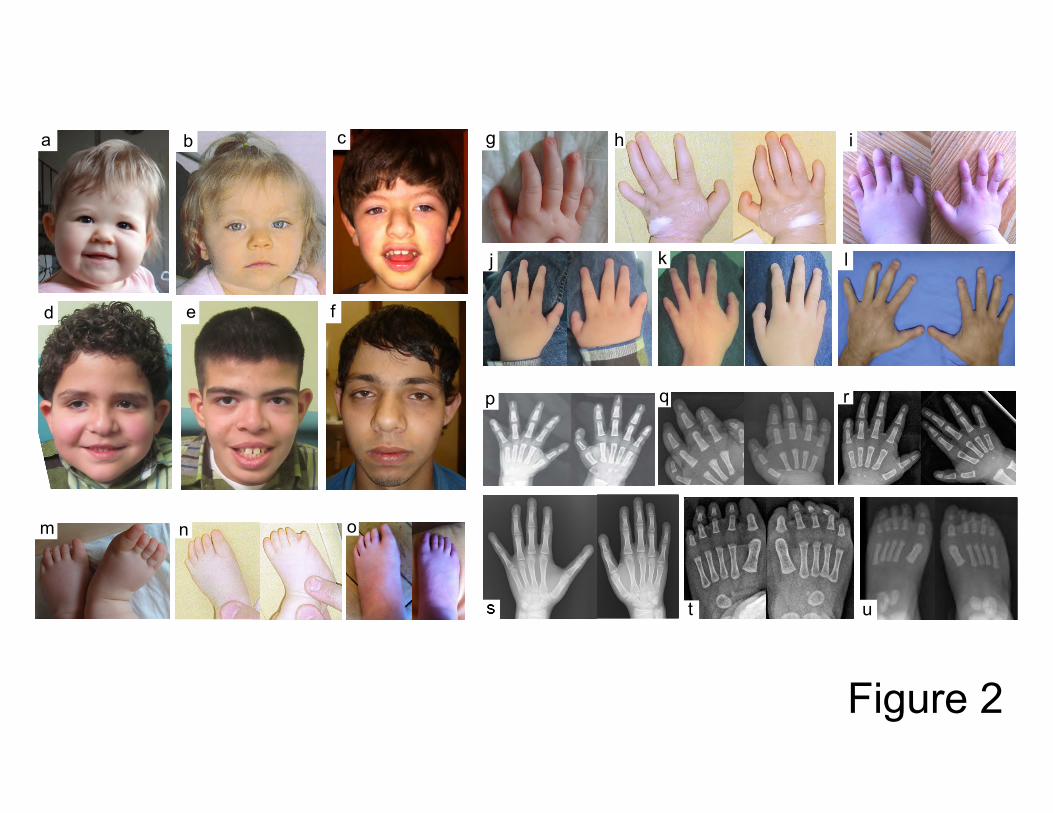
a
d e
b c
f
ig h
l
m on
p r
t
q
u
j k
Figure 2