
Portrait of a canine probiotic Bifidobacterium—From gut to gut
Upload
independentCategory
view
1download
0
JOURNAL OF BACTERIOLOGY, Dec. 2005, p. 8411–8426 Vol. 187, No. 240021-9193/05/$08.00�0 doi:10.1128/JB.187.24.8411–8426.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
The ClgR Protein Regulates Transcription of the clpP Operonin Bifidobacterium breve UCC 2003†
Marco Ventura,* Ziding Zhang,‡ Michelle Cronin, Carlos Canchaya, John G. Kenny,Gerald F. Fitzgerald, and Douwe van Sinderen
Alimentary Pharmabiotic Centre and Department of Microbiology, Bioscience Institute,National University of Ireland, Western Road, Cork, Ireland
Received 11 July 2005/Accepted 21 September 2005
Five clp genes (clpC, clpB, clpP1, clpP2, and clpX), representing chaperone- and protease-encoding genes, werepreviously identified in Bifidobacterium breve UCC 2003. In the present study, we characterize the B. breve UCC2003 clpP locus, which consists of two paralogous genes, designated clpP1 and clpP2, whose deduced proteinproducts display significant similarity to characterized ClpP peptidases. Transcriptional analyses showed thatthe clpP1 and clpP2 genes are transcribed in response to moderate heat shock as a bicistronic unit with a singlepromoter. The role of a clgR homologue, known to control the regulation of clpP gene expression in Streptomyceslividans and Corynebacterium glutamicum, was investigated by gel mobility shift assays and DNase I footprintexperiments. We show that ClgR, which in its purified form appears to exist as a dimer, requires a protein-aceous cofactor to assist in specific binding to a 30-bp region of the clpP promoter region. In pull-downexperiments, a 56-kDa protein copurified with ClgR, providing evidence that the two proteins also interact invivo and that the copurified protein represents the cofactor required for ClgR activity. The prediction of theClgR three-dimensional structure provides further insights into the binding mode of this protein to the clpP1promoter region and highlights the key amino acid residues believed to be involved in the protein-DNAinteraction.
Bifidobacteria are some of the most common inhabitants ofmammalian and animal gastrointestinal tracts (37). In the hu-man gastrointestinal tract, their presence has been associatedwith beneficial health effects, such as the prevention of diar-rhea, amelioration of lactose intolerance, and immunomodu-lation (21, 28, 37, 45). The preparation of Bifidobacteriumcontaining products may require that the microbes surviveindustrial food manufacturing processes, such as freeze-drying,freezing, and spray drying, while remaining viable during stor-age. Such probiotic products reinforce the need for robustbifidobacteria to survive passage through the upper parts of thedigestive tract, compete with the resident intestinal flora, pref-erably colonize the digestive tract, and express specific func-tions, probably under suboptimal growth conditions. Despitethe commercial, and consequently, scientific interest, the ge-netics of bifidobacteria has not developed to a degree that canbe compared to that of other high-G�C gram-positive bacteria(45), and therefore molecular tools that, for example, allowgene inactivation or in vivo transcription analysis are not yetfeasible for bifidobacteria.
In order to resist stressful environmental challenges, cellssynthesize protective proteins, including both chaperones andsubstrate-specific proteases, that primarily act to prevent theaccumulation of misfolded proteins by performing various
roles, such as protein folding, stabilization, renaturation, andresolubilization (22, 35, 39). Some of the genetic elementsencoding these chaperones have been identified in bifidobac-teria, including the groEL-groES (43) and dnaK (47) genes.
Recent studies of bacteria have focused on the expression ofthe Clp protein family, members of which are well conserved inboth eukaryotic and prokaryotic organisms (30, 52). Many Clpproteins contain ATPase activity, and the number of ATPnucleotide-binding domains in such proteins has been usedfor classification purposes. It is widely accepted that ClpATPases can function both as molecular chaperones and asregulator components of the proteolytic complex (52). In Es-cherichia coli, the Clp complex consists of two functionallydistinct subunits: the larger ClpA protein functions as theATP-binding regulatory subunit (25), conferring substratespecificity, whereas the smaller ClpP protein provides the pro-teolytic activity (25). On its own, ClpP possesses only peptidaseactivity, and it requires an association with ClpA to degradepolypeptides of longer than six amino acids. The Clp proteasecomplex consists of two central heptameric rings of ClpPflanked by two hexameric rings of ClpA. In Firmicutes, with theexception of Bacillus thuringiensis (8), only a single chromo-somal copy of the clpP gene has been found, whereas in Actino-bacteridae up to five clpP paralogous genes have been identi-fied (49, 51). So far, it is not known if the presence of multiplecopies of the clpP gene is correlated with enhanced protectionagainst certain stressful conditions. In Streptomyces lividans,the five identified clpP-like genes are organized into two oper-ons, one that includes the clpP1 and clpP2 genes and one thatencompasses the clpP3 and clpP4 genes, and a third monocis-tronic transcription unit harbors clpP5 (51).
In eubacteria, expression of the genes belonging to the clp
* Corresponding author. Present address: Department of Genetics,Evolution and Anthropology, University of Parma, Parma, Italy.Phone: 39 0521 905479. Fax: 39 0521 905609. E-mail: [email protected] .
† Supplemental material for this article may be found at http://jb.asm.org/.
‡ Present address: Bioinformatics Center, College of Biological Sci-ences, China Agricultural University, Beijing, 100094 China.
8411
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

family is subject to multiple modes of regulation. In Esche-richia coli, the clpP, clpB, and clpX genes are controlled bythe general heat shock sigma factor �32 (11, 18). In contrast,in several gram-positive bacteria, including Bacillus subtilis,Listeria monocytogenes, Streptococcus salivarius, Enterococ-cus faecalis, Lactobacillus sakei, Lactococcus lactis, Oenococ-cus oeni, and Clostridium acetobutylicum, transcription ofthe single clpP gene is directed by the vegetative sigmafactor �A and controlled by the CtsR repressor, which bindsa heptanucleotide repeat that overlaps the �10 and �35hexamers (6).
In high-G�C gram-positive bacteria like S. lividans, expres-sion of the clpP1 clpP2 operon, as well as that of the clpC gene,is regulated by a transcriptional activator, ClgR (for clp generegulator), which binds an imperfect consensus motif (CGCT-4N-GCGNAC) (2, 7). The expression of the second clpP operon(clpP3 clpP4) in Streptomyces lividans is regulated by a secondtranscriptional activator, designated PopR (49).
In this report, the clpP operon of Bifidobacterium breve UCC2003 is described. The transcriptional induction of this operonupon exposure to stressful conditions was investigated, whilethe role of a ClgR homologue and an as-yet-unidentified co-factor protein in the regulation of the clpP operon was ex-plored, revealing evidence for a novel heat shock-controlledregulatory mechanism in the genus Bifidobacterium.
MATERIALS AND METHODS
Bacterial strains and culture conditions. All Bifidobacterium strains used forthis study are described in Table 1. They were grown anaerobically in MRS(Difco, Detroit, MI) supplemented with 0.05% L-cysteine–HCl and wereincubated at 37°C for 16 h. Escherichia coli was grown aerobically on a rotaryshaker (150 rpm) at 37°C in LB medium or plated onto LB agar (Difco,Detroit, MI) plates when appropriate. Antibiotics were used at the followingconcentrations: ampicillin, 100 �g/ml; kanamycin, 25 �g/ml; and chloram-phenicol, 2 �g/ml.
DNA amplification. Genomic DNAs used as templates for PCRs were ex-tracted following a protocol described in a previous study (46).
PCR was used to amplify a DNA fragment corresponding to a 1,000-bp internalfragment of the clpP operon from all investigated Bifidobacterium strains, using theoligonucleotides clpP1-UNIV and clpP1-REV (Table 2). PCRs were carried outaccording to the standard procedure described by Sambrook et al. (32). The result-ing amplicons were separated in a 1.5% agarose gel, followed by ethidium bromidestaining. PCR fragments were purified using a PCR purification spin kit (QIAGEN,West Sussex, United Kingdom) and were subsequently sequenced.
Phylogenetic analysis. Phylogeny calculations, including distance calculationsand the generation of phylogenetic trees, were performed using the PHYLIPpackage (9) and the ClustalX program. The numbers of synonymous substitu-tions between all possible pairs of the clpP1 and clpP2 genes were determined byapplying the method of Nei and Gojobori (27), using the MEGA computerprogram (19). Correction for multiple substitutions was done according to theJukes-Cantor formula (17).
Plasmids and plasmid constructions. The E. coli pQE-30 vector (QIAGEN)was used for overproduction and purification of an N-terminally six-histidine-tagged bifidobacterial ClgR protein (h-ClgR). The clgR gene from B. breve UCC2003 was amplified using the primers 903-uni and 903-rev, which contain aBamHI and a HindIII restriction site, respectively. The resultant 566-bp PCR
TABLE 1. Strains used for this study, with clpP1 and clpP2 sequence accession numbers
Species Straina clpP1 accession no.b clpP2 accession no.b
Bifidobacterium animalis subsp. animalis ATCC 25527 AY955244 AY955233Bifidobacterium animalis subsp. lactis DSM 10140 AY955250 AY955241Bifidobacterium breve UCC 2003 AY955251 AY955251Bifidobacterium adolescentis LMG 10502 AY955247 AY955239Bifidobacterium longum NCC 2705 NC_004307 NC_004307Bifidobacterium infantis JCM 1222 AY955248 AY955238Bifidobacterium suis LMG 21814 AY955245 AY955236Bifidobacterium dentium JCM 1195 AY955243 AY955234Bifidobacterium cuniculi LMG 10738 AY955249 AY955240Bifidobacterium pullorum LMG 21816 AY955246 AY955235Bifidobacterium magnum DSM 20222 AY955242 AY955237Lactobacillus plantarum WCFS1 AL935263 AL935263Lactobacillus johnsonii NCC 533 NC_002662 NC_002662L. lactis subsp. lactis IL1403 NC_002662 NC_002662Streptococcus pyogenes MGAS315 NC_006086 NC_006086Streptococcus thermophilus CNRZ 1066 CP000024 CP000024Enterococcus faecalis V583 NC_004668 NC_004668Clostridium acetobutylicum ATCC 824 NC_003030 NC_003030Mycobacterium tuberculosis H37Rv NC_000962 NC_000962Streptomyces coelicolor A3 NC_003888 NC_003888Streptomyces avermitilis MA-4680 NC_003155 NC_003155Corynebacterium efficiens YS-314 NC_004369 NC_004369Listeria innocua Clip 11262 AL592022 AL592022Escherichia coli K12 U00096 U00096Thermobifida fusca YX NZ_AAAQ00000000 NZ_AAAQ00000000Mycobacterium avium subsp. paratuberculosis K10 NC_002944 NC_002944Mycobacterium leprae TN NC_002677 NC_002677Mycobacterium bovis AF2122/97 NC_002945 NC_002945Leifsonia xyli subsp. xyli CTCB07 NC_006087 NC_006087Tropheryma whipplei TW08/27 NC_004551 NC_004551
a ATCC, American Type Culture Collection; DSM, Deutsche Sammlung von Mikroorganismen; JCM, Japanese Collection of Microorganisms; LMG, BacteriaCollection Universiteit Gent.
b For those strains whose genome sequences are available, the clpP1 and clpP2 sequences were retrieved from the complete bacterial genome, and their accessionnumbers are provided.
8412 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

fragment was digested with BamHI and HindIII and ligated into similarly re-stricted pQE30, using the T4 DNA ligase enzyme (Roche, Sussex, United King-dom), to generate plasmid pQE-ClgR, which was introduced into E. coli M15(QIAGEN, United Kingdom) as described by Sambrook et al. (32).
Plasmid pNZ272 (29), which contains a promoterless gusA gene system, wasused as a reporter system. Various portions of the clpP promoter region weregenerated by PCR, using one fixed primer for the 3� end (clp-Rev [complemen-tary to sequences 9 bp upstream of the start codon of the B. breve UCC 2003clpP1 gene]) and various primers for the 5� end of this promoter region (Table 2).The resultant PCR amplicons were digested with BglII and PstI and ligated intosimilarly restricted pNZ272, which was used to transform E. coli M15 (QIAGEN,United Kingdom). From these transformant plasmids, pclp1, pclp6, pclp7, pclp8,and pclp3 were isolated, which carry the whole clpP promoter region (pclp1) ordecreasing portions of it (pclp6, pclp7, pclp8, and pclp3). All of the above-mentioned plasmids were then introduced into B. breve UCC 2003 by electro-transformation. DNA sequences of all genetic constructs were confirmed byDNA sequencing (MWG Biotech, Ebersberg, Germany) and restriction analysis.
GUS assay. B. breve UCC 2003 cultures with an inoculum level of 2% con-taining pNZ272 or a derivative thereof were grown exponentially at 37°C until anoptical density at 600 nm (OD600) of 0.3 was reached, after which the temper-ature was shifted to 43°C or 50°C or NaCl was added to a final concentration of0.7 M, and the cultures were incubated for 180 min. One milliliter of cells wasthen centrifuged at 7,000 rpm for 5 min, and �-glucuronidase (GUS) activity wasdetermined as described by Platteeuw et al. (29).
Overproduction of h-ClgR in E. coli and protein purification. A 300-ml cultureof an E. coli M15 strain containing the pQE-ClgR plasmid was grown to anOD600 of 0.6 prior to induction by the addition of 1 mM IPTG (isopropyl-�-D-thiogalactopyranoside; Fluka, Germany). Three hours following induction, cellswere harvested by centrifugation at 10,000 rpm for 10 min. Cell pellets wereresuspended in lysis buffer (50 mM NaH2PO4, 10 mM Tris-HCl, 30 mM imida-zole, pH 8.0) as recommended by the supplier (QIAGEN) and allowed to lyse bybeing shaken gently at 27°C for 2 h. Cell debris was eliminated from the lysate bycentrifugation at 13,000 rpm for 10 min. The resulting supernatant was passedthrough a column containing 4 ml of Ni-nitrilotriacetic acid (Ni-NTA) agarose(QIAGEN), which had been preequilibrated with 10 ml of lysis buffer. Thecolumn was washed two times with 10 ml of wash buffer (50 mM NaH2PO4, 1 MNaCl, 30 mM imidazole, 0.5% [vol/vol] Triton X-100, 5 mM �-mercaptoethanol,pH 8.0) and then eluted using 10 ml of elution buffer (50 mM NaH2PO4, 1 MNaCl, 250 mM imidazole, pH 8.0). Protein concentrations were determined usinga Bio-Rad protein assay in conjunction with a bovine serum albumin standard
curve. The expected size and purity of the eluted h-ClgR protein were verified bysodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
h-ClgR cross-linking. Cross-linking experiments were performed according toa previously published procedure (1). All samples were fractionated in loadingbuffer by SDS-PAGE.
RNA isolation and Northern blot analysis. B. breve UCC 2003 cells weregrown to an OD600 of 0.6. Temperature stress was applied by transferring theculture to either 20°C, 37°C, 43°C, 47°C, or 50°C, while osmotic stress was appliedby the addition of 5 M NaCl-containing prewarmed medium to give a finalconcentration of either 0.5 M or 0.7 M. At various time points, 30-ml aliquots ofculture were collected and briefly centrifuged to harvest cells. Total RNA wasisolated using the macaloid acid method and then treated with DNase (Roche,United Kingdom). Briefly, cell pellets were resuspended in 0.5 ml of phenol, pH7.5, and placed in a tube containing 0.18 g of macaloid acid (Sigma) and 0.8 g ofglass beads (diameter, 106 �m; Sigma). The cells were lysed by shaking the mixat the maximum setting on a BioSpec homogenizer at 4°C for 2 min. The mixturewas then centrifuged at 12,000 rpm for 15 min, and the upper phase containingthe RNA sample was recovered. The RNA sample was further purified by phenoland ethanol precipitated according to the method described by Sambrook et al.(32). Slot blot hybridizations were carried out following a previously describedprotocol (48). RNA electrophoresis and Northern blot hybridization were car-ried out as described previously (42, 44). All slot blot and Northern hybridizationexperiments were performed at least twice.
Primer extension analysis. The 5� end of the clpP1 RNA transcript wasdetermined using a protocol described in a previous study (44). The syntheticoligonucleotide used was named clpP1-prom (Table 2).
Gel mobility shift DNA binding assays. A 377-bp DNA fragment correspond-ing to the clpP1 promoter region (from position �249 to position �128 withrespect to the putative transcription start site) was amplified by PCR withprimers P1-uni and P1-rev. The resultant amplicon was purified using a G50-Spincolumn (Amersham, Little Chalfont, United Kingdom) and then labeled using[�-32P]dATP and T4 polynucleotide kinase (New England Biolabs, MA). Thelevel of radioactive labeling was measured using a Beckman LS multipurposescintillation counter (Fullerton, CA).
Binding reactions were performed according to a previously described proto-col (48). Bands were visualized by autoradiography at �70°C, using KodakBiomax MR film (Eastman-Kodak).
All gel retardation assays were performed at least twice.Protease treatment of crude cell extract from B. breve UCC 2003. Ten micro-
grams of crude cell extract from B. breve UCC 2003 was incubated with 20 U ofpronase enzyme (Roche, United Kingdom) for 4 h at 37°C, as recommended bythe supplier. The pronase was subsequently heat inactivated and/or chemicallyinactivated by incubating the mixture with 3 �l of 1� protease inhibitor cocktail(Roche, United Kingdom), as recommended by the supplier. The control samplewas treated identically, except that no pronase was added.
Protein pull-down procedure. The h-ClgR or h-HspR (48) protein was at-tached to a column containing 4 ml of Ni-NTA agarose (QIAGEN) which hadbeen preequilibrated with 10 ml of lysis buffer. A crude extract from a UCC 2003culture grown at 43°C or 50°C was then passed through the h-ClgR- or h-HspR-containing column. The column was treated as described above, except that thewash buffer did not contain Triton X-100 or �-mercaptoethanol and the elutionbuffer contained 500 mM imidazole. The expected size and purity of the coelutedprotein were verified by SDS-PAGE.
DNase I footprint assays. A 377-bp DNA fragment covering the clpP1 pro-moter region (from position �249 to position �128 with respect to the putativetranscription start site) was obtained by PCRs using two primer combinations, inwhich one primer was an IRD800-labeled oligonucleotide (MWG Biotech,Germany) and the second primer was unlabeled in order to effect strand-specificlabeling. The two primer combinations used for this purpose were P1-TOPIRD-800/P1-REV and P1-BOT/P1-uni (IRD800) (Table 2).
Binding reactions were performed as described above in the section outlininggel mobility shift DNA binding assays, using 0.15 pmol of labeled fragments, 25to 100 pmol of h-ClgR, and 1 �g of crude lysate from B. breve UCC 2003 culturesgrown at 43°C. The resulting reaction mixtures were then incubated for 30 minat 37°C, followed by the addition of 5 �l of 0.25-�g/ml DNase I in DNase I buffer(10 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 5 mM CaCl2, 50 mM KCl, and 1 mMdithiothreitol), after which digestion was allowed to proceed for exactly 5 min at37°C and stopped by the addition of 0.5 �l of 0.5 M EDTA, pH 8.0. After theaddition of 500 �l of 95% ethanol, the DNA was precipitated overnight at �20°Cand recovered by centrifugation (10,000 rpm for 15 min). The resulting DNApellet was washed with 500 �l of 70% ethanol, air dried, and dissolved in 2.5 �lof water and 2.5 �l of loading solution (LicoR, Cambridge, United Kingdom).The reactions were separated as described for primer extension analysis (7).
TABLE 2. Oligonucleotides used for this study
Oligonucleotide Sequence (5�33�) (restriction enzyme)a
clpP1-UNIV ............................... GAGGTCAAGGACGATATGclpP1-REV ................................. CTTGCCGAAGCCCTGGTC903-uni ........................................ CGCGGATCCGGTCATCGTGTT
GTACTAAG (BamHI)903-rev ........................................ CCCAAGCTTACGTTCGACGGA
CTCGAG (HindIII)Clp1-f .......................................... AAAAGATCTGCTCGACGGAGT
CGAGAC (BlgII)Clp6-f .......................................... AAAAGATCTCTGGCCGCCAAA
AAGCGTG (BlgII)Clp7-f .......................................... AAAAGATCTCGCCAAAAAGCG
TGAA (BlgII)Clp8-f .......................................... AAAAGATCTGCGTGAAAAGCC
GTCTTC (BlgII)Clp3-f .......................................... AAAAGATCTGTCTTCAGTCGC
TTC (BlgII)Clp-Rev....................................... AAACTGCAGGACCGGCAAGGT
CGCAAAGGC (PstI)P1-uni .......................................... GATGCGGTCTTTGAGCAGP1-rev .......................................... CTAGTCCAGCCAATGCAATGclpP1-prom ................................. CTTGACCTCTTCACCCATCP1-TOP ....................................... GATGCGGTCTTTGAGCAGP1-BOT....................................... CTAGTCCAGCCAATGCAATG
a In some cases, oligonucleotides were designed to introduce recognition sitesfor restriction endonucleases (restriction sites are underlined, and restrictionendonucleases are indicated in parentheses).
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8413
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 1. Comparison of the clpP operon in B. breve UCC 2003 and corresponding loci in various other bacteria. Each arrow indicates an openreading frame. The lengths of the arrows are proportional to the lengths of the predicted open reading frames. Orthologs are marked with the samecolor. The putative function of the protein is indicated above each arrow. The amino acid identity of each protein with respect to B. breve UCC2003 is indicated as a percentage.
8414 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

ClgR 3D prediction. To derive structure templates for the B. breve UCC 2003ClgR protein, fold recognition techniques were carried out via the protein struc-ture prediction Meta server (http://bioinfo.pl/Meta/). Several structure templateswere used, including the tertiary structure of the DNA binding domain of SinRfrom Bacillus subtilis (20) (PDB entry 1b0n; chain A) and that of the CI repressorfrom bacteriophage (PDB entry 1rio; chain A). An alignment between theC-terminal portion (residues 94 to 170) of ClgR, 1b0n, and 1rio was generated byusing the Fold and Function Assignment System (FFAS) (31), a profile-profilealignment algorithm. Finally, a three-dimensional (3D) model of this C-terminaldomain of ClgR was obtained via the Swiss Model server (34).
Nucleotide sequence accession numbers. The GenBank accession numbers forthe partial clpP1 and clpP2 gene sequences generated in this study are reportedin Table 1. Nucleotide sequence data regarding the clpP operon of B. breve UCC2003 have been deposited in GenBank under accession number AY955251. Thenucleotide sequence of the B. breve UCC 2003 clgR gene has been deposited inGenBank under accession number AY837843.
RESULTS
Identification of the clpP operon in B. breve UCC 2003.An analysis of the B. breve UCC 2003 genome sequence (S.Leahy, M. O’Connell-Motherway, J. A. Moreno-Munoz, G. F.Fitzgerald, D. G. Higgins, and D. van Sinderen, unpublisheddata) revealed the presence of adjacent genes, designatedclpP1 and clpP2, whose protein products displayed 53% and48% identity to ClpP1 and ClpP2 of Streptomyces coelicolor A3,respectively. The residues Ser, His, and Asp, which constitutethe catalytic triad of the serine protease ClpP in E. coli (25),are conserved in the ClpP1 sequences of B. breve UCC 2003(positions 102, 121, and 172, respectively).
The structural organization and locations of the clpP1 andclpP2 genes in the chromosomes of B. breve UCC 2003 andother bacteria are schematically displayed in Fig. 1, where thededuced amino acid sequences of the B. breve UCC 2003 clpPoperon are aligned with those of high-G�C gram-positive bac-teria and low-G�C gram-positive bacteria. This comparativeanalysis showed that the most similar proteins to the predictedB. breve ClpP1/ClpP2 proteases were the assumed ClpP1/ClpP2 proteins from Bifidobacterium longum (33). However,identity levels of �46% were still observed between the B.breve ClpP1 protein and the ClpP proteins of less-related bac-terial taxa, such as Bacillus clausii. In contrast, the flankingDNA regions of the clpP1 and clpP2 genes were shown to behighly variable with respect to gene synteny, except betweenthe two bifidobacterial species.
The clpP1 and clpP2 genes in bifidobacteria are preceded bythe eriC gene, which putatively encodes a chloride channelprotein, and are followed by clpX, a gene predicted to encodea ClpX protease (Fig. 1).
Phylogenetic analysis of the clpP operon in bifidobacteria.The clpP1 and clpP2 DNA sequences from B. breve and B.longum were aligned and compared. Two identical regionscorresponding to the 5� and 3� ends of the clpP1 and clpP2genes were identified, and a pair of primers (clpP1-UNIV andclpP1-REV) was designed. These primers allowed the ampli-fication of a 1,000-bp region encompassing part of the clpP1and clpP2 sequences of nine Bifidobacterium species. Align-ments of clpP1 and clpP2 DNA sequences were used to gen-erate a phylogenetic tree by the neighbor-joining method(Fig. 2). These data were supported by the indicated bootstrapvalues (9). For completeness, we included in the analysis ho-mologous DNA sequences from other strains belonging todifferent genera representing gram-positive bacteria with high
and low G�C contents. This tree showed a clear separationinto two major clusters representing the Firmicutes and theActinobacteridae taxa. Furthermore, the two ActinobacteridaeclpP paralogs separated into two distinct phylogenetic groups(Fig. 2).
A comparison of the phylogenetic trees based on the clpP1and clpP2 sequences shows very similar phylogenetic topolo-gies in the short term of evolution, whereas some discrepanciesin the branching order were depicted in bifidobacterial genusevolution (e.g., for the B. breve-Bifidobacterium suis-Bifidobac-terium infantis subcluster and B. longum).
A phylogenetic tree which was constructed on the basis ofthe 16S rRNA gene sequences available in public databaseswas mostly similar to the clpP1- and clpP2-based phylogenies(data not shown). Moreover, the correlations (r2) between thepairwise distances for the 16S rRNAs and the synonymousdistances for the clpP1 and clpP2 sequences were 0.795 and0.823, respectively. Therefore, it can be concluded that thebase substitutions occurring in the clpP1 and clpP2 sequencesduring the evolutionary process render these genes reliablemolecular evolutionary clocks. Interestingly, closely relatedstrains exhibit nearly identical 16S rRNA sequences, e.g., Bi-fidobacterium animalis subsp. animalis and B. animalis subsp.lactis occupy separate branches in the clpP1 and clpP2 se-quence-based tree (Fig. 2).
Heat induction of the clpP operon in B. breve UCC 2003.Expression of the clpP locus in high-G�C gram-positive bac-teria such as Corynebacterium glutamicum (7) and Mycobacte-rium tuberculosis (36) is induced by a number of protein-dena-turing stress treatments, such as heat and osmotic stress. Todetermine if the induction of the clpP operon occurs uponexposure to stressful conditions in B. breve UCC 2003, slot blothybridization was used to analyze total RNAs isolated from B.breve cultures following exposure for up to 150 min to temper-atures ranging from 20°C to 50°C and to NaCl concentrationsof 0.5 M and 0.7 M (Fig. 3a).
Based on the strength of the hybridization signal, the highestexpression levels of the clpP1 gene occurred at 43°C followinga 150-min exposure, whereas exposure to higher temperatures,high NaCl concentrations, or a low temperature (20°C) did notappear to significantly increase the level of clpP1 transcription(Fig. 3a). Densitometric analysis of Northern slot blots re-vealed that the levels of clpP1 mRNA were increased 15-fold incells that were subjected to heat shock at 43°C for 150 mincompared to those in unstressed cells (Fig. 3b).
Characterization of clpP1 and clpP2 gene transcription ac-tivity by Northern blotting. Northern hybridization experi-ments were performed in order to determine whether theclpP1 and clpP2 genes, and perhaps the downstream gene, arecotranscribed. Total mRNA was isolated from B. breve UCC2003 grown at 37°C, following heat shock at 43°C or 50°C, orupon osmotic shock with 0.7 M NaCl. Transcription of theclpP1 gene was investigated by Northern blotting using aninternal clpP1 probe. A 1.4-kb transcript was detected in RNAsextracted from 37°C and 43°C samples (Fig. 3c and d). Theshift to heat shock conditions (43°C) strongly increased thestrength of expression of the 1.4-kb transcript (Fig. 3d). Also,when a probe spanning the flanking clpP2 gene was used, asignal of 1.4 kb was detected (data not shown). The transcrip-tional kinetics of the clpP2 gene were found to be identical to
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8415
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG
.2.
Phyl
ogen
etic
tree
obta
ined
usin
gth
ecl
pP1
and
clpP
2ge
nese
quen
ces.
Bar
,phy
loge
netic
dist
ance
s.B
oots
trap
valu
esar
ere
port
edfo
ra
tota
lof
1,00
0re
plic
ates
.The
clpP
1an
dcl
pP2
gene
sequ
ence
sar
ein
dica
ted.
Bac
teri
abe
long
ing
toth
eF
irmic
utes
and
Act
inob
acte
ridae
grou
psar
ein
dica
ted.
8416 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 3. Transcriptional analysis of the B. breve UCC 2003 clpP operon. (a) Slot blot hybridization using RNAs extracted from cells incubatedfor up to 150 min at various temperatures or with various NaCl concentrations (indicated in the left-hand margin). (b) Schematic representationof mRNA levels of induction. The different colors and filling of the pillars correspond to the various times for which heat and osmotic shocks wereapplied, as indicated in the figure. (c) Position of transcript with respect to the clpP locus map. The estimated size of the transcript is indicated.Hairpin symbols indicate possible rho-independent terminators. (d) Northern blot analysis of the B. breve UCC 2003 clpP operon performed usingclpP1 as a probe and total mRNA isolated from cultures exposed to 37°C or under heat or hyperosmotic conditions for the times indicated.
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8417
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

those of the clpP1 gene. Both genes increased their transcriptionlevel upon temperature shift and reached their maximum tran-scriptional level at 150 min. This result clearly demonstratedthat the clpP1 and clpP2 genes form a bicistronic transcrip-tional unit. When Northern hybridization was performed witha probe corresponding to the clpX gene, no transcripts weredetected (data not shown), showing that this gene is not part ofthe clpP operon and that its transcription is not induced byheat or osmotic shock. The latter finding is in contrast to thesituation in E. coli, where clpP and clpX are part of a singletranscriptional unit (11). In B. breve, two stem-loop structures(G � �20.3 kcal and �12.9 kcal), which represent possiblerho-independent transcription terminators, are present down-stream of clpP2 and upstream of clpP1, respectively (Fig. 3c).
Identification of the clpP1 transcriptional start site. To de-termine the transcriptional start point of the clpP1 gene,primer extension analyses were performed using RNAs ex-tracted from B. breve cells which had been subjected to heatshock (see Fig. S1 in the supplemental material). An extensionproduct was identified 30 nucleotides 5� of the predicted trans-lational start site of the clpP1 gene (see Fig. S1a and b in thesupplemental material). The transcription start site was in thesame position at 43°C and at 47°C (see Fig. S1a in the supple-mental material). The analysis of the putative promoter regionof clpP1 revealed a potential promoter-like sequence weaklyresembling the previously found �10 and �35 bifidobacterialhexamers (42, 43, 47, 48). The predicted translational start siteis preceded by a typical ribosome-binding-site sequence(AAGGAG) located eight nucleotides upstream of the puta-tive translational start site.
The sequences of the region upstream of the clpP1 genes ofboth B. breve UCC 2003 and B. longum NCC 2705 were alignedin an attempt to identify putative regulatory elements. Forcompleteness, we also identified by PCR analysis the putativepromoter regions of the clpP1 genes from the closely related B.suis and B. infantis taxa and from two more distantly relatedBifidobacterium species (B. dentium and B. globosum). Asshown in Fig. S1c in the supplemental material, a large con-sensus promoter sequence can be deduced from the six se-
quences, which includes the putative �10 hexamers, �35hexamers, the ribosome-binding-site region, and the transcrip-tional start site, which were conserved in all bifidobacterialsequences examined. Moreover, a number of other DNA mo-tifs were shown to be conserved in all of these strains, includinga 4-bp partially inverted repeat (IR; CGCT-4N-GCCNA)which is almost identical to a clpP operator site for the ClgRprotein (CGCT-4N-GCGNAC) found in other members of theActinobacteridae group (2, 7).
Regulation of clpP operon: h-ClgR binds to the clpP1 pro-moter region. In several other members of the Actinobacteridaegroup, the ClgR protein was shown to bind the clpP1 promoterregion, indicating that it acts as a transcriptional regulator ofthe clpP operon (2, 7).
An analysis of the B. breve UCC 2003 genome sequenceshowed a gene homologous to clgR from C. glutamicum andS. lividans (7), which in a similar manner, could be respon-sible for the regulation of some clp genes. We identified thatin B. breve UCC 2003 and B. longum NCC 2705, the clgRgene and clpP operon are located in different chromosomalregions (data not shown). The clgR gene is located down-stream of a predicted diacylglycerol-glycerol-3-phosphate-3-phosphatidyltransferase-encoding gene (pgsA3) and upstream ofthe presumed recA gene (Fig. 4).
In order to determine if ClgR of B. breve UCC 2003 binds tothe promoter region of the clpP1 gene, the UCC 2003 ClgRprotein was overproduced in E. coli as a His-tagged version,purified, and designated h-ClgR (Fig. 5). The h-ClgR proteinwas then used in gel mobility shift DNA binding assays with377-bp radiolabeled DNA fragments corresponding to the pro-moter region of the clpP1 gene (clpP1p). These experimentsshowed that 100 pmol of purified h-ClgR protein did not affectthe mobility of the clpP1p fragment (Fig. 5b). In contrast, whenthe binding assay was performed using 50 or 100 pmol ofpurified h-ClgR protein combined with 1 �g of crude cellextract from B. breve UCC 2003 cells, designated CX, that hadbeen subjected to heat stress (43°C for 150 min), the mobilityof the clpP1p fragment was clearly reduced (Fig. 5b). Further-more, no difference in migration was observed when the clpP1p
FIG. 4. Organization of the clgR locus in different members of the Actinobacteridae. Each arrow indicates an open reading frame. The lengthsof the arrows are proportional to the lengths of the predicted open reading frames. Orthologs are marked with the same color. The putativefunction of the protein is indicated above each arrow. The amino acid identity of each protein with respect to B. breve UCC 2003 is indicated asa percentage.
8418 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

fragment was incubated with 1 �g of CX without the purifiedh-ClgR protein (Fig. 5b).
No binding activity was observed when 50 pmol or 100 pmolof h-ClgR protein was used in the presence of 1 �g of a crudeextract of B. breve UCC 2003 cultures exposed to 50°C (Fig. 5b)or grown in the presence of 0.7 M NaCl (Fig. 5b). This suggeststhat the CX extract obtained from cells exposed to 43°C con-tains one or more cofactors required for ClgR binding activity.
In order to characterize the nature of this cofactor(s), pro-tease treatment (with pronase) of the CX followed by pronaseinactivation by thermal and/or chemical means was performed,and the reactions were subsequently analyzed in gel mobilityshift assays (Fig. 5b). Interestingly, when the equivalentamount of 1 �g of pronase-treated CX was incubated with100 pmol of h-ClgR, no displacement of clpP1p was observed(Fig. 5b). However, a clear retardation of the clpP1p fragment
was observed in a control experiment employing the sameamount of protein which had been treated in an identicalmanner but without the addition of pronase (Fig. 5b).
ClgR is a dimer. To determine whether the purified ClgRprotein exists in a multimeric form, we carried out in vitrocross-linking assays in the absence and presence of glutar-aldehyde. Using SDS-PAGE, it was shown that in the absenceof glutaraldehyde, the h-ClgR protein migrated as a singleband at a position which corresponds to the molecular mass ofthe monomeric form. However, in the presence of glutaralde-hyde, a fraction of the h-ClgR protein was shifted, with amolecular weight corresponding to a dimer (Fig. 6). The ap-pearance of a cross-linked species with the apparent mobility ofa dimer occurred as soon as 10 min after the addition of glutar-aldehyde. After 80 min of glutaraldehyde treatment, the cross-linking process appeared to be completed.
FIG. 5. Detection of ClgR binding to the clpP1 promoter of B. breve UCC 2003. (a) Overproduction and purification of ClgR. SDS-PAGEanalysis was performed with the purified ClgR protein (lanes 1 to 3, containing 15, 20, and 8 �g of protein, respectively) and with crude extractsfrom E. coli M15 carrying pEQ-ClgR upon IPTG induction (lane 4). A molecular weight standard (Bio-Rad, United Kingdom) was loaded in laneMK. (b) Gel retardation assays were performed with the clpP1p fragment as a probe. CX indicates a crude lysate from B. breve UCC 2003 culturesgrown at 43°C or 50°C or in medium with 0.7 M NaCl. The amounts of h-ClgR and CX used are indicated above each lane. Gel retardation resultsfor the clpP1p fragment as a probe and h-ClgR plus CX grown at 43°C, which prior to the binding experiment had been treated with pronase (�)or incubated in its absence, are presented in the rightmost panel. The arrows indicate the positions of the wells.
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8419
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

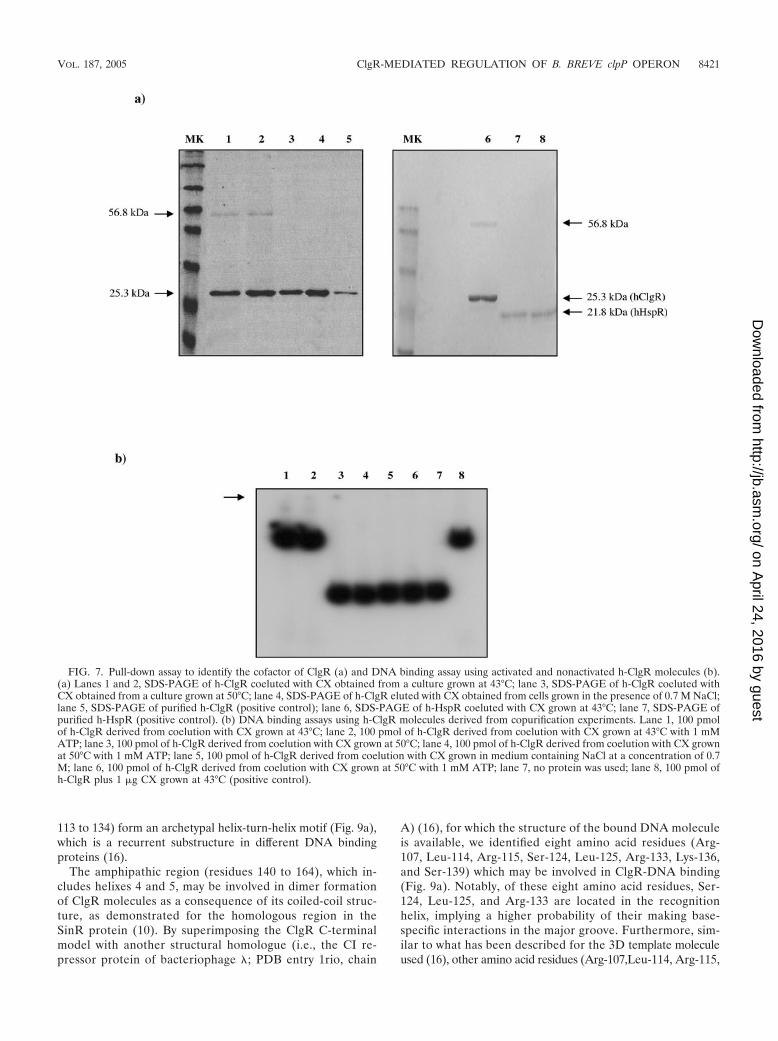
Purification of putative cofactor protein. In order to deter-mine whether the cofactor protein(s) required for ClgR bind-ing to the clpP1 promoter region could form stable complexeswith ClgR, a Ni-NTA column was first loaded with h-ClgR,after which the column was flooded with a crude cell extractfrom B. breve UCC 2003 cells that had been subjected to heatstress (43°C for 150 min) or with a crude cell extract from B.breve UCC 2003 cultures exposed to 50°C. The column wasthen washed, h-ClgR was finally eluted from the column, andthe eluted fractions containing h-ClgR were analyzed by SDS-PAGE for coeluted proteins (Fig. 7a). In the fraction whereh-ClgR had been immersed with the UCC 2003 extract ob-tained from cells exposed to 43°C, h-ClgR was shown to coe-lute with a protein of �56 kDa (Fig. 7a). This protein wasabsent in equivalent fractions from control experiments whereClgR was coeluted with UCC 2003 extract obtained from cellsexposed to 50°C and/or in equivalent fractions where ClgR wascoeluted with CX extract obtained from cells grown in thepresence of 0.7 M NaCl (Fig. 7a). Moreover, in a controlexperiment using another bifidobacterial transcription regula-tor, h-HspR (48) was subjected to the same experimental pro-cedure and subsequently analyzed by SDS-PAGE. Only oneband, of 21 kDa, which corresponds to the h-HspR protein,was detected (Fig. 7a).
Since we had shown that a proteinaceous cofactor containedin the UCC 2003 cell extract obtained from cultures grown at43°C promotes the binding of h-ClgR to the clpP promoterregion, we wanted to determine whether the coeluted 56-kDaprotein and ClgR were able to interact with the clpP1 promoterregion (clpP1p) in gel mobility shift assays. These experimentsshowed that h-ClgR plus the 56-kDa coeluate was capable of acomplete mobility shift of the clpP1p fragment (in either thepresence or absence of ATP), while no displacement of clpP1pwas observed when a control h-ClgR coeluate, obtained with aUCC 2003 crude extract of cells exposed to 50°C or grown inthe presence of 0.7 M NaCl, was used (Fig. 7b). This resultstrongly suggests that the 56-kDa coeluted protein, which is
uniquely present in UCC 2003 cell extracts exposed to 43°C, isin fact the cofactor required for ClgR binding activity.
Operator site of ClgR. To precisely delineate the sequencesthat constitute the ClgR binding site, DNase I footprint assayswere performed on the 377-bp DNA fragment that encom-passes the clpP1 promoter region. As shown in Fig. 7a and b,using end-labeled template or nontemplate strands, the puri-fied h-ClgR protein, in the presence of 1 �g of crude extractfrom B. breve UCC 2003 cultures exposed to 43°C, protects aregion extending from positions �79 and �99 on the template(Fig. 8a) and nontemplate (Fig. 8b) strands, respectively. Theprotected region contains the partially inverted repeat (CGCT-4N-GCCNA) (Fig. 7c), which resembles the ClgR operator site(CGCT-4N-GCGNAC) reported for the clpP1 promoter re-gion of other members of the Actinobacteridae group (2, 7).The palindromic structure of the putative operator site ofclpP1 in B. breve UCC 2003 is consistent with ClgR beingpresent as a dimer in solution. Moreover, DNase I hypersen-sitivity sites, which are sites that become more susceptible toDNase I cleavage upon protein binding, were detected in theprotected DNA region, which suggests that a distortion of thenormal DNA structure had occurred as a result of ClgRbinding.
The involvement of the ClgR protected region, including theIR element, in gene expression of the B. breve UCC 2003 clpPoperon was assessed by means of a number of transcriptionalfusions between various clpP promoter fragments and a pro-moterless gusA reporter gene in the promoter probe vectorpNZ272 (Fig. 8d). The GUS activity from cells harboring themore complete clpP1 promoter region (pclp1) and grown at43°C represented the same expression pattern as that deter-mined by slot blot hybridization experiments. This heat shockregulation of �-glucuronidase is partially or completely lost inclones containing plasmid pclp6 or pclp7 or plasmid pclp8 orpclp3, respectively, where the DNA encompassing the bindingsite of ClgR, as determined by footprinting experiments, ispartially (pclp6, pclp7, and pclp8) or completely (pclp3) re-moved (Fig. 8c and d). These data show that ClgR, possibly inassociation with a cofactor, can bind to the IR in a sequence-specific manner, thereby positively regulating the expression ofthe clpP operon at 43°C.
Structural investigation of ClgR of B. breve UCC 2003.Using fold recognition prediction, it was possible to identifystructural homologues of the ClgR protein that were limited tothe C-terminal region only (from residues 94 to 170 of B. breveUCC 2003 ClgR). In contrast, the N-terminal part of ClgR didnot display any significant matches. The most significant struc-tural homologue for the C-terminal part of UCC 2003 ClgR isthe SinR protein of B. subtilis (PDB entry 1b0nA) (20). Thisprotein shares the highest sequence identity (31% identicalresidues for 65 matched positions) as well as the most signifi-cant sequence fold compatible score (FFAS score, �36.1).Therefore, the 1b0nA protein was employed as a structuraltemplate for 3D modeling. Moreover, it should be emphasizedthat the sequence alignment between ClgR and SinR gener-ated from FFAS was highly consistent with those based onother methods, which implies that the alignment was highlyreliable (data not shown).
The predicted C-terminal region of B. breve UCC 2003 ClgRcontains five helices. Interestingly, helix 2 and helix 3 (residues
FIG. 6. Subunit composition of ClgR. An SDS-PAGE gel of glut-araldehyde-cross-linked and non-cross-linked h-ClgR is shown. Theabsence (�) or presence (�) of the glutaraldehyde cross-linking re-agent and the time of the cross-linking reaction are shown at the topof each lane. The positions of the h-ClgR monomer and dimer formsare indicated.
8420 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
113 to 134) form an archetypal helix-turn-helix motif (Fig. 9a),which is a recurrent substructure in different DNA bindingproteins (16).
The amphipathic region (residues 140 to 164), which in-cludes helixes 4 and 5, may be involved in dimer formationof ClgR molecules as a consequence of its coiled-coil struc-ture, as demonstrated for the homologous region in theSinR protein (10). By superimposing the ClgR C-terminalmodel with another structural homologue (i.e., the CI re-pressor protein of bacteriophage ; PDB entry 1rio, chain
A) (16), for which the structure of the bound DNA moleculeis available, we identified eight amino acid residues (Arg-107, Leu-114, Arg-115, Ser-124, Leu-125, Arg-133, Lys-136,and Ser-139) which may be involved in ClgR-DNA binding(Fig. 9a). Notably, of these eight amino acid residues, Ser-124, Leu-125, and Arg-133 are located in the recognitionhelix, implying a higher probability of their making base-specific interactions in the major groove. Furthermore, sim-ilar to what has been described for the 3D template moleculeused (16), other amino acid residues (Arg-107,Leu-114, Arg-115,
FIG. 7. Pull-down assay to identify the cofactor of ClgR (a) and DNA binding assay using activated and nonactivated h-ClgR molecules (b).(a) Lanes 1 and 2, SDS-PAGE of h-ClgR coeluted with CX obtained from a culture grown at 43°C; lane 3, SDS-PAGE of h-ClgR coeluted withCX obtained from a culture grown at 50°C; lane 4, SDS-PAGE of h-ClgR eluted with CX obtained from cells grown in the presence of 0.7 M NaCl;lane 5, SDS-PAGE of purified h-ClgR (positive control); lane 6, SDS-PAGE of h-HspR coeluted with CX grown at 43°C; lane 7, SDS-PAGE ofpurified h-HspR (positive control). (b) DNA binding assays using h-ClgR molecules derived from copurification experiments. Lane 1, 100 pmolof h-ClgR derived from coelution with CX grown at 43°C; lane 2, 100 pmol of h-ClgR derived from coelution with CX grown at 43°C with 1 mMATP; lane 3, 100 pmol of h-ClgR derived from coelution with CX grown at 50°C; lane 4, 100 pmol of h-ClgR derived from coelution with CX grownat 50°C with 1 mM ATP; lane 5, 100 pmol of h-ClgR derived from coelution with CX grown in medium containing NaCl at a concentration of 0.7M; lane 6, 100 pmol of h-ClgR derived from coelution with CX grown at 50°C with 1 mM ATP; lane 7, no protein was used; lane 8, 100 pmol ofh-ClgR plus 1 �g CX grown at 43°C (positive control).
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8421
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG
.8.
DN
ase
Ifo
otpr
ints
ofth
e34
7-bp
frag
men
tfr
omth
eB
.bre
veU
CC
2003
clpP
1pr
omot
erre
gion
for
the
tem
plat
est
rand
(a)
and
the
nont
empl
ate
stra
nd(b
).(c
and
d)�
-Glu
curo
nida
seac
tiviti
esof
vari
ous
clpP
1-gu
sAfu
sion
sin
B.b
reve
UC
C20
03gr
own
ata
rang
eof
tem
pera
ture
sor
with
0.7
MN
aCl.
(aan
db)
Lan
e1,
nopr
otei
nex
trac
t;la
ne2,
100
pmol
ofh-
Clg
R;l
ane
3,25
pmol
ofh-
Clg
Rpl
us1
�g
ofcr
ude
lysa
tefr
omB
.bre
veU
CC
2003
cultu
res
(CX
)gr
own
at43
°C;l
ane
4,50
pmol
ofh-
Clg
Ran
d1
�g
CX
grow
nat
43°C
;lan
e5,
100
pmol
ofh-
Clg
Ran
d1
�g
CX
grow
nat
43°C
;lan
e6,
1�
gC
Xgr
own
at43
°C.P
r,C
lgR
-pro
tect
edre
gion
.Arr
ows
indi
cate
hype
rsen
sitiv
itysi
tes.
(c)
Tem
plat
e(c
lpP
1t)
and
nont
empl
ate
(clp
P1n
t)cl
pP1
prom
oter
sequ
ence
s.IR
,put
ativ
ere
gula
tor
sequ
ence
s.T
he�
10an
d�
35he
xam
ers
are
unde
rlin
ed,l
ette
rssh
owin
bold
deno
teth
etr
ansc
ript
ion
star
tsi
te,a
ndar
row
sin
dica
tehy
pers
ensi
tivity
site
s.B
ars
indi
cate
the
posi
tions
ofth
ege
netic
cons
truc
tsw
ithre
spec
tto
the
clpP
1pr
omot
erre
gion
.(d)
GU
Sac
tiviti
esof
the
pclp
1,pc
lp6,
pclp
7,pc
lp8,
and
pclp
3tr
ansc
ript
ion
fusi
ons
and
the
empt
yve
ctor
pNZ
272
grow
nat
37°C
,43°
C,o
r50
°Cor
inm
ediu
mco
ntai
ning
0.7
MN
aCl.
8422 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

FIG
.9.
Predicted3D
modelfor
theC
-terminalend
ofC
lgR.(a)
Inorder
topredict
them
odeof
bindingbetw
eenC
lgRand
theD
NA
molecule,the
DN
Am
olecule(the
ligand)from
thecrystalstructure
of1rio
was
superimposed
ontothe
predictedC
lgRm
odelandis
depictedby
magenta
tubes.Potentialresiduesin
ClgR
involvedin
theinteraction
with
theD
NA
molecule
areindicated.T
heatom
sin
thesehighlighted
residuesare
coloredand
arerepresented
assticks.C
olorcode:green,carbon;red,oxygen;blue,nitrogen.(b)
Alignm
entofClgR
proteinsfrom
severalhigh-G
�C
gram-positive
bacteria.Shadingindicates
conservationat
agiven
positionin
atleast
50%of
theproteins
inthe
alignment
aseither
identical(black)or
similar
(lightgray)
residues.T
heam
inoacid
residuesinvolved
inD
NA
bindingare
indicatedw
itharrow
s,theturns
areindicated
with
“T,”
andthe
helicesare
indicatedw
ith“H
.”
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8423
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

Lys-136, and Ser-139) could also be involved in DNA interac-tions by nonspecific contacts (e.g., contacts with the DNAbackbone). The high conservation of this set of amino acidresidues between various ClgR homologues (Fig. 9b) may sup-port this hypothesis.
DISCUSSION
This report describes the characterization of a bifidobacte-rial clpP operon which is activated upon heat stress treatmentsin bifidobacteria and gives evidence for the first time of the roleof the transcriptional activator ClgR and a proteinaceous co-factor molecule in the regulation of such an operon in thisgroup of bacteria. The clpP operon of B. breve UCC 2003 wasidentified on the basis of high levels of homology to previouslydescribed clpP operons (25, 49–51). Our results clearly indicatethat the B. breve clpP operon consists of two genes, clpP1 andclpP2, in an organization which is identical to that of presumedclpP regions of other sequenced Bifidobacterium species and toother available sequences of members of the Actinobacteridaegroup, such as S. coelicolor A3 (4).
Phylogenetic analysis of the clpP1 and clpP2 paralogs re-vealed that the observed genetic constellation may have beenderived from a duplication of the clpP gene during the evolu-tionary process from Firmicutes, representing a “true” paral-ogy; alternatively, they may have arisen as a consequence of ahidden paralogy, implying that the ancestor of the Firmicutesand Actinobacteria taxa once had multiple copies of the genesand that a differential loss of gene copies occurred, resulting inthe current clpP gene distribution.
The clpP1 and clpP2 genes constitute ideal target candidatesfor diagnostic purposes because they are highly conserved andubiquitous in bacteria (12–14, 23, 39). Thus, these genes maybe used as alternative molecular markers to the rRNA se-quences, which could be added to the current databases ofalternative molecular markers such as the tuf, recA, groEL,atpD, grpE, and dnaK genes (24, 40–44, 47) and might corrob-orate and help to complete the evolutionary history of variousbifidobacterial species (38, 41, 42).
Although molecular chaperone-encoding genes such as clpB,clpC, clpP, dnaK, groEL, and groES have been demonstrated tobe induced by heat stress in bifidobacteria (39, 43, 47, 48; thisstudy), nothing is known about a global regulation of this stressresponse in these bacteria.
The expression of the B. breve UCC 2003 clpP operon isinduced at its highest level upon moderate heat shock regi-mens (T of 6 K, where T means the temperature differencebetween 37°C and the applied stress temperature) but notupon severe heat stress (T of 10 to 13 K) or osmotic stress.
Notably, similar results have been found for another mem-ber of the B. breve UCC 2003 clp gene family, i.e., the clpCgene. The similarities between the transcription patterns of theclpP1/clpP2 and clpC genes are striking, not only with respectto stimulus induction but also with respect to the tuning sys-tems which govern their expression, thus suggesting that inbifidobacteria the clpP operon and the clpC gene belong to thesame regulon.
The clpP1 promoter region was only bound by the purifiedClgR protein in the presence of a crude lysate of heat-stressedB. breve UCC 2003 cells, and this binding activity did not occur
upon protease treatment of the crude lysate of heat-stressed B.breve UCC 2003 cells. In pull-down assays using whole-cellextracts from heat-stressed B. breve UCC 2003 cultures, a pro-tein of 56 kDa was shown to copurify with ClgR. Moreover, weshowed that the h-ClgR–56-kDa protein coeluate mixture wasable to bind to the clpP1 promoter region without the assis-tance of any other cofactor, which would generate an h-ClgR-activated molecule. Taken together, these results represent anovel type of positive cofactor-mediated regulation of geneexpression in high-G�C gram-positive bacteria. Preliminaryresults from mass spectrometric analysis identified the 56-kDaprotein to be the B. breve UCC 2003 GroEL chaperone, andfuture experiments involving the overproduction and purifica-tion of B. breve UCC 2003 GroEL will be carried out in orderto better elucidate the ClgR-GroEL interactions and to inves-tigate if other molecules may be involved in this process.
We speculate that this cofactor is involved in assisting theproper folding of ClgR. To our knowledge, two other heat-induced transcriptional regulators which require the presenceof a molecular chaperone as a cofactor to function properlyhave been previously described for B. subtilis. (26) and S.coelicolor (3). In B. subtilis, the GroEL chaperone machinemodulates the activity of the transcriptional repressor HrcA(26), whereas in S. coelicolor the DnaK protein coregulates theactivity of the heat-induced transcriptional repressor HspR (3).Thus, the ClgR-mediated regulation in bifidobacteria repre-sents a third and novel cofactor-mediated activation of tran-scriptional acts involved in the heat response.
In order to provide evidence that, similar to other membersof the Actinobacteridae group (2, 7), the bifidobacterial ClgRhomologue is a transcriptional activator of the clpP operon, areporter system which includes different portions of the clpP1promoter region was used. Only clones harboring plasmidsencompassing the putative ClgR operator site were shown toinduce �-glucuronidase activity when subjected to moderateheat shock regimens. On the other hand, the levels of �-glu-curonidase activity were drastically reduced in B. breve strainsharboring a promoter probe vector in which the ClgR bindingsite was removed, thus suggesting that ClgR acts as a transcrip-tional activator of the clpP operon in B. breve UCC 2003 andother bifidobacteria.
The clpP1 promoter regions protected in DNase I foot-print experiments contain a palindromic motif (CGCT-4N-GCCNA) which may represent the operator site for the ClgRprotein in B. breve UCC 2003. This motif is highly similar to theconsensus operator site (CGCT-4N-GCGNAC) of the ClgRregulons in Streptomyces, Mycobacterium, and Corynebacterium(2, 7). For all of these organisms, it has been shown that ClgRregulates many stress-induced genes, such as clpP1, clpP2,clpC, and lon (2). However, we have shown that in contrastto those of other high-G�C gram-positive bacteria, the bi-fidobacterial ClgR homologue requires a proteinaceous cofac-tor in order to acquire binding activity.
According to DNase I footprint analysis, the ClgR bindingsite in the clpP1 promoter region of B. breve UCC 2003 islocated upstream of the transcription start site. Thus, we canhypothesize that ClgR activates expression by altering the pro-moter conformation. However, we cannot exclude that ClgRmay alter the interaction with the RNA polymerase. Moreover,binding of ClgR may induce DNA bending, since several en-
8424 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

hanced DNase I cleavage sites appear within the protectedregion of the clpP1 gene.
By analogy to other DNA binding proteins (10, 16, 53), thestructure of B. breve UCC 2003 ClgR suggests that it may beactive as a dimer or even as a multimer. Chemical cross-linkingwith glutaraldehyde (5) indicated that B. breve UCC 2003 ClgRis present as a dimer in solution and may thus bind to the clpP1operator as a dimer. Although in vitro evidence of higher-order protein complexes was not obtained, it remains a possi-bility that the protein may bind DNA to form a higher-ordermultimeric complex.
There are very strong similarities between ClgR of B. breveUCC 2003 and its homologues in Streptomyces (2) and Coryne-bacterium (7), with 46% identity in the central region contain-ing the predicted helix-turn-helix motif. Moreover, the predic-tion of the ClgR fold structure highlights the importance ofeight amino acid residues, five of which are located within thehelix-turn-helix motif, which supports its functional relation-ship with other transcriptional regulators (15, 53), althoughthis requires experimental verification by nuclear magnetic res-onance and/or crystallography structural studies. Prediction ofa 3D model for B. breve UCC 2003 ClgR was possible only forthe C-terminal part of the protein; the N-terminal domain didnot reveal any homologous structures. Interestingly, the N-terminal domain of the bifidobacterial ClgR protein appears tobe 72 to 92 amino acid residues longer than those of homolo-gous proteins in other members of the Actinobacteridae group.Taken together, this finding along with the fact that the ClgRhomologues in other members of the Actinobacteridae groupdo not require any cofactor molecules to bind to the clpP1promoter region (2, 7) might suggest that this N-terminal ex-tension found only in bifidobacterial ClgR interacts with thecofactor molecule. Therefore, this would represent a novelmechanism which is apparently unique to bifidobacteria ofClgR-mediated heat shock transcriptional regulation. One ofthe main tasks to be addressed in future studies will be gainingan understanding of ClgR regulation and activation in bi-fidobacteria. In fact, in other high-G�C gram-positive bacte-ria, ClgR was found to be regulated by its degradation by as yetunknown stimuli using the ClpP proteolytic complex (7). In S.lividans, the degradation of ClgR as well as another transcrip-tional regulator, PopR, by the ClpP proteolytic complex re-quires the C-terminal Ala-Ala residues (49). Since the B. breveUCC 2003 ClgR protein possesses C-terminal Ala-Ala resi-dues, we speculate that bifidobacterial ClgR might also bedegraded by ClpP. Thus, we postulate that UCC 2003 ClgRactivity itself is controlled via regulated proteolysis by Clpprotease.
The topic of stress responses in bifidobacteria is highly rel-evant to the food industry. Crucial aspects related to industrialapplications, such as the preparation of cells using freeze-drying technologies and cell survival in products which presenta hostile environment for bifidobacteria, make it essential toincrease our knowledge of the molecular mechanisms and mo-lecular actors (e.g., ClpP) involved in the heat stress response.More investigations on such molecular mechanisms are re-quired in order to better understand the molecular basis ofheat protection for bifidobacterial cells during food manufac-turing and to select new heat stress-tolerant bifidobacterialstrains.
ACKNOWLEDGMENTS
This work was financially supported by the Higher Education Au-thority Programme for Research in Third-Level Institutions, by theScience Foundation Ireland Alimentary Pharmabiotic Centre locatedat University College Cork, and by a Marie Curie Development hostfellowship (HPMD-2000-00027) to M.V.
We thank the members of the B. longum DJO10A genome sequenc-ing project funded by the U.S. Department of Energy Joint GenomeInstitute for making available the sequence of the clpP locus. Finally,we thank Valentina Bernini for helpful and constructive discussions.
REFERENCES
1. Ashok, K. D., C. S. Baker, K. Suzuki, A. D. Jones, P. Pandit, T. Romero, andP. Babitzke. 2003. CsrA regulates translation of the Escherichia coli carbonstarvation gene cstA by blocking ribosome access to the cstA transcript. J.Bacteriol. 15:4450–4460.
2. Bellier, A., and P. Mazodier. 2004. ClgR, a novel regulator of clp and lonexpression in Streptomyces. J. Bacteriol. 186:3238–3248.
3. Bucca, G., A. M. E. Brassington, H. J. Schonfeld, and C. P. Smith. 2000. TheHspR regulon of Streptomyces coelicolor: a role for the DnaK chaperone asa transcriptional co-repressor. Mol. Microbiol. 38:1093–1103.
4. Crecy-Lagard, V., P. Servant-Moisson, J. Viala, C. Grandvalet, and P.Mazodier. 1999. Alteration of the synthesis of the Clp ATP-dependent pro-tease affects morphological and physiological differentiation in Streptomyces.Mol. Microbiol. 32:505–517.
5. Derre, I., G. Rapoport, and T. Msadek. 1999. CtsR, a novel regulator ofstress and heat shock response, controls clp and molecular chaperone geneexpression in gram-positive bacteria. Mol. Microbiol. 31:117–131.
6. Derre, I., G. Rapoport, and T. Msadek. 2000. The CtsR regulator of stressresponse is active as a dimer and specifically degraded in vivo at 37°C. Mol.Microbiol. 38:335–347.
7. Engels, S., J. E. Schweitzer, C. Ludwig, M. Bott, and S. Schaffer. 2004. clpCand clpP1P2 gene expression in Corynebacterium glutamicum is controlled byregulatory network involving the transcriptional regulators ClgR and HspRas well as the ECF sigma factor �H. Mol. Microbiol. 52:285–302.
8. Fedhila, S., T. Msadek, P. Nel, and D. Lereclus. 2002. Distinct clpP genescontrol specific adaptive responses in Bacillus thuringiensis. J. Bacteriol.184:5554–5562.
9. Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package), version 3.5c.Distributed by the author. University of Washington, Seattle, Wash.
10. Gaur, N. K., J. Oppenheim, and I. Smith. 1991. The Bacillus subtilis sin gene,a regulator of alternative developmental processes, codes for a DNA-bindingprotein. J. Bacteriol. 173:678–686.
11. Gottesman, S. 1996. Proteases and their targets in Escherichia coli. Annu.Rev. Genet. 30:465–506.
12. Gupta, R. S. 1995. Evolution of the chaperonin families (Hsp60, Hsp10 andTcp-1) of proteins and origin of eukaryotic cells. Mol. Microbiol. 15:1–11.
13. Gupta, R. S. 2001. The branching order and phylogenetic placement ofspecies from completed bacterial genomes, based on conserved indels foundin various proteins. Int. Microbiol. 4:187–202.
14. Gupta, R. S. 2002. Phylogeny of bacteria: are we now close to understandingit? ASM News 68:284–291.
15. Harrison, S. C., and A. K. Aggarwal. 1990. DNA recognition by proteins withhelix-turn-helix motif. Annu. Rev. Biochem. 59:933–969.
16. Jain, D., B. E. Nickels, L. Sun, A. Hochschild, and S. A. Darst. 2004.Structures of a ternary transcription activation complex. Mol. Cell 13:45–53.
17. Jukes, T. H., and C. R. Cantor. 1969. Evolution of protein molecules, p.21–132. In H. N. Munro (ed.), Mammalian protein metabolism. AcademicPress, New York, N.Y.
18. Kroh, H. E., and L. D. Simon. 1990. The ClpP component of the Clpprotease is the sigma 32-dependent heat shock protein. J. Bacteriol. 172:6026–6034.
19. Kumar, S., K. Tamura, and M. Nei. 1993. MEGA: molecular evolutionarygenetics analysis, version 1.01. Pennsylvania State University, College Park,Pa.
20. Lewis, R. J., J. A. Brannigan, W. A. Offen, I. Smith, and A. J. Wilkinson.1998. An evolutionary link between sporulation and prophage induction inthe structure of a repressor:anti-repressor complex. J. Mol. Biol. 283:907–912.
21. Lievin, V., I. Pfeiffer, S. Hudault, F. Rochat, D. Brassart, J. R. Neeser, andA. L. Servin. 2000. Bifidobacterium strains from resident infant human gas-trointestinal microflora exert antimicrobial activity. Gut 47:646–652.
22. Lindquist, S., and E. A. Craig. 1988. The heat-shock proteins. Annu. Rev.Genet. 22:631–677.
23. Ludwig, W., and K. H. Schleifer. 1999. Phylogeny of bacteria beyond the 16SrRNA standard. ASM News 65:752–757.
24. Masco, L., M. Ventura, R. Zink, G. Huys, and J. Swings. 2004. Polyphasictaxonomic analysis of Bifidobacterium animalis and Bifidobacterium lactisreveals relatedness at subspecies level: reclassification of Bifidobacteriumanimalis as Bifidobacterium animalis subsp. animalis comb. nov. and Bi-
VOL. 187, 2005 ClgR-MEDIATED REGULATION OF B. BREVE clpP OPERON 8425
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from

fidobacterium lactis as Bifidobacterium animalis subsp. lactis comb. nov. Int.J. Syst. Evol. Microbiol. 54:1137–1143.
25. Maurizi, M. R., W. P. Clark, S. H. Kim, and S. Gottesman. 1990. ClpPrepresents a unique family of serine proteases. J. Biol. Chem. 265:12546–12552.
26. Mogk, A., G. Homuth, C. Scholz, L. Kim, F. X. Schmid, and W. Schumann.1997. The GroE chaperonin machine is a major modulator of the CIRCEheat shock regulon of Bacillus subtilis. EMBO J. 16:4579–4590.
27. Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbersof synonymous and nonsynonymous nucleotide substitutions. Mol. Biol.Evol. 3:418–426.
28. Ouwehand, A. C., S. Salminen, and E. Isolauri. 2002. Probiotics: an overviewof beneficial effects. Antonie Leeuwenhoek 82:279–289.
29. Platteeuw, C., G. Simons, and W. de Vos. 1994. Use of the Escherichia coli�-glucuronidase (gusA) gene as a reporter gene for analyzing promoters inlactic acid bacteria. Appl. Environ. Microbiol. 60:587–593.
30. Porankiewicz, J., J. Wang, and A. K. Clarke. 1999. New insights into theATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol.21:449–458.
31. Rychlewski, L., L. Jaroszewski, W. Li, and A. Godzik. 2000. Comparison ofsequence profiles. Strategies for structural predictions using sequence infor-mation. Protein Sci. 9:232–241.
32. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
33. Schell, M. A., M. Karmirantzou, B. Snel, D. Vilanova, B. Berger, G. Pessi,M. C. Zwahlen, F. Desiere, P. Bork, M. Delley, R. D. Pridmore, and F.Arigoni. 2002. The genome sequence of Bifidobacterium longum reflects itsadaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. USA99:14422–14427.
34. Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: anautomated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385.
35. Squires, C., and C. L. Squires. 1992. The Clp proteins: proteolysis regulatorsor molecular chaperones? J. Bacteriol. 174:1081–1085.
36. Stewart, G. R., L. Wernisch, R. Stabler, J. A. Mangan, J. Hinds, and K. G.Laing. 2002. Dissection of the heat-shock response in Mycobacterium tuber-culosis using mutants and microarrays. Microbiology 148:3129–3138.
37. Tannock, G. W. 1994. The acquisition of the normal microflora of thegastrointestinal tract, p. 1–16. In S. A. Gibson (ed.), Human health: thecontribution of microorganisms. Springer, London, United Kingdom.
38. Vandamme, P., B. Pot, M. Gillis, P. de Vos, K. Kersters, and J. Swings. 1996.Polyphasic taxonomy, a consensus approach to bacterial systematics. Micro-biol. Rev. 60:407–438.
39. Van de Guchte, M., P. Serror, C. Chervaux, T. Smokvina, S. D. Ehrlich, andE. Manguin. 2002. Stress responses in lactic acid bacteria. Antonie Leeu-wenhoek 82:187–216.
40. Ventura, M., and R. Zink. 2002. Rapid identification, differentiation, and
proposed new taxonomic classification of Bifidobacterium lactis. Appl. Envi-ron. Microbiol. 68:6429–6434.
41. Ventura, M., and R. Zink. 2003. Comparative sequence analysis of the tufand recA genes and restriction fragment length polymorphism of the internaltranscribed spacer region sequences supply additional tools for discriminat-ing Bifidobacterium lactis from Bifidobacterium animalis. Appl. Environ. Mi-crobiol. 69:7517–7522.
42. Ventura, M., C. Canchaya, D. van Sinderen, G. F. Fitzgerald, and R. Zink. 2004.Bifidobacterium lactis DSM 10140: identification of the atp (atpBEFHAGDC)operon and analysis of its genetic structure, characteristics, and phylogeny. Appl.Environ. Microbiol. 70:3110–3121.
43. Ventura, M., C. Canchaya, R. Zink, G. F. Fitzgerald, and D. van Sinderen.2004. Characterization of the groEL and groES loci in Bifidobacterium breveUCC 2003: genetic, transcriptional and phylogenetic analysis. Appl. Environ.Microbiol. 70:6197–6209.
44. Ventura, M., C. Canchaya, V. Meylan, T. R. Klaenhammer, and R. Zink.2003. Analysis, characterization, and loci of the tuf genes in Lactobacillus andBifidobacterium and their direct application for species identification. Appl.Environ. Microbiol. 69:6908–6922.
45. Ventura, M., D. van Sinderen, G. F. Fitzgerald, and R. Zink. 2004. Insightsinto the taxonomy, genetics and physiology of bifidobacteria. Antonie Leeu-wenhoek 86:205–223.
46. Ventura, M., R. Reniero, and R. Zink. 2001. Specific identification andtargeted characterization of Bifidobacterium lactis from different environ-mental isolates by a combined multiplex-PCR approach. Appl. Environ.Microbiol. 67:2760–2765.
47. Ventura, M., R. Zink, G. F. Fitzgerald, and D. van Sinderen. 2005. Genestructure and transcriptional organization of the dnaK operon of Bifidobac-terium breve UCC 2003 and its application in bifidobacterial tracing. Appl.Environ. Microbiol. 71:487–500.
48. Ventura, M., J. G. Kenny, Z. Zhang, G. F. Fitzgerald, and D. van Sinderen.2005. The clpB gene of Bifidobacterium breve UCC 2003: transcriptionalanalysis and first insights into stress induction. Microbiology 151:2861–2872.
49. Viala, J., and P. Mazodier. 2002. ClpP-dependent degradation of PopRallows tightly regulated expression of the clpP3clpP4 operon in Streptomyceslividans. Mol. Microbiol. 44:633–643.
50. Viala, J., and P. Mazodier. 2003. The ATPase ClpX is conditionally involvedin the morphological differentiation of Streptomyces lividans. Mol. Genet.Genomics 268:563–569.
51. Viala, J., G. Rapoport, and P. Mazodier. 2000. The ClpP multigenic familyin Streptomyces lividans: conditional expression of the clpP3 clpP4 operon iscontrolled by PopR, a novel transcriptional activator. Mol. Microbiol. 38:602–612.
52. Wawrzynow, A., B. Banecki, and M. Zylicz. 1996. The Clp ATPases define anovel class of molecular chaperones. Mol. Microbiol. 21:895–899.
53. Wintjens, R., and M. Rooman. 1996. Structural classification of HTX DNAbinding domains and protein-DNA interaction modes. J. Mol. Biol. 262:294–313.
8426 VENTURA ET AL. J. BACTERIOL.
on April 24, 2016 by guest
http://jb.asm.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















