
Anxiolytic-like effects of 8-acetylene imidazobenzodiazepines in a rhesus monkey conflict procedure

Applied Catalysis B: Environmental 29 (2001) 195–205
The chemical state of palladium in alkene and acetylene oxidationA study by XRD, electron microscopy and TD-DTG analysis
Zohra Ferhat-Hamida1, Jacques Barbier Jr., Sandrine Labruquere, Daniel Duprez∗Laboratoire de Catalyse en Chimie Organique, UMR6503, CNRS et Université de Poitiers,
40 Av. Recteur Pineau, 86022 Poitiers Cedex, France
Received 12 March 2000; received in revised form 3 July 2000; accepted 3 July 2000
Abstract
Oxidation of propene, but-1-ene or acetylene in O2 sub-stoichiometry was studied over two alumina-supported catalysts(1 and 3% Pd) and over an unsupported palladium oxide. Structural changes of palladium in these reactions was investigatedby XRD, TEM and electron diffraction as well as by TG-DTG analysis. Palladium, initially in the form of PdO, reduces toa new cubic phase denoted PdX with a lattice parameter (0.398 nm) higher than in metallic palladium obtained by reductionin H2 (0.389 nm). PdX can be observed by XRD at the very beginning of propene oxidation, which is confirmed by electrondiffraction patterns taken on several Pd particles after reaction. This new phase of palladium is specifically formed in alkeneand alkyne oxidation, which contrasts with what was observed in propane oxidation during which PdO reduced into Pd◦.TG-DTG analysis and XRD results obtained at various O2/propene ratios support the hypothesis that PdX is formed by Oinsertion into the cubic structure of metallic palladium. © 2001 Elsevier Science B.V. All rights reserved.
Keywords:Palladium catalysts; Oxidation of propene; Butene and acetylene; New cubic phase of Pd (in catalytic oxidation)
1. Introduction
Palladium catalysts are now widely used in manyenvironmental applications, particularly in three-waycatalysts [1]. Moreover, owing to its high activity inmethane combustion [2–13], palladium is expectedto be employed as an active component in catalyticcombustion gas turbine [14]. Some studies were de-voted to the oxidation of other hydrocarbons: ethane[2,15], propane [2,16–21], butane [2], heptane [22],propene and other olefins [16,17,20,23–26],o-xylene[27] and naphthalene [28]. Most of these previous
∗ Corresponding author. Fax:+33-549-4534-99.E-mail address:[email protected](D. Duprez).
1 On leave from the University H. Boumediene, Algers, Algeria.
studies showed that the oxidation activity of palla-dium was largely dependent on its chemical stateunder oxygen/fuel atmosphere. Various techniqueswere used to characterize the different forms of pal-ladium present in reaction: XPS [27,29–32], XRDand grazing-incidence-XRD [18–20], TPO and O2TPD as well as TGA of O2 sorption and release[10,15,33–37], electron microscopy [38–40] or com-bined techniques [41].
It is generally thought that a palladium oxide formis required to develop a good activity in alkane oxida-tion [42], which explains why hysteresis phenomenawere observed when palladium oxide decomposedinto palladium metal and oxygen [10,34] or whenpalladium metal is oxidized into PdO [8,38]. Manyauthors have proposed that two types of palladiumoxide could co-exist on the support surface: bulk PdO
0926-3373/01/$ – see front matter © 2001 Elsevier Science B.V. All rights reserved.PII: S0926-3373(00)00203-4

196 Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205
crystallites and fully dispersed palladium oxide spe-cies interacting strongly with the support [29,40–44].The formation of inactive Pd(OH)2 species has alsobeen proposed to explain the deactivating effect ofsteam added to the HC/O2 mixtures [12,17,19,45].
Recently, we have investigated the palladium be-havior in oxidation of various pollutants (methane,CO, propene, and propane) under rich conditions[19,20]. We concluded that palladium, initially inthe PdO form, reduced at a temperature denotedTRduring the temperature-programmed heating underthe reducer/O2 flow. If Tox designates the light-offtemperature observed over Pd/Al2O3 catalysts (tem-perature corresponding to a 50% O2 conversion), weshowed the following:1. with CH4, T R � T ox : PdO reduced far beyond
methane oxidation;2. with CO and C3H6 T R � T ox : Pd was already
reduced before CO and propene oxidation started;3. with C3H8, T R ∼ T ox : PdO reducedduringC3H8
oxidation, which led to an apparent deactivation ofcatalyst.XRD carried out on the used Pd/Al2O3 catalyst con-
firmed that PdO was reduced into Pd◦ with CO andC3H8 while, with C3H6, amodified phasewith a latticeparameter (0.399 nm) higher than in Pd◦ (0.389 nm)was formed.
The peculiar behavior of Pd in propene oxidationprompted us to study the changes of palladium statein oxidation of various alkenes and acetylene. XRD,TG-DTG, electron microscopy and selected area elec-tron diffraction (SAED) were used to investigate thestructural modifications of palladium.
2. Experimental
2.1. Catalysts
The support was ag-Al2O3 (E1633) provided bythe French Institute of Petroleum (100 m2 g−1; porevolume: 1.15 cm3 g–1; main impurities: Na, 460 ppm,Ca, 560 ppm, Si, 310 ppm, Fe, 210 ppm). The aluminabeads were crushed, sieved at 0.1–0.2 mm and cal-cined at 500◦C for 10 h. Most of the experiments werecarried out over a 1% Pd/Al2O3 catalyst, while a 3%Pd catalyst was used in some instances. The catalysts(1 and 3% Pd) were prepared by impregnation of the
support with solutions of palladium acetylacetonatein acetone. They were dried at 120◦C overnight. Allthe experiments were carried out over catalyst sam-ples sintered at 900◦C in a 3% O2/N2 mixture (spacevelocity: 44 000 h−1; temperature ramp: 10◦C min−1,samples hold at 900◦C for 1 h and cooled down at am-bient in 3% O2 (−10◦C min−1). These cooling condi-tions are efficient to assure PdO genesis. These oxidecatalysts were denoted ‘catalyst before reaction’. Themean particle size measured by H2–O2 titration was10 nm (9% dispersion) in the 1% Pd catalyst and19.5 nm (4.6% dispersion) in the 3% Pd catalyst.
To obtain supported Pd◦ particles, aliquot portionsof the sintered catalysts were reduced in diluted hy-drogen (3% H2/N2) at 300◦C for 5 h. They are denoted‘reduced catalyst’. Their characterizations were per-formed immediately after the reducing pretreatment toavoid sample re-oxidation.
Propene, but-1-ene or acetylene oxidation was car-ried out over oxide catalyst samples (see Section2.2). The temperature-programmed reaction was in-terrupted at different temperatures below or aboveTox(light-off temperature corresponding, here, to a 50%O2 conversion). The oven heating was cut off and,after cooling down to ambient under the reactant mix-ture, the catalyst sample was immediately analyzed.We denote this catalyst ‘catalyst after reaction’.
To evaluate possible support effects on the structuralchanges of Pd, certain experiments were carried outover an unsupported PdO sample (Johnson–Matthey,anhydrous, 99.9%,< 20mesh). Before use, the powderwas pretreated at 400◦C for 1 h in a 3% O2 + N2mixture. This catalyst is denoted ‘bulk palladium’.
2.2. Oxidation reactions
Oxidation reactions of propene (Eq. (1)), but-1-ene(Eq. (2)) and acetylene (Eq. (3)) have been investigatedover the 1% Pd catalyst.
C3H6 + 92O2 → 3CO2 + 3H2O (1)
C4H8 + 6O2 → 4CO2 + 4H2O (2)
C2H2 + 52O2 → 2CO2 + H2O (3)
Some reactions were carried out with propene overthe 3% Pd/Al2O3 catalyst or over the unsupported PdOsample. Each run was carried out in a 12 mm i.d. quartz

Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205 197
reactor of 40 mg weight catalyst sample. The gas flowrate (HC+O2 +N2) was 340 cm3 min−1 and the totalpressure was 1 atm. The hydrocarbon concentrationwas 0.4 vol.% and the O2 concentration was adjustedto change the stoichiometry ratioSdefined as the ratiobetween the factual concentration of O2 in the feedand the concentration stoichiometrically required fortotal oxidation
S = [O2]
(n + m/4)[CnHm](4)
Most experiments were carried out in hydrocarbon ex-cess (S = 0.4). Propene oxidation was also investi-gated at differentS ratios (0–1.1).
2.3. X-ray diffraction
All the analyses were performed in a SIEMENSD500 diffractometer using the Cu Ka radiation (λ =0.15406 nm) as a source (current intensity, 30 mA;voltage, 40 kV). The catalyst powder (40 mg) was gen-tly spread on a flat sample holder but care was takento put all the sample under the X-ray beam of theapparatus. A 20–90◦ range of 2θ was investigated,the diffraction angle being increased by step of 0.02◦every 5 s.
XRD pattern were compared to ICDD files43–1024 for metallic palladium (a = 0.389 nm) and46–1043 for tetragonal PdO (a = b = 0.304 nm,c = 0.534 nm). In fact, only the most intense peaksemerging from the alumina background were used asa reference: the peaks at 2θ = 40.118◦ (I = 100%)and at 2θ = 82.098◦ (I = 55%) for cubic Pd◦ and thepeak at 2θ = 33.888◦ (I = 100%) for PdO. The linepositions were checked by using an internal standard(Si monocrystal).
2.4. Electron microscopy and electron diffraction
Analysis by transmission electron microscopy wasperformed on a Philips CM120 apparatus. A smallamount of catalyst was crushed into fine powder andmixed to alcohol. A microdrop of the suspension wasdeposited on a Cu grid covered with a carbon layerand the solvent was evaporated so as to leave somecatalyst micrograins on the carbon layer.
Electron diffraction patterns were obtained in thesame electron microscope. Interreticular distancesdhkl
as well as lattice parameters were calculated fromthe distances between the spots and from the anglesformed by the lines passing through these spots. Aprogram [46] allows to check the calculated values bydrawing the theoretical pattern along the same zoneaxis and by comparing it to the experimental pattern.
2.5. TG and DTG analysis
TG and DTG measurements were carried out in aTA instruments (SDT2960) thermal analyzer equippedwith two horizontal arms. About 20 mg of catalystsample were laid down in one of the two small Pt cru-cibles of the analyzer, the other one being filled withan equivalent amount ofa-alumina. Depending on thetype of analysis, the apparatus was swept with a flowof nitrogen or air at 50 cm3 min−1. When equilibriumat 30◦C was reached, the temperature was increased to800◦C with a ramp of 4◦C min−1. Weight changes andweight derivatives (dm, dm/dT or dm/dt) expressed, re-spectively, in %, %◦C−1 and % min−1 were recordedsimultaneously.
Four TG-DTG experiments were carried out on the3% Pd/Al2O3 catalyst to study the effect of heatingin oxygen or inert atmosphere (i) thermogravimetry inN2 of the catalyst after propene oxidation, (ii) TG inair of the same catalyst, (iii) TG in N2 of the fresh,calcined catalyst and finally (iv) TG in air of the fresh,reduced catalyst.
3. Results and discussion
3.1. Chemical state of palladium in propeneoxidation atS = 0.4
3.1.1. X-ray diffraction studyThe curve ‘conversion %’ versus ‘temperature’ is
very close to the curve obtained by Maillet et al. [20]on a 1% Pd/Al2O3 catalyst with similar characteristics:the oxidation reaction starts at about 200◦C and theconversion increases regularly up to 40% reached by360◦C. Above this temperature, a pseudo-plateau canbe observed with a slow increase of conversion due toa steam reforming reaction with the water formed inoxidation.
Fig. 1 shows the XRD diagram of the calcined1% Pd/Al2O3 catalyst before reaction, after propene

198 Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205
Fig. 1. (a) X-ray diffractograms of the 1% Pd/Al2O3 catalyst before reaction, after reaction (propene oxidation at 220◦C, S = 0.4) andafter reduction in H2 at 300◦C ((r) PdO, (j) Pd◦, (.) PdX); (b) XRD of (a), after correction of the alumina background.
oxidation (stopped at 360◦C) and of the fresh cata-lyst reduced in H2 at 300◦C. Several peaks, amongthe most intense ones, can be assigned to the supportbut the peaks assigned to Pd depend strongly on thecatalyst pretreatment. This is evidenced in Fig. 1bwhere the alumina diagram has been subtracted fromeach catalyst diffractogram. The corrected pattern ofthe catalyst before reaction shows an intense peak at33.90◦, which shows the presence of tetragonal PdO,
no other palladium structure was detected in this sam-ple. The diffractogram of the reduced catalyst displaysa strong peak at 40.14◦ and a smaller one at 82.16◦.These values are unambiguously ascribed to metal-lic fcc palladium (Pd◦). The structure of the catalystafter reaction is quite different from that of palladiumoxide, which shows that PdO has been transformedin the reacting medium; it resembles a metallic palla-dium but the peaks corresponding to the catalyst after

Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205 199
Fig. 2. XRD of the 1 and 3% Pd catalysts after propene oxidationat 220◦C (S = 0.4).
reaction are clearly shifted to lower 2θ values than inPd◦. The most important peaks corresponding to thisnew fcc phase appear at 39.14◦ (I = 100%), 45.37◦(I = 60%) and 79.62◦(I = 55%). The shift observedin 2θ (1.00◦ for the peak at 39.14◦) is indicative ofan increase of the lattice parameter: 0.398 nm insteadof 0.389 nm in metallic palladium.
A similar study was carried out with the 3%Pd/Al2O3 catalyst. The diffractogram of this catalystafter propene oxidation at 220◦C (Fig. 2) shows thatthe new cubic phase of palladium is formed as it waswith the 1% Pd catalyst.
We confirm that a new fcc phase of palladium isformed in the C3H6 + O2 reaction medium with a lat-tice less dense than in Pd◦. In the following, we pro-pose to precise the reaction parameters which promoteor inhibit the construction of this new palladium phasedenoted PdX.
3.1.2. Electron microscopyThe 1% Pd/Al2O3 catalyst was studied by TEM
and electron diffraction at different stages of itspretreatment or of the reaction: preoxidized beforereaction (PdO), after reaction at 220◦C (PdX) andreduced under hydrogen (Pd◦). TEM pictures andelectron diffraction patterns of the catalyst in thesethree states are shown on Fig. 3. Large particles ofpalladium oxide (≈20–50 nm) can be seen on Fig.3a. Besides relatively small particles of about 10 nm,the catalyst contain very large clusters of palladium.
The specific structure of the alumina support withquasi-spherical grains but also needle-shape parti-cles is easily observed on this TEM picture. Electrondiffraction showed the presence of tetragonal PdO inall the particles but without any preferential orien-tation: for instance, particles growing preferentiallyalong (13 1), (1 13) and(33 1) have been observed.Fig. 3a presents an electron diffraction pattern of a(13 1) orientated particle. The lattice parameter de-duced from this ED image is in good agreement withthe ICDD file of PdO.
Fig. 3b and c represent micrographs of the catalystafter reaction and of the catalyst reduced in H2. A dualdistribution of particle size can again be observed butthe particles are slightly smaller than in the oxidizedcatalyst. Electron diffraction patterns clearly indicatethe presence of fcc structure. The values of the mea-sured parameters (angles and distances) are reportedin Fig. 3b and c. There is a very good agreement ofthe calculated parameter for metal in the reduced state(0.389± 0.003 nm) with the ICCD file of Pd◦ whilefor the catalyst after reaction, the calculated lattice pa-rameter is unambiguously higher (0.401± 0.003 nm)than in metallic palladium.
In conclusion, there is a good accordance betweenelectron diffraction and XRD measurements. Beforereaction, the palladium phase is made of tetragonalPdO particles which are reduced into Pd◦ after treat-ment in H2 at 350◦C. The same cubic structure can beobserved after propene oxidation at 220◦C but the Pdparticles have a lattice parameter larger than in metal-lic palladium (PdX phase). A likely hypothesis is thatthis phase is formed by O insertion in the lattice ofmetallic palladium. PdX would thus be a ‘quasi-metal’not totally reduced.
3.1.3. TG and DTG analysisThis study was performed on the 3% Pd catalyst to
enhance the weight changes which can be observedduring PdO or PdX decomposition or genesis. At lowtemperature, weight losses due to water desorptionare systematically recorded while the most interestingfeatures of the TG profiles can be observed in the650–750◦C temperature range.
TG profiles obtained under N2 flow for the cat-alyst ‘before reaction’ and ‘after reaction’ showthat both catalyst samples decompose around 700◦C(Fig. 4). However, the decomposition is rather slow

200 Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205
Fig. 3. TEM pictures and electron diffraction patterns of the 1% Pd/Al2O3 catalyst: (a) before reaction; (b) after reaction (propene oxidationat 220◦C, S = 0.4) and (c) after reduction in H2 at 300◦C.
for the catalyst after reaction (PdX) while the catalystbefore reaction (PdO) decomposes in two steps with arapid process superimposed to a slow one. The deriva-tive curves dm/dt on Fig. 5 displays the phenomenonmore accurately and show that PdX decomposes
at a lower temperature (705◦C) than PdO (725◦C).Integrating these curves lead to a total weight lossof 0.39% for the preoxidized catalyst and of 0.21%for the catalyst after reaction. The former value isin agreement with the palladium content (3%) if we

Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205 201
Fig. 3. (Continued).
assume that all palladium in the form of PdO decom-poses to give Pd◦ (theoretical weight loss: 0.44%).The value obtained with the catalyst after reactionconfirms that the O content of PdX is significantlylower than in PdO, almost 0.5.
TG analysis in air of the catalyst after reaction (notrepresented here) shows an O2 uptake by 450◦C fol-
Fig. 4. TG analysis in N2 of the 3% Pd/Al2O3 catalyst before andafter reaction.
lowed by a very sharp weight loss at 720◦C. PdXseems to be transformed into PdO at 450◦C (weightincrease), which decomposes at 720◦C, practically atthe same temperature as in N2. A similar TG profilewas obtained with the reduced catalyst, with a verysharp weight loss at 720◦C. Nevertheless, the trans-
Fig. 5. DTG analysis in N2 of the 3% Pd/Al2O3 catalyst beforeand after reaction.

202 Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205
formation of Pd◦ into PdO was not observed in a tem-perature range as narrow as for PdX.
3.2. Effect of the reaction temperature atS = 0.4
Three experiments were carried out with the 1%Pd catalyst by interrupting the oxidation reaction atdifferent temperatures: 150◦C (no conversion), 200◦C(≈1% conversion) and 220◦C (≈5% conversion).XRD spectra corresponding to these three experi-ments are reported on Fig. 6. At 150◦C, PdO is notyet reduced; the corresponding peak at 33.9◦ is stillintense while the peak corresponding to the new phasedoes not appear. Temperature 200◦C seems to be anintermediate in the formation of the PdX structure.There remains a small amount of palladium oxidewhile the peak corresponding to the new PdX phaseis becoming to be seen as a shoulder of alumina peak.At 220◦C all the palladium oxide is transformed intothe new PdX phase. We can note that the diffrac-togram at 220◦C appears much more regular than at200◦C, which indicates that Pd particles at 220◦C arewell crystallized.
3.3. Effect of the oxygen/propene ratio
Oxygen/alkene ratio is a major factor for control-ling conversion and selectivity of palladium catalysts.As reported by many authors [12,34,43] oxygen pres-
Fig. 6. XRD of the 1% Pd/Al2O3 catalyst after propene oxidationat different temperatures.
Fig. 7. Effect of the O2/propene ratio on the XRD of the 1%Pd/Al2O3 catalyst after reaction at 220◦C.
sure at the catalyst surface could modify the chemicalstate of palladium. Some authors suggested that sur-face PdOx species could be the active phase in someoxidation reaction on Pd. Therefore, we have investi-gated the possible changes in the construction of thenew PdX phase that could be induced by an increaseof O2 partial pressure. The reaction of propene oxida-tion was carried out and interrupted at 220◦C at vari-ous oxygen/propene ratios corresponding toS = 0 (noO2), S = 0.4, 0.8 and 1.1. (excess of oxygen) XRDspectra of the catalyst after each reaction are shownon Fig. 7. In the absence of oxygen in the reactantflow mixture (S = 0), Pd◦ and PdO cannot be ob-served but the new PdX phase can hardly be formed.In all other cases (S = 0.4, 0.8 or 1.1), XRD spectraare similar and show clearly the presence of the PdXphase (peaks at 39.1 and 79.6◦). We can conclude thatoxygen favors the construction of this new phase. Asalready assumed, oxygen is likely to be inserted in thefcc lattice of palladium which induces an increase ofthe lattice parameter of Pd◦.
3.4. Effect of the nature of hydrocarbon (S = 0.4,220◦C)
The results obtained in propene oxidation have beenextended to other hydrocarbons with C=C double bond(but-1-ene) and C≡C triple bond (acetylene). Thereactions have been carried out under the same con-ditions as for propene oxidation.
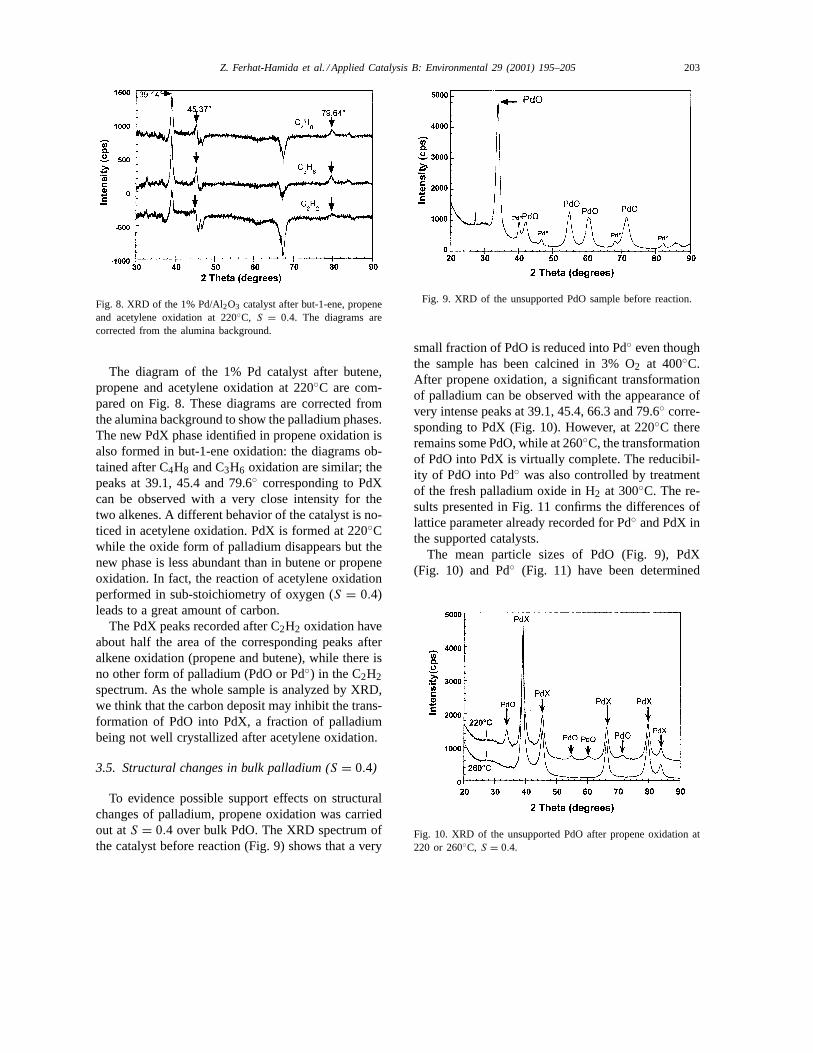
Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205 203
Fig. 8. XRD of the 1% Pd/Al2O3 catalyst after but-1-ene, propeneand acetylene oxidation at 220◦C, S = 0.4. The diagrams arecorrected from the alumina background.
The diagram of the 1% Pd catalyst after butene,propene and acetylene oxidation at 220◦C are com-pared on Fig. 8. These diagrams are corrected fromthe alumina background to show the palladium phases.The new PdX phase identified in propene oxidation isalso formed in but-1-ene oxidation: the diagrams ob-tained after C4H8 and C3H6 oxidation are similar; thepeaks at 39.1, 45.4 and 79.6◦ corresponding to PdXcan be observed with a very close intensity for thetwo alkenes. A different behavior of the catalyst is no-ticed in acetylene oxidation. PdX is formed at 220◦Cwhile the oxide form of palladium disappears but thenew phase is less abundant than in butene or propeneoxidation. In fact, the reaction of acetylene oxidationperformed in sub-stoichiometry of oxygen (S = 0.4)leads to a great amount of carbon.
The PdX peaks recorded after C2H2 oxidation haveabout half the area of the corresponding peaks afteralkene oxidation (propene and butene), while there isno other form of palladium (PdO or Pd◦) in the C2H2spectrum. As the whole sample is analyzed by XRD,we think that the carbon deposit may inhibit the trans-formation of PdO into PdX, a fraction of palladiumbeing not well crystallized after acetylene oxidation.
3.5. Structural changes in bulk palladium (S = 0.4)
To evidence possible support effects on structuralchanges of palladium, propene oxidation was carriedout atS = 0.4 over bulk PdO. The XRD spectrum ofthe catalyst before reaction (Fig. 9) shows that a very
Fig. 9. XRD of the unsupported PdO sample before reaction.
small fraction of PdO is reduced into Pd◦ even thoughthe sample has been calcined in 3% O2 at 400◦C.After propene oxidation, a significant transformationof palladium can be observed with the appearance ofvery intense peaks at 39.1, 45.4, 66.3 and 79.6◦ corre-sponding to PdX (Fig. 10). However, at 220◦C thereremains some PdO, while at 260◦C, the transformationof PdO into PdX is virtually complete. The reducibil-ity of PdO into Pd◦ was also controlled by treatmentof the fresh palladium oxide in H2 at 300◦C. The re-sults presented in Fig. 11 confirms the differences oflattice parameter already recorded for Pd◦ and PdX inthe supported catalysts.
The mean particle sizes of PdO (Fig. 9), PdX(Fig. 10) and Pd◦ (Fig. 11) have been determined
Fig. 10. XRD of the unsupported PdO after propene oxidation at220 or 260◦C, S = 0.4.

204 Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205
Fig. 11. XRD of the unsupported PdO: (a) fresh PdO reduced in H2 at 300◦C; (b) PdO contacted with propene at 260◦C (S = 0.4).
by the Debye–Scherrer equation. While particles ofPdO (7.7 nm) or PdX (15.2 nm) are relatively small,very big particles of Pd◦ are formed (148 nm) uponreduction of PdO in H2. This is a reversible process:re-oxidation of Pd◦ particles leads to smaller PdOmicrocrystals (XRD patterns not shown). It seems asif PdO particles agglomerate upon reduction whilePd◦ crystals are fragmented upon re-oxidation. Theseresults are in agreement with the model recently pro-posed by Lyubovsky and coworkers for the Pd↔PdO transformation on alumina supports [47,48].
3.6. Nature of the PdX phase
All the results obtained in this study are coherentwith a PdX phase exhibiting a cubic structure with alattice parameter higher than in Pd◦. This new palla-dium phase is formed in olefin or acetylene oxidation[20] and this study, butnot in alkane or CO oxidation[20]. Four hypotheses can be put forward to explainthese results.1. Stressed Pd◦ particles, in epitaxy with the support,
could be formed during the oxidation reaction. Thishypothesis reasonably may be ruled out becausePdX is formed even in the absence of support (seeSection 3.4).
2. The increase of the lattice parameter could be dueto an insertion of hydrogen species. There exist sev-eral palladium hydrides, one of them, PdH0.64 hav-ing a fcc structure (ICDD file 84–448). However,
the lattice parameter of this compound (0.403 nm)is higher than in PdX. Moreover, palladium hy-drides are not stable above 120◦C and should havebeen formed during sample cooling and not at thetemperature of reaction. The fact that PdX cannotbe formed if the reaction is carried out at too lowa temperature (see Section 3.2) or in the presenceof pure H2 does not support this hypothesis.
3. Insertion of carbon atoms is a likely hypothesis.Olefin and acetylene are strongly bonded to palla-dium and may lead to carbon species as reactionintermediates. However, there is no definite palla-dium carbide materials referenced in the literature.We may also remark that CO, a possible precursorof Pd carbides, does not lead to PdX.
4. Finally, insertion of oxygen atoms seems to be themost likely hypothesis. PdX can be formed over awide range of O2 to olefin ratio but rather under richconditions. However, PdX is virtually not formedin the absence of oxygen, which supports the ideathat O species should intervene in the constructionof this new Pd phase. Nevertheless, further worksare needed to elucidate completely structure andchemical composition of PdX.
4. Conclusions
A new cubic phase of palladium is formed dur-ing alkene and alkyne oxidation carried out in O2sub-stoichiometry. This phase, denoted PdX, is char-

Z. Ferhat-Hamida et al. / Applied Catalysis B: Environmental 29 (2001) 195–205 205
acterized by a larger lattice parameter than in metallicpalladium. It has been detected by XRD and electrondiffraction under various experimental conditions bothover unsupported or alumina-supported palladium cat-alysts. Oxygen favors the construction of PdX whichmight be formed by O insertion into the cubic struc-ture of metallic palladium.
References
[1] R. Van Yperen, D. Linder, L. Mussmann, E.S. Lox, T.Kreuzer, Catalysis and automotive pollution control IV, Stud.Surface Sci. Catal. 116 (1998) 51.
[2] Y.F. Yu Yao, Ind. Eng. Chem. Prod. Res. 19 (1980) 293.[3] C.F. Cullis, B.M. Willatt, J. Catal. 83 (1983) 267.[4] T.R. Baldwin, R. Burch, Catal. Lett. 6 (1990) 131.[5] T.R. Baldwin, R. Burch, Appl. Catal. 66 (1990) 337.[6] R.F. Hicks, H. Qi, M.L. Young, R.G. Lee, J. Catal.0 122
(1990) 295.[7] R.J. Farrauto, T. Kennelly, E.M. Watermann, US Patent No.
4,893,465 (1990).[8] P. Briot, M. Primet, Appl. Catal. 68 (1991) 301.[9] N. Mouaddib, C. Feumi-Jantou, E. Garbowski, M. Primet,
Appl. Catal. A 87 (1992) 129.[10] R.J. Farrauto, M.C. Hobson, T. Kennelly, E.M. Watermann,
Appl. Catal. 81 (1992) 227.[11] K. Sekizawa, M. Machida, K. Eguchi, H. Arai, J. Catal. 142
(1993) 655.[12] F.H. Ribeiro, M. Chow, R.A. Dalla Betta, J. Catal. 146 (1994)
537.[13] K.-I. Muto, N. Katada, M. Niwa, Appl. Catal. A 134 (1996)
203.[14] R.A. Dalla Betta, Catal. Today 35 (1997) 129.[15] A.M. Pisanu, C.E. Gigola, Appl. Catal. B 11 (1996) L37.[16] H. Shinjoh, H. Muraki, Y. Fujitani, Appl. Catal. 49 (1989)
195.[17] P. Marécot, A. Fakche, B. Kellali, G. Mabilon, M. Prigent,
J. Barbier, Appl. Catal. B 3 (1994) 283.[18] C. Hoang-Van, R. Harivololona, S. Fayeulle, Catalysis and
automotive pollution control III, Stud. Surface Sci. Catal. 96(1995) 249.
[19] T. Maillet, J. Barbier Jr., D. Duprez, Appl. Catal. B 9 (1996)251.
[20] T. Maillet, C. Soleau, J. Barbier Jr., D. Duprez, Appl. Catal.B 14 (1997) 85.
[21] Y. Yazawa, H. Yoshida, N. Tagaki, S.-I. Komai, A. Satsuma,T. Hattori, Appl. Catal. B 19 (1998) 261.
[22] A.B. Kooh, W.-J. Han, R.G. Lee, R.F. Hicks, J. Catal. 130(1991) 374.
[23] W.R. Patterson, C. Kemball, J. Catal. 2 (1963) 465.[24] N.W. Cant, W. Keith Hall, J. Catal. 16 (1970) 220.[25] Y.F. Yu. Yao, J. Catal. 87 (1984) 152.[26] A.L. Boehman, S. Niksa, Appl. Catal. B 8 (1996) 41.[27] L. Borkó, I. Nagy, Z. Schay, L. Guczi, Appl. Catal. A 147
(1996) 95.[28] M. Ferrandon, J. Carnö, S. Järås, E. Björnbom, Appl. Catal.
A 180 (1999) 153.[29] K. Otto, L.P. Haack, J.E. deVries, Appl. Catal. B 1 (1992) 1.[30] P.J. Schmitz, K. Otto, J.E. deVries, Appl. Catal. 92 (1992)
59.[31] B.H. Engler, D. Lindler, E.S. Lox, A. Schäfer-Sindlinger, K.
Ostgathe, Catalysis and automotive pollution control III, Stud.Surface Sci. Catal. 96 (1995 ) 441.
[32] L.P. Haack, K. Otto, Catal. Lett. 34 (1995) 31.[33] T.E. Hoost, K. Otto, Appl. Catal. A 92 (1992) 39.[34] P. Salomonsson, S. Johansson, B. Kasemo, Catal. Lett. 33
(1995) 1.[35] R.J. Farrauto, J.K. Lampert, M.C. Hobson, E.M. Waterman,
Appl. Catal. B 6 (1995) 263.[36] Y.-S. Ho, C.-B. Wang, C.-T. Yeh, J. Mol. Catal. A 112 (1996)
287.[37] M. Haneda, T. Mizushima, N. Kakuta, J. Phys. Chem. B 102
(1998) 6579.[38] E. Garbowski, C. Feumi-Jantou, N. Mouaddib, M. Primet,
Appl. Catal. A 109 (1994) 277.[39] N.M. Rodriguez, S.G. Oh, R.A. Dalla-Betta, R.T.K. Baker,
Catalyst deactivation 1994, Stud. Surface Sci. Catal. 88 (1994)417.
[40] K.-I. Fujimoto, F.H. Ribeiro, M. Avalos-Borja, E. Iglesia, J.Catal. 179 (1998) 431.
[41] K. Furuta, A. Nishida, H. Yata, K. Narui, Y. Kohtoku, Appl.Catal. A 164 (1997) 149.
[42] R. Burch, M.J. Hayes, J. Mol. Catal. 100 (1995) 13.[43] J.J. Chen, E. Ruckenstein, J. Phys. Chem. 85 (1981) 1606.[44] H. Lieske, J. Völter, J. Phys. Chem 89 (1985) 1841.[45] C.F. Cullis, T.G. Nevell, D.L. Trimm, J. Chem. Soc., Faraday
Trans. 1 68 (1972) 1406.[46] J.P. Morniroli, D. Vankieken, L. Winter, in: U.S.T. Lille
(Publ.), Computer Program, Version 3.6.[47] M. Lyubovsky, L. Pfefferle, A. Datye, J. Bravo, T. Nelson,
J. Catal. 187 (1999) 275.[48] A.K. Datye, J. Bravo, T.R. Nelson, P. Atanasova, M.
Lyubovsky, L. Pfefferle, Appl. Catal. A 198 (2000) 179.
Copyright © 2022 FDOKUMEN




















