
Evaluación de materiales compuestos resistentes a impacto ...
Upload
independentCategory
view
0download
0


PRÓLOGO
Poco podría decir si no hubiese tenido la posibilidad de participar
en uno de los laboratorios de investigación del Consejo Superior de
Investigaciones Científicas (CSIC) en la Isla de la Cartuja, Sevilla, y jamás
por tanto, hubiera tenido la idea, ni el conocimiento suficiente, como para
escribir este breve resumen de las técnicas más usadas en un laboratorio de
Química Organometálica. Nada más entrar en el recinto y poner el pie en el
lugar de trabajo, me impuse poner por escrito cualquier método
desconocido que viniese a encontrarme, al principio sin otra intención más
que el hecho de que me fuese útil a mí mismo. Sin embargo no hubo de
transcurrir mucho tiempo para que me quedase claro que el volumen de
información, relativa a nuevos conceptos y técnicas, era tan elevado que a
la hora de apuntar había que realizar un serio y concienzudo trabajo de
selección y de síntesis. Fue por tanto una labor continua la de apuntar y
describir situaciones, lo cual devoró mucho de mi tiempo, aunque fue grato
de igual modo. De la misma manera realicé una gran cantidad de dibujos,
que pretenden ser aclaratorios, y que espero sirvan para comprender mejor
las diferentes explicaciones concernientes a los métodos.
Tuve la suerte entonces de abordar el campo de la química práctica,
no existiendo parangón con sea cual fuere la práctica realizada durante el
periodo de aprendizaje en la Facultad de Químicas. La naturaleza tan
particular del trabajo con compuestos organometálicos y la línea admirable
de investigación del propio centro, supusieron desde el principio una
venturosa etapa de aprendizaje, tan o más importante si cabe que la propia
enseñanza universitaria.
A medida que fui confeccionando el manual me fui dando cuenta de que
podría ser útil a otras personas que, estudiando química en general, vinieren

a toparse con esos tan traídos compuestos, y que se preguntaren cómo es
posible trabajar con ellos sin que exista exposición alguna al aire o a la
humedad. Espero que este volumen sirva para eso y logre interesar a
alguien y créanme ustedes si digo que me vi con el deber moral de escribir
todo aquel caudal de conocimiento fugaz que pronto habría de quedar
obsoleto, superado por nuevas técnicas. Por otra parte, estando como estoy,
en pleno acuerdo con la opinión no muy generalizada de que la propia
Ciencia debe ser la encargada de informar, ya no tan sólo a los compañeros
más o menos cercanos de otras ramas, sino también a cualquier persona; no
creo haber perdido el tiempo presentando este sucinto volumen que, aun
estando dirigido a futuros químicos, en particular químicos
organometálicos, se encuentra desarrollado, o así lo espero, de un modo
claro y ameno. He huido de los párrafos largos, apostando por los dibujos
para una mejor comprensión del hecho. De todas maneras desearía pedir
disculpas por aquellas ideas que se encuentren difusas y que, a pesar de que
en el momento de la escritura, me parecieran suficientemente claras y
plausibles es posible no haya logrado desarrollar con precisión.
Finalmente querría agradecer a las personas que, directa o
indirectamente, me han condicionado, en uno u otro sentido, a escribir este
manual.
La práctica totalidad de los métodos, conceptos, técnicas y
argumentos me han sido proporcionados de modo ejemplar, tanto su
calidad científica como por la diligencia y cordialidad en el trato, por mis
dos compañeros y amigos de laboratorio, Celia Maya y Diego del Río, no
sólo pues se han impuesto como ejemplos de capacidad investigadora sino
también de compañerismo, por lo cual me considero muy honrado con la
mera circunstancia de haber podido aprender de ellos. Igualmente desearía
agradecer la siempre bien tenida amistad, con la que se me ha recibido y

ayudado, por parte de los Directores del laboratorio, Pilar Palma y Juan
Cámpora. No puedo en modo alguno olvidar en esta letanía de
agradecimientos a mi buen amigo y eterno compañero de laboratorio
durante la Facultad, Raúl Martínez Salazar, con el que las más veces
peleando y algunas otras trabajando, aprendí los principios básicos del
trabajo en equipo y de la química del laboratorio. Para acabar, es mi mayor
placer agradecer especialmente a Ernesto Carmona la disposición favorable
que ha mantenido y mantiene para con cualquier propuesta que implique un
desarrollo científico, sea en su más pura significación, sea en la expansión
de los conocimientos como vehículo formativo. Sin la concurrencia de
todas estas personas, especialmente de mis compañeros de laboratorio a los
que debo mucho más que el conocimiento adquirido, este trabajo no
hubiese llegado a concretarse.
Sevilla, 2000

ÍNDICE
PARTE TEÓRICA
1. LOS COMPUESTOS ORGANOMETÁLICOS 1
2. EL ENLACE 2
2.1 UNA VISIÓN COMPARATIVA DEL ENLACE 2
2.2 ENLACES DEFICIENTES EN ELECTRONES 6
2.3 CARACTERÍSTICAS ESPECIALES 9
2.4 ENLACE IÓNICO 10
3. TIPOS DE COPUESTOS ORGANOMETÁLICOS 11
3.1 SEGÚN EL ENLACE 11
3.2 SEGÚN EL LIGANDO 12
3.2.1 LIGANDO ALQUÍLICO 12
3.2.1.1 SÍNTESIS 12
3.2.1.2 APLICACIONES 13
3.2.2 LIGANDO OLEFÍNICO 13
3.2.2.1 SÍNTESIS 13
3.2.2.2 APLICACIONES 14
4. OTROS COMPUESTOS CARACTERÍSTICOS 14
4.1 LOS COMPUESTOS DE GRIGNARD 15
4.2 LOS CARBONILOS METÁLICOS 16
4.3 LAS FOSFINAS 17
4.4 LOS COMPUESTOS TIPO SANDWICH 18

PARTE PRÁCTICA
1. LOS COMPUESTOS ORGANOMETÁLICOS 20
1.1 ORGANOMETÁLICOS AL AIRE 20
1.2 O2 Y H2O. LOS ENEMIGOS 20
2. LA LÍNEA 21
2.1. DESCRIPCIÓN 21
2.2. LIMPIEZA DE LA LÍNEA 28
2.2.1. QUITAMOS LAS GOMAS 28
2.2.2. LIMPIAMOS LA CÁMARA DE VACÍO 29
2.2.3. LIMPIAMOS LAS LLAVES DE VIDRIO 30
2.2.4. LIMPIAPIPAS 31
2.2.5. LIMPIEZA DE LA TRAMPA DE VACÍO 31
2.2.6. LIMPIEZA DEL MERCURIO 32
2.2.7. ACETONA Y ÉTER PARA LAVAR 34
2.3. MONTAJE DE LA LÍNEA 35
2.3.1. UNIÓN DE LA RÓTULA 35
2.3.2. COLOCAMOS LOS TUBOS 37
2.3.3. ENROSCAMOS LOS ALAMBRES 38
2.3.4. ABRAZADERAS 39
2.3.5. SOBRE LAS GOMILLAS 40
2.3.6. GRASA DE ALTO VACÍO 40
2.3.7. LA RIGIDEZ DE LA ENCIMERA DE TRABAJO 40
2.4. PUESTA EN MARCHA DE LA LÍNEA 41

2.4.1. ANTES DE TOCAR LA LÍNEA 41
2.4.2. COLOCAMOS LA AMPOLLA 42
2.4.3. PREVIO A LA CONEXIÓN DE LA BOMBA 45
2.4.4. CONECTAMOS LA BOMBA DE VACÍO 45
2.4.5. MANORREDUCTORES 46
2.4.6. INYECTAMOS NITRÓGENO GASEOSO 46
2.5. QUITAMOS LA LÍNEA 47
2.5.1. PARAMOS LA BOMBA 47
2.5.2. TERMINAMOS EL PROCESO 47
3. INSTRUMENTAL DE TRABAJO (I) 49
3.1. MATRACES Y TUBOS SCHLENK 49
3.1.1. MATRACES DE REACCIÓN 49
3.1.2. TUBOS SCHLENK 50
3.1.3. COLOCAMOS LA LLAVE DE VIDRIO 51
3.1.4. PONEMOS LAS GOMILLAS 53
3.2. TAPONES SUBA-SEAL Y DE VIDRIO 56
3.2.1. TAPONES SUBA-SEAL 56
3.2.1.1. PINCHADO O NO PINCHADO 58
3.2.1.2. COLOCAMOS GOMILLAS AL TAPÓN 59
3.2.2. TAPONES DE VIDRIO 60
3.3. TAPONES DE TEFLÓN 61
4. IMPONEMOS ATMÓSFERA INERTE 64
5. DISOLVENTES 65

5.1. TIPOS Y NATURALEZA 65
5.1.1. ÉTER DIETÍLICO 66
5.1.2. THF 66
5.1.3. ÉTER DE PETRÓLEO 67
5.1.4. DICLOROMETANO 67
5.1.5. TOLUENO 67
5.1.6. ACETONITRILO 68
5.1.7. ACETONA 68
5.1.8. ETANOL 68
5.2. ELIMINACIÓN DE AGUA Y DE OXÍGENO 69
5.2.1. EL SODIO 69
5.2.2. EL SODIO Y EL AGUA 70
5.2.3. EL REFLUJO 71
5.2.4. PROBLEMAS CON EL REFLUJO 73
5.2.5. CONECTAMOS EN SERIE LOS DISOLVENTES 74
5.2.6. SISTEMAS DE SEGURIDAD 74
5.2.7. DESTRUIMOS EL SODIO REMANENTE 75
5.2.8. VALIDEZ DEL MÉTODO DEL SODIO 76
5.2.9. SECADO DE LA ACETONA 77
5.2.10. ACETONITRILO Y DICLOROMETANO 77
5.2.11. INDICADORES DE HUMEDAD 77
5.2.12. ATENCIÓN FINAL 78
6. INSTRUMENTAL DE TRABAJO (II) 78

6.1. AMPOLLAS Y SUS TAPONES DE TEFLÓN 78
6.1.1. AMPOLLAS 78
6.1.2. TAPONES PARA AMPOLLAS 80
6.2. JERINGAS Y AGUJAS 82
6.2.1. JERINGAS 82
6.2.1.1. JERINGAS DE VIDRIO 82
6.2.1.2. AGUJAS-CÁNULA 83
6.2.1.3. LIMPIEZA DE JERINGAS DE VIDRIO 84
6.2.1.4. JERINGAS DE PLÁSTICO 85
6.2.1.5. MICROJERINGAS 86
7. REACTIVOS SÓLIDOS 87
7.1. USO DE LA LÍNEA 87
7.2. PREPARATIVOS PARA LA REACCIÓN 88
7.2.1. PESADA 88
7.2.1.1. REACTIVOS ESTABLES AL AIRE 88
7.2.1.2. CON DESCOMPOSICIÓN LENTA 88
7.2.1.3. CON DESCOMPOSICIÓN RÁPIDA 89
7.2.1.4. CON DESCOMPOSICIÓN MUY RÁPIDA 91
7.2.2. ESCALA DE LAS REACCIONES 92
7.2.3. CONSEJOS AL PONER UN TAMPÓN 93
7.2.4. TARAMOS UN SCHLENK O UN MATRAZ 94
8. INYECCIÓN DE DISOLVENTES 95
8.1. RECOGEMOS DISOLVENTES EN LA JERINGA 95

8.2. PONEMOS LA AGUJA E INYECTAMOS 96
8.3. UN MAYOR VOLUMEN DE DISOLVENTE 98
9. INSTRUMENTAL DE TRABAJO (III) 98
9.1. SISTEMAS DE AGITACIÓN 98
9.1.1. RATONES 98
9.1.2. AGITADORES MAGNÉTICOS 99
9.2. TUBOS DE CENTRÍFUGA Y PINCHO 100
9.2.1. TUBOS DE CENTRÍFUGA 100
9.2.2. PINCHO 101
9.3. AGUJAS 102
9.4. CÁNULAS 103
9.4.1. CÁNULAS 103
9.4.2. LIMPIEZA DE LAS CÁNULAS 103
9.4.3. ALMACENAMIENTO 105
10. INTRODUCCIÓN Y EXTRACCIÓN DE RATONES 106
10.1. INTRODUCCIÓN DE UN RATÓN 106
10.2. EXTRACCIÓN DE UN RATÓN 107
11. SE TRANSVASA EN GRAN CANTIDAD 107
11.1. DESOXIGENACIÓN EN TUBO DE CENTRÍFUGA 108
11.2. PASAMOS DISOLVENTE A UN MATRAZ 109
12. EL FRÍO 111
12.1. EL DEWAR 112
12.2. TOMAMOS NITRÓGENO LÍQUIDO DEL TANQUE 113

12.3. LLENAMOS EL TANQUE 116
1.2.4. EL NITRÓGENO COMO LÍQUIDO 118
1.2.5. OBTENCIÓN DEL DIÓXIDO DE CARBONO EN PASTILLA 120
12.6. EL DIÓXIDO DE CARBONO COMO SÓLIDO 122
12.7. BAÑO FRÍO DE ACETONA Y DIÓXIDO DE CARBONO 123
12.8. BAÑO FRÍO DE ACETONA Y NITRÓGENO LÍQUIDO 124
12.9. TERMÓMETROS DE BAJA TEMPERATURA 125
12.10. PROBLEMAS CON LA PRESIÓN 125
13. ADICIÓN DE REACTIVOS 126
13.1. ADICIÓN DE SUSTANCIAS SÓLIDAS 126
13.2. ADICIÓN DE LÍQUIDOS PUROS O DISOLUCIONES 127
13.2.1. DISOLUCIÓN PROCEDENTE DE UN SÓLIDO 127
13.2.2. DISOLUCIONES Y LÍQUIDOS 129
13.2.3. DISPENSAMOS REACTIVOS GOTA A GOTA 133
13.2.4. ADICIÓN CON JERINGA A BAJA TEMPERATURA 134
13.2.5. ADICIÓN CON CÁNULA 135
13.2.6. MEDICIÓN DE UN VOLUMEN BAJO PRESIÓN DE
NITRÓGENO 136
13.3. REACTIVOS GASEOSOS 137
13.3.1. USAMOS EL MONÓXIDO DE CARBONO 137
13.3.2. OTROS GASES 139
13.3.3. REACTORES DE ALTA PRESIÓN 140
13.4. DEJAMOS QUE LA REACCIÓN SE COMPLETE 140

13.5. LA GOMILLA DE SEGURIDAD 141
13.6. BOTES CON TAPÓN DE SEGURIDAD 142
13.7. LAVADO DE LAS AGUJAS-CÁNULA 144
14. EL CALOR 144
14.1. ELIMINACIÓN DEL BAÑO FRÍO 145
14.2. USO DE SECADORES 145
14.3. CALENTADORES DE AGITACIÓN 146
14.4. CALENTAMIENTO EN BAÑO DE AGUA 148
14.5. CALENTAMIENTO EN BAÑOS DE ACEITE 149
14.5.1. BAÑOS DISEÑADOS PARA ACEITE 149
14.5.2. BAÑOS INDIVIDUALES 150
14.6. TERMÓMETROS 151
14.7. AMPOLLAS O MATRACES 152
14.8. ESTUFAS 153
15. ELIMINACIÓN DE DISOLVENTES A VACÍO 154
15.1. EN EL PROPIO MATRAZ DE REACCIÓN 155
15.2. EN UN SCHLENK 156
15.3. EVAPORACIÓN DE GRANDES CANTIDADES 159
15.4. EL ROTAVAPOR 161
16. SEPARACIÓN POR CENTRIFUGACIÓN 162
16.1. DESOXIGENACIÓN DE UN TUBO DE CENTRÍFUGA 162
16.2. PASAMOS EL LÍQUIDO DEL MATRAZ AL TUBO 163
16.3. EL PARAFILM 165

16.4. SOPORTES PARA TUBOS DE CENTRÍFUGA 166
16.5. EL CONTRAPESO DE LA CENTRÍFUGA 167
16.6. LA CENTRÍFUGA 168
16.7. TRANSVASE DEL LÍQUIDO 169
17. FILTRADO POR CÁNULA 170
17.1. CON CÁNULA DE FILTRADO 171
17.2. SIN APLIQUE DE FILTRADO 173
17.3. LOS DISCOS DE FILTRO 174
17.4. USAMOS LOS FILTROS 175
18. FILTRACIÓN EN PLACA O LECHO POROSO 177
18.1. MATERIALES PARA LECHO 177
18.2. LA PLACA 179
18.3. MONTAJE Y PREPARACIÓN DEL SISTEMA 179
18.4. DESOXIGENACIÓN DEL MONTAJE 182
18.5. LA OPERACIÓN DE FILTRADO 183
18.6. FILTRACIÓN PARA RECOGER EL SÓLIDO 186
18.7. FILTRACIÓN EN PLACA AL AIRE 191
18.8. OTROS FILTRADOS Y CROMATOGRAFÍAS 196
18.9. LIMPIEZA DE PLACAS 197
19. CROMATOGRAFÍA EN CAPA FINA 197
19.1. LA PLACA 198
19.2. PREPARAMOS LA PLACA 199
19.3. DISPENSAMOS LAS ALÍCUOTAS EN LA PLACA 200

19.4. LA FASE MÓVIL 202
19.5. LA SEPARACIÓN 203
19.6. INTERPRETACIÓN DE UNA PLACA 204
20. EL CROMATOTRÓN 207
20.1. EL APARATO 207
20.2. ELECCIÓN Y PREPARACIÓN DE LA FASE MÓVIL 210
20.3. LA FASE ESTACIONARIA 211
20.4. ACONDICIONAMIENTO DEL CROMATÓGRAFO 212
20.5. INYECCIÓN DE LA DISOLUCIÓN 213
20.6. SEGUIMIENTO DE LAS BANDAS 214
20.7. RECOLECCIÓN DE LAS DISTINTAS FRACCIONES 216
20.8. LIMPIEZA DE LA PLACA 217
21. LA RECRISTALIZACIÓN 217
21.1. BUSCAMOS LAS CONDICIONES ÓPTIMAS 218
21.2. ELIMINACIÓN DE LOS RESTOS EN LA PARTE SUPERIOR DEL
SCHLENK 219
21.3. METEMOS EL SCHLENK EN LA NEVERA 220
21.4. CRISTALIZACIÓN POR ELIMINACIÓN DE DISOLVENTE 221
21.5. SACAMOS EL SCHLENK DE LA NEVERA 222
21.6. SEPARAMOS LOS CRISTALES DE LAS AGUAS MADRES 222
22. SECADO DE LOS CRISTALES 223
22.1. SECADO A VACÍO 224
22.2. SECADO CON CORRIENTE DE NITRÓGENO 224

23. CRISTALES PARA DIFRACCIÓN 225
23.1. ELECCIÓN DE CRISTALES PARA DIFRACCIÓN 225
23.2. PREPARAMOS UNA PIPETA PASTEUR 227
23.3. GUARDAMOS LOS CRISTALITOS EN LA PIPETA 227
24. RMN 229
24.1. SU BASE CIENTÍFICA 230
24.2. LOS DISOLVENTES DEUTERADOS 232
24.3. TUBOS DE RMN Y SUS TAPONES DE GOMA 234
24.4. DESOXIGENACIÓN DE UN TUBO DE RMN 236
24.5. ARREGLAMOS LA BOCA DE LOS TUBOS 237
24.6. SE TOMA MUESTRA PARA SEGUIMIENTO DE REACCIÓN 238
24.7. PREPARAR MUESTRA PARA ANÁLISIS DE CRISTALES 241
24.8. RUPTURA DE UN TUBO DE RMN EN LA OPERACIÓN 244
24.9. NOS CORTAMOS CON EL VIDRIO 245
24.10. LA SALA DE RMN 245
24.11. EL APARATO DE RMN 246
24.12. HACEMOS EL ESPECTRO 248
24.13. INTERPRETACIÓN DE UN ESPECTRO 254
24.14. REFLEXIÓN FINAL SOBRE EL TRABAJO EN RMN 255
24.15. LIMPIAMOS TUBOS DE RMN 256
25. ESPECTROSCOPÍA DE INFRARROJOS 256
25.1. LA BASE CIENTÍFICA 257
25.2. MUESTRAS SÓLIDAS 258

25.3. EL APARATO 260
25.4. INTERPRETACIÓN DE LOS ESPECTROS 261
25.5. OTROS SISTEMAS DE ANÁLISIS 261
26. ALMACENAMIENTO 262
26.1 .EL ALMACÉN 262
26.2. EL FRIGORÍFICO COMO ALMACÉN 265
26.3. ALMACENAMIENTO DE AMPOLLAS 266
26.4. ALMACENAMIENTO EN BOTES 267
26.5. ALMACENAMIENTO TEMPORAL EN MATRACES 268
26.6. ALMACENAMIENTO EN TUBOS DE SELLADO 268
27. LA CÁMARA SECA 271
27.1. DESCRIPCIÓN FÍSICA Y FUNCIONAL 272
27.2. INTRODUCCIÓN DE OBJETOS 275
27.3. CICLOS EN LA PRECÁMARA 277
27.4. PASAMOS LOS OBJETOS AL INTERIOR 280
27.5. PESAMOS UN COMPUESTO 281
27.6. LIMPIEZA DE LA CÁMARA SECA 282
27.7. SACAMOS EL PRODUCTO YA PESADO 283
27.8. USO SIMULTÁNEO DE LAS DOS PRECÁMARAS 283
28. DESECHOS 284
28.1. LOS DISOLVENTES 285
28.2. DISOLUCIONES 286
28.3. SÓLIDOS 287

28.4. RECUPERACIÓN POR DESTILACIÓN 287
28.5. COMPUESTOS PELIGROSOS 287
28.6. COMPUESTOS GASEOSOS 289
28.7. MATERIAL DE VIDRIO ROTO 289
29. OPERACIONES DE LIMPIEZA 290
29.1. LAVADO DE MATERIAL SIN GRASA 290
29.2. LAVADO DE MATERIAL CON GRASA 291
29.2.1. BAÑO DE POTASA ALCOHÓLICA 291
29.2.2. TRAS EL BAÑO 292
29.3. LAVAMOS RESTOS DE METALES NOBLES 294
29.4. SOBRE LOS DISOLVENTES EMPLEADOS 295
29.5. LIMPIEZA DE LAS VITRINAS 295
29.6. LIMPIEZA DEL LABORATORIO 296
30. SE PONE UNA REACCIÓN 296
31. INSTRUMENTAL DE TRABAJO (IV) 301
31.1. MATRACES DE DOS Y DE TRES BOCAS 301
31.2. SOPORTES DE MATRACES DE FONDO REDONDO 302
31.3. LLAVES DE CONEXIÓN INDEPENDIENTE 303
31.4. COLOCACIÓN DE LAS GOMILLAS 304
31.5. EL EMBUDO DE ADICIÓN DE REACTIVOS 305
31.6. COLOCAMOS LAS GOMILLAS 306
31.7. EMBUDO DE PRESIÓN COMPENSADA 307
31.8. LAS GOMILLAS 309

31.9. HACEMOS LA ABRAZADERA DE ALAMBRE 310
32. SÍNTESIS DE COMPUESTOS DE GRIGNARD 311
32.1. LA REACCIÓN 311
32.2. SE PONE LA REACCIÓN 312
32.3. FUGAS EN EL SISTEMA 319
32.4. LA CINTA DE TEFLÓN 320
32.5. EL REFLUJO 320
32.6. VALORACIÓN DE UN GRIGNARD 321
33. FOSFINAS 323
33.1. LAS FOSFINAS 323
33.2. SÍNTESIS DE FOSFINAS 324
33.3. CALENTAMOS DISOLUCIONES CON FOSFINAS 325
33.4 .EL OLOR DE LAS FOSFINAS 325
34. SE COMIENZA UNA INVESTIGACIÓN 326
34.1. CATALOGACIÓN DE COMPUESTOS 326
34.2. LA QUÍMICA DE LA COORDINACIÓN 328
34.3. LA QUÍMICA ORGANOMETÁLICA 329
34.4. LAS PUBLICACIONES 330
35. CONCLUSIÓN 332


1
TEORÍA DE COMPUESTOS
ORGANOMETÁLICOS
1 LOS COMPUESTOS ORGANOMETÁLICOS
No quisiera que esta parte se considerase más que como unas notas básicas,
que sólo pretenden perfilar de algún modo el amplio y difuso mundo de los
compuestos organometálicos. Teniendo pues en cuenta cuál es el objetivo,
se espera una lectura benevolente, pues para análisis en profundidad están
ya otros manuales correspondientes a personas infinitamente más
cualificadas.
Se consideran compuestos organometálicos a todos aquellos complejos que
presentan uno o varios enlaces metal–carbono. Este carbono puede ser de
tipo carbonílico (formando los carbonilos metálicos) o de tipo orgánico
(olefínicos, arílicos o alquílicos). Existen compuestos de naturaleza muy
semejante a los anteriores que, sin ser estrictamente compuestos
organometálicos, suelen contemplarse como familias hermanas a éstas y
por tanto considerables desde el punto de vista de la organometálica. Entre
estos encontramos los compuestos con enlace M–B, o B–B (igualmente
podemos encontrar estructuras análogas para P o N), y a su vez el H,
entendido como hidruro, que se presenta unido a metales.
La variedad en la naturaleza de los enlaces es tal vez la característica más
importante de estos compuestos, existiendo situaciones en las que no se
puede definir un enlace tradicional de 2 centros – 2 electrones (enlace

2
covalente típico). La descripción más hábil, a mi entender es la que permite
hacer la teoría de orbitales moleculares (T.O.M.), que requiere
conocimientos de simetría.
2 EL ENLACE
2. 1 UNA VISIÓN COMPARATIVA DEL ENLACE
En una molécula se pueden diferenciar básicamente dos tipos de orbitales,
los enlazantes y los antienlazantes.
Cuando hay dos núcleos, éstos sufren una repulsión electrostática mutua,
tal que provoca el alejamiento entre ellos (es una visión simplificada en
exceso pero que me parece interesante). Si los electrones pasan de vez en
cuando entre los dos, el efecto es parecido a si se neutralizasen las cargas,
siendo por tanto una situación muy favorable. A más veces se sitúen en la
zona intermedia, más estable será la relación y se percibirán menos ambos
núcleos.
En los orbitales moleculares enlazantes, la probabilidad de encontrar al
electrón entre los dos núcleos es muy grande, de ahí que tengamos la
máxima capacidad enlazante y la mínima repulsión internuclear.
Esta misma razón se puede aplicar para la discriminación de los distintos
tipos de interacciones enlazantes. Debido a la existencia de nodos podemos
encontrarnos con barreras energéticas que impidan que los electrones se
coloquen entre los núcleos con una completa permisividad.

3
En este dibujo se representa la interacción en fase de dos orbitales p.
Obsérvese como los electrones pueden permanecer sin problemas entre
ambos núcleos, ya que no presenta ningún plano nodal en la región
internuclear.
A este tipo de enlace donde no existe ningún plano nodal que contenga a
ambos núcleos se le denomina enlace , y puesto que los electrones se
sitúan preferentemente entre los núcleos, éstos se sienten muy poco y por
tanto el enlace es el más fuerte.
En las representaciones se indican ahora la
interacción entre dos orbitales p de manera
lateral (a la derecha) y la interacción entre dos
orbitales tipo d (más abajo). En ambos casos se
percibe que los electrones no pueden circular
por el plano que contiene a ambos núcleos
(perpendicular al papel). Se apreciará por tanto cómo la densidad
electrónica, aún situándose entre
los núcleos, evita precisamente esa
zona. El enlace por tanto será más
débil que el anterior, ya que los
electrones no forman una pantalla
tan efectiva como en el caso

4
primero. A este enlace, con un plano nodal que contiene a ambos núcleos
se le denomina .
Existe un tercer tipo de enlace, mucho
menos común, consistente en una
interacción lateral entre dos orbitales d.
Este enlace es el más débil de los tres ya
que presenta dos planos nodales que
contienen a ambos núcleos (aquel que es
perpendicular al papel y el papel mismo).
A este enlace se le conoce por el nombre de enlace .
Los lóbulos representados en las figuras poseen tonalidades bien distintas,
aquellos más claros equivalen a un signo de la función de onda, en tanto los
oscuros el otro. De este modo oscuro–oscuro o claro–claro muestran
interacciones en fase, en tanto oscuro–claro o claro–oscuro, en antifase.
Los orbitales moleculares enlazantes surgen como consecuencia de la
interacción en fase de los orbitales atómicos, en tanto que los orbitales
moleculares de antienlace lo hacen a partir de interacciones en antifase.
En los orbitales moleculares de antienlace la posibilidad de encontrar a los
electrones en la región internuclear es mínima por lo que actúan en contra
de la formación de la molécula, puesto que la repulsión internuclear es
entonces muy grande.
Todo lo visto con anterioridad se puede entender también con una
comparación que resulta especialmente indicada para explicar el punto
inmediatamente posterior a este.

5
La situación es la siguiente: imaginemos que cada núcleo es una ciudad,
ambas situadas una cerca de otra. La interacción entre las dos fuerza a la
construcción de caminos que las conecten. Se construyen así varios
caminos: uno en línea recta (), otros formando una pequeña curva
alrededor del anterior () y otros con una separación mucho mayor (). No
obstante también se han hecho otros caminos que desarrollan su recorrido
en direcciones contrarias a la ciudad opuesta, y que por tanto, llevan a otros
lugares como podrían ser posesiones de la misma ciudad a las afueras
(serían vías análogas a las de antienlace y no enlace). Existen por otra parte
una serie de restricciones:
1. Por cada camino sólo podrán ir únicamente dos personas, no importan de
donde sean, si de la ciudad A o de la ciudad B.
2. Dependiendo de lo enérgicas que sean las personas se elegirán unos
caminos u otros.
3. La relación y unidad entre las ciudades será mayor a más comunicación
exista entre ambas.
Podemos entonces comprender que dos personas de la primera ciudad
puedan visitar a la segunda, pero también al revés. Igualmente puede partir
una de una ciudad A y otra de la ciudad B.
La frecuencia de tránsito por las carreteras determina la relación entrambas,
por tanto un enlace más estable a más personas se encuentren en dichos
caminos por unidad de tiempo. La vía de comunicación más efectiva será la
llamada , pues conecta las ciudades en el menor tiempo posible. Los otros
caminos son más largos, y por tanto más lentos, no agilizan la

6
comunicación y por ello resultan menos efectivos que los de tipo . Pero
sólo habrá 2 personas (2 electrones) que transiten por , y el resto deberá
obligadamente tomar las otras vías ( y ) si existen. Lo más importante es
notar cómo en estos movimientos, las personas que entran en los caminos
no pertenecen a una u otra ciudad sino que simplemente circulan por los
caminos, vengan de donde vengan. Del mismo modo no hay electrones
fijos que se mantienen encerrados en un orbital, sino que pueden ser
sustituidos por otros cuando las condiciones energéticas lo requieran.
2. 2 ENLACES DEFICIENTES EN ELECTRONES
En algunos casos la interacción enlazante se puede dar entre varios núcleos
simultáneamente donde algunos de los núcleos ponen solamente orbitales,
creándose por ello un déficit electrónico.
Resultará obvio que aquí también se
pueden analizar la existencia de planos
nodales, que surgen como consecuencia de
la simetría e interacción de varios
orbitales. En este dibujo se ha
representado una aproximación al orbital
molecular enlazante resultante en función
de los tres orbitales atómicos implicados en el proceso. Puesto que no
existe ningún plano nodal que contenga a los tres núcleos, este enlace será
de tipo .

7
Considérese sin más la existencia de los otros dos tipos, así como los
correspondientes de antienlace (recuérdese que tres orbitales atómicos
interaccionan para formar tres orbitales moleculares: uno de enlace, uno de
no enlace y uno de antienlace).
Para entender mejor cómo es posible que tres núcleos puedan compartir dos
electrones (enlace de 3 centros–2 electrones) haremos referencia a la
comparación con las ciudades.
Si al lado de la ciudad A y B ponemos en nuestra imaginación una tercera,
C, surgirán necesariamente nuevos caminos. Consideraremos a las ciudades
muy parecidas entre sí (lo que equivale a decir que en términos energéticos
los átomos que se consideran son semejantes entre sí, lo cual no tiene por
que ser obligado), por ello los caminos que harán serán muy parecidos. El
camino que antes existía entre A y B se amplía y se conecta a C,
llamándose ahora camino ABC. De este modo se crea un nuevo camino que
pasa por B y C para concluir volviendo a A. Lo mismo ocurrirá con el resto
de caminos si las tres ciudades son iguales. Al margen se crearán otros
caminos completamente nuevos que tendrán una longitud entre la más corta
(ABC) y la más larga, al igual que caminos que no aprecien para nada la
comunicación con las otras ciudades.
Considerando que ABC es un único camino se deduce que podrá ser
recorrido por dos personas solamente por la ley impuesta al hacer los
caminos. Las personas que circulen por ABC pueden ser de cualquier
ciudad y llevar el sentido que deseen, sólo que no puede haber en el
camino más de dos.
Puede ocurrir que la tercera ciudad sea algo diferente a las dos anteriores y
que permita solamente un tipo determinado de caminos (o ). En este
caso únicamente los caminos de tal carácter serán modificados y serán

8
compartidos por las tres ciudades, en tanto los caminos del tipo restante
permanecerán uniendo sólo las dos ciudades previas.
Sea cual fuere el caso, al haber aumentado la longitud de los caminos, la
frecuencia de transito por un punto determinado en un tiempo marcado
disminuirá y con ello la capacidad de relación (lo que se compara con una
pérdida en la fuerza del enlace).
Pero si además consideramos que C está abandonada, sólo los habitantes de
B y A podrán circular por los nuevos caminos, lo cual es perfectamente
viable, aunque con la consiguiente disminución de la comunicación (orden
de enlace) entre A y B.
Muchos compuestos organometálicos tienen enlaces deficientes en
electrones, donde en lugar de átomos tenemos moléculas que interaccionan
entre sí y forman moléculas más complejas. Una de las moléculas suele
disponer de orbitales más o menos vacíos (generalmente de un metal que
va a resultar el centro de coordinación de la nueva molécula), en tanto la
otra (u otras) pondrá no sólo orbitales, sino también electrones (se
denominarán los ligandos).
Este tipo de enlaces se puede dar entre un número indeterminado de átomos
metálicos, de tal modo que conforman poliedros de coordinación (muy
relacionados con los compuestos organometálicos) formando los
denominados clusters, lo que se traduce por “racimos” a causa de que los
metales unidos de esta manera forman asociaciones semejantes a estos
conjuntos de frutas.

9
2. 3 CARACTERÍSTICAS ESPECIALES
Bajo este nombre se pretende encabezar un apartado que se destina a la
presentación de enlaces revolucionarios desde el punto de vista del
concepto, enlaces que en la química anterior al desarrollo de la química de
coordinación o a la química organometálica, eran del todo imposibles de
concebir.
El concepto de enlace covalente que se viene estudiando desde la
Secundaria establece que dos orbitales, con la misma forma (o
aproximadamente la misma) y pareja energía, interaccionan y generan un
orbital molecular de enlace y un orbital molecular de antienlace. Este
primer orbital recién creado se verá ocupado formalmente por los
electrones que ponen en juego cada uno de los orbitales atómicos
implicados en el enlace.
La primera variación que encontramos ahora, con respecto a lo anterior, es
que en realidad ambos átomos (o grupos atómicos) no tienen por qué
participar con electrones, y como se dijo en la sección anterior, uno puede
presentar orbitales vacíos en tanto el otro interacciona con éstos
mediante los suyos, que van a ser los que cedan también los electrones. De
este modo se establece una interacción de carácter dativo en la que una
molécula cede densidad electrónica (electrones) a otra, que presenta
orbitales vacíos.

10
Así este nuevo concepto de enlace covalente debe ser tenido en cuenta en la
química organometálica pues gran parte de sus compuestos se forman
como consecuencia de esta interacción.
Así podemos encontrar cesiones electrónicas desde un orbital de tipo a
un orbital metálico de simetría comparable
Aunque también podemos encontrar cesiones de densidad electrónica desde
un orbital tipo del ligando al metálico que corresponda.
Estos dos ejemplos sirven para representar un bloque bastante extenso ya
que, piénsese, basta con que la simetría de ambos orbitales sea comparable,
así como su energía, para que podamos encontrarnos un enlace dativo.
2. 4 EL ENLACE IÓNICO
Es bien conocido por todos que el enlace iónico puro no existe, y que se
debe más bien hablar del tanto por ciento de ionicidad que posee un enlace.
Esta componente electrostática por tanto estará presente en cualquier enlace
donde la electronegatividad no sea exactamente la misma, y los compuestos
organometálicos no van a ser por ello una excepción. Los enlaces donde
esta componente alcanza valores de importancia (en torno al 50%) son

11
aquellos en los que participa algún metal alcalinotérreo o de transición con
alta electropositividad (Ti).
3 TIPOS DE COMPUESTOS ORGANOMETÁLICOS
3. 1 SEGÚN EL ENLACE
Intentar clasificar los compuestos organometálicos de una manera eficiente
y sencilla atendiendo al enlace es del todo imposible. Sin embargo se
tratará de hacer una clasificación guía, orientadora, tal que sirva para
generalizar y resaltar aspectos generales (que es lo que aquí nos interesa).
Los metales alcalinos y alcalinotérreos presentan enlaces con fuerte
carácter iónico, lo cual resulta su principal característica. Dentro de los
mismos podemos encontrar que algunos de ellos tienen tendencia a formar
“clusters” y por tanto estructuras con varios átomos metálicos enlazados
entre sí.
Los metales de transición presentan normalmente enlaces dativos donde la
donación puede ser tanto de tipo como de tipo .
Para los metales de postransición el enlace es predominantemente de tipo
dativo (en la forma ).
Podríamos indicar también que tanto el Al como el B (boranos) presentan
tendencia a formar enlaces consigo mismos, generando enlaces
multicéntricos (clusters).

12
3. 2 SEGÚN EL LIGANDO
3. 2. 1 LIGANDO ALQUÍLICO
3. 2. 1. 1 SÍNTESIS
La síntesis de los metal–alquilos se puede llevar a cabo de muchas
maneras, pero en el intento de resumir al máximo y de lograr una
exposición concisa y asimilable, marcaremos solamente la más importante.
En este caso las alquilaciones se producen por la reacción directa entre el
metal y el haluro de alquilo correspondiente. La reacción suele llevarse a
cabo usando como medio el éter.
RX (l) + 2M (s) RM (l) + MX (s)
éter
Dentro de este tipo de reacciones se habrá reconocido la síntesis de los
compuestos de Grignard, que se trató en la parte anterior y que por su
importancia, serán analizados aquí posteriormente puesto que sirven para
alquilar metales con gran efectividad y son, por tanto, compuestos
fundamentales a la hora de formar organometálicos.

13
3. 2. 1. 2 APLICACIONES
Los compuestos organometálicos con radicales alquílicos se usan
principalmente como agentes alquilantes de gran capacidad en reacciones
de síntesis orgánica y organometálica (puesto que pueden servir para
alquilar a otros metales o a otros compuestos organometálicos).
Al margen de esto muchos poseen aplicaciones particulares, destacando
entre ellos los derivados alquílicos de Al que se usan como catalizadores en
la síntesis de polímeros de olefinas. También el tetraetilplomo, usado hace
algunos años como antidetonante en las gasolinas.
3. 2. 2 LIGANDO OLEFÍNICO
3. 2. 2. 1 SÍNTESIS
Normalmente se producen compuestos organometálicos con ligandos
olefínicos mediante la sustitución de ligandos en compuestos
organometálicos o complejos de coordinación. Estos ligandos a ser
sustituidos deben ser ligandos que salgan fácilmente y permitan por tanto
que la olefina ocupe su puesto.
Un ejemplo se puede encontrar en la formación de la sal de Zeise:
K2(PtCl4) (solv.)+ C2H4(l) K(C2H4PtCl3)·H2O(solv.) + KCl(s)

14
Al margen de este método se pueden encontrar otros muchos donde el
enlace de la olefina al metal se puede lograr sin que necesariamente tenga
ésta que sustituir a ningún otro ligando en el transcurso de la reacción, sino
por aumento del número de coordinación (número de átomos directamente
unidos al átomo metálico considerado como central).
3. 2. 2. 2. APLICACIONES
La gran mayoría de los compuestos organometálicos con ligandos
olefínicos tienen utilidad en tanto en cuanto son intermediarios en la
síntesis de compuestos orgánicos, donde un compuesto organometálico o
de coordinación es capaz de aceptar como ligando a una olefina,
cambiando entonces las características electrónicas de la misma (dado que
cede densidad electrónica al metal) y haciéndola atacable por otros
reactivos que, en un principio hubiesen resultado completamente
insuficientes.
Igualmente podemos destacar la gran importancia de estos compuestos en
la síntesis de polímeros cuyo monómero provenga de una olefina.
4 OTROS COMPUESTOS CARACTERÍSTICOS
Un poco al margen de la clasificación anterior podemos encontrar algunas
familias de compuestos organometálicos que han resultado y resultan ser de
una inestimable importancia a la hora de conocer ya no sólo la naturaleza
de los compuestos organometálicos sino también el enlace en general, así

15
como la síntesis de otros compuestos más complicados e inaccesibles. En
esta sección trataremos muy brevemente de presentar algunas de estas
series de compuestos y de resaltar algunas de sus características más
importantes.
4. 1 LOS COMPUESTOS DE GRIGNARD
Los compuestos de Grignard se vieron en la unidad 32 de la primera parte.
De este modo debe resultar bien conocida su síntesis:
Mg (s) + RX (l) “Mg(R) X” (solv.)
En ella el Mg sólido se pone en contacto con el haluro de alquilo en medio
etéreo tras tratarlo con I2 (s), que actúa de catalizador. El compuesto que se
forma Mg(R)X es el Grignard, que al estar en disolución sufre una serie de
reacciones en equilibrio consigo mismo denominados equilibrios de
Schlenk (de ahí que entrecomillemos la molécula). Muchos de estos
procesos, así como su formación, mecanismos de reacción y aquellas
reacciones en las que intervienen son aún materia de investigación.
Este tipo de reactivos resultó crucial para disparar la investigación
organometálica. Se usan principalmente para alquilar y arilar compuestos,
formándose así nuevos compuestos organometálicos o modificaciones de
los anteriores.

16
4. 2 LOS CARBONILOS METÁLICOS
Este grupo de compuestos, de una tremenda importancia histórica en el
campo de la naturaleza del enlace no es propiamente hablando un grupo
perteneciente a la química organometálica, puesto que su carbono no es
orgánico. Sin embargo resulta imposible separarlo de ella, de tal modo que
le dedicamos aquí un obligado apartado.
Los carbonilos metálicos están formados por un metal al que, por enlace de
carácter donador, se unen moléculas de CO. Este CO cede densidad
electrónica al metal (cesión tipo ) por un orbital molecular que se
encuentra centrado principalmente sobre el C.
El fenómeno más característico de su enlace es que, el metal, al recibir la
densidad electrónica del CO, reacciona devolviendo parte de la misma a
través de otros orbitales en lo que se denomina retrodonación (de tipo ).
Esta retrodonación suele encontrarse en los compuestos con ligandos
olefínicos (lo cual fue obviado en su momento por motivos de
simplificación). Este enlace de dos componentes sirve para discriminar
todo un gran grupo de compuestos organometálicos denominados “no
clásicos”, precisamente por esta doble componente.
La síntesis de los carbonilos metálicos se realiza normalmente por
exposición directa sobre el metal del CO (s). El metal normalmente se halla
muy finamente dividido y la reacción se fuerza aumentando la temperatura.
Suelen aparecen como intermedios en reacciones de sustitución de
ligandos. Muchos de los compuestos pertenecientes a esta categoría tienen
propiedades que les hacen intervenir como catalizadores en reacciones
orgánicas importantes.

17
4. 3 LAS FOSFINAS
Un ligando muy parecido al ligando carbonilo es la fosfina (y derivados).
Sus características son las de una base blanda de Lewis y por tanto
estabilizan mejor a metales de bajo estado de oxidación.
Todas las fosfinas se pueden considerar ligandos formalmente derivados de
PH3. Sustituyendo los hidrógenos por radicales alquílicos o arílicos,
podemos tener una vasta lista de ligandos a nuestra disposición.
Las fosfinas son ligandos que, como se ha dicho, se parecen al CO, es
decir, tienen capacidad donadora () y capacidad de aceptar retrodonación
(). Pero presentan la ventaja con respecto al CO que, en este caso
podemos ajustar su basicidad en función de los radicales que empleemos.
Si los radicales tienen cierta avidez electrónica y retiran densidad
electrónica del P (sea por su alta electronegatividad o por cualquier otro
factor), la fosfina será menos básica, pero de modo contrario ocurrirá si los
radicales presentan un marcado carácter donador. Estas características
interfieren no sólo en la capacidad de donación de la fosfina, sino también
en su capacidad de acepción .
Otro parámetro que podemos manejar a conveniencia es el impedimento
estérico. Los factores estéricos, junto con los electrónicos, condicionan las
reacciones y ambos deben ser analizados y tenidos en cuenta. Ahora,
puesto que las fosfinas pueden tener cualquier tipo de radical, se puede
seleccionar aquella que estéricamente nos ofrezca más ventajas.
En definitiva, puede comprobarse cómo los ligandos fosfina resultan de una
enorme importancia a la hora de sintetizar compuestos que sean
particularmente inestables, puesto que se puede encontrar una fosfina

18
adecuada para cada complejo organometálico. La dificultad la mayoría de
las veces no es otra que la de sintetizar la propia fosfina, lo cual suele ser
caro, tedioso y largo.
4. 4 LOS COMPUESTOS TIPO SANDWICH
El primer compuesto organometálico tipo sandwich fue el ferroceno,
sintetizado en 1951. En este compuesto encontramos cómo el Fe se
encuentra encerrado en dos anillos de ciclopentadieno, a modo de
bocadillo.
Véase en la figura la aparentemente extraña forma en la que los
ciclopentadienos se enlazan al hierro, quedando el conjunto con esta
estructura tan llamativa e ilustrativa del nuevo concepto de enlace que se
empezaba a gestar. El enlace de donación desde los ciclopentadienos al
hierro es análogo a los detallados con anterioridad, y por supuesto un hecho
la existencia de la retrodonación. Este grupo de compuestos, forma una
serie muy interesante e importante de compuestos organometálicos. La
síntesis de los mismos se realiza mediante la reacción con ciclopentadieno.
El ciclopentadieno se hace reaccionar con sodio para formar el anión
reactivo:

19
2C5H6 (l) + 2Na (s) 2Na+ + 2C5H5
- + H2 (g)
Es este anión el que reacciona con el hierro en el seno de un disolvente
orgánico.
2 C5H5Na + FeCl2 Cp2Fe + 2 NaCl
La abreviatura Cp es usualmente empleada para representar al
ciclopentadienilo.
El uso de los compuestos organometálicos tipo sandwich no es diferente al
de los anteriores. Son usados como catalizadores en procesos de síntesis
orgánica donde participan en multitud de procesos.

20
PRÁCTICA CON COMPUESTOS
ORGANOMETÁLICOS
1 LOS COMPUESTOS ORGANOMETÁLICOS
Para comenzar este manual sobre la experimentación con compuestos
organometálicos es necesario definir previamente qué tipo de compuestos
tenemos como objeto de estudio.
Los compuestos organometálicos son definidos como aquellos compuestos
con enlaces directos entre el C y un metal M donde existe cierto grado de
covalencia al mismo tiempo que cierto grado de polarización debido a la
diferencia entre la electronegatividad de ambos átomos.
1. 1 ORGANOMETÁLICOS AL AIRE
Muchos compuestos organometálicos, como veremos más adelante, son
inestables en atmósfera normal. Esto es, reaccionan con algunos de sus
componentes. Así pues, si tenemos intención de trabajar con ellos, de
aislarlos, de almacenarlos y de volver a utilizarlos en otras reacciones es
preciso usar métodos especiales para protegerlos de los componentes de la
atmósfera. Estos métodos fueron desarrollados por Schlenk, y resultan

21
verdaderamente sorprendentes e ingeniosos como iremos descubriendo a
medida que vayamos introduciéndonos en el tema, ingenio que, por otra
parte, no ha menguado con las mejoras que con el tiempo se han realizado.
1. 2 O2 Y H2O. LOS ENEMIGOS
Como se podía prever, estas dos moléculas son las que hay que eliminar
hasta la última traza, ya que destruyen el enlace C-M.
Puede comprenderse la dificultad del asunto, pues el agua se encuentra,
como el dioxígeno, en estado gaseoso en el aire, así hay que idear sistemas
que puedan montarse fácilmente, al aire libre, y después, también sin
dificultad, cambiar el aire por una atmósfera inerte, usualmente de
dinitrógeno; pero por otra parte lograr que, una vez hecho esto, no haya
entrada de aire por ningún sitio y se mantenga el conjunto estable en el
tiempo. No hay que ocultar, puesto que resulta evidente, lo arduo del
problema.
2 LA LÍNEA
2. 1 DESCRIPCIÓN
La línea de vacío es el sistema más importante dentro del laboratorio. Su
buen uso en todo momento asegura y salva aquellos reactivos o productos
que participan en la reacción o son generados a partir de la misma. Si bien
al principio familiarizarse con este conjunto, un tanto aparatoso, de tubos,
ya sean de vidrio o de goma, resulta complicado e incluso difícil de lograr,
se acaba uno acostumbrando, de modo que al final, todo movimiento es
firme y mecánico.

22
Tengamos previamente un esquema de la misma, comentando a
continuación cada una de las características y su función.
Esta compleja estructura de tubos y accesorios juega con la capacidad de
hacer vacío y de introducir N2 (g). Para tal menester se han usado los
siguientes elementos.
1. Es una de las salidas de la línea, que se conecta a un tubo que se fija a su
vez mediante una abrazadera atornillada a la boquilla de vidrio. Este tubo
llega a la bomba de vacío y por lo tanto, a lo largo de todo el sistema
conectado a este tubo, encontraremos el vacío oportuno.

23
2. La llave que se señala suele ser de teflón, tiene la misión de abrir al aire
el interior del sistema. La razón de esto es sencilla. Una vez se termina de
trabajar, el sistema se halla a vacío, pero la trampa de vidrio (5) hay que
quitarla tirando con suavidad (más adelante veremos la razón de esto) y si
tenemos el interior a vacío, es imposible retirar la ampolla.
3. Esta llave es a su vez generalmente de teflón. Su uso es el de conectar el
sistema de succión de la bomba con el interior de la línea. Una vez que se
conecta la bomba, esta llave debe quedar abierta si se quiere que el resto de
la línea se encuentre a vacío. También es útil cuando tras evaporar gran
cantidad de disolvente se colma la trampa (5), corriéndose el riesgo de que
parte del disolvente pase a la bomba. Si se cierra esta llave y también la
número 7 se puede cambiar la ampolla sin afectar a las condiciones del
producto sobre el que se está trabajando (el cual debe esperar bajo
atmósfera de N2 a que la operación pueda ser reanudada). Estos detalles se
comprenderán mejor a medida que se vayan desarrollando los distintos
temas.
4. Este elemento se denomina cabeza de línea y entronca los distintos
cuerpos, como son la ampolla (5) y la rótula (6) del sistema de distribución
de vacío (8).
5. La ampolla de la trampa fría tiene la finalidad de acumular los
disolventes orgánicos que se eliminan al llevar a sequedad una disolución,
junto con todas las materias volátiles que puedan acompañar a los mismos.
Como pasan en estado gaseoso basta un golpe de frío para que queden en
forma sólida y no pasen a la bomba.

24
6. La rótula es una pieza cuya misión es análoga a la propia rótula humana,
de donde le viene el nombre por su semejanza física. Tiene una gran
importancia.
La línea se fija a una celosía de barras con el concurso de pinzas, que son
idénticas a las que se encuentran en los laboratorios de cualquier Facultad.
Lógicamente, esta fijación debe ser muy firme, pues si se nos cayese el
montaje estaríamos en un gran aprieto. Sin embargo, cuando ponemos la
línea es necesario llevar la ampolla de la trampa fría (que generalmente se
suele quitar tras trabajar) contra la cabeza de la línea, con cuidado pero con
fuerza para que ajuste bien. Pues bien, si nos fijamos atentamente se puede
observar lo fino que es el tubito que conecta el cuerpo con las dos llaves a
su izquierda. Si la rótula no estuviera, para dar cierta libertad de
movimiento a la cabeza, muchas veces se partiría ese tubito por la tensión
originada al poner la trampa.
7. Es la llave que, como ya se ha dicho con anterioridad, sirve para separar
el resto del sistema de la cabeza. Así se puede dejar a vacío el interior de la
línea durante un tiempo razonable con independencia de la bomba. Si la
cerramos, el sistema queda a vacío, lógicamente no hay sistema perfecto en
el sellado de los ajustes y poco a poco va entrando aire, pero podemos
dejarle una noche a vacío si fuera menester, ya que no podemos dejar
conectada la bomba sin echar un vistazo al N2 (l) de la trampa. Es una
llave de cristal como el resto de las que se van a comentar.
8. Es una cámara de la que parten las llaves correspondientes a todos los
tubos que posteriormente servirán para conectar los matraces y tubos de
Schlenk, que se utilizarán durante las operaciones que vayan a realizarse en
la línea.

25
9. Es un tapón de vidrio que cierra el sistema herméticamente. Otras líneas
no poseen ahí nada, sino que la cámara se cierra suavemente como si fuese
un ampolla (obsérvese la cámara 10).
10. Nos encontramos ante la cámara destinada a repartir el N2 (g) gracias a
los tubos que la conectan con las llaves de la cámara de vacío.
11. Son dos boquillas de vidrio donde se conectan dos tubos de goma. Uno
para la entrada de N2 (g) y otro para el argón.
12. Esta llave de vidrio tiene una estructura análoga a la de las llaves de
manejo de la línea, o sea, aquellas que se encuentran bajo la cámara de
vacío. Conecta la entrada de gas con el resto del tubo, sin embargo
encontramos dos entradas de gases (11), una para el dinitrógeno que es el
que se usa habitualmente y la otra, como se indicó en el apartado anterior
para el gas noble. La misma llave es una llave doble, es decir que en su
interior posee 2 canales, de modo que, dependiendo de qué gas queramos
introducir su posición será una u otra. Véase el esquema de la llave análoga
en la página siguiente.
13. Una nueva llave que conecta el sistema de distribución del gas con un
conjunto de regulación de presión, debiéndose por tanto mantener abierta
para que no haya problemas de sobrepresión.
14. Los tres elementos señalados constituyen el sistema de seguridad que
logra una presión homogénea en las salidas de los tubos (16). El primer
elemento es una válvula, el segundo un bulbo de vidrio y el tercero un
burbujeador de parafina que sustituye al tradicional de mercurio.

26
Anteriormente existía (y aún hoy coexiste con los de parafina) un
burbujeador de mercurio donde, cuando la presión de N2 (g) era alta, se
lograba hacer borbotear al metal líquido. Dada la peligrosidad de los
vapores de mercurio se opta por ir cambiándolos en favor de los de
parafina. La salida conduce por un tubo el gas sobrante.
15. Ahora tratamos del elemento, si no más importante, sí el más usado y
característico del trabajo con compuestos organometálicos, las llaves de
manejo. Estas permiten que exista una succión a través de los tubos de
goma (14) o una corriente de N2 (g). Van a ser el elemento clave a la hora
de trabajar y su dominio va a resultar indispensable. Tienen dos canales
internos para conectar bien la entrada de gas, bien la succión con los tubos
correspondientes.
Con los siguientes dibujos se puede entender el funcionamiento de las
mismas.
Se podrá apreciar fácilmente ambos canales y su oportuna alineación con la
salida al tubo.
En la posición 1 la llave conecta la salida (S)
con la cámara de vacío, con lo que el matraz o el
Schlenk que se encuentre conectado al tubo se
vacía de aire, el cual es succionado por la bomba.

27
En la posición 2 ambos canales se apartan,
quedando perpendiculares a toda abertura y por
tanto, en esta posición no existe ni succión ni
entrada de gas posible.
En la posición 3 obsérvese cómo el giro de la llave
ahora conecta la otra entrada (N) con la salida (S),
realizándose el aporte de N2 (g) correspondiente.
Es un hecho importante el que no debamos confundir la posición de la
llave, por propio interés ya que un error podría destruir cualquier
compuesto o experimento. Para ello se pueden hacer dos cosas, bien
cerciorarse cada vez que se va a usar la llave, mediante inspección directa
del lateral de la misma, determinando qué movimiento conecta el canal que
interesa con la salida (S) o, lo que es más usual, debido al continuo uso de
estas llaves, pintar uno de los extremos de la llave para recordar una de las
posiciones. En mis dibujos la posición hacia arriba del extremo pintado
indica salida de dinitrógeno y por tanto la posición hacia abajo hace vacío.
16. Los tubos de salida sirven para conectar las llaves de manejo con los
distintos elementos que vayan a usarse, tales como matraces o tubos
Schlenk. Son de goma gruesa y así de un diámetro mucho mayor que el de
los otros tubos que se observan en la línea, los cuales son de goma más fina
y flexible.

28
2. 2 LIMPIEZA DE LA LÍNEA
Imaginemos que llegamos al laboratorio el primer día y nos encontramos
una línea instalada, pero que hace mucho tiempo que no se usa. Se corre el
riesgo de que la grasa de los ajustes no esté en perfectas condiciones, así
como que las gomas se hayan deformado, perdiendo elasticidad con el
tiempo y picándose. Lo mismo con las gomillas elásticas que sirven para
ajustar las llaves en cada uno de los correspondientes asientos. Entonces
hay que cambiarlo todo, eliminar la grasa vieja y dispensar nueva.
Al igual se concibe cuando la línea lleva ya mucho tiempo funcionando,
sometida a un trabajo diario que la ensucia y la desgasta. Si se trabaja al día
unas 9 horas, la línea debe ser limpiada de nuevo en torno a los 3 o 4 meses
aproximadamente. Aunque claro, todo ello dependerá del tipo de material
con el que trabajemos y de lo cuidadosos que seamos con nuestra
herramienta de investigación.
2. 2. 1 QUITAMOS LAS GOMAS
Para empezar a limpiar la línea y quedarnos con su parte de vidrio
exclusivamente debemos quitar las gomas (tanto gruesas como finas) y así
poder cambiar aquellas que se puedan encontrar dañadas o en estado
dudoso. Las gomas gruesas (16) no suelen cambiarse, dado que son muy
resistentes y aguantan mucho tiempo por lo que su vida útil es mucho más
prolongada que la de las gomas más finas. Estas últimas se van
resquebrajando y van perdiendo elasticidad por lo que hay que sustituirlas
cada cierto tiempo (que en cualquier caso siempre es mucho más corto que
el de las anteriores). Estos tiempos son muy relativos y el cambio se hace
cuando se observa alguna anomalía, sea en el aspecto de la goma, sea en el
funcionamiento.

29
Las gomas gruesas se quitan con cuidado mediante rotación de la goma
sobre la boquilla de vidrio que se quiere librar. Las gomas finas suelen
estar adheridas, pues su mayor flexibilidad y adaptabilidad las hace
amoldarse firmemente sobre el vidrio. Lo más apropiado pues, es cortar la
goma con una cuchilla muy afilada en la dirección en la que sale el tubito
de vidrio al cual rodea la goma. Así se van eliminando todas y cada una de
las distintas gomas finas.
2.2.2 LIMPIAMOS LA CÁMARA DE VACÍO
Esta cámara con el tiempo, y debido a la grasa que existe en las llaves de
manejo, se va ensuciando poco a poco. Muchas veces, al hacer vacío,
arrastramos un poco de grasa de ajuste que está, por accidente, en el canal,
y que se pega en las paredes de vidrio del tubo. Tras muchos experimentos
la grasa va absorbiendo compuestos y se colorea, mostrando su presencia
con toda nitidez y haciendo precisa la limpieza correspondiente.
Para eliminarla se deben quitar primero las gomas que están unidas a la
misma. También hay que quitar, para limpiarlas individualmente, las llaves
de manejo, la llave número 7 y el tapón.
Llegado el momento se coge el tubo y se agarra con dos pinzas normales en
un pie de columna, colocándolo en posición vertical en la campana (es
mejor lavarlo ahí ya que usaremos disolventes orgánicos). Tras situar un
contenedor adecuado donde se recojan los líquidos usados para la limpieza
se van dispensando éstos por turnos, de manera que cuelen desde la parte
superior del tubo hasta la inferior, cayendo por gravedad. Se utiliza primero
el diclorometano, que disuelve y arrastra bien este tipo de sustancias grasas,
también se prueba con algo de acetato de etilo que acaba por arrancar todo
lo remanente. Al final se lava con acetona y éter, en este orden (por

30
razones que ya se indicarán en otros apartados), y ya se tendrá en perfectas
condiciones para el trabajo.
2. 2. 3 LIMPIAMOS LAS LLAVES DE VIDRIO
Antes de quitar la cámara de vacío hay que extraer las llaves de vidrio de
los huecos en donde se encuentran. Estas llaves no son intercambiables,
cada llave está hecha para ajustar perfectamente en un hueco, de ahí que
sea de una extrema importancia el que no coloquemos una llave en el
agujero de la otra. Por tanto, antes de sacarlas y proceder a su limpieza se
enumeran con un rotulador especial para vidrio.
Una vez numeradas se procede al lavado de las mismas. Con papel, acetona
y éter se limpian, observando este orden escrupulosamente. Una vez
lavadas con el éter vemos que la superficie de ajuste tiene esa apariencia
esmerilada de un cristal pulido, pero que no es brillante. Cuidémonos de no
eliminar el número de la llave, escrito con rotulador, pues la acetona lo
disuelve rápidamente.
Para concluir hay que limpiar los canalitos de las llaves. Esto se hace con
un limpiapipas. Este se pasa hasta que no deja rastro de grasa. Se le aplica
finalmente un chorro de éter, arrastrando las pelusas que puedan haber
quedado del limpiapipas.

31
2. 2. 4 LIMPIAPIPAS
Brevemente comentaremos qué son, aunque debe intuirse sin mucho
problema. Son instrumentos de limpieza constituidos por un alambre de
longitud relativamente corta (15 cm) que está forrado de un material en
forma de pelusa que sirve para arrastrar y eliminar la suciedad. Pueden
doblarse formando arcos o codos, para facilitar el trabajo de limpieza,
debido a lo finos que son. Podemos decir que son análogos a los
limpiatubos que se hallan en las piletas de los laboratorios de las
Facultades, sólo que más pequeños y finos.
2. 2. 5 LIMPIEZA DE LA TRAMPA DE VACÍO
Lo que se pasa a describir forma parte del trabajo diario de la puesta en
funcionamiento de la línea. Se realiza siempre que se termina la sesión de
trabajo o justo cuando se va a comenzar (la grasa y los disolventes a
eliminar serían del día anterior). Es preferible, en mi opinión, limpiarla
justo cuando se deja la investigación por la tarde para ser reanudada al día
siguiente y así encontrarla en perfecto estado.
Se debe eliminar la grasa del macho de la cabeza de la línea con un poco de
acetona y éter (en realidad se suele limpiar únicamente con papel, no
siendo necesario el uso de disolventes). También se quita la grasa a la boca
de la ampolla (la trampa) y se limpia bien su interior con acetona y éter.
Una de las razones por las que se deja muchas veces la trampa sin limpiar,
apoyada en alguna de las piletas de lavado, hasta el día siguiente, es porque
al retirarla de la línea, los disolventes capturados en su interior están
solidificados (a causa del foco frío que se le aplica) y se gasta mucha más

32
cantidad de disolvente para fundirlo y eliminarlo. Si los dejamos entonces
en la pileta, hasta el día siguiente, nos ahorramos disolvente y tiempo.
2. 2. 6 LIMPIEZA DEL MERCURIO
Llegados a este punto tenemos la línea desmontada y todos los elementos
limpios. Obviamente aquellos correspondientes a la conducción del N2 (g)
no requieren un especial tratamiento y si no se
limpian (al margen de las llaves) tampoco pasa nada. Sin embargo tenemos
los burbujeadores que, poco a poco, se van contaminando de distintas
impurezas. Si el burbujeador es de parafina, ésta se retira y se pone nueva.
Pero si es de mercurio hay que reutilizarlo.
Bien para amalgamas, bien para su uso en burbujeadores, el mercurio debe
estar más o menos puro, dependiendo del uso que pretendamos darle.
Cualquier trabajo que se realice con este metal debe de ser en la campana
dado su carácter nocivo. No obstante, el mercurio es una sustancia
espectacular bajo muchos aspectos y siempre es interesante una operación
con tal metal.
El mercurio suele encontrarse en el burbujeador cubierto con una cantidad
de parafina. La parafina es lo suficientemente densa como para impedir que
el mercurio borbotee, haciéndolo ella misma e impidiendo que sean los
vapores de mercurio los que salgan a la atmósfera.
La parafina se elimina por decantación pues sobrenada al mercurio. La
primera gran sorpresa que nos llevamos ocurre al coger el recipiente donde
se contiene el metal y levantarlo.
Todo el mundo sabe que el mercurio es muy denso, pero hasta que no lo
comprueba físicamente, realmente no se percata de ello. Es curioso cómo la
propia conciencia llega a negar la evidencia, pues parece imposible que un

33
recipiente de unos 250 ml de mercurio pueda dar la sensación de pesar
tanto, por muy denso que sepamos que sea. Repito, no es un problema
conceptual, sino de experiencia sensorial directa.
Cuando tenemos el mercurio sin parafina pasamos a lavarlo. Esto se hace
añadiendo un poco de éter y agitándolo ligeramente. Hecho esto, el éter se
elimina por decantación. Se lava varias veces hasta que el mercurio esté lo
suficientemente limpio. Algunas veces, cuando está muy sucio, es preciso
lavarlo con otros disolventes como metanol, acetona, agua,... aunque en
cualquier caso el proceso es el mismo.
Hecho esto hay que secar el mercurio, lo cual se hace con papel de filtro.
Así se introduce papel de filtro en el mercurio con cuidado de no
derramarlo. Los disolventes pasan al filtro en tanto que el mercurio debido
a las fuerzas de cohesión permanece en el recipiente. Se repite las veces
que se considere oportuno. Repetimos, se trata de secarlo y para ello lo que
se hace es mojar en él tiras de papel de filtro. Aquí tenemos otra sorpresa
visual, pues la vista espera que al introducir el filtro en el mercurio aquel
impregne el papel y acabe manchándolo o humedeciéndolo al menos, y
aunque la razón ponga las cosas en su sitio, es curioso ver cómo sale el
papel impoluto del interior del líquido.
Una vez seco hay dos opciones: destilar el mercurio o filtrarlo en embudo
alemán.
Se destilaría si fuese preciso usarlo muy puro (amalgamas o reactivo), pero
como no suele ser así, el proceso habitual es el del filtrado. Por este
procedimiento se elimina todo residuo que permanezca en el metal.
Tomamos pues un embudo alemán y hacemos un filtro en forma de cono,
con el tamaño adecuado. El mercurio es tan denso que no pasa a través del
filtro, por ello se practican, antes de poner el metal, unos agujeritos en el
filtro con una aguja.

34
El mercurio obtenido está totalmente limpio y se guarda en recipientes
adecuados, perfectamente herméticos, o se pone en el burbujeador de
nuevo
2. 2. 7 ACETONA Y ÉTER PARA LAVAR
A lo largo de todos los procesos anteriores hemos recurrido a la acetona y
al éter para lavar los distintos elementos. La acetona es un disolvente
suficientemente bueno como para ser usado a gran escala: es barato, fácil
de destilar, disuelve gran cantidad de sustancias orgánicas,... siendo su
única desventaja, la de tardar en evaporarse un poco más de lo que sería
deseable.
Precisamente para eliminar este último defecto se usa el éter, que al margen
de poder ser usado como disolvente se usa para secar el vidrio. Su punto de
ebullición es mucho más bajo que el de la acetona y esto se aprovecha para
secar más rápidamente. Por ello se lava siempre primero con acetona y
luego con éter.
Cuando se lava es conveniente hacerlo repetidas veces con pequeños
volúmenes, pues la inversa no es muy apropiada para el adecuado
transporte de materia que debe producirse.
El éter y la acetona se encuentran en el laboratorio en dos dispensadores
(absolutamente análogos a los que contienen el agua destilada en las
Facultades). La diferencia radica en que en un laboratorio de
organometálica es el agua destilada la que requiere un dispensador con una
etiqueta que la identifique claramente, en tanto que en la Facultad hubiese
sido al contrario. En cualquier laboratorio de esta naturaleza se usan ambos
disolventes orgánicos en cantidades enormes y, démonos cuenta, sustituyen
al agua en casi todas las tareas de limpieza (no en todas como ya

35
mostraremos más adelante). Es tan alto el uso que se hace de estos
dispensadores que en lugar de una etiqueta, que requiere leerla y desde
lejos no se aprecia si es el éter o la acetona, se usan dispensadores con
tapones de color. De este modo un tapón rojo podría corresponder a la
acetona, en tanto que otro azul serviría para identificar el éter.
Estos dos botes se gastan rápidamente y han de ser llenados conforme se
agotan. Para ello suele haber dos bidones grandes, con un grifo en su parte
inferior, uno de éter y otro de acetona; los cuales también se agotan, pero es
labor de otra persona el llenarlos, y no del investigador.
2. 3 MONTAJE DE LA LÍNEA
Para proceder al montaje de la línea se deben también tener una serie de
cuidados específicos que hacen que su funcionamiento final sea óptimo.
Sobre todo con respecto a los ajustes.
2. 3. 1 UNIÓN DE LA RÓTULA
Como ya se ha dicho, la rótula une el tubo de vacío con la cabeza de la
línea. Todas las piezas de ajuste deben estar perfectamente engrasadas con
grasa de alto vacío antes de ser instaladas. La grasa se coloca en la rótula,
sin excesos, y se adapta con

36
una serie de movimiento circulares hasta que toda se ha repartido
uniformemente por la superficie de contacto.
Una vez afirmadas las dos piezas por pinzas a la celosía del fondo de la
campana, colocamos una pinza de cocodrilo sobre las dos partes de la
articulación antes lograda, de modo que queden unidos sin tensiones, pero
de manera adecuada para que no haya entrada del aire exterior.
En el dibujo se ha intentado representar el ensamblaje de las dos piezas. A
la derecha se aprecia la “bola” de la rótula, que es donde se aplica la grasa
y, a la izquierda, el otro miembro de la articulación, correspondiente a la
cabeza de la línea.
La pinza logra la presión adecuada para que esté lo suficientemente sellada,
pero no muy rígida. Se han dibujado sólo los dos brazos superiores de la
pinza para favorecer la visión.

37
2. 3. 2 COLOCAMOS LOS TUBOS
Y he aquí una de las operaciones más complejas para el novato sin
experiencia en el ejercicio. Introducir los tubos de goma gruesos (16) por
las boquillas de la cámara de vacío es sencillo y no requiere ningún consejo
adicional. Sin embargo la colocación de los tubos más finos, en algunos
sitios, se presenta como una buena prueba. Los tubos que parten de la
cámara (10) se instalan sin problemas, al igual que el que llega y el que sale
del borboteador. Pero aquel que une la llave (13) con la válvula (14) y
aquel que va de la válvula al bulbo, constituyen un auténtico reto de
habilidad (a causa de las formas de las boquillas).
Los tubos deben ser lo más cortos posibles, para que no se doblen ni tengan
estrangulamientos y deben quedar estancos. Fijar las bocas de las gomas a
las bocas de la válvula y al ensanchamiento en que termina el tubo de
vidrio del N2 (g) es lo más difícil, pues la boca elástica de la goma, aun
siendo elástica, no se desliza fácilmente, al tener cierto carácter adhesivo.
Para evitar este problema hay dos soluciones:
A) Ponemos en el interior de la boca del tubo de goma un poco de grasa.
¡Sólo un poco!
B) Con una gotita de agua humedecemos el interior de la boca del
tubo y se trata de colocar con rapidez, para aprovechar el efecto deslizante
del agua.
Ambos métodos y una mano hábil bastan para el logro. Pero todavía
tenemos que afirmar las uniones con alambre fino, lo cual es también
complicado pues hay que hacerlo sin que se salga la goma.

38
2. 3. 3 ENROSCAMOS LOS ALAMBRES
Generalmente en el laboratorio hay dos tipos de alambre, uno grueso y otro
fino, bastante diferenciados. Aquí, en estas uniones se usa primeramente el
alambre fino.
Son puntos en los que, debido a la forma de las boquillas, a la situación del
tubo de goma y a la conducción de gas a presión, es posible que se suelten
los tubos de goma del sistema si no se amarran con alambre. Por ello se
dirá que los alambres se colocan siempre que haya una tensión clara que
desestabilice la unión de la goma y el aplique, en el resto de casos no es
necesario.
En los siguientes dibujos se pretende dar una visión de cómo se disponen
los alambres, aunque la lógica lo hace evidente.
Como puede apreciarse en la posición 1,
hacemos que el alambre rodee lo que
queremos abrazar. Esto se hace con la
mano, sin usar todavía los alicates.
En 2 forzamos a que el lazo se cierre en torno a
la mitad de la boquilla, cruzando los alambres
tal y como indica la figura y dando algunas
vueltas con los dedos, para que quede estable y
pueda aplicarse el alicate sin temor a que pueda
salirse la goma.

39
Una vez hecho podemos usar el alicate y
vamos retorciendo con mucho cuidado
hasta lograr una presión eficaz por parte
del alambre en torno a la boquilla. Cuando
consideremos que es suficiente cortamos
las dos hebras de acero con cuidado con el
mismo instrumento.
Tampoco es conveniente apretar mucho la goma dado que pudiera llegar a
cortarla el alambre y se invalidaría por tanto todo lo hecho por culpa de una
tensión excesiva.
Esto, sobre el papel, parece tremendamente sencillo, pero en la práctica es
de una gran dificultad.
Una vez afirmado con el alambre fino se procede de la misma manera con
el de mayor grosor, contemplando los mismos consejos anteriores.
2. 3. 4 ABRAZADERAS
A veces es necesario fijar las gomas gruesas (16) a las correspondientes
boquillas de vidrio. Para tal finalidad se usan las abrazaderas. Un ejemplo
se puede apreciar en el dibujo general de la línea donde el tubo 1 se aprieta
contra la boquilla de vidrio que da comienzo a la línea.
Se cierran o abren enroscando o desenroscando un tornillo que poseen. No
son otra cosa que las abrazaderas que se encuentran en cualquier ferretería
y que el mismo lector puede tener en su casa en la misma goma de riego,
por lo que no daré más explicaciones.

40
2. 3. 5 SOBRE LAS GOMILLAS
Las gomillas elásticas son muy usadas, y son iguales a las que se usan en
las cajas de zapatos para sostener la tapadera. Sirven para afirmar la llave
de vidrio en su asiento y también para mejorar el contacto entre los ajustes.
El modo en el que se colocan es idéntico al de las llaves de los tubos de
Schlenk y matraces, por lo que se detallará más adelante.
2. 3. 6 GRASA DE ALTO VACÍO
Se diferencia de la grasa normal en que es mucho más densa, lo cual se
nota perfectamente al contacto con los dedos. Posee un color grisáceo
translúcido, más oscuro por tanto que la grasa habitual, que es blanca. Se
presenta generalmente en tubos, como si fuese una pomada.
Esta grasa logra lubricar y sellar las llaves de la línea y de todo elemento de
ajuste. Se aplica igual que la otra, lo que se verá en la sección de llaves de
matraces y de Schlenk.
Con esta grasa se logran vacíos aceptables sin peligro de entrada de aire.
Aunque en la línea no es muy importante, posee una alta estabilidad
térmica, manteniendo sus características entre –40º y 200º C.
2. 3. 7 LA RIGIDEZ DE LA ENCIMERA DE TRABAJO
Puede parecer algo poco importante, e incluso fuera de lugar aquí, en el
bloque correspondiente a la línea. Sin embargo, el material del que está
hecha la superficie de la encimera de la campana debe ser considerado

41
ahora, ya que podemos ahorrarnos bastante dinero con unas indicaciones al
respecto. Sobre todo antes de comenzar a desmontar la línea.
El material que se usa es caro, por lo que deben extremarse los cuidados
para que no se parta. Curiosamente la superficie de algunos bancos de
trabajo y de las campanas puede estar hecha con material muy rígido
(cerámico), que quiebra rápidamente cualquier cosa que cae aunque sea
desde muy poca altura, o aquello que se golpea, por muy leve y ligero que
sea el impacto. La razón es bien sencilla, al ser rígido comunica
rápidamente las vibraciones que ocasionan los al otro objeto (mucho más
frágil) que se rompe inmediatamente.
De todas maneras sea cual fuere el material de la superficie de la mesa es
interesante desplegar un plástico de burbujas (esos que encontramos
envolviendo algunos electrodomésticos), tal que sirva de protección.
Encima del mismo colocaremos un papel de filtro, que por otra parte es
obligatorio, por las normas de limpieza más elemental.
Considero estas prevenciones como algo necesario y como una costumbre
muy rentable.
2. 4 PUESTA EN MARCHA DE LA LÍNEA
2. 4. 1 ANTES DE TOCAR LA LÍNEA
Cuando se llega al laboratorio el primero, lo cual es un sano hábito de
trabajo, se deben poner a reflujo los disolventes para que cuando vayan a
ser usados los encontremos a punto (más adelante comprenderemos en qué

42
consiste esto exactamente y su razón), e inmediatamente después tenemos
que activar la línea de trabajo.
2. 4. 2 COLOCAMOS LA AMPOLLA
La trampa (5) suele dejarse aparte una vez que se desconecta la línea y se
deja de trabajar. Cuando se quita, como ha estado en N2 (l), los disolventes
que se han evaporado usando la bomba, se habrán acumulado en el fondo
de la ampolla, quedando un residuo sólido. Así muchas personas la dejan
en algún sitio seguro para que se fundan y al día siguiente se procede a
tirarlos y a limpiar la ampolla con un poco de acetona y éter.
Hecho esto se toma un poco de papel y se limpia la grasa que queda en el
macho de la cabeza de la línea. Después se esparce grasa nueva sobre el
mismo de manera que nos aseguremos un sellado perfecto. Aquí algunos
ponen la grasa en cantidades normales, como si fuese una llave (al colocar
la ampolla no debe, por tanto, formarse un borde de grasa sobrante sobre la
circunferencia de contacto), pero parece ser lo más conveniente y por tal lo
más extendido es dispensarla en buena cantidad, de modo que se forme una
superficie de grasa protectora en torno al ajuste.
A continuación se toma la gomilla y se pone doble, teniéndola preparada,
para asegurar la trampa en la cabeza.
Todo el mundo sabe girar una gomilla para que pueda usarse doble,
aumentando así su capacidad para afirmar por compresión y tensión del
mismo modo que si fuese sencilla. Es por ello por lo que no acompaño el
proceso con los dibujos correspondientes. Aún así y por el carácter
sistemático y exhaustivo de la presente obra paso a detallar el proceso de
palabra.

43
Basta coger la gomilla manteniendo fijo uno de sus extremos al
mismo tiempo que se gira el otro creándose una figura con la goma
semejante a la del número ocho.
Tras este sencillo movimiento llevamos un extremo sobre el otro lo
que acaba plegando ambos lazos el uno sobre el otro, generando una nueva
gomilla doble.
Esta nueva gomilla es mucho más resistente que la anterior y por
tanto más útil a la hora de afirmar la ampolla sobre la cabeza de la línea.
Es interesante notar que muchas veces es conveniente forzar elásticamente
la gomilla mediante pequeños tirones, para que después no nos resulte
imposible colocarla donde queremos. Una gomilla doble nueva es tan
resistente que al intentar colocarla podemos partir en un esfuerzo
desmedido los finos tubos de la línea. Forzarla ligeramente para que pierda
parte de su resistencia es muy conveniente.
Antes de colocar la gomilla se coger la ampolla y se lleva cuidadosamente
contra la cabeza de la línea. Se comprime entonces como si se fuese a
atornillar. Con este lento movimiento la grasa se extiende por toda la
superficie e impide que entre aire.
Una vez hecho esto, y sin soltar la ampolla, se coloca la gomilla doble.
Mucha atención con la ampolla, a veces parece que queda estanca con la
presión a la que la hemos sometido y que si la soltamos para coger la
gomilla no caerá por gravedad. No pocas ampollas se han roto de este
modo. También se pueden poner dos gomillas simples como alternativa.
Además de lo anterior jamás se debe soltar la ampolla para poner la gomilla
con las dos manos, ya que como hemos dicho, aunque parezca haberse
quedado sujeta con la grasa, se corre el peligro de que resbale y se parta.
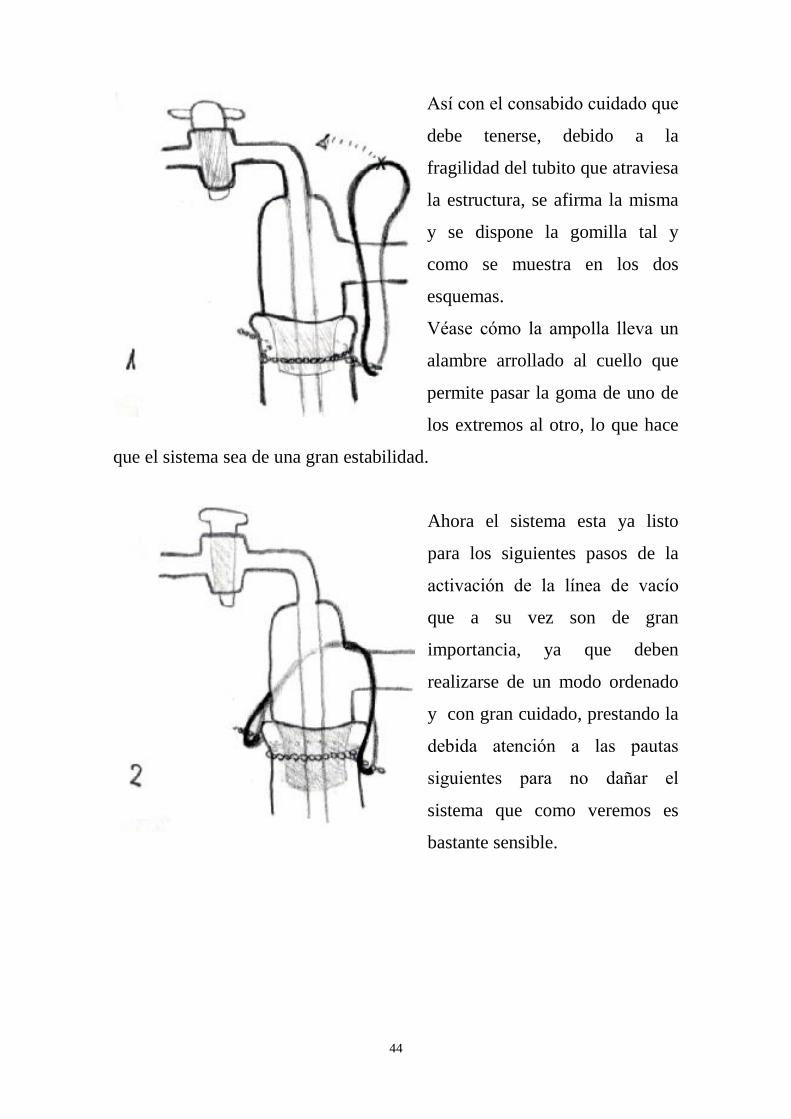
44
Así con el consabido cuidado que
debe tenerse, debido a la
fragilidad del tubito que atraviesa
la estructura, se afirma la misma
y se dispone la gomilla tal y
como se muestra en los dos
esquemas.
Véase cómo la ampolla lleva un
alambre arrollado al cuello que
permite pasar la goma de uno de
los extremos al otro, lo que hace
que el sistema sea de una gran estabilidad.
Ahora el sistema esta ya listo
para los siguientes pasos de la
activación de la línea de vacío
que a su vez son de gran
importancia, ya que deben
realizarse de un modo ordenado
y con gran cuidado, prestando la
debida atención a las pautas
siguientes para no dañar el
sistema que como veremos es
bastante sensible.

45
2. 4. 3 PREVIO A LA CONEXIÓN DE LA BOMBA
Nos cercioraremos de que las llaves 3 y 7 están abiertas y que la llave 2
está cerrada (si está abierta cuando se encienda la bomba oiremos un
sonido estridente que corresponde a la vibración que produce el aire
aspirado al pasar por el tubo).
También precisaremos tener preparado el N2 (l) en el Dewar
correspondiente (es un recipiente adecuado para recibir N2 (l) que impide lo
más posible el intercambio de calor con el medio). El Dewar que sirve para
traer el N2 (l) desde su fuente hasta la campana es de mayor radio y por
tanto de mayor volumen que el que acoge a la ampolla.
Este último Dewar se coloca debajo de la ampolla conteniéndola,
aprovechando un soporte plano que hay debajo de la misma, destinado a tal
necesidad (aún el Dewar debe mantenerse vacío y sin añadirle el N2 (l), que
deberá esperar contenido en el otro recipiente hasta que esta operación de
transvase pueda realizarse).
2. 4. 4 CONECTAMOS LA BOMBA DE VACÍO
Debemos seguir rigurosamente los pasos señalados para no tener
problemas. Así se conecta la bomba de vacío y después se añade el N2 (l),
transvasándolo de un Dewar a otro. Si se hiciese al revés, o sea, añadiendo
primero el nitrógeno líquido, pudiera ocurrir que solidificase el oxígeno,
sólido azul violáceo, que es explosivo.
Al añadir el N2 (l) al Dewar de la trampa el líquido bulle y se evapora hasta
que el recipiente alcanza la temperatura del líquido y queda establecido el
equilibrio. Al entrar en ebullición el nitrógeno líquido salpica hacia fuera y
resulta muy espectacular, como lo es en sí todo experimento en donde

46
interviene. El sonido que hace al evaporarse turbulentamente también es
digno de ser mencionado y cesa cuando lo hace la diferencia de
temperatura. Como consecuencia de esta evaporación súbita, se produce un
gasto de líquido considerable. Para evitar que la propia ebullición, en su
vehemencia, derrame N2 (l) lo que se hace es añadirlo poco a poco, hasta
que cese de bullir y después completar de una vez.
2. 4. 5 MANORREDUCTORES
Existen en el laboratorio dos tipos de manorreductores, los de baja presión
y los normales. Ambos miden en bares. Se usan de modo distinto, ya que la
información que presentan los normales no es de mucha utilidad.
En el de baja presión se mide desde una presión nula hasta 1.6 bares. Para
inyectar N2 (g) se suele poner siempre la presión en torno a 0.3 bares. Así
se mide la presión fácilmente con la aguja.
En los de presión normal, el rango de medición va desde los 0 a los 10
bares, con lo que no sirve de nada la señalización de la agujita. Para ello
colocamos el dedo sobre la salida de algún tubo de la línea (16),
apoyándolo suavemente, y damos paso al gas con la llave de manejo,
abriendo entonces el manómetro lentamente hasta alcanzar el flujo
adecuado (lo que se sabe por la oposición del dedo, aunque esto requiere
cierta experiencia).
2. 4. 6 INYECTAMOS N2 (g)
Cuidando de que las llaves 12 y 13 estén abiertas, se abre el gas con el
manorreductor tal y como se ha indicado antes.

47
El manorreductor puede estar en cualquier sitio del sistema a presión, pero
siempre se localiza en un tubo que conduzca el nitrógeno y que proceda del
contenedor general de gas. Si alguna vez este nitrógeno falta en ese
depósito, suena una alarma indicando la carestía.
Siempre que pongamos el nitrógeno en la línea usando el manorreductor de
baja presión, debemos comprobar que verdaderamente sale gas por el
extremo de los tubos, no vaya a ser que alguna irregularidad nos estropee el
experimento.
2. 5 QUITAMOS LA LÍNEA
2. 5. 1 PARAMOS LA BOMBA
Lo más importante al quitar la línea es parar primero la bomba de
extracción. Inmediatamente después se aparta el Dewar con el N2 (l) usado
como foco frío. El orden es muy importante, y por eso lo he separado en un
punto aparte. Si lo hiciera al revés, eliminando primero el nitrógeno
líquido, podría ocurrir que parte de las materias solidificadas de la trampa
se evaporasen por la pérdida inmediata del foco, pasando a la bomba y
dañándola gravemente.
2. 5. 2 TERMINAMOS EL PROCESO
Hecho lo anterior, se abre la llave 2, para que se pueda separar la ampolla
que hace de trampa. Al abrir la llave suena un agudo silbido provocado por
la entrada de aire en el sistema que está a vacío. Después se quitan las
gomillas y se tira de la ampolla. Esto último requiere mucha atención por
dos motivos:

48
A) En el intento, puede ceder de golpe y chocar el fondo de la trampa con
el soporte metálico del Dewar y causar un estropicio.
B) A causa de la varilla, que sale de la cabeza y llega hasta la mitad de la
trampa. Es bastante frágil y suele partirse a la menor oportunidad. Nótese
que cuando tenemos que retirar la ampolla, se encuentra llena de escarcha
(el vapor de agua se condensa sobre su superficie) y así la varilla hueca del
interior se encuentra totalmente oculta, no acordándonos muchas veces de
su existencia.
Se deja entonces la ampolla con los sólidos, en un sitio seguro, para que al
día siguiente todo vuelva a comenzar.
Se cierra también el suministro de nitrógeno con el manómetro y
desalojamos el gas que hay dentro del sistema abriendo el nitrógeno en
alguna de las llaves de manejo del sistema.
El paso de cerrar la corriente del gas se olvida con facilidad y conviene
prestarle mucha atención desde el principio, aunque sus consecuencias no
serían tan graves como la de olvidar desconectar la bomba (se agotaría el
N2 (l) tarde o temprano y los sólidos se fundirían y se marcharían por la
línea).

49
3 INSTRUMENTAL DE TRABAJO ( I )
3. 1 MATRACES Y TUBOS SCHLENK
3. 1. 1 MATRACES DE REACCIÓN
Los matraces de reacción pueden ser de dos tipos, dependiendo del tipo de
llave que se use para conectarlos al tubo de la línea: de llave de vidrio y de
llave de teflón.
Los primeros son los más abundantes. Tanto uno como otro están
constituidos por un bulbo de vidrio cuya capacidad varía según la escala de
la reacción. Los más usados son los de 250 ml, pero se encuentran con
facilidad otros de distinto volumen.
La diferencia fundamental entre uno de llave de vidrio y otro de llave de
teflón radica en que si usamos uno de llave de vidrio es preciso, tras usar el
matraz, meterlo en el baño alcalino (líquido de limpieza que se hace en el
propio laboratorio), para eliminarle la grasa que hemos untado en el ajuste.
En cuanto al de llave de teflón, que no usa grasa, es reutilizable tras lavarlo
con un poco de acetona y éter (en la gran mayoría de los casos).
Los matraces se tapan con unos tapones de goma (en esta misma sección) y
teniendo en cuenta que poseen un diámetro de boca muy variable,
requieren tapones de diversos grosores.
En este primer dibujo pretendo mostrar cómo
es, en líneas generales un matraz con llave de
vidrio. Véase cómo se usa, para conectar el
interior del tubo con la línea, una llave
(análoga a algunas de la línea) de un solo
canal, que dependiendo de la posición de la

50
llave permite el paso del gas o su salida si lo que existe es una succión. La
llave se ajusta por medio de grasa.
No comentaré más sobre esta llave, puesto que todos los estudiantes de
química la conocen bastante bien, pues no es otra que la que controla la
caída de la gota durante las valoraciones en las buretas.
A la derecha podemos observar el correspondiente
matraz de teflón (se llama de teflón porque usa llave
de teflón, no obstante, como es lógico, el propio
matraz es de vidrio). La diferencia visual con el
anterior está en la ausencia de llave en el brazo de
vidrio. En su lugar encontramos un brazo terminado
en rosca, donde se va a enroscar la llave de teflón.
Esta llave posee un funcionamiento peculiar que
veremos más adelante
Se usan para contener la reacción que se lleva a cabo, siendo así los
reactores del químico organometálico, aunque a lo largo del manual
veremos cómo también pueden tener otros usos para los que se encuentran
perfectamente capacitados, a pesar de que no fueron diseñados para ello.
3. 1. 2 TUBOS SCHLENK
En los matraces anteriormente descritos se llevan a cabo las reacciones que,
tarde o temprano darán productos a analizar. A estos productos se les
purifica mediante técnicas de separación y se concluye por hacerlos, o más
bien, querer hacerlos cristalizar. Esto último se hace en estos tubos,
conjuntamente con otras operaciones relacionadas con el aislamiento y

51
purificación. Sin embargo, muchas veces son usados también para disolver
algunos reactivos, momentos antes de ser necesitados, o incluso como
contenedores temporales de productos. En definitiva, para operar con los
compuestos inestables al aire.
Un Schlenk no es más que un tubo de vidrio con una llave para conectarlo a
los tubos de goma de la línea. Al igual que en el caso de los matraces, hay
dos tipos, según lleven llave de vidrio o llave de teflón. Las diferencias
entre ambos modelos son las mismas que las que existían entre los
matraces.
3.1.3 COLOCAMOS LA LLAVE DE VIDRIO
En principio al colocar una llave de vidrio, siempre debemos cerciorarnos
de que esa llave pertenece al tubo o al matraz que se considera. Para que no
existan confusiones cada matraz o tubo posee una señal rayada en su
superficie, que puede ser un número o una letra o una combinación de
ambas generalmente formada por las iniciales del investigador y el número
que ese matraz o tubo posee entre los considerados como suyos. El mismo
símbolo se encuentra en la llave, de modo que la pareja es ya indisoluble.

52
Así que si estrenamos uno de estos elementos, lo primero que debemos
hacer es marcarlos con una cuchilla.
Para poner la llave se coge un poco de grasa con el dedo y se extiende
sobre el macho de la llave. No debemos usar mucha cantidad, todo lo
contrario, mientras menos mejor; pero debe ser suficiente para cubrir toda
la superficie. Este tacto necesario se va aprendiendo con la experiencia.
Si se pone mucha grasa, ésta acabará por introducirse en el canal de la llave
y al hacer vacío puede llenar el extremo de vidrio de la boquilla, o si
metemos N2 (g), puede ensuciar las paredes del matraz o del Schlenk. Por
otra parte, un exceso de grasa nos interferirá después a la hora de la
interpretación de los espectros de resonancia magnética nuclear (RMN).
Para homogeneizar perfectamente la grasa se coge la llave y se aprieta con
firmeza hacia abajo, como si se estuviese atornillándola. Cuando se mire la
superficie cilíndrica de la hembra desde fuera no deben quedar estrías
(marcas que indiquen que la grasa no se encuentra por toda la superficie,
quedando pequeñas bolsas de aire). Si las hubiera tenemos las siguientes
respuestas:
A) La superficie está rayada o la pareja llave-tubo es incorrecta.
B) Falta grasa y no cubre la superficie en su completitud.
C) No se ha apretado lo suficiente.
Es muy importante que no existan estas estrías y que la grasa
impermeabilice correctamente el ajuste.

53
3. 1. 4 PONEMOS LAS GOMILLAS
Quizás la operación más curiosa y tal vez la que requiere más maña sea la
de colocar las gomillas. La función de las mismas no es otra que la de hacer
estanca la llave, de modo que no gire por un accidente o golpe, y
permanezca en la posición que se desee; pero también que exista una
presión efectiva y continua contra el hueco, para así impedir cualquier
entrada de aire al interior o salida del nitrógeno interior. Algunas veces, por
la tensión de la gomilla, la llave puede sin ningún impedimento girar en
algún sentido (logrando el efecto contrario al pretendido) y para impedir
esto se recurre a otra gomilla, llamada de seguridad (más adelante veremos
cómo se pone).
Para colocar la gomilla podemos hacerlo usando dos alternativas:
A) Uso la gomilla sin ponerla doble, con lo que la situación es difícil pero
abarcable.
B) Gomilla doble, con lo que resulta poco menos que imposible (aunque
hay gente que lo hace).
Sea como sea los pasos a dar son los que
siguen:
Se toma la gomilla y se pasa por la parte
de atrás de la llave, tirando un poco para
empezar a tensar la goma elástica y que

54
nos resulte más sencillo completar el
proceso.
En la figura 2 vemos cómo el siguiente punto
es pasar la gomilla hacia la parte de atrás de
la llave.
Al representar los
distintos pasos me he
decidido usar diferente
trazo dependiendo de si la gomilla está por
delante de la llave o si, por el contrario, se encuentra
por detrás. El motivo no es otro que el de clarificar
una operación trascendental, que merece la pena
conocer con detalle para llevarla a la práctica con
exactitud.
Una vez se encuentra la gomilla en la parte
de atrás de la llave se gira de modo que se
forme un lazo análogo al que se representa
en la figura siguiente, que va a servir para
apresar la llave en su asiento.
Paso ahora de nuevo la gomilla hacia delante,
quedando en la parte trasera un cruce idéntico
al que generaremos en la parte delantera.
En la quinta figura volvemos a hacer un giro,
buscando formar un nuevo bucle en la
gomilla. Hay que cuidar mucho estos

55
movimientos, no vaya a ocurrir que la llave, gomilla mediante, salga
disparada hacia arriba y se parta. Esto último que se ha indicado ocurre más
veces de las que uno podría en principio esperar, debido sobre todo a la
falta de pericia en las primeras ocasiones en las que se acomete el proceso.
Al estar la goma muy tirante, un mal movimiento puede hacerla funcionar
como un arco, tomando la llave por su traviesa y lanzándola fuera de su
hueco. No obstante, a medida que se practica, se logra una habilidad tan
grande, que al final se hace sin mirar, y sin pensar siquiera cuál es el
movimiento que se debe hacer de seguido.
En la figura 6 se muestra cómo, habiendo
concluido el cruce anterior se vuelve a pasar
a la parte de atrás, con los consejos
anteriores bien vigentes. Se ve ya el cruce
que anteriormente citamos y que es igual al
existente en la otra cara. Cuando la gomilla
ha pasado a la otra parte de la llave, ésta
queda afirmada. Un tanto tensionada la
gomilla por tantos dobleces resulta difícil
exigirle un último esfuerzo para rebasar el punto inferior de la llave. A tal
posición se fuerza el elástico para formar una camisa perfecta que aprese la
llave, quedando a prueba de fugas.
Al final puede verse que queda rodeada. No
puede entonces, bajo ninguna causa, salirse
del asiento ni girar, a no ser que la propia
tensión de la gomilla la fuerce.

56
Concluyendo esta sección aprovecho para
decir que éste es uno de los ejercicios más
reiterados que se harán y que al principio,
antes de comenzar a trabajar por primera
vez en un laboratorio de organometálica
conviene pasarse algunas horas (si no algún
día que otro) con estos ejercicios básicos, de
modo que adquiramos la presteza necesaria
para llevar a buen puerto las reacciones. Igualmente habrá de hacerse con
otros ejercicios parejos como la colocación de tapones, las inyecciones de
disolventes,...
3. 2 TAPONES SUBA-SEAL Y DE VIDRIO
Se puede hablar de dos tipos de tapones usados para cerrar los matraces y
los tubos Schlenk. Los tapones llamados suba-seal y los de vidrio (los
cuales se usan mucho menos).
3. 2. 1 TAPONES SUBA-SEAL
Los tapones suba-seal también se denominan tapones de goma, atendiendo
a su naturaleza. Poseen distintos diámetros, acoplándose así a las variadas
aberturas de las bocas de los elementos de vidrio. Estas diferencias en el
tamaño del tapón vienen dadas por un número inscrito en el propio tapón.
Entre estos números encontramos frecuentemente los que siguen: 17, 25,
29, 33, 37, 41, 45, 49, 53 y 61, siendo los más utilizados con gran
diferencia los del 29, 33, 37 y 49.

57
Estos tapones de goma son extraordinariamente usados y conviene saber
cómo se colocan correctamente. Están constituidos por un material blando,
un tanto elástico y de color rojo (lo cual es característico de estos tapones).
Se introducen a presión por la boca del tubo, forzándolo un poco como si
fuese una rosca. Posteriormente, cuando toda la parte con forma roscada
del tapón ha entrado, se abate el ala, plegándose sobre la boca,
imposibilitando aún más si cabe la entrada de aire (o la salida del
nitrógeno).
En esta figura tenemos el tapón en su posición normal,
tal y como lo encontramos sobre la mesa de trabajo.
Podemos distinguir la parte roscada, que se introduce en
la boca del tubo, y el ala, que en este dibujo se encuentra
sin plegar formando una especie de cáliz en torno al
tapón propiamente dicho.
El mismo tapón viene a ser representado en este dibujo
en el cual se ha tomado una sección del mismo cuando
se ha introducido en el tubo y se ha plegado su ala.
Cada químico suele tener en su mesa de trabajo una cajita donde guarda los
tapones. Los tapones pueden encontrarse pinchados o nuevos, por lo que es
aconsejable tener 2 compartimentos: uno para los nuevos y otro para los
pinchados. Sin embargo, aunque se extremen los cuidados por segregarlos
al final siempre acaban mezclándose y debemos cerciorarnos antes de
usarlo de cuál es cuál (ya se entenderá por qué se pinchan y cómo se
pueden distinguir pues al ser gomosos la cosa no es nada sencilla). Al
inyectar a través de los tapones se prefiere utilizar aquellos ya usados

58
(pinchados por tanto), pero con el tiempo los nuevos vienen a ser
atravesados también, por lo que llega el momento en que la búsqueda de un
tapón nuevo y de un número en concreto llega a ser un tanto angustiosa.
3. 2. 1. 1 PINCHADO O NO PINCHADO
Son tapones de goma lo suficientemente elástica y densa como para que un
pinchazo no deje marca clara.
Aunque en muchos tapones, las huellas de las agujas son visibles a causa
de las veces que se les ha castigado, otros carecen de esas evidencias que
externamente no muestran la perforación.
Es de crasa importancia usar tapones nuevos cuando dejamos
reaccionando los reactivos en el matraz, o cuando tenemos intención de
cristalizar el producto, o cuando lo guardamos para usarlo con
posterioridad. Si el tapón se halla pinchado se escapará la sobrepresión de
nitrógeno existente en el interior del recipiente y acabará por entrar aire.
Para distinguir entre un tapón pinchado y uno que no lo está, se coge el
tapón por su parte media, justo donde termina la parte cilíndrica roscada
(valga esta expresión aunque no sea una verdadera rosca), y apretamos
fuertemente. Así en la plana superior del tapón, donde se le pincha para
introducir líquidos en los tubos, aparecerá el correspondiente agujero, en el
caso de estar picado.

59
3. 2. 1. 2 COLOCAMOS GOMILLAS AL TAPÓN
Cuando una reacción se deja evolucionando, se ponen gomillas al tapón,
ora dos simples ora una doble, puesto que durante la reacción pueden
generarse gases que, aumentando la presión, podrían lanzar el tapón al aire
y dejar la disolución al descubierto.
Igualmente se hace cuando se coloca un producto a cristalizar en un
Schlenk o cuando se guarda un producto en almacén. La
impermeabilización lograda entonces es muy superior y efectiva.
Los pasos a seguir son extremadamente
sencillos, pero se comentarán brevemente para
un mejor entendimiento.
Una vez introducido el tapón y plegado sobre la
boca del tubo se arrolla la gomilla, doble o simple,
pasándola primero por la parte de atrás del tapón. Si
queremos que el ajuste sea bueno, es aquí donde
debemos forzar al máximo (sin tampoco
excedernos) la tensión.
Tras la maniobra anterior giramos la goma para lograr un lazo comparable
a los ya visto en el engomillado de llaves.

60
En esta tercera figura podemos observar
que el proceso se repite, ahora en sentido
opuesto, de modo que logremos un nuevo
lazo en la posición oportuna. Este proceso
de girar y pasar al otro lado se realiza
tantas veces como permita la gomilla, la
cual acabará por no poder estirarse más y pondrá fin a nuestro ejercicio.
El resultado final debe ser muy semejante a la
ilustración de la derecha. En este caso ya no hay nada
que temer respecto a lo que a fugas o entrada de aire se
refiere.
En principio esta operación es muy simple, pero pensemos que, en algún
momento será preciso hacerlo con los guantes de látex puestos, y entonces
nos encontraremos ante problemas menos asequibles. Es pues una
recomendación que, antes de hacer reacciones se ejercite uno en este
método con y sin guantes, para que llegado el momento no tengamos
contrariedades.
3. 2. 2 TAPONES DE VIDRIO
Presentan la forma de un tapón normal y se ajustan con un poco de cinta
de teflón. La cinta de teflón es muy fina y se halla arrollada en una rueda a
modo de esparadrapo, sirve para aislar, es deformable y muy poco reactiva.
Esta cinta envuelve al apéndice del tapón y después se encaja en la boca.

61
Otros modelos tienen forma de ampollas y se cierran con gomillas.
Sin embargo ninguno de ellos se puede usar en la nevera como tapones de
tubos Schlenk, porque la contracción a la que se somete el vidrio desvirtúa
el ajuste (los dos vidrios son distintos y se contraerán en diferente grado).
En general su uso es muy limitado y suele preferirse el de goma, salvo en
casos particulares que dependen de los reactivos, productos o del propio
programa de la reacción.
3. 3 TAPONES DE TEFLÓN
Me ha parecido conveniente separar este tipo de los dos anteriores a causa
de que su funcionalidad difiere de la de un mero tapón. Se utilizan con una
alta frecuencia, aunque sólo en aquellos tubos y matraces que están
especialmente diseñados, como ya se ha visto en 3. 1. 1 y 3. 1. 2.
Estos tapones pues, no bastan a la hora de cerrar el tubo, pues la boca
principal requiere siempre un suba-seal, pero la boquilla roscada, donde se
inserta queda firmemente cerrada.
No obstante, dependiendo de su posición puede servir también como llave
para que pueda entrar N2 (g) o hacer vacío, de ahí que, más bien, nos
encontremos ante una llave de teflón.
El tapón tiene dos ajustes, uno primero impide que, una vez conectado a la
goma, entre aire del exterior; pero pueda entrar N2 (g) o hacer vacío. Un
segundo ajuste cierra el tubo para cualquier gas. El tapón se enrosca en la
boquilla de vidrio hasta que alcanza el primer ajuste, lo cual se hace patente
a la vista a causa de la aparición de una circunferencia de contacto en la
superficie del vidrio. Si queremos sellar completamente el recipiente de
vidrio sólo hay que enroscar aún más hasta que aparezca la segunda
circunferencia de contacto en el cuello de la boquilla.

62
1. Por este extremo se conecta la
goma que parte de la línea. En
general queda bien sujeta a la
goma, pero suele ponerse una
abrazadera si no se quiere correr
el peligro de que se salga. Es un tubo de teflón (de color blanco por tanto)
hueco en su interior para permitir a los gases pasar a su través.
2. El número 2 representa la carcasa amarilla que sirve para enroscar la
boquilla del tubo. Su interior es una espiral roscada de tal modo que encaja
y enrosca perfectamente. Es una pieza que gira independientemente a la
espiga hueca de teflón.
3. Es el extremo de la espiga, no posee orificio frontal sino dos laterales
(dado que si fuese terminal jamás llegaría a poder tapar completamente la
abertura, en caso de necesidad).
Vamos en cualquier caso a detenernos con más detalle en el análisis de esto
último y cómo realmente la espiga de teflón puede cerrar la entrada,
primero al aire exterior y luego a la propia línea.
Con este fin vamos a representar
en mayor tamaño las partes
implicadas en el proceso y así
podremos comprender mejor el
ingenioso sistema de cierre.
En el dibujo de la parte izquierda
se observa que se logra un aislamiento en todo lo que no sea el canal
interno que recorre el eje de la espiga. El arco de circunferencia que vemos

63
en negro es en realidad blanco, pero para resaltarlo se ha pintado en ese
color. En esta posición puede hacerse vacío o meterse gas.
Si enrosco aún más, hasta
llegar a la posición segunda, se
obtiene un sellado perfecto. No
puede entrar ni salir nada, con
lo cual se convierte en un
tapón de garantía. En este caso
el sellado se produce por la fuerza con la que la propia rosca impele el
émbolo contra la entrada de la boquilla del tubo. Nótese la formación de la
segunda circunferencia de sellado.
Hay que considerar que la forma de la boquilla de este tipo de matraces y
de Schlenk viene a ser poco más o menos como refiere el dibujo, es decir,
parecida a una copa tubular con una rosca en su borde.
Igualmente hay que hacer ver que este tipo de tapones no debe cerrarse con
fuerza desmedida, pues el material se deforma y la llave queda inútil. Se
aconseja entonces que se cierre suavemente hasta que, progresivamente
vayan apareciendo las dos señales de ajuste.
Cuando las señales pertinentes aparecen no hay que forzar más la llave por
que aunque parezca que no está completamente cerrada, el interior del tubo
está ya aislado, pudiéndose trabajar adecuadamente sin temor a una entrada
de aire o a pérdida de la atmósfera inerte.

64
4. IMPONEMOS ATMÓSFERA INERTE
El proceso que se detallará a continuación es de una trascendencia vital y
posee dos etapas principalmente: la desoxigenación y la inyección del gas
inerte (normalmente N2 (g)).
En principio supongamos que tenemos un Schlenk o un matraz de fondo
redondo conectado a la línea por uno de los tubos de goma (16). La
conexión puede ser regulada por una llave de vidrio o por una llave de
teflón totalmente cerrada al inicio. Al mismo tiempo el matraz se
encontrará tapado con un tapón de goma nuevo. Estas operaciones deben
resultar ya familiares debido a los capítulos precedentes.
La primera operación es la desoxigenación. En la llave de manejo de la
línea abrimos en el sentido del vacío. Al hacer esto se logra vacío a lo largo
de todo el tubo de goma hasta la llave del matraz. Una vez llegados a este
punto se abre lentamente la llave del recipiente. Un sonido de aspiración
rápido viene a oírse con nitidez, hasta que acaba cesando, cuando el vacío
es suficiente.
Se espera en torno a 5 minutos (en realidad dependiendo del volumen del
elemento que se desoxigena) y se cierra la llave que conecta el tubo de la
línea con el Schlenk. Acto seguido se introduce N2 (g) con la llave de
manejo, llenándose el tubo de goma con el gas inerte. Se abre
inmediatamente la llave y otro sonido, ahora de expansión, se oye con
claridad.
Como la velocidad de expansión de un gas en el vacío es muy alta,
repetimos la operación de desoxigenación sin esperar.
Cada par desoxigenación–inyección de N2 (g) debe repetirse 3 veces,
constituyendo lo que se denomina los 3 ciclos. Cada uno de los cuales
requiere menos tiempo que el anterior en la fase de vaciado.

65
En este instante tenemos una atmósfera de nitrógeno, que es inerte para la
gran mayoría de los compuestos. Sólo queda indicar que, a veces se lleva a
cabo la reacción bajo atmósfera de Ar, para lo que basta con cambiar la
llave 12 de posición, del nitrógeno al argón.
La presión a la que la nueva atmósfera se encuentra en el interior del
matraz es superior a la atmosférica, con lo que la entrada de aire no es ya
cuestionable y cualquier producto que se encuentre en el interior está a
salvo de la descomposición a la que pueda ser susceptible al contacto con el
aire.
5 DISOLVENTES
5. 1 TIPOS Y NATURALEZA
Los disolventes que se usan son de naturaleza, evidentemente, orgánica. En
general interesan aquellos que sean suficientemente baratos y poco
nocivos, aunque se debe hacer hincapié en que lo más importante es su
naturaleza química y su capacidad de coordinación. Los más usados con
diferencia son: diclorometano, tolueno, éter de petróleo, éter dietílico, THF
(tetrahidrofurano), acetonitrilo, acetona y alcoholes (a los cuales se recurre
raramente).
Al usar estos disolventes debemos estar seguros de que se hallan exentos de
toda traza de humedad o de oxígeno, ya que se incorporan ambos al
disolvente con una rapidez sorprendente.
El modo en el que la humedad y el oxígeno se eliminan varía de unos
disolventes a otros, dependiendo de sus características intrínsecas, sin

66
embargo, el reflujo es una técnica aceptable de mantenerlos en perfecto
estado y poder ser utilizados en cualquier momento.
5. 1. 1 ÉTER DIETÍLICO
El éter es tal vez el disolvente más usado en este tipo de reacciones, debido
a las grandes ventajas que presenta. Es un solvente muy bueno, tanto para
compuestos polares como para apolares. A su vez es poco miscible con el
agua, lo que siempre es una suerte. Finalmente, y es esto lo que condiciona
que se use, posee un punto de ebullición de 35 ºC, por lo que se evapora
fácilmente y con una rapidez que el siempre acuciante trabajo del
laboratorio agradece.
5. 1. 2 TETRAHIDROFURANO
El tetrahidrofurano es, como el anterior, un éter, lo cual suele ser sinónimo
de ventajas.
No obstante el THF posee una temperatura de ebullición de 65 ºC, por lo
que tarda más en irse por la línea a la hora de eliminarlo.
Tanto el éter como el THF son moléculas con cierta capacidad coordinante,
lo cual resulta muy importante en la química de compuestos
organometálicos, donde la mayoría de las estructuras son
electrodeficientes, y la coordinación, como agente estabilizante, resulta
clave para que la reacción marche.

67
5. 1. 3 ÉTER DE PETRÓLEO
El éter de petróleo consiste en una mezcla de hidrocarburos ligeros, sobre
todo pentanos y hexanos, es decir líquidos de bajo peso molecular,
incoloros, que se obtienen como producto entre las principales fracciones
del petróleo. Disuelven muy bien compuestos orgánicos apolares, para los
que se usan principalmente. Su temperatura media de ebullición se
encuentra en una horquilla bastante variable, dependiendo del porcentaje
relativo entre hexanos y octanos, de 30 a 60 ºC.
5. 1. 4 DICLOROMETANO
Es un compuesto muy usado. Agente solvente muy bueno, disuelve
compuestos que los anteriores no lograr solvatar. Se marcha bien a la hora
de evaporarlo, siendo su punto de evaporación de 40 ºC. El principal
problema es su peligrosidad, ya que es potencialmente cancerígeno;
además de esta toxicidad, puede disolver aceites y grasas de la piel.
Debemos, por todo ello, usar guantes de látex cada vez que lo manejemos.
5. 1. 5 TOLUENO
Un grave problema presenta al tolueno como un disolvente verdaderamente
tedioso. Su temperatura de ebullición es de 111 ºC, lo que lo hace muy
lento de evaporar, si lo comparamos con el éter.
Si a esto añadimos su carácter tóxico se completa un cuadro muy poco
atractivo. Así pues, si se puede evitar, se evita. Se usa sólo en aquellas
reacciones en las que cualquier otro disolvente no funciona.

68
El consejo a seguir es claro, úsese cuando no halla otro remedio y, como en
el caso anterior, los guantes de látex son una prevención absolutamente
necesaria.
5. 1. 6 ACETONITRILO
Este derivado de ácido se encuentra también, usualmente entre los
disolventes habituales de un laboratorio de organometálica. Disuelve
especialmente a sustancias polares sin el peligro que representa, para la
reacción, la existencia de protones con carácter ácido. Es miscible con el
agua. El principal inconveniente en su uso es su temperatura de ebullición,
que es de 82 ºC.
5. 1. 7 ACETONA
Como debe saberse, es miscible con el agua en cualquier proporción,
siendo una traba a resolver. Es un buen solvente de sustancias polares,
gracias a su grupo carbonilo, lo que la hace ser muy completa.
Esta ambivalencia a la hora de disolver la hace muy útil como medio de
reacción, por lo que siempre debemos tenerla preparada para ser usada.
Su punto de ebullición es de tan sólo 56º C, lo que la hace fácil de eliminar.
5. 1. 8 ETANOL
Es útil como disolvente de algunos reactivos que se usan para formar
compuestos organometálicos a partir de sales inorgánicas.

69
Eso justifica su inclusión en este apartado, sin embargo no suele usarse
nunca en reacciones de síntesis directa de compuestos organometálicos
dada su reactividad con los mismos (no se olvide su analogía química con
el agua).
Es miscible con el agua en cualquier proporción y posee un punto de
ebullición de 78.3 ºC debido a la formación de puentes de hidrógeno.
Pero, como ya se ha dicho, resulta insustituible cuando se trata de disolver
sales (NiCl2 · 6 H2O) para realizar las primeras reacciones que acabarán
dando un compuesto organometálico. Es por tanto el disolvente por
excelencia en el primer peldaño de la química que analizamos.
Con este compuesto termino la serie de disolventes que se ha considerado
oportuno señalar. No es que con ellos se tenga la gama completa, pero sí
que el resto de disolventes son menos habituales, y por tanto menos
considerables en una breve exposición como la que aquí se realiza. Es
verdad que también esta mayor o menor importancia depende de la
naturaleza de los compuestos que manejemos, y que para algunos habré
olvidado disolventes que ellos usan con asiduidad. Aún así creo haber sido
suficientemente general en el análisis.
5. 2 ELIMINACIÓN DE AGUA Y DIOXÍGENO
5. 2. 1 EL SODIO
Desde las enseñanzas básicas se sabe que el sodio es un metal alcalino.
Siendo metal presenta una tonalidad gris de brillo metálico, que tan bien le
conocemos al hierro, al plomo o al estaño. Se guarda en pequeñas
cantidades, en forma de cilindros, embutido en parafina líquida

70
completamente, para que no le afecte el aire, dentro de tarros cerrados. El
Na reacciona al aire dando el peróxido correspondiente, lo que se puede
observar por la formación de una capa blancuzca que recubre la superficie
metálica.
El sodio reacciona explosivamente con el agua (y con la humedad del aire
por tanto), así que se debe manejar con cuidado.
Cuando se va a usar para secar disolventes, se saca del recipiente y se corta
con el cuchillo en pequeñas laminitas. Esto no debe extrañar ya que es un
material blando y maleable (la fuerza de enlace en el enlace metálico
depende del número de electrones que participan en el mismo, y en el caso
del sodio cada átomo da formalmente 1 electrón). Al cortar el metal
aparece una superficie brillante y limpia.
Al tener los trocitos de sodio cortados, el resto vuelve a protegerse bajo la
parafina. Nunca debe almacenarse en grandes cantidades pues un accidente
sería entonces agravado por la cantidad.
El método del secado por sodio sirve para el éter, el THF, el éter de
petróleo y el tolueno.
5. 2. 2 EL SODIO Y EL AGUA
La reacción entre el sodio y el agua es aplicada a la eliminación de la
humedad existente en los disolventes. Como ésta se encuentra en una
cantidad muy pequeña no hay peligro de que haya explosión.
Es una reacción de oxidorreducción, en la que el sodio se oxida para
reducir al agua a dihidrógeno, formándose el hidróxido de sodio.

71
Na (s) + H2O ( l ) Na (OH) (s) + H2 (g)
La sosa formada se va al fondo, a causa de que el disolvente es orgánico e
incapaz de solvatar iones.
5. 2. 3 EL REFLUJO
Para lograr un efectivo contacto entre el sodio y el disolvente, que permita
eliminar los restos de agua, se hace refluir al disolvente. Este proceso
consiste en calentar el disolvente y ponerlo a temperatura de ebullición, de
modo que se comience a evaporar.
Para que no se escape se coloca el refrigerante de reflujo que condensa el
disolvente y lo devuelve al matraz de fondo redondo. Este proceso también
elimina el oxígeno del medio, pues la ebullición, poco a poco lo va
desplazando del disolvente ya que el oxígeno no va a condensar. Aún con
todo, eliminar el oxígeno es bastante más complicado pues lo que
eliminamos en esta etapa lo reintegramos cuando lo recogemos para usarlo
y hay que desoxigenarlo por otros medios como se verá más adelante. Sin
embargo es cierto que el proceso elimina una gran cantidad del oxígeno
que debería estar presente en cualquier otro caso.
El montaje del reflujo para los disolventes es el siguiente:

72
1. La manta eléctrica permite calentar un
matraz de fondo redondo de gran tamaño,
alojándolo en el hueco que le sirve de
asiento. Con este calentamiento,
eléctrico, se anula el peligro de cualquier
otro sistema (basado en llama), que hubiera
sido insensato. Se regulan por un pequeño
dial que generalmente se encuentra al lado
de ellos.
2. Un matraz de fondo redondo de 3 bocas
sirve para contener el disolvente, siendo de
un volumen determinado, de acuerdo a las
necesidades del uso. Dentro se añade el
sodio que secará al disolvente. Los tapones
laterales son de vidrio y se quitan cuando
hay que añadir un poco de disolvente
nuevo, para renovarlo. Como se comentó en
su momento, se ajustan utilizando cinta de
teflón (como ocurre con el resto de ajustes de este montaje).
3. Aquí representamos el refrigerante de reflujo. Consiste en un tubo
donde en su interior serpentea otro, más fino, también de vidrio. Debe sin
duda resultar conocido para todos, por lo que ahorraré cualquier
descripción adicional. Un error muy común al principio es el de conectar
las gomas de agua al refrigerante al revés, por lo que no llega a llenarse el
tubo interno de agua. El agua se debe introducir por la boquilla que lleva
directamente al fondo del reflujo, que después asciende hasta salir por la
otra boquilla. La función del reflujo no viene a ser más que la de impedir
que el disolvente se pierda, al encontrarse en estado gaseoso. El gas va

73
condensándose en el refrigerante y gota a gota
vuelve al matraz previo paso por 4.
4. Este elemento sirve para recoger disolvente
apto para uso inmediato, al cual resta la
desoxigenación. Según la posición de la llave hay
3 alternativas, como ocurría con las llaves de
manejo de la línea.
a) Posición que conecta la cámara de recogida
con el matraz de fondo redondo, con lo cual todo
el disolvente que se licua pasa de nuevo al matraz.
b) Posición que cierra el paso entre 2 y 4, por lo
que el disolvente se acumula en 4, para después
poder ser utilizado.
c) La última posición permite vaciar el contenido de la cámara, a través de
grifo, en el recipiente deseado.
En esta perspectiva puede observarse que, cuando el disolvente se
almacena hasta un cierto límite, baja por el camino paralelo hasta encontrar
de nuevo el matraz. De este modo se elimina el peligro de que el disolvente
ascienda y se salga por la parte superior del reflujo.
5. 2. 4 PROBLEMAS CON EL REFLUJO
Algunas veces ocurre que se está calentando el disolvente por encima de la
temperatura que se aconseja (que siempre es algo superior a la temperatura
de vaporización) por lo que la cantidad de disolvente que recircula es
mayor que la que puede bajar por el brazo de seguridad. De este modo

74
comienza a subir más arriba del límite estimado, señalado por la
conjunción de la vía lateral con el recogedor. Si no nos percatamos a
tiempo ocurrirá que el disolvente se acumulará en el reflujo y acabará
saliendo por la parte superior del mismo, en tanto el matraz de fondo
redondo se queda seco.
La solución pasa por cuidar bien las temperaturas. Aún bien, si ocurriere,
no hay más que, con un giro de llave, bajar todo el líquido recogido al
matraz y, posteriormente disminuir la temperatura de calentamiento.
5. 2. 5 CONECTAMOS EN SERIE LOS DISOLVENTES
Para abreviar el trabajo y ahorrar espacio se conectan los equipos de reflujo
entre sí, de modo que con un solo botón, todas las mantas calientan, y con
abrir el grifo designado para llenar los reflujos, todos se llenan igualmente.
Este sistema es sencillo de hacer: el agua va por una tubería principal de la
cual, ramificándose, salen otras que llegan a cada reflujo.
Para las mantas eléctricas se hace otro tanto, de modo que se controlen
todas desde un interruptor, aunque el regulador de cada una permanecerá
independiente.
5. 2. 6 SISTEMAS DE SEGURIDAD
Los disolventes orgánicos son muy peligrosos si no se usan y almacenan
debidamente. Uno de los peligros es que al conectar el agua, su flujo no sea
suficiente, y sin saberlo se escape poco a poco disolvente en estado
gaseoso, que se acumularía en el laboratorio.

75
También puede cortarse el agua de repente y llevarnos la misma
desagradable sorpresa.
Para impedir esto hay dos elementos, uno de los cuales es indispensable,
como se comprenderá.
A) Al salir la goma del grifo del agua, se la intercepta con una válvula, que
indica el flujo de la misma (con el giro que produce el agua sobre una rueda
pequeña). Con la velocidad de rotación puede uno hacerse la idea del
caudal de agua y así, basándonos en la experiencia, saber si es o no
suficiente. Resulta a su vez útil para no abrir el grifo más de lo necesario, lo
cual podría hacer peligrar toda la instalación.
B) El verdadero sistema de seguridad consiste en un aparato que conecta el
sistema eléctrico que permite el calentamiento, con un medidor de flujo. Si
el caudal es suficiente, el sistema cierra el circuito y empieza a calentar.
Pero si el flujo es deficiente, el circuito no se cierra y las mantas
permanecen inactivas. Este sistema garantiza la seguridad de los procesos
de reflujo.
5. 2. 7 DESTRUIMOS EL SODIO REMANENTE
Cuando se desea cambiar el sodio del matraz, porque existan demasiadas
impurezas y la cantidad de sodio sea ya pequeña, se debe considerar su
desactivación pues es un elemento muy reactivo.
Este proceso se hace con alcoholes. En general usamos alcohol etílico, que
es sin embargo lo suficientemente ácido como para que la reacción vaya
demasiado deprisa. Por esta razón, cuando se sospecha que la cantidad de

76
sodio es considerable, se recurre a alcoholes secundarios o terciarios, que
enlentecen la reacción, pero la hacen más segura.
En general la reacción marcha bien:
CH3CH2OH (l) + Na (s) CH3CH2ONa (sol) + ½ H2 (g)
Una vez que se ha dejado el tiempo suficiente, con un poco de agua,
añadida muy lentamente, acabamos por neutralizar cualquier traza de sodio
activo que nos pudiera ocasionar algún problema.
5. 2. 8 VALIDEZ DEL MÉTODO DEL SODIO
Esta propuesta anterior para secar con sodio es aplicable para aquellos
disolventes que no presentan enlaces muy polares. Por tanto resulta
apropiado para el éter, éter de petróleo, tolueno y THF. Sin embargo, por
ejemplo para el diclorometano no resulta válido, y hay que cuidarse muy
bien de no equivocarse porque su reacción con el sodio es explosiva (dado
el carácter exotérmico de la misma). Así para cualquier alcalino:
2 M (s) + n RX (l) RnM (sol.) + MXn (s)
Para este tipo de elementos se usan adsorbentes de humedad, los cuales se
ponen en el reflujo de la misma manera, e igualmente hay que cambiarlos
cada cierto tiempo (dependiendo del gasto que se haga del disolvente).

77
5. 2. 9 SECADO DE LA ACETONA
La acetona no se puede poner a refluir con sosa y necesita condiciones de
secado más suaves. Con tal idea se usa el CaH2 (s) que reacciona con el
agua y la elimina del medio.
Un problema es que la acetona, en caliente, produce consigo misma
reacciones de condensación y llega a formar polímeros. Así al ser destilada
se descompone, y por tanto, tal método no es conveniente.
De este modo la acetona se encuentra guardada en atmósfera de N2 (g) en
una ampolla en el laboratorio, con el hidruro añadido. En la parte central
de la ampolla se debe pegar una etiqueta que indique la fecha de cuando se
llenó la ampolla con esa acetona seca. Esta ampolla estará en un lugar bien
a mano en el laboratorio pues es de mucho uso.
Cada vez que se utilice deberá dejarse en el mismo lugar y bajo atmósfera
de nitrógeno, como se comentará más adelante.
5. 2. 10 ACETONITRILO Y DICLOROMETANO
Al igual que la acetona no permiten el secado con sodio, pero sí con CaH2
(s), lo que se hace en el matraz de fondo redondo y con el sistema de
reflujo; a diferencia de la acetona, no hay peligro al ponerlos en reflujo.
5. 2. 11 INDICADORES DE HUMEDAD
En el éter, éter de petróleo y THF se añade una pequeña cantidad de un
agente indicador que posee dos colores completamente distintos cuando
hay trazas de agua y cuando no se registra cantidad alguna.

78
Este compuesto de naturaleza orgánica posee un color azul intenso cuando
el disolvente está completamente seco, en tanto un color pardo anaranjado
muestra con claridad que el disolvente contiene humedad y no es apto para
el uso. Para los otros compuestos no se usan indicadores.
5. 2. 12 ATENCIÓN FINAL
Aunque ya se ha anotado, preferimos llamar la atención una segunda vez
sobre el peligro inherente al equívoco entre disolventes. Si por cualquier
razón tomamos diclorometano por cualquiera de los que se secan con
sodio, y lo añadimos al matraz (creyendo que por ejemplo es éter) para
rellenarlo, la posibilidad de una explosión es muy grande.
Por tanto, cuidado. Es una gran cosa el familiarizarse con los olores de cada
disolvente y, sin hacer mucho caso a la etiqueta, cerciorarnos por el olfato
de cuál es exactamente el disolvente que queremos añadir. Esto evitará
cualquier desliz que tendría consecuencias terribles.
6 INSTRUMENTAL DE TRABAJO (II)
6. 1 AMPOLLAS Y SUS TAPONES DE TEFLÓN
6. 1. 1 AMPOLLAS
Estos recipientes son de los más usados en el laboratorio y tienen funciones
características que los hacen insustituibles en algunas situaciones.
Conviene entonces conocerlos adecuadamente.

79
Las funciones de estos recipientes se pueden estructurar en:
A) Almacenamiento de disolventes deuterados, acetona seca, y en general,
reactivos en disolución. Todos estos compuestos gozarán de atmósfera
inerte para asegurar su buena conservación.
B) Como reactor para reacciones que han de realizarse a una alta
temperatura (lo cual se detallará más adelante).
En la ampollas podemos encontrar una gran gama de tamaños, lo que
permite almacenar todo tipo de sustancias. De modo que los deuterados,
que son muy caros, se almacenan en pequeñas cantidades (por ello
ampollas de pequeño volumen) para minimizar el coste si se nos rompiese
el recipiente por cualquier fallo. Pero otros menos valiosos (o que pueden
disolverse para ser manejados con mayores volúmenes) se encierran en
ampollas de tamaño mucho más considerable.
1. Puede observarse cómo la parte superior tiene una
forma adecuada (roscada) para que un tapón de teflón
tipo ampolla (distinto del visto anteriormente) se
enrosque en el vidrio.
2. Es la boquilla por la que se conecta a la línea. Se
introduce en el orificio del tubo de la línea a presión.
3. Depósito de vidrio (ampolla) donde se deposita el
líquido.

80
6. 1. 2 TAPONES PARA AMPOLLA
Estos tapones son en realidad muy parecidos a los analizados con
anterioridad. Son igualmente de teflón, y permiten un perfecto aislamiento
de la disolución contenida en la ampolla. Poseen una rosca en la carcasa
que permite (al enroscarse) hacer bajar la espiga maciza central, de tal
modo que sella el pasaje.
1. Carcasa amarilla de plástico
con interior roscado que permite,
gracias a la rosca, el
desplazamiento del pistón.
2. Espiga maciza de teflón, con
dos ajustes a distinta altura (líneas negras), que sellan el conducto y que
son visibles solamente al encajar y cerrarse sobre la boquilla.
Podemos apreciar mejor, cómo se realiza este sellado, por los siguientes
dibujos.
A la derecha se aprecia el cierre perfecto
que logra el émbolo del tapón en el primer
estrechamiento de la ampolla. Este ajuste
impide la entrada o salida de cualquier gas
desde el interior de la ampolla al exterior.
Sin embargo la comunicación con la línea a
través del brazo lateral se encuentra libre,
pudiéndose introducir nitrógeno o hacer
vacío si nos fuera preciso.

81
Mostramos a la derecha cómo puede
cerrarse y dejarse completamente
aislado del exterior el contenido de la
ampolla cuando los dos ajustes operan
convenientemente sobre el vidrio del
recipiente.
Es tan seguro que una vez bajo
nitrógeno, se puede cerrar enroscando
hasta el tope (siempre sin forzar) y
retirar la ampolla de la línea, pudiendo
almacenarse sin problema mucho
tiempo.
Para verificar que los ajustes son seguros, en cada uno de los casos
aparecerá una circunferencia blanca que corresponde al contacto entre el
vidrio y el teflón.
Si queremos hacer vacío se abre la llave de manejo en el sentido adecuado
y después se gira el tapón para eliminar el ajuste interno. Para introducir
nitrógeno, se cierra de nuevo ese paso y se da vía al nitrógeno con la llave,
para acabar abriendo de nuevo el ajuste.

82
6. 2 JERINGAS Y AGUJAS
6. 2. 1 JERINGAS
6. 2. 1. 1 JERINGAS DE VIDRIO
Antes, todas las jeringas eran de vidrio, desde aquellas que se usaban para
la adición de reactivos hasta las que se usaban para añadir disolventes. En
cambio ahora, la presencia del plástico (tan importante en el campo
organometálico) permite la aparición de las jeringas de este material. El uso
de estas últimas es mucho más cómodo y sencillo, por lo que han relegado
a las primeras a la toma e inyección de disolventes orgánicos.
Trabajar con jeringas de vidrio es un engorro. Al ser de cristal el cuidado
debe ser extremo, pues son bastantes caras. El disolvente que se recoge en
el sistema de destilación se echa en la jeringa y rápidamente se le pone el
émbolo. Al darle la vuelta, y quedar el disolvente sobre el émbolo, se va
lentamente por el reducido espacio que queda entre ambas piezas (carcasa y
émbolo), llenándonos las manos y perdiendo disolvente.
Otra dificultad, como ya veremos, es el manejo de la jeringa en sí, que es
bastante problemático si no estamos habituados.
Existe una gama de jeringas de vidrio que va desde los más pequeños
volúmenes hasta los más grandes, pero las que se usan regularmente son,
fuera de dudas, las de 20 ml, conjuntamente con las de 50 ml (mucho más
caras) que se utilizan de vez en cuando.
En cuanto a la forma y al uso no queda decir sino que son iguales a las
jeringas de plástico que conocemos. La única diferencia es que en su parte
frontal poseen un aplique metálico con rosca interna, donde se pueden
insertar las agujas especiales que existen en esta rama de la química. Este

83
aplique es cilíndrico, de pequeño tamaño y perforado para que el disolvente
pueda pasar.
6. 2. 1. 2 AGUJAS – CÁNULA
Como se comentó en el punto anterior vienen a utilizarse unas agujas
especiales en la inyección de disolventes. Estas agujas son de tres tipos
principalmente. Se usan tanto para las jeringas de vidrio como para las de
plástico.
A) Cortas y gruesas (unos 15cm)
B) Largas y finas (unos 25cm)
C) De RMN (largas y muy finas, unos 25cm)
Las primeras se usan para la inyección de disolventes con jeringas de
vidrio, ya que el volumen a dispensar es grande y no hay necesidad de
llevar la aguja hasta el fondo del matraz.
Las segundas se utilizan para tomar reactivos de las ampollas (a los cuales
hay que alcanzar muchas veces en el mismo fondo de la ampolla) y para
inyectar reactivos (los cuales deben ser añadidos justo sobre la disolución,
para que no queden sobre las paredes y es preciso llegar hasta la superficie
de la misma).
Las terceras se requieren para tomar disolvente deuterado (en un volumen
estimado en 0.6 ml) de las ampollas correspondientes, con jeringas de
plástico de 1 ml.
Las agujas–cánula más finas llevan en su interior un pelo de acero,
extremadamente fino, que sirve para desatascarlas cuando se diere el caso.
Cuando se van a usar, el pelo se saca y se guarda con cuidado, pues su

84
utilidad es muy grande. En caso de perderlo, lo cual no es difícil, nos
veremos en aprieto demasiadas veces.
La aguja en sí no es otra cosa que un tubo de acero hueco muy fino,
terminado en una punta muy aguda. En su otro extremo posee un aplique
complementario al de la jeringa de vidrio, tal que, mediante una ligera
rotación, queda firme y listo para ser usado.
He aquí un dibujo representando la aguja descrita en el texto. En el extremo
de la derecha de la aguja puede apreciarse el aplique metálico que se une al
de la jeringa.
Los dos apliques de metal encajan y
enroscan el uno en el otro. Primero se
ajustan apretando con los dedos y luego se
afirma la unión levemente con los alicates
(sin excedernos ya que podríamos
pasarnos de rosca y dañar la jeringa). Si no están lo suficientemente bien
unidos, el disolvente puede salirse por ahí al hacer frente a la presión del
nitrógeno del matraz (más adelante se tratará).
6. 2. 1. 3 LIMPIEZA DE JERINGAS DE VIDRIO
Cada vez que usemos una jeringa de vidrio debemos limpiarla. Siempre se
utiliza para ello acetona y éter, en este mismo orden. No obstante, resulta
evidente que si el disolvente es éter no hay que limpiarla y nos limitaremos

85
a esperar a que se seque (no ha lugar que sea acetona, pues está en ampolla
y el método es bien distinto).
Una vez limpia la debemos secar, para esto se coge el émbolo, no por la
parte diseñada para ello, sino algo más adelante, por la superficie
esmerilada. De este modo, al entrar en la carcasa no golpeará la parte
interior frontal, donde se halla el aplique metálico.
Esto es fundamental pues un golpe podría quebrar la jeringa. Se comienza
entonces a mover el émbolo una y otra vez con fuerza, llevándolo fuera
para volverlo a conducir a la carcasa. Cada vez que se saca el émbolo se
produce un sonido semejante a un chasquido de la lengua. La jeringa se
seca rápidamente y puede volverse a usar. Incluso hay personas que la
depositan en la estufa.
Vemos en la figura por dónde se coge el
émbolo para impedir que en los movimientos
de secado golpee contra el frontal de la jeringa.
La razón de esto es obvia, simplemente no
llega.
6. 2. 1. 4 JERINGAS DE PLÁSTICO
Hoy en día existe un surtido bastante completo de jeringas de plástico, en
lo que a su capacidad se refiere. Se usan siempre con preferencia a las de
vidrio cuando esto es posible.
Sus ventajas son:
1) Son baratas, tanto, que muchos no pierden el tiempo en
limpiarlas. Pero si se desea se pueden limpiar con acetona y éter,

86
teniendo en cuenta que destruyen el plástico y que llegará un
momento en que no serán ya reutilizables.
2) Muy fáciles de manejar.
3) Gran variedad de volúmenes.
4) Requieren el mismo tipo de aguja que la usada por las de vidrio
por lo que no hay que invertir en un nuevo modelo de aguja. Se
adaptan al aplique metálico de la aguja-cánula por presión,
ajustándose tan bien que no requieren ningún cuidado
alternativo.
Sus inconvenientes son:
1) Atacables por los disolventes orgánicos.
2) Medidas de volumen no tan exactas como las de vidrio (en las
tomas de cantidades mínimas).
6. 2. 1. 5 MICROJERINGAS
Merecedoras de un atención aparte están las microjeringas. Son de vidrio, y
se usan para tomar pequeños volúmenes. Las hay distintas, según el rango
para el que fueron diseñadas (por debajo del mililitro). Son
excepcionalmente finas y delicadas, debiéndose guardar inmediatamente
después de ser usadas y lavadas, en su caja de corcho.
Puede suponerse, por estos cuidados, que son bastante caras. Llevan una
aguja acoplada e inseparable de mediana longitud, lo que a veces resulta
incómodo a la hora de coger y adicionar los reactivos.
El limpiado de las microjeringas es muy importante ya que se ocluyen con
facilidad. Se limpian con acetona y éter.

87
Se coge un vaso de precipitados y se le añade un poco de acetona. Luego,
con la jeringa se realizan tomas reiteradas al mismo tiempo que se tira el
volumen adquirido. Con unas 20 veces que se haga esto, es suficiente. A
continuación se repite con éter otras tantas, quedando limpia la
microjeringa.
No se meten en la estufa para secarlas, pues se deforman.
7 REACTIVOS SÓLIDOS
7. 1 USO DE LA LÍNEA
El uso de la línea debe ser ya conocido a estas alturas del texto: cómo se
hace vacío, cómo se introduce nitrógeno,... sin embargo conviene hacer una
anotación preliminar antes de pasar a desarrollar distintos métodos
mediante los cuales se pueden llevar a cabo reacciones u operaciones
diversas.
Siempre que destapemos un Schlenk donde halla un compuesto afectable
por el aire, la corriente de N2 (g) debe estar abierta, para que, al salir a una
presión efectiva mayor que la atmosférica, imposibilite la entrada de aire al
matraz.
Igualmente si se inyecta a través de un tapón pinchado un líquido
cualquiera, debe existir una corriente que impida la entrada de aire al
interior.
Este método de gran ingenio salvaguarda de modo absoluto la integridad
del producto. Un olvido al quitar un tapón, o al conectar (por cualquier
vía) el interior con la atmósfera, sin abrir la corriente de gas inerte, podría
resultar fatal para el fin de la reacción o del propio producto.

88
7. 2 PREPARATIVOS PREVIOS A LA REACCIÓN
7. 2. 1 LA PESADA
7. 2. 1. 1 REACTIVOS ESTABLES AL AIRE
Los reactivos sólidos que son estables al aire se pueden pesar usando las
balanzas electrónicas existentes en los laboratorios. Generalmente hay
siempre una diferente cuya precisión es mayor que la de las otras y por
tanto es la que se suele usar para pesar las cantidades para la reacción.
Como norma general las pesadas son pequeñas y suelen situarse por debajo
del gramo.
Para pesar no es necesario sino un papel encerado que se usa como soporte
y sobre el cual se pone el producto una vez tarado el sistema. Suele
colocarse el papel plegado, es decir, formando un ángulo obtuso entre una
parte y otra, para facilitar su descarga en el matraz de reacción. Este papel
tiene la enorme ventaja de que retiene muy poco sólido, no perdiéndose
nada en este transporte.
Una recomendación extensible a toda pesada es que se haga en ausencia de
corrientes de aire, que arrastran parte del producto y afectan la sensibilidad
de la balanza.
7. 2. 1. 2 CON DESCOMPOSICIÓN LENTA
Muchas veces el material pesado no es estable al aire, pero la reacción de
descomposición es lo suficientemente lenta como para que pueda pesarse al
aire.

89
Este tipo de productos se guarda en recipientes bajo atmósfera de
nitrógeno. Cuando hacen falta se destapan y se pesan, como si fuesen
estables, velozmente. Inmediatamente después se vuelven a tapar y se
llevan a la línea, donde se les hace los 3 ciclos y se vuelven a guardar bajo
atmósfera inerte. Lo mismo con el producto que se haya puesto en el
matraz, con el que vamos a hacer la reacción; debe salvarse bajo N2 (g).
7. 2. 1. 3 CON DESCOMPOSICIÓN RÁPIDA
Se contienen bajo este título general todos aquellos productos que
quedarían destruidos parcial o totalmente al intentar pesarlos por el método
anterior. Se descomponen rápido, pero permiten el contacto con la
atmósfera durante unos segundos, y son esos pocos segundos los que se
aprovechan para realizar la operación de pesado.
Para lograr esto es necesario destreza y mucha práctica. Este método, no
obstante, no sirve para pesar una cantidad exacta, sino que se usa para
poner en un matraz tarado una cierta cantidad, y a partir de ahí (sabiendo
lo que pesa) adicionar a escala el resto de reactivos.
Se colocan los dos recipientes que van a ser utilizados en este traspaso (uno
previamente tarado y otro que contiene el producto) muy cerca el uno del
otro. Ambos destapados y con la corriente de nitrógeno preservando la
entrada de aire. El sonido del gas saliendo a presión por la boca de los
tubos es profundo y llega a ensordecer el laboratorio. Se inclina hacia
delante ambos recipientes, para favorecer la introducción de la espátula.
La operación en su conjunto es delicada, debe realizarse con rapidez y con
pulso firme:

90
1) Se quitan los tapones de goma al Schlenk (suponemos que es el
que contiene al producto del cual queremos una porción) y al
matraz tarado (en el cual va a llevarse a efecto la reacción).
2) Se coge la espátula y se introduce en el Schlenk. La inclinación
del mismo debe permitir que pueda tomarse una cantidad con
facilidad sin que se derrame nada más subirla. Este punto, con
algunos compuestos de consistencia grasa puede resultar
molesto.
3) Al ir levantando la espátula con su contenido se cierra la llave
del Schlenk un segundo, al tiempo que se tarda en sacar la carga.
Inmediatamente se vuelve a abrir, tal que se reinicie la corriente
de gas.
4) Sin dilación se lleva a la boca del matraz y se cierra su llave para
poder introducir la espátula. Una vez esté abajo la espátula, se
abre de nuevo la entrada del N2 (g). Cuando el material presenta
un aspecto cristalino se deja en el fondo sin problema alguno,
pero cuando tiene consistencia oleosa (lo cual ocurre muy a
menudo) es una real complicación.
En un compuesto purulento si no hubiese cerrado las llaves, tanto en este
punto como en el anterior, el producto hubiese salido aventado por el gas y
nos hubiésemos percatado de su sabor y olor, siendo esto poco conveniente
por la toxicidad que suelen presentar.
5) Estas operaciones se repiten hasta que se tenga una cantidad de
producto razonable. Tras lo cual se vuelven a colocar los tapones
de goma sobre las bocas.
6) Se hace entonces vacío en uno y otro para eliminar cualquier
posible entrada de aire que hubiera habido y se hacen los tres

91
ciclos para asegurarnos de tener una atmósfera inerte en ambos
recintos.
7) Al hacer vacío se debe girar la llave con suavidad, sin vaciar de
golpe el recipiente. El producto pudiera ser ligero y ser
arrastrado al hacer vacío. Igualmente ocurre al meter N2 (g),
interesándonos que el sólido quede abajo y que no se esparza por
las paredes. De este modo la inyección de gas tras el vacío debe
hacerse con cuidado, abriendo la llave lentamente.
Es importante considerar que la inclinación que hay que dar a los dos tubos
Schlenk para proceder a un intercambio efectivo de reactivo debe ser
adecuada, a veces pueden quedar casi horizontales, manteniéndolos en esta
posición gracias a la acción de las pinzas. Con una mayor verticalidad el
producto recogido con la espátula se derramaría al sacarlo, y lo mismo
ocurriría con el otro tubo, que debe recibir a la espátula.
7. 2. 1. 4 CON DESCOMPOSICIÓN MUY RÁPIDA
Para este grupo ni siquiera es válido el método anterior pues se
descompondrían en esos escasos segundos de exposición al aire. Estas
medidas de masa se hacen en la balanza existente en la cámara seca. Esta
cámara, como se explicará más adelante, contiene una atmósfera de
nitrógeno que permite realizar cualquier operación en su interior sin que
haya que temer lo más mínimo por la seguridad de nuestro compuesto.

92
Estas actividades que se llevan a cabo en su interior, son dirigidas desde
fuera, con la ayuda de unos guantes que penetran en la cámara. Se tratará
en capítulos posteriores.
7. 2. 2 ESCALA DE LAS REACCIONES
Considerando que los productos de partida son caros, difíciles de elaborar
o al menos lentos de sintetizar, necesitando algunos de ellos días enteros,
hay que ser muy precavidos y sensatos al planificar las reacciones y no
dilapidar lo que con tanto esfuerzo se ha obtenido antes. De este modo las
reacciones se ponen usando pequeñas cantidades la práctica totalidad de las
veces.
Cuando se pone una reacción por primera vez, ignorando por tanto si lo que
uno espera va a producirse o cuál va a ser su rendimiento (si será
cuantitativo o no), se hace a escala de 0.5 mmoles.
Si se ha obtenido un éxito sobre el plan establecido, se repite al menos una
vez más la reacción con las siguientes intenciones:
1) Observar que la experiencia es repetible, anotando las
condiciones de reacción y el porcentaje de rendimiento.
2) Obtener una cantidad necesaria para los análisis del producto por
los diversos sistemas de determinación estructural.
En esta segunda experiencia se pretende conseguir una cantidad útil para el
análisis, por eso la escala pasa a ser habitualmente de 1 mmol.
Finalmente cuando se comprueba que le reacción va bien en las
condiciones predichas y que el producto es un punto de partida importante

93
para otra serie de reacciones, se piensa en aumentar la escala de la reacción
a varios mmoles, dependiendo de lo que se estime necesario.
En general las reacciones que tienen como objetivo obtener productos de
partida muy generales se realizan siempre a gran escala.
De este modo, atendiendo a nuestras intenciones, elegiremos una escala u
otra, y en función a esta escala, pesaremos las cantidades que vayamos a
necesitar.
7. 2. 3 CONSEJOS AL PONER UN TAPÓN
Bien que puedan parecer triviales, resultan interesantes algunas
disposiciones previas que garanticen la operación.
A) Sujétese fuertemente el Schlenk o el matraz al introducir el tapón, para
que no resbale y no se parta al chocar su fondo con la superficie de la
mesa.
B) Asegúrese de que el Schlenk o el matraz no se encuentra justo encima
de uno de los brazos del pie metálico, pues al más mínimo fallo de la pinza
(cosa que no es extraña) el vidrio estará roto.
C) Antes de reinsertar el tapón tras haberlo quitado previamente con alguna
intención y estando el interior bajo atmósfera inerte, se debe eliminar el
aire que queda ocluido en el hueco inferior del tapón. Esta bolsa de aire se
elimina por exposición a la corriente de nitrógeno que sale del recipiente y
por torsiones al tapón (en esa misma parte del objeto) que contribuyen a la
salida del aire. Esto último hay gente que no lo hace, pero al existir una
cierta cantidad de aire hay que eliminarlo por el bien de la reacción.

94
7. 2. 4 TARAMOS UN SCHLENK O MATRAZ
Tarar significa descartar el peso del vidrio del Schlenk, así como todo lo
que no sea producto, para que al volver a pesar, con la sustancia en su
interior, tengamos exactamente el peso del compuesto por diferencia.
En principio sólo se taran tubos de Schlenk pues, aunque los productos
aparezcan en el matraz, éstos pueden disolverse y transvasarse (por un
método que se detallará más adelante) a un Schlenk. Allí se elimina el
disolvente, aplicando vacío y nos quedamos con el producto únicamente (si
la reacción es cuantitativa y al pasar del matraz al Schlenk hemos
aprovechado para separar las impurezas).
Lo más cómodo es tarar un Schlenk al principio y tenerlo preparado para
cuando venga a ser necesario. Los pasos para tararlo son los siguientes:
A) Se toma un Schlenk con tapón nuevo y se lleva a vacío.
B) Se marca el tapón con un rotulador, de tal modo que sepamos cuál es el
tapón con el que se taró el Schlenk (puede ocurrir que necesitemos el tapón
o el Schlenk para otra cosa o que en una de las veces que tengamos que
quitar o poner el tapón lo confundamos con otro, desvirtuándose la
medida).
C) Se cierra la llave y se separa al Schlenk de la goma, pesándose. Esto hay
que hacerlo rápido pues, poco a poco, va entrando algo de aire, dado que
los ajustes no son perfectos y el alto vacío en el interior tiende a eliminarse
a través de la más mínima irregularidad.
D) Se anota en el mismo vidrio el número obtenido y se vuelve a poner
bajo nitrógeno. El peso de la tinta que hemos incorporado sobre el vidrio es
despreciable, y en absoluto afecta el valor de nuestra medida.

95
El plato de la balanza es más pequeño que la longitud del Schlenk (o del
matraz correspondiente) y suele ocurrir que toque fuera de dicho plato,
reposando parte de su peso. Debe cuidarse de que esto no ocurra, y que el
tubo descanse sobre el plato sin apoyarse en ningún punto externo al
mismo.
8 INYECCIÓN DE DISOLVENTES
8. 1 RECOGEMOS DISOLVENTE EN LA JERINGA
Como se comentó, el sistema de reflujo tiene un embudo donde se recoge
el disolvente destilado, cuando la llave se coloca en una posición concreta,
en espera de ser utilizado. De este modo, cuando nos vaya a hacer falta
disolvente lo pondremos a recoger al menos media hora antes (depende en
realidad del disolvente en concreto, el éter va muy rápido en tanto que el
tolueno hace lo opuesto) para que en el momento justo de ser utilizado
tengamos una buena cantidad a nuestra disposición.
Se coge la carcasa de la jeringa, sin el émbolo, y se añade en su interior el
volumen deseado de disolvente. Para que no se salga se tapa el orificio con
un dedo.
Inmediatamente se pone el émbolo y
se invierte la jeringa de modo que el
disolvente quede retenido por el
émbolo. Suele ocurrir que algo del
disolvente se derrame en el proceso.
Algunos disolventes, que se
encuentran a alta temperatura

96
pueden llegar a quemar (tolueno, acetonitrilo,...) por lo que, a causa de esto
y de lo anterior, es aconsejable usar los guantes de látex.
La operación en sí no es muy difícil, pero al hacerla con guantes se debe
tener cuidado pues si la jeringa o su émbolo se nos escapa de las manos
tendremos disgusto asegurado. Se ha creído necesaria la inclusión de este
dibujo que permite ver cómo se coge la jeringa al llenarla de disolvente.
Véase que el meñique sirve de barrera al agujero del aplique metálico de la
carcasa de la jeringa, impidiendo cualquier pérdida por ese camino.
8. 2 PONEMOS LA AGUJA E INYECTAMOS
Con la mano libre se coge la aguja–cánula corta y se enrosca en el aplique.
Con unos alicates la afirmamos y nos disponemos a inyectar.
Como medida previa, antes de coger el disolvente, hemos tenido que
cambiar el tapón, poniendo uno pinchado en su lugar. Para tal cambio se
debe tener, obviamente, la corriente de nitrógeno abierta, y dejarla así
durante la operación de inyección. Un fallo típico es precisamente olvidarse
de este cambio, y cuando se tiene la jeringa en las manos se cae en la
cuenta, debiéndose volver a comenzar el proceso. Si alguien piensa que
basta con pinchar el tapón nuevo, se contestará que resulta harto difícil si
no se pica previamente con agujitas más finas.
Con la jeringa debemos tener cuidado de no impulsar el émbolo hacia
delante o saldrá disolvente y lo perderemos, así hay que sostener el pistón
con los dedos para que no deslice en ningún sentido.
La inyección se puede estructurar en las siguientes etapas:

97
A) Se coge la jeringa, sosteniéndola en la mano y cogiendo el pistón con
los dedos índice y pulgar. Con la otra mano se pinza la punta de la aguja
metálica.
B) Se lleva directamente la punta sobre el tapón pero curvándola, de tal
modo que la aguja se combe desde su unión con la jeringa hasta su punta.
Esto impide que el disolvente caiga rápidamente, al mantenerse la jeringa
lo más horizontal posible.
C) Al mismo tiempo que se hace lo anterior y se pincha el tapón (clavando
la aguja en el tapón y traspasándolo) se lleva con los dedos al émbolo de la
jeringa hacia atrás, de modo que la succión que se crea impide caer por
gravedad al disolvente antes de llegar la punta al tapón, y así se derrame
fuera del matraz.
D) Una vez que se ha pinchado, se coge la jeringa con la mano libre
(aquella que se usó para sostener la punta) y con la otra se retira total o
parcialmente el émbolo. Como hay una alta presión de N2 (g) el disolvente
no cae en el matraz, sino que burbujea por el paso de gas a su través. Esto
permite que el O2 (g) absorbido durante el traslado se elimine.
El oxígeno absorbido es muy elevado, al contrario de lo que pudiera
parecer, pues la velocidad de incorporación al disolvente seco es muy alta.
Por ello es muy importante dejar borbotear el disolvente un tiempo (en
torno a un minuto) dependiendo de la cantidad. Este proceso se llama
desoxigenación del disolvente.
E) Ya desoxigenado, se lleva lentamente el pistón contra el disolvente y
éste se vacía en el matraz. Tras esto se coloca de nuevo el tapón sin
pinchar. Se desenrosca la aguja – cánula de la jeringa y se limpia la jeringa
y la aguja sin demora.

98
Cuando se pincha en el tapón es importante no soltar el pistón de la jeringa,
pues la presión de N2 (g) es tan grande que lo lanzaría como si fuese una
bala en un cañón, partiéndose donde viniera a chocar.
Al apretar el émbolo contra el disolvente cuesta trabajo vaciar el líquido
debido a la presión que se debe vencer correspondiente al gas inerte.
La operación, en su conjunto, puede parecer de lo más complejo cuando se
hace las primeras veces, pero una vez nos acostumbramos a ello podemos
realizarla sin siquiera pensar, llegando a lograr inyectar perfectamente.
8. 3 UN MAYOR VOLUMEN DE DISOLVENTE
Si el volumen requerido sobrepasa el que nos proporciona una jeringa,
usamos otra técnica, en la que nos encontramos nuevos elementos cuyo
uso es habitual en el laboratorio y que precisamos su conocimiento. Serán
presentados en la siguiente sección.
9 INSTRUMENTAL DE TRABAJO (III)
9. 1 SISTEMAS DE AGITACIÓN
9. 1. 1 RATONES
Estas famosas piezas son imanes de tamaño alargado y fusiforme,
envueltos en una coraza de teflón, lo que los hace ser elementos neutros y
seguros en el seno de la reacción (aún así se han visto ratones con
polímero plástico unido de modo inquebrantable). Reciben también el
nombre de moscas, y sirven para mover la reacción continuamente desde su

99
interior. Se mueven en círculos a causa del campo magnético que bajo el
matraz impone un agitador magnético.
Los ratones pueden tener tamaños muy variados: desde algunos
sorprendentemente pequeños, hasta otros más grandes que nuestro pulgar.
Se dice que el secreto de una buena reacción es una agitación eficiente, por
lo que al poner la reacción se debe imponer una rotación fuerte, mezclando
adecuadamente los distintos reactivos. El tamaño del ratón no debe ser
desdeñado: para disoluciones de gran volumen, ratones grandes, y para
aquellas de menor volumen, ratones pequeños.
9. 1. 2 AGITADORES MAGNÉTICOS
Llamamos agitadores magnéticos a unos dispositivos que poseen un motor
que provoca el giro de unos brazos imantados, haciendo estos girar al ratón.
Estos agitadores poseen también una segunda función, son calentadores.
No tiene sentido que podamos agitar y no calentar, cuando es la
combinación de los dos factores lo que permite que se den las reacciones.
Sin embargo, esta característica será estudiada en el bloque correspondiente
a los sistemas de calentamiento.
Las partes de un agitador son a analizadas a continuación. Todo estudiante
de química ha usado alguna que otra vez este aparato durante su
preparación, por lo que no incluiré ninguna representación de mismo.
Podemos reducir a cuatro los elementos de interés de un agitador
magnético y que se distribuyen de una u otra manera según el modelo con
el que trabajemos.
1. Botón de conexión del sistema de calefacción. Al conectarse y calentar
se enciende un piloto que se encuentra habitualmente al lado del mando.
Suele disponer el aparato de un sistema de seguridad que impide que

100
caliente la placa cuando, accidentalmente, se pulsa tal botón. En el
lateral opuesto se encuentra el botón que corresponde a la agitación y
que suele ser una copia exacta del interruptor de calefacción.
2. Control de la temperatura de calefacción que nos permite elegir la
temperatura a la que va a desarrollarse la reacción química.
3. Control de agitación que se encuentra graduado de 0 a 10 y que nos
permite al igual que en el caso anterior, elegir la agitación de la
disolución, la cual suele ser muy alta (nivel 8).
4. Placa de calefacción. Debajo está el motor que hace girar al ratón.
9. 2 TUBOS DE CENTRÍFUGA Y PINCHO
9. 2. 1 TUBOS DE CENTRÍFUGA
La centrífuga es una máquina que sirve para sedimentar mediante la fuerza
que le da nombre al aparato (provocando la traslación de la disolución
alrededor de un eje) la materia dispersada en un líquido. Para tal fin se
usan unos recipientes especiales llamados tubos de centrífuga.
Son tubos cilíndricos de vidrio extremadamente grueso, con una boca
ancha que no posee sistema alguno de sellado. Son pues, muy simples en su
constitución. La justificación del grosor del vidrio está relacionada con la
necesidad de que resista las vibraciones de la centrífuga.
Los hay de diferentes tamaños, aunque en el laboratorio se llaman según las
categorías usuales: grandes, medianos y pequeños.
Al margen de su uso principal, sirven para desoxigenar grandes cantidades
de disolvente.

101
9. 2. 2 EL PINCHO
Este es el nombre con el que se conoce a un conjunto de elementos, los
cuales, unidos, forman un instrumento muy útil, que sirve para desoxigenar
aquellos recipientes que no tienen una llave ni boquilla para unirse a las
gomas de la línea.
Hay varios tipos, dependiendo del material con el que se haga, pero la
función de todos es la misma.
1. Es una varilla hueca de vidrio, que encaja perfectamente en la goma
gruesa de la línea. Servirá por tanto para conectar al pincho con la línea.
2. Goma fina análoga a la que sirve para conducir el N2 (g) desde la
cámara 10 a las llaves de manejo. Se encuentra sólidamente cogida por una
atadura de alambre, que impide una salida inoportuna.
3. En la otra extremidad de la goma se introduce una aguja-cánula, que
puede ser de cualquier tipo según convenga. Generalmente esta aguja va
igualmente afirmada por alambre.
En algunos pinchos las partes 1 y 2 están sustituidas por una pieza metálica
cilíndrica en cuyo extremo la aguja–cánula encaja perfectamente (por un
sistema análogo al de la jeringa) y donde el otro es tan grueso como la
abertura de la goma de la línea, ajustando perfectamente.

102
De un modelo o del otro siempre encontraremos un pincho para RMN
donde la aguja–cánula es larga y muy fina.
9. 3 AGUJAS
Nos referiremos ahora a un tipo de agujas que nada tienen que ver con las
vistas anteriormente. Son agujas muy cortas y pequeñas, de gran utilidad en
las operaciones cotidianas del laboratorio.
Son agujas huecas, con una corona de plástico en el extremo no agudo, que
sirve para conectarlas en el extremo de la jeringuilla a la que pertenecía
(vienen conjuntamente con las jeringas de 1 ml, empaquetadas y con una
capucha de plástico, de aquí se cogen y se guardan, para una posterior
utilización). Sin embargo como ya hemos visto en organometálica se usan
las agujas–cánulas para la funcionalidad para la que se diseñaron por lo que
han perdido su tradicional uso.
La utilidad real por las que se usan de un modo continuo consiste en poner
a presión atmosférica el interior de un tubo con atmósfera de N2 (g) en su
interior, con la corriente de gas lógicamente abierta. Ya veremos qué
importancia puede tener esta circunstancia en nuestras experiencias.
Las agujitas se conservan clavadas en un trozo de esponja que encontramos
usualmente pegada a las paredes de la campana, es el mejor modo de
conservarlas y tenerlas siempre a mano.
Este es uno de los rasgos más característicos de un laboratorio de esta
rama, siendo curioso ver cómo semejan erizos agarrados a la pared.

103
9. 4 CÁNULAS
9. 4. 1 LAS CÁNULAS
Lo más novedoso en un laboratorio de esta índole tal vez sea este
instrumento. Básicamente es un tubo de acero de un diámetro muy pequeño
y de longitud variable. Es flexible, pudiéndose doblar en arco con facilidad,
lo cual favorece su función. Sirve para conducir un líquido desde un
recipiente a otro con facilidad, sin que entre en contacto con el aire.
El método es muy ingenioso y será estudiado en la siguiente parte. Toda
cánula posee una punta roma (usada para repasar la zona en donde está el
líquido y agotarlo) y otra afilada en punta, para pinchar el tapón del otro
recipiente.
Pueden ser gruesas, especialmente indicadas para grandes volúmenes, o
bien más finas, señaladas para cuando la cantidad de líquido no es
demasiado importante. Existe un tipo de cánula muy fina, usada para RMN,
que es en todo lo demás igual al resto.
En algunos casos, para cantidades de líquido inusitadas (por ejemplo al
hacer material de partida en masa) se usan cánulas de teflón que son muy
anchas y flexibles.
9. 4. 2 LIMPIEZA DE LAS CÁNULAS
De nuevo la imaginación ofrece su ayuda a la hora de limpiar por dentro
una cánula. Recuérdese cómo en algunas prácticas de Facultad se filtra a
vacío en un embudo Buchner, pues bien, un sistema parecido permite la
limpieza de las cánulas.

104
En lugar de un Venturi conectado a un grifo (en realidad perfectamente
válido), se usa ahora una bomba de aire comprimido o cualquier método
que permita hacer vacío en el interior de un Kitasato. La boca del Kitasato
se encuentra tapada por un tapón de goma pinchado (de gran tamaño) y
anudado con una gomilla. Evidentemente aquí no importa que su interior
esté expuesto al aire, y la única función del tapón es la de hacer efectiva la
succión sobre la cánula. Cuando se conecta el sistema y se introduce el
extremo puntiagudo de la cánula a través del tapón, en el Kitasato, se
produce una absorción por el otro extremo de la cánula. Así, no tenemos
más que sumergir ese extremo en el disolvente adecuado para limpiar el
interior de la cánula.
Detalladamente:
1. Conectar el sistema de succión (en
este caso un Venturi y una bomba de
aire comprimido).
2. Introducir la punta afilada de la cánula
en el Kitasato.
3. Llevar la punta roma al disolvente de limpieza.
La naturaleza del disolvente es algo a tener en cuenta. Si es un compuesto
orgánico normal en disolución lo que hemos hecho pasar por la cánula,
basta limpiarlo con acetona y éter. Para ello existen al lado del Kitasato dos
botellas de tales disolventes.
Si lo que se ha hecho pasar es de carácter iónico, en forma de sal, lo
anterior ya no es adecuado. Se lava primero con un poco de agua (usando el
método anterior), poniendo el agua en un vaso de precipitados para ser
absorbida. Este agua puede ser acompañada de un poco de HCl (ac.) lo que
permite una disolución más eficaz de las sales. Si se usa el ácido, después

105
se lavará con un poco de agua para eliminar los restos y la acidez. Al final
se vuelve a lavar con acetona y éter.
La secuencia lógica de lavado es:
1. Agua + HCl (ac.) para disolver las sales.
2. Agua para arrastrar restos y quitar la acidez.
3. Acetona para eliminar posibles compuestos orgánicos y arrastrar al agua.
4. Éter para secar.
El modo de lavado consiste en la realización de un continuo de pequeñas
inmersiones de la punta roma en el disolvente, tal que poco a poco lavemos
el interior del tubo. Esto es preferible a hacer una larga inmersión ya que el
gradiente de concentraciones es máximo en el primer caso.
9. 4. 3 ALMACENAMIENTO
Una vez lavadas las cánulas se colocan en una estufa. Esta estufa se
encuentra algo por encima de los 100 ºC (en general 105 ºC), lo que
facilita la completa eliminación de la humedad en el interior de las cánulas.
En toda estufa generalmente 2 bandejas, además del propio suelo del
recinto.
En el suelo se colocan todas las cánulas ya secas (aquellas que llevan de
una a dos horas). En la bandeja inmediatamente superior se colocan
aquellas que se están secando. En la bandeja tercera se encuentran algunas
sustancias que deben conservarse en condiciones anhidras (celita).
Es evidente que esta segregación de cánulas secas y húmedas es totalmente
arbitraria. Me he limitado a poner un ejemplo de cómo podría ser, pero lo
importante es señalar la distinción que debe hacerse entre unas y otras.

106
Cuando se llega al laboratorio, todas las cánulas están en la bandeja de
secado, pero como han pasado toda la noche en la estufa, están secas. Así
se pasan a la parte destinada a las mismas. Lo mismo ocurre tras el
almuerzo, ya que habiendo estado largo tiempo en la bandeja de secado
habrán de pasarse al otro lugar ya que en un par de horas, la humedad las
ha abandonado. Debe atenderse a esta ordenación y no confundirse. Dado
lo aparatoso de su longitud, las cánulas se suelen introducir en la estufa
doblándolas sobre sí formando una especie de lazo. Se fuerzan y se
entrelazan, formando con ellas un círculo, lo que permite depositarlas y
volver a sacarlas con facilidad. No obstante y a pesar de enlazarlas sobre sí
muchas veces se enredan entre ellas dificultando su extracción de la parte
inferior de la estufa.
Aunque están a 105º C las cánulas no queman puesto que son muy finas.
Las que poseen un mayor grosor pueden llegar a ser molestas, pero no lo
suficiente como para tener que coger los guantes. Hay que cuidarse sin
embargo de unas cánulas que poseen un cilindro metálico en su extremo
(son cánulas que sirven para filtrar como ya veremos), el cual quema
verdaderamente cuando se coge con firmeza.
10 INTRODUCCIÓN Y EXTRACCIÓN DE RATONES
10. 1 INTRODUCCIÓN DE RATONES
Para introducir un ratón se deben considerar las condiciones bajo las cuales
se va a realizar la operación. Puede que no hayamos hecho aún vacío y
tengamos el matraz al aire. A causa de ello no tenemos que cuidarnos de la
entrada de aire. Se coge el ratón con una de las pinzas (análogas a las que
se usan en la depilación pero de mayor longitud) y se lleva a la boca del

107
matraz. La otra mano se coloca como sostén, bajo el fondo redondo del
matraz. Se suelta entonces el ratón y se termina el asunto. La utilidad de
cubrir el fondo con la mano es la de absorber las vibraciones del choque,
que podrían llegar a quebrar el vidrio.
Si ya se hubiese impuesto la atmósfera inerte habría que quitar el tapón con
el N2 (g) abierto e introducir un poco el ratón con las pinzas en el matraz.
Se mantiene así un tiempo para que el gas inerte arrastre el aire que pueda
haber quedado ocluido en su superficie. Después y protegiendo el fondo
con la mano, se suelta el ratón.
10. 2 EXTRACCIÓN DE UN RATÓN
Puede ocurrir que al término de una reacción, por algún motivo nos veamos
precisados a sacar el ratón del medio, sin que deba entrar aire. Para tal
menester existe un cazarratones (al que yo preferiría llamar gato por
coherencia). Es una varilla de material plástico inerte, lo suficientemente
larga como para llegar al fondo de cualquier tubo, y tan estrecho que cabe
por toda boca de un diámetro mediano. En su extremidad inferior se oculta,
rodeado de plástico, un imán. Haciendo bajar lentamente esta punta de la
varilla por el matraz, se logra atrapar el ratón por magnetismo.
11 SE TRANSVASA EN GRAN CANTIDAD
Cuando se requiere un volumen de disolvente mayor que el que se puede
contener en una jeringa, por comodidad se usa un sistema que permite
disponer de disolvente seco en grandes cantidades.

108
En estos casos se pide auxilio a los tubos de centrífuga y al pincho cuya
principal misión es la de desoxigenar
11.1 DESOXIGENACIÓN EN TUBO DE CENTRÍFUGA
Tomamos en un tubo de centrífuga la cantidad de disolvente necesaria, que
siempre es más de la que realmente necesitaríamos, de no tener que
desoxigenar. Esto es razonable, ya que parte del disolvente, como veremos,
va a ser arrastrado por el gas.
El tubo de centrífuga se fija con una pinza y se le coloca un tapón de goma
pinchado. A un tubo de goma de la línea le encajamos el pincho, y damos
paso al N2 (g). Tras esto, por el pincho saldrá una fina corriente del gas
inerte. Es conveniente elegir uno de los pinchos más gruesos para que la
velocidad en la desoxigenación sea mayor. Se clava en el tapón pinchado y
se introduce hasta el fondo del tubo. Allí comenzará a burbujear,
arrastrando el oxígeno que haya absorbido durante el traslado.
Para forzar una salida del gas, cargado con el oxígeno y parte del
disolvente, se clavan varias agujitas en el tapón. Estas conectan interior con
exterior. Como hay una diferencia de presión a favor del interior, se
produce una tumultuosa salida del gas hacia fuera con un silbido agudo.
La situación se mantiene un tiempo, dependiendo del volumen del
disolvente, pero siempre superior a 5 minutos.
Podemos incluso recurrir a colocar, en lugar de las agujitas, una de las
agujas–cánula, lo que permite una salida de gas en mayor cantidad y así
una desoxigenación más rápida.

109
11. 2 PASAMOS EL DISOLVENTE A UN MATRAZ
Una vez desoxigenado, tenemos que pasarlo al matraz para que sirva de
medio de reacción. De este modo podemos tener en el matraz esperando un
sólido y el correspondiente ratón.
En el matraz debe haber atmósfera inerte antes de trasvasar el líquido.
Se toma una cánula y se pincha por su extremo romo en el tapón de goma
del tubo de centrífuga, sin eliminar el pincho y sin llegar a sumergirla. Se
espera unos segundos a que el gas, gracias a la presión ocasionada por el
gas del pincho, salga por la cánula, arrastrando el aire contenido en la
misma. En tanto quito las agujitas del tapón para que la presión sea más
efectiva y aprovecho para cambiar el tapón nuevo del matraz por uno
pinchado.
Lo siguiente es llevar la punta de la cánula contra el tapón del matraz,
atravesarlo y situarla hacia la mitad del recipiente.
Con ambas manos se coge la cánula justo por el trozo que se encuentra
inmediatamente por encima de la superficie de cada tapón, para controlar la
cantidad de líquido que vamos a añadir, así como para repartirlo
uniformemente por las paredes (ya que al añadir el sólido suele quedarse
cierta cantidad sobre las paredes del matraz).
Se baja la cánula por su punta roma hasta sumergirla en el disolvente. Al
principio no pasará ningún líquido, ya que las presiones en ambos
recipientes son las mismas. Para que se produzca el trasvase deseado se
debe establecer una diferencia de presión entre un recinto y otro, sin que
ello conlleve una entrada de aire.
Para facilitar la comprensión del proceso he aquí un esquema de las
posiciones generales que tienen los diferentes elementos indicados con
anterioridad, y cómo se conectan entre sí de modo adecuado.
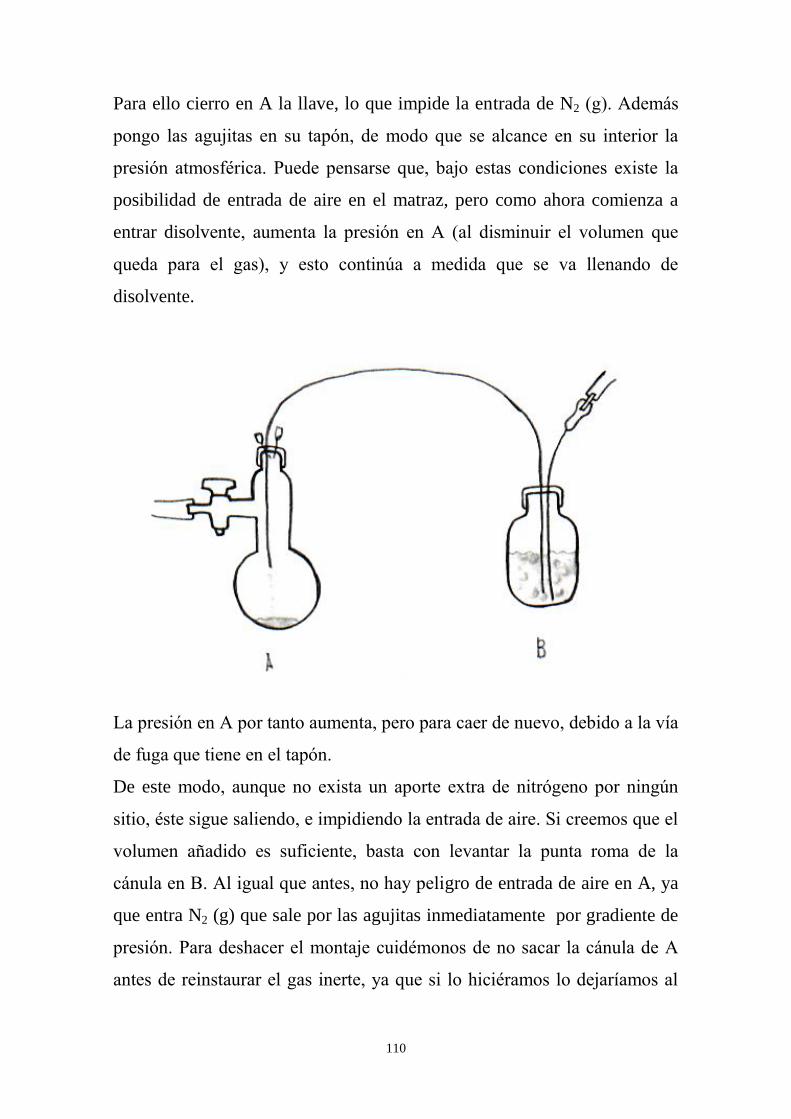
110
Para ello cierro en A la llave, lo que impide la entrada de N2 (g). Además
pongo las agujitas en su tapón, de modo que se alcance en su interior la
presión atmosférica. Puede pensarse que, bajo estas condiciones existe la
posibilidad de entrada de aire en el matraz, pero como ahora comienza a
entrar disolvente, aumenta la presión en A (al disminuir el volumen que
queda para el gas), y esto continúa a medida que se va llenando de
disolvente.
La presión en A por tanto aumenta, pero para caer de nuevo, debido a la vía
de fuga que tiene en el tapón.
De este modo, aunque no exista un aporte extra de nitrógeno por ningún
sitio, éste sigue saliendo, e impidiendo la entrada de aire. Si creemos que el
volumen añadido es suficiente, basta con levantar la punta roma de la
cánula en B. Al igual que antes, no hay peligro de entrada de aire en A, ya
que entra N2 (g) que sale por las agujitas inmediatamente por gradiente de
presión. Para deshacer el montaje cuidémonos de no sacar la cánula de A
antes de reinstaurar el gas inerte, ya que si lo hiciéramos lo dejaríamos al

111
descubierto. Por tanto, se abre la llave del gas y se retira la cánula. Se
cambia el tapón por uno nuevo, y el proceso queda concluido.
En cuanto al tubo de centrífuga con el disolvente que reste se le retira la
cánula y el pincho, después se le puede poner un tapón nuevo
(exponiéndolo un segundo al aire) para usar más adelante si hiciere falta
(debiendo desoxigenar de nuevo).
Este método visto para trasvasar líquidos sin que el aire venga a afectarlos,
se usará mucho tanto para disolventes como para reactivos, e incluso para
las operaciones de separación y purificación de los productos obtenidos,
por lo que conviene conocerlo bien.
12 EL FRÍO
Al hacer por primera vez una reacción no sabemos qué puede ocurrir al
mezclar los dos reactivos. Generalmente se intuye cuando se tiene
experiencia lo que sucederá, pero nadie apostaría gran cosa si las
reacciones no se han sacado de otras paralelas donde ya se conocen los
resultados.
Para evitar cualquier mal innecesario se recurre a enlentecer las reacciones
desde el principio e irlas poco a poco acelerando, observando atentamente
cualquier cambio en la coloración de la disolución, en su aspecto, densidad
o alguna referencia que pueda controlarse por la vista.
Para lograr conocer todas las etapas de la reacción, se adicionan los
reactivos en frío. Enfriamos uno de ellos (o los dos) y se ponen en contacto.
Una vez hecha la adición se deja enfriando un tiempo para luego retirar el
foco frío, por lo que la temperatura asciende paulatinamente hasta la
ambiente. Si en tal caso se observa que la reacción necesita más calor,

112
tendríamos que acudir a las técnicas de calentamiento, que son siempre
posteriores a la adición de reactivos.
12. 1 EL DEWAR
Un Dewar es un recipiente diseñado para mantener el mínimo intercambio
de calor con el medio, sirviendo por ello para contener al líquido
congelante más utilizado, el N2 (l).
Tienen diversas formas dependiendo del uso que se vaya a hacer de ellos,
pero en general, podemos ver que son cilíndricos y con una muy gruesa
capa de material aislante.
Ambos modelos son muy comunes,
prefiriéndose A para el transporte y B
para los baños fríos.
Los Dewar de tipo A, como se ha
indicado, se usan para el transporte
de N2 (l), portándolo desde la fuente
del mismo hasta su lugar de uso. Entre ellos hay 2 tipos que se diferencian
en el radio. Los de mayor radio tienen la función indicada, sirven para
transportar el líquido, en tanto los de menor radio se utilizan como trampas
de frío para la línea, conteniendo la ampolla que se une a la cabeza de la
línea (visto ya en el tema 1).
En esta clase de recipientes el N2 (l) puede mantenerse durante horas sin
sufrir una excesiva evaporación. Sin embargo esto último depende en
sobremanera de la temperatura del laboratorio. En invierno pues, soporta
muchas horas, pero en verano se consume más rápidamente. Para
solucionar en parte la evaporación del N2 (l) en cualquier época de año, se
opta por poner un paño tupido sobre la boca del recipiente. El gas

113
evaporado sube muy frío y fuerza la condensación del vapor de agua sobre
la tela, formando una capa de escarcha que impide el intercambio de calor
debido a su carácter aislante.
Los Dewar de tipo B son utilizados para contener las mezclas frías que se
usan para ralentizar las reacciones, de ahí su uso como baños. Su poca
altura y su gran diámetro y capacidad, los hacen perfectos para ello. Su
fondo puede ser plano o cóncavo, sin embargo son los segundos los más
utilizados.
Los Dewar están formados por una cubierta exterior de aluminio (pintada
de azul), en tanto el interior es una superficie especular opalescente de
vidrio de borosilicato.
Hay otros tipos semejantes a termos, donde la apertura está cerrada por una
superficie abatible, a guisa de puerta, pero no son muy habituales.
12. 2 TOMAMOS N2 (l) DEL TANQUE
El nitrógeno líquido, en un laboratorio de organometálica se gasta con una
velocidad muy alta; se consume en litros y siempre con la intención de
usarlo como foco frío.
Para su distribución, a partir de un gran contenedor que se encuentra fuera
del recinto destinado a los laboratorios generalmente, se usan unos
cilindros o bombonas metálicas de gran tamaño que depositados sobre una
base con ruedas, o con estas mismas ruedas incorporadas a su estructura,
permiten transportar sin problema una gran cantidad de nitrógeno líquido.
Estos tanques poseen una boca en su parte superior, que sirve para el
llenado y posterior salida del N2 (l). Allí encontramos encajada y bien

114
sujeta, una varilla (la lanza), a través de la cual circula el N2 (l) cuando es
requerido.
Por lo general este tanque se halla
situado en el laboratorio donde se
trabaja, de tal modo que cada uno
de los laboratorios dispone de uno
de ellos. Los encargados de su
mantenimiento y de su llenado
cuando esto es necesario son los
propios investigadores. Como el
tanque es bastante grande se sitúa
en algún rincón del recinto donde
se pueda acceder con facilidad
puesto que los Dewar se llenan muy a menudo, pero también de fácil salida
porque el tanque se debe sacar para llenarlo, y es preciso que tenga vía
libre.
Una representación del mismo la encontramos arriba, detallándose sus
partes principales:
1. Encontramos el recipiente con forma de bala, donde se contiene el
nitrógeno líquido.
2. Constituye un accesorio que se adapta a la boca del tanque. Permite
un cierre perfecto. A él se fija una goma que lo conecta a una corriente
de N2 (g) que sirve para impulsar, por presión al N2 (l) por la lanza.
3. La lanza. Es el canal por donde mana el nitrógeno. Se encuentra unido a
2 y continúa su curso hacia abajo, para llegar a contactar con el líquido
del interior.

115
Este sistema de presión que hace salir al N2 (l) se encuentra regulado por
una llave que permite que entre al interior N2 (g) y aumente la presión. El
líquido se ve forzado a ascender por la lanza, hasta caer al recipiente que
coloquemos al otro extremo. Nótese que este sistema es idéntico al usado
anteriormente para pasar el disolvente del tubo de centrífuga al matraz,
mediante la cánula.
Tenemos el dibujo de la llave que conecta la salida de N2 (g) del
suministrador con la bala de N2 (l). La goma b lleva N2 (g) hasta la llave.
Esta llave posee tres posiciones:
1. Permite la entrada del N2 (g).
2. Cierra la entrada al gas.
3. Conecta la goma del gas con el
aire.
De este modo, 1 hace que salga el líquido, 2 aísla el sistema, y 3 elimina el
gradiente de presión e impide que salga más nitrógeno líquido cuando ya
hemos llenado nuestro Dewar. En esta posición debemos siempre dejar la
llave, para que no se produzcan sobrepresiones inesperadas.
Detallaremos a continuación las acciones principales para extraer el líquido
del contenedor:
1. Colocar el Dewar debajo de la lanza, sobre un banco o soporte alguno.
2. Desplazar el brazo de la llave de la válvula en la posición que permite el
paso de la corriente de gas inerte.

116
3. Se da paso al N2 (g) con el manorreductor, lentamente, para no provocar
una salida vehemente del líquido. Si el volumen de N2 (l) existente en el
tanque es elevado, saldrá casi inmediatamente, mas si queda poco tardará
más en salir, hasta que al final sólo salga por la lanza la corriente de gas.
4. Antes de que se llene completamente el Dewar se cierra el flujo gaseoso
con el manorreductor. La presión remanente opera durante un tiempo y
sigue saliendo N2 (l) hasta llenar completamente el Dewar.
Hay que notar que al principio, cuando el N2 (l) cae sobre el Dewar aparece
una gran humareda blanca muy espectacular, correspondiente al vapor de
agua condensado. Al mismo tiempo que sobre la superficie metálica de la
lanza se forma escarcha.
Para coger el N2 (l) se aconseja usar guantes, pero no es necesario si se
tiene el suficiente cuidado. Suelen caer gotitas muy menudas del líquido en
la piel durante el transporte, pero no hacen verdadero daño, sintiéndose la
sensación de ser aguijoneado por diminutos alfileres. De todos modos los
guantes tienen su función y conviene acostumbrarse a su uso, más que a
despreciarlos. Los guantes que se utilizan en estos menesteres no son los de
látex, obviamente, sino otros de tela gruesa que se emplean también para
sacar instrumentos y recipientes de vidrio de la estufa.
12. 3 LLENAMOS EL TANQUE
El tanque anterior, como promedio, suele agotarse antes de una semana de
uso. Es costumbre que aquel que lo encuentre vacío vaya a llenarlo (aunque
tal vez sería más conveniente establecer un turno). Se desencaja
previamente la lanza, lo cual se hace quitando las palometas que fijan la
pieza 2 y haciéndola rotar a ésta sobre su base hasta que se libera.

117
Se suelta el conjunto formado por la pieza 2 y se deja la lanza en un lugar
conveniente del laboratorio mientras se lleva el recipiente vacío
arrastrándolo sobre sus ruedas hasta la caseta de gases. Esta caseta de gases
se construye siempre en un lugar muy ventilado por razones obvias,
encontrándose por tanto en patios o lugares abiertos. Realmente, por caseta
de gases se conocen varias dependencias donde se almacenan balas de
gases, aunque claro, aquí nos referimos a la correspondiente al N2 (l).
Al margen de lo contado antes, debemos llevarnos una pareja de guantes
gruesos y un tapón para la boca del tanque, que puede ser de goma o de
corcho, y que debe encontrarse siempre al lado de la localización del
tanque en el laboratorio.
Cada centro tiene un tanque de grandes dimensiones de N2 (l) el cual se ve
muchas veces desde el exterior y se puede reconocer sin dificultad. Al lado
de estos depósitos se encuentran las habitaciones pertinentes donde poder
llenar los tanques del laboratorio. Del depósito baja un tubo que suele
entrar en el recinto a través de una abertura adecuada en la pared,
terminando en una lanza semejante a la de nuestro tanque. Esta lanza tiene
un elemento cónico que sirve para soportarla cuando introduzcamos su
apéndice por la boca del tanque, de modo que su punta no toque el fondo.
De este modo, se apoya la lanza en la boca de la bala y se abre la llave que
da paso al nitrógeno líquido. Lentamente, además del sonido habitual, se
puede observar cómo va apareciendo una densa niebla que se forma
alrededor de nosotros y que sale hacia el exterior en grandes volutas. Es
conveniente salir de allí a respirar el aire fuera, ya que la gran producción
de N2 (g) desplaza al oxígeno en un cierto grado. Se comprende entonces
que la localización de la instalación se realice en un espacio abierto
(cuando uno se encuentra dentro de la caseta se experimenta cierto ahogo,
seguramente por sugestión).

118
En torno a los 15 minutos (hay modelos de capacidades distintas por lo que
el tiempo de llenado oscila entre 10 y 15 minutos) se puede regresar al
interior para cerrar la llave del N2 (l). No se puede llegar tarde, pues el
líquido rebosaría del recipiente, con el consecuente peligro y gasto
económico.
Con los guantes se cierra la llave, la cual se ha enfriado tanto, que no
permite ser tocada sino con la protección adecuada. Se coloca el tapón y se
regresa al laboratorio.
En la caseta suele haber una tabla de control que informa cuándo los
diferentes grupos han llenado su tanque mediante el registro que hacen los
propios usuarios, por lo que, antes de marcharse, se resuelve la formalidad
apuntando la hora, día y grupo al que se pertenece.
Dado que la caseta está en el exterior, suele haber un tramo largo hasta
alcanzar el laboratorio de nuevo. Durante este trayecto hay que tener
mucho cuidado con no volcar el tanque (cosa que no es imposible pues he
notado alguna inestabilidad en el equilibrio de algunos diseños). Si
sucediere, circunstancia lamentable, no puede aconsejarse otra salida que la
de correr y abrir cuantas ventanas y puertas sean necesarias, avisando a los
compañeros para que hagan lo mismo. El volumen de aire que desalojaría
sería bastante grande como para tener problemas.
Ya en el laboratorio se monta el sistema teniendo la precaución de situar
debajo de la lanza un Dewar, pues al insertar ésta, parte del N2 (l) sale por
su punta y no está de más recogerlo para aprovecharlo posteriormente.
12. 4 EL NITRÓGENO COMO LÍQUIDO
Hasta ahora nos hemos referido al nitrógeno en este estado de agregación,
como si fuese algo conocido y no requiriese un apartado propio. Sin

119
embargo me parece demasiado singular y característico como para dejarlo
pasar sin dedicarle al menos unas líneas.
El N2 (l) es sin dudas una sustancia curiosa y sorprendente a pesar de hablar
desde el conocimiento que da el tener una fuerte formación científica.
Es líquido cuando debiera ser gas, y la mente, que comprende la razón por
la cual es y se mantiene líquido, involuntariamente espera que pase
rápidamente a gas; pero persiste, casi inmutable, desafiando la aparente
lógica de los sentidos.
Al ser un líquido transparente e incoloro, uno no puede sino compararlo
con el agua, pero desde el principio, su comportamiento es radicalmente
distinto.
Si derramamos un poco de N2 (l) sobre la mesa observamos que las gotitas
que se forman no se quedan quietas y estancas como el agua, sino que
corren a una velocidad sorprendente debido a la evaporación de las capas
que rozan la superficie lo que suspende a la gotita en un colchón gaseoso
que elimina el rozamiento. Podemos de esta manera darnos cuenta de la
inferior fuerza de cohesión que presentan sus moléculas entre sí. Aún así es
evidente que su evaporación impide que la comparación pueda resultar
plausible.
Si lanzásemos un poco desde una cierta altura, teniendo intención de que
caiga al suelo, no llega a caer, se evapora antes; lo cual nos deja pasmados,
pues (de nuevo los sentidos) se esperaba que, como el agua, chocase contra
el suelo y salpicase en todas direcciones, y lo que tenemos a cambio es una
bruma que se deshace a nuestros pies.
En terreno más serio diremos que el N2 (l), ora líquido ora gas, es muy
poco reactivo y su uso se ve limitado al de líquido térmico o atmósfera
inerte. En este segundo caso, a veces puede reaccionar como lo haría un
carbonilo, pero su naturaleza le hace ser la atmósfera artificial más

120
fidedigna para salvaguardar los compuestos inestables al aire (conjugando
esto con la vertiente económica).
Finalmente citemos que la temperatura del N2 (l) es de –196 ºC, o sea de 77
K, lo que le hace ser muy útil en todas aquellas funciones en las que lo
hemos utilizado, antes de haberle presentado.
Me he extendido un poco, haciendo referencia a elementos tal vez fuera del
propósito de este trabajo, pero que me han parecido interesantes,
sorprendiéndome mucho en su tiempo y resultándome gratos de observar y
analizar por lo que he decidido incluirlos en el texto. Las curiosas
características químicas que se exhiben en el nitrógeno líquido son tan
numerosas como fascinantes y merecerían un análisis aún más extenso que
éste que he realizado en este apartado.
12. 5 OBTENCIÓN DE CO2 (s) EN PASTILLA
El otro gran aliado para lograr una eficaz disminución de la temperatura es
el CO2 (s). El método para obtener una pastilla del compuesto es muy hábil,
usando el efecto Joule a su conveniencia.
En una bala grande se contiene a presión una gran cantidad de CO2 (l) del
cual formaremos la pastilla. Se abre la llave que da
paso al líquido a través de un tubo conductor que
desemboca en una caja en cuyo interior se formará la
pieza sólida. Al llegar a la caja se produce una
expansión repentina con el consecuente enfriamiento
y el CO2 pasa a estado sólido en forma de polvo.
Lentamente se va acomodando en el espacio interior y
se va formando la pastilla.

121
Una vez que la cámara se copa con todo sólido que puede contener,
empieza a expulsar por las rendijas el que entra en exceso. Este sale a gran
presión provocando un silbido al tiempo que se ven finos chorros
partiendo de la caja. Es tiempo entonces de cerrar la llave.
El siguiente paso es desmontar la cámara, lo que se
hace con guantes gruesos (hay personas que lo hacen
sin guantes y no sufren molestia alguna por la baja
temperatura). Lo podemos describir del siguiente
modo:
La cámara desmontable es de madera y
tiene la forma que indica el dibujo. Se ve
cómo el tubo entra por cara cenital. Esta
cara está unida al tubo y es la primera que
se quita. Nótense los broches que la unen
a las caras laterales.
Se abren las 4 hebillas de la tapa superior con los guantes, de modo que
queda tal como muestra la figura. El tubo que apresa la cara desmontada
está muy frío, al igual que los cierres, por lo que los guantes se hacen
necesarios. Ya se puede apreciar en la parte de arriba la superficie blanca
de la pastilla.
Las paredes laterales donde se atornillaban los broches se abaten en virtud
a las bisagras que se encuentran en la parte inferior de las mismas y como
consecuencia de ello, dos nuevas superficies blancas aparecen en su lugar.
Tal y como se puede apreciar en la tercera imagen la pastilla de dióxido de
carbono está ya perfectamente formada en el interior de la caja. La pareja
de paredes restantes están encajadas en rieles. Se desencajan y se libera el
bloque de hielo seco. La pastilla de hielo seco se puede coger entonces con
los guantes y se envuelve en un paño de tela gruesa, donde se conservará
convenientemente hasta que llegue la hora de utilizarlo. Es también muy

122
útil el paño ya que durante el transporte la pastilla se puede fragmentar o
desmoronar.
Para montar el sistema no hay más que invertir cada uno de los pasos
descritos.
En realidad el hielo seco en forma de pastilla obtenido por este
procedimiento posee gran cohesión pero al ser la pastilla de tamaño
considerable una vibración la puede fragmentar sin esfuerzo.
Hay otras maneras de obtener el hielo seco que lo generan en forma más o
menos granulada (en forma de granalla), pero el sistema es básicamente el
mismo.
12. 6 EL CO2 SÓLIDO
Tan útil como el N2 (l) es el CO2 (s) para hacer baños fríos. Dadas sus
características especiales es muy adecuado y manejable. Los baños fríos
preparados con él duran más que aquellos preparados con N2 (l), aunque su
temperatura no es tan baja. Es justo pues utilizarlo cuando la temperatura a
exigir no sea extrema (esto último ocurre rara vez) y por tanto se usa más a
menudo el hielo seco.
Pasa directamente del estado sólido al gas, esto es, sublima, por lo se
explica el nombre anteriormente dado de hielo seco. De hecho si se
introduce un trozo en un vaso de precipitados con agua, vemos que
alrededor del mismo se forma una coraza de agua congelada, pero detrás se
ve perfectamente seco (efecto sorprendente para la vista) y siempre
congelando el agua que toca al mismo tiempo que sublima y su vapor frío
provoca que condense el vapor de agua de la atmósfera formando una
neblina.

123
La temperatura de sublimación es de - 80 ºC, y este es su límite de
enfriamiento cuando se utiliza en los baños fríos. Durante horas se puede
dejar la pastilla al aire libre sin casi afectarse, aunque como siempre,
depende de la temperatura de la sala.
12. 7 BAÑO FRÍO DE ACETONA Y CO2 (s)
Es la combinación más habitual. Logra enfriamientos cercanos a los - 80 ºC
durante largo tiempo, pudiéndose mantener la baja temperatura por la
adición continuada de hielo seco.
Cogemos uno de los Dewar indicados previamente para su uso como baño
y lo llenamos hasta la mitad con acetona (en todo laboratorio hay un bidón
con acetona para estas necesidades y para llenar el dispensador). La pastilla
de CO2 (s) se corta con la espátula si se quiere, pues no requiere fuerza
alguna para trocearla. Protegidos con un guante machacamos en la mano
uno de los trozos de CO2 (s) y lo añadimos desgranado. Se produce un
burbujeo por la sublimación del hielo seco que se reduce a medida que
vamos añadiéndolo. Al final, la temperatura de la acetona es la del CO2 (s)
y se establece un equilibrio térmico entre ambos compuestos. Esta mezcla
es muy estable y la temperatura no asciende sino muy lentamente.
Esta mezcla absorbe gran cantidad de agua de la atmósfera por lo que la
temperatura a la que desciende es superior al punto mínimo, no
superándose el - 78 ºC, sino quedándose bastantes grados por encima.
Se presentan a su vez problemas cuando la acetona se ha utilizado varias
veces y lleva algún tiempo en el Dewar. En realidad no tendría por qué ser
un inconveniente, pero la acetona puede encontrarse saturada de CO2 (g) y
no admite más, por lo que el enfriamiento se ralentiza sobremanera.
Aunque se añada de nuevo hielo seco, la temperatura puede incluso quedar

124
estancada entre los - 20 ºC y los - 30 ºC. La solución es coger acetona
nueva y empezar los preparativos.
Una última ventaja resulta del análisis económico, pues el CO2 (s) es más
barato que el N2 (l), por tanto se prefiere el primero a la hora de hacer un
baño frío si la temperatura no es inferior a los 78ºC bajo cero.
12. 8 BAÑO FRÍO DE ACETONA Y N2 (l)
Puede ocurrir que la reacción requiera un enfriamiento por debajo de los
80ºC bajo cero que logra la pastilla de hielo seco. En tal caso usamos el
nitrógeno líquido.
Llenamos un Dewar con acetona hasta su mitad y añadimos N2 (l) con
cuidado. Un humo blanco muy denso borbotea y se extiende como una
densa niebla por la superficie de la mesa. Llama la atención la opacidad y
la blancura tan intensa que exhibe. Poco a poco deja de humear. Si
vertimos de nuevo otra cantidad de N2 (l), la acetona se hace más densa y
comienza a congelar en forma de grandes escamas o conchas blancas.
Una vez se congela la parte de arriba, podemos romperla con una espátula
y emplear el baño ya, pues un enfriamiento mayor se hace inasequible. La
acetona se funde casi inmediatamente, siendo muy difícil mantener este
límite de temperatura.
El rango de trabajo de este tipo de baños por tanto va desde los -79ºC hasta
los -95ºC (donde se solidifica la acetona). Una temperatura inferior no es
necesaria.

125
12. 9 TERMÓMETROS DE BAJA TEMPERATURA
Quizás tengamos la forzosa imposición de hacer una reacción en un rango
estricto de temperatura, inferior al cero centígrado. Para determinar, por
tanto, la cantidad de CO2 (s) que se añade, se utiliza un termómetro en
baja. Es un termómetro normal, cuyo rango oscila entre los 20 o 30 ºC y los
- 100 ºC.
Cuando se meten en la mezcla fría es razonable no usarlos como si fuesen
varillas de vidrio para agitar. Tampoco es conveniente dejarlos dentro de la
mezcla sin un apoyo externo que impida que se caigan, pues son bastante
largos, y resbalan, partiéndose al golpe.
12. 10 PROBLEMAS CON LA PRESIÓN
Imaginemos que en el proceso de reacción quitamos el baño frío tras añadir
el reactivo para que vaya alcanzando progresivamente la temperatura
ambiente y la reacción se vaya completando.
Al añadir el reactivo, se ha abierto la llave de N2 (g) para cambiar el tapón
y proteger la posterior adición. El nitrógeno que entra se enfría rápidamente
y su volumen disminuye notablemente. Cuando se vuelve a poner el tapón
nuevo y después se cierra la entrada al gas, la cantidad de gas que
permanece en el interior del matraz es muy superior a la que habría en
condiciones normales. Al alcanzar finalmente la temperatura ambiente, la
presión será mucho más elevada que la que conviene, pudiendo hacer saltar
el tapón o sufrir la línea una sobrepresión al volver a abrir la llave del gas.
Para solucionar este problema, antes de dejar al matraz que alcance la
temperatura ambiente damos un golpe de vacío (con la llave de manejo se
abre el vacío y a continuación, con la llave del matraz, se hace un giro muy

126
rápido de manera que se produzca una succión durante una fracción de
tiempo mínima). Así se disminuyen los moles de gas existentes en el
recipiente y al retomar la temperatura del laboratorio la presión será
aproximadamente la de siempre.
No debe olvidarse esta circunstancia que podría, por pérdida del tapón,
destruir la reacción desde el principio.
13 ADICIÓN DE REACTIVOS
Cuando la disolución se ha enfriado (lo cual se logra dejándola en agitación
en el baño durante unos 15 minutos) debemos mezclar los reactivos, uno de
los cuales suele encontrarse ya en el disolvente, en tanto el otro debe ser
aún adicionado.
En general, una vez se adiciona, el matraz permanece otro tiempo igual al
anterior en agitación dentro del baño, para que la nueva sustancia adquiera
la temperatura establecida. Transcurrido el tiempo, quitamos el baño y
dejamos que la temperatura ascienda.
13. 1 ADICIÓN DE SUSTANCIAS SÓLIDAS
Una sustancia sólida, como tal, no suele adicionarse en esta rama de la
química que tratamos. Siempre se trata de disolver previamente el
compuesto en el disolvente adecuado y posteriormente añadirlo a la
disolución del matraz en donde se encuentra el otro producto. Por esta
razón trataremos este punto como una parte dentro de las adiciones de
líquidos en el bloque siguiente.

127
13. 2 ADICIÓN DE LÍQUIDOS
La gran mayoría de las reacciones requiere reactivos en estado líquido o
en disolución. Dependiendo de las características de la reacción y de los
propios reactivos, encontraremos uno u otro tipo de adición.
13. 2. 1 DISOLUCIÓN PROCEDENTE DE UN SÓLIDO
Aquí el reactivo al principio se encontraba en estado sólido pero para
proceder a la adición se debe disolver adecuadamente, usando para ello el
mismo disolvente que se utiliza como medio de reacción.
Se pesa la cantidad exacta de reactivo, usando las técnicas ya conocidas y
se introduce en un Schlenk, donde hacemos vacío y salvamos el compuesto
bajo atmósfera de N2 (g) aunque sea estable al aire, puesto que lo debemos
añadir en ausencia completa de oxígeno y de humedad.
El compuesto se disuelve en la menor cantidad de disolvente posible (no
olvidemos que después habrá que eliminarlo) y sabiendo que no podemos
excedernos mucho en la carga al matraz (no debemos llenarlo por encima
de las ¾ partes de su volumen porque el espacio disponible para el gas
inerte y para otros posibles que se puedan generar es muy pequeño y
aumentaría la presión).
En este proceso de disolución podemos necesitar el concurso de un ratón,
que vendría a acelerarlo enormemente.
Tenemos pues el matraz bajo N2 (g) y también el reactivo en disolución en
el Schlenk bajo atmósfera inerte. Para pasar la disolución al matraz se
siguen los siguientes pasos:

128
1) Se quitan ambos tapones y se sustituyen por tapones pinchados.
2) Se toma una cánula y se inserta su punta roma en el Schlenk,
dejando transcurrir unos segundos para que se desoxigene su
interior. Su punta aguda se pasa al matraz, de modo que no
toque las paredes, sino que caiga directamente encima de la
superficie del medio de reacción. Si tocase las paredes,
perderíamos parte del producto.
3) Se cierra la llave de N2 (g) en el matraz y se coloca una aguja en
el tapón del mismo. Así pasará el líquido con el reactivo a la
disolución del matraz, donde se dará lentamente la reacción.
4) Se abre de nuevo el N2 (g) del matraz y se retira la aguja.
5) Se lava con un poco de disolvente el fondo del Schlenk para no
perder nada del producto que pudiera haber quedado por las
paredes, y se vuelve a repetir la operación anterior.
6) Ahora sí se concluye abriendo en N2 (g) en el matraz y
cambiando de tapón, colocándose uno nuevo.
7) Se da el golpe de vacío, para eliminar el riesgo de la
sobrepresión.
8) Se fijan gomillas en el tapón y se pone la gomilla de seguridad
como se indicará más adelante, tras cerrar la llave del matraz.
El Schlenk que se use para disolver vale la pena que sea el de llave de
teflón, pues nos ahorraremos mucho tiempo. Basta con limpiarlo con
acetona y éter para poderlo utilizar una vez seco. Si usamos el otro hay que
meterlo en la mezcla de potasa alcohólica y esperar al día siguiente.

129
13. 2. 2 DISOLUCIONES Y LÍQUIDOS
En este grupo encontramos aquellos compuestos que se encuentran
guardados en ampollas, bajo atmósfera de N2 (g), en forma de disoluciones
cuya molaridad es conocida y clave para saber qué volumen debemos
adicionar. A su vez engloban a aquellos líquidos que se guardan en
recipientes comunes y cuyo interior está constituido por aire normal.
Debido a que ahora el volumen es la magnitud a medir, es preciso obrar
con jeringas, usando las de plástico del modo que ya es conocido.
Lógicamente las microjeringas también se emplean para estos reactivos.
Hay por tanto dos métodos de actuación bien diferenciados, según se tenga
que añadir reactivos almacenados en ampollas o reactivos guardados en
botes comunes.
A) Para ampollas bajo N2 (g).
Los compuestos que se contienen en ampollas, ya sean puros o en
disolución, se guardan protegidos bajo atmósfera inerte. Suelen contener
reactivos delicados, por lo que o son caros o son de síntesis compleja y
engorrosa, debiéndose mantener a salvo durante todo el tiempo que dure su
manipulación.
Se coge la ampolla y se une al tubo de la línea por su salida lateral. Como
en la goma hay aire, se desoxigena haciendo los 3 ciclos consabidos.
Aquí, la duración de las etapas es mínima, porque el volumen interno del
tubo es muy pequeño y con unos cuantos segundos basta para el objetivo.
Cuidémonos de no abrir la llave cuando hagamos vacío, pues el disolvente
(y el producto con él) se marcharían, y esta negligencia costaría cara.

130
Ya con el N2 (g) llenando el tubo, se abre la llave y se cambia el tapón de
teflón por uno de goma pinchado. La boca de las ampollas es muy pequeña
y la puesta del tapón se puede hacer de dos maneras:
1) Usando un tapón pequeño, del diámetro
justo, como hasta ahora se ha ido haciendo.
2) Empleando un tapón grande (del 49) cuyo
hueco inferior recibe a modo de hembra, a la boca del
tubo, ajustando perfectamente.
La boca de la ampolla es tan estrecha que se puede acoplar al tapón. Este
sistema, que no se ha comentado con anterioridad es el que más se utiliza,
y resulta seguro, no temiéndose ninguna entrada de aire ni una excesiva
salida de nitrógeno.
Cuando interviene la jeringa, lo que uno no puede olvidar es que su aguja–
cánula se halla llena de aire, debiéndose desoxigenar completamente. La
desoxigenación de la jeringa se logra tomando N2 (g) de la ampolla y
expulsándolo tras retirarla del interior. Estas toman se repiten unas cuantas
veces (no menos de cinco) y a continuación se absorbe el líquido.
El volumen debe ser cuidadosamente medido por lo que ha de tenerse en
cuenta la cantidad que queda en la aguja–cánula, que no se registra en la
regla de la carcasa. Si no consideramos pues, ese volumen, lo añadido a la
reacción será mayor que lo determinado de antemano con el gasto
económico que conlleva y con la falsedad de los datos.
La solución pasa por coger algo menos de lo que pretendemos según marca
la graduación y levantar la jeringa hasta dejar de tocar la disolución de
reactivo. Sin que la punta salga de la ampolla, combándose por tanto la

131
aguja–cánula, se lleva la jeringa con su boca hacia arriba, hasta la altura del
cuello de la ampolla. Se tira entonces del émbolo hacia atrás, de manera
que lo recogido en la aguja–cánula pase al depósito graduado de la jeringa,
midiéndose entonces el volumen real (si aspira algo del interior será tan
sólo gas puesto que ahora la punta no está inmersa).
En este dibujo se ilustra la operación arriba descrita.
Se debe agarrar el aplique de la aguja–cánula para que
no se salga en tanto operamos con la jeringa.
Este método, repetido con un poco de pericia,
asegura que la toma de reactivo sea exacta y válida.
Debemos cerciorarnos de que en todos los movimientos no haya peligro de
que la aguja–cánula se salga del tapón, al igual de cuidar, como se ha
comentado en el cuadro, que la propia aguja-cánula no se suelte de la
jeringa.
Tomada la disolución se lleva la aguja hacia arriba, con intención de
sacarla de la ampolla. Sin embargo nos detenemos justo cuando vaya a salir
del tapón. Por ahí se coge con los dedos (casi por la punta para que sea
firme el pinchazo sobre el tapón del matraz) y rápidamente se saca y se
clava en el otro tapón (se debe hacer con velocidad para que no entre aire
en la disolución de la jeringa).
Se hace bajar la punta hasta la superficie, y se descarga con atención para
que no roce las paredes. Algunas veces es aconsejable sumergir la punta en
el líquido de reacción y tomar una cantidad, para arrastrar los restos de
reactivo que pudieran haber quedado en la jeringa.
Finalmente se saca la aguja y se cambia el tapón.

132
Debemos reinsertar el tapón de teflón en la ampolla y guardarla en su sitio.
Se termina con el lavado de la aguja–cánula.
B) Para botes normales.
Es muy común usar botes que suelen encontrarse guardados en el
frigorífico a baja temperatura o en el almacén a temperatura ambiente. En
su interior no hay N2 (g) sino aire. Esta atmósfera se debe esquivar al
realizar la toma, pero la solución no resulta fácil en absoluto. De hecho se
prefiere tomar directamente del bote y añadirlo sin buscar la protección de
una atmósfera inerte. La desoxigenación de la jeringa únicamente es
posible usando el propio matraz para realizar las tomas de N2 (g), pero
como después hay que sacarla de nuevo para coger el producto, no resulta
muy efectivo. No obstante tampoco es inútil, pues es bastante poco el aire
que puede afectar la reacción y se considera válido con o sin desoxigenar el
pequeño volumen de la aguja. En el caso anterior era trascendental, pues el
reactivo mismo se debía salvar del aire y utilizarlo varias veces sin
desoxigenar la jeringa acabaría por descomponerlo.
Es costumbre también rodear el tapón y la boca de estos botes con un trozo
de parafilm (es una película protectora plástica especial, usada muy a
menudo para aislar el interior de los recipientes de la humedad, que se
analizará en capítulos posteriores), para impedir la entrada de humedad o la
salida de los propios vapores generados, que suelen resultar tóxicos. Antes
de devolverlos a su sitio se rodean con un trozo de parafilm nuevo.
Igual que en el caso anterior debemos considerar la cantidad adicional que
queda en la aguja-cánula, resolviéndose por el mismo proceso que antes
(aunque se tome un poco de aire que aquí es inevitable). El resto de la
adición es idéntica.

133
13. 2. 3 DISPENSAMOS REACTIVO GOTA A GOTA
En el caso en que cojamos el reactivo con jeringa basta con que nosotros
determinemos la velocidad de goteo con nuestras propias manos, sin
embargo cuando se utiliza una cánula para realizar la adición, esto no es
posible y hay que recurrir a algún método que nos permita adicionar a
voluntad.
Nos encontraremos verdaderamente, muchas reacciones en las que
adicionar el reactivo por goteo es imprescindible. Se obtiene un éxito en
esto mediante las dos estrategias siguientes:
A) Por control de la presión de N2 (g).
Encontramos el montaje a punto, con el matraz y el Schlenk (donde se
contiene el reactivo a dispensar) bajo presión de nitrógeno y con la cánula
conectándolos. El borde romo aún no toca la superficie del reactivo a
transvasar.
1. Se introduce la punta plana en la disolución del Schlenk. Nada pasará
pues aún tenemos abierto el N2 (g) en el matraz.
2. Se cierra un poco la llave que da paso al gas en el matraz y se clava una
agujita. Ahora la presión baja con respecto a la del Schlenk, pero no tanto
como para hacerse igual a la atmosférica.
3. Abriendo o cerrando la llave, el flujo de caída se reducirá hasta ser el
adecuado, con un paso de gotas al minuto.
B) Por gravedad.
Es muy parecido al anterior, sólo que la precisión en el goteo se logra
subiendo o bajando el Schlenk en la barra del pie. Este método es más

134
seguro que el anterior, pues la llave del matraz tiende a girar, lentamente, a
causa de la tensión de la gomilla y alterar el flujo de caída: puede cerrar el
flujo o hacer que caiga todo de repente.
1 y 2. Son iguales que el caso anterior.
3. Al subir o bajar se logra concretar el goteo según la velocidad apetecida.
A veces no caen las gotas a no ser que se dé un ligerísimo golpe de vacío
en el matraz de reacción (parecido a cuando se extrae gasolina de un
recipiente con una goma).
13. 2. 4 ADICIÓN A BAJA TEMPERATURA
La adición de determinados reactivos puede ser un verdadero problema
cuando la temperatura del laboratorio es alta. En caso del acetaldehído,
cuya temperatura de ebullición es de 21 ºC, podemos encontrarnos con que
cuando cogemos el volumen requerido en el bote que lo contiene, al ir a
añadirlo, todo o gran parte se ha evaporado, quedando la reacción mermada
o completamente impedida.
Para solucionar este problema el baño frío no basta pues el calor ya está
presente antes de la adición, y es en la toma del producto donde tenemos la
complicación. Lo más usual es hacer la toma en el congelador de uno de los
frigoríficos existentes para almacenar productos. Sin embargo, hay
dificultades de espacio y se debe ser ciertamente hábil. Con experiencia
acaba saliendo bien.
Las etapas serían:

135
1. Dejamos un tiempo la jeringa con su aguja–cánula en el congelador
rodeada de una tela impermeable (sirve un guante de látex) para impedir
que se acumule agua sobre la misma.
2. Se lleva el bote de reactivo también a la parte superior del frigorífico y
se hace la toma cuidadosamente.
3. Con mucha rapidez la inyectamos en el matraz (el cual debe encontrarse
preparado y con un tapón pinchado).
Como el líquido del matraz se encuentra a baja temperatura (por el baño) la
reacción no debe tener más complicaciones.
13. 2. 5 ADICIÓN CON CÁNULA EN FRÍO
La naturaleza de los reactivos condiciona los métodos que vienen a usarse
para poner en marcha las reacciones, debido a que cada uno tiene unas
características naturales que le hacen ser útil en unas condiciones, y todo lo
contrario en otras.
Así algunos reactivos deben mezclarse a baja temperatura, debiendo estar
ellos mismos en las mismas condiciones de frío antes de ser mezclados.
En este caso tenemos la disolución a añadir en un Schlenk con llave de
teflón en un baño frío a la temperatura indicada. Igualmente el matraz en
otro baño frío.
Pasar un líquido del uno al otro es cosa ya bien conocida, mas ahora
encontramos un problema adicional. El reactivo se debe transvasar del
Schlenk al matraz, pero ambos líquidos deben mezclarse a la temperatura
deseada. Sin embargo, al atravesar la cánula (que está a temperatura
ambiente) el reactivo se calienta, invalidando la reacción.
Con el siguiente montaje nos deshacemos del problema:

136
1. Se rodea con algodón la cánula, pero al mismo tiempo que se van
colocando los algodones se enroscan y afirman con un hilo alámbrico,
desde un extremo hasta el otro.
2. Se empapan con N2 (l), teniendo cuidado de no derramar demasiado.
3. Se arropan con un paño alrededor de la cánula para que guarde la
temperatura el mayor tiempo posible.
4. Se prueba entonces a hacer pasar el líquido de un recipiente al otro, sin
peligro de que cambie de temperatura.
Existe un problema intrínseco a este montaje, y no es otro que la
solidificación del líquido en la cánula, lo que impediría su salida. O
también que el líquido, al enfriarse, aumente su densidad y viscosidad,
siendo un obstáculo para la consecución del experimento. La única
solución en tal caso es la de esperar que la cánula se temple un poco.
13. 2. 6 MEDICIÓN DE UN VOLUMEN BAJO N2 (g)
Puede ocurrir que se deba usar un reactivo líquido o en disolución en un
volumen tal que una jeringa sea totalmente impropia para medir el
volumen.
En principio la respuesta es directa, usamos una probeta. Pero si esa
medición debe realizarse en condiciones de atmósfera inerte porque el
reactivo es inestable al aire, se nos presenta una complejidad que hay que
salvar.
También se usará la probeta, pero trabajando del modo oportuno. La
probeta tiene una boca amplia, pero siempre encontraremos un tapón de
goma que se le pueda adaptar. Hay que cuidar que el tapón no se salga, lo

137
cual es fácil que ocurra, ya que estos tapones están diseñados para otro tipo
de cuellos.
Tras colocar dicho tapón pinchado, se desoxigena la probeta con el pincho,
tal y como se vio para el tubo de centrífuga (16. 1). Se mantiene un tiempo
la desoxigenación usando agujitas sobre el tapón para una eficiente
evacuación del aire.
Mediante una cánula se pasa el líquido a la probeta y se mide su volumen.
De ahí, con otra cánula, se pasará al matraz de reacción. El modo en que se
desmonta el sistema ya no debe significar problema alguno, debiendo
cuidarse que el aire no llegue jamás a afectar los compuestos.
13. 3 REACTIVOS GASEOSOS
Aunque mucho menos habituales que los otros, entre ellos encontramos
algunos de los más característicos del mundo organometálico, por lo que no
deben ser obviados. Contemos así al CO (g) y al CO2 (g) principalmente,
que han contribuido al desarrollo de esta disciplina de un modo muy
notable.
13. 3. 1 USAMOS EL CO (g)
Es bien evidente el cuidado con que debe abordarse cualquier experimento
en el que intervenga este gas. Siendo como es, un gas en extremo peligroso
y absolutamente inodoro, resulta siempre un compuesto inquietante.

138
Como todo gas suele emplearse en bala, lo que hace que tengamos que
trasladarla desde el almacén de gases a la vitrina. Podemos igualmente
encontrar que existen algunos laboratorios con sistemas de tubos que
conectan la bombona de suministro con una salida dentro de la línea (una
goma a la que se le adapta un pincho). En este segundo caso la bombona
que sirve de fuente del gas se encuentra en un patio o al aire libre, para que
un fallo no perjudique gravemente a los investigadores.
Para usar así el CO (g) debemos abrir la llave que cierra la bala en el patio.
En el sistema de tuberías encontraremos una segunda llave y un
manómetro, que asegura el control de la presión. En el extremo de la goma
de salida se introduce el pincho, a través del cual vamos a insuflar el gas.
El matraz de reacción se cierra con la llave y se deja bajo nitrógeno. Esta
vez no se realiza el cambio de tapón por uno pinchado porque nos interesa
una presión más alta de lo habitual, y para asegurarnos un ajuste perfecto
entre el pincho y la goma se usará uno nuevo. Perforar un tapón nuevo con
la aguja – cánula de la jeringa es muy difícil porque al mismo tiempo hay
que cuidar que no se salga el disolvente, pero con el pincho (aún siendo en
realidad una aguja – cánula) sí es posible. También interesa usar un tapón
nuevo porque antes de introducir el CO (g) se debe quitar la goma que lo
une a la línea, y no podríamos hacerlo si el tapón está pinchado.
Se abre la segunda llave en la tubería del CO (g) y con el manorreductor se
ajusta la presión. Tras salir CO (g) unos segundos, se pincha el tapón nuevo
y se baja el pincho hasta acercarlo a la superficie del líquido.
Ahora hay que lograr el cambio de atmósfera, pasando de la de nitrógeno a
la de monóxido. Para ello se abre a intervalos de 30 segundos la llave de
vidrio, por lo que sale N2 (g) y CO (g) al exterior. Al abrir esta llave como
no hay tubo de goma, se conecta el interior con la atmósfera, y se desaloja
la atmósfera interna, reemplazándose por CO (g).

139
Una vez se ha repetido esto 5 veces estaremos seguros de que la atmósfera
se ha cambiado completamente en CO (g).
Para que la reacción se complete se deja en torno a unos 20 minutos (estas
reacciones de carbonilación o inserción de carbonilos son muy rápidas en
general).
De cuando en cuando en este tiempo se renueva la atmósfera de CO (g) por
abertura de la llave.
Una vez terminada la reacción se conecta el matraz a la línea por medio de
la goma, y se desoxigena ésta antes de abrir nuevamente la llave. Hecho lo
anterior, se mete N2 (g) al interior, pudiéndose entonces retirar el pincho y
cambiar el tapón por uno nuevo.
Es imprescindible que a lo largo del experimento no salga CO (g) fuera de
la campana, para ello se baja todo lo posible el vidrio de la vitrina. Esta una
medida fundamental, así como la eliminación del gas que pueda quedar en
la tubería. Para evacuarlo se cierra la llave de la bala de suministro y
después se vuelve a abrir la segunda llave. De este modo el gas remanente
saldrá completamente y no tendremos que preocuparnos de que pueda
escaparse por algún fallo.
13. 3. 2 OTROS GASES
Usados de modo más esporádico tenemos otros gases, los cuales se
encuentran todos contenidos en balas iguales a la que puede contener al gas
anterior.
Las balas se almacenan en la parte exterior del edificio, en un patio abierto,
para impedir que una posible fuga sea especialmente grave. Son
normalmente de gran tamaño, trasladándose con ayuda de un pequeño carro

140
con dos ruedas y suficientemente alto como para equilibrar la bala, que va
fijada al respaldar del carrito por su parte media con una cadena.
Cuando tenemos la bala al lado de la vitrina, la encadenamos a la misma
(posee un hueco la estructura de la vitrina) y hacemos uso del gas uniendo
una goma con manómetro a la boca de la bombona. El uso posterior es
igual al descrito para el CO (g).
13. 3. 3 REACTORES DE ALTA PRESIÓN
Si las presiones a lograr son muy altas (del orden de varias atmósferas) se
usan unos recipientes especiales, bien diseñados para soportar la presión
del gas.
Son muy parecidos a los tubos de centrífuga, aunque más grandes y con un
grosor de vidrio mayor. En su parte superior poseen una boca donde se
ajusta un manómetro que indicará la presión existente. Este manómetro
está, en realidad montado sobre una llave de conexión con entradas y
salidas. Se le puede insuflar gas inerte o gas reactivo, siempre a presiones
elevadas.
13. 4 DEJAMOS QUE LA REACCIÓN SE COMPLETE
Como hemos indicado con anterioridad, las reacciones se van completando
con el tiempo y con el aumento paulatino de la temperatura tras quitar el
baño frío. Esto puede ser seguido mediante la inspección de los cambios
físicos que pueden surgir con los cambios químicos, tales como el color, la
densidad...

141
La duración de las reacciones oscila entre una hora y un día, aunque la
media podría estimarse en unas 3 horas, dentro de lo delicado que es hacer
una estimación en estos casos.
Para que se complete dejamos el matraz en una posición estable, tal que no
pueda sobrevenir altercado alguno. Cuando el baño frío es innecesario tras
cumplir su misión, se deja que la temperatura vaya ascendiendo hasta
alcanzar la ambiente. Pero antes de abandonar la reacción para hacer otras
labores, es preciso cerciorarnos de los siguientes puntos:
1. Colocar las gomillas al tapón de goma.
2. Agitación adecuada de la disolución, pues si es pobre la reacción tardará
mucho más en completarse o no lo hará en absoluto..
3. Golpe de vacío para prevenir la sobrepresión.
4. Cierre de la entrada de gas y colocación de la gomilla de seguridad.
13. 5 LA GOMILLA DE SEGURIDAD
Impide que la llave, mientras dure la reacción gire en uno u otro sentido. Su
buena disposición asegura por tanto que la llave permanezca en posición de
cerrado.
También es muy útil cuando guardamos un producto en el frigorífico en su
recipiente (protegido con gas inerte) pues si la llave gira por cualquier
razón, se destruiría el compuesto.
En las siguientes representaciones se ha obviado la gomilla de la llave, que
la mantiene firmemente contra su asiento, para una mayor claridad en el
dibujo.

142
En esta primera imagen vemos que la gomilla se
comienza a colocar (doble), colgándola de uno de
los brazos de la llave, que se encuentra en posición
de cierre.
Se pasa por debajo de la llave de modo que se
enganche en el brazo restante de la misma,
quedando una situación parecida a la que se
representa en la figura de la derecha.
En esta tercera figura vemos cómo se debe cruzar el
enganche de un extremo con el del otro, de manera
que la rotación de la llave esté impedida por la
elasticidad de las gomillas. Obsérvese cómo la llave
se opondrá a cualquier cambio de posición
ocasionado por algún golpe o giro.
13. 6 BOTES CON TAPÓN DE SEGURIDAD
Si un producto es inestable al aire, se expende en botes con un tapón
especial que asegura su mantenimiento a lo largo del tiempo. Este tapón es
análogo a una chapa de cerveza, encontrándose ajustado de la misma
manera en su boca. Esta chapa tiene en el centro una especie de tela
(constituida en realidad por un conjunto de fibras distintas que
impermeabilizan el interior con respecto a la entrada de humedad o aire)
por donde lograremos extraer al reactivo.

143
La primera obligación es la de pasar su contenido a una ampolla, pues una
vez pinchado el tapón resulta inservible y se afectaría el interior por entrada
de aire.
Para trasvasar el líquido el sistema es muy sencillo:
1. Preparamos una ampolla y la dejamos bajo N2 (g) en espera de contener
el reactivo del bote. Nos debemos asegurar de que el volumen de la
ampolla es superior a aquel del frasco comercial. Se le pone un tapón de
goma pinchado.
2. Se coloca un pincho a una de las gomas de la línea y se introduce a
través de la malla de la chapa tras abrir la corriente de gas.
3. Con una cánula se atraviesa también la tela, pasando su extremo
punzante por el tapón de goma pinchado de la ampolla. Como el tejido es
nuevo, será muy difícil clavar la cánula por su parte roma, aconsejándose
practicar varias punzadas con su extremo agudo y luego probar con el
romo, de manera que resulte viable.
4. Usando unas agujas y cerrando la presión de N2 (g) en la ampolla, se
logra que el reactivo pase de un recipiente al otro. Allí quedará todo y se
reinstalará la corriente de N2 (g). Finalmente cambiaremos el tapón por el
de teflón y guardaremos la ampolla.
Cuando pasemos un reactivo a una ampolla, hay que hacer una etiqueta
donde claramente se indique qué reactivo es, su peso molecular, la
concentración en la que se encuentra y la fecha en la que se transvasó desde
el bote a la ampolla, garantizándose así el reconocimiento del reactivo y sus
características más importantes.

144
13. 7 LAVADO DE LAS AGUJAS–CÁNULA
Se han usado para la inyección de reactivos con jeringas de plástico, que
son de un solo uso, pero las agujas–cánula deben ser lavadas antes de
volver a utilizarse.
En principio usaremos las mismas jeringas para limpiarlas, aunque a
continuación desechemos estas jeringas. Como siempre, el lavado se hace
con absorciones de acetona primero y de éter después; realizadas varias
veces de modo que se asegure la completa eliminación de cualquier resto.
Es obvio que si la sustancia posee un carácter salino, será más indicado
lavarla primero con ácido clorhídrico diluido, después con agua y
finalmente con los dos disolventes orgánicos.
14 EL CALOR
Las reacciones se ralentizan con la baja temperatura y a medida que ésta
aumenta la reacción se desencadena hasta completarse. Así los distintos
elementos mediante los cuales se eleva la temperatura de la disolución son
muy importantes para el buen fin de las mismas, y merecen un bloque
propio que se encargue de analizar sus características.
Cuando se usa algún sistema que permita calentar a temperatura mucho
mayor que la ambiente, resulta crucial recordar que sólo se pueden usar
ampollas como reactores, dado que los matraces no están diseñados para
resistir las presiones que un disolvente volátil pudiera generar en su
interior. Para ello es preciso realizar la mezcla, sea en la ampolla, sea en el
matraz, y posteriormente pasar la mezcla de reacción a la ampolla.
Igualmente no deben usarse tapones de goma para tapar la boca de la
ampolla (lo que por otra parte sería impropio) pues al aumentar la

145
temperatura podemos forzar la evaporación de algún reactivo volátil
(fosfinas) que el tapón absorbería, con la consecuente parada o pérdida de
rendimiento en la reacción.
14. 1 ELIMINACIÓN DEL BAÑO FRÍO
La mayoría de las reacciones no requieren un aporte suplementario de
temperatura más allá de la ambiente. Las reacciones pues se dejan
completar tras alcanzar la temperatura del laboratorio, sin aplicar foco
alguno. En este rango de temperaturas el matraz no tiene ningún problema
para acoger la reacción dado que la presión interior no es demasiado
grande.
Este método en sí es un calentamiento, y por eso se ha introducido en este
tema, lo que ocurre es que el foco de temperatura es el propio medio y por
lo que no usamos ningún calefactor específico.
14. 2 USO DE SECADORES
Para elevar la temperatura ligeramente, sin alcanzar por tanto valores altos,
se utilizan los secadores. Son iguales a aquellos domésticos, por lo que no
necesitan descripción alguna. Su principal virtud es dirigir el calor hacia el
punto que se necesita, pero el mismo calor puede hacerse cesar
inmediatamente con tan sólo apagarlo o girarlo. Estas cualidades permiten
un control excelente del calentamiento, usándose en aquellas reacciones en
las que el calentamiento sea una etapa delicada por la propia exotermia de
la reacción (las de síntesis de Grignard).

146
Los secadores pueden quemarse si están mucho tiempo conectados, por lo
cual se usan generalmente en parejas (cuando su misión es calentar una
reacción) encendiéndolos alternativamente.
Otra función del secador, seguramente la más extendida, es la de eliminar
las trazas de humedad de la boquilla de los Schlenk que se introducen en el
congelador del frigorífico para que cristalicen los compuestos. Así, antes de
conectarlos a la línea, hay que evaporar el agua que se haya condensado en
las boquillas y resquicios de las llaves.
Esta función hace que siempre encontremos un secador en un rincón de la
vitrina del químico organometálico.
Para finalizar diremos que hay un modelo especialmente diseñado para el
laboratorio y cuya horquilla de calentamiento es mayor que la de un
secador convencional, así como la potencia del aire caliente que expulsa.
Se llama cañón, y raramente se usa.
14. 3 CALENTADORES DE AGITACIÓN
Completo aquí las funciones de los agitadores magnéticos, que ya se
analizaron en la sección 9.1.2 y que constituyen un elemento
importantísimo en la vida diaria del científico.
El sistema de calentamiento se basa en el calor que produce la circulación
de la electricidad por una serie de
resistencias. El rango de calentamiento
va desde el 0 hasta los 300ºC, que se
despliegan alrededor del botón
izquierdo de la figura.

147
1. Botón que da paso al calentamiento. La lucecita de la derecha del mando
indica si realmente se está produciendo la acción.
2. Dial que permite señalar la temperatura aproximada que se pretende
lograr.
3. Plataforma metálica que irradia el calor de forma que un matraz situado
arriba sea calentado efectivamente.
Recuérdese que, si la temperatura va a ser alta, se pasará el contenido del
matraz a una ampolla y se calentará en ella.
En esta vista posterior del aparato
señalaremos primero la clavija
hembra donde se conectan las
patillas del aplique que se halla a su
lado, unido por un cordón plástico
(para que no se extravíe). Este
conjunto clavija-aplique es un sistema de seguridad que impide el
calentamiento si el aplique correspondiente no se encuentra insertado en la
clavija. Al encajarse se cierra el circuito y el calentamiento se lleva cabo
sin impedimento. Siempre que no vaya a usarse el aparato como calefactor,
la clavija debe retirarse, pues un golpe en el lateral la activaría, con el
consiguiente peligro, pues hay multitud de productos que se descomponen
por la temperatura y no pocos podrían resultar explosivos.
En este tipo de sistemas de seguridad la conexión suele realizarse de
manera que las patillas del aplique entren en las ranuras correspondientes
(normalmente la 1 y la 3, aunque esto puede variar según el modelo). No
obstante tampoco es posible confundirse ya que hay unos topes que
impiden que las dos espigas conecten cualquier otro par de ranuras.

148
Este conjunto de hendiduras sirve también para insertar el termómetro
electrónico, que se usa muy a menudo para controlar la temperatura del
calefactor, pero a su vez, para llevar a cabo complejos sistemas de
calentamiento por fases a diferentes temperaturas, que veremos en páginas
sucesivas.
A veces encontramos placas que no disponen de dispositivos de seguridad
que impidan un calentamiento por accidente. En un laboratorio de química
organometálica son indispensables estos elementos de seguridad, dada la
inflamabilidad de los disolventes que se usan a diario. Además, no basta
con tenerlos en el laboratorio, hay que acostumbrarse a usarlos
correctamente, desconectando el aplique de la clavija hembra una vez que
no es necesario calentar.
14. 4 CALENTAMIENTO EN BAÑO DE AGUA
Puede ser necesario que el calentamiento sea homogéneo a lo largo de
toda la superficie del matraz para lo cual se usa este tipo de baños. El agua
se mantiene, por resistencias eléctricas, a una temperatura uniforme, en un
recipiente que puede ser de naturaleza varia, pero en general tiene forma de
acuario de vidrio.
La temperatura se controla por una rueda graduada que indica la cifra
centígrada a la que se calienta el agua. La magnitud exacta se mide por un
termómetro que en algunos modelos viene incorporado en la estructura.
Algunos tienen la posibilidad de realizar un programa de calentamiento (un
conjunto de pasos donde la temperatura varía con el tiempo).
Estos baños sólo son apropiados para calentar por debajo de los 100ºC, por
motivos evidentes.

149
14. 5 CALENTAMIENTO EN BAÑOS DE ACEITE
Cuando se requieren temperaturas superiores a 100ºC para llevar a cabo la
reacción, pero una uniformidad en el calentamiento, se emplean los baños
de aceite.
En ellos el agua es sustituida por una parafina que soporta bien las altas
temperaturas sin descomponerse.
Hay 2 tipos de modelos:
A) Baños de vidrio, donde se contiene la parafina y que se colocan en el
agitador magnético. Son simples recipientes de vidrio con forma varia,
usualmente cilíndrica u ortoédrica.
B) Baños diseñados especialmente para contener la parafina y que son
mucho más complejos como veremos a continuación.
14. 5. 1 BAÑOS DISEÑADOS PARA ACEITE
Es un recipiente con resistencias internas que permiten calentar el aceite
que se deposita en un contenedor específico. Poseen un termómetro
incorporado que facilita la lectura de la temperatura exacta, pudiéndose en
algunos modelos introducir un programa de calentamiento. Son en
definitiva, análogos a los baños de agua comentados con anterioridad,
únicamente que permiten un rango muy superior de calentamiento.
Las partes más importantes de estos baños son:
1. Cavidad donde se aloja el aceite a calentar y donde se sumergirá
parcialmente la ampolla que contiene la disolución.

150
2. Panel de control, donde las funciones de calentamiento se encuentran
expresas pueden ser controladas por los botones correspondientes.
Como en el tradicional baño de agua, las ampollas se calientan sostenidas
por pinzas que a su vez se fijan a los pies metálicos que actúan de soporte
tradicionalmente.
La temperatura no debe elevarse excesivamente, cuidando siempre que el
aceite no humee, lo que sería evidencia de su descomposición.
Los modelos vienen equipados con una especie de rejilla de madera
taladrada con pequeños agujeros que se coloca a modo de tapa sobre el
baño de aceite.
Su función es la de servir de sostén a los tubos de RMN (que son muy
finos) para lograr un seguimiento de la reacción a lo largo de una amplia
gama de temperaturas.
14. 5. 2 BAÑOS INDIVIDUALES
Estos baños son simples recipientes de vidrio (vasos de precipitados) que
contienen parafina y se calientan por medio de la placa calefactora. Los hay
de diferentes tamaños, aplicándose aquel que convenga al tamaño de la
ampolla.
Para controlar la temperatura en su calentamiento debemos usar
termómetros que tendremos que incorporar externamente. Entre éstos el
más utilizado por su versatilidad y fácil manejo es el electrónico.

151
14. 6 TERMÓMETROS
Existen dos tipos de termómetros cuyo uso es obligado en un laboratorio.
Los tradicionales y los electrónicos.
Los termómetros tradicionales son aquellos que se basan en la dilatación
del líquido contenido en su parte baja por el aumento de la temperatura, y
no merecen más descripción, ya que se usan en los laboratorios de
cualquier facultad o en nuestras mismas casas. Lo único a referir es que los
termómetros utilizados en los laboratorios tienen un rango de temperatura
mucho mayor que estos últimos.
Los termómetros electrónicos sirven, no sólo para medir la
temperatura, sino también para controlar el calefactor, impidiendo que la
temperatura suba por encima del nivel establecido, manteniéndose fijo el
valor al que queremos hacer la reacción. Incluso permite la ejecución de
programas de temperatura más complejos.
Están especialmente diseñados para su utilización en baños independientes
(vasos de precipitados), ya que los otros suelen tener los sistemas de
medición incorporados, además necesita la clavija hembra que poseen los
agitadores magnéticos en su parte de detrás.
Las partes del termómetro deben ser
bien conocidas para sacarle el máximo
partido y cuidarse de algunas acciones
que podrían comprometer seriamente no
sólo la reacción sino incluso la
seguridad del laboratorio.

152
1. Esta varilla metálica sirve para indicar la temperatura en el seno del
baño que se calienta. Cuando la temperatura alcanza el nivel exigido, el
agitador-calefactor cesa el calentamiento por las indicaciones del
termómetro. Lo más importante es que la varilla esté siempre dentro del
aceite, ya que de no ser así no registraría aumento alguno de temperatura y
el termómetro ordenaría el aumento del calentamiento hasta que la placa
estuviese al límite. Se comprende el peligro que puede representar esto,
para la placa, para la reacción y para nosotros mismos.
2. Es el cuerpo del termómetro, donde se albergan sus elementos
constitutivos. En su cara anterior presenta los botones de control que sirven
para activarlo y para introducir el valor de temperatura deseado.
3. Este aplique constituye la pieza de unión entre el termómetro y la placa
calefactora pues, insertándose en la parte de atrás del agitador magnético,
no sólo cierra el circuito sino que permite el control de la temperatura
según nuestro deseo.
Así esta modalidad de termómetro constituye un elemento indispensable
cuando se tiene intención de calentar reacciones en baños de aceite, permite
trabajar con una mayor tranquilidad, pudiéndose abandonar la reacción
para realizar otras experiencias.
Una vez nos hemos servido de él, limpiamos la varilla con papel y lo
guardamos en su caja de corcho, siendo un instrumento delicado.
14. 7 AMPOLLAS O MATRACES
A lo largo del desarrollo de este tema hemos comentado a veces la
necesidad de pasar el contenido del matraz a una ampolla para calentar.

153
Las situaciones en las que debe hacerse esto son las que surgen como
consecuencia de un peligro por sobrepresión en el interior del matraz:
A) Cuando la temperatura a lograr sea especialmente alta. Aquellas
temperaturas en torno a 150ºC resultan ya poco apropiadas para matraces
(pero decir esto sin tener en cuenta la naturaleza del disolvente es un tanto
arriesgado).
B) Cuando el disolvente es muy volátil. Así el éter, a poco que se caliente,
genera una presión muy alta, que crearía un exceso de presión de no
cambiar el reactor de matraz a ampolla.
Finalmente hay un caso que es también muy comprometido. En aquellas
disoluciones con reactivos muy volátiles a calentar, es conveniente usar la
ampolla. Aquí el peligro no es el calentamiento en sí ni el aumento de
presión, sino que, durante el proceso el reactivo volátil se nos va y se
absorbe en el tapón de goma. Siendo así, la reacción se parará y
esperaríamos inútilmente algún cambio en la misma (un análisis de RMN
de la disolución nos advertirá que el reactivo volátil ha desaparecido). En
una ampolla, el tapón de teflón no absorbe y no se corre ese riesgo.
14. 8 ESTUFAS
Aunque no es, propiamente hablando, un sistema de calentamiento de
reacción, sí es un elemento de calefacción, por lo que creo oportuno
incluirlo como último punto de este bloque.
En cualquier laboratorio de esta disciplina encontramos siempre estufas de
mayor o menor tamaño, cuya misión es la de eliminar la humedad de
aquellos objetos que se encuentran en su interior.

154
Una estufa es un recinto amplio, parecido a un horno, donde un
mecanismo eléctrico genera calor en el interior.
Encontramos al menos dos estufas por laboratorio, una destinada a las
cánulas (como se vio con anterioridad) y la otra para los objetos de vidrio
que acaban de lavarse.
Tanto una como otra se encuentran a 105ºC, temperatura suficiente para
eliminar los restos de agua que puedan hallarse en los distintos utensilios.
Sobre ambas estufas se deben encontrar siempre los guantes apropiados
para sacar los vidrios de la estufa.
Cada vez que se abre una estufa, sobre todo la que contiene el material de
vidrio recién lavado, debemos intentar no respirar los vapores que emanan
del interior, puesto que contienen, además de los gases orgánicos usuales,
HCl (g) que procede del ácido clorhídrico usado en la limpieza de los
vidrios. Esta desagradable presencia aconseja no colocar ningún tipo de
llave o manorreductor cerca de la estufa o al revés, pues se corroe a una
velocidad mucho mayor que la que cabría esperar si no estuviese en
atmósfera ácida.
15. ELIMINACIÓN DE DISOLVENTES A VACÍO
Cuando se concluye una reacción suele ocurrir que la jornada de trabajo
también llega a su fin y sea preciso marchar a casa, o bien que no tengamos
tiempo para proceder a los trabajos de purificación del producto, porque
algún otro asunto urge más. Para ello hay que dejar el producto obtenido de
un modo seguro en el frigorífico, para que al día siguiente podamos trabajar
con él.
Dejar los productos obtenidos, en disolución, puede acarrear
consecuencias poco afortunadas, por la posibilidad de reacciones

155
posteriores indeseadas o por descomposición del producto. El medio
permite la movilidad de las moléculas y así su reacción, de modo que si
eliminamos el disolvente (llevando a sequedad) estabilizaremos el
compuesto durante mucho más tiempo, puesto que esas reacciones no
podrán ocurrir.
15. 1 EN EL PROPIO MATRAZ DE REACCIÓN
Aprovechando que tenemos un ratón en el interior del matraz, colocamos
un baño de agua sobre el agitador y sumergimos el reactor. Tras poner una
agitación vigorosa se abre lentamente el vacío. La agitación provocada por
el ratón impide que el disolvente se evapore a borbotones, dando saltos
incontrolados. Al disminuir la presión, la temperatura de vaporización baja
y el líquido bulle a temperatura ambiente. Sin embargo se tiene que cuidar
que el borboteo no sea excesivo, pues de ser así se podría marchar la
disolución aspirada por la línea. Para impedirlo se controla la llave con la
mano, y en el momento en que la espuma del líquido que se evapora sube
demasiado, se cierra la succión. Se continúa de este modo hasta que el
volumen del líquido ha bajado lo suficiente como para dejar fija la llave del
matraz (abierta en su totalidad), logrando entonces un vacío máximo.
Finalmente llega a evaporarse todo el disolvente y el producto, junto a
subproductos y restos, queda en forma de aceite (suele estar mezclado con
impurezas que son difíciles de separar) o en forma de espuma (las veces en
las que está bastante puro).
La colocación de un baño de agua durante el proceso es algo inteligente. Si
no se hiciese perderíamos tiempo debido a que, al bajar la temperatura por
la evaporación, las paredes del matraz se enfrían mucho y sobre ellas
comienza a formarse escarcha. Esta escarcha impide que el exterior ceda

156
calor a la evaporación, y así el ritmo de eliminación de disolventes se frena.
Incluso con el baño de agua, llega a formarse una capa de hielo sobre el
fondo redondo, con forma de lentilla gigante, que hay que quitar de vez en
cuando.
Teniendo el disolvente evaporado se prepara para meterlo en el frigorífico,
como si fuese un Schlenk (lo cual ya se analizará). Con un rotulador para
vidrio se indica la reacción con un número (todas las reacciones deberán ir
numeradas para impedir la posibilidad de equivocarse respecto a la
naturaleza del compuesto contenido).
Allí quedará hasta que se tenga tiempo para trabajar el producto,
purificándolo y cristalizándolo. Como puede deducirse se almacenará bajo
atmósfera de N2 (g) y nunca a vacío.
Consideremos que los procedimientos de eliminación de disolvente a vacío
forman también parte de esa fase de purificación de los productos
obtenidos, y que no sólo es utilizado para dejar los productos en depósito.
Para concluir debemos hacer ver que éstas técnicas son útiles cuando los
productos obtenidos no son volátiles, en cuyo caso se marcharían con el
vacío y no tendríamos nada.
15. 2 EN UN SCHLENK
La evaporación en un Schlenk se usa para concentrar el producto por
eliminación de parte del disolvente, con intención de cristalizar el producto
en el congelador; o bien para cambiar el disolvente (llevando a sequedad) y
volviendo a añadir otro más conveniente para eliminar impurezas solubles
en él, ya sea porque el compuesto deseado sea más soluble en el nuevo
disolvente que las impurezas, o porque va a cristalizar mejor en él.

157
También puede usarse para guardar durante algún tiempo la sustancia para
después trabajar con ella. No obstante, un Schlenk es un recipiente muy
útil, y no se debería prescindir de él a causa de haberlo usado para
almacenar un producto recién obtenido.
La operación de eliminación de disolvente se puede hacer de dos maneras:
agitando con un ratón o manualmente.
A) Con el ratón.
La técnica a seguir es sencilla. Es una operación semejante a la realizada
para el matraz aunque, a causa del menor volumen del Schlenk, hay que
inclinarlo un poco ya que un pequeño salto del disolvente provocaría que se
marchase por la línea.
1. Se introduce el Schlenk con un ratón en el baño de agua, sobre la placa
de agitación.
2. Se inclina el tubo de modo que si viniese alguna subida repentina del
líquido, la boca de la llave quede lo más lejos posible del nivel alcanzado,
ayudando de este modo a una correcta evaporación. Evidentemente la
inclinación se hace en el sentido opuesto a la llave del Schlenk de modo
que cualquier ebullición súbita no alcance la apertura de la llave sino la
zona opuesta a ésta.
3. Si se pretende sólo reducir el volumen, el método apropiado sería el
manual, pero si el disolvente posee una alta temperatura de ebullición es
preferible hacerlo de esta manera. Así, una vez llegado al punto de
reducción exigido se corta la aspiración, se incorpora N2 (g) y se prepara la
cristalización. Antes de ponerlo a cristalizar se extraerá el ratón del seno de
la disolución.

158
B) A mano.
Este método es muy usado aunque, dependiendo de la cantidad de
disolvente y su naturaleza, uno pueda quedar físicamente agotado cuando la
temperatura de ebullición no es baja. Está especialmente indicado para
reducir el volumen de líquidos o para llevar a sequedad cuando el
disolvente es muy volátil.
La diferencia de volatilidad entre los disolventes se percibe perfectamente
en este procedimiento, ya que su temperatura de ebullición es proporcional
al tiempo que tenemos que emplear en llevar a sequedad el compuesto.
Se coge el tubo Schlenk con una mano, en tanto la otra se extiende de tal
manera que, con giros de muñeca de la primera, la parte inferior del tubo
golpee en la palma, estableciendo así una agitación continuada que
favorece la ebullición sin saltos.
Poco a poco se abre la llave que permite hacer vacío y se controla su
apertura sin dejar de agitar lo más mínimo para impedir que borbotee
violentamente.
Cada cual tiene un modo particular de coger el Schlenk, pero el brazo de la
llave siempre debe quedar hacia arriba, de modo que una eyección no entre
directamente por la boca de la llave (como se vio en el caso anterior).
Muchas veces, en lugar de un salto, lo que se produce es la ascensión
continua de espuma y habrá que estar igualmente atentos a este caso.
Al ir evaporando se va formando escarcha alrededor del tubo y se debe
eliminar aplicando la mano desocupada, calentando. La necesidad de
eliminar la escarcha hace que continuamente apliquemos calor mediante
frotaciones a la parte inferior del tubo Schlenk lo que debe hacerse de un
modo rápido, ya que no se puede dejar de agitar lo más mínimo.
Hecha la reducción pretendida en el volumen del disolvente, hay que
guardar el tubo convenientemente en el congelador del frigorífico en el
caso de que nuestra intención fuese cristalizarlo. Si en cambio queremos

159
usarlo para hacer una muestra de RMN el producto debe llevarse a
sequedad y quedar a vacío durante unos cuantos minutos, agitándose de vez
en cuando para que se elimine también el disolvente retenido.
Este método de eliminación de disolvente es una de las operaciones más
características del laboratorio y se llega a dominar de un modo absoluto
gracias al enorme número de veces que debe llevarse a cabo.
15. 3 EVAPORACIÓN DE GRANDES CANTIDADES
Cuando evaporamos disolvente en gran cantidad, puede ocurrir que
saturemos la trampa de frío de la propia línea. Esto es muy peligroso pues
el disolvente podría comenzar a burbujear y pasar a la bomba de vacío. Si
ocurriese eso, lo mejor es cerrar la llave que hace vacío y meter N2 (g) en
el matraz del que se evapora el disolvente. Tras lo cual se cierra la llave
número 7 de la línea y se corta la comunicación entre un lado y otro. A
continuación se apaga la bomba y se quita el N2 (l) de la ampolla. Abriendo
la llave 2 se consigue colocar a presión atmosférica la cabeza de la línea y
así separar la trampa. Con un secador se evapora el disolvente que se haya
depositado en la parte superior de la cabeza.
Con la trampa en la mano podemos ver cómo se halla llena de disolventes.
Cuando nos hemos deshecho de los mismos en los bidones adecuados,
lavamos la ampolla con acetona y éter, eliminando los restos congelados
que pudieran quedar por dentro. Una vez concluido, se espera a que se
evapore el éter de limpieza y se vuelve a situar la trampa. Igualmente que si
pusiésemos la línea de nuevo se hacen las operaciones ya acostumbradas,
recordando que debemos reabrir la llave 7 y que el N2 (g) se encuentra en
funcionamiento.

160
Sin embargo, cuando se vaya a evaporar mucho disolvente es interesante
eliminar cualquier posibilidad de que se nos ciegue la trampa por exceso
del mismo, utilizando un colector intermedio usado también a modo de
trampa fría.
Ese recipiente es una ampolla de gran capacidad que se instala entre el
matraz y la línea. Se introduce en un Dewar con N2 (l) y allí se recoge el
volumen del disolvente. Aquí podemos ver un dibujo del mismo.
A) Extremo que se conecta al tubo de la
línea, a través del cual se succiona,
abriendo la llave del colector.
B) Es el cuerpo del recipiente, donde se
solidificará el disolvente evaporado
gracias al foco frío. Se introduce en el N2
(l), permitiendo así una completa
eliminación del gas.
C) Boquilla que se conecta a una goma accesoria que se une a su vez al
matraz. Abriendo la llave del matraz se produce la evaporación a vacío del
líquido.
Concluido esto, lo único que queda es volver a conectar el matraz a la línea
(tras ponerlo bajo N2 (g)) y limpiar la trampa, lo cual se hace como ya se ha
explicado.
Si se necesitase evaporar disolvente de más de un matraz y no sufrir la
pérdida de tiempo que implica el hacerlo de uno en uno, podemos emplear
un accesorio muy útil que consiste en un tubo ramificado, gracias al cual
podemos conectar dos o más recipientes a la goma de vacío (dependiendo

161
de la ramificación). Con este elemento se ahorra mucho tiempo y su
simplicidad obliga a que se encuentre en todo laboratorio puesto que lo
podemos construir nosotros mismos.
Aquí se dibuja uno de estos tubos donde encontramos una bifurcación que
capacita para eliminar el disolvente de dos
matraces. Con aumentar la ramificación de los
tubos, empleando en cada brazo una “Y” como
la del centro, tendremos aún más variedad
(aunque eso sí, se perderá capacidad de vacío a
más matraces conectemos).
15. 4 EL ROTAVAPOR
Con el rotavapor se pretende evaporar a vacío todos aquellos disolventes
cuya volatilidad no es muy alta por lo que el proceso sería muy largo por
otros métodos, aunque, por supuesto un disolvente volátil a su vez, podría
evaporarse sin problemas en el rotavapor.
Este dispositivo se conocerá ya al emplearse habitualmente en la
Facultades de Química sobre todo en la rama de orgánica, por lo que no
serán necesarias muchas descripciones.
Básicamente está constituido por una bomba de vacío, un motor de rotación
y un matraz de fondo redondo, donde se instala la disolución a evaporar. Se
usa preferentemente para aquellos compuestos orgánicos estables al aire
(aunque hay modelos que permiten la evaporación de disolventes cuando
hay productos inestables al aire, mediante la conexión al sistema de
nitrógeno).

162
El líquido se vierte en el matraz, el cual se instala en una boca
especialmente diseñada para él. Existe un baño de agua en el que se
introduce parte del matraz (con la misma finalidad que antes). Al
conectarse, el motor hace rotar al matraz, permitiendo esto una agitación
continua y muy rápida, para que el líquido no salte al ser evaporado. Un
refrigerante de reflujo interno (de serpentín, como los usados en los
destiladores) licua los vapores del disolvente y los recoge en otro matraz de
fondo redondo.
Como puede observarse el mecanismo es casi el mismo que se emplea en la
línea, lo que ocurre es que, aquí se encuentra optimizado y por tanto es más
rápido.
Al manejarlo se debe cuidar que el flujo de agua que circula por el
refrigerante no sea excesivo, tanto por la conveniencia del propio aparato,
como por los problemas que una salida de agua puede provocar en un
laboratorio.
Igualmente debe extremarse la atención al encajar el matraz en el
rotavapor, de manera que quede bien fijo y que no se suelte mientras rota a
gran velocidad.
16 SEPARACIÓN POR CENTRIFUGACIÓN
16. 1 DESOXIGENACIÓN DE UN TUBO CENTRÍFUGA
Para centrifugar se debe pasar la disolución a un tubo de centrífuga
previamente desoxigenado, donde coexistirán productos, residuos y restos
de reactivos.

163
El proceso de desoxigenación es sencillo y en gran medida presentado ya
atrás para situaciones análogas. Requeriremos solamente la participación de
agujitas, un tapón de goma pinchado y el pincho.
Se coge el tubo de centrífuga cuyo volumen permita contener
holgadamente la disolución del matraz y se le coloca un tapón pinchado.
Después se le inserta el pincho con la corriente de gas inerte, llevando la
punta hasta el fondo. Se colocan varias agujas y se deja un rato hasta que se
considere desoxigenado.
La corriente de N2 (g) arrastra lentamente al oxígeno que se encuentra
contenido en el recipiente, y siendo evacuado de este modo, termina por
ceder completamente su sitio al N2 (g).
16. 2 PASAMOS EL LÍQUIDO DEL MATRAZ AL TUBO
La centrifugación es una técnica muy interesante cuando existen sales en el
medio que impiden una filtración adecuada a causa de su depósito sobre los
poros de los filtros. En algunos casos impide la aplicación de estos
procedimientos de separación basados en filtro, siendo necesario recurrir a
la separación por centrifugación. De todas maneras hay personas que
prefieren la centrifugación en primer lugar por ser más efectiva y la utilizan
siempre que en la disolución hay materias sólidas.
En principio debemos hacer pasar todo el líquido al tubo de centrífuga, sin
preocuparnos por el hecho de que haya restos sólidos en la disolución (si no
los hubiera no habría lógica al utilizar este método).
Como los restos sólidos suelen tener poco tamaño y existe una agitación
continua por parte del ratón, se encuentran suspendidos en el líquido, y esto
beneficia a la hora de pasarlos al tubo de centrífuga.

164
Se coge una cánula gruesa y se pincha por su extremo romo en el tapón del
matraz (recordemos que se hace así pues el extremo plano permite absorber
eficientemente los restos que quedan en el fondo de los tubos y matraces).
Se espera a que el N2 (g) elimine el aire que puede haber en la cánula y se
clava posteriormente en el tapón pinchado del tubo de centrífuga,
llevándolo hasta una posición media.
Como el tubo ya se encuentra protegido por la corriente de gas inerte que
llega a través de la cánula, se puede retirar el pincho sin temer una entrada
de aire. Después se comienza a bajar el extremo romo de la cánula y,
clavando un par de agujas en el tapón del tubo, pasará disolvente de un sitio
al otro.
Hay que lograr que la agitación sea buena, para que pasen todas las
partículas, incluidas las que quedan adheridas en las paredes. Es costumbre,
una vez que se ha trasvasado el líquido, tomar un poco del disolvente de los
destiladores con una jeringa, y añadirlo al matraz, de modo que nos
aseguremos que todo nuestro producto, pasará al tubo de centrífuga. De
este modo se acaba con toda la disolución en el tubo, y es ahora donde
comienza la operación más delicada, pues tenemos que quitar la cánula sin
desproteger al líquido que queda dentro. Para ello se toma un trocito de
parafilm y se comienza a colocar alrededor del tapón, estirándolo de modo
que se adapte al mismo. En un segundo se retira la cánula y con el parafilm
se tapa el hueco dejado, terminándose por rodearlo con tantas vueltas como
sea posible.
Suele ocurrir que, como el interior está a una presión superior a la de
afuera, el parafilm se empiece a hinchar (como si fuese un globo),
pudiendo acabar reventando. Para salvaguardar esto, se colocan varias
capas del parafilm sobre el agujero.
Así queda listo el tubo para introducirse en la centrífuga.

165
16. 3 EL PARAFILM
El parafilm es una película formada por una sustancia plástica flexible,
moldeable, aislante, resistente a la humedad y sin olor, con un color
grisáceo translúcido. Se encuentra guardado en una caja y para usarlo se
corta con unas tijeras, que habitualmente se encontrarán al lado, un trozo de
tamaño adecuado a la necesidad. Un trozo pequeño sirve para sellar un
tapón de goma pinchado, a causa del carácter extensible de la lámina.
Se usa muy a menudo, con la finalidad de aislar del medio el contenido de
un recipiente. Así lo encontraremos sellando tubos con tapones pinchados,
o alrededor de los tapones de algunos productos comerciales, impidiendo
que vapores salgan al exterior y que la humedad penetre dentro.
Las desventajas que poseen radican en lo fácil que se rompen al estirarlos
por encima del límite al intentar aprovecharlos al máximo. Por lo mismo
una presión desde dentro del bote o tubo puede hacer un estrago como ya
se vio anteriormente.
Para colocarlo basta un poco de práctica, siendo en sí una operación
sencilla, que nos permite asegurar el contenido de ciertos recipientes:
En 1 se muestra cómo se toma el parafilm
por uno de sus extremos, fijándolo con el
pulgar y estirándolo con paciencia hacia el
otro lado.
Al mismo tiempo que se va estirando, se
intenta expandirlo de manera que cubra toda

166
la superficie del tapón, donde, por ejemplo,
podríamos tener el agujero de un pinchazo.
En 3 se lleva sobre el lateral buscando que
quede lo más firme posible, para que no
haya problema en cuanto a posteriores
fugas.
El parafilm se va rodeando en torno al tapón,
pasándose varias veces si se puede por la parte
superior, para que la zona más débil quede
suficientemente protegida.
16. 4 SOPORTES PARA TUBOS DE CENTRÍFUGA
En realidad sirven para sostener cualquier tipo de tubo con forma de
ampolla cuyo diámetro sea inferior al de la boca del soporte.
Las ampollas y tubos con un fondo redondeado se almacenan sin problema
fijándolos con pinzas a las celosías del banco de trabajo o a un pie de la
línea, o conjuntamente en latas grandes de forma cilíndrica (incluso en
cajas de cartón de tamaño medio, tal que no les permita tumbarse). Sin
embargo, para pesar los contenidos de las mismas, los de ampollas o tubos
de centrífuga, se deben utilizar soportes especiales que no les permitan
moverse pero que no los sostengan parcialmente ni sean impropios para
colocarse en el plato de la balanza.
Estos soportes son cilindros huecos de gran diámetro de boca, donde, a la
altura del ensanchamiento (aproximadamente a la mitad) poseen en el
interior un tope, para que el tubo de vidrio que se aloje no resbale hasta

167
abajo. Gracias a este tope pueden servir para mantener erectos tubos de
distinto tamaño, aunque siempre de radio inferior al suyo.
Son de material plástico y usualmente son de color rojo, por lo que se
distinguen inmediatamente en el laboratorio.
A la izquierda se muestra uno de estos soportes. El
ensanchamiento en su parte media se puede observar aquí sin
dificultad. Es a esa altura donde se desarrolla un tope interno en
forma cóncava, que a su ver está hueco en el centro.
16. 5 EL CONTRAPESO DE LA CENTRÍFUGA
Como se indicará en este punto, la centrífuga necesita un contrapeso que
equilibre el peso de la disolución y el tubo a centrifugar.
Para tomar tal contrapeso pesamos primero nuestro tubo centrífuga con la
disolución, tapón y parafilm puesto. Después cogemos otro tubo de
centrífuga lleno de agua hasta cierta altura. En ese tubo añadiremos o
retiraremos agua para que su peso llegue a ser exactamente el del tubo con
el producto.
Al lado de la balanza debe haber para estos menesteres un recipiente con
agua y una pipeta Pasteur. Este tipo de pipetas consiste en un tubo muy fino
que se va ensanchando a medida que ascendemos hasta un cierto límite
(véase el punto 19. 3). Se succiona o expulsa el líquido gracias a una
especie de perilla que se inserta en la parte de arriba. Así, ayudándonos de
esta pipeta, vamos dando o quitando agua, hasta igualar ambos pesos.
Cuando el contrapeso se coloca en la centrífuga no necesita tapón, pues por
la traslación a la que se ve sometido su masa se pega al fondo del frasco y
nada de líquido se sale del recipiente.

168
16. 6 LA CENTRÍFUGA
La centrífuga es un aparato diseñado para sedimentar suspensiones por
traslación del frasco alrededor de un eje. El número de brazos de una
centrífuga puede ser muy variable.
En la química organometálica las centrífugas suelen tener pocos brazos,
normalmente cuatro, opuestos dos a dos, en contraposición del alto número
que suele emplearse en otras ramas, como es el caso de la bioquímica.
El conjunto gira, gracias a un motor, en torno a la espiga donde se unen los
cuatro brazos. Al girar, todo elemento se ve arrastrado hacia el fondo a
causa de la fuerza centrífuga y así, los cuerpos más densos (mayor masa en
menor volumen) se aplastan contra el fondo. Este método por tanto es
adecuado para separar el producto deseado, que permanece en disolución,
de los residuos generados en la reacción que sean de características iónicas
y por tanto en forma de precipitado salino. Debido a la menor masa
atómica de los compuestos sólidos en suspensión con los que se trabaja en
esta disciplina, en relación a las estructuras proteínicas y de alto peso
molecular propias de la bioquímica, la centrífuga suele requerir un motor
más potente, ser de mayor tamaño, y estar provista de menor número de
brazos.
Los brazos de la centrífuga son metálicos y como los tubos son de vidrio
debemos aislarlos de las vibraciones del movimiento, que se propagan
fácilmente por el metal. Para ello hay unos cilindros huecos de plástico (los
hay de varios tamaños dependiendo del diámetro del tubo de centrífuga que
se vaya a utilizar) que se ajustan en los alvéolos metálicos. Además hay un
taco de goma que se introduce en el cilindro hasta el fondo. Estos dos
elementos absorben las vibraciones producidas, por lo que el peligro se
minimiza notablemente.

169
Cuando introducimos el tubo donde se encuentra nuestra disolución lo
haremos de tal modo que quede tumbado ligeramente en la misma
dirección y sentido en la que crece el brazo. Igualmente haremos con el
contrapeso, que debe colocarse en la parte opuesta. Si ambas masas no se
alinean de esta manera se generarán momentos de fuerza que acabarían por
doblar el eje de rotación e inutilizar la centrífuga. No obstante las últimas
centrífugas vienen ya con un sistema de seguridad que frena el proceso e
impide que siga acelerando si hay algún tipo de anomalía, sea en el peso de
los tubos, o en la disposición de los mismos.
Para conectar la centrífuga hay que cerrar la tapa de la misma y ponerla
después en marcha. Si la tapa no está bien cerrada, por seguridad, el motor
no actúa, aunque la activemos.
Las velocidades angulares más usuales utilizadas en la centrífuga se sitúan
alrededor de 2600r.p.m., en tanto que los tiempos de giro en torno a los 5
minutos.
Una vez se ha concluido el proceso una alarma sonora indica el final. Se
encuentra entonces que se ha formado un bloque compacto en el fondo del
tubo (su cohesión depende de la naturaleza del compuesto), en tanto un
líquido transparente lo sobrenada.
El tubo de centrífuga se traslada a la campana, con cuidado de no turbar por
agitación la sedimentación de los residuos.
16. 7 TRANSVASE DEL LÍQUIDO
Con la atención precisada para que no se levante el lecho sólido se lleva el
tubo de centrífuga a la vitrina y se fija a una pinza. Luego se coloca el
pincho en alguno de los tubos de la línea y se da vía al nitrógeno. Se clava
entonces en el tapón del tubo-centrífuga, pues será necesaria la presión de

170
N2 (g) para salvar el compuesto durante la operación. Se introduce ahora
una cánula por su parte roma en tanto que su punta aguda se lleva a un
Schlenk adecuadamente acondicionado para contener el producto.
El proceso se ha realizado ya muchas veces, por lo
que no es necesario comentar nada a su respecto,
excepto algunas anotaciones someras. Durante la
aspiración no se debe tocar el lecho sólido, e
intentar, dentro de lo posible, que la turbulencia
sea mínima. Cuando quede una pequeña cantidad,
debemos tumbar el tubo con las pinzas, tal y como muestra la figura, de
manera que sea más fácil agotarlo. Se baja entonces la cánula hasta que
toque el líquido y de este modo se logra transvasar todo.
El tubo de centrífuga se lava al final, tras eliminar el residuo sólido en una
papelera.
17 FILTRADO POR CÁNULA
Se usa para separar nuestro compuesto en disolución de restos no solubles
que quedan en el fondo del matraz o en suspensión. Estos restos
acostumbran a ser de carácter salino pero deben ser de tal naturaleza que no
formen placas sobre el papel de filtro, de modo que lo cieguen y hagan
imposible la filtración al hallarse adheridas al filtro.
Es una alternativa a la centrifugación que hemos visto en el apartado
anterior, sin embargo se realiza en menos tiempo, por lo que es muy
empleada.

171
17. 1 CON CÁNULA DE FILTRADO
Este tipo de cánulas presenta en lugar del extremo romo habitual un
cilindro hueco, que es la continuación de la cánula y no un accesorio
encajado terminado en forma cónica.
En este cilindro va a hacerse el filtro de modo que permita filtrar la
disolución lo más eficientemente posible.
El cilindro de la izquierda forma parte
de la estructura de la cánula, y será la
base sólida sobre la que se fijará el
filtro. Nótese cómo su cara exterior
tiene una forma cóncava para que la filtración se realice correctamente.
Al sacar este tipo de cánulas de la estufa hay que esperar a que el cilindro
se enfríe pues si no es inmanejable. Por tanto antes de hacer el filtro se
debe esperar a que alcance la temperatura normal. Al tiempo debemos
darnos cuenta que introducir una cánula caliente en una disolución es
arriesgarnos inútilmente a descomponer el producto.
Para montar el filtro en la base se debe obrar con delicadeza, pues el papel
de filtro no debe arrugarse, ni rasgarse ni ensuciarse.
Pasemos entonces a la operación:
El manejo en sí requiere mucha destreza,
por lo cual se deduce que en esta
operación la experiencia resulta
indispensable para un aceptable ejercicio.
Se toma el disco de papel de filtro y se
coloca sobre la circunferencia del extremo

172
cóncavo de la cánula, situándolo concéntricamente.
Inmediatamente se coge por cada extremo y se abate
sobre los bordes, llevándolos hacia abajo, hasta que
queden sobre la parte inferior del cilindro, fijándolos allí
con los dedos. A continuación se hace lo mismo con los
otros dos salientes que quedan.
De este modo tras fijar los dos últimos cabos nos
encontramos con una figura parecida a la de la izquierda.
A lo largo del proceso conviene tocar sólo las puntas del
filtro, para no ensuciarlo en su superficie.
Los cuatro salientes que se han formado de esta manera
cuando hemos plegado el filtro, se cruzan uno sobre otro,
quedando como describe el propio dibujo. Obsérvese cómo
queda el papel, rodeando perfectamente toda la superficie
para lograr un filtrado perfecto.
Lo único que resta es amarrarlo con un hilo de alambre para
que no se salga durante la operación.
Al principio de hacer el montaje del filtro se corta de la
bobina un trozo de alambre apropiado, para tenerlo
preparado en este momento. El alambre se pone,
procurando dejarlo lo más firme posible. A continuación
será preciso emplear los alicates con mucho cuidado, ya
que cualquier roce del metal de la herramienta puede
ocasionar desgarros al papel, o cuando menos ensuciarlo.

173
Se giran los dos cabos de alambre hasta que se partan por la tensión.
17. 2 SIN APLIQUE DE FILTRADO
Igualmente podemos usar cánulas normales donde no existe el cilindro
sobre el que se ha de montar el filtro. En ese caso el proceso es análogo al
anterior, salvo que más complejo debido a la menor extensión de la
superficie donde se aloja el papel. Esta indicado para volúmenes a filtrar
más pequeños.
Ahora debemos cortar el filtro, al contrario que en el caso anterior, donde
utilizábamos el círculo de papel completo. Se corta un rectángulo que suele
tener como base el diámetro del círculo de papel y una altura igual a 1.5cm.
Como la superficie frontal sobre la que se instala el filtro es más pequeña,
la presión que se realiza contra el papel de filtro es mucho mayor, de modo
que muchas veces, de no ser cuidadosos, se perfora el papel de filtro,
acabando por salir el extremo metálico a su través.
Los pasos a seguir son parecidos a los anteriores, tal y como se ha
comentado, pero presentan variaciones claras a causa de la estrechez de la
cánula, que impide realizar todos los puntos dados en el apartado anterior.
Al igual que antes se sitúa el filtro sobre la
boca roma de la cánula, procurando no
apretarla contra la misma por peligro de
perforación. Aquí como las dimensiones son
más reducidas que las de antes, lo que
entonces era difícil, ahora es aún más difícil

174
y se debe considerar que la mejor manera de llegar a hacer montajes
aceptables es la experiencia directa.
Se pliegan las dos alas más grandes de modo que se
lleven sobre la superficie de la cánula. Es importante
hacer notar que es aquí, y en las operaciones sucesivas
donde se perfora el papel habitualmente. Tras fijar con
los dedos los bordes del papel se procede al siguiente
movimiento.
Cada una de las dos alas que quedan tras doblar como se
indica en el segundo dibujo, se pliegan a su vez en sentido de
giro idéntico, de modo que la cánula quede completamente
encerrada en el papel de filtrado y no permita la entrada a
ninguna sustancia sólida durante el proceso de separación.
Para forzar la inmovilidad del filtro se rodea con un hilo de
alambre fino del mismo modo que se hizo antes. Aquí hay
que cuidar especialmente cualquier tipo de torsión hacia
abajo, que conllevaría la ruptura del filtro en su parte
cenital.
17. 3 LOS DISCOS DE FILTRO
Son círculos de papel de filtro con una porosidad determinada que dejan
pasar unas partículas de tamaño mínimo dependiendo del tipo de filtro que
utilicemos. Se guardan en la caja donde se expenden, la cual es cuadrada y

175
tiene un tamaño ligeramente superior a la de éstos. Suele guardarse esta
caja conjuntamente con los alambres y alicates.
El papel es muy parecido al papel de filtro que se emplea normalmente,
sólo que se puede elegir más o menos tupido según sea el tamaño de
partícula a filtrar. Otra virtud de no mucha utilidad en organometálica y que
es más propia de laboratorios de análisis es que al quemar este tipo de
filtros, no dejan ceniza.
Deben conservarse siempre con mucha limpieza, para que no estén en
contacto con ningún elemento extraño en su superficie, lo cual podría
perturbar la reacción.
17. 4 USAMOS LOS FILTROS
El uso de estas cánulas con filtro en su extremo no es distinto al normal
para cualquier tipo de cánula, dado que la función de la cánula no cambia,
y tan sólo se limita a conducir el líquido de un lugar a otro.
Sin embargo, al instalar un papel filtrante en uno de los extremos, es cierto
que dificultamos el proceso, que a veces resulta lento debido a la
viscosidad de la disolución o a la deposición de sales sobre la superficie del
filtro que resulten poco porosas.
Para introducir el extremo de la cánula que posee el filtro en la disolución a
filtrar, previamente debemos insertar la cánula en el tapón pinchado que va
a usarse para tapar al matraz en tanto dure el proceso de separación.
Esta operación se lleva a cabo de modo que el extremo agudo pase por el
tapón desde dentro, para que así quede el filtro en la posición interior
cuando el tapón se coloque en el matraz.
Hecho esto, con el N2 (g) abierto, cambiamos el tapón. Esperamos que
poco a poco se elimine el oxígeno que pueda haber en la cánula y entre los

176
pliegues del filtro. Tras ello podemos pinchar la cánula en el Schlenk
preparado para contener el producto.
El filtrado se debe hacer agitando la disolución del matraz para que no
quede nada de producto capturado entre los residuos sólidos. Si todo va
bien, realizado el primer filtrado, se toma un poco de disolvente seco (de
los recogedores de los destiladores) y se lava varias veces el matraz y los
residuos sólidos (pasándose el líquido cada vez al tubo nuevo vía cánula).
Para lavar se sube un poco el filtro y se agita bien, para después volver a
bajarlo y succionar el líquido. Seguidamente se vuelve a abrir el N2 (g) en
el Schlenk (donde se recoge la disolución filtrada) y se retira la cánula.
El tejido poroso queda recubierto por el sólido retenido. Se retira el
alambre con el alicate y junto con el filtro se arroja a la papelera. La cánula
se lava exactamente igual que las cánulas normales, depositándose después
en la bandeja apropiada de la estufa.
Tenemos problemas con el filtrado cuando la cánula es demasiado fina,
igualmente cuando la disolución es viscosa o cuando el residuo tapa los
poros de modo infranqueable, debido a su cohesión. Estos problemas se
pueden resolver total o parcialmente mediante algunos recursos:
1. El más usado es golpear la cánula en la zona cercana al tapón de goma
del Schlenk, mediante martilleos con los dedos, estimulando así la caída del
líquido.
2. Si esto no es suficiente, se agita con intensidad la disolución con el filtro,
sacando y sumergiendo el filtro rápidamente e intentando que pierda la
materia que pudiera ocluir los poros.
3. En último caso se pueden quitar las agujas que igualan la presión en el
Schlenk, con la ambiente, y dar un golpe de vacío lo más breve posible, de
modo que se tire de la disolución que se halla en el matraz. Sin embargo

177
este método no es muy aconsejable para aquellos compuestos que sean muy
reactivos al aire.
18 FILTRACIÓN EN PLACA O LECHO POROSO
Este tipo de método de separación cromatográfico está especialmente
indicado para separar sustancias con polaridades claramente diferenciadas,
donde la más polar va a quedar atrapada en el lecho poroso y la otra
sustancia va a pasar al recipiente adecuado. A continuación veremos cómo
se realiza la operación y cuáles son los lechos más usados.
18. 1 MATERIALES PARA LECHOS
Principalmente se usan dos tipos de materias que sirven para separar
sustancias: la sílice y la tierra de diatomeas o celita. Además de éstas se
pueden usar otros tipos de tierras, pero las dos indicadas son las que se
emplean más a menudo en el laboratorio.
A) Sílice o gel de sílice.
Es un polvo muy fino y blanco, que se maneja con facilidad gracias a que
se presenta en granos muy pequeños y sueltos, que no quedan apelmazados.
Está formado por vidrio y cuarzo pulverizados, tratando con ello de
aumentar su superficie específica. Retiene preferentemente sustancias
polares, gracias a los grupos silanoles de su superficie.
Se guarda en el laboratorio en bote del cual se toma a voluntad,
dependiendo de la masa a filtrar, con una espátula grande.

178
Hay que cuidarse de no respirar el polvillo que durante su manejo puede
levantarse, puesto que es dañino para la salud.
B) Celita, tierra de diatomeas o Kieselgur.
Este otro adsorbente tiene como constituyente principal esqueletos silíceos
de foraminíferos fósiles, con multitud de orificios que retienen
especialmente el agua. Los Foraminíferos son tanto microorganismos del
suelo, como del plancton, que se remontan al Paleozoico y poseen una
concha de caliza.
Su tamaño suele oscilar entre los microscópicos hasta los 5cm. Retienen
sustancias fuertemente polares (más polares que aquellas para las que se
utiliza la sílice).
Es una arena muy fina y blanca que, a diferencia de la anterior, se
compacta mucho consigo misma, levantándose menos por la agitación de la
corriente de gas, por lo que su uso, dentro de la placa va a resultar más
sencillo. Aún así es tan peligrosa para las vías respiratorias como la otra,
por lo que debe tenerse cuidado al trabajar con ella.
Se guarda en una de las estufas, en un erlenmeyer donde se escribe la fecha
en la que se ha cogido del recipiente existente en el almacén. Es importante
conocer la fecha en que se ha cogido del almacén por la afinidad que tiene
por el agua, siendo necesario usarla muy seca, por el bien de los
compuestos.

179
18. 2 LA PLACA
Es un cilindro de vidrio hueco donde en su parte más
baja se halla una placa de cerámica que constituye la
base sobre la que se va depositando la masa de
material filtrado. Su extremo inferior está diseñado
para encajar en un matraz de fondo redondo (se usa
el de dos bocas), donde el producto se irá
acumulando a medida que se va filtrando.
1. Boca de entrada, por donde se añade la sílice o celita y donde se instala
un tapón de goma.
2. Boca roscada para tapón de teflón que servirá para unir el embudo de
filtración con la goma de la línea.
3. Placa de cerámica porosa que será la base para el lecho.
4. Alambre que rodea y abraza con fuerza el estrangulamiento del embudo,
cuya finalidad es servir de apoyo a las gomillas de sujeción.
5. Extremo de vidrio en cuyo interior se puede observar un embudo
colector que sale de la cerámica.
18. 3 MONTAJE Y PREPARACIÓN DEL SISTEMA
Para montar el sistema lo primero que hacemos es fijar con una pinza el
matraz de fondo redondo y colocarle un tapón de teflón. Sobre él, con otra
pinza se afirma el embudo de filtración. Es más cómodo dejar inmóvil el de
abajo y ajustar el de arriba que al revés.
La adición de la tierra porosa se hace con la espátula o mediante un papel
de pesada (donde se ha puesto una cantidad de tierra). Recordemos que no
se debe respirar sobre el producto por el riesgo que supone para la salud.

180
Se intentará aplanar lo más posible la superficie del lecho, de modo que
quede igualado por todos lados. En cuanto al grosor del lecho dependerá de
la cantidad de disolución que vayamos a hacer pasar, manteniendo una
proporcionalidad directa.
Una vez añadido se coloca el tapón rojo arriba y uno de teflón en el lateral.
Aquí podemos ver cómo el conjunto se ofrece a la vista. Para una mejor
comprensión de los capítulos posteriores se han representado las llaves con
letras:
A) Llave que une el tubo de la línea con el
embudo de filtración.
B) Llave que une el tubo de la línea con el
matraz de fondo redondo.
No se ha dibujado el sistema de gomillas para una mejor visión del dibujo.
Aunque ambas piezas ajusten perfectamente, al ser materiales de vidrio
necesitan grasa en sus ajustes. Se pone muy poca grasa, para que después
se pueda limpiar rápidamente, sin tener que recurrir al baño. Sin embargo
debe quedar reforzado el ajuste por gomillas, que se engancharán en los dos
cinturones metálicos existentes en los dos cuerpos.
Ese sistema de gomillas es muy importante dado que se utiliza para afirmar
la unión entre elementos de vidrio de características similares. Así cuando
se habló de los destiladores, sus piezas también se unían mediante el
sistema de engomillado que pasaremos a describir en este punto. De la
misma manera se puede ahora comprender por qué la gran mayoría de los
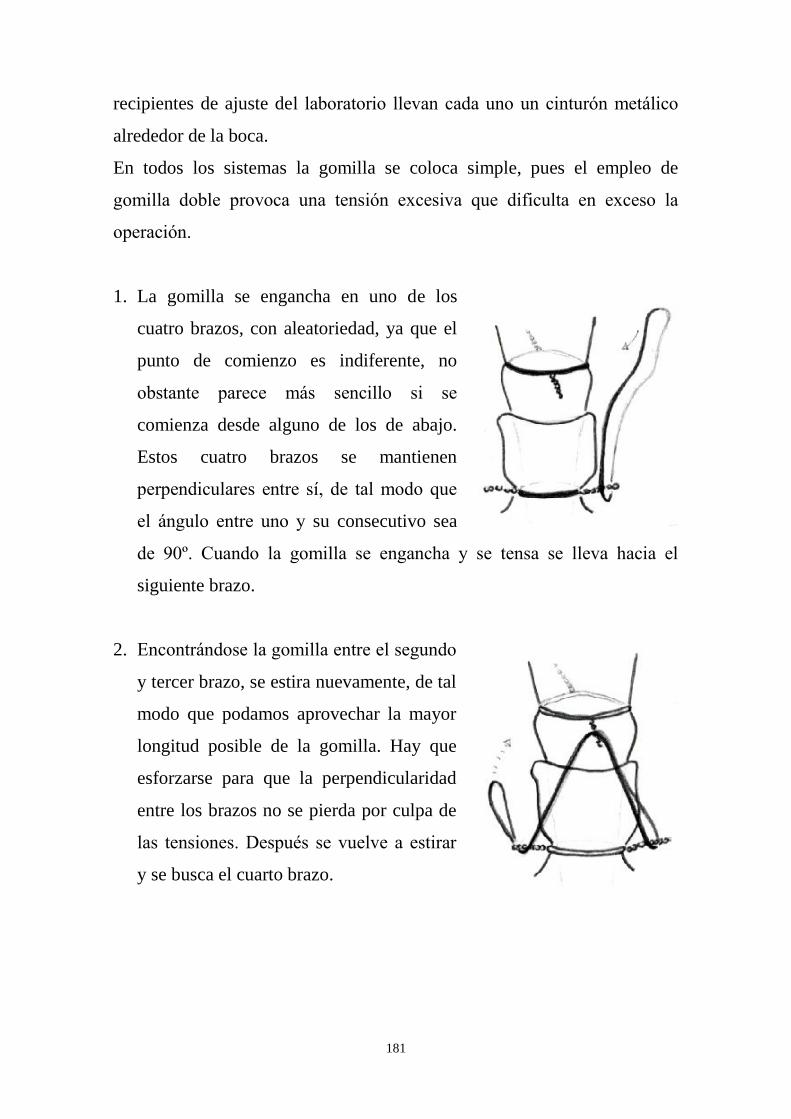
181
recipientes de ajuste del laboratorio llevan cada uno un cinturón metálico
alrededor de la boca.
En todos los sistemas la gomilla se coloca simple, pues el empleo de
gomilla doble provoca una tensión excesiva que dificulta en exceso la
operación.
1. La gomilla se engancha en uno de los
cuatro brazos, con aleatoriedad, ya que el
punto de comienzo es indiferente, no
obstante parece más sencillo si se
comienza desde alguno de los de abajo.
Estos cuatro brazos se mantienen
perpendiculares entre sí, de tal modo que
el ángulo entre uno y su consecutivo sea
de 90º. Cuando la gomilla se engancha y se tensa se lleva hacia el
siguiente brazo.
2. Encontrándose la gomilla entre el segundo
y tercer brazo, se estira nuevamente, de tal
modo que podamos aprovechar la mayor
longitud posible de la gomilla. Hay que
esforzarse para que la perpendicularidad
entre los brazos no se pierda por culpa de
las tensiones. Después se vuelve a estirar
y se busca el cuarto brazo.

182
3. Tras pasar la gomilla por el cuarto brazo se
estira otra vez y se repite el proceso tantas
veces como se pueda, hasta que la gomilla no
ceda más, enganchándose entonces al brazo más
cercano. En este dibujo la gomilla se ha dejado
sobre el primer brazo, pero no es más que un
ejemplo.
Toda esta manipulación se hace con ambos elementos cogidos por las
pinzas, pero el embudo suele dejarse algo suelto, para permitir el
movimiento mientras ajustamos las dos piezas y que sea efectivo el
proceso, pues de no poderse mover un poco la placa, sería imposible
fijarlos adecuadamente.
18. 4 DESOXIGENACIÓN DEL MONTAJE
Para la correcta desoxigenación del sistema hay que comenzar cerrando la
llave A y la B. La llave A estará conectada a la línea en N2 (g), en tanto B
lo estará al vacío. De este modo cuando se abra cualquiera de las dos
lograremos hacer inmediatamente vacío e introducir N2 (g). Si fuese al
revés levantaría toda la capa de tierra y se iría por la línea.
1. Se abre B. Consiguientemente se logra vacío en el matraz de fondo
redondo, pero como el embudo está conectado, poco a poco el vacío se irá
logrando también en su interior. Es necesario dejarlo unos minutos, para
que el vacío sea aceptable y eliminemos todo el aire posible.

183
2. Se cierra B y se abre A. Esta llave debe ser abierta lentamente para no
turbar la superficie del lecho. Generalmente en el lecho de sílice se crea un
momentáneo torbellino, en tanto que en el de la celita suele conmoverse
desde abajo y agrietarse. Casi al tiempo de abrir, el embudo y el matraz se
llenan de gas.
3. Se repite el proceso hasta que se cumplan los consabidos 3 ciclos.
Se nos puede ocurrir no obstante que podríamos desoxigenar más rápido
abriendo A y B, de modo que una corriente continua de N2 (g) pase,
arrastrando cualquier resto de aire a través de todo el sistema, eliminando
toda traza indeseada. Sin embargo se presenta el siguiente problema: el N2
(g) al llegar a la trampa condensa (pues la temperatura del N2 (l) es
justamente la que hace falta para hacerle condensar) de modo que la trampa
se colapsa y se acumula escarcha en la parte superior de la cabeza de la
línea. Como no debe ocurrir esto el proceso descrito en el primer caso es el
más conveniente a seguir.
18. 5 LA OPERACIÓN DE FILTRADO
Para realizarla será necesario tomar en un tubo de centrífuga un poco de
disolvente y ponerlo a desoxigenar. Se usará para mojar el lecho
previamente y después lavar. Su desoxigenación se hace por el método ya
conocido.
En el matraz de reacción tendremos la disolución donde coexisten los dos
componentes a separar, ambos en un disolvente que es igual al que se ha
puesto a desoxigenar en un tubo de centrífuga aparte, para mojar el lecho.
La complicación más evidente es la de cambiar el tapón en el embudo de
filtración. Debemos bajar la corriente de N2 (g), aunque no totalmente, de

184
modo que se agite lo menos posible la superficie del lecho. Mas aún
disminuyendo el flujo de gas, se produce un torbellino que lanza partículas
de sílice hacia arriba, de ahí la importancia de no respirar sobre la boca del
embudo. Lo mismo, aunque menos marcado, ocurre con la celita, que más
bien tiende a resquebrajarse.
Se trata pues de colocar lo más velozmente posible el tapón pinchado. A
continuación se pasará una cánula desde el tubo de centrífuga al embudo y
se mojará la capa filtrante. Al principio este disolvente añadido cae al
matraz de fondo redondo gota a gota, pero es preferible así y no de golpe,
para que se moje bien el lecho y no retenga el producto que debe eluir.
Al añadir el disolvente para mojar el lecho, lo más conveniente es hacerlo
apoyando el extremo agudo en la pared de vidrio. Esto impide que la
potencia de salida del líquido haga agujeros y canales en el lecho, lo cual es
poco deseable a la hora de una buena filtración.
Se abre la llave del N2 (g) en el embudo de filtrado y se introduce la cánula
en el matraz donde se encuentra el producto que vamos a pasar por la placa,
de este modo se conectará el tubo de centrífuga, donde hay disolvente seco,
y el matraz, para después lavar varias veces y no perder parte del producto.
Acto seguido con otra cánula se conecta el matraz de reacción con el
embudo y se comienza a hacer pasar disolución de la manera conocida,
buscando siempre apoyar el extremo agudo en el vidrio.
Cuando se ha añadido el producto, se espera a que baje por la capa filtrante
y pase al matraz de fondo redondo. Si el proceso es demasiado lento se
intentará solucionar mediante los consejos descritos en el punto 17.4. Aquí,
obviamente, el golpe de vacío se dará desde el matraz de dos bocas que
cierra el conjunto de filtración, siendo esta estrategia muy efectiva, aunque
tiene la desventaja de acelerar demasiado el proceso, por lo que puede
arrastrar parte de compuestos indeseables que deberían haber quedado en la
placa.

185
Se procede después a lavar el lecho poroso con el disolvente del tubo de
centrífuga para que no quede retenido nuestro producto. Así se añade
disolvente desde el tubo de centrífuga al matraz de reacción, así se
aprovecha también para lavar su interior y no dejar nada de producto atrás;
y a continuación se pasa al embudo y se filtra por gravedad o por golpe de
vacío. Hecha esta operación varias veces, se abre el N2 (g) en el embudo y
se retiran las cánulas. Siempre es mejor que la filtración se produzca por
gravedad pero si es demasiado lenta tendremos que recurrir a un ligerísimo
golpe de vacío.
Para separar los dos cuerpos del montaje se abren las llaves A y B con
salida de N2 (g). La sílice no se levantará dado que la corriente que la
impulsa desde abajo se contrarresta con la del embudo. Se quita la gomilla
y se separa el embudo con cuidado. Tras esto se coloca en el matraz de
recolección un tapón nuevo y se cierra el flujo de gas inerte en los dos
elementos.
El embudo se limpia tirando la tierra de filtración en la papelera. Como está
húmeda y compacta se extrae golpeando el cilindro contra la mano, lo que
hace que se suelte el material y el embudo quede en condiciones de ser
limpiado con acetona y éter.
Como conclusión, se comentará que la operación más difícil es, sin lugar a
dudas, la de cambiar el tapón. El lecho reacciona siempre de modo
inconveniente, y en el caso de la sílice resulta desesperante si no se
disminuye sustancialmente la fuerza de la corriente, ya que el remolino
puede sin dificultad expulsar hacia arriba la mitad del lecho.

186
18. 6 FILTRACIÓN PARA RECOGER EL SÓLIDO
En el caso anterior el montaje estaba aplicado a la separación de dos
solutos, de modo que uno se adsorbiese en el lecho y el otro pasase por él
sin ser retenido. Sin embargo podemos también usarlo cuando queramos
quedarnos con un sólido el cual no puede ser filtrado al aire al ser inestable
en esas condiciones. Se usa cuando la cantidad de producto es muy grande
y el uso de los filtros de papel por tanto es poco recomendable.
El proceso ahora es análogo al de filtración en embudo Buchner, sólo que
ahora se realiza bajo atmósfera de N2 (g).
El montaje del sistema es el mismo que el del apartado anterior,
diferenciándose únicamente en la ausencia de material adsorbente. Esto es
comprensible puesto que si pretendemos recoger un sólido para separarlo
de las aguas madres, basta con usar el tamiz constituido por una placa de
cerámica de características apropiadas sin precisarse un lecho de tierra que
posteriormente tendríamos que separarla del producto.
Para realizar la operación sin demasiados contratiempos, los cuales siempre
surgen inesperadamente, debemos tener en tubos de centrífuga, dos
disolventes que vayan desoxigenándose. Uno de los disolventes (D1)
servirá para lavar los cristales, y es el mismo disolvente que sirvió para
hacer la reacción donde se obtuvieron los cristales (si aparecieron como
tales en el seno de la misma) o que se usó para cristalizar posteriormente.
El producto será soluble en él, pero se disolverán preferentemente las
impurezas (debe ser así para que el procedimiento tenga sentido) por lo que
es útil como líquido de lavado, aplicado varias veces en pequeños
volúmenes. El segundo (D2) debe ser capaz de disolver el compuesto
cristalizado completamente y será usado para llevarnos los cristalitos,
pasándolos a un Schlenk. Es evidente que aquí no se puede raspar con la
espátula y pasarlo alegremente a un tubo.

187
Con todo dispuesto pasaremos a detallar el proceso y sus etapas más
características:
1. Con una cánula muy gruesa (generalmente de teflón) pasamos el
producto del matraz al embudo de filtración. Al pasarlo debemos agitar
muy bien la disolución del matraz, para que los cristales pasen sin
problema a través de la cánula y no se queden sedimentando en el fondo
del matraz.
2. Al llegar a la placa del embudo, se produce la filtración, y en la parte
superior se depositan los cristales. El matraz de reacción por tanto, se
encuentra bajo N2 (g) y manda su corriente a través de la cánula, en tanto
que el embudo tiene cerrada la llave de gas y unas agujitas en el tapón
hacen caer la presión en su interior. Si fuese necesario, un golpe de vacío
forzaría el paso del líquido a través de la porcelana.
3. Se pincha con una cánula en el tubo de centrífuga que contiene a D1 y se
pasa una cierta cantidad al matraz de reacción. Aquí se busca recoger, no
disolviendo, sino por arrastre, los pequeños cristalitos que permanezcan y
pasándolos al embudo. No se indican los movimientos de apertura y
cerrado de las llaves, para hacer pasar la disolución del matraz al embudo,
pues debe ser ya bien conocido de todos tras los capítulos iniciales, donde
se ha insistido mucho al respecto. Se produce una nueva filtración, al
mismo tiempo que se lavan los cristales. Esta operación se reiterará con
volúmenes muy pequeños de D1.
4. A veces puede incluso ser interesante lavar con algún otro disolvente que
disuelva exclusivamente las impurezas y en el cual los cristales sean
completamente insolubles. Esto en sí suele ser muy poco usual
desgraciadamente, por lo que lo acostumbrado es que el líquido disuelva
mayoritariamente a uno, pero en cierta medida, también al otro.

188
5. Cuando la operación de filtrado se da por concluida y no pasa más
líquido a través de la porcelana se llega a la fase más delicada. Debemos
cambiar el matraz de fondo redondo donde se han colectado las aguas
madres por un tubo de Schlenk donde se recogerán los cristalitos, o mejor
dicho, la disolución de los mismos en D2. El Schlenk a usar tendrá boca
hembra, para que se pueda encajar en el embudo de filtración.
Para separar los dos elementos de vidrio se procede exactamente igual que
en el caso anterior. Lo primero será abrir el N2 (g) en ambos recintos.
Eliminar las gomillas y separarlos con cuidado a continuación.
El matraz con las aguas madres puede ser guardado, si aún contiene una
cantidad de producto interesante (lo cual es lo más natural) y tras
concentración y posterior cristalización cederá otra cosecha de cristales. El
número de cosechas de cristales que se obtienen de esta manera, depende
enormemente de la naturaleza de los componentes, pero en líneas generales
no es extraño encontrarnos con cristales en una tercera o cuarta
cristalización.
El tubo que sustituye al matraz será, como ya se ha dicho, un tubo Schlenk
con boca hembra al cual se le habrá colocado un collar de alambre, que
permita poner gomillas (al igual que antes). Este tubo deberá haber sido
desoxigenado previamente y salvado bajo atmósfera de nitrógeno. Con la
corriente de gas inerte abierta en ambos recipientes se ajustan de modo
adecuado.
Como el aire no puede vencer la fuerza de salida del N2 (g) no podrá entrar
en ninguno de los dos recipientes durante el proceso y no hay peligro de
que se destruya el compuesto cristalizado. Tras el ajuste referido se colocan
las gomillas y no se cierra el N2 (g) en ninguno de los dos elementos (se
entenderá en el siguiente punto).

189
6. Se vendrá a disolver el producto y a hacerlo pasar al Schlenk. Con una
cánula se transvasa una cierta cantidad de D2 al embudo, que al estar
abierta la corriente de N2 (g) en el Schlenk, no podrá caer dado que a la
gravedad se le opone el flujo del gas. De este modo el contacto entre los
cristales y D2 será más conveniente para que en ese volumen de disolvente
se disuelva la mayor cantidad posible de producto (no olvidemos que
después habrá que concentrar, llevando a vacío la disolución hasta
acercarnos a la saturación).
7. Como no se dispone de agitación, el proceso de disolución sería largo de
no servirnos de dos artimañas de gran efecto:
A) Se cierra la llave del embudo y se colocan agujas. Esto provoca que el
N2 (g) que sale de la llave del Schlenk se vea obligado a atravesar el lecho
de porcelana y a hacer burbujear al disolvente entre los cristalitos, logrando
una agitación eficiente que lentamente logra disolver el producto.
B) Con un secador se aplica calor en las paredes del embudo, facilitando la
disolución por aumento de temperatura.
8. Cuando se cree que el volumen empleado de D2 está saturado, se abre el
N2 (g) en el embudo y se da un golpe de vacío para que pase la disolución.
Aún en la parte superior quedará producto sin disolver. La operación se
repite hasta que no quede resto de producto sobre la placa porosa.
9. Se separan el embudo y el Schlenk como se sabe por los análogos
anteriores.
10. El líquido con los cristales disueltos se concentra usando la línea. A
continuación se lleva al frigorífico y se deja que recristalice. Los cristalitos
se recuperarán de nuevo como se hace para cualquier otro compuesto (lo
cual se explicará posteriormente).

190
11. Las aguas madres darán nuevos cristales que tendrán que ser separados
por el mismo procedimiento tras concentrar la disolución nuevamente.
Este largo y tedioso sistema puede variarse aplicándolo a casos
particulares, pero aquí se ha intentado hacer un tratamiento general. Está
especialmente indicado para separar grandes cantidades de producto, que
por otro proceso sería inviable debido a su masa, por lo que puede
deducirse que es una operación propia de la preparación de materiales de
partida.
No se ha hecho constar una anotación fundamental y merece la pena que se
indique. Esta operación podría muy bien haber sido computada entre los
puntos dedicados a la cristalización de productos, sin embargo se ha
considerado apropiado entenderla como una estrategia de separación donde
la placa filtrante se constituía en elemento clave.
Toda separación de cristales se realiza manteniendo el matraz de reacción,
donde coexisten los cristales con las aguas madres, a baja temperatura. Esto
se logra por el uso de baños fríos de acetona (sin hielo seco y sin N2 (l))
que se guardan en el congelador del frigorífico. Mediante su uso se impide
que la mayor parte de los cristales se redisuelvan durante el proceso de
filtrado a causa del aumento de la temperatura tras ser sacados del
congelador. Por tanto durante toda la maniobra el matraz estará introducido
en el baño de acetona.
Igualmente, por la misma razón, cuando se lavan los cristales con D1, es
importante enfriar el disolvente con otro baño de acetona.
Estos baños de acetona que se guardan en el congelador no suelen ser vasos
de precipitados, como los usados para calentar el aceite, sino que pueden
obtenerse recortando las partes de debajo de botellas de plástico. Es en
estos recipientes donde se añade la acetona, enfriándose en el congelador y

191
usándose siempre que tengamos intención de separar los cristales del resto
de la disolución.
18. 7 FILTRACIÓN EN PLACA AL AIRE
Para separar un producto sólido que nos interesa, de una matriz líquida se
usan placas como ya se estudiaron en el apartado anterior, sin embargo
aquellas se usan cuando el compuesto a filtrar es inestable al aire. Si
tenemos la suerte de que el compuesto es estable en atmósfera normal,
podemos a su vez filtrarlo usando medios similares. Al igual que antes, son
operaciones que se hacen con productos en gran cantidad y de ahí que estén
vinculadas en su mayoría a reacciones de formación de productos de
partida. Si la cantidad fuese menor se podría separar de su fracción líquida
mediante filtrado con cánula, lo cual sería más rápido.
Hay dos tipos de embudos que se usan en este caso: el Buchner normal y el
de placa de cerámica.
A) Por embudo Buchner.
No hay diferencia con el otro más que en
su estructura, lo cual condiciona su modo
de empleo, pero manteniendo las mismas
funciones.
El embudo es de porcelana normalmente,
y como puede apreciarse, la base sobre la que se forma la torta de producto
sólido, está agujereada. El resto del diseño es análogo a un embudo normal
(llamado embudo “alemán”), salvo diferencias poco significativas.

192
Para emplear este embudo se debe recortar un círculo de papel de filtro con
un tamaño de poro determinado. Para obtener un filtro regular y apropiado
se obra del siguiente modo: con la propia circunferencia del embudo se
marca, mediante el concurso de la punta de unas tijeras, un círculo de filtro,
que después se recorta tal que quede dentro del embudo para lo cual el
corte se realiza unos cuantos milímetros por debajo de la circunferencia a la
que se sigue. El papel no debe tocar las paredes para que no haya ninguna
deformación del mismo, sino que quede adherido sobre la base con
perfección.
El embudo se ajusta al Kitasato usando unas cuñas de
goma que se adaptan, tanto al embudo como al otro
recipiente. Este último se conecta a un sistema de
succión que generalmente está basado en el efecto
Venturi, y usa una corriente de aire comprimido o la
presión de salida del agua del grifo abierto al máximo.
B) Por placa de cerámica.
En este caso el embudo es de vidrio y en vez de poseer un fondo perforado
posee una placa de cerámica porosa, análoga a la del embudo de filtración
visto en 18.2. Gracias a esta placa se separa el sólido de la matriz líquida.
No requiere papel filtrante, bastando su superficie para la separación. Esta
superficie será al final del proceso raspada con la espátula para recuperar el
producto. Su uso, como puede comprenderse, es idéntico al anterior y así
serán tratados conjuntamente en la exposición al respecto.

193
Obsérvese aquí cómo el embudo está
provisto de una placa en su parte
media, que impide el paso del sólido y
lo retiene en forma de torta sobre su
superficie.
Cada placa tiene un tamaño de poro distinto, existiendo varios tipos según
los compuestos que podamos tener necesidad de filtrar.
La conexión del embudo con el Kitasato se hace gracias a un ajuste
existente en el cuello del embudo y en la boca de aquel, que requiere la
colocación de un poco de grasa para que sea una unión fiable.
En principio hay que montar el sistema de succión. Al fondo de cada
vitrina hay una boquilla que se controla desde la parte de fuera con una
llave. Al accionarla sale una gran corriente de aire comprimido por la
tubería. Si se conecta a ella un Venturi se podrá hacer vacío dentro del
Kitasato que se conecte con una goma al propio Venturi.
El sistema completo puede analizarse en la siguiente imagen:
1. Salida del aire comprimido que
se encuentra en la pared que
cierra la campana.
2. Tubo de goma muy gruesa que
conecta a 1 con el Kitasato.
3. Cajita donde se halla la cámara
donde por efecto Venturi se
extrae el aire del Kitasato.

194
4. Goma que conecta el Kitasato con el Venturi.
5. Sistema de filtración.
El tubo 2 va conectado a la salida de aire comprimido mediante una
abrazadera. Esta abrazadera se aprieta con un destornillador, de modo que,
con la propulsión del aire no salga despedida, pues al hacerlo provocaría un
silbido terrible, un sonido tan agudo como potente. Como consecuencia, el
Venturi junto con la goma saldría despedido y se podría partir contra el
suelo. Por tal motivo, resulta obligado apretar la abrazadera de la boquilla
con toda la fuerza posible.
Se ha nombrado varias veces en el transcurso de los capítulos al Venturi sin
explicar en qué consiste exactamente. Va siendo hora de que lo tratemos
aunque con brevedad. Se trata simplemente de hacer conducir el aire o un
fluido en general por un tubo a una velocidad muy alta. En algún lugar, ese
tubo tiene una pequeña abertura que lo conecta mediante otra goma a la
cámara donde se quiere hacer vacío. El fluido va tan rápido que arrastra al
aire que se encuentra cerca de la abertura, dentro del tubo de conexión. De
este modo acaba por lograr un vacío considerable en el interior del
recipiente.
Otro consejo a tener en cuenta es el de fijar el Kitasato a un pie mediante
una pinza, esto evitará que cualquier golpe venga a desequilibrar el montaje
y estrellarlo con el suelo.
Antes de comenzar la verdadera operación de filtrado nos aseguraremos de
tener el vaso de precipitados con el producto a filtrar al lado del montaje,
con una varilla de vidrio para agitarlo cuando sea necesario. También la
botella del disolvente en el que se ha precipitado el producto, para al
terminar la filtración, lavar el interior del vaso de precipitados y arrastrar
los restos que pudieran quedar.

195
Cuando se tiene todo dispuesto se abre la llave del aire comprimido
lentamente hasta que no pueda dar más de sí y el gas salga con la máxima
potencia. El filtro se debe mojar previamente, usando para ello un poco del
disolvente puro y sirviéndonos de la varilla de vidrio, tratando con ello de
que no se arrugue o se desplace al añadir el producto.
Se agita la suspensión con la varilla de vidrio, y se va añadiendo la
suspensión sobre el filtro. A medida que el filtrado se va llevando a efecto
va apareciendo una torta de producto sobre el embudo.
La manera más correcta de verter el producto sobre
el lecho poroso o sobre el filtro es valiéndonos de la
varilla. Esta se apoya en la acanaladura del vaso. Se
debe cuidar que su parte inferior se sitúe encima de
la torta, pero sin tocarla. A medida que cae la
mezcla deslizándose por la varilla, se deposita en el
lugar elegido. Con un movimiento de muñeca se
cambia la posición inferior de la varilla y se reparte
uniformemente la masa del producto. Así pues se
tratará de evitar que el producto forme un montículo en el centro, en tanto
por los alrededores no haya nada.
La varilla posee en su extremo una goma que rodea al vidrio e impide que,
agitando la disolución, se quiebre el vaso de precipitados por un mal golpe.
La varilla así dispuesta se denomina también policía. Una manera
alternativa de agitar la disolución con la varilla, sin peligro, es la de coger
el matraz por la boca, con la misma mano tapando toda su abertura y, con
la varilla encajada inmóvil entre los dedos de la mano, comenzar a girar el
vaso entero, de manera que el líquido es el que gira y rompe contra la
varilla y no al revés.
Cuando se haya pasado todo por el filtro, debemos lavar el producto.
Entonces tomaremos un poco de disolvente (en el cual el producto sea

196
insoluble) y lo pondremos en el vaso de precipitados (así arrastraremos el
producto que haya quedado por las paredes). Lo haremos varias veces, con
pequeños volúmenes, hasta que pase todo el producto.
Tras lavar se deja que la succión seque suficientemente el producto, para
concluir por despegar el Kitasato y por cortar la corriente de aire
comprimido.
Una vez se tiene terminada la operación hay que almacenar el producto.
Tanto usando uno como otro embudo, hay que raspar con la espátula e ir
depositando el producto en el envase que se precise. Para no perder nada
durante el proceso, éste se hace sobre un papel encerado y los restos que
caigan se podrán añadir al final.
18. 8 OTROS FILTRADOS Y CROMATOGRAFÍAS
Existen otras técnicas, al margen de las mencionadas que de vez en cuando
son necesarias. Así encontramos cromatografías en columnas pequeñas,
incluso en pipetas tipo Pasteur (cuando las cantidades de producto son
mínimas) o con otros lechos de filtración. No obstante, la mayoría de las
técnicas más usadas han sido aquí detalladas y expuestas.
Hemos dejado, eso sí es verdad, una última técnica cromatográfica para
desarrollarla aparte, pues su importancia es tal, que merece esta
consideración. Se trata de una cromatografía especial, donde la fase móvil
se acelera, aprovechando la fuerza centrífuga, en un lecho poroso en
rotación. El instrumento usado se denomina cromatotrón, y es tan
espectacular como su nombre hace suponer.

197
18. 9 LIMPIEZA DE PLACAS
Una conclusión para este bloque no puede sino encargarse de la limpieza
de las placas, lo cual es incluso más engorroso que el mismo proceso de
limpiado.
El primer disolvente que se usa es la acetona, para eliminar aquellos
residuos orgánicos u organometálicos que puedan reaccionar con el agua.
Así se añade acetona al embudo y se espera a que, gota a gota se vaya
vaciando. De este modo se limpiará la cerámica. A continuación se
empleará ácido clorhídrico y tras él agua. Finalmente se deberá lavar con
acetona y éter.
Si tuviésemos zonas oscuras de color grisáceo, se deduce que han quedado
restos metálicos nobles. Estos desaparecerán con el agua regia (mezcla de
HCl (ac.) y HNO3). Más adelante nos referiremos al lavado con agua regia
y a los cuidados que se ha de tener.
19 CROMATOGRAFÍA PLANA EN CAPA FINA
Esta técnica de separación es muy usada con la finalidad de determinar la
pureza de una muestra, para saber cuántos compuestos hemos obtenido al
final de la reacción o para conocer cuántos permanecen todavía tras las
separaciones que hemos llevado a cabo.
La separación se basa en colocar una gota de la muestra sobre la
cromatoplaca, que posee una capa adsorbente por la que se hace pasar a
continuación una mezcla de disolventes o disolvente puro que se llama fase
móvil. Dependiendo de la polaridad de los compuestos, éstos serán más o
menos arrastrados por la placa, distribuyéndose así en zonas bien
diferenciadas unos de otros.

198
Como no es mi intención desarrollar las bases de la cromatografía en este
trabajo, baste saber lo anterior con respecto al fundamento de esta técnica.
19. 1 LA PLACA
Está constituida por una lámina metálica sobre la que se deposita una capa
de alúmina o de sílice (esto las diferencia) y también una cierta proporción
de CaSO4 (s), que ayuda a dar consistencia. A la mezcla anterior se le ha
adicionado algo de fluoresceína, que bajo la lámpara ultravioleta emite una
luz fluorescente de una tonalidad amarillo verdosa. El conjunto de la masa
es de color blanco.
El tamaño de partícula de estos elementos debe ser muy fino, lo cual ayuda
a abreviar la técnica por efectos de capilaridad.
Hay dos tipos de placas que se usan en el laboratorio para estos menesteres:
las de sílica-gel y las de alúmina.
Las primeras se usan para sustancias con cierto carácter ácido o neutro, en
tanto las segundas se prefieren para sustancias básicas. Se usan más las de
sílice que las otras, pero esta afirmación debe ser matizada, ya que
dependiendo del tipo de productos con los que se trabaje en el laboratorio
se usarán más unas u otras.
Se compran ya hechas a una casa comercial, encontrándose por tanto en
forma de láminas de tamaño folio, encerradas en una caja plana donde se
indica claramente el material a usar como fase estacionaria.
Para su uso, se recortan con unas tijeras unos trozos rectangulares cuyas
dimensiones oscilan alrededor de los 5cm de alto y 2cm de ancho.

199
19. 2 PREPARAMOS LA PLACA
La preparación de la placa no es algo complejo, pero deben tenerse en
cuenta una serie de precauciones importantes:
1. Intentemos no ensuciar la placa cuando la recortemos pues esto podría
interferir en el camino que hacen los compuestos, una vez atrapados en la
fase móvil.
2. Recortada pues, se coge un lápiz y en cualquiera de sus extremos se hace
una raya algunos milímetros por encima del borde.
3. Con el lápiz señalamos tantos puntos como muestras vayamos a colocar,
contando siempre con la muestra correspondiente a la mezcla del conjunto
de sustancias. Para entender esto es preciso señalar dos situaciones:
A) La placa la podemos hacer para ver el grado de pureza de un producto.
En este caso un único punto basta para comprobarlo, pues poniendo una
gota de muestra veremos si ese punto se abre en más o permanece como tal.
B) En cambio, puede usarse para analizar cómo se han separado los
productos (que inicialmente estaban en la misma disolución) tras un
proceso de separación (de cualquier tipo en general, pero especialmente
aquellos cromatográficos). Entonces tendremos varias fracciones, y a cada
una le asignaremos un punto en la placa, considerando también aquel que
sirve como patrón (conteniendo el conjunto completo de compuestos que
pretendemos haber separado mediante el método cromatográfico). Cada
punto lo numeramos, dependiendo de a qué fracción pertenece:
P = patrón
1 = primera fracción
2 = segunda fracción

200
3 = tercera fracción
4. En estos momentos la placa se encuentra en disposición de recibir las
porciones correspondientes, cada una de las cuales se coloca sobre el punto
señalado.
Algunos ejemplos de placas los encontramos más adelante, en la sección
19.6 donde puede verse con claridad cómo deben distribuirse los puntos
antes de poder realizar el experimento de manera adecuada. En esos
dibujos se puede observar la línea que se ha trazado con lápiz en su parte
inferior. Esta línea servirá para saber hasta donde tenemos que añadir el
disolvente de la fase móvil, ya que si se supera podríamos disolver las
muestras en el líquido y por tanto después no habría mancha que observar
dado que el producto se habría disuelto en el disolvente al ir avanzando.
Cada uno de los puntos está suficientemente separado el uno del otro, para
que no haya interferencias cuando corra la fase móvil.
Un último consejo es no cometer la torpeza de hacer los puntos y la línea
con bolígrafo, pues la tinta correría arrastrada por el disolvente.
19. 3 DISPENSAMOS LAS ALÍCUOTAS EN LA PLACA
Puede ser que lo más curioso del método sea el medio por el cual
extraemos de cada matraz, en donde se contienen las distintas fracciones
del proceso de separación, las pequeñas cantidades que se usan para hacer
la cromatografía plana.
Nos valemos para ello de unos elementos llamados capilares. Son tubos
finísimos de vidrio que pueden tener una longitud variable. Al introducirlos
en una disolución absorben por capilaridad una cierta cantidad de líquido
(con lo que podemos hacernos una idea de su diminuto diámetro).

201
Los capilares se fabrican por los propios usuarios y es una operación para
manos hábiles. No obstante, sobre el papel, la manera es muy simple.
A una pipeta Pasteur como la representada en 1, se la calienta con un
mechero de llama (se verá posteriormente en qué consiste) a lo largo del
extremo fino. Véase en el dibujo dónde se aplica la llama. Al instante,
empieza a enrojecer y a hacerse elástico el vidrio. Vamos entonces
estirando y se logra un alargamiento del vidrio al tiempo que disminuye el
radio de su grosor (2). Finalmente, cuando se alcanza la longitud adecuada,
se deja enfriar y se parte por donde se une al grueso de la pipeta.
Con este capilar cogemos una cantidad de la disolución. De este modo, con
el N2 (g) abierto, se introduce un extremo del capilar en el líquido. Este
asciende por capilaridad hasta una determinada altura, a la cual se corta la
ascensión y el volumen recogido no varía.
Todas las maniobras deben hacerse con sumo cuidado, pues el capilar es
demasiado frágil, y cualquier tensión lo quiebra. También debemos tener
cuidado de que no gotee, lo cual ocurre algunas veces, sobre la placa ya
que si esto ocurre tendríamos que recortar una nueva.
Cuando ponemos la punta del capilar sobre la placa en el punto
correspondiente, ésta absorbe lentamente el contenido del capilar. Hay que
lograr que el producto así depositado no se expanda, y que permanezca lo
más recogido posible sobre el punto marcado. Esto se consigue llevando el
extremo del capilar sobre el punto y dejando que absorba una parte. Justo

202
entonces se levanta y se espera a que se seque, volviéndose a depositar una
nueva porción de líquido. Así se hace hasta que todo el líquido se haya
transferido. La cantidad de líquido que se debe depositar dependerá de la
concentración de la disolución, pero en general se puede decir que unos dos
volúmenes de capilar son suficientes.
Cada vez que hay que cambiar de fracción para coger una nueva cantidad
con el capilar se tiene la obligación de lavarlo previamente. En dos tubos
de ensayo o dos vasos de precipitado de pequeño tamaño ponemos acetona
y éter. Mojando el capilar en la acetona se llena y a continuación llevamos
su punta sobre un trozo de papel absorbente que aspira el líquido. Tras
varios lavados con la acetona, emulamos con el éter. Una vez seco
podemos tomar alícuotas de otro matraz.
19. 4 LA FASE MÓVIL
Para elegir una fase móvil adecuada es preciso conocer la naturaleza de los
compuestos. Esto es relativamente complejo ya que no siempre se intuye la
polaridad de las moléculas a separar. En principio nos regiremos por la
regla general que dice que sustancias de polaridad afín son miscibles, de
donde extraemos que, compuestos polares se disolverán bien en disolventes
polares, en tanto los apolares requerirán disolventes apolares.
Las fases móviles que se usan con más asiduidad en el laboratorio están
constituidas por mezclas de los disolventes más usuales, de donde
destacamos los tres siguientes:
Eo
Tolueno 2.4
Éter 4.3
Acetato de etilo 6

203
De este modo, como los compuestos organometálicos suelen tener una
polaridad marcada, debemos eluir con mezclas adecuadas para polares. Así
el porcentaje de acetato de etilo se incrementará a mayor velocidad de
elución queramos conseguir.
La velocidad de elución debe ser lo suficientemente rápida como para que
no se pierda el tiempo inútilmente, pero no tan grande como para impedir
que las bandas se separen bien (en este caso los puntos correspondientes a
los distintos compuestos).
Conociendo la serie eluotrópica anterior (donde se ordenan los disolventes
según su capacidad para eluir) podemos confeccionar una fase móvil que se
adecue a nuestros compuestos y lograr una velocidad aceptable de
separación.
Al principio no se sabrá exactamente cuál será la proporción oportuna, pero
tras hacer varias placas de prueba, viendo cómo se separan los compuestos
entre sí, encontraremos alguna que se considere buena para realizar la
experiencia a todas las fracciones. Esta combinación se escribirá en la placa
por detrás o en la parte inferior, debajo de la raya.
19. 5 LA SEPARACIÓN
Para llevar a cabo la separación tomamos un recipiente, que suele ser una
cajita pequeña de vidrio. En su interior se añade una cantidad de fase móvil
de modo que quede siempre por debajo de la línea a lápiz de la placa, pues
si se superase (como se comentó anteriormente) las pequeñas cantidades
que se encuentran sobre los puntos se disolverían en este volumen y no
serían arrastradas placa arriba.
Con unas pinzas cogemos la placa y la colocamos con cuidado en el suelo
del recipiente, en tanto la apoyamos en una de las paredes de vidrio de la

204
cajita. De este modo la placa queda como si fuese una rampa o la superficie
de un plano inclinado.
Con el tiempo el disolvente asciende por la placa por capilaridad, puesto
que el granulado genera pasillos muy estrechos en el interior, y disuelve los
compuestos distintamente, dependiendo de su polaridad. Aquellos que sean
más solubles partirán antes y serán arrastrados hacia la parte más alta de la
placa. Los otros se irán repartiendo a medida que desciende la polaridad de
modo que, el menos polar queda más abajo cuando el disolvente alcance el
borde superior y cese de pasar.
19. 6 INTERPRETACIÓN DE UNA PLACA
La placa, como hemos indicado, sirve para corroborar que el método
elegido para separar las sustancias ha sido el adecuado o por el contrario
tendremos que intentarlo por una vía diferente. También, en caso de
separación, nos indica el grado real de pureza que se ha logrado.
Si por el contrario nos preguntábamos simplemente si el compuesto que
tenemos está o no puro, la placa también responde adecuadamente.
Para analizar el resultado es necesaria una lámpara de rayos ultravioleta.
Esta radiación logra que la fluoresceína de la masa fluorezca con luz
verdosa.
Sin embargo, cuando tenemos un producto, éste se interpone entre la
radiación estimulante y la placa, por lo que en ese lugar no aparece emisión
fluorescente. Esos puntos oscuros, a lo largo de la placa, se corresponderán
con nuestros compuestos.
Dependiendo de la distribución de esos puntos oscuros se deducirán una
serie de informaciones. Cuando se enciende la lámpara se desvelan las

205
zonas oscuras, y entonces se aprovecha para marcar con un círculo a lápiz
su posición.
Posibles situaciones:
A) Separación correcta.
En el dibujo de la izquierda se ven los 4 puntos
correspondientes a las 4 alícuotas tomadas de los distintos
matraces o recipientes en general usados para recoger las
fracciones en el método de separación.
Las manchas oscuras se han representado por zonas a
lápiz.
La información ofrecida por inspección de la placa se
puede resumir rápidamente. Al principio en el reactor había 3 sustancias
distintas que se aprecian sin problemas en la columna del patrón. La
primera fracción contiene exclusivamente el compuesto más polar
(suponiendo que la fase móvil esté buscada para este tipo de sustancias). En
las dos fracciones siguientes se han obtenido puros los otros dos
compuestos.
Se concluye que la fase móvil es adecuada para separar las sustancias
mediante un método cromatográfico, a priori, viendo cómo se han separado
los tres compuestos en la columna patrón. Pero también, estudiando los
otros tres, que el método empleado ha sido efectivo en verdad. Muchas
veces es impredecible si las sustancias se van a separar aceptablemente
aunque la placa de prueba haya mostrado una alentadora respuesta previa
(que se hace con un solo punto tomado directamente de la disolución a
separar).

206
B) Separación incorrecta.
Al mismo tiempo podemos encontrar la situación opuesta, en la que los
productos apenas se han separado, a causa de una errónea elección del
método de separación o de la mezcla efluente. De nuevo tenemos 4 puntos
correspondientes a 4 muestras provenientes del proceso de separación. Al
igual que antes, las zonas oscuras se representan con tono grisáceo.
Puede determinarse la mala elección de la fase móvil pues
en la columna correspondiente al patrón los puntos se han
extendido de modo que algunos llegan a tocar a los otros.
Esto predice que la separación va a ser insuficiente, lo que
se verifica en las otras tres columnas donde únicamente el
último producto se encuentra puro.
No queda entonces más que agrupar las fracciones de
nuevo y volver a intentar separarlas usando otro método u
otra fase móvil.
Conviene volver a comentar, aunque ya se haya dicho bastantes veces, que
antes de hacer una cromatografía con la intención de separar el compuesto
que nos interesa del resto, la elección de la fase móvil se realiza mediante
las placas. Así, para una mezcla de proporciones definidas hacemos una
placa con un punto (una alícuota) correspondiente al conjunto.
Dependiendo de cómo se vayan separando cambiaremos las proporciones
hasta que observemos una separación correcta.

207
20. EL CROMATOTRÓN
Los métodos de separación o purificación vistos en los bloques 16, 17 y 18
son usados continuamente en el laboratorio. También se puede llevar a
sequedad un compuesto y extraer con un disolvente selectivo el que nos
interesa o aquel que nos incordia.
Mas el método de separación más efectivo cuando no existen solubilidades
suficientemente diferenciadas o las polaridades de los mismos son
demasiado parecidas para una cromatografía convencional, es el
cromatotrón. Este método cromatográfico logra una efectividad
sorprendente mediante la aceleración de la fase móvil por fuerza centrífuga
al hacerse rotar la fase móvil a gran velocidad. Cada eluato tendrá una
velocidad determinada a la hora de recorrer un espacio mucho mayor que el
de una placa tradicional, precisamente este aumento de la distancia a
recorrer es la causa de que se separen y salgan a distintos tiempos del
aparato, pudiéndose recoger en matraces distintos, ya que diferencias
mínimas se acentúan ante distancias tan largas. La rotación lo que viene a
hacer es acelerar el proceso.
20. 1 EL APARATO
Al ser de un tamaño relativamente grande puesto que ocupa
aproximadamente el espacio de un televisor de 14 pulgadas, y al tener una
forma característica resulta muy llamativo cuando se encuentra en la
campana de trabajo, reconociéndose inmediatamente.

208
1. Abultamiento donde se encuentra el motor que hace girar el disco con la
placa porosa que sirve de base fija. La rotación es a gran velocidad,
permitiendo así que la elución se acelere gracias a la fuerza centrífuga.
2. Es la caja que contiene el disco de rotación. El punto central indica el
pivote del eje de giro en torno al cual da vueltas el disco de vidrio sobre el
que se ha dispuesto la capa adsorbente.
3. Son las dos patas metálicas sobre las que se apoya el cromatotrón, en las
mismas recae el centro de masa para que el aparato no se caiga hacia
delante, aunque las patas se acomoden en la parte trasera.
4. Disco propiamente dicho donde está la fase porosa. Sobre él se añadirá la
fase móvil y el reactivo. Es una parte fundamental y la pasta que constituye
la placa deberá ser hecha de vez en cuando (es la única parte que tendremos
que cambiar con el tiempo, debido a que se va ensuciando). Existen discos
de vidrio de radios distintos, que se usan en función de la cantidad de
muestra que tengamos que eluir.
5. Es el sifón por donde se va eluyendo la fase móvil con los
correspondientes compuestos. El ritmo de salida suele ser de goteo, por lo

209
que se requiere algún tiempo para la separación completa de todos los
constituyentes.
6. Tapa plástica que se usa para impedir que agentes extraños caigan al
interior y para mantener la presión de N2 (g) que se precisa en el interior
para proteger los compuestos a separar. Se encuentra horadada cerca del
centro (al lado izquierdo con respecto al operante) para recibir el sistema de
goteo (7). Las grapas que tenemos en la caja (2) se encargarán de fijarla
fuertemente sobre la placa, de modo que no haya fugas.
7. Extremo metálico hueco de la sonda, que se une mediante un tubo al
embudo de decantación, donde se almacena la fase móvil. Así se une el
embudo de decantación con el plato poroso. Posee un reborde que impide
que toque el disco, y se soporta en la abertura justo encima de unos pelitos
que dispensan homogéneamente el disolvente procedente del embudo.
8. Tubo de material plástico que se usa para conducir la fase móvil desde el
embudo hasta el plato. Hay que tratarlo con cuidado pues puede romperse
por su punto más débil, que es la unión con su terminal metálico, o la salida
del embudo.
9. Embudo de decantación. Se maneja con la llave que se muestra en su
parte inferior. Se mantiene en una posición alta, sostenido lo más arriba
posible de un pie gracias a un soporte apropiado que posee (el cual no se ha
dibujado), para que el líquido caiga por gravedad.
10. Entrada del N2 (g) procedente de la línea para mantener el interior
protegido bajo atmósfera inerte.
11. Válvula usada como medidor del flujo del gas. Si éste fuese excesivo
sería peligroso para el aparato, por lo que una estimación del mismo resulta
indispensable.
12. Mordaza estranguladora cuya finalidad no es otra que la de regular el
flujo mediante la compresión de la goma.

210
20. 2 ELECCIÓN Y PREPARACIÓN DE FASE MÓVIL
Una vez que se ha descrito el sistema de separación pasemos a detallar las
etapas características de su manejo.
En principio debemos buscar la fase móvil idónea, o lo que es lo mismo, la
mezcla apropiada para separar los compuestos existentes en la disolución
de reacción. Debe resultar ya evidente que esto se logra haciendo una
plaquita y buscando la mejor combinación de disolventes para eluir
gradualmente nuestros compuestos.
Los más usados en la técnica, alineados en razón a su polaridad creciente
(serie eluotrópica) son:
Éter de petróleo < Éter < Acetato de Etilo
La combinación de los mismos basta para la mayoría de los compuestos
organometálicos solubles en los mismos. Cada compuesto requerirá unas
proporciones determinadas, no obstante esta proporción se puede cambiar a
medida que transcurre el proceso si nos conviene para acelerar o retardar la
elución de algunos de los compuestos.
Cuando encontramos la proporción correcta pasamos a gran escala y
hacemos el cálculo del volumen que nos va a ser necesario (suele superar
los 750 ml). Para tal finalidad existen probetas de 1 litro que resultan muy
útiles.
La fase móvil se echa en el embudo de decantación, con la llave inferior
cerrada, el cual puede alojar un volumen de 250 ml.

211
20. 3 LA FASE ESTACIONARIA
Generalmente la fase estacionaria no suele cambiarse antes de pasados los
5 años de uso medio. Sin embargo, como todo, depende de la frecuencia
con la que el cromatotrón sea utilizado.
El cambio del lecho poroso se describirá a continuación. En principio
tenemos que quitar la tapa plástica que oculta la placa de sílice. A
continuación hay que extraer el plato de vidrio que sirve de soporte a la
fase fija. Con la espátula despegamos el lecho del vidrio hasta que éste
quede libre de la masa adsorbente. Hecho esto hay que usar guantes pues la
operación de limpieza del vidrio debe ser tan eficaz que no se pueden dejar
huellas siquiera sobre su superficie. Se lava con un detergente y un cepillo
especial (que viene con el aparato) y finalmente se aplica éter para secar
completamente la superficie. El vidrio debe dejarse secar un buen rato.
Mientras esperamos su secado procedemos a preparar la masa que
constituirá la fase estacionaria. Para ello se dispone de unos instrumentos
especialmente diseñados que facilitan la operación. Esta pasta es análoga a
la de las placas de capa fina y la cantidad a hacer dependerá del diámetro
del disco de vidrio a emplear. Se pueden contar 4 diámetros distintos, y por
tanto tendremos que elegir las cantidades atendiendo a esto, lo cual no es
problema ya que vienen bien indicadas en la documentación del propio
aparato.
La masa se compone de sílica-gel mayoritariamente, algo de yeso para dar
consistencia y estabilidad mecánica, y fluoresceína. Tras hacer la pasta, a
modo de yeseros, se dispone sobre el disco de vidrio uniformemente,
usando las herramientas adecuadas para ello y finalmente se deja
endurecer.
En la confección de la fase estacionaria se pueden emplear otro tipo de
sustancias, dependiendo de la naturaleza de las sustancias a separar, pero en

212
el caso de los compuestos organometálicos, los componentes usados son
los que acabamos de citar.
Una vez la masa se ha solidificado, se vuelve a colocar en su sitio, dentro
de la caja del cromatotrón, y se cierra el aparato. En esos momentos se
puede ya usar con normalidad para las labores de separación.
20. 4 ACONDICIONAMIENTO DEL CROMATOTRÓN
Una serie de disposiciones son necesarias antes de llevar a cabo la adición
de la mezcla a separar.
1. En primer lugar debemos poner en marcha el motor y hacer que el disco
comience a girar. El giro debe ser suave, sin vibración, y para ello es
fundamental que el cromatotrón esté asentado con firmeza sobre la mesa de
la campana.
2. A continuación se conectará el N2 (g) de modo que comience a pasar al
interior del aparato. Regularemos su flujo con la ayuda de la mordaza, de
modo que no sea muy alto.
3. Tras dejar un tiempo el aparato en estas condiciones hacemos pasar la
mezcla de la fase móvil por el tubo fino, hasta la zona del disco apropiada.
Debe graduarse tal que se establezca un goteo rápido, no un continuo de
líquido. Las fibras existentes en la hendidura hacen que las gotas se
dispensen homogéneamente por el disco. Puede darse el caso de que si
colocamos mal el aplique metálico, o si la tapa no se ha cerrado
adecuadamente, o incluso, que el flujo sea demasiado grande, el disolvente
se salga, y en lugar de ser absorbido por la placa porosa, se extienda por su
superficie, debiéndose reabrir el sistema y evaporar el disolvente con el
secador.

213
4. A medida que la fase móvil se va extendiendo, va mojando al lecho
completamente, para acabar al final saliendo por el sifón. En previsión de
esto situaremos debajo un vaso de precipitados para recoger el disolvente.
5. Debemos ser precavidos y considerar que, a lo largo del proceso
debemos siempre tener lleno el embudo, ya que si llega a agotarse habrá un
trozo de placa que se secará. Esta zona avanzará como banda seca que
puede interferir en la separación dado que tras ella puede venir producto en
disolución y no conviene que corra sin fase móvil delante. Téngase así
siempre suficiente reserva de mezcla de disolvente para ser añadida cuando
empiece a faltar.
6. Como no conocemos el número de fracciones a recoger será necesario
preparar un buen número de tubos de Schlenk y matraces desoxigenados y
bajo atmósfera de nitrógeno para contener las fracciones que se considere
oportuno, dado que sería una lástima que tuviésemos que repetir la
cromatografía por no tener suficientes recipientes preparados.
20. 5 INYECCIÓN DE LA DISOLUCIÓN
El matraz donde tenemos la disolución de reacción, libre de elementos
sólidos, se pone bajo N2 (g) con un tapón pinchado. Luego se inserta una
cánula de las más finas, tipo RMN, en el tapón y se acerca al lugar donde la
sonda está encajada en la tapadera.
Se toma la sonda sin que deje de gotear en el interior, colocándola algo
separada, de modo que se apoye en el borde y deje espacio para la cánula.
Se puede llamar a algún compañero para que nos ayude en el proceso, pero
uno mismo debe afrontar las incomodidades mediante la habilidad, y esto
es preferible.

214
La adición se hace adoptando las medidas usuales. Bajamos el extremo
romo de la cánula a la disolución. Por el otro lado saldrá una cantidad muy
pequeña de la misma lo que nos conviene. Como el sistema es muy grande,
y el flujo de nitrógeno reducido, la presión dentro del matraz será mayor,
por lo que no hay que quitar la corriente de N2 (g) del cromatotrón. Las
gotas de la disolución de productos a separar son libradas lentamente por
las hebras consabidas, y su paso al lecho se realiza uniformemente.
Tras la adición se encaja de nuevo la sonda y todo queda en su sitio, en
espera de que la separación sea completa.
20. 6 SEGUIMIENTO DE LAS BANDAS
Cuando se hace una placa lo que se observa es una mancha más o menos
redondeada. Pero si, a esta placa, la hacemos girar a una velocidad elevada
lo que apreciaremos será una banda (una circunferencia de un grosor
determinado).
Con la lámpara de rayos ultravioletas se sigue la marcha de estas
circunferencias, opacas sobre el fondo verde fluorescente, que se van
haciendo más grandes a medida que van corriendo por la placa adsorbente.
La primera banda en detectarse será aquella con una polaridad más pareja a
la de la fase móvil.
Al avanzar la banda se va ensanchando y va perdiendo nitidez a causa de
varios factores entre los que se destaca el hecho de que el compuesto debe
extenderse por una superficie mayor y así se opone menos al paso de la
radiación ultravioleta. No obstante resulta también interesante comentar los
factores clásicos que provocan un ensanchamiento de las bandas:

215
A) Efecto de múltiple recorrido.
Producido principalmente por el mero hecho de que la estructura interna de
la placa adsorbente no es un retículo perfecto, y que en su carrera hacia el
extremo de la placa (en este caso del disco) las moléculas realizan
recorridos distintos, aunque todos en torno a un valor considerado como
medio. Pero por lo mismo unas avanzarán más rápido al recorrer menos
tramo y otras vendrán a aparecer ligeramente retrasadas. Su importancia se
reduce por el mínimo tamaño de partícula del material del lecho, pero sigue
siendo considerable
B) Difusión en gradiente.
Causada por la tendencia de los solutos a marchar desde los puntos donde
la concentración es mayor a aquellos con concentración inferior.
En este caso no es muy importante, pues a mayor velocidad de elución,
menos se manifiesta este efecto.
C) Resistencia a la transferencia de masa.
Provocada por la velocidad limitada de los procesos de transferencia, lo
que provoca que a veces la fase móvil arrastre al soluto sin que el equilibrio
se haya alcanzado completamente. Aquí es un término importante pues las
velocidades de elución son mucho más elevadas que en el caso de las
columnas normales.
Conociendo pues esta tendencia suele ser necesario apagar las luces del
laboratorio y hacer un seguimiento intensivo con la lámpara ultravioleta en
la mano, siguiendo las evoluciones de las bandas para recogerlas cuando
nos parezca que van a salir. De no hacer esto, resulta imposible saber por

216
dónde marchan en verdad las bandas y se hace muy complicado acertar a
recogerlas. Si la luz ultravioleta se hace incidir sesgadamente y no de modo
directo se aprecian mejor las bandas ya que se obliga al haz a atravesar una
capa de mayor grosor.
20. 7 RECOLECCIÓN DE LAS FRACCIONES
Cada banda que se observa es un producto distinto. Debemos recoger cada
una en un matraz diferente, teniendo cuidado de comenzar la recolección
antes de que comience a caer la banda por el sifón y de retirarla cuando
estamos completamente seguros de que todo el producto se ha eluido.
Para recoger el producto usamos un Schlenk o matraz al que previamente le
hemos quitado el tapón, por lo que el N2 (g) sale por la boca del mismo sin
ningún impedimento, protegiendo al producto que gotea en el interior. Lo
mejor es fijar de algún modo (con una pinza preferentemente) el matraz en
esa posición, con el problema de que es una posición muy baja, y se hace
verdaderamente difícil. Lo más recurrido es sostener el matraz con una
mano, en tanto que con la otra se sostiene la lámpara de ultravioleta.
Cuando se cree que el producto está cercano a salir se coge un nuevo tubo
para sustituir al anterior, y es aquí donde nos damos cuenta de las
limitaciones de sólo tener dos brazos. El proceso es complicado pues
debemos quitarle el tapón con una mano, soltarlo de la pinza y, con
rapidez, intercambiarlo con el otro sin que una gota caiga sobre la mesa de
trabajo. Generalmente el proceso se hace entre dos, aunque con la maña
suficiente, uno puede afrontar todos los problemas y llegar a buen puerto.
Cuando se ha cambiado el tubo, el que se ha retirado se vuelve a tapar con
el tapón de goma y se numera con un rotulador, para saber qué fracción

217
exactamente tenemos en nuestras manos. El proceso se continúa hasta tener
cada compuesto en un matraz diferente.
20. 8 LIMPIEZA DE LA PLACA
Cuando concluye la separación se deja que fluya la fase móvil,
recogiéndola en un vaso de precipitados. En la placa porosa quedará una
cantidad indeterminada de sustancias adheridas, que la fase móvil no puede
arrastrar. Debido a que la misma placa debe ser usada aún muchas veces,
nos será necesario lavarla y eliminar todas las materias remanentes. Así,
antes de que toda la fase móvil se haya marchado, llenamos el embudo con
acetato de etilo y lo dejamos eluir. Este disolvente es muy polar y arrancará
todas aquellas sustancias (que tendrán un marcado carácter polar),
adsorbidas en la capa de sílice. Con una segunda adición del disolvente se
logra una limpieza aceptable. Cuando deja de gotear se desconecta tanto el
N2 (g) como la corriente eléctrica, guardándose inmediatamente el
instrumento.
El acetato de etilo tiene un olor muy fuerte y para muchos, desagradable,
por lo que su manejo se debe realizar en la campana de extracción y
protegidos con guantes.
21 LA RECRISTALIZACIÓN
Esta fase es siempre la última parte de la purificación de un compuesto. Si
un compuesto se logra cristalizar, sus cristales estarán normalmente en un
elevado grado de pureza, debido a que un compuesto no cristaliza en
combinación con otro, sino solo y por tanto puro. Cada compuesto requiere
un disolvente particular o una mezcla determinada de disolventes para una

218
eficiente cristalización, pero también debe encontrarse disuelto en pequeño
volumen por lo que hay que reducirlo un poco a vacío.
21. 1 BUSCAMOS LAS CONDICIONES ÓPTIMAS
Para cristalizar un compuesto lo primero que debemos hacer es eliminar el
disolvente que le quede tras los métodos de separación anteriores y después
tratar de disolverlo en el mínimo volumen posible, con la mezcla adecuada.
Esta mezcla dependerá principalmente de la solubilidad del producto, pues
no debe ser insoluble en ella, pero tampoco tan soluble que dificulte el
proceso de cristalización. En general se compone por una parte de
disolvente en el cual el producto es soluble y una pequeña cantidad de otro
disolvente que disminuya la solubilidad del compuesto en la mezcla. La
experiencia dará las proporciones buscadas.
Ya disuelto debemos buscar saturar el disolvente. Así reducimos a vacío su
volumen como se vio en la sección 15.2. La reducción no debe ser
excesiva, deberemos parar justo antes de que se formen los primeros
cristales de producto como consecuencia de la evaporación. Será entonces
cuando tendremos el compuesto disuelto en la mínima cantidad de
volumen, lo que implica saturación a la temperatura del laboratorio. La
cristalización será un hecho al disminuir la temperatura al introducir el tubo
en el frigorífico.
Toda operación de cristalización debe ser hecha en un tubo Schlenk, por lo
que si al principio el compuesto se encuentra en cualquier otro tipo de
recipiente deberá ser pasado mediante cánula a uno de estos tubos.

219
21. 2 RESTOS EN LA BOCA DEL SCHLENK
En la acción de reducción de volumen puede ocurrir que el borboteo y la
agitación provoquen la subida de la disolución hasta la parte superior de las
paredes del Schlenk. Allí cristaliza sobre las mismas algo de producto que
se pierde y no pasa por el lento proceso de cristalización que conlleva su
purificación.
Debemos entonces disolverlos de nuevo para que no se desaprovechen,
pero sin añadir nuevamente disolvente, y sin usar la espátula para
desprender los restos de manera mecánica. El método que se pasa a
describir está restringido a aquellos disolventes de bajo punto de ebullición
tales como el éter (preferentemente) o la acetona.
Dejamos el vacío que ha sido necesario para la reducción de volumen y con
las manos se calienta la disolución del tubo, rodeándolo con las mismas.
Muy lentamente se produce la evaporación de parte del disolvente y al
llegar arriba vuelve a licuarse, cayendo por las paredes y arrastrando los
cristalitos que se han formado sobre éstas.
Véase que si no hay vacío la evaporación no sería muy efectiva, y se
perdería un tiempo precioso calentando con las manos y sin evaporar gran
cosa.
A veces ocurre que tenemos demasiado producto en las paredes y se va
disolviendo lentamente, o que el producto no es muy soluble, o que la
temperatura del laboratorio es alta y condensa poco disolvente, por lo que
el arrastre es insuficiente. Entonces la solución llega de la mano del N2 (l).
Ataviados con guantes gruesos mojamos un paño con un poco de N2 (l) y lo
ponemos (cuando se ha calentado el tubo durante un tiempo con las manos)
en la parte de arriba del Schlenk. La condensación se produce entonces en
gran cantidad y en unos segundos todo el producto se encuentra abajo de
nuevo. Solamente dos problemas encontraremos con esta maña: hay que

220
tener cuidado con las quemaduras que el líquido pudiera hacer, pero al
mismo tiempo considerar que al estar a vacío el disolvente, la bajada
repentina de la temperatura puede hacer que hierva por la caída de la
presión y vuelva a borbotear, ensuciando nuevamente las paredes del tubo.
Este método aquí explicado se usa mucho y resulta indispensable a la hora
de no perder producto alguno.
21. 3 METEMOS EL SCHLENK EN LA NEVERA
Para cristalizar un producto saturado es un recurso conocido ayudarse de
una bajada de la temperatura. De este modo se introducen habitualmente en
el congelador del frigorífico el tubo de Schlenk con el producto a
cristalizar. Sin embargo para ello debemos usar unas medidas de seguridad
especialmente aplicadas contra la humedad.
A) En la boquilla del Schlenk se colocará un tapón de goma
introduciéndolo por el agujero que el tapón de goma posee en su parte
inferior. También podemos cortar el ala de un tapón viejo, quedando sólo el
cilindro roscado y usarlo en su lugar. De este modo se impide que condense
agua en el interior de la boquilla que es la terminación por la que se conecta
el Schlenk a la línea.
B) En la llave tenemos que colocar las gomillas, pero no únicamente las
que fijan la llave, sino también la de seguridad, que impide que rote.
C) En el tapón también pondremos la gomilla acostumbrada.

221
No se debe olvidar jamás que, siempre que se ponga a cristalizar un
producto, o que quede en estado de reserva en el frigorífico, tendrá que
quedar bajo atmósfera de N2 (g) y nunca dejarse a vacío el interior del tubo.
En el enrejado superior del congelador los tubos de Schlenk se colocan de
manera que quedan inclinados, apoyándose en la parte superior (justo por
debajo del tapón) y en el arco de la llave. En muchos laboratorios existen
soportes plásticos en forma de caja que son a su vez recipientes adecuados
para dejar reposar las disoluciones en el interior del frigorífico.
Así permanecerán hasta que se logre cristalizar. El plazo es de varios días,
pero si transcurren más de cinco sin que aparezcan cristalitos más vale que
perdamos la esperanza en cuanto a la obtención de éstos. Lo más juicioso
es llevar a sequedad y probar con otra mezcla de disolventes o volver a
purificar.
21. 4 CRISTALIZACIÓN POR EVAPORACIÓN
Si el disolvente es éter existe también una alternativa al método anterior.
En lugar de hacer menos soluble al compuesto, disminuyendo la
temperatura, lo hacemos por evaporación lenta del disolvente.
La idea es la siguiente: se deja el Schlenk preparado como si lo fuésemos a
meter en el congelador, pero en la vitrina. Allí lo mantendremos unos días.
A medida que el tiempo vaya pasando, el disolvente se irá evaporando muy
despacio y se absorberá en el tapón. Pasado el tiempo conveniente quedará
tan sólo una pequeñísima parte de líquido y se habrán formado los cristales.
A los 5 días aproximadamente, dependiendo de la cantidad de éter y de la
temperatura del laboratorio casi todo el disolvente habrá desaparecido. No
debe evaporarse completamente, pues de ser así la operación no serviría de
nada, pues las impurezas no pueden marcharse con el éter y habrán

222
quedado sobre los cristales puros. Lo mejor es pasar a la siguiente fase
cuando aún quede algo de disolución.
21. 5 SACAMOS UN SCHLENK DE LA NEVERA
Al sacar un tubo de Schlenk de la nevera para cualquier uso posterior
debemos proceder con cuidado, debido a la humedad que se habrá
depositado en la boquilla que sirve de conexión entre el interior del
Schlenk y el tubo de goma de la línea. De este modo, antes de conectar la
goma hay que eliminar la humedad que se pueda encontrar en la boquilla.
Esta operación se hace quitando primero el tapón cortado que protege la
boquilla y luego aplicando calor con un secador.
Con unos 10 segundos de aplicación de calor basta, no obstante se aprecia
fácilmente si la humedad ha desaparecido o no por completo, calentando el
tiempo que hiciese falta. Cuando no queda resto alguno, se conecta con la
goma y se desoxigena la misma, para a continuación, proceder a la
operación deseada.
21. 6 SEPARAMOS CRISTALES DE AGUAS MADRES
En la operación de separación lo primero será sacar los cristales del
frigorífico. Como se ha cristalizado el producto disminuyendo la
temperatura, habrá que mantenerla, o al ascender ésta, los cristales se
redisolverían y perderíamos parte de la cosecha.
Con la idea de impedir esto último existe en la nevera un baño lleno de
acetona a la temperatura del Schlenk recién sacado. Este baño suele ser el
fondo de una botella de plástico, como se dijo con anterioridad. El Schlenk,

223
durante esta operación, se introducirá en el baño, para que el calentamiento
se retarde lo más posible.
Para que el Schlenk esté el menor tiempo posible esperando ser empleado,
fuera del frigorífico, todos los elementos necesarios habrán de ser
preparados con anterioridad.
La separación de cristales se hace de 2 modos:
A) A través de una cánula muy fina (de RMN). Los cristales deben ser
gruesos y bien formados para que no se marchen vía cánula. La disolución
de aguas madres se lleva a otro Schlenk, pues habitualmente en una sola
cosecha, no se recoge todo el producto, y será necesario ponerlo de nuevo a
cristalizar. La ventaja de usar una cánula fina es que algunos cristales de
pequeñísimo tamaño pasarán al nuevo tubo y participarán como núcleos de
cristalización en la segunda cosecha. De aquí que muchas veces no se tenga
la obligación de reducir el volumen de disolvente nuevamente.
B) A través de una cánula con filtro (sin cilindro). Se usa siempre que los
cristales sean finos y pequeños. En el otro lado de la cánula colocaremos
también un Schlenk nuevo, pues al igual que en el caso anterior nos
conviene hacer una segunda cristalización.
22 SECADO DE LOS CRISTALES
Aunque los hayamos separado en la etapa anterior del disolvente y
aparentemente presenten una superficie de aspecto seco, todavía tienen
mucho disolvente retenido. Por tanto el proceso de secado es importante y
prepara los cristales para posteriores usos en técnicas de análisis.

224
22. 1 SECADO A VACÍO
Es excelente para eliminar cualquier resto de disolvente, mas los cristales
deben ser robustos y no quebrarse y fragmentarse ante el cambio de
presión. Cuando los cristales son frágiles este método es demasiado
enérgico pues se parten, no siendo útiles para las técnicas de rayos X que
necesitan monocristales de cierto tamaño (que en realidad son más
pequeños de lo que en un principio se espera).
Se coge el Schlenk con una mano y se comienza a hacer vacío lentamente
pues el cambio de presión no debe ser muy rápido, lo cual podría ocasionar
la ruptura de los cristales. A continuación se golpea, como si estuviésemos
evaporando una cantidad grande de disolvente, para que los cristalitos se
muevan y liberen cualquier traza que pueda permanecer entre ellos. Se
aplican golpes fuertes y firmes, sin cuidarse de que el tamaño de los
cristales pueda verse reducido por la agitación mecánica ya que las
dimensiones requeridas para el análisis son lo suficientemente pequeñas
como para temer que la agitación reduzca los cristales por debajo de esos
niveles.
Se deja unos minutos a vacío y al final se restituye el N2 (g).
22. 2 SECADO CON CORRIENTE DE N2 (g)
Si los cristales son muy frágiles hay que tratarlos con cuidado y el secado
debe hacerse por otros medios. Una vez se tengan los cristales en el tubo,
separados de las aguas madres, se conecta a la línea el pincho y se abre la
corriente de gas inerte de modo que salga un chorro a presión por la punta
del mismo. A continuación se atraviesa el tapón del tubo y se lleva su punta
hacia abajo.

225
Cerrando el N2 (g) en el tubo se Schlenk y colocando algunas agujas, se
renueva continuamente la atmósfera interior. Con el pincho se dirige la
corriente hacia el producto y el N2 (g) arrastra las moléculas de disolvente
que no fueron eliminadas en la operación de separación. No debemos bajar
mucho la punta, pues al secarse completamente el compuesto, si fuese poco
denso, se levantaría y se pegaría por las paredes.
23 CRISTALES PARA DIFRACCIÓN
23. 1 ELECCIÓN DE CRISTALES PARA DIFRACCIÓN
Uno de los objetivos, cuando se purifica una sustancia es lograr una
aceptable pureza para poder determinar su estructura. En difracción de
rayos X es pues necesario un monocristal con características determinadas,
que fuerzan la necesidad de buscarlo entre otros muchos.
Teniendo ya los cristales separados de las aguas madres por alguno de los
métodos anteriores tenemos que encontrar un cristal de algunos milímetros
de longitud (un par de milímetros a lo sumo) y que no presente ni fisuras,
ni grietas, ni maclas.
Los cristales, como regla general, son mucho más estables que las
disoluciones del compuesto, debido a la rigidez a que los compuestos están
sujetos en la red cristalina. Así muchos productos que en estado disuelto
son atacables por el aire, en estado sólido son perfectamente estables
(durante un periodo determinado). Por otra parte, al estar incluidos en una
red, sólo resultan atacadas las moléculas (considero que el sólido formado

226
siempre es de características covalentes) de la superficie, por lo que la
descomposición es mucho más lenta.
Teniendo esto en cuenta, se abre el Schlenk y se esparcen las agujas del
producto en un papel encerado (de aquellos que sirven para pesar). Sobre
este papel se elegirán aquellos trozos que, sin ser demasiado largos,
presenten una disposición que a priori parezca ventajosa. El manejo de los
cristalitos es un problema añadido. Durante el proceso de secado, a causa
de los roces entre ellos (o con el N2 (g) que se les haya podido dirigir) se
han cargado, y se quedan pegados en las distintas superficies. Este
fenómeno, aun no siendo extensible a todos los productos, se presenta muy
habitualmente.
Cuando los cristales son demasiado largos deben ser cortados. Para ello hay
unas cuchillas especiales (que en verdad, debido a la fragilidad de los
mismos no son de mucha utilidad) que ayudan en tal sentido, aunque
usualmente es más fácil partirlos que cortarlos.
De entre todos los cristalitos que vamos eligiendo sólo habrá unos cuantos
que valgan. Para escoger entre ellos de un modo apropiado se usa una lente
para estudiarlos más detenidamente. Esta lente es un instrumento análogo a
un microscopio, que permite alojar los cristales en una plataforma irradiada
con luz y utilizar un juego de lentes y ocular para el estudio de su
superficie. Así se eligen aquellos cristales que carezcan de grietas y que
posean una forma mejor desarrollada.
Concluido este punto se comentará cómo se guardan para ser transportados
al lugar de análisis.

227
23. 2 ACONDICIONAMOS UNA PIPETA PASTEUR
Como los cristales son extremadamente pequeños, no se pueden guardar en
un recipiente de gran volumen, sino que se prefiere uno reducido, donde
será más fácil encontrarlos. La pipeta Pasteur sirve a tal efecto.
Fijamos la pistola de llama (la cual presentaremos más adelante) con su haz
de fuego dirigido verticalmente en la campana. Cogiendo la pipeta por sus
dos extremos con ambas manos calentamos por el punto medio de su parte
más estrecha. A la tercera pasada se desprende uno de los trozos.
Proseguimos pasando la llama sobre el muñón de tal modo que se tapen los
poros y se redondee, no permitiendo que queden picos. Se espera a que la
temperatura descienda y podamos introducir los cristales.
Si estos son grandes se pueden guardar en un tubo de sellado, lo cual será
detallado en la sección de almacenamiento de productos.
23. 3 GUARDAMOS LOS CRISTALITOS EN LA PIPETA
Cuando la pipeta se ha enfriado, y no presenta ya ninguna salida por su
punta, se deben introducir los cristales en su interior. Es un laborioso
ejercicio ya que, como se dijo, los cristales se pegan en las superficies,
impidiendo una ágil conclusión del proceso. Todo lo contrario, con
paciencia y lentitud, vamos introduciendo los cristales aunque seamos
auxiliados por las cuchillas en el traspaso.
Manejar los cristalitos sin partirlos ni dañarlos e introducirlos por la boca
de la pipeta es difícil pero tras un tiempo todos los cristales estarán
guardados.
Generalmente se hacen dos pipetas y en cada una se ponen de cinco a seis
cristales. Si por cualquier razón se cae una pipeta y se rompe, habríamos

228
perdido un trabajo costoso, por lo que siempre es aconsejable que exista
una segunda que nos salve de tener que empezar de nuevo.
Una vez hecho lo anterior, hay que sellar el tubo por arriba. Esto se hace
exactamente igual a como se describirá más adelante para los tubos de
sellado. Aquí no hay moldura o muesca que indique donde calentar con el
mechero, pero el vidrio es tan fino que se sella al primer contacto. Para
comprender el proceso exactamente léase el punto 26.6.
La pipeta Pasteur así sellada, sigue siendo demasiado frágil para que su
transporte se considere seguro, aunque resulte efectiva para confinar los
diminutos cristales en un volumen reducido. Para aumentar la seguridad
tendremos que usar algún nuevo ingenio.
La pipeta cabe exactamente en una varilla de vidrio hueca, cuyo cristal
presenta un grosor mayor y puede servir como camisa de protección para la
pipeta que se albergue en su interior. Para ello cortamos un trozo de varilla
con un tamaño algo mayor que el de la propia pipeta. Las varillas se venden
con una longitud que las hace inadecuadas para todo, siendo necesario
partirlas para que resulten manejables. Para cortar se utiliza una cuchilla
llamada cortavidrio. Es una cuchilla fijada a dos cachas que pueden ser de
material vario, pero que mediante tornillos apresan de manera efectiva la
hoja entre sí y permiten un manejo seguro de la misma.
Con una mano se afirma la varilla sobre la mesa, en tanto con la otra se
intenta hacer una muesca en la superficie de la varilla para que sirva de
apoyo en un corte continuo, firme y lo más uniforme posible. Al llegar a la
mitad del radio de la varilla podemos abreviar el proceso mediante un
golpe seco que escinda en dos partes la varilla. También podemos, con los
guantes gruesos puestos, tensionar la varilla en la hendidura, para que se
parta por donde ha sido atacada. Ambos recursos son adecuados y permiten
obtener un corte preciso.

229
Tras esto resulta interesante, con vistas a no cortarse, limar los bordes de la
varilla por ambos extremos usando papel de lija.
A continuación se coloca en uno de los dos extremos un trozo de fixo, al
tiempo que se introduce un poco de algodón hasta el mismo fondo, que
servirá de colchón amortiguador. Después insertamos la pipeta de modo
que repose sobre el algodón e introducimos sobre ella otro algodón,
cerrándose finalmente con otro trozo de fixo. En este momento la pipeta se
encuentra perfectamente protegida contra algún posible accidente.
Antes de guardar el conjunto se hace una etiqueta con el número de
reacción del producto obtenido o con un nombre apropiado para conocer de
qué reacción procede.
El resto del producto desechado en la elección de los cristales debe
almacenarse de manera segura, tal que no se descomponga ni se destruya.
24 RMN
La Resonancia Magnética Nuclear es el método más potente, tras los
métodos de difracción, para la elucidación de estructuras, que se encuentra
a disposición del químico organometálico. Su uso es casi continuo y la vida
del químico transcurre a caballo entre la sala del RMN y la vitrina del
laboratorio. Se usa tanto para determinar estructuras como para seguir las
reacciones.
Cuando se pretende conocer la estructura de un compuesto los espectros se
hacen una vez se tiene el producto cristalizado y puro. Con tal intención se
llevan a cabo todas aquellas modalidades que tengan sentido (1H,
13C,
31P...).
Si pretendemos seguir la evolución de una reacción no hay nada más
efectivo que ver cómo cambian estructuralmente sus reactivos y verificar

230
cómo en el espectro hay picos que van desapareciendo y otros que se van
formando de la nada.
Así pues el RMN es una prueba bien estimada para apoyar la propuesta de
una determinada estructura, aunque no es tan determinante como lo sería
un difractograma. Sin embargo puede demostrarse la estructura de un
compuesto sintetizado en base al historial del mismo, observando los
espectros de los reactivos y los espectros correspondientes a los cambios
que se producen a lo largo de la reacción, comparándolos a los del producto
obtenido.
24. 1 SU BASE CIENTÍFICA
No entra dentro del propósito de este compendio un análisis sobre la
naturaleza del fenómeno, no obstante algunas nociones básicas sí son
exigibles por lo que dedicaremos a ello los siguientes párrafos.
Se puede decir que un átomo con número másico impar, presenta espín
nuclear distinto de cero. Así el más simple de todos los átomos, el H, está
constituido por un único protón, presentando comportamiento magnético a
causa del espín nuclear.
Como en la química organometálica hay restos o fragmentos orgánicos,
ricos por tanto en hidrógeno, podremos hacer un seguimiento de la reacción
o conocer su estructura analizando un RMN 1H.
No obstante, al ser el átomo central, un átomo metálico, podemos tener la
posibilidad de que su núcleo sea impar y así, estudiar también su
comportamiento. Igualmente otros ligandos muy usados como las fosfinas
pueden ser seguidos, ya que el isótopo de fósforo 31
P, activo en RMN.
Cada núcleo por tanto tendrá un momento magnético, cuyo vector se
orientará al azar. Pero al encontrarse en un campo más intenso (impuesto

231
por nosotros) existirán dos posiciones extremas en energía: (favorable
energéticamente) en la que los vectores se orientan en el mismo sentido que
el campo, y (desfavorable energéticamente) donde los vectores apuntan
en sentido opuesto.
De este modo encontraremos dos tipos de núcleos activos dependiendo de
la orientación que tengan sus vectores.
Al someter la muestra a un campo magnético elevado, sus núcleos activos
se abrirán en dos niveles, uno muy poblado y otro escasamente poblado
. Si se busca una radiación de la misma energía que la diferencia entre
ambos niveles, se excitan los núcleos de un nivel al otro. Se dice entonces
que los núcleos resuenan, absorbiendo energía. La frecuencia a la que
absorben puede ser registrada, obteniéndose así una información.
Pero según lo anterior no podríamos emplear este fenómeno para distinguir
moléculas entre sí, por lo que es preciso hacer ver que el entorno
electrónico afecta a cada núcleo activo. Sin esta puntualización, todos los
núcleos serían iguales, aunque químicamente presentasen características
distintas.
Cada núcleo tiene una nube electrónica que depende de la naturaleza
estructural y química de su entorno, es decir que un H de alcano no es igual
electrónicamente hablando que uno de alqueno. Así su nube electrónica es
distinta y su comportamiento en RMN desigual por la razón que se
explicará inmediatamente.
La atmósfera electrónica reacciona formando un campo magnético
inducido (por el campo externo) de modo que se opone a éste. De tal modo
que el campo que recibe el núcleo está menguado y hay que hacerlo más
intenso para que el núcleo reciba la magnitud que lo hace resonar.
Como los H pertenecientes a grupos químicos distintos son diferentes
electrónicamente, cada uno resonará a un valor de campo determinado y así

232
distinguiremos entre unos y otros. Sin embargo también hay otros
fenómenos importantes como son:
- Número de absorciones diferentes, que indica los distintos tipos
de núcleos existentes.
- Intensidad de las señales, que da el número de H de cada clase.
- Multiplicidad de las señales, que informa acerca de otros núcleos
activos cercanos.
Sin embargo ahondar más en estos asuntos correspondería a un volumen
de otra naturaleza y no es la intención del presente sino exponer el trabajo
cotidiano del laboratorio.
Este método presenta muchas ventajas, entre las cuales se destacan el
hecho de no destruir la muestra (por lo que una vez analizada se puede
devolver al matraz si lo que hacemos es seguir una reacción), la rapidez en
la obtención de un espectro, su versatilidad (pues es aplicable a muchos
núcleos) y que requiere poca cantidad de muestra (en general).
24. 2 LOS DISOLVENTES DEUTERADOS
Para hacer los espectros debemos disolver la muestra en un disolvente
apropiado, dado que los disolventes comunes poseen al hidrógeno en su
variedad Protio (cuyo número de masa es impar y tiene comportamiento
magnético) y generan señales que dificultan el análisis del espectro.
En cambio si lo que queremos es seguir el desarrollo de una reacción lo que
tomamos es la propia disolución de reacción, analizando los espectros
obtenidos a tiempos diferentes, y así podemos determinar cómo se produce
la reacción y sobre todo, cuándo se puede considerar concluida. El

233
problema principal es siempre que el disolvente da señal y complica la
lectura.
En el primer caso pues, nos servimos de disolventes deuterados, ya que el
Deuterio posee número másico par y no da señal. En estos disolventes
todos los átomos de hidrógeno son del tipo deuterio, con lo que los
espectros son muy limpios y debidos únicamente a los hidrógenos del
compuesto analizado.
Los disolventes deuterados se guardan en ampollas del tipo visto en 6.1.1
donde se almacenan bajo N2 (g) en condiciones anhidras. Cada ampolla
lleva una etiqueta con su contenido, encontrándose todas en un lugar
protegido pero a mano, para que puedan utilizarse rápidamente. Se suelen
guardar agrupadas en un recipiente de boca ancha y almohadillado. Las
ampollas son siempre pequeñas conteniendo poco disolvente, ya que son
muy caros y un accidente ocasionaría gran pérdida.
Para notar cuánto sentido tiene esto último, copiaremos a continuación un
cuadro donde se determina el valor relativo de cada espectro realizado en
un determinado disolvente. Se le ha dado el valor 1 al cloroformo. El
volumen usado para cada muestra es de aproximadamente 0.6 ml, que se
toma con la jeringa de 1 ml.
Cloroformo 1
Agua 2.7
DMSO (dimetilsulfóxido) 5
Acetona 5.4
Benceno 8.
Acetonitrilo 12.2
Metanol (guantes) 12.2
Tolueno (muy usado, guantes) 14.4
Diclorometano (muy usado) 19.4

234
Ciclohexano 36.7
THF 66.7
Los precios correspondientes son relativos pero podemos hacernos una idea
bastante clara de la importancia que tiene una buena elección al respecto, y
del cuidadoso manejo al que debe someterse la ampolla, teniendo en cuenta
que la toma de cloroformo puede costar en torno a 50 céntimos de euro en
el 2006.
24. 3 TUBOS DE RMN Y SUS TAPONES DE GOMA
Los tubos de RMN son tubos de vidrio muy finos que se
hacen en el propio centro de trabajo a partir de varillas
huecas de borosilicato (que es lo que en el laboratorio se
acostumbra a llamar vidrio). Mediante una pistola de
llama se calientan en un punto hasta lograr escindir poco
a poco por fusión un trocito de la varilla. Después se le
da forma con la llama y se calienta hasta hacer
desaparecer todos los poros.
En la figura A se ha representado uno de estos tubos. No se ha respetado la
escala, y el tubo debería ser mucho más estilizado, ya que en realidad es
mucho más largo que ancho. Por otra parte los tapones son algo más
pequeños en relación al tubo de lo que muestra la figura. En realidad este
tipo de tubos tiene una longitud media de 12 a 14cm por un diámetro de
boca en torno a los 5mm. Se almacenan todos juntos en un vaso de
precipitados dentro de la estufa donde se seca el material de vidrio. El

235
grosor del vidrio es muy poco considerable, partiéndose las más veces
cuando se manejan, debiéndose tratar con cuidado.
Los tapones de goma que se usan son análogos a los correspondientes para
matraces y Schlenk, aunque con las dimensiones mucho más reducidas.
Están formados por un material blando, elástico y deformable, más fino que
en los tapones del otro tipo, pero suficiente para asegurar la hermeticidad.
Se reconocen por su color blanco. Presentan de igual modo dos posiciones
idénticas a las señaladas en la sección 3.2.1 para los otros. En este caso
para doblar las alas y alcanzar la posición 2 se usan unas pinzas.
Se apresa uno de los laterales del ala con la pinza,
mientras que el otro se coge con la punta de los
dedos de la mano restante. Justo después se hace
un movimiento con los dedos y con la pinza, que
lleva al ala entera a plegarse hacia abajo, quedando
como se indica la figura de más abajo.
Este cambio en la forma del tapón se hace con la práctica, consiguiéndose
una destreza suficiente con el tiempo.
Hay que plegar todos los tapones de RMN antes de ponerlos en el
correspondiente tubo, al contrario de como se hace con los grandes, pues
intentar hacerlo sin plegarlos previamente sería sinónimo a romper el vidrio
de la boca del tubo.
Pero no sólo se debe tener cuidado con lo anterior sino que además tienen
que atravesarse antes con el pincho de desoxigenar, porque igualmente, el
intento de clavar el pincho generaría tensiones que podrían quebrar el tubo.

236
Aquí se representa un tapón de RMN. Compárese con
los normales y se comprenderá la analogía existente
entre ellos y estos. Estos tapones poseen un ala que se
abate e incomunica el interior del tubo, en tanto su
tronco obtura la salida del mismo.
A veces encontramos un tapón nuevo que ha sido poco
agujereado, por lo que hincar el pincho resulta difícil, o bien clavar la
posterior cánula que lleva la disolución a analizar desde un recipiente al
tubo. Cualquier dificultad al insertar, si ya tenemos el tapón encajado en el
tubo, aumenta enormemente las posibilidades de partir el vidrio. Para
impedirlo, cuando encontremos cierta resistencia en el tapón, se cogerá una
agujita y se picará la superficie unas cuantas veces, hasta que permita el
paso de la cánula sin oposición.
24. 4 DESOXIGENACIÓN DE UN TUBO DE RMN
Para usar uno de estos tubos hay que eliminar todo el aire que existe en su
interior. El método es muy parecido al utilizado en el caso de un tubo de
centrífuga.
En principio se toma uno de los tapones y se atraviesa por un pincho de
RMN (que es mucho más fino que una aguja–cánula habitual) visto en la
sección 9.2.2. El tapón se lleva hasta una altura de la aguja–cánula tal que
cuando se introduce en el tubo, alcanzando con su punta hasta el fondo, el
tapón queda cerca de la boca y se logra el cierre con facilidad.
Tras cerrar la abertura con el tapón se abre el N2 (g) del pincho. Es más
seguro poner el tapón como si lo estuviésemos enroscando, dado que se
resiste menos y por tanto hay menos riesgo de ruptura del borde.

237
Después ponemos una agujita, permitiendo salir así el N2 (g), arrastrando al
aire que haya dentro. Pasados unos minutos el tubo se halla ya en
disposición de ser utilizado.
Debemos fijar este conjunto, colocándolo con cuidado en un rincón, pues el
tubo pesa tan poco que queda sostenido por el pincho en el aire, a merced
de cualquier mal golpe que lo parta. Lo mejor es llevarlo a un rincón,
sujetando la goma de la línea con algún objeto lo suficientemente pesado
como para que no se mueva.
24. 5 ARREGLAMOS LA BOCA DE LOS TUBOS
Aún observando todas estas precauciones se romperán muchos tubos de
RMN, algunos de modo irreparable, pero la gran mayoría se romperán por
la boca. Así nos quedaremos con tubos partidos en cuya parte superior
encontramos peligrosos dientes que habrá que eliminar con cuidado si no
queremos cortarnos.
Debemos arrancar con unos alicates dichos picos y buscar hacer roma la
superficie. Los pequeños cristalitos que se desprendan irán cayendo sobre
un depósito para vidrio que debe existir en el laboratorio, sobre el cual
haremos esta operación de tosco nivelado.
Enrasando el borde en sus desigualdades principales eliminaremos también
las más pequeñas de modo que el resultado sea una superficie lisa. Para ello
contamos con el papel de lijar.
Extendemos el papel sobre una superficie dura y cogemos el tubo como si
fuese un lápiz, con la abertura hacia abajo. Frotándola convenientemente
lograremos pulir sus irregularidades.
Aunque la longitud del tubo haya descendido, pueden encontrarse muchos
por debajo de los 12cm y seguir siendo válidos.

238
24. 6 TOMAMOS LA MUESTRA EN SEGUIMIENTO
Como ya hemos indicado podemos seguir la muestra mediante RMN y
analizar sus cambios estructurales viendo cómo éstos afectan al espectro. El
proceso a seguir para ello es muy ajetreado, y requiere un continuo ir y
venir entre el laboratorio y la sala del RMN que dependerá del intervalo de
tiempo en el que decidamos hacer las mediciones.
Hay tipos dependiendo de si la reacción se va calentando desde la retirada
del foco frío hasta la temperatura del laboratorio, o si por el contrario
calentamos en un baño de aceite. Se concibe también un tercer caso en el
cual la reacción marcha a una temperatura constante.
A) Primer caso.
El matraz sigue conectado y en agitación a la línea, por lo que podemos
hacer tantas tomas como nos parezca oportuno, sin impedimento de ningún
tipo.
Al principio cogemos un trozo de parafilm y lo reservamos para la parte
final de la operación. Las etapas de la misma son:
1. Se tiene desoxigenado el tubo de RMN. Se le quita la aguja y se toma el
tubo por la boca con los dedos, procurando apretar el tapón para que no se
salga en lo que dure el ejercicio, pero también con cuidado de no quebrar el
cristal.
2. Con una cánula de RMN se pincha el tapón del matraz (que deberá haber
sido cambiado por uno pinchado) y se espera a que se desoxigene su
volumen interno.

239
3. Se lleva el extremo libre de la cánula sobre el tapón del tubo y se hace
bajar hasta el fondo. Aquí hay que ser muy diestro y cuidadoso, si no se
romperá la parte de arriba del tubo y acabaría el experimento.
4. Otro problema muy serio es el de sacar el pincho, que es la operación
siguiente. Con los dedos se fuerza al tapón a permanecer en su sitio en
tanto se procura retirar el pincho con lentitud. Si no se cuidan estos
consejos ocurre habitualmente que el tapón se sale, arrastrado por el
pincho. En todo este movimiento el peligro de que se rompa el vidrio es
constante.
5. Una vez sin el pincho, se sumerge la cánula en la disolución a analizar
que, tras clavar una aguja en el tapón blanco, se pasará al tubo de RMN. Se
toman algo menos de tres dedos de disolución en el tubo (entre 4 y 5cm
desde el fondo del tubo hasta la línea del líquido), o lo que es lo mismo, un
volumen en torno a los 6 o 7ml. Si el volumen empleado es menor no
registraremos el espectro, por las razones técnicas que se expondrán más
adelante.
6. Hecho esto se elimina la aguja y dejará de caer muestra al elevar el otro
extremo por encima de la disolución. Entonces será el momento de sacar la
cánula. Cuando se extrae la cánula el volumen recién tomado quedará
desprotegido, por ello se habrán de tomar las medidas pertinentes. Como en
la situación de la filtración en tubo centrífuga se enrollará el tapón con
parafilm de modo que los agujeros se tapen mediante la lámina plástica. Si
se hace convenientemente el tiempo de contacto entre el aire y la muestra
desprotegida es mínimo.
Este último proceso es delicado pues, al tiempo que se fija el parafilm y se
estira para una buena adherencia y sellado, se debe sacar la cánula, con los
riesgos ya comentados. Si en este momento se parte la boca del tubo
estaríamos en un verdadero aprieto, perdiendo la muestra preparada.
Para terminar se retira la cánula del matraz y se pone un tapón nuevo.

240
7. Hecho el espectro de RMN se devolverá la muestra tomada al matraz ya
que si pretendemos hacer un seguimiento completo sin devolver las
porciones, al final acabaríamos por darnos cuenta que quedaría bien poco
del producto.
Se clava para ello con cuidado el pincho, con corriente de gas abierta, y
después con una cánula se pasa el líquido del interior al matraz.
8. La operación descrita se repetirá cada vez que se crea conveniente para
realizar un seguimiento completo de la reacción.
Estos seguimientos requerirán tomas dependiendo del rango de
temperaturas a desarrollar y dependiendo de la velocidad con la que ese
calentamiento se produzca, pero en cualquier caso deben mostrar cómo
evoluciona la reacción desde el principio hasta el final de un modo
suficiente.
B) Segundo caso.
Dado que la ampolla de reacción, donde se calienta, también está conectada
a la línea, el primer método puede ser utilizado, sin embargo resulta más
conveniente el que se pasa a detallar.
En un tiempo determinado de reacción se toman unas cuantas muestras en
tubos de RMN (una para cada tiempo deseado). Estos tubos se colocan
después en el baño de aceite visto en la sección 14.5.1, donde existe un tipo
de rejilla especialmente diseñada para sostener los tubos de RMN en el
baño (esta rejilla se puede fabricar con panel de madera).
El baño debe estar en funcionamiento de antemano, de modo que esté a la
misma temperatura que la reacción del matraz. A medida que se calienta el
matraz, se va calentando el baño de aceite, por lo que la reacción del
matraz y aquella de los tubos marcharán paralelamente. Cada vez que se
haga un RMN a una temperatura, se subirá la misma tanto en el matraz

241
como en los tubos y el baño. Tras las mediciones, las muestran se vuelven a
pasar al matraz para que no se pierda producto de ningún modo.
C) Tercer caso.
Cuando la temperatura no varía se suele usar el primer método, ya que el
manejo del tapón de goma del matraz es más sencillo y rápido que el de la
ampolla (al margen de que no es lo mismo abrir un matraz a temperatura
ambiente para cambiarle el tapón que una ampolla caliente con, tal vez,
reactivos que se evaporan y vuelven a caer, en una especie de reflujo).
Simplemente realizaremos tomas a distintos tiempos, sin variación de la
temperatura. Aquí no se busca ya a qué temperatura comenzará la reacción,
sino a qué tiempo se puede dar por concluida. En los dos casos anteriores
ambos parámetros se consideraban objetivos.
También hay que indicar que con el tiempo, tanto el primer como el
segundo caso acaban por convertirse en el tercero, ya que la temperatura no
se hace subir indefinidamente sino que, una vez que comienza la reacción
se suele mantener constante en espera de que se complete.
24. 7 RMN DE PRODUCTO CRISTALIZADO
La otra posibilidad que tenemos es la de hacerle un espectro a un producto
ya cristalizado. El proceso es mucho más relajado, pues con un tubo de
RMN se tiene suficiente y no se necesitan hacer varias tomas.
Las etapas por las que se debe pasar son las que se indican a continuación:

242
1. Existe un tubo Schlenk de un tamaño muy reducido pero absolutamente
idéntico a los de mayor tamaño, con salida para conexión de la goma de la
línea mediante un tapón de teflón. Se usa para preparar las muestras,
disolverlas en el disolvente deuterado y pasarlas al tubo de vidrio de RMN.
Para trabajar con él se cierra la boca principal mediante un tapón normal de
goma rojo. Este tapón puede ser, al igual que en el caso de las ampollas,
uno pequeño o uno grande (49), prefiriéndose este último. Al mismo
tiempo en el brazo lateral que lo une a la goma de la línea se enrosca un
tapón de teflón cuyo uso es ya perfectamente conocido por el lector.
2. El Schlenk donde se guarda el producto se debe conectar a la línea. Allí
se le prepara para añadir el producto sólido al Schlenk de pequeño tamaño
mediante una espátula tal y como se vio en la sección 7.2.1 (si el producto
es reactivo al aire) o bien se pone en el tubo al aire y después se le hace
vacío (si estable a la atmósfera) y se logran los 3 ciclos restantes.
Para un espectro de RMN1H la cantidad de muestra requerida es
sorprendentemente pequeña, y basta tomar una ligera cantidad con la
espátula (lo que se llama una punta de espátula). No obstante, para
espectros de 13
C se debe tomar una cantidad muy superior, debido a que el
número de átomos que dan señal es mínimo.
Un resumen de los pasos a dar en este transvase es realizado abajo:
- Se inclina el tubo de Schlenk y se mete la espátula, tomando la
cantidad establecida.
- Al ir a sacar la espátula, se cierra la corriente de N2 (g) durante
unos segundos para después abrirla de nuevo.
- Se cierra la corriente de gas en el otro tubo y se introduce el
producto, abriendo a continuación el N2 (g).

243
- Se colocan los tapones de nuevo en ambos recipientes y se hacen
los 3 ciclos para eliminar cualquier resto de aire que pudiera
haber penetrado.
En general esto vale para cualquier producto, pues los cristales son más
estables que sus disoluciones, y el contacto con el aire es tan breve que no
hay temor en el manejo.
3. A continuación tenemos que disolver la mezcla con alguno de los
disolventes deuterados. La elección del disolvente está vinculada a criterios
químicos y económicos, seleccionándose así el más barato posible de entre
todos los que puedan disolver la muestra.
Hecho esto se coge la ampolla y se conecta a la línea. En principio hay que
desoxigenar la goma y después se pasa a inyectarle el N2 (g), de modo que
se pueda abrir la llave de teflón sin peligro. Colocando el tapón pinchado
conveniente la tenemos dispuesta para realizar la toma de disolvente.
Esta toma se realiza mediante la jeringa de plástico de 1 ml, succionando
un volumen de entre 0.6 y 0.65 ml. La operación exacta se analizó en la
sección 13. 2. 2.
4. Disuelta la muestra en el disolvente deuterado, nos será preciso pasarla a
través de una cánula de RMN al tubo que se ha dejado desoxigenando hasta
este momento. Se transvasa como ya se sabe y se hermetiza mediante un
parafilm.
No se debe olvidar que una vez usada, la ampolla se habrá de guardar
cuidadosamente bajo atmósfera de nitrógeno.

244
24. 8 RUPTURA DE TUBO DE RMN EN OPERACIÓN
La posibilidad de que se nos rompa un tubo o se nos parta el borde del
mismo es una continua preocupación siempre que nos encontramos
trabajando con esta clase de tubos. Hay dos tipos de ruptura, dependiendo
del momento en que nos sucede el accidente: antes de pasar la disolución y
después de haberla conducido.
En el primer caso no hay mayor problema que tener que reiniciar toda la
operación de desoxigenación con un tubo nuevo, retrasando todo el
programa de trabajo que nos habíamos impuesto.
En el segundo caso la circunstancia es más grave, ya que no debemos
perder el producto al que queremos hacerle el espectro.
A) Si nos hallamos ante una toma directamente desde el matraz de
reacción, no hay solución posible, ya que hacer retornar al líquido a la
disolución de reacción es peligroso (porque ha podido absorber cierta
cantidad de aire y puede interferir la reacción).
B) Si hacemos la toma desde el Schlenk de RMN, podemos invertir el
camino de la disolución y volver a verterla en el tubo de Schlenk. Ahí
hacemos vacío y eliminamos el disolvente, para a continuación intentar
nuevamente el trabajo.
Cuando el tubo de RMN se rompe por la boca, suele tener el pincho dentro
(es al sacarlo cuando suele romperse ésta). De modo que quitando la agujita
y bajando la cánula hasta tocar el líquido, se puede devolver al Schlenk de
RMN si se cierra en aquel la entrada de N2 (g) y se coloca la aguja.
La utilidad de evaporar el disolvente en el pequeño Schlenk (so pena de su
coste) es la de eliminar cualquier traza de aire que se haya podido colar, y
salvar entonces el compuesto.

245
24. 9 NOS CORTAMOS CON EL VIDRIO
A lo largo de este bloque hemos amenazado una y otra vez con la ruptura
de los tubos de RMN debido a su fragilidad. El presagio puede ir
acompañado por otro todavía más desagradable. Como es evidente, cuando
se rompe el tubo, solemos estar apretando con los dedos cerca de la boca
del mismo, y podemos cortarnos muy fácilmente.
Para que el accidente sea lo menos grave posible en el caso de que ocurra,
nos dirigiremos inmediatamente a la pileta y, colocando el dedo debajo del
grifo, apretaremos la herida haciéndola sangrar para expulsar cualquier
sustancia tóxica que pudiera tener. A continuación se tomarán las medidas
usuales de curación.
24. 10 LA SALA DE RMN
Esta habitación está destinada a albergar los distintos tipos de equipos de
RMN existentes en un centro. Cada equipo se encontrará suficientemente
alejado el uno del otro para que no se interfieran mutuamente.
Como los equipos se controlan por ordenador encontraremos, a cierta
distancia uno de ellos, junto con un panel de control propio del RMN.
La necesidad de imprimir los espectros, fuerza también la presencia de
impresoras, conectadas a los ordenadores y repartidas por la sala.
La nota más característica de la habitación se halla en su puerta, donde un
cartel avisa a todo el que lleva marcapasos del peligro que corre en el
interior (habida cuenta de la presencia de campos magnéticos altos en la
habitación, que pueden alterar su funcionamiento).

246
Finalmente hay que resaltar la necesidad de mantener la temperatura del
recinto a 25 ºC, para que las mediciones se realicen a temperatura
constante durante todo el año, por lo que hay un sistema de refrigeración-
calefacción muy efectivo.
24. 11 EL APARATO DE RMN
El sistema de RMN consiste básicamente en la combinación de cuatro
elementos:
1. Un generador de campo magnético.
2. Una fuente de radiación que emita a la adecuada.
3. Un detector con capacidad para medir las absorciones.
4. Un ordenador encargado de trabajar con la señal y hacer las gráficas.
Estos bloques resultan indispensables para obtener un espectro, aunque no
son los únicos que se usan; de hecho existen muchos más accesorios que
mejoran y hacen más versátil la funcionalidad del aparato.
Los tres primeros se encuentran dentro de lo que, externamente, parece una
bombona de gran tamaño, en cuyo punto más alto se observa la abertura
por donde se introduce el tubo de RMN.
Algo más abajo (dentro de la bombona) un haz de radiofrecuencias es
emitido, debiendo atravesar la parte inferior del tubo con la muestra. En su
interior, el campo magnético alcanza los valores más altos, siendo entonces
efectivo para realizar la gráfica con la mayor nitidez posible.
En la zona baja del aparato hay un dispositivo que sirve para cambiar de
sonda cuando queremos hacer un espectro de RMN distinto al de 1H. Con

247
un levísimo giro de tornillo se pasa de una sonda a otra, sin otra
preocupación que la de no forzar las llaves.
En el mismo lugar existe también un elemento (con forma de tornillo) que
sirve para ajustar la orientación del campo magnético. La correcta
orientación del mismo se logrará groseramente observando
las señales de un nuevo aparato que se halla a los pies del RMN y que
marca el desajuste mediante puntos de color (hay muchos modelos no
obstante). La indicación de este aparato es muy fácil de analizar. Posee una
pequeña pantalla rectangular muy alargada, donde una luz muestra en qué
sentido se da el desajuste. Cuando la luz está en el centro (verde) el sistema
está ajustado, pero a medida que se aleja a izquierda o derecha (siendo
entonces rojiza) se muestra el desajuste pertinente. Girando el tornillo de
ajuste convenientemente, en uno u otro sentido logramos que la luz del
accesorio sea verde, y por tanto consideraremos calibrado al aparato. Un
ajuste mucho más fino y definitivo se hace desde el ordenador de control.
El tornillo de ajuste del RMN suele estar en una posición tan baja que es
preciso tenderse en el suelo para manejarlo.
El ordenador, que constituye el cuarto elemento, junto con el panel de
control del RMN (que se halla a su lado) gobierna las funciones del
aparato, por lo que con tan sólo apretar teclas obtenemos los distintos
espectros, los modificamos a voluntad y los optimizamos de un modo
sencillo y rápido.
Debemos considerar finalmente que es posible también hacer espectros a
baja temperatura, existiendo un dispositivo adicional que capacita al
aparato para dicha función.

248
24. 12 SE HACE EL ESPECTRO
No es objeto del manual detallar exactamente cómo se maneja el programa
informático para la adquisición del espectro, sin embargo se darán notas
sobre algunas características generales, mas de un modo somero y poco
exhaustivo.
Para poder usar el aparato de RMN hay que coger hora, dado que la
investigación requiere mucha gente y todos tienen las mismas necesidades.
Así encontraremos que, por cada aparato de RMN hay una hoja donde
podemos apuntarnos. El tiempo a repartir a lo largo del día es mucho, pero
sólo tenemos a disposición un lapso muy concreto del cuál no podemos
excedernos. Los intervalos de tiempo son normalmente de media hora, y
como máximo se ocupará una hora entera.
Antes de comenzar nos aseguraremos que todo objeto metálico que
poseamos quede sobre la mesa, al lado del ordenador,
pues al acercarnos a introducir la muestra en el aparato, podría afectarse su
campo. Recuérdese que las tarjetas con bandas magnéticas pueden sufrir
modificaciones y resultar dañadas por los altos campos magnéticos en
torno al aparato. Los relojes de pulsera que generalmente se usan también
deben ser considerados y dejados sobre la mesa.
Considero que el programa de control del RMN contiene las mismas
funciones con una nomenclatura análoga en todas partes.
Es importante verificar que ningún compañero haya dejado una muestra en
el aparato de RMN que esté siendo medida en algún experimento. Existen
métodos de espectroscopia que requieren tiempos prolongados y así,
pudiera ocurrir que tengamos algún compañero invadiendo nuestro tiempo.
Para ello tecleamos en el ordenador el siguiente comando:
acqu: que indicará el tiempo que resta para que acabe el experimento.

249
Para continuar en el caso de que haya un experimento, lo que se hace es
parar momentáneamente éste, pero de manera que más adelante pueda ser
continuado. Así se usa:
halt: usado para detener un experimento, el cual se volverá a retomar
posteriormente.
Si hubiésemos empleado stop el compañero habría perdido todo el trabajo
realizado.
Verificado este punto se pasa a introducir el tubo de RMN en el aparato
(tras sacar si fuera necesario el del experimento que se está completando
como se indicará más adelante). Esta operación se debe hacer con cuidado,
ya que el aparato es muy sensible, y cualquier alteración en su campo
magnético acabaría desajustando el sistema.
Para seguir paso a paso los movimientos a realizar, se considerará una
exposición con puntos, como ya se ha hecho otras veces. Los botones que
deben apretarse se encuentran al lado del ordenador en un cuadro de
mando.
- Se desconecta el botón llamado lock on/off. Al observar la luz
encendida se entenderá on y al verla apagada off. Este botón
cierra o abre el sistema, permitiendo así la introducción o
extracción de un tubo.
- Apagamos spin on/off. Botón que regula la rotación de la
muestra, lo que es necesario para que el campo que reciba sea lo
más homogéneo posible.
Resulta obvio que si una u otra función está activada sería imposible
introducir la muestra.

250
- Se enciende lift on/off, el cual se ocupa de levantar o bajar el
soporte donde acomodaremos la muestra. De no estar en esta
posición, no habría soporte donde colocar el tubo.
Al subir el asiento del tubo de RMN nos encontraremos con el tubo de
referencia patrón, el cual deberá dejarse siempre en este lugar tras concluir
las medidas.
Al lado del ordenador debe haber un recipiente para depositar los tubos de
RMN en espera de ser usados. Allí se puede también dejar el patrón en
tanto se hace el experimento de medida.
Ese tubo de referencia lleva una peana de color azul que se ajusta a una
altura muy concreta del tubo, indicando perfectamente por dónde va a pasar
el haz que la muestra debe de absorber. Esta peana se fija a semejante
altura mediante el concurso de un cilindro hueco numerado. Al insertar el
tubo de RMN con la peana en este último, llevándolo hasta el fondo, la
peana se desliza hasta la altura correcta, indicándonos su posición exacta,
asegurando así una lectura adecuada. De este modo se quita el cilindro de
indicación y se deposita el tubo con peana en el hueco superior del aparato
de RMN.
Muchas veces es necesario limpiar el tubito de muestra con un paño de
papel (que suele encontrarse sobre la mesa).
- Se desconecta lift on/off. Se esperará por tanto a que la muestra
descienda. Se oye un crujido dentro de la máquina
correspondiente al acoplamiento con el propio mecanismo.
- Se vuelve a conectar spin on/off y lock on/off para que la
muestra gire.

251
En estos momentos no hay más que usar el ordenador para poder obtener el
espectro.
- Una vez se escribe lock con el teclado podemos acceder en la
pantalla a todos los disolventes deuterados en los que se pueden
hacer los espectros. Se indica cuál es el que se usa en la muestra
a analizar, simplificando esto el trabajo enormemente ya que los
nuevos parámetros se establecerán por sí mismos. Si el
programa no tuviese esa aplicación cada vez que se hace un
espectro con un disolvente distinto al usado con anterioridad hay
que cambiar todos los parámetros del programa, aplicándolos al
nuevo disolvente.
- Tras lo anterior se busca en directory y se determina el aparato
de RMN en el que se va a trabajar. Los aparatos de RMN más
comunes que se pueden encontrar son los de: 300, 400 y
500MHz.
- Hecho esto se señala el grupo de investigación al que se
pertenece y se busca el archivo propio al investigador, para que
los espectros se almacenen en esa dirección.
Obviamente se han eliminado algunos pasos que están destinados a
conectar los diferentes procesos entre sí. También se han dejado a un lado
los dispositivos de seguridad que impiden que se escriba en archivos
correspondientes a otros compañeros. Como estos elementos se escapan a
la idea originaria de este volumen, no creo conveniente extenderme más
sobre estas cuestiones.
- Cuando todo lo anterior se ha llevado a cabo se precisa una
operación que se denomina “ajuste del lock” que consiste en
calibrar el aparato con mayor precisión de la que se hizo al

252
principio. Anteriormente se vio cómo se mantenía el campo
magnético uniformemente, dentro de unos parámetros muy
restringidos, usando un tornillo en la parte baja del aparato.
Ahora el calibrado se realiza con mayor efectividad mediante las
órdenes que se mandan desde el ordenador y la tabla de mandos.
El campo es generado por una serie de bobinas cuya orientación relativa
depende de las características de la muestra, de modo que debe ser
reajustada a cada cambio de la misma. Pequeños movimientos de estas
bobinas se pueden dirigir desde la tabla de mandos con unos botones, en
tanto en el ordenador se aprecia la evolución y conveniencia de uno u otro
movimiento.
En la parte baja de la tabla hay una serie de botones que sirven
precisamente para ajustar el campo (lo que hemos dicho que se llama hacer
lock en el aparato). De entre todos los botones sólo hay dos que deban ser
usados: Z y Z2
- Se conecta Z y posteriormente se gira un dial existente en la
tabla tal que se mueven con precisión absoluta las bobinas del
aparato de RMN. Entre tanto, en la pantalla del ordenador se
puede observar una línea que tiende a ser más o menos
horizontal. Se tratará, girando el dial a derecha o izquierda, de
llevarla siempre lo más arriba posible. Cada giro provoca un
movimiento ascendente o descendente de la línea que se dibuja
en el monitor. Cuando parezca que la línea no puede ascender
más, se prueba entonces con el otro botón.
- Se enciende Z2 en lugar de Z y se reproducen los pasos
anteriores tal que logremos una línea lo más arriba posible y
horizontal.

253
El proceso descrito por los dos puntos anteriores se reitera hasta que
parezca que no se puede mejorar más la disposición de la línea y por tanto
el ajuste óptimo de las bobinas. Entonces se considera que el aparato está
calibrado y en perfecto estado para obtener un espectro aceptable.
- A continuación se accionan autoshim y stand by (posición de
espera) cuya finalidad es la de mantener el ajuste hasta que
termine la medida.
Hay otros medios de verificar si el aparato se ha calibrado correctamente,
pero con los que se ha citado basta como ejemplo.
- Tras el calibrado comienza el verdadero proceso de obtención
del espectro. Para lograrlo se teclean comandos seguidos cuya
función es precisamente la de obtener el espectro con sus picos
definidos y con una línea base perfectamente definida (lo que se
denomina “fasear” el espectro). Estos pasos consecutivos son:
rga, zg, fp, fase, biggest, pH0 y pH1.
- Para poner límites al espectro y que se mantenga en el rango que
interesa tenemos dp1 y para cifrar los picos xwp_pp. Finalmente
se utiliza un programa de dibujo para poner una etiqueta al
espectro con el nombre del compuesto o el número del
experimento del que proviene. Se guarda dentro del archivo del
investigador y se imprime.
- Para extraer el tubo de RMN se hace igual que con el tubo
patrón al principio, volviéndose a insertar éste en el aparato.
Debemos hacer notar que el patrón es benceno, siendo por tanto
obligado marcar el tipo de disolvente en el programa tal y como
se hizo para indicar aquel que se había empleado para disolver
nuestra mezcla.

254
Téngase con esto una idea de lo que es el trabajo en la sala del RMN. Si ha
sido demasiado extenso para algunos, me temo que por el contrario, habrá
sido muy corto e impreciso para otros. No obstante lo que pretendía era dar
una visión somera de un proceso que constituye una de las tareas más
importantes del químico organometálico.
24. 13 INTERPRETACIÓN DE UN ESPECTRO
Se acostumbra uno en la Facultad a enfrentarse a los espectros de RMN de
compuestos desconocidos. La dificultad entonces es máxima y conlleva un
esfuerzo enorme en el que la experiencia del estudio diario resulta
insustituible.
Aquí sin embargo, el análisis no es tan grave ni tan abstruso. Si bien es
verdad que la experiencia es fundamental, no se deducen de la nada las
estructuras sino que éstas son parcialmente conocidas. Dicha estructura se
intuye generalmente, y lo que se hace es verificar esta intuición o
desestimarla en función de los datos obtenidos. Mas la gran mayoría de las
veces no difiere sustancialmente de lo que se espera. La ciencia, ante todo
es lógica, y por tanto no suele sorprender a un razonamiento prudente. Las
reacciones no se hacen a tontas y a locas, sino con la idea de obtener algo,
y tras el apoyo de largas sesiones de lectura sobre reacciones similares.
Cuando se mezclan dos productos, lo más normal es que la reacción no
vaya, pero si lo hace, suele seguir los pasos marcados por la lógica, no
separándose mucho del objetivo. De este modo, el problema no es
preguntarse de qué sustancia es el espectro, sino si el espectro se adapta a la
sustancia que tenemos en mente.

255
Al margen de todo, siempre hay un espectro previo, que sirve para trazar
una evolución. Este seguimiento resulta fundamental y es una inestimable
ayuda a la hora de demostrar que nuestro compuesto tiene una estructura
determinada.
24. 14 REFLEXIÓN FINAL SOBRE EL RMN
Generalmente se piensa que el trabajo del laboratorio absorbe
completamente el tiempo del investigador, y que por consiguiente, es lo
único que el químico hace en el centro en el que trabaja. Se cree
erróneamente que el laboratorio es el lugar donde siempre se va a encontrar
al científico, pegado a su vitrina, y que la investigación por tanto no es más
que mezclar sustancias y obtener unos productos al final.
Se pretende desde aquí anotar que el trabajo verdadero del químico está, no
sólo vinculado a una síntesis afortunada de un producto, sino a la
demostración de su estructura al mismo tiempo. La primera parte de este
proceso se hace, es verdad, en el laboratorio, pero la segunda es motivo de
visita continua al RMN, de inspección de espectros y análisis de resultados.
Es también un trabajo que se lleva a casa, pues la duda se fija en la mente y
hasta que no se resuelve no se deja.
El químico organometálico pasa entonces casi el mismo tiempo en la sala
del RMN que en el laboratorio ante su vitrina. No quiero por tanto mostrar
otra realidad más que aquella que encontramos y ser justos en cuanto a este
hecho.
Por último queda decir que lo anterior no es sino la primera planta de una
investigación, que conlleva una dedicación plena, pero una investigación
dirigida, como es necesario que sea. Es esta dirección la que decide qué
reacción se hace y cuál se considera inapropiada. En este otro nivel, o

256
segunda planta de la investigación, el tiempo se emplea en leer y en pensar.
El contacto con el laboratorio se difumina lentamente, al igual que con la
sala de RMN, pero es entonces cuando la investigación alcanza su
verdadera dimensión.
24. 15 SE LIMPIAN LOS TUBOS DE RMN
La limpieza de los tubos de RMN es obligada al final de las mediciones.
Algunas veces pueden acumularse un buen número de los mismos ya que
es más cómodo limpiarlos en grupo que de uno en uno. Si esto último
ocurre conviene protegerse con guantes, no fuera que alguno contenga
benceno como disolvente deuterado, del cual siempre conviene cuidarse.
Al igual que otras veces el lavado se hace con acetona y éter, pudiéndose
en caso de necesidad usar otros disolventes como el agua o ácidos. Incluso
podemos utilizar los limpiapipas para eliminar del interior alguna sustancia
que permanezca adherida.
Tras lavarlos se meten en la estufa en un vaso de precipitados u otro
recipiente que se utilice para recogerlos, permaneciendo siempre en ese
lugar hasta que deban ser empleados nuevamente.
25 ESPECTROSCOPIA DE INFRARROJOS
No siempre es posible usar el RMN para verificar la estructura de la
molécula. Así cuando hay muchos núcleos activos o cuando contrariamente
no hay ninguno, debemos recurrir a otro poderoso auxiliar en la
determinación de estructuras orgánicas e inorgánicas. Se tiene en la

257
espectroscopia de IR una inestimable ayuda que, en su propia extensión
(notablemente inferior al RMN), capacita para deducir estructuras. Como
su propio nombre indica, se usa la radiación infrarroja para obtener un
espectro de absorción indicativo de los grupos existentes, así como de su
distribución espacial.
25. 1 LA BASE CIENTÍFICA
La banda de infrarrojos se divide en tres zonas más o menos diferenciadas:
cercano, medio y lejano. Dentro de estas zonas se encuentran las
diferencias entre los distintos estados vibracionales de los enlaces de una
molécula. La irradiación con la energía adecuada puede hacer que se de una
transición desde el estado vibracional elemental al primer estado de
vibración excitado. Esta energía cuantizada será absorbida y por tanto se
podrá registrar su ausencia en un espectro.
Las energías de los enlaces dependen precisamente de los átomos
constituyentes del enlace y de los grupos más cercanos. De este modo, cada
grupo químico distinto afecta a un enlace en diferente grado, y la energía
entre el estado vibracional y el excitado variará consecuentemente. Se
puede considerar que cada tipo de compuesto presentará un conjunto de
bandas de absorción vibracional que le será propio y que se juntas se
conciben como si fuesen la huella dactilar del compuesto, por lo que la
identificación de un compuesto es accesible.
En general el IR no es una prueba irrefutable de la estructura de un
compuesto, sino que es más bien un método complementario a otras
técnicas más capaces, como un espectro de RMN. Pero cuando esta última
no es viable, es una ayuda fundamental.

258
No todos los enlaces absorben radiación, sino aquellos en los que la
vibración provoca un cambio en el momento dipolar de la molécula. Estos
análisis no se corresponden con los objetivos de este trabajo por lo que no
nos detendremos más en ello.
Las bandas esperadas para un compuesto pueden deducirse a partir de los
grados de libertad del mismo, teniendo en mente una estructura previa, que
es la que se desea corroborar. Sin embargo hay que considerar que no
siempre se encuentran todas las bandas que se aguardan. Las razones son
las siguientes:
1. Algunas vibraciones, como ya se ha comentado, pueden no
provocar cambios en el momento dipolar.
2. Aquellas vibraciones que posean la misma energía solaparán en
el espectro.
3. Bandas con muy baja intensidad pueden pasar desapercibidas.
4. Algunas frecuencias de vibración pueden quedar fuera del rango
de trabajo del instrumento.
Las frecuencias a las que se producen las absorciones en un enlace concreto
pueden deducirse por la mecánica cuántica; y su presencia o no en el
espectro, en razón al punto número 1, se extrae a partir de la Teoría de
Grupos de simetría.
25. 2 MUESTRAS SÓLIDAS
El IR se puede usar tanto para muestras sólidas como para líquidas o en
disolución. Las más utilizadas son las primeras, por lo que nos ceñiremos a

259
ellas. Estas dos últimas, se debe señalar, son generalmente introducidas por
una jeringa en unas celdas especiales con ventanas de haluros alcalinos
(obviamente las disoluciones no serán acuosas).
En las muestras sólidas el compuesto se dispersa en KCl (s). Con la mezcla
se formará una pastilla que servirá para la realización del espectro. Tanto el
producto como el KCl (s) deberán estar finamente divididos y mezclados.
Con tal fin se usa un mortero de ágata (variedad microcristalina del cuarzo,
muy poco porosa, por lo que apenas retiene material) con el que molturar
ambos mediante una maja del mismo material.
La mezcla se pasa a un soporte rectangular que posteriormente se colocará
en una prensa de gran eficiencia que compacta ambos constituyentes entre
sí formando un disco muy fino y traslúcido. En unos segundos se extrae la
pastilla formada de la máquina de presión y se coloca en un nuevo soporte
para el aparato de IR. La pastilla sorprende por su fragilidad, debido a la
considerable extensión en la que se ha expandido la sal con el compuesto,
debiéndose, por todo ello, manejar con sumo cuidado.
Esta operación se puede también realizar usando, en lugar del KCl (s), el
mujol. El mujol es una olefina líquida excelente para dispersar la muestra,
la cual se debe majar en el mortero. Es muy importante que las partículas
de la muestra sean lo más pequeñas posible, menores que la longitud de
onda de la radiación incidente. Si son mayores que , dispersan mucha
radiación y el espectro no es fidedigno.
Tanto el mujol como el KCl (s) se guardan en un desecador para que no
retengan agua, que sin duda resultaría dañina para el compuesto.

260
25. 3 EL APARATO
Como puede observarse el proceso de preparación de la muestra es mucho
más sencillo que el correspondiente al RMN, y en el uso del aparato, la
simplicidad será también una de sus características.
El espectrofotómetro de infrarrojos mide las frecuencias de absorción de
los compuestos en el espectro de infrarrojos, y se compone de los
siguientes elementos:
1. Fuente de radiación infrarroja, que es la que posteriormente
analizará un detector, verificando qué frecuencias faltan por
comparación con un rayo patrón.
2. Monocromador, responsable de que a la muestra llegue una
banda estrecha de longitudes de onda.
3. Detector, que se encarga de analizar la señal y de encontrar las
absorciones.
4. Ordenador, que sirve para graficar la disminución de la
transmitancia frente al número de onda.
El ordenador se encuentra integrado en el propio aparato, aunque claro, ésta
es la versión más moderna del espectrofotómetro pues antiguamente era
una plumilla la que sobre un papel normal (que se hacía pasar bajo la
pluma) dibujaba una línea representativa de la transmitancia. Ahora la
imagen aparece en el monitor y se imprime a voluntad.
El sistema de infrarrojos se suele encontrar en una sala dedicada a medidas,
junto con otros aparatos, donde se encuentra a disposición también la

261
prensa que permite la obtención de la pastilla, para que el manejo y
transporte de la misma sea lo más breve posible.
Véase cómo, en relación al RMN, el espacio que se le dedica es mínimo.
En la realidad ocurre algo parejo, pues el aparato es pequeño y compacto,
pudiéndose colocar en el rincón de alguna salita. Por otro lado no es
necesario siquiera tomar hora de antemano, siendo pocas las veces que el
químico organometálico recurre a él para obtener alguna información.
25. 4 INTERPRETACIÓN DE LOS ESPECTROS
Lo que se dijo para el RMN tiene igual cabida en este apartado. No se
busca simplemente a ciegas un compuesto estudiando las frecuencias a las
que aparecen las bandas, sino que se intuye siempre qué grupos tenemos,
por lo que se trata simplemente de identificar su marca en el espectro a la
frecuencia esperada. Es un ejercicio de corroboración de una hipótesis y no
la elucidación de una estructura a partir del desconocimiento más absoluto.
Encontrar algunas frecuencias concretas permite descartar determinadas
opciones y afirmar otras, lo que en una investigación científica es
fundamental, debido a la dificultad con la que habitualmente se obtiene la
información.
25. 5 OTROS SISTEMAS DE ANÁLISIS
Al margen del IR y de la técnica de RMN existen otros procedimientos
mediante los cuales podemos obtener información acerca de las estructuras
moleculares. Sin contar los diversos tipos de

262
RMN a disposición del investigador, se tienen también otros analizadores.
Sin embargo estos últimos instrumentos son usados por personal
cualificado, y el químico organometálico suele entregar las muestras a
éstos, para que sean ellos los que se encarguen de obtener los datos. De ahí
que no se reflejen en el manual estas técnicas, que se salen de lo que sería
el trabajo habitual del químico organometálico.
La técnica más importante de todas las que dejamos sin analizar es sin duda
la de difracción electrónica. Los difractogramas se obtienen en centros
particulares, debiéndose mandar las muestras allí donde estos se
encuentren.
26 ALMACENAMIENTO
En esta sección examinaremos los distintos recursos que tiene el científico
a mano para almacenar sus compuestos, tanto los obtenidos mediante
reacción, como aquellos que se usan como reactivos base.
26. 1 EL ALMACÉN
Para guardar los reactivos más comunes de uso en la química que se trata
se suele disponer de una pequeña habitación bien acondicionada y aireada.
En ella se almacenan de manera catalogada los compuestos en haber, cada
uno según su tipo y naturaleza química.
La habitación está, como se ha dicho, ventilada gracias a un extractor que
elimina el aire viciado (a causa de las posibles fugas o volatilización en los
productos), resultando así una atmósfera inocua. Este punto es
importantísimo a la hora de construir un almacén, pues la presencia de

263
muchos compuestos ocasiona atmósferas tóxicas si no se toman las
medidas adecuadas.
La puerta del almacén se cierra con llave por motivos obvios de seguridad,
aunque exista confianza plena en los compañeros del propio centro no
puede decirse lo mismo de los que vengan de fuera. La llave de la misma se
debe controlar siempre y es necesario saber dónde se encuentra en todo
momento. Generalmente va insertada en el llavero que reúne las llaves
útiles para el investigador, el cual se guarda normalmente en el laboratorio
para emplearlo cuando sea oportuno.
A continuación se pasa a enumerar los distintos bloques de productos que
podemos encontrar en el almacén. Al ser necesaria una búsqueda, ésta va a
estar siempre dirigida en razón a la naturaleza química de los compuestos.
- Hidrocarburos alifáticos
- Hidrocarburos aromáticos
- Alquenos (muy importantes para la coordinación con centros
metálicos en la química organometálica)
- Alcoholes
- Fenoles
- Aldehídos
- Cetonas
- Monocloruros (usados en la síntesis de compuestos de Grignard)
- Policloruros (usados en la síntesis de compuestos de Grignard)
- Cloruros funcionalizados (también de gran importancia)
- Monobromuros
- Polibromuros
- Hidruros
- Fluoruros
- Éteres

264
- Éteres corona (éteres en forma de anillo de varios miembros que
estabilizan muy bien a los cationes de los elementos alcalinos)
- Ésteres
- Haluros de ácido
- Poliaminas alifáticas
- Aminas aromáticas
- Heterociclos de 5 miembros
- Amidas
- Nitrilos
- N-derivados
- Heterociclos de 6 miembros
- Ácidos orgánicos
- Anhídridos
- Compuestos organometálicos (estables a temperatura ambiente y
a la atmósfera aunque pueden también guardarse bajo N2 (g) en
el mismo almacén)
- Sulfonatos
- Sulfuros
- Fosfinas (compuestos de gran importancia en la síntesis de los
compuestos organometálicos)
- Fosfitas
- Elementos (tanto metales como no metales)
- Sales (en general ordenadas según el metal)
- Lechos de cromatografía (tales como la celita, la sílice, el carbón
activo, arenas,...)
- Ácidos inorgánicos
Todos estos compuestos habrán de ser estables a temperatura ambiente y no
ser demasiado volátiles. Si se presentase esto último y se decide guardarlos

265
en el almacén deberán ser sellados correctamente con parafilm, de modo
que no pierdan producto por la evaporación. Los reactivos que son poco
estables a dicha temperatura o son demasiado volátiles se depositan en los
frigoríficos existentes en almacenes análogos.
Para controlar tantos productos, así como para saber su localización exacta,
existe un archivo en el almacén que indica desde los datos fisicoquímicos
del compuesto hasta el lugar que ocupa en el almacén (en la estantería
donde se ha de buscar o en el frigorífico correspondiente). Si alguien toma
uno de los productos, y lo va a usar durante un determinado tiempo, es
conveniente que lo indique en la ficha del producto, pues el almacén suele
ser compartido por todos los grupos de investigación de la disciplina.
26. 2 EL FRIGORÍFICO COMO ALMACÉN
En cada laboratorio hay al menos un frigorífico cuya principal misión (al
margen de facilitar la cristalización de productos en el congelador) es la de
almacenar reactivos y sustancias a baja temperatura.
Es en estos frigoríficos, además de los que se deben encontrar en un
almacén general del centro, donde el químico guarda aquellos reactivos que
le son más útiles. A su vez se protegen ahí muchos de los productos
sintetizados en espera de ser analizados, o que servirán como base a
reacciones futuras. Por todo ello las bandejas están atestadas de tubos de
Schlenk y matraces con compuestos organometálicos, bien cerrados y
protegidos bajo atmósfera inerte.
En la parte inferior del frigorífico se colocan ampollas contenidas en latas,
donde se mantienen compuestos de Grignard o fosfinas a baja temperatura.

266
Aún más abajo, donde en cualquier hogar se conservan las hortalizas, se
almacenan también ampollas con fosfina u otros compuestos afectables por
la temperatura.
Los compartimentos existentes en la puerta son usados igualmente como
almacén. En ellos se encuentran botes con productos comerciales para los
cuales la alta temperatura ambiente resulta peligrosa.
El contenido de estos frigoríficos es particular para cada laboratorio, de
modo que los productos que contienen pertenecen a cada grupo de
investigación en concreto y no pueden considerarse como reactivos de
consumo común.
La descripción que se ha realizado de los frigoríficos es, lógicamente, algo
muy relativo tanto en la disposición como en los contenidos de los mismos
pero se ha hecho aquí un análisis de lo que resulta más habitual.
26. 3 ALMACENAMIENTO DE AMPOLLAS
Las ampollas se usan para guardar líquidos o disoluciones de reactivos.
Estos reactivos podrán ser más o menos sensibles a la temperatura, y así
muchos habrán de ser guardados en el frigorífico, mientras otros se
almacenarán en algún cajón o rincón del laboratorio.
A causa de su forma deberán mantenerse erguidas, usando un recipiente
adecuado que las proteja de cualquier choque. Estos recipientes suelen ser
cajas de cartón pequeñas o cilindros de lata, que se llenan de esponjitas de
corcho, de modo que no sean de temer golpes o vibraciones que quiebren la
frágil estructura de la ampolla.
Siempre los compuestos se guardarán bajo atmósfera de N2 (g) en la
ampollas, cuidándose cerrar completamente el tapón de la misma,
impidiendo una destrucción del producto.

267
26. 4 ALMACENAMIENTO EN BOTES
Si un producto es estable al aire, se preferirá siempre guardarlo en un bote
típico, con tapón de rosca. Se emplean con preferencia aquellos botes
comerciales cuyo contenido se ha agotado con el tiempo, quedando tan
sólo el envase. Esto facilita su manejo y permite que lo coloquemos sobre
una estantería con otros compuestos análogos, lo que impide
acumulaciones absurdas en el frigorífico.
Lamentablemente, este caso es extraño pues la mayoría de las veces los
compuestos obtenidos serán reactivos al aire y por otra parte la cantidad
obtenida es tan pequeña que sería ridículo emplear un bote. Sin embargo es
un método perfectamente válido para almacenar algunos productos de
partida que se sintetizan en masa.
Muchos productos de partida exclusivamente comprados para el uso de un
grupo de investigación concreto (por lo cual no están en el almacén
general), se almacenan dentro del propio laboratorio, situándose bien en las
repisas del laboratorio, bien en las bandejas del frigorífico. Estos productos
se expenden en botes normales, de ahí su presencia en este apartado.
Igualmente, aunque el diseño del recipiente no sea equiparable a los aquí
presentados, podemos considerar los botes con tapón de seguridad, vistos
en la sección 13.6 que se guardan, dependiendo de su naturaleza, en las
repisas o en el frigorífico, antes de pasar su contenido a una ampolla.
Cada vez que se decida guardar un producto en cualquiera de estos botes,
es preciso hacer una etiqueta donde se especifique claramente el nombre
del compuesto, su peso molecular, su densidad en caso de que sea líquido y
la fecha en la que el depósito se ha realizado. Como se ha dicho con
anterioridad es también aconsejable proteger la boca con parafilm para que
no se produzca entrada de humedad.

268
26. 5 ALMACENAMIENTO TEMPORAL EN SCHLENK
Sirva tan sólo este punto para completar la visión acerca de los distintos
recipientes empleados en el almacenamiento de los compuestos,
considerando a los tubos como meros recipientes de almacenamiento. Por
su utilidad y por el continuo uso que se hace de ellos resultan
absolutamente impropios para emplearse como contenedores, por lo que
serán recipientes temporales.
Cuando se hace una reacción y se lleva a sequedad, los matraces suelen
dejarse en el frigorífico bajo atmósfera inerte hasta que les llega la hora de
pasar a la fase de separación y purificación del compuesto. Entre tanto se
almacenan temporalmente en el frigorífico, en los matraces donde la
reacción se ha producido.
Del mismo modo, una vez que se ha llevado a cabo la cristalización de un
producto, y se ha separado de las aguas madres, se suele dejar en el
Schlenk, dentro del frigorífico hasta que se tiene tiempo de preparar las
muestras para los diferentes análisis.
Sea cual fuere el caso, en esta etapa transitoria los recipientes se
mantendrán a presión de N2 (g) y con todas las gomillas bien colocadas, así
como el tapón protector contra la humedad en la boquilla.
26. 6 ALMACENAMIENTO EN TUBOS DE SELLADO
Cuando el compuesto a guardar es reactivo al aire tras un cierto tiempo de
contacto y su cantidad no es muy grande, se almacena en los llamados
tubos de sellado. Estos compuestos son sustancias ya analizadas que
conviene guardar para posteriores usos sin que ocupen los tubos de
Schlenk, que serán necesarios para otras sustancias.

269
Un tubo de sellado es una varilla hueca de borosilicato, cerrada por un
extremo. En la otra punta hay un estrechamiento que posteriormente
resultará clave a la hora de lograr el sellado, pues es el punto por el cual
vamos a cerrar la abertura.
Antes de explicar el procedimiento mediante el cual se va a aislar el
producto en el interior del tubo pasaremos a describir la pistola de llama
usada para calentar el vidrio.
El artefacto presenta una gran variedad en su presentación, y alguno de sus
modelos tiene verdaderamente aspecto de pistola, lo que le ha dado el
nombre. Se carga con gas como combustible (tal y como se hace con los
mecheros recargables), que viene a salir en forma de llama azul, estilizada
y dirigida. Posee un regulador de la entrada de aire, justo donde se procede
a quemar el gas, lo que posibilita el control de la temperatura de la llama.
Lo primero es llenar la varilla con el producto lo más rápido posible. Esto
se hace con la espátula, procurando que no quede por las paredes internas
del tubo (esto es muy importante). Una vez dentro se conectará a la línea
introduciendo la parte superior de la varilla por uno de los tubos de goma y
se le practicará vacío. Esto se podrá hacer con todo compuesto que
reaccione con relativa lentitud al aire, ya que los que se descomponen con
demasiada velocidad tendrán que almacenarse en la cámara seca.
Tras hacer vacío durante un tiempo, se comienza a aplicar la llama
manteniéndola en su posición menos energética (llama más ancha y
amarilla) en la zona del estrechamiento. Hay que proceder primero
calentando lentamente con esta llama de baja energía para que el cambio de
temperatura no quiebre el vidrio. También hay que considerar que la llama
nunca debe tocar el lateral de la pared en la que se encuentre el producto
pegado (por ello se debe cuidar que no se deposite por las paredes) pues
éste se descompondría por el calor y podría actuar de catalizador en la
descomposición del resto del producto (aunque la llama no se haya

270
aplicado a la masa del mismo). Lo mejor para impedir esta inconveniencia
es limpiar con papel el interior del tubo en torno al estrangulamiento,
incluso unos centímetros por debajo de éste, y cuidarse bien de no bajar el
haz.
Después aplicamos la llama azul sobre el cuello. Al existir vacío en el
interior de la ampolla, las paredes, reblandeciéndose, se pegan una con la
otra, sellando perfectamente la abertura. Poco a poco el vidrio cede y se va
estirando como una goma elástica. Al final se tira y se separa la ampolla
formada de la varilla que queda cogida del tubo de la línea. Se sigue
aplicando calor con la pistola para eliminar las puntas y cerrar poros que
puedan aparecer en la superficie de la varilla. Es muy importante repasar
una y otra vez con la llama la zona que se acaba de crear debido a que un
sellado insuficiente puede provocar la descomposición del producto en el
tiempo.
Para concluir la operación se vuelve a cambiar la llama, buscando entonces
una menos energética, para que el cambio de temperatura sea menos
brusco. Como regla general, siempre que se aplica una llama a un vidrio
conviene hacerlo gradualmente para que la dilatación sea homogénea en
todo el cuerpo y no se quiebre; igualmente ocurre cuando se retira con la
contracción del mismo. Si retiramos la llama azul sin pasar antes por la
amarilla, el cambio de temperatura puede generar rupturas microscópicas
que provocarán igualmente la descomposición del compuesto.
Durante el proceso se debe estar atento para que no se den los siguientes
errores, que a pesar de su obviedad, suelen resultar habituales:
1. Olvidando que el extremo del cilindro está todavía caliente se maneja sin
cuidado, y acaba descomponiéndose el producto al tocar la parte superior
del receptáculo.

271
2. Al intentar quitar el trozo del tubo que queda en la goma de conexión a
la línea, olvidamos que sigue estando caliente y nos podemos quemar los
dedos.
3. Al probar a sacar el predicho trozo de vidrio de la goma, se olvida que
sigue habiendo vacío, y nuestros esfuerzos serán tan baldíos como cómicos.
Para volver a usar el producto basta con romper la ampolla y pasarlo rápido
a un tubo de Schlenk.
27 LA CÁMARA SECA
Puede concebirse la cámara seca como un recinto donde se almacenan
compuestos que en cualquier otro lugar serían inestables y se
descompondrían con el tiempo. No obstante la visión que debe tenerse de la
cámara seca ha de ser mucho más amplia. Este compartimento cerrado es el
lugar ideal para llevar a cabo operaciones en las que el aire resulta un
obstáculo insalvable, no pudiéndose realizar más que bajo atmósfera inerte.
Sus funciones principales son:
1. Almacén para compuestos inestables en condiciones de atmósfera
normal.
2. Pesada de compuestos muy reactivos al aire.
3. Purificación de pequeñas cantidades de compuestos inestables al aire
usando columnas de tamaño reducido (incluso pipetas Pasteur).
4. Cualquier labor que sea inadecuada para la línea y que requiera
condiciones de ausencia completa de oxígeno y agua.

272
27. 1 DESCRIPCIÓN FÍSICA Y FUNCIONAL
Está formada por cuatro paredes: las dos laterales y la de detrás son
metálicas, en tanto la frontal es de vidrio, permitiendo ver el interior
claramente. Las tapas superior e inferior son también metálicas. El armazón
está construido sólidamente y remachado para impedir cualquier desunión.
La impermeabilización es perfecta por lo que no puede entrar aire dentro,
ni salir el gas inerte.
El vidrio de la parte frontal se encuentra perforado con dos agujeros, donde
se han colocado guantes de goma gruesa, unidos con eficiencia al cristal.
Como la presión interior es mayor que la exterior, ambos guantes están
inflados desde dentro y se extienden hacia fuera como si fuesen dos brazos.
Se debe hacer notar que los guantes tienen una longitud tal que permiten
dar cabida al brazo entero.
En la pared de la izquierda encontramos una pequeña cámara frigorífica
que contiene aquellos productos que deben no sólo ser protegidos del
aire sino también de una temperatura demasiado elevada como la
ambiente. Esta cámara se abre desde dentro mediante una llave que posee
en su interior.
En la pared de la derecha, se puede observar la manija de la precámara
pequeña y de la precámara grande. El resto de sus volúmenes se desarrolla
hacia el exterior de la cámara.
Es en el exterior donde encontramos el panel de control de la cámara, justo
en la parte superior derecha.
Un esquema de la cámara es el que se presenta debajo:

273
1. Cara frontal de la cámara seca, donde un cristal permite ver el interior de
la misma.
2. Lateral que muestra el volumen de la cámara frigorífica cuya puerta se
abre al interior del recinto.
3. Los dos círculos representan sendos guantes de goma negra que salen
hacia fuera de la cámara, los cuales no se dibujan por motivos de claridad.
4. Antecámara pequeña cuyo uso es el de permitir la entrada y salida de la
cámara a objetos y recipientes de pequeño tamaño.
5. Antecámara grande que posee la misma finalidad que la anterior pero
para objetos de mayores dimensiones.
6. Panel de control desde donde las funciones de la cámara seca se
establecen y regulan.
7. Analizador de O2 (g) que indica el nivel de dicho gas en la atmósfera
inerte.
8. Analizador de H2O (g) con pareja función al anteriormente señalado.
Tanto el detector de O2 (g) como el de agua son indispensables para
verificar el perfecto estado de la atmósfera de N2 (g) interior. Esta
atmósfera se mantiene renovada casi de modo constante por un sistema de
recirculación de gas muy eficaz que debe estar siempre conectado (así su
botón pulsado y encendido en el panel). Cualquier elemento que se

274
introduzca en la cámara deberá estar seco y no contener oxígeno, mas si la
eliminación de ambas sustancias no es completa aumentará la
concentración de agua y O2 (g) dentro de la cámara y será registrado por
los analizadores, mostrando una luz roja de alarma y el valor alcanzado.
Merece también una descripción el interior de la cámara, donde se
encuentra todo lo necesario para las operaciones de pesada, aunque las más
veces se precise introducir algunos elementos que nos resultan
imprescindibles.
1. Manivela de la portezuela que abre la cámara frigorífica. Posee una
cerradura con llave, la cual suele colgar de la cerradura.
2. Repisas que se suceden unas sobre otras, sobre las que se colocan los
botes con los productos a almacenar en condiciones de máxima seguridad.
También suele encontrarse sobre ellas viales vacíos y otros instrumentos
útiles.
3. Gran cantidad de botes y viales se hallan en el suelo de la cámara seca,
estén llenos o vacíos, donde puedan ser usados con comodidad (lo cual es
muy importante en este recinto, como veremos).
4. Un pie con pinzas también se encuentra en el interior, para soportar
tubos, matraces,... y así poder pasar reactivo de un bote al matraz sin
peligro de que el aire descomponga el producto.

275
5. Rollos de papel que sirven para limpiar espátulas y todo cuanto haya
podido ensuciarse dentro. La limpieza en este recinto es más que una ley.
6. Juego de espátulas de diferentes tamaños y características, que resultan
indispensables para pesar. En el mismo lugar se puede encontrar una
brocha para ayudarnos con ella en las labores de limpieza.
7. Peso electrónico que se sitúa en el centro, para permitir las maniobras en
su derredor.
8. Papeles encerados para pesar, unidos por una pinza o mordaza.
9. Vaso de precipitados con pipetas Pasteur y tubos de RMN que pueden
ser necesarios en algunas operaciones en la cámara.
10. Mango de la tapa que abre desde dentro la antecámara pequeña.
11. Ídem de la antecámara grande.
27. 2 INTRODUCCIÓN DE OBJETOS
Cuando se precisa pesar en la cámara seca, o bien realizar cualquier
operación en la misma, hay que introducir previamente aquellos elementos
que nos resultan necesarios. Dependiendo del tamaño de los objetos a
ingresar se empleará la grande o la pequeña. No existe diferencia en el
funcionamiento de ambas, diferenciándose únicamente en que la pequeña
se maneja mediante un mando situado debajo de ella, mientras que la
grande está regida por el panel de control.
Para la descripción del proceso usaremos la primera, pues la de mayor
capacidad se utiliza raramente.
Para introducir los objetos habremos de quitar la tapa que cierra la boca
exterior del cilindro que conforma el cuerpo de la cámara. Los pasos son
los siguientes:

276
1. Al abrir la precámara debemos considerar primero el estado en el que se
encuentra su interior. La precámara siempre debe dejarse a vacío durante
los periodos en los que no esté siendo usada, por lo que así nos la
encontraremos cuando queremos usarla, e intentar abrirla en tal estado sería
iluso. Por tanto será obligado meter N2 (g) con la llave que está debajo y
después colocar la llave en posición de cerrado.
2. Tomamos el pomo y lo giramos en el sentido de apertura durante unos
instantes. El pomo gira sin que lo haga el plato metálico al que está unido,
justo como lo hace un tornillo. Esto logra abrir un canal donde se alojan
dos salientes que hay en la tapa y permite que, ahora sí, gire la plataforma
entera.
3. Para desprender la tapa hay que girar el disco para que los dos apliques
salgan de las guías ocultas, cuyas entradas son idénticas y se encuentran
opuestas la una a la otra. Cuando sea necesario cerrar la abertura se harán
coincidir los dos salientes de la tapa con los dos rebajes del dintel metálico,
volviendo a girar convenientemente.
Una vez se ha logrado extraer la portezuela, se colocarán los elementos en
el interior de la precámara cuidando los puntos que siguen:
1. Nunca se introducirá un tubo, matraz, bote o recipiente, cerrado, puesto
que al hacer vacío en el interior de la precámara, la presión exterior será
menor que la interior y el vidrio del recipiente reventaría. Si se hubiese
hecho vacío en el interior del recipiente,
entonces sí podría meterse cerrado dado que las dos presiones serían
iguales.
2. Jamás deben meterse agujitas sueltas, pues al cogerlas desde dentro
podríamos pinchar los guantes y pondríamos entonces en peligro toda la

277
hermeticidad de la cámara. Cuando vayan a ser necesarias se introducirán
pinchadas en un tapón de goma, para que su uso resulte totalmente seguro.
3. Al introducir papeles para pesar se fijarán previamente todos entre sí con
una pinza o mordaza, para que al evacuar el gas de la antecámara no se
vuelen por las corrientes de aire (vaciado– llenado).
4. Se pondrán los objetos lo más al fondo posible, para que después, al
recuperarlos desde dentro, se puedan alcanzar con facilidad. Resultará
imposible si los dejamos a mitad de cámara.
5. Antes de cerrar se debe verificar que no falta nada y que se han previsto
todos los elementos necesarios para la operación a realizar porque, una vez
dentro, habría que repetir todo el proceso para meter aquello que se nos ha
quedado atrás.
Al cerrar la tapa de la precámara sólo hay que repetir las operaciones vistas
para su apertura, mas a la hora de hacer rotar el pomo en el sentido de
cierre se pondrá atención para no forzarlo. Se gira hasta que quede
apresado sin necesidad de apretar el pomo, lo cual sería contraproducente
pues provocaría deformaciones que originarían problemas subsecuentes.
Finalmente se debe hacer vacío, para lo cual será necesario conocer el
funcionamiento de la llave que regula estas funciones. Estos elementos se
analizan en la sección inmediata.
27. 3 CICLOS EN LA PRECÁMARA
Como cualquier Schlenk a desoxigenar, el aire de la precámara debe ser
desoxigenado y cambiado por N2 (g). Esto se hace mediante tres ciclos
análogos a los de la línea, pero de una duración muy diferente. La llave se
encuentra generalmente debajo de dicha antecámara y es con esta llave con

278
la que el proceso se ejecuta tal y como se conoce en los experimentos
realizados en la línea.
Las posiciones que rigen las etapas de los 3 ciclos son las que se marcan en
el dibujo:
A) Posición de vacío.
B) Posición de cierre.
C) Posición de llenado
Las precámaras, tanto una como otra, deben dejarse siempre a vacío con lo
que impediremos un contacto directo entre en gas de la cámara seca y el
aire. Al introducir los distintos objetos hemos pasado de la posición A a la
C, como se vio en el apartado anterior. Este llenado se hace lentamente, sin
girar con excesiva rapidez la llave, esperando a que el gas ocupe todo el
recinto. Tras ello se pasa a la posición B, logrando abrir la cámara sin
dificultad. De este procedimiento se deduce que siempre que se comienzan
los 3 ciclos la posición de partida será B.
El primer ciclo comienza con un vacío de unos 10 a 15 minutos, tras lo cual
se pasa a la posición C. Los dos ciclos consecutivos siguientes tendrán
duraciones en su etapa de vacío inferiores a 15, pero siempre por encima de
5 minutos. Realizado el tercer ciclo, hay que tener en cuenta que los
objetos, así como el volumen del recinto, habrán de estar exentos de aire y
ser aptos para ser introducidos en la cámara seca sin peligro para ésta.
Los ciclos de la cámara grande se realizarán empleando los botones del
tablero de mandos. Allí se encuentra uno para hacer vacío y otro para
introducir N2 (g). El tiempo empleado en este caso para hacer el vacío es
mucho mayor que antes, estando en torno a la media hora cada fase
(aunque es evidente que en las dos últimas este tiempo puede reducirse un
tanto).

279
Hecho lo anterior la llave se deja en posición de cerrado B, pudiéndose
proceder a sacar los elementos de la antecámara desde el interior de la
cámara seca.
Dado que los ciclos son largos, uno no puede permanecer delante de la
cámara esperando a que se completen, sino que debe proseguir con otras
tareas. Para indicar a los compañeros que se está usando la precámara, y
por tanto, que no la deben utilizar, se puede seguir un protocolo muy
sencillo y eficaz que consiste en escribir sobre una hoja de papel todos los
nombres de las personas que usan la cámara seca habitualmente, y al lado
se escriben las palabras que indican el ciclo en el que se encuentra el
proceso. Un ejemplo de esto sería el siguiente esquema:
USUARIO 1 PRIMER CICLO
USUARIO 2 ---SEGUNDO CICLO
USUARIO3 TERCER CICLO
USUARIO 4 ---CUARTO CICLO
Sobre los bordes del papel se ajustan dos clips, por ejemplo, uno en cada
lateral. De este modo se marcará qué persona está usando la cámara y en
qué ciclo concreto se encuentra.
En el ejemplo de arriba las líneas hacen el efecto de los clips. La
interpretación del mismo sería que el usuario 4 se encuentra usando la
precámara y que de los tres ciclos obligados es el segundo el que se está
llevando a cabo.
Si sucediese que estuviese en el primero y nos fuese urgente introducir un
objeto en la cámara seca podríamos parar el vacío y abrir la precámara tras
meter el gas inerte, para a continuación volver a poner a vacío el recinto.
Lógicamente conviene siempre avisar al compañero.

280
27. 4 PASAMOS LOS OBJETOS AL INTERIOR
El paso que nos disponemos a explicar resulta una de las experiencias más
interesantes que uno puede tener en un laboratorio de esta naturaleza. La
sensación de aprisionamiento, de presión y falta de presteza, al introducir
los brazos en el espacio de la cámara, es realmente sorprendente.
La protección de los guantes de la cámara seca resulta prioritario por lo que
debemos colocarnos guantes de látex antes de introducir las manos en los
guantes de goma negra que se desarrollan hacia fuera por efecto de la alta
presión interna. Incluso a veces se colocan otros guantes gruesos sobre los
negros, desde el interior, y se dejan allí puestos para mayor protección.
Los guantes son largos como brazos y están vueltos del revés, de modo que
al colocárnoslos quedan al derecho cuando tenemos las manos en su
interior, dentro de la cámara.
El gran problema es precisamente cómo colocarnos los guantes sin
pasarnos más tiempo del debido y sin provocar ningún desajuste en un
sistema que si bien está perfectamente sellado, no está preparado para sufrir
grandes tensiones mecánicas. El proceso es ya difícil por naturaleza, ya que
estos ceden a la menor presión ejercida sobre ellos desde fuera y así al
intentar meter los dedos el guante huye bajo nuestras manos. Sin embargo
aunemos a esto el hecho de que estamos en cierto modo impedidos por los
guantes de látex, por lo que la maniobra es aún más compleja. No queda
otro consejo que el de intentarlo, primero tratando de ajustar los dedos, y
después como nos parezca oportuno.
Al llevar los dos brazos hacia el interior puede notarse cómo la temperatura
es más baja que fuera. La sensación de frío, unida a la de presión, es una
circunstancia desconcertante. Para poder manejar las cosas debemos se
deben ajustar los guantes en el interior de la cámara, ayudándonos con las
propias manos. Lo más cabal es estirar al límite los brazos hacia el fondo,

281
sin miedo a que pueda romperse la goma, lo que contribuirá plausiblemente
a una suficiente adaptación de la goma a la mano y al brazo.
Logrado esto tendremos la sensación de estar tocando un medio denso,
extraño y curioso, por lo que merece la pena pasar por los problemas
anteriores.
Se debe vigilar siempre para no volcar ni romper nada que pueda
encontrarse dentro, ya que la escasa habilidad a la que uno queda reducido
hace que nuestra atención tenga que agudizarse. A continuación se abrirá la
llave de la puerta de la precámara y la tapa se desencaja de la boca. El
proceso es idéntico al descrito anteriormente, sólo que aquí, la escorada
posición de la portezuela hace que nos contorsionemos. Véase al mismo
tiempo que lo que es difícil para un diestro, resulta una hazaña para un
zurdo y esta gran contrariedad debe ser solucionada con paciencia y mucha
destreza adicional.
Se sacan lentamente los recipientes y objetos, volviéndose a colocar la tapa.
Puede ocurrir que en tanto hacemos el intento de introducir ambos brazos,
la circulación de la cámara se pare. Si algún compañero estuviese cerca
debemos pedirle que la conecte de nuevo mediante el botón de la tabla de
mandos. Si no fuese así esperaremos a terminar y después la conectaríamos
nosotros.
27. 5 PESAMOS UN COMPUESTO
La balanza que hay en el interior de la cámara seca está destinada a pesar
compuestos tan reactivos que ninguno de los métodos observados en la
sección 7.2.1 es apropiado. El método de pesada no es diferente al usado en
atmósfera normal, sólo que aquí se extremará el cuidado, para no derramar
el sólido sobre la mesa de trabajo.

282
La principal dificultad es la electricidad estática que aparece en la
superficie de los objetos, dificultando el manejo, sobre todo de los papeles
de pesar. Los papeles encerados se adhieren entre sí y tratar de separarlos
enfundados en guantes es complejo. Al mismo tiempo se constata que
también los granos de compuestos se pegan entre sí y son mucho menos
manejables que antes, por lo que si bien la pesada es más sencilla desde el
punto de vista operacional no lo es en lo que a su ejecución se refiere.
27. 6 LIMPIEZA DE LA CÁMARA SECA
Ya se ha comentado que la cámara debe mantenerse limpia, por lo que tras
su uso es obligado recoger cualquier producto que se haya podido derramar
en la misma; así como ordenar su interior, procurando dejarlo en el mismo
estado en el que se encontró.
La cámara seca es común para todos los investigadores del laboratorio, a
veces incluso común para todos los investigadores del centro, lo que hace
que esté sometida a un uso continuo. Si las medidas de limpieza no se
extreman, pronto se acumularía la suciedad en un lugar tan habitual para
todos.
Para lograrlo suele encontrarse en el interior un rollo de papel y una
brocha, que no resulta muy útil como consecuencia de la electricidad
estática, pero que se deben emplear constantemente para mantener la
cámara en un estado aceptable. Después, todos los papeles sucios se
sacarán por la precámara, junto con el producto pesado.

283
27. 7 SACAMOS EL PRODUCTO PESADO
Cuando el producto se ha pesado y se concibe la operación como
terminada, se coloca un tapón de goma nuevo, que se debe haber
introducido de antemano, en el matraz o Schlenk en el que se haya
depositado el producto. Se vuelve a continuación a abrir la antecámara
desde dentro (operación en todo idéntica a la ya explicada) y a alojar en su
interior todos aquellos elementos que han se sacarse. Se cierra y se procede
a sacar los brazos de los guantes, lentamente.
Al ir extendiendo los guantes, observaremos cómo éstos quedan
impregnados de gotitas, a causa de la transpiración cutánea, dada su
absoluta impermeabilidad. Ya librados los brazos conviene, con un poco
de papel, secar la superficie de los guantes (aunque únicamente sea para
que no queden con mala presencia).
Igualmente nos podemos deshacer de los guantes de látex y podemos abrir
la precámara para extraer los objetos. Tras ello, imponemos vacío por
medio de la llave y se cumple así con la obligatoriedad de dejar la
antecámara a vacío.
Finalmente nos cercioraremos de si la cámara seca está con la recirculación
activada, conectándola en caso contrario. También habrá que mover los
clips de los bordes de la hoja, indicando que no está siendo usada.
27. 8 USO SIMULTÁNEO DE LAS DOS PRECÁMARAS
Puede ocurrir que encontramos la antecámara grande desoxigenándose.
Debido a su gran volumen, el tiempo que se tarda en lograr la completa
eliminación del aire es muy largo, por lo que si vamos a usar la cámara

284
pequeña debemos saber cómo manejar esta última sin afectar al proceso de
vaciado de la otra.
Los botones que controlan la precámara grande están en el tablero de
mandos, por lo que tendremos que recurrir a ellos.
La situación será la siguiente: ambas cámaras están a vacío, empero se
quiere que la de mayor volumen continúe igual, en tanto la pequeña se
llena con N2 (g) para abrirla posteriormente.
1. En primer lugar se tiene que cerrar la conexión existente entre
las cámaras. Esto se hace apretando el botón llamado
“antecámara” con intención de desconectarlo.
2. Ahora se puede abrir el N2 (g) con la llave y llenar la precámara
pequeña, sin que por ello se llene la grande también.
3. Se coloca la llave en posición de cerrado y se abre la tapa,
introduciendo posteriormente lo que sea menester. Tras cerrar se
repone el vacío.
4. Se conecta nuevamente el botón “antecámara”, volviendo a la
situación de partida y haciendo nuevamente vacío en ambos
recintos.
5. Como deben realizarse 3 ciclos, cada vez que se deba introducir
gas inerte, se cerrará la conexión entre las cámaras, tal y como se
ha visto.
28 DESECHOS
Sobre aquellos materiales que dejan de ser útiles debemos ser cuidadosos,
de modo que su eliminación sea adecuada, económica y respetuosa con el
medio ambiente. Con tal intención se busca clasificar los desechos para

285
poder recuperar la mayor parte posible de los mismos que un proceso de
reutilización.
28. 1 LOS DISOLVENTES
En disolventes se establecen 4 categorías básicas que se pueden observar en
cualquier laboratorio de química: orgánicos no clorados, orgánicos
clorados, ácidos y bases.
En el caso particular de un laboratorio de compuestos organometálicos la
presencia de los dos primeros es indispensable, ya que los otros apenas se
usan.
Hay pues dos garrafas o bidones de gran tamaño, cada uno con un letrero
indicativo de su contenido. En ellos tenemos la obligación de echar
aquellos disolventes que han sido usados según corresponda.
Es importante cerciorarse bien de la naturaleza de lo que se procede a tirar,
y no confundirnos entre unos y otros. Si no recordamos qué era
exactamente, es más conveniente arrojarlo en el bidón de clorados que en el
otro.
Los contenedores suelen estar en el suelo, por lo que al echar el líquido en
el interior, nuestra cabeza suele situarse justo encima de la abertura.
Cuidémonos de que esto no sea así y apartar la cabeza oportunamente, dado
que las emanaciones no pueden ser muy beneficiosas (además de impedir
hipotéticos accidentes por reacciones violentas e inesperadas entre el
contenido y el líquido que se elimina).
En cuanto a los disolventes de carácter ácido o básico se pueden arrojar por
el sumidero. Sea o no conveniente así se hace, porque su cantidad es
mínima y siempre se emplea gran volumen de agua para disminuir en lo

286
posible la concentración de los mismos con lo que la peligrosidad
disminuye enormemente.
28. 2 DISOLUCIONES
Las disoluciones que resultan inútiles también se hacen desaparecer en
función del disolvente. Aquellas disoluciones con solventes clorados se
tirarán en el bidón respectivo, en tanto aquellas cuyo disolvente sea no
clorado se vaciarán en el paralelo.
Algunas veces nos vemos precisados a tirar una pequeña cantidad de aguas
madres (a la hora de quedarnos con los cristales, como se vio en 21.6)
cuando se considera que en ellas nada queda del producto, tras varias
cosechas de cristales. Dicho volumen se elimina mediante una cánula filtro
cuyo extremo agudo se clava en el tapón de goma de un erlenmeyer
destinado a la recolección de tales residuos.
El erlenmeyer citado está tapado por un tapón rojo de goma de gran
tamaño, afirmado por gomillas al estilo conocido, y en su interior almacena
día tras día pequeños volúmenes de desechos hasta que se completa su
capacidad. Este erlenmeyer se reconoce fácilmente en toda vitrina de un
organometálico, a causa de la pátina especular de metal, proveniente de los
productos descompuestos, que se observa sobre las paredes de vidrio del
recipiente.
Una vez lleno se tira su contenido en el bidón de clorados, pues es probable
que tenga cierta cantidad de disolvente halogenado.

287
28. 3 SÓLIDOS
En general son pocos los productos sólidos que se eliminan. Normalmente
son sustancias que no han cristalizado y que permanecen en el fondo del
Schlenk con un aspecto a veces polvoriento, a veces oleoso y denso. Lo que
se hace habitualmente es disolverlos con el disolvente apropiado y después
arrojarlos en el bidón correspondiente al disolvente empleado.
No obstante tenemos que indicar que los lechos usados para filtrar, tales
como la celita, la sílice,... se desechan y se tiran directamente en las
papeleras del laboratorio.
28. 4 RECUPERACIÓN POR DESTILACIÓN
La gran mayoría de los disolventes que constituyen la mezcla del bidón de
disolventes no clorados no son otra cosa que acetona y éter. Resultará pues
absurdo no recuperarlos mediante una destilación sencilla que separaría
fácilmente ambos compuestos, ya que sus temperaturas de ebullición distan
más de 20º C.
Igualmente podemos afirmar que la casi totalidad de disolvente clorado del
otro contenedor será diclorometano, por lo que se debe recuperar y se
puede volver a emplear.
28. 5 COMPUESTOS PELIGROSOS
Considérese que hay compuestos cuya toxicidad implica un trato especial
en cuanto a su eliminación. Según su naturaleza podemos definir dos líneas
de trabajo:

288
A) Previamente hay una desactivación o neutralización del peligro
potencial, para que posteriormente se pueda eliminar
convenientemente (esto tendrá sentido para aquellos compuestos
que se puedan realmente destruir).
B) Se procede a un confinamiento del producto (ya sea para
posterior transformación o para un almacenamiento
prolongado).
Entre otros compuestos tenemos los metales alcalinos usados en el secado
de productos, los cuales deben ser desactivados, transformándolos en
sustancias manejables que puedan desecharse sin problema (sección 5.2.7).
El mercurio también debe ser tenido en cuenta, y en lugar de tirarse vale
más purificarlo por destilación cuando éste deje de encontrarse a la pureza
necesaria (sección 2.2.6).
Un ejemplo de compuestos confinados para almacenamiento son los
derivados de los metales radiactivos. El trabajo con uranio es muy delicado,
y la limpieza será la línea central a lo largo de todo experimento. No se
puede derramar producto, que podría quedar en
los rincones durante largo tiempo y resultar peligroso allá donde esté, a
causa de su radiactividad. Esto conlleva la periódica limpieza de la vitrina y
de la línea, que han de montarse y desmontarse, de modo incansable una y
otra vez. Los residuos obtenidos se confinan y almacenan por las vías
legales pertinentes.

289
28. 6 COMPUESTOS GASEOSOS
La extracción del aire existente en las vitrinas, que protege al científico de
las emanaciones de los productos, marcha por tubos especialmente
diseñados que acaban abocando a la atmósfera.
28. 7 MATERIAL DE VIDRIO ROTO
Resulta interesante esta última anotación sobre desechos, referida a
cristales y vidrios rotos. Durante el trabajo cotidiano suelen romperse
desgraciadamente algunos materiales de vidrio. Para su almacenaje hay un
recipiente exclusivo, en el cual se arroja cualquier vidrio que se parta.
Sin embargo, muchas veces se rompen tubos, matraces e instrumentos, de
modo que quedan intactas las bocas de los mismos u otro elemento sobre el
cual se puede volver a formar el resto del objeto. En ese caso se guardarán
dichos trozos importantes para que puedan volver a emplearse tras el
arreglo correspondiente.
Estas reparaciones de reconstrucción son realizadas por empresas
especializadas. Cuando se tiene un instrumento de vidrio roto se les envía y
éstos se encargan de dejarlo en condiciones óptimas para ser reutilizado.
Esta opción es mucho más rentable que comprar el instrumento o el
recipiente de nuevo.

290
29 OPERACIONES DE LIMPIEZA
A lo largo de las experiencias el material de vidrio se va ensuciando, siendo
necesario un lavado al final de los experimentos que elimine la suciedad y
puedan ser usados de nuevo. Esta operación es cotidiana y cada día obliga
al químico a unos minutos de limpieza frente a la pileta.
Las operaciones de limpieza no sólo son necesarias para los matraces y los
Schlenk, sino también para todo el laboratorio, y como se verá se requiere
un continuo esfuerzo en el que el orden constituye uno de los principales
argumentos.
29. 1 LAVADO DEL MATERIAL SIN GRASA
En general, es un lavado de materiales sin mucha suciedad y sin ajustes
engrasados. El lavado se hace en este caso como se ha venido haciendo a lo
largo del manual.
1. Se enjuaga con un poco de acetona para eliminar cualquier
residuo que pudiera ser reactivo con el agua.
2. Se friega con algo de agua y jabón si se quiere, incluso con
estropajo o con un limpiatubos (es una varilla de alambre de la
que salen hebras y fibras radialmente). Al usar el limpiatubos
hay que tener en cuenta su extremo inferior, ya que muchas
veces no tiene fibrilla, y un mal golpe puede desfondar un
matraz. Tras esta operación se aplica agua para eliminar la
espuma.

291
3. Si se quiere eliminar alguna sal que se encuentre pegada en las
paredes del matraz o del recipiente se empleará ácido
clorhídrico. Posteriormente se lava nuevamente con agua.
4. Se seca externamente con papel de laboratorio (el cual suele
encontrarse colocado en un armazón sobre la pileta) y se lava
con acetona y éter. Con la acetona se elimina cualquier
compuesto orgánico que aún quede en el matraz, y con el éter, al
margen de ser un buen disolvente orgánico, se logra un secado
más efectivo del interior del tubo.
5. Finalmente se deposita el matraz en la estufa para que se seque.
29. 2 LAVADO DE MATERIAL CON GRASA
Aplicable también a todos aquellos objetos que se encuentren muy sucios o
que no se disponga del tiempo suficiente para limpiarlos. En este proceso
se requiere un baño alcalino, que se prepara y se usa durante meses, sin
necesidad de tener que preparar uno nuevo. Este baño sirve para eliminar
cualquier resto de grasa de ajuste de las llaves, y su efectividad para
disolver grasas es tan elevada que un contacto con la piel, aunque breve,
puede ocasionar quemaduras graves y dejar cicatrices en las manos de por
vida.
29. 2. 1 EL BAÑO DE POTASA ALCOHÓLICA
En un cubo grande con tapadera, se dispone un lecho de lentejas de potasa
(KOH) y sobre él se añade una pequeña cantidad de agua. Después se

292
dispensa generosamente el alcohol hasta cubrir el volumen exigido. Las
proporciones adecuadas por litro de etanol son:
120g de KOH o de NaOH
120ml de agua
El agua es necesaria para poder solubilizar la base en el alcohol con mayor
facilidad. Las cantidades están calculadas para mantener una disolución
saturada de potasa, lo que conviene al proceso de eliminación de grasas.
La reacción de limpieza no es más que una saponificación o hidrólisis
básica de las grasas, que pasan al medio disueltas en alcohol. El uso del
alcohol es requerido para permitir una mejor asimilación de los productos
obtenidos en su seno en comparación con el agua.
El principal problema del etanol es su volatilidad, lo que fuerza que se deba
renovar continuamente la parte evaporada, para que no se tenga un
volumen excesivamente bajo. Una solución es el uso del isopropanol como
sustituto, lo cual se hace en algunos sitios, pero por el contrario tiene la
desventaja de dejar los vidrios jabonosos y resbaladizos.
El material de vidrio no se debe dejar más de una noche en la mezcla
alcohólica pues es atacado con el tiempo por los agentes alcalinos. Así se
deberá proceder diariamente al lavado de los objetos, cada mañana.
Con el tiempo, la coloración de la disolución cambia de la tonalidad
blancuzca, algo translúcida del principio, hasta un color rojizo casi negro.
29. 2. 2 TRAS EL BAÑO
Debido a que en el baño se introducen objetos correspondientes a todos los
investigadores es importante establecer un orden o un turno para lavar cada

293
mañana nada más se llega al laboratorio. Lo habitual es establecer turnos de
una semana. De este modo durante toda una semana es una persona la
encargada de limpiar los objetos que están dentro de la mezcla, y a la
siguiente otra persona llevará a cabo el proceso, entonces la operación no
se hace demasiado pesada cuando el número de investigadores es alto.
Para introducir o coger los recipientes del baño hay que protegerse
debidamente las manos mediante guantes de plástico grueso. Sin embargo,
con el uso cotidiano acaban picándose y pueden darnos una sorpresa muy
desagradable al permitir el paso de la disolución a través de alguno de los
agujeros. Se aconseja que nos pongamos previamente guantes de látex, y
encima los otros, porque así podremos darnos cuenta a tiempo si hubiere
alguna picadura. Repito que la disolución es más dañina de lo que parece a
priori.
Una vez sacados todos los objetos y depositados sobre la pileta, se procede
a lavarlos con profusión con agua del grifo. El grifo debe poseer una goma
conectada que permita dirigir el chorro hacia donde parezca más
conveniente, siendo muy útil esto a la hora de limpiar el interior de los
tubos y matraces.
Cuando ya están lavados se tratan con ácido clorhídrico al 32%, para
neutralizar la basicidad anterior. Se recuerda que todo el proceso de lavado
se lleva a cabo con guantes, por el peligro que conllevan los disolventes
usados.
Hecho esto se vuelven a enjuagar con agua para quitar los restos ácidos que
permanezcan en el vidrio.
Tras ello se secan con el papel y se lavan con acetona y éter, para
finalmente ponerlos a secar en las estufas.
En el proceso se puede hacer uso del jabón, estropajo y limpiatubos si su
concurso fuere necesario para una limpieza completa.

294
29. 3 LAVAMOS RESTOS DE METALES NOBLES
Los metales nobles que se usan normalmente para formar gran cantidad de
compuestos organometálicos pueden quedar fijados al descomponerse los
compuestos en donde están, en las paredes de los matraces de reacción, en
forma de pátina metálica cuya eliminación es imposible por los métodos ya
comentados. En general son residuos negruzcos, que se encuentran en el
fondo de los matraces y de los Schlenk, y que algunas veces dejan su marca
en forma de mancha oscura la cual es importante eliminar, pues puede
catalizar la descomposición de otros compuestos que puedan ser contenidos
con posterioridad. Los metales más usados en la química organometálica
que requieren este tratamiento son el Pd y el Pt.
El agua regia es el reactivo más efectivo para disolver estos metales gracias
a la formación de complejos de coordinación donde participa el ion cloruro,
que permite la oxidación del metal en estado elemental.
Para hacer agua regia se ponen HCl (ac) y HNO3 (ac) en proporción 3:1 en
volumen, en un recipiente que se guardará en un rincón de la campana para
ser usado cuando corresponda.
Esta disolución es peligrosa y debe ser manejada con guantes gruesos. Se
dispensa por medio de una pipeta Pasteur que posee una pequeña perilla
que sirve para succionar y expulsar el líquido. Se lava con pequeñas
cantidades, agitándose con tranquilidad y firmeza, siempre dentro de la
vitrina.
Para no mezclar el agua regia limpia con aquella que ya se ha usado, y que
por tanto está contaminada, suele disponerse de otro botecito que se debe
encontrar junto al que contiene la disolución de limpieza, para recibir el
ácido empleado.

295
Cuando al recipiente a limpiar se le ha eliminado la capa de metal noble se
lava bien con agua y se incorpora entonces a la cadena de lavado habitual
que concluye con el uso de la acetona y el éter.
29. 4 SOBRE LOS DISOLVENTES EMPLEADOS
1. El agua utilizada es agua del grifo, no se considera necesario usar agua
destilada.
2. El ácido clorhídrico al 32%, aludido en todas las secciones de lavado no
es sino agua fuerte tradicional, también llamado salfumant, que se expende
en las tiendas como artículo de limpieza. Obviamente también puede usarse
uno de casa comercial especializada, pero resulta más económico el
anterior.
3. La acetona y el éter de lavar son suministrados por el mismo servicio que
los retira, destila y recupera. No son de gran calidad, es decir, no pueden
usarse como reactivos, pero para lavar son suficientes.
4. El agua regia es preparada por el propio químico, haciéndose en
pequeñas cantidades, que suelen durar mucho tiempo en un ritmo normal
de trabajo.
29. 5 LIMPIEZA DE LAS VITRINAS
Como puede suponerse, de cuando en cuando es necesario limpiar la
vitrina. Esto consiste en la ordenación de los tapones de goma, gomillas,
tapones de teflón y todos los adminículos necesarios para las operaciones
normales del laboratorio.

296
Se cambiará también el pliego de papel secante que suele cubrir la mesa de
la campana y que retiene la suciedad y los productos esparcidos en su
interior. Al hacer esto se aprovecha para eliminar con una brocha aquellos
restos que puedan haber quedado en los resquicios de la propia vitrina.
El lavado de la línea propiamente dicha se realiza cada vez que presente
suciedad patente mediante los métodos detallados en 2.2, así como el
cambio de gomas que será precisado cada cierto tiempo.
29. 6 LIMPIEZA DEL LABORATORIO
Al margen del buen hacer del personal de limpieza, es conveniente ordenar
el laboratorio tras una semana de trabajo. Por tanto es una costumbre muy
sana dejar el laboratorio en perfecto estado cada viernes por la tarde al
cesar nuestro trabajo diario.
Objetos de especial cuidado son las balanzas, sobre las que los productos
pueden derramarse, quedando en su superficie. Aún cuando veamos este
hecho y lo limpiemos, siempre habrá otros tantos que se nos pasen
inadvertidos, siendo obligado hábito la limpieza de las mismas un día por
semana.
30 SE PONE UNA REACCIÓN
En los capítulos precedentes no hay ejemplos concretos relativos a
reacciones químicas. Esto se ha hecho a conciencia, para ahora presentar
una reacción, que en modo alguno pretende ser prototípica, pero que
permite ilustrar punto por punto las distintas fases por las que atraviesa el
químico organometálico durante su ejecución.

297
Cada reacción es distinta a cualquier otra, y debe estudiarse siempre
independientemente, aunque existen reacciones análogas gracias a las que
se pueden extraer informaciones muy interesantes para desarrollar el campo
de investigación en compuestos afines. Sin embargo, en líneas generales, lo
que para una reacción resulta válido, para una segunda puede ser
inadecuado, de modo que no se puede hablar de un modelo general de
reacción, ni de condiciones generales absolutas. Teniéndose en cuenta todo
ello, describiré a continuación los pasos necesarios para la obtención de un
producto a partir de los reactivos en una reacción determinada que servirá
dentro de lo que cabe, para hacernos una idea del proceso conjunto.
La reacción que nos interesa es la siguiente:
K[(η2-Tp)Pd(CH2)4] + NClS ------- (η
3-Tp)ClPd(CH2)4
Donde el compuesto de partida es un metalaciclo algo más complicado del
que he escrito en la reacción. Un metalaciclo es un metal coordinado en
forma de quelato con una cadena carbonada, de modo que se establece una
estructura especialmente estable. Coordinado también al paladio en el
compuesto de partida se encuentra el ligando hidrotrispirazolilborato o Tp
unido como ligando bidentado (dihapto). El agente halogenante es la N-
clorosuccinimida, que se usa habitualmente en reacciones de sustitución y
que en la reacción se representa por NClS. El compuesto resultante es muy
parecido al de partida sólo que aumenta su coordinación a octaédrica en
una adición oxidante, pasando el cloro a ser un ligando más y el
hidrotrispirazolilborato a coordinarse como trihapto.

298
Para llegar a este proceso tendremos que partir de un compuesto muy
usado, que es la base para un gran número de compuestos posteriores. Este
compuesto se denomina comúnmente Pd neofilcod o neofilcod (no se darán
los nombres sistemáticos de los compuestos dado que no se usan en el
laboratorio sino tan sólo en la propia tesis, que está sometida al rigor
científico, cosa que no ocurre con el lenguaje del químico en el trabajo
cotidiano). En el laboratorio el nombre de los compuestos suele
apocoparse, usando fragmentos del mismo, que aluden a los ligandos la
mayoría de las veces. El neofilcod posee un ciclooctadieno coordinado por
enlace π al Pd. Es este ciclooctadieno el que hay que desplazar para que en
su lugar entre el hidrotrispirazolilborato.
Con la intención de formar el reactivo base de la reacción expresada
anteriormente se toma el neofilcod y el ligando, que como se ha detallado
en la reacción anterior es el hidrotrispirazolilborato.
El hidrotrispirazolilborato tiene como átomos donadores nitrógenos con
capacidad de coordinación, pudiéndose unir por dos o tres de los mismos,
dependiendo de la deficiencia electrónica del centro metálico.
La formación del reactivo de nuestra reacción se llevará a cabo mediante la
sustitución del ciclooctadieno del neofilcod por un hidrotrispirazolilborato:
1. El Pd neofilcod es un compuesto organometálico de aspecto polvoriento
y color pardo grisáceo. Se guarda a baja temperatura en un tubo de Schlenk
de gran cabida, bajo atmósfera de N2 (g). Se puede pesar al aire, siempre y
cuando se vuelva a almacenar bajo atmósfera inerte a continuación. Se
pesará por tanto y se dispondrá en el matraz de reacción conjuntamente con
un ratón. El matraz se protegerá con nitrógeno tras los ya conocidos 3
ciclos. Para este punto nos remitimos a la sección 7.2.1.
2. El ligando Tp se encuentra en forma de sal potásica, formando cristales
blancos que se manejan con facilidad al aire, sin necesidad de almacenarlos

299
bajo atmósfera artificial. Una vez pesada la cantidad requerida se deposita
en un Schlenk (a ser posible de los de llave de teflón) y se le pasarán los 3
ciclos. Hecho esto se dejará bajo N2 (g).
3. Nunca se mezclan dos sólidos para después disolverlos conjuntamente,
sino que se disuelve uno (o los dos separadamente) y después se mezclan
(en frío o a temperatura ambiente, según sea la reacción). Teniendo en
cuenta lo anterior se disolverá la sal potásica en acetona (usando un ratón
igualmente) en el Schlenk.
4. En tanto se disuelve se van cambiando los tapones del Schlenk y del
matraz, para después pasar mediante una cánula el contenido del tubo al
reactor donde la primera de las reacciones dará lugar bajo agitación.
5. Hay que considerar que en este caso no se ha empleado baño frío, pues
ya se conoce el mecanismo de la reacción y se sabe que no es necesario. Si
lo fuese se disolverían ambos reactivos (al tiempo que se aprovecha para
hacer el baño frío) y después se pondría a enfriar el matraz.
6. La reacción es de sustitución que apoyada por la agitación, se viene a
terminar rápidamente. De este modo, tras una hora esta fase puede darse
por concluida. La sal del metalaciclo se habrá entonces obtenido sin
excesivos problemas. La disolución habrá experimentado un levísimo
cambio de color que va del tono translúcido del inicio hasta una tonalidad
algo más pardusca.
7. En tanto evoluciona la disolución anterior vamos preparando la siguiente
parte que consiste en la disolución de la cantidad adecuada de N-
clorosuccinimida. Este compuesto es estable al aire, posee una tonalidad
blanca, un tanto amarillenta, y presenta aspecto cristalino.
Tras pesarla se disuelve en un poco de acetona y se espera a que la reacción
anterior se concluya.
8. Ya acabada la reacción de sustitución se pone en un baño frío de
acetona-CO2 durante unos 15 minutos. A continuación se procede a hacer

300
gotear el otro reactivo (que debe dispensarse muy lentamente) por
cualquiera de los métodos explicados en 13.2.3.
Esta reacción es una adición oxidante en la que la esfera de coordinación
del Pd pasa de 4 a 6 en el número de coordinación, y así el compuesto de
16 a 18 electrones.
9. El baño frío se mantiene durante otro cuarto de hora. La reacción se
continúa durante unas tres horas en la que la disolución se torna de un
amarillo limón muy hermoso.
10. Al obtener el TpCl (así es como se llama familiarmente al producto
formado, que posee los ligandos Cl y Tp en su esfera de coordinación) se
lleva la disolución a sequedad tal y como se vio en la sección 15.1.
11. Para extraer el producto lavaremos con diclorometano (donde el
producto es muy soluble) y filtramos según se hizo en 16.
12. Se vuelve a hacer vacío y se lleva a sequedad, pesando para calcular el
rendimiento (se supone que no se ha generado ningún producto paralelo
que permanezca en disolución; si lo hubiese habría que separarlo y volver a
empezar de nuevo con las operaciones de separación).
13. De no estar puro suficientemente, se puede pasar por columnas o por el
cromatotrón, según convenga.
14. Se trata finalmente de cristalizar el compuesto para los análisis
posteriores tal y como se vio en 21.
Datos cuantitativos para una reacción a escala de medio mmol:
0.5 mmoles Pd neofilcod ---------------------------- 173 mg.
0.5 mmoles KTp -------------------------------------- 126 mg.
0.5 mmoles N-clorosuccinimida -------------------- 67 mg.

301
Se obtienen 0.5 mmoles que resultan 230 mg de TpCl, o sea, un
rendimiento cuantitativo.
31 INSTRUMENTAL DE TRABAJO (IV)
31. 1 MATRACES DE DOS Y TRES BOCAS
Aunque ya han aparecido a lo largo del volumen estos recipientes, es justo
tratarlos de manera independiente pues su importancia es grande en el
trabajo diario del laboratorio. Se utilizan para llevar a cabo reacciones a
gran escala, aunque también existen de pequeño volumen, o en aquellas
reacciones donde se tenga que añadir cada cierto tiempo reactivos por una
de sus bocas, colocar un reflujo,... Se usan también en procesos tales como
las destilaciones.
El matraz de tres bocas es un matraz de fondo
redondo donde se han dispuesto dos aberturas
adicionales a los lados de la principal. Esta apertura
central se destina generalmente al refrigerante de
reflujo, en tanto que las otras dos se usan con otras
finalidades o bien se cierran mediante tapones de goma.
El matraz de dos bocas es también muy útil, usándose
en procesos donde es necesario reflujo y
administración de algún reactivo por el lateral. Se
suelen usar matraces de reducido volumen, en tanto los
de mayor capacidad tienden a ser de tres bocas.

302
En definitiva son matraces de reacción cuya aplicación varía con respecto
al matraz usual en tanto en cuanto aquí se permite instalar un reflujo e
incluso trabajar con volúmenes de reacción claramente superiores que los
empleados en los matraces habituales. Las condiciones de control de
atmósfera se logran con accesorios nuevos que todavía no han sido
detallados y que vienen a analizarse en este mismo bloque. Otras funciones
interesantes ya han sido vistas en 18.3, donde se ha empleado una variante
de 2 aberturas, cuya boca lateral poseía rosca para permitirle la conexión
con un tapón de teflón.
31. 2 SOPORTES PARA FONDO REDONDO
Son elementos que sirven para hacer estanco un matraz y que no se rompa
ya que debido a su forma, los matraces son susceptibles de rodar y
quebrarse al primer golpe. Para evitar esto existen soportes cuyo modelado
cóncavo complementa perfectamente la convexidad del matraz. Los hay de
diferentes tamaños para cubrir toda la gama de matraces.
Los más comunes son de corcho y aquí se pretende representarlos en una
vista lateral y otra cenital. El corcho presenta el color marrón típico, por lo
que no resultará difícil identificarlos en un laboratorio.

303
Nótese cómo el soporte es un cilindro cuya superficie superior se va
rebajando de manera que se hace cóncava, hasta terminar en un hueco
cilíndrico, coaxial con el anterior.
En este segundo hueco un instrumento con fondo redondeado y del tamaño
adecuado (ampolla) se podrá estabilizar sin problemas.
Además de este tipo de soportes existen otros de distintos materiales: los
hay de goma, que tienen una buena capacidad de adhesión y la ventaja de
aislar térmica y eléctricamente (al igual que el corcho); igualmente
encontramos los de silicona, con las mismas prestaciones, pero con un
rango de temperaturas más elevado:
Goma: -20 a 70ºC (calor seco) y -20 a 120ºC (calor húmedo)
Silicona: -20 a 200ºC
31. 3 LLAVES DE CONEXIÓN INDEPENDIENTE
Las llamo independientes porque su uso no está restringido a un matraz en
concreto sino que puede extenderse a todos aquellos de 2 o 3 bocas cuyo
diámetro de apertura sea adecuado para el macho de la llave.
Este tipo de elementos, como ya
se ha podido intuir, sirve para
conectar a la línea los matraces
tipo 31.1. Es, así pues, una
alternativa cuando el matraz no
dispone de boca roscada donde
pueda colocarse una llave de
teflón (véase 18.3).

304
En el dibujo puede verse que el aplique está constituido por un cuerpo
hueco que se conecta mediante una llave a la boquilla, la cual se introduce
por el tubo de goma de la línea.
La conexión con el matraz se logra poniendo un poco de grasa en el
elemento de ajuste, para después afirmarlo como es habitual por rotación y
concluir por apresarlo gracias al concurso de las gomillas. Sin embargo hay
otros elementos que permiten una unión segura tal y como se verá más
adelante.
Los modelos existentes varían su tamaño y forma, aunque básicamente son
iguales al descrito, mas lo verdaderamente importante es el tamaño del
ajuste que determinará en qué matraz podrá o no ser usada.
31. 4 COLOCACIÓN DE LAS GOMILLAS
Esta operación, siempre delicada, debe realizarse con una gomilla doble o
con dos simples, dependiendo también del tamaño de la llave. En el cuello
de la salida del matraz suele haber arrollado un alambre tal que en sus dos
brazos alineados puedan afirmarse las gomillas que abrazan la llave.
La gomilla se engancha primero a uno
de los dos brazos de alambre y se estira
con fuerza para hacerla pasar por la
parte superior de la boquilla.
A continuación, llevando la goma
elástica por detrás de la llave se
alcanzará el otro brazo, donde se

305
colgará la goma nuevamente.
Con este sistema la llave queda fija y sin peligro de que pueda salirse en el
transcurso de la operación o de que existan fugas por un mal acoplamiento.
31. 5 EL EMBUDO DE ADICIÓN DE REACTIVOS
Muy a menudo hay que añadir un reactivo líquido a un matraz de 2 o 3
bocas. En general este reactivo puede adicionarse por los métodos
indicados en el bloque correspondiente, pero pudiera ser interesante una
alternativa en algunos casos, por motivos de comodidad.
El reactivo deberá ser estable al aire para que el método se pueda usar.
También está indicada esta clase de adición cuando se tenga que realizar a
goteo, no de una vez, y el volumen sea tal que resulte impráctico realizarlo
con una jeringa.
Hay que considerar que las reacciones pueden exigir distinta frecuencia de
goteo de reactivos durante el transcurso de la reacción, por lo que un
embudo que permita esta regulación resulta muy útil.
El líquido a dispensar se añade por la boca del embudo, teniendo el brazo1
cerrado por un tapón especial, largo y delgado, que sella perfectamente la
salida del contenido. A medida que se desatornilla el tapón, el líquido cae
por el sifón diseñado para ello hasta que acaba en el matraz. La frecuencia
de goteo, y por tanto la de caída de reactivo, estará determinada por el
espacio libre que deja la espiga del tapón.
La conexión con el matraz es idéntica a la vista para el elemento anterior.
1. Brazo de vidrio que conecta el depósito del reactivo con el matraz
mediante el canal que se aprecia en la figura.

306
2. Tapón de teflón cuyo
cilindro es largo y más fino que
el del tapón de teflón visto para
la ampolla. Este tapón se
conecta a la boca del brazo 1
por roscado y a medida que se
atornilla cierra la salida de
reactivo, lográndose así un
control del flujo de caída del
mismo.
3. Es la boca del depósito donde se coloca el reactivo. Es un cilindro de
vidrio cerrado por debajo, excepto por la salida al brazo 1, que será por
donde escaparán las gotas de reactivo a la frecuencia necesaria.
4. Brazo de vidrio que conecta el matraz con el depósito (3). Su misión es
la de impedir que el aire entre en el sistema y que el líquido contenido en 3
absorba aire en tanto se agota. Aunque el reactivo a adicionar sea estable al
aire, no se puede permitir que con él baje al seno de la disolución una cierta
cantidad de oxígeno y vapor de agua. De modo que este canal aporta el
flujo de N2 (g), que proviene del matraz de reacción, impidiendo que entre
aire dentro del sistema.
5. Ajuste de conexión entre la boca del matraz y el extremo inferior del
embudo. Sobre él será donde extendamos la grasa de ajuste.
31. 6 COLOCAMOS LAS GOMILLAS
Al manejar el embudo hay que tener mucho cuidado con el brazo 4 ya que
por su fragilidad puede partirse a la menor tensión.

307
La gomilla se enlaza simple, ya que
doble no alcanza al otro extremo
dadas las dimensiones del embudo.
Se engancha en un extremo del
alambre para después hacerla llegar
al segundo brazo tras rodear la
unión entre el depósito y el brazo 1.
En la imagen puede apreciarse
cómo la gomilla pasa por ese
segmento de vidrio.
Si se pretendiera hacer lo mismo, pasando el elástico por el espacio que
deja la vía 4 al unirse con 3, podría quebrarse el fino conducto, dada la
tensión de la gomilla.
Esta colocación de las gomillas sirve para afirmar el embudo en el hueco
del matraz y permite una adecuada dosificación del reactivo, asegurando
así el buen éxito de la reacción.
31. 7 EMBUDO DE PRESIÓN COMPENSADA
Cuando se desea poner bajo N2 (g) un sistema voluminoso con salidas
varias de gas inerte (véase el montaje en la formación de un Grignard) es
indispensable colocar un embudo de presión compensada para que el gasto
de gas sea mínimo, así como para mantener una presión interior aceptable.
La presión interna se controla mediante una parafina, que se coloca en uno
de los bulbos donde burbujeará el N2 (g). Cuando la presión es demasiado
alta el burbujeo impide que los elementos del resto del sistema peligren, ya

308
que la parafina limita la presión a un valor fijo. A veces cuando la corriente
es muy grande se aconseja incluso colocar en el extremo 7 un tapón de
goma con una aguja que reduce la salida torrencial del gas.
Se suelen emplear colocados sobre reflujos, dejando a presión alta de gas
inerte el conjunto del sistema. Son indispensables en aquellas reacciones
organometálicas que conllevan utilización de refrigerantes de reflujo y que
deban realizarse en condiciones de absoluta ausencia de O2 (g) y de H2O
(g).
1. Entrada del N2 (g) y así conexión con la goma de la línea. El tubo de
goma se debe insertar con cuidado, ya que el embudo es muy frágil y
delicado.
2. Cámara donde entra el gas proveniente de la línea y que lo dirige hacia
abajo, de tal modo que se extiende por el reflujo e impide la entrada de aire
por su parte superior.
3. Salida principal del embudo donde se colecta el gas inerte y abastece al
interior de la estructura con una presión alta de N2 (g). Su ajuste se realiza
untando un poco de grasa en su superficie y apretando como siempre. Esta
operación se completará mediante la colocación de gomillas.
4. Primera de las dos cámaras donde se trata de controlar la presión del N2
(g) debido a la amplificación del conducto (el cual se convierte en cámara)
y a que la única vía de continuación es un pequeño canal (con abertura

309
mínima en cada uno de los extremos). Esta disposición impide al N2 (g)
salir hacia 7 en gran cantidad, por lo que preferentemente marcha por 3.
Aún así, un cierto volumen logra pasar por el canal, y es misión de la
parafina retenerlo bajo una presión constante (por encima de la cual se
produce el burbujeo de la parafina contenida en 6).
5. Canal de conexión entre los dos bulbos. En sus extremos, el diámetro del
conducto se ha reducido al límite, dejando un pequeñísimo agujero para la
entrada y salida del gas en cada terminal.
6. Segunda ampolla donde se controla la presión interior. La corriente debe
tener siempre una presión determinada, constante a lo largo de todo el
interior del montaje. El conjunto está construido de tal modo que la
parafina dispuesta en 6 no burbujee más que cuando la presión sea mayor
que la crítica, dejando escapar el exceso de la misma mediante burbujeo
(como ya se ha dicho). Puede entonces comprenderse que lo más
conveniente es que no haya burbujeo o que de existir, éste sea pequeño (lo
cual indicaría una alta presión pero no excesiva).
Por supuesto el volumen de parafina debe ser tal que la extremidad del tubo
5 se halle inmersa en ella, pues de no ser así no tendría sentido. La cantidad
de parafina tampoco debe ser muy grande, y basta con que esté algo por
encima de esta salida del conducto. Para introducir la parafina en la
ampolla se toma con una jeringa y se inyecta a través de 7 con una aguja-
cánula del grosor apropiado.
7. Salida del gas.
31. 8 LAS GOMILLAS
Aquí debemos cuestionarnos acerca de cuál es la disposición más ventajosa
de las gomillas en un sistema tan complejo.

310
El método más usado, por otra
parte, es el más obvio:
El refrigerante de reflujo, donde
normalmente viene a insertarse el
embudo, lleva una abrazadera de
alambre dispuesta como se ve en la
figura. La goma elástica se puede
enganchar en uno de los brazos, de modo simple. Se estirará al tiempo que
se abre, de manera que pase cada parte de la goma por los laterales de la
cámara número 2. Finalmente se colocará de nuevo en el alambre.
Esta gomilla puede verse reforzada por una segunda cuya instalación será
idéntica. A veces podemos encontrar 2 gomillas dispuestas de modo que
una afirma un lado en tanto la otra asegura el opuesto.
31. 9 HACEMOS LA ABRAZADERA DE ALAMBRE
Es importante saber hacer el lazo de alambre sobre el que se enganchan las
gomillas y aunque pudiera parecer evidente se señalarán unas notas
prácticas a continuación.
Se toman dos alambres de la misma longitud y se fijan con la ayuda de
unos alicates por un punto determinado que permita ir girando y enlazando
con los dedos los hilos remanentes. De este modo se obtendrá una horquilla
que puede adaptarse al cuello del matraz o del recipiente correspondiente.
Una vez que se ha hecho la horquilla se rodea con ella el cuello del
recipiente y con los alicates se giran ambos hilos de modo que se trencen
entre sí. Tras unas vueltas el cuello quedará bien apresado por la abrazadera
de alambre. No se debe retorcer en exceso el alambre ya que podría partirse

311
cerca de la superficie del vidrio y habría que volver a empezar con todo el
proceso.
Se debe colocar estas abrazaderas de alambre a cada recipiente que
se utilice por vez primera ya que son indispensables para sujetar los ajustes
mediante el concurso de gomillas.
32 SÍNTESIS DE COMPUESTOS DE GRIGNARD
32. 1 LA REACCIÓN
Los compuestos de Grignard son sustancias importantes en la síntesis de
los compuestos organometálicos, por lo que conocerlos y ser capaces de
formarlos resulta imprescindible a la hora de crear posteriormente los
productos deseados. En su estructura podemos encontrar un Mg como
átomo central, en tanto que un halógeno y un grupo alquílico o arílico serán
ligandos.
Para sintetizar uno de estos compuestos se usa Mg en granalla. Como
disolvente se emplea éter dietílico, donde se irá solubilizando el Grignard a
medida que se vaya formando. El segundo reactivo a usar es el haluro de
alquilo correspondiente que es el elemento diferenciador en la estructura
química de los organomagnesianos.
Mg (s) + RX (dis. o l.) “Mg(R)X” (dis.)
Et2O
La reacción es exotérmica y va bien desde el punto de vista termodinámico,
pero debemos considerar dos factores esenciales para que marche:

312
A) El magnesio en contacto con el aire se recubre de una capa de MgO que
lo preserva y lo hace estable, así que para que reaccione hay que activarlo.
Se dice que, en esta situación el Mg se ha pasivado. Se usan como
activador unos cristalitos de I2 (s) que, no se sabe exactamente cómo,
eliminan la capa de óxido, dejando limpia la superficie, en estado óptimo
para reaccionar.
B) Aunque la reacción termodinámicamente sea favorable hay que vencer
la energía de activación (problema cinético) mediante un calentamiento
previo. Es exactamente esta energía a dar lo que hace que unos compuestos
de Grignard sean muy fáciles de preparar en tanto otros sean
verdaderamente difíciles.
32. 2 SE PONE LA REACCIÓN
Los montajes usados en la síntesis de un Grignard son muy espectaculares
tanto por la cantidad de elementos que usan y como por su aparatosidad
que sin duda constituye parte del encanto que posee la preparación de estos
compuestos.
Para que la explicación de la síntesis de estos organomagnesianos sea lo
más práctica posible la aplicaré a la síntesis de un Grignard lento o lo que
es lo mismo, de elevada energía de activación, concretamente al Grignard
de neofilo.
Ph-CH2C(CH3)2Cl + Mg Ph-CH2C(CH3)2MgCl

313
1) La operación que se hace al principio es la pesada del Mg en virutas para
realizar la reacción. Como se ha dicho el Mg (s) debe usarse en granalla,
expendiéndose embolsado y dentro de cajas cilíndricas de metal. La pesada
se realiza al aire, libremente, y a continuación se echa en un matraz de tres
bocas con el volumen adecuado.
2) El uso de ratón es obligado ya que precisamente en la síntesis de
Grignard la agitación de la disolución es muy importante para la
consecución final del compuesto. Por ello su tamaño es clave en la reacción
y se debe estimar con tacto buscándose un ratón lo suficientemente grande,
como para que pueda agitar sobradamente toda la disolución existente.
3) En las bocas del matraz se suelen poner tapones de ajuste de teflón, que
se encajan a presión y garantizan la hermeticidad de la conexión. Si se hace
esto no se colocará grasa a los apéndices de los distintos elementos que
deben encajar en ellos. Estos tapones de ajuste son apropiados para las
bocas de los matraces de fondo redondo y tienen las siguientes
características:
Estos tapones más bien debieran ser llamados “juntas”,
debido a que hacen de moldura entre las dos piezas a
encajar. Ya se ha dicho que son de teflón, blancos en
color, y huecos (para permitir alojar la parte inferior del
objeto que se coloca en la parte de arriba). Tienen gran
capacidad de aislamiento, no permitiendo fugas ni
entradas de aire.

314
Los elementos que se ajustan
mediante estos tapones lo hacen
por presión, haciendo un
movimiento de roscado (aunque
no haya rosca) para así
favorecer el máximo contacto
entre las superficies y lograr la
mayor capacidad de asiento.
4) Se conecta la llave independiente en una de las bocas laterales y el
refrigerante de reflujo en la boca central, procurando que no queden fisuras
en su unión con el matraz de tres bocas. Las gomillas se instalarán como se
ha indicado en las secciones anteriores, y las que afirman el refrigerante de
reflujo tal y como se mostró en 18.3.
5) Al tiempo se pondrá sobre el reflujo un embudo de presión compensada
firme mediante la gomilla pertinente.
6) En la boca que queda libre del matraz se encajará un tapón pinchado de
los acostumbrados.
7) Se dará paso al agua de modo que entre por la boquilla del refrigerante
de reflujo que conduce directamente hasta el fondo y que después sube
hasta la salida.
Es evidente que la numeración de pasos anterior es absolutamente aleatoria,
y se debe considerar tan sólo como un conjunto de puntos que pueden
llevarse a cabo con un orden tal que podemos alterarlo sin que por ello se
afecte la reacción lo más mínimo.
8) Para eliminar el aire del interior se conecta un tubo de la línea a la llave
independiente y al extremo adecuado (1) del embudo de presión

315
concertada. Nótese que no se han dibujado las gomillas para no entorpecer
el dibujo. La corriente de N2 (g) se abrirá en las dos entradas (E), por lo que
el aire saldrá por los tapones agujereados con las agujitas. Así poco a poco
el volumen interior se irá evacuando de aire en tanto se llena con
dinitrógeno. Se puede considerar el sistema desoxigenado cuando pasen de
5 a 10 minutos.
9) Entre tanto se desoxigena el sistema podemos proceder a hacer lo mismo
con el éter dietílico que se va a usar como disolvente. Aún siendo el éter
recogido en los sistemas de destilación habrá que someterlo a
desoxigenación mediante el pase de una corriente de N2 (g), como se vio en
la sección 11.1. Como se necesita un volumen grande se tomará el tubo
centrífuga de mayor capacidad.
10) Una vez se ha desoxigenado el sistema se añade I2 (s) con la espátula,
tomándose lo que se llama una punta de espátula. Se echan los cristalitos al
interior por la boca lateral del matraz de tres bocas, cerrando previamente
la corriente de gas para que no se vuele el yodo. Este yodo servirá para
activar al Mg. Tras la adición se reabre la entrada de gas y se procede a
cambiar el tapón pinchado por uno nuevo.
11) Si se añadiese ahora el disolvente, el I2 (s) no se solubilizaría con la
rapidez precisa y la activación del Mg (s) sería muy lenta. Para lograr que
el yodo se esparza por todos los lugares previamente, lo que se hace es
calentar. Al calentar, el yodo sublima y pasa a ser I2 (g) que presenta una
hermosa coloración violeta, que junto al tono verdoso del Cl2 (g) y al pardo
del Br2 (g) hace a los halógenos tan característicos. Al enfriarse se deposita
sobre la superficie metálica del Mg de un modo homogéneo.
Este calentamiento se lleva a cabo con dos secadores, ambos a máxima
potencia, cuidando siempre las indicaciones presentadas en 14.2. El
calentamiento se detendrá cuando comience a apreciarse una leve tonalidad
rosácea en el interior del matraz.

316
Es trascendental que en este proceso E2 esté cerrada y no exista corriente
de N2 (g) de abajo a arriba, ya que arrastraría al I2 (g), el cual se marcharía
por la salida del embudo de presión. Al hacer esto, colorea la parafina con
una tonalidad rojiza, lo cual es indicativo de la pérdida de I2 (g) que el
sistema está sufriendo. De suceder así no habría más remedio que arrojar
otros cristalitos al matraz y empezar otra vez a calentar.
Cuando el color violáceo haya desaparecido, al enfriarse el interior y
alcanzar la temperatura ambiente, se considerará la operación conclusa y a
todo el I2 (s) diseminado sobre la superficie del Mg.
12) Sólo cuando no tengamos ya traza alguna del halógeno gaseoso, se
pasará a añadir el disolvente desoxigenado. Se abrirá E2 y se quitará el
tapón nuevo, cambiándolo por uno pinchado. Pasaremos entonces con una
cánula el disolvente del tubo centrífuga al matraz. Se concluye por volver a
conectar E2 y reinsertar el tapón nuevo, para cerrar convenientemente la
llave (ahora que se va a calentar de nuevo no tiene sentido que afectemos el
reflujo con una corriente de gas hacia arriba).
Cuando cae el Et2O (l) lo primero que llama la atención es la coloración
parda que aparece. Esta tonalidad se explica por la generación de aductos o
complejos de transferencia entre el I2 (solvatado) y los oxígenos del éter
que poseen capacidad de donación de densidad electrónica. En ese instante
se aplicará al ratón un movimiento de giro acusado.
13) Se cerrará E2 como se ha dicho en el punto anterior y se conectan los
secadores para hacer refluir el éter. La coloración parda va desapareciendo
poco a poco, a causa de que el I2 en disolución va reaccionando y
eliminando la capa de MgO existente sobre el Mg (s). Cuando todo el color
haya pasado del pardo al gris claro podemos dejar de calentar y se esperará
a que la disolución alcance la temperatura ambiente. Todo este proceso
dura en torno a una hora.

317
Siempre que se caliente un disolvente en un montaje como este hay que
tener la llave E2 cerrada para no complicar el proceso de reflujo.
14) Mientras se espera que el éter alcance la temperatura del laboratorio, se
preparará la disolución del reactivo que falta en un tubo aparte. Es
conveniente que sea una concentración baja, ya que esto permite un mayor
control sobre el ritmo de reacción. La disolución se hace con éter y se
añade después al embudo de reactivos. Como se dijo con anterioridad este
compuesto no se afecta con el aire por lo que la disolución se lleva a cabo
en el laboratorio sin el concurso de la atmósfera inerte.
15) Acto seguido se conecta el embudo al matraz. Se abre entonces el N2
(g) en E2 y se quita el tapón de goma para permitir conectar el embudo. A
continuación se coloca un tapón pinchado en la boca de éste y una agujita,
para que se logre una desoxigenación completa a través del canal diseñado
para ello.
16) Ahora comienza la verdadera reacción, siendo además éste el momento
más delicado de todos por la razón que se comentará acto seguido. Al
principio se adiciona mediante el embudo un buen chorro de reactivo, con
él tendremos que ser capaces
de lograr que la reacción se
inicie. Conectando ambos
secadores desde el inicio se
calentará intensamente nuestra
disolución hasta que el éter
refluya. Se dice que la reacción
ha empezado cuando ésta
refluye por sí sola, lo que a
veces requiere práctica en la
observación pues el ritmo de
goteo, en algunos casos es lento.

318
Esto logrado, basta con añadir de cuando en cuando pequeñas cantidades
de disolución de haluro, o mejor imponer un goteo de reactivo muy lento,
logrando así que el haluro no se acumule y se consuma al caer en el medio.
Sobre el papel esto resulta muy fácil, sin embargo en la práctica esta
reacción cuesta iniciarla, aunque claro, esto depende de la reactividad del
haluro. Para haluros primarios la velocidad de reacción es alta,
disminuyendo en secundarios y terciarios. Este es entonces el problema.
Tratando de que la reacción se inicie se puede tener la tentación de añadir
otro poco de haluro, y luego otra porción, hasta que la acumulación del
reactivo en la disolución es mayor de lo aconsejable. Entonces puede
ocurrir que la reacción se inicie de repente, y al haber gran cantidad de
haluro, la exotermia haga que se descontrole y acabe explotando.
En tal caso lo más favorable es desconectar el par de secadores y bajar el
cristal de la vitrina, esperando que no se tengan más problemas que el de
recoger los trazos de vidrio y el de limpiar la vitrina.
Conseguir que refluya por sí mismo el éter (es decir con el calor generado
por la reacción) puede llevar horas, por tanto lo mejor es armarse de
paciencia y controlar que la mano no se vaya con demasiada frecuencia al
embudo.
17) La reacción una vez activada marcha por sí misma, el Mg se va
agotando lentamente y la disolución se hace cada vez más grisácea y
oscura. Llega un tiempo en el que no queda Mg (s) en el fondo del matraz y
la reacción se enfría dejando de refluir cuando ha concluido
completamente.
18) La mezcla se pasa mediante una cánula a un tubo de centrífuga del
modo establecido en 16.7 y se centrifuga.
19) A continuación se pasa a una ampolla que servirá para contener al
Grignard. Se anotará en ella el tipo de Grignard sintetizado, su
concentración y la fecha de la síntesis.

319
32. 3 FUGAS EN EL SISTEMA
Puede ocurrir que una vez montado el sistema se detecten fugas en las
bocas de unión entre el matraz y los distintos accesorios.
La solución no es fácil pues hay que levantar los elementos uno por uno y
mediante cinta de teflón proteger el ajuste, haciéndolo de modo que no
entre aire. La fuga en sí suele venir a consecuencia de del mal estado de
alguno de los tapones de teflón, o incluso por la falta de pericia al colocar
las piezas de nuestro montaje. Al principio la fuga no se nota, hasta que
comienza a ser evidente por la disminución del volumen del disolvente
(éter) en el matraz.
Analicemos el problema por partes:
A) Si la fuga se registra en el embudo de dispensar reactivos, lo que se hace
es abrir en un momento determinado E2 y quitar el embudo. En su cono de
ajuste arrollaremos unas vueltas de cinta de teflón, que actuará como
aislante y volveremos a encajarlo en el hueco quitando el tapón de ajuste.
Tras lo cual se cierra E2 para que no afecte al reflujo.
B) Si la fuga acontece en el ajuste correspondiente al refrigerante de reflujo
se procede exactamente igual, aprovechando que los dos se encuentran con
presión positiva de N2 (g).
C) En el tercer caso, con el N2 (g) abierto en ambas partes, separaremos el
accesorio con cuidado para que no entre aire. Si no lo separamos mucho, el
cono de acción del gas inerte que parte desde la llave protegerá la boca del
matraz de la posible entrada de aire. Piénsese que aunque la entrada 1 esté
abierta hay una gran distancia y la caída de presión debe acusarse.

320
El único problema que debe mencionarse es que, a medida que vaya
transcurriendo la reacción, el teflón se vaya empapando con el disolvente
de abajo a arriba y éste acabe por volver a salirse y perderse.
Finalmente hay que considerar el hecho de que puede hacer falta volver a
añadir éter, en el caso de que su pérdida fuese excesiva. Así habrá que
desoxigenar otra cantidad y añadirla mediante la ayuda de una cánula.
32. 4 LA CINTA DE TEFLÓN
El teflón es un polímero de flúor cuyo monómero es una unidad “CF2” que
se repite según la estructura (CF2)n. Se usa como aislante gracias a su
capacidad impermeabilizante y a su escasa reactividad frente a los
compuestos químicos.
Se vende en tiras muy finas y delicadas, de color blanco, con tacto suave y
plástico, enrolladas en forma de rulo (como si fuese un rollo de fixo).
Debido a su adaptabilidad puede modificarse con los dedos, estrujándolo o
extendiéndolo, por lo que necesita una carcasa de protección. Para cortarlo
no hacen falta tijeras sino que basta con un simple tirón, no obstante, este
tirón deja arrugada la cinta y debemos volver a extenderla por el lugar por
el cual se ha roto.
32. 5 EL REFLUJO
Al calentar un disolvente volátil, una gran cantidad del mismo pasa a
estado vapor, perdiéndose si el recipiente donde se hace la experiencia está
abierto. Precisamente para corregir este fallo encontramos los refrigerantes

321
de reflujo. Estos elementos son tubos de vidrio hueco donde en su interior
hay canales estrechos por los que circula agua. La gran cantidad de bucles
de estos canales se explica por la necesidad de que exista la mayor
superficie de contacto posible entre el gas y la pared que conduce el agua.
El agua es un intercambiador de calor y el vidrio hace de pared de
separación entre ambas fases. Cuando el gas toca la superficie de vidrio, el
agua absorbe el calor de éste, que se vuelve a enfriar, condensando y
cayendo en forma de gotas que se acumulan y se precipitan desde el último
bucle del tubo conductor de agua al matraz. Mediante este ingenioso
sistema se pueden calentar disolventes muy volátiles sin peligro de que se
vayan.
Poner a reflujo un disolvente es pues esto último, es decir calentarlo hasta
que el gas se condense en líquido (por el refrigerante) y vuelva a caer al
matraz. Este hecho continuo se conoce como refluir.
32. 6 VALORACIÓN DE UN GRIGNARD
El método que se pasa a comentar es extensible para los compuestos
organometálicos en general y no exclusivo para los organomagnesianos.
Con una jeringa de 1 ml se toma una porción del compuesto de Grignard
sintetizado (conociendo así perfectamente el volumen usado) y se hidroliza
echándolo sobre agua en un erlenmeyer. Durante la caída del Grignard se
pueden oír pequeñas explosiones en el interior del recipiente que se
corresponden con la vigorosidad de la reacción. Agitando el erlenmeyer
durante unos segundos se completará la hidrólisis y la valoración podrá
comenzar.

322
Mg(R)X (dis.) + H2O (l) RH (dis.) + Mg(OH)X (dis.)
El Grignard en disolución con éter reacciona con el agua líquida para
convertirse en el alcano o compuesto aromático correspondiente,
formándose al mismo tiempo una sal, que queda en disolución, donde el
Mg+2
(ac.) tiene como contraiones al OH- (ac.) y al X
- (ac.). El compuesto
orgánico formado de este modo permanecerá en forma de micelas o en fase
extendida, pero al ser tan pequeña la cantidad formada, lo más probable es
que quede formando micelas suspendidas en el agua a causa de la agitación
(las cuales tardarán en homogeneizarse para formar una superficie continua
que termodinámicamente es más estable, pero que requiere una energía de
activación considerable.
El OH- (ac.) se valora con HCl (ac.), lo cual se realizará a continuación. El
indicador usado es la fenolftaleína, de la que se pondrán en el erlenmeyer
un par de gotas (dependiendo esto último de su concentración). El HCl
(ac.) se prepara generalmente a una molaridad igual a la unidad, ya que el
Grignard se sintetiza con cantidades que al final logran una concentración
del mismo entorno a 1M (que son las más manejables).
La operación de valoración se repite varias veces, tomando como valor
verdadero la media de todos los volúmenes obtenidos. La bureta se lava al
final con agua destilada, dejándola lista para ser usada cuando vuelva a ser
necesario.
La concentración obtenida se escribe en una etiqueta junto con el nombre
del compuesto y la fecha de síntesis, como se ha indicado ya, pegándola
con fixo al vientre de la ampolla. Este tipo de reacciones casi nunca son
cuantitativas por lo que no se debe esperar una conversión completa.

323
33 FOSFINAS
Resulta interesante dedicar unas breves notas a uno de los ligandos más
importantes del mundo organometálico ya que en el trabajo de laboratorio
la síntesis de ligandos es tan determinante como la de los propios
compuestos organometálicos. En el caso concreto de las fosfinas sus
características las hacen ser excepcionales entre los ligandos y la variedad
de estos compuestos hace que su síntesis sea un verdadero arte,
dependiendo de los grupos orgánicos que se unan al fósforo.
33. 1 LAS FOSFINAS
Son un grupo de sustancias que derivan formalmente de la fosfina PH3,
presentando enlaces P–C. La capacidad donadora del fósforo (donación
electrónica de carácter ) permite la coordinación de la fosfina con el
metal, estabilizando de este modo a los compuestos organometálicos en
donde toma parte.
La síntesis de las mismas es un proceso complejo, lento y tedioso, siendo
en muchos casos más difícil lograr la fosfina que el compuesto
organometálico final. La gran ventaja es que prácticamente hay un tipo de
fosfina adecuada para estabilizar cada compuesto organometálico.
Variando sus radicales desde alquílicos a arílicos tenemos una ingente
variedad de fosfinas a nuestra disposición, permitiéndonos usar criterios
electrónicos y estéricos para dirigir una síntesis determinada. Existen así
pues muchas fosfinas que teóricamente estabilizarán uno u a otro
compuesto. De este modo una vez elegida la fosfina sobre el papel sólo
queda sintetizarla, lo que es el asunto más grave.

324
A todo esto no debemos olvidar que podemos tener también fosfinas unidas
por cadenas de carbono (es decir, una cadena de carbono con dos fosfinas
terminales), análogo estructural a la etilendiamina, que a todos los efectos
estabilizadores propios une el efecto quelato.
Las fosfinas son, en general, líquidos incoloros que se guardan en ampollas
bajo N2 (g) dada su reactividad al aire en el frigorífico, puesto que la
temperatura también las desestabiliza. Su volatilidad es doblemente
preocupante: en primer lugar porque son caras y difíciles de obtener y en
segundo lugar porque presentan un olor repugnante.
33. 2 SÍNTESIS DE FOSFINAS
La síntesis de las fosfinas resulta especialmente importante por la cantidad
de compuestos organometálicos que originan y por la bondad de las
mismas como ligandos, que estabilizan los compuestos en los que
participan de una manera muy significativa.
Básicamente las fosfinas se forman a partir de los reactivos de Grignard, y
aunque pueden también formarse de algunas otras maneras esta es la más
sencilla, por lo que se puede comprender la gran utilidad de estos últimos,
que ya hemos aprendido a sintetizar. Lo más fácil es hacerlos reaccionar
con halogenuros de alquilo en medio éter dietílico. La reacción se puede
llevar a cabo en un matraz de reacción análogo al que usamos para formar
el Grignard.
PX3 + 3RMgX3 PR3 + 3MgX2

325
Las fosfinas así obtenidas se utilizarán en la síntesis de compuestos
organometálicos. Tal vez la más conocida de todas ellas sea la
trifenilfosfina, que debido a su alto peso molecular es cristalizable,
llegando a tener una estabilidad tan grande que se vende en forma de sólido
blanco cristalino.
33. 3 DISOLUCIONES CON FOSFINAS
Las fosfinas, ya se ha indicado, son muy volátiles. Esto es un problema a la
hora de hacer reacciones a temperatura más elevada que la ambiente (e
incluso a la propia temperatura ambiente). Para ello deben usarse tapones
de teflón y ampollas apropiadas, ya que la fosfina se va de la disolución y
se absorbe en el tapón, quedando sin reaccionar. Este suceso es a tener en
cuenta, pues no se debe perder fosfina inútilmente pudiéndolo impedir de
un modo tan sencillo.
33. 4 EL OLOR DE LAS FOSFINAS
Esta es la característica más singular de cualquier fosfina. A causa de su
volatilidad, la fosfina se marcha al instante que se usa dejando un olor
rabioso, insistente y repugnante, que se aferra a la nariz y no la deja hasta
pasado un buen tiempo. De hecho, tras usar fosfina (o tras haberla sufrido
por la acción de algún compañero) se conserva el olor de la misma durante
horas, y no sabría explicar si es debido a su presencia real o a alguna
imposición del subconsciente.
Eliminar el olor del ambiente es imposible, pero su alcance puede
minimizarse procurando hacer un uso correcto de la misma. Con tal

326
intención, cada vez que se tome una cantidad de su disolución se procederá
de inmediato a lavar la jeringa, y los recipientes que la hayan contenido
momentáneamente, o las cánulas que la hayan transportado. Se lava con
acetona y éter, como es usual, pero siempre posteriormente al empleo de
HCl (ac.) y agua. El ácido forma el clorhidrato de fosfina, que no es volátil
y que por tanto no huele.
34 SE COMIENZA UNA INVESTIGACIÓN
La investigación atañe a materias desconocidas o estudios que aún han de
completarse, por tanto, será obligado conocer qué se ha hecho en un
determinado campo dentro de la química de compuestos organometálicos
para poder avanzar al respecto.
Con ese fin existen gran número de publicaciones y bibliografías que
pretenden mantener informados a los científicos en cuanto a cuáles son los
últimos logros en una u otra disciplina.
Resumir cuáles son las fuentes de información más importantes no es tarea
fácil por lo que me limitaré a aquellas que resultan ser las más usadas.
34. 1 CATALOGACIÓN DE COMPUESTOS
El tratado encargado de la clasificación de los compuestos organometálicos
es el Dictionary of organometallic compounds (como resultará obvio toda
la bibliografía está en inglés).
Su propósito es el de seleccionar aquellos compuestos organometálicos
que, en opinión de los especialistas, son los más representativos,
interesantes y útiles, presentando sus propiedades y las referencias

327
particulares de cada uno de ellos, para que puedan ser encontrados en las
revistas donde se publicó su síntesis y estudio.
Los compuestos vienen ordenados por secciones, cada una asignada a un
metal. Dentro de cada metal encontraremos los compuestos seleccionados,
ordenados según reglas que no vamos a considerar. Cada compuesto posee
una ficha encabezada por la fórmula molecular.
La ficha se puede describir del modo siguiente:
- Fórmula molecular: Usada para ordenar el compuesto dentro del
conjunto, a su lado encontraremos un metal y un número, M–nº,
que muestra el código asignado al compuesto en el diccionario.
- Nombre I.U.P.A.C.
- Nombres alternativos.
Estos dos puntos refieren todos aquellos nombres por los que se conoce al
compuesto, primero aquel que se considera oficial, en tanto que el segundo
punto muestra los nombres comunes del producto.
- Estructura y descripción estereoquímica: Se representa la
estructura del compuesto mediante un dibujo o bien se hace
referencia a la estructura modelo en la que se basa.
- Peso molecular.
- Uso: Se comentan aquellas reacciones en las que interviene, y
también su función dentro de dichas reacciones.
- Toxicidad: Aquellas precauciones que deben ser tenidas en
cuenta a la hora de manejarlo.
- Datos físicos.
- Referencias bibliográficas: Referencias de los artículos
publicados en las revistas de mayor prestigio para acceder a una
información más detallada del compuesto.

328
Todos los compuestos vienen acompañados por un número entre corchetes
que es asociado al compuesto en el registro internacional. Si no tuviese
número el compuesto se consideraría inexistente. Este registro
internacional responde a las siglas C.A.S. (Chemical Abstract Service).
En este diccionario, cada sección (destinada a un metal en concreto) viene
encabezada por un análisis muy completo del metal y de los tipos de
compuestos en los que se presenta normalmente. Notando desde los estados
de oxidación, los colores más comunes de los productos, la disponibilidad
de los reactivos y materiales de partida, cómo han de manejarse los
compuestos, hasta una visión de la espectroscopia.
El diccionario se va ampliando poco a poco gracias a la aparición sucesiva
de anexos que van recogiendo los nuevos compuestos. Como consecuencia
del desarrollo informático e Internet, este tipo de fuente en papel pierde
cada vez más importancia en razón a la rapidez y accesibilidad de los
últimos soportes digitales mucho más cómodos y ventajosos.
34. 2 LA QUÍMICA DE LA COORDINACIÓN
Tan unida se encuentra la química organometálica a la química de la
coordinación que no son entendibles la una sin la otra. Este es el motivo
por el que introducimos aquí la fuente principal básica de toda la química
de la coordinación al igual que posteriormente haremos para la química
organometálica. Esta fuente se denomina Comprehensive coordination
chemistry. Es un conjunto de volúmenes que pormenoriza los distintos
aspectos de esta rama de la química. En ella se siguen los mismos
parámetros que se encuentran en su paralelo organometálico, que se
describirá a continuación en la siguiente sección.

329
La división realizada en esta enciclopedia es la que sigue:
Primer volumen Theory and Background
Segundo volumen Ligands
Tercer volumen Main group and early transition elements
Cuarto volumen Middle transition elements
Quinto volumen Late transition elements
Sexto volumen Applications
Séptimo volumen Indexes
34. 3 LA QUÍMICA ORGANOMETÁLICA
Se presentan aquí las dos series I y II de Comprehensive Organometallic
Chemistry, siendo en gran medida mutuamente excluyentes, por lo que
siguen siendo útiles los volúmenes de la serie más antigua.
Es la fuente básica a la cual hay que recurrir a la hora de investigar. Desde
aquí nos remitirán a las publicaciones adecuadas que se hacen en las
revistas más importantes. Es quizás el primer escalón de la investigación
pues de sus páginas tendremos que marchar a las publicaciones y desde
éstas al laboratorio.
La información se encuentra distribuida en catorce tomos donde se
estructura toda la información requerida.
v.1 Lithium, Beryllium and Boron Groups
v.2 Silicon Group, Arsenic, Antimony, and Bismuth
v.3 Copper and Zinc Groups
v.4 Scandium, Yttrium, Lanthanides and Actinides, and Titanium Group
v.5 Vanadium and Chromium Groups

330
v.6 Manganese Group
v.7 Iron, Ruthenium, and Osmium
v.8 Cobalt, Rhodium, and Iridium
v.9 Nickel, Palladium, and Platinum
v.10 Heteronuclear Metal-Metal Bonds
v.11 Main-group Metal Organometallics in Organic Synthesis
v.12 Transition Metal Organometallics in Organic Synthesis
v.13 Structure Index
v.14 Cumulative Indexes
34. 4 LAS PUBLICACIONES
He aquí el verdadero puntal de la investigación mundial. Lo que se publica
es lo que se conoce y se adjudica internacionalmente al grupo de
investigadores que lo ha publicado. De esto se deduce que en tanto se
publica y no, las ideas o resultados queden más bien en secreto, ya que
aquel que publique antes será considerado el primero antes los ojos del
mundo, quedando el trabajo como suyo, sea o no el primero que lo haya
considerado o incluso, que haya obtenido resultados interesantes al
respecto.
Cada publicación o artículo se distingue del resto por la referencia, donde
se contiene la información suficiente para poder encontrar al artículo en
medio de un maremagno de iguales.
Un ejemplo de referencia es:
J. G. Kester, J. C. Huffman and L. J. Todd, Inorg. Chem., 1988, 27, 4528

331
Los tres primeros nombres hacen alusión a los autores del experimento
referido, mientras que las dos palabras apocopadas muestran la revista en la
que se ha publicado el experimento o la discusión. A continuación se da la
fecha anual, el número del boletín dentro del año y el número asignado al
artículo en dicho boletín.
El artículo refiere el experimento o el análisis de un modo serio y riguroso,
demostrando sus aseveraciones y dando suficientes reseñas como para que
el experimento pueda ser repetido.
Existen revistas de más o menos prestigio, dependiendo de la naturaleza y
calidad de los artículos que aceptar para publicar. Esta aceptación se hace
en base a un extenso comité de expertos que garantizan la veracidad y
rigurosidad de las experiencias.
Las revistas más consultadas en razón a su prestigio e importancia son las
que siguen:
Organometallics
J. Am. Chem. Soc.
J. Am. Chem. Commun
Polyhedron
Inorg. Chem.
J. Chem. Soc., Dalton Trans.

332
35 CONCLUSIÓN
Aquí finaliza este manual cuyos objetivos fundamentales consisten en
realizar un análisis de la química organometálica experimental a un nivel
elemental y acercar esta admirable rama del conocimiento a aquellos
compañeros que, estando interesados en ella, aún no hayan tenido la
oportunidad de satisfacer por experiencia directa las inquietudes al
respecto.
No puedo asegurar que haya logrado un tratamiento claro de las distintas
nociones que he intentado explicar, pero sí que lo he intentado con
denuedo. Quizás lo más difícil haya sido precisamente elegir qué es
importante y qué puede ser obviado, para con ello poner una extensión
razonable a este trabajo.
A lo largo de la exposición he tratado de presentar la materia de un modo
racional, ilustrando primero aquellos conceptos que son precisos al
comienzo de la reacción, cuando ésta se monta, en tanto el resto se ha ido
distribuyendo a medida que las distintas fases de la reacción lo han
requerido.
Ha sido por mi parte un enorme esfuerzo el empleado, durante muchos
meses, para poner las técnicas aprendidas por escrito y a continuación
darles una forma aceptable para el lector. Así, mi única recompensa válida
sería que alguien se sintiese atraído, primero por este volumen y después
como consecuencia, por el campo de trabajo que pretende representar.
Copyright © 2022 FDOKUMEN




















