Synthesis, structural and magnetic properties of spin ladder compound Ca 1− x Co x Cu 2O 3
11
Hyperfine Interact DOI 10.1007/s10751-012-0640-5 Synthesis, structural and magnetic properties of La 1−x Ca x FeO 3 prepared by the co-precipitation method E. K. Abdel-Khalek · Hany M. Mohamed © Springer Science+Business Media B.V. 2012 Abstract Lanthanum calcium ferrite La 1−x Ca x FeO 3 (where x = 0.05, 0.10 and 0.15) were synthesized by co-precipitation method. The structural refinement confirmed that all samples formed with the orthorhombic (Pbmn) structure with small impuri- ties (LaFeO 3 and Fe 2 O 3 ). Fourier transformed infrared spectroscopy (FT-IR) results indicated that the electronic imbalance caused by the partial substitution for La 3+ by Ca 2+ is compensated by an oxidation state of iron ions. Mössbauer spectra at room temperature (RT) show five 6-line sub-spectra corresponding to Fe 3+ (superposition of 3 spectra), Fe 4+ and Fe 5+ in all samples. Magnetic hysteresis loops of these samples showed a significant weak ferromagnetic component at RT. Keywords Co-precipitation synthesis · Mössbauer spectroscopy · X-ray diffraction · Magnetic properties 1 Introduction Lanthanum calcium ferrite La 1−x Ca x FeO 3 have found applications in several tech- nologies, such as in solid oxide fuel cells, catalysis and gas sensors, mainly due to their unique structural features [1–3]. The properties of La 1−x Ca x FeO 3 are influenced by the percentage of Ca ions, as well as the synthesis procedure employed to process these materials [4, 5]. Accordingly, the literature describes several known procedures to make the lanthanum calcium ferrite in general, such as solid-state reaction [6], sol–gel [5] and citrate [1, 2] methods. Ya-Qiong Liang et al. [7, 8] have employed solid-state method to obtain La 0.5 Ca 0.5 FeO 3 in air, sample (SA), and flowing 100 % E. K. Abdel-Khalek (B ) Physics Department, Faculty of Science, Al Azhar University, Nasr City 11884, Cairo, Egypt e-mail: [email protected] H. M. Mohamed Chemistry Department, Faculty of Science, Al Azhar University, Nasr City 11884, Cairo, Egypt
Transcript of Synthesis, structural and magnetic properties of spin ladder compound Ca 1− x Co x Cu 2O 3

Hyperfine InteractDOI 10.1007/s10751-012-0640-5
Synthesis, structural and magnetic properties ofLa1−xCaxFeO3 prepared by the co-precipitation method
E. K. Abdel-Khalek · Hany M. Mohamed
© Springer Science+Business Media B.V. 2012
Abstract Lanthanum calcium ferrite La1−xCaxFeO3 (where x = 0.05, 0.10 and 0.15)were synthesized by co-precipitation method. The structural refinement confirmedthat all samples formed with the orthorhombic (Pbmn) structure with small impuri-ties (LaFeO3 and Fe2O3). Fourier transformed infrared spectroscopy (FT-IR) resultsindicated that the electronic imbalance caused by the partial substitution for La3+ byCa2+ is compensated by an oxidation state of iron ions. Mössbauer spectra at roomtemperature (RT) show five 6-line sub-spectra corresponding to Fe3+ (superpositionof 3 spectra), Fe4+ and Fe5+ in all samples. Magnetic hysteresis loops of these samplesshowed a significant weak ferromagnetic component at RT.
Keywords Co-precipitation synthesis · Mössbauer spectroscopy ·X-ray diffraction · Magnetic properties
1 Introduction
Lanthanum calcium ferrite La1−xCaxFeO3 have found applications in several tech-nologies, such as in solid oxide fuel cells, catalysis and gas sensors, mainly due to theirunique structural features [1–3]. The properties of La1−xCaxFeO3 are influenced bythe percentage of Ca ions, as well as the synthesis procedure employed to processthese materials [4, 5]. Accordingly, the literature describes several known proceduresto make the lanthanum calcium ferrite in general, such as solid-state reaction [6],sol–gel [5] and citrate [1, 2] methods. Ya-Qiong Liang et al. [7, 8] have employedsolid-state method to obtain La0.5Ca0.5FeO3 in air, sample (SA), and flowing 100 %
E. K. Abdel-Khalek (B)Physics Department, Faculty of Science, Al Azhar University, Nasr City 11884, Cairo, Egypte-mail: [email protected]
H. M. MohamedChemistry Department, Faculty of Science, Al Azhar University, Nasr City 11884, Cairo, Egypt

E.K. Abdel-Khalek, H.M. Mohamed
oxygen of 1 atm to obtain another sample (SO), respectively. They found that theMössbauer spectrum at RT for SO exhibits a typical paramagnetic singlet withcenter shift (CS) of 0.20 mm/s. This singlet is corresponding to the average-valencecharge state between Fe4+ and Fe3+ ions. For SA sample, in addition to a similarsinglet with that of SO, two additional sextets with hyperfine fields of 364 kOe and205 kOe, center shifts of 0.39 mm/s, and −0.20 mm/s, exist in the spectrum, whichare assigned to be Fe3+ and Fe4+ ions, respectively. Mössbauer spectra also indicatethat TN of SA is above RT and that of SO is below RT. Hudspeth et al. [5] haveemployed sol–gel method to obtain La1−xCaxFeO3 where x = 0.2 and 0.33. Theyfound that the Mössbauer spectra at RT consist of magnetic sextets identified withFe3+, and a weak paramagnetic doublet at x = 0.33 is observed. Barbero et al. [2]have employed a citrate method to obtain La1−xCaxFeO3 where x = 0, 0.2 and 0.4.They found that the catalytic behavior has been related to Fe4+ species existence,which has been suggested from the XRD, TPR, and Fourier transform infrared (FT-IR) results. The Mössbauer spectra for La1−xCaxFeO3 where x = 0 and 0.4 havebeen reported by Barbero et al. also [9]. They found that the Mössbauer spectrumfor LaFeO3 at 298 K consisted of a magnetic sextet which corresponded to thetypical value of octahedrally coordinated Fe3+ ions with a high spin configuration.Moreover, a central signal with a contribution of approximately 2 % is detected.The Mössbauer spectrum for La0.6Ca0.4FeO3 at 298 K consist of six lines highlyoverlapped and a very intense central peak with isomeric shift of 0.12 mm/s whichindicates that it corresponds to Fe4+. This behavior indicates that the sample is nearto its magnetic order–disorder transition [9]. From these earlier researches we foundthat the Mössbauer spectra at RT are different with the preparation techniques but atliquid helium temperature the spectra are very similar regardless of the preparationtechnique. On the basis of these results, Fe4+ is present as a central peak, a doublet orsextet at room temperature depending on the method of preparation [9]. In view ofthe aforementioned aspects La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15) samples weresynthesized by the co-precipitation method [10, 11]. This preparation method isquite simple and leads to homogeneous, highly reactive and nanosized powders.Accordingly single-phase perovskite powders can be produced at low temperature[10, 11]. We selected the Ca substitution range of 0.05 ≤ x ≤ 0.15 to get the maximumcontent of Fe3+ ions in order to obtain the competition between the ferromagneticinteraction of Fe3+–Fe5+ and antiferromagnetic interaction of Fe3+–Fe3+ in theexperiment at room temperature. Structural and magnetic properties analysis at RTwere characterized by means of the Rietveld refinement using XRD, FT-IR, MS, andvibrating sample magnetometer.
2 Experimental
The La1−xCaxFeO3 (where x = 0.05, 0.10 and 0.15) perovskites were prepared byco-precipitation method. The final oxide product was prepared by dissolution ofnitrate salts of the desired metal. In the present case, this involve the metal nitratesCa(NO3)2·4H2O and Fe(NO3)3.9H2O, which were dissolved in distilled water. Onthe other hand, LaCl3.7H2O was dissolved in distilled H2O and a concentrated HNO3
was added to the solution which was heated to dryness. The resulting solid La(NO3)3
was dissolved in distilled H2O. Metal nitrate solutions were mixed in appropriate

Synthesis, structural and magnetic properties of La1−xCaxFeO3...
proportions depending on the target composition. The cations were coprecipitated byslow addition of a saturated (NH4)2CO3 solution. Finally, few grams of the obtainedpowder were pressed into pellet which was sintered in air for 24 h at 1273 K to obtaina highly dense final product.
X-ray diffraction analysis was carried out using a SIEMENS D5000 diffractometerusing Cu Kα radiation. The structural refinements were carried out using theFULLPROF Rietveld program. The crystallite size (L) was evaluated from thebroadening of the X-ray diffraction peaks using Scherrer’s formula. The IR trans-mission spectra of the perovskites in the wavenumber range of 410–1000 cm−1 wererecorded at room temperature on a Bruker (Vector 22), single beam spectrometerwith a resolution of 2 cm−1. The IR absorption spectra were recorded using KBrpellets. The Mössbauer absorption spectra were recorded at room temperature(RT) by using a conventional constant acceleration spectrometer with 57Co (Cr)radioactive source and using metal Fe foil for calibration. The absorbers wereobtained by pressing the powdered (70 mg) samples into holders of cross sectionalarea of 1.77 cm2. The Mössbauer spectra were fitted using the MOSF program [12].The magnetic properties of the perovskite samples were measured using vibratingsample magnetometer (VSM) model 9600-1 LDJ at RT. These properties includedsaturation Bs, remanent Br and coercive field HC.
3 Results and discussion
3.1 X-ray measurements
Figure 1 illustrates the observed and fitted XRD patterns of La1−xCaxFeO3 (wherex = 0.05, 0.10 and 0.15) perovskites. Rietveld refinement of the data for major phase(Fig. 1) reveals that the samples crystallize in orthorhombically distorted perovskitestructure (Pbnm space group). From Fig. 1 it can been seen that the differencebetween the experimental and theoretical lines is consistent with the presence ofsmall amounts of LaFeO3 and Fe2O3 impurities [13, 14]. In the orthorhombicstructure, oxygen ions occupy two nonequivalent sites, namely OI(4c) and OII(8d),with a population ratio of 1:2. Six O2− ions (two OI and four OII) surround eachFe ion and form an octahedron [7]. The refined lattice parameters, atomic positionparameters, unit cell volume and selected bond lengths and angles are summarizedfor all three samples in Table 1. The lattice parameters for x = 0.15 are found tobe within the experimental error with the lattice parameters reported in ref [15]while for x = 0.05 and 0.1 these new parameters are close to the values reported forundoped LaFeO3 [16]. The increase of Ca2+ at the expense of La3+ from 0.05 up to0.15, causes an electronic imbalance in the perovskite lattice that can be compensatedby the generation of oxygen vacancies and increased formation of Fe4+ ions. Thusthe decrease of the lattice parameters and unit-cell volume is not only due to thedifference between the ionic radii of the Ca2+ (1.34 Å) and La3+ (1.36 Å), sincethey are quite similar [17–19], but can be attributed to the existence of oxygenvacancies and Fe4+ where the ionic radius of Fe4+ (0.585 Å) is significantly smallerthan that of Fe3+ (0.645 Å) [18, 19]. Moreover, the FeO6 octahedron is observedto become increasingly distorted as more Fe4+ ions are formed. The decrease inaverage bond length <Fe—O> and the bond angle Fe—OII—Fe with the increase

E.K. Abdel-Khalek, H.M. Mohamed
Fig. 1 Rietveld plot of X-raydiffraction pattern forLa1−xCaxFeO3 Red dotsindicate the experimental dataand the black lines overlappingthem indicate calculatedprofiles. The lowest curveshows the difference betweenexperimental and calculatedpatterns. The LaFeO3 (©),Fe2O3 (∇), peaks areattributed to the smallimpurities

Synthesis, structural and magnetic properties of La1−xCaxFeO3...
Table 1 Structural parameters for La1−xCaxFeO3 perovskite samples obtained from the structuralrefinement using X-ray powder diffraction data at room temperature
Lattice constants Lattice coordinate Fe-O-Fe (◦) Fe-O(Å) Cry Size
Atom (Site) x y z Fe-OI-Fe Fe-OII-Fe Fe-OI Fe-OII L(nm)
La0.95Ca0.05FeO3 (Pbnm)
a = 5.536 Å La/Ca (4c) 0.515 −0.024 0.250 172.74 127.03 1.964 1.987 39b = 5.552 Å Fe(4a) 0.000 0.000 0.000 1.951c = 7.832 Å OI(4c) 0.526 0.507 0.250V(Å3) = 240.73 OII(8d) −0.224 −0.271 1.001
La0.90Ca0.10FeO3 (Pbnm)
a = 5.530 Å La/Ca(4c) 0.515 −0.025 0.250 171.72 126.81 1.961 1.984 40b = 5.539 Å Fe(4a) 0.000 0.000 0.000 1.947c = 7.824 Å OI(4c) 0.527 0.507 0.250V(Å3) = 239.66 OII(8d) −0.223 −0.271 1.001
La0.85Ca0.15FeO3 (Pbnm)
a = 5.500 Å La/Ca (4c) 0.515 −0.024 0.250 169.72 126.02 1.951 1.973 51b = 5.511 Å Fe (4a) 0.000 0.000 0.000 1.937c = 7.780 Å OI(4c) 0.528 0.507 0.250V(Å3) = 235.82 OII(8d) −0.224 −0.271 1.001
of Ca2+ due to the formation of Fe4+ ions [20]. The average bond length <Fe—O>is decreased due to increased covalency between the Fe and oxygen ions also [21].Thus when the number of Ca2+ ions increases, the angle Fe—OII—Fe is distortedand the hybridization tp−d between the Fe(3d) and O(2p) decreases [9].
The crystallite size was estimated using Scherrer’s equation; L = kλ/βcosθ whereL is the mean dimension of the crystallite, k is a constant approximately equal tounity, λ is the wavelength used (1.5406 Å), β is the full-width at half-maximumin radians (obtained for the high intensity peak in the present studies) and θ isthe related Bragg angle. It is known that in nanocrystals many factors such aslattice distortion, defect concentration and cation ordering may influence the unitcell volume. Thus the increase in average crystallite size with the increase of Ca2+(Table 1) may be creates positive pressure on the lattice leading to a lattice cellvolume contraction [22].
3.2 FTIR measurements
Figure 2 shows the FTIR spectra of the La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15)perovskite samples. The vibration modes of the perovskite ABO3 are mainly activein three IR spectral regions (i) in the region 500–730 cm−1; (ii) in the region 250–500 cm−1 and (iii) at about 200 cm−1 (external) [23]. The first region of bands isattributed to the stretching modes of the BO6 octahedra while the second regionof bands is attributed to the bending modes of these same polyhedra. Becausethe Fe–O bonds of the BO6 octahedral units are undoubtedly stronger than thoseof the 12-coordinated La–O units, one may predict that the BO6 units dominatethe spectroscopic behaviour [24]. The FTIR spectrum of La1−xCaxFeO3 at x = 0.05(Fig. 2) exhibits the bands around 469,574 cm−1 with shoulders at 619, 684,722 and802 cm−1. The band at 469 cm−1 is assigned to the stretching vibration of Ca2+–O2−bond. The main band at 574 cm−1 is assigned to antisymmetric stretching vibrations

E.K. Abdel-Khalek, H.M. Mohamed
Fig. 2 The IR spectra ofLa1−xCaxFeO3 perovskitesamples
1000 900 800 700 600 500
869
793
717
671 63
9 549
451
Ca0.15
Wavenumber (cm-1)
% T
ran
smis
sio
n
796
574
719
678
617
570
468
Ca0.10
802
722
684
619
469
Ca0.05
of Fe–O–Fe bonds in FeO6 octahedra. The shoulder bands at 619, 684 and 722 cm−1
are assigned to the Fe–O symmetric stretching vibration of these octahedra whichinvolves internal motion of a change in Fe–O bond length [16, 24, 25]. The shoulderband at 802 cm−1 is assigned to the stretching vibration of La–O [15]. The FTIRspectrum of La1−xCaxFeO3 at x = 0.10 exhibits the same bands as for x = 0.05 with aslight decrease in the wavenumber. The FTIR spectrum of La1−xCaxFeO3 at x = 0.15(Fig. 2) exhibits the bands around 869, 793, 717, 671, 549, 451 cm−1 and a shoulderat 639 cm−1. It is known that in GdFeO3 [26] type structure (8 A–O) bonds amongthe 12 bonds become shorter and the other (4 A–O) bonds become longer graduallyas the size of the A cation decreases. Thus we can write the coordination numberfor the La3+ ion; in La1−xCaxFeO3; as 4 + 4 + 4 which is equivalent to 8 + 4 [15].For this reason the two bands at 869 and 793 cm−1 can be assigned to the stretchingvibration of La–O [15]. The appearance of band at around 717 cm−1 is due to theincrease of Fe–O constant force as a result of charge increase of iron ions (Fe4+ ionsformation) to conserve the electroneutrality of the perovskite structure in agreementwith Berger et al. [27]. The increase in the intensity of bands and decrease in thewavenumber (Fig. 2) with increasing Ca2+ ion concentration. This agreement withdecreasing in the unit cell volume as shown in Table 1. The decrease in the unit cellvolume is accompanied also by a decrease in the frequency of vibration which resultsfrom the difference in the atomic weight between the two ions (La and Ca) [15].
3.3 Mössbauer measurements
In order to get the detailed information about the charge states and local environ-ments of Fe nuclei, we carried out the Mössbauer spectroscopy measurements for

Synthesis, structural and magnetic properties of La1−xCaxFeO3...
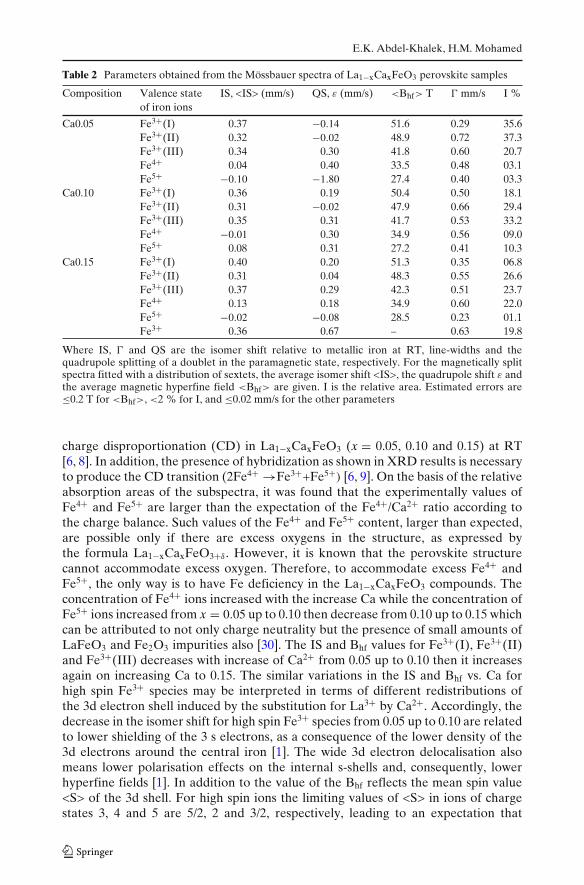
Fig. 3 The Mössbauer spectraof La1−xCaxFeO3 perovskitesamples recorded at RT
-10 -5 0 5 10
Tra
nsm
issi
on
(%
)
Velocity(mm/s)
Ca0.15
Ca0.10
Ca0.05
La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15) at RT, as shown in Fig. 3. From Fig. 3it can be seen that the spectra are composed of broad magnetically split sextets,which confirmed that all specimens are magnetically ordered (antiferromagnetic),in addition to the appearance of an intense paramagnetic doublet at x = 0.15. Thusthe Néel temperatures (TN) for these samples at x = 0.05 and 0.10 lie above RTwhile the quite blurred lines of x = 0.15 suggest that its Néel temperature is onlya little above room temperature [7, 28]. The hyperfine parameters of the samples,obtained from the best fitting results for these spectra of La1−xCaxFeO3 are shown inTable 2. From this table it can be seen that the Mössbauer spectra for La1−xCaxFeO3
are composed of five 6-line sub-spectra corresponding to Fe3+ (superposition of 3spectra due to different near-neighbour environments), Fe4+ and Fe5+ [8, 28]. Thefitted three sextets at x = 0.05 with isomer shift (IS) in the range of 0.32–0.37 mm/sand magnetic hyperfine field Bhf = 51.6, 48.9 and 41.8 T are assigned to the Fe3+(I),Fe3+(II) and Fe3+(III)) respectively with octahedral oxygen coordination and a highspin configuration [5, 9]. The fourth sextet with IS of 0.04 mm/s and Bhf = 33.5 T isassigned to be Fe4+ localized ions with a high spin configuration which is higher thanthe value of Bhf found in ref. [7] at RT. The fifth sextet with IS of −0.10 mm/s and Bhf
of 27.4 T is assigned to be Fe5+ ions which is in agreement with the parameters in ref.[1, 9] at 15 and 24 K respectively. According to the model proposed by Russo et al.[1] the existence of three sextets (Fe3+) is due to calcium ions substituting randomlyfor their near-neighbour La3+ ions to produce different magnetic environments. Thevery high values of the magnetic hyperfine field near TN result from charge, spin,and orbital ordering phases. In this case, however, the electron transfer is not asfast as in La1−xCaxFeO3, since the different oxidation states (3+, 4+ and 5+) areobserved as resolved magnetic sextets [29]. These results confirm the existence of
E.K. Abdel-Khalek, H.M. Mohamed
Table 2 Parameters obtained from the Mössbauer spectra of La1−xCaxFeO3 perovskite samples
Composition Valence state IS, <IS> (mm/s) QS, ε (mm/s) <Bhf> T � mm/s I %of iron ions
Ca0.05 Fe3+(I) 0.37 −0.14 51.6 0.29 35.6Fe3+(II) 0.32 −0.02 48.9 0.72 37.3Fe3+(III) 0.34 0.30 41.8 0.60 20.7Fe4+ 0.04 0.40 33.5 0.48 03.1Fe5+ −0.10 −1.80 27.4 0.40 03.3
Ca0.10 Fe3+(I) 0.36 0.19 50.4 0.50 18.1Fe3+(II) 0.31 −0.02 47.9 0.66 29.4Fe3+(III) 0.35 0.31 41.7 0.53 33.2Fe4+ −0.01 0.30 34.9 0.56 09.0Fe5+ 0.08 0.31 27.2 0.41 10.3
Ca0.15 Fe3+(I) 0.40 0.20 51.3 0.35 06.8Fe3+(II) 0.31 0.04 48.3 0.55 26.6Fe3+(III) 0.37 0.29 42.3 0.51 23.7Fe4+ 0.13 0.18 34.9 0.60 22.0Fe5+ −0.02 −0.08 28.5 0.23 01.1Fe3+ 0.36 0.67 – 0.63 19.8
Where IS, � and QS are the isomer shift relative to metallic iron at RT, line-widths and thequadrupole splitting of a doublet in the paramagnetic state, respectively. For the magnetically splitspectra fitted with a distribution of sextets, the average isomer shift <IS>, the quadrupole shift ε andthe average magnetic hyperfine field <Bhf> are given. I is the relative area. Estimated errors are≤0.2 T for <Bhf>, <2 % for I, and ≤0.02 mm/s for the other parameters
charge disproportionation (CD) in La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15) at RT[6, 8]. In addition, the presence of hybridization as shown in XRD results is necessaryto produce the CD transition (2Fe4+ →Fe3++Fe5+) [6, 9]. On the basis of the relativeabsorption areas of the subspectra, it was found that the experimentally values ofFe4+ and Fe5+ are larger than the expectation of the Fe4+/Ca2+ ratio according tothe charge balance. Such values of the Fe4+ and Fe5+ content, larger than expected,are possible only if there are excess oxygens in the structure, as expressed bythe formula La1−xCaxFeO3+δ . However, it is known that the perovskite structurecannot accommodate excess oxygen. Therefore, to accommodate excess Fe4+ andFe5+, the only way is to have Fe deficiency in the La1−xCaxFeO3 compounds. Theconcentration of Fe4+ ions increased with the increase Ca while the concentration ofFe5+ ions increased from x = 0.05 up to 0.10 then decrease from 0.10 up to 0.15 whichcan be attributed to not only charge neutrality but the presence of small amounts ofLaFeO3 and Fe2O3 impurities also [30]. The IS and Bhf values for Fe3+(I), Fe3+(II)and Fe3+(III) decreases with increase of Ca2+ from 0.05 up to 0.10 then it increasesagain on increasing Ca to 0.15. The similar variations in the IS and Bhf vs. Ca forhigh spin Fe3+ species may be interpreted in terms of different redistributions ofthe 3d electron shell induced by the substitution for La3+ by Ca2+. Accordingly, thedecrease in the isomer shift for high spin Fe3+ species from 0.05 up to 0.10 are relatedto lower shielding of the 3 s electrons, as a consequence of the lower density of the3d electrons around the central iron [1]. The wide 3d electron delocalisation alsomeans lower polarisation effects on the internal s-shells and, consequently, lowerhyperfine fields [1]. In addition to the value of the Bhf reflects the mean spin value<S> of the 3d shell. For high spin ions the limiting values of <S> in ions of chargestates 3, 4 and 5 are 5/2, 2 and 3/2, respectively, leading to an expectation that
Synthesis, structural and magnetic properties of La1−xCaxFeO3...
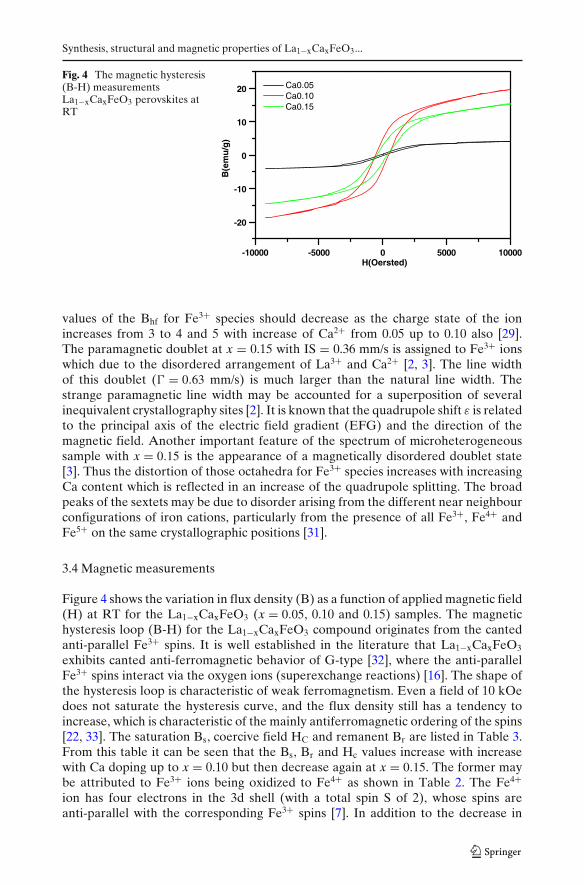
Fig. 4 The magnetic hysteresis(B-H) measurementsLa1−xCaxFeO3 perovskites atRT
-10000 -5000 0 5000 10000
-20
-10
0
10
20
H(Oersted)
B(e
mu
/g)
Ca0.05 Ca0.10 Ca0.15
values of the Bhf for Fe3+ species should decrease as the charge state of the ionincreases from 3 to 4 and 5 with increase of Ca2+ from 0.05 up to 0.10 also [29].The paramagnetic doublet at x = 0.15 with IS = 0.36 mm/s is assigned to Fe3+ ionswhich due to the disordered arrangement of La3+ and Ca2+ [2, 3]. The line widthof this doublet (� = 0.63 mm/s) is much larger than the natural line width. Thestrange paramagnetic line width may be accounted for a superposition of severalinequivalent crystallography sites [2]. It is known that the quadrupole shift ε is relatedto the principal axis of the electric field gradient (EFG) and the direction of themagnetic field. Another important feature of the spectrum of microheterogeneoussample with x = 0.15 is the appearance of a magnetically disordered doublet state[3]. Thus the distortion of those octahedra for Fe3+ species increases with increasingCa content which is reflected in an increase of the quadrupole splitting. The broadpeaks of the sextets may be due to disorder arising from the different near neighbourconfigurations of iron cations, particularly from the presence of all Fe3+, Fe4+ andFe5+ on the same crystallographic positions [31].
3.4 Magnetic measurements
Figure 4 shows the variation in flux density (B) as a function of applied magnetic field(H) at RT for the La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15) samples. The magnetichysteresis loop (B-H) for the La1−xCaxFeO3 compound originates from the cantedanti-parallel Fe3+ spins. It is well established in the literature that La1−xCaxFeO3
exhibits canted anti-ferromagnetic behavior of G-type [32], where the anti-parallelFe3+ spins interact via the oxygen ions (superexchange reactions) [16]. The shape ofthe hysteresis loop is characteristic of weak ferromagnetism. Even a field of 10 kOedoes not saturate the hysteresis curve, and the flux density still has a tendency toincrease, which is characteristic of the mainly antiferromagnetic ordering of the spins[22, 33]. The saturation Bs, coercive field HC and remanent Br are listed in Table 3.From this table it can be seen that the Bs, Br and Hc values increase with increasewith Ca doping up to x = 0.10 but then decrease again at x = 0.15. The former maybe attributed to Fe3+ ions being oxidized to Fe4+ as shown in Table 2. The Fe4+ion has four electrons in the 3d shell (with a total spin S of 2), whose spins areanti-parallel with the corresponding Fe3+ spins [7]. In addition to the decrease in

E.K. Abdel-Khalek, H.M. Mohamed
Table 3 Bs, Br and Hc forLa1−xCaxFeO3 perovskitesamples
Ca Bs(emu/g) Br(emu/g) Hc(Oe)
0.05 4.254 0.203 142.10.10 19.41 4.354 497.30.15 15.29 2.746 413.2
the Fe-O-Fe bond angle decreases the overlap between the Fe-O atomic states andleads to a low super-exchange interaction between Fe-Fe [34]. The decrease in Bs
from x = 0.10 up to 0.15 may be attributed to the decrease of the ferromagnetic(FM) interaction in Fe3+–Fe5+ as shown in Table 2. The coercivity (Hc; listed inTable 3) of a magnetic material is related to magnetic anisotropy [32]. Thus the largeincrease of coercive field (Hc) with increase Ca ions up to 0.10 can be attributed tothe interaction between ferromagnetic Fe3+–Fe5+ and antiferromagnetic Fe3+–Fe3+.
4 Conclusion
La1−xCaxFeO3 (x = 0.05, 0.10 and 0.15) nanostructural orthorhombic perovskitesynthesis has been achieved by co-precipitation process. XRD and FT-IR resultsare consistent with the structure electroneutrality of La1−xCaxFeO3 perovskites isachieved by an oxidation state of iron ions. The magnetic measurement shows theexistence of weak ferromagnetism and the mainly antiferromagnetic ordering of thespins in these samples. In this paper, we conclude for the first time the appearance ofa superposition of three distinct sextets which corresponded to the typical hyperfinefield values of Fe3+, Fe4+ and Fe5+ components at RT. The CD was observed also byMS. These results are related to the co-precipitation approach.
References
1. Russo, U., Nodari, L., Faticanti, M., Kuncser, V., Filoti, G.: Local interactions and electronicphenomena in substituted LaFeO3 perovskites. Solid State Ion. 176, 97 (2005)
2. Barbero, B.P., Andrade Gamboa, J., Cadús, L.E.: Synthesis and characterisation ofLa1−xCaxFeO3 perovskite-type oxide catalysts for total oxidation of volatile organic compounds.Appl. Catal. B 65, 21 (2006)
3. Isupova, L.A., Yakovleva, I.S., Gainutdinov, I.I., Pavlyukhin, Y.T., Sadykov, V.A.: Mössbauerstudies of the phase composition and microstructure of the La1−xCaxFeO3−y system as relatedto the reactivity of surface and bulk oxygen. React. Kinet. Catal. Lett. 81, 373 (2004)
4. Pecchi, G., Reyes, P., Zamora, R., Campos, C., Cadus, L.E., Barbero, B.P.: Effect of the prepa-ration method on the catalytic activity of La1−xCaxFeO3 perovskite-type oxides. Catal. Today133–135, 420 (2008)
5. Hudspeth, J.M., Stewart, G.A., Studer, A.J., Goossens, D.J.: Crystal and magnetic structures inPerovskite-related La1−xCaxFeO3−δ (x = 0.2, 0.33). J. Phys. Chem. Sol. 72, 1543 (2011)
6. Mogi, M., Inoue, Y., Arao, M., Koyama, Y.: Features of structural phase transition inLa1−xSrFeO3. Physica C 392–396, 295 (2003)
7. Liang, Y.-Q., Di, N.-l., Cheng, Z.-h.: Charge-disproportionation-induced magnetic glassy behav-ior in La0.5Ca0.5FeO3. Phys. Rev. B 72, 134416 (2005)
8. Liang, Y.-Q., Di, N.-L., Cheng, Z.-H.: Mössbauer spectroscopy as a probe of magnetic states inLa0.5Ca0.5FeO3−δ perovskite. J. Magn. Magn. Mater. 306, 35 (2006)
9. Barbero, B.P., Cadús, L.E., Marchetti, S.G.: Determination of Fe(IV) species in partially substi-tuted perovskite La0.6Ca0.4FeO3. Hyperfine Interact. 194, 367 (2009)

Synthesis, structural and magnetic properties of La1−xCaxFeO3...
10. Abdel-Khalek, E.K., EL-Meligy, W.M., Mohamed, E.A., Amer, T.Z., Sallam, H.A.: Studyof the relationship between electrical and magnetic properties and Jahn–Teller distortion inR0.7Ca0.3Mn0.95Fe0.05O3 perovskites. J. Phys. Condens. Matter 21, 026003 (2009)
11. Mostafa, A.G., Abdel-Khalek, E.K., Daoush, W.M., Hassaan, M.Y.: Structural, magnetic andelectrical transport properties of the La0.70Sr0.30Mn57
0.96Fe0.04O3+δ perovskite. Hyperfine Inter-act. 184, 167 (2008)
12. Vandenberghe, R.E., De Grave, E., de Bakker, P.M.A.: On the methodology of the analysis ofMössbauer spectra. Hyperfine Interact. 83, 29 (1994)
13. Geller, S., Wood, E.A.: Crystallographic studies of perovskite-like compounds. I. Rare earthorthoferrites and YFeO3, YCrO3, YAlO3. Acta Crystallogr. 9, 563 (1956)
14. Sadykov, V.A., Isupova, L.A., Tsybulya, S.V., Cherepanova, S.V., Litvak, G.S., Burgina, E.B.,Kustova, G.N., Kolomiichuk, V.N., Ivanov, V.P., Paukshtis, E.A., Golovin, A.V., Avvakumov,E.G.: Effect of mechanical activation on the real structure and reactivity of iron (III) oxide withcorundum-type structure. J. Solid State Chem. 123, 191 (1996)
15. Ahmed, M.A., Seoudi, R., El-dek, S.I.: Spectroscopic and structural analysis of Ca substituted Laorthoferrite. J. Mol. Struct. 754, 41 (2005)
16. Bellakki, M.B., Kelly, B.J., Manivannan, V.: Synthesis, characterization, and property studies of(La, Ag)FeO3 (0.0 ≤ x ≤ 0.3) perovskites. J. Alloys Compd. 489, 64 (2010)
17. Barbero, B.P., Gamboa, J.A., Cadus, L.E.: Synthesis and characterisation of La1−xCaxFeO3perovskite-type oxide catalysts for total oxidation of volatile organic compounds. Appl. Catal.B Environ. 65, 21 (2006)
18. Ciambelli, P., Cimino, S., De Rossi, S., Lisi, L., Minelli, G., Porta, P., Russo, G.: AFeO3 (A=La,Nd, Sm) and LaFe1−xMgxO3 perovskites as methane combustion and CO oxidation catalysts:structural, redox and catalytic properties. Appl. Catal. B: Environmental 29, 239 (2001)
19. Shannon, R.D.: Crystal physics, diffraction, theoretical and general crystallography. Acta Cryst.A32, 751 (1976)
20. Shimony, U., Knudsen, J.M.: Mössbauer Studies on Iron in the Perovskites La1-xSrxFeO3 (0 <
x < 1). Phys. Rev. 144, 361 (1966)21. Yo, C.H., Jung, I.Y., Ryu, K.H., Ryu, K.S., Choy, J.H.: A study of the nonstoichiometry and
physical properties of the perovskite Nd1−xCaxFeO3−y system. J. Solid State Chem. 114, 265(1995)
22. Li, J., Xinli, K., Yong, Q., He, H.: Microstructure and magnetic properties of La1−xSrxFeO3nanoparticles. Phys. Stat. Sol. 191, 255 (2002)
23. Ross, S.D.: Inorganic Infrared and Raman Spectra. McGraw-Hill, London (1972)24. Lavat, A.E., Baran, E.J.: IR-spectroscopic characterization of A2BB′O6 perovskites. Vibrat.
Spectrosc. 32, 167 (2003)25. Davydov, A.: Infrared Spectroscopy of Adsorbed Species on the Surface of Transition Metal
Oxides, Chapter 1. Wiley, England (1990)26. Kim, J., Demazeau, G., Presniakov, I., Choy, J.H.: Structural distortion and chemical bonding in
TlFeO3: comparison with AFeO3 (A=rare earth). J. Solid State Chem. 161, 197 (2001)27. Berger, D., Fruth, V., Jitaru, I., Schoonman, J.: Synthesis and characterisation of La1−xSrxCoO3
with large surface area. Mater. Lett. 58, 2418 (2004)28. Li, J.: Investigation of Orthorhombic Perovskite La1−xCaxFeO3−y (0<=x<=0.50). Phys. Scr. 45, 62
(1992)29. Al-Rawwas, A.D., Johnson, C.E., Thomas, M.F., Dann, S.E., Weller, M.F.: Mössbauer studies on
the series La1−xSrxFeO3. Hyperfine Interact. 93, 1521 (1994)30. Morimoto, S., Yamanaka, T., Tanaka, M.: Structure and electron density distribution of CaFeO3
in the “charge disproportionate” state. Phys. B 67, 237 (1997)31. Dann, S.E., Currie, D.B., Weller, M.T., Thomas, M.F., Al-Rawwas, A.D.: The effect of oxygen
stoichiometry on phase relations and structure in the system La1−xSrxFeO3−δ (0 ≤ x ≤ 1, 0 ≤ δ ≤0.5). J. Solid State Chem. 109, 134 (1994)
32. Shein, I.R., Shein, K.I., Kozhevnikov, V.L., Ivanovskii, A.L.: Band structure and the magneticand elastic properties of SrFeO3 and LaFeO3 perovskites. Phys. Solid State 47, 2082 (2005)
33. Treves, D.: Studies on orthoferrites at the Weizmann Institute of Science. J. Appl. Phys. 36, 1033(1965)
34. Yang, J.B., Yelon, W.B., James, W.J., Chu, Z., Kornecki, M., Xie, Y.X., Zhou, X.D., Anderson,H.U., Joshi, A.G., Malik, S.K.: Crystal structure, magnetic properties, and Mössbauer studiesof La0.6Sr0.4FeO3−δ prepared by quenching in different atmospheres. Phys. Rev. B 66, 184415(2002)