
Posttranslational processing of concanavalin A precursors in jackbean cotyledons

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Synthesis and characterization of Li(I)–M(II) (M = Co, Ni) heterometalliccomplexes as molecular precursors for LiMO2
Zhanna Dobrokhotova a, Anna Emelina b, Aleksei Sidorov a, Grigory Aleksandrov a, Mikhail Kiskin a,⇑,Pavel Koroteev a, Mikhail Bykov b, Marat Fazylbekov b, Artem Bogomyakov c, Vladimir Novotortsev a,Igor Eremenko a
a N.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninsky Prosp. 31, 119991 Moscow, GSP-1, Russian Federationb Department of Chemistry, M.V. Lomonosov Moscow State University, 1 Leninskie Gory, 119992 Moscow, Russian Federationc Institute International Tomography Centre, Siberian Branch of the Russian Academy of Sciences, Institutskaya Str. 3a, 630090 Novosibirsk, Russian Federation
a r t i c l e i n f o
Article history:Received 21 August 2010Accepted 30 September 2010Available online 17 November 2010
Keywords:Heterometallic complexCarboxylate ligandsX-ray analysisThermal decomposition
a b s t r a c t
Lithium-containing heterometallic complexes with cobalt (Li2Co2(Piv)6(2,4-Lut)2 (2, Piv is the pivalateanion) and Li2Co2(O2CCH2But)6(2,4-Lut)2 (3)) and with nickel (Li2Ni2(Piv)6(DME)2 (4) and Li2Ni2(Piv)6
(2,20-bpy)2 (5)) were synthesized. The structures of the complexes were established by X-ray diffraction.The magnetic properties of complexes 2 and 4 were studied. The thermal behavior of compounds 2, 3, and5 was investigated. It was shown that the compounds under study can be used as molecular precursorsfor the synthesis of lithium cobaltate and nickelate.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
It is only in the recent past that polynuclear coordination com-pounds have attracted attention of chemists working on the designof new materials. Investigations of the methods for the chemicalassembly of target molecules of a particular composition and eventhe structures not only have a great potential for the solutionof fundamental problems of chemistry and physics of this classof complex compounds but also provide a route to the design ofnew materials based on these compounds [1].
In the synthesis of complex oxide systems with the use of het-erometallic coordination compounds, the composition of the targetoxide and, probably, its properties, can be specified in the step ofthe formation of a molecular precursor yielding the oxide underrather mild conditions (at temperatures below 500 �C) after the re-moval of the ‘‘organic moiety” of the molecule. This approach hasattracted particular attention. Thus, the thermal behavior of thecoordination compounds [Fe2Ni(C4H4O5)2.5(OH)2]NO3�5H2O, [Fe2-
Ni(C4H8O3N2)4](NO3)8�24H2O, and (NH4)[Fe2Ni(C4H4O5)3(OH)3]�3H2O, which are formally potential precursors for nickel ferrite,was investigated [2]. Heteropolynuclear copper and zinc com-plexes with ethylenediamine [3] and the heterometallic CuII/MnII
oxalate complex with ethylenediamine [4] are the starting com-pounds for the synthesis of inorganic ceramics, which are used as
electrocatalysts for the reduction of oxygen. It was shown thatmixed-metal Cu–Co and Ni–Cu oxide thin films can be preparedstarting from molecular heterometallic cubane-type precursors [5].
An important requirement for heterometallic molecules used asthe starting compounds for the generation of the target materials isthat the ratio between metals should remain unchanged duringthermolysis. The complexes Li(H2O)M(N2H3CO2)3]�0.5H2O (M = Coor Ni), whose thermolysis affords materials of the compositionLiMO2 [6], are of interest in this respect. Recently, we have shownthat the thermal decomposition (400–500 �C) of the heterometalliccoordination polymer [Li2Co2(Piv)6(l-L)2]n (1, L = 2-amino-5-methylpyridine, Piv is the pivalate anion) and the molecularcomplex Li2Co2(Piv)6(NEt3)2, which were used as the starting pre-cursors, gave LiCoO2 [7] in almost quantitative yield.
The aim of the present study was to search for new molecularsystems, which can be used as molecular precursors for lithiumcobaltate and nickelate LiMO2 in the mild thermolysis. It is neces-sary that the organic components of the selected heterometalliccomplexes should be easily eliminated with the ratio betweenmetals remaining unchanged in the final product.
2. Experimental
2.1. Synthesis
Commercial reagents and solvents (MeCN, EtOH, THF, and DME)were used without additional purification. The starting complexes
0277-5387/$ - see front matter � 2010 Elsevier Ltd. All rights reserved.doi:10.1016/j.poly.2010.09.040
⇑ Corresponding author. Tel.: +7 495 955 4817; fax: +7 495 952 1279.E-mail address: [email protected] (M. Kiskin).
Polyhedron 30 (2011) 132–141
Contents lists available at ScienceDirect
Polyhedron
journal homepage: www.elsevier .com/locate /poly

Author's personal copy
Co2(Piv)4(2,4-Lut)2, Ni9(OH)6(Piv)12(HPiv)4, and Ni2(H2O)(Piv)4
(2,20-bpy)2 were synthesized according to procedures describedpreviously [8–10]. Commercial tert-butylacetic acid (Acros organ-ics), LiOH (Alfa Aesar), 2,20-dipyridyl (Alfa Aesar), and 2,4-lutidine(Acros organics) were used for the synthesis of new compounds.
2.1.1. Li2Co2(Piv)6(2,4-Lut)2 (2)Tetrahydrofuran (30 mL) was added to a mixture of
Co2(Piv)4(2,4-Lut)2 (0.5 g, 0.68 mmol) and LiPiv (0.15 g, 1.35 mmol)(LiPiv was synthesized by the metathesis reaction of LiOH withHPiv). The reaction mixture was heated at 60 �C for 30 min. Theresulting violet solution was filtered, concentrated to 10 mL, andcooled to room temperature. The violet crystals suitable for X-raydiffraction that precipitated after 24 h were separated from the solu-tion by decantation, washed with cold THF, and dried in air. The yieldof compound 2 was 0.62 g (95%). Anal. Calc. for C44H72Co2Li2N2O12
(%): C, 55.5; H, 7.6; N, 2.9. Found: C, 55.5; H, 7.5; N, 3.0%. IR (KBr,cm�1): 2964 s, 2924 m, 2872 m, 1652 m, 1608 s, 1576 s, 1564 s,1484 s, 1460 m, 1420 s, 1400 s, 1360 s, 1300 m, 1260 m, 1224 s,1176 v.w, 1100 w, 1028 m, 928 w, 904 w, 892 w, 832 m, 792 s, 756w, 668 v.w, 616 s, 576 m, 544 v.w, 472 s, 448 s, 432 s.
2.1.2. Li2Co2(O2CCH2But)6(2,4-Lut)2 (3)A solution of 2,4-lutidine (0.18 g, 1.68 mmol) in ethanol (50 mL)
was added to a mixture of CoCl2�6H2O (0.4 g, 1.68 mmol) andLiO2CCH2But (0.41 g, 3.36 mmol) (LiO2CCH2But was synthesizedby the metathesis reaction of LiOH with HO2CCH2But). The reactionmixture was heated at 80 �C for 40 min. The resulting green–bluesolution was filtered and concentrated to dryness. Acetonitrile(30 mL) was added to the precipitate, and then LiO2CCH2But
(0.205 g, 1.68 mmol) was added. The reaction mixture was heatedat 80 �C for 30 min. The resulting violet solution was filtered, con-centrated to 20 mL, and cooled to room temperature. The violetcrystals suitable for X-ray diffraction that precipitated after 24 hwere separated from the solution by decantation, washed withcold MeCN, and dried in air. The yield of compound 3 was 0.69 g(80%). Anal. Calc. for C50H84Co2Li2N2O12 (%): C, 57.9; H, 8.2; N,2.7. Found: C, 57.8; H, 8.0; N, 2.7%. IR (KBr, cm�1): 2960 s, 2904m, 2868 m, 1668 w, 1624 s, 1616 s, 1564 s, 1508 w, 1476 m,1452 m, 1432 m, 1420 m, 1932 s, 1364 m, 1296 m, 1272 w,1232 w, 1196 w, 1176 v.w, 1152 v.w, 1136 v.w, 1044 w, 1024w, 928 v.w, 912 v.w, 836 w, 804 w, 736 w, 648 w, 632 w, 548v.w, 464 m, 444 w, 416 v.w.
2.1.3. Li2Ni2(Piv)6(DME)2 (4)Dimethoxyethane (20 mL) was added to a mixture of Ni9(OH)6(-
Piv)12(HPiv) (0.4 g, 0.17 mmol) and LiPiv (0.17 g, 1.54 mmol). Thereaction mixture was heated at 80 �C for 30 min. The resultinggreen solution was filtered, concentrated to 10 mL, and cooled toroom temperature. The green crystals suitable for X-ray diffractionthat precipitated after 24 h were separated from the solution bydecantation, washed with cold DME, and dried in air. The yield ofcompound 4 was 0.64 g (90%). Anal. Calc. for C38H74Li2Ni2O16 (%):C, 49.7; H, 8.1. Found: C, 49.7; H, 8.0%. IR (KBr, cm�1): 2956 s,2928 m, 2864 m, 2836 w, 1684 w, 1664 m, 1624 s, 1584 s, 1572s, 1536 s, 1484 s, 1460 m, 1412 s, 1356 s, 1280 w, 1228 s, 1196w, 1168 v.w, 1120 m, 1096 s, 1076 s, 1028 m, 1016 m, 940 w,908 m, 892 m, 868 s, 836 w, 812 w, 796 s, 612 s, 576 w, 536 w,468 s, 428 s, 408 s.
2.1.4. Li2Ni2(Piv)6(2,20-bpy)2 (5)2.1.4.1. Method A. Acetonitrile (30 mL) was added to a mixture ofLi2Ni2(Piv)6(DME)2 (4) (0.5 g, 0.54 mmol) and 2,20-dipyridyl(0.17 g, 1.08 mmol). The reaction mixture was heated at 80 �C for30 min. The resulting green solution was filtered, concentrated to10 mL, and cooled to room temperature. The green crystals suitable
for X-ray diffraction that precipitated after 24 h were separatedfrom the solution by decantation, washed with cold MeCN, anddried in air. The yield of compound 5 was 0.51 g (90%). Anal. Calc.for C50H70Li2N4Ni2O12 (%): C, 57.2; H, 6.7; N, 5.3. Found: C, 57.2; H,6.5; N, 5.2%. IR (KBr, cm�1): 2960 s, 2928 m, 2868 m, 1608 s, 1560 s,1536 m, 1484 s, 1476 s, 1460 m, 1444 m, 1432 s, 1416 s, 1372 m1356 m, 1312 w, 1284 v.w, 1224 s, 1172 w, 1156 w, 1104 v.w,1056 w, 1024 w, 908 m, 888 m, 816 w, 796 m, 768 s, 736 m, 656m, 636 w, 604 s, 564 w, 536 v.w, 464 s, 448 s, 416 s.
2.1.4.2. Method B. Acetonitrile (35 mL) was added to a mixture ofNi2(H2O)(Piv)4(2,20-bpy)2 (0.4 g, 0.55 mmol) and LiPiv (0.12 g,1.1 mmol). The reaction mixture was heated at 80 �C for 30 min.The resulting green solution was filtered, concentrated to 15 mL,and cooled to room temperature. The green crystals suitable forX-ray diffraction that precipitated after 24 h were separated fromthe solution by decantation, washed with cold MeCN, and driedin air. The yield of compound 5 was 0.52 g (90%). Anal. Calc. forC50H70Li2N4Ni2O12 (%): C, 57.2; H, 6.7; N, 5.3. Found: C, 57.1; H,6.6; N, 5.2%. IR (KBr, cm�1): 2960 s, 2928 m, 2868 m, 1608 s,1560 s, 1536 m, 1484 s, 1476 s, 1460 m, 1444 m, 1432 s, 1416 s,1372 m 1356 m, 1312 w, 1284 v.w, 1224 s, 1172 w, 1156 w,1104 v.w, 1056 w, 1024 w, 908 m, 888 m, 816 w, 796 m, 768 s,736 m, 656 m, 636 w, 604 s, 564 w, 536 v.w, 464 s, 448 s, 416 s.
2.2. Methods
The spectra were measured on a Specord M-80 IR spectrometerin KBr pellets. The elemental analysis was carried out on a CarloErba instrument. The magnetochemical measurements were per-formed on a Quantum Design MPMS-5S SQUID magnetometer inthe temperature range of 2–300 K in a magnetic field of up to5 kOe. The calculated molar magnetic susceptibility v was cor-rected for the diamagnetic contribution. The effective magneticmoment was calculated by the formula leff = (8vT)1/2.
2.3. X-ray data collection
The X-ray data sets for complexes 2 and 3 were collected on aBruker SMART APEX II diffractometer equipped with a CCD cameraand a graphite monochromated Mo Ka radiation source(k = 0.71073 Å) [11]. The X-ray diffraction study of complexes 4and 5 was carried out on an Enraf Nonius CAD-4 diffractometer(graphite monochromator, k = 0.71073 Å, x-scanning technique,the scan step was 0.3�, the exposure time per frame was 10 s)according to a standard technique [12]. Semi-empirical absorptioncorrections for all complexes were applied [13]. The structureswere solved by direct methods and using Fourier techniques andwere refined by the full-matrix least squares against F2 with aniso-tropic thermal parameters for all non-hydrogen atoms. The tert-butyl substituents at the carboxylate groups in 4 and 5 are partiallydisordered. The positions of all methyl carbon atoms in the disor-dered CMe3 fragments were located in difference Fourier maps andrefined with occupancies of 0.660(9) and 0.340(9) for the tert-butylgroup at the atom C(2) and 0.611(5) and 0.389(5) at the atom C(7)for 4 and with occupancies of 0.809(7) and 0.191(7) for the tert-bu-tyl group at the atom C(7) for 5. The hydrogen atoms of the carbon-containing ligands in compounds 2–5 were positioned geometri-cally and refined using the riding model. All calculations were car-ried out with the use of the SHELX97 program package [14]. Thecrystallographic parameters and the refinement statistics are givenin Table 1.
The X-ray powder diffraction analysis of the solid products ofthe decomposition in air was carried out on a G670 (HUBER) Gui-nier camera using Cu Ka1 radiation. The X-ray powder diffractionanalysis of the solid products of the decomposition under an inert
Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141 133

Author's personal copy
atmosphere was performed on a FR-552 monochromator chamber(Cu Ka1 radiation) using germanium as the internal standard(X-ray diffraction patterns were processed on an IZA-2 comparatorwith an accuracy of ±0.01 mm).
2.4. Thermal analysis
Thermoanalytical measurements were carried out on NETZSCHTG 209 F1 and NETZSCH DSC 204 F1 instruments under a flow(20 mL/min) of artificial air (O2, (20.9 ± 0.5) vol.%; N2, (79.1 ± 0.5)vol.%; CH4, CO, CO2, <0.005 vol.%) and argon (Ar, >99.998%; O2,0.0002%; N2, <0.001%; aqueous vapor, <0.0003%; CH4, <0.0001%).The samples were weighed on a SARTORIUS RESEARCH R 160Panalytical balance with an accuracy of 1 � 10�2 mg. The calorime-ter was calibrated against the ISO 11357-1 standard.
The thermal decomposition of the compounds was studied bydifferential scanning calorimetry (DSC) and thermogravimetricanalysis (TGA) at a heating rate of 10 K/min. The composition ofthe gas phase at temperatures below 250 �C was studied on anAëolos QMS 403C mass spectrometer under TGA conditions; theionizing electron energy was 70 eV; the largest determined massnumber (the mass-to-charge ratio) was 300 amu. Each experimentwas repeated at least three times.
The relaxation parameters of the glassy crystals formed by thecomplex Li2Ni2(Piv)6(2,20-bpy)2 (5) were measured by DSC in astream of dry argon (10 mL/min). The cooling and heating ratesof the HR/CR measurement system were varied from 2 to 20 K/min; the final temperature of cooling Tmin was varied from 120to 220 K. The characteristics of the anomalies were measured dur-ing heating. Both the starting polycrystalline samples (the graindiameter was about 10�4 m) and the samples subjected to themechanical treatment (samples were ground in an agate mortarto a grain diameter of about 10�6 m and pressed under a pressureof 5 atm to (1–2) � 10�3 m thick pellets 4 � 10�3 m in diameter)were used.
The experimental data for the estimation of the activation en-ergy of the relaxation were obtained from the pressed samplesaccording to the following scheme. Samples were annealed at
420 K for 15 min, cooled at a rate of 20 K/min to Ta (240, 250,255, 260, 270 K), and kept at this temperature during differentperiods of time ta (5, 10, 15, 20, 30, 60 min). All measurementswere carried out during heating. Each measurement was repeatedat least three times. The results were statistically processed. Theconfidence interval for the temperature anomaly was 2–6 �C; theconfidence interval for the jump in the heat capacity was at most10%.
2.5. Calculations
The data derived from thermal analysis experiments were pro-cessed according to the ISO 11357-1, ISO 11357-2, ISO 11358, andASTM E 1269-95 standards with the use of the NETZSCH ProteusThermal Analysis software.
The fictive glass transition temperature TF was determined bythe Moynihan method [15].
The equations for the estimation of the relaxation parameterswere taken from the publication [16]. The average activation en-ergy of the relaxation EA was calculated by the Arrhenius-Frenkelequation
s0ðTaÞ ¼ s0;rðTaÞ expEA
RTa
� �ð1Þ
where s0(Ta) is the relaxation time of the system in an arbitraryequilibrium state (ta ?1). In this case, by the equilibrium state ismeant the metastable phase of the supercooled high-temperaturemodification of the complex. The points s0 for each Ta were deter-mined by the extrapolation of the plot of the relaxation time ofthe system s(ta, Ta) versus dH according to Eq. (2), which directly fol-lows from the KAHR (Kovacs-Aklonis-Hutchinson-Ramos) model.
ln1
sðta; TaÞ¼ ln
1s0ðTaÞ
þ C � dHðta; TaÞ ð2Þ
The parameter C can be considered as a constant in a narrow ta
range.The parameter dH(ta, Ta) was calculated from the experimental
heat capacity curves using the following equations:
Table 1Crystallographic parameters and the structure refinement statistics for complexes 2–5.
Complex/parameter 2 3 4 5
Formula C44H72Co2Li2N2O12 C50H84Co2Li2N2O12 C38H74Li2Ni2O16 C50H70Li2N4Ni2O12
Formula weight (g mol�1) 952.78 1036.93 918.27 1050.4T (K) 143(2) 293(2) 293(2) 293(2)Wavelength (Å) 0.71073 0.71073 0.71073 1.5418Crystal system Triclinic Monoclinic Monoclinic TriclinicSpace group P�1 P21/c P21/n P�1a (Å) 10.4861(15) 14.7417(18) 13.5510(15) 11.2909(10)b (Å) 11.2572(15) 16.665(2) 13.1504(16) 11.5246(10)c (Å) 12.6195(16) 13.7199(17) 14.2354(17) 12.5884(10)a (�) 109.769(4) 90 90 90.076(10)b (�) 91.021(4) 115.9720(10) 94.592(8) 102.299(10)c (�) 108.859(4) 90 90 119.028(10)V (Å3) 1313.0(3) 3030.2(7) 2528.6(5) 1388.9(2)Z 1 2 2 1Dcalc (g cm�3) 1.205 1.136 1.206 1.256l (mm�1) 0.686 0.599 0.803 1.326Total no. of reflections/unique 7112/5106 26671/9051 4909/4909 5590/5590Rint 0.0884 0.0541 0 0Tmin/max 0.7953/0.9041 0.7826/0.9050 0.6666/0.8959 0.5995/0.7328hmax (�) 26.37 30.54 26 74.96Goodness-of-fit (GOF) 0.963 1.062 1.048 0.97R1 (I > 2r(I)) 0.0725 0.0623 0.0392 0.0699wR2 (I > 2r(I)) 0.1495 0.1379 0.1255 0.2075R1 (all data) 0.1842 0.1618 0.0495 0.0805wR2 (all data) 0.1707 0.1592 0.1361 0.224
134 Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141

Author's personal copy
dHðta; TaÞ ¼ DH1ðta !1; TaÞ � DHðta; TaÞ ð3ÞDHðta; TaÞ ¼ Hun � Hag ð4ÞDH1ðta !1; TaÞ ¼ Hun � HHT ð5Þ
DHðta; TaÞ ¼Z T>Tg
Ta
ðCp;agðTÞ � Cp;unðTÞÞdT ð6Þ
DH1ðta !1; TaÞ ¼Z T>Tg
Ta
ðCp;HTðTÞ � Cp;unðTÞÞdT ð7Þ
where the indices ag, un, and HT correspond to the phase obtainedafter the annealing at Ta for ta, the phase obtained at ta = 0, and thesupercooled high-temperature phase at Ta, respectively. The plotCp,HT(T) was obtained by the extrapolation of the experimentalcurve for the high-temperature phase to the low-temperatureregion.
The left part of Eq. (2) was determined by the approximation ofthe experimental data with the use of the Williams–Watts functionon the assumption that the relaxation time depends only slightlyon the annealing time of the sample, and the non-exponentialityparameter is close to the unity.
The sums of the barriers to the rotation of the tert-butyl groupswere calculated by quantum chemical methods. The quantumchemical calculations were carried out by the density functionaltheory (DFT) method with the use of the PBE [17] and B3LYP[18] exchange–correlation functionals. The calculations employingthe PBE functional were carried out with the use of the PRIRODA pro-gram [19]. The calculations employing the B3LYP functional wereperformed with the use of the PCGamess/Firefly QC package [20],which is partially based on the GAMESS (US) [21] source code.The preliminary geometry optimization of the molecule was per-formed using the PBE functional and the VDZP basis set [22]. Thestructure determined by X-ray diffraction was used as the initialapproximation. For the determined equilibrium configuration ofthe nuclear subsystem, the matrix of the second derivatives ofthe energy with respect to the coordinates was calculated analyti-cally. The absence of the negative eigenvalues indicates that theresulting configuration corresponds to a minimum on the potentialenergy surface. The further optimization was carried out using theB3LYP functional and the TZVPP basis set for lithium and nickelatoms [23] and the 6-311G** basis set for the other atoms [24].The barriers to the rotation were calculated by the relaxed poten-tial energy surface scan along the normal coordinates that corre-spond to the rotation of the tert-butyl groups (the dihedral anglebetween the plane passing through the carboxyl oxygen atom,the carboxyl carbon atom, and the quaternary carbon atom andthe plane passing through the carboxyl carbon atom, the quater-nary carbon atom, and the methyl carbon atom). The relaxed scaninvolves the search for the equilibrium configuration of the nuclearsubsystem at several fixed values of the scan coordinate. In spite ofthe absence of the full local symmetry of the tert-butyl groups, thepreliminary calculations showed that the difference between thebarriers to the rotation as the torsion angle changes by 120�,240�, and 360� is negligibly small. Hence, the subsequent scanwas carried out by changing the angle by 8� in a range of 0–120�.
3. Results and discussion
3.1. Synthesis
We chose the compounds Li2Co2(Piv)6(2,4-Lut)2 (2) and Li2-
Co2(O2CCH2But)6(2,4-Lut)2 (3) as the possible molecular precursorsfor the synthesis of LiCoO2. Compound 2 was synthesized in virtu-ally quantitative yield by the insertion of lithium pivalate into thedinuclear tetrabridged complex Co2(Piv)4(2,4-Lut)2, and relatedcomplex 3 with tert-butylacetate anions was synthesized by the
self-assembly from cobalt chloride and an excess of the corre-sponding lithium carboxylate.
Attempts to synthesize nickel-containing analogues of Li–Cocomplexes 2 and 3 according to the above-described proceduresfailed. One of the possible factors destabilizing the heteronuclearnickel-containing molecules analogous to cobalt-containing com-pounds 2 and 3 is that the d metal in these structures is in a trigo-nal–bipyramidal ligand environment, whereas the coordinationnumber of Ni in most of NiII carboxylates (except for the knowndinuclear tetrabridged complexes LNi(l-OOCR)4NiL) is 6 [9,10,25].
It appeared that the octahedral environment of the nickel atomscan easily be achieved with the use of chelating bidentate ligandsin the Li2M2(Piv)6L2 molecule (M = Ni, L = is a bidentate chelatingligand). Thus, the reaction of Ni9(OH)6(Piv)12(HPiv)4 with an excessof LiPiv in dimethoxyethane affords Li2Ni2(Piv)6(DME)2 (4), whichreacts with 2,20-bpy to give its analogue, Li2Ni2(Piv)6(2,20-bpy)2
(5). It should be noted that compound 5 can be alternativelysynthesized by the insertion of lithium pivalate into the knowndinuclear complex Ni2(H2O)(Piv)4(2,20-bpy)2 [10].
3.2. Structural studies
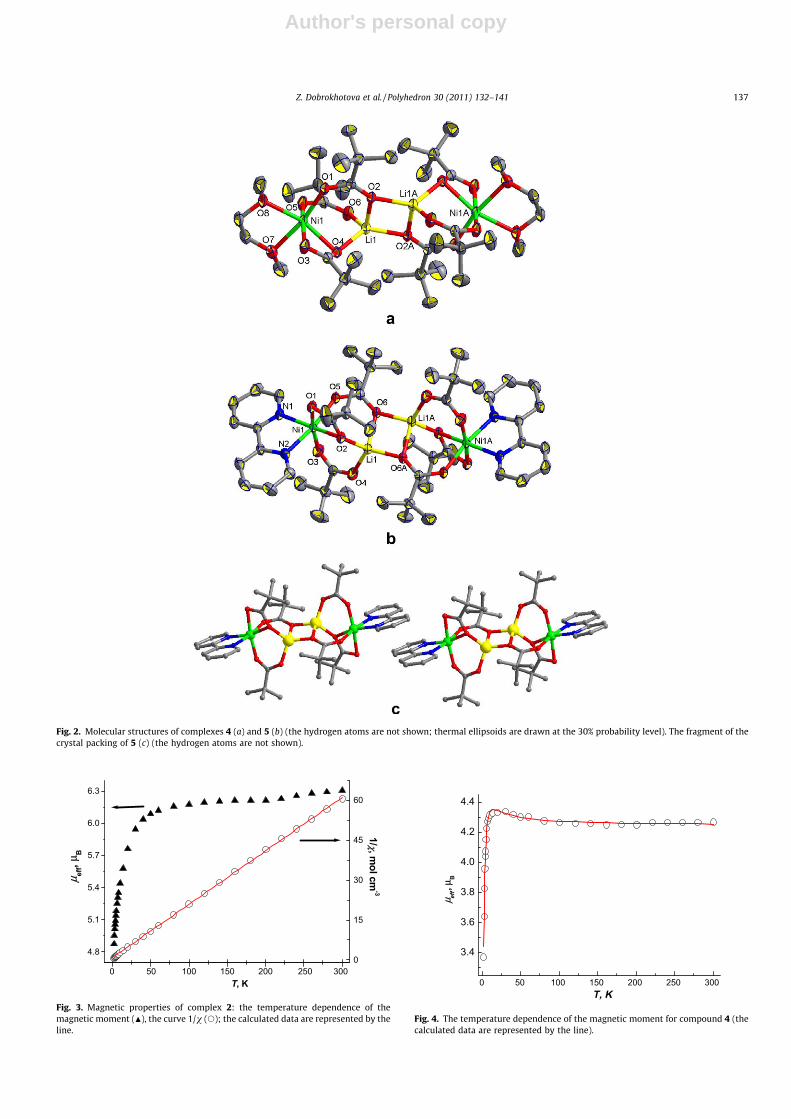
The molecules of both complexes are centrosymmetric and arestructurally similar. The center of symmetry of the molecules is atthe intersection of the diagonals of the almost square O2Li2 moiety.The CoLiLiCo metal core has a structure of a zigzag chain, whoseatoms are in the same plane. Molecules 2 and 3 consist of twoidentical LiCo(O2CR)3(2,4-Lut) moieties (Fig. 1), in which cobaltatoms are bound to the nitrogen atoms of the pyridine ligands (se-lected geometric parameters for 2 and 3 are given in Table 2). Thecobalt and lithium atoms are linked together by three carboxylatebridges of the anion. The bidentate-bridging anions are bound to
Fig. 1. Molecular structures of complexes 2 (a) and 3 (b) (the hydrogen atoms arenot shown; thermal ellipsoids are drawn at the 30% probability level).
Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141 135

Author's personal copy
the metals in a different fashion. Two anions act as bridges be-tween one Co atom and one Li atom, and one anion is l3-biden-tate-bridging and links two Li atoms through one O atom. Thisprovides the binding of two {Li–Co} moieties by two Li–O bonds.In complex 2, each cobalt atom is in a strongly distorted tetrahe-dral environment formed by three oxygen atoms of the bridgingpivalate anions and the nitrogen atom of the neutral N-donor li-gand. The N–Co–O angles (97.0(2)�, 98.3(2)�, and 100.9(2)�) aresubstantially smaller than the O–Co–O angles (105.3(2)�,117.0(2)�, and 130.4(2)�), which is indicative of the strong distor-tion of the O3N tetrahedron almost toward a trigonal pyramid.The tetrahedral environment of the cobalt atoms is completed bythe weakly coordinated O atom of one of the bridging groups(Co� � �O(6), 2.634(5) Å). As a result of the chelate-ring closure ofone of the bridging groups, the adjacent Co and Li atoms are rigidlyfixed. In addition, the coordination of one of the bridging anions tothe second lithium atom additionally restricts its conformationalflexibility, resulting in the unexpectedly high rigidity of the geom-etry of the Co–Li–Li–Co zigzag chain. As opposed to complex 2, thecoordination environment of the Co atoms in complex 3 can be de-scribed as a distorted tetrahedron; the coordination environmentof the Li atoms is also tetrahedral.
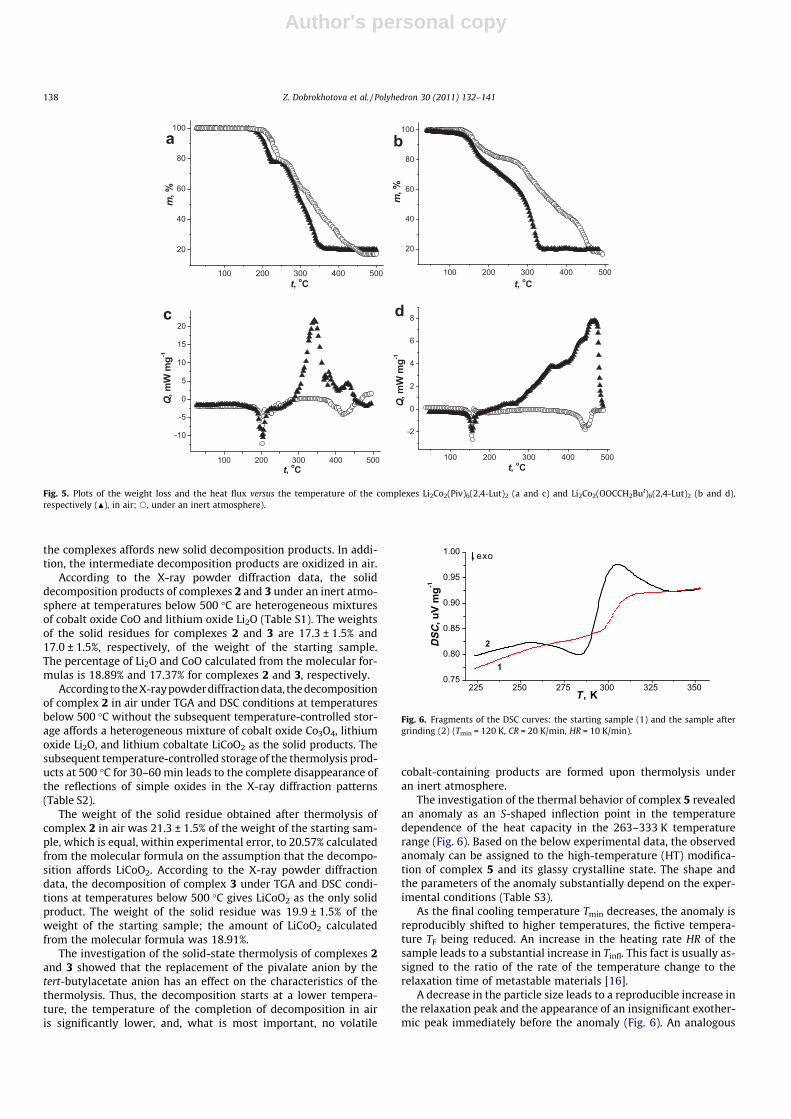
According to the X-ray diffraction data, compounds 4 and 5 arestructurally similar. The metal core of the nickel-containing com-plexes {Li2Ni2(Piv)6} is similar to {Li2Co2(O2CR)6} in tetranuclearcomplexes 2 and 3 (Fig. 2, selected geometric parameters of com-plexes 4 and 5 are given in Table 2). The only difference is thatthe carboxylate group acting as a bridge has a pronounced chelat-ing function. It should also be noted that the neutral donor ligands(DME and 2,20-bpy) are coordinated in a chelating mode, resultingin a distorted octahedral environment of the Ni atoms. In the crys-tal structure of 5, there are stacking interactions between the coor-dinated 2,20-bpy ligands of adjacent complexes (d � 3.5 Å) (Fig. 2c).
3.3. Magnetic properties
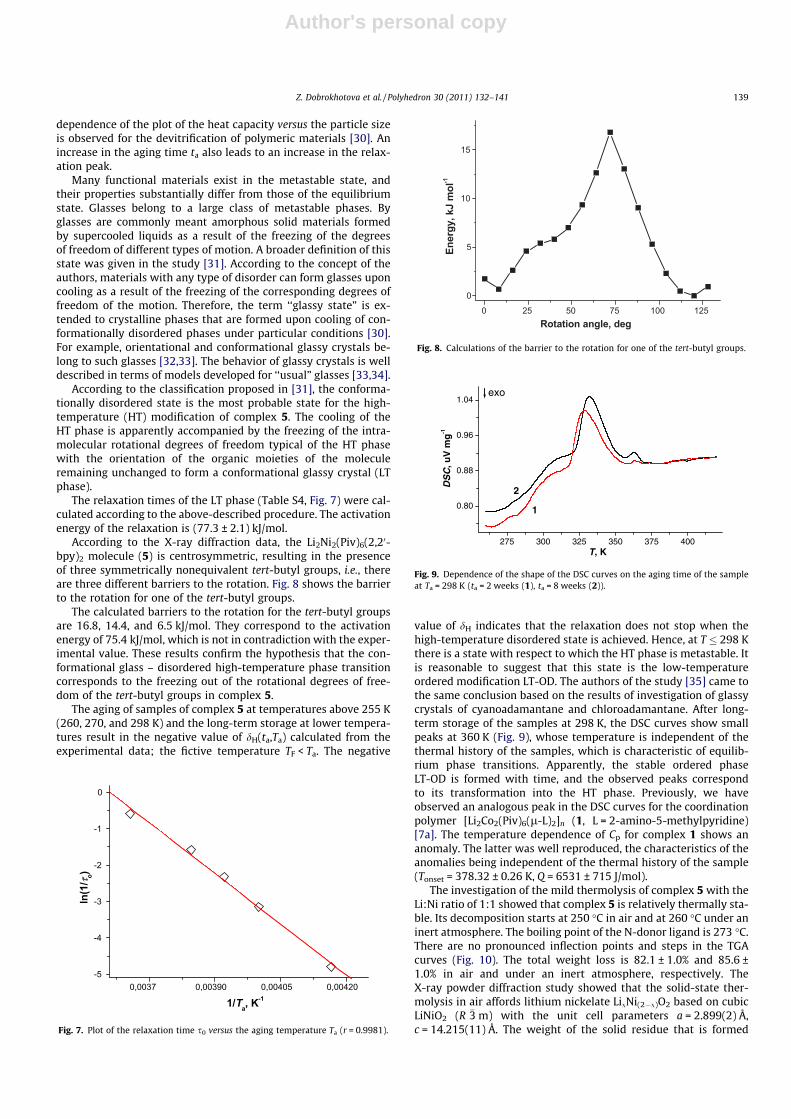
Magnetic measurements showed that the effective magneticmoment of compound 2 at room temperature is 6.30 lB per mole-cule (Fig. 3) or 4.45 lB per cobalt atom. These values are close tothe experimental values observed for the high-spin CoII atoms(S = 3/2) taking into account the spin–orbital interaction (4.4–5.2 lB) [26]. A decrease in the temperature leads to a decrease in leff
down to 4.87 lB at 2 K. The temperature dependence of the inversemagnetic susceptibility (1/v) for compound 2 in the 10–300 K tem-perature range obeys the Curie–Weiss law with the parametersC = 4.99 K cm3/mol and h = �4.3 K (Fig. 3). The Curie constant Ccorresponds to two CoII ions with the g factor of 2.3. The negativevalue of the parameter h is indicative of weak antiferromagneticexchange interactions between the paramagnetic centers, but it
can also be attributed to the spin–orbital interaction typical of CoII
ions in an octahedral environment.The temperature dependence of leff for compound 4 is pre-
sented in Fig. 4. The high-temperature value of the effective mag-netic moment (4.27 lB) is in good agreement with the theoreticalvalue for two non-interacting NiII ions with the g factor of 2.13. Adecrease in the temperature to 100 K results in almost no changesin the magnetic moment leff. Then leff slightly increases to 4.33 lB
at 30 K and strongly decreases at temperatures below 10 K. Thisbehavior of the plot leff(T) indicates that there are ferromagneticand weaker antiferromagnetic exchange interactions between theparamagnetic centers of the complex in the solid state. An analysisof the plot leff(T) in terms of the model of exchange-coupled di-mers [27] taking into account the interactions between the dimersin the molecular field approximation [28] gave the followingparameters: g = 2.12, J/k = 2.4 K, zJ0/k = �0.57 K (Fig. 4).
3.4. Thermal behavior of complexes
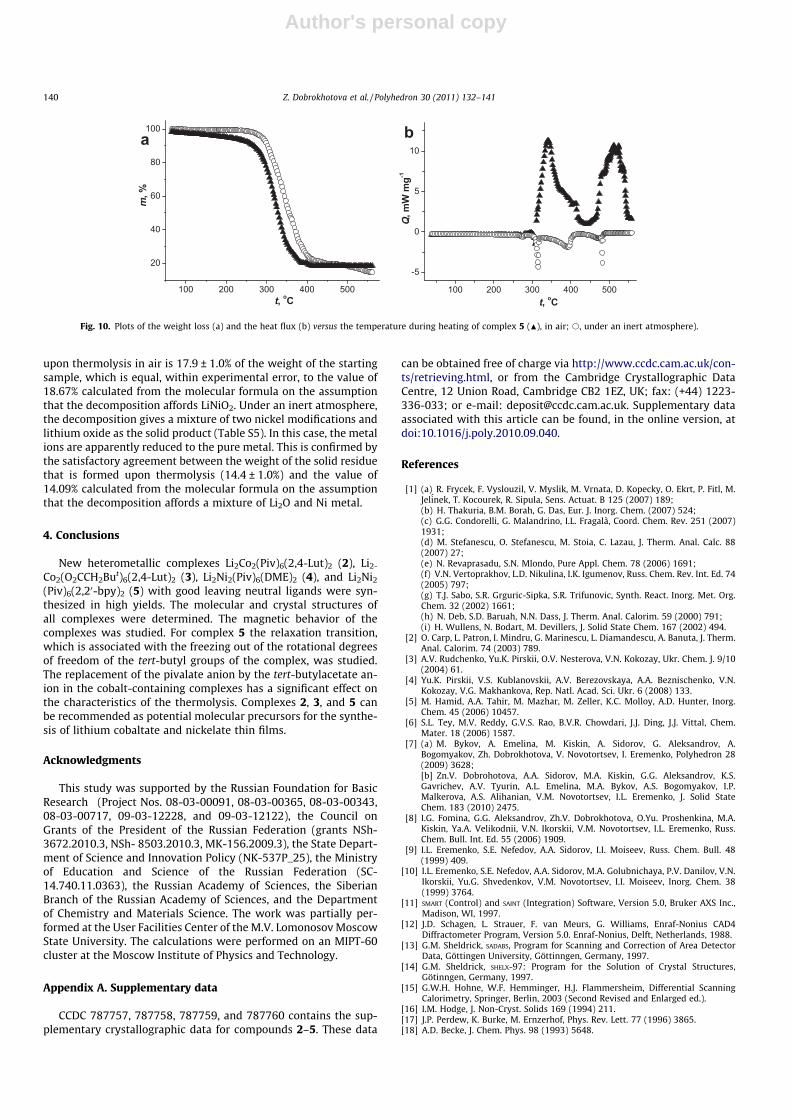
We investigated the mild thermolysis of complexes 2 and 3with the Li:Co ratio of 1:1, which have similar molecular structuresbut contain anions of different nature (the pivalate anion in com-plex 2 and the tert-butylacetate anion in complex 3). It appearedthat, according to the TGA data, the decomposition of both com-plexes both in air and under an inert atmosphere does not affordstable intermediates (Fig. 5a and b). The experimental DSC curvesobtained in air and under an inert atmosphere have features incommon (Fig. 5c and d). Thus, the first endothermic low-tempera-ture step is clearly observed in the DSC curves. The endothermiceffects in the DSC curves for pivalate complex 2 (Fig. 5c) (Tonset =150 �C (in air) and Tonset = 170 �C (under argon), Q = 128.5 ± 3.5(in air) and Q = 129.5 ± 3.5 (under argon) kJ/mol of the complex)correspond to the weight loss of 21.0 ± 1.0% and 23.2 ± 1.0% in airand under argon, respectively. For tert-butylacetate complex 3(Fig. 5d), the endothermic effects (Tonset = 136 �C (in air) and Tonset =140 �C (under argon), Q = 134.4 ± 4.0 (in air) and Q = 133.5 ± 4.0(under argon) kJ/mol of the complex) are accompanied by theweight low of 21.3 ± 1.5% and 20.0 ± 1.5% in air and under argon,respectively.
In the first decomposition step (T = 130–250 �C) of both com-plexes, only 2,4-lutidine molecules are present in the gas phase[29]. The percentage of the N-donor ligand in complexes 2 and 3calculated from the molecular formulas is 22.45% and 20.66%,respectively, and the heat of the evaporation of complex 3 at theboiling point is 47.8 kJ/mol [29]. Therefore, the first step of thethermal decomposition of complexes 2 and 3 both in air and underan inert atmosphere in the temperature range of 130–250 �C isaccompanied by the elimination of two N-donor ligands. At tem-peratures above 250 �C, the further destructive decomposition of
Table 2Selected bond lengths (Å) and bond angles (�) for complexes 2–5.
2 3 4 5
Li–Li–M (�) 123.3(6) 117.7(3) 124.8(2) 125.8(3)Li–O–Li (�) 87.0(5) 89.1(3) 88.69(18) 83.7(2)O–C–O (�) 122.2(8), 123.7(6), 124.8(6) 123.5(4), 123.7(4), 124.0(4) 119.8(2), 124.9(2), 125.4(2) 119.9(2), 124.0(3), 124.2(3)M� � �Li (Å) 3.157(10) 3.151(6) 3.031(4) 2.997(5)Li� � �Li (Å) 2.69(2) 2.742(12) 2.745(8) 2.625(9)M–N (Å) 2.097(5) 2.059(3) 2.054(2), 2.086(3)M–O(DME) (Å) 2.1157(17), 2.1675(19)M–O(O2CR) (Å) 1.949(5), 1.964(4), 1.980(6) 1.936(3), 1.939(3), 1.951(3) 1.9595(18), 2.0005(19), 2.0859(18),
2.1675(19)2.006(2), 2.020(2), 2.1288(19),2.169(2)
Li–O (Å) 1.815(13), 1.929(13), 1.934(11),1.979(12)
1.875(6), 1.885(7), 1.941(6),1.966(7)
1.889(5), 1.937(4), 1.984(5), 1.990(5) 1.898(5), 1.943(5), 1.921(5),2.013(5)
C–O(O2CR) (Å) 1.233(7)–1.266(8) 1.223(5)–1.276(5) 1.234(3)–1.269(3) 1.245(4)–1.271(3)C–O(DME) (Å) 1.414(3)–1.433(4)
136 Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141
Author's personal copy
0 50 100 150 200 250 3000
15
30
45
60
μ eff,
μ B
T, K
1/ χ, mol cm
-3
4.8
5.1
5.4
5.7
6.0
6.3
Fig. 3. Magnetic properties of complex 2: the temperature dependence of themagnetic moment (N), the curve 1/v (s); the calculated data are represented by theline.
0 50 100 150 200 250 300
3.4
3.6
3.8
4.0
4.2
4.4
T, K
μ eff,
μ B
Fig. 4. The temperature dependence of the magnetic moment for compound 4 (thecalculated data are represented by the line).
Fig. 2. Molecular structures of complexes 4 (a) and 5 (b) (the hydrogen atoms are not shown; thermal ellipsoids are drawn at the 30% probability level). The fragment of thecrystal packing of 5 (c) (the hydrogen atoms are not shown).
Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141 137
Author's personal copy
the complexes affords new solid decomposition products. In addi-tion, the intermediate decomposition products are oxidized in air.
According to the X-ray powder diffraction data, the soliddecomposition products of complexes 2 and 3 under an inert atmo-sphere at temperatures below 500 �C are heterogeneous mixturesof cobalt oxide CoO and lithium oxide Li2O (Table S1). The weightsof the solid residues for complexes 2 and 3 are 17.3 ± 1.5% and17.0 ± 1.5%, respectively, of the weight of the starting sample.The percentage of Li2O and CoO calculated from the molecular for-mulas is 18.89% and 17.37% for complexes 2 and 3, respectively.
According to the X-ray powder diffraction data, the decompositionof complex 2 in air under TGA and DSC conditions at temperaturesbelow 500 �C without the subsequent temperature-controlled stor-age affords a heterogeneous mixture of cobalt oxide Co3O4, lithiumoxide Li2O, and lithium cobaltate LiCoO2 as the solid products. Thesubsequent temperature-controlled storage of the thermolysis prod-ucts at 500 �C for 30–60 min leads to the complete disappearance ofthe reflections of simple oxides in the X-ray diffraction patterns(Table S2).
The weight of the solid residue obtained after thermolysis ofcomplex 2 in air was 21.3 ± 1.5% of the weight of the starting sam-ple, which is equal, within experimental error, to 20.57% calculatedfrom the molecular formula on the assumption that the decompo-sition affords LiCoO2. According to the X-ray powder diffractiondata, the decomposition of complex 3 under TGA and DSC condi-tions at temperatures below 500 �C gives LiCoO2 as the only solidproduct. The weight of the solid residue was 19.9 ± 1.5% of theweight of the starting sample; the amount of LiCoO2 calculatedfrom the molecular formula was 18.91%.
The investigation of the solid-state thermolysis of complexes 2and 3 showed that the replacement of the pivalate anion by thetert-butylacetate anion has an effect on the characteristics of thethermolysis. Thus, the decomposition starts at a lower tempera-ture, the temperature of the completion of decomposition in airis significantly lower, and, what is most important, no volatile
cobalt-containing products are formed upon thermolysis underan inert atmosphere.
The investigation of the thermal behavior of complex 5 revealedan anomaly as an S-shaped inflection point in the temperaturedependence of the heat capacity in the 263–333 K temperaturerange (Fig. 6). Based on the below experimental data, the observedanomaly can be assigned to the high-temperature (HT) modifica-tion of complex 5 and its glassy crystalline state. The shape andthe parameters of the anomaly substantially depend on the exper-imental conditions (Table S3).
As the final cooling temperature Tmin decreases, the anomaly isreproducibly shifted to higher temperatures, the fictive tempera-ture TF being reduced. An increase in the heating rate HR of thesample leads to a substantial increase in Tinfl. This fact is usually as-signed to the ratio of the rate of the temperature change to therelaxation time of metastable materials [16].
A decrease in the particle size leads to a reproducible increase inthe relaxation peak and the appearance of an insignificant exother-mic peak immediately before the anomaly (Fig. 6). An analogous
100 200 300 400 500
20
40
60
80
100
m, %
t, oC100 200 300 400 500
20
40
60
80
100
m, %
t, oC
100 200 300 400 500
-10
-5
0
5
10
15
20
Q, m
W m
g-1
t, oC100 200 300 400 500
-2
0
2
4
6
8
Q, m
W m
g-1
t, oC
a b
c d
Fig. 5. Plots of the weight loss and the heat flux versus the temperature of the complexes Li2Co2(Piv)6(2,4-Lut)2 (a and c) and Li2Co2(OOCCH2But)6(2,4-Lut)2 (b and d),respectively (N), in air; s, under an inert atmosphere).
225 250 275 300 325 3500.75
0.80
0.85
0.90
0.95
1.00 exo
1
2DSC,
uV
mg-1
T , K
Fig. 6. Fragments of the DSC curves: the starting sample (1) and the sample aftergrinding (2) (Tmin = 120 K, CR = 20 K/min, HR = 10 K/min).
138 Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141
Author's personal copy
dependence of the plot of the heat capacity versus the particle sizeis observed for the devitrification of polymeric materials [30]. Anincrease in the aging time ta also leads to an increase in the relax-ation peak.
Many functional materials exist in the metastable state, andtheir properties substantially differ from those of the equilibriumstate. Glasses belong to a large class of metastable phases. Byglasses are commonly meant amorphous solid materials formedby supercooled liquids as a result of the freezing of the degreesof freedom of different types of motion. A broader definition of thisstate was given in the study [31]. According to the concept of theauthors, materials with any type of disorder can form glasses uponcooling as a result of the freezing of the corresponding degrees offreedom of the motion. Therefore, the term ‘‘glassy state” is ex-tended to crystalline phases that are formed upon cooling of con-formationally disordered phases under particular conditions [30].For example, orientational and conformational glassy crystals be-long to such glasses [32,33]. The behavior of glassy crystals is welldescribed in terms of models developed for ‘‘usual” glasses [33,34].
According to the classification proposed in [31], the conforma-tionally disordered state is the most probable state for the high-temperature (HT) modification of complex 5. The cooling of theHT phase is apparently accompanied by the freezing of the intra-molecular rotational degrees of freedom typical of the HT phasewith the orientation of the organic moieties of the moleculeremaining unchanged to form a conformational glassy crystal (LTphase).
The relaxation times of the LT phase (Table S4, Fig. 7) were cal-culated according to the above-described procedure. The activationenergy of the relaxation is (77.3 ± 2.1) kJ/mol.
According to the X-ray diffraction data, the Li2Ni2(Piv)6(2,20-bpy)2 molecule (5) is centrosymmetric, resulting in the presenceof three symmetrically nonequivalent tert-butyl groups, i.e., thereare three different barriers to the rotation. Fig. 8 shows the barrierto the rotation for one of the tert-butyl groups.
The calculated barriers to the rotation for the tert-butyl groupsare 16.8, 14.4, and 6.5 kJ/mol. They correspond to the activationenergy of 75.4 kJ/mol, which is not in contradiction with the exper-imental value. These results confirm the hypothesis that the con-formational glass – disordered high-temperature phase transitioncorresponds to the freezing out of the rotational degrees of free-dom of the tert-butyl groups in complex 5.
The aging of samples of complex 5 at temperatures above 255 K(260, 270, and 298 K) and the long-term storage at lower tempera-tures result in the negative value of dH(ta,Ta) calculated from theexperimental data; the fictive temperature TF < Ta. The negative
value of dH indicates that the relaxation does not stop when thehigh-temperature disordered state is achieved. Hence, at T � 298 Kthere is a state with respect to which the HT phase is metastable. Itis reasonable to suggest that this state is the low-temperatureordered modification LT-OD. The authors of the study [35] came tothe same conclusion based on the results of investigation of glassycrystals of cyanoadamantane and chloroadamantane. After long-term storage of the samples at 298 K, the DSC curves show smallpeaks at 360 K (Fig. 9), whose temperature is independent of thethermal history of the samples, which is characteristic of equilib-rium phase transitions. Apparently, the stable ordered phaseLT-OD is formed with time, and the observed peaks correspondto its transformation into the HT phase. Previously, we haveobserved an analogous peak in the DSC curves for the coordinationpolymer [Li2Co2(Piv)6(l-L)2]n (1, L = 2-amino-5-methylpyridine)[7a]. The temperature dependence of Cp for complex 1 shows ananomaly. The latter was well reproduced, the characteristics of theanomalies being independent of the thermal history of the sample(Tonset = 378.32 ± 0.26 K, Q = 6531 ± 715 J/mol).
The investigation of the mild thermolysis of complex 5 with theLi:Ni ratio of 1:1 showed that complex 5 is relatively thermally sta-ble. Its decomposition starts at 250 �C in air and at 260 �C under aninert atmosphere. The boiling point of the N-donor ligand is 273 �C.There are no pronounced inflection points and steps in the TGAcurves (Fig. 10). The total weight loss is 82.1 ± 1.0% and 85.6 ±1.0% in air and under an inert atmosphere, respectively. TheX-ray powder diffraction study showed that the solid-state ther-molysis in air affords lithium nickelate Li[Ni(2�[)O2 based on cubicLiNiO2 (R �3 m) with the unit cell parameters a = 2.899(2) Å,c = 14.215(11) Å. The weight of the solid residue that is formed
0,0037 0,00390 0,00405 0,00420-5
-4
-3
-2
-1
0
ln(1
/ τ o)
1/Ta, K-1
Fig. 7. Plot of the relaxation time s0 versus the aging temperature Ta (r = 0.9981).
0 25 50 75 100 125
0
5
10
15
Ener
gy, k
J m
ol-1
Rotation angle, deg
Fig. 8. Calculations of the barrier to the rotation for one of the tert-butyl groups.
275 300 325 350 375 400
0.80
0.88
0.96
1.04exo
1
2
DS
C, u
V m
g-1
T, K
Fig. 9. Dependence of the shape of the DSC curves on the aging time of the sampleat Ta = 298 K (ta = 2 weeks (1), ta = 8 weeks (2)).
Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141 139
Author's personal copy
upon thermolysis in air is 17.9 ± 1.0% of the weight of the startingsample, which is equal, within experimental error, to the value of18.67% calculated from the molecular formula on the assumptionthat the decomposition affords LiNiO2. Under an inert atmosphere,the decomposition gives a mixture of two nickel modifications andlithium oxide as the solid product (Table S5). In this case, the metalions are apparently reduced to the pure metal. This is confirmed bythe satisfactory agreement between the weight of the solid residuethat is formed upon thermolysis (14.4 ± 1.0%) and the value of14.09% calculated from the molecular formula on the assumptionthat the decomposition affords a mixture of Li2O and Ni metal.
4. Conclusions
New heterometallic complexes Li2Co2(Piv)6(2,4-Lut)2 (2), Li2-
Co2(O2CCH2But)6(2,4-Lut)2 (3), Li2Ni2(Piv)6(DME)2 (4), and Li2Ni2
(Piv)6(2,20-bpy)2 (5) with good leaving neutral ligands were syn-thesized in high yields. The molecular and crystal structures ofall complexes were determined. The magnetic behavior of thecomplexes was studied. For complex 5 the relaxation transition,which is associated with the freezing out of the rotational degreesof freedom of the tert-butyl groups of the complex, was studied.The replacement of the pivalate anion by the tert-butylacetate an-ion in the cobalt-containing complexes has a significant effect onthe characteristics of the thermolysis. Complexes 2, 3, and 5 canbe recommended as potential molecular precursors for the synthe-sis of lithium cobaltate and nickelate thin films.
Acknowledgments
This study was supported by the Russian Foundation for BasicResearch (Project Nos. 08-03-00091, 08-03-00365, 08-03-00343,08-03-00717, 09-03-12228, and 09-03-12122), the Council onGrants of the President of the Russian Federation (grants NSh-3672.2010.3, NSh- 8503.2010.3, MK-156.2009.3), the State Depart-ment of Science and Innovation Policy (NK-537P_25), the Ministryof Education and Science of the Russian Federation (SC-14.740.11.0363), the Russian Academy of Sciences, the SiberianBranch of the Russian Academy of Sciences, and the Departmentof Chemistry and Materials Science. The work was partially per-formed at the User Facilities Center of the M.V. Lomonosov MoscowState University. The calculations were performed on an MIPT-60cluster at the Moscow Institute of Physics and Technology.
Appendix A. Supplementary data
CCDC 787757, 787758, 787759, and 787760 contains the sup-plementary crystallographic data for compounds 2–5. These data
can be obtained free of charge via http://www.ccdc.cam.ac.uk/con-ts/retrieving.html, or from the Cambridge Crystallographic DataCentre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: [email protected]. Supplementary dataassociated with this article can be found, in the online version, atdoi:10.1016/j.poly.2010.09.040.
References
[1] (a) R. Frycek, F. Vyslouzil, V. Myslik, M. Vrnata, D. Kopecky, O. Ekrt, P. Fitl, M.Jelinek, T. Kocourek, R. Sipula, Sens. Actuat. B 125 (2007) 189;(b) H. Thakuria, B.M. Borah, G. Das, Eur. J. Inorg. Chem. (2007) 524;(c) G.G. Condorelli, G. Malandrino, I.L. Fragala, Coord. Chem. Rev. 251 (2007)1931;(d) M. Stefanescu, O. Stefanescu, M. Stoia, C. Lazau, J. Therm. Anal. Calc. 88(2007) 27;(e) N. Revaprasadu, S.N. Mlondo, Pure Appl. Chem. 78 (2006) 1691;(f) V.N. Vertoprakhov, L.D. Nikulina, I.K. Igumenov, Russ. Chem. Rev. Int. Ed. 74(2005) 797;(g) T.J. Sabo, S.R. Grguric-Sipka, S.R. Trifunovic, Synth. React. Inorg. Met. Org.Chem. 32 (2002) 1661;(h) N. Deb, S.D. Baruah, N.N. Dass, J. Therm. Anal. Calorim. 59 (2000) 791;(i) H. Wullens, N. Bodart, M. Devillers, J. Solid State Chem. 167 (2002) 494.
[2] O. Carp, L. Patron, I. Mindru, G. Marinescu, L. Diamandescu, A. Banuta, J. Therm.Anal. Calorim. 74 (2003) 789.
[3] A.V. Rudchenko, Yu.K. Pirskii, O.V. Nesterova, V.N. Kokozay, Ukr. Chem. J. 9/10(2004) 61.
[4] Yu.K. Pirskii, V.S. Kublanovskii, A.V. Berezovskaya, A.A. Beznischenko, V.N.Kokozay, V.G. Makhankova, Rep. Natl. Acad. Sci. Ukr. 6 (2008) 133.
[5] M. Hamid, A.A. Tahir, M. Mazhar, M. Zeller, K.C. Molloy, A.D. Hunter, Inorg.Chem. 45 (2006) 10457.
[6] S.L. Tey, M.V. Reddy, G.V.S. Rao, B.V.R. Chowdari, J.J. Ding, J.J. Vittal, Chem.Mater. 18 (2006) 1587.
[7] (a) M. Bykov, A. Emelina, M. Kiskin, A. Sidorov, G. Aleksandrov, A.Bogomyakov, Zh. Dobrokhotova, V. Novotortsev, I. Eremenko, Polyhedron 28(2009) 3628;[b] Zn.V. Dobrohotova, A.A. Sidorov, M.A. Kiskin, G.G. Aleksandrov, K.S.Gavrichev, A.V. Tyurin, A.L. Emelina, M.A. Bykov, A.S. Bogomyakov, I.P.Malkerova, A.S. Alihanian, V.M. Novotortsev, I.L. Eremenko, J. Solid StateChem. 183 (2010) 2475.
[8] I.G. Fomina, G.G. Aleksandrov, Zh.V. Dobrokhotova, O.Yu. Proshenkina, M.A.Kiskin, Ya.A. Velikodnii, V.N. Ikorskii, V.M. Novotortsev, I.L. Eremenko, Russ.Chem. Bull. Int. Ed. 55 (2006) 1909.
[9] I.L. Eremenko, S.E. Nefedov, A.A. Sidorov, I.I. Moiseev, Russ. Chem. Bull. 48(1999) 409.
[10] I.L. Eremenko, S.E. Nefedov, A.A. Sidorov, M.A. Golubnichaya, P.V. Danilov, V.N.Ikorskii, Yu.G. Shvedenkov, V.M. Novotortsev, I.I. Moiseev, Inorg. Chem. 38(1999) 3764.
[11] SMART (Control) and SAINT (Integration) Software, Version 5.0, Bruker AXS Inc.,Madison, WI, 1997.
[12] J.D. Schagen, L. Strauer, F. van Meurs, G. Williams, Enraf-Nonius CAD4Diffractometer Program, Version 5.0. Enraf-Nonius, Delft, Netherlands, 1988.
[13] G.M. Sheldrick, SADABS, Program for Scanning and Correction of Area DetectorData, Göttingen University, Göttinngen, Germany, 1997.
[14] G.M. Sheldrick, SHELX-97: Program for the Solution of Crystal Structures,Götinngen, Germany, 1997.
[15] G.W.H. Hohne, W.F. Hemminger, H.J. Flammersheim, Differential ScanningCalorimetry, Springer, Berlin, 2003 (Second Revised and Enlarged ed.).
[16] I.M. Hodge, J. Non-Cryst. Solids 169 (1994) 211.[17] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[18] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
100 200 300 400 500
20
40
60
80
100m
, %
t, oC100 200 300 400 500
-5
0
5
10
t, oC
Q, m
W m
g-1
ba
Fig. 10. Plots of the weight loss (a) and the heat flux (b) versus the temperature during heating of complex 5 (N), in air; s, under an inert atmosphere).
140 Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141

Author's personal copy
[19] D.N. Laikov, Chem. Phys. Lett. 281 (1997) 151.[20] A.A. Granovsky, PCGamess/Firefly, Version 7.1.F, http://classic.chem.msu.su/
gran/firefly/index.html.[21] M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert, M.S. Gordon, J.H. Jensen, S.
Koseki, N. Matsunaga, K.A. Nguyen, S. Su, T.L. Windus, M. Dupuis, J.A.Montgomery, J. Comput. Chem. 14 (1993) 1347.
[22] D.N. Laikov, Chem. Phys. Lett. 416 (2005) 116.[23] F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7 (2005) 3297.[24] R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72 (1980) 650.[25] (a) I.G. Fomina, A.A. Sidorov, G.G. Aleksandrov, V.I. Zhilov, V.N. Ikorskii, V.M.
Novotortsev, I.L. Eremenko, I.I. Moiseev, Russ. Chem. Bull. Int. Ed. 53 (2004)116;(b) J.-H. Liao, C.-T. Su, C.-C. Hsu, Acta Crystallogr., Sect. E: Struct. Rep. Online57 (2001) m501;(c) M. Kurmoo, C. Estournes, Y. Oka, H. Kumagai, K. Inoue, Inorg. Chem. 44(2005) 217;(d) I.S. Evstifeev, M.A. Kiskin, V.S. Mironov, A.S. Bogomyakov, A.A. Sidorov,V.M. Novotortsev, I.L. Eremenko, Inorg. Chem. Commun. 13 (2010) 498.
[26] Yu.V. Rakitin, V.T. Kalinnikov, Sovremennaya magnetokhimiya (Nauka. St-Petersburg. 1994) (in Russian) [Modern magnetochemistry (Science. St.Petersburg. 1994)].
[27] Yu.V. Rakitin, V.T. Kalinnikov, Introduction to Magnetochemistry [in Russian],Nauka, Moscow, 1980.
[28] A.P. Ginsberg, M.E. Lines, Inorg. Chem. 11 (1972) 2289.[29] <http://webbook.nist.gov>.[30] (a) B. Wunderlich, Thermal Analysis of Polymeric Materials, Springer, 2005;
(b) B. Wunderlich, Thermochim. Acta 340–341 (1999) 37.[31] H. Suga, S. Seki, J. Non-Cryst. Solids 16 (2) (1974) 171.[32] (a) H. Szwarc, C. Bessada, Thermochim. Acta 266 (1995) 1;
(b) J.C. Van Miltenberg, A. Alvarez-Larena, M. Labrador, L. Palacios, J.Rodriguez-Romero, E. Tauler, E. Estop, Thermochim. Acta 273 (1996) 31.
[33] M. Oguni, T.M. Yoshida, K. Wada, Y. Fukuda, N. Ogino, T. Ito, H. Miyamae, K.Sato, J. Phys. Chem. Solids 62 (2001) 613.
[34] N.T. Correia, J.J. Moura Ramos, H.P. Diogo, J. Phys. Chem. Solids 63 (2002) 1717.[35] O. Delcourt, M. Descamps, J. Even, M. Bertault, J.F. Willart, Chem. Phys. 215
(1997) 51.
Z. Dobrokhotova et al. / Polyhedron 30 (2011) 132–141 141
Copyright © 2022 FDOKUMEN




















