
Synthesis and characterization of high-pressure and high-temperature sphene (CaTiSiO5)
10
1 23 Physics and Chemistry of Minerals ISSN 0342-1791 Volume 41 Number 10 Phys Chem Minerals (2014) 41:775-782 DOI 10.1007/s00269-014-0693-x Synthesis and characterization of high- pressure and high-temperature sphene (CaTiSiO 5 ) Jelena Pantić, Vladimir Urbanovich, Vesna Poharc-Logar, Bojan Jokić, Marija Stojmenović, Aleksandar Kremenović & Branko Matović
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Synthesis and characterization of high-pressure and high-temperature sphene (CaTiSiO5)

1 23
Physics and Chemistry of Minerals ISSN 0342-1791Volume 41Number 10 Phys Chem Minerals (2014) 41:775-782DOI 10.1007/s00269-014-0693-x
Synthesis and characterization of high-pressure and high-temperature sphene(CaTiSiO5)
Jelena Pantić, Vladimir Urbanovich,Vesna Poharc-Logar, Bojan Jokić, MarijaStojmenović, Aleksandar Kremenović &Branko Matović

1 23
Your article is protected by copyright and
all rights are held exclusively by Springer-
Verlag Berlin Heidelberg. This e-offprint is
for personal use only and shall not be self-
archived in electronic repositories. If you wish
to self-archive your article, please use the
accepted manuscript version for posting on
your own website. You may further deposit
the accepted manuscript version in any
repository, provided it is only made publicly
available 12 months after official publication
or later and provided acknowledgement is
given to the original source of publication
and a link is inserted to the published article
on Springer's website. The link must be
accompanied by the following text: "The final
publication is available at link.springer.com”.

1 3
Phys Chem Minerals (2014) 41:775–782DOI 10.1007/s00269-014-0693-x
ORIGINAL PAPER
Synthesis and characterization of high‑pressure and high‑temperature sphene (CaTiSiO5)
Jelena Pantic · Vladimir Urbanovich · Vesna Poharc‑Logar · Bojan Jokic · Marija Stojmenovic · Aleksandar Kremenovic · Branko Matovic
Received: 1 April 2014 / Accepted: 13 June 2014 / Published online: 28 June 2014 © Springer-Verlag Berlin Heidelberg 2014
compaction. Microstructure and particle size of both phases were analyzed by scanning electron microscopy.
Keywords Sphene · Phase transition · Raman spectroscopy · X-ray diffraction · SEM · IR
Introduction
Sphene or titanite belongs to the group of nesosilicate min-erals with monoclinic symmetry. It can crystallize in mono-clinic prismatic class 2/m both as primary (space group A2/a) and lower T phase (space group P21/a). The ideal chemical formula of sphene can be written as CaTiSiO5 or CaTiO(SiO4) (Dana 1959). The structure was first solved by Zachariasen (1930), who proved that this mineral is composed of isolated tetrahedra [SiO4] and groups [CaO7] and [TiO6]. The main difference between these two space groups is position of Ti atoms in octahedrons. In space group P21/a, the TiO6 octahedra are strongly distorted. All the atoms of titanium in octahedral chain are moved in the same direction along the axis a (Ribbe 1982). These atoms are moved out from their geometric center in octahedrons, which is reflected in structure geometry, i.e., longer and shorter Ti–O bonds (Speer and Gibbs 1976). Additional shifts of Ca atoms to their high-temperature positions along a and c can occur. The sphene structure undergoes a phase transition at 496 K and room pressure, where no Ti off-center distortion can be observed (Speer and Gibbs 1976; Ghose et al. 1991; Higgins and Ribbe 1976; Oberti et al. 1991; Taylor and Brown 1976; Bismayer et al. 1992; Zhang et al. 1995). Also, at room temperature and high pressure around 3.5 GPa, phase transition is present (Kunz et al. 1996; Angel et al. 1999; Kunz et al. 2000). Another high-temperature phase is observed at 825 K and correlates with
Abstract Sphene (CaTiSiO5), a calcium titanosilicate ceramic has been prepared from a powder mixture of CaCO3, TiO2 and SiO2 using vibro-milling for homog-enization and activation of precursors. During the high-pressure and high-temperature synthesis (HPS) process at 4 GPa and 1,200 °C, sphene undergoes into phase transi-tion, from room-temperature phase P21/a to high-tempera-ture phase A2/a. Evidence of that structural phase transition is given in this paper using infrared, Raman spectroscopy and X-ray powder diffraction. Rietveld refinement was employed to get the structural information of the synthe-sized powder. The most important structural change due to phase transition, the disappearance of the characteristic out-of-center distortion of the Ti atom and moving to the center of octahedra, was confirmed. HPS is an effective method for producing full-dense ceramics without any additives. Reduction of particle size occurred during high-pressure
Electronic supplementary material The online version of this article (doi:10.1007/s00269-014-0693-x) contains supplementary material, which is available to authorized users.
J. Pantic (*) · M. Stojmenovic · B. Matovic Vinca Institute of Nuclear Sciences, University of Belgrade, P. O. Box 522, Belgrade, Serbiae-mail: [email protected]
V. Urbanovich Scientific-Practical and Materials Research Center, NAS of Belarus, 19 P. Brovka Street, 220072 Minsk, Belarus
V. Poharc-Logar · A. Kremenovic Faculty of Mining and Geology, University of Belgrade, Djušina 7, Belgrade, Serbia
B. Jokic Faculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, 11000 Belgrade, Serbia
Author's personal copy

776 Phys Chem Minerals (2014) 41:775–782
1 3
high-pressure phase (Kunz et al. 2000). In P21/a, as low symmetry phase, loss of the centering translational symme-try [0, ½, ½] is present. The size of the unit cell is almost the same in both space groups (Ghose et al. 1991).
To obtain high-pressure and high-temperature phase, we used the high-pressure sintering processing (HPS) that has been considered as an effective method for producing full-dense ceramics without any additives while duration of sinter-ing can be reduced (Urbanovich 1996). The main features of HPS are a low sintering temperature and high sintering rate. The sphene samples used in this experiment are obtained pre-viously by the mechanochemistry method (Pantic et al. 2013).
The aim of this work was to see whether our previ-ously reported synthesized sphene (Pantic et al. 2013) can undergo phase transition at high-pressure and high-temper-ature conditions and to investigate the difference between these two phases. IR spectroscopy can detect small struc-tural changes correspond to strong changes in the phonon spectra (Zhang et al. 1995, 1997). Also, Raman spectros-copy was used in the determination of bond parameters, especially in Ti–O octahedral (Zhang et al. 2000a, b, 2008). These two methods proved to be useful tool for detect-ing subtle atomic rearrangement on a local length scale in sphene.
In this manuscript, all the data obtained from high-pres-sure sintering process are compared with the data obtained from previously reported synthesized sphene (Pantic et al. 2013).
Experimental
Powder preparation
Reactants used in the synthesis are commercially obtained powders: TiO2 (Lab. Art. 808 E. Merck), SiO2 (ASP-K-amorphous, Prahovo) and CaCO3 (pro analysis, 11490, Kemika, Zagreb). The powder mixtures were homoge-nized in the vibratory mill (Fritsch Pulverisette Analysette Laborette, type 09 003, no. 155, 380 V). The vibratory pulverizer uses ring (ø = 5.3 cm, h = 4.3 cm) and a disk (ø = 10.3 cm, h = 4.3 cm) inside a hard metal tungsten carbide grinding bowl (ø = 13 cm, h = 6.3 cm). Volume of the container is 100 ml with m = 3,200 g. The device can operate at two speeds of vibration: 750 and 1,000 min−1. The best experimental condition for preparing sphene for further calcination and sintering process was grinding time of 30 min in air atmosphere with speed of 750 min−1. The highest achieved density was obtained at 1,200 °C (Pan-tic et al. 2013). After sintering of compacted powders at 1,200 °C in the air at a heating rate of 10 °C min−1 and a soaking period of 2 h in alumina crucibles, samples were grinded in agate mortal.
This powder with the average particle size of 3–5 μm was used as a starting material in the present investigation. Ten gray pellets with a diameter of ~7.5 mm and thickness of ~4 mm were formed using a Bridgmann-type high-pres-sure apparatus at 3.5 and 4.0 ± 0.2 GPa and at tempera-ture of 1,100 and 1,200 ± 50 °C (Urbanovich and Shkatulo 2003). Samples (sphene1–10) were heated in an internal graphite heater (in a stainless steel die). A modified high-pressure anvil-type apparatus was used for high-pressure sintering (Mazurenko et al. 1994). Such type of apparatus is simple to manufacture and widely used for both indus-trial synthesis of hard materials and research. The sam-ples were subjected to the pressure of 3.50 and 4 GPa at room temperature and then heated to 1,100 and 1,200 °C (at the same pressure) during 60 and 180 s. The sintering process was controlled and monitored using previously developed automated control system for thermobaric sin-tering (Urbanovich and Shkatulo 2003). Relative density was calculated in accordance with the theoretical density of 3.5 g cm−3 (Dana 1922).
Characterization
All of the powders were characterized at room tempera-ture by X-ray powder diffraction (XRPD) using Ultima IV Rigaku diffractometer, equipped with Cu Kα1,2 radiation, using a generator voltage (40.0 kV) and a generator current (40.0 mA), without monochromator. The range of 10°–90° 2θ was used for all powders in a continuous scan mode with a scanning step size of 0.02° and at a scan rate of 2° min−1.
Data for structural refinement were taken in the 2θ range 10°–90° 2θ, with a scanning step size of 0.02° and at a scan rate of 0.5° min−1. The refinement was performed with the FullProf computer program which adopts the Rietveld calculation method (Rodríguez-Carvajal 1998, 2001). The TCH pseudo-Voigt profile function was used (Rodriguez-Carvajal 1993).
Infrared spectroscopy method was used to identify dif-ference between two phases in the samples. Pellets were made of both phases and KBr and subjected to analysis on IR spectrophotometer Perkin Elmer Model 597. Experi-ments were performed between 400 and 4,000 cm−1 at the recording time of 4 min.
The sintered pellets were polished and thermally etched at temperature of 1,150 °C for 30 min. Prior to the SEM analysis, the powder samples were coated with Au–Pd alloy using a spatter coater. The morphology of the obtained powders was studied by field emission scanning electron microscopy (FESEM) TESCAN Mira3 XMU at 20 kV.
Raman spectra excited with a diode-pumped solid-state high-brightness laser (532 nm) were collected on a DXR Raman microscope (Thermo Scientific, USA), equipped with an Olympus optical microscope and a CCD detector.
Author's personal copy
777Phys Chem Minerals (2014) 41:775–782
1 3
The measurements were made at room temperature in the spectral range 50–1,500 cm−1. The powdered sample was placed on X–Y motorized sample stage. The laser beam was focused on the sample using an objective magnifica-tion 10×. The scattered light was analyzed by the spectro-graph with a grating 900 lines mm−1. Laser power was kept at 1 mW.
Results and discussion
Relative density was calculated for each sample after sin-tering at high pressure and temperature. It was shown that the highest density have samples sintered with pressure of 4.0 ± 0.2 GPa at temperature 1,200 ± 50 °C (Table 1). Samples sphene1–3 and sphene6–7 were sintered without temperature regime, and the relative density of those sam-ples was below 90 %. Samples sphene4–5 and sphene8–9 were sintered with pressure of 4.0 ± 0.2 GPa at tempera-ture 1,200 ± 50 °C. Sample sphene10 was sintered with pressure of 4.0 ± 0.2 GPa at temperature 1,100 ± 50 °C. It was shown that the highest density have samples sin-tered with pressure of 4.0 ± 0.2 GPa at temperature 1,200 ± 50 °C (Table 1). The density of sphene5 sample is 3.539 g cm−2, which corresponds to density of sphene monocrystal (100 % of the theoretical value). This sample was used for further investigations.
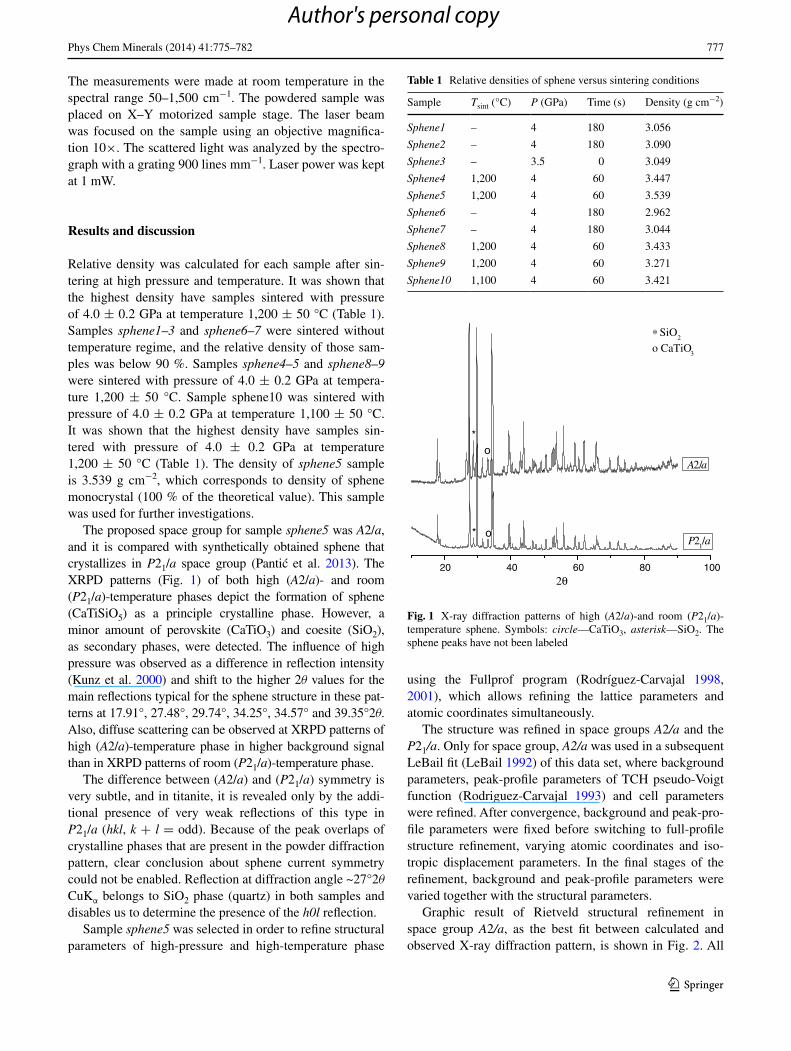
The proposed space group for sample sphene5 was A2/a, and it is compared with synthetically obtained sphene that crystallizes in P21/a space group (Pantic et al. 2013). The XRPD patterns (Fig. 1) of both high (A2/a)- and room (P21/a)-temperature phases depict the formation of sphene (CaTiSiO5) as a principle crystalline phase. However, a minor amount of perovskite (CaTiO3) and coesite (SiO2), as secondary phases, were detected. The influence of high pressure was observed as a difference in reflection intensity (Kunz et al. 2000) and shift to the higher 2θ values for the main reflections typical for the sphene structure in these pat-terns at 17.91°, 27.48°, 29.74°, 34.25°, 34.57° and 39.35°2θ. Also, diffuse scattering can be observed at XRPD patterns of high (A2/a)-temperature phase in higher background signal than in XRPD patterns of room (P21/a)-temperature phase.
The difference between (A2/a) and (P21/a) symmetry is very subtle, and in titanite, it is revealed only by the addi-tional presence of very weak reflections of this type in P21/a (hkl, k + l = odd). Because of the peak overlaps of crystalline phases that are present in the powder diffraction pattern, clear conclusion about sphene current symmetry could not be enabled. Reflection at diffraction angle ~27°2θ CuKα belongs to SiO2 phase (quartz) in both samples and disables us to determine the presence of the h0l reflection.
Sample sphene5 was selected in order to refine structural parameters of high-pressure and high-temperature phase
using the Fullprof program (Rodríguez-Carvajal 1998, 2001), which allows refining the lattice parameters and atomic coordinates simultaneously.
The structure was refined in space groups A2/a and the P21/a. Only for space group, A2/a was used in a subsequent LeBail fit (LeBail 1992) of this data set, where background parameters, peak-profile parameters of TCH pseudo-Voigt function (Rodriguez-Carvajal 1993) and cell parameters were refined. After convergence, background and peak-pro-file parameters were fixed before switching to full-profile structure refinement, varying atomic coordinates and iso-tropic displacement parameters. In the final stages of the refinement, background and peak-profile parameters were varied together with the structural parameters.
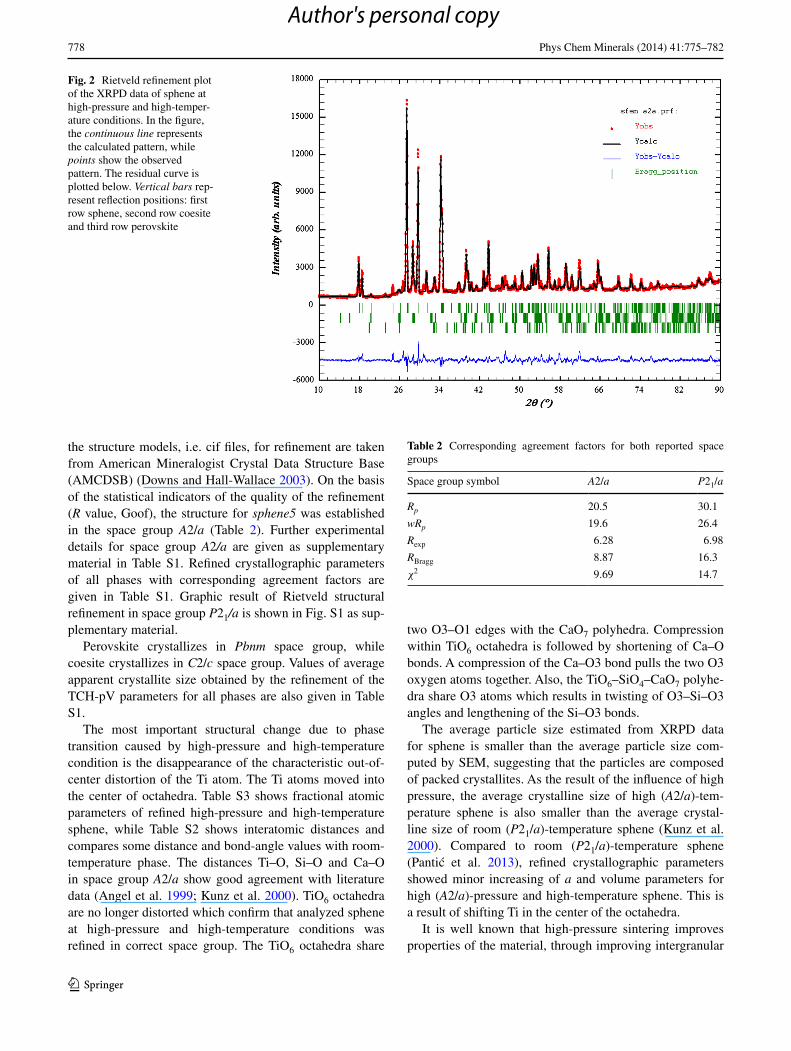
Graphic result of Rietveld structural refinement in space group A2/a, as the best fit between calculated and observed X-ray diffraction pattern, is shown in Fig. 2. All
Table 1 Relative densities of sphene versus sintering conditions
Sample Tsint (°C) P (GPa) Time (s) Density (g cm−2)
Sphene1 – 4 180 3.056
Sphene2 – 4 180 3.090
Sphene3 – 3.5 0 3.049
Sphene4 1,200 4 60 3.447
Sphene5 1,200 4 60 3.539
Sphene6 – 4 180 2.962
Sphene7 – 4 180 3.044
Sphene8 1,200 4 60 3.433
Sphene9 1,200 4 60 3.271
Sphene10 1,100 4 60 3.421
40 60 80 10020
∗ SiO2
o CaTiO3
A2/ao
o*
*
2θ
P21/a
Fig. 1 X-ray diffraction patterns of high (A2/a)-and room (P21/a)-temperature sphene. Symbols: circle—CaTiO3, asterisk—SiO2. The sphene peaks have not been labeled
Author's personal copy
778 Phys Chem Minerals (2014) 41:775–782
1 3
the structure models, i.e. cif files, for refinement are taken from American Mineralogist Crystal Data Structure Base (AMCDSB) (Downs and Hall-Wallace 2003). On the basis of the statistical indicators of the quality of the refinement (R value, Goof), the structure for sphene5 was established in the space group A2/a (Table 2). Further experimental details for space group A2/a are given as supplementary material in Table S1. Refined crystallographic parameters of all phases with corresponding agreement factors are given in Table S1. Graphic result of Rietveld structural refinement in space group P21/a is shown in Fig. S1 as sup-plementary material.
Perovskite crystallizes in Pbnm space group, while coesite crystallizes in C2/c space group. Values of average apparent crystallite size obtained by the refinement of the TCH-pV parameters for all phases are also given in Table S1.
The most important structural change due to phase transition caused by high-pressure and high-temperature condition is the disappearance of the characteristic out-of-center distortion of the Ti atom. The Ti atoms moved into the center of octahedra. Table S3 shows fractional atomic parameters of refined high-pressure and high-temperature sphene, while Table S2 shows interatomic distances and compares some distance and bond-angle values with room-temperature phase. The distances Ti–O, Si–O and Ca–O in space group A2/a show good agreement with literature data (Angel et al. 1999; Kunz et al. 2000). TiO6 octahedra are no longer distorted which confirm that analyzed sphene at high-pressure and high-temperature conditions was refined in correct space group. The TiO6 octahedra share
two O3–O1 edges with the CaO7 polyhedra. Compression within TiO6 octahedra is followed by shortening of Ca–O bonds. A compression of the Ca–O3 bond pulls the two O3 oxygen atoms together. Also, the TiO6–SiO4–CaO7 polyhe-dra share O3 atoms which results in twisting of O3–Si–O3 angles and lengthening of the Si–O3 bonds.
The average particle size estimated from XRPD data for sphene is smaller than the average particle size com-puted by SEM, suggesting that the particles are composed of packed crystallites. As the result of the influence of high pressure, the average crystalline size of high (A2/a)-tem-perature sphene is also smaller than the average crystal-line size of room (P21/a)-temperature sphene (Kunz et al. 2000). Compared to room (P21/a)-temperature sphene (Pantic et al. 2013), refined crystallographic parameters showed minor increasing of a and volume parameters for high (A2/a)-pressure and high-temperature sphene. This is a result of shifting Ti in the center of the octahedra.
It is well known that high-pressure sintering improves properties of the material, through improving intergranular
Fig. 2 Rietveld refinement plot of the XRPD data of sphene at high-pressure and high-temper-ature conditions. In the figure, the continuous line represents the calculated pattern, while points show the observed pattern. The residual curve is plotted below. Vertical bars rep-resent reflection positions: first row sphene, second row coesite and third row perovskite
Table 2 Corresponding agreement factors for both reported space groups
Space group symbol A2/a P21/a
Rp 20.5 30.1
wRp 19.6 26.4
Rexp 6.28 6.98
RBragg 8.87 16.3
χ2 9.69 14.7
Author's personal copy

779Phys Chem Minerals (2014) 41:775–782
1 3
bonding (Benjamin 1992). Densification by compaction processes involves particle deformation and is covered with attention to pressure transmission and friction effects. Voids that exist between the particles have a low coordina-tion number (number of touching neighbor particles). Pres-sure process includes particle size reduction (comminution) and particle shape changes (Fig. 3). As pressure is applied, the first response is rearrangement of the particles with fill-ing of large pores, giving a higher packing coordination. Increasing pressure provides better packing and leads to
decreasing porosity with the formation of new particle con-tacts. Since sphene is a brittle material, densification can occur by fragmentation at ambient temperature. However, a full densification is hard to attain due to a small particle size, which hinders compaction because of the higher inter-particle friction and higher particle work hardening rate. To achieve full densification, beside to applied pressure, a thermal treatment is also necessary.
The microstructure of the both, high- and room-tem-perature phases, is shown in Fig. 4a–c. All samples exhibit similar structural and morphological characteristics on thermal treatments. The analysis shows bimodal grain structure. The micrographs revealed that the compact sin-tered at high pressure and high temperature is irregularly shaped with elongated grains and no porosity is observed. The average particle size for high-temperature phase esti-mated from SEM images is 1 and 3 µm. Reduction of par-ticle size, Fig. 4, due to pressure applied confirmed the above-mentioned phenomena.
Initial mechanochemical treatment leads to a decrease of the particle size and an increase of the contribution of superficial energy to the Gibbs energy of the phases (Avva-kumov 1986). Additionally, pressure process includes parti-cle size reduction.
The IR and Raman spectra of high (A2/a)- temperature phase are compared with those of room (P21/a)-tempera-ture phase.
Infrared spectroscopy confirmed the presence of sphene in all samples. The IR spectra of high (A2/a)-and room (P21/a)-temperature sphenes are shown in Fig. 5. Vibrations centered at about 862, 705, 675, 562, 435 and 373 cm−1 are in good agreement with published data (Moenke 1962; Gadsden 1975). Previously reported single-crystal sphene from Lešnica river is used as a reference material (Pantic et al. 2011).
Fig. 3 Particle fragmentation due to a high-pressure compaction
Fig. 4 SEM micrographs of sphene of a pressureless sintered, b high-pressure compaction and c high-pressure and high-temperature treatment
Author's personal copy

780 Phys Chem Minerals (2014) 41:775–782
1 3
The spectra are dominated by the IR band near 860 cm−1 which is attributed mainly to SiO4 stretching modes. The broad and intense band near 675 cm−1 is associated with TiO6 octahedral stretching modes. The P21/a sample exhib-its much broader band at 675 cm−1 which may be mainly due to local disorder.
The effect of pressure and/or temperature increase, associated with structural phase transition from (P21/a) to (A2/a) phase, is clearly seen as an increase in band width, a decrease in band intensity and a softening of phonon bands associated with Si–O bending at 562 cm−1 and the Si–O stretching band at about 862 cm−1 (Salje and Bismayer 1997; Zhang et al. 1997). The behavior of the TiO6 octa-hedron is very different from that of the SiO4 tetrahedra. While the SiO4 tetrahedra behave as a rigid body in P21/a, both O–Si–O bending and stretching modes become softer at high pressure (A2/a), due to additional shifts of Ca.
The Raman spectra collected from both samples are shown in Fig. 6. The characteristic bands of sphene that occur in Raman spectra are centered around 71, 251, 464, 540 and 600 cm−1 (Salje and Bismayer 1997). All peaks shift to higher wave numbers with increasing pressure and temperature.
The position of the peak near 600 cm−1 probably arises from a symmetrical mode involving Ti–O bond stretching and Ti–O–Ti bond bending (Heyns et al. 2000; Su et al. 2000; Kostov-Kytin et al. 2005). Peak intensity increases with increasing pressure and temperature. The FWHM of this peak generally decreases with increasing annealing temperature (Beirau et al. 2012).
The peaks near 422 and 464 cm−1 related to SiO4 bend-ing modes as well as the external SiO4 mode near 282 cm−1 exhibit the same dependence, an increasing wave number and a decreasing FWHM, on increasing pressure and tem-perature. Peak near 464 cm−1 arises from the SiO4 bending
mode (Heyns et al. 2000). The Raman peaks near 873 cm−1 correspond to Si–O stretching modes in orthosilicates (Dowty 1987).
The effect of pressure and/or temperature increase, asso-ciated with structural phase transition from (P21/a) to (A2/a) phase, is clearly seen in shifting of bands. Ti–O bond stretch-ing band shifts from 598 ± 0.5 to 602 ± 0.5 cm−1. O–Si–O bending modes at 462 ± 0.5 cm−1 shift to 464 ± 0.5, 420 ± 0.5 cm−1 shift to 424 ± 0.5 and 280 ± 0.5 cm−1 shift to 284 ± 0.5 cm−1. Si–O stretching modes at 867 ± 0.8 cm−1 shift to a position at 873 ± 0.8 cm−1 (Fig. 6). The low-est energy mode occurring near 71 cm−1 also shows a slight shift to higher wave numbers, from 69 ± 0.5 shift to 73 ± 0.5 cm−1.
Conclusions
Low- and high-pressure sphene ceramics are obtained by pressureless and high-pressure and high-temperature syn-thesis process (at 4 GPa, 1,200 °C). Initial mechanochemi-cal treatment leads to a decrease of the particle size and an increase of the contribution of superficial energy to the Gibbs energy of the phases. Because of the thermodynamics of the phase, phase transition is changed. Additionally, pres-sure process includes particle size reduction. The resulting room (P21/a)-temperature phase is thermally unstable and it transforms to high (A2/a)-pressure and high-temperature phase. The most important structural change due to phase transition, the disappearance of the characteristic out-of-center distortion of the Ti atom and moving to the center of octahedra, was confirmed in Rietveld refinement. Phase transition was also verified in the IR spectra. It relates to the TiO6 octahedra and SiO4 tetrahedra. While the Ti–O band
4000 3500 3000 2500 2000 1500 1000 5000
10
20
30
40
50
60
70
80
90
100
435
562705
675862
T%
cm-1
373
A2/a
P21/a
Fig. 5 The IR spectra of both, high (A2/a)- and room-temperature (P21/a) sphene
0 500 1000 15000
50
100
150
200
250
300
350
Wavenumber (cm-1)
P21/a
A2/a
Ram
an in
tesi
ty
600
71
422
873
464
282
160
251
540
908
Fig. 6 The Raman spectra of both, high (A2/a)- and room-tempera-ture (P21/a) sphene
Author's personal copy

781Phys Chem Minerals (2014) 41:775–782
1 3
shows softening at high-pressure and high-temperature con-ditions, the Si–O modes harden. In the Raman spectra, the intensities of the measured spectra distinctly increase, peaks shift to higher wave numbers and the Raman peaks sharpen (decreasing FWHM) after annealing. Changes of the Ti–O bond stretching modes (Raman signal near 600 cm−1 gener-ated by Ti–O bond stretching) and O–Si–O bond bending vibrations indicate the occurrence of phase transition which leads to a recovery of the octahedral coordination of Ti atoms in the sphene structure. Further eduction of particle size occurred during high-pressure compaction. However, fully densitified sphene ceramic is obtained by using simul-taneously high-pressure and high-thermal treatment, which is promising technique for low sinterable powders.
Acknowledgments Financial support from the Serbian Education and Science Ministry in the Framework of Projects No. 45012 and 176016 is gratefully acknowledged.
References
Angel RJ, Kunz M, Miletich R, Woodland AB, Koch M, Xirouchakis D (1999) High pressure phase transition in CaTiOSiO4 titanite. Phase Trans 68:533–543
Avvakumov E (1986) Mekhanicheskiye metody aktivatsii khimich-eskikh protsessov. Nauka, Novosibirsk
Beirau T, Mihailova B, Matveeva G, Kolb U, Malcherek T, Groat LA, Bismayer U (2012) Structural anisotropy and annealing-induced nanoscale atomic rearrangements in metamict titanite. Am Miner 97:1354–1365
Benjamin JS (1992) Fundamentals of mechanical alloying. Mater Sci Forum 1:88–90
Bismayer U, Schmahl W, Schmidt C, Groat LA (1992) Linear bire-fringence and X-ray diffraction studies of the structural phase transition in titanite CaTiSiO5. Phys Chem Miner 19:260–266
Dana ES (1922) A text-book of mineralogy: with an extended trea-tise on crystallography and physical mineralogy, vol 316. Wiley, Chapman Hall, New York, London, pp 195–200
Dana ES (1959) Manual of mineralogy, 17th edn. Wiley, New York, pp 412–413
Downs RT, Hall-Wallace M (2003) The American Mineralogist Crys-tal Structure Database. Am Mineral 88:247–250
Dowty E (1987) Vibrational interactions of tetrahedra in silicate glasses and crystals: I. Calculations on ideal silicate–aluminate–germanate structural units. Phys Chem Miner 14:80–93
Gadsden JA (1975) Infrared spectra of minerals and related inorganic compounds, Butterworth, Group addresses, England, Australia, Canada, New Zealand, South Africa, USA
Ghose S, Ito Y, Hatch DM (1991) Paraelectric–antiferroelectric phase transition in titanite, CaTiSiO5: a high temperature X-ray diffrac-tion study of the order parameter and transition mechanism. Phys Chem Miner 17:591–603
Heyns AM, Harden PM, Prinsloo LC (2000) Resonance Raman study of the high-pressure phase transition in chromium-doped titanite, CaTiOSiO4. J Raman Spectrosc 31:837–841
Higgins B, Ribbe PH (1976) The crystal chemistry and space groups of natural and synthetic titanites. Am Miner 61:878–888
Kostov-Kytin V, Mihailova B, Kalvachev Y, Tarassov M (2005) Atomic arrangements in amorphous sodium titanosilicate precur-sor powders. Microporous Mesoporous Mater 86:223–230
Kunz M, Xirouchakis D, Lindsley DH, Hauserman D (1996) High-pressure phase transition in titanite (CaTiOSiO4). Am Miner 81:1527–1530
Kunz M, Arlt T, Stolz J (2000) In situ powder diffraction study of titanite (CaTiOSiO4) at high pressure and high temperature. Am Miner 85:1465–1473
LeBail A (1992) Extracting structure factors from powder diffraction data by iterating full pattern profile fitting. In: Prince E, Stalick JK (eds) Accuracy in powder diffraction II, special publication, vol 846. National Institute of Standards and Technology, Gaith-ersburg, p 213
Mazurenko AM, Urbanovich VS, Kuchinski VM (1994) High pres-sure apparatus for sintering ceramics based on high-melting point compounds. News of the Academy of Science, ser. Phys Tech Sci 1:42–46 (in Russian)
Moenke H (1962) Mineralspectren. Deutsche Akademie der Wissen-schaften zu Berlin, Berlin
Oberti R, Smith DC, Rossi G, Caucia F (1991) The crystal chemistry of high-aluminum titanites. Eur J Miner 3:777–792
Pantic J, Kahlenberg V, Poharc-Logar V, Kremenovic A (2011) Natural CaO–TiO2–SiO2 based ceramics. Process Appl Ceram 5:79–84
Pantic J, Kremenovic A, Došen A, Prekajski M, Stankovic N, Bašcarevic Z, Matovic B (2013) Influence of mechanical acti-vation on sphene based ceramic material synthesis. Ceram Int 39:483–488
Ribbe PH (1982) Titanite. In: Ribbe PH (ed) Orthosilicates, Mineral-ogical Society of America. Rev Miner 5:137–155
Rodriguez-Carvajal J (1993) Recent advances in magnetic structure determination by neutron powder diffraction. Phys B 192:55–69
Rodriguez-Carvajal J (1998) FullProf computer program. ftp://charybde.saclay.cea.fr/pub/divers/fullprof.98/windows/winfp98.zip
Rodríguez-Carvajal J (2001) Recent developments of the program FULLPROF. In: Commission on Powder Diffraction (IUCr). Newsletter 26:12-19. http://journals.iucr.org/iucr-top/comm/cpd/Newsletters/
Salje EKH, Bismayer U (1997) Hard mode spectroscopy: the concept and applications. Phase Trans 63:1–75
Speer JA, Gibbs GV (1976) The crystal structure of synthetic titan-ite, CaTiOSiO4 and the domain textures of natural titanites. Am Miner 1976(61):238–247
Su Y, Balmer ML, Bunker BC (2000) Raman spectroscopic studies of silicotitanates. J Phys Chem B 104:8160–8169
Taylor M, Brown GE (1976) High-temperature structural study of the P21/a ↔A2/a phase transition in synthetic titanite, CaTiSiO5. Am Miner 61:435–447
Urbanovich VS (1996) Sintering at high pressures and properties of aluminum nitride ceramics. In: Trzeciakowski WA (ed) High pressure science and technology. World Scientific Publishing, Singapore, pp 112–114
Urbanovich VS, Shkatulo GG (2003) Computerized system for the sintering of nanoceramics at high pressures. Powder Metall Met Ceram 42:19–23
Zachariasen WHZ (1930) The crystal structure of titanite. Krystallog-raphy 73:7–16
Zhang M, Salje E, Bismayer U, Unruh H, Wruck B, Schmidt C (1995) Phase transition(s) in titanite CaTiSiO5: an infrared spectroscopy, dielectric response and heat capacity study. Phys Chem Miner 22:41–49
Zhang M, Salje EKH, Bismayer U (1997) Structural phase transi-tion near 825 K in titanite: evidence from infrared spectroscopic observations. Am Miner 82:30–35
Zhang M, Salje EKH, Capitani GC, Leroux H, Clark AM, Schluter J, Ewing RC (2000a) Annealing of α-decay damage in zir-con: a Raman spectroscopic study. J Phys: Condens Matter 12:3131–3148
Author's personal copy

782 Phys Chem Minerals (2014) 41:775–782
1 3
Zhang M, Salje EKH, Farnan I, Graem-Barber A, Danial P, Ewing RC, Clark AM, Leroux H (2000b) Metamictisation of cir-con: Raman spectroscopic study. J Phys: Condens Matter 12:1915–1925
Zhang M, Boatner L, Salje EKH, Ewing RC, Daniel P, Weber WJ, Zhang Y, Farnan I (2008) Micro-Raman and micro-infrared spectroscopic studies of Pb- and Au-irradiated ZrSiO4: optical properties, struc-tural damage and amorpization. Phys Rev B 77:144110–1444113
Author's personal copy




















