
Synthesis and characterization of 3-hexyl multi-substituted α,ω-diformyl-α-oligothiophenes (n =...
17
Macromol. Chem. Phys. 198,1091-1107 (1997) 1091 Synthesis and characterization of 3-hexyl multi-substituted a,o-diformyl-a-oligothiophenes (n = 3,6,8) Thomas Olingaa), Silvia Destri*, William Porzio Istituto di Chimica delle Macromolecole del C.N.R., via E. Bassini 15,I-20133 Milan, Italy Antonio Selva Centro di Studio sulle Sostanze Organiche Naturali del C.N.R., via Mancinelli 7,1-20131 Milan, Italy (Received: August 29, 1996; revised manuscript of October 21, 1996) SUMMARY: The synthesis and characterization of a series of thienylenic a,o-diformyl-a-oligothio- phenes with 3, 6 and 8 residues is reported. The chemical structure was determined by H NMR, FT-IR, mass and GPC analyses, while the electronic properties in the neutral state were studied by UV-Vis spectroscopy both in solution and in the solid state. In the UV-Us spectra of thin films, the vibronic structure is observed. The pronounced red-shift of the absorption maximum, with respect to spectra of unsubstituted a-oligothiophenes,is indicative of a contribution of carbonyl n-electrons to the overall delocalization via a mesomeric effect leading to a more planar structure. The solid state aggregation was investigated by XRD spectroscopy and optical microscopy. We observed for the longest terms a smectic arrangement typical of liquid crystalline compounds and a tendency to self-assembling onto hydrophilic substrates, forming angles ranging from 40-50" with respect to the normal to the surface. Introduction In the last few years a-oligothiophenes have been extensively studied from opti- cal, electronic and structural points of view, because they constitute both a useful model for the study of polythiophene (PT) and suitable materials for many applica- tions in electronic devices like LED, FET etc. 1-3). In fact, as compared with PT, they are soluble and do not show chemical defects due to the polymerization reaction. To increase the solubility of the oligomers, parti- cularly for terms containing more than six thienylene residues, compounds having alkyl chains in position 3 have been synthesized4) Many different molecules having alkyl side chains either in defined positions or randomly distributed have been prepared (n = 8, 125) and n = 16@, which is the long- est substituted term reported up to now in the literature). Moreover larger analogues can be doped and, in order to stabilize the quinoidic form, end-capped oligomers have been prepared7). Sexithiophenes a,o-substituted with alkyl chains have been a) Present address: Institut Charles Sadron (CNRS), 6 rue Boussingault, 67083 Stras- bourg, Cedex-France. 0 1997, Huthig & Wepf Verlag, Zug ccc 1022-1352/97/$10.00
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis and characterization of 3-hexyl multi-substituted α,ω-diformyl-α-oligothiophenes (n =...

Macromol. Chem. Phys. 198,1091-1107 (1997) 1091
Synthesis and characterization of 3-hexyl multi-substituted a,o-diformyl-a-oligothiophenes (n = 3,6,8)
Thomas Olingaa), Silvia Destri*, William Porzio
Istituto di Chimica delle Macromolecole del C.N.R., via E. Bassini 15,I-20133 Milan, Italy
Antonio Selva
Centro di Studio sulle Sostanze Organiche Naturali del C.N.R., via Mancinelli 7,1-20131 Milan, Italy
(Received: August 29, 1996; revised manuscript of October 21, 1996)
SUMMARY: The synthesis and characterization of a series of thienylenic a,o-diformyl-a-oligothio-
phenes with 3, 6 and 8 residues is reported. The chemical structure was determined by H NMR, FT-IR, mass and GPC analyses, while the electronic properties in the neutral
state were studied by UV-Vis spectroscopy both in solution and in the solid state. In the UV-Us spectra of thin films, the vibronic structure is observed. The pronounced red-shift of the absorption maximum, with respect to spectra of unsubstituted a-oligothiophenes, is indicative of a contribution of carbonyl n-electrons to the overall delocalization via a mesomeric effect leading to a more planar structure. The solid state aggregation was investigated by XRD spectroscopy and optical microscopy. We observed for the longest terms a smectic arrangement typical of liquid crystalline compounds and a tendency to self-assembling onto hydrophilic substrates, forming angles ranging from 40-50" with respect to the normal to the surface.
Introduction
In the last few years a-oligothiophenes have been extensively studied from opti- cal, electronic and structural points of view, because they constitute both a useful model for the study of polythiophene (PT) and suitable materials for many applica- tions in electronic devices like LED, FET etc. 1-3).
In fact, as compared with PT, they are soluble and do not show chemical defects due to the polymerization reaction. To increase the solubility of the oligomers, parti- cularly for terms containing more than six thienylene residues, compounds having alkyl chains in position 3 have been synthesized4)
Many different molecules having alkyl side chains either in defined positions or randomly distributed have been prepared (n = 8, 125) and n = 16@, which is the long- est substituted term reported up to now in the literature). Moreover larger analogues can be doped and, in order to stabilize the quinoidic form, end-capped oligomers have been prepared7). Sexithiophenes a,o-substituted with alkyl chains have been
a) Present address: Institut Charles Sadron (CNRS), 6 rue Boussingault, 67083 Stras- bourg, Cedex-France.
0 1997, Huthig & Wepf Verlag, Zug ccc 1022-1352/97/$10.00

1092 T. Olinga, S. Destri, W. Porzio, A. Selva
used by Gamier*) in order to decrease the sublimation temperature of the materials and to force the orientation (parallel versus orthogonal) of the molecules with respect to the substrate. a,o-Silyl derivatives have been synthesized by Tour et al.", and silyl derivatives of bithienylsilanes have been prepared by Garnier et al. in order to obtain polysiloxythienylenesl'). Other polymers containing thiophene moieties can be obtained using a,@-dialdehydes, namely, polyazomethines and polyazines").
Here, we detail the synthetic route to prepare some derivatives of 3-substituted oligo(a-thiophene)s (n = 3, 6, 8) having aldehyde groups in both a and w positions. The aldehyde function provides the opportunity for both the introduction of other substituents by condensation reactions and the polymer preparation by polyconden- sation as usual in polyazomethine ~ynthesis'~-'~).
Results and discussion
The synthesis of larger 3-substituted oligo(a-thiophene)s (n 2 4) requires some intermediate steps, outlined in Scheme 1. The starting material 3-hexylthiophene (I) was synthesized by coupling of hexylmagnesium bromide with 3-bromothiophene in the presence of diphenylphosphinopropanenickel chloride (NiC12dppp) according to the Kumada meth~d'~) . The iodination of I or of other 3-substituted oligo(a-thio- phene)s, i.e. (n = 3), can be realized by using - as oxidizing agents to reform iodine - nitric acid, iodic acid, and periodic acid'@, or by the Unlenbroek method, using iodine with yellow mercuric oxide in benzene") or other solvents'@. We used the last method because of its ease of realization and its very good yields. 3,3"- Dihexyl-2,2' : 5',2"-terthiophene (111) was synthesized using the Kumada method starting from 3-hexyl-2-iodothiophene (11). The introduction of a carbaldehyde group in a position of I11 by the Vilsmeier reaction'*) provided the a-monoaldehyde derivative (IVa), while the a,o-dialdqhyde derivative (IVb) was obtained by using an excess of two equivalents of dimethylformamide-phosphoryl chloride reagentlg).
Due to the fact that aldehydes and ketones are very sensitive to metallic reagents such as lithium, magnesium and copper etc., they require protection via transforma- tion into their corresponding acetal or keta12" before use in the.presence of metallic reagents. However, the aldehyde carbonyl group tolerates the presence of mercuric oxide at the iodination stage15) and in this way we obtained 5-formylJ"-iod0-3,3"- dihexyL2,2':5',2"-terthiophene (V) in good yields without byproducts.
The a,o-dialdehyde of 3,3",3"',3""-a-sexithiophene (VI) was prepared following Tiecco's synthesis2') of halopyridine homocoupling, as reported in Scheme I. For the synthesis of the n = 8 term, two routes were attempted: firstly, the palladium- catalyzed coupling of organostannanes with arylhalides following the Stille reac- tion22), secondly, the Suzuki method23).
To apply the first reaction, we tried to prepare organostannanes from the 5-acetal of IVa using butyllithum or lithiumdiisopropylamide (LDA). Our attempt was unsuccessful, producing mostly by-products. These results can be explained by assuming that the aldehydic hydrogen becomes more acid in the acetal form, prob- ably due to the great polarizability and electron-withdrawing inductive-mesomeric effect of a-terthienyl residue.
Synthesis and characterization of 3-hexyl multi-substituted ... 1093
Considering the hydrogen atoms of the acetal group in thienyl, bithienyl and terthienyl acetal derivatives and comparing them with the a-protons of the thieny- lenic ring, one can infer that their acidity increases in the order:
where Kacm, Kacb and Kact are the acid constants of the acetalic protons in thienyl, bithienyl and terthienyl derivatives respectively and K, is the acid constant of the
Scheme 1:

1094 T. Olinga, S. Destri, W. Porzio, A. Selva
Scheme 1 (continued):
CHO
V
'&HI 3 c%H3
NU2. 6HzO
DMF
a-protons in terthiophene compounds. According to these considerations, in terthio- phene the hydrogen could react with hard bases such as n-butyl(nBu-) or diisopropy- lamide ( i -pr~p)~N- anions. To overcome this situation, we used the palladium' cata- lyzed reaction of arylhalides with boronic acid derivatives starting from V and the diboronic acid of a-bithiophene. This reaction tolerates on both the boron and halide compound many functional groups including ester, ketone and aldehydeZ3).

Synthesis and characterization of 3-hexyl multi-substituted ... 1095
As mentioned above, the synthesis of soluble 3-substituted a-thiophene oligomers silylated at a,w-positions has been described by Tour et aL9’. 3-Substitution and a,w- silyl end groups allowed an increase of the solubility of large oligothiophene sequences. Instead of silyl end groups, we adopted carbonyl residues in order to use these oligomers as starting materials in the synthesis of organic multiple quantum wells by polycondensation of dialdehyde with diarninel2,. 14). Tour’s synthesis of large oligomer sequences (n 2 4 ) was not applicable in our case because of the great sensitivity of carbonyl groups which require protection according to the synthesis outlined above.
The compounds I, 11, I11 and IV were prepared as described in the literature. The compound I11 presents an UV-Vis spectrum, in chloroform, whose absorption max- imum (A,,,) is centered at 337 nm. This value is in agreement with Gallazzi’s mea- surement~’~). The formylation of I11 in the a position induces a bathochromic effect in UV-Vis spectrum of the molecule IVa, in fact the A,,, is shifted to 386 nm. This effect is due to the increase of the polarizability induced by carbonyl group. In the FTIR spectrum of IVa and V two important stretching modes of thienylic CH at 3 102-3 107 cm-‘ and at 3060-3066 cm-’ are observed, and they are assigned to the a and p positions, respectively. On this basis we can follow the iodination reac- tion in the a position by the disappearance of CH stretching mode at 3 102 cm-’ (see Fig. 1).
$ 100
5 80 E
- ln
60
; 10
20
0 1000 3200 2LOO 1600 800
C 0
Wavenumber in crn-’ FTIR spectra of IVa and V. The arrow indicates the band attributed to the Fig. 1.
stretching mode of thienylic C-H in a-position, which disappears after the reaction
Using nickel chloride, zinc and triphenylphosphine in dimethylformamide, the compound VI is obtained (60% yield) from V. In Fig. 2a the UV-Vis spectrum of VI is reported (A,,, is 446 nm). The A,,, shift observed for VI with respect to V matches the increase of the conjugation length. In fact, this A,= value is very high as compared with the unsubstituted a-sexithiophene without a,o-aldehyde groups (Amax,c~cl3 438 nm25)). A similar behaviour is shown by IVb (see Fig. 2a) and is probably due to the electron-withdrawing effect of the polar carbonyl group with respect to thienyl compound, leading to a more planar conformation of the chain.

1096 T. Olinga, S. Destri, W. Porzio, A. Selva
$ 2 8 0 C 0
0 u)
2 2.21
9 1.68
112
0.56
0 0- 230 3LL 158 572 686 230 311 L58 572 686
Wavelength in nrn Wavelength in nm
UV-Vis spectra of IVb, VI, VII in CHC13 solution (a) and of IVb, VI, VII spin- Fig. 2. coated films (b)
The synthesis of VII, the a,o-dialdehyde of the octithienyl derivative by cross- coupling reaction between 5,5'-diboronic acid of bithiophene with V, was tried under different experimental conditions, as reported in Tab. 1.
Tab. 1. hydroxybory1)-a-bithiophene using tetrakis-Pd(tripheny1phospine) as catalysta3 b,
Reaction of 5-formyl-5"-iodo-3,3"-dihexyl-a-terthiophene (V) with 5,5'-bis(di-
Products Reagents
toluene/H,O DME/H,O DME/H20 DMF/NEt, Na2C03 Na2CO3 Na2C03 ref.26'
low stirring speed high stirring speed
Sexithienyl VI 20% traces traces 510% Octithienyl VII traces 6-10% 25 - 30% 50% Starting materials 80% 90% 70% 40%
a) The products were checked by mass spectrometry and confirmed by GPC and NMR analyses.
b, The separation of VII from VI by flash chromatography turned out to be quite diffi- cult, however the slightly different solubility in hexane allowed us to perform a rather satisfactory separation by fractional precipitation.
The UV-Vis spectrum of VII in chloroform shows a pronounced red shift ( ~ m a x , c ~ c l , 462 nm) with respect to the starting material v (see Fig. 2a), showing in this case, as for molecule VI, an increase of the polarizability and the coplanarity of VII. This planarity is significantly increased in the solid state, as shown in the UV- Vis spectra of cast films of the series (see Fig. 2b), which also shows vibronic struc- tures. The vibronic spacings are about 0.18 eV, a value already observed in oli- gothienyl spectroscopic studies26).
Field ionization (FI) and/or field desorption (FD) mass spectrometry (MS)27' allowed the detection of the molecular ions [M'.] of compounds IVb, V, VI and VII
Synthesis and characterization of 3-hexyl multi-substituted ... 1097
100 t IJ -fl g 0.80 s
0.60
0.40
0.20
0 1 I 1 I I I I 1 I I I 4000 3000 2000 1600 1200 800 400
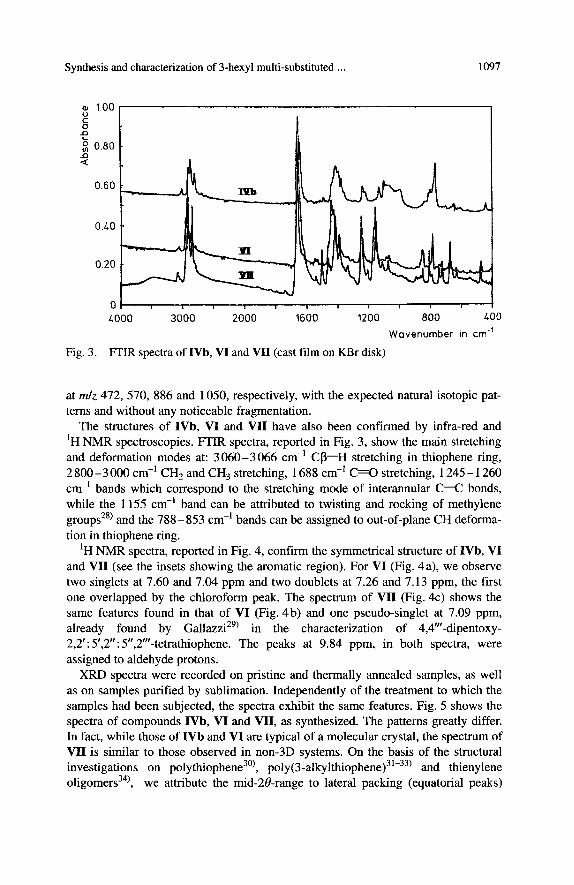
Fig. 3. WIR spectra of IVb, VI and VII (cast film on KBr disk) Wavenumber in cm-'
at mlz 472, 570, 886 and 1050, respectively, with the expected natural isotopic pat- terns and without any noticeable fragmentation.
The structures of IVb, VI and VII have also been confirmed by infra-red and 'H NMR spectroscopies. FTIR spectra, reported in Fig. 3, show the main stretching and deformation modes at: 3060-3066 cm-' CP-H stretching in thiophene ring, 2800-3000 cm-' CH, and CH, stretching, 1688 cm-' C = O stretching, 1245- 1260 cm-' bands which correspond to the stretching mode of interannular C--C bonds, while the 1155 cm-' band can be attributed to twisting and rocking of methylene groups2@ and the 788-853 cm-' bands can be assigned to out-of-plane CH deforma- tion in thiophene ring.
'H NMR spectra, reported in Fig. 4, confirm the symmetrical structure of IVb, VI and VII (see the insets showing the aromatic region). For VI (Fig. 4a), we observe two singlets at 7.60 and 7.04 ppm and two doublets at 7.26 and 7.13 ppm, the first one overlapped by the chloroform peak. The spectrum of VII (Fig. 4c) shows the same features found in that of VI (Fig. 4b) and one pseudo-singlet at 7.09 ppm, already found by Gallazzi") in the characterization of 4,4"'-dipentoxy- 2,2': 5',2": 5",2"'-tetrathiophene. The peaks at 9.84 ppm, in both spectra, were assigned to aldehyde protons.
XRD spectra were recorded on pristine and thermally annealed samples, as well as on samples purified by sublimation. Independently of the treatment to which the samples had been subjected, the spectra exhibit the same features. Fig. 5 shows the spectra of compounds IVb, VI and VII, as synthesized. The patterns greatly differ. In fact, while those of IVb and VI are typical of a molecular crystal, the spectrum of VII is similar to those observed in non-3D systems. On the basis of the structural investigations on p~lythiophene~'), poly(3-alkyIthi0phene)~'-~~) and thienylene o l igomer~~~) , we attribute the mid-28-range to lateral packing (equatorial peaks)

1098
-
T. Olinga, S. Destri, W. Porzio, A. Selva
1 L
I..
2 3 L 3
Y
6 in ppm Fig. 4a. 'H NMR spectrum (CDC13) of IVb
75 70
i 110 100 90 8 0 7 0 60 50 L O 30 2 0 10 0
6 in ppm
Fig. 4b. 'H NMR spectrum (CDCl,) of VI. (The inlet reports the aromatic region)

Synthesis and characterization of 3-hexyl multi-substituted ...
2.3 10.11 I EC8 7.5 7.0
1099
10.0 9.0 8.0 7.0 60 5.0 1.0 3.0 2.0 1.0 0 6 in ppm
Fig. 4c. 'H NMR spectrum (CDC13) of VII. (The inlet reports the aromatic region)
10" 20"
Fig. 5. XRD powder patterns of IVb, VI and VII, as synthesized
30" 20

1100 T. Olinga, S . Destri, W. Porzio, A. Selva
and the low-angle region to in-chain reflection (repeating-unit length). By assuming conventional bond distances and angles, we adopted a reasonable model for all the molecules, i.e., an anti-planar conformation for adjacent thienylene residues and an extended planar conformation for alkyl side chains. While the detailed analysis is still in progress, we report here on some of the main features that have emerged.
The spectra of compounds IVb and VI strictly resemble that of the corresponding oligomer (see Fig. 5): - large number of diffraction effects, indicating a low-symme- try unit cell (monoclinic or triclinic); - several peaks belonging to the same crystal- lographic direction (long edge), suggesting that the molecular axis and this edge form a small angle; - the presence of a second long axis due to the interdigitation commonly found in p~lyalkythiophene~~-~~).
On the contrary, the spectrum of compound VII is characterized by two diffrac- tion peaks, probably indicating a smectic C arrangement; in fact, the model described above, minimized using MACROMODEL35', fits well with an attribution of the long spacing (=26 A) to the repetition of main chains. As the molecular length is about 34.5 A, we infer that the molecules are inclined with respect to the normal to the substrate, the angle so formed being about 48" (see below). Moreover the short spacing (-3.9 A) is attributed to the distance between adjacent facing molecules, allowing a n-electron overlap.
Therefore, we carried out a detailed analysis on VII to reveal unambiguously its liquid-crystal nature. Different sublimation conditions, namely, temperature increased from 200°C up to 300°C and vacuum from lo4 to mmHg , led to samples having different crystallite dimensions and in which the molecules are con- sistently inclined to the normal to the substrate, albeit at a different angle of inclina- tion.
Quite similar results were obtained with a proper thermal treatment on films cast from chloroform and tetrachloroethane. In fact, by annealing such samples at 170°C and then cooling down to room temperature (cooling rate =0.2"C/min), we obtained samples with long spacing close to 26 A and large crystallite size (-300 A).
We observed that upon casting or sublimating thin films of VII onto hydrophilic substrates a slightly different orientation of main chains could be obtained as a func- tion of the deposition speed: the faster the evaporatiordsublimation, the shorter the detected spacing. This means that the long spacing could range from 22.5 A to 26 A, and the derived angle of inclination of the chains to the normal ranges from 40" up to 50". In Fig. 6 the low-angle XRD spectra of both slowly sublimated and thermally treated samples are reported (see the experimental section for details). It is evident that both the spacings (22.6 A vs 24 A) and the full width at half peak (0.24" vs 0.14') are remarkably different. From these data the coherence length along this crystallographic direction can be derived, being - 150 A and -3 10 A for sublimated and annealed samples, respectively.
Such a remarkable molecular orientation (self-assembly) is confirmed by the dis- appearance of the short-spacing peak (3.9 A), similarly to the XRD observations on Langmuir-Blodgett films3? Therefore we could derive an approximate number of coherent layers, along the large spacing direction, i. e., normally to the substrate, and we found for the best samples about 12 (300 h 2 4 ) layers. The self-assembly of

Synthesis and characterization of 3-hexyl multi-substituted ... 1101
Fig. 6. XRD patterns of VII. Sublimated film (top) and cast film annealed (bottom)
II)
C - - 3 300 e 0
200 x c UI C 0) - 5 100
0 30" 3 5 O L O 0 1 5 " 5 0 " 55O 6 0 "
28
the molecules onto hydrophilic substrates can be readily accounted for by consider- ing the possibility that VII forms hydrogen bonds via the aldehyde residues. Attempts to obtain molecule orientation onto hydrophobic substrates are still in pro- gress.
The smectic C nature of VII has been clearly shown by optical microscopy obser- vations, as shown in Fig. 7. The liquid crystalline phase formed on cooling the iso- tropic liquid is identified as smectic C by optical analysis on the basis of its distinc- tive natural Schlieren texture, which does not show Brownian motion or tendency to flash when submitted to mechanical stress. Since the point singularities and their related black bands are quite small and clouded by the red color of the sample, it is difficult to perceive the number of Schlieren brushes coming from each point singu-
GPC measurements have been performed on purified samples of VI and VII and also on the product obtained in the first experiment of Tab. 1. Using both UV-Vis spectrophotometer (A = 254 nm and 430 nm) and refractometer as detectors, VI and
larity37'.
Fig. 7. Optical microscopy photo- graph of sample of VII cooled from melt (1 cmg4Opm)

1102 T. Olinga, S. Destri, W. Porzio, A. Selva
also the material obtained in the first experiment were found to be a pure a-sexithio- phene derivative, while, on the contrary, product VII, obtained in the last experiment of Tab. 1 , was shown to be an a-octithiophene derivative containing some of a-sexi- thiophene.
These findings are in agreement with the ‘H NMR results obtained by comparison of the area of pseudo-singlet peak (attributed to unsubstituted central ring protons) to that of protons H4 and H, in the spectrum of VII (see Fig. 4c). In fact, the first signal is typical of the a-octithiophene molecule, while the second signal is common to both a-thienylene derivatives.
Conclusions
The preparation of soluble a,o-diformyl-a-oligothiophenes has been described. These compounds are both starting materials for the synthesis and useful models for the study of soluble polyazomethines. These latter polymeric systems synthesized either in bulk or via molecular deposition in ultra high vacuum (OMBE technique) can be used in the assembly of devices for telecommunications and optoelectronics, due to their remarkable third order nonlinearities”). The larger terms of the series (n = 6, 8) show noticeable non linear optical properties, which are presently under investigation.
The octithienyl dialdehyde shows liquid-crystal behaviour (smectic C) and also self-assembly onto hydrophilic substrates, probably due to the formation of hydro- gen bonding among aldehyde residues. This property makes this compound promis- ing in nanochemistry applications.
Experimental part
glassware under nitrogen. All reactions involving air- and water-sensitive materials were performed in dried
Materials and measurements
Diethyl ether was purified over sodium benzophenone, toluene (Fluka) was used as received, DMF was purified over KOH and distilled; 3-bromothiophene (Aldrich), 2,5- dibromothiophene (Aldrich) and I-bromohexane (Fluka) were used as received. Magne- sium (Jansen), yellow mercuric oxide (Aldrich), zinc (Aldrich), tetrakis(tripheny1pho- sphine)palladium(O) (Jansen), triphenylphosphine (Aldrich), nickel chloride hexahydrate (Car10 Erba), triethylamine (Hoechst), and *H-chloroform (Acro.organics) were also used as received.
Flash chromatography was performed on silica gel 60, 230-400 mesh ASTM (Merck or Macherey-Nagel) column.
A gas chromatographic apparatus (DAN1 3865) with capillary column (RSL 200 RP 15 m) was used for GC analysis. FI’IR spectra were recorded on a Bruker IFS 48 instru- ment, DSC experiments were performed on a Perkin Elmer DSC I1 apparatus. UV-Vis spectra were obtained using a Varian-Cary 2400 instrument.
Optical observations and melting point determinations were carried out on a Reichert microscope coupled with a Mettler FP5 hot stage. ‘H NMR spectra were obtained at 270 MHz on a Bruker instrument. Mass spectroscopic (MS) measurements of compounds IVb, V, VI and VII were performed on a VG ZAB-2F spectrometer equipped with a FI/

Synthesis and characterization of 3-hexyl multi-substituted ... 1103
FD ion source kept at ca. 200 "C operating at 8 kV accelerating and 4 kV output potentials and with the emitter activated according to the standard VG-micromass procedure. For FI MS of compounds IVb and V, the samples were introduced directly into the ion source. For FD MS of compounds VI and VII, the emitter, dipped in CH2Clz solutions of the samples, was then heated by applying a suitable filament current. MS measurements of compounds 11, III and IVa were carried out on a GC-MS Hewlett-Packard 5985B instrument, operating in electron impact mode (EI) at 70eV.
X-ray diffraction (XRD) experiments were carried out with a Siemens D-500 computer controlled apparatus, with Soller slits, graphite monochromator (002 direction), using CuK, radiation.
Size exclusion chromatography was carried out on a Waters SEC system consisting of a 600E pump, a 717 autosampler, a 410 different@ refractometer and a 490 UV detector. The column was from Waters (Ultrastyragel, 100 A pore size).
Sublimation experiments were performed by using an Edwards-Diffstak diffusion pump and a vertical tube furnace, inside a Pyrex cylinder equipped with sample holder cooled at 20°C. Typical conditions for VII have been 200°C for 44 h at 2 - lo-' mmHg and 280 "C for 90 min at 5 -
Spin-coated films were prepared using an Electron mec PRS 5V apparatus, spinning chloroform or 1,1,2,2-tetrachloroethane solutions (= 1 - 2 mg/mL) at 1 500 rpm.
mmHg.
3- Heql-2- iodoth iophene (11) To a solution of 3-hexylthiophene (I) (6.57 g, 39.1 mmol) in toluene (50 mL) at 0°C
were alternatively added while stirring, in small portions, mercuric oxide (9 g, 41.6 mmol) and iodine (5.3 g, 41.6 mmol). The mixture was stirred at room temperature for 2 h. The yellow mercuric oxide became orange and was removed by filtration, the precipitate was washed with ether. The filtered solution and the washings were poured into an aqueous sodium thiosulfate solution. The organic layer was recovered and dried over magnesium sulfate. Ether was removed by rotary evaporation to provide 11.10 g (97%) of colourless oil, containing 2-3% of disubstituted (2,s) compound. After distilla- tion, the purity of compound wasgreater than 99%, checked by gas chromatography. MS (EI): mlz 194 [M'.].
3,3"-Diheql-2,2' : 5',2"-terthiophene (111) To a suspension of magnesium (0.7 g, 29 mmol) in ether (30 mL), a solution of 3-
hexyl-2-iodothiophene (11) (5.51 g, 28.4 mmol) in ether (40 mL) was added dropwise at 0°C and stirred. The reaction was then allowed to proceed at room temperature until all of the halide was consumed. To a mixture of 2.5-dibromothiophene (3.44 g, 14.2 mmol) and diphenylphosphinopropanenickel chloride (0.1 g, 0.18 mmol) in ether (30 mL), the Grignard reagent of II was then added dropwise at 0 "C in 30 min. The reaction was car- ried out under reflux overnight, then quenched with an aqueous solution of ammonium chloride. The aqueous layer was extracted with ether, and the combined organic extracts were washed with brine and dried over magnesium sulfate. Solvent removal by evapora- tion provided a light yellow oil which was purified by flash chromatography on silica gel using hexane as eluent, to give 5.56 g of pure product (94%).
MS (EI): mlz 416 [M+']. UV: A,, 337 nm in CHCI, FTIR (cm-I): 3102, 3066, 2926, 2855, 1465, 1377, 1244, 1086, 834,797, 723, 692,
656. H NMR: 6 (ppm) = 7.17 (d, H5, H5", J = 5.20 Hz, 2H), 6.93 (d, H4, H4", J = 5.20 Hz,
2H), 7.05 (s, 2H), 2.78 (t, CH2 a-thiophene, 4H), 1.67-1.26 (2m, --CH2-, 16H), 0.88 1
(t. CH3--, 6H).

1104 T. Olinga, S. Destri, W. Porzio, A. Selva
5- Formyl-3,3”-dihexyl-2,2’:5’, 2”-terthiophene (IVa) and 5,5”-diformyl-3,3”-dihexyl- 2,2’: 5’,2’’-terthiophene (IVb)
Dimethylformamide (DMF) (0.965 g, 13.2 mmol) and phosphorous oxychloride (POCI,) (1.84 g, 12 mmol) were mixed together at 0°C and stirred for 30-40 min at room temperature. To the pale red DMF-POCl, complex, a solution of I11 (5 g, 12 mmol) in DMF (20 mL) was added dropwise at 0°C. The reaction was maintained at about 70°C for 2 h and then at room temperature for 1 h. The solution became dark red, and the reaction was followed by TLC using 80120 (v/v) methylene chlorideihexane solution as eluent. The complete conversion of the starting material could be detected both by using UV lamp at 366 nm and 2,4-dinitrophenylhydrazine reagent. Finally the reaction mixture was poured into dilute HCl solution (150 mL). After 30 min, the organic layer was extracted with ether, which was washed with an aqueous solution of sodium bicarbonate and subsequently with water. After drying over sodium sulfate and removal of ether by evaporation, the crude product was purified by flash chromatography on silica gel. The separation provided two materials: 4.696 g (88% yield) of liquid monoaldehyde deriva- tive (IVa), purer than 99% by gas chromatography, and 0.6 g (10.5% yield) of dialdehyde derivative (IVb).
IVa: MS (EI): mlz 444 [M+’]. UVCHC~~: A,, 386 nm. FTIR (cm-’) 3102, 3066, 2926, 2855, 1667, 1516, 1455, 1240, 1155, 1088, 855,
H NMR: 6 (ppm) = 9.83 (s, aldehyde proton), 7.59 (s, &, 1 H), 7.24 (d, H,,, J = 4.00
5.16 Hz, IH), 2.7-2.8 (m, CH, a-thiophene, 4H), 1.67-126 (2m, CH,, 16H), 0.91-0.85 (m. CH,, 6H).
834,798,726,673,476. 1
Hz, 1H),7.21(d,Hy1J=5.16Hz, lH),7.10(d,H4~,J=4.00Hz, lH),6.95(d,H4.,J=
IVb: MS (FI): mlz 472 [M’.]; m.p. 76-77°C at 2”C/min, UVCHQ: A,, 401 nm. FTIR (cm-I): 3084, 2917, 2849, 1656, 1505, 1451, 1419, 1250, 1150, 1075, 851,
H NMR: 6 (ppm) = 9.84 (s, aldehyde proton, 2H), 7.55 (s, H,, &”, 2H), 7.21 (s, H,,, 810,791,746,675,475.
a’, 2H), 2.78 (t. CH, a-thiophene, 4H), 1.67-1.22 (2m, CH2, 16H), 0.84 (t, CH,, 6H).
C26H3202S3 (472.71) Calc. C 66.08 H 6.82 0 6.76 S 20.34 Found C 66.38 H 6.97 0 6.30 S 20.29
I
5- Fomyl-5”- iodo-3,3”-diheql-2,2’ : 5’,2”-terthiophene (V) 4 g (9 mmol) of IVa were treated according to the method described for the preparation
of 11. The reaction was followed by TLC (70130 (v/v) CH,Cl,/hexane) observing the complete disappearance of the starting compound to provide 5-formyl-5”-iodo-3,3”- dihexyl-a-terthiophene without by-products. Moreover, throughout all the reaction, the formation of 2,4-dinitrophenylhydrazone derivatives on TLC sample was observed. The purity of the liquid compound was confirmed by spectroscopical analyses.
MS (FI): mlz 570 [M+‘]. UVCHQ: A,, 392 nm. FTIR (cm-’): 3066 (PH-C thienyl stretching), 2926, 2855, 1667 ( C 3 0 stretching),
1513, 1455,1240, 1155, 1088,855,834,798,726,673,476. H NMR: 6 (ppm) = 9.82 (aldehyde proton, 1 H), 7.59 (s, H4. 1 H), 7.21 (d, H3#, J =
3.84 Hz, lH), 7.03 (d, H4#, J = 3.84 Hz, IH), 7.08 (s, b t t , lH), 2.83-2.69 (m, CH, a- thiophene, 4H), 1.67-1.2 (2m, CH,, 16H), 0.92-0.86 (m, CH3, 6H).
1

Synthesis and characterization of 3-hexyl multi-substituted ... 1105
5,5””’-Difonnyl-3,3”4”’, 3’””-tetrahexyl-2,2’ : 5’, 2” : 5”, 2”’ : 5’”, 2’”’ : 5’”’, 2””’-sexith iophene (W
To a deep blue solution of nickel(I1) chloride hexahydrate (0.83 g, 3.5 mmol) and tri- phenylphosphine (3.67 g, 14 mmol) in DMF (40 mL) at 50-55”C, zinc powder (0.23 g, 3.5 mmol) was added while stirring. After 1 h of reaction, the colour of the mixture passed from deep blue to reddish brown. Then V (2.0 g, 3.5 mmol) in DMF (40 mL) was added, and the reaction was carried out overnight. The red-orange mixture was added by a syringe to methanol (600 mL) to provide a dark red precipitate which was collected by filtration and washed with a large amount of methanol in order to remove impurities. The dark red solid was dried under vacuum at 60°C overnight, to give 0.92 g (60%) of pro- duct; m.p. 115.5-117°C at 2”C/min.
MS (FD): mlz 886 [M+’], UVCHC~~: A,, 446 nm. mIR (cm-’): 3060,2951,2855, 1667, 1428, 1392, 1249, 1155, 1071,853,834,788,
726,672,472. ‘H NMR: 6 (ppm) = 9.84 (s, aldehyde proton, 2H), 7.6 (s, H4. H 4 ~ ~ ~ ~ ~ , 2H), 7.26 (d, H38,
H4,,,,, J = 3.86 Hz, 2H, masked by chloroform), 7.13 (d, H4,, H3*“, J = 3.86 Hz, 2H), 7.04 (s, H4”, H3!t#, 2H), 2.87-2.75 (CH, a-thiophene, 8H), 1.69-1.33 (CH,, 32H), 0.90 (CH3, 12H).
C5862s602 (887,43) Calc. C 67.67 H7.04 03.61 S 21.67 Found C67.64 H7.16 04.04 S20.99
5,5””N’-DifonnyE-3,3”,4””’,3”””’-tetrahexyl-2,2’ : 51,211 : 511,2”‘ : 5 ” ~ , ~ ~ ” : 5~”’,2””’ : 51~1l~,2~~l111:
5”””,2””’”-octithiophene (VII) To 5,5’-bis(dihydroxyboryl)-2,2’-bithiophene (0.293 g, 1.156 mmol) in DMF (20 mL),
triethylamine (14.89 g, 147.12 mmol) was added at room temperature. The blue green solution of the diboronic acid became a pale blue suspension after amine addition. 1.59 g (2.779 mmol) of V in DMF (40 mL) and 0.016 g (0.014 mmol) of tetrakis(tripheny1phos- phine)palladium(O) were added to the mixture, which was kept at 100°C overnight and then at room temperature for 1 h. The solution was poured into a large amount of acet- onehydrochloric acid mixture (90: lo), to give a dark red precipitate which was filtered off, dissolved in chloroform, then reprecipitated from methanol/HCl to provide a dark red solid which was dried under vacuum at 60°C overnight (yield 0.61 g, 50%); m.p. 143-145°C at 1 “C/min,.
MS (FD): mlz 1050 [M”], UVCHC~,: ,I,,,= 462 nm. FT-IR (cm-’): 3062,2956,2927,2857, 1667, 1516, 1435, 1249, 1155, 1123, 1023,
853, 834,791,675,470. ‘H NMR: 6 (ppm) = 9.84 (s, aldehyde protons, 2H), 7.6 (s, &, H4*”<er,, 2H), 7.26 (d, H3t,
&””. J = 3.86 Hz, 2H, masked by chloroform), 7.13 (d, H4t, H 3 ~ ~ ~ ~ , J = 3.86 Hz, 2H), 7.09 (pseudo singlet H31, H4f, H3, &, 4H), 7.04 (s, H4”, H3~1, 2H), 2.87-2.75 (m, CH, a-thio- phene, 8H), 1.69-1.33 (2m, CH2, 32H), 0.90 (m, CH3, 12H).
C58&6S802 (1051,68) Calc. C 66.24 H 6.32 0 3.05 S 24.39 Found C65.86 H6.46 03.92 S 23.84
5,5’-Bis(dihydroxyboryl)-2,2’-bithiophene A solution of butylithium (1.6 M, 19 mL) in hexane was added dropwise to a solution
of 2.21 g (13.3 mmol) of a-bithiophene in ether (30 mL) at 0°C in 10-15 min. A fine yellow suspension appeared and was stirred for 2 h under reflux and then at room tem- perature for 1 h. The mixture was cooled to -78 “C, and a solution (12.5 g, 66.5 mmol) of

1106 T. Olinga, S. Destri, W. Porzio, A. Selva
triisopropyl borate in THF (30 mL) was quickly added via a cannula. The reaction was carried out at 0°C for 1 h and then at room temperature overnight. 7% aqueous hydro- chloric acid (90 mL) was added and the mixture stirred for another 30 min; the blue eth- eta1 solution was separated, washed with water and dried over magnesium sulfate. The recovered blue powder was re-dissolved in ether and crystallized by adding petroleum ether. Yield 2.83 g (84%), m.p. (on cyclic ester, through reaction with ethylene glycol) 170- 172°C at 5 “C/min.
UVmethanol: A,, 321 nm. FT-IR (cm-’): 3288, 1510, 1433, 1339, 1200, 1 154, 1093, 1062,971,883, 804,702,
H NMR (on cyclic ester, in CDC13): 6 (ppm) = 7.55 (d, H4. H40, J = 3.59, 2H), 7.33 (d, 640,566,475.
1
H3, H3,. J = 3.59, 2H), 4.38 (s, 17H,+, 8H).
Acknowledgements: This research has been supported in part by Italian Oriented Pro- ject “Materiali Innovativi” of C. N. R. and by the HCM-SELMAT project of EEC. We are indebted to Dr. L. Zetta and Dr. R. Cunsonni for helpful discussions on NMR. We also thank Dr. R. Mendichi and Mr. A. Schieroni for GPC analyses. We finally thank Prof. B. Valenti and Dr. G. Costa for kindly performing the optical microscopy experiments and Mr. M . Mascherpa for carrying out sublimation experiments.
’) a) G. Zotti, G. Schiavon, A. Berlin, G. Pagani, Chem. Matel: 5,430 (1993); b) F. Gei- ger, M. Stoldt, H. Schweizer, P. Bauerle, E. Umbach, Adv. Matel: 5,922 (1993); c) K. Uchiyama, H. Akimichi, S. Hotta, H. Noge, H. Sakaki, Synth. Met. 6,57 (1994); d) G. Horowitz, G. Dellanoy, H. Bouchriha, F. Deloffre, J. L. Fave, F. Gamier, R. Haylaoui, M. Heyman, F. Kouki, P. Valat, V. Wintgens, A. Yassar, Adv. Matel: 6,752 (1994)
2, a) G. Horowitz, D. Fichou, X. Peng, B. Xu, F. Gamier, Solid State Commun. 2, 381 (1989); b) P. Ostoja, S. Guerri, M. Impronta, P. Zabberoni, R. Danieli, S. Rossini, C. Taliani, R. Zamboni, Adv. Matel: Opt. Electron. 1, 127 (1992)
3, D. Fichou, G. Horowitz, Y. Nishikitani, F. Gamier, Chemtronics 3, 176 (1993) 4, a) C. Van Pham, A. Burkhardt, R. Shabanal, D. D. Cunnigham, H. B. Mark, Jr.,
H. Zimmer, Phosphorus, Sulfur and Silicon 46, 153 (1989); b) G. Barbarella, A. Bon- gini, M. Zambianchi, Tetrahedron 48,6701 (1992)
5 , a) W. ten Hoeve, H. Wynberg, E. W. Meijer, E. E. Havinga, J. Am. Chem. SOC. 113, 5887 (1991); b) A. Yassar, D. Delabouglise, M. Hrnyene, B. Nessak, G. Horowitz, F. Gamier, Adv. Matel: 4,490 (1992); c) J. Liao, M. Benz, E. LeGoff, M. G. Kanatzi- dis, Adv. Mate,: 6, 135 (1994)
6, P. Bauerle, T. Fisher, B. Bidlingmeier, A. Stabel, J. P. Rabe, Ang. Chem., Int. Ed. Engl. 34,303 (1995)
7, P. Bauerle, Adv. Matel: 4, 102 (1992); b) S. Hotta, K. Waragai, Synth. Met. 41, 519 (1991)
’) F. Gamier, A. Yassar, R. Hajlaoui, G. Horowitz, F. Deloffre, in “Frontiers of polymers andadvanced materials”, P. Prasad, Ed., Plenum Press, New York, 1994, p. 263-271
9, J. M. Tour, R. Wu, Macromolecules 25, 1901 (1992) lo) P. Chicart, R. J. P. Corriu, J. J. E. Moreau, F. Gamier, A. Yassar, Chem. Matel: 3, 8
(1991) a) G. Kossmehl, Bel: Bunsenges. Phys. Chem. 83,417 (1979); b) C. Amari, C. Pelizzi, G. Predieri, S. Destri, W. Porzio, Synth. Met. 72,7 (1995)

Synthesis and characterization of 3-hexyl multi-substituted ... 1107
1 2 ) a) C. J. Yang, S. A. Jenekhe, Chem. Mate,: 3, 878 (1991); b) K. Lee, J. Chan Won, J. C. Jung, Makromol. Ckem. 190, 1547 (1989); c) P. W. Morgan, S. L. Kwolek, T. C. Pletcher, Macromolecules 20,729 (1987)
13) a) S. B. Park, H. Kim, W. C. Zin, J. C. Jung, Macromolecules 26, 1627 (1993); b) K. Li, C. Li, S. Li, Synth. Met. 60,285 (1993)
14) S. Destri, W. Porzio, R. Tubino, in “Frontiers of polymers and advanced materials”, P. Prasad Ed., Plenum Press, New York, 1994, p. 281-287
15) K. Tamao, S. Kodama, I. Nakajima, M. Kumada, Tetrahedron, 38,3347 (1982) M. G. Reinecke, P. Pedaja, in “Thiophene and its derivatives”, Part 11, S. Gronowitz, Ed., J. Wiley and Sons, New York 1986, p. 246
17) Unlenbroek, J. H. Bijloo, J. D. Reel, Recl. Trav. Chim. Pays-Bas 79, 1181 (1960) 18) a) J. Nakayama, I. Nakamura, T. Tajiri, M. Yoshino, Heterocycles 24, 637 (1986);
b) J. Nakayama, S. Murabayashi, M. Hoshino, Heterocycles 24,2639 (1986) 19) a) S. Destri, W. Porzio, Y. Dubitsky, Synth. Met. 791, 25 (1995); b) A. Mac Eachern,
C. Soucy, L. C. Leitch, J. T. Amason, P. Morand, Tetrahedron 44,2403 (1988) 20) A. Turkauf, H. E. Jacobson, K. C. Rice Ho, Synth. Commun. 4,265 (1974) ’l) M. Tiecco, L. Testaferri, Synthesis 9,736 (1984) 22) J. K. Stille, Ang. Chem., Int. Ed. Engl. 25,508 (1986) 23) T. Oh-e, N. Miyaura, A. Suzuki, J. Org. Chem. 58, 2201 (1993), and references
24) M. C. Gallazzi, L. Castellani, G. Zerbi, Synth. Met. 41,495 (1991) 25) J. Nakayama, T. Konishi, M. Hoshino, Heterocycles 27, 1731 (1988) 26) D. Fichou, G. Horowitz, B. Yu, F. Gamier, Synth. Met. 48, 167 (1992) 27) H. D. Beckey, “Field Ionization Mass Spectrometry”, Pergamon Press, Oxford 1970 28) A. Huth, I. Beetz, I. Schumann, Tetrahedron 45,6679 (1989) 29) G. Zotti, M. C. Gallazzi, S. V. Meille, Syntk. Met. 73,217 (1995) 30) S. Briickner, W. Porzio, Makromol. Chem. 189,961 (1988) 31) A. Bolognesi, W. Porzio, F. Provasoli, T. A. Ezquerra, Makromol. Chem. 194, 817
32) A. Bolognesi, W. Porzio, G. Zhuo, T. Ezquerra, Eu,: Polym. J. 32, 1097 (1996) 33) S. Destri, W. Porzio, S. Briickner, Macromol. Rapid Commun. 16,297 (1995) 34) W. Porzio, S. Destri, M. Mascherpa, S. Briickner, Acta Polym. 44,266 (1985) 35) F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Carfield,
36) See, for example, G. Bajo, A. Bolognesi, S. Destri, Z. Geng, W. Porzio, Mol. Cryst.
37) G. W. Gray, J. W. G. Goodby, “Smectic Liquid Crystals”, Leonard Hill, Glasgow
therein
( 1993)
G. Chang, T. Hendrickson, W. C. Still, J. Comp. Chem. 11,440 (1990)
Liq. Cryst. 229,91 (1993)
1984




















