
In situ kinetic analysis of glyoxalase I and glyoxalase II in Saccharomyces cerevisiae
Upload
independentCategory
view
1download
0
d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
avai lab le at www.sc iencedi rec t .com
journa l homepage: www.e lsev ier .com/ locate /dnarepai r
Suppression of genomic instability by SLX5 and SLX8 inSaccharomyces cerevisiae
Chaoying Zhang, Tania M. Roberts, Jay Yang, Ridhdhi Desai, Grant W. Brown ∗
Department of Biochemistry, University of Toronto, Toronto, Ont., Canada M5S 1A8
a r t i c l e i n f o
Article history:
Received 23 July 2005
Received in revised form 20 October
2005
a b s t r a c t
Replication forks can stall spontaneously at specific sites in the genome, and upon encoun-
tering DNA lesions resulting from chemical or radiation damage. In Saccharomyces cerevisiae
proteins implicated in processing of stalled replication forks include those encoded by the
SGS1, TOP3, MUS81, MMS4, SLX1, SLX4, SLX5/HEX3, and SLX8 genes. We tested the roles
Accepted 27 October 2005Available online 1 December 2005
Keywords:
Gross chromosomal rearrangements
SLX genes
Genomic instability
Spontaneous DNA damage
of these genes in suppressing gross chromosomal rearrangements (GCRs), which include
translocations, large interstitial deletions, and loss of a chromosome arm with de novo
telomere addition. We found that mus81, mms4, slx1, slx4, slx5, and slx8 mutants all have
elevated levels of spontaneous GCRs, and that SLX5 and SLX8 are particularly critical sup-
pressors of GCRs during normal cell cycle progression. In addition to increased GCRs, dele-
tion of SLX5 or SLX8 resulted in increased relocalization of the DNA damage checkpoint
protein Ddc2 and activation of the checkpoint kinase Rad53, indicating the accumulation
of spontaneous DNA damage. Surprisingly, mutants in slx5 or slx8 were not sensitive to
transient replication fork stalling induced by hydroxyurea, nor were they sensitive to repli-
cation dependent double-strand breaks induced by camptothecin. This suggested that Slx8
and Slx8 played limited roles in stabilizing, restarting, or resolving transiently stalled replica-
tion forks, but were critical for preventing the accumulation of DNA damage during normal
cell cycle progression.
© 2005 Elsevier B.V. All rights reserved.
1. Introduction
Efficient completion of S phase requires that replication forksproceed unimpeded through the entire genome. In addition tostalling at lesions caused by exogenous sources, such as chem-ical and radiation damage, DNA replication forks can stallupon encountering secondary structures, topological stress,and protein–DNA complexes. Such stalling during an other-wise unperturbed S phase, and the subsequent collapse andbreakage of the stalled replication forks, could represent asignificant source of genomic instability in normal cyclingcells. Indeed, in the budding yeast Saccharomyces cerevisiaeseveral lines of evidence suggest that DNA replication errors
∗ Corresponding author. Tel.: +1 416 946 5733; fax: +1 416 978 8548.E-mail address: [email protected] (G.W. Brown).
cause spontaneous genome rearrangements. Recombinationrepair foci assemble at a significant frequency during nor-mal S phase [1]. Deregulation of origin firing causes genomeinstability [2–4], as does mutation in checkpoint genes thatmonitor S phase [5–7]. Mutations in a number of DNA repli-cation genes cause genome rearrangements [8,5]. Addition-ally, recent data in fission yeast indicates that replication forkstalling caused by non-nucleosomal protein binding causesformation of recombination repair foci and induces recombi-nation events [9].
A number of pathways have been described that arethought to contribute to the stability of replication forks andthat aid in restarting forks that have stalled. Among these
1568-7864/$ – see front matter © 2005 Elsevier B.V. All rights reserved.doi:10.1016/j.dnarep.2005.10.010

d n a r e p a i r 5 ( 2 0 0 6 ) 336–346 337
pathways, RecQ DNA helicases are thought to play a centralrole in restarting replication forks (reviewed in [10]). RecQ heli-cases are conserved throughout evolution. In bacteria RecQis important for resolving stalled replication forks that haveregressed to form Holliday junction-like structures [11]. Sim-ilarly, the budding yeast RecQ homologue Sgs1 prevents theaccumulation of abnormal recombination structures at dam-aged replication forks [12]. The precise role of Sgs1 in forkrestart is unclear, but may involve the ability of this DNA heli-case to resolve Holliday junctions [13]. In addition to its puta-tive role in fork restart, Sgs1 has other functions that couldbe important for maintaining genome integrity. Sgs1 is impor-tant for full checkpoint activation in response to replicationfork stalling [14], for replication fork integrity during replica-tion stress [15], and for timely replication of the rDNA repeats[16,17], which are rich in replication fork barriers. Of particularinterest, sgs1 mutants show elevated levels of gross chromoso-mal rearrangements (GCRs) in the absence of exogenous DNAdamage [18]. This suggests that Sgs1 plays an important rolein suppressing genomic instability during normal S phase pro-gression.
In the absence of Sgs1 function, the MMS4, MUS81, SLX1,SLX4, SLX5/HEX3, and SLX8 genes are essential, and so weretermed SLX genes (for Synthetic Lethal of unknown func-tion) [19]. One model that accounts for these synthetic lethalgenetic interactions is that these genes function in a redun-dant pathway or pathways that can stabilize and restartsTttMafitwaascMaataludd
mbrnHSrgtc
accumulation of spontaneous DNA lesions. Finally, althoughslx5 and slx8 mutants were sensitive to prolonged exposureto hydroxyurea (HU), they were not sensitive to transient HUexposure or to prolonged exposure to the topoisomerase Ipoison camptothecin (CPT). Thus the Slx5/8 complex func-tioned primarily in the suppression of spontaneous DNAdamage.
2. Materials and methods
2.1. Yeast strains and media
Yeast strains used in this study are listed in Table 1, andare derivatives of S288C [25]. Non-essential haploid deletionstrains were made by the Saccharomyces Gene Deletion Project[26]. Standard yeast media and growth conditions were used[27]. Strains for the assay of GCR rates were constructed bycrossing to the GCR starter strain CZY105. Following sporula-tion, MATa haploids carrying the relevant deletion mutationand the CAN1 and hxt13::URA3 genes were selected on SD-ura-his-lys containing 50 �g/ml 5-(2-aminoethyl)-l-cysteine (AEC;Sigma) and 100 �g/ml G418 (Sigma). Double mutant strainswere made by crossing the relevant single mutant strainswith MAT� strains carrying the relevant gene replaced bythe NAT resistance gene. After sporulation, double mutantstrains were selected on SD-his-ura-lys plus 50 �g/ml AEC plus
talled replication forks when Sgs1 (or its binding partnersop3 and Rmil) are absent [19]. In addition to their func-ional relationship with SGS1/TOP3 subsets of these genes arehought to function in concert with each other. Mus81 andms4 are subunits of an endonuclease [20,21], as are Slx1
nd Slx4 [22]. These nucleases have different substrate speci-cities in vitro, and can act to resolve branched DNA struc-ures in a manner consistent with their redundant functionith Sgs1/Top3 in vivo. The biochemical functions of Slx5
nd Slx8 remain unclear, but the proteins interact in vivond deletion mutants of either are phenotypically similar,uggesting that they may act together in a manner reminis-ent of Mus81/Mms4 or Slx1/Slx4 [19]. It is not clear whetherus81/Mms4, Slx1/Slx4, and Slx5/Slx8 function together insingle pathway, or separately in multiple pathways, par-
llel to Sgs1/Top3. Furthermore, while a role for these pro-eins in restarting or stabilizing stalled replication forks is anttractive hypothesis, the mechanistic basis for the syntheticethal interactions between the SLX genes and SGS1 remainsnknown with the exception of slx4� which causes a lethalefect in rDNA replication when combined with an sgs1 con-itional mutation [16].
Pathways that contribute to the suppression of gross chro-osomal rearrangements have been studied extensively in
udding yeast, and include S phase checkpoint genes [6,7],eplication and recombination genes [5], telomere mainte-ance genes [23], and chromatin remodeling genes [24].ere we describe roles for the Mus81/Mms4, Slx1/Slx4, andlx5/Slx8 proteins in suppressing gross chromosomal rear-angements during normal cell cycle progression. Of the SLXenes, we found that SLX5 and SLX8 are particularly impor-ant for suppressing GCRs during an otherwise unperturbedell cycle. SLX5 and SLX8 also suppressed mutations and the
200 �g/ml G418 plus 100 �g/ml clonNAT (Hans-Knoll Institutefur Naturstoff-Forschung, Jena, Germany). The presence ofCAN1 was confirmed by assessing sensitivity to 60 �g/ml cana-vanine (Sigma).
2.2. GCR, mutation rate, and recombination rateassays
GCR assays were carried out essentially as described [5]. Allstrains were isogenic with CZY106, which was used as the wildtype control. MMS induced GCR rates were measured followingtreatment with 0.01% MMS for 2 h at 30 ◦C. GCR rates were cal-culated using the method of the median [28], and are averagesof at least two fluctuation tests of three to five independentassays. Mutation rates were determined by measuring for-ward mutation rates to canavanine resistance, as described[29]. Mutation rates were calculated using the method of themedian [28], and are averages of three fluctuation tests of 10independent assays. Direct repeat recombination rates weredetermined using a leu2�EcoRI-URA3-leu2�BstEII marker [30],essentially as described [31]. Leucine prototrophs were scoredirrespective of URA3 status such that both gene conversionand intrachromatid recombination were included in the rate.Recombination rates were calculated using the method of themedian [28], and are averages of three fluctuation tests of fiveindependent assays.
2.3. CHEF gel electrophoresis
Canavanine resistant, 5-fluoroorotic acid resistant coloniesthat had undergone a GCR event were grown to satura-tion in YPD medium. 2 × 108 cells were harvested and pro-cessed for CHEF gel electrophoresis as described [2]. Chromo-
338 d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
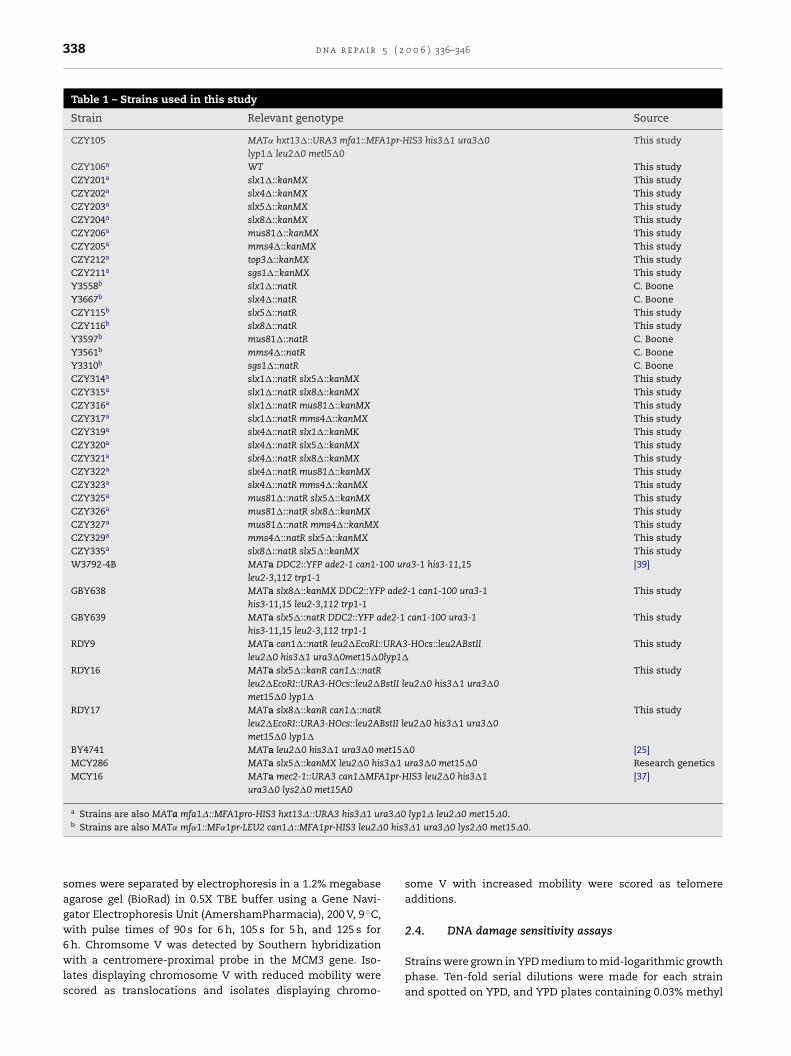
Table 1 – Strains used in this study
Strain Relevant genotype Source
CZY105 MAT˛ hxt13�::URA3 mfa1::MFA1pr-HIS3 his3�1 ura3�0lyp1� leu2�0 metl5�0
This study
CZY106a WT This studyCZY201a slx1�::kanMX This studyCZY202a slx4�::kanMX This studyCZY203a slx5�::kanMX This studyCZY204a slx8�::kanMX This studyCZY206a mus81�::kanMX This studyCZY205a mms4�::kanMX This studyCZY212a top3�::kanMX This studyCZY211a sgs1�::kanMX This studyY3558b slx1�::natR C. BooneY3667b slx4�::natR C. BooneCZY115b slx5�::natR This studyCZY116b slx8�::natR This studyY3597b mus81�::natR C. BooneY3561b mms4�::natR C. BooneY3310b sgs1�::natR C. BooneCZY314a slx1�::natR slx5�::kanMX This studyCZY315a slx1�::natR slx8�::kanMX This studyCZY316a slx1�::natR mus81�::kanMX This studyCZY317a slx1�::natR mms4�::kanMX This studyCZY319a slx4�::natR slx1�::kanMK This studyCZY320a slx4�::natR slx5�::kanMX This studyCZY321a slx4�::natR slx8�::kanMX This studyCZY322a slx4�::natR mus81�::kanMX This studyCZY323a slx4�::natR mms4�::kanMX This studyCZY325a mus81�::natR slx5�::kanMX This studyCZY326a mus81�::natR slx8�::kanMX This studyCZY327a mus81�::natR mms4�::kanMX This studyCZY329a mms4�::natR slx5�::kanMX This studyCZY335a slx8�::natR slx5�::kanMX This studyW3792-4B MATa DDC2::YFP ade2-1 can1-100 ura3-1 his3-11,15
leu2-3,112 trp1-1[39]
GBY638 MATa slx8�::kanMX DDC2::YFP ade2-1 can1-100 ura3-1his3-11,15 leu2-3,112 trp1-1
This study
GBY639 MATa slx5�::natR DDC2::YFP ade2-1 can1-100 ura3-1his3-11,15 leu2-3,112 trp1-1
This study
RDY9 MATa can1�::natR leu2�EcoRI::URA3-HOcs::leu2ABstIIleu2�0 his3�1 ura3�0met15�0lyp1�
This study
RDY16 MATa slx5�::kanR can1�::natRleu2�EcoRI::URA3-HOcs::leu2�BstII leu2�0 his3�1 ura3�0met15�0 lyp1�
This study
RDY17 MATa slx8�::kanR can1�::natRleu2�EcoRI::URA3-HOcs::leu2ABstII leu2�0 his3�1 ura3�0met15�0 lyp1�
This study
BY4741 MATa leu2�0 his3�1 ura3�0 met15�0 [25]MCY286 MATa slx5�::kanMX leu2�0 his3�1 ura3�0 met15�0 Research geneticsMCY16 MATa mec2-1::URA3 can1�MFA1pr-HIS3 leu2�0 his3�1
ura3�0 lys2�0 met15A0[37]
a Strains are also MATa mfa1�::MFA1pro-HIS3 hxt13�::URA3 his3�1 ura3�0 lyp1� leu2�0 met15�0.b Strains are also MAT˛ mf˛1::MF˛1pr-LEU2 can1�::MFA1pr-HIS3 leu2�0 his3�1 ura3�0 lys2�0 met15�0.
somes were separated by electrophoresis in a 1.2% megabaseagarose gel (BioRad) in 0.5X TBE buffer using a Gene Navi-gator Electrophoresis Unit (AmershamPharmacia), 200 V, 9 ◦C,with pulse times of 90 s for 6 h, 105 s for 5 h, and 125 s for6 h. Chromsome V was detected by Southern hybridizationwith a centromere-proximal probe in the MCM3 gene. Iso-lates displaying chromosome V with reduced mobility werescored as translocations and isolates displaying chromo-
some V with increased mobility were scored as telomereadditions.
2.4. DNA damage sensitivity assays
Strains were grown in YPD medium to mid-logarithmic growthphase. Ten-fold serial dilutions were made for each strainand spotted on YPD, and YPD plates containing 0.03% methyl

d n a r e p a i r 5 ( 2 0 0 6 ) 336–346 339
methanesulfonate (MMS, Sigma–Aldrich), 0.2 M Hydroxyurea(HU, Sigma–Aldrich), 0.05 M HU, or 15 �g/ml camptothecin(CPT, Sigma–Aldrich). Plates were incubated at 25 ◦C for 3 days.Viability assays were performed in liquid YPD medium in thepresence of 150 mM HU. At the indicated times samples wereremoved from HU and plated on YPD. The viability relative tothe culture at time 0 was determined.
2.5. Microscopy
To examine cellular and nuclear morphology, cells were har-vested, washed with phosphate-buffered saline, resuspendedin 70% ethanol, and stored at −20 ◦C. Prior to examination,cells were resuspended in Vectashield mounting media withDAPI (Vector Laboratories Inc.). At least 200 cells were countedfor each sample. Intracellular localization of Ddc2-YFP wasdetermined as previously described for Rad52-YFP [32].
2.6. Rad53 in situ kinase assay
Rad53 in situ kinase assays of samples of asynchronouslygrowing, logarithmic phase cultures were carried out essen-tially as described [33]. Briefly, following fixation with TCAcells were lysed and resuspended in SDS-PAGE sample buffer.These extracts were fractionated on SDS-PAGE and transferredto PVDF membrane. Following renaturation in situ the immo-bilized proteins were incubated with [�-32P]ATP. Under thesecrsDlbbt
3
3
IwfcsesovhmSsfaccwG
Fig. 1 – SLX genes are suppressors of gross chromosomalrearrangements. GCR rate was measured in the indicateddeletion mutant strains by selecting for simultaneous lossof the CAN1 and URA3 genes on chromosome V. Theaverages and standard deviations of at least threefluctuation tests are shown. The GCR rate for the wild typewas (0.76 ± 0.18) × l0−9.
When we examined mutants in the six genes originally identi-fied as synthetic lethal with sgs1� we found that all displayedan increase in GCRs, but to different extents (Fig. 1). Deletionof SLX1 or SLX4 resulted in a very modest increase in GCRrate of approximately 7–10-fold that seen in wild type. In con-trast, deletion of SLX5 or SLX8 caused a dramatic increase inGCR rate, to 152–296-fold over wild type. Deletion of MUS81 orMMS4 resulted in a GCR rate similar to that seen in sgs1� andtop3�, 36–41-fold wild type levels. The similarity of GCR ratesin the functional pairs of SLX genes (SLX1/SLX4, SLX5/SLX8,MUS81/MMS4, and SGS1/TOP3) was consistent with previ-ous phenotypic and biochemical analysis [19,18,20,22,35], andwith evidence that each pair forms a complex in vivo [19,35].
3.2. GCR spectrum of SLX mutants
We analysed the spectrum of rearrangements in SLX mutantsthat had undergone a GCR event by contour-clamped homo-geneous electric field (CHEF) gel electrophoresis followed bySouthern blotting to detect chromosome V [2] (Table 2). TheGCRs detected in the budding yeast chromosome V assayused here are typically either the loss of the chromosome armfollowed by de novo telomere addition (telomere additions) ornonreciprocal translocations with or without microhomology(translocations) [8,6]. Although the CHEF gel assay does notactually map the breakpoints, it can readily distinguishtelomere additions from translocations in a large number
onditions, activated Rad53 will autophosphorylate. Phospho-ylated Rad53 was detected by exposure to a storage phosphorcreen, subsequent scanning on a Storm scanner (Molecularynamics), and analysis with ImageQuant software (Molecu-
ar Dynamics). Equivalent loading in each lane was confirmedy ponceau S staining of the membrane and was normalizedy comparison of the intensity of a non-specific phosphopro-ein band between samples.
. Results
.1. SLX genes suppress GCRs
n budding yeast gross chromosomal rearrangements (GCRs),hich include interstitial deletions, loss of a chromosome arm
ollowed by de novo telomere addition, non-reciprocal translo-ations, and chromosome fusions, can be detected by theimultaneous loss of two dominant counter-selectable mark-rs located on a non-essential arm of a specialized chromo-ome V [5]. Strains carrying wild type CAN1 and URA3 genesn the specialized chromosome V are sensitive to both cana-anine and 5-fluoroorotic acid (5-FOA), whereas strains thatave undergone a GCR event that results in the loss of botharkers will grow in the presence of canavanine and 5-FOA.
GS1, TOP3, and more recently MMS4, have been found touppress GCRs [18,34]. We tested whether other genes thatunction in pathways that are genetically parallel to SGS1/TOP3re also suppressors of GCRs (Fig. 1). To assay for GCRs weonstructed strains bearing the relevant mutation and the spe-ialized chromosome V. Consistent with previous results [18],e found that sgs1� and top3� both caused an increase inCRs, of 41- and 34-fold wild type levels, respectively (Fig. 1).
of isolates. We found that mus81�, slx4�, slx5�, slx8�, andsgs1� GCRs were all mostly telomere additions (Table 2),with the percent of telomere additions ranging from 53% to100%. This profile was similar to that previously noted forboth MMS treatment, which is known to cause replicationfork stalling, and for S phase checkpoint genes, which areimportant for detecting and stabilizing stalled replicationforks [6]. The difference observed between slx5� and slx8�
was not statistically significant.

340 d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
Table 2 – Structure of rearrangements in independentisolates following a GCR event
Gene # Translocation/fusion (%) # Deletion (%)
sgs1� 2 (11%) 17 (89%)top3�a 8 (61%) 5 (39%)mus81� 0 (0%) 19 (100%)mms4� n.d. n.d.slx1� n.d. n.d.slx4�b 6 (43%) 8 (57%)slx5� 4 (25%) 12 (75%)slx8�c 9 (47%) 10 (53%)rad52� 1 (5%) 18 (95%)Wild type 3 (16%) 16 (84%)
a Karyotype contained six isolates that could not be classifiedunambiguously.
b Karyotype contained five isolates that could not be classifiedunambiguously.
c Karyotype contained one isolate that could not be classifiedunambiguously.
3.3. SLX genes do not function as redundantsuppressors of GCRs
To determine whether the SLX genes make redundant con-tributions to the suppression of spontaneous GCRs we con-structed all possible double mutants between slx1�, slx4�,slx5�, slx8�, mus81�, and mms4�. GCR rates were determinedfor all double mutants and compared with the rates observedin single mutants. The results of this analysis for slx1�, slx5�,and mus81� are shown in Fig. 2. We found that in every casethe GCR rate of the double mutant was not significantly dif-ferent than the highest GCR rate observed in the relevantsingle mutant strains. This suggested that these genes do notmake redundant contributions to suppression of spontaneousGCRs as no significant synergistic or even additive effects wereobserved, although in some cases additive effects could bemasked by the large differences in the GCR rates of the singlemutants. The lack of additive effects in slx1� slx4�, mus81�
mms4�, and slx5� slx8� was again consistent with each pairforming a complex [19]. Additionally, the elevated GCR ratesobserved in each of these mutants did not depend entirelyon any of the other genes, as no significant suppression ofGCRs was observed in any of the double mutants. For example,the approximately 200-fold increase in GCRs observed in slx5�
was not reduced significantly in slx5� mus81�, slx5� mms4�,slx5� slx1�, slx5� slx4�, or slx5� slx8� double mutants(Fig. 2B). These data were consistent with the SLX mutants
Fig. 2 – SLX genes do not make redundant contributions tosuppression of gross chromosomal rearrangements. GCRrate was measured for all possible double mutantcombinations of (A) slx1�, (B) slx5�, and (C) mus81� withthe other SLX genes. The averages and standard deviationsof at least two fluctuation tests are shown.
functioning in a non-redundant fashion to suppress GCRs.
3.4. Induction of GCRs by DNA damage in SLXmutants
MMS is an efficient inducer of GCRs when used at levels knownto cause replication fork stalling, but not activation of Gl or G2DNA damage checkpoint pathways [36]. Treatment with 0.01%MMS for 2 h caused a 51-fold induction of GCRs in the wild typestrain in our assay (Fig. 3A and B). We found that MMS treat-ment caused a similar induction of GCRs in slxl� and slx4�,with induction of 37- and 38-fold (Fig. 3A and B). The inductionof GCRs by MMS in mus81� and mms4� was slightly reduced
d n a r e p a i r 5 ( 2 0 0 6 ) 336–346 341
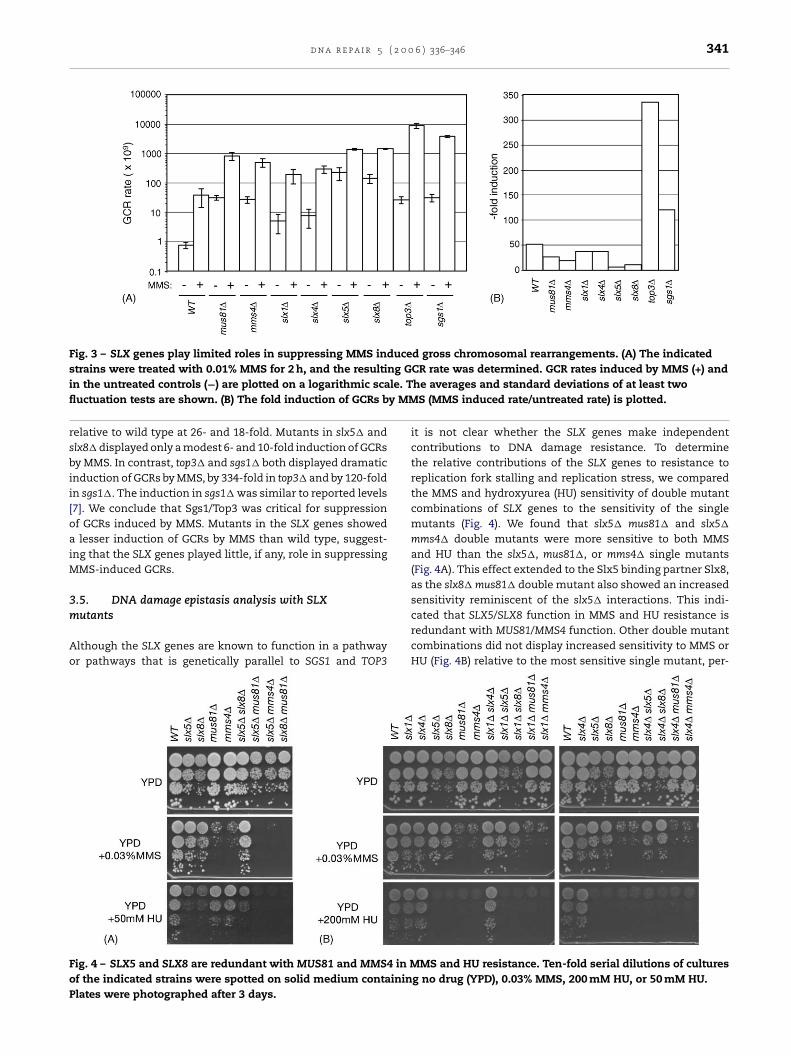
Fig. 3 – SLX genes play limited roles in suppressing MMS induced gross chromosomal rearrangements. (A) The indicatedstrains were treated with 0.01% MMS for 2 h, and the resulting GCR rate was determined. GCR rates induced by MMS (+) andin the untreated controls (−) are plotted on a logarithmic scale. The averages and standard deviations of at least twofluctuation tests are shown. (B) The fold induction of GCRs by MMS (MMS induced rate/untreated rate) is plotted.
relative to wild type at 26- and 18-fold. Mutants in slx5� andslx8� displayed only a modest 6- and 10-fold induction of GCRsby MMS. In contrast, top3� and sgs1� both displayed dramaticinduction of GCRs by MMS, by 334-fold in top3� and by 120-foldin sgs1�. The induction in sgs1� was similar to reported levels[7]. We conclude that Sgs1/Top3 was critical for suppressionof GCRs induced by MMS. Mutants in the SLX genes showeda lesser induction of GCRs by MMS than wild type, suggest-ing that the SLX genes played little, if any, role in suppressingMMS-induced GCRs.
3.5. DNA damage epistasis analysis with SLXmutants
Although the SLX genes are known to function in a pathwayor pathways that is genetically parallel to SGS1 and TOP3
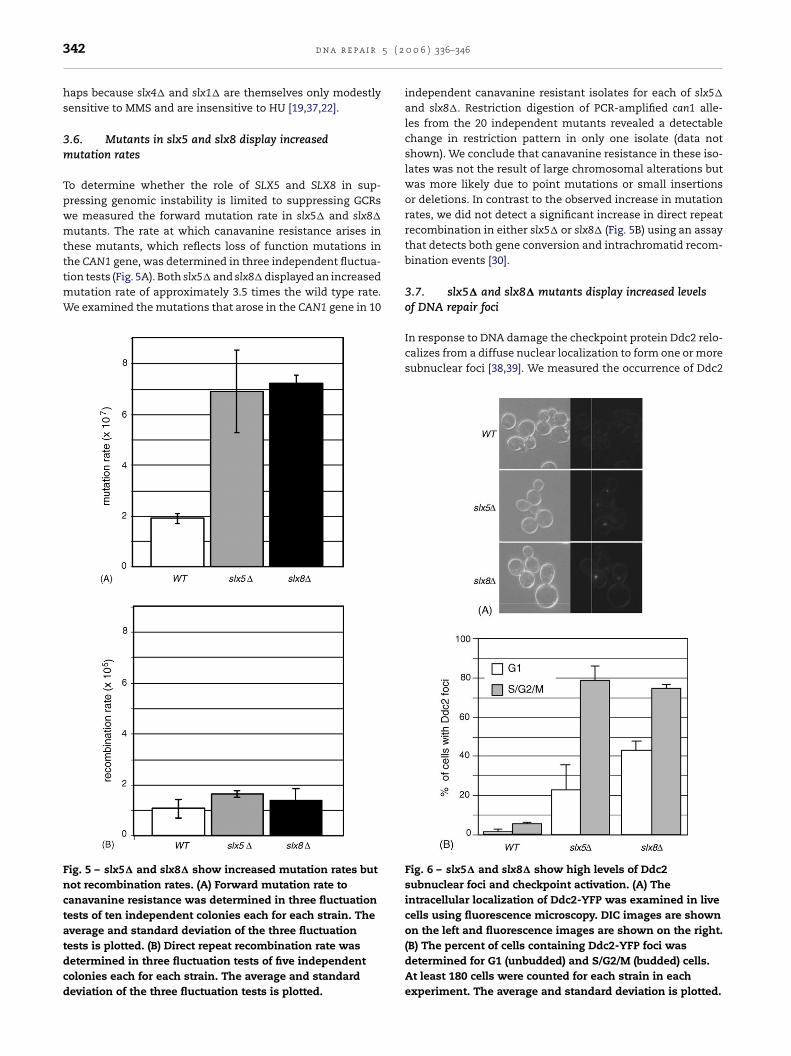
it is not clear whether the SLX genes make independentcontributions to DNA damage resistance. To determinethe relative contributions of the SLX genes to resistance toreplication fork stalling and replication stress, we comparedthe MMS and hydroxyurea (HU) sensitivity of double mutantcombinations of SLX genes to the sensitivity of the singlemutants (Fig. 4). We found that slx5� mus81� and slx5�
mms4� double mutants were more sensitive to both MMSand HU than the slx5�, mus81�, or mms4� single mutants(Fig. 4A). This effect extended to the Slx5 binding partner Slx8,as the slx8� mus81� double mutant also showed an increasedsensitivity reminiscent of the slx5� interactions. This indi-cated that SLX5/SLX8 function in MMS and HU resistance isredundant with MUS81/MMS4 function. Other double mutantcombinations did not display increased sensitivity to MMS orHU (Fig. 4B) relative to the most sensitive single mutant, per-
F in Mo ininP
ig. 4 – SLX5 and SLX8 are redundant with MUS81 and MMS4f the indicated strains were spotted on solid medium contalates were photographed after 3 days.
MS and HU resistance. Ten-fold serial dilutions of culturesg no drug (YPD), 0.03% MMS, 200 mM HU, or 50 mM HU.
342 d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
haps because slx4� and slx1� are themselves only modestlysensitive to MMS and are insensitive to HU [19,37,22].
3.6. Mutants in slx5 and slx8 display increasedmutation rates
To determine whether the role of SLX5 and SLX8 in sup-pressing genomic instability is limited to suppressing GCRswe measured the forward mutation rate in slx5� and slx8�
mutants. The rate at which canavanine resistance arises inthese mutants, which reflects loss of function mutations inthe CAN1 gene, was determined in three independent fluctua-tion tests (Fig. 5A). Both slx5� and slx8� displayed an increasedmutation rate of approximately 3.5 times the wild type rate.We examined the mutations that arose in the CAN1 gene in 10
independent canavanine resistant isolates for each of slx5�
and slx8�. Restriction digestion of PCR-amplified can1 alle-les from the 20 independent mutants revealed a detectablechange in restriction pattern in only one isolate (data notshown). We conclude that canavanine resistance in these iso-lates was not the result of large chromosomal alterations butwas more likely due to point mutations or small insertionsor deletions. In contrast to the observed increase in mutationrates, we did not detect a significant increase in direct repeatrecombination in either slx5� or slx8� (Fig. 5B) using an assaythat detects both gene conversion and intrachromatid recom-bination events [30].
3.7. slx5� and slx8� mutants display increased levelsof DNA repair foci
In response to DNA damage the checkpoint protein Ddc2 relo-calizes from a diffuse nuclear localization to form one or moresubnuclear foci [38,39]. We measured the occurrence of Ddc2
Fig. 5 – slx5� and slx8� show increased mutation rates butnot recombination rates. (A) Forward mutation rate tocanavanine resistance was determined in three fluctuationtests of ten independent colonies each for each strain. Theaverage and standard deviation of the three fluctuationtests is plotted. (B) Direct repeat recombination rate wasdetermined in three fluctuation tests of five independentcolonies each for each strain. The average and standarddeviation of the three fluctuation tests is plotted.
Fig. 6 – slx5� and slx8� show high levels of Ddc2subnuclear foci and checkpoint activation. (A) Theintracellular localization of Ddc2-YFP was examined in livecells using fluorescence microscopy. DIC images are shownon the left and fluorescence images are shown on the right.(B) The percent of cells containing Ddc2-YFP foci wasdetermined for G1 (unbudded) and S/G2/M (budded) cells.At least 180 cells were counted for each strain in eachexperiment. The average and standard deviation is plotted.

d n a r e p a i r 5 ( 2 0 0 6 ) 336–346 343
foci in cells expressing a Ddc2-YFP fusion protein during nor-mal cell cycle progression (Fig. 6). Mutants in slx5 and slx8displayed dramatically increased levels of Ddc2 foci comparedto wild type, with almost 80% of cells having Ddc2 foci in theabsence of any DNA damaging agent. By contrast, only 7%of wild type cells had Ddc2 foci. This suggests spontaneousDNA damage accumulated in most cells when Slx5 or Slx8was absent.
3.8. slx5� and slx8� mutants display a cell cycledelay and constitutive checkpoint activation
DNA damage results in the activation of cell cycle checkpoints.We examined asynchronous cultures in mid-logarithmicphase and found that both slx5� and slx8� mutants accu-mulated in mitosis, with a single nucleus at the bud neck(Fig. 7A). This accumulation could be the result of check-point activation, so we measured the activity of the checkpointkinase Rad53 in slx5� and slx8� using an in situ kinase assay
Fclambleoaa
(Fig. 7B and C). Consistent with the presence of spontaneousDNA damage, Rad53 was activated in both slx5� and slx8�
mutants.
3.9. SLX5 and SLX8 are not critical for viability duringtransient replication fork stalling
slx5� and slx8� mutants are sensitive to continuous exposureto HU, but are not sensitive to levels of MMS known to causereplication fork stalling ([19] and Fig. 4). We tested the sensitiv-ity of slx5� and slx8� to transient exposures (up to 6 h) to HU(Fig. 8A). We found that slx5� and slx8� were no more sensitivethan wild-type to this exposure, indicating that Slx5 and Slx8did not play critical roles in stabilizing, resolving, or restart-ing replication forks stalled transiently by nucleotide deple-tion. The topoisomerase I poison camptothecin (CPT) causesdouble strand DNA breaks when replication forks encountertopo I-CPT induced DNA lesions [40,41]. When slx5� and slx8�
mutants were treated with camptothecin (CPT) they did not
ig. 7 – slx5� and slx8� accumulate in mitosis and displayonstitutive checkpoint activation. (A) Cells fromogarithmically growing cultures were stained with DAPInd examined by phase-contrast and fluorescenceicroscopy. The percent of cells with no bud (G1), a small
ud (S), or a large bud (G2/M) is plotted. (B) Cells fromogarithmically growing cultures were fixed with TCA andqual amounts of the resulting extracts were fractionatedn SDS-PAGE for in situ kinase assay of Rad53. (C) Rad53ctivity in the in situ kinase assay is plotted relative to thectivity in the wild type sample.
Fig. 8 – slx5� and slx8� mutants are not sensitive totransient HU exposure or to CPT. (A) Viability was measuredfollowing growth in liquid culture in the presence of150 mM HU for the indicated times. The checkpointdefective rad53-11 strain is included as a positive control.(B) Sensitivity to CPT was determined by spotting 10-foldserial dilutions of the indicated strains on solid mediumcontaining 15 �g/ml CPT. The control plate contains DMSO,which is the solvent in which the CPT is dissolved.

344 d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
display detectable sensitivity, either during continuous expo-sure (Fig. 8B) or during transient exposure (data not shown).Thus Slx5 and Slx8 did not appear to play important rolesin resistance to replication dependent double strand breaksinduced by CPT.
4. Discussion
We have investigated the role of the SLX genes SLX1, SLX4,MUS81, MMS4, SLX5, and SLX8 in suppression of gross chromo-somal rearrangements. Of these, we found that SLX5 and SLX8are particularly critical suppressors of GCRs during normal cellcycle progression. The molecular role of Slx5 and Slx8 in DNAdamage resistance is currently unclear, although they likelyfunction as a complex [19]. Our finding that deletion of eithergene resulted in 100–200-fold increase in GCRs suggests thatboth are important suppressors of genomic instability duringnormal cell cycle progression, and not just when the SGS1 orTOP3 gene products are absent.
Genetic interactions between the SLX genes are complex.We did not detect any additive or synergistic effects on GCRrates in any of the double mutant combinations, suggestingthat the SLX genes do not function redundantly in the sup-pression of GCRs. We did, however, observe very different GCRrates in each pair of SLX genes, ranging from ∼6-fold wild typein slxl� or slx4�, to ∼29-fold wild type in mus81� or mms4�,
could explain our data, and that of others [19], that indicatethat SLX5/8 plays a role in the DNA damage response and insuppressing GCRs that is distinct from that played by SLX1/4,MUS81/MMS4, and SGS1/TOP3.
Of particular interest, we found that slx5� and slx8�
mutants display high levels of subnuclear Ddc2 foci. SinceDdc2 relocalizes to foci in response to DNA damage, this sug-gests that there was a great deal of spontaneous DNA dam-age present in cells lacking Slx5 or Slx8. Consistent with thispossibility, we observed a mitotic delay and activation of thecheckpoint kinase Rad53 in both slx5� and slx8� mutants.Spontaneous DNA damage could certainly be the cause of theincreased GCR rates and increased mutation rates that weobserved in slx5� and slx8� mutants. This raises the possi-bility that the synthetic lethality exhibited by slx5� and slx8�
mutants when combined with an sgs1� mutation could bedue to the dramatically elevated levels of spontaneous DNAdamage in slx5� and slx8� mutants, particularly since SGS1 isimportant in mounting a checkpoint response to DNA damage[14,43]. Alternatively, Sgs1 could be essential for the repair ofthe lesions that arise in the absence of Slx5 or Slx8.
We examined the role of Slx5 and Slx8 in stabilizing,restarting, or resolving stalled replication forks by comparingthe sensitivity of slx5� and slx8� mutants to prolonged andtransient exposure to agents known to stall forks. In particular,we hypothesized that proteins with critical roles in stabiliz-ing, restarting, or resolving stalled replication forks would be
to ∼170-fold wild type in slx5� or slx8�. This suggests that thegenes do not function in a simple linear pathway to suppressGCRs. We also noted that when we combined a mutant witha large GCR rate with a mutant with a small one (for example,slx5� with slx1�) it did not result in significant suppression ofthe GCR rate. Thus the GCRs observed in the absence of oneSLX gene do not depend on the action of the other SLX genes,and the action of each SLX gene in suppressing GCRs seems tobe independent of the action of the other SLX genes. However,one caveat to this analysis is that the variation inherent in theGCR assay makes it difficult to detect small additive effects, asmight be expected when a mutant with a modest increase inGCR rate is combined with a mutant with a large increase inGCR rate.
DNA damage epistasis analysis indicated that SLX5 andSLX8 function in resistance to MMS and HU in a pathway thatis redundant with MUS81/MMS4 function. We did not observeany increased DNA damage sensitivity of double mutantsbetween the slx5/slx8 and slx1/slx4 genes, likely because slx1�
and slx4� mutants display only modest MMS sensitivity andare not HU sensitive [37,22]. Thus an additive effect mightnot result in a detectable increase in sensitivity in the dou-ble mutants. One interesting possibility is that the SLX geneproducts that function as nucleases, Slx1/4 and Mus81/Mms4,play damage resistance roles that are mechanistically distinctfrom those played by Slx5/8. The biochemical activity of Slx5/8remains to be determined, but neither protein possesses aknown nuclease domain. Both Slx5 and Slx8 contain a RINGfinger domain [19], which often functions in protein ubiquiti-nation [42]. It will be of great interest to determine whetherthe RING finger domains of Slx5 and/or Slx8 mediate inter-actions with ubiquitin conjugating enzymes and function insignaling or proteolysis via ubiquitination. Such an activity
important for viability following transient exposure to DNAdamaging agents known to cause replication fork stalling.Mutants in slx5� and slx8� were not MMS sensitive, so Slx5and Slx8 did not appear to play a major role when replica-tion fork progression is slowed by alkylation damage. slx5�
and slx8� mutants were sensitive to prolonged exposure toHU, but did not lose viability during transient HU exposure.This contrasts with mutants in checkpoint pathways, and sug-gests that Slx5 and Slx8 did not play a critical, non-redundantrole during transient replication fork stalling. The sensitivityof slx5� and slx8� mutants to prolonged HU exposure couldreflect a requirement for Slx5 and Slx8 when replication forkscollapse. However, slx5� and slx8� mutants were not sensitiveto CPT exposure, a treatment that results in DSBs when CPTinduced damage is encountered by a replication fork [41,40,44].Thus the clearest in vivo function of Slx5 and Slx8 was in pre-venting the accumulation of DNA damage during normal cellcycle progression.
With the exception of MMS4, none of the SLX genes weredetected by a recent genome-wide GCR screen of the S. cere-visiae viable haploid gene deletion collection [34]. This sug-gests that genes with critical roles in suppressing GCRs remainundiscovered, as mus81�, slx5�, and slx8� all displayed ahigher rate of GCRs than mms4� in our analysis, and so werenot likely to be below some threshold value in the genome-wide screen. Further analysis of the gene deletion collectionwill likely identify additional suppressors of GCRs.
Gross chromosomal rearrangements detected in yeastresemble those observed frequently in cancerous cells [45].Identification of genes with roles in suppressing GCRs willbe important in understanding the mechanisms by whichthese rearrangements arise. Although SLX5 and SLX8 lack clearsequence homologues in human cells, it is possible that as yet

d n a r e p a i r 5 ( 2 0 0 6 ) 336–346 345
unidentified functional homologues exist which play similarroles in suppressing GCRs in humans, a view that is encour-aged by the existence of homologues of SLX5 and SLX8 in thedistantly related yeast Schizosaccharomyces pombe [19].
Acknowledgements
We thank Richard Kolodner, Charlie Boone, and Rodney Roth-stein for yeast strains, Kyungjae Myung and Richard Kolodnerfor advice on the GCR assay, and Michael Chang for construc-tive comments on the manuscript. This work was supportedby the National Cancer Institute of Canada. GWB is a ResearchScientist of the National Cancer Institute of Canada.
r e f e r e n c e s
[1] M. Lisby, R. Rothstein, U.H. Mortensen, Rad52 forms DNArepair and recombination centers during S phase, Proc.Natl. Acad. Sci. U.S.A. 98 (2001) 8276–8282.
[2] A. Lengronne, E. Schwob, The yeast CDK inhibitor Sic1prevents genomic instability by promoting replicationorigin licensing in late G(1), Mol. Cell 9 (2002) 1067–1078.
[3] S. Tanaka, J.F. Diffley, Deregulated G1-cyclin expressioninduces genomic instability by preventing efficient pre-RCformation, Genes Dev. 16 (2002) 2639–2649.
[4] D. Huang, D. Koshland, Chromosome integrity in
[14] C. Frei, S.M. Gasser, The yeast Sgs1p helicase actsupstream of Rad53p in the DNA replication checkpointand colocalizes with Rad53p in S-phase-specific foci,Genes Dev. 14 (2000) 81–96.
[15] J.A. Cobb, L. Bjergbaek, K. Shimada, C. Frei, S.M. Gasser,DNA polymerase stabilization at stalled replication forksrequires Mec1 and the RecQ helicase Sgs1, EMBO. J. 22(2003) 4325–4336.
[16] V. Kaliraman, S.J. Brill, Role of SGS1 and SLX4 inmaintaining rDNA structure in Saccharomyces cerevisiae,Curr. Genet. 41 (2002) 389–400.
[17] G. Versini, I. Comet, M. Wu, L. Hoopes, E. Schwob, P.Pasero, The yeast Sgs1 helicase is differentially requiredfor genomic and ribosomal DNA replication, EMBO J. 22(2003) 1939–1949.
[18] K. Myung, A. Datta, C. Chen, R.D. Kolodner, SGS1, theSaccharomyces cerevisiae homologue of BLM and WRN,suppresses genome instability and homeologousrecombination, Nat. Genet. 27 (2001) 113–116.
[19] J.R. Mullen, V. Kaliraman, S.S. Ibrahim, S.J. Brill,Requirement for three novel protein complexes in theabsence of the Sgs1 DNA helicase in Saccharomycescerevisiae, Genetics 157 (2001) 103–118.
[20] V. Kaliraman, J.R. Mullen, W.M. Fricke, S.A.Bastin-Shanower, S.J. Brill, Functional overlap betweenSgs1–Top3 and the Mms4–Mus81 endonuclease, Genes Dev.15 (2001) 2730–2740.
[21] S.A. Bastin-Shanower, W.M. Fricke, J.R. Mullen, S.J. Brill,The mechanism of Mus81–Mms4 cleavage site selectiondistinguishes it from the homologous endonucleaseRad1–Rad10, Mol. Cell Biol. 23 (2003) 3487–3496.
[22] W.M. Fricke, S.J. Brill, Slx1–Slx4 is a secondstructure-specific endonuclease functionally redundantwith Sgs1–Top3, Genes Dev. 17 (2003) 1768–1778.
[23] K. Myung, C. Chen, R.D. Kolodner, Multiple pathwayscooperate in the suppression of genome instability inSaccharomyces cerevisiae, Nature 411 (2001) 1073–1076.
[24] K. Myung, V. Pennaneach, E.S. Kats, R.D. Kolodner,Saccharomyces cerevisiae chromatin-assembly factors thatact during DNA replication function in the maintenance ofgenome stability, Proc. Natl. Acad. Sci. U.S.A. 100 (2003)6640–6645.
[25] C.B. Brachmann, A. Davies, G.J. Cost, E. Caputo, J. Li, P.Hieter, J.D. Boeke, Designer deletion strains derived fromSaccharomyces cerevisiae S288C: a useful set of strains andplasmids for PCR-mediated gene disruption and otherapplications, Yeast 14 (1998) 115–132.
[26] E.A. Winzeler, D.D. Shoemaker, A. Astromoff, H. Liang, K.Anderson, B. Andre, R. Bangham, R. Benito, J.D. Boeke, H.Bussey, A.M. Chu, C. Connelly, K. Davis, F. Dietrich, S.W.Dow, M. El Bakkoury, F. Foury, S.H. Friend, E. Gentalen, G.Giaever, J.H. Hegemann, T. Jones, M. Laub, H. Liao, R.W.Davis, et al., Functional characterization of the S. cerevisiaegenome by gene deletion and parallel analysis, Science285 (1999) 901–906.
[27] F. Sherman, Getting started with yeast, Meth. Enzymol.194 (1991) 3–21.
[28] D.E. Lea, C.A. Coulson, The distribution of the numbers ofmutants in bacterial populations, J. Genet. 49 (1949)264–285.
[29] M. Bellaoui, M. Chang, J. Ou, H. Xu, C. Boone, G.W. Brown,Elg1 forms an alternative RFC complex important for DNAreplication and genome integrity, EMBO J. 22 (2003)4304–4313.
[30] J. Smith, R. Rothstein, An allele of RFA1 suppressesRAD52-dependent double-strand break repair inSaccharomyces cerevisiae, Genetics 151 (1999)447–458.
Saccharomyces cerevisiae: the interplay of DNA replicationinitiation factors, elongation factors, and origins, GenesDev. 17 (2003) 1741–1754.
[5] C. Chen, R.D. Kolodner, Gross chromosomalrearrangements in Saccharomyces cerevisiae replication andrecombination defective mutants, Nat. Genet. 23 (1999)81–85.
[6] K. Myung, A. Datta, R.D. Kolodner, Suppression ofspontaneous chromosomal rearrangements by S phasecheckpoint functions in Saccharomyces cerevisiae, Cell 104(2001) 397–408.
[7] K. Myung, R.D. Kolodner, Suppression of genomeinstability by redundant S-phase checkpoint pathways inSaccharomyces cerevisiae, Proc. Natl. Acad. Sci. U.S.A. 99(2002) 4500–4507.
[8] C. Chen, K. Umezu, R.D. Kolodner, Chromosomalrearrangements occur in S. cerevisiae rfa1 mutator mutantsdue to mutagenic lesions processed bydouble-strand-break repair, Mol. Cell 2 (1998) 9–22.
[9] S. Lambert, A. Watson, D.M. Sheedy, B. Martin, A.M. Carr,Gross chromosomal rearrangements and elevatedrecombination at an inducible site-specific replication forkbarrier, Cell 121 (2005) 689–702.
[10] I.D. Hickson, RecQ helicases: caretakers of the genome,Nat. Rev. Cancer 3 (2003) 169–178.
[11] J. Courcelle, P.C. Hanawalt, RecQ and RecJ process blockedreplication forks prior to the resumption of replication inUV-irradiated Escherichia coli, Mol. Gen. Genet. 262 (1999)543–551.
[12] G. Liberi, G. Maffioletti, C. Lucca, I. Chiolo, A.Baryshnikova, C. Cotta-Ramusino, M. Lopes, A. Pellicioli,J.E. Haber, M. Foiani, Rad51-dependent DNA structuresaccumulate at damaged replication forks in sgs1 mutantsdefective in the yeast ortholog of BLM RecQ helicase,Genes Dev. 19 (2005) 339–350.
[13] R.J. Bennett, J.L. Keck, J.C. Wang, Binding specificitydetermines polarity of DNA unwinding by the Sgs1 proteinof S. cerevisiae, J. Mol. Biol. 289 (1999) 235–248.

346 d n a r e p a i r 5 ( 2 0 0 6 ) 336–346
[31] R.M. Spell, S. Jinks-Robertson, Determination of mitoticrecombination rates by fluctuation analysis inSaccharomyces cerevisiae, Meth. Mol. Biol. 262 (2004) 3–12.
[32] M. Chang, M. Bellaoui, C. Zhang, R. Desai, P. Morozov, L.Delgado-Cruzata, R. Rothstein, G.A. Freyer, C. Boone, G.W.Brown, RMI1/NCE4 a suppressor of genome instability,encodes a member of the RecQ helicase/Topo III complex,EMBO J. 24 (2005) 2024–2033.
[33] A. Pellicioli, C. Lucca, G. Liberi, F. Marini, M. Lopes, P.Plevani, A. Romano, P.P. Di Fiore, M. Foiani, Activation ofRad53 kinase in response to DNA damage and its effect inmodulating phosphorylation of the lagging strand DNApolymerase, EMBO J. 18 (1999) 6561–6572.
[34] S. Smith, J.Y. Hwang, S. Banerjee, A. Majeed, A. Gupta, K.Myung, Mutator genes for suppression of grosschromosomal rearrangements identified by agenome-wide screening in Saccharomyces cerevisiae, Proc.Natl. Acad. Sci. U.S.A. 101 (2004) 9039–9044.
[35] Y. Fu, W. Xiao, Functional domains required for theSaccharomyces cerevisiae Mus81–Mms4 endonucleasecomplex formation and nuclear localization, DNA Rep.(Amst.) 2 (2003) 1435–1447.
[36] K. Myung, R.D. Kolodner, Induction of genome instabilityby DNA damage in Saccharomyces cerevisiae, DNA Rep.(Amst.) 2 (2003) 243–258.
[37] M. Chang, M. Bellaoui, C. Boone, G.W. Brown, Agenome-wide screen for methylmethanesulfonate-sensitive mutants reveals genesrequired for S phase progression in the presence of DNAdamage, Proc. Natl. Acad. Sci. U.S.A. 99 (2002) 16934–16939.
[38] J.A. Melo, J. Cohen, D.P. Toczyski, Two checkpointcomplexes are independently recruited to sites of DNAdamage in vivo, Genes Dev. 15 (2001) 2809–2821.
[39] M. Lisby, J.H. Barlow, R.C. Burgess, R. Rothstein,Choreography of the DNA damage response;spatiotemporal relationships among checkpoint and repairproteins, Cell 118 (2004) 699–713.
[40] L.F. Liu, P. Duann, C.T. Lin, P. D’Arpa, J. Wu, Mechanism ofaction of camptothecin, Ann. NY Acad. Sci. 803 (1996)44–49.
[41] J.L. Nitiss, J.C. Wang, Mechanisms of cell killing by drugsthat trap covalent complexes between DNAtopoisomerases and DNA, Mol. Pharmacol. 50 (1996)1095–1102.
[42] K.L. Lorick, J.P. Jensen, S. Fang, A.M. Ong, S. Hatakeyama,A.M. Weissman, RING fingers mediateubiquitin-conjugating enzyme (E2)-dependentubiquitination, Proc. Natl. Acad. Sci. U.S.A. 96 (1999)11364–11369.
[43] L. Bjergbaek, J.A. Cobb, M. Tsai-Pflugfelder, S.M. Gasser,Mechanistically distinct roles for Sgs1p in checkpointactivation and replication fork maintenance, EMBO J.(2004).
[44] L.F. Liu, S.D. Desai, T.K. Li, Y. Mao, M. Sun, S.P. Sim,Mechanism of action of camptothecin, Ann. NY Acad. Sci.922 (2000) 1–10.
[45] R.D. Kolodner, C.D. Putnam, K. Myung, Maintenance ofgenome stability in Saccharomyces cerevisiae, Science 297(2002) 552–557.
Copyright © 2022 FDOKUMEN




















