
Transalkylation of m-diethylbenzene over large-pore zeolites

Structure and nuclearity of active sites in Fe-zeolites: comparison with
iron sites in enzymes and homogeneous catalysts
Adriano Zecchina,*a Mickael Rivallan,ab Gloria Berlier,a Carlo Lambertia and
Gabriele Ricchiardia
Received 6th March 2007, Accepted 12th April 2007
First published as an Advance Article on the web 16th May 2007
DOI: 10.1039/b703445h
Fe-ZSM-5 and Fe-silicalite zeolites efficiently catalyse several oxidation reactions which find close
analogues in the oxidation reactions catalyzed by homogeneous and enzymatic compounds. The
iron centres are highly dispersed in the crystalline matrix and on highly diluted samples,
mononuclear and dinuclear structures are expected to become predominant. The crystalline and
robust character of the MFI framework has allowed to hypothesize that the catalytic sites are
located in well defined crystallographic positions. For this reason these catalysts have been
considered as the closest and best defined heterogeneous counterparts of heme and non heme iron
complexes and of Fenton type Fe2+ homogeneous counterparts. On this basis, an analogy with
the methane monooxygenase has been advanced several times. In this review we have examined
the abundant literature on the subject and summarized the most widely accepted views on the
structure, nuclearity and catalytic activity of the iron species. By comparing the results obtained
with the various characterization techniques, we conclude that Fe-ZSM-5 and Fe-silicalite are not
the ideal samples conceived before and that many types of species are present, some active and
some other silent from adsorptive and catalytic point of view. The relative concentration of these
species changes with thermal treatments, preparation procedures and loading. Only at lowest
loadings the catalytically active species become the dominant fraction of the iron species. On the
basis of the spectroscopic titration of the active sites by using NO as a probe, we conclude that
the active species on very diluted samples are isolated and highly coordinatively unsaturated Fe2+
grafted to the crystalline matrix. Indication of the constant presence of a smaller fraction of Fe2+
presumably located on small clusters is also obtained. The nitrosyl species formed upon dosing
NO from the gas phase on activated Fe-ZSM-5 and Fe-silicalite, have been analyzed in detail and
the similarities and differences with the cationic, heme and non heme homogeneous counterparts
have been evidenced. The same has been done for the oxygen species formed by N2O
decomposition on isolated sites, whose properties are more similar to those of the (FeO)2+ in
cationic complexes (included the [(H2O)5FeO]2+ ‘‘brown ring’’ complex active in Fenton reaction)
than to those of ferryl groups in heme and non heme counterparts.
1. Introduction
Several gaps are encountered in heterogeneous catalysis: some
‘‘internal’’ and some other ‘‘external’’ (because they concern
the relations with other branches of catalysis like homoge-
neous and enzymatic catalysis).
Concerning the ‘‘internal gaps’’, the difference in the experi-
mental conditions employed in surface characterization and in
catalytic reactions can be considered as a ‘‘pressure gap’’.
Besides, the discrepancy between the composition of model
surfaces (investigated with surface science approaches) and
that involved in real catalysis is the ‘‘material gap’’.1–3
As for the ‘‘external’’ gaps, special mention must be made of
the gap between heterogeneous and homogeneous catalysis, i.e.
between branches of science which, although both devoted to
the study of the common function of promoting the assembly of
molecules, are often supposed to deal, on one hand, with well
defined systems (homogeneous complexes) and, on the other
hand, with ill defined and disordered materials difficult to
characterize (heterogeneous catalysts). Another similar gap
can be hypothesized to hold for the relations between hetero-
geneous and enzymatic catalysis, the latter showing an enor-
mous selectivity and activity advantage over the former.
A further gap, which cannot be classified either internally or
externally (because it is common to all catalysis branches) is
that between existing precursor structures (either constituted
by the structures of surface sites or by the structure of
homogeneous complexes) and the active sites really operating
under reaction conditions.
Scientists involved in heterogeneous catalysis and surface
characterization have been challenged by these gaps. One of
aUniversita di Torino, NIS Centre of Excellence, University ofTorino, Dipartimento di Chimica Inorganica, Fisica e dei Materiali,Via P. Giuria 7, 10125 Torino, Italy. E-mail:[email protected]; Fax: +39-011-6707855; Tel: +39-011-6707860
bUniversite de Rennes 1, Sciences Chimiques de Rennes, UMR-6226,Inorganic Materials: Soft Chemistry and Reactivity of Solids,F-35042 Rennes, France
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3483
INVITED ARTICLE www.rsc.org/pccp | Physical Chemistry Chemical Physics

the most effective efforts to bridge the selectivity gap with
respect to enzymes is the development of microporous cata-
lysts. Crystalline microporous materials like zeolites, charac-
terized by an extraordinary variety of crystalline structures, by
a great ability to exchange cationic species, by the controlled
nuclearity of the active sites, and by the presence of accessible
nanosized cavities, have appeared as an ideal playground. It is
outside the scope of this review to give a general view of the
catalytic and structural properties of the zeolites and the
interested reader is referred to some reviews in the field and
to the references cited therein.4–10
Similarly, the fundamental studies involving the structure,
distribution and reactivity of metal centres like Al3+,11–13
Ga3+,14–17 B3+,17 Ti4+,18–22 Ge4+,23,24 Fe3+25 in zeolite
frameworks and of metal centres in extraframework positions
(Cu+,26–28 Cu2+,29–32 Rh+,33,34 Ni2+,35 Zn2+,36 Co2+ 37),
being outside the scope of the review, will not be dealt with in
detail.
In this review we shall concentrate only on zeolitic materials
containing Fe sites (with special emphasis on Fe-silicalite and
Fe-ZSM-5) and on the possible links between the chemistry of
Fe2+ and Fe3+ ions in extraframework position and the
chemistry of homogeneous analogues, like iron based metallo-
enzymes38–41 and other homogeneous Fe2+, 42 Fe3+ 43 and
(FeO)2+ complexes.44–48
The choice to limit the comparison only between Fe-based
heterogeneous and homogeneous systems is not decreasing the
generality of the approach because:
(i) Fe-zeolites are active in several important reactions (vide
infra) which find close analogues in a reaction catalyzed by
homogeneous and enzymatic complexes.49,50 For this reason
they are ideal solids for discussing analogies and differences
between heterogeneous and homogeneous catalysts and pos-
sibly for finding a link with enzymatic catalysis.
(ii) The crystalline structure of zeolites is expected to greatly
increase the insight into the structure of the catalytic centres (a
property lacking when amorphous supports are considered
and which is at the origin of many characterization difficulties)
and hence to help the characterization of the species formed by
interaction with incoming reactants.
(iii) Fe-zeolites are a family of catalysts where very diluted
metal ions appear to be extremely active, suggesting a com-
parison with the same ions in enzymes.
(iv) The ability of zeolites to exchange or incorporate single
cations in well defined crystallographic positions should allow
to control the nuclearity and structure of the metal centre, so
favouring the investigation of the analogies between homo-
geneous, enzymatic and heterogeneous counterparts.
(v) The known structure of the channels and cavities where
the sites are located is expected to facilitate the comprehension
of the size and shape effects influencing selectivity, a property
very pronounced in enzymatic catalysts.
(vi) Fe-ZSM-5 and Fe-silicalite have been extensively stu-
died and hence the number of experimental data useful to
perform a comparison with the homogeneous and enzymatic
compounds is abundant. However in spite of abundant studies
the exact structure of the active sites in iron zeolites remains
unclear, although some well supported hypothesis can be
made.
In conclusion, Fe-silicalite and Fe-ZSM-5 can be assumed
as models of catalysts in which diluted single sites of unknown
structure display enzyme-like activity. Therefore, we believe
that a careful comparison of the manifestations of iron ions in
zeolites, homogeneous catalysts and enzymes can bring some
contributions to our understanding of these catalysts.
2. The reactions catalysed by Fe-ZSM-5 and
Fe-silicalite
Fe-ZSM-5 and Fe-silicalite are active catalysts in many rele-
vant oxidation reactions, which find close analogues in homo-
geneous and enzymatic catalysis. For this reason in the
following the most important reactions are presented together
with information on surface sites obtained from catalytic
experiments.
2.1. N2O decomposition into N2 and O2
This simple reaction
2N2O! 2N2 þO2 ð1Þ
has been extensively studied.51–63 It is thought that the reac-
tion is proceeding via the initial deposition of one oxygen atom
per iron atom (the famous ‘‘a oxygen’’).64 This species (or
family of species) is active in many oxidation reactions
(vide infra).
As for the reactivity of ‘‘a oxygen’’ species, it must be taken
into account that not all the oxygen species formed by N2O
decomposition show the same activity. For instance it has been
found that only a part of them are reacting with CO at
523 K.61
Because of its simplicity, the N2O decomposition reaction
has been widely considered as ideal for basic studies on the
mechanism of active oxygen formation and migration on
Fe-zeolites. This reaction has also been proposed for the
titration of the surface active iron species.
The most frequently proposed mechanisms are:
(i) N2O + * - N2 + *O then *O + O - O2 + 2 *, see
ref. 54 and 61
(ii) N2O + * - N2 + *O then *O + N2O - N2 + O*O
O*O - *O2 - * + O2, see ref. 56
(iii) N2O + * - N2 + *O then *O + N2O - N2 + *O2
N2O + *O2 - *O3 + N2 then *O3 - *O + O2, see ref. 65
In mechanism (i), the migration of oxygen from one active
site (*) to the other is the rate determining step.54,61 This
reaction mechanism needs the active participation of at least
two iron centres not necessarily located in adjacent position.
In mechanisms (ii)56 and (iii)65 N2O decomposition and
oxygen evolution occur at the same isolated sites (*). Mechan-
isms (i)–(iii) are all in agreement with transient response
experiments, showing that N2 appears before O2 upon direct
N2O decomposition pulses in the 773–848 K interval (Fig. 1).
This is because in all cases the global decomposition reaction is
limited by reaction pathways leading to gas phase oxygen.
More recent accurate temporal analysis of products56 in-
dicates that mechanism (ii) is the most likely one.
From the point of view of the oxygen species formed on iron
active sites (*), the three mechanisms are associated with
3484 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

increasingly complex oxygen species: *O for (i); *O, O*O and
*O2 for (ii); and *O, *O2 and *O3 for (iii).
Other very relevant results coming from these catalytic
experiments are:
(a) Fe-ZSM-5 is always more active than Fe-silicalite con-
taining the same amount of Fe.56,61–63
(b) The active sites are formed during activation in inert
atmosphere.56,61–63 There is a widespread agreement that after
this treatment a large fraction of iron is in the divalent state.
(c) The activity augments with increasing activation
temperature in inert gas; the same happens with the number
of active sites.56,61–63
(d) In zeolites with low Fe content, the number of Fe sites
where adsorbed oxygen species are formed is roughly identical
in Fe-ZSM-5 and Fe-silicalite.56,61–63
(e) The activity (calculated per Fe centre) increases with
dilution.61 From this, it can be safely concluded that the active
sites contain a very small number of Fe atoms or, more likely,
a single atom. From this it is also inferred that the concentra-
tion of clustered species becomes relevant at the highest Fe
contents and that their catalytic activity in N2O decomposition
is lower than that of isolated species. The debate concerning
the nuclearity of the catalytic sites (mono- or dinuclear) is still
alive. The number of ‘‘a-sites’’ grows with the concentration,
this growth being smaller than that of clustered species.66
(f) All other factors being equal, the number of active sites is
maximum on catalysts formed by high temperature activation
of isomorphously substituted zeolites,61 as compared for ex-
ample with catalysts obtained with post synthesis impregna-
tion.
(g) An interesting point is the positive effect of NO,67–71
which is enhancing the decomposition rate of N2O. This
observation has relevant mechanistic implications.
2.2. N2O reduction by CO
This simple reaction
N2Oþ CO! N2 þ CO2 ð2Þ
is important mainly for mechanistic investigations.61,72,73 Its
simplicity is associated also with the fact that the product
(CO2) is not adsorbed on the catalyst surface, a fact which
simplifies the kinetic investigations and surface characteriza-
tion. From transient response studies it has been concluded
that not all the centres carrying adsorbed oxygen species are
active at 523 K and that the species active in CO oxidation are
also active in benzene hydroxylation.
2.3. Hydroxylation of benzene to phenol with N2O
Among the reactions catalyzed by Fe-zeolites, the reaction:
C6H6 þN2O! C6H5OHþN2 ð3Þ
is certainly the most studied51,61,64,74–85 because of its potential
industrial application.86 This reaction is also very important
Fig. 1 Normalized O2 and N2 transient responses upon direct N2O decomposition over iron-containing zeolites, showing that N2 appears before
O2.. Reprinted with permission from ref. 56. Copyright 2006 American Chemical Society.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3485

because it can be conducted on Fe-ZSM-5 with a selectivity
near to 100% (a fact which is not common in catalysis).
Perhaps the most relevant conclusion coming from kinetic
experiments is that the rate limiting step of this reaction does
not involve the cleavage of the C–H bond.85 The reaction
reported in Scheme 1 has been proposed:(where a dimeric
species is involved). This mechanism, which is different from
that hypothesized for hydrocarbon hydroxylation and oxida-
tion reactions (vide infra), has been explained also on the basis
of theoretical calculations involving a single metal atom.87
2.4. Oxidation of methane and higher hydrocarbons with N2O
Due to its simplicity and its potential practical interest, the
reaction
CH4 þN2O! CH3OHþN2 ð4Þ
has received great attention.88–94 Unfortunately, unlike the
oxidation of CO by N2O, which generates a product that is not
adsorbed on the surface, in this case the methanol formed by
interaction of methane with adsorbed oxygen, remains
strongly adsorbed with formation of �OCH3. This fact is
limiting the practical application to methanol synthesis. When
the reaction is conducted at 723 K,91 oxidation products like
CO and CO2 are appearing in the gas phase. Similar oxidation
products are formed during oxidation of higher hydrocar-
bons.56,92,95–98
The formation of adsorbed methanol has stimulated the
hypothesis that the active site contains a pair of Fe centres like
in methane monooxygenase (MMO).99
2.5. Selective catalytic reduction (SCR) of NO by ammonia
and hydrocarbons
This reaction is of considerable practical importance and
is supposed to be catalysed by the same sites discussed
before.100–104 The catalytic reduction of nitrogen oxides can
be carried out selectively by using ammonia or urea. SCR of
NO by hydrocarbons is another important reaction which is
believed to be the most promising way to eliminate NO from
traffic exhausts.105,106
Some mechanistic studies were reported, showing that
nitrile groups derived from nitroso compounds as intermedi-
ates are formed when hydrocarbons are used as reduc-
tants.107–110
Most of the catalysts used in SCR with hydrocarbons were
initially prepared by post-synthesis iron exchange. The method
based on FeCl3 sublimation has been extensively used by the
group of Sachtler, as testified by copious scientific produc-
tion111–116 and by van Santen.117 The use of MFI zeolites
exchanged with FeCl3118 or FeCl2 was also reported for the
SCR of NO with ammonia.119–121
Generally, these preparation methods lead to high Fe load-
ings, so that many authors suggested that dimeric or clustered
Fe ions were responsible for the catalytic activ-
ity.104,108,111,112,122 However, careful titration of active sites
showed that the active sites in these samples usually represent
a very small fraction of total iron.123 The more recent
improvement appears to be the preparation of diluted samples,
because there is a compelling evidence that isolated sites are
the active centres, while clusters were found to be detrimental
to the catalytic activity (favoring total oxidation of the
reductant)124–126 or inactive.100
In conclusion, from the above-described results it is inferred
that the same sites active in the model N2O decomposition
reaction are also likely to be active in reduction of NO.
2.6. Oxidation of substrates with H2O2
Iron based zeolites (Fe-ZSM-5, Fe-silicalite and Fe-TS-1) have
been tested as Fenton-type catalysts in oxidation reactions
in solution.127,128 Iron was introduced by ion exchange or
directly during zeolite synthesis. Prior to their use in catalytic
tests, the samples were calcined at 773 K in air. Although the
results are somewhat influenced by leaching, all samples show
activity in phenol and propionic acid degradation with H2O2.
The reaction mechanism is supposed to be the same for the
Fenton reaction in solution.
3. Preparation methods
Several methods have been proposed for zeolite functionalisa-
tion with iron. The different procedures were initially designed
to insert Fex+ ions in definite structural position of the zeolite
framework with the hope to obtain crystalline and well defined
catalysts. In the following a concise description of the methods
will be given.
3.1. Exchange of H-ZSM-5 with Fe2+ and Fe3+ salts
Even if some authors reported solid state ion exchange synth-
esis using FeCl2 salts,122,129,130 the vast majority of studies on
iron loading methods deal with exchange in aqueous solu-
tions.126 The most frequently used salts are FeCl2,129,131
FeSO4,111,122,131,132 (NH4)2Fe(SO4)2,
133 FeCl3134 and
Fe(NO3)3.61,122,133,135 Since ferric salts have some tendency
to form dinuclear clusters in solution and then to precipitate as
hydroxides and because of insufficient local charge balance in
the MFI framework, ferrous salts are preferred. However, due
to the low stability of the ferrous state, synthesis in controlled
atmosphere or in the presence of reducing agents is often used
to avoid oxidation of Fe2+ to Fe3+.106 The final iron content
inside the MFI edifice is usually less than 1 wt%. To transform
the system into an active catalyst, it is calcined in air at 773 K
and activated in vacuo or in inert flow at T 4 773 K.
3.2. Exchange of H-ZSM-5 with Fe3+ oxalate
In the past some methods based on organic salts like Fe3+
acetylacetonate, Fe2+ oxalate106,136 have been reported. Even
if problems of precipitation during synthesis were observed,
some interesting innovations have been discovered. In
Scheme 1
3486 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

particular, Nechita et al. have recently proposed an exchange
method using [Fe(COO)2]+1 cations.137 This method has the
advantage, with respect to the famous report of Feng and Hall
about samples prepared by ferrous oxalate,106 to be reprodu-
cible also on a large scale. The resulting Fe-containing zeolites
(o0.5 wt%) exhibit a remarkable iron dispersion. Also in this
case its transformation into an active catalyst requires calcina-
tion and activation.
3.3. Impregnation of silicalite with Fe2+
and Fe3+
salts
The impregnation method has been used to introduce Fe in
Al-free silicalite (i.e. a structure not containing exchangeable
cations). It was observed that, also in the absence of Brønsted
sites, some Fe could be introduced in the silicalite channels
and that the interaction with silanol was the main mechanism
of the anchoring process as evidenced by FTIR spectroscopy
(unpublished results from this laboratory). This interaction,
leading to formation of grafted Fex+(O�Si)x species is im-
portant because the resulting samples are catalytically active in
oxidation reactions. In other words, grafted sites in a fully
siliceous framework must also be considered as active sites.138
3.4. Sublimation of FeCl3 into H-ZSM-5
Ion-exchange can also be done in vapour phase. This kind of
exchange, also called chemical vapour deposition (CVD), has
been extensively reported in literature as testified by the
number of papers.111,129,139–149 The CVD process uses iron
salts that are volatile at elevated temperatures like FeCl3. This
method offers the possibility, after elimination of the FeCl3excess by means of successive high temperature treatment, to
obtain fully and over-exchanged zeolites. The final iron con-
tent is generally higher compared to other ion exchange
techniques (3.0–5.0 wt% in Fe). The system is then calcined
in air at 773 K and activated in inert gas at T 4 773 K to
transform into an active catalyst.
3.5. Isomorphously substituted Fe-ZSM-5 and Fe-silicalite
In isomorphously substituted Fe-MFI catalysts, iron is gen-
erally incorporated into the lattice during hydrothermal synth-
esis.82,150 The amount of iron that can be introduced in MFI
framework is very limited so that this technique usually leads
to diluted samples (less than 0.6 wt%). After synthesis, various
treatments can convert the as-synthesized sample into the
H-form. In this preparation the Fe3+ ion is located in a
tetrahedral position on the lattice.151–153 The method allows
to prepare both isomorphously substituted Fe-ZSM-5 and
Fe-silicalite. The active form is reached after a thermal treat-
ment at high temperature (more than 773 K) during which the
majority of the iron is dislodged from framework positions
due to the low stability of Fe in the framework. Many studies
were devoted to understanding the migration phenomenon of
Fe.61,69,72,153–157
4. The activation procedures
To summarize, it can be concluded that the methods described
in sections 3.1, 3.2 and 3.4 give iron species in extraframework
position, a method as in section 3.3 gives grafted species, while
isomorphous substitution (section 3.5) produces Fe3+ in
framework positions. The samples as prepared are not active.
To induce activity, the samples have to be calcined at TE 773 K
in air and treated in a flow of inert gas (or in vacuo) at
T 4 773 K.
High temperature treatments in air and inert flow or in
vacuo, of samples prepared by employing methods described in
sections 3.1, 3.2 and 3.4 can induce migration of Fe from the
initial position with subsequent clustering and grafting. Ana-
logously it has been demonstrated that high temperature
treatments of isomorphously substituted zeolites (section 3.5)
induces the removal of Fe3+ from the framework to extra-
framework positions, with subsequent migration and graft-
ing.151–153 All these processes are highly dependent upon the
presence of H2O impurities in the channels, which are known
to favour both hydrolysis of SiOSi, SiOAl, SiOFe and FeOFe
bridges (with formation of hydroxylated sites suitable for
anchoring migrating species) and clustering of extraframe-
work species.158 It is so concluded that the final (active) state
of the catalyst is completely different from that of the starting
material.
Perhaps method 3.5 appears to be the most convenient as it
allows to start from the best defined precursors, with higher Fe
dilution and because migration of iron can be better con-
trolled.61,72,153,159
A general scheme involving dislodgment, migration, graft-
ing and clustering is reported in Fig. 2a, giving a pictorial
representation of the structure of the Fe2+ species formed by
migration into extraframework positions and successive graft-
ing to different reactive centers located on the walls of the
zeolite channels. Two structures with regard to different
anchoring sites have been considered: Brønsted sites (structure
a) associated with residual M3+ in the lattice (for Fe-ZSM-5
M = Al or Fe; for Fe-silicalite M = Fe) and silanol nests
(structure b).
The Fex+ species resulting from this complex mechanism
form a distribution of isolated, dimeric or clustered sites, and
Fe2O3 particles as graphically represented in Fig. 2b.
From these considerations it can be concluded that what-
ever is the origin of the samples, the successive thermal
treatments at high temperature needed to induce catalytic
activity, tend to modify the catalysts in such a way that
the resulting distribution of sites is not dependent much
on the preparation method. Hereafter we assume the rea-
listic view that this distribution is substantially unknown.
The consequence of this assumption is that sensitive and
appropriate characterization methods are needed. On the
basis of the scheme presented in Fig. 2a it can be expected
that after high temperature thermal treatments, the following
groups of atoms can be present on the samples in various
proportions:
Fe-silicalite
� isolated Fe2+ and Fe3+ chemically anchored to the frame-
work via SiOFe bridges;
� species containing Fe3+OFe3+, Fe2+OFe2+ and
Fe3+OFe2+ ion pairs, oligomeric, clustered sites and Fe2O3
particles chemically anchored to the framework via SiOFe
bridges.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3487
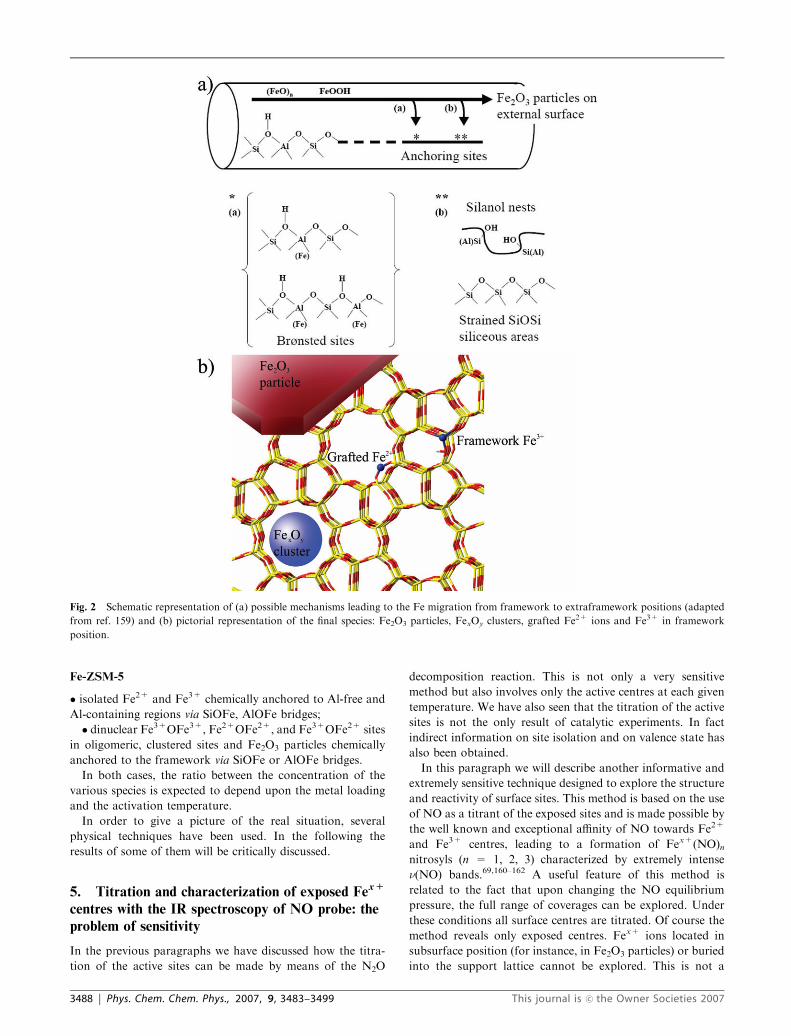
Fe-ZSM-5
� isolated Fe2+ and Fe3+ chemically anchored to Al-free and
Al-containing regions via SiOFe, AlOFe bridges;
� dinuclear Fe3+OFe3+, Fe2+OFe2+, and Fe3+OFe2+ sites
in oligomeric, clustered sites and Fe2O3 particles chemically
anchored to the framework via SiOFe or AlOFe bridges.
In both cases, the ratio between the concentration of the
various species is expected to depend upon the metal loading
and the activation temperature.
In order to give a picture of the real situation, several
physical techniques have been used. In the following the
results of some of them will be critically discussed.
5. Titration and characterization of exposed Fex+
centres with the IR spectroscopy of NO probe: the
problem of sensitivity
In the previous paragraphs we have discussed how the titra-
tion of the active sites can be made by means of the N2O
decomposition reaction. This is not only a very sensitive
method but also involves only the active centres at each given
temperature. We have also seen that the titration of the active
sites is not the only result of catalytic experiments. In fact
indirect information on site isolation and on valence state has
also been obtained.
In this paragraph we will describe another informative and
extremely sensitive technique designed to explore the structure
and reactivity of surface sites. This method is based on the use
of NO as a titrant of the exposed sites and is made possible by
the well known and exceptional affinity of NO towards Fe2+
and Fe3+ centres, leading to a formation of Fex+(NO)nnitrosyls (n = 1, 2, 3) characterized by extremely intense
n(NO) bands.69,160–162 A useful feature of this method is
related to the fact that upon changing the NO equilibrium
pressure, the full range of coverages can be explored. Under
these conditions all surface centres are titrated. Of course the
method reveals only exposed centres. Fex+ ions located in
subsurface position (for instance, in Fe2O3 particles) or buried
into the support lattice cannot be explored. This is not a
Fig. 2 Schematic representation of (a) possible mechanisms leading to the Fe migration from framework to extraframework positions (adapted
from ref. 159) and (b) pictorial representation of the final species: Fe2O3 particles, FexOy clusters, grafted Fe2+ ions and Fe3+ in framework
position.
3488 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

negative factor since we are interested only in the species
accessible to reactants. A further advantage of the method
lies in the fact that the stretching frequency of formed nitrosyls
strongly depends upon the valence state of the adsorbing
centre (Fe2+ or Fe3+). Furthermore, as the sites with single
or multiple coordinative unsaturation can show mono- or
polyaddition and form Fex+(NO), Fex+(NO)2 and Fex+(NO)3complexes upon increasing the equilibrium pressure, it is
evident that the method is also informative about the structure
(ligands sphere) of the adsorbing sites.153,158,161 In the follow-
ing we shall show that while Fe3+ forms only mononitrosyls,
isolated Fe2+ can bind up to three NO molecules.
An example of the results of this titration method obtained
on two samples of different origin and composition (Fe-ZSM-5
ex-oxalate, prepared by the ferric oxalate method and Fe-
silicalite prepared by thermal removal of Fe3+ from a tetra-
hedral position of isomorphously substituted silicalite) and
activated in the same way (in vacuum at 773 K) is illustrated in
Fig. 3. On the basis of our experience, this figure is represen-
tative of all Fe-zeolites.
Without discussing in detail the attribution of the various
bands two facts are evident. Firstly, the spectra are very
similar. This similarity demonstrates that after the treatment
(activation) at high temperature, the distribution of Fe2+ and
Fe3+ sites is similar in Fe-ZSM-5 and Fe-silicalite. This result
is quite surprising because one of the two matrices (Fe-ZSM-5)
contains Al ions (the framework) while the other does not
(Fe-silicalite). In the following we shall however draw atten-
tion to some small but significant differences which are
influenced by the Al content (see discussion of Fig. 6).
Secondly, when the spectra are normalized with respect to
the Fe content, it comes out that the intensity of the NO
spectra on Fe-ZSM-5 is about 2.5 times larger than that of Fe-
silicalite. From this it is inferred that on Fe-silicalite a larger
fraction of Fe cannot be titrated by NO. This fact is a clear
indication of clustering. An indirect deduction is that the
presence of Al in the structure is favouring dispersion.
Concerning the sensitivity of the method, Fig. 4 reports the
normalized IR spectra of NO adsorbed on Fe-ZSM-5 samples
prepared by different methodologies137 and containing different
Fe loading content down to 0.08 wt% Fe. It is a fact that the IR
spectra of NO adsorbed on samples containing less than 0.1%Fe
can be clearly detected. This result highlights the high sensitivity
of the method. A second striking aspect is related to the
impressive decrease of the fraction of surface Fe sites (propor-
tional to the nitrosyl band intensity) with increasing Fe loading.
Fig. 3 FTIR spectra (background subtracted) of NO dosed at RT on
Fe-ZSM-5 (post synthesis exchange by ferric oxalate, left part) and Fe-
silicalite (isomorphous substitution, right part) samples previously
activated in vacuum at 773 K. Spectra were collected by reducing
NO equilibrium pressure (PNO) from 15 Torr (dashed spectrum) to
10�3 Torr (dotted spectrum). Unpublished.
Fig. 4 FTIR spectra (background subtracted) of NO dosed at room temperature (decreasing PNO from 15 Torr, dashed line spectrum, to 10�3
Torr, dotted line spectrum) on Fe-ZSM-5 samples obtained with different preparation methods and iron content, previously activated in vacuo at
773 K. Spectra were normalized with respect to the pellet thickness and with respect to the iron content. Top spectrum of part (a): 15 Torr of NO
on Fe2O3 sample (ex-goethite) outgassed and oxidized at 773 K. Springer Catal. Lett., 2005, 103, 33, Nechita et al., Fig. 3. Copyright 2005. With
kind permission from Springer Science and Business Media.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3489

From the same figure it can also be clearly inferred that the
normalized intensity grows in the order: 0.08 4 0.38 4 3.30
wt%. If we assume, quite arbitrarily, that the spectra of NO on
the most diluted sample are due mainly to atomically dispersed
species (which is not: vide infra), it is deduced that even in the
0.38 wt% sample, clusters and Fe2O3 particles are present and
that aggregation is dominating in the sample containing
3.3 wt% Fe. In relation to the problem of formation of
Fe2O3 particles on the 3.3 wt% sample, in the inset of Fig. 4
the peak of NO adsorbed on a pure Fe2O3 sample is reported
for the sake of comparison. The complete coincidence of the
frequency of this peak with the frequency of one of the peaks
observed on the various samples is again a clear indication
that iron oxide particles are formed.
The detailed assignment of the IR peaks shown in Fig. 3 and
4 is schematically illustrated in Fig. 5b. This assignment has
been thoroughly discussed in ref. 137. The stretching frequen-
cies of the Fex+(NO)n peaks (x = 2, 3 and n = 1, 2, 3) are
strongly influenced by the iron oxidation state and by the
electron donating ability of the surrounding ligands.42,163
Comparison with homogeneous metal–organic complexes
shows that stretching frequencies in the 1800–1900 cm�1 range
are normally observed in Fe2+ cationic complexes
[FeLn(NO)]+X�42,163,164 and [Fe(H2O)5NO]2+,165 (Fig. 5a)
while bands in a lower frequency range (1600–1700 cm�1)
are found when Fe2+(NO) groups are bonded to strong
electron donor ligands such as porphyrins,166–168 imidazole169
or dithiolene.163 From this it is inferred that NO probing
Fe-ZSM-5 and Fe-silicalite reveals the presence of Fe2+ with
reduced d–p-bonding ability.
From the pressure dependence of the peaks the reversible
Fe2+(NO) $ Fe2+(NO)2 $ Fe2+(NO)3 transformation is
also documented for a large fraction of sites. Notice that the
Fe2+(NO) and Fe2+(NO)3 complexes are EPR active.153,170
From all the data reported in Fig. 3, 4 and 5b the following
conclusions are safely derived. (i) A fraction of Fe2+ sites
(very relevant in the most diluted samples) adsorbs up to 3 NO
ligands. This means that the adsorbing sites are highly un-
saturated (i.e. the coordination sphere is incomplete). Com-
plexes with the same stoichiometry are obtained on both
Al-containing and Al-free samples (Fig. 3). (ii) The Fe2+ in
zeolites sites are characterized by reduced d–p back-donation
ability and are therefore different from those present in heme
and non-heme organic complexes. (iii) As the frequency of the
associated peaks is not changing with coverage it is also
concluded that there is no dipole–dipole coupling2,3 or solvent
type effects between the NO oscillators. In other words, the
Fe2+ sites behave as isolated centres. (iv) A minor fraction of
Fe2+ sites inserts only one NO ligand into its coordination
sphere. Whether this site is isolated, paired or located at the
surface of a small cluster is difficult to infer. Considering the
tendency of dislodged iron to form clusters, we are more in
favor of the second hypothesis. (v) A fraction of sites is in the
trivalent state and it inserts only one NO ligand. The similarity
of the stretching frequency of these species with that measured
for the Fe2O3/NO system (see vertically shifted curve in
Fig. 4a) suggests that the involved sites are located on nano-
particles formed by aggregation.
In conclusion, the picture emerging for all investigated
samples with a Fe concentration in the 0.08–3.3% range is
that in all cases the nature of accessible sites is not dramati-
cally changing with passing from one sample to the other.
What is really changing upon increasing the Fe concentration
is the sites distribution and the fraction of non accessible sites
(located in oxide particles and clusters) which become defi-
nitely predominant in the sample containing 3.3 wt% Fe.
On the basis of these results we are forced to consider the
following questions: (i) Why is Fe-ZSM-5 always more active
than Fe-silicalite in N2O decomposition? (ii) Is this greater
activity related to the superior dispersion of sites (plausibly
containing a single Fe2+) observed on Fe-ZSM-5? (iii) Is the
titration method sufficiently sensitive to explore the really
active sites? (iv) Which is the real structure (coordination
sphere) of the highly coordinatively unsaturated Fe2+ sites?
(v) If grafted Fe2+ species are responsible for the catalytic
activity, why Fe/silica and Fe/MCM41 are less active?138
To answer these questions the results illustrated in Fig. 6 are
of extreme utility. The analysis of the results of a large number
of NO titration experiments on Fe-ZSM-5 and Fe-silicalite
clearly demonstrates that:
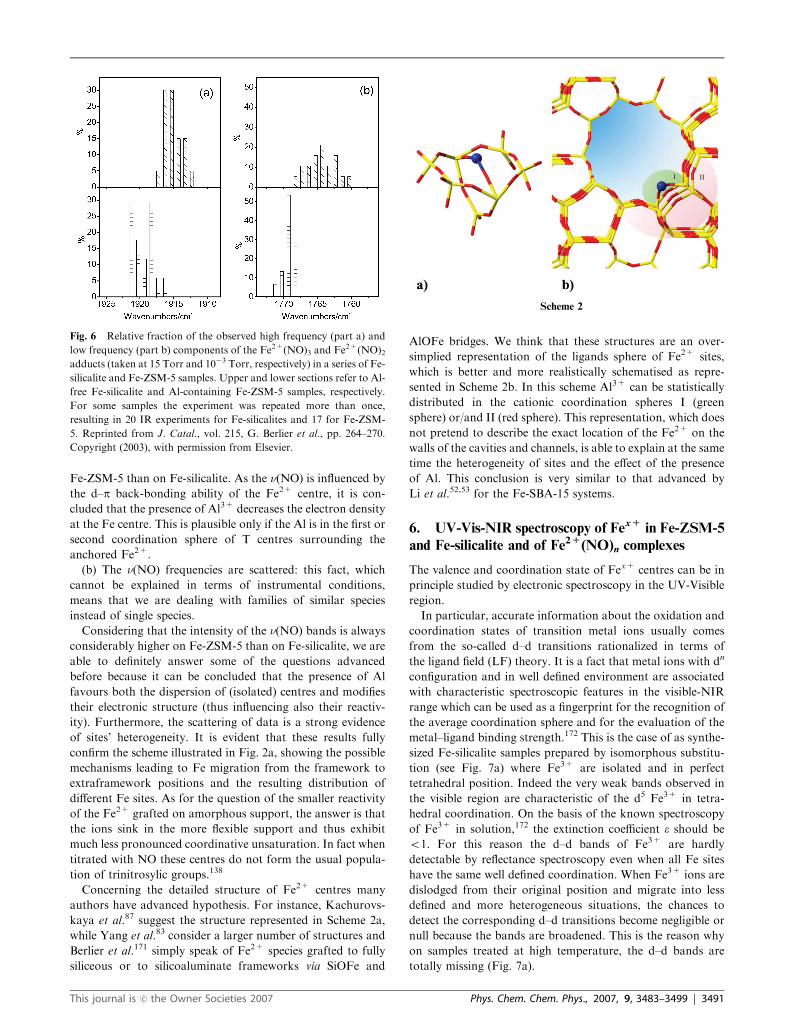
(a) The frequency of the NO stretching modes of the
equivalent Fe2+(NO)n complexes is statistically higher on
Fig. 5 (a) Frequencies of n(NO) in different Fe nitrosyl complexes
formed in mononuclear homogeneous systems or on supported oxides,
as detailed in ref. 137. Fe2+(NO) heme and non-heme nitrosyls absorb
at lower frequencies as indicated by the arrow. (b) FTIR spectra
(background subtracted) of NO dosed at room temperature (decreas-
ing PNO from 15 Torr, dashed line spectrum to 10�3 Torr, dotted line
spectrum) on Fe-ZSM-5oxa sample previously activated in vacuum at
773 K. Springer Catal. Lett., 2005, 103, 33, Nechita et al., Fig. 2.
Copyright 2005. With kind permission from Springer Science and
Business Media.
3490 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007
Fe-ZSM-5 than on Fe-silicalite. As the n(NO) is influenced by
the d–p back-bonding ability of the Fe2+ centre, it is con-
cluded that the presence of Al3+ decreases the electron density
at the Fe centre. This is plausible only if the Al is in the first or
second coordination sphere of T centres surrounding the
anchored Fe2+.
(b) The n(NO) frequencies are scattered: this fact, which
cannot be explained in terms of instrumental conditions,
means that we are dealing with families of similar species
instead of single species.
Considering that the intensity of the n(NO) bands is always
considerably higher on Fe-ZSM-5 than on Fe-silicalite, we are
able to definitely answer some of the questions advanced
before because it can be concluded that the presence of Al
favours both the dispersion of (isolated) centres and modifies
their electronic structure (thus influencing also their reactiv-
ity). Furthermore, the scattering of data is a strong evidence
of sites’ heterogeneity. It is evident that these results fully
confirm the scheme illustrated in Fig. 2a, showing the possible
mechanisms leading to Fe migration from the framework to
extraframework positions and the resulting distribution of
different Fe sites. As for the question of the smaller reactivity
of the Fe2+ grafted on amorphous support, the answer is that
the ions sink in the more flexible support and thus exhibit
much less pronounced coordinative unsaturation. In fact when
titrated with NO these centres do not form the usual popula-
tion of trinitrosylic groups.138
Concerning the detailed structure of Fe2+ centres many
authors have advanced hypothesis. For instance, Kachurovs-
kaya et al.87 suggest the structure represented in Scheme 2a,
while Yang et al.83 consider a larger number of structures and
Berlier et al.171 simply speak of Fe2+ species grafted to fully
siliceous or to silicoaluminate frameworks via SiOFe and
AlOFe bridges. We think that these structures are an over-
simplied representation of the ligands sphere of Fe2+ sites,
which is better and more realistically schematised as repre-
sented in Scheme 2b. In this scheme Al3+ can be statistically
distributed in the cationic coordination spheres I (green
sphere) or/and II (red sphere). This representation, which does
not pretend to describe the exact location of the Fe2+ on the
walls of the cavities and channels, is able to explain at the same
time the heterogeneity of sites and the effect of the presence
of Al. This conclusion is very similar to that advanced by
Li et al.52,53 for the Fe-SBA-15 systems.
6. UV-Vis-NIR spectroscopy of Fex+
in Fe-ZSM-5
and Fe-silicalite and of Fe2+
(NO)n complexes
The valence and coordination state of Fex+ centres can be in
principle studied by electronic spectroscopy in the UV-Visible
region.
In particular, accurate information about the oxidation and
coordination states of transition metal ions usually comes
from the so-called d–d transitions rationalized in terms of
the ligand field (LF) theory. It is a fact that metal ions with dn
configuration and in well defined environment are associated
with characteristic spectroscopic features in the visible-NIR
range which can be used as a fingerprint for the recognition of
the average coordination sphere and for the evaluation of the
metal–ligand binding strength.172 This is the case of as synthe-
sized Fe-silicalite samples prepared by isomorphous substitu-
tion (see Fig. 7a) where Fe3+ are isolated and in perfect
tetrahedral position. Indeed the very weak bands observed in
the visible region are characteristic of the d5 Fe3+ in tetra-
hedral coordination. On the basis of the known spectroscopy
of Fe3+ in solution,172 the extinction coefficient e should be
o1. For this reason the d–d bands of Fe3+ are hardly
detectable by reflectance spectroscopy even when all Fe sites
have the same well defined coordination. When Fe3+ ions are
dislodged from their original position and migrate into less
defined and more heterogeneous situations, the chances to
detect the corresponding d–d transitions become negligible or
null because the bands are broadened. This is the reason why
on samples treated at high temperature, the d–d bands are
totally missing (Fig. 7a).
Fig. 6 Relative fraction of the observed high frequency (part a) and
low frequency (part b) components of the Fe2+(NO)3 and Fe2+(NO)2adducts (taken at 15 Torr and 10�3 Torr, respectively) in a series of Fe-
silicalite and Fe-ZSM-5 samples. Upper and lower sections refer to Al-
free Fe-silicalite and Al-containing Fe-ZSM-5 samples, respectively.
For some samples the experiment was repeated more than once,
resulting in 20 IR experiments for Fe-silicalites and 17 for Fe-ZSM-
5. Reprinted from J. Catal., vol. 215, G. Berlier et al., pp. 264–270.
Copyright (2003), with permission from Elsevier.
Scheme 2
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3491
Substitutional Fe3+ ions in the silicalite matrix are sur-
rounded by a sphere of 4 SiOd� ligands and hence LMCT
transition are also expected in the 40 000–50 000 cm�1 range
characterized by an extinction coefficient which is about two
or three order of magnitude larger than that of the d–d
bands.151,152
The Fe-silicalite spectra do indeed show (Fig. 7a), two
intense bands in the UV region which were assigned to LMCT
transitions in the [FeO4] tetrahedral group.151,152 These bands,
although very intense, cannot be safely considered as an
evidence of tetrahedral coordination. In fact, also octahedral
complexes of Fe3+ are characterized by two strong bands in
the same energy range.173 The frequency of LMCT bands is
affected by clustering and in Fe2O3 they are shifted down to
the visible region. Consequently they can overlap the d–d
bands. The consequence of this is that when we are in the
presence of both isolated, clustered species and Fe2O3 parti-
cles, the spectrum in the visible becomes very complex and
broad and the interpretation is troublesome or impossi-
ble.126,130,151 The formation of clusters and of Fe2O3 particles
by removal of isolated Fe3+ from substitutional positions
induced by high temperature treatments is, at least partially,
responsible of the shift of the LMCT band towards the lower
frequency of Fe-silicalite (Fig. 7a).
As we have discussed in the previous paragraphs, the
treatment at high temperature in vacuo or in inert gas should
cause the formation of Fe2+ species either isolated or clus-
tered. So the question arises whether it can be possible to
detect them by UV-Vis-NIR spectroscopy. We can so start by
considering first the d–d bands of isolated Fe2+ ions in low
coordination. On the basis of the smaller ligand field and of
the lower charge of the metal ion we expect weak d–d transi-
tions shifted to the lower wavenumbers (in the NIR region)38
where they should not overlap with any other ligand field
transition. So far we were not able to single out d–d bands
surely assigned to Fe2+ ions: this is likely due to the low
intensity. We think that more accurate measurements in the
NIR region are needed. As for the LMCT associated with
Fe2+ they are expected to fall at high energy, so that it is hard
to observe them in the UV range especially in the presence of
Fe3+ species.
In conclusion, the information which can be extracted from
the reflectance spectra in the UV-Vis- NIS range of the
Fe-ZSM-5 and Fe-silicalite samples activated at high T is very
modest, especially because of the low intensity of the d–d
bands and the heterogeneity of sites.
From the IR experiments of the titration of the Fe2+ sites
we have learnt that Fe2+(NO)n complexes are formed by
interaction with NO. Homogeneous nitrosylic complexes
of Fe2+ are known for their intense absorptions in the visible
range. For instance, the ‘‘brown ring’’ [Fe(H2O)5NO]2+
complex is characterized by three strong absorptions (A, B,
C) in the 10 000–30 000 cm�1 range with extinction coefficients
two or three order of magnitude larger that those of the d–d
bands of the parent [Fe(H2O)6]2+ complex.
We have thus investigated by reflectance spectroscopy the
formation of surface nitrosyls on Fe-silicalite and Fe-ZSM-5
by NO adsorption from the gas phase and compared the
obtained spectra with those obtained by dosing NO in a
solution containing [Fe(H2O)6]2+.165 The results are shown
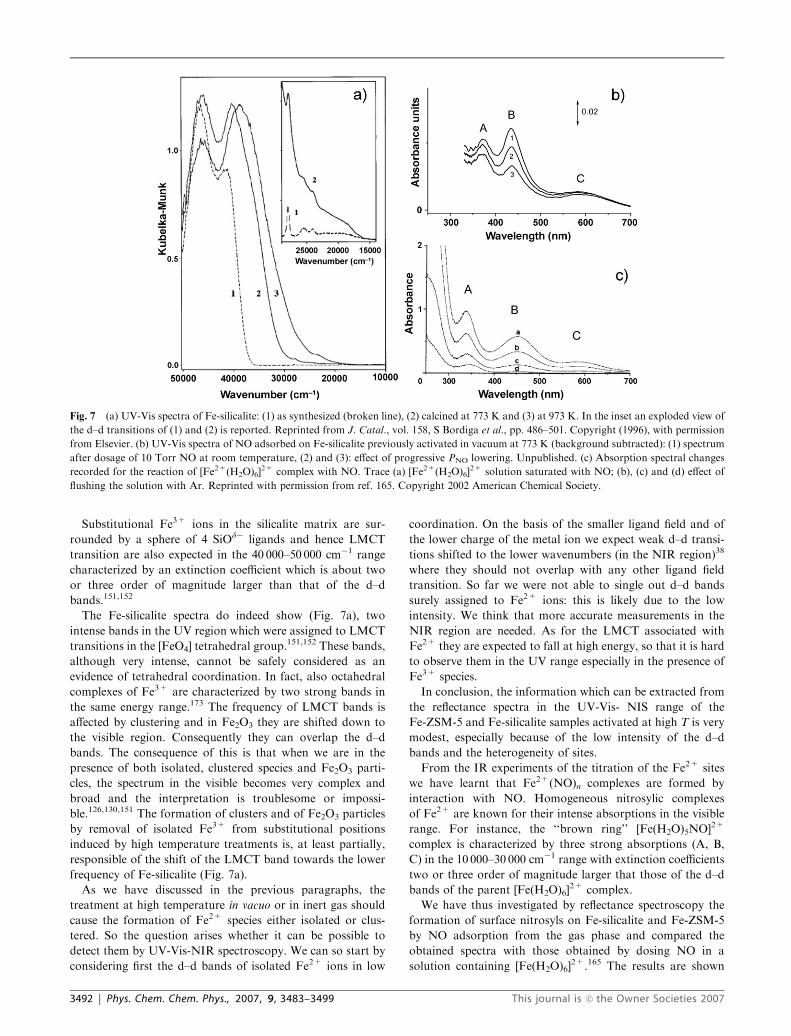
Fig. 7 (a) UV-Vis spectra of Fe-silicalite: (1) as synthesized (broken line), (2) calcined at 773 K and (3) at 973 K. In the inset an exploded view of
the d–d transitions of (1) and (2) is reported. Reprinted from J. Catal., vol. 158, S Bordiga et al., pp. 486–501. Copyright (1996), with permission
from Elsevier. (b) UV-Vis spectra of NO adsorbed on Fe-silicalite previously activated in vacuum at 773 K (background subtracted): (1) spectrum
after dosage of 10 Torr NO at room temperature, (2) and (3): effect of progressive PNO lowering. Unpublished. (c) Absorption spectral changes
recorded for the reaction of [Fe2+(H2O)6]2+ complex with NO. Trace (a) [Fe2+(H2O)6]
2+ solution saturated with NO; (b), (c) and (d) effect of
flushing the solution with Ar. Reprinted with permission from ref. 165. Copyright 2002 American Chemical Society.
3492 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

in Fig. 7b and c for the nitrosyl complexes formed on
Fe-silicalite and for [Fe(H2O)6]2+, respectively.
The similarity of the UV-Vis-NIR spectra is stringent. It is
thus inferred that UV-Vis-NIR experiments shown in Fig. 7b
are the analogues of the IR experiments shown in Fig. 3b, 4, 5.
However, a detailed assignment of the UV-Vis bands in terms
of mono-, di- and trinitrosyl was not possible. What is clear
(and this is the main message coming from this comparison) is
that the Fe site interacting with NO molecules, responsible for
the intense UV-Vis bands (Fig. 7b and c), is very similar to the
Fe2+ ion of the Fentom complex, so that the environment
formed by the water molecules in the latter must be similar to
that formed by the silicate matrix.
Regarding the nature of the UV-Vis nitrosyl bands, on the
basis of their position we propose that they are related to d–d
transitions with mixed p character. The high intensity of the
bands indicates an important mixing with the empty ligand
p-orbitals. In the case when there is an extensive mixing of the
metal and ligand wave functions in the molecular orbitals of
the complex, the distinction between ligand field and charge
transfer cannot be made sharply.172
From this we conclude that UV-Vis-NIR spectroscopy of
the NO/Fe2+ interaction can be used to titrate the isolated
Fe2+ species on Fe-ZSM-5 and Fe-silicalite, exactly as it is
done in solution. Furthermore it is a very sensitive and
diagnostic method.
7. The sites structure and distribution emerging
from XAS investigations
Usual structural investigation methods such as X-ray diffrac-
tion (XRD) are limited in determining the structure of iron
active sites inside MFI-zeolite because of the low concentra-
tion of heteroatoms and because of the lack of long-range
order after the substitution process. Few XRD studies just
report on the iron locations in the framework, which are not
the active sites in catalysts.25 XAS absorption spectroscopies
can in principle overcome these problems. In particular,
extended X-ray absorption fine structure (EXAFS) is emer-
ging as one of the preferred techniques for probing active site
structures in catalysis.54,122,131,140,151,152,174–181 While the
EXAFS part of the spectrum allows to gain information on
the first and second coordination sphere of the metal species,
X-ray absorption near edge structure (XANES) displays its
great utility in clarifying the average valence state of the
absorbing metal centre.
Looking at the XANES results that have appeared in
the literature on Fe-ZSM-5 and Fe-silicalite it can be con-
cluded that they confirm that after high temperature activation
a part of the iron is in a divalent state.62,152,153,174,176 As far
as the EXAFS results are concerned, a summary of the
diverse Fe–O, Fe–Fe and Fe–Si/Al distances reported in the
literature are plotted as a function of iron content in
Fig. 8.54,122,131,140,151,152,174–181
As clearly displayed in Fig. 8, the results are heavily
scattered and at low iron concentration (from 0.2 to 1.0
wt%), the Fe–O distances are found in the 1.80–2.25 A range.
Concerning other distances measured in XAS experiments,
most of the Fe–Fe distances in iron containing MFI appear
centred on those of a-Fe2O3 (2.96 and 3.49 A). Moreover the
Fe–Fe distances are superimposed to those of Fe–Si/Al in the
2.80 to 3.20 A range.
This impressive scattering of the 1st shell distances can be
explained assuming two main causes. The first cause concerns
the fact that, in most of the cases, different groups investigate
significantly different samples, as preparation and post synth-
esis procedures strongly affect the final form of iron species
(vide supra sections 3 and 4). Secondly, notwithstanding the
fact that the accuracy of a first shell distance determination
may be as good as �0.01 or �0.02 A, these error bars are
statistical and systematic errors are not accounted for. In the
specific case of Fe-zeolites systematic errors may have a double
origin. Usually phase-shifts and amplitude functions, crucial
in determining bond distances and coordination numbers, are
theoretically generated from a given guessed cluster. As the
actual geometry of the active Fe species is still unknown,
phases and amplitudes generated in that way can be question-
able. The second source of possible systematic errors concerns
the assumption of Gaussian distance distribution done in the
standard EXAFS formula, usually used in most of the cited
papers. It is known that in systems characterized by a high
degree of heterogeneity, like liquids or amorphous systems,
this assumption is no longer valid. In such cases, EXAFS data
should be analyzed according to the cumulant approach, as
elegantly shown recently in the field of catalysis by the ETH
group of Prof. van Bokhoven.182
These observations suggest that the low concentration of
iron and, more importantly, the heterogeneity of the sites are
at the basis of the scattering of data. This result does once
more confirm that after calcination and activation a broad
distribution of iron centres is present on Fe-ZSM-5 and
Fe-silicalite systems.
A few results in the figure merit a specific comment. In
particular, we think that the distances centred at about 2.5 A
are likely due to Fe–Cl groups on samples prepared from
Fig. 8 Summary of the diverse Fe–O (open square), Fe–Fe (open
circle) and Fe–Si/Al (open triangle) distances calculated in the litera-
ture on the basis of EXAFS data. Distances are reported as a function
of iron content. Full and dashed vertical lines indicate average Fe–O
and Fe–Fe distances, respectively, calculated from XRD of a-Fe2O3.
Data have been collected from ref. 54, 122, 131, 140, 151, 152,
174–181.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3493

FeCl3 exchanged systems. The distance at about 1.4 A found
by Choi et al. in oxidized samples could be consistent with the
formation of Fe4+QO species.177,178
Coming to the important problem of nuclearity of the
extraframework iron species EXAFS in principle could be
the technique of choice to face this problem. By fitting the
EXAFS contribution in the 2–4 A interval with a Fe–Fe model
one could obtain the average Fe–Fe coordination number
(NFe�Fe).174,179,180 Unfortunately, the relationship between
such NFe�Fe value and the average Fe nuclearity if far from
straightforward, because complexity is introduced by the huge
heterogeneity of extraframework species. As a consequence,
an average Fe–Fe coordination number of e.g. NFe�Fe = 1.0
could be interpreted as 100% of dimers, as 50% of isolated
monomers and 50% of trimers (having two Fe neighbours)
etc. The situation is even more complex, because the 2–4 A
interval is the region where also the backscattering of frame-
work atoms (Al or Si) is potentially expected. In this regard,
the group of Bell178 showed that the peak at 2.5 A, previously
ascribed to Fe–Fe scattering and used to argue for the
presence of di-iron-oxo species was actually due to Fe–Al
contributions.
8. What is known about the ‘‘active’’ oxygen
Active oxygen on Fe-ZSM-5 and Fe-silicalite is formed upon
decomposition of N2O at T Z 523 K. It is widely accepted
that active oxygen is formed predominantly on Fe 2+ sites.
The fraction of these sites has recently been evaluated with
several methods by Yuranov et al.61 and it has been found to
vary from 50–70% for isomorphously substituted samples
with less than 0.035 wt% Fe and treated at high T (1323 K
in He) to less than 10–15% for post-synthesis exchanged
samples. On the basis of catalytic experiments, the same
authors have found an indication of the presence of at least
two types of Fe2+ sites characterized by different reactivity. It
has been hypothesized that these sites are isolated and paired,
respectively. In the first case the structure of the oxidized
centre is Fe4+QO (or Fe3+–O�), while in the second case an
oxo-bridged Fe3+O2�Fe3+ structure is more likely. All struc-
tures are EPR silent.183
On the basis of an elegant resonant inelastic X-ray scatter-
ing (RIXS) experiment on a post-synthesis exchanged sample
containing 0.36% and treated at 1218 K, Pirngruber et al.184
have concluded that the Fe4+QO is not present after oxida-
tion with N2O. Although they are favouring the dinuclear
structure, they do not exclude the formation of Fe3+–O�. The
interpretation of the spectra has been given by assuming that
the concentration of the active sites is about 0.01 mmol g�1 (i.e.
6.1018 atoms g�1). However the data from Yuranov et al.61
regarding post-synthesis samples with similar iron content
(0.55% corresponding to about 60.1018 atoms g�1) give smal-
ler values (about 3.1018 atoms g�1), the remaining iron frac-
tion (more than 90%) being presumably in the form of iron
oxide clusters and particles: thus the question arises whether
the method is sensitive enough to allow clear-cut conclusions
especially in presence of a large fraction of spectator species.
A dimeric structure containing two active Fe2+ sites is
favoured by Dubkov et al.156 on the basis of Mossbauer
spectroscopy. However as the authors have found that each
atom in the complex is able to adsorb one active oxygen, the
Fe2+ sites behaving as monoatomic entities in a paired
arrangement are registered by Mossbauer spectroscopy as
dinuclear complexes. This in turn suggests that active oxygen
is not an oxo-bridged species and that two adjacent Fe4+QO
are formed upon N2O decomposition.
Concerning the nature of adsorbed oxygen it has to be
pointed out that the first proposed structure was the dimeric
one.185 This hypothesis was substantially based on the
observed reactivity of oxygen species formed by N2O dissocia-
tion towards methane, this reactivity being similar to that of
the enzyme methane oxygenase MMO (which indeed contains
two metal centres).99 Today we know that many other mono-
nuclear non-heme iron complexes are active in the same
reaction.40 For this reason other structures have to be
considered.
From the above discussion it can be seen that nuclearity of
sites and structure of adsorbed oxygen are closely connected.
From our IR results concerning the titration of Fe2+ sites on
very diluted samples, we have concluded that a relevant
fraction is associated with isolated sites characterized by high
coordinative unsaturation (and hence highly reactive). This
means that isolated (FeO)2+ structure is emerging as our
preferred candidate for active adsorbed oxygen in these sam-
ples. This does not exclude the presence of a minor fraction of
paired Fe2+–Fe2+ active species, in particular located on the
surface of small, partially reduced FexOy clusters entrapped
into the framework cavities, where the formation of oxo-
bridged species is more likely. However as we have no direct
information on the vibrational properties of adsorbed oxygen
(like on the contrary obtained by resonant Raman spectro-
scopy on homogeneous counterparts and for oxygen adsorbed
on Fe-SBA-15)53 we cannot reach a completely safe conclu-
sion. This is not unexpected because we have seen in the
previous chapters that the Fe-ZSM-5 and Fe-silicalite catalysts
are not the simple model systems imagined initially.
9. Are the structure and reactivity of active sites in
Fe-zeolites and in homogeneous compounds really
comparable?
Before starting the discussion of this point, let us first empha-
sise that Fe2+ in Fe-ZSM-5 and Fe-silicalite formed by
activation in vacuo or in helium flow at high temperature are
coordinatively unsaturated species, i.e. their coordination site
is not complete. The formation of surface species by interac-
tion with gases is, to a first approximation, well represented by
a ligand insertion into a vacant sphere as shown below:
LnFe2 þ þ nNO! LnFe
2 þðNOÞn ðn ¼ 1; 2; 3Þ ð5Þ
LnFe2 þ þOðex-N2OÞ ! LnðFeOÞ2 þðL ¼ SiO� or AlO�Þ ð6Þ
This situation is never found in homogeneous conditions
because the ions are always surrounded by a full coordination
sphere of ligands.
For instance, the formation of the simple homogeneous
[Fe(H2O)5NO]2+ nitrosyl (the ‘‘brown ring complex’’ of Fe2+)
3494 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

in solution is a ligand exchange reaction:186
½FeðH2OÞ6�2 þ þNO! ½FeðH2OÞ5NO�2 þ þH2O ð7Þ
For this reason in the following we will compare only the
structure and properties of (coordinatively saturated) Fe2+-
(NO)n (n = 1, 2, 3) and (FeO)2+ groups anchored, on the one
hand, to the internal surfaces of ZSM-5 and silicalite or
located in the ligands sphere of homogeneous complexes, on
the other hand. This comparison together with the evaluation
of the catalytic properties of the systems will form the basis of
our considerations.
The first comparison to be made is between the spectro-
scopic properties of nitrosylic complexes formed on Fe2+ ions
anchored on Fe-ZSM-5 and Fe-silicalite and the [Fe(H2O)5-NO]2+ and [LFe(NO)]2+ (L = polyamine carboxylate) cat-
ionic complexes.165 In a second step the spectroscopic properties
of heme and non-heme nitrosyls167,187–189 will be considered.
From the first comparison it is emerging that the n(NO) of
[Fe(H2O)5NO]2+ and of [LFe(NO)]2+ complexes are in the
1780–1810 cm�1 range. This range coincides with the range
where the n(NO) of Fe2+(NO)n complexes in Fe-ZSM-5 and
Fe-silicalite are observed: it is indicative of a strong similarity
between the two situations.
When the n(NO) of heme and non-heme nitrosyls are
considered, it is noticed that the frequency is consistently
downward-shifted with respect to the complexes discussed
before.
This means that the electron donating ligand sphere of heme
and non-heme complexes greatly enhances the d–p backdona-
tion ability of the metal centre with respect to the ligand sphere
present in [Fe(H2O)5NO]2+ and [LFe(NO)]2+ and in Fe2+-
zeolites.
The consequence is that NO bonded to heme and non-heme
compounds is better described as a bent NO� while the charge
back-donated to NO on Fe2+ ions on Fe-ZSM-5 and Fe-
silicalite is definitely smaller (so that the NO ligand is more
likely to be linear, or only slightly bent). In other words the
electron donating character of the ligand sphere modulates
the electron density at the Fe2+: a fact which of course induces
different reactivities. The different reactivity of Fe2+ in
Fe-zeolites, on the one hand, and in heme and non-heme
compounds, on the other hand, is testified by the different
affinity towards CO and O2. In fact the interaction of CO with
Fe-ZSM-5 and Fe-silicalite is very weak and O2 is not ad-
sorbed at all at RT (while it readily forms Fe3+O2� groups
with Fe2+ in heme compounds). From this it is concluded that
a simple parallelism between Fe2+ ions on Fe-ZSM-5 and
Fe-silicalite, on the one hand, and heme and non-heme (en-
zymatic) complexes, on the other hand, is not straightforward
and that the most correct and simple homogeneous counter-
part is the [(H2O)5FeO]2+ complex.
On the basis of these considerations we can now move to the
next step by comparing the electronic and vibrational proper-
ties of ferryl (FeO)2+ groups grafted on the internal surfaces
of Fe-ZSM-5 and Fe-silicalite with ferryl groups in the sim-
plest homogeneous complex [(H2O)5FeO]2+. Then we will
continue considering ferryl groups in heme (cytochrome
P450) and non-heme complexes, which are very active in CH
hydroxylation.41,190
The simple [(H2O)5FeO]2+ complex recently characterized
by Mossbauer and XAS spectroscopy48 (Scheme 3) is thought
to be the active species in the Fenton reaction158 and is
characterized by a quite covalent FeQO bond (d = 0.162
nm) which reacts with benzene to produce phenol but does not
react with methane at RT.44 The first reaction is the same
catalyzed by Fe-ZSM-5 and Fe-silicalite, so the analogy
between this compound and the species formed by the inter-
action with N2O is relevant. The XANES spectrum of this
compound48 shows an edge at 7126 eV, i.e. 3 eV downwards
shifted with respect to that of the [(H2O)6Fe]3+ structure. It is
interesting to note that a similar shift has been observed on
passing from Fe-silicalite contacted with N2O at 523 K (7123.1
eV) to the same sample outgassed at 973 K in vacuo.62,152,153
Unfortunately the samples prepared by activation at high
temperature always contain a large fraction of Fe2O3 particles
and FexOy clusters, so the comparison between the two cases is
not fully satisfactory.
As for the n(FeQO) of the ferryl group in Fe-ZSM-5 and
Fe-silicalite nothing is known.
Scheme 4 presents a ferryl group formed on isolated grafted
Fe2+ sites in Fe-zeolites after contact with N2O at 523 K, as
proposed by Berlier et al. on the basis of XANES and EXAFS
measurements.152 This structure is indeed similar to that of ‘‘a-oxygen’’ suggested by Kachurovskaya et al.87 formed by the
addiction of a ‘‘QO’’ ligand to the Fe complex presented in
Scheme 2a. We think that resonant Raman experiments which
have demonstrated their utility in determining the vibrational
properties of ferryl groups in heme and non-heme
Scheme 3
Scheme 4
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3495

complexes,38,40 could be of high utility. About the ferryl
groups in heme and non-heme complexes we can say that they
are more polar and that the FeQO bond is slightly longer with
respect to the same groups in [(H2O)5FeO]2+. The increased
polarity is associated with the high electron donating ability of
the ligands present in heme and non-heme complexes. This
increased polarity is making the ferryl group of these com-
pounds more exposed to an electrophilic attack and this fact is
at the basis of the reactivity of these species with C–H bonds of
aliphatic and aromatic hydrocarbons even under very mild
conditions.
In conclusion, the ferryl groups in Fe-ZSM-5 and
Fe-silicalite, should show reactivity similar to that of the
Fenton complex.191 However, as they are associated with a
ligand sphere with donating character smaller than that of the
enzymatic complexes, they are also expected to be definitely
less active. This expectation is in agreement with the observa-
tion that the two types of catalysts operate in entirely different
temperature regimes. These considerations also suggest that
the search of analogies between heterogeneous and homoge-
neous complexes although representing an important area of
investigation,192 must be made with caution.
All the conclusions reached before are valid only for the
isolated Fe2+, Fe2+(NO)n and (FeO)2+ groups. As we know
that these species are predominant only in very diluted samples
activated at high temperature, these considerations cannot be
taken for all Fe loadings. In particular we cannot exclude that
dimeric and other type of aggregated species present in the
channels and cavities can contribute to the catalytic properties
of more concentrated samples.
10. Conclusions
In this review we have examined the abundant literature on
Fe-ZSM-5 and Fe-silicalite and summarized the most widely
accepted views on the structure, nuclearity and catalytic
activity of the iron species. By comparing the results obtained
with the various characterization techniques with the results
derived from catalytic experiments, it is concluded that
Fe-ZSM-5 and Fe-silicalite are not the ideal samples conceived
before and that many type of species are present, some active
and some other silent from adsorptive and catalytic point of
view. The relative concentration of these species changes with
thermal treatments, preparation procedures and Fe loading.
Only at the lowest loadings the catalytically active species
become the dominant fraction of the iron species. On the basis
of the spectroscopic titration of the active sites by NO, we
conclude that the active species on extremely diluted samples
are isolated and highly coordinatively unsaturated Fe2+
grafted to the crystalline matrix. We have also found the proof
of the presence of a minor fraction of Fe2+ ions characterized
by a more complete coordination sphere, likely located on
small clusters entrapped in the framework. The nitrosylic
species have been analyzed in detail and the similarities and
differences with the cationic, heme and non-heme homoge-
neous counterparts have been put into evidence. The same has
been done for the oxygen species formed on very diluted
samples by N2O decomposition, whose properties are more
similar to those of the (FeO)2+ in cationic complexes (includ-
ing the [(H2O)5FeO]2+ ‘‘brown ring’’ complex considered
active in the Fenton reaction) than to those of ferryl groups
in heme and non-heme counterparts. The formation of bridged
oxospecies on Fe2+ pairs located on small clusters is not
excluded.
Acknowledgements
Progetto Regionale NANOMAT DOCUP 2000-2006 Ob. 2
Reg. (CE) 1260/99 and EC NoE IDECAT are acknowledged
for financial support.
References
1 O. Seiferth, K. Wolter, B. Dillmann, G. Klivenyi, H.-J. Freund,D. Scarano and A. Zecchina, Surf. Sci., 1999, 421, 176.
2 A. Zecchina, D. Scarano, S. Bordiga, G. Spoto and C. Lamberti,Adv. Catal., 2001, 46, 265.
3 G. Spoto, E. Gribov, G. Ricchiardi, A. Damin, D. Scarano,S. Bordiga, C. Lamberti and A. Zecchina, Prog. Surf. Sci.,2004, 76, 71.
4 R. M. Szostak, Molecular Sieves, Van Nostrand Reinhold, NewYork, 1989.
5 A. Corma, Chem. Rev., 1995, 95, 559.6 A. Corma, Chem. Rev., 1997, 97, 2373.7 A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307.8 J. M. Thomas, Angew. Chem., Int. Ed., 1999, 38, 3589.9 J. M. Thomas, R. Raja, G. Sankar and R. G. Bell, Acc. Chem.Res., 2001, 34, 191.
10 J. M. Thomas and R. Raja, Annu. Rev. Mater. Res., 2005, 35, 315.11 K. D. Schmitt, J. Haase and E. Oldfield, Zeolites, 1994, 14, 89.12 A. Zecchina and C. O. Arean, Chem. Soc. Rev., 1996, 25, 187.13 J. Sivaguru, A. Natarajan, L. S. Kaanumalle, J. Shailaja,
S. Uppili, A. Joy and V. Ramamurthy, Acc. Chem. Res., 2003,36, 509.
14 R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev.,2000, 100, 2303.
15 A. K. Cheetham, G. Ferey and T. Loiseau, Angew. Chem., Int.Ed., 1999, 38, 3268.
16 B. Sulikowski, Heterogeneous Chemistry Reviews, 1996, 3, 203.17 L. Palin, C. Lamberti, A. Kvick, F. Testa, R. Aiello, M. Milanesio
and D. Viterbo, J. Phys. Chem. B, 2003, 107, 4034.18 C. Lamberti, S. Bordiga, A. Zecchina, G. Artioli, G. Marra and
G. Spano, J. Am. Chem. Soc., 2001, 123, 2204.19 S. Bordiga, A. Damin, F. Bonino, G. Ricchiardi, C. Lamberti and
A. Zecchina, Angew. Chem., Int. Ed., 2002, 41, 4734.20 S. Bordiga, A. Damin, F. Bonino, G. Ricchiardi, A. Zecchina,
R. Tagliapietra and C. Lamberti, Phys. Chem. Chem. Phys., 2003,5, 4390.
21 F. Bonino, A. Damin, G. Ricchiardi, M. Ricci, G. Spano,R. D’Aloisio, A. Zecchina, C. Lamberti, C. Prestipino and S.Bordiga, J. Phys. Chem. B, 2004, 108, 3573.
22 C. Prestipino, F. Bonino, S. Usseglio, A. Damin, A. Tasso, M. G.Clerici, S. Bordiga, F. D’Acapito, A. Zecchina and C. Lamberti,ChemPhysChem, 2004, 5, 1799.
23 X. H. Bu, P. Y. Feng, T. E. Gier, D. Y. Zhao and G. D. Stucky,J. Am. Chem. Soc., 1998, 120, 13389.
24 A. Corma, F. Rey, S. Valencia, J. L. Jorda and J. Rius, Nat.Mater., 2003, 2, 493.
25 M. Milanesio, C. Lamberti, R. Aiello, F. Testa, M. Piana andD. Viterbo, J. Phys. Chem. B, 2000, 104, 9951.
26 C. Prestipino, G. Berlier, F. Xamena, G. Spoto, S. Bordiga,A. Zecchina, G. T. Palomino, T. Yamamoto and C. Lamberti,Chem. Phys. Lett., 2002, 363, 389.
27 G. T. Palomino, S. Bordiga, A. Zecchina, G. L. Marra andC. Lamberti, J. Phys. Chem. B, 2000, 104, 8641.
28 V. Bolis, A. Barbaglia, S. Bordiga, C. Lamberti and A. Zecchina,J. Phys. Chem. B, 2004, 108, 9970.
29 G. T. Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamelloand C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064.
3496 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

30 S. Bordiga, C. Paze, G. Berlier, D. Scarano, G. Spoto, A. Zec-china and C. Lamberti, Catal. Today, 2001, 70, 91.
31 A. Delabie, K. Pierloot, M. H. Groothaert, R. A. Schoonheydtand L. G. Vanquickenborne, Eur. J. Inorg. Chem., 2002, 515.
32 D. Berthomieu and G. Delahay,Catal. Rev. Sci. Eng., 2006, 48, 269.33 H. Miessner, I. Burkhardt, D. Gutschick, A. Zecchina, C. Mor-
terra and G. Spoto, J. Chem. Soc., Faraday Trans., 1990, 86, 2321.34 K. Hadjiivanov, E. Ivanova, L. Dimitrov and H. Knozinger,
J. Mol. Struct., 2003, 661, 459.35 Z. M. Liu and S. I. Woo, Catal. Rev. Sci. Eng., 2006, 48, 43.36 N. A. Kachurovskaya, G. M. Zhidomirov and R. A. Van Santen,
Res. Chem. Intermed., 2004, 30, 99.37 B. Wichterlova, Z. Sobalik and J. Dedecek, Appl. Catal., B, 2003,
41, 97.38 E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K.
Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou,Chem. Rev., 2000, 100, 235.
39 M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev.,2004, 104, 939.
40 S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem.Rev., 2005, 105, 2175.
41 J. U. Rohde, M. R. Bukowski and L. Que, Curr. Opin. Chem.Biol., 2003, 7, 674.
42 P. N. Hawker and M. V. Twigg, Iron(II) and lower states, inComprehensive coordination chemistry: the synthesis reactions,properties and applications of coordination compounds, ed. G.Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press,Oxford, 1987, vol. 4, p. 1179.
43 S. Martin Nelson, Iron(III) and higher states, in Comprehensivecoordination chemistry: the synthesis, reactions, properties andapplications of coordination compounds, ed. G. Wilkinson, R. D.Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol.4, p. 217.
44 M. J. Louwerse and E. J. Baerends, Phys. Chem. Chem. Phys.,2007, 9, 156.
45 Y. Suh, M. S. Seo, K. M. Kim, Y. S. Kim, H. G. Jang, T. Tosha,T. Kitagawa, J. Kim and W. Nam, J. Inorg. Biochem., 2006, 100,627.
46 M. P. Mehn, K. Fujisawa, E. L. Hegg and L. Que, J. Am. Chem.Soc., 2003, 125, 7828.
47 A. Zingg, B. Felber, V. Gramlich, L. Fu, J. P. Collman andF. Diederich, Helv. Chim. Acta, 2002, 85, 333.
48 O. Pestovsky, S. Stoian, E. L. Bominaar, X. P. Shan, E. Munck,L. Que and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871.
49 K. Yoshizawa, Y. Shiota and T. Kamachi, J. Phys. Chem. B,2003, 107, 11404.
50 A. Corma and H. Garcia, Chem. Rev., 2002, 102, 3837.51 E. Hensen, Q. J. Zhu, P. H. Liu, K. J. Chao and R. van Santen,
J. Catal., 2004, 226, 466.52 K. Q. Sun, H. Xia, E. Hensen, R. van Santen and C. Li, J. Catal.,
2006, 238, 186.53 Y. Li, Z. C. Feng, H. C. Xin, F. T. Fan, J. Zhang, P. Magusin,
E. J. M. Hensen, R. A. van Santen, Q. H. Yang and C. Li, J.Phys. Chem. B, 2006, 110, 26114.
54 G. D. Pirngruber, M. Luechinger, P. K. Roy, A. Cecchetto andP. Smirniotis, J. Catal., 2004, 224, 429.
55 J. Perez-Ramirez, F. Kapteijn and A. Bruckner, J. Catal., 2003,218, 234.
56 E. V. Kondratenko and J. Perez-Ramirez, J. Phys. Chem. B, 2006,110, 22586.
57 A. Heyden, N. Hansen, A. T. Bell and F. J. Keil, J. Phys. Chem.B, 2006, 110, 17096.
58 I. Melian-Cabrera, S. Espinosa, J. C. Groen, B. van de Linden,F. Kapteijn and J. A. Moulijn, J. Catal., 2006, 238, 250.
59 G. D. Pirngruber and P. K. Roy, Catal. Today, 2005, 110, 199.60 A. Heyden, B. Peters, A. T. Bell and F. J. Keil, J. Phys. Chem. B,
2005, 109, 4801.61 I. Yuranov, D. A. Bulushev, A. Renken and L. Kiwi-Minsker,
J. Catal., 2004, 227, 138.62 G. Berlier, C. Prestipino, M. Rivallan, S. Bordiga, C. Lamberti
and A. Zecchina, J. Phys. Chem. B, 2005, 109, 22377.63 E. V. Kondratenko and J. Perez-Ramirez, Appl. Catal., B, 2006,
64, 35.64 G. I. Panov, G. A. Sheveleva, A. S. Kharitonov, V. N. Roman-
nikov and L. A. Vostrikova, Appl. Catal., A, 1992, 82, 31.
65 A. Heyden, B. Peters, A. T. Bell and F. J. Keil, J. Phys. Chem. B,2005, 109, 1857.
66 N. Hansen, A. Heyden, A. T. Bell and F. J. Keil, J. Phys. Chem.C, 2007, 111, 2092.
67 G. Grubert, M. J. Hudson, R. W. Joyner and M. Stockenhuber,J. Catal., 2000, 196, 126.
68 G. Mul, J. Perez-Ramirez, F. Kapteijn and J. A. Moulijn, Catal.Lett., 2001, 77, 7.
69 G. Mul, J. Perez-Ramirez, F. Kapteijn and J. A. Moulijn, Catal.Lett., 2002, 80, 129.
70 J. Perez-Ramirez, F. Kapteijn, G. Mul and J. A. Moulijn,J. Catal., 2002, 208, 211.
71 C. M. Sang, B. H. Kim and C. R. F. Lund, J. Phys. Chem. B,2005, 109, 2295.
72 L. Kiwi-Minsker, D. A. Bulushev and A. Renken, J. Catal., 2003,219, 273.
73 J. Perez-Ramirez, M. S. Kumar and A. Bruckner, J. Catal., 2004,223, 13.
74 A. S. Kharitonov, G. A. Sheveleva, G. I. Panov, V. I. Sobolev,Y. A. Paukshtis and V. N. Romannikov, Appl. Catal., A, 1993,98, 33.
75 V. I. Sobolev, G. I. Panov, A. S. Kharitonov, V. N. Romannikov,A. M. Volodin and K. G. Ione, J. Catal., 1993, 139, 435.
76 G. Centi, S. Perathoner, R. Arrigo, G. Giordano, A. Katovic andV. Pedula, Appl. Catal., A, 2006, 307, 30.
77 G. Centi, S. Perathoner, F. Pino, R. Arrigo, G. Giordano,A. Katovic and V. Pedula, Catal. Today, 2005, 110, 211.
78 E. J. M. Hensen, Q. Zhu, R. A. J. Janssen, P. Magusin, P. J.Kooyman and R. A. van Santen, J. Catal., 2005, 233, 123.
79 E. J. M. Hensen, Q. Zhu and R. A. van Santen, J. Catal., 2005,233, 136.
80 S. Perathoner, F. Pino, G. Centi, G. Giordano, A. Katovic andJ. B. Nagy, Top. Catal., 2003, 23, 125.
81 L. V. Pirutko, V. S. Chernyavsky, A. K. Uriarte and G. I. Panov,Appl. Catal., A, 2002, 227, 143.
82 R. Leanza, I. Rossetti, I. Mazzola and L. Forni, Appl. Catal., A,2001, 205, 93.
83 G. Yang, D. H. Zhou, X. C. Liu, X. W. Han and X. H. Bao,J. Mol. Struct., 2006, 797, 131.
84 D. Meloni, R. Monaci, V. Solinas, G. Berlier, S. Bordiga, I.Rossetti, C. Oliva and L. Forni, J. Catal., 2003, 214, 169.
85 G. I. Panov, K. A. Dubkov and E. V. Starokon, Catal. Today,2006, 117, 148.
86 A. K. Uriarte, M. A. Rodkin, M. J. Gross, A. S. Kharitonov andG. I. Panov, Stud. Surf. Sci. Catal., 1997, 110, 857.
87 N. A. Kachurovskaya, G. M. Zhidomirov, E. J. M. Hensen andR. A. van Santen, Catal. Lett., 2003, 86, 25.
88 N. S. Ovanesyan, A. A. Shteinman, K. A. Dubkov, V. I. Sobolevand G. I. Panov, Kinet. Catal., 1998, 39, 792.
89 K. A. Dubkov, V. A. Sobolev and G. I. Panov, Kinet. Catal.,1998, 39, 299.
90 V. I. Sobolev, K. A. Dubkov, O. V. Panna and G. I. Panov,Catal.Today, 1995, 24, 251.
91 E. V. Kondratenko and J. Perez-Ramirez, Catal. Today, 2007,119, 243.
92 B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C.Ott, J. Catal., 2004, 225, 300.
93 F. Arena, G. Gatti, G. Martra, S. Coluccia, L. Stievano,L. Spadaro, P. Famulari and A. Parmaliana, J. Catal., 2005,231, 365.
94 P. Pantu, S. Pabchanda and J. Limtrakul, ChemPhysChem, 2004,5, 1901.
95 J. Perez-Ramirez, E. V. Kondratenko and M. N. Debbagh,J. Catal., 2005, 233, 442.
96 J. Perez-Ramirez, A. Gallardo-Llamas, C. Daniel and C. Miro-datos, Chem. Eng. Sci., 2004, 59, 5535.
97 E. V. Kondratenko and J. Perez-Ramirez, Appl. Catal., A, 2004,267, 181.
98 R. Bulanek, B. Wichterlova, K. Novoveska and V. Kreibich,Appl. Catal., A, 2004, 264, 13.
99 L. J. Shu, J. C. Nesheim, K. Kauffmann, E. Munck, J. D.Lipscomb and L. Que, Science, 1997, 275, 515.
100 M. Devadas, O. Krocher, M. Elsener, A. Wokaun, G. Mitrikas,N. Soger, M. Pfeifer, Y. Demel and L. Mussmann, Catal. Today,2007, 119, 137.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3497

101 M. Devadas, O. Krocher, M. Elsener, A. Wokaun, N. Soger, M.Pfeifer, Y. Demel and L. Mussmann, Appl. Catal., B, 2006, 67,187.
102 O. Krocher, M. Devadas, M. Elsener, A. Wokaun, N. Soger, M.Pfeifer, Y. Demel and L. Mussmann, Appl. Catal., B, 2006, 66,208.
103 M. Devadas, O. Krocher and A. Wokaun, React. Kinet. Catal.Lett., 2005, 86, 347.
104 P. Marturano, A. Kogelbauer and R. Prins, Stud. Surf. Sci.Catal., 1999, 125, 619.
105 E. Kikuchi, K. Yogo, S. Tanaka and M. Abe, Chem. Lett., 1991,1063.
106 X. B. Feng and W. K. Hall, J. Catal., 1997, 166, 368.107 N. W. Cant and I. O. Y. Liu, Catal. Today, 2000, 63, 133.108 H. Y. Chen, T. Voskoboinikov and W. M. H. Sachtler, Catal.
Today, 1999, 54, 483.109 H. H. Ingelsten, A. Palmqvist and M. Skoglundh, J. Phys. Chem.
B, 2006, 110, 18392.110 G. Piazzesi, M. Devadas, O. Krocher, M. Elsener and A.
Wokaun, Catal. Commun., 2006, 7, 600.111 H. Y. Chen and W. M. H. Sachtler, Catal. Today, 1998, 42, 73.112 H. Y. Chen, T. Voskoboinikov and W. M. H. Sachtler, J. Catal.,
1998, 180, 171.113 H. Y. Chen, T. Voskoboinikov and W. M. H. Sachtler, J. Catal.,
1999, 186, 91.114 E. M. El-Malki, R. A. van Santen and W. M. H. Sachtler,
J. Catal., 2000, 196, 212.115 Z. X. Gao, S. Qi and W. M. H. Sachtler, Appl. Catal., B, 2001, 33,
9.116 Q. Sun, Z. X. Gao, B. Wen and W. M. H. Sachtler, Catal. Lett.,
2002, 78, 1.117 Q. Zhu, R. M. van Teeffelen, R. A. van Santen and E. J. M.
Hensen, J. Catal., 2004, 221, 575.118 A. Z. Ma and W. Grunert, Chem. Commun., 1999, 71.119 R. Q. Long and R. T. Yang, J. Am. Chem. Soc., 1999, 121, 5595.120 R. Q. Long and R. T. Yang, J. Catal., 2002, 207, 224.121 G. S. Qi and R. T. Yang, Appl. Catal., A, 2005, 287, 25.122 R. Joyner andM. Stockenhuber, J. Phys. Chem. B, 1999, 103, 5963.123 R. Q. Long and R. T. Yang, Chem. Commun., 2000, 1651.124 M. S. Kumar, M. Schwidder, W. Grunert and A. Bruckner,
J. Catal., 2004, 227, 384.125 M. Schwidder, M. S. Kumar, A. Bruckner and W. Grunert,
Chem. Commun., 2005, 805.126 M. Santhosh Kumar, M. Schwidder, W. Grunert, U. Bentrup and
A. Bruckner, J. Catal., 2006, 239, 173.127 G. Ovejero, J. L. Sotelo, F. Martinez, J. A. Melero and L. Gordo,
Ind. Eng. Chem. Res., 2001, 40, 3921.128 G. Centi, S. Perathoner, T. Torre and M. G. Verduna, Catal.
Today, 2000, 55, 61.129 R. Q. Long and R. T. Yang, Catal. Lett., 2001, 74, 201.130 L. Capek, V. Kreibich, J. Dedecek, T. Grygar, B. Wichterlova, Z.
Sobalik, J. A. Martens, R. Brosius and V. Tokarova,MicroporousMesoporous Mater., 2005, 80, 279.
131 G. D. Pirngruber, P. K. Roy and R. Prins, Phys. Chem. Chem.Phys., 2006, 8, 3939.
132 G. S. Qi and R. T. Yang, Appl. Catal., B, 2005, 60, 13.133 G. Centi, C. Genovese, G. Giordano, A. Katovic and S. Per-
athoner, Catal. Today, 2004, 91–92, 17.134 A. A. Battiston, J. H. Bitter, F. M. F. de Groot, A. R. Overweg,
O. Stephan, J. A. van Bokhoven, P. J. Kooyman, C. van der Spek,G. Vanko and D. C. Koningsberger, J. Catal., 2003, 213, 251.
135 G. Delahay, D. Valade, A. Guzman-Vargas and B. Coq, Appl.Catal., B, 2005, 55, 149.
136 P. Marturano, A. Kogelbauer and R. Prins, J. Catal., 2000, 190,460.
137 M. T. Nechita, G. Berlier, G. Ricchiardi, S. Bordiga and A.Zecchina, Catal. Lett., 2005, 103, 33.
138 G. Berlier, F. Bonino, A. Zecchina, S. Bordiga and C. Lamberti,ChemPhysChem, 2003, 4, 1073.
139 K. Krishna, G. B. F. Seijger, C. M. van den Bleek, M. Makkee,G. Mul and H. P. A. Calis, Catal. Lett., 2003, 86, 121.
140 P. Marturano, L. Drozdova, A. Kogelbauer and R. Prins,J. Catal., 2000, 192, 236.
141 P. Marturano, L. Drozdova, G. D. Pirngruber, A. Kogelbauerand R. Prins, Phys. Chem. Chem. Phys., 2001, 3, 5585.
142 A. A. Battiston, J. H. Bitter, W. M. Heijboer, F. M. F. de Grootand D. C. Koningsberger, J. Catal., 2003, 215, 279.
143 J. A. Z. Pieterse, S. Booneveld and R. W. van den Brink, Appl.Catal., B, 2004, 51, 215.
144 P. K. Roy and G. D. Pirngruber, J. Catal., 2004, 227, 164.145 G. Centi and F. Vazzana, Catal. Today, 1999, 53, 683.146 M. Schwidder, F. Heinrich, M. S. Kumar, A. Bruckner and W.
Grunert, Stud. Surf. Sci. Catal., 2004, 154, 2484.147 J. D. Henao, B. Wen and W. M. H. Sachtler, J. Phys. Chem. B,
2005, 109, 2055.148 G. D. Pirngruber and J. A. Z. Pieterse, J. Catal., 2006, 237, 237.149 A. Guzman-Vargas, E. Lima, G. Delahay, B. Coq and V. Lara,
Ind. Eng. Chem. Res., 2006, 45, 4163.150 P. Ratnasamy and R. Kumar, Catal. Today, 1991, 9, 329.151 S. Bordiga, R. Buzzoni, F. Geobaldo, C. Lamberti, E. Giamello,
A. Zecchina, G. Leofanti, G. Petrini, G. Tozzola and G. Vlaic,J. Catal., 1996, 158, 486.
152 G. Berlier, G. Spoto, P. Fisicaro, S. Bordiga, A. Zecchina, E.Giamello and C. Lamberti, Microchem. J., 2002, 71, 101.
153 G. Berlier, G. Spoto, S. Bordiga, G. Ricchiardi, P. Fisicaro, A.Zecchina, I. Rossetti, E. Selli, L. Forni, E. Giamello and C.Lamberti, J. Catal., 2002, 208, 64.
154 J. Perez-Ramirez, F. Kapteijn, J. C. Groen, A. Domenech, G.Mul and J. A. Moulijn, J. Catal., 2003, 214, 33.
155 P. Fejes, K. Lazar, I. Marsi, A. Rockenbauer, L. Korecz,J. B. Nagy, S. Perathoner and G. Centi, Appl. Catal., A, 2003,252, 75.
156 K. A. Dubkov, N. S. Ovanesyan, A. A. Shteinman, E. V.Starokon and G. I. Panov, J. Catal., 2002, 207, 341.
157 S. Faggian, P. Fisicaro, E. Giamello, R. Gobetto, A. Viale, G.Berlier, C. Lamberti and I. Rossetti, J. Phys. Chem. B, 2003, 107,8922.
158 A. M. Ferretti, C. Oliva, L. Forni, G. Berlier, A. Zecchina and C.Lamberti, J. Catal., 2002, 208, 83.
159 G. Berlier, A. Zecchina, G. Spoto, G. Ricchiardi, S. Bordiga andC. Lamberti, J. Catal., 2003, 215, 264.
160 L. J. Lobree, I. C. Hwang, J. A. Reimer and A. T. Bell, J. Catal.,1999, 186, 242.
161 G. Spoto, A. Zecchina, G. Berlier, S. Bordiga, M. G. Clerici andL. Basini, J. Mol. Catal. A: Chem., 2000, 158, 107.
162 G. Berlier, M. Pourny, S. Bordiga, G. Spoto, A. Zecchina and C.Lamberti, J. Catal., 2005, 229, 45.
163 J. A. McCleverty, N. M. Atherton, J. Locke, E. J. Wharton andC. J. Winscom, J. Am. Chem. Soc., 1967, 89, 6082.
164 R. K. Afshar, A. K. Patra, M. M. Olmstead and P. K. Maschar-ak, Inorg. Chem., 2004, 43, 5736.
165 A. Wanat, T. Schneppensieper, G. Stochel, R. van Eldik, E. Billand K. Wieghardt, Inorg. Chem., 2002, 41, 4.
166 B. B. Wayland and L. W. Olson, J. Am. Chem. Soc., 1974, 96,6037.
167 D. S. Bohle and C. H. Hung, J. Am. Chem. Soc., 1995, 117, 9584.168 M. Hoshino, L. Laverman and P. C. Ford, Coord. Chem. Rev.,
1999, 187, 75.169 N. Reginato, C. T. C. McCrory, D. Pervitsky and L. J. Li, J. Am.
Chem. Soc., 1999, 121, 10217.170 P. Fisicaro, E. Giamello, G. Berlier and C. Lamberti, Res. Chem.
Intermed., 2003, 29, 805.171 G. Berlier, G. Ricchiardi, S. Bordiga and A. Zecchina, J. Catal.,
2005, 229, 127.172 B. N. Figgis, Introduction to ligand fields, John Wiley & Sons,
New York, 1967.173 G. Lehmann, Z. Phys. Chem., Neue Folge, 1970, 72, 279.174 E. J. M. Hensen, Q. Zhu, M. Hendrix, A. R. Overweg, P. J.
Kooyman, M. V. Sychev and R. A. van Santen, J. Catal., 2004,221, 560.
175 W. M. Heijboer, D. C. Koningsberger, B. M. Weckhuysen and F.M. F. de Groot, Catal. Today, 2005, 110, 228.
176 J. F. Jia, Q. Sun, B. Wen, L. X. Chen and W. M. H. Sachtler,Catal. Lett., 2002, 82, 7.
177 S. H. Choi, B. R. Wood, A. T. Bell, M. T. Janicke and K. C. Ott,J. Phys. Chem. B, 2004, 108, 8970.
178 S. H. Choi, B. R. Wood, J. A. Ryder and A. T. Bell, J. Phys.Chem. B, 2003, 107, 11843.
179 M. Schwidder, M. S. Kumar, K. Klementiev, M. M. Pohl, A.Bruckner and W. Grunert, J. Catal., 2005, 231, 314.
3498 | Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 This journal is �c the Owner Societies 2007

180 F. Heinrich, C. Schmidt, E. Loffler, M. Menzel and W. Grunert,J. Catal., 2002, 212, 157.
181 G. D. Pirngruber, P. K. Roy and N. Weiher, J. Phys. Chem. B,2004, 108, 13746.
182 E. Bus, J. T. Miller, A. J. Kropf, R. Prins and J. A. vanBokhoven, Phys. Chem. Chem. Phys., 2006, 8, 3248.
183 A. M. Volodin, G. M. Zhidomirov, K. A. Dubkov, E. J. M.Hensen and R. A. van Santen, Catal. Today, 2005, 110, 247.
184 G. D. Pirngruber, J. D. Grunwaldt, J. A. van Bokhoven, A.Kalytta, A. Reller, O. V. Safonova and P. Glatzel, J. Phys. Chem.B, 2006, 110, 18104.
185 G. I. Panov, V. I. Sobolev, K. A. Dubkov, V. N. Parmon, N. S.Ovanesyan, A. E. Shilov and A. A. Shteinman, React. Kinet.Catal. Lett., 1997, 61, 251.
186 P. C. Ford and L. E. Laverman, Coord. Chem. Rev., 2005, 249,391.
187 R. K. Afshar, A. A. Eroy-Reveles, M. M. Olmstead and P. K.Mascharak, Inorg. Chem., 2006, 45, 10347.
188 T. W. Hayton, W. S. McNeil, B. O. Patrick and P. Legzdins,J. Am. Chem. Soc., 2003, 125, 12935.
189 M. Ibrahim, C. L. Xu and T. G. Spiro, J. Am. Chem. Soc., 2006,128, 16834.
190 P. E. M. Siegbahn and T. Borowski, Acc. Chem. Res., 2006, 39,729.
191 F. Buda, B. Ensing, M. C. M. Gribnau and E. J. Baerends,Chem.–Eur. J., 2003, 9, 3436.
192 A. Zecchina, E. Groppo and S. Bordiga, Chem.–Eur. J., 2007, 13,2440.
This journal is �c the Owner Societies 2007 Phys. Chem. Chem. Phys., 2007, 9, 3483–3499 | 3499
Copyright © 2022 FDOKUMEN




















