
Homogeneous Catalysts with a Mechanical (“Machine-like”) Action
14
Homogeneous Catalysts with a Mechanical (“Machine-like”) Action Gerhard F. Swiegers,* [a] Junhua Huang, [b] Robin Brimblecombe, [c] Jun Chen, [a] G. Charles Dismukes, [d] Ulrich T. Mueller-Westerhoff, [e] Leone Spiccia, [c] and Gordon G. Wallace [a] # 2009 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 4759 4746 DOI: 10.1002/chem.200802396
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Homogeneous Catalysts with a Mechanical (“Machine-like”) Action
Homogeneous Catalysts with a Mechanical (“Machine-like”) Action
Gerhard F. Swiegers,*[a] Junhua Huang,[b] Robin Brimblecombe,[c] Jun Chen,[a]
G. Charles Dismukes,[d] Ulrich T. Mueller-Westerhoff,[e] Leone Spiccia,[c] andGordon G. Wallace[a]
� 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594746
DOI: 10.1002/chem.200802396

Introduction
According to collision theory, when two or more reactantmolecules or atoms collide with each other in an uncata-lyzed process, they momentarily form an unstable, high-energy species known as the transition state (Figure 1 a).[1]
This moiety exists for only an exceedingly brief period oftime, after which its constituent atoms or molecules disen-gage to yield either the original reactants back (“elastic col-lision”), or they generate new chemical species known asthe products of the reaction (“reactive collision”). The out-come of such a collision is determined by its energy. Prod-ucts are formed if the collision is sufficiently energetic toovercome a certain threshold energy, known as the activa-tion energy (Ea ; Figure 1b).[1] If this threshold is not over-come, the collision is unsuccessful and the reactants are re-formed. The lower the threshold energy (Ea), the greater
will be the proportion of reactive collisions. If Ea is close tozero, then virtually every collision will result in a reaction.
The overall rate of a chemical reaction is given by the Ar-rhenius equation [Eq. (1)], which relates reaction rate (k) tothe frequency with which the reactant molecules collidewith each other (the pre-exponential term A, known as thecollision frequency), and the proportion of those collisionsthat are sufficiently energetic to result in product formation(the exponential term, �Ea/RT).
k ¼ A exp��Ea
RT
�ð1Þ
Figure 1 a schematically depicts the sequential steps in-volved in a reaction, namely, step i: reactant collision, andstep ii : overcoming the activation energy of the collision(Ea) to form products.
The least-favored of these steps will determine the charac-ter and course of the reaction. In reactions where the activa-tion energy is large, there will be many collisions, of whichonly a small proportion will be successful. That is, step ii willbe disfavored relative to step i, meaning that the reactionwill be dominated by the exponential term in Equation (1).The overall rate, k, will then depend on the thermodynamicsof the collision and the activation energy that must be over-come in the transition state (expressed in kcal mol�1 orkJ mol�1). The reaction could be considered to display anenergy-dependent character and course.
Abstract: Chemical reactions may be controlled byeither: 1) the minimum threshold energy that must beovercome during collisions between reactant molecules/atoms (the activation energy, Ea), or: 2) the rate atwhich reactant collisions occur (the collision frequency,A)—for reactions with low Ea. Reactions of type 2 aregoverned by the physical, mechanical interaction of thereactants. Such mechanical processes are unusual, butnot unknown in molecular catalysts. In this work we ex-amine the machine-like nature of the action in variousabiological mechanical catalysts and consider the impli-cations for mimicry of biological catalysts.
Keywords: biomimetic synthesis · collision frequency ·homogeneous catalysis · mechanical reaction ·nonequilibrium
[a] Dr. G. F. Swiegers, Dr. J. Chen, Prof. Dr. G. G. WallaceARC Centre of Excellence for Electromaterials ScienceIntelligent Polymer Research InstituteUniversity of WollongongWollongong, NSW 2522 (Australia)Fax: (+61) 2 4221-3114E-mail : [email protected]
[b] Dr. J. HuangCSIRO Energy TechnologyBag 10, Clayton, Victoria 3169 (Australia)
[c] R. Brimblecombe, Prof. Dr. L. SpicciaSchool of Chemistry, Monash UniversityWellington Road, Clayton, VIC 3800 (Australia)
[d] Prof. Dr. G. C. DismukesDepartment of Chemistry and Princeton Environmental InstitutePrinceton University, Princeton, NJ 08540 (USA)
[e] Prof. Dr. U. T. Mueller-WesterhoffDepartment of Chemistry, University of ConnecticutStorrs, CT 06268 (USA)
Figure 1. Schematic depiction of an uncatalyzed chemical reaction as:a) a collision between two molecules, A and B, leading to a chemical re-action in which products are formed, and b) the energy profile followedduring the collision, showing the minimum threshold energy needed forproduct formation (termed: the activation energy, Ea).
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4747
CONCEPT

However, in reactions in which the activation energy issmall, virtually every collision will result in a reaction. Thatis, step ii will be favored relative to step i, meaning that thereaction will be dominated by the pre-exponential term inEquation (1). The overall rate, k, will then depend on thecollision frequency of the reactants. In other words, the reac-tion rate will depend most fundamentally on the mechanicsof reactant collision (expressed in s�1 or s). Since frequencyis intrinsically a measure of time and not of energy, we willterm such reactions time-dependent so as to distinguish themfrom reactions governed by Ea.
These distinctions reflect, in broad terms, the two funda-mental transformational processes that are recognized in sci-ence: 1) thermodynamics, which describes transformationsdue to an energy differential, and 2) mechanics, which de-scribes transformations due to physical cause-and-effect se-quences that play out over time.[2] While the field of thermo-dynamics has been extensively developed in chemistry, me-chanics is substantially less developed.[2]
Most uncatalyzed reactions in the liquid phase at ambienttemperature are undoubtedly energy-dependent. However,time-dependent processes are known. One example in thisrespect is the reaction of H+ and OH�. When an H+ ion en-counters an OH� ion, they react rapidly to form H2O (k=
ca. 10�10 s). This occurs, essentially, without regard to theenergy of the encounter, because the Ea threshold for thisreaction is exceedingly low. Such reactions are said to be dif-fusion-controlled, because they are governed by the rate atwhich the reactant species diffuse to each other in solution.In other words, the overall reaction rate is dependent on thefrequency with which H+ and OH� encounter each other(“collide”) in solution and this is a function of their speed ofdiffusion.
Catalysts are species that intervene in, and acceleratechemical reactions without themselves being changed. Cata-lytic action involves, firstly, binding the reactants and, sec-ondly, bringing them into collision while bound to the cata-lyst. In binding the transition state, catalysts decrease the ac-tivation energy of the reaction.
Figure 2 schematically illustrates how a catalyst wouldtypically intervene in the processes depicted in Figure 1. Ascan be seen, the catalyst will generally first bind the two re-actants A and B, and then bring them into collision witheach other. The transition state, thus formed, is stabilized bybinding to the catalyst, thereby lowering the threshold acti-vation energy that must be overcome, Ecat
a , relative to its un-catalyzed equivalent, Euncat
a (Figure 2 b).How does a catalyst lower the activation energy? Upon
binding, a catalyst typically polarizes or stretches bondswithin the reactant, thereby making these bonds more con-ducive to reaction. This process is known as activation andis illustrated in Figure 3 for a representative reactant, dioxy-gen O2. As can be seen, catalysts usually stretch bonds byeither withdrawing electron density from reactant bondingmolecular orbitals or increasing it in anti-bonding molecularorbitals.
A typical catalyst then further facilitates collisions be-tween the bound reactants in which the orientations of theactivated reactants are relatively ideal for reaction; that is,the threshold activation energy of the collision (Ea) is rela-tively low. The activation provided to the reactants by thecatalyst is then sufficient for them to react with each otherwhen brought into physical contact. The catalyst may createsuch “low-threshold-energy” collisions by constraining thereactants to approach each other along trajectories that areenergetically favorable. By contrast, collisions in uncata-lyzed reactions typically involve random approach trajecto-ries, including some highly inopportune ones that have veryhigh energy thresholds. Reaction is therefore less likelywithout a catalyst than with one.
Figure 2. Schematic depiction of a catalyzed chemical reaction as: a) acollision between two catalyst-bound molecules, A and B, leading to achemical reaction in which products are formed, and b) the energy pro-file followed during the collision, showing the minimum threshold energyneeded for product formation, Ecat
a , relative to the threshold in the unca-talyzed reaction, Euncat
a .
Figure 3. Reactant activation by a catalyst. Schematic illustrating how acatalyst (CAT) activates a representative reactant (O2) upon binding, byincreasing the O�O bond length in order to facilitate its cleavage. Theactivation is achieved by either or both of withdrawing electron densityfrom its s-bonding molecular orbital (left), and increasing electron densi-ty in its p*-antibonding orbital (right).
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594748
G. F. Swiegers et al.

As in an uncatalyzed reaction, the overall rate of the cata-lyzed reaction depicted in Figure 2 a is also dependent onthe collision frequency (A) and the rate at which collisionsovercome the activation energy (Ea) to form products. How-ever, the reaction process differs in two important aspects,from the uncatalyzed process depicted in Figure 1 a:
1) As shown in Figure 2 b, the activation energy (Ea) is typi-cally much lower in a catalyzed reaction than in itsequivalent uncatalyzed counterpart. This makes step ii inFigure 2 a more favorable than it would be in the equiva-lent uncatalyzed reaction, meaning that the reaction ismore likely to depend on step i. In other words, the factthat catalysts diminish Ea increases the likelihood thatthe reaction will be controlled by the catalyst-mediatedcollision frequency. If some un-catalyzed reactions areknown to be time-dependent in liquid phases at ambienttemperature, then a comparatively greater number ofsuch reactions should be governed by their collision fre-quency when catalyzed.
2) As shown in step i of Figure 2 a, the collision frequency(A) in a catalyzed system is dependent on the rate of re-versible catalyst–reactant binding and the rate at whichthe catalyst then mediates collisions between bound re-actants. In a molecular catalyst, collisions would typicallybe created when the catalyst framework undergoes aconformational change. This is different to the uncata-lyzed reaction shown in step i of Figure 1 a, the collisionfrequency of which depends only on the rate at whichthe reactants diffuse to, and collide with each other in so-lution. In other words, time-dependent catalytic reactionsneed not be diffusion-controlled. Moreover, they mustnecessarily display a catalyst–reactant intermediate intheir rate expression, since the formation and processingof this intermediate comprises the slowest step of the re-action process.
While a dependence on collision frequency is formallyrecognized in uncatalyzed chemical reactions, it is less wellknown in homogeneous catalysis. Indeed, to the best of ourknowledge, the distinction that must necessarily exist be-tween energy- and time-dependent actions in molecular cat-alysis has not been explored in any detail. Such processesshould be very different. A time-dependent action should,for example, rely on the spatial and temporal factors in-volved in achieving reactant collision. By contrast, anenergy-dependent catalytic action will not be limited by thecollision frequency and will, instead, be governed by thethermodynamic efficiency of the collisions it mediates; thatis, by the extent to which it diminishes the activation energyof reactant collision (Figure 2 b).
In this work we will consider this distinction. We will ex-amine several non-biological molecular catalysts, the actionsof which appear to be controlled by their collision frequen-cy, including [1.1]ferrocenophane hydrogen reduction cata-lysts, Co diporphyrin dioxygen reduction catalysts, and Mn�O cubane water oxidation catalysts. Drawing on these exam-
ples, we will discuss the nature of “mechanical” catalytic ac-tions. The incidence of “mechanical” actions in the catalystsof biology, enzymes, will be considered here only insofar asthey may influence the development of bioinspired molecu-lar catalysts. A recent edited volume by the authors consid-ers mechanical actions in biological catalysis in greaterdetail.[2]
For convenience and continuity in argument, we will usethe terms collision and collision frequency to describe reac-tive contact between reactant functionalities in the liquidphase. We acknowledge that collision is a gas-phase termand the liquid-phase equivalent is, more correctly, termedan encounter.[1]
Reactions Governed by their Collision Frequencyare not Formally Subject to Transition-State
Theory
Perhaps the most critical distinction between reactions(both catalyzed and uncatalyzed) that are governed by theircollision frequency rather than by their activation energy re-lates to their fundamental character. The latter are formallydescribed by transition-state theory, while the former arenot. To explain this aspect, we need to go back in history.
In the 1930s, Henry Eyring of Princeton University devel-oped transition-state theory by applying quantum mechanicsto the general theory of gas phase reactions, known at thetime as Hinshelwood–RRK theory.[3] It is not well knownthat Hinshelwood–RRK theory, which was an elaboration ofcollision theory, involves two so-called “limits” and thattransition-state theory, in fact, conforms to only one of theselimits. The limits are:[3]
1) The high-pressure limit, in which the rate of reaction isgoverned by the activation energy of the transition stateand the system is subject to a thermodynamic equilibri-um, and
2) The low pressure limit, in which the rate of reaction isgoverned by the frequency of reactant collisions and thesystem operates under non-equilibrium conditions (asdemonstrated by a depletion in the Boltzmann distribu-tions during reaction[3e]).
In order to employ quantum mechanics, Eyring had tomake the assumption that the reactants, the products, andthe transition state were all in equilibrium with each other.However, this situation only exists in the high-pressure limitof Hinshelwood–RRK theory, at which the rate of reactionis governed by the activation energy of the transition state.It does not apply in the low-pressure limit, at which reac-tions are governed by the collision frequency.
Thus, a critical distinction between catalyzed or uncata-lyzed reactions governed by their collision frequency ratherthan by their Ea, is that they are not formally subject to tran-sition-state theory. This is because the key issue involves
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4749
CONCEPTHomogeneous Catalysis

achieving collision and this need not be influenced by theenergy landscape about the collision.
We will discuss the meaning of this later. Suffice to say atthis stage, that these implications are potentially significant,especially since nonequilibrium processes have been rela-tively unknown and unstudied in chemistry to date. More-over, the distinction between a time- and an energy-depen-dent process is demonstrated to be more than mere intellec-tual semantics. It has a real and a rather important basis.
Dynamism in Reactant Binding Induces aMechanical Action in Heterogeneous Catalysis
How, then, does one recognize and distinguish a catalyticaction that is mechanical from one that is not? What is ma-terially different between them? In particular, what physicalfeature causes the action of a catalyst to be governed by thecollision frequency not Ea?
A possible insight into this question can be found in atechnique employed in heterogeneous catalysis to distin-guish catalysts governed by their collision frequency fromthose controlled by their Ea. In the 1950s, Balandin andothers noticed a distinctive relationship between the relativerates at which formic acid is decomposed in the gas phaseby various metal catalysts and the strength of catalyst–for-mate binding.[4–6] This relationship is depicted in Figure 4.
As can be seen, the reaction rate initially rises with in-creasing binding strength (quantified as the heat of adsorp-tion of formate), after which it falls, giving a characteristicvolcano-like shape. Such graphs are therefore termed “vol-cano plots”. They are common to a wide range of catalyzedreactions and are routinely used to optimize the heterogene-ous catalysts employed in industrial chemical transforma-tions.
Based on various physical studies,[4–6] it was concludedthat the formate ion is an intermediate in this reaction,which has the mechanism (K=k1/k�1) shown in Equa-tions (2) and (3).
HCOOHGK
HHCOO�adsorbed þHðþÞ
adsorbed ð2Þ
HðþÞadsorbed þHCOO�adsorbed
k2�!CO2 þH2 ð3Þ
The importance of these equations is that they, effectively,depict the two general steps involved in a catalyzed reac-tion: steps i and ii of Figure 2 a. Note that the right-handside of Equation (2) is identical to the left-hand side ofEquation (3). Thus, Equation (2) describes the rate at whichthe reactants are bound and brought into collision with eachother on the catalyst (step i in Figure 2 a). That is, Equa-tion (2) depicts all of the processes involved in catalyst-mediated collision. Equation (3) describes the rate at whichsuch collisions, once achieved, are converted into products(step ii in Figure 2 a). That is, Equation (3) depicts the pro-cess of overcoming the threshold activation energy (Ea).
The trends shown in Figure 4 represent these underlyingprocesses. Thomson and Webb summarize it as follows:[7] onthe left side of the plot, Equation (2) is rate-limiting (reac-tant binding and collision) and its rate increases with bind-ing strength (since this increases the likelihood that the re-actants will be bound and available for catalyst-mediatedcollisions). On the right side of the plot, Equation (3) israte-limiting (overcoming Ea to form products) and its ratedecreases with binding strength (because stronger bindingincreases Ea). At the peak of the volcano plot the two pro-cesses are balanced and their rates are identical.
Fahrenfort, van Reyen, and Sachtler have put it thus:[6]
“[the reason for the shape of the volcano plot is that] onone side of the maximum [the right-hand side], the activa-tion energy becomes unfavorably high; on the other side[the left-hand side], the frequency factor [collision frequen-cy] drops to low values”.
The critical point that comes out of volcano plots is thatcatalytic reactions are governed by their collision frequencywhen the individual catalyst–reactant binding contacts areweak and dynamic. In other words, when the reactant func-tionalities bind and release the catalyst rapidly and dynami-cally, they are attached so briefly that there is little time forthem to collide with each other. Achieving collision then be-comes the key impediment that must be overcome in thecatalytic process; that is, the collision frequency becomeslow and rate-determining.
In such time-dependent catalysts, binding and collision ofthe type shown in Equation (2) is the slower process andtherefore rate-limiting overall. Thus, their rate expressionsmust necessarily contain a rapidly equilibrating catalyst–re-actant intermediate of the type shown in Equation (2), sincethe formation and processing of this intermediate is theslowest step.
The key implication is that a mechanical action in a cata-lyst originates in the physical feature of dynamic reactant
Figure 4. Volcano plot for the decomposition of formic acid by a range ofheterogeneous metal catalysts. The constants K and k2 are those of Equa-tions (2) and (3). The labels “binding too weak” and “binding toostrong” are taken from reference [8].
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594750
G. F. Swiegers et al.

binding. To find homogeneous catalysts with a mechanicalaction, one should therefore look for examples in which thereactants dynamically interact with the catalyst.
Nonbiological Homogeneous Catalysts
Ferrocenophane hydrogen reduction catalysts : To check theabove deduction, we reviewed the scientific literature insearch of non-biological homogeneous species throughwhich the use of dynamic, weak binding interactions leadsto time-dependent catalysis. Such catalysts are extremelyrare; only a few potential examples could be found.[2] Whilea dependence on collision frequency is highly probable inseveral of them, data is not available to unequivocally dem-onstrate it in most of them. A clear example of an abiologi-cal time-dependent catalyst was found, however, in theproton reduction catalyst [1.1]ferrocenophane 1 (Scheme 1).
This species converts strong acids (H+) to dihydrogen (H2)in the presence of a sacrificial reductant.[9,10] The mechanismof the catalysis involves simultaneous homolytic cleavage oftransient Fe�H species during conformational flexing of thecatalyst as shown in Scheme 1.[10] Theoretical calculations in-dicate that the bound H+ ions in intermediate 2 are effec-tively atomic hydrogen (HC) with most of their positivecharge resident on the metal.[10–12] A collision between twosuch highly reactive atomic hydrogen atoms, of the typeshown in [2]TSC, must, consequently, involve an exceedinglylow Ea for H2 formation.
The key feature of catalysis by 1 is weak and dynamicproton binding. The ferrocenes in 1 are very weakly basic(pKa1 =�6.5, pKa2 =�7.1).[12] During the infrequent periodsof ferrocene�H+ binding, the protons participate in a rapid,dynamic equilibrium involving Fe�H and agostic Cp ringC�H isomers as shown in Scheme 2.[10,11] Density functionaltheory (DFT) calculations indicate that the +Fe�HC state is
of higher energy and its lifetime is exceedingly short; tooshort to be measured at ambient temperature.[11] As such, itmust be considered to be the activated form of the boundreactant. Recently, 2 could be directly observed by 1H NMRspectroscopy at �122 8C.[11]
This confirmed that both iron atoms became protonatedand that the exchange processes were too rapid to be re-solved by NMR spectroscopy. The effect of this dynamicand transient reactant binding is to make it difficult for thecatalyst to create collisions between bound reactants.Indeed, this becomes the major obstacle and the controllingfactor in the catalytic process. The reasons for this are asACHTUNGTRENNUNGfollows.
Catalysis by 1 involves, in effect, two highly dynamic pro-cesses: 1) reactant binding by the catalyst and 2) conforma-tional flexing by the catalyst. Reactive collisions can onlyoccur if protons are simultaneously bound and activated atboth ferrocene Fe ions (event I) at the instant that confor-mational flexing brings the catalyst into a structure (even-t II), which complements the transition state (shown in anexaggerated form as [2]TSC in Scheme 1). However, events Iand II occur only very briefly and with relatively long timeintervals between them. Moreover, they occur independent-ly of each other. To have any chance of a reactive collision,catalyst flexing must therefore be synchronized with reac-tant binding. In other words: the ferrocene groups must un-dertake rapid conformational flexing about a structure thatcomplements the reaction transition state. That is, the ar-rangement of the ferrocene catalytic groups must be suchthat vigorous conformational motion is available to bring si-multaneously activated HC substrates into reactive collisionswith each other in the very brief time periods that both Featoms bear activated protons. For this reason, the structureand conformational dynamics of this class of catalyst arecritical to achieving a catalytic effect.
Species like 1 and 4, which rapidly and dynamically oscil-late about a structure that complements the transition state,are therefore highly active catalysts of proton reduction(Schemes 1 and 3).[10] However, complex 5, which also flexesabout a suitable structure but does so only slowly because of
Scheme 1. Hydrogen reduction catalysis by [1.1]ferrocenophane, 1.
Scheme 2. Dynamic proton binding by ferrocene. Scheme 3. Hydrogen reduction catalysis by other ferrocenophanes.
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4751
CONCEPTHomogeneous Catalysis

the steric constraints of the additional Me groups, is entirelyinactive as a catalyst (Scheme 3).[10] Moreover, 6 and 7,which do not flex about a structure that complements thetransition state, yield no catalytic effect (Scheme 3).[10] Thesame is true for diferrocenylethane 8 (Scheme 3) and mono-meric ferrocene in open solution;[10] they also have a lowlikelihood of mediating a collision between simultaneouslybound protons. Their collision frequency is, consequently,zero.
Catalyst 1 is therefore governed by the spatial and tempo-ral fluctuations that lead to reactive collisions. In otherwords, it is a mechanical catalyst. This terminology is partic-ularly apt, since 1 exhibits numerous characteristics commonto mechanical processes in general. For example, like essen-tially all mechanical devices, the movements and actions ofthe components of 1 must be synchronized in order to real-ize an effect. That is, the catalytic groups must act in a con-certed, coordinated manner or, more correctly, a convergentmanner.[13] In other words, their actions must converge inorder to achieve a catalytic effect.[2,13] Moreover, catalysisby 1 is driven by a mechanical impulse, namely conforma-tional flexing.
The catalytic action of 1 is, indeed, intrinsically machine-like. That is, when the processes of catalyst flexing and bind-ing are synchronized, protons are dynamically bound, acti-vated, and carried, repeatedly, along specific, unchanging,near-optimum pathways into reactive collisions with eachother. These collisions occur within a structure that comple-ments the desired outcome, namely the transition state. Theprocess is driven by the mechanical impulse of conforma-tional flexing. The products are then dynamically ejectedand new reactants taken up for the next cycle. This binding,collision, and ejection process is arguably analogous to amechanical device.
The action of 1 therefore generates products because itsmechanical action and spatial arrangement at the point ofcollision is such that H2 is formed. This is not the case for 5–8 and free ferrocene in open solution.
In terms of its general catalytic properties, [1.1]ferroce-nophane can be, firstly, characterized as being a highly struc-ture-sensitive catalyst. That is, small changes in its structuredrastically affect its catalytic properties. In other words,structural modifications that decrease the rate of flexing (asin 5) or that diminish the population of the required confor-mational exchange during flexing (as in 6–8) rapidly andnonlinearly destroy the catalytic effect. This is because theyseverely diminish the extent of synchronization. In the sameway, minor changes in the structure of a single cog within amachine may cause disproportionate, nonlinear losses in itsefficiency.
A second feature is that the ferrocene Fe atom, which isnot known to be a catalytic species in any other reaction, istransformed into a potent catalyst in 1.[10] This transforma-tion is wholly the result of the optimum conformational dy-namics of 1. Without that, the ferrocene Fe atom cannot cat-alyze proton reduction because it binds and activates pro-tons too transiently. This is confirmed by the fact that 1 pro-
gressively loses its activity in solution as it is cooled. At�122 8C, Fe�proton binding is still rapid and dynamic, butconformational flexing is halted on the NMR timescale.[11]
In conclusion, we should note that [1.1]ferrocenophane isremarkably active and long-lived for a homogeneous cata-lyst. Individual molecules of 1 have been shown to turn over1 000 000 H2 molecules, on average, without any noticeableloss of activity.[14a] When bound to polystyrene and coatedon a p-type silicon photocathode, 1 turns over an estimatedfive molecules of H2 per second, over five days of continu-ous operation.[14b] The sheer durability of 1 can only be dueto its excellent selectivity, which prevents the formation ofnonfunctional intermediates and avoids deactivation. Pro-duction of such vigor and high fidelity is typical of a ma-chine—in this case, a “molecular machine”.1
The physical chemistry of complex 1 during catalysis : Final-ly, it is worthwhile asking: is the activation energy of 1 lowand does it display a catalyst–reactant intermediate in itsrate expression? These features were said to be characteris-tic of mechanical catalysis.
The former question is not simple to answer. The problemis that the standard assumptions which underlie most meth-ods for determining Ea are not appropriate to reactions gov-erned by the collision frequency. For example, the Eyringequation that is widely used in this respect, is based on tran-sition-state theory, which, as mentioned earlier, does not for-mally apply in these cases.
An alternative approach is to use an Arrhenius plot, inwhich the natural logarithm of the reaction rate (ln k) isplotted against the reciprocal of the temperature (1/T).[1]
According to Equation (1), such a plot should yield a linegraph, the slope (�Ea/R) of which gives the activationenergy and the intercept of which corresponds to the loga-rithm of the collision frequency, ln A.[1] Implicit in thismethod, however, is an assumption that the collision fre-
1 A referee has argued that all of the above points regarding ideal spatialarrangement, the bringing together of reaction partners, and conforma-tional movements in 1, can be described using the Eyring equation incombination with Michaelis–Menten kinetics (involving substrate–cata-lyst binding as a pre-equilibrium). For this reason, the referee arguesthat it is redundant to invoke a mechanical description of catalysis by1. The authors respectfully disagree with this assertion. Transition-statetheory may, indeed, describe the optimum equilibrium thermodynamicsof reactant collision. That is, it may elucidate the ideal approach path-ways and trajectories leading to the energetically most optimum colli-sion between the bound reactants. However, it does not describe thenonequilibrium actions of a molecular catalyst that consistently and re-peatedly achieves highly optimum collision thermodynamics. In suchcases, the course of the reaction becomes determined by other, externalfactors that are unrelated to the thermodynamic efficiency of collision,including: 1) the frequency and rapidity of catalyst conformational flex-ing and 2) the extent to which it is synchronized with dynamic reactantbinding. These factors control the frequency of collision, which be-comes the chief determinant of reaction. Turnover in such species isthen a function of the mechanics of reactant encounter, not its thermo-dynamics. In mapping the equilibrium energy hypersurface about reac-tant collision, transition-state theory does not describe nonequilibriumsystems which, under an external impulse, repeatedly track only alongthe valleys on the surface.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594752
G. F. Swiegers et al.

quency (A) is unaffected by, and independent of, tempera-ture and, moreover, that Ea constitutes the most significantbarrier to reaction.[1] This is, of course, not true in a reactionthat depends on the collision frequency. For example, an Ar-rhenius plot for the reaction of H+ and OH� will yield thetemperature dependence of the collision frequency, not theactivation energy. As such, techniques based on the Arrhe-nius relationship generally describe the temperature de-pendence of the slowest step of the reaction. In a mechani-cal molecular catalyst, this is not the step of overcoming thethreshold Ea in the transition state. It would, instead, be thestep of binding and subsequent flexing of the catalyst lead-ing to reactant collision.
How then does one determine the Ea for a mechanicalcatalyst? At this stage, we are, frankly, unaware of a suitablemethod for doing so.
The second question above relates to the presence of acatalyst–reactant intermediate in the rate expression of 1.This is expected for catalysts governed by their collision fre-quency, since they are limited by the formation and process-ing of this intermediate.
The kinetics of homogeneous catalysts involving a rate-limiting, rapidly equilibrating, catalyst–reactant intermediatehas been extensively developed in the field of biochemistry,which terms it Michaelis–Menten kinetics.[15] One method ofconfirming the presence of such kinetics in a system involvesseeing whether the experimental data fits a so-called Line-weaver–Burke plot, that is, a graph of 1/k versus 1/ ACHTUNGTRENNUNG[reactant]as a straight line with a slope Km/kmax and an intercept 1/kmax
(k= rate, kmax =maximum rate, Km = the Michaelis con-stant).[15]
To the best of our knowledge, only one kinetic study of 1has been reported.[12] This study employed the acid BF3
.H2O,and produced kinetic data that does indeed conform to aLineweaver–Burke plot (with Km for 1�0.2 mm).[12] Whilethe significance of this result was not appreciated until veryrecently, 1 does therefore appear to display a kinetically ob-servable rapidly equilibrating catalyst–reactant intermediate.
Cobalt–diporphyrin oxygen reduction catalysts : Anotherlikely example of a time-dependent homogeneous catalyst isthe co-facial dicobalt–diporphyrin 9 first described by Coll-man (Scheme 4).[16] Compound 9 catalyzes the four-electronreduction of O2 to H2O at potentials negative of 0.71 V (vs.NHE) and below pH 3.5, when adsorbed on a graphite elec-trode.[16] Under identical conditions, the equivalent dipor-phyrin 10, which contains a single extra -CH2- group in itstethers, catalyzes the two-electron formation of H2O2.
[16,17]
The same is true for the corresponding and other Co–por-phyrin monomers, which also generate H2O2.
[16,17]
A similar effect is observed for the dicobalt porphyrins 12and 13, which also facilitate exclusively 4-electron dioxygenreduction despite a difference of almost 1 � in their averageCo�Co separations (Scheme 5).[16,17] The aryl linkers in 12and 13 constrain the Co ions to rapidly and repeatedly flexlongitudinally along a binding pocket in which the metalions eclipse each other (Scheme 6). This is critically necessa-
ry in this class of catalyst, since a capacity for “slippage”away from an eclipsed arrangement during flexing, diminish-es the extent of four-electron reduction.[16, 17] The importantrole of rapid, longitudinal flexing in these catalysts has ledto the proposal of a so-called “Pac-Man” catalytic mecha-nism, which is named after a 1980s video game icon that em-ployed a rapid and repeated biting motion (Scheme 6).[16,17d]
Extensive studies have revealed that, for O�O bondcleavage to occur in this class of catalyst, the O=O moleculemust be simultaneously bound to, and activated by both Cocenters in the cleft between the porphyrins.[16] One of theCo centers needs only to act as a Lewis acid, as demonstrat-ed by the fact that 11, the mono-Co analogue of 9, is also afour-electron reduction catalyst (Scheme 4).[18] During thetime that the diporphyrin binds dioxygen, the activation pre-ferred by the Co ions is likely ideal for spontaneous O�Ocleavage. This presumably occurs via a transition state inwhich the two O atoms are pulled apart along longitudinal
Scheme 4. Co di- and monoporphyrin dioxygen reduction catalysts.
Scheme 5. Co di- and monoporphyrin dioxygen reduction catalysts.
Scheme 6. “Pac-Man” mechanism of conformational flexing in Co-dipor-phyrins during dioxygen reduction.
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4753
CONCEPTHomogeneous Catalysis

trajectories that are optimum for O�O scission. The struc-ture of the diporphyrin conformer that binds the O2 duringthis process must necessarily complement the transitionstate for O�O bond cleavage. The population of this confor-mer at any one time will depend on the conformationalproperties of the Co diporphyrin. It is clearly high for 9, 11,12, and 13, but low for 10.
It therefore appears that four-electron dioxygen reductionin the Co diporphyrins requires a very specific and ratherunique confluence of circumstances. Each O atom in the O=
O molecule must be simultaneously bound to a different Cocenter at the instant that conformational flexing of the di-porphyrin framework pulls them apart.
The aryl linkers in 12 and 13 are ideal for maintaining aneclipsed structure, whilst allowing for the necessary oscilla-tion in the Co�Co distance during flexing. In contrast, 9 de-pends on the shortness of its linkers to achieve satisfactoryflexing about a face-to-face orientation. Lengthening thelinkers, as in 10, increases the flexibility, thereby diminishingthe population of the face-to-face conformer and destroyingits selectivity for the necessary, longitudinal conformationalchange. The likelihood of the required, coordinated,synchronized interplay is then too low in 10 (or in mono-meric Co porphyrins), to yield substantial four-electron re-duction. Instead, a slower two-electron reduction, involvingO2 bound to a single Co center at the instant of reduction, isfavored.[16]
The extreme sensitivity of the catalytic effect to thisminor structural change is consistent with dynamism in oneor both of the individual Co�O binding contacts. In 10, thedioxygen (or its peroxide intermediate) must be attachedtoo briefly to both Co centers simultaneously to be presentwhen they are also correctly arrayed and on the optimumtrajectory for O�O bond cleavage. At any one instant there-fore, effectively no molecules of 10 simultaneously have thedioxygen attached to both Co ions and the porphyrins flex-ing out of a face-to-face arrangement. That is, the collisionfrequency is zero. The term collision frequency refers hereto the rate per unit time at which the dioxygen O atoms dis-engage from each other.
By contrast, the catalytic rates for 9, 11, 12, and 13, whichalso depend upon and are set by the collision frequency, areclearly substantial. This is undoubtedly due to the fact thatthese catalysts vigorously oscillate in a longitudinal manner,about the ideal, face-to-face conformer. In other words, theyflex rapidly about a structure that complements the transi-tion state for O�O cleavage.
In four-electron dioxygen reduction by 9, 11, 12, and 13we again see the distinctive features of a mechanical action,albeit in the form of the reactant being pulled apart, ratherthan being put together into a new moiety.
Thus, we again have two dynamic and independent pro-cesses that must be synchronized: 1) transient O2 binding atboth Co ions simultaneously and 2) catalyst conformationalflexing along a very specific and well-defined pathway.These processes can only be synchronized by constrainingthe catalyst to flex, vigorously and longitudinally, about a
face-to-face structure that complements the desired transi-tion state. When this is achieved, as in 9, 11, 12, and 13, thecatalytic groups act in a concerted, coordinated, and conver-gent manner.[13] When it is not, as in 10 or in monomeric Coporphyrins, the collision frequency declines rapidly and non-linearly to zero.
The whole process is driven by the mechanical impulse ofconformational flexing. The system is machine-like in that itdynamically takes up reactant molecules, and then mechani-cally pulls them apart within a structure that complementsthe desired outcome. It does so in a highly repeatable andvery specific way.
The process therefore involves synchronized mechanicalactions the spatial and temporal features of which lead tothe formation of products. These products, effectively,derive from an advantageous confluence of circumstances inwhich everything that was needed to create them occurredsimultaneously and in the necessary spatial arrangement andorientation. These conditions originate in the conformation-al and binding constraints of the Co diporphyrins.
A final important point is that, in common with [1.1]ferro-cenophane 1, the need for synchronized, convergent (me-chanical) actions in the Co diporphyrins derives, fundamen-tally, from dynamism in the individual Co�O binding con-tacts. These contacts are clearly weak and transient, formingand releasing constantly. That does not mean however, thatthe overall strength of dioxygen binding by 9, 11, 12, or 13need be weak. In fact, cooperative molecular recognition ef-fects can cause overall binding to be much stronger than theindividual binding contacts. Thus, for example, the dioxygenbinding affinity of 9 (KO2
= 103.0�0.3 atm�1), is several ordersof magnitude larger than that of comparable monomeric Coporphyrins.[17e] This undoubtedly reflects the cooperative in-fluence of molecular recognition.
Biological Homogeneous Catalysts
A question that arises in light of the above discussions iswhether any of the homogeneous catalysts of biology, thatis, enzymes, employ a mechanical action? This topic will beexamined in detail elsewhere,[2] so that we will consider ithere only in respect of the possible implications for develop-ing bio-inspired molecular catalysts.
Several characteristics of catalysis by certain enzymes arereminiscent of “mechanical” homogeneous catalysts. For ex-ample, enzymes are known to generally employ the lowestactivation energies of any class of catalyst.[19] Of course, wedo not know whether the reported Ea for each enzyme trulyreflects the energy threshold that must be overcome in sub-strate collision; this would be the case only if the enzymewas an energy-dependent catalyst. If an enzyme were atime-dependent catalyst, its reported Ea would actuallyrelate to the energy requirement of conformational flexingleading to substrate collision. Nevertheless, the reporteddata for enzymatic Ea�s are generally notably lower thanthose of abiological catalysts.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594754
G. F. Swiegers et al.

Some recent studies also indicate that the activities of cer-tain enzymes are governed by conformational fluctuationsin their protein framework,[20] rather than by the activationenergy involved in substrate collision. For example, NMRstudies on cyclophilin A and triosephosphate isomerasehave detected conformational fluctuations within the activesite that occur on a timescale that correlates with the micro-scopic rate of substrate turnover.[21,22] These conformationalchanges are currently believed to facilitate other parallelprocesses, such as gating and solvent exclusion of the activesite,[22,23]
Enzymes are, moreover, widely considered to employ con-certed (convergent) catalytic actions involving very particu-lar spatial arrangements and conformational movements intheir active site.[2,20] As we have seen, this is also true forabiological homogeneous catalysts that employ a mechanicalaction.
Indeed, as first noted by Pauling,[24] a characteristic fea-ture of enzymes is that their active sites are often structural-ly complementary to their reaction transition states. This ap-pears to also be true for non-biological, homogeneous cata-lysts that involve a mechanical action, such as 1, 4, 9, and11–13. That property arose in those cases, because the indi-vidual catalyst–reactant binding contacts were weak andtransient. Reactive collisions could therefore only everoccur if the catalyst vigorously flexed about a structure thatcomplements the transition state. The individual bindingcontacts between enzymes and their substrates are, however,also often weak and transient, comprising of hydrogen-bonding, ion-pairing, hydrophobic–hydrophilic, or van derWaals interactions. A mechanical action therefore offers apotential explanation for the fact that enzyme active sitesoften structurally complement their transition states.
There seem to be many more such correlations. For exam-ple, many enzymes are extremely structure-sensitive cata-lysts, with even minor changes in the spatial arrangement oftheir active site leading to large, nonlinear decreases (>108-fold) in their catalytic activity.[20e,f,25] An extreme structuresensitivity is also a feature of non-biological mechanical ho-mogeneous catalysts that depend on vigorous and very spe-cific conformational dynamics for their catalytic effect.
As noted earlier, highly efficient conformational dynamicsin abiological mechanical systems may transform transientlybinding and activating groups into potent catalysts (e.g., theFe ions in 1 or the free porphyrin ring in 11). In the absenceof the necessary conformational dynamics, such groups aretypically entirely inactive in open solution, because theybind and activate their reactants too briefly.
This could conceivably also be the case with the aminoacid residues that serve as powerful catalytic groups in manyenzymes. Of all of the available monomers and oligomers ofamino acids, only artificial l-prolines[26] have been reportedto be catalytically active outside of enzymology, in openACHTUNGTRENNUNGsolution.
Another distinctive and characteristic feature of many en-zymes is that they display Michaelis–Menten kinetics, whichinvolves a rate-limiting, rapidly equilibrating, catalyst–reac-
tant intermediate (known as the Michaelis complex).[15] Asnoted earlier, abiological mechanical homogeneous catalystsnecessarily also display such a catalyst–reactant intermediatein their rate expressions. A mechanical action thereforeoffers a potential explanation for the origin of Michaelis–Menten kinetics in biology.
Finally: the concept of a “mechanical” catalytic action hasbeen raised before in enzymology, but not explored indetail. For example, Moss and others have explicitly de-scribed enzymes as machine-like “specialized combiningcenters”,[27,28] while Williams has characterized enzymaticaction as being akin to a mechanical device.[29] In othercases, more oblique allusions have been used. For example,Benkovic and Hammes-Shiffer describe catalysis by en-zymes in terms of “coupled protein motions”,[20] whileMenger has proposed a spatiotemporal hypothesis for enzy-mic action.[30] The archetypal spatiotemporal system is, how-ever, a mechanical device, which operates by coupling oneaction to another (e.g., the interlocking cogs in a mechanicalwatch). Machines are critically dependent on the spatialpathways followed by their components and the precisetimes at which they do so. The high activities and specifici-ties of enzymes are consistent with “molecular machines”.
Bioinspired Homogeneous Catalysts
While detailed, confirmatory experimentation is thereforestill required, the evidence available at present suggests thatthe catalytic action of at least some enzymes may be deter-mined by their collision frequency. In that case, we won-dered whether it would be possible to emulate the catalyticfeats of an enzyme using a model species that employed amechanical action? Such a model would necessarily have toexhibit :
I) An active site capable of facilitating reactive collisionsby rapid conformational flexing about a shape that iscomplementary to the transition state of the reaction.
II) Highly dynamic individual catalyst–reactant bindingcontacts, involving a continuous attachment and releaseof reactant functionalities.
Mn-Oxo cubanes—a model of the photosystem II water-oxi-dizing complex (PSII-WOC): Only one homogeneous cata-lyst is known to sustainably facilitate water oxidation: thewater-oxidizing complex (WOC) of photosystem II (PSII),which is found in all photosynthetic organisms and operatesat 1.00 V versus Ag/AgCl (1.20 V vs. SHE). An atomicmodel of the resting oxidation state of the PSII-WOC core,isolated from the cyanobacterium Thermosynechococcus sp. ,has recently been derived from single-crystal X-ray diffrac-tion (XRD) data.[31] Although the low-XRD resolution (3.2to 3.6 �) lead to differences in data interpretation, onemodel[32] indicates an inorganic core comprised of a cubicalMn4Ca cluster, with four metal atoms in a symmetrical het-
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4755
CONCEPTHomogeneous Catalysis

erocubane arrangement of Mn3Ca, connected to a single Mnatom (Figure 5).[33] Four O atoms have been proposed tolink the CaMn4O4 cluster, which is compatible with MnEXAFS and EPR studies.[34]
The absence of evolutionary diversity in the PSII-WOCimplies that combinatorial biosynthesis in nature has pro-duced a single catalyst capable of oxidizing water. This con-clusion has implications for research and has informed at-tempts to model this catalyst.
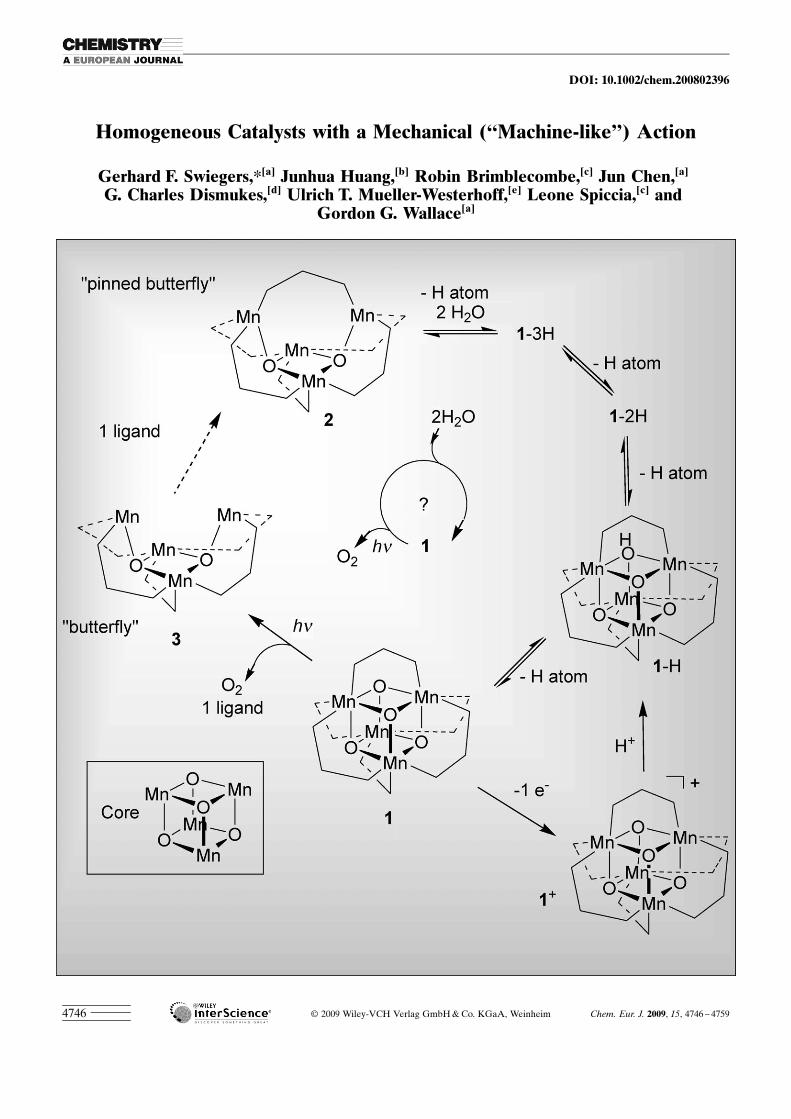
One model system of the PSII-WOC are the cubaneMn4O4L6 complexes 14 (L�= (p-R-C6H4)2PO2
� ; R=H,OMe; Scheme 7). These species comprise a flexible, cubicalMn�O arrangement, the structural motif of which is analo-gous to that proposed to be present in the active site of thePSII-WOC (Figure 5).[35] They also spontaneously self-as-semble from Mn2+ .[35] Under suitable conditions, 14 will,moreover, sequentially abstract 4 H atoms (4 H+ +4 e�) toyield the “pinned butterfly” 15, with associated release oftwo H2O molecules (Scheme 7).[36] This process is readily re-versible, indicating that the oxygen bridges in 14 are dynam-ically formed from water.
UV illumination of 14 in the gas phase leads to the spon-taneous release of O2 (and a Ph2PO2
� ligand, thereby form-ing the “butterfly” complex 16 ; Scheme 7).[37] The quantumefficiency for this process at 350 nm was found to approach100 % for 14 (R= OMe).
A potential catalytic cycle therefore exists for 14 (theframed question mark in the middle of Scheme 7). Such acycle had, however, never been realized in practice.
Of particular interest to us was the likely mechanism forthe step of photolytic O2 release (14!16), which is depictedin Scheme 8.[38] Theoretical calculations suggested that the
key catalytic step involves thephotoinduced loss of aligand.[38] The remainingcubane framework is then ableto flex, causing the two highlyreactive, coordinatively unsatu-rated, corner O atoms to col-lide with each other(Scheme 8).[38] Given the highquantum efficiency of this pro-cess for 14 (R=OMe), this col-lision must readily overcomethe activation energy thresh-old, thereby resulting in reac-tion. However, the flexing pro-cess that creates the collisionand subsequently sees the re-lease of the O2 product, wascalculated to require 96–126 kJ mol�1 of energy over-all.[38]
Thus, in the gas phase atleast, indications existed tosuggest that 14 may facilitate amechanical process, albeit only
for a single turnover.[38] Moreover, 14 appeared to displayboth of the required features I and II described earlier, andneeded for a mechanical catalyst, namely: it creates success-ful, reactive collisions between the corner O atoms by rapidconformational flexing after photorelease of a Ph2PO2
�
ligand (as observed in the gas phase), and in its self-assem-
Figure 5. XRD model of photosystem II water-oxidizing complex (PSII-WOC) showing a proposed model ofthe core active site.
Scheme 7. Interconversion of water and O2 during redox reactions of themodel complex 14 (R=OMe)
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594756
G. F. Swiegers et al.

bly and other properties, it interacts dynamically with reac-tant water molecules (as observed in liquid solution).
Based on these considerations, it seemed to us that 14 of-fered a potential bio-inspired catalyst that may employ amechanical action. In that case, we wondered whether itwould be capable of facilitating active water oxidation inaqueous solution, if suitable conditions for continuous turn-over could be found. Such conditions would necessarily pro-vide for the disassembly of the cubane by photolytic ligandrelease prior to O2 formation (14!16), as well as its subse-quent reassembly to take up the free ligand, along with twowater molecules (16!15!14 ; Scheme 7). As 14 (and 14+)are strongly hydrophobic and therefore entirely insoluble inaqueous medium, we examined the use of Nafion as a cata-lyst support. Nafion offers an interface of hydrophobic andhydrophilic domains.
Nafion-doped with Mn–Oxo cubane is an efficient photo-electrocatalyst of water oxidation : In experiments we foundthat when 14+ (R= OMe) is doped, by ion exchange, into athin Nafion layer deposited on a glassy carbon disk elec-trode that is then immersed in H2O, poised at 1.00 V (vs Ag/AgCl), and illuminated with light (EM 1.5), a substantialand sustained photocurrent is generated (Figure 6).[39] Thephotocurrent appears when the light is turned on (up arrowsin Figure 6). It disappears when the light is turned off (downarrows in Figure 6). Under constant illumination, the currentpersists over a continuous 65 h of testing.
The following evidence supported the contention that theobserved photocurrent derived from water oxidation cataly-sis by the cubane.[39] Gas bubbles generated as a result of
the current were collected and shown to be pure O2. Thevolume of O2 matched the cumulative current passingthrough the electrode. Isotopic-labeling studies confirmedthat the O2 originated in the H2O electrolyte. When an elec-trolyte of pure CH3CN was used, no photocurrent was ob-served. However, the addition of H2O to the CH3CN led toan increasing photocurrent up to 8 % water/CH3CN, atwhich point saturation occurred (Michaelis–Menten kinet-ics). The photocurrent varied with pH in accord with thatexpected for H2O oxidation, the E0 of which changes by59 mV per pH unit. There was no photocurrent without 14+
or with a range of other Mn species in the Nafion layer. Thephotocurrent declined when 14+ was progressively leachedout of the Nafion by competitive ion-exchange. EPR spec-troscopy of the doped Nafion indicated that 14+ was takenup intact and disassembled and reassembled during turnoverunder illumination at 1.00 V (vs. Ag/AgCl).[40]
The reduction in overpotential for H2O oxidation catalysiswas found to be 0.38 V (plus light). A typical Nafion layeron the 3 mm diameter disk electrode was about 7 mm thickand contained mere nanograms of 14+ . The average turn-over frequency per 14+ at 1.00 V (vs. Ag/AgCl) was, atleast, 24 molecules O2 h�1 over 65 h of continuous opera-tion.[39] Peak turnover frequencies of up to 270 moleculesO2 h�1 were measured.[40] These data make 14+ one of themost active and durable known abiological Mn homogene-ous catalysts of H2O oxidation, to the best of ourACHTUNGTRENNUNGknowledge.[2]
The action spectrum indicated that the maximum catalyticefficiency occurred at 350 nm illumination, which corre-sponds to the main MLCT absorption peak of 14. Catalysiswas facilitated however upon illumination up to 600 nm inthe visible spectrum.[39]
These results are, we believe, significant. They representone of the first times, as far as we know, that a model com-plex (of the PSII-WOC in this case) has demonstrated sus-tained and active catalysis that is similar to its enzyme. Thecatalysis cannot be said to be identical to the enzyme, sincethe PSII-WOC is known to turn over without illuminationby sunlight, whereas 14+ in Nafion requires illumination.The observed 0.38 V decline in the overpotential for water
Scheme 8. Likely mechanism of O2 release by cubane 14 in the gas phase.
Figure 6. Photocurrent of 14+/Nafion/glassy carbon. Amperogram at1.00 V (vs Ag/AgCl) of doped Nafion (a) and undoped Nafion (d)on a 3 mm diameter glassy carbon electrode in 0.1 m aq. Na2SO4 at 22 8C(Nafion thickness: 7 mm). The samples were exposed to a filtered Xelight source (275–750 nm) (EM 1.5), up arrows indicate: light on, downarrows indicate: light off.
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4757
CONCEPTHomogeneous Catalysis

oxidation may reflect, in part, the added influence of thelight.
While 14+ is critical to the catalysis, we cannot yet pro-nounce unequivocally upon the mechanism (nor, certainly,on its relationship to that in PSII-WOC). However, it islikely that, during the process of doping Nafion from an or-ganic solution of 14+ , the hydrophobic 14+ ions are drawninto the water channels of the membrane, attracted by thesulfonate anions. Subsequent immersion of the electrode inan aqueous electrolyte may then cause the cubane cations tomigrate into the hydrophobic pockets of the Nafion poly-mer. Within these hydrophobic domains, electrochemical,EPR, and other evidence suggests that illumination resultsin spontaneous photodissociation of one of the phosphinateligands from the cubane core.[40] This facilitates the releaseof dioxygen and the formation of the butterfly cubane 16 ac-cording to the gas-phase mechanism shown in Scheme 8.Under the applied potential of 1.00 V (vs. Ag/AgCl), thebutterfly 16 appears to take up two water molecules andcycle through an EPR-detectable pinned butterfly inter-mediate 15, to regenerate the cubane by four successiveproton-coupled electron transfer steps (4 H+ +4 e�). The H+
ions thus generated, are likely transported away by theproton channels in the Nafion in much the same way that isbelieved to occur in the PSII-WOC.
In summary, we have shown that a Nafion membranedoped with miniscule quantities of the cubical model com-plex 14+ displays a significant photocatalytic effect in theoxidation of water. While we have not demonstrated that14+ undertakes mechanical catalysis of water oxidation, theevidence available at present does not contradict such a pos-sibility. Moreover, the assumption of a mechanical catalyticaction in 14/14+ has led us to develop a new and efficientwater oxidation photoelectrocatalyst.
Conclusions
Two fundamental transformational processes are recognizedin science: thermodynamics, which describes transformationsdue to an energy differential, and mechanics, which de-scribes transformations due to physical cause-and-effect se-quences, which play out over time.
Thermodynamics and its associated realm of transforma-tions under equilibrium conditions, is, historically, a highlydeveloped field of endeavor in chemistry and catalysis. Ki-netics describes the physical interactions that mediate ther-modynamic imperatives.
Mechanics, and its associated realm of transformationsunder non-equilibrium conditions, is, by contrast, under-developed.
In this work we have sought to address this imbalance.We have examined the incidence, properties, and fundamen-tal character of mechanical reactions facilitated by selectednon-biological homogeneous catalysts. We have shown that,in being governed by their collision frequency rather thanby their activation energy, such catalysts rely on spatial and
temporal fluctuations in their molecular framework. Suchfluctuations must be narrowly constrained to the optimumin order to yield an observable catalytic effect. Necessaryconditions include: 1) vigorous conformational oscillationabout a molecular structure that complements the transitionstate of the catalyzed reaction and 2) highly dynamic cata-lyst–reactant binding contacts. These constraints arise in thefact that multiple, weak, and transient binding contactsallow for catalyst-mediated collisions only if reactant bind-ing is synchronized with catalyst flexing.
When a molecular catalyst employs a mechanical action,it appears to operate somewhat like a machine that is drivenby regular conformational flexing to facilitate chemicaltransformations. Products are generated as a response to therepeated and optimally configured spatial and temporal(mechanical) impulses present.
In seeking to understand, define, and test the principles ofmechanical catalysis, we have discovered a new ground-breaking catalyst, Nafion–14+ , with potentially importantimplications for the field of biomimetic chemistry and for at-tempts to mimic biological catalysis in general.[41] This dis-covery has been made by working towards homogeneouscatalysts with mechanical actions. The fact that it has led toa new catalyst suggests that the approach has potentialmerit and that we and others should continue with it.
Acknowledgements
The authors thank the Australian Research Council, Monash University,Princeton University, and the University of Wollongong for financial sup-port. G.F.S. thanks the Australian Academy of Science for a travel fellow-ship. Wolf Sasse (CSIRO) and Bob Williams (Oxford University) arethanked for their patience and insightful comments.
[1] P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford,1978, pp. 52 –782; Collision theory: P. W. Atkins, Physical Chemistry,Oxford University Press, Oxford, 1978, pp. 897 –928.
[2] Mechanical Catalysis: Methods of Heterogeneous, Homogeneous,and Enzymatic Catalysis (Ed.: G. F. Swiegers), Wiley, New York,2008 ; a listing of known, probable, and possible time-dependent mo-lecular catalysts is given in Chapter 9 of this book.
[3] M. J. Pilling, P. W. Seakins, Reaction Kinetics, Oxford UniversityPress, Oxford, 1996, pp. 3 –142; a) Collision theory: M. J. Pilling,P. W. Seakins, Reaction Kinetics, Oxford University Press, Oxford,1996, pp. 61– 66; b) transition-state theory: M. J. Pilling, P. W. Sea-kins, Reaction Kinetics, Oxford University Press, Oxford, 1996,pp. 66 –85; c) reaction dynamics and potential-energy surfaces: M. J.Pilling, P. W. Seakins, Reaction Kinetics, Oxford University Press,Oxford, 1996, pp. 106 –120; d) Lindemann and Hinshelwood–RRKtheory: M. J. Pilling, P. W. Seakins, Reaction Kinetics, Oxford Uni-versity Press, Oxford, 1996, pp. 121 – 138; e) Boltzmann distributionsin the high- and low-pressure limit of Hinshelwood–RRK theory:M. J. Pilling, P. W. Seakins, Reaction Kinetics, Oxford UniversityPress, Oxford, 1996, pp. 128 –130.
[4] A. A. Balandin, Adv. Catal. 1958, 10, 120, and references therein.[5] G. C. Bond, Heterogeneous Catalysis: Principles and Applications,
2nd ed., Oxford Science Publications, Oxford, 1987, pp. 62– 64, andreferences therein; see also: G. C. Bond, Catalysis by Metals, Aca-demic Press, London, 1962, pp. 476 – 478.
[6] J. Fahrenfort, L. L. van Reyen, W. M. H. Sachtler in The Mechanismof Heterogeneous Catalysis (Ed: J. H. De Boer), Elsevier, Amster-
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 4746 – 47594758
G. F. Swiegers et al.

dam, 1960, pp. 23 –48, and references therein; see also: W. M. H.Sachtler, Z. Phys. Chem. 1960, 26, 16; and J. Fahrenfort, L. L. vanRiegen, W. H. M. Sachtler, Z. Elektrochem. 1960, 64, 216.
[7] S. J. Thomson, G. Webb, Heterogeneous Catalysis, Oliver and Boyd,London, 1968, pp. 184 –188.
[8] M. W. Roberts, C. S. McKee, Chemistry of the Metal–Gas Interface,Clarendon Press, Oxford, 1978, p. 30.
[9] T. E. Bitterwolf, A. C. Ling, J. Organomet. Chem. 1973, 57, C15.[10] U. T. Mueller-Westerhoff, Angew. Chem. 1986, 98, 700; Angew.
Chem. Int. Ed. Engl. 1986, 25, 702, and references therein; see also:U. T. Mueller-Westerhoff, T. J. Haas, G. F. Swiegers, T. K. Leipert, J.Organomet. Chem. 1994, 472, 229.
[11] a) A. Karlsson, A. Broo, P. Ahlberg, Can. J. Chem. 1999, 77, 628;b) A. Karlsson, G. Himersson, P. Ahlberg, J. Phys. Org. Chem. 1997,10, 590.
[12] M. Hillman, S. Michalle, S. Felberg, Organometallics 1985, 4, 1258;Figure 4 in this paper is effectively a Lineweaver–Burke plot.
[13] J. Chen, W. Zhang, D. Officer, G. F. Swiegers, G. G. Wallace, Chem.Commun. 2007, 3353.
[14] a) Complex 1 (10 mg) will dissolve lead (at least 5 g) as sacrificial re-ductant during H2 generation; see reference [10]; b) a coating con-taining 2 m [1] over an area of 0.16 cm2 and a response thickness esti-mated to be 1 mm, generates a saturation current of 37 mA duringcatalysis ; see U. T. Mueller-Westerhoff, A. Nazzal, J. Am. Chem.Soc. 1984, 106, 5381.
[15] L. Stryer, Biochemistry, 3rd ed., W. H. Freeman, New York, 1988,pp. 177 –200.
[16] J. P. Collman, P. S. Wagenknecht, J. E. Hutchison, Angew. Chem.1994, 106, 1620; Angew. Chem. Int. Ed. Engl. 1994, 33, 1537, and ref-erences therein.
[17] a) J. P. Collman, P. Denisevich, Y. Konai, M. Marrocco, C. Koval,F. C. Anson, J. Am. Chem. Soc. 1980, 102, 6027; b) J. P. Collman, M.Marrocco, P. Denisevich, C. Koval, F. C. Anson, J. Electroanal.Chem. Interfacial Electrochem. 1979, 101, 117; c) C. K. Chang, H. Y.Abdalmuhdi, J. Am. Chem. Soc. 1984, 106, 2725; d) C. J. Chang, Y.Deng, C. Shi, C. K. Chang, F. C. Anson, D. G. Nocera, Chem.Commun. 2000, 1355; e) Y. Le Mest, M. L�Her, J. P. Collman, N. H.Hendricks, L. McElwee-White, J. Am. Chem. Soc. 1986, 108, 533.
[18] a) J. P. Collman, N. H. Hendricks, K. Kim, C. S. Bencosme, J. Chem.Soc. Chem. Commun. 1987, 1537; b) H. Y. Liu, I. Abdalmuhdi, K. C.Chang, F. C. Anson, J. Phys. Chem. 1985, 89, 665; c) J. P. Collman,K. Kim, J. Am. Chem. Soc. 1986, 108, 7847.
[19] See for example, for catalase: a) Ea =7 kcal mol�1: A. L. Lehninger,Biochemistry, Worth Publishing, New York, 1975, p. 189; b) Ea =
2.5 kcal mol�1: N. J. Brown-Peterson, M. L. Salin, J. Bacteriol. 1993,175, 4197; some other examples of low energy barriers in enzymaticcatalysis : c) A. O. Kolawole, D. M. Sanni, J. O. Ajele, J. Plant Sci.2007, 2, 425; d) A. O. Kolawole, D. M. Sanni, J. O. Ajele, J. PlantSci. 2006, 1, 273; e) P. Mirjafari, K. Aghari, N. Mahinpey, Ind. Eng.Chem. Res. 2007, 46, 921.
[20] a) S. Hammes-Schiffer, S. J. Benkovic, Annu. Rev. Biochem. 2006,75, 519, and references therein; b) S. J. Benkovic, S. Hammes-Schiff-er, Science 2003, 301, 1196, and references therein; c) P. T. Rajago-palan, S. J. Benkovic, Chem. Rec. 2002, 2, 24; d) K. O. Alper, M.
Singla, J. L. Stone, C. K. Bagdassarian, Protein Sci. 2001, 10, 1319;e) A. Tousignant, J. N. Pelletier, Chem. Biol. 2004, 11, 1037; f) S.Hammes-Schiffer, Biochemistry 2002, 41, 13335.
[21] a) E. Z. Eisenmesser, D. A. Bosco, M. Akke, D. Kern, Science 2002,295, 1520; b) E. Z. Eisenmesser, O. Millet, W. Labeikovsky, D. M.Korzhnev, M. Wolf-Watz, D. Bosco, J. J. Skalicky, L. E. Kay, D.Kern, Nature 2005, 438, 117.
[22] G. Jogl, S. Rozovsky, A. E. McDermott, L. Tong, Proc. Natl. Acad.Sci. USA 2003, 100, 50, and references therein.
[23] a) T. C. Bruice, F. C. Lightstone, Acc. Chem. Res. 1999, 32, 127; b) E.Lau, T. C. Bruice, J. Am. Chem. Soc. 2000, 122, 7165; c) T. C.Bruice, S. J. Benkovic, Biochemistry 2000, 39, 6267; d) P. K. Agarwal,Microb. Cell Fact. 2006, 5, 2, and references therein; e) P. K. Agar-wal, J. Am. Chem. Soc. 2005, 127, 15248; f) Workshop on future di-rections of catalysis science, Catal. Lett. 2001, 76, 111.
[24] a) L. Pauling, Chem. Eng. News 1946, 24, 1375; b) L. Pauling,Nature 1948, 161, 707.
[25] W. N. Lipscomb in Structural and Functional Aspects of EnzymaticCatalysis (Eds: H. Eggerer, R. Huber), Springer, Berlin, 1981, p. 17,and references therein.
[26] a) A. J. Cobb, D. M. Shaw, D. A. Longbottom, J. Gold, S. V. Ley,Org. Biomol. Chem. 2005, 3, 84 and references therein; b) H.Grçger, J. Wilken, Angew. Chem. 2001, 113, 545; Angew. Chem. Int.Ed. 2001, 40, 529; c) D. R. Kelly, S. Roberts, Chem. Commun. 2004,2018; d) D. R. Kelly, A. Meek, S. M. Roverts, Chem. Commun. 2004,2021; e) A. C�rdova, W. Zou, I. Ibrahem, E. Reyes, M. Engqvist,W.-W. Liao, Chem. Commun. 2005, 3586; f) S. G. Davies, R. L. Shep-pard, A. D. Smith, J. E. Thomson, Chem. Commun. 2005, 3802.
[27] D. W. Moss, Enzymes, Oliver and Boyd, London, 1968, p. 68.[28] P. K. Agarwal, Microb. Cell Fact. 2006, 5, 2, and references therein.[29] R. J. P. Williams, Trends Biochem. Sci. 1993, 18, 115.[30] F. M. Menger, Acc. Chem. Res. 1993, 26, 206, and references therein.[31] K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Sci-
ence 2004, 303, 1831.[32] N. Kamiya, J. R. Shen, Proc. Natl. Acad. Sci. USA 2003, 100, 98.[33] G. C. Dismukes, Phys. Chem. Chem. Phys. 2004, 6, 4793.[34] G. C. Dismukes, J. Biol. Inorg. Chem. 2002, 7, 2.[35] W. Ruettinger, C. Campana, G. C. Dismukes, J. Am. Chem. Soc.
1997, 119, 6670.[36] M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek, G. C. Dismukes,
Angew. Chem. 2001, 113, 3009; Angew. Chem. Int. Ed. 2001, 40,2925.
[37] W. Ruettinger, C. Campana, G. C. Dismukes, J. Am. Chem. Soc.1997, 119, 6670.
[38] G. C. Dismukes, R. Carr, P. De Angelis, unpublished results.[39] R. Brimblecombe, G. C. Dismukes, L. Spiccia, G. F. Swiegers,
Angew. Chem. 2008, 120, 7445; Angew. Chem. Int. Ed. Angew.Chem. Int. Ed. Engl. 2008, 47, 7335.
[40] A. Bond, R. Brimblecombe, G. C. Dismukes, L. Spiccia, G. F. Swieg-ers, unpublished results.
[41] R. Breslow, Acc. Chem. Res. 1995, 28, 146.
Published online: April 6, 2009
Chem. Eur. J. 2009, 15, 4746 – 4759 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 4759
CONCEPTHomogeneous Catalysis




















