
Predicting the Morphologies of γ Precipitates in Cobalt-Based ...
Upload
independentCategory
view
10download
0
Journal of Molecular Structure 1076 (2014) 713–718
Contents lists available at ScienceDirect
Journal of Molecular Structure
journal homepage: www.elsevier .com/ locate /molst ruc
Structural and magnetic properties of cobalt(II) complexeswith pyridinecarboxamide ligands
http://dx.doi.org/10.1016/j.molstruc.2014.08.0310022-2860/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author.E-mail address: [email protected] (B. Dojer).
Brina Dojer a,⇑, Andrej Pevec b, Ferdinand Belaj c, Zvonko Jaglicic d,e, Matjaz Kristl f, Miha Drofenik f,g
a Faculty of Natural Sciences and Mathematics, University of Maribor, Koroška cesta 160, SI-2000 Maribor, Sloveniab Faculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerceva 5, SI-1000 Ljubljana, Sloveniac Karl-Franzens-Universität Graz, Institut für Chemie, Schubertstr. 1, A-8010 Graz, Austriad Institute of Mathematics, Physics and Mechanics, Jadranska 19, SI-1000 Ljubljana, Sloveniae Faculty of Civil and Geodetic Engineering, Jamova 2, SI-1000 Ljubljana, Sloveniaf Faculty of Chemistry and Chemical Engineering, University of Maribor, Smetanova 17, SI-2000 Maribor, Sloveniag Jozef Stefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
h i g h l i g h t s
� Two novel coordination compounds of cobalt(II) acetate with pyridinecarboxamides were synthesized.� Both of the compounds possess trans-distorted octahedral geometry around the Co center.� Magnetic measurements reveal the 2+ ionic state of cobalt and paramagnetic behavior.� IR spectral studies show differences in CONH2 stretching region and COO� stretching vibrations.
a r t i c l e i n f o
Article history:Received 24 June 2014Received in revised form 18 August 2014Accepted 18 August 2014Available online 26 August 2014
Keywords:Cobalt(II) acetate tetrahydrateNicotinamideIsonicotinamideX-ray structure determinationMagnetic propertiesHydrogen bonds
a b s t r a c t
The synthesis and characterization of two new cobalt(II) coordination compounds with nicotinamide(nia) and isonicotinamide (isn) are reported. The products were characterized magnetically, structurallyby single-crystal X-ray diffraction analysis and spectrally by FT-IR spectroscopy. Using the reaction ofcobalt(II) acetate tetrahydrate and nicotinamide in methanol we obtained light-red crystals of the mono-nuclear complex [Co(nia)2(H2O)4](CH3COO)2�2H2O (1). The synthesis in a system cobalt(II) acetatedihydrathe, isonicotinamide and dimethylformamide–methanol mixture gave a new dinuclear coordina-tion compound with the formula [Co2(CH3COO)4(isn)4]�2C3H7NO (2). In both compounds a trans arrange-ment of pyridinecarboxamide ligands was found. Intermolecular hydrogen bonds in the crystal structuresof both complexes are discussed. The magnetic properties were studied between 2 K and 300 K giving theresult leff = 4.6 BM for 1 and leff = 4.7 BM for 2 in the paramagnetic region.
� 2014 Elsevier B.V. All rights reserved.
Introduction
In reviewing the literature of cobalt coordination compoundswith nicotinamide and carboxylate ligands we found a triclinicmodification of [Co(nia)2(H2O)4](CH3COO)2�2H2O [1] and manyother mononuclear complexes with benzoate [2,3], 4-fluoro-,chloro- and bromobenzoate [4–7], 3-hydroxybenzoate [8],4-methylamino- [9] and 4-dimethylaminobenzoate [10], 3-amino-benzoate [11], 4-methoxybenzoate [12], 2- and 4-nitrobenzoate[13,14] and salicylate [15] ligands. There are also some complexeswith the same ligands (nicotinamide and water molecules) bonded
to Co2+ ion and different uncoordinated ions like chloride [16], suc-cinate [17] and phthalate [18] in the structure. These anions arehydrogen bonded to the complex. Furthermore, one dinuclearcomplex can also be found in the literature [19], where each Co(II)is coordinated by two nicotinamide molecules, one monodentatevalerate and two bridging valerate ligands and a bridging watermolecule. There are also two polymorphic modifications of thepolynuclear compound with Co(II) central ions and nicotinamide,water and succinate ligands [20,21].
In the literature we have also found some mononuclear com-plexes of Co2+ ion with isonicotinamide ligands and terephthalate[22], saccharinate [23], 3-hydroxybenzoate [24] and 4-form-ylbenzoate [25] ions. Polynuclear compounds of cobalt with isonic-otinamide and carboxylate ligands (phthalato [26] and benzoato

Table 1Experimental data for the X-ray diffraction studies on compounds 1 and 2.
1 2
Formula C16H30CoN4O12 C38H50Co2N10O14
Fw (g mol�1) 529.37 988.74Crystal size (mm) 0.34 � 0.08 � 0.08 0.23 � 0.10 � 0.05Crystal color Colorless RedCrystal system Monoclinic TriclinicSpace group C2/c P � 1a (Å) 16.0490(16) 8.9837(3)b (Å) 6.8528(6) 9.2077(3)c (Å) 21.267(2) 14.4506(5)a (�) 90 72.564(2)b (�) 90.748(5) 89.418(2)c (�) 90 78.555(2)
714 B. Dojer et al. / Journal of Molecular Structure 1076 (2014) 713–718
[3]) have been reported. Besides we also found a report of a dinu-clear copper compound, [Cu2(CH3COO)4(isn)4] [27].
The main aim of our interest was to find the structural and mag-netical differences for the compounds with the same central ion,acetate ligands and different pyridinecarboxamide ligands. Twotypes of cobalt(II) carboxylates with pyridinecarboxamide ligands,mononuclear and dinuclear, respectively, were investigated. Inorder to develop the structural and magnetic diversity of somenew Co(II) compounds with pyridine derivatives with a differentposition of the amide substituent, we extended our investigationsto nicotinamide and isonicotinamide ligands. The stabilisation ofthis kind of compounds increases with intra- and intermolecularhydrogen bonds and p–p interactions.
V (Å3) 2338.7(4) 1116.19(6)Z 4 1Calcd. density (g cm�3) 1.503 1.471F(000) 1108 514h range (�) 1.92–26.00 3.18–27.48No. of collected reflns 11,799 8877No. of independent reflns 2263 5051Rint 0.0572 0.0240No. of reflns observed 1811 4124No. parameters 157 308R[I > 2r(I)]a 0.0501 0.0341wR2 (all data)b 0.1339 0.0807Goof, Sc 1.056 1.044Maximum/minimum residual
electron density (e �3)+1.47/�0.70 +0.30/�0.37
a R =P
||Fo| � |Fc||/P
Fo|.b wR2 = {
P[w(Fo
2 � Fc2)2]/
P[w(Fo
2)2]}1/2.c S = {
P[w(Fo
2 � Fc2)2]/(n/p}1/2, where n is the number of reflections and p is the
total number of parameters refined.
Experimental procedure
General experimental procedures
All the starting compounds and solvents were used aspurchased. Infrared spectra were recorded on a SHIMADZUIRAffinty-1 FTIR spectrometer. Elemental analyses were carriedout on a Perkin-Elmer 2400 CHN analyzer at the University ofLjubljana. Magnetic properties of both modifications were studiedwith a Quantum Design MPMS-XL-5 SQUID magnetometer at theUniversity of Ljubljana (Institute of Mathematics, Physics andMechanics).
Synthesis of tetra-aqua-bis(nicotinamide-N)-cobalt(ii)diacetatedihydrate, [Co(nia)2(H2O)4](CH3COO)2�2H2O (1)
Solid cobalt(II) acetate tetrahydrate (0.249 g; 1.0 mmol) wasadded to a solution of nicotinamide (0.244 g; 2.0 mmol) in 20 mLof methanol. The solution was refluxed for about 2 h. After filtra-tion and slow cooling, the solution was left in the air until mostof the solvent was evaporated. The sample was put in a refrigeratorand left there for 3 days. A light-red crystalline product of 1 wasdried in a desiccator above KOH. Yield: 0.288 g (54.44%). Anal. Calc.for C16H30CoN4O12 (Mr = 529.37): C, 36.30; H, 5.71; N, 10.58.Found: C, 36.77; H, 5.67; N, 10.44.
Synthesis of di-acetato-bis[(acetato-O,O0)bis(isonicotinamide-N)cobalt(II)] dimethilformamide, [Co2(CH3COO)4(isn)4]�2C3H7NO (2)
We prepared cobalt(II) acetate dihydrate by the already knownprocedure [28].
The mentioned product (0.107 g; 0.5 mmol) was then dissolvedin 10 mL of methanol and added to the solution of isonicotinamidein dimethylformamide (10 mL). The solution was refluxed forabout 4 h. After filtration and slow cooling, it was necessary to fil-ter it once more because a white layer appeared. The next day thesample was put in the refrigerator and left there for a week. Redcrystalline product of 2 was dried in a desiccator above KOH. Yield:0.465 g (47.03%). Anal. Calc. for C38H50Co2N10O14 (Mr = 988.74):C, 46.16; H, 5.11; N, 14.16. Found: C, 46.02; H, 5.23; N, 14.34.
X-ray crystallography
The molecular structures of complexes 1 and 2 were deter-mined by single-crystal X-ray diffraction methods. Crystallo-graphic data and refinement details are given in Table 1.
X-ray crystallographic data for 1 were collected on a BrukerAPEX-II CCD diffractometer with graphite-monochromated MoKa radiation (k = 0.71072 Å) at 100 K. The structure was solvedby direct methods (SHELXS-97) [29] and refined by full-matrixleast-squares techniques against F2 (SHELXL-97) [29]. The
non-hydrogen atoms were refined with anisotropic displacementparameters, without any constraints. The H atoms of the pyridinering were put at the external bisectors of the C–C–C angles atC–H distances of 0.95 Å and a common isotropic displacementparameter was refined. The H atoms of the methyl group wererefined with common isotropic displacement parameters and ide-alized geometries with tetrahedral angles, enabling rotationaround the C–C bond, and C–H distances of 0.98 Å. The positionsof the H atoms of the water molecules and of the NH2 group weretaken from a difference Fourier map; the O–H and the N–H dis-tances were fixed to 0.84 Å and to 0.91 Å, respectively, and the Hatoms were refined without any constraints to the bond angleswith common isotropic displacement parameters for the H atomsbonded to the same atom.
X-ray crystallographic data for 2 were collected on a NoniusKappa CCD diffractometer with graphite-monochromated Mo Karadiation (k = 0.71072 Å) at 150 K. The structure was solved bydirect methods (SIR-92) [30] and refined by full-matrixleast-squares techniques against F2 (SHELXL-97) [29]. All of thenon-hydrogen atoms were refined anisotropically. All of the C–Hhydrogen atoms, except those bonded to the C17 of DMF molecule,were included in the model at geometrically calculated positionsand refined using a riding model. The hydrogen atoms bonded toamide nitrogen N3 and N4 or DMF carbon C17 were visible inthe last stages of the refinement process and were refined with aconstrained bond length (0.88 Å for N–H and 1.00 for C–H) andisotropic thermal parameters (1.2 times the thermal parameter ofthe attached nitrogen or carbon atom).
Magnetic measurements
Magnetic properties of both modifications were studiedbetween 2 K and 300 K in a magnetic field H = 1000 Oe and at a

B. Dojer et al. / Journal of Molecular Structure 1076 (2014) 713–718 715
constant temperature of 5 K between l0H = ±5 T with a QuantumDesign MPMS-XL-5 SQUID magnetometer.
Results and discussion
By changing the ligands (nicotinamide, isonicotinamide) wesynthesized and characterized two compounds, one polymorphicmodification and one new, in different solutions of cobalt acetatedi- and tetrahydrate.
Structure of tetra-aqua-bis(nicotinamide-N)-cobalt(ii)diacetatedihydrate, [Co(nia)2(H2O)4](CH3COO)2�2H2O (1)
The crystal structure analysis of 1 showed the compound to bethe monoclinic modification of tetra-aqua-bis(nicotinamide-N)-cobalt(II) diacetate dihydrate. The Co atom is situated on a
Fig. 1. Molecular structures of compound 1 (ellipsoids set to 50%).
Table 2Selected bond lengths (Å) and angles (�) for 1 and 2.
1 2
Co1–O1 2.085(2) 2.1970(13)Co1–O2 2.081(2) 2.1722(13)Co1–O3 2.0239(13)Co1–O4i 2.0195(13)Co1–N1 2.159(2) 2.1507(14)Co1–N2 2.1699(14)
N1–Co1–O1 87.12(9) 88.58(5)N1–Co1–O2 88.53(9) 85.78(5)N1–Co1–O3 87.69(6)N1–Co1–O4i 88.13(5)N1–Co1–N1ii 177.83(13)N1–Co1–N2 176.66(6)O1–Co1–O2 90.50(9) 59.96(5)O1–Co1–O1ii 90.02(12)O1–Co1–O2ii 179.46(9)O1–Co1–O3 87.01(5)O1–Co1–O4i 149.73(5)
Symmetry codes: (i) �x + 2, �y, �z; (ii) �x + 1, y, �z + 3/2.
crystallographic twofold rotation axis, all the other atoms lie ongeneral positions (Fig. 1).
In the triclinic modification [1] the Co atom lies on an inversioncenter; therefore, the ligands in trans position enclose with the Coatom angles of 180�. In the monoclinic modification (1) the angleN1–Co1–N10 is 177.83(13)�, and the angle N7� � �Co1� � �N70 is168.83(4)�, due to a slight rotation of the amide group towardsthe same side (obviously this angle is also 180� in the triclinic mod-ification) (Table 2).
The acetate ions are not coordinated to Co, but presumably havestrong hydrogen bonds to the aqua ligands. Taking the amide
Fig. 2. Part of the crystal structure of the compound 1, showing hydrogen bondsand p–p stacking interactions. Hydrogen atoms not involved in hydrogen bondinghave been omitted for clarity. [Symmetry codes: (i) x � 1/2, y � 1/2, z; (ii) x � 1/2,y + 1/2, z; (iii) �x + 3/2, �y + 1/2, �z + 1; (iv) �x + 3/2, �y + 3/2, �z + 1.]

Table 3Hydrogen bonding geometry for 1 and 2.
D–H� � �A d(D–H)/Å d(H� � �A)/Å d(D� � �A)/Å <(DHA)/� Symmetry transformation for acceptors
1O1–H11� � �O11 0.84 1.85 2.681(3) 169.3O1–H12 � � �O7 0.84 1.89 2.725(3) 173.5 x � 1/2, y � 1/2, zO2–H21 � � �O12 0.84 1.80 2.630(3) 172.0O2–H22� � �O7 0.84 1.91 2.751(3) 174.7 x � 1/2, y + 1/2, zN7–H71� � �O3 0.91 1.92 2.810(3) 164.9N7–H72� � �O11 0.91 2.17 3.061(3) 167.9O3–H31� � �O11 0.84 1.98 2.794(3) 164.7 �x + 3/2, �y + 1/2, �z + 1O3–H32� � �O12 0.84 2.01 2.791(3) 153.2 �x + 3/2, �y + 3/2, �z + 1
2N3–H20� � �O6 0.882(17) 2.006(18) 2.870(2) 166(2) x + 1, y, z + 1N3–H21� � �O7 0.866(17) 2.131(19) 2.930(2) 153(2)N4–H22� � �O1 0.862(17) 2.013(17) 2.865(2) 169(3) �x + 1, �y, �zN4–H23� � �O7 0.877(17) 2.146(18) 2.995(2) 163(2) x � 1, y, z � 1
Fig. 3. Molecular structures of compound 2 (ellipsoids set to 50%). [Symmetry code:(i) �x + 2, �y, �z.] Fig. 4. Part of the crystal structure of the compound 2, showing hydrogen bonds
and p–p stacking interactions. Hydrogen atoms not involved in hydrogen bondinghave been omitted for clarity. [Symmetry codes: (i) x + 1, y, z + 1; (ii) �x + 1, �y, �z.]
716 B. Dojer et al. / Journal of Molecular Structure 1076 (2014) 713–718
groups and the water molecules into account a three-dimensionalhydrogen bonded network is formed (Fig. 2 and Table 3). The ori-entation of the pyridine rings suggests the aromatic stacking alongthe b-axis with an inter-ring distance of 3.628 Å. The planes ofthese rings are inclined to each other with an angle of 5.80�.
Structure of di-acetato-bis[(acetato-O,O’)bis(isonicotinamide-N)cobalt(II)] dimethilformamide, [Co2(CH3COO)4(isn)4]�2C3H7NO (2)
The centrosymmetric binuclear complex, [Co2(CH3COO)4(isn)4]�2C3H7ON, is made up of two cobalt(II) central ions, four acetateand four isonicotinamide ligands and two dimethylformamide sol-vent molecules. Each cobalt(II) ion is trans-coordinated by twonitrogen atoms from two isonicotinamide ligands and four oxygenatoms from three acetate ligands, one of them is bidentate chelate
and two monodentate (Fig. 3). The coordination geometry aroundCo ion could be defined as lightly distorted trigonal bipyramid.The chelate bond lengths Co–O1 (2.1970(13) Å) and Co–O2(2.1722(13) Å are quite longer than the distances Co–O3(2.0239(13) Å) and Co–O4i (2.0195(13) Å) of unidentate bindingligand. In the isostructural copper complex[Cu2(O2CCH3)4(isn)4][27], non-bridging acetate units bind asymmetrically, one withthe bond distance 2.740(2) Å and the other with 2.020(2) Å, so thisacetate group is unidentate, rather than chelate. Cobalt coordi-nated acetate oxygen atoms define an equatorial plane to whichthe Co–N bonds of isonicotinamide ligands are almost perpendicu-lar. The double acetate bridge between the cobalt ions is notcoplanar with the CoO4 equatorial planes. The dihedral angle
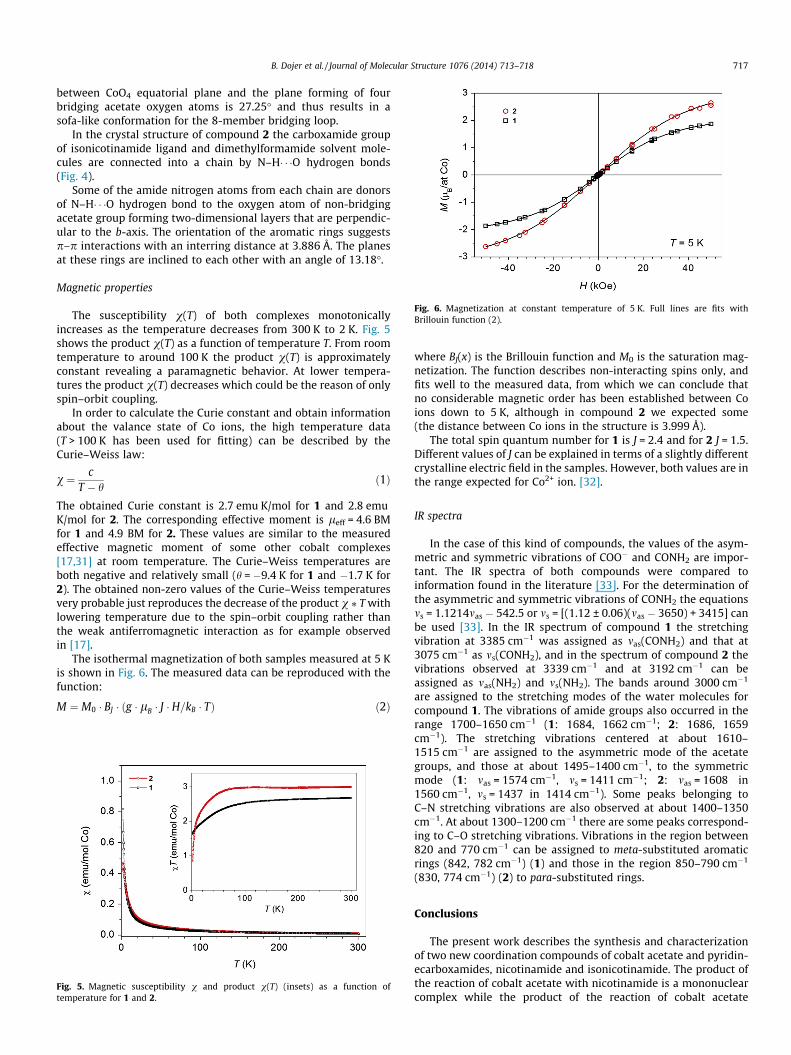
Fig. 6. Magnetization at constant temperature of 5 K. Full lines are fits withBrillouin function (2).
B. Dojer et al. / Journal of Molecular Structure 1076 (2014) 713–718 717
between CoO4 equatorial plane and the plane forming of fourbridging acetate oxygen atoms is 27.25� and thus results in asofa-like conformation for the 8-member bridging loop.
In the crystal structure of compound 2 the carboxamide groupof isonicotinamide ligand and dimethylformamide solvent mole-cules are connected into a chain by N–H� � �O hydrogen bonds(Fig. 4).
Some of the amide nitrogen atoms from each chain are donorsof N–H� � �O hydrogen bond to the oxygen atom of non-bridgingacetate group forming two-dimensional layers that are perpendic-ular to the b-axis. The orientation of the aromatic rings suggestsp–p interactions with an interring distance at 3.886 Å. The planesat these rings are inclined to each other with an angle of 13.18�.
Magnetic properties
The susceptibility v(T) of both complexes monotonicallyincreases as the temperature decreases from 300 K to 2 K. Fig. 5shows the product v(T) as a function of temperature T. From roomtemperature to around 100 K the product v(T) is approximatelyconstant revealing a paramagnetic behavior. At lower tempera-tures the product v(T) decreases which could be the reason of onlyspin–orbit coupling.
In order to calculate the Curie constant and obtain informationabout the valance state of Co ions, the high temperature data(T > 100 K has been used for fitting) can be described by theCurie–Weiss law:
v ¼ cT � h
ð1Þ
The obtained Curie constant is 2.7 emu K/mol for 1 and 2.8 emuK/mol for 2. The corresponding effective moment is leff = 4.6 BMfor 1 and 4.9 BM for 2. These values are similar to the measuredeffective magnetic moment of some other cobalt complexes[17,31] at room temperature. The Curie–Weiss temperatures areboth negative and relatively small (h = �9.4 K for 1 and �1.7 K for2). The obtained non-zero values of the Curie–Weiss temperaturesvery probable just reproduces the decrease of the product v ⁄ T withlowering temperature due to the spin–orbit coupling rather thanthe weak antiferromagnetic interaction as for example observedin [17].
The isothermal magnetization of both samples measured at 5 Kis shown in Fig. 6. The measured data can be reproduced with thefunction:
M ¼ M0 � BJ � ðg � lB � J � H=kB � TÞ ð2Þ
Fig. 5. Magnetic susceptibility v and product v(T) (insets) as a function oftemperature for 1 and 2.
where BJ(x) is the Brillouin function and M0 is the saturation mag-netization. The function describes non-interacting spins only, andfits well to the measured data, from which we can conclude thatno considerable magnetic order has been established between Coions down to 5 K, although in compound 2 we expected some(the distance between Co ions in the structure is 3.999 Å).
The total spin quantum number for 1 is J = 2.4 and for 2 J = 1.5.Different values of J can be explained in terms of a slightly differentcrystalline electric field in the samples. However, both values are inthe range expected for Co2+ ion. [32].
IR spectra
In the case of this kind of compounds, the values of the asym-metric and symmetric vibrations of COO� and CONH2 are impor-tant. The IR spectra of both compounds were compared toinformation found in the literature [33]. For the determination ofthe asymmetric and symmetric vibrations of CONH2 the equationsms = 1.1214mas � 542.5 or ms = [(1.12 ± 0.06)(mas � 3650) + 3415] canbe used [33]. In the IR spectrum of compound 1 the stretchingvibration at 3385 cm�1 was assigned as mas(CONH2) and that at3075 cm�1 as ms(CONH2), and in the spectrum of compound 2 thevibrations observed at 3339 cm�1 and at 3192 cm�1 can beassigned as mas(NH2) and ms(NH2). The bands around 3000 cm�1
are assigned to the stretching modes of the water molecules forcompound 1. The vibrations of amide groups also occurred in therange 1700–1650 cm�1 (1: 1684, 1662 cm�1; 2: 1686, 1659cm�1). The stretching vibrations centered at about 1610–1515 cm�1 are assigned to the asymmetric mode of the acetategroups, and those at about 1495–1400 cm�1, to the symmetricmode (1: mas = 1574 cm�1, ms = 1411 cm�1; 2: mas = 1608 in1560 cm�1, ms = 1437 in 1414 cm�1). Some peaks belonging toC–N stretching vibrations are also observed at about 1400–1350cm�1. At about 1300–1200 cm�1 there are some peaks correspond-ing to C–O stretching vibrations. Vibrations in the region between820 and 770 cm�1 can be assigned to meta-substituted aromaticrings (842, 782 cm�1) (1) and those in the region 850–790 cm�1
(830, 774 cm�1) (2) to para-substituted rings.
Conclusions
The present work describes the synthesis and characterizationof two new coordination compounds of cobalt acetate and pyridin-ecarboxamides, nicotinamide and isonicotinamide. The product ofthe reaction of cobalt acetate with nicotinamide is a mononuclearcomplex while the product of the reaction of cobalt acetate

718 B. Dojer et al. / Journal of Molecular Structure 1076 (2014) 713–718
dihydrate and isonicotinamide is a dinuclear complex. The stabili-sation of both compounds increases with hydrogen-bonding andp–p stacking interactions. Magnetic measurements reveal the 2+ionic state of cobalt and paramagnetic behavior. IR spectral studiesshow differences between the compounds, especially in CONH2
stretching region and COO� stretching vibrations.
Supplementary material
CCDC 995508 for 1 and 998372 for 2 contains the supplemen-tary crystallographic data. These data can be obtained free ofcharge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Acknowledgements
This work was supported by Grant P1-0175-103 from the Min-istry of Higher Education, Science and Technology of the Republicof Slovenia and the Slovenian Research Agency. We also thankMr. Paul McGuiness for language editing.
References
[1] M.I. Matsaberidze, A.S. Batsanov, R.G. Gerr, Yu.T. Struchkov, G.V. Tsintsadze,Koord. Khim. 11 (1985) 411.
[2] T. Hokelek, H. Necefoglu, Acta Crystallogr. C Sect. C Cryst. Struct. Commun. 55(1999) 545.
[3] J. Hudak, R. Boca, L. Dlhan, J. Kozisek, J. Moncol, Polyhedron 30 (2011) 1367.[4] O. Sahin, O. Buyukgungor, D.A. Kose, E.F. Ozturkkan, H. Necefoglu, Acta
Crystallogr. C 63 (2007) 243.[5] N. Caylak, T. Hokelek, H. Necefoglu, Acta Crystallogr. Sect. E: Struct. Rep. Online
63 (2007) 1341.[6] N. Caylak, T. Hokelek, F.E. Ozturkkan, H. Necefoglu, Acta Crystallogr. Sect. E:
Struct. Rep. Online 63 (2007) 1344.[7] T. Hokelek, N. Caylak, H. Necefoglu, Acta Crystallogr. Sect. E: Struct. Rep. Online
63 (2007) 1873.[8] T. Hokelek, H. Necefoglu, Acta Crystallogr. C 55 (1999) 1438.[9] H. Necefoglu, O. Aybirdi, B. Tercan, E. Ermis, T. Hokelek, Acta Crystallogr. Sect. E
Struc. Rep. Online 66 (2010) 448.
[10] T. Hokelek, H. Necefoglu, Acta Crystallogr. Sect. E: Struct. Rep. Online 63 (2007)1078.
[11] T. Hokelek, H. Necefoglu, Anal. Sci. 15 (1999) 1043.[12] T. Hokelek, H. Dal, B. Tercan, E. Tenlik, H. Necefoglu, Acta Crystallogr. Sect. E:
Struct. Rep. Online 66 (2010) 910.[13] J.G. Lin, L. Qiu, W. Cheng, S.N. Luo, K. Wang, Q.J. Meng, Inorg. Chem. Commun.
13 (2010) 855.[14] T. Hokelek, H. Necefoglu, Acta Crystallogr. Sect. C Cryst. Struct. Commun. 54
(1998) 1242.[15] O. Sahin, O. Buyukgungor, D.A. Kose, H. Necefoglu, Acta Crystallogr. Sect. C
Cryst. Struct. Commun. 64 (2008) 317.[16] A. Vegas, A. Perez-Salazar, F. Silio, Acta Crystallogr. Sect.B: Struct. Crystallogr.
Cryst. Chem. 37 (1981) 1916.[17] S. Demir, V.T. Yilmaz, J. Mrozinski, T. Lis, M. Holynska, Z. Naturforsch. 65b
(2010) 695.[18] A.S. Antsyshkina, G.G. Sadikov, T.V. Koksharova, I.S. Gritsenko, V.S. Sergienko,
Zh. Neorg. Khim. (Russ.) (Russ. J. Inorg. Chem.) 54 (2009) 1310.[19] G.G. Sadikov, A.S. Antsyshkina, T.V. Koksharova, I.S. Gritsenko, V.S. Sergienko,
Crystallogr. Rep. 52 (2007) 812.[20] T.A. Azizov, Kh.T. Sharipov, N.A. Parpiev, Z. Karimov, M. Askarov, A.B.
Khudoyarov, Russ. J. Inorg. Chem. 36 (1991) 1722.[21] S. Demir, V.T. Yilmaz, F. Yilmaz, O. Buyukgungor, J. Inorg. Organomet. Polym
Mater. 19 (2009) 342.[22] Guang-Gang Gao, Bo Liu, Chuan-Bi Li, Guang-Bo Che, Acta Crystallogr. Sect. E:
Struct. Rep. Online 62 (2006) m3357.[23] I. Ucar, B. Karabulut, H. Pasaoglu, O. Buyukgungor, A. Bulut, J. Mol. Struct. 787
(2006) 38.[24] I.G. Zaman, N.C. Delibas, H. Necefoglu, T. Hokelek, Acta Crystallogr. Sect. E:
Struct. Rep. Online 68 (2012) m249.[25] T. Hokelek, F. Yilmaz, B. Tercan, M. Sertcelik, H. Necefoglu, Acta Crystallogr.
Sect. E: Struct. Rep. Online 65 (2009) m1130.[26] D.K. Kumar, A. Das, P. Dastidar, J. Mol. Struct. 796 (2006) 139.[27] M. Perec, R. Baggio, Acta Crystallogr. Sect. E: Struct. Rep. Online 66 (2010)
m275.[28] C. Balarew, D. Stoilova, L. Demirev, Z. Anorg. Allg. Chem. 410 (1974) 75.[29] G.M. Sheldrick, Acta Crystallogr. A64 (2008) 112.[30] A. Altomare, M.C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi,
A.G.G. Moliterni, G. Polidori, R. Spagna, J. Appl. Cryst. 32 (1999) 115.[31] Z. Jaglicic, M. Zentkova, M. Mihalik, Z. Arnold, M. Drofenik, M. Kristl, B. Dojer,
M. Kasunic, A. Golobic, M. Jagodic, J. Phys.: Condens. Matter 24 (2012) 056002.[32] N.W. Ashcroft, N.D. Mermin, Solid State Physics, Saunders College Publishing,
USA, 1976. p. 657, 658.[33] L.J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall,
London, 1975.
Copyright © 2022 FDOKUMEN




















