
Identification of a Linear Epitope in Sortilin That Partakes in Pro-neurotrophin Binding
Upload
independentCategory
view
0download
0
SRNR
NA
LCN1
AnrsnusifeisctNrsTbdHcdnclpvb
1
C*4EAseFvtRslrd
Neuroscience 154 (2008) 978–993
0d
TATUS EPILEPTICUS INDUCES A TrkB TO p75 NEUROTROPHINECEPTOR SWITCH AND INCREASES BRAIN-DERIVEDEUROTROPHIC FACTOR INTERACTION WITH p75 NEUROTROPHIN
ECEPTOR: AN INITIAL EVENT IN NEURONAL INJURY INDUCTIONKh
Btsrtpppbcctas
araiBs1ctf(c(aftspFpctpo2lia
. UNSAIN, N. NUÑEZ,1 A. ANASTASÍAND D. H. MASCÓ*
aboratorio de Neurobiología, Centro de Biología Celular y Molecular,átedra de Biología Celular, Facultad de Ciencias Exactas, Físicas yaturales, Universidad Nacional de Córdoba, Av. Vélez Sarsfield611, X5016 GCA, Córdoba, Argentina
bstract—In neuronal cultures it has been demonstrated thateurotrophins can elicit neuronal death through the p75 neu-otrophic receptor (p75ntr) in the absence of concomitant Trkignaling. However, it was suggested that p75ntr induceseuronal death after status epilepticus (SE) in neuronal pop-lations that express relatively high quantities of tropomyo-in receptor kinase B (TrkB). Here, using Western blot andmmunohistochemistry analyses in the hippocampus, weound that 3-h SE caused a remarkable decrease in TrkBxpression and phosphorylation, and a significant increasen p75ntr. TrkB modification occurs before the overexpres-ion of the tumor suppressor protein p53, accompanies theell damage taking place in the dentate gyrus, and precedeshe CA1 neuronal injury as assessed by Fluoro-Jade B andissl staining. Co-immunoprecipitation of brain-derived neu-
otrophic factor (BDNF) or its immature form proBDNFhowed increased interaction with p75ntr after its binding torkB was reduced. Interestingly, proBDNF also increases itsinding with p75ntr after seizures that do not cause neuronaleath (animals injected with pilocarpine that fail to enter SE).owever, in those animals, TrkB protein levels remained un-hanged and its phosphorylation increased. Our results in-icate an intrinsic capacity of neurons in vivo to modify finaleurotrophin output by changing the proportion of their re-eptors’ expression and the receptors’ interaction with theirigands. These early events support the idea that neurotro-hins may be involved in the induction of neuronal death inivo under pathological conditions. © 2008 IBRO. Publishedy Elsevier Ltd. All rights reserved.
Present address: Natural Sciences Department, Hostos Communityollege, City University of New York.
Corresponding author. Tel: �54-351-4334141x403; fax: �54-351-332097.-mail address: [email protected] (D. H. Mascó).bbreviations: BDNF, brain-derived neurotrophic factor; BSA, bovineerum albumin; EDTA, ethylenediaminetetraacetic acid; EGTA, ethyl-ne glycol-bis(2-aminoethylether)-N,N,N=,N= tetraacetic acid; FJ-B,luoro-Jade B; IP, immunoprecipitation; LES, low-intensity electrocon-ulsive shock; PBS, phosphate buffer saline; p-TrkB, phosphorylatedropomyosin receptor kinase B; p75ntr, p75 neurotrophic receptor;IPA, radio-immunoprecipitation assay; RT, room temperature; SE,tatus epilepticus; TrkB, tropomyosin receptor kinase B; TrkB-fl, full-ength tropomyosin receptor kinase B; TrkB-IR, full-length tropomyosin
receptor kinase B immunoreactivity; TUNEL, terminal deoxynucleoti-yl transferase-mediated dUTP nick-end labeling.
306-4522/08$32.00�0.00 © 2008 IBRO. Published by Elsevier Ltd. All rights reseroi:10.1016/j.neuroscience.2008.04.038
978
ey words: seizures, pilocarpine, neurotrophin receptors,ippocampus, co-immunoprecipitation.
rain-derived neurotrophic factor (BDNF) is a member ofhe neurotrophin family that plays an important role in theurvival, differentiation and growth of many types of neu-ons (Bibel and Barde, 2000; Chao, 2003). BDNF binds thewo receptors, tropomyosin receptor kinase B (TrkB) and75 neurotrophic receptor (p75ntr). Studies of neurotro-hin receptor biology are challenging because of the com-lex physical and functional interactions that take placeetween BDNF and its receptors (Bibel et al., 1999; Zac-aro et al., 2001), and also because the biological out-omes vary, sometimes in diverse directions that includerophic, growth/survival, and neuritogenic differentiationnd, paradoxically, apoptotic cell death which is cell/tissue-pecific (Chao, 2003; Nykjaer et al., 2005; Lu et al., 2005).
BDNF effects on survival and differentiation have beenssociated with activation of the TrkB receptor, implicatingeceptor auto-phosphorylation after ligand binding (Kaplannd Stephens, 1994; Barbacid, 1994). On the other hand,
t has been demonstrated that the activation of p75ntr byDNF induces cell death only in the absence of TrkBignaling or when this is diminished (Davey and Davies,998; Friedman, 2000). The differential maturation of se-reted neurotrophins adds another level of complexity tohe system. All neurotrophins are synthesized as precursororms (proneurotrophins), which dimerize after translationKolbeck et al., 1994). Proneurotrophins, including the pre-ursor of BDNF (proBDNF), may be secreted from cellsHeymach and Shooter, 1995; Lee et al., 2001; Mowla etl., 2001; Chen et al., 2004) or cleaved intracellularly byurin or proconvertases to yield C-terminal mature neuro-rophin dimers. Until recently, only mature BDNF was con-idered as the biologically active form that elicited TrkB- or75ntr dependent signaling (Huang and Reichardt, 2001;riedman, 2000). However, recent studies indicate that therecursor form of BDNF, proBDNF, can be secreted byells and act via a dual receptor system of p75ntr and theype I transmembrane protein sortilin to mediate cell apo-tosis (Teng et al., 2005). This phenomenon was alsobserved for proNGF (Lee et al., 2001; Nykjaer et al.,004). Despite our knowledge of functionally distinct bio-
ogical effects mediated by the TrkB and p75ntr receptorsn vitro (Poo, 2001; Chao, 2003; Lu et al., 2005; Zweifel etl., 2005), it does seem to be the balance between both
eceptors on the cell surface modulates BDNF activity.ved.
Tpt
cnacrFleHTfiS
wtw
A
AahcltC1ofsmCkHoisjoscbsOps
W
AfmpeRipET
pwlcttippgtIsMio(TtprrEaIbag(eapl(e
I
AoOs4wswiewbnseSRTw�Tfpd0aws1
N. Unsain et al. / Neuroscience 154 (2008) 978–993 979
his possibility has only occasionally been explored underathological conditions leading to neuronal death (Costan-ini et al., 2006).
Prolonged seizures induced by pilocarpine lead to aondition called status epilepticus (SE), and result in bothecrotic and apoptotic cell death (Fujikawa, 1996; Covolannd Mello, 2000). At the same time, SE induces an in-rease in BDNF protein expression in the same brainegions (Schmidt-Kastner et al., 1996; Rudge et al., 1998).or many years, this rise in the neurotrophin was postu-
ated as playing a protective role against seizure-mediatedxcitotoxicity (Gall, 1993; Schmidt-Kastner et al., 1996).owever, the finding that p75ntr is upregulated after SE inUNEL-positive neurons suggests a potential apoptotic
unction of BDNF after seizures (Roux et al., 1999). Thenteraction of BDNF/proBDNF with TrkB and p75ntr afterE has not yet been determined.
Therefore in the present study we aimed to determinehether pilocarpine-induced SE shifts the TrkB/p75ntr ra-
io and how this modification correlates spatiotemporallyith neuronal death.
EXPERIMENTAL PROCEDURES
nimals and induction of SE
dult male Wistar rats (Instituto Ferreyra, Córdoba, Argentina)ged 2–2.5 months and weighing 250–320 g were used andoused under environmentally controlled conditions. Animals re-eived water and food ad libitum, and were maintained in a 12-hight/dark cycle. The experimental protocol for this study followedhe guidelines of the USA National Research Council Guide for theare and Use of Laboratory Animals (National Research Council,996) and was approved by the Animal Care and Use Committeef the National University of Córdoba. All experiments were per-ormed in order to minimize the number of animals used and theiruffering. SE was induced by the administration of scopolamineethyl-bromide (1 mg/kg, i.p.; ICN Biomedical Inc., Costa Mesa,A, USA) and 30 min later of pilocarpine hydrochloride (380 mg/g, i.p.; Sigma, St. Louis, MO, USA). Diazepam (10 mg/kg, i.p.;offmann-La Roche Ltd., Switzerland) was injected 3 h after thenset of SE to stop seizure activity. Control animals were treated
dentically to the experimental group, except that they receivedaline instead of pilocarpine. Following the initial pilocarpine in-ection, the animals were closely monitored to determine the onsetf SE. The validity of relying on behavioral monitoring to assesseizure activity has been demonstrated previously by studiesorrelating electrophysiological measures with observed seizureehaviors (Tremblay et al., 1984). By a close observation ofeizure behavior we were able to identify two clear-cut groups.ne of them was named “R2” and the other “SE.” Behavioralatterns of each group are described in detail in the Resultsection.
estern blot analyses
nimals were killed by decapitation at the indicated time pointsollowing diazepam administration in SE-induced animals or im-ediately after diazepam in saline-treated controls. The hip-ocampus was immediately removed from the skull and homog-nized in radio-immunoprecipitation assay (RIPA) buffer or inIPA-modified buffer containing protease and phosphatase inhib-
tors (phenylmethyl sulfonyl fluoride 100 �g/ml; leupeptin 5 �g/ml;epstatin 10 �M; aprotinin 2 �g/ml; sodium orthovanadate 1 mM;GTA 5 mM; EDTA 5 mM; NaF 50 mM; phenanthroline 1 mM).
he results obtained in the quantification of proteins from hip- wocampal homogenates by Western blot were indistinguishablehen using either RIPA or RIPA-modified buffer (described be-
ow). RIPA-modified buffer was always used for co-immunopre-ipitation. Homogenates were cleared by centrifugation at 500�gwice for 7 min and protein concentration was determined usinghe Bradford protocol (Bradford, 1976); samples were then boiledn gel-loading buffer and separated using sodium dodecyl sulfate–olyacrylamide gel electrophoresis (SDS-PAGE, 10% for TrkB,75ntr and p53 and 15% for BDNF and proBDNF). Forty micro-rams of total proteins were loaded in each line. Proteins wereransferred to nitrocellulose membranes (Bio-Rad Laboratoriesnc., PA, USA) and blocked with 5% non-fat milk in Tris-bufferedaline with 0.05% Tween, for 3 h at room temperature (RT).embranes were then incubated with the primary antibody of
nterest. Rabbit polyclonal antibodies against TrkB (sc:8316 rec-gnizing all isoforms and sc:012 recognizing full length), BDNFsc:546 recognizing pro- and mature-BDNF), p75ntr (sc:8317),rkC (sc:14025) and p53 (sc:6243) were from Santa Cruz Bio-
echnology, CA, USA. An alternative antibody against BDNF androBDNF (ANT-010, Alomone Laboratories Ltd., Jerusalem, Is-ael) was also used. A rabbit monoclonal antibody that specificallyecognizes TrkB phosphorylated in Y817 was a kind gift frompitomics, Inc., CA, USA, and was used to detect phospho-TrkB;nd a rabbit polyclonal antibody �-human NTR3 (Alpha Diagnosticnternational, SA, TX, USA) was used to detect sortilin. Mem-ranes were then incubated with appropriate biotinylated second-ry antibodies, followed by incubation with streptavidin-conju-ated peroxidase and enhanced chemiluminescence detectionECL), and then exposed to Kodak X-OMAT films (Kodak, Roch-ster, NY, USA). Membranes were re-probed with a monoclonalntibody against �-tubulin (Santa Cruz Biotechnology) to controlrotein loading. Images were scanned and gel bands were ana-
yzed using gel-scanning integrated optical density softwareScion Imaging 4.0.2., NIH, USA). Values for treatments werexpressed as a percentage of control animals.
mmunohistochemistry
nimals were anesthetized at the indicated time points by injectionf chloral hydrate (16%, 400 mg/kg, i.p.; MTC Pharmaceuticals,ntario, Canada), and perfused transcardially with physiologicalolution (0.8% NaCl, 0.4% glucose and 0.8% sucrose) followed by% paraformaldehyde in borate buffer. After perfusion, brainsere left in situ for 24 h, then removed and cryoprotected in 30%ucrose at 4 °C for 3 day before sectioning. When frozen, theyere sectioned with a microtome and 40 �m-sections were stored
n cryoprotectant solution at �20 °C (30% sucrose and 30%thylene glycol in phosphate buffer saline, PBS) and assayedithin 2 weeks of sectioning. Free-floating sections were incu-ated for 1 h at RT in 0.3% hydrogen peroxide to block endoge-ous peroxidase, washed and blocked for 2 h at RT in 5% bovineerum albumin (BSA). Afterward, sections were incubated withither the polyclonal antibody �-TrkB-full-length (sc:012 1:100,anta Cruz Biotechnology) or �-p53 (OP03L 1:50, Oncogeneesearch Products, MA, USA), diluted in PBS, 1% BSA and 0.3%riton X-100 for a period of 2 day at 4 °C, followed by threeashes in PBS 0.01 M for 5 min and incubated with biotinylated-rabbit secondary antibody (1:100 in PBS, 1% BSA and 1%riton X-100) for 60 min at RT. After three washes in PBS 0.01 M
or 5 min, sections were incubated with streptavidin-conjugatederoxidase for 90 min at RT, and finally exposed to a 3–3=-iaminobenzidine (Sigma) solution containing 0.03% NH4Cl,.0075% Cl2Ni, 0.0095% Cl2Co, 0.0375% 3–3=-diaminobenzidinend 0.6 �l/ml H2O2. After 10 min incubation, sections wereashed four times in PBS at RT, mounted in alcohol-gelatinolution on gelatinized slides, dried overnight, cleared in xylene forh and coverslipped with Canadian balsam.
For the analyses of TrkB-immunoreactivity labeled sections
ere measured in four different sections of the dorsal hippocam-
pffCIisspt
F
Tap2tdarcafiepnd
C
Bod0i
Idl
TrU
C
Aw7mwpt4cnLwTRl(8iubtiw
Lt
LpUrnmfs(S
S
RPglhetic
Sl
IiptSacz3pTetfUad(oTivdsapr1fads
N. Unsain et al. / Neuroscience 154 (2008) 978–993980
us. Images were captured digitally. Four square fields each fromour different sections per animal were analyzed for each of theollowing areas: hilus, granule cell layer of the dentate gyrus andA1. Square fields are equivalent to those shown in Figs. 2 and 3.
n the hilus, measurements were done with the number of TrkB-mmunoreactive cells. For the other regions, a densitometry mea-urement was performed in delineated areas containing exclu-ively the granule cell layer of the dentate gyrus or the stratumyramidale of CA1. Values of controls were set at 100%, andreatment values are expressed as a percentage of controls.
luoro Jade B staining
he number of dying neurons after pilocarpine-induced SE wasssessed by labeling with Fluoro-Jade B (FJ-B) according to theublished protocol (Schmued et al., 1997; Schmued and Hopkins,000). Labeled neurons were counted in four different sections ofhe dorsal hippocampus. Epifluorescent images were capturedigitally. Four square fields each from four different sections pernimal were analyzed for each of the following areas: hilus (neu-ons within the polymorphic layer of the dentate gyrus), granuleell layer of the dentate gyrus, and CA1. Square fields are equiv-lent to those shown in Figs. 2 and 3. Total neurons counted pereld were normalized taking into account the size of the areavaluated in a particular image for a given field. The quantificationerformed did not aim to estimate the total number of injuredeurons in a given brain area, but to quantitatively compare theegree of injury among different treatments and brain areas.
resyl Violet staining
riefly, 40 �m-sections were mounted in alcohol–gelatin solutionn gelatinized slides, dried overnight at 37 °C and hydrated toistilled water. Slides were then incubated in Cresyl Violet acetate.5% for 2 min, differentiated in 70% alcohol, dehydrated, cleared
n xylene and coverslipped.
n situ DNA nick-end labeling (terminaleoxynucleotidyl transferase mediated nick end
abeling (TUNEL) staining)
UNEL staining was carried out using the DeadEnd™ Fluoromet-ic TUNEL System (G3250, Promega Corporation, Madison, MI,SA) following the protocol provided by the manufacturer.
o-immunoprecipitation
t the indicated time points after SE termination, the hippocampusas homogenized with RIPA-modified buffer (50 mM Tris–HCl, pH.5, 150 mM NaCl, 10 mM EDTA and 1% Triton X-100) supple-ented with protease and phosphatase inhibitors. Homogenatesere centrifuged at 14,000�g for 10 min and 500 �g of totalrotein from the supernatants was pre-cleared with 10 �l of Pro-ein A/G PLUS-agarose (Santa Cruz Biotechnology) for 3 h at
°C. After centrifugation, the pre-cleared supernatants were in-ubated with either �-BDNF (sc:546, 2 �g, Santa Cruz Biotech-ology) or �-proBDNF (ANT-006, 2 �g Alomone Laboratoriestd.) at 4 °C for 13 h. Then 12.5 �l Protein A/G PLUS-Agaroseas added, pre-blocked in 1% BSA and incubated at 4 °C for 5 h.he immunoprecipitates were washed four times with ice-coldIPA-modified buffer, eluted with SDS sample buffer, and ana-
yzed by Western blot. Membranes where then probed with �-TrkBsc:8316 or sc:012, Santa Cruz Biotechnology) or �-p75ntr (sc:317, Santa Cruz Biotechnology). Given that the primary antibod-
es used were made in rabbit, and a secondary �-rabbit-IgG wassed, we first incubated the membranes with the secondary anti-ody alone to control for equal protein loading between wells (dueo the presence of �-BDNF IgG in the membranes) and to betternterpret the positive bands when �-TrkB and �-p75ntr antibodies
ere used. iow-intensity electroconvulsive shock (LES)reatment
ES were administered via corneal electrodes (200 pulses/s;ulse width: 0.1 ms; shock duration: 0.2 s; 40 mA) using an ECTnit 7801 (Ugo Basile, Comerio, Italy). Control (sham) animals
eceived the same handling and contact with the electrodes, buto current was passed. Animals were monitored to ensure thatinimal limbic motor seizures (clonic movements of the face and
orelimbs) lasting 5–10 s occurred after each LES. Each daily LESession consisted of three LES seizures, given at 30 min intervalsi.e. at 0, 30 and 60 min). The whole LES treatment lasted 7 days.E was induced 24 h after the last LES.
tatistical analysis
esults are expressed as percentage of control (means�SEM).rotein levels were quantified in a minimum of three animals perroup. A one-way ANOVA was used to compare relative protein
evels between groups. The ANOVA was followed by Tukey postoc comparisons, with P�0.05 considered significant. When nec-ssary, the Kruskal-Wallis test was used for non-normal distribu-ions. In the quantification of Fluoro Jade B positive cells and TrkBmmunoreactivity, an ANOVA test with a nested model was used,onsidering each slice of the same animal as a pseudo-replicate.
RESULTS
E decreases TrkB and increase p75ntr proteinevels
n order to make inferences on the possible roles of BDNFn vivo after CNS injury, it is necessary to determine theossible modifications of the whole BDNF signaling sys-em (TrkB, p75ntr, sortilin, mature BDNF and proBDNF).ystemic administration of pilocarpine leads to SE, char-cterized by intense seizures of a sustained nature, typi-ally starting with violent, generalized clonic–tonic sei-ures. Animals were allowed to remain in this condition forh, exhibiting continuous myoclonus of head and fore-
aws and at least four generalized clonic–tonic seizures.o determine if SE modifies BDNF and its receptor levelxpression, groups of animals were killed immediately af-er 3-h SE (0 h time) or 12, 24, and 72 h later, and proteinsrom the hippocampus were analyzed by Western blot.sing an antibody that recognizes the full-length (TrkB-fl)nd truncated variants of TrkB (TrkB-total), a remarkableecrease in TrkB-fl was observed 12, 24 and 72 h after SEFig. 1a and 1c, 63.3�3.1%, 55.1�3.1% and 43.4�5.6%f control, respectively). However, truncated variants ofrkB did not undergo any significant modifications, indicat-
ng that SE specifically downregulates the full length spliceariant under this condition (Fig. 1a). In contrast, SE in-uced a sustained increase in p75ntr receptor that reachedtatistical significance by 72 h (Fig. 1b and 1c). Moreover,n increase in mature BDNF was observed at all timeoints determined, while the increase in proBDNF onlyeached statistical significance 72 h after SE (Fig. 1d ande). Immunoblotting of BDNF and proBDNF was per-ormed with similar results using two commercially-avail-ble antibodies, as specified in the Experimental Proce-ures section. These results show that there is a receptorwitch after SE, as TrkB began to decrease soon after the
nsult, and p75ntr increased later.

Fd(SmA
N. Unsain et al. / Neuroscience 154 (2008) 978–993 981
ig. 1. SE decreases TrkB-fl protein and increases p75ntr, mature BDNF and proBDNF in the hippocampus. (a, c) TrkB-fl was significantlyownregulated by SE as early as 12 h after seizures and continued to 72 h. On the other hand, truncated variants were not significantly modified.b, c) p75ntr Protein was slightly increased at 12 and 24 h, and raised significantly 72 h after SE. (d, e) Mature BDNF increased from 0 to 72 h afterE, while pro-BDNF was significantly different from controls 72 h after SE. (f) SE also decreased the levels of p-TrkB at 0 and 24 h without inducingodifications in the levels of the co-receptor sortilin. (g) Furthermore, SE did not modify the levels of TrkC, P�0.4273. Means�SEM are indicated.
sterisks indicate significant differences compared with control animals, P�0.05.
vfwpplhser
raaabcal
bar
T
GesPcoeTttecoae
plawhTsst1pwmrtts
aaptdaVdtpo
sMrTstlsnttFtndr
a(baTdpcm
cpcpNdbCucgodhNuF
p
N. Unsain et al. / Neuroscience 154 (2008) 978–993982
The TrkB-fl receptor is phosphorylated (and thus acti-ated) after ligand binding. Then, to further assess theunctional significance of TrkB-fl protein decrease after SEe sought to determine the levels of phosphorylated tro-omyosin receptor kinase B (p-TrkB) in the rat hippocam-us after SE. Interestingly, we found a decrease in the
evels of p-TrkB that began to be significant after 3-h SE (0time point), and continued for 24 h (Fig. 1f). This finding
hows that the decline in BDNF-TrkB signaling takes placearlier than the decrease in TrkB-fl protein (0 h vs. 12 h,espectively).
Sortilin1 (95 kDa) is a member of the Vps10p-domaineceptors family and has been described as a co-receptornd molecular switch governing the p75ntr-mediated pro-poptotic signal induced by pro-neurotrophins (Nykjaer etl., 2004; Teng et al., 2005). Then, as an important mem-er of proBDNF signaling, we sought to evaluate possiblehanges of sortilin protein levels in the rat hippocampusfter SE. We found no significant changes in sortilin protein
evels after SE (Fig. 1f).Finally, we asked whether the levels of another mem-
er of the Trk receptor family, TrkC, were also modifiedfter SE. We found that SE did not modify the levels of thiseceptor (P�0.4273, Fig. 1g).
rkB modifications and cell damage after SE
iven that previous works indicate that at 24 h, but notarlier, cell loss (i.e. reduction in cell number) begins to beignificant (Friedman et al., 1994, 2003; Friedman, 1998;oirier et al., 2000; Wall et al., 2000; Roch et al., 2002) aentral concern was to know if the decrease in TrkB proteinbserved was a simple consequence of the loss of TrkB-xpressing neurons or an authentic downregulation ofrkB protein. To answer this question, and to determine
he localization and extent of SE-induced TrkB-fl modifica-ions within the hippocampus and to corroborate the West-rn blot studies, we examined full-length tropomyosin re-eptor kinase B immunoreactivity (TrkB-IR) in immunoper-xidase-stained sections from animals killed at 0, 12, 24nd 72 h after SE. In addition, consecutive sections werexamined for cell damage using FJ-B and Nissl staining.
The staining pattern obtained with omission of therimary antibody resulted in a complete absence of TrkB
abeling. We used an antibody that recognizes only TrkB-flnd not its truncated forms. In control animals, TrkB-IRas distinguished in the CA1, CA2 and CA3 regions of theippocampus and to a lesser extent in the dentate gyrus.rkB-IR was prominent in pyramidal cell bodies and mild inhort, smooth processes extending from somata to thetratum radiatum. The staining pattern was the same ashat obtained by others (Fryer et al., 1996; Drake et al.,999). A mild decrease in TrkB-IR occurred in all hip-ocampal areas at 0 h following the excitotoxic insult, andas more pronounced in dentate gyrus (Figs. 2 and 3). Aoderate increase was evident in the stratum oriens and
adiatum but not confirmed by the quantification, becausehe densitometry analyses were performed exclusively inhe stratum pyramidale. A substantial reduction was ob-
erved from 12 h after SE onwards in CA1 and CA3 and an 2lmost complete loss of TrkB-IR at 72 h (Fig. 3). In allnimals developing SE the prominent decrease in hip-ocampal TrkB-IR was evident along the entire septo-emporal axis. Interestingly, in the same sections, TrkB-IRid not change in the CA2 area at any time tested, nor inpical dendrites of the frontal cortex ascending from layer
to layer I, indicative of the regional specificity of theecrease obtained. Taken together, these results indicatehat the decrease shown by Western blot of total hip-ocampus homogenates is indeed composed of a mixturef region-specific changes.
Although neuronal injury after SE has been extensivelytudied (Liu et al., 1994; Fujikawa, 1996; Covolan andello, 2000), it was important to further correlate time and
egional patterns of cell damage with the downregulation ofrkB under the same experimental conditions and in theame animals used for immunohistochemical studies. Forhat purpose, immediate consecutive sections were ana-yzed by FJ-B staining to evaluate neuronal injury. Aftereizures, an important and similar number of FJ-B-positiveeurons were found in the hippocampal hilus and fewer inhe granule cell layer from SE termination (“0 h”) throughhe last time point observed (72 h, Fig. 2). The increase inJ-B stain in dentate gyrus micrographs at 24 and 72 h is
he result of the increase in brightness of hilar neurons andeurons of the CA3 that lies within the blades of theentate gyrus. However, the actual number of hilar neu-ons stained remained unchanged.
On the other hand, the few FJ-B-positive cells in CA1t 0 and 12 h after SE were restricted to the stratum oriensFig. 3, second and third row). However, at 24 h the num-er of stained neurons in the pyramidal CA1 area showedremarkable increase, reaching a peak by 72 h (Fig. 3).
his shows that a local decrease in TrkB accompanies cellamage taking place in the dentate gyrus (Fig. 2), butrecedes that occurring in the CA1 region (Fig. 3). Noellular damage was observed at any time in control ani-als injected with saline.
In order to find out if cell loss or damage was a majorontributing factor to changes observed in TrkB-IR, weerformed a Nissl stain as another way to assess the timeourse of neuronal damage or loss in the different hip-ocampal areas. As can be observed in Figs. 2 and 3, thisissl stain yielded similar results to that of FJ-B. In theentate hilus, the loss of Nissl staining became significanty 0 h, while in the CA1 area it was evident by 24 h.hanges in Nissl staining were not significant in the gran-le cell layer of the dentate gyrus. Hence, TrkB-IR de-rease in CA1, observed from 12 h, took place before anyross morphological alteration or FJ-B staining (Fig. 3). Inur setting of SE (3 h of SE before the administration ofiazepam i.p. 10 mg/kg), the loss of neurons in CA1 andilus was outstanding, as can be observed in FJ-B andissl stains of 72 h sections and as reported elsewheresing a similar protocol to induce SE (Fujikawa, 1996;abene et al., 2004; Rangel et al., 2005).
The tumor suppressor protein p53 has been involved in75ntr signaling (Aloyz et al., 1998; Kaplan and Miller,
000; Wen et al., 2004; Linggi et al., 2005) and in different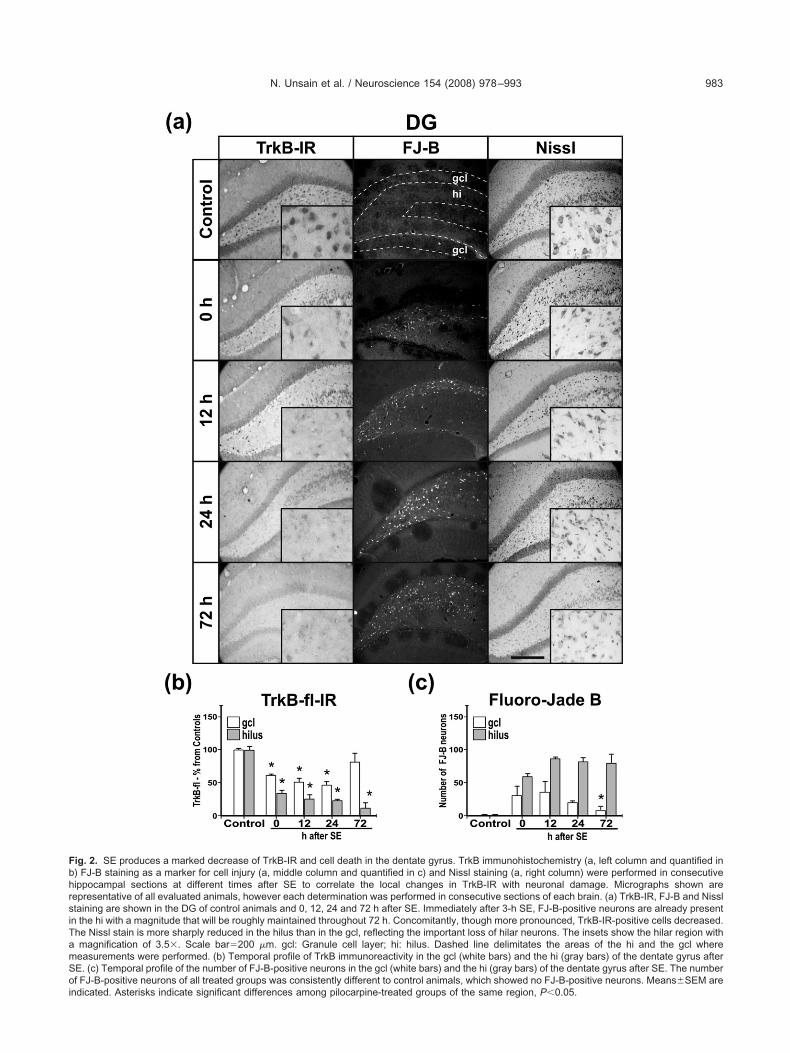
FbhrsiTamSoi
N. Unsain et al. / Neuroscience 154 (2008) 978–993 983
ig. 2. SE produces a marked decrease of TrkB-IR and cell death in the dentate gyrus. TrkB immunohistochemistry (a, left column and quantified in) FJ-B staining as a marker for cell injury (a, middle column and quantified in c) and Nissl staining (a, right column) were performed in consecutiveippocampal sections at different times after SE to correlate the local changes in TrkB-IR with neuronal damage. Micrographs shown areepresentative of all evaluated animals, however each determination was performed in consecutive sections of each brain. (a) TrkB-IR, FJ-B and Nissltaining are shown in the DG of control animals and 0, 12, 24 and 72 h after SE. Immediately after 3-h SE, FJ-B-positive neurons are already presentn the hi with a magnitude that will be roughly maintained throughout 72 h. Concomitantly, though more pronounced, TrkB-IR-positive cells decreased.he Nissl stain is more sharply reduced in the hilus than in the gcl, reflecting the important loss of hilar neurons. The insets show the hilar region withmagnification of 3.5�. Scale bar�200 �m. gcl: Granule cell layer; hi: hilus. Dashed line delimitates the areas of the hi and the gcl whereeasurements were performed. (b) Temporal profile of TrkB immunoreactivity in the gcl (white bars) and the hi (gray bars) of the dentate gyrus afterE. (c) Temporal profile of the number of FJ-B-positive neurons in the gcl (white bars) and the hi (gray bars) of the dentate gyrus after SE. The numberf FJ-B-positive neurons of all treated groups was consistently different to control animals, which showed no FJ-B-positive neurons. Means�SEM are
ndicated. Asterisks indicate significant differences among pilocarpine-treated groups of the same region, P�0.05.

FcabSpMFwt
N. Unsain et al. / Neuroscience 154 (2008) 978–993984
ig. 3. SE produces a marked decrease of TrkB-IR previous to cell death in the CA1. (a) TrkB-IR, FJ-B and Nissl staining is shown in the CA1 ofontrol animals and 0, 12, 24 and 72 h after SE. Immediately after 3-h SE, there is a sustained reduction in TrkB-IR. However, this modification is notccompanied at early times (0 and 12 h) by FJ-B-positive neurons neither by a Nissl staining showing gross morphological alterations. On the contrary,y 24 and 72 h after SE there are an important number of FJ-B neurons and the corresponding morphological alterations evidenced by Nissl staining.cale bars�200 �m (white) and 50 �m (black). Arrows indicate the sp of CA1. Dashed line delimitate the area of the CA1 where measurements wereerformed. So: stratum oriens; sp: stratum pyramidale; sr: stratum radiatum. (b) Temporal profile of TrkB immunoreactivity in the CA1-sp after SE.eans�SEM are indicated. Asterisks denote significant differences compared with control animals, P�0.05. (c) Temporal profile of the number ofJ-B-positive neurons in the CA1-sp after SE. The number of FJ-B-positive neurons of all treated groups was consistently different to control animals,hich showed no FJ-B-positive neurons. Means�SEM are indicated. Asterisks indicate significant differences between pilocarpine-treated groups of
he same region, P�0.05.

m1amqpfF2ri(MaiTTpwi
St
IderOc1p2m
pdd1iwpihptdb
bwlwssabww
rtTaaci
F7cc t to apop( L-positive
N. Unsain et al. / Neuroscience 154 (2008) 978–993 985
odels of SE-induced neuronal death (Hughes et al.,996; Morrison et al., 1996; Culmsee et al., 2001; Tan etl., 2002a). In light of this evidence, and to further deter-ine if the TrkB decrease occurs before, or as a conse-uence of, apoptosis-induced neuronal loss in the hip-ocampus, we analyzed the temporal expression of p53ollowing pilocarpine-induced SE. As can be observed inig. 4a, SE induced a drastic increase in p53 expression at4 and 72 h (191.8�16.4% and 260.0�26.4% of controls,espectively). Interestingly, at 12 h after SE, when the dropn TrkB was evident, the p53 expression was not modified111.8�10 of controls, compare Fig. 1a with Fig. 4a).oreover, immunohistochemical analysis showed that p53nd TUNEL staining immunoreactive cells were localized
n the same areas in which TrkB was diminished (Fig. 4b).aken together these results show that the decrease inrkB-fl protein in whole hippocampus homogenates takeslace before the increase of p53, the upregulation of whichas correlated with TUNEL staining and has been involved
n the induction of apoptosis in neurons.
E modifies BDNF and proBDNF interactions withheir receptors
t has been suggested that p75ntr mediates neuronaleath after SE (Roux et al., 1999; Troy et al., 2002; Volosint al., 2006), but the interaction of the receptor with neu-otrophins in such conditions has never been addressed.wing to the abundant antecedents showing that SE in-
reases BDNF protein expression (Schmidt-Kastner et al.,996; Rudge et al., 1998; Nawa et al., 2004) and itsarticular activity-dependent secretion (Lessmann et al.,003), we considered BDNF and as proBDNF to be the
ig. 4. SE increases hippocampal p53 protein levels following TrkB d2 h. Interestingly, at 12 h after SE (when the drop in TrkB was evidenhanges in the hippocampus indicate that major modifications takeorrelate p53 changes with DNA cleavage, an indicator of commitmennot shown). Arrows show p53-positive pyramidal cells (B) and TUNE
ost suitable candidates to bind, and perhaps to activate w
75ntr in the hippocampus after SE. As it has already beenemonstrated in vitro that p75ntr activation induces celleath in the absence of TrkB signaling (Davey and Davies,998; Friedman, 2000), we sought to address whether the
nteraction of BDNF with TrkB decreases as its interactionith p75ntr rises while SE-induced neuronal death is inrogress. For that purpose, groups of animals were killed
mmediately after 3-h SE (0 h time), 12 or 72 h later, andippocampal protein samples were subjected to immuno-recipitation (IP), using an antibody that recognizes ma-ure and/or pro-BDNF. Immunoprecipitated proteins pulledown together with BDNF were then analyzed by Westernlot using antibodies against TrkB or p75ntr.
For internal control, we determined the presence of tu-ulin in the immunoprecipitates as an indicator of improperashes and the consequent contamination of the IPs with
oading sample. As can be seen in Fig. 5a (top blot), tubulinas not present in the IPs, but was found in the loadingample and supernatant of the IPs. The presence of IPamples in the blots was confirmed by detecting the primaryntibody used in the IP assay (rabbit IgGs �-BDNF, Fig. 5a,ottom blot). No positive bands for TrkB or p75ntr were foundhen purified rabbit IgGs were used as primary antibodies orith omission of primary antibodies from the assay.
When the IP was performed with an antibody thatecognizes both mature and proBDNF, SE induced a no-iceable decrease (�25–60%) in co-immunoprecipitatedrkB at 0 and 12 h, reaching statistical significance at 72 hfter SE (Fig. 5b). Analyses were conducted taking intoccount three independent experiments (Fig. 5c). We thenonfirmed the presence and modifications of TrkB-fl by
mmunoblotting IP samples from another group of animals
lation. (a) SE induced a drastic increase in p53 expression at 24 andpression was not modified. (b) Immunohistochemical analyses of p53he CA1 subfield (A, B). TUNEL staining was conducted in order totosis. TUNEL-positive nuclei were evident in CA1 (C and D) and CA3
nuclei (D). Scale bars�500 �m (black) and 100 �m (white).
ownregut) p53 ex
place in t
ith an antibody that recognizes TrkB-fl as well as trun-

cdBTmBaccia
sIpstofpc
F4sltsssw
N. Unsain et al. / Neuroscience 154 (2008) 978–993986
ated TrkB. Using this antibody it was not possible toetect truncated TrkB proteins co-immunoprecipitated withDNF (data not shown). Neither antibody used to detectrkB cross-reacts with TrkA or TrkC, as determined by theanufacturer and our own analyses. The decreasedDNF-TrkB-fl interaction found in these experimentsgrees with our finding that p-TrkB is significantly de-reased at 0 and 24 h after SE. In contrast to the de-reased interaction with TrkB, SE induced a remarkablencrease in p75ntr co-immunoprecipitated with BDNF 12 h
ig. 5. Changes in the interaction of mature and pro-BDNF with their) indicating no contamination from total homogenates (lysates), whupernatants (lane 5). The presence of �-BDNF-IgGs in the IP was c
anes 2, 3 and 4). (b) TrkB-fl, co-immunoprecipitated along with totalo diminish 12 h after SE and was very low at 72 h. (c) Percentage oignificant at 72 h. (d) Levels of p75ntr co-immunoprecipitated with toignificantly up-regulated at 0 and 72 h. (e) When proBDNF was speignificantly increased at all time points determined (0, 12 and 72 h). Mith control animals, P�0.05.
fter SE and then returned to control values (Fig. 5d). t
We next determined the interaction of p75ntr exclu-ively with proBDNF. We subjected the animal samples toP with �-proBDNF, a specific polyclonal antibody, and therecipitated proteins were probed by Western blot analy-es using the �-p75ntr antibody. It should be pointed outhat this antibody does not cross-react with mature BDNFr other pro-neurotrophins, as determined by the manu-acturer and our own analyses. Interestingly, the amount of75ntr co-immunoprecipitated together with proBDNF in-reased immediately after SE (0 h) and was maintained at
s after SE. (a) Lack of tubulin (top blot) in IP samples (lanes 2, 3 andn was detected at normal levels in total homogenates (lane 1) andby subsequent incubation with �-rabbit-IgG antibodies (bottom blot,ing an antibody that recognizes both mature and pro-BDNF), beganunoprecipitation of TrkB/BDNF showed a drop at 12 h that became
F reached a significant peak 12 h after SE and was slightly, but notmunoprecipitated, the levels of co-immunoprecipitated p75ntr were
EM are indicated. Asterisks indicate significant differences compared
receptorile tubulionfirmedBDNF (usf co-immtal BDN
cifically imeans�S
his level 12 and 72 h later (Fig. 5e).

Ti
Aipdioimrs3sTSeasow
jdwacpc(ibwtiS(Byaas
ctn(awnLcrtwbL
tw
Snr1ssifto
EBeMtFab1ttlrmaoeiertsargSrtlenmSpbestusti
N. Unsain et al. / Neuroscience 154 (2008) 978–993 987
rkB does not decrease when neuronal injurys prevented
major point was to establish if the drop in TrkB would benduced by seizures in a case where cell damage is notresent. During the induction of SE we distinguished twoifferent groups of seizure behavior, despite having admin-
stered the same dose of pilocarpine. One of them devel-ped SE, approximately 70% of the animals, and was used
n all experiments described. The other group had inter-ittent behavioral seizures, typically stood up in a kanga-
oo posture about five times, and continuously manifestedtages 1–3 of the Racine scale (Racine, 1972) during the-h period, but lacked generalized clonic–tonic convul-ions and did not develop SE. This group was named “R2.”he presence of animals that have seizures but fail to enterE is normally observed in the pilocarpine model (Biaginit al., 2001; Fujikawa, 1996; Fujikawa et al., 1999; Covolannd Mello, 2000). Interestingly, R2 animals continued withuch behavior despite being re-injected with pilocarpine inrder to make them enter SE. In these animals, diazepamas administered 3 h after the first kangaroo posture.
We found, in agreement with previous reports (Fu-ikawa, 1996), that this kind of seizures did not induce cellamage (Fig. 6a). So it was important to determinehether in these animals the modifications observed innimals with SE were also present. No significant modifi-ations were observed in TrkB (Fig. 6b), p75ntr, BDNF orroBDNF expression (data not shown). However, the in-reased interaction of proBDNF with p75ntr also occurredFig. 6c). Taken together, these results indicate that thencrease in the interaction of proBDNF with p75ntr occursoth in the damage-inducing SE and in harmless seizures,hile the rapid decrease in TrkB-fl was exclusively found in
he injury-inducing SE. Moreover, R2 animals showed anncreased TrkB phosphorylation (Fig. 6d), in contrast toE, which induced a decrease in the activation of TrkB
Fig. 1f). In R2 animals there were no modifications inDNF or TrkB protein levels, thus the increased phosphor-lation of TrkB in these animals is most likely due to anctivity-dependent release of BDNF. In such animals welso assessed the levels of sortilin (Fig. 6d) and found noignificant changes.
We next asked whether changes in TrkB occur whenell damage after SE could be prevented by the use of LESreatment, which was previously demonstrated to have aeuroprotective effect against SE-induced neuronal deathKondratyev et al., 2001). We therefore analyzed cell deathnd TrkB protein levels in animals after SE, with andithout previous LES treatment. As in previous reports,umerous cells were TUNEL positive 72 h after SE (Fig. 7a).ES treatment per se did not induce TUNEL-positiveells, but when administered before SE it prevented neu-onal death (i.e. TUNEL staining). Interestingly, we foundhat hippocampal TrkB-fl downregulation induced by SEas totally prevented when neuronal death was impededy LES treatment (Fig. 7b). It is important to remark that
ES treatment alone did not induce changes in TrkB pro- rein levels at times when SE was induced (LES�24 h) norhen cell death was assessed (LES�96 h; Fig. 7c).
DISCUSSION
ince the discovery that neurotrophins (NT) can regulateot only neuronal survival and differentiation but also neu-onal apoptosis (Barret and Bartlet, 1994; Frade et al.,996, 1997), many questions have arisen regarding howuch opposite functions can be regulated by the sameignaling protein. In the present study we show that SE
nduces a switch between TrkB and p75ntr receptor levels,orcing BDNF to interact largely with one receptor ratherhan the other, an event that might result in a distinct andpposite message.
How does SE induce TrkB-fl downregulation in vivo?xperiments in cell culture demonstrated that exposure toDNF itself induces downregulation of TrkB protein (Frankt al., 1996; Knusel et al., 1997; Sommerfeld et al., 2000).oreover, TrkB truncated variants were not changed in
hose reports, resembling our in vivo observation after SE.urthermore, the infusion in vivo of BDNF in retina (Chennd Weber, 2004), midbrain (Frank et al., 1997), olfactoryulb (Frank et al., 1997) and hippocampus (Frank et al.,996; Xu et al., 2004) also downregulates the TrkB recep-or. Taken together, this evidence supports the hypothesishat the overproduction and release of BDNF after pro-onged seizures might be the signal leading to the down-egulation of hippocampal TrkB after SE. Observationsade in cell cultures by Sommerfeld et al. (2000) suggestmechanism underlying ligand-dependent downregulationf the receptor. Under their conditions and in our in vivoxperiments, the down-regulation of TrkB was rapid, point-
ng to a regulation at the post-transcriptional level. How-ver, it is also possible that longer exposures to the neu-otrophin would also induce regulation at the transcrip-ional level (Frank et al., 1996; Knusel et al., 1997). Asuch, ligand-mediated receptor downregulation is commonmong receptor tyrosine kinases, the activated ligand-eceptor complexes being internalized and either de-raded or recycled to the cell surface (for review, seeorkin and Waters, 1993). A prerequisite for the process is
eceptor activation and this probably explains why theruncated kinase inactive form of TrkB is not downregu-ated after SE (Fig. 1a) or by BDNF exposure (Sommerfeldt al., 2000). Typically, the internalized receptors that areot recycled to the cell surface are targeted to the lysoso-al degradation system. However, in the study made byommerfeld et al. (2000), lysosomal inhibitors could notrevent BDNF-dependent degradation of TrkB, whereaslockers of the ubiquitin–proteasome pathway were veryfficient in stabilizing the receptor. The ubiquitin–protea-ome pathway usually requires ubiquitination of the proteino be degraded (for review, see Hicke, 1999), and TrkBbiquitination after BDNF exposure has indeed been de-cribed (Makkerh et al., 2005; Arevalo et al., 2006). It isempting to speculate that similar mechanisms may occurn vivo after SE. In this regard, the activity-dependent
elease and huge increase of BDNF during and after SE
co
st2r
1tftit
F(scatad
N. Unsain et al. / Neuroscience 154 (2008) 978–993988
ould have the same consequences as the long exposuref cell cultures with a high concentration of BDNF.
The downregulation of TrkB induced by SE is indeedurprising. Previous works have not found TrkB modifica-ions using Western blot (Rudge et al., 1998; Danzer et al.,004). However, other reports have found the TrkB down-
ig. 6. Seizures that do not induce cell death do not downregulate Tra) R2 animals do not show neuronal degeneration as assessed by Fhown at high magnification from control, R2 and SE animals, resorresponding to sp. so: Stratum oriens; sp: stratum pyramidale; sr: strs demonstrated by Western blot analyses of hippocampal samples tahe interaction of proBDNF with p75ntr, as assessed by co-immunoprefter seizures in R2 animals. The levels of sortilin, however, remaifferences compared with control animals, P�0.05.
egulation using immunohistochemistry (Goutan et al., s
998; Ampuero et al., 2007). Protocols used to induce SE,he duration of SE and/or the animal’s age, may accountor the differences among results. These results indicatehe importance of variables such as seizure duration andntensity in determining cell death and TrkB downregula-ion. Moreover, we showed that rats experiencing Racine
but increase proBDNF/p75ntr interaction and TrkB phosphorylation.ing. Pictures of the hilus (top row) and the CA1 sp (bottom row) are. Scale bar�50 �m. Dashed line delimitate the area of the CA1iatum. (b) Seizures in R2 animals do not downregulated TrkB protein,nd 24 h after SE. (c) Interestingly, R2 animals showed an increase in. (d) Moreover, there was an increase in the phosphorylation of TrkBhanged. Means�SEM are indicated. Asterisks indicate significant
kB proteinJ-B stainpectivelyatum radken 12 acipitationined unc
tages 1–3 intermittently for 3 h (R2 animals) did not show

assiKoja
cp
tHei
Fsantai trol anim
N. Unsain et al. / Neuroscience 154 (2008) 978–993 989
ny TrkB downregulation or cell damage. Conflicting re-ults have been obtained regarding TrkB mRNA expres-ion after SE. Some of them show a generalized increasen the whole hippocampus (Kokaia et al., 1993; Schmidt-astner et al., 1996; Simonato et al., 2002), while othersbserved an increase in the dentate gyrus (Dugich-Djord-
evic et al., 1995; Mudo et al., 1996; Poulsen et al., 2004)
ig. 7. LES treatment, which protects neurons from dying after SE,taining) and TrkB protein levels were assessed in control animals, inlone did not produce neuronal death (B, C), it prevented SE-induceduclei in the CA1 of SE-treated animals. Scale bars�100 �m (A–E)reatment before SE prevented the downregulation of TrkB induced bys analyzed at the times when SE was induced (LES�24 h) or when
ndicated. Asterisks indicate significant differences compared with con
nd one observed no changes (Rudge et al., 1998). In any p
ase, TrkB mRNA expression may not necessarily mirrorrotein levels.
It is possible that an early loss of neurons contributeso the effect observed in TrkB decrease at early times.owever, we showed that in the case of FJB staining atarly times (0 and 12 h after SE termination) neuronal
njury was found only in the hilus when neurons were still
ents the downregulation of TrkB. Neuronal apoptosis (using TUNELith LES alone, with SE alone and with LES before SE. (a) While LESinjury (D, E, magnified in F, G). Arrows in D indicate TUNEL positivem (F, G). (b) Western blot analyses of TrkB protein show that LESn the other hand, LES per se does not increase TrkB protein levels,
ced neuronal death was determined (LES�96 h). Means�SEM areals, P�0.05.
also prevanimals wneuronaland 50 �SE. (c) OSE-indu
resent. Although it is possible that TrkB downregulation is

araFtshngpcd
tcaCttsvat(ipme
zop2siteiolcWrpw(
iliBibpveate
ltipoasttpiattt
f2plmccfipc(STbai
mpBitsl
oictpsrtgi
tMhaw
N. Unsain et al. / Neuroscience 154 (2008) 978–993990
consequence of cell damage, it is not probable that itesults from neuronal loss. TrkB-fl downregulation showedn overlapping temporal profile with a higher increase inJB positive neurons in the dentate gyrus, and preceded
he appearance of neuronal injury in the CA1 subfield. Theequence of the appearance of neuronal loss in eachippocampal area is very similar to that obtained by us foreuronal injury using FJB staining (i.e. hilar neurons¡ranule cells¡CA1), which fits the suggestion that FJBositive cells will then be removed from the injury site (i.e.ontributing to cell loss, see Poirier et al., 2000, for aetailed discussion).
TrkB downregulation preceded the upregulation of theumor suppressor protein p53, the involvement of which inell death induced by SE has been well described (Sakhi etl., 1994, 1996; Morrison et al., 1996; Liu et al., 1999;ulmsee et al., 2001; Tan et al., 2002a,b). Furthermore,
he fact that p53 is upregulated after the switch from TrkBo p75ntr is very important because it has been demon-trated that apoptosis due to p75ntr depends on p53 acti-ation (Aloyz et al., 1998; Kaplan and Miller, 2000; Wen etl., 2004; Linggi et al., 2005). The unexpected delay be-ween the binding of p75ntr to proBDNF (0 h) and BDNF12 h) and the increase in p53 protein (24 h) found in ourn vivo experiments can be explained by the need of p53rotein synthesis and the need for an important recruit-ent of responding neurons to detect the change by West-rn blot of whole hippocampal homogenates.
The interaction of BDNF with its receptors after sei-ures has been essentially determined by phosphorylationf TrkB (Binder et al., 1999) and the activation of signalingroteins downstream of TrkB or p75ntr (Berkeley et al.,002; Troy et al., 2002). In the present report, we alsotudied direct BDNF interaction with its receptors by co-mmunoprecipitation analyses. The switch in the interac-ion of BDNF with each receptor induced by SE providesvidence for a prime functional consequence of the switch
n the protein levels of the receptors per se. The variabilityf co-immunoprecipitation studies in vivo could explain the
ack of a strict coincidence between the temporal profile ofo-immunoprecipitated TrkB-fl and that determined byestern blot. The strength of the interaction between neu-
otrophins and their receptors has proved to be enough toull down ligand–receptor complexes by direct IP in vivoithout the need of crosslinkers, as described by others
Harrington et al., 2004).The common neurotrophin receptor, p75ntr, has been
mplicated in cell death, but the identity of the physiologicaligand has remained unclear. Interestingly, we found anncrease of the receptor that co-immunoprecipitates withDNF. However, it cannot be excluded that the pathophys-
ological ligand for this p75ntr is proBDNF, since the anti-ody used in these assays recognizes both mature andro-BDNF. Released, unprocessed neurotrophins were in-olved in inducing neuronal death after CNS injury (Beattiet al., 2002; Harrington et al., 2004) and are strongerctivators of p75ntr-induced apoptosis than mature neuro-rophins possibly due to their greater affinity to p75ntr (Lee
t al., 2001; Nykjaer et al., 2004). Considering this new pevel of complexity, we demonstrated an increase in pro-ein levels of proBDNF at 72 h and, more important, anmmediate and sustained (from 0 to 72 h) increase inroBDNF interacting with p75ntr. However, the occurrencef a similar increase in proBDNF/p75ntr complexes in R2nimals in a scenario where TrkB was not downregulateduggests that both processes might occur at the same timeo induce cell death. At this point it is important to highlighthat while the increased interaction between proBDNF and75ntr took place in both SE and R2 animals, the decrease
n TrkB-fl protein and in p-TrkB was exclusive of the dam-ge-inducing SE. Moreover, it is reasonable to speculatehat the raise in p-TrkB in R2 animals may have a protec-ive effect against the increased proBDNF-p75ntr interac-ion observed.
Recently, the notion that proBDNF can be releasedrom CNS neurons has been challenged (Matsumoto et al.,008). In their report, Matsumoto et al. have shown thatroBDNF is present in such small amounts in mice brain
ysates that it cannot be detected without previous enrich-ent of the extract. They show evidence for a rapid intra-
ellular processing of proBDNF that explains its low intra-ellular levels and prevents its release unprocessed. Theirndings contrast with previous reports which detectedroBDNF without any particular methodology conducted tooncentrate the extracts, including the present reportMichalski and Fahnestock, 2003; Peng et al., 2005;chnydrig et al., 2007; Silhol et al., 2007; Ullal et al., 2007).he techniques used by Matsumoto et al. (a conditionaldnf knockout mice and a bdnf-myc knock-in mice) make itconvincing report, though still waiting for further support-
ng evidence.Interestingly, one of the conclusions drawn by Matsu-
oto et al. (2008) is that given that transfected cells ex-ressing BDNF accumulate and release unprocessedDNF, the efficiency of the cellular processing machinery
s limited. Then, it is possible that after a prolonged exci-atory stimulation, such as SE, excessive proBDNF couldurpass the processing machinery, thus being accumu-
ated and even released from CNS neurons.Given that the studies quoted above were carried out
n rats and the one by Matsumoto et al. (2008) on mice, its reasonable to speculate that a slight difference in pro-essing efficiency of immature BDNF can exist betweenhese species. On the other hand, the fact that we couldull down p75ntr in co-immunoprecipitation assaystrongly suggests that the antibody used in the presenteport is actually binding to proBDNF. In all, the details ofhe issue have little impact on our main conclusions re-arding the possible role of neurotrophin receptor’s switch
n neuronal injury after SE.Although the reactive band that appears by using the
wo BDNF antibodies does not exactly co-migrate with aW marker of 14.4 kDa, a similar MW for mature BDNFas been shown using one of those antibodies (Silhol etl., 2007). Moreover, the same band, with no background,as found with two widely used antibodies and we could
ull down TrkB and p75ntr in co-immunoprecipitation as-
sB
aoitrr
dvrnpOcmwdirpTwll
aqasmpmrsdns
ACadh
A
A
A
B
B
B
B
B
B
B
B
B
B
C
C
C
C
C
C
D
D
D
D
N. Unsain et al. / Neuroscience 154 (2008) 978–993 991
ays, indicating that the antibody is actually binding toDNF.
The fact that the levels of sortilin after SE or in R2nimals did not change is not enough to determine its lackf participation in the mechanism of SE-induced neuronal
njury. Perhaps the determination of possible changes inhe sub-cellular localization of sortilin in hippocampal neu-ons after SE is needed for a better understanding of itsole.
The concept that Trk signaling can abrogate p75ntr-ependent apoptosis (Yoon et al., 1998; Davey and Da-ies, 1998; Bamji et al., 1998; Teng et al., 2005) has beenecently challenged by the finding that in basal forebraineurons the activation of Trk signaling could not prevent75ntr-dependent neuronal death (Volosin et al., 2006).ne explanation for the difference between those reportsould be the neuronal type used in cell cultures experi-ents. In a previous report, using hippocampal culture, itas shown that Trk signaling could prevent p75ntr-depen-ent neuronal death, and so the same event could happen
n the hippocampus after SE in vivo. Thus, an apoptoticesponse would be activated only by BDNF and/orroBDNF in cells expressing p75ntr in the absence ofrkB. Moreover, the demonstration that blocking cell deathith LES treatment also prevents changes in TrkB protein
evels further support the hypothesis that TrkB downregu-ation might be involved in cell death.
Because hippocampal neurons express both p75ntrnd TrkB receptors and mature BDNF and proBDNF, theuestion arises of which might be the factor or the inter-ction event driving the apoptosis signal. An importantuggestion from this work is that survival/death decisionsay depend on a balance between different signalingathways. The presence or absence of receptor signalingay convert a survival to an apoptotic outcome. The neu-
otrophin receptors may have opposite effects on neuronalurvival. According to this view, whether a neuron lives ories depends on a balancing act wherein the key determi-ant is the predominance of signaling from one receptorubtype over that from the other.
cknowledgments—This work was supported by grants fromONICET, FONCYT (PICT 5-14398) and SECyT-UNC. D.H.M. iscareer member of CONICET; N.U. and A.A. are recipients of
octoral fellowships from CONICET. We thank Laura Montroull forer help with some experiments.
REFERENCES
loyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, MillerFD (1998) P53 is essential for development neuronal death asregulated by the TrkA and p75ntr receptors. J Cell Biol 143:1691–1703.
mpuero E, Dagnino-Subiabre A, Sandoval R, Zepeda-Carreño R,Sandoval S, Viedma A, Aboitiz F, Orrego F, Wyneken U (2007)Status epilepticus induces region-specific changes in dendriticspines, dendritic length and TrkB protein content of rat brain cortex.Brain Res 1150:225–238.
revalo JC, Waite J, Rajagopal R, Beyna M Chen ZY, Lee FS, ChaoMV (2006) Cell survival through Trk neurotrophin receptors is
differentially regulated by ubiquitination. Neuron 50:549–559.amji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz RS, Kohn J,Causing CG, Miller FD (1998) The p75ntr mediates neuronal ap-optosis and is essential for naturally occurring sympathetic neurondeath. J Cell Biol 140:911–923.
arbacid M (1994) The Trk family of neurotrophin receptors. J Neurbiol25:1386–1403.
arret GL, Bartlet PF (1994) The p75 nerve growth factor receptormediates survival or death depending on the stage of sensoryneuron development. Proc Natl Acad Sci U S A 91:6501–6505.
eattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM,Bresnahan JC, Hempstead BL, Yoon SO (2002) ProNGF inducesp75-mediated death of oligodendrocytes following spinal cord in-jury. Neuron 36:375–386.
erkeley JL, Decker MJ, Levey AL (2002) The role of muscarinicacetylcholine receptor-mediated activation of ERK 1/2 in pilo-carpine-induced seizures. J Neurochem 82:192–201.
iagini G, Avoli M, Marcinkiewicz J, Marcinkiewicz M (2001) Brain-derived neurotrophic factor superinduction parallels anti-epileptic-neuroprotective treatment in the pilocarpine epilepsy model. J Neu-rochem 76:1814–1822.
ibel M, Barde YA (2000) Neurotrophins: Key regulators of cell fateand cell shape in the vertebrate nervous system. Genes Dev14:2919–2937.
ibel M, Hoppe E, Barde YA (1999) Biochemical and functional inter-actions between the neurotrophin receptors trk and p75NTR.EMBO J 18:616–622.
inder DK, Routbort MJ, McNamara JO (1999) Immunohistochemicalevidence of seizure-induced activation of trk receptors in themossy fiber pathway of adult rat hippocampus. J Neurosci 19:4616–4626.
radford MM (1976) A rapid and sensitive method for the quantitationof microgram quantities of protein utilizing the principle of proteindye binding. Anal Biochem 72:248–254.
hao MV (2003) Neurotrophins and their receptors: a convergencepoint for many signalling pathways. Nat Rev Neurosci 4:299–309.
hen H, Weber AJ (2004) Brain-derived neurotrophic factor reducesTrkB protein and mRNA in the normal retina and following opticnerve crush in adult rats. Brain Res 1011:99–106.
hen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, LeeFS (2004) Variant brain-derived neurotrophic factor (BDNF)(Met66) alters the intracellular trafficking and activity-dependentsecretion of wildtype BDNF in neurosecretory cells and corticalneurons. J Neurosci 24:4401–4411.
ostantini C, Scrable H, Puglielli L (2006) An aging pathway controlsthe TrkA to p75NTR receptor switch and amyloid beta-peptidegeneration. EMBO J 25:1997–2006.
ovolan L, Mello LE (2000) Temporal profile of neuronal injury follow-ing pilocarpine or kainic acid-induced status epilepticus. EpilepsyRes 39:133–152.
ulmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH,Mattson MP (2001) A synthetic inhibitor of p53 protects neuronsagainst death induced by ischemic and excitotoxic insults, andamyloid beta-peptide. J Neurochem 77:220–228.
anzer SC, He X, McNamara JO (2004) Ontogeny of seizure-inducedincreases in BDNF immunoreactivity and TrkB receptor activationin rat hippocampus. Hippocampus 14:345–355.
avey F, Davies AM (1998) TrkB signalling inhibits p75-mediatedapoptosis induced by nerve growth factor in embryonic propriocep-tive neurons. Curr Biol 8:915–918.
ugich-Djordjevic MM, Ohsawa F, Okazaki T, Mori N, Day JR, BeckKD, Hefti F (1995) Differential regulation of catalytic and non-catalytic TrkB messenger RNAs in the rat hippocampus followingseizures induced by systemic administration of kainate. Neuro-science 66:861–877.
rake CT, Milner TA, Patterson SL (1999) Ultrastructural localizationof full-length trkB immunoreactivity in rat hippocampus suggestsmultiple roles in modulating activity-dependent synaptic plasticity.
J Neurosci 19:8009–8026.
F
F
F
F
F
F
F
F
F
F
F
F
G
G
H
H
H
H
H
K
K
K
K
K
K
L
L
L
L
L
L
M
M
M
M
M
M
N
N
N
N
P
N. Unsain et al. / Neuroscience 154 (2008) 978–993992
abene PF, Andrioli A, Priel MR, Cavalheiro EA, Bentivoglio M (2004)Fos induction and persistence, neurodegeneration, and interneu-ron activation in the hippocampus of epilepsy-resistant versusepilepsy-prone rats after pilocarpine-induced seizures. Hippocam-pus 14:895–907.
rade JM, Bovolenta P, Martinez-Morales JR, Arribas A, Barbas JA,Rodriguez-Tebar A (1997) Control of early cell death by BDNF inthe chick retina. Development 124:3313–3320.
rade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell deathby endogenous nerve growth factor through its p75 receptor. Na-ture 383:166–168.
rank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS (1996)BDNF down-regulates neurotrophin responsiveness, TrkB proteinand TrkB mRNA levels in cultured rat hippocampal neurons. EurJ Neurosci 8:1220–1230.
rank L, Wiegand SJ, Siuciak JA, Lindsay RM, Rudge JS (1997)Effects of BDNF infusion on the regulation of trkB protein andmessage in adult rat brain. Exp Neurol 145:62–70.
riedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennet MVL,Moshé SL, Zukin RS (1994) Kainate-induced status epilepticusalters glutamate and GABAa receptor gene expression in adultrat hippocampus: An in situ hybridization study. J Neurosci 14:2697–2707.
riedman LK (1998) Selective reduction of GluR2 protein in adulthippocampal CA3 neurons following status epilepticus but prior tocell loss. Hippocampus 89:511–525.
riedman WJ (2000) Neurotrophins induce death of hippocampal neu-rons via the p75 receptor. J Neurosci 20:6340–6346.
riedman LK, Veliskova J, Kaur J, Magrys BW, Liu H (2003) GluR2(B)knockdown accelerates CA3 injury after kainate seizures. J Neu-ropathol Exp Neurol 62:733–750.
ryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Kromer LF (1996)Developmental and mature expression of full-length and truncatedtrkB receptors in the rat forebrain. J Comp Neurol 374:21–40.
ujikawa DG (1996) The temporal evolution of neuronal damage frompilocarpine-induced status epilepticus. Brain Res 725:11–22.
ujikawa DG, Shinmei SS, Cai B (1999) Lithium-pilocarpine-inducedstatus epilepticus produces necrotic neurons with internucleoso-mal fragmentation in adult rats. Eur J Neurosci 11:1605–1614.
all CM (1993) Seizures-induced changes in neurotrophin expression:implications for epilepsy. Exp Neurol 124:150–166.
outan E, Marti E, Ferrer I (1998) BDNF, and full length and trunkatedTrkB expression in the hippocampus of rat following kainic acidexcitotoxic damage. Evidence of complex time-dependent andcell-specific responses. Mol Brain Res 59:154–164.
arrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Morl K,Meyer M, Hempstead BL, Yoon SO, Giehl KM (2004) SecretedproNGF is a pathophysiological death-inducing ligand after adultCNS injury. Proc Natl Acad Sci U S A 101:6226–6230.
eymach JV Jr, Shooter EM (1995) The biosynthesis of neurotrophinheterodimers by transfected mammalian cells. J Biol Chem270:12297–12304.
icke L (1999) Gettin’ down with ubiquitin: turning off cell-surfacereceptors, transporters and channels. Trends Cell Biol 9:107–112.
uang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal de-velopment and function. Annu Rev Neurosci 24:677–736.
ughes PE, Alexi T, Yoshida T, Schreiber SS, Knusel B (1996) Exci-totoxic lesion of rat brain with quinolinic acid induces expression ofp53 messenger RNA and protein and p53-inducible genes Bax andGadd-45 in brain areas showing DNA fragmentation. Neuro-science 74:1143–1160.
aplan DR, Miller FD (2000) Neurotrophin signal transduction in thenervous system. Curr Opin Neurobiol 10:381–391.
aplan DR, Stephens RM (1994) Neurotrophin signal transduction bythe Trk receptor. J Neurobiol 25:1404–1417.
nusel B, Gao H, Okazaki T, Yoshida T, Mori N, Hefti F, Kaplan DR
(1997) Ligand-induced down-regulation of trk messenger RNA,protein and tyrosine phosphorylation in rat cortical neurons.Neuroscience 78:951–962.
okaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O(1993) Coexpression of neurotrophins and their receptors in neu-rons of the central nervous system. Proc Natl Acad Sci U S A90:6711–6715.
olbeck R, Jungbluth S, Barde YA (1994) Characterisation of neuro-trophin dimers and monomers. Eur J Biochem 225:995–1003.
ondratyev A, Sahibzada N, Gale K (2001) Electroconvulsive shockexposure prevents neuronal apoptosis after kainic acid-evokedstatus epilepticus. Mol Brain Res 91:1–13.
ee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cellsurvival by secreted proneurotrophins. Science 294:1945–1948.
essmann V, Gottmann K, Malcangio M (2003) Neurotrophin secre-tion: current facts and future prospects. Prog Neurobiol 69:341–374.
inggi MS, Burke TL, Williams BB, Harrington A, Kraemer R, Hemp-stead BL, Yoon SO, Carter BD (2005) Neurotrophin receptor inter-acting factor (NRIF) is an essential mediator of apoptotic signalingby the p75 neurotrophin receptor. J Biol Chem 280:13801–13808.
iu W, Rong Y, Baudry M, Schreiber SS (1999) Status epilepticusinduces p53 sequence-specific DNA binding in mature rat brain.Mol Brain Res 63:248–253.
iu Z, Nagao T, Desjardins GC, Gloor P, Avoli M (1994) Quantitativeevaluation of neuronal loss in the dorsal hippocampus in rats withlong-term pilocarpine seizures. Epilepsy Res 17:237–247.
u B, Pang PT, Woo NH (2005) The yin and yang of neurotrophinaction. Nat Rev Neurosci 6:603–614.
akkerh JP, Ceni C, Auld DS, Vaillancourt F, Dorval G, Barker PA(2005) p75 Neurotrophin receptor reduces ligand-induced Trk re-ceptor ubiquitination and delays Trk receptor internalization anddegradation. EMBO Rep 6:936–941.
atsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M,Barde YA (2008) Biosynthesis and processing of endogenousBDNF: CNS neurons store and secrete BDNF, not pro-BDNF. NatNeurosci 11:131–133.
ichalski B, Fahnestock M (2003) Pro-brain-derived neurotrophic fac-tor is decreased in parietal cortex in Alzheimer’s disease. Mol BrainRes 111:148–154.
orrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA,Schwartzkroin PA (1996) Loss of the p53 tumor suppressor geneprotects neurons form kainate-induced cell death. J Neurosci16:1337–1345.
owla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG,Murphy RA (2001) Biosynthesis and post-translational processingof the precursor to brain-derived neurotrophic factor. J Biol Chem276:12660–12666.
udo G, Jiang XH, Timmusk T, Bindoni M, Belluardo N (1996) Changein neurotrophins and their receptor mRNAs in the rat forebrain afterstatus epilepticus induced by pilocarpine. Epilepsia 37:198–207.
ational Research Council (1996) Guide for the use and care oflaboratory animals. National Academy of Sciences, Washington,D.C.: The National Academies Press.
awa H, Carnahan J, Gall C (2004) BDNF protein measured by anovel enzyme immunoassay in normal brain and after seizures:partial disagreement with mRNA levels. Eur J Neurosci 7:1527–1535.
ykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS,Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, HempsteadBL, Petersen CM (2004) Sortilin is essential for proNGF-inducedneuronal cell death. Nature 427:843–848.
ykjaer A, Willnow TE, Petersen CM (2005) p75NTR: live or let die.Curr Opin Neurobiol 15:49–57.
eng S, Wuu J, Mufson EJ, Fahnestock M (2005) Precursor form ofbrain-derived neurotrophic factor and mature brain-derived neu-rotrophic factor are decreased in the pre-clinical stages of Alz-
heimer’s disease. J Neurochem 93:1412–1421.
P
P
P
R
R
R
R
R
S
S
S
S
S
S
S
S
S
S
T
T
T
T
T
U
V
W
W
X
Y
Z
Z
N. Unsain et al. / Neuroscience 154 (2008) 978–993 993
oirier JL, Capek R, De Koninck Y (2000) Differential progression ofdark neuron and Fluoro-Jade labelling in the rat hippocampusfollowing pilocarpine-induced status epilepticus. Neuroscience97:59–68.
oo MM (2001) Neurotrophins as synaptic modulators. Nat Rev Neu-rosci 2:24–32.
oulsen FR, Lauterborn J, Zimmer J, Gall CM (2004) Differentialexpression of brain-derived neurotrophic factor transcripts afterpilocarpine-induced seizure-like activity is related to mode of Ca2�
entry. Neuroscience 126:665–676.acine RJ (1972) Modifications of seizure activity by electrical stimu-
lation. II. Motor seizure. Electroencephalogr Clin Neurophysiol32:281–294.
angel P, Cysneiros RM, Arida RM, de Albuquerque M, Colugnati DB,Scorza CA, Cavalheiro EA, Scorza FA (2005) Lovastatin reducesneuronal cell death in hippocampal CA1 subfield after pilocarpine-induced status epilepticus: preliminary results. Arq Neuropsiquiatr63:972–976.
och C, Leroy C, Nehlig A, Namer IJ (2002) Magnetic resonanceimaging in the study of the lithium-pilocarpine model of temporallobe epilepsy in adult rats. Epilepsia 43:325–335.
oux PP, Colicos MA, Barker PA, Kennedy TE (1999) p75 Neurotro-phin receptor expression is induced in apoptotic neurons afterseizure. J Neurosci 19:6887–6896.
udge JS, Pasnikowski E, Corcoran T, Cai N, Achenson A, AndersonK, Lindsay RM, Wiegand SJ (1998) Endogenous BDNF protein isincreased in adult rat hippocampus after a kainic acid inducedexcitotoxic insult but exogenous BDNF is not neuroprotective. ExpNeurol 149:398–410.
akhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS (1994)p53 Induction is associated with neuronal damage in the centralnervous system. Proc Natl Acad Sci U S A 91:7525–7529.
akhi S, Sun N, Wing LL, Mehta P, Schreiber SS (1996) Nuclearaccumulation of p53 protein following KA-induced seizures. Neu-roreport 7:493–496.
chmidt-Kastner R, Humpel C, Wetmore C, Olson L (1996) Cellularhybridization for BDNF, trkB, and NGF mRNAs and BDNF-immu-noreactivity in rat forebrain after pilocarpine-induced status epilep-ticus. Exp Brain Res 108:331–347.
chmued LC, Albertson C, Slikker W Jr (1997) Fluoro-Jade: a novelfluorochrome for the sensitive and reliable histochemical localiza-tion of neuronal degeneration. Brain Res 751:37–46.
chmued LC, Hopkins KJ (2000) Fluoro-Jade: novel fluorochromes fordetecting toxicant-induced neuronal degeneration. Toxicol Pathol28:91–99.
chnydrig S, Korner L, Landweer S, Ernst B, Walker G, Otten U, KunzD (2007) Peripheral lipopolysaccharide administration transientlyaffects expression of brain-derived neurotrophic factor, cortico-tropin and proopiomelanocortin in mouse brain. Neurosci Lett429:69–73.
ilhol M, Arancibia S, Maurice T, Tapia-Arancibia L (2007) Spatialmemory training modifies the expression of brain-derived neuro-trophic factor tyrosine kinase receptors in young and aged rats.Neuroscience 146:962–973.
imonato M, Bregola G, Armellin M, Del Piccolo P, Rodi D, Zucchini S,
Tongiorgi E (2002) Dendritic targeting of mRNAs for plasticitygenes in experimental models of temporal lobe epilepsy. Epilepsia43:153–158.
ommerfeld MT, Schweigreiter R, Barde YA, Hoppe E (2000) Down-regulation of the neurotrophin receptor TrkB following ligand bind-ing. Evidence for an involvement of the proteasome and differentialregulation of TrkA and TrkB. J Biol Chem 275:8982–8990.
orkin A, Waters M (1993) Endocytosis of growth factor receptors.Bioessays 15:375–382.
an Z, Sankar R, Shin D, Sun N, Liu H, Wasterlain CG, Schreiber SS(2002a) Differential induction of p53 in immature and adult rat brainfollowing lithium-pilocarpine status epilepticus. Brain Res 928:187–193.
an Z, Sankar R, Tu W, Shin D, Liu H, Wasterlain CG, Schreiber SS(2002b) Immunohistochemical study of p53-associated proteins inrat brain following lithium-pilocarpine status epilepticus. Brain Res929:129–138.
eng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, KermaniP, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, HempsteadBL (2005) ProBDNF induces neuronal apoptosis via activation of areceptor complex of p75NTR and sortilin. J Neurosci 25:5455–5463.
remblay E, Nitecka L, Berger ML, Ben-Ari Y (1984) Maturation ofkainic acid seizure-brain damage syndrome in the rat. I. Clinical,electrographic and metabolic observations. Neuroscience 13:1051–1072.
roy CM, Friedman JE, Friedman WJ (2002) Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J BiolChem 277:34295–34302.
llal GR, Michalski B, Xu B, Racine RJ, Fahnestock M (2007) NT-3modulates BDNF and proBDNF levels in naïve and kindled rathippocampus. Neurochem Int 50:866–871.
olosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Fried-man WJ (2006) Interaction of survival and death signaling in basalforebrain neurons: roles of neurotrophins and proneurotrophins.J Neurosci 26:7756–7766.
all CJ, Kendall EJ, Obenaus A (2000) Rapid alterations in diffusion-weighted images with anatomic correlates in a rodent model ofstatus epilepticus. AJNR Am J Neuroradiol 21:1841–1852.
en CJ, Xue B, Qin WX, Yu M, Zhang MY, Zhao DH, Gao X, Gu JR,Li CH (2004) hNRAGE, a human neurotrophin receptor interactingMAGE homologue, regulates p53 transcriptional activity and inhib-its cell proliferation. FEBS Lett 564:171–176.
u B, Michalski B, Racine RJ, Fahnestock M (2004) The effects ofbrain-derived neurotrophic factor (BDNF) administration on kin-dling induction, Trk expression and seizure-related morphologicalchanges. Neuroscience 126:521–531.
oon SO, Carter BD, Casaccia-Bonnefil P, Chao MV (1998) Compet-itive signaling between TrkA and p75 nerve growth factor receptorsdetermines cell survival. J Neurosci 18:3273–3281.
accaro MC, Ivanisevic L, Perez PO, Meakin S, Saragovi HU (2001)p75 Co-receptors regulate ligand-dependent and ligand-indepen-dent Trk receptor activation, in part by altering Trk docking subdo-mains. J Biol Chem 276:31023–31029.
weifel LS, Kuruvilla R, Ginty DD (2005) Functions and mechanismsof retrograde neurotrophin signalling. Nat Rev Neurosci 6:615–
625.(Accepted 4 April 2008)(Available online 2 May 2008)
Copyright © 2022 FDOKUMEN




















