
The tricyclic antidepressant amitriptyline is cytotoxic to HTB114 human leiomyosarcoma and induces...
12
The tricyclic antidepressant amitriptyline is cytotoxic to HTB114 human leiomyosarcoma and induces p75 NTR -dependent apoptosis Grazia Pula * , Alessandra Pistilli * , Claudia Montagnoli, Anna M. Stabile, Maria G. Rambotti and Mario Rende Nerve growth factor (NGF) receptors, TrKA and p75 NTR , are being investigated in cancer therapy. Our previous data show that, in HTB114 uterine leiomyosarcoma cells, p75 NTR -dependent apoptosis is inducible by cytotoxic drugs and can suppress nerve growth factor-dependent growth. Although amitriptyline can kill cancer cells and bind TrKA/B, its effects on p75 NTR -dependent apoptosis are unknown. The aim of this paper was to evaluate the antineoplastic potential of amitriptyline, and the role of p75 NTR -dependent apoptosis in the chemoresistant uterine HTB114 leiomyosarcoma. Using proliferation assays and fluorescence-activated cell sorting analysis, we found that amitriptyline caused a marked reduction in HTB114 cell viability, associated with the parallel upregulation of p75 NTR expression. This converted the TrKA + -proliferating cells into TrKA + /p75 NTR + , leading to downregulation of TrKA-prosurvival signaling (AKT) and activation of p75 NTR -dependent apoptosis (through caspase-3). Overall, we provide novel evidence that HTB114 uterine leiomyosarcoma cells are highly sensitive to amitriptyline, supporting the role of p75 NTR -dependent apoptosis as a novel cytotoxic mechanism of this drug and of p75 NTR as an inducible stress receptor and a novel target in clinical oncology. Anti-Cancer Drugs 00:000–000 c 2013 Wolters Kluwer Health | Lippincott Williams & Wilkins. Anti-Cancer Drugs 2013, 00:000–000 Keywords: amitriptyline, apoptosis, cancer, cancer therapy, neurotrophins, nerve growth factor, p75 NTR , TrKA, uterine leiomyosarcoma Anatomy Section, Department of Medico-Surgical Specialties and Public Health, School of Medicine, University of Perugia, Perugia, Italy Correspondence to Grazia Pula, MD, PhD, Anatomy Section, Department of Medico-Surgical Specialties and Public Health, School of Medicine, University of Perugia, Via del Giochetto, 06126 Perugia, Italy Tel: +39 075 585 7449; fax: +39 075 5857454; e-mail: [email protected] *Grazia Pula and Alessandra Pistilli contributed equally to this work. Received 15 January 2013 Revised 6 June 2013 Revised form accepted 12 June 2013 Introduction Uterine leiomyosarcoma is an aggressive tumor, which is radioresistant and chemoresistant. Generally, resistance to chemotherapy is linked to the ability of cancer cells to repair DNA damage and develop adaptations, which promote survival in toxic environments. Thus, finding alternative strategies to ‘switch off ’ the survival pathways of these cells may provide a clinical benefit. Furthermore, investigation of existing drugs that are currently pre- scribed for other indications can prove cost-effective. Like other developmental factors, neurotrophins (NTs) have been increasingly linked to cancer [1–6]. In fact, accumulating evidences show that tumors can develop autonomous secretion of NTs, especially nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF). This triggers an autocrine loop, leading to constitutive activation of their cognate receptors, unrest- ricted growth, and resistance to apoptosis. Thus, neurotrophin receptors (NTRs) have become promising targets in cancer therapy [7,8]. There are two type of NTRs, namely, the NT-selective tropomyosin receptor kinases (TrKA, TrKB, and TrKC) and the nonselective p75 NTR , which binds all NTs [9]. The TrKs are tyrosine- kinase receptors. They are encoded by proto-oncogenes and activate the classic signaling pathways of growth factor receptors, including (a) the mitogenic p38 mito- gen-activated protein kinase pathway and (b) the prosurvival phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which also inhibits apoptosis [10]. They can also trigger Ca 2+ release through the phospholipase C-g/ phosphatidylinositol bisphosphate/inositol trisphosphate/ diacylglycerol pathway [9]. In contrast, p75 NTR is a death receptor of the tumor necrosis factor (TNF) receptor superfamily, with a functional death domain [5,11,12]. However, this receptor is also a stem cell marker (CD271) [13], associated with cancer growth [13,14], and can form complexes with different coreceptors activating multiple pathways. Therefore, whereas the TrKs are generally prosurvival, the role of p75 NTR is more complex and enigmatic [11–15] and it can induce either (a) apoptosis, through c-jun N-terminal kinase (JNK)/ caspase-3, -6, and -9, or (b) survival, through NF-kB (NF- kB), depending on crosstalk with the TrKs and other coreceptors [13,15,16]. This ambiguity has been de- scribed in other death receptors [17] and reflects the different signaling molecules expressed by differentiated Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (www.anti-cancerdrugs.com). Preclinical report 1 0959-4973 c 2013 Wolters Kluwer Health | Lippincott Williams & Wilkins DOI: 10.1097/CAD.0b013e328364312f
Transcript of The tricyclic antidepressant amitriptyline is cytotoxic to HTB114 human leiomyosarcoma and induces...

The tricyclic antidepressant amitriptyline is cytotoxicto HTB114 human leiomyosarcoma and inducesp75NTR-dependent apoptosisGrazia Pula*, Alessandra Pistilli*, Claudia Montagnoli, Anna M. Stabile,Maria G. Rambotti and Mario Rende
Nerve growth factor (NGF) receptors, TrKA and p75NTR,
are being investigated in cancer therapy. Our previous data
show that, in HTB114 uterine leiomyosarcoma cells,
p75NTR-dependent apoptosis is inducible by cytotoxic
drugs and can suppress nerve growth factor-dependent
growth. Although amitriptyline can kill cancer cells and
bind TrKA/B, its effects on p75NTR-dependent apoptosis
are unknown. The aim of this paper was to evaluate the
antineoplastic potential of amitriptyline, and the role
of p75NTR-dependent apoptosis in the chemoresistant uterine
HTB114 leiomyosarcoma. Using proliferation assays and
fluorescence-activated cell sorting analysis, we found
that amitriptyline caused a marked reduction in HTB114
cell viability, associated with the parallel upregulation
of p75NTR expression. This converted the TrKA+-proliferating
cells into TrKA + /p75NTR + , leading to downregulation
of TrKA-prosurvival signaling (AKT) and activation of
p75NTR-dependent apoptosis (through caspase-3).
Overall, we provide novel evidence that HTB114 uterine
leiomyosarcoma cells are highly sensitive to amitriptyline,
supporting the role of p75NTR-dependent apoptosis as
a novel cytotoxic mechanism of this drug and of p75NTR as
an inducible stress receptor and a novel target in clinical
oncology. Anti-Cancer Drugs 00:000–000 �c 2013 Wolters
Kluwer Health | Lippincott Williams & Wilkins.
Anti-Cancer Drugs 2013, 00:000–000
Keywords: amitriptyline, apoptosis, cancer, cancer therapy, neurotrophins,nerve growth factor, p75NTR, TrKA, uterine leiomyosarcoma
Anatomy Section, Department of Medico-Surgical Specialties and Public Health,School of Medicine, University of Perugia, Perugia, Italy
Correspondence to Grazia Pula, MD, PhD, Anatomy Section, Departmentof Medico-Surgical Specialties and Public Health, School of Medicine,University of Perugia, Via del Giochetto, 06126 Perugia, ItalyTel: + 39 075 585 7449; fax: + 39 075 5857454; e-mail: [email protected]
*Grazia Pula and Alessandra Pistilli contributed equally to this work.
Received 15 January 2013 Revised 6 June 2013Revised form accepted 12 June 2013
IntroductionUterine leiomyosarcoma is an aggressive tumor, which is
radioresistant and chemoresistant. Generally, resistance
to chemotherapy is linked to the ability of cancer cells to
repair DNA damage and develop adaptations, which
promote survival in toxic environments. Thus, finding
alternative strategies to ‘switch off ’ the survival pathways
of these cells may provide a clinical benefit. Furthermore,
investigation of existing drugs that are currently pre-
scribed for other indications can prove cost-effective.
Like other developmental factors, neurotrophins (NTs)
have been increasingly linked to cancer [1–6]. In fact,
accumulating evidences show that tumors can develop
autonomous secretion of NTs, especially nerve growth
factor (NGF) and brain derived neurotrophic factor
(BDNF). This triggers an autocrine loop, leading to
constitutive activation of their cognate receptors, unrest-
ricted growth, and resistance to apoptosis. Thus,
neurotrophin receptors (NTRs) have become promising
targets in cancer therapy [7,8]. There are two type of
NTRs, namely, the NT-selective tropomyosin receptor
kinases (TrKA, TrKB, and TrKC) and the nonselective
p75NTR, which binds all NTs [9]. The TrKs are tyrosine-
kinase receptors. They are encoded by proto-oncogenes
and activate the classic signaling pathways of growth
factor receptors, including (a) the mitogenic p38 mito-
gen-activated protein kinase pathway and (b) the
prosurvival phosphatidylinositol 3-kinase (PI3K)/AKT
pathway, which also inhibits apoptosis [10]. They can
also trigger Ca2 + release through the phospholipase C-g/
phosphatidylinositol bisphosphate/inositol trisphosphate/
diacylglycerol pathway [9]. In contrast, p75NTR is a death
receptor of the tumor necrosis factor (TNF) receptor
superfamily, with a functional death domain [5,11,12].
However, this receptor is also a stem cell marker
(CD271) [13], associated with cancer growth [13,14],
and can form complexes with different coreceptors
activating multiple pathways. Therefore, whereas the
TrKs are generally prosurvival, the role of p75NTR is more
complex and enigmatic [11–15] and it can induce either
(a) apoptosis, through c-jun N-terminal kinase (JNK)/
caspase-3, -6, and -9, or (b) survival, through NF-kB (NF-
kB), depending on crosstalk with the TrKs and other
coreceptors [13,15,16]. This ambiguity has been de-
scribed in other death receptors [17] and reflects the
different signaling molecules expressed by differentiated
Supplemental digital content is available for this article. Direct URL citationsappear in the printed text and are provided in the HTML and PDF versions of thisarticle on the journal’s website (www.anti-cancerdrugs.com).
Preclinical report 1
0959-4973 �c 2013 Wolters Kluwer Health | Lippincott Williams & Wilkins DOI: 10.1097/CAD.0b013e328364312f

and cancer (or stem) cells. In fact, the final cellular
response results from the integration of multiple
variables, including the presence of mature/immature
NTs, TrK isoforms, transactivating receptors, coreceptors
and adapters, and the balance between TrKs/p75NTR, all
of which can be significantly dysregulated by the
oncogenic transformation [6].
Generally, because of the higher Kd of the TrKs versus
p75NTR (pmol/l vs. nmol/l, respectively), NTs bind the
TrKs preferentially. Activating mutations, upregulation or
ectopic expression of the TrKs, have been linked to
cancer [6,18]. TrK activation can induce growth, invasion,
metastasis, ankoisis resistance, and angiogenesis [6], all of
which promote survival under limiting conditions, and
multidrug resistance [6,18,19]. In addition, overexpres-
sion of the TrKs and constitutive activation of their
prosurvival pathways (especially AKT) can inhibit apop-
tosis [2–4,20], causing apoptosis resistance. TrK inhibitors
(i.e. CEP-701, AZ7451) [7] are therefore investigated in
clinical trials for cancer therapy (http://clinicaltrialsfeeds.org).
However, previous work from our group and others has
shown that upregulation of p75NTR can restore apoptosis
in susceptible cancers and could represent an alternative
antineoplastic strategy [2–4,20].
Sarcomas are among the tumors characterized by auto-
crine activation of the NGF/TrKA/AKT axis [2–4,21].
In the human HTB114 uterine leiomyosarcoma, this self-
sustaining loop leads to a constitutive receptor imbalance
– that is high-TrKA/low-p75NTR - promoting malignant
growth and survival [3,4]. Recently, the BDNF/TrKB axis
has also been linked to uterine leiomyosarcoma growth
[22]. In HTB114 cells, p75NTR-dependent apoptosis is
inducible by both TrK inhibitors and gene therapy.
Interestingly, we observed that the proapoptotic effect
of TrK blockade was associated with the upregulation of
p75NTR/caspase-3, rather than AKT inhibition [3,4],
confirming the key role of crosstalk in NGF receptor
signaling. Upregulation of p75NTR by injury and trau-
ma [23] is known to trigger apoptosis in the nervous
system (see the Discussion section). We therefore
postulated that the same mechanism could be used to
kill susceptible cancers. In fact, p75NTR may act as an
inducible stress receptor [24] in dysfunctional cells,
leading to p75NTR-dependent apoptosis as a final common
pathway for both cellular injuries and drug cytotoxicity.
We therefore sought to investigate whether other
cytotoxic drugs could induce p75NTR-dependent apopto-
sis in cancer cells. Tricyclic antidepressants (TCAs) are
currently prescribed to control depression and neuro-
pathic pain. However, although their potential relation
with cancer is controversial in epidemiological stu-
dies [25–30], they possess documented antineoplastic
effects. These drugs are cytotoxic to cancer cell lines
in vitro [31–34] and can reverse chemoresistance in
animal models [35–37]; furthermore, a preliminary
clinical trial has investigated the TCA chlorimipramine
as a potential therapy for glioma patients [38]. Although
the antidepressive effects of the TCAs have been
attributed to the inhibition of monoamine transporters,
their exact mechanism of action is unknown. On the basis
of their cationic amphiphilic properties, accumulation
into intracellular organelles is an off-target effect leading
to acute oxidant damage, through inhibition of the
mitochondrial complex III [33,39,40]. However, cytotoxi-
city has also been attributed to nonmitochondrial damage,
through cell cycle arrest [41], and autophagy [42]. These
diverse mechanisms may confer different sensitivities
to TCAs.
Although TCA cytotoxicity has been documented in
different cancers [31,39–46], these drugs have never
been tested in uterine leiomyosarcoma. Furthermore,
their potential modulation of p75NTR-dependent apop-
tosis has never been explored. Because of its known
antineoplastic activity [38–41] and as it has recently been
shown to bind TrK-A/B receptors [47], amitriptyline
appeared to be the ideal candidate.
The aim of this report was to investigate p75NTR-
dependent apoptosis and the NGF/TrKA/p75NTR axis in
HTB114 human uterine leiomyosarcoma cells exposed to
cytotoxic concentrations of amitriptyline. Our data show
for the first time the antineoplastic potential of
amitriptyline in human uterine leiomyosarcoma. Further-
more, we provide novel and interesting evidence that the
drug induces the membrane expression of p75NTR, which
has already been shown to trigger apoptosis in this cancer
cell model [2–4].
Materials and methodsReagents
Human recombinant b-NGF was obtained from Alomone
Labs (Zion, Israel). Amitriptyline was from Sigma-Aldrich
(Milan, Italy) and was solubilized in dimethylsulfoxide
(DMSO; Sigma) before adding to cells. Trypan blue and
the CellTrace CSFE Cell Proliferation kit (C34554) were
from Gibco BRL (Invitrogen, Monza, Italy). BSA was
from Santa Cruz Biotechnology (Santa Cruz, California,
USA). The fluorescein isothiocyanate (FITC) Annexin V
apoptosis detection kit and the cytofix/cytoperm kit were
from BD (Milan, Italy).
The following antibodies were used: phycoerythrin (PE)-
conjugated anti-human TrKA mouse monoclonal antibody
(R&D Systems, Abingdon, Virginia, USA); FITC-con-
jugated anti-human p75NTR mouse monoclonal antibody
(Alomone Labs); PE-conjugated anti-AKT mouse mono-
clonal antibody; and PE-conjugated anti-active caspase-3
rabbit monoclonal antibody (BD).
Cells were cultured in minimum essential medium
(MEM), containing 1 g/l glucose and supplemented with
antibiotic/antimycotic, L-glutamine (2 mmol/l), MEM
2 Anti-Cancer Drugs 2013, Vol 00 No 00

nonessential amino acid solutions (100�), sodium pyr-
uvate, and 10% fetal bovine serum. Cells were harvested
in Dulbecco’s phosphate-buffered saline (D-PBS) con-
taining 2 mmol/l EDTA. All culture media and supple-
ments were from Gibco BRL (Invitrogen).
Cancer cell lines
The HTB114 human leiomyosarcoma cell line, derived
from human uterine leiomyosarcoma, was from the
American Type Culture Collection (ATCC, Manassas,
Virginia, USA) and was cultured in MEM medium
according to ATCC instructions.
Amitriptyline treatment
For all experiments, cells were seeded in duplicate in six-
multiwell plates (Corning, Milan, Italy), at a density of
100 000 cells/well, and cultured for 24 h at 371C in a
humidified atmosphere of 5% CO2 to promote adhesion.
Amitriptyline, or its vehicle DMSO, was then added to
the culture medium at the indicated concentrations, and
cells were further incubated for the indicated times (see
below). Cells were then harvested, using 2 mmol/l EDTA
in D-PBS (D-PBS/EDTA), and analyzed, as described
below. All experiments included positive controls, treated
in parallel with b-NGF (10 ng/ml).
CFSE proliferation assay and vital counts
Cell cultures were set up in parallel for the carboxy-
fluorescein succinimidyl ester (CFSE) proliferation assay
and vital counts, as described, to have comparable
conditions. For the proliferation assay, cells were pre-
labeled with the CSFE probe, according to the manu-
facturer’s instructions, before plating, and then seeded
into multiwells. After 24 h culture, serial concentrations
of amitriptyline (0.5, 50, 100, 500, and 1000 mmol/l), or its
vehicle DMSO, were added to the culture medium and
cells were further incubated for 24, 48, 72 h, and up to 7
days. Cells were then harvested in D-PBS/EDTA and
analyzed by cytofluorimetric [fluorescence-activated cell
sorting (FACS)] scan. Trypan blue vital count was
performed as described in Rende et al. [2].
Determination of apoptosis by Annexin V labeling
Cells were seeded as described. After 24 h, serial
concentrations (0.5, 50, 100, 500, and 1000 mmol/l) of
amitriptyline, or its vehicle DMSO, were then added to
the culture medium and the cells were further incubated
for 24, 48, and 72 h. Cells were then harvested, labeled
using the FITC Annexin V apoptosis detection kit,
according to the manufacturer’s instructions, and ana-
lyzed by FACS scan.
Determination of NGF-receptor expression
Cells were seeded as described. After 24 h, 50 mmol/l
amitriptyline, or its vehicle DMSO, was added to the
culture medium, and cells were further incubated for 24,
48, and 72 h. Cells were then harvested, preincubated
with 0.5% BSA in D-PBS for 15 min at room temperature
to minimize unspecific staining, followed by centrifuga-
tion, and 1-h incubation at 41C with either the PE-
conjugated anti-human TrKA antibody (1:10) or the
FITC-conjugated anti-human p75NTR antibody (1:10),
in 0.5% BSA in D-PBS (D-PBS/BSA). Cells were then
washed in D-PBS/BSA and analyzed by FACS scan. All
experiments included negative controls, incubated with
human nonimmune immunoglobulins G (1:10).
Determination of caspase-3 and AKT activation
Cells were seeded as described. After 24 h, serial concentra-
tions (0.5, 50, 100, 500, and 1000mmol/l) of amitriptyline, or
its vehicle DMSO, were added to the culture medium, and
cells were further incubated for 24, 48, and 72 h. Cells were
then harvested in D-PBS/EDTA and fixed-permeabilized
using the cytofix/cytoperm kit according to the manufac-
turer’s instructions. Cells were then incubated for 1 h at 41C
with either the PE-conjugated anti-AKTantibody or the PE-
conjugated anti-active caspase-3 antibody in perm/wash
buffer (from the same kit). Samples were then rinsed in
the same buffer and analyzed by FACS scan.
Statistical analysis
Duplicate samples were used in each experiment. Data
are expressed as mean±SD of three separate experi-
ments. Student’s t-test was used to compare two groups.
A probability value of P less than 0.05 indicates statistical
significance.
ResultsAmitriptyline reduces cell viability and proliferation,
and induces apoptosis in HTB114 cells.
Cell viability and proliferation were analyzed by cell counts
and CSFE assay, respectively (Fig. 1). As shown in Fig. 1a,
a trend toward reduced cell viability was already apparent at
24 h in cells treated with 50mmol/l amitriptyline. The cell
viability decreased below 15% of controls (13%, P < 0.05)
at 48 h and approached 0% (2%, P < 0.05) at 72 h (Fig. 1b).
The IC50 was B4mmol/l at 48 h (Fig. 1b).
Cytotoxicity was confirmed by parallel CFSE proliferation
assays. The proliferative index is calculated by measuring
the reduction in the mean fluorescence intensity of cells
labeled with the fluorescent dye CSFE, which is
consequent to the dilution of the dye after cell division.
Measurement of the duplication rate of viable cells allows
a better estimate of the antiproliferative effect, without
interference of cell death. As shown in Fig. 1c, amitripty-
line significantly reduced proliferation at 48 h versus
controls and almost suppressed it (– 92.5%, P < 0.05) in
cells exposed to 500 mmol/l drug for 72 h. The dose–
response curves seem to show a more prominent effect on
cell viability as compared with cell proliferation (see the
Discussion section).
The analysis of apoptosis, using the Annexin V assay
(Fig. 2), confirmed the antineoplastic effect. Apoptosis was
Antineoplastic activity of amitriptyline Pula et al. 3

time and dose dependent, starting at 48 h and reaching
a plateau at 72 h, when greater than 80% of the cells exposed
to 100mmol/l drug were apoptotic. The dose–response
curves are in agreement with proliferation data.
In contrast, exogenous b-NGF (10 ng/ml) (Supplemental
Fig. 1, http://links.lww.com/ACD/A12) slightly increased cell
proliferation (+ 29% vs. controls at 48 h, P < 0.05%, data
not shown) and a trend toward increased cell counts
(+ 15% vs. controls at 48 h) was also observed, confirming
its mitogenic role in HTB114 cells. The limited effect
suggests that autocrine secretion is already sufficient to
sustain growth and survival in these cells. In agreement
with our previous data, no apoptosis was observed in cells
exposed to exogenous b-NGF for up to 7 days.
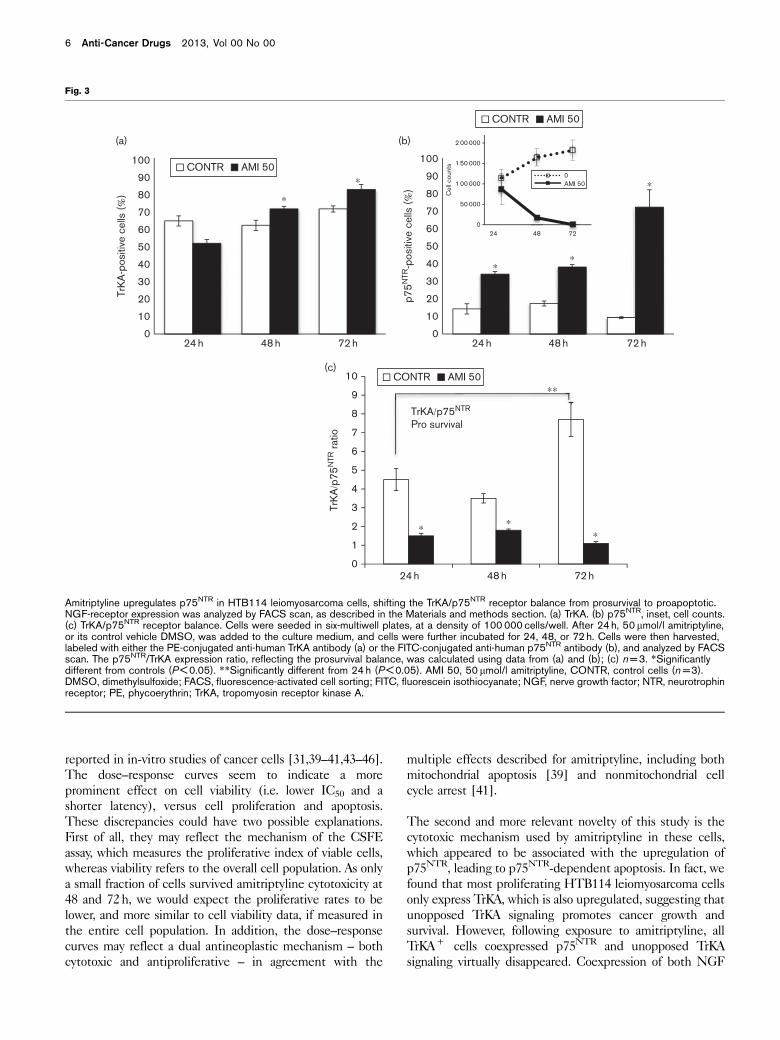
Amitriptyline upregulates p75NTR expression, converting
the TrKA + HTB114 cells into TrKA + /p75NTR + ,
and inducing cell death.
Our previous work has shown that, although expressing
both NGF receptors, HTB114 cells constitutively upre-
gulate TrKA while downregulating p75NTR [2–4]. FACS
analysis confirmed the high TrKA/p75NTR prosurvival ratio
(64/14% at 24 h) of proliferating control cells (Fig. 3a and
b), which was further increased as cells progressed along
the log-growth phase (Fig. 3c). In contrast, exposure to
50mmol/l amitriptyline upregulated p75NTR time depen-
dently (Fig. 3). The effect was already significant at 24 h,
versus controls (34 vs. 14%, P < 0.05, respectively), and
was further increased at 72 h, both versus 24 h (72 vs. 34%,
P < 0.05, respectively) and versus controls (72 vs. 9.3%,
P < 0.05, respectively). Thus, whereas proliferating cells
‘switched off ’ their p75NTR (9.9%, at 72 h), the cells
exposed to amitriptyline ‘switched it on’ to almost the
same levels of TrKA (72 vs. 82% at 72 h, respectively).
This ‘switch’ led to a marked reduction in the TrKA/
p75NTR prosurvival ratio at 72 h versus controls (1.1 vs.
7.8, P < 0.05, respectively, Fig. 3c).
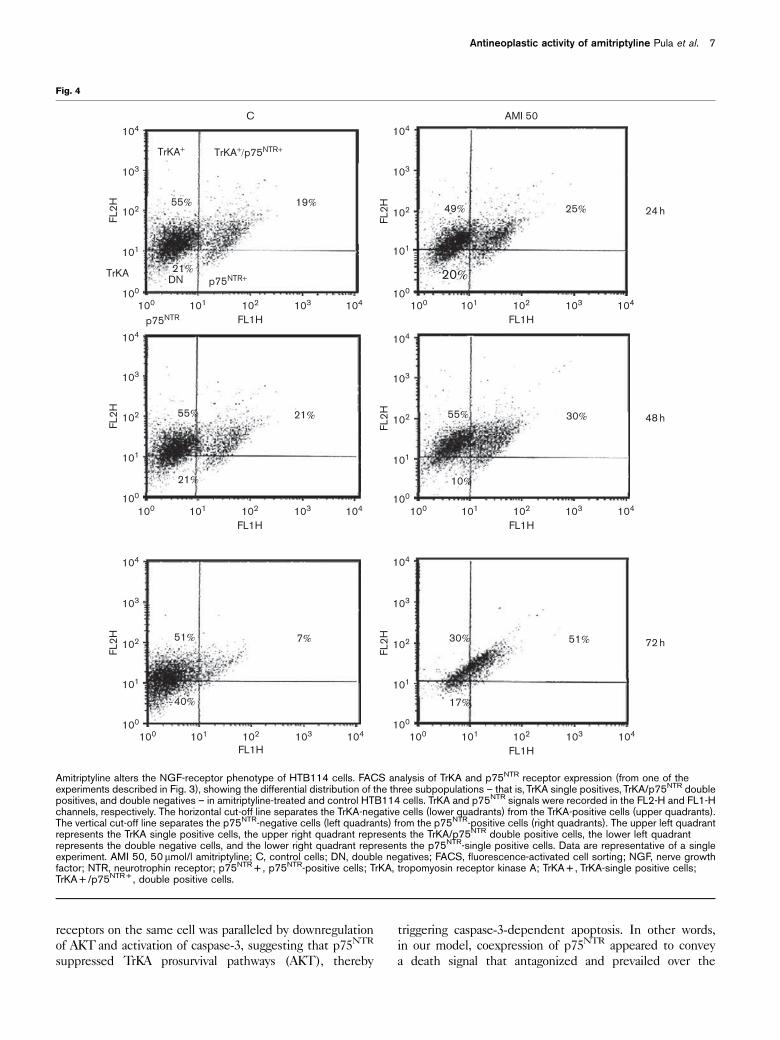
Interestingly, FACS analysis also showed that p75NTR is
always coexpressed with TrKA (Fig. 4), in HTB114 cells,
and therefore the double positives (TrKA + /p75NTR + )
Fig. 1
∗
∗
∗∗
∗
∗
∗
0
50 000
100 000
150 000
200 000
0 h 24 h 48 h 72 h
cell
coun
ts (c
ells
/ml)
CONTRAMI 50NGF
∗∗
0.010.020.030.040.050.060.070.080.090.0
100.0
0 50 500 1000
prol
ifera
tive
inde
x (%
of c
ontr
ols)
Amitriptyline (μmol/l)
48 h72 h
0.010.020.030.040.050.060.070.080.090.0
100.0
1 10 100 1000
Via
bilit
y (%
of c
ontr
ols)
Amitriptyline (μmol/l)
24 h
48 h
72 h
∗
∗
IC50
0
(a)
(b)
(c)
Amitriptyline reduces HTB114 leiomyosarcoma cell viability and proliferation. Cell viability and proliferation were analyzed in parallel by vital counts(a, b) and the CFSE proliferation assay (c). Cells were seeded in six-multiwell plates, at a density of 100 000 cells/well. After 24 h culture, serialconcentrations of amitriptyline, or its control vehicle DMSO, were added to the culture medium and cells were further incubated for 24, 48, or 72 h.Cells were then harvested and analyzed as described in the Materials and methods section. Cells treated with b-NGF (10 ng/ml) were used aspositive controls. Data in (b) and (c) have been normalized to controls (n = 3). *Significantly different from controls (P < 0.05). **Significantly differentfrom NGF (P < 0.05). AMI 50, 50 mmol/l amitriptyline; CONTR, control cells; CFSE, carboxyfluorescein succinimidyl ester; DMSO, dimethylsulfoxide;IC50, drug concentration reducing cell viability by 50%; NGF, nerve growth factor.
4 Anti-Cancer Drugs 2013, Vol 00 No 00

represent a subpopulation of TrKA + cells. This led to the
identification of three different subpopulations (or
phenotypes), on the basis of the expression of different
combinations of NGF receptors (Fig. 5), namely: (a) the
double positives (TrKA + /p75NTR + ); (b) the TrKA + -
single positives; and (c) the double negatives (TrKA – /
p75NTR – ). The distribution of the three phenotypes in
the 24 h-control population was B14/50/35%, respec-
tively. However, these figures changed significantly in
cells exposed to amitriptyline and b-NGF, as shown
in Table 1 and Fig. 5.
The high-TrKA/p75NTR ratio of proliferating control cells,
reflecting the high proportion of TrKA + -single positives
(Fig. 5), suggests that unopposed TrKA signaling can
boost cell growth. In fact, proliferating control cells
progressively upregulated their overall TrKA (71%, at
72 h) while further suppressing their already low p75NTR
(9.3%, at 72 h). Therefore, the TrKA + -single positives
increased accordingly (61.7 vs. 50%, P < 0.05, at 72 vs.
24 h, respectively), at the expense of the other two
subpopulations, namely, the double positives (9 vs. 14%,
at 72 vs. 24 h, respectively) and the double negatives (29
vs. 36%, P < 0.05, at 72 vs. 24 h, respectively). In contrast,
amitriptyline reduced the TrKA/p75NTR ratio to B1 : 1
(1.1) at 72 h, meaning that almost all TrKA + cells
coexpressed p75NTR (Fig. 5). This phenotype switch
occurred at the expense of the TrKA + -single positives
(the proliferating cells), which decreased to 10% at 72 h,
from 61.7% of controls (P < 0.05). In other words,
amitriptyline converted the HTB114 cells from prolifer-
ating TrKA + -single positives into ‘agonizing’ double
positives (72 vs. 9.3%, P < 0.05, amitriptyline-treated
vs. control cells double positives at 72 h, respectively),
suggesting that coexpression of p75NTR interferes with
TrKA prosurvival signaling, inducing cell death.
As expected, exogenous b-NGF upregulated both the
overall TrKA expression (81.7%, P < 0.05, at 24 h,
Supplemental Fig. 1, http://links.lww.com/ACD/A12) and
the proportion of TrKA + -single positives, thereby con-
firming its mitogenic role. Because of the rapid extra-
cellular degradation of b-NGF, the effect was transient
and peaked at 24 h.
Coexpression of p75NTR in TrKA + -HTB114 cells, induced
by amitriptyline, downregulated AKT and activated
caspase-3
To elucidate the mechanism of action of amitriptyline, we
next analyzed the downstream effectors of TrKA and
p75NTR receptors by FACS analysis (Fig. 6). As already
mentioned, in HTB114 cells, TrKA-dependent survival
involves AKT, whereas p75NTR-dependent apoptosis is
associated with the activation of caspase-3 [3,4]. We
found that, in the proliferating control cell population,
composed of about 50% of TrKA + -single positives, AKT
was constitutively ‘on’ (38%), whereas caspase-3 was
almost suppressed (4%). As expected, the coexpression of
p75NTR, induced by amitriptyline, dose dependently
‘switched on’ caspase-3 (25% at 500 mmol/l) while
‘switching off ’ AKT (15% at 500 mmol/l). These effects
started at 24 h and reflected the upregulation of p75NTR,
supporting the hypothesis that coexpression of this
receptor negatively regulates TrKA signaling, switching
off its survival pathways. These data confirm our previous
observations of caspase-3-dependent apoptosis induced
by upregulation of p75NTR in HTB114 cells.
As expected, exogenous b-NGF (10 ng/ml) significantly
upregulated AKT at 1 h and up to 24 h (Supplemental
Figs 1 and 2, http://links.lww.com/ACD/A12, http://links.lww.com/ACD/A13), reflecting TrKA expression and further
confirming its prosurvival role in these cells. The effect
was transient because of the rapid degradation of NGF
in the culture medium (data not shown).
DiscussionOur study shows considerable in-vitro activity of the TCA
amitriptyline in the HTB114 uterine leiomyosarcoma cell
line, confirming the antineoplastic potential of this drug.
This is the first report of amitriptyline cytotoxicity to
human uterine leiomyosarcoma, which is an aggressive
cancer, and highly resistant to nonsurgical therapies.
In our in-vitro model, amitriptyline (50 mmol/l) markedly
suppressed cell viability and proliferation, and induced
apoptosis. Cytotoxicity was dose and time dependent.
The concentrations used in this report are in the range
Fig. 2
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
90.0
100.0
10 100 1000
Apo
ptos
is (%
)
Amitriptyline (μmol/l)
24 h48 h72 h96 h7 days
0
Amitriptyline induces time-dependent and dose-dependent apoptosisin HTB114 leiomyosarcoma cells. Apoptosis was determined by theAnnexin V assay, as described in the Materials and methods section.Cells were seeded in six-multiwell plates, at a density of 100 000 cells/well. After 24 h, serial concentrations of amitriptyline (50, 100, 500, and1000mmol/l), or its vehicle DMSO, were added to the culture mediumand cells were further incubated for 24, 48, 72, 96 h, and up to 7 days.Cells were then harvested, labeled with FITC-conjugated Annexin V,and analyzed by FACS scan (n = 3). DMSO, dimethylsulfoxide;FACS, fluorescence-activated cell sorting; FITC, fluorescein isothiocyanate.
Antineoplastic activity of amitriptyline Pula et al. 5
reported in in-vitro studies of cancer cells [31,39–41,43–46].
The dose–response curves seem to indicate a more
prominent effect on cell viability (i.e. lower IC50 and a
shorter latency), versus cell proliferation and apoptosis.
These discrepancies could have two possible explanations.
First of all, they may reflect the mechanism of the CSFE
assay, which measures the proliferative index of viable cells,
whereas viability refers to the overall cell population. As only
a small fraction of cells survived amitriptyline cytotoxicity at
48 and 72 h, we would expect the proliferative rates to be
lower, and more similar to cell viability data, if measured in
the entire cell population. In addition, the dose–response
curves may reflect a dual antineoplastic mechanism – both
cytotoxic and antiproliferative – in agreement with the
multiple effects described for amitriptyline, including both
mitochondrial apoptosis [39] and nonmitochondrial cell
cycle arrest [41].
The second and more relevant novelty of this study is the
cytotoxic mechanism used by amitriptyline in these cells,
which appeared to be associated with the upregulation of
p75NTR, leading to p75NTR-dependent apoptosis. In fact, we
found that most proliferating HTB114 leiomyosarcoma cells
only express TrKA, which is also upregulated, suggesting that
unopposed TrKA signaling promotes cancer growth and
survival. However, following exposure to amitriptyline, all
TrKA+ cells coexpressed p75NTR and unopposed TrKA
signaling virtually disappeared. Coexpression of both NGF
Fig. 3
0
10
20
30
40
50
60
70
80
90
100
0
10
20
30
40
50
60
70
80
90
100
2 00 000
0AMI 50
1 50 000
1 00 000
50 000
024 48 72
24 h 48 h 72 h
24 h 48 h 72 h
TrK
A-p
ositi
ve c
ells
(%)
p75N
TR-p
ositi
ve c
ells
(%)
CONTR AMI 50
3438
24 h 48 h 72 h
∗
∗∗
∗
∗
Cel
l cou
nts
0
1
2
3
4
5
6
7
8
9
10
TrKA/p75NTR
Pro survival
TrK
A/p
75N
TR ra
tio
∗ ∗∗
∗∗
CONTR AMI 50
CONTR AMI 50
(a) (b)
(c)
Amitriptyline upregulates p75NTR in HTB114 leiomyosarcoma cells, shifting the TrKA/p75NTR receptor balance from prosurvival to proapoptotic.NGF-receptor expression was analyzed by FACS scan, as described in the Materials and methods section. (a) TrKA. (b) p75NTR, inset, cell counts.(c) TrKA/p75NTR receptor balance. Cells were seeded in six-multiwell plates, at a density of 100 000 cells/well. After 24 h, 50mmol/l amitriptyline,or its control vehicle DMSO, was added to the culture medium, and cells were further incubated for 24, 48, or 72 h. Cells were then harvested,labeled with either the PE-conjugated anti-human TrKA antibody (a) or the FITC-conjugated anti-human p75NTR antibody (b), and analyzed by FACSscan. The p75NTR/TrKA expression ratio, reflecting the prosurvival balance, was calculated using data from (a) and (b); (c) n = 3. *Significantlydifferent from controls (P < 0.05). **Significantly different from 24 h (P < 0.05). AMI 50, 50 mmol/l amitriptyline, CONTR, control cells (n = 3).DMSO, dimethylsulfoxide; FACS, fluorescence-activated cell sorting; FITC, fluorescein isothiocyanate; NGF, nerve growth factor; NTR, neurotrophinreceptor; PE, phycoerythrin; TrKA, tropomyosin receptor kinase A.
6 Anti-Cancer Drugs 2013, Vol 00 No 00
receptors on the same cell was paralleled by downregulation
of AKT and activation of caspase-3, suggesting that p75NTR
suppressed TrKA prosurvival pathways (AKT), thereby
triggering caspase-3-dependent apoptosis. In other words,
in our model, coexpression of p75NTR appeared to convey
a death signal that antagonized and prevailed over the
Fig. 4
TrKA+
21%
55%
21%
55%
19%
21%
49% 25%
20%TrKA
FL1H FL1H
FL2H
FL2H
FL2H
DN
TrKA+/p75NTR+
p75NTR+
p75NTR
C AMI 50104
103
102
101
100
100100
100101 102 103 104
FL1H100
100
101
101
102
102
103
103
104
104
10%
55% 30%
FL2H
FL1H100
100
101
101
102
102
103
103
104
104
40%
51% 7%
FL2H
FL1H100
100
101
101
102
102
103
103
104
104
17%
30% 51% 72 h
48 h
24 h
FL2H
FL1H100
100
101
101
102
102
103
103
104
104
104
103
102
101
101 102 103 104
Amitriptyline alters the NGF-receptor phenotype of HTB114 cells. FACS analysis of TrKA and p75NTR receptor expression (from one of theexperiments described in Fig. 3), showing the differential distribution of the three subpopulations – that is, TrKA single positives, TrKA/p75NTR doublepositives, and double negatives – in amitriptyline-treated and control HTB114 cells. TrKA and p75NTR signals were recorded in the FL2-H and FL1-Hchannels, respectively. The horizontal cut-off line separates the TrKA-negative cells (lower quadrants) from the TrKA-positive cells (upper quadrants).The vertical cut-off line separates the p75NTR-negative cells (left quadrants) from the p75NTR-positive cells (right quadrants). The upper left quadrantrepresents the TrKA single positive cells, the upper right quadrant represents the TrKA/p75NTR double positive cells, the lower left quadrantrepresents the double negative cells, and the lower right quadrant represents the p75NTR-single positive cells. Data are representative of a singleexperiment. AMI 50, 50mmol/l amitriptyline; C, control cells; DN, double negatives; FACS, fluorescence-activated cell sorting; NGF, nerve growthfactor; NTR, neurotrophin receptor; p75NTR + , p75NTR-positive cells; TrKA, tropomyosin receptor kinase A; TrKA + , TrKA-single positive cells;TrKA + /p75NTR + , double positive cells.
Antineoplastic activity of amitriptyline Pula et al. 7

prosurvival pathways of NGF/TrKA, providing further insight
into NGF receptor crosstalk. Although we have already
described inducible p75NTR-dependent apoptosis in
HTB114 uterine leiomyosarcoma [2–4], this mechanism
has never been proposed for amitriptyline.
In support of our hypothesis, we also found that
coadministration of noncytotoxic concentrations of ami-
triptyline and exogenous b-NGF suppressed the mito-
genic effects of the latter in HTB114 cells. This
antiproliferative effect was again associated with down-
regulation of AKT and upregulation of both p75NTR
expression and apoptosis (Supplemental Fig. 2, http://links.lww.com/ACD/A13), suggesting functional antagonism.
Although this scenario is different from that described in
PC12 cells [47], distinct intracellular pathways may be
involved.
Overall, our data suggest that p75NTR-dependent apop-
tosis may represent a novel cytotoxic mechanism of
amitriptyline in HTB114 cells and possibly in human
uterine leiomyosarcomas in general (and other cancers
alike, Supplemental Fig. 3, http://links.lww.com/ACD/A14).
As p75NTR-dependent apoptosis is also activated by
Fig. 5
TrKA p75NTR
Survival and growth
p75NTR−
dependentapoptosis
24 h48 h
72 h
0
10
20
30
40
50
60
70
80
90
100
C AMI 50 C AMI 50 C AMI 50
TrKA+ TrKA+/p75NTR+ TrkA−/p75NTR−
Pos
itive
cel
ls (%
)
24 h
48 h
72 h
∗
∗
∗
∗ ∗
∗
∗
∗∗
∗∗ ∗∗
Amitriptyline converts HTB114 cells from proliferating TrKA + -singlepositives into apoptotic TrKA + /p75NTR + -double positives.NGF-receptor expression in amitriptyline-treated and control cells.Data from Table 1 have been plotted to show the effects of amitriptylineon TrKA + cells. *Significantly different from controls (P < 0.05).**Significantly different from 24 h (P < 0.05). AMI 50, 50mmol/lamitriptyline; C, control cells (n = 3); NGF, nerve growth factor;NTR, neurotrophin receptor; TrKA, tropomyosin receptor kinase A.
Table 1 Expression of NGF receptors in HTB114 cells exposed to 50 lmol/l amitriptyline
TrKA + TrKA + /p75NTR + TrKA-/p75NTR –
C AMI 50 C AMI 50 C AMI 50
24 h 50.0 (±2.9) 17.5 (±3.5)* 14.2 (±1.7) 34.0 (±1.4)* 35.7 (±2.9)** 48.5 (±2.1)*48 h 44.3 (±3.2) 33.0 (±2.8)* 17.3 (±0.6) 38.0 (±1.4)* 38.3 (±2.9)** 29.0 (±1.4)*72 h 61.7 (±1.5)** 10.0 (±7.1)*, ** 9.3 (±1.15)** 72.0 (±9.9)* 29.0 (±1.7)** 18.0 (±2.8)*, **
Expression of NGF receptors in control and amitriptyline-treated HTB114 human uterine leiomyosarcoma cells. Cells were seeded into six-multiwell plates, at a density of100 000 cells/well. After 24 h culture, 50 mmol/l amitriptyline, or its control vehicle DMSO, was added to the culture medium. Cells were further incubated for theindicated times and analyzed by FACS scan, as described in the Materials and methods section.Data are expressed as percentage of positive cells in the overall population±SD.AMI 50, amitriptyline 50 mmol/l; C, controls (n = 3); DMSO, dimethylsulfoxide; FACS, fluorescence-activated cell sorting; NGF, nerve growth factor; NTR, neurotrophinreceptor; TrKA, tropomyosin receptor kinase A.*P < 0.05 versus controls.**P < 0.05 versus 24 h.
Fig. 6
0
10
20
30
40
50
60
70
80
90
100
0 0.5 50 500 1000
Pos
itive
cel
ls (%
)
Amitriptyline (μmol/l)
24 h
AKTCASPASE-3
∗ ∗∗∗
∗ ∗
The coexpression of p75NTR and TrKA downregulates AKT andactivates caspase-3, inducing death in HTB114 cells. The expressionof AKT and active caspase-3 was analyzed by FACS scan, as describedin the Materials and methods section. Cells were seeded in six-multiwellplates, at a density of 100 000 cells/well. After 24 h, 50 mmol/lamitriptyline, or its control vehicle DMSO, was added to the culturemedium and cells were further cultured for 24 h. Cells were thenharvested, fixed-permeabilized, incubated with either anti-AKT oranti-active caspase-3 PE-conjugated antibodies, and analyzed byFACS scan (n = 3). *Significantly different from controls (P < 0.05).DMSO, dimethylsulfoxide; FACS, fluorescence-activated cell sorting;NTR, neurotrophin receptor; PE, phycoerythrin; TrKA, tropomyosinreceptor kinase A.
8 Anti-Cancer Drugs 2013, Vol 00 No 00

different cytotoxic agents (Trk inhibitors) [3,4],
we propose that the activation of this inducible death
receptor (activated by cellular stressors) may represent
a potential therapeutic strategy in susceptible cancers
(Fig. 7).
As already mentioned, the role of p75NTR is still
enigmatic, as it can modulate divergent signaling path-
ways by forming complexes with different coreceptors to
induce specific responses [11,12]. Depending on the cell
type, p75NTR can act synergistically, antagonistically, or
independently from TrKs. In the presence of mature
NTs, it can function as a coreceptor for the TrKs,
increasing their selectivity and prosurvival signaling. In
central neurons, it can form trimeric complexes with the
Nogo receptor-1 (NgR1) and the leucine-rich repeat and
Ig domain containing 1 (Lingo-1) receptor, which can
suppress neurite outgrowth through the Ras homolog
gene family member A (RhoA) [15]. Apoptosis can be
induced in different ways, including forming complexes
with sortilin, which are activated by pro-NTs [15], or
acting as a dependence receptor activated by the absence
of ligand (e.g. apoptosis by NTs withdrawal), or binding
non-NT ligands produced by pathogenic processes (e.g.
prion peptides, viruses, b-amyloid peptides). Thus,
p75NTR-dependent apoptosis can be triggered either by
its ligands (i.e. pro-NTs, mature NTs, or non-NT ligands)
or by their absence (e.g. NTs withdrawal) depending on
the extracellular signals and on the signaling apparatus
expressed by the cell.
In the nervous system, p75NTR-dependent apoptosis is
induced in response to damage. This receptor is ex-
pressed during neural development and ‘switched off ’ in
adult life, but it is re-expressed with neuronal injury and
death [23], suggesting the reactivation of a develop-
mental program by lesion-induced plasticity. Multiple
cellular stressors can induce re-expression of p75NTR in
neuronal and glial cells, which is sometimes associated
with the upregulation of pro-NTs [48]. In this setting,
the ‘toxic’ effects of pro-NTs seem to predominate over
the ‘trophic’ effect of mature NTs, despite the presence
of functional TrKs. These observations have led to the
hypothesis that p75NTR-dependent apoptosis could
represent a homeostatic mechanism to eliminate da-
maged cells – similar to Fas-dependent apoptosis
associated with inflammation – and possibly a ‘class
effect’ of death receptors [23].
Fig. 7
TrKA p75NTR
Autocrine NGF release
AmitriptylineTrK blockade
p75NTR gene transfer
TrKA
TRKA - dependentproliferation and survival
p75NTR - dependent apoptosisand cell death
AKT ‘on’caspase -3 ‘off’
Oncogenictransformation
Suppressionof cancer growth
Caspase-3 ‘on’
TrKA
NGF
HTB114 human uterine leiomyosarcoma
In HTB114 uterine leiomyosarcoma cells, p75NTR behaves as a stress/death receptor, which is inducible by cytotoxic drugs to trigger cell death.HTB114 cells are characterized by autocrine activation of the NGF/TrKA/AKT axis, leading to constitutive upregulation of TrKA and downregulationof p75NTR, which promotes unrestricted growth and survival. In these cells, the upregulation of p75NTR by different cytotoxic drugs, includingamitriptyline or TrK inhibitors [2,3], or by gene transfer [2], induces apoptosis and cell death. We therefore suggest that p75NTR acts as a stressreceptor, inducible by cellular injury, which can be pharmacologically upregulated to target the survival pathways of susceptible cancer cells.NGF, nerve growth factor; NTR, neurotrophin receptor; TrKA, tropomyosin receptor kinase A.
Antineoplastic activity of amitriptyline Pula et al. 9

We propose that this same ‘homeostatic’ mechanism of
p75NTR-dependent apoptosis could also be used to kill
cancer cells. In fact, re-expression of p75NTR can suppress
growth in tumors characterized by constitutive upregula-
tion of the TrKs and downregulation of p75NTR (e.g.
uterine leiomyosarcoma, prostatic adenocarcinoma, gas-
tric cancer, hepatocellular carcinoma, and bladder can-
cer [2–4,20,49–51]). Indeed, we found that PC3 prostatic
carcinoma was also sensitive to amitriptyline (Supple-
mental Fig. 3, http://links.lww.com/ACD/A14). As the same
mechanism of p75NTR-dependent apoptosis seems to be
inducible by both cellular injury and unrelated cytotoxic
agents, in neural and extraneural cells, it might represent
a conserved ‘final common pathway’ to trigger cell suicide
in dysfunctional cells.
Our data provide a rationale for investigating p75NTR-
dependent apoptosis in susceptible cancers. However, this
strategy represents both an opportunity and a challenge.
First of all, the molecular mechanisms leading to the
upregulation of p75NTR warrant further investigation. In
neurons, the hypo-osmotic stress can upregulate p75NTR
through Sp1 [52], a transcription factor expressed in
development and cancer, and regulating genes that control
differentiation, proliferation, and apoptosis [53]. Brain
injury has recently been shown to upregulate p75NTR
through the proinflammatory cytokines interleukin-1b and
tumor necrosis factor-alpha (TNF-alpha) [54]; the effect
is both cytokine and cell type specific. Cytokine secretion
has been documented in the activated myometrium,
where it seems to trigger delivery [55,56] (which is also
a cellular stress). In HTB114 cells, p75NTR can be
upregulated by both TrK blockade [3,4] and PI3K
inhibitors (our unpublished observations). Further, pro-
NGF can also upregulate p75NTR and induce apoptosis in
these cells (our unpublished observations). Secretion of
both pro-NGF and b-NGF has been documented in rat
myometrium and pro-NGF release increases at the end of
pregnancy (also a stressful condition), paralleled by the
decrease of b-NGF [57], thereby promoting delivery.
Thus, although the inhibition of TrK signaling and the
activation of caspase-3 can explain the p75NTR-dependent
apoptosis induced by amitriptyline in leiomyosarcoma
cells, we cannot rule out the possibility that other stress-
inducible factors, such as pro-NGF and/or cytokines,
might be released following exposure to the drug, further
contributing toward p75NTR-dependent apoptosis.
Second, as for any antineoplastic chemotherapy, concerns
might be raised about the potential side effects caused by
the pharmacological modulation of p75NTR in non-
neoplastic tissues. For example, the activation of the
NgR/Lingo-1/p75NTR receptor complex has been linked
to neurodegeneration. As already mentioned, this path-
way inhibits neurite outgrowth in the central nervous
system (CNS) and seems to be one of the reasons why
central neurons cannot regenerate following injury [58].
However, the role of p75NTR in neurodegenerative
diseases is still unclear. For example, in the C57BL/6
experimental autoimmune encephalomyelitis model – a
murine model for the human multiple sclerosis – p75NTR
knockout mice developed a more severe (or even lethal)
disease and concomitant CNS inflammation, compared
with wild-type mice [59]. Thus, despite the initial
enthusiasm for p75NTR, the studies targeting this path-
way in neurodegenerative diseases are currently focused
on Lingo and NgR [60–62]. In fact, while binding of
myelin inhibitory proteins to NgR1/Lingo-1 activates
RhoA, leading to inhibition of neurite outgrowth, binding
of NTs to p75NTR seems to suppress this effect [58,63],
suggesting that the functional role of this pathway may
depend on the extracellular ligands interacting with the
complex [64]. Moreover, functional redundancy has been
shown for p75NTR [65], which is generally absent in adult
neurons, where most NgR1/Lingo-1 complexes contain
its functional homologue TAJ/TROY – also a member of
the TNF receptor family [66]. Although we did not test
the effects of amitriptyline on TROY, this receptor does
not interact with the TrKs, whereas the upregulation of
p75NTR by cytotoxic drugs is apparently associated with
TrK inhibition ([4] and our unpublished observations).
Therefore, amitriptyline should not impact significantly
on this pathway in the adult CNS. In fact, neurodegen-
eration has never been reported in the postmarketing
experience with this drug.
Another potential safety concern, however, might be
associated with the risk of unwanted apoptosis in non-
neoplastic tissues. A potential pathway could be the
formation of p75NTR/sortilin complexes, which are
activated by pro-NTs. Sortilin-dependent apoptosis has
been studied in the nervous system. Our preliminary data
show that, despite a functional p75NTR/sortilin apparatus,
pro-NGF-dependent apoptosis is low in HTB114 cells,
especially compared with apoptosis associated with the
mere overexpression of p75NTR; furthermore, the two
pathways do not seem to be synergistic (our unpublished
observations). Although we did not test the effects of
amitriptyline on sortilin, we could find no evidence in the
literature of a potential modulation of sortilin by
antidepressants.
Although we have no direct evidence of amitriptyline (or
other antidepressants) cytotoxicity in non-neoplastic cells
through upregulation of p75NTR, unwanted apoptosis could
be a possibility, as for any systemic chemotherapy. The
animal studies investigating the antineoplastic effects of
antidepressants did not report specific organ toxicity;
however, in-vitro cytotoxicity to non-neoplastic fibroblasts
is described at the concentrations used in our report [67].
The interactions between antidepressants and NTs have
only been studied in the brain, where they have been
shown to affect NT levels. The only report investigating
the potential modulation of NT receptors [47] was limited
10 Anti-Cancer Drugs 2013, Vol 00 No 00

to mouse brain and found that amitriptyline binds neuronal
TrK-A/B, but does not bind p75NTR. The drugs were used
at noncytotoxic concentrations (i.e. 500mmol/l in vitro), and
apoptosis was not observed. Otherwise, there are no data on
a potential modulation of p75NTR by these drugs. However,
it is also possible that p75NTR-dependent apoptosis might
be a ‘homeostatic’ mechanism related to development or
cellular dysfunction, such as cancer. In fact, we have
preliminary evidence that, in certain human pathologies,
associated with cellular stress and severe inflammation,
p75NTR is upregulated but does not induce apoptosis (our
unpublished observations). This has also been shown in
oligodendrocytes, where p75NTR induces apoptosis under
specific conditions (i.e. after spinal cord injury), but does
not invariably lead to cell death, suggesting that the cellular
response to p75NTR may depend on the state of maturation
or activation of the cell [15]. In fact, although we did not
test non-neoplastic cells, we also found variable in-vitro
modulation of p75NTR expression and apoptosis by
amitriptyline in other cancer cell lines, suggesting that
the effects of the drug are dependent on the cell line (our
unpublished observations).
Conclusion
Our study shows considerable in-vitro antineoplastic
activity of the TCA amitriptyline in the HTB114 uterine
leiomyosarcoma cell line and provides novel evidence that
cytotoxicity is mediated by p75NTR-dependent apoptosis.
Our data suggest that p75NTR-dependent apoptosis may
be a novel strategy in cancer therapy in uterine
leiomyosarcoma, and possibly other cancers, characterized
by autocrine activation of TrKs. Further research in
xenograft models is warranted to confirm these findings
and to assess the safety profile of the drug for this
indication.
AcknowledgementsThis study was funded by Fondazione Cassa di Risparmio
di Terni (CARIT), Italy.
Conflicts of interest
There are no conflicts of interest.
References1 Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and
neural development. Cancer Lett 2001; 169:107–114.2 Rende M, Brizi E, Conner J, Provenzano C, Sanna PP. NGF influences
differentiation and proliferation of myogenic cells in vitro via TrKA. Int J DevNeurosci 2000; 18:869–885.
3 Rende M, Pistilli A, Stabile AM, Terenzi A, Cattaneo A, Ugolini G, Sanna P.Role of NGF and its receptors in non-nervous cancer growth: efficacy of atyrosine kinase inhibitor (AG879) and neutralizing antibodies anti-TrKA andanti-NGF: an in vitro and in vivo study. Anticancer Drugs 2006; 17:929–941.
4 Stabile AM, Montagnoli C, Pistilli A, Rambotti MG, Pula G, Rende M.Antiproliferative and pro-apoptotic effects of the Trk-inhibitor GW441756 inhuman myosarcomas and prostatic carcinoma. Curr Signal Transduct Ther2013; 8:74–83.
5 Molloy NH, Read DE, Gorman AM. Nerve growth factor in cancer cell deathand survival. Cancers 2011; 3:510–530.
6 Thiele CJ, Li Z, McKee AE. On Trk – the TrkB signal transduction pathway isan increasingly important target in cancer biology. Clin Cancer Res 2009;15:5962–5967.
7 Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosinekinases: targets for cancer therapy. Nat Rev Cancer 2004; 4:361–370.
8 Wang T, Yu D, Lamb ML. Trk kinase inhibitors as new treatments for cancerand pain. Expert Opin Ther Pat 2009; 3:305–319.
9 Skaper SD. The biology of neurotrophins: signaling pathways, and functionalpeptide mimetics of neurotrophins and their receptors. CNS Neurol DisordDrug Targets 2008; 7:46–62.
10 Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ.Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activationand the promotion of cell survival by nerve growth factor. Proc Natl Acad SciUSA 1997; 94:12419–12424.
11 Barrett GL. The p75 neurotrophin receptor and neuronal apoptosis. ProgNeurobiol 2000; 61:205–229.
12 Arevalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! CellMol Life Sci 2006; 63:1523–1537.
13 Kuci S, Kuci Z, Kreyenberg H, Deak E, Putsch K, Huenecke S, et al. CD271antigen defines a subset of multipotent stromal cells withimmunosuppressive and lympho-hematopoietic engraftment-promotingproperties. Haematologica 2010; 95:651–659.
14 Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP,et al. Human melanoma-initiating cells express neural crest nerve growthfactor receptor CD271. Nature 2010; 466:133–137.
15 Cragnolini A, Friedman WJ. The function of p75NTR in glia. Trends Neurosci2008; 31:99–104.
16 Segal RA, Greenberg ME. Intracellular signaling pathways activated byneurotrophic factors. Annu Rev Neurosci 1996; 19:463–489.
17 Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK,et al. The CD95 receptor: apoptosis revisited. Cell 2007; 129:447–450.
18 Lagadec C, Meignan S, Adriaenssens E, Foveau B, Vanhecke E, Romon R,et al. TrkA overexpression enhances growth and metastasis of breastcancer cells. Oncogene 2009; 28:1960–1970.
19 Lee J, Jiffar T, Kupferman ME. A novel role for BDNF-TrkB in the regulation ofchemotherapy resistance in head and neck squamous cell carcinoma. PLoSOne 2012; 7:e30246.
20 Papatsoris AG, Liolitsa D, Deliveliotis C. Manipulation of the nerve growthfactor network in prostate cancer. Expert Opin Investig Drugs 2007;44:303–309.
21 Werrbach-Perez K, Perez-Polo JR. De novo synthesis of NGF subunits inS-180 mouse sarcoma cell line. Neurochem Res 1987; 12:875–883.
22 Makino K, Kawamura K, Sato W, Kawamura N, Fujimoto T, Terada Y.Inhibition of uterine sarcoma cell growth through suppression ofendogenous tyrosine kinase B signaling. PLoS One 2012; 7:e41049.
23 Ibanez C, Simi A. p75 neurotrophin receptor signaling in nervous systeminjury and degeneration: paradox and opportunity. Trends Neurosci 2012;35:431–440.
24 Bhakar AL, Roux PP, Lachance C, Kryl D, Zeindler C, Barkeri PA. The p75neurotrophin receptor (p75NTR) alters tumor necrosis factor-mediated NF-kB activity under physiological conditions, but direct p75NTR-mediated NF-kB activation requires cell stress. J Biol Chem 1999; 274:21443–21449.
25 Tamim HM, Mahmud S, Hanley JA, Boivin JF, Stang MR, Collet JP.Antidepressants and risk of prostate cancer: a nested case–control study.Prostate Cancer Prostatic Dis 2007; 11:53–60.
26 Toh S, Garcia Rodriguez L, Hernandez-Dıaz S. Use of antidepressants andrisk of lung cancer. CCC 2007; 18:1055–1064.
27 Xu W, Tamim H, Shapiro S, Stang MR, Collet JP. Use of antidepressants andrisk of colorectal cancer: a nested case–control study. Lancet Oncol 2006;7:301–308.
28 Fulton-Kehoe D, Rossing MA, Rutter C, Mandelson MT, Weiss NS. Use ofantidepressant medications in relation to the incidence of breast cancer.Br J Cancer 2006; 94:1071–1078.
29 Walker AJ, Card T, Bates TE, Muir K. Tricyclic antidepressants and theincidence of certain cancers: a study using the GPRD. Br J Cancer 2011;104:193–197.
30 Chubak J, Boudreau DM, Rulyak SJ, Mandelson MT. Colorectal cancer riskin relation to antidepressant medication use. Int J Cancer 2011; 128:227–232.
31 Arimochi H, Morita K. Characterization of cytotoxic actions of tricyclicantidepressants on human HT29 colon carcinoma cells. Eur J Pharmacol2006; 541:17–23.
32 Xia Z, Bergstrand A, DePierre JW, Nassberger L. The antidepressantsimipramine, clomipramine, and citalopram induce apoptosis in human acutemyeloid leukemia HL-60 cells via caspase-3 activation. J Biochem MolToxicol 1999; 13:338–347.
Antineoplastic activity of amitriptyline Pula et al. 11

33 Daley E, Wilkie D, Loesch A, Hargreaves IP, Kendall DA, Pilkington GJ, et al.Chlorimipramine: a novel anticancer agent with a mitochondrial target.Biochem Biophys Res Commun 2005; 328:623–632.
34 Levkovitz Y, Gil-Ad I, Zeldich E, Dayag M, Weizman A. Differential inductionof apoptosis by antidepressants in glioma and neuroblastoma cell lines:evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J MolNeurosci 2005; 27:29–42.
35 Tsuruo T, Iida H, Nojiri M, Tsukagoshi S, Sakurai Y. Potentiation ofchemotherapeutic effect of vincristine in vincristine resistant tumor bearing miceby calmodulin inhibitor clomipramine. J Pharmacobiodyn 1983; 6:145–147.
36 Merry S, Hamilton TG, Flanigan P, Freshney RI, Kaye SB. Circumventionof pleiotropic drug resistance in subcutaneous tumors in vivo with verapamiland clomipramine. Eur J Cancer 1991; 27:31–34.
37 Pommerenke EW, Volm M. Reversal of doxorubicin-resistance in solidtumors by clomipramine. In Vivo 1995; 9:99–101.
38 Beaney RP, Gullan RW, Pilkington GJ. Therapeutic potential ofantidepressants in malignant glioma: clinical experience with clomipramine[abstract]. J Clin Oncol 2005; 23:1535.
39 Cordero MD, Sanchez-Alcazar JA, Bautista-Ferrufino MR, Carmona-Lopez MI,Illane M, Rıos MJ, et al. Acute oxidant damage promoted on cancer cells byamitriptyline in comparison with some common chemotherapeutic drugs.Anticancer Drugs 2010; 21:932–944.
40 Higgins SC, Pilkington GJ. The in vitro effects of tricyclic drugs anddexamethasone on cellular respiration of malignant glioma. Anticancer Res2010; 30:391–397.
41 Mao X, Hou T, Cao B, Wang W, Li Z, Chen S, et al. The tricyclicantidepressant amitriptyline inhibits D-cyclin transactivation and inducesmyeloma cell apoptosis by inhibiting histone deacetylases: in vitro and insilico evidence. Mol Pharmacol 2011; 79:672–680.
42 Jeon SH, Kim SH, Kim Y, Kim YS, Lim Y, Lee YH, et al. The tricyclicantidepressant imipramine induces autophagic cell death in U-87MGglioma cells. Biochem Biophys Res Commun 2011; 413:311–317.
43 Varga A, Nugel H, Baehr R, Marx U, Hever A, Nacsa J, et al. Reversal of multidrugresistance by amitriptyline in vitro. Anticancer Res 1996; 16:209–211.
44 Pilkington GJ, Parker K, Murray SA. Approaches to mitochondrially mediatedcancer therapy. Semin Cancer Biol 2008; 18:226–235.
45 Pilkington GJ, Akinwunmi J, Amar S. The role of tricyclic drugs in selectivetriggering of mitochondrially-mediated apoptosis in neoplastic glia: atherapeutic option in malignant glioma? Radiol Oncol 2006; 40:73–85.
46 Parker KA, Glaysher S, Hurren J, Knight LA, McCormick D, Suovouri A, et al.The effect of tricyclic antidepressants on cutaneous melanoma cell lines andprimary cell cultures. Anticancer Drugs 2012; 23:65–69.
47 Jang SW, Liu X, Chan CB, Weinshenker D, Hall RA, Xiao G, et al.Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkBheterodimerization and has potent neurotrophic activity. Chem Biol 2009;6:644–656.
48 Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR,Di Polo A, et al. ProNGF induces TNFa-dependent death of retinal ganglioncells through a p75NTR non-cell-autonomous signaling pathway. Proc NatlAcad Sci USA 2010; 107:3817–3822.
49 Jin H, Pan Y, Zhao L, Zhai H, Li X, Sun L, et al. P75 neurotrophin receptorsuppresses the proliferation of human gastric cancer cells. Neoplasia 2007;9:471–478.
50 Yuanlong H, Haifeng J, Xiaoyin Z, Jialin S, Jie L, Li Y, et al. The inhibitoryeffect of p75 neurotrophin receptor on growth of human hepatocellularcarcinoma cells. Cancer Lett 2008; 268:110–119.
51 Khwaja F, Djakiew D. Inhibition of cell-cycle effectors of proliferation inbladder tumor epithelial cells by the p75NTR tumor suppressor. MolCarcinog 2003; 36:153–160.
52 Ramos A, Ho WC, Forte S, Dickson K, Boutilier J, Favell K, et al. Hypo-osmolar stress induces p75NTR expression by activating Sp1-dependenttranscription. J Neurosci 2007; 27:1498–1506.
53 Deniaud E, Baguet J, Chalard R, Blanquier B, Brinza L, Meunier J, et al.Overexpression of transcription factor Sp1 leads to gene expressionperturbations and cell cycle inhibition. PLoS One 2009; 4:e7035.
54 Choi S, Friedman WJ. Inflammatory cytokines IL-1b and TNF-a regulatep75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signaling mechanisms. ASN Neuro 2009; 1:e00010.
55 Mittal P, Romero R, Tarca AL, Gonzalez J, Draghici S, Xu Y, et al.Characterization of the myometrial transcriptome and biological pathwaysof spontaneous human labor at term. J Perinat Med 2010; 38:617–643.
56 Shynlova O, Lee YH, Srikhajon K, Lye S. Physiologic uterine inflammationand labor onset: integration of endocrine and mechanical signals. ReprodSci 2013; 2:154–167.
57 Lobos E, Gebhardt C, Kluge A, Spanel-Borowski K. Expression of nervegrowth factor (NGF) isoforms in the rat uterus during pregnancy:accumulation of precursor proNGF. Endocrinology 2005; 146:1922–1929.
58 Mathew SJ, Haubert D, Kronke M, Leptin M. Looking beyond death: amorphogenetic role for the TNF signalling pathway. J Cell Sci 2009;122:1939–1946.
59 Kust B, Mantingh-Otter I, Boddeke E, Copray S. Deficient p75 low-affinityneurotrophin receptor expression does alter the composition of cellularinfiltrate in experimental autoimmune encephalomyelitis in C57BL/6 mice.J Neuroimmunol 2006; 174:92–100.
60 Jepson S, Vought B, Gross CH, Gan L, Austen D, Frantz JD, et al. LINGO-1,a transmembrane signaling protein, inhibits oligodendrocyte differentiationand myelination through intercellular self-interactions. J Biol Chem 2012;287:22184–22195.
61 Mi S, Sandrock A, Miller RH. LINGO-1 and its role in CNS repair. Int JBiochem Cell Biol 2008; 40:1971–1978.
62 McDonald CL, Bandtlow C, Reindl M. Targeting the Nogo receptor complexin diseases of the central nervous system. Curr Med Chem 2011; 18:234–244.
63 Barker PA. P75NTR is positively promiscuous: novel partners and newinsights. Neuron 2004; 42:529–533.
64 Blaise S, Kneib M, Rousseau A, Gambino F, Chenard MP, Messadeq N,et al. In vivo evidence that TRAF4 is required for central nervous systemmyelin homeostasis. PLoS One 2012; 7:e30917.
65 Mandemakers WJ, Barres BA. Axon regeneration: it’s getting crowded at thegates of TROY. Curr Biol 2005; 15:R302–R305.
66 Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, et al.TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor1 and regulates axonal regeneration. Neuron 2005; 45:353–359.
67 Moreno-Fernandez AM, Cordero MD, De Miguel M, Delgado-Rufino MD,Sanchez-Alcazar JA, Navas P. Cytotoxic effects of amitriptyline in humanfibroblasts. Toxicology 2008; 243:51–58.
12 Anti-Cancer Drugs 2013, Vol 00 No 00




















