
Membrane microdomains, caveolae, and caveolar endocytosis of sphingolipids (Review)
Upload
independentCategory
view
1download
0
MOLECULAR AND CELLULAR BIOLOGY, July 2006, p. 4970–4981 Vol. 26, No. 130270-7306/06/$08.00�0 doi:10.1128/MCB.00308-06Copyright © 2006, American Society for Microbiology. All Rights Reserved.
ST6Gal-I Restrains CD22-Dependent Antigen Receptor Endocytosis andShp-1 Recruitment in Normal and Pathogenic Immune Signaling†
Prabhjit K. Grewal,1 Mark Boton,1 Kevin Ramirez,1 Brian E. Collins,2 Akira Saito,1Ryan S. Green,1 Kazuaki Ohtsubo,1 Daniel Chui,1 and Jamey D. Marth1*
Department of Cellular and Molecular Medicine and Howard Hughes Medical Institute, 9500 Gilman Drive MC0625,University of California—San Diego, La Jolla, California 92093,1 and Departments of Molecular Biology and
Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, California 920372
Received 18 February 2006/Returned for modification 5 April 2006/Accepted 19 April 2006
The ST6Gal-I sialyltransferase produces Siglec ligands for the B-cell-specific CD22 lectin and sustainshumoral immune responses. Using multiple experimental approaches to elucidate the mechanisms involved,we report that ST6Gal-I deficiency induces immunoglobulin M (IgM) antigen receptor endocytosis in theabsence of immune stimulation. This coincides with increased antigen receptor colocalization with CD22 inboth clathrin-deficient and clathrin-enriched membrane microdomains concurrent with diminished tyrosinephosphorylation of Ig�/�, Syk, and phospholipase C-�2 upon immune activation. Codeficiency with CD22restores IgM antigen receptor half-life at the cell surface in addition to reversing alterations in membranetrafficking and immune signaling. Diminished immune responses due to ST6Gal-I deficiency further correlatewith constitutive recruitment of Shp-1 to CD22 in unstimulated B cells independent of Lyn tyrosine kinaseactivity and prevent autoimmune disease pathogenesis in the Lyn-deficient model of systemic lupus erythem-atosus, resulting in a significant extension of life span. Protein glycosylation by ST6Gal-I restricts access ofantigen receptors and Shp-1 to CD22 and operates by a CD22-dependent mechanism that decreases the basalrate of IgM antigen receptor endocytosis in altering the threshold of B-cell immune activation.
Cells of the mammalian immune system undergo selectivechanges in protein glycosylation during differentiation, im-mune activation, and autoimmune disease (2, 22, 23). A subsetof Golgi glycosyltransferases involved in these posttransla-tional glycan modifications are termed sialyltransferases andproduce sialic acid linkages on glycoproteins. Sialyltransferasesvary in their substrate specificities and expression profiles andmay thereby participate uniquely or collaboratively in differentphysiologic processes (20, 42, 45). For example, sialic acidscontribute to the glycan ligands of the selectins that modulateleukocyte trafficking (31, 37). A subset of sialyltransferases areinvolved in selectin ligand formation, and each may uniquelymodulate leukocyte and endothelial physiology (12). A distinctsialic acid linkage formed by the ST3Gal-I sialyltransferasedoes not participate in construction of selectin ligands butinstead inhibits caspase activation mechanisms associated withCD8� T-cell apoptosis (34). In contrast, the ST6Gal-I sialyl-transferase promotes B-cell immune activation responses andserum immunoglobulin (Ig) expression. Loss of ST6Gal-I func-tion does not grossly alter B-cell development or trafficking butattenuates IgM antigen receptor (B-cell receptor [BCR]) acti-vation, depresses Ca2� mobilization, decreases mature B-cellproliferation, and results in low antibody titer induction uponimmunization (16). How protein glycosylation by ST6Gal-Imaintains B-cell immune responses is not resolved but may
involve the functions of endogenous lectins that bind to glycanligands produced by ST6Gal-I.
Mammalian immune receptors of the Siglec family are trans-membrane lectins that exhibit extracellular binding specificitiesfor sialic acid linkages and often encode cytoplasmic immuno-receptor tyrosine inhibitory motif (ITIM) domains (8). Thesehallmarks of Siglec structure suggest that binding to cell sur-face glycan ligands may in some contexts modify intracellularsignal transduction pathways involving protein tyrosine phos-phorylation. The B-cell-specific CD22 (Siglec-2) glycoproteincontains cytoplasmic domain ITIMs that are tyrosine phosphor-ylated upon activation and which recruit the Shp-1 tyrosinephosphatase to dampen immune signaling (7, 9, 11, 30, 43).The extracellular domain of CD22 bears Siglec ligand bindingactivity toward �2–6–linked sialic acid to underlying �1–4–linked galactose (33, 39). CD22 Siglec ligands are produced byST6Gal-I in the Golgi apparatus and are expressed on theB-cell surface, where they typically occupy (or “mask”) cellsurface CD22 Siglec binding activity (5, 16, 36). The masking ofCD22 Siglec binding activity is reduced among postactivatedsplenic and bone marrow B cells, and thus ST6Gal-I functionmay be regulated in some contexts (5, 10, 13, 36). These find-ings infer a role for ST6Gal-I in modulating immune functionby constructing Siglec ligands for CD22. Indeed, previous stud-ies have shown that mutant CD22 molecules bearing mutations inthe Siglec binding domain impart altered B-cell activation re-sponses in vitro and in vivo (18, 19, 32). More recently, increasedcolocalization of cell surface IgM with CD22 and clathrin do-mains was observed among mice lacking ST6Gal-I, while resto-ration of normal IgM colocalization frequency with clathrin oc-curred coincident with elevated B-cell immune activationresponses in the absence of both CD22 and ST6Gal-I (6).
* Corresponding author. Mailing address: Department of Cellularand Molecular Medicine and Howard Hughes Medical Institute, 9500Gilman Drive MC0625, University of California—San Diego, La Jolla,CA 92093. Phone: (858) 534-6526. Fax: (858) 534-6724. E-mail: [email protected].
† Supplemental material for this article may be found at http://mcb.asm.org/.
4970
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

We have further investigated the mechanisms and glycopro-tein specificity by which ST6Gal-I modulates B-cell immunefunction among mice lacking combinations of ST6Gal-I, CD22,and the Lyn protein tyrosine kinase. Our findings are consis-tent with those recently published (6) and further show thatST6Gal-I function normally restricts Shp-1 recruitment toCD22 independent of Lyn activity. We also find that ST6Gal-Ideficiency selectively increases the rate of IgM BCR and CD22endocytosis, reducing the cell surface BCR half-life on un-stimulated B cells by a CD22-dependent mechanism, and fur-ther attenuates autoimmune disease pathogenesis, yielding asignificant increase in the life span of Lyn-deficient mice.
MATERIALS AND METHODS
Mice, cells, and antibodies. ST6Gal-I-deficient mice (16) and sex-matchedwild-type littermates were used to harvest splenic and lymph node B lympho-cytes. The ST6Gal-I mutation was bred for more than 10 generations into theC57BL/6 background prior to experimentation. Mice bearing null mutations inCD22 and the Lyn-encoded tyrosine kinase were previously described (3, 25).MD4 transgenic mice (14) were kindly provided by R. Rickert (Burnham Insti-tute, San Diego, CA) with permission from C. C. Goodnow (Australian NationalUniversity, Canberra, Australia). These mice were bred to generate littermateswith and without ST6Gal-I function. CD22 or Lyn mutant mice were bred withST6Gal-I-null mates to produce compound heterozygous offspring used for gen-erating littermates with the indicated genotypes in experimentation. B lympho-cytes were enriched to �90% from spleen and lymph nodes using a negativesorting strategy with biotin-conjugated anti-Thy-1.2, CD43 (clone S7), Ter-119,NK-1.1, Gr-1, and Mac-1 (PharMingen) antibodies and streptavidin-conjugatedmagnetic beads (Dynal, Great Neck, NY).
Protein half-lives and endocytosis. Isolated B cells were incubated with EZ-Link Sulfo-NHS-biotin (Pierce) for 30 min at 4°C to label cell surface proteins.Following two washes in ice-cold phosphate-buffered saline (PBS) with 100 mMglycine to quench excess biotin, 1 � 106 aliquots of cells were incubated at 37°Cin RPMI 1640 containing 0.1 mM 2-mercaptoethanol, 10% fetal calf serum(FCS), and 2 mM L-glutamine. Cells were harvested at indicated times and lysedat 4°C in lysis buffer containing 0.1% sodium dodecyl sulfate (SDS), 50 mMTris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.25% deoxycholate, 1% NP-40,and protease inhibitor cocktail and then sonicated for two 30-s pulses. Cells wereincubated for the indicated times in the absence or presence of methyl-�-cyclo-dextrin (10 mM) as described previously (41). Lysates were incubated withmonoavidin-agarose (Pierce) for 4 h. at 4°C. Precipitates were washed in lysisbuffer, and biotinylated proteins were eluted by boiling in SDS-polyacrylamidegel electrophoresis (PAGE) sample buffer. Total B-cell membrane proteinscomprising 1 � 106 cell equivalents were loaded in each lane of 5 to 15%gradient SDS-PAGE gels, and the resulting gel blots were probed with antibodiesincluding horseradish peroxidase (HRP)-conjugated anti-IgM and goat antibod-ies recognizing CD22 (clone Y19), Shp-1 (clone C19), and CD45 (clone M20),followed by HRP-conjugated anti-goat IgG (Santa Cruz). Blots were visualizedby enhanced chemiluminescence (Amersham Biosciences), and the signals werequantified (UVP; BioImaging Systems).
Colocalization measurements. Isolated B cells (1 � 106 cells/ml) maintained at4°C in RPMI 1640 with 2% FCS were treated with 2% paraformaldehyde priorto antibody labeling. Identical results were obtained when cells were insteadmaintained at 4°C and incubated with antibodies prior to treatment with 2%paraformaldehyde. No effect of cell isolation was observed as B cells among totalsplenocytes and lymph node cells treated as described above gave identicalresults. The antibodies used included CD22-fluorescein isothiocyanate (FITC),CD45-FITC (Pharmingen), anti-IgM–Texas red (Jackson, West Grove, PA), andCTB(GM1)-FITC (Sigma) with treatment for 30 min. Following washing, cellswere rendered permeable by treatment with CytoFix/CytoPerm (BD Bio-sciences) for 20 min before incubation with antibody to clathrin (clone X22;Affinity BioReagents) followed by aminomethylcuoumarin acetate (AMCA)-conjugated secondary antibody (Jackson, West Grove, PA). Cells were allowedto settle on glass slides and mounted with antifade aqueous mount medium(BioMedia Corp., CA). Deconvolution images were obtained using a Delta-Vision Restoration microscope system (Applied Precision Inc., Issaquah, WA)and analyzed using DeltaVision SoftWorx (version 2.50) in cell sections of 0.2�m. Quantification of fluorescent signals and their localization were accom-plished for each 0.2-�m section in the linear range of the digital camera by
object-based analysis algorithms using MetaMorph software (Universal ImagingCorporation, Downington, PA). Maximum projection views of cells are shownwhich combine all 0.2-�m images (z-stacks). A range of fluorescent signal ex-clusion thresholds was also applied in these analyses to validate the default valueotherwise input by the software and as used for visualization.
Protein phosphotyrosine and coprecipitation. Isolated B lymphocytes (1 �106) were suspended in 50 �l of RPMI 1640 with 5% FCS and 10 mM HEPES.Cells were warmed to 37°C before stimulation with 1.2 �g/ml of goat F(ab�)2
anti-mouse IgM. At the indicated times, 450 �l of lysis buffer was added (withsubstitution of Triton X-100 for NP-40). Antibodies to Ig� and CD22, as well asLyn, Syk, phospholipase C-�2 (PLC-�2), Shp-1, or Vav (10 �l) (Santa CruzBiotechnology) were added to the lysates with 25 �l of protein A-Sepharose. Thelysates were incubated for 4 h and washed three times in lysis buffer. Immunecomplexes were subjected to SDS-PAGE on a 5 to 15% gradient gel after elutionin sample buffer. Gels were transferred to nitrocellulose, and tyrosine phosphor-ylation was detected by blotting with monoclonal antibody 4G10 (Santa CruzBiotechnology). Blots were then stripped and reprobed with antibodies to Lyn,Syk, PLC-�2, Shp-1, Ig�, Ig�, and CD22.
BrdU pulse-chase and flow cytometry. Mice were given drinking water adlibitum containing 0.8 mg bromodeoxyuridine (BrdU)/ml for 3 weeks (pulse) andreturned to water lacking BrdU (chase) for 2 weeks. Animals were sacrificed atspecified times, and lymphocytes were isolated and analyzed for BrdU incorpo-ration. Single-cell suspensions from spleen, lymph node, or bone marrow wereharvested and subjected to red blood cell lysis with NH4Cl. Cell surface bindingof anti-CD4-phycoerythrin, CD8-allophycocyanin (APC), and B220-Cychromeantibodies (PharMingen) was carried out in 100 �l of fluorescence-activated cellsorter (FACS) buffer (2% FCS in PBS) with 1 � 106 cells on ice for 10 min. Cellswere then resuspended in 0.5 ml of ice-cold PBS, and 1.2 ml ice-cold 95%ethanol was added dropwise under gentle mixing to fix the cells. After 30 min ofincubation, cells were centrifuged and washed with 2 ml of PBS and thenincubated in of 1 ml of PBS, 1% paraformaldehyde, and 0.01% Tween 20 for 30min at 22°C. Cells were again centrifuged and resuspended in 1 ml of 0.15 MNaCl, 5 mM MgCl2, 10 �M HCl, and 50 Kunitz units/ml of DNase-1 for 10 min.at 22°C. After centrifugation, cells were washed with 2 ml of PBS, incubated in10 �l of FITC-conjugated anti-BrdU antibody (Becton Dickinson) for 30 min,and then again sedimented, washed in 2 ml PBS, and finally resuspended in 0.5ml FACS buffer for fluorescence-activated cell sorting analyses, performed on aFACScalibur flow cytometer using CellQuest software (Becton Dickinson). Flowcytometry using lectin conjugates was accomplished as described previously (16).
Calcium mobilization and protein phosphotyrosine analyses. Isolated B cellswere measured for calcium flux following stimulation by Indo-1 loading followedby FACS analysis, as previously described (16). Radiolabeled thymidine incor-poration was used to measure B-cell proliferation responses to goat F(ab�)2
anti-mouse IgM (Jackson) as described previously (16).Histology. Examinations of frozen embedded kidney, liver, or spleen (O.C.T.
medium; Sakura Finetek, Torrance, CA) were performed on 5-�m sections fixedin formalin and rehydrated in PBS. Sections were incubated with anti-IgM–rhodamine or anti-IgG-FITC (Jackson ImmunoResearch Labs, West Grove,PA). Washed and stained sections were mounted in aqueous gel mount(Biomeda Corp., Foster City, CA) and observed by fluorescent microscopy at amagnification of �200 (Zeiss, Gottingen, Germany).
Lyn-deficient autoimmune disease. Antinuclear antibody (ANA) detectionand serum antibody titers to double-stranded DNA (dsDNA), histones, Smantigen, and total kidney protein were assayed as previously described (4).
Statistical analysis. Analyses of variance and Student’s t test were used asdescribed in the figure legends to compare the groups shown in each experiment.Results are expressed as means and standard errors of the mean unless otherwiseindicated.
RESULTS
Glycoprotein-selective acceleration of IgM antigen receptorand CD22 endocytosis in ST6Gal-I deficiency. B cells lackingST6Gal-I exhibit reductions in total expression of IgM andCD22 that appear to reflect decreased levels at the cell surface(16). Splenic and lymph node B cells were placed under cellculture conditions following cell surface biotinylation to deter-mine the cell surface half-lives of various glycoproteins. Nor-mally, the half-lives of IgM, CD22, and CD45 on the wild-typeunstimulated B-cell surface were 8 h or longer. In contrast, the
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4971
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
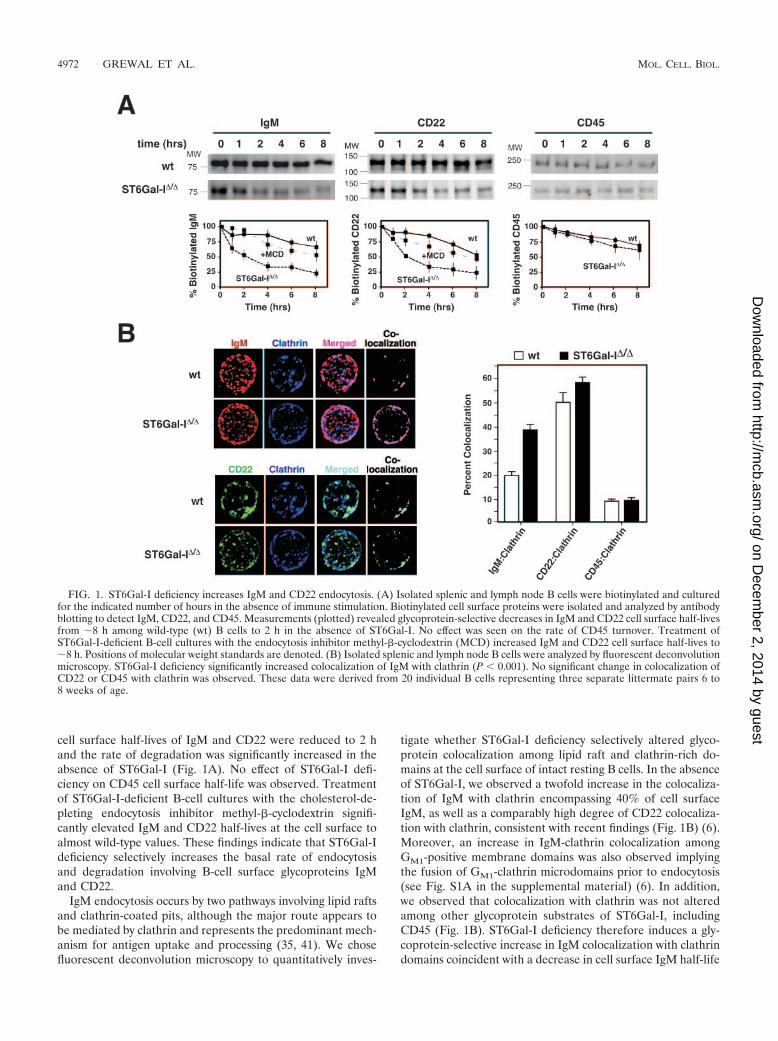
cell surface half-lives of IgM and CD22 were reduced to 2 hand the rate of degradation was significantly increased in theabsence of ST6Gal-I (Fig. 1A). No effect of ST6Gal-I defi-ciency on CD45 cell surface half-life was observed. Treatmentof ST6Gal-I-deficient B-cell cultures with the cholesterol-de-pleting endocytosis inhibitor methyl-�-cyclodextrin signifi-cantly elevated IgM and CD22 half-lives at the cell surface toalmost wild-type values. These findings indicate that ST6Gal-Ideficiency selectively increases the basal rate of endocytosisand degradation involving B-cell surface glycoproteins IgMand CD22.
IgM endocytosis occurs by two pathways involving lipid raftsand clathrin-coated pits, although the major route appears tobe mediated by clathrin and represents the predominant mech-anism for antigen uptake and processing (35, 41). We chosefluorescent deconvolution microscopy to quantitatively inves-
tigate whether ST6Gal-I deficiency selectively altered glyco-protein colocalization among lipid raft and clathrin-rich do-mains at the cell surface of intact resting B cells. In the absenceof ST6Gal-I, we observed a twofold increase in the colocaliza-tion of IgM with clathrin encompassing 40% of cell surfaceIgM, as well as a comparably high degree of CD22 colocaliza-tion with clathrin, consistent with recent findings (Fig. 1B) (6).Moreover, an increase in IgM-clathrin colocalization amongGM1-positive membrane domains was also observed implyingthe fusion of GM1-clathrin microdomains prior to endocytosis(see Fig. S1A in the supplemental material) (6). In addition,we observed that colocalization with clathrin was not alteredamong other glycoprotein substrates of ST6Gal-I, includingCD45 (Fig. 1B). ST6Gal-I deficiency therefore induces a gly-coprotein-selective increase in IgM colocalization with clathrindomains coincident with a decrease in cell surface IgM half-life
FIG. 1. ST6Gal-I deficiency increases IgM and CD22 endocytosis. (A) Isolated splenic and lymph node B cells were biotinylated and culturedfor the indicated number of hours in the absence of immune stimulation. Biotinylated cell surface proteins were isolated and analyzed by antibodyblotting to detect IgM, CD22, and CD45. Measurements (plotted) revealed glycoprotein-selective decreases in IgM and CD22 cell surface half-livesfrom 8 h among wild-type (wt) B cells to 2 h in the absence of ST6Gal-I. No effect was seen on the rate of CD45 turnover. Treatment ofST6Gal-I-deficient B-cell cultures with the endocytosis inhibitor methyl-�-cyclodextrin (MCD) increased IgM and CD22 cell surface half-lives to8 h. Positions of molecular weight standards are denoted. (B) Isolated splenic and lymph node B cells were analyzed by fluorescent deconvolutionmicroscopy. ST6Gal-I deficiency significantly increased colocalization of IgM with clathrin (P 0.001). No significant change in colocalization ofCD22 or CD45 with clathrin was observed. These data were derived from 20 individual B cells representing three separate littermate pairs 6 to8 weeks of age.
4972 GREWAL ET AL. MOL. CELL. BIOL.
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
among resting B cells due to an increase in the basal rate ofIgM endocytosis. Normally, IgM endocytosis is induced follow-ing BCR stimulation and correlates with attenuated immunesignaling (41). We analyzed IgM immune activation responsesamong ST6Gal-I-deficient B cells with emphasis on detectingmembrane-proximal positive and negative signaling compo-nents.
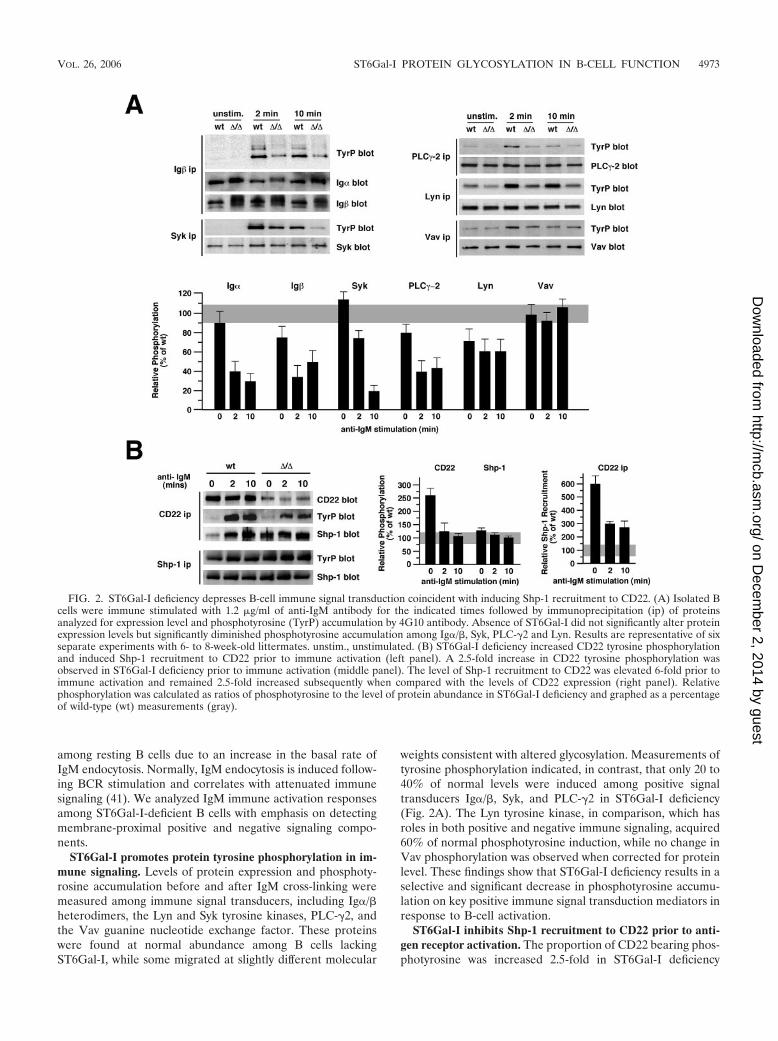
ST6Gal-I promotes protein tyrosine phosphorylation in im-mune signaling. Levels of protein expression and phosphoty-rosine accumulation before and after IgM cross-linking weremeasured among immune signal transducers, including Ig�/�heterodimers, the Lyn and Syk tyrosine kinases, PLC-�2, andthe Vav guanine nucleotide exchange factor. These proteinswere found at normal abundance among B cells lackingST6Gal-I, while some migrated at slightly different molecular
weights consistent with altered glycosylation. Measurements oftyrosine phosphorylation indicated, in contrast, that only 20 to40% of normal levels were induced among positive signaltransducers Ig�/�, Syk, and PLC-�2 in ST6Gal-I deficiency(Fig. 2A). The Lyn tyrosine kinase, in comparison, which hasroles in both positive and negative immune signaling, acquired60% of normal phosphotyrosine induction, while no change inVav phosphorylation was observed when corrected for proteinlevel. These findings show that ST6Gal-I deficiency results in aselective and significant decrease in phosphotyrosine accumu-lation on key positive immune signal transduction mediators inresponse to B-cell activation.
ST6Gal-I inhibits Shp-1 recruitment to CD22 prior to anti-gen receptor activation. The proportion of CD22 bearing phos-photyrosine was increased 2.5-fold in ST6Gal-I deficiency
FIG. 2. ST6Gal-I deficiency depresses B-cell immune signal transduction coincident with inducing Shp-1 recruitment to CD22. (A) Isolated Bcells were immune stimulated with 1.2 �g/ml of anti-IgM antibody for the indicated times followed by immunoprecipitation (ip) of proteinsanalyzed for expression level and phosphotyrosine (TyrP) accumulation by 4G10 antibody. Absence of ST6Gal-I did not significantly alter proteinexpression levels but significantly diminished phosphotyrosine accumulation among Ig�/�, Syk, PLC-�2 and Lyn. Results are representative of sixseparate experiments with 6- to 8-week-old littermates. unstim., unstimulated. (B) ST6Gal-I deficiency increased CD22 tyrosine phosphorylationand induced Shp-1 recruitment to CD22 prior to immune activation (left panel). A 2.5-fold increase in CD22 tyrosine phosphorylation wasobserved in ST6Gal-I deficiency prior to immune activation (middle panel). The level of Shp-1 recruitment to CD22 was elevated 6-fold prior toimmune activation and remained 2.5-fold increased subsequently when compared with the levels of CD22 expression (right panel). Relativephosphorylation was calculated as ratios of phosphotyrosine to the level of protein abundance in ST6Gal-I deficiency and graphed as a percentageof wild-type (wt) measurements (gray).
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4973
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
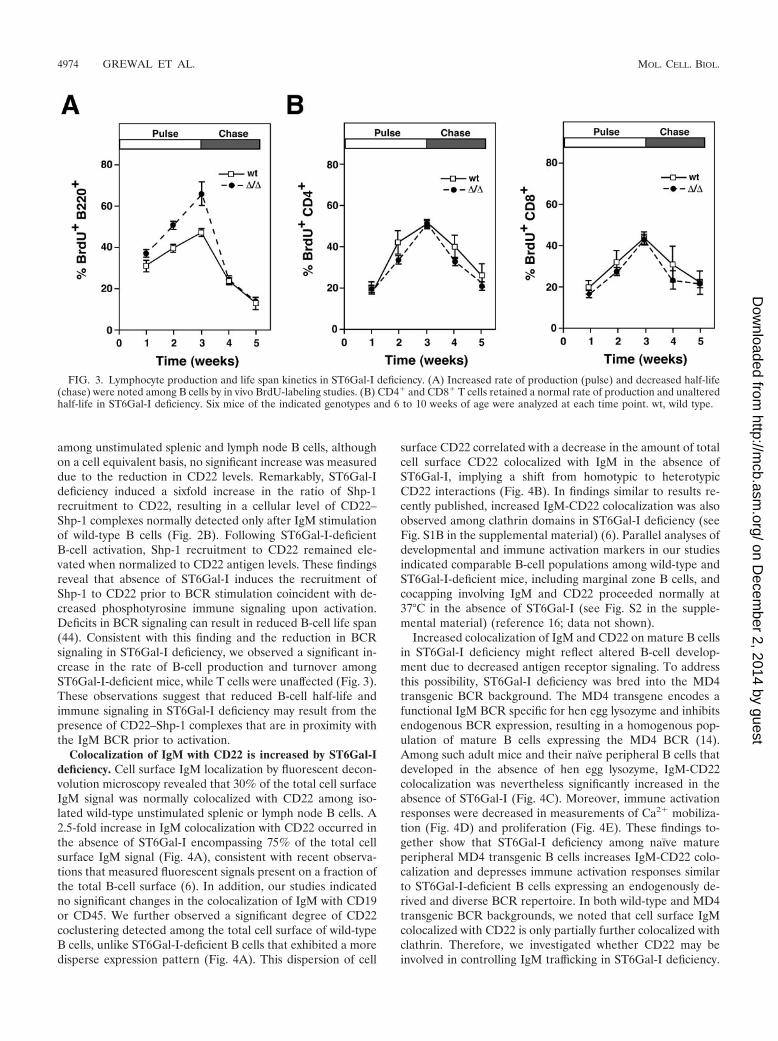
among unstimulated splenic and lymph node B cells, althoughon a cell equivalent basis, no significant increase was measureddue to the reduction in CD22 levels. Remarkably, ST6Gal-Ideficiency induced a sixfold increase in the ratio of Shp-1recruitment to CD22, resulting in a cellular level of CD22–Shp-1 complexes normally detected only after IgM stimulationof wild-type B cells (Fig. 2B). Following ST6Gal-I-deficientB-cell activation, Shp-1 recruitment to CD22 remained ele-vated when normalized to CD22 antigen levels. These findingsreveal that absence of ST6Gal-I induces the recruitment ofShp-1 to CD22 prior to BCR stimulation coincident with de-creased phosphotyrosine immune signaling upon activation.Deficits in BCR signaling can result in reduced B-cell life span(44). Consistent with this finding and the reduction in BCRsignaling in ST6Gal-I deficiency, we observed a significant in-crease in the rate of B-cell production and turnover amongST6Gal-I-deficient mice, while T cells were unaffected (Fig. 3).These observations suggest that reduced B-cell half-life andimmune signaling in ST6Gal-I deficiency may result from thepresence of CD22–Shp-1 complexes that are in proximity withthe IgM BCR prior to activation.
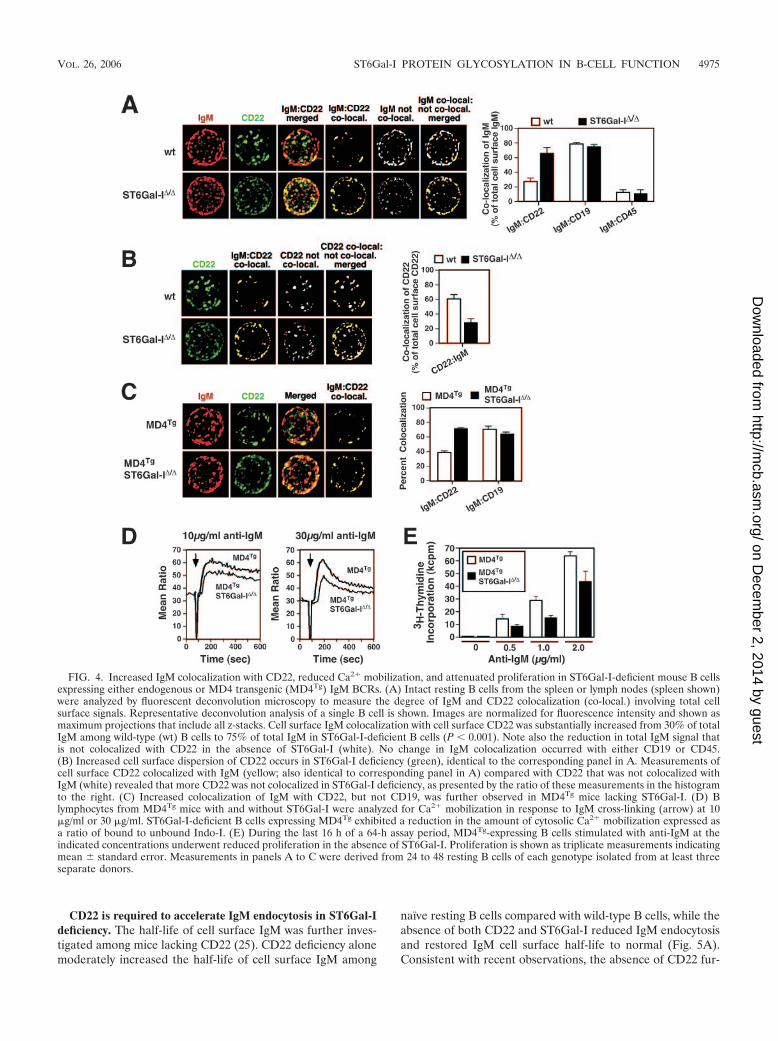
Colocalization of IgM with CD22 is increased by ST6Gal-Ideficiency. Cell surface IgM localization by fluorescent decon-volution microscopy revealed that 30% of the total cell surfaceIgM signal was normally colocalized with CD22 among iso-lated wild-type unstimulated splenic or lymph node B cells. A2.5-fold increase in IgM colocalization with CD22 occurred inthe absence of ST6Gal-I encompassing 75% of the total cellsurface IgM signal (Fig. 4A), consistent with recent observa-tions that measured fluorescent signals present on a fraction ofthe total B-cell surface (6). In addition, our studies indicatedno significant changes in the colocalization of IgM with CD19or CD45. We further observed a significant degree of CD22coclustering detected among the total cell surface of wild-typeB cells, unlike ST6Gal-I-deficient B cells that exhibited a moredisperse expression pattern (Fig. 4A). This dispersion of cell
surface CD22 correlated with a decrease in the amount of totalcell surface CD22 colocalized with IgM in the absence ofST6Gal-I, implying a shift from homotypic to heterotypicCD22 interactions (Fig. 4B). In findings similar to results re-cently published, increased IgM-CD22 colocalization was alsoobserved among clathrin domains in ST6Gal-I deficiency (seeFig. S1B in the supplemental material) (6). Parallel analyses ofdevelopmental and immune activation markers in our studiesindicated comparable B-cell populations among wild-type andST6Gal-I-deficient mice, including marginal zone B cells, andcocapping involving IgM and CD22 proceeded normally at37°C in the absence of ST6Gal-I (see Fig. S2 in the supple-mental material) (reference 16; data not shown).
Increased colocalization of IgM and CD22 on mature B cellsin ST6Gal-I deficiency might reflect altered B-cell develop-ment due to decreased antigen receptor signaling. To addressthis possibility, ST6Gal-I deficiency was bred into the MD4transgenic BCR background. The MD4 transgene encodes afunctional IgM BCR specific for hen egg lysozyme and inhibitsendogenous BCR expression, resulting in a homogenous pop-ulation of mature B cells expressing the MD4 BCR (14).Among such adult mice and their naıve peripheral B cells thatdeveloped in the absence of hen egg lysozyme, IgM-CD22colocalization was nevertheless significantly increased in theabsence of ST6Gal-I (Fig. 4C). Moreover, immune activationresponses were decreased in measurements of Ca2� mobiliza-tion (Fig. 4D) and proliferation (Fig. 4E). These findings to-gether show that ST6Gal-I deficiency among naıve matureperipheral MD4 transgenic B cells increases IgM-CD22 colo-calization and depresses immune activation responses similarto ST6Gal-I-deficient B cells expressing an endogenously de-rived and diverse BCR repertoire. In both wild-type and MD4transgenic BCR backgrounds, we noted that cell surface IgMcolocalized with CD22 is only partially further colocalized withclathrin. Therefore, we investigated whether CD22 may beinvolved in controlling IgM trafficking in ST6Gal-I deficiency.
FIG. 3. Lymphocyte production and life span kinetics in ST6Gal-I deficiency. (A) Increased rate of production (pulse) and decreased half-life(chase) were noted among B cells by in vivo BrdU-labeling studies. (B) CD4� and CD8� T cells retained a normal rate of production and unalteredhalf-life in ST6Gal-I deficiency. Six mice of the indicated genotypes and 6 to 10 weeks of age were analyzed at each time point. wt, wild type.
4974 GREWAL ET AL. MOL. CELL. BIOL.
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
CD22 is required to accelerate IgM endocytosis in ST6Gal-Ideficiency. The half-life of cell surface IgM was further inves-tigated among mice lacking CD22 (25). CD22 deficiency alonemoderately increased the half-life of cell surface IgM among
naıve resting B cells compared with wild-type B cells, while theabsence of both CD22 and ST6Gal-I reduced IgM endocytosisand restored IgM cell surface half-life to normal (Fig. 5A).Consistent with recent observations, the absence of CD22 fur-
FIG. 4. Increased IgM colocalization with CD22, reduced Ca2� mobilization, and attenuated proliferation in ST6Gal-I-deficient mouse B cellsexpressing either endogenous or MD4 transgenic (MD4Tg) IgM BCRs. (A) Intact resting B cells from the spleen or lymph nodes (spleen shown)were analyzed by fluorescent deconvolution microscopy to measure the degree of IgM and CD22 colocalization (co-local.) involving total cellsurface signals. Representative deconvolution analysis of a single B cell is shown. Images are normalized for fluorescence intensity and shown asmaximum projections that include all z-stacks. Cell surface IgM colocalization with cell surface CD22 was substantially increased from 30% of totalIgM among wild-type (wt) B cells to 75% of total IgM in ST6Gal-I-deficient B cells (P 0.001). Note also the reduction in total IgM signal thatis not colocalized with CD22 in the absence of ST6Gal-I (white). No change in IgM colocalization occurred with either CD19 or CD45.(B) Increased cell surface dispersion of CD22 occurs in ST6Gal-I deficiency (green), identical to the corresponding panel in A. Measurements ofcell surface CD22 colocalized with IgM (yellow; also identical to corresponding panel in A) compared with CD22 that was not colocalized withIgM (white) revealed that more CD22 was not colocalized in ST6Gal-I deficiency, as presented by the ratio of these measurements in the histogramto the right. (C) Increased colocalization of IgM with CD22, but not CD19, was further observed in MD4Tg mice lacking ST6Gal-I. (D) Blymphocytes from MD4Tg mice with and without ST6Gal-I were analyzed for Ca2� mobilization in response to IgM cross-linking (arrow) at 10�g/ml or 30 �g/ml. ST6Gal-I-deficient B cells expressing MD4Tg exhibited a reduction in the amount of cytosolic Ca2� mobilization expressed asa ratio of bound to unbound Indo-I. (E) During the last 16 h of a 64-h assay period, MD4Tg-expressing B cells stimulated with anti-IgM at theindicated concentrations underwent reduced proliferation in the absence of ST6Gal-I. Proliferation is shown as triplicate measurements indicatingmean � standard error. Measurements in panels A to C were derived from 24 to 48 resting B cells of each genotype isolated from at least threeseparate donors.
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4975
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
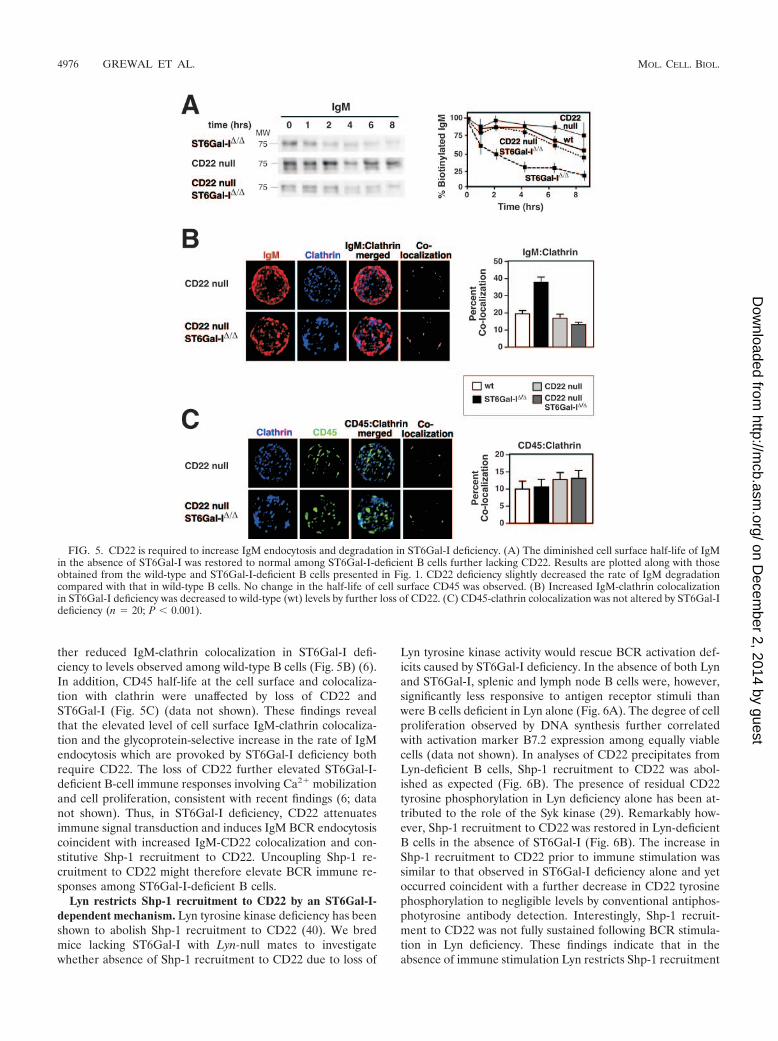
ther reduced IgM-clathrin colocalization in ST6Gal-I defi-ciency to levels observed among wild-type B cells (Fig. 5B) (6).In addition, CD45 half-life at the cell surface and colocaliza-tion with clathrin were unaffected by loss of CD22 andST6Gal-I (Fig. 5C) (data not shown). These findings revealthat the elevated level of cell surface IgM-clathrin colocaliza-tion and the glycoprotein-selective increase in the rate of IgMendocytosis which are provoked by ST6Gal-I deficiency bothrequire CD22. The loss of CD22 further elevated ST6Gal-I-deficient B-cell immune responses involving Ca2� mobilizationand cell proliferation, consistent with recent findings (6; datanot shown). Thus, in ST6Gal-I deficiency, CD22 attenuatesimmune signal transduction and induces IgM BCR endocytosiscoincident with increased IgM-CD22 colocalization and con-stitutive Shp-1 recruitment to CD22. Uncoupling Shp-1 re-cruitment to CD22 might therefore elevate BCR immune re-sponses among ST6Gal-I-deficient B cells.
Lyn restricts Shp-1 recruitment to CD22 by an ST6Gal-I-dependent mechanism. Lyn tyrosine kinase deficiency has beenshown to abolish Shp-1 recruitment to CD22 (40). We bredmice lacking ST6Gal-I with Lyn-null mates to investigatewhether absence of Shp-1 recruitment to CD22 due to loss of
Lyn tyrosine kinase activity would rescue BCR activation def-icits caused by ST6Gal-I deficiency. In the absence of both Lynand ST6Gal-I, splenic and lymph node B cells were, however,significantly less responsive to antigen receptor stimuli thanwere B cells deficient in Lyn alone (Fig. 6A). The degree of cellproliferation observed by DNA synthesis further correlatedwith activation marker B7.2 expression among equally viablecells (data not shown). In analyses of CD22 precipitates fromLyn-deficient B cells, Shp-1 recruitment to CD22 was abol-ished as expected (Fig. 6B). The presence of residual CD22tyrosine phosphorylation in Lyn deficiency alone has been at-tributed to the role of the Syk kinase (29). Remarkably how-ever, Shp-1 recruitment to CD22 was restored in Lyn-deficientB cells in the absence of ST6Gal-I (Fig. 6B). The increase inShp-1 recruitment to CD22 prior to immune stimulation wassimilar to that observed in ST6Gal-I deficiency alone and yetoccurred coincident with a further decrease in CD22 tyrosinephosphorylation to negligible levels by conventional antiphos-photyrosine antibody detection. Interestingly, Shp-1 recruit-ment to CD22 was not fully sustained following BCR stimula-tion in Lyn deficiency. These findings indicate that in theabsence of immune stimulation Lyn restricts Shp-1 recruitment
FIG. 5. CD22 is required to increase IgM endocytosis and degradation in ST6Gal-I deficiency. (A) The diminished cell surface half-life of IgMin the absence of ST6Gal-I was restored to normal among ST6Gal-I-deficient B cells further lacking CD22. Results are plotted along with thoseobtained from the wild-type and ST6Gal-I-deficient B cells presented in Fig. 1. CD22 deficiency slightly decreased the rate of IgM degradationcompared with that in wild-type B cells. No change in the half-life of cell surface CD45 was observed. (B) Increased IgM-clathrin colocalizationin ST6Gal-I deficiency was decreased to wild-type (wt) levels by further loss of CD22. (C) CD45-clathrin colocalization was not altered by ST6Gal-Ideficiency (n � 20; P 0.001).
4976 GREWAL ET AL. MOL. CELL. BIOL.
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
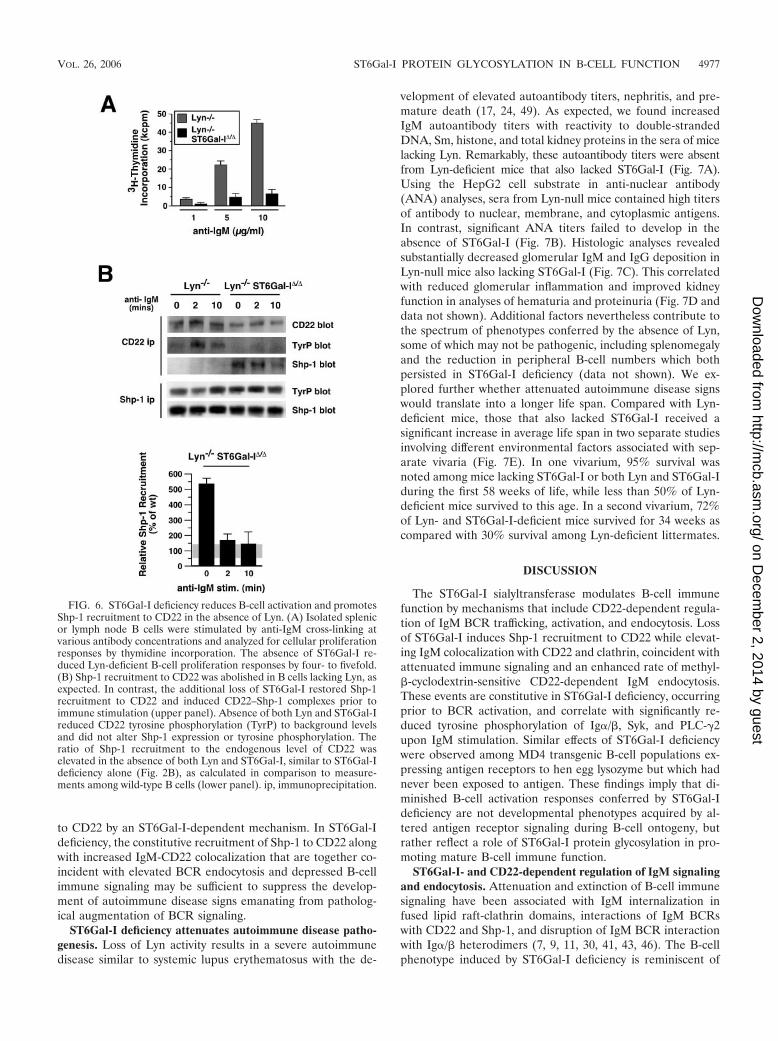
to CD22 by an ST6Gal-I-dependent mechanism. In ST6Gal-Ideficiency, the constitutive recruitment of Shp-1 to CD22 alongwith increased IgM-CD22 colocalization that are together co-incident with elevated BCR endocytosis and depressed B-cellimmune signaling may be sufficient to suppress the develop-ment of autoimmune disease signs emanating from patholog-ical augmentation of BCR signaling.
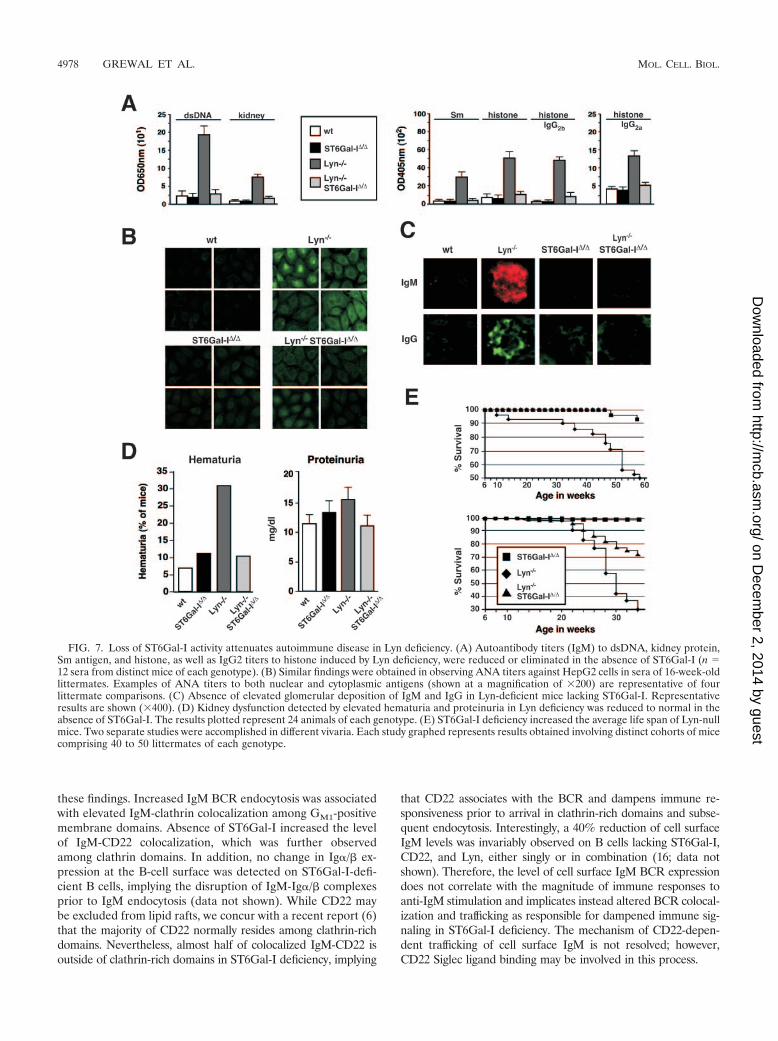
ST6Gal-I deficiency attenuates autoimmune disease patho-genesis. Loss of Lyn activity results in a severe autoimmunedisease similar to systemic lupus erythematosus with the de-
velopment of elevated autoantibody titers, nephritis, and pre-mature death (17, 24, 49). As expected, we found increasedIgM autoantibody titers with reactivity to double-strandedDNA, Sm, histone, and total kidney proteins in the sera of micelacking Lyn. Remarkably, these autoantibody titers were absentfrom Lyn-deficient mice that also lacked ST6Gal-I (Fig. 7A).Using the HepG2 cell substrate in anti-nuclear antibody(ANA) analyses, sera from Lyn-null mice contained high titersof antibody to nuclear, membrane, and cytoplasmic antigens.In contrast, significant ANA titers failed to develop in theabsence of ST6Gal-I (Fig. 7B). Histologic analyses revealedsubstantially decreased glomerular IgM and IgG deposition inLyn-null mice also lacking ST6Gal-I (Fig. 7C). This correlatedwith reduced glomerular inflammation and improved kidneyfunction in analyses of hematuria and proteinuria (Fig. 7D anddata not shown). Additional factors nevertheless contribute tothe spectrum of phenotypes conferred by the absence of Lyn,some of which may not be pathogenic, including splenomegalyand the reduction in peripheral B-cell numbers which bothpersisted in ST6Gal-I deficiency (data not shown). We ex-plored further whether attenuated autoimmune disease signswould translate into a longer life span. Compared with Lyn-deficient mice, those that also lacked ST6Gal-I received asignificant increase in average life span in two separate studiesinvolving different environmental factors associated with sep-arate vivaria (Fig. 7E). In one vivarium, 95% survival wasnoted among mice lacking ST6Gal-I or both Lyn and ST6Gal-Iduring the first 58 weeks of life, while less than 50% of Lyn-deficient mice survived to this age. In a second vivarium, 72%of Lyn- and ST6Gal-I-deficient mice survived for 34 weeks ascompared with 30% survival among Lyn-deficient littermates.
DISCUSSION
The ST6Gal-I sialyltransferase modulates B-cell immunefunction by mechanisms that include CD22-dependent regula-tion of IgM BCR trafficking, activation, and endocytosis. Lossof ST6Gal-I induces Shp-1 recruitment to CD22 while elevat-ing IgM colocalization with CD22 and clathrin, coincident withattenuated immune signaling and an enhanced rate of methyl-�-cyclodextrin-sensitive CD22-dependent IgM endocytosis.These events are constitutive in ST6Gal-I deficiency, occurringprior to BCR activation, and correlate with significantly re-duced tyrosine phosphorylation of Ig�/�, Syk, and PLC-�2upon IgM stimulation. Similar effects of ST6Gal-I deficiencywere observed among MD4 transgenic B-cell populations ex-pressing antigen receptors to hen egg lysozyme but which hadnever been exposed to antigen. These findings imply that di-minished B-cell activation responses conferred by ST6Gal-Ideficiency are not developmental phenotypes acquired by al-tered antigen receptor signaling during B-cell ontogeny, butrather reflect a role of ST6Gal-I protein glycosylation in pro-moting mature B-cell immune function.
ST6Gal-I- and CD22-dependent regulation of IgM signalingand endocytosis. Attenuation and extinction of B-cell immunesignaling have been associated with IgM internalization infused lipid raft-clathrin domains, interactions of IgM BCRswith CD22 and Shp-1, and disruption of IgM BCR interactionwith Ig�/� heterodimers (7, 9, 11, 30, 41, 43, 46). The B-cellphenotype induced by ST6Gal-I deficiency is reminiscent of
FIG. 6. ST6Gal-I deficiency reduces B-cell activation and promotesShp-1 recruitment to CD22 in the absence of Lyn. (A) Isolated splenicor lymph node B cells were stimulated by anti-IgM cross-linking atvarious antibody concentrations and analyzed for cellular proliferationresponses by thymidine incorporation. The absence of ST6Gal-I re-duced Lyn-deficient B-cell proliferation responses by four- to fivefold.(B) Shp-1 recruitment to CD22 was abolished in B cells lacking Lyn, asexpected. In contrast, the additional loss of ST6Gal-I restored Shp-1recruitment to CD22 and induced CD22–Shp-1 complexes prior toimmune stimulation (upper panel). Absence of both Lyn and ST6Gal-Ireduced CD22 tyrosine phosphorylation (TyrP) to background levelsand did not alter Shp-1 expression or tyrosine phosphorylation. Theratio of Shp-1 recruitment to the endogenous level of CD22 waselevated in the absence of both Lyn and ST6Gal-I, similar to ST6Gal-Ideficiency alone (Fig. 2B), as calculated in comparison to measure-ments among wild-type B cells (lower panel). ip, immunoprecipitation.
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4977
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
these findings. Increased IgM BCR endocytosis was associatedwith elevated IgM-clathrin colocalization among GM1-positivemembrane domains. Absence of ST6Gal-I increased the levelof IgM-CD22 colocalization, which was further observedamong clathrin domains. In addition, no change in Ig�/� ex-pression at the B-cell surface was detected on ST6Gal-I-defi-cient B cells, implying the disruption of IgM-Ig�/� complexesprior to IgM endocytosis (data not shown). While CD22 maybe excluded from lipid rafts, we concur with a recent report (6)that the majority of CD22 normally resides among clathrin-richdomains. Nevertheless, almost half of colocalized IgM-CD22 isoutside of clathrin-rich domains in ST6Gal-I deficiency, implying
that CD22 associates with the BCR and dampens immune re-sponsiveness prior to arrival in clathrin-rich domains and subse-quent endocytosis. Interestingly, a 40% reduction of cell surfaceIgM levels was invariably observed on B cells lacking ST6Gal-I,CD22, and Lyn, either singly or in combination (16; data notshown). Therefore, the level of cell surface IgM BCR expressiondoes not correlate with the magnitude of immune responses toanti-IgM stimulation and implicates instead altered BCR colocal-ization and trafficking as responsible for dampened immune sig-naling in ST6Gal-I deficiency. The mechanism of CD22-depen-dent trafficking of cell surface IgM is not resolved; however,CD22 Siglec ligand binding may be involved in this process.
FIG. 7. Loss of ST6Gal-I activity attenuates autoimmune disease in Lyn deficiency. (A) Autoantibody titers (IgM) to dsDNA, kidney protein,Sm antigen, and histone, as well as IgG2 titers to histone induced by Lyn deficiency, were reduced or eliminated in the absence of ST6Gal-I (n �12 sera from distinct mice of each genotype). (B) Similar findings were obtained in observing ANA titers against HepG2 cells in sera of 16-week-oldlittermates. Examples of ANA titers to both nuclear and cytoplasmic antigens (shown at a magnification of �200) are representative of fourlittermate comparisons. (C) Absence of elevated glomerular deposition of IgM and IgG in Lyn-deficient mice lacking ST6Gal-I. Representativeresults are shown (�400). (D) Kidney dysfunction detected by elevated hematuria and proteinuria in Lyn deficiency was reduced to normal in theabsence of ST6Gal-I. The results plotted represent 24 animals of each genotype. (E) ST6Gal-I deficiency increased the average life span of Lyn-nullmice. Two separate studies were accomplished in different vivaria. Each study graphed represents results obtained involving distinct cohorts of micecomprising 40 to 50 littermates of each genotype.
4978 GREWAL ET AL. MOL. CELL. BIOL.
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

ST6Gal-I sialylation in CD22 Siglec-dependent and -inde-pendent binding. The physiologic role of CD22 Siglec bindinghas been difficult to perceive. As CD22 is the only moleculedetected on the B-cell surface with Siglec binding activity forcounter-receptors produced by ST6Gal-I (5, 19), it would beexpected that CD22 mutations specifically disabling Siglecbinding would recapitulate phenotypes observed in ST6Gal-Ideficiency. CD22 Siglec mutations targeted to the mouse germline and studied among primary B cells have indeed producedphenotypes similar to those observed in ST6Gal-I deficiency,including attenuated BCR-induced proliferation (32). In con-trast, an exogenous approach to disrupt CD22 Siglec ligandsusing glycan ligand mimetics increased antigen receptor-stim-ulated Ca2� mobilization when added to established B-celllines, suggesting a reduction of CD22 Siglec-dependent asso-ciation with IgM complexes (19). Moreover, study of a B-cellline derived from CD22-deficient mice revealed augmentedCa2� mobilization upon expression of mutant CD22 moleculesdisabled in Siglec binding (18). Such findings appear difficult toreconcile. From the earliest discordances observed amongCD22 mutant mice, it seems likely that mouse strain differ-ences may contribute to alterations in the outcome involvingCD22 function in BCR-induced proliferation (25, 27, 28, 32,38). In addition, the use of primary B cells may be relevant, asa study using the Daudi B-cell line reported no increase inCD22 endocytosis in the absence of CD22 Siglec ligands (50).
The identities of CD22 binding partners depend upon themethodologies employed. IgM-CD22 interaction has been re-ported in ranges from 0.5% to 18% by coprecipitation andprotein cross-linking (21, 30, 50). Those methods can detectclosely adjacent and even direct interactions but may also dis-rupt selective in situ binding and can rely upon strict intermo-lecular distances. Quantitative fluorescent deconvolution mi-croscopy cannot achieve this degree of spatial resolution;however, this technique can analyze native and intact B-cellsurfaces. Our findings by this method are consistent with re-cent studies (6) that report a substantial although minor frac-tion of total cell surface IgM (30%) is typically colocalizedwith CD22 in wild-type unstimulated peripheral B cells. Asignificant basal level of IgM-CD22 colocalization is consistentwith the normal role of CD22 expression in modulating pe-ripheral naıve B-cell activation thresholds. It is not clear, how-ever, whether IgM-CD22 colocalization on B cells expressingST6Gal-I involves a subset of total cell surface glycoproteinslacking CD22 Siglec ligands due to the phenomenon of micro-heterogeneity of glycan linkage formation in the Golgi appa-ratus. It remains possible that increased colocalization of IgMwith CD22 in ST6Gal-I deficiency results from the absence ofCD22 Siglec binding.
Many proteins are glycosylated by ST6Gal-I in B cells, andall would appear to be CD22 Siglec ligands by lectin copre-cipitation from homogenized cell extracts (16; data not shown).However, CD22 interactions are likely more selective in situ.On the resting intact B-cell surface, CD22 Siglec binding wasrecently shown to predominantly involve cis ligands resulting inhomotypic multimeric CD22 complexes (15). Our results byquantitative fluorescent deconvolution microscopy of entireintact as well as naıve B-cell surfaces appear consistent withthose findings. We observed more dispersed CD22 expressionat the cell surface in ST6Gal-I deficiency as well as a reduced
fraction of total cell surface CD22 colocalized with IgM. Thesefindings suggest that reduced homotypic CD22 binding occursin the absence of Siglec ligands concurrent with increasedheterotypic CD22 interactions among other cell surface mole-cules, including the IgM BCR, possibly by enabling CD22Siglec-independent binding contributed by the conserved ex-tracellular non-Siglec Ig-like domains. This may explain thesignificant amount of IgM cross-linking to CD22 observed onintact cell surfaces that lack Siglec ligands (50).
Control of Shp-1 recruitment to CD22 by ST6Gal-I proteinglycosylation. The attenuation of BCR signaling by CD22 isattributed to Shp-1 recruitment following activation (7, 9, 11,43). In wild-type B cells, a low level of Shp-1 coprecipitationwith CD22 is observed prior to IgM cross-linking, with maxi-mal Shp-1 recruitment occurring after several minutes of BCRstimulation. In contrast, ST6Gal-I deficiency induced maximalShp-1 recruitment to CD22 prior to IgM stimulation and theresultant increase in phosphotyrosine on CD22. Nevertheless,constitutive Shp-1 recruitment to CD22 in ST6Gal-I deficiencymay not simply reflect the absence of CD22 Siglec ligands. Nosimilar increase in Shp-1 recruitment to CD22 was reportedamong B cells bearing mutant CD22 molecules with eitherdeleted or likely inactive Siglec binding domains (32). It ispossible that the absence of CD22 sialylation by ST6Gal-Ialters CD22 conformation in promoting Shp-1 recruitmentwhen CD22 is colocalized with IgM BCRs. Or perhapsST6Gal-I deficiency increases access of Shp-1 to CD22 byaltering glycoprotein trafficking and residence among mem-brane microdomains. It is widely accepted that Shp-1 recruit-ment to CD22 reflects binding of Shp-1 src homology 2 (SH2)domain sequences to phosphotyrosine within CD22 ITIM do-mains. Remarkably, the level of phosphotyrosine detected onCD22 in ST6Gal-I deficiency was reduced to negligible levelsin the further absence of Lyn, and yet the constitutively highlevel of Shp-1 recruitment to CD22 remained in the absence ofimmune stimulation. Shp-1 recruitment was not sustained fol-lowing BCR stimulation of ST6Gal-I-deficient B cells, reveal-ing that Lyn function increases the half-life of CD22–Shp-1complexes following BCR activation. While the mechanism ofCD22–Shp-1 complex formation in ST6Gal-I deficiency re-mains to be established, it is possible that detection of phos-photyrosine by antibody binding is thwarted by ST6Gal-I de-ficiency or a mechanism exists by which CD22 and Shp-1complexes can form independent of CD22 tyrosine phospho-rylation.
Endogenous control of ST6Gal-I function. The immunemodulatory capability of ST6Gal-I may be normally regulatedamong B cells, perhaps as indicated by unmasking of CD22Siglec binding following immune stimulation. Although nochange in ST6Gal-I RNA levels occurs subsequent to BCRactivation of wild-type B cells (data not shown), multiplechanges in B-cell surface glycan linkages occur which mimicthose observed on ST6Gal-I-deficient B cells bearing un-masked CD22 (10, 36) (see Fig. S3 in the supplemental mate-rial). Decreased ST6Gal-I function may result from the induc-tion of glycosyltransferases that compete for glycoproteinsubstrates, by the expression or activation of endogenous siali-dases, or perhaps from proteolysis in the stem region abolish-ing glycosyltransferase substrate access in the Golgi apparatusby releasing the catalytic domain as a secreted fragment. Ex-
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4979
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

cept in the instance of a rapid desialylation at the cell surface,it appears unlikely that these mechanisms are involved duringthe early postactivation responses that result in rapid Shp-1recruitment to CD22 and IgM endocytosis. Interestingly, thenormal induction of BCR endocytosis in response to IgM stim-ulation among wild-type B cells, which also results in a cellsurface IgM half-life of less than 2 h, was not affected by theloss of CD22 or Lyn (data not shown). The similar decrease ofIgM BCR cell surface half-life in ST6Gal-I deficiency, which incontrast is CD22 dependent and occurs in the absence of IgMstimulation, likely represents a distinct pathway to endocytosisthat typically employs ST6Gal-I and CD22 function to restrainIgM BCR internalization in promoting naive mature B-cellimmune function.
Posttranslational modifications contributing to intracellularimmune signal transduction pathways include kinase and phos-phatase enzymes that regulate protein phosphorylation. Lessevident has been whether protein glycosylation, confined toextracellular compartments and topologically separated fromprotein phosphorylation, may participate in the formation ofthese intracellular signals. We have observed humoral immunedeficits in the absence of protein glycosylation by ST6Gal-Ithat result in diminished tyrosine phosphorylation of proteinsassociated with the BCR complex, in findings that reflect bothcell-type-specific and glycoprotein-selective roles for ST6Gal-I.Such focused purpose is increasingly evident among the dis-cerned functions of mammalian glycosyltransferases, as furtherexemplified by the specificity of ST8Sia-II and -IV in neuralNCAM function, FT-8 in lung epithelial transforming growthfactor � receptor signaling, and GnT-4a in pancreatic �-cellglucose transporter expression (1, 26, 47, 48). Diminished B-cell immune responses observed in ST6Gal-I deficiency reflectconstitutive disruption of a CD22-dependent mechanism thatcontrols glycoprotein-selective cell surface modulation of IgMtrafficking and colocalization with CD22–Shp-1 complexesamong unstimulated mature B cells coincident with enhancingthe rate of IgM endocytosis. Therefore, ST6Gal-I operateswith CD22 in an immune regulatory pathway that convergeswith clathrin-dependent IgM endocytosis in establishingthresholds for naıve mature peripheral BCR activation. Dimin-ishing ST6Gal-I function spares B-cell viability yet has a ther-apeutic effect in the context of the severe systemic lupus ery-thematosus phenotype due to Lyn deficiency and possiblyother autoimmune disease syndromes arising from abnormaland hyperimmune B-cell activity.
ACKNOWLEDGMENTS
We thank Linda Walker and Maria Conejo for excellent technicalsupport. Fluorescent microscopy was performed with the UCSD Can-cer Center Digital Imaging Shared Resource Facility directed by JamesFeramisco. CD22-deficient and ST6Gal-I/CD22 doubly deficient micewere provided by James C. Paulson of The Scripps Research Institute(La Jolla, Calif.).
J.D.M. is a founder of Abaron Biosciences, Inc., a company that isdeveloping drugs related to the research described in the manuscript.The University of California—San Diego is also an equity holder. Theterms of this arrangement have been reviewed and approved by theUniversity of California—San Diego, in accordance with its conflict ofinterest policies.
This research was funded by NIH grants HL57345 (J.D.M.),AI050143 (J.C.P.), and GM25042 (B.E.C.). J.D.M. is supported as anInvestigator of the Howard Hughes Medical Institute.
REFERENCES
1. Angata, K., J. M. Long, O. Bukalo, W. Lee, A. Ditayev, A. Wynshaw-Boris, M.Schachner, M. Fukuda, and J. D. Marth. 2004. Sialyltransferase ST8Sia-IIassembles a subset of polysialic acid that directs hippocampal axonal target-ing and promotes fear behavior. J. Biol. Chem. 279:32603–32613.
2. Baum, L. 2002. Developing a taste for sweets. Immunity 16:5–8.3. Chan, V. W. F., F. Meng, P. Soriano, A. L. DeFranco, and C. A. Lowell. 1997.
Characterization of the B lymphocyte populations in Lyn-deficient mice andthe role of Lyn in signal transduction and down-regulation. Immunity 7:69–81.
4. Chui, D., G. Sellakumar, R. Green, M. Sutton-Smith, T. McQuistan, K.Marek, H. Morris, A. Dell, and J. D. Marth. 2001. Genetic remodeling ofprotein glycosylation in vivo induces autoimmune disease. Proc. Natl. Acad.Sci. USA 98:1142–1147.
5. Collins, B. E., O. Blixt, N. V. Bovin, C. P. Danzer, D. Chui, J. D. Marth, L.Nitschke, and J. C. Paulson. 2002. Constitutively unmasked CD22 on B cellsof ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glyco-biology 12:563–571.
6. Collins, B. E., B. A. Smith, P. Bengston, and J. C. Paulson. 2006. Ablationof CD22 in ligand-deficient mice restores B cell receptor signaling. Nat.Immunol. 7:199–206.
7. Cornall, R. J., J. G. Cyster, M. L. Hibbs, A. R. Dunn, K. L. Otipoby, E. A.Clark, and C. C. Goodnow. 1998. Polygenic autoimmune traits: Lyn, CD22,and SHP-1 are limiting elements of a biochemical pathway regulating B cellsignaling and selection. Immunity 8:497–508.
8. Crocker, P. R., and A. Varki. 2001. Siglecs in the immune system. Immunol-ogy 103:137–145.
9. Cyster, J., and C. C. Goodnow. 1997. Tuning antigen receptor signaling byCD22: integrating cues from antigens and the microenvironment. Immunity6:509–517.
10. Danzer, C. P., B. E. Collins, O. Blixt, J. C. Paulson, and L. Nitschke. 2003.Transitional and marginal zone B cells have a high proportion of unmaskedCD22: implications for BCR signaling. Int. Immunol. 15:1137–1147.
11. Doody, G. M., L. B. Justement, C. C. Delibrias, R. J. Matthews, J. Lin, M. L.Thomas, and D. T. Fearon. 1995. A role in B cell activation for CD22 and theprotein tyrosine phosphatase SHP. Science 269:242–244.
12. Ellies, L. G., D. Ditto, G. G. Levy, M. Wahrenbrock, D. Ginsburg, A. Varki,D. T. Le, and J. D. Marth. 2002. Sialyltransferase ST3Gal-IV operates as adominant modifier of hemostasis by concealing asialoglycoprotein receptorligands. Proc. Natl. Acad. Sci. USA 99:10042–10047.
13. Floyd, H., L. Nitschke, and P. R. Crocker. 2000. A novel subset of murine Bcells that expresses unmasked forms of CD22 is enriched in bone marrow:implications for B-cell homing to the bone marrow. Immunology 101:342–347.
14. Goodnow, C. C., J. Crosbie, S. Adelstein, T. B. Lavoie, S. J. Smith-Gill, R. A.Brink, H. Pritchard-Briscoe, J. S. Wotherspoon, R. H. Loblay, and K. Raphael.1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334:676–682.
15. Han, S., B. E. Collins, P. Bengston, and J. C. Paulson. 2005. Homomulti-meric complexes of CD22 in B cells revealed by protein-glycan crosslinking.Nat. Chem. Biol. 1:93–97.
16. Hennet, T., D. Chui, J. C. Paulson, and J. D. Marth. 1998. Immune regu-lation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. USA 95:4504–4509.
17. Hibbs, M. L., D. M. Darlinton, J. Armes, D. Grail, G. Hodgson, R. Maglitto,S. A. Stacker, and A. R. Dunn. 1995. Multiple defects in the immune systemof Lyn-deficient mice, culminating in autoimmune disease. Cell 83:301–311.
18. Jin, L., P. A. McLean, B. G. Neel, and H. H. Wortis. 2002. Sialic acid bindingdomains of CD22 are required for negative regulation of B cell receptorsignaling. J. Exp. Med. 195:1199–1205.
19. Kelm, S., J. Gerlach, R. Brossmer, C. P. Danzer, and L. Nitschke. 2002. Theligand-binding domain of CD22 is needed for inhibition of the B cell recep-tor signal, as demonstrated by a novel human CD22-specific inhibitor com-pound. J. Exp. Med. 195:1207–1213.
20. Kitagawa, H., and J. C. Paulson. 1994. Differential expression of five sialyl-transferase genes in human tissues. J. Biol. Chem. 269:17872–17878.
21. Leprince, C., K. E. Draves, R. L. Geahlen, J. A. Ledbetter, and E. A. Clark.1993. CD22 associates with the human surface IgM-B-cell antigen receptorcomplex. Proc. Natl. Acad. Sci. USA 90:3236–3240.
22. Lowe, J. B. 2001. Glycosylation, immunity, and autoimmunity. Cell 104:809–812.
23. Lowe, J. B., and J. D. Marth. 2003. A genetic approach to mammalian glycanfunction. Annu. Rev. Biochem. 72:643–691.
24. Nishizumi, H., I. Taniuchi, Y. Yamanashi, D. Kitamura, D. Ilic, S. Mori, T.Watanabe, and T. Yamamoto. 1995. Impaired proliferation of peripheral Bcells and indication of autoimmune disease in lyn-deficient mice. Immunity3:549–560.
25. Nitschke, L., R. Carsetti, B. Ocker, G. Kohler, and M. C. Lamers. 1997.CD22 is a negative regulator of B-cell receptor signaling. Curr. Biol. 7:133–143.
26. Ohtsubo, K., S. Takamatsu, M. T. Minowa, A. Yoshida, M. Takeuchi, and
4980 GREWAL ET AL. MOL. CELL. BIOL.
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

J. D. Marth. 2005. Dietary and genetic control of glucose transporter-2glycosylation promotes insulin secretion in suppressing diabetes. Cell 123:1307–1321.
27. O’Keefe, T. L., G. T. Williams, S. L. Davies, and M. S. Neuberger. 1996.Hyperresponsive B cells in CD22-deficient mice. Science 274:798–801.
28. Otipoby, K. L., K. B. Andersson, K. E. Draves, S. J. Klaus, A. G. Farr, J. D.Kerner, R. M. Perlmutter, C. L. Law, and E. A. Clark. 1996. CD22 regulatesthymus-independent responses and the lifespan of B cells. Nature 384:634–637.
29. Otipoby, K. L., K. E. Draves, and E. A. Clark. 2001. CD22 regulates B cellreceptor-mediated signals via two domains that independently recruit Grb2and SHP-1. J. Biol. Chem. 276:44315–44322.
30. Peaker, C., J. G., and M. S. Neuberger. 1993. Association of CD22 with theB cell antigen receptor. Eur. J. Immunol. 23:1358–1363.
31. Phillips, M. L., E. Nudelman, F. C. Gaeta, M. Perez, A. K. Singhal, S.Hakomori, and J. C. Paulson. 1990. ELAM-1 mediates cell adhesion byrecognition of a carbohydrate ligand, sialyl-LeX. Science 250:1130–1132.
32. Poe, J. C., Y. Fujimoto, M. Hasegawa, K. M. Haas, A. S. Miller, I. G.Sanford, C. B. Bock, M. Fujimoto, and T. F. Tedder. 2004. CD22 regulatesB lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat. Immunol. 5:1078–1087.
33. Powell, L. D., R. K. Jain, K. L. Matta, S. Sabesan, and A. Varki. 1995.Characterization of sialoligosaccharide binding by recombinant soluble andnative cell-associated CD22. J. Biol. Chem. 270:7523–7532.
34. Priatel, J. J., D. Chui, N. Hiraoka, C. J. T. Simmons, K. B. Richardson,D. M. Page, M. Fukuda, N. M. Varki, and J. D. Marth. 2000. The ST3Gal-Isialyltransferase controls CD8� T cell homeostasis by modulating O-glycanbiosynthesis. Immunity 12:273–283.
35. Putnam, M. A., A. E. Moquin, M. Merrihew, C. Outcalt, E. Sorge, A.Caballero, T. A. Gondre-Lewis, and J. R. Drake. 2003. Lipid raft-indepen-dent B cell receptor-mediated antigen internalization and intracellular traf-ficking. J. Immunol. 170:905–912.
36. Razi, N., and A. Varki. 1998. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc. Natl.Acad. Sci. USA 95:7469–7474.
37. Rosen, S. D. 2004. Ligands for L-selectin: homing, inflammation and beyond.Annu. Rev. Immunol. 22:129–156.
38. Sato, S., A. S. Miller, M. Inaoki, C. B. Bock, P. J. Jansen, M. L. Tang, andT. F. Tedder. 1996. CD22 is both a positive and negative regulator of Blymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity 5:551–562.
39. Sgroi, D., A. Varki, S. Braesch-Andersen, and I. Stamenkovic. 1993. CD22,a B cell-specific immunoglobulin superfamily member, is a sialic acid-bindinglectin. J. Biol. Chem. 268:7011–7018.
40. Smith, K. G. C., K. M. Tarlinton, G. M. Doody, M. L. Hibbs, and D. T.Fearon. 1998. Inhibition of the B cell by CD22: a requirement for Lyn. J.Exp. Med. 187:807–811.
41. Stoddart, A., A. P. Jackson, and F. M. Brodsky. 2005. Plasticity of B cellreceptor internalization upon conditional depletion of clathrin. Mol. Biol.Cell 16:2339–2348.
42. Takashima, S., Y. Tachida, T. Nakagawa, T. Hamamoto, and S. Tsuji. 1999.Quantitative analysis of expression of mouse sialyltransferase genes by com-petitive PCR. Biochem. Biophys. Res. Commun. 260:23–27.
43. Tedder, T. F., J. Tuscano, S. Sato, and J. H. Kehrl. 1997. CD22, a Blymphocyte-specific adhesion molecule that regulates antigen receptor sig-naling. Annu. Rev. Immunol. 15:481–504.
44. Torres, R. M., H. Flaswinkel, M. Reth, and K. Rajewsky. 1996. Aberrant Bcell development and immune response in mice with a compromised BCRcomplex. Science 272:1804–1808.
45. Tsuji, S., A. K. Datta, and J. C. Paulson. 1996. Systematic nomenclature forsialyltransferases. Glycobiology 6:v–vii.
46. Vilen, B. J., T. Nakamura, and J. C. Cambier. 1999. Antigen-stimulateddissociation of BCR mIg from Ig-alpha/Ig-beta: implications for receptordesensitization. Immunity 10:239–248.
47. Wang, X., S. Inoue, J. Gu, E. Miyoshi, K. Noda, W. Li, Y. Mizuno-Horikawa,M. Nakano, M. Asahi, M. Takahashi, N. Uozumi, S. Ihara, S. H. Lee, Y.Ikeda, Y. Yamaguchi, Y. Aze, Y. Tomiyama, J. Fujji, K. Suzuki, A. Kondo,S. D. Shapiro, C. Lopez-Otin, T. Kuwaki, M. Okabe, K. Honke, and N.Taniguchi. 2005. Dysrregulation of TGF-�1 receptor activation leads toabnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. USA 102:15791–15796.
48. Weinhold, B., R. Seidenfaden, I. Rockle, M. Muhlenhoff, F. Schertzinger, S.Conzelmann, J. D. Marth, R. Gerardy-Schahn, and H. Hildebrandt. 2005.Genetic ablation of polysialic acid causes severe neurodevelopmental defectsrescued by NCAM deletion. J. Biol. Chem. 280:42971–42977.
49. Xu, Y., K. W. Harder, N. D. Huntington, M. L. Hibbs, and D. M. Tarlinton.2005. Lyn tyrosine kinase: accentuating the positive and the negative. Im-munity 22:9–18.
50. Zhang, M., and A. Varki. 2004. Cell surface sialic acids do not affect primaryCD22 interactions with CD45 and surface IgM nor the rate of constitutiveCD22 endocytosis. Glycobiology 14:939–949.
VOL. 26, 2006 ST6Gal-I PROTEIN GLYCOSYLATION IN B-CELL FUNCTION 4981
on Decem
ber 2, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















