
The glycosphingolipid, lactosylceramide, regulates beta1-integrin clustering and endocytosis
9
The Glycosphingolipid, Lactosylceramide, Regulates B 1 -Integrin Clustering and Endocytosis Deepak K. Sharma, Jennifer C. Brown, Zhijie Cheng, Eileen L. Holicky, David L. Marks, and Richard E. Pagano Department of Biochemistry and Molecular Biology, Thoracic Diseases Research Unit, and Mayo Clinic Cancer Center, Mayo Clinic College of Medicine, Rochester, Minnesota Abstract Glycosphingolipids are known to play roles in integrin- mediated cell adhesion and migration; however, the mecha- nisms by which glycosphingolipids affect integrins are unknown. Here, we show that addition of the glycosphingolipid, C8-lactosylceramide (C8-LacCer), or free cholesterol to human fibroblasts at 10°C causes the formation of glycosphingolipid- enriched plasma membrane domains as shown by visualizing a fluorescent glycosphingolipid probe, BODIPY-LacCer, incorpo- rated into the plasma membrane of living cells. Addition of C8- LacCer or cholesterol to cells initiated the clustering of B 1 -integrins within these glycosphingolipid-enriched domains and the activation of the B 1 -integrins as assessed using a HUTS antibody that only binds activated integrin. On warming to 37°C, B 1 -integrins were rapidly internalized via caveolar endocytosis in cells treated with C8-LacCer or cholesterol, whereas little B 1 -integrin was endocytosed in untreated fibroblasts. Incubation of cells with C8-LacCer or cholesterol followed by warm-up caused src activation, a reorganization of the actin cytoskeleton, translocation of RhoA GTPase away from the plasma membrane as visualized using total internal reflection fluorescence microscopy, and transient cell detach- ment. These studies show that LacCer can regulate integrin function both by modulating integrin clustering in micro- domains and by regulating integrin endocytosis via caveolae. Our findings suggest the possibility that aberrant levels of glycosphingolipids found in cancer cells may influence cell attachment events by direct effects on integrin clustering and internalization. (Cancer Res 2005; 65(18): 8233-41) Introduction Glycosphingolipids are plasma membrane constituents in mam- malian cells, which play important roles in normal cell adhesion, migration, and proliferation as well as in pathologic conditions, such as tumorigenesis and atherosclerosis (1–3). Glycosphingolipids are present mainly in the outer leaflet of the plasma membrane where they interact closely with cholesterol to form lipid micro- domains (4, 5). These glycosphingolipid-enriched microdomains have been shown to act as organizing centers for some cellular signaling complexes (3, 5) and are also initiation sites for clathrin- independent endocytic events (3, 5, 6). One mechanism by which glycosphingolipids could affect cell adhesion and migration is via their interaction with integrins. Integrins are a family of ah heterodimeric, integral membrane proteins at the plasma membrane, which bind to extracellular matrix (ECM) proteins and cell surface ligands, and are responsible for many types of cell adhesion events (7, 8). Glycosphingolipids have been shown to directly modulate integrin-based cell attachment. For example, gangliosides (sialic acid–terminated glycosphingolipids) extracted from neuroblastoma cells or atherosclerotic plaques enhance platelet adhesion via integrin binding to collagen (9–11). Gangliosides also enhance binding of integrins to the ECM in mouse mammary carcinoma, melanoma, and neuroblastoma cells (12–14). Several models have been proposed for the mechanisms by which glycosphingolipids or glycosphingolipid-enriched microdomains may regulate integrin function (11, 15, 16). First, glycosphingolipids could initiate signaling events, which cause downstream activation of integrins. Indeed, addition of exogenous glycosphingolipids to cells has been shown to have significant effects on signaling cascades. Another possibility is that glycosphingolipids promote the clustering of integrins in glycosphingolipid-enriched microdomains, thus increasing their avidity for ligand. The cross-linking of integrins with certain integrin antibodies is an established method for integrin activation (17, 18). Similarly, integrin function can be modulated by antibody cross-linking of cholera toxin B subunit bound to GM1 ganglioside or glycophosphatidylinositol-linked proteins (15, 16). However, no studies have provided direct evidence that glycosphingolipids modulate integrin clustering in glycosphingolipid-enriched micro- domains in the absence of cross-linking agents. An additional mechanism by which glycosphingolipids could regulate integrins is by affecting their endocytosis from the plasma membrane. Recent studies have shown that some integrins can be internalized via caveolae (18, 19), a subset of glycosphingolipid- enriched microdomains defined as invaginations at the plasma membrane enriched in caveolin-1 (Cav1; refs. 20, 21). Caveolae are sites for clathrin-independent endocytosis of glycosphingolipids as well as some viruses and bacterial toxins (22–27). We reported recently that the addition of glycosphingolipids or cholesterol to the plasma membrane of cells stimulates caveolar endocytosis via activation of src kinase (25). During these studies, we noted that on treatment with exogenous sphingolipids the cells began to reorganize their actin cytoskeleton and retract, 1 suggesting a link between plasma membrane glycosphingolipid and cholesterol composition and cell adhesion via integrins. Note: Joint first authors: D.K. Sharma and J.C. Brown. D.K. Sharma is currently at Photometrics, Inc., 3440 East Britannia Drive, Tucson, AZ 85706. Supplementary data for this article are available at Cancer Research Online (http:// cancerres.aacrjournals.org/). Requests for reprints: Richard E. Pagano, Mayo Clinic and Foundation, Stabile 8, 200 First Street, Southwest, Rochester, MN 55905-0001. Phone: 507-284-8754; Fax: 507- 266-4413; E-mail: [email protected]. I2005 American Association for Cancer Research. doi:10.1158/0008-5472.CAN-05-0803 1 Unpublished data. www.aacrjournals.org 8233 Cancer Res 2005; 65: (18). September 15, 2005 Research Article
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The glycosphingolipid, lactosylceramide, regulates beta1-integrin clustering and endocytosis

The Glycosphingolipid, Lactosylceramide, Regulates
B1-Integrin Clustering and Endocytosis
Deepak K. Sharma, Jennifer C. Brown, Zhijie Cheng, Eileen L. Holicky,David L. Marks, and Richard E. Pagano
Department of Biochemistry and Molecular Biology, Thoracic Diseases Research Unit, and Mayo Clinic Cancer Center,Mayo Clinic College of Medicine, Rochester, Minnesota
Abstract
Glycosphingolipids are known to play roles in integrin-mediated cell adhesion and migration; however, the mecha-nisms by which glycosphingolipids affect integrins areunknown. Here, we show that addition of the glycosphingolipid,C8-lactosylceramide (C8-LacCer), or free cholesterol to humanfibroblasts at 10�C causes the formation of glycosphingolipid-enriched plasma membrane domains as shown by visualizing afluorescent glycosphingolipid probe, BODIPY-LacCer, incorpo-rated into the plasma membrane of living cells. Addition of C8-LacCer or cholesterol to cells initiated the clustering ofB1-integrins within these glycosphingolipid-enriched domainsand the activation of the B1-integrins as assessed using a HUTSantibody that only binds activated integrin. On warming to37�C, B1-integrins were rapidly internalized via caveolarendocytosis in cells treated with C8-LacCer or cholesterol,whereas little B1-integrin was endocytosed in untreatedfibroblasts. Incubation of cells with C8-LacCer or cholesterolfollowed by warm-up caused src activation, a reorganization ofthe actin cytoskeleton, translocation of RhoA GTPase awayfrom the plasma membrane as visualized using total internalreflection fluorescence microscopy, and transient cell detach-ment. These studies show that LacCer can regulate integrinfunction both by modulating integrin clustering in micro-domains and by regulating integrin endocytosis via caveolae.Our findings suggest the possibility that aberrant levels ofglycosphingolipids found in cancer cells may influence cellattachment events by direct effects on integrin clustering andinternalization. (Cancer Res 2005; 65(18): 8233-41)
Introduction
Glycosphingolipids are plasma membrane constituents in mam-malian cells, which play important roles in normal cell adhesion,migration, and proliferation as well as in pathologic conditions,such as tumorigenesis and atherosclerosis (1–3). Glycosphingolipidsare present mainly in the outer leaflet of the plasma membranewhere they interact closely with cholesterol to form lipid micro-domains (4, 5). These glycosphingolipid-enriched microdomainshave been shown to act as organizing centers for some cellular
signaling complexes (3, 5) and are also initiation sites for clathrin-independent endocytic events (3, 5, 6).One mechanism by which glycosphingolipids could affect cell
adhesion and migration is via their interaction with integrins.Integrins are a family of ah heterodimeric, integral membraneproteins at the plasma membrane, which bind to extracellularmatrix (ECM) proteins and cell surface ligands, and are responsiblefor many types of cell adhesion events (7, 8). Glycosphingolipids havebeen shown to directly modulate integrin-based cell attachment. Forexample, gangliosides (sialic acid–terminated glycosphingolipids)extracted from neuroblastoma cells or atherosclerotic plaquesenhance platelet adhesion via integrin binding to collagen (9–11).Gangliosides also enhance binding of integrins to the ECM in mousemammary carcinoma, melanoma, and neuroblastoma cells (12–14).Several models have been proposed for the mechanisms
by which glycosphingolipids or glycosphingolipid-enrichedmicrodomains may regulate integrin function (11, 15, 16).First, glycosphingolipids could initiate signaling events, whichcause downstream activation of integrins. Indeed, addition ofexogenous glycosphingolipids to cells has been shown to havesignificant effects on signaling cascades. Another possibility isthat glycosphingolipids promote the clustering of integrins inglycosphingolipid-enriched microdomains, thus increasing theiravidity for ligand. The cross-linking of integrins with certainintegrin antibodies is an established method for integrin activation(17, 18). Similarly, integrin function can be modulated by antibodycross-linking of cholera toxin B subunit bound to GM1 gangliosideor glycophosphatidylinositol-linked proteins (15, 16). However, nostudies have provided direct evidence that glycosphingolipidsmodulate integrin clustering in glycosphingolipid-enriched micro-domains in the absence of cross-linking agents.An additional mechanism by which glycosphingolipids could
regulate integrins is by affecting their endocytosis from the plasmamembrane. Recent studies have shown that some integrins can beinternalized via caveolae (18, 19), a subset of glycosphingolipid-enriched microdomains defined as invaginations at the plasmamembrane enriched in caveolin-1 (Cav1; refs. 20, 21). Caveolae aresites for clathrin-independent endocytosis of glycosphingolipids aswell as some viruses and bacterial toxins (22–27). We reportedrecently that the addition of glycosphingolipids or cholesterol tothe plasma membrane of cells stimulates caveolar endocytosis viaactivation of src kinase (25). During these studies, we noted that ontreatment with exogenous sphingolipids the cells began toreorganize their actin cytoskeleton and retract,1 suggesting a linkbetween plasma membrane glycosphingolipid and cholesterolcomposition and cell adhesion via integrins.
Note: Joint first authors: D.K. Sharma and J.C. Brown.D.K. Sharma is currently at Photometrics, Inc., 3440 East Britannia Drive, Tucson,
AZ 85706.Supplementary data for this article are available at Cancer Research Online (http://
cancerres.aacrjournals.org/).Requests for reprints: Richard E. Pagano, Mayo Clinic and Foundation, Stabile 8,
200 First Street, Southwest, Rochester, MN 55905-0001. Phone: 507-284-8754; Fax: 507-266-4413; E-mail: [email protected].
I2005 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-0803 1 Unpublished data.
www.aacrjournals.org 8233 Cancer Res 2005; 65: (18). September 15, 2005
Research Article

Here, we investigated the possibility that addition of glyco-sphingolipid or cholesterol to cells might affect the function of h1-integrin, a key protein that mediates adhesion in many cell types(28, 29). These studies revealed that exogenously added C8-lactosylceramide (C8-LacCer) or cholesterol induced the clusteringof h1-integrins together with BODIPY-LacCer in glycosphingolipid-enriched microdomains that were visible by fluorescence micros-copy in living cells. In addition, the endocytosis of h1-integrin wassignificantly stimulated by both C8-LacCer and cholesteroladdition. Finally, the clustering and internalization of h1-integrinby C8-LacCer and cholesterol initiated downstream signalingevents that resulted in changes in cytoskeletal organization andadhesion consistent with the modulation of integrin function.These studies have important implications for understanding howplasma membrane lipid composition may affect integrin-mediatedcellular processes.
Materials and Methods
Lipids, fluorescent probes, and miscellaneous reagents. BODIPY-LacCer (30) or nonfluorescent C8-LacCer (D-lactosyl-h1-1V-N-octanoyl-D-erythro-sphingosine; Avanti Polar Lipids, Alabaster, AL) were complexed
with defatted bovine serum albumin (BSA; Serologicals Corp., Norcross, GA)
and used as described (25). Alexa Fluor 594 (AF594)–labeled transferrin andalbumin were fromMolecular Probes (Eugene, OR). Tyrosine kinase (PP2) and
protein kinase C (PKC) inhibitors (Go 6976)were fromCalbiochem (SanDiego,
CA). Cav1 small interfering RNA (siRNA) was from Super Array (Frederick,
MD). A RhoA GTPase plasmid was kindly provided by Dr. D. Billadeau (MayoClinic, Rochester, MN); GFP-RhoA was generated by subcloning into the
EGFP-C3 vector (BD Biosciences Clontech, Palo Alto, CA). GFP-actin was also
from BD Biosciences Clontech.Anti-h1-integrin (IgG) and anti-phospho-Tyr14 Cav1 antibodies were from
BD Biosciences PharMingen (San Diego, CA). Fab anti-h1-integrin fragments
were generated from this IgG using immobilized ficin, purified with protein
A-Sepharose, and labeled with Alexa Fluor 647 (AF647) succinimidyl ester(Molecular Probes). The HUTS-4 antibody, which recognizes activated h1-
integrin (31), was from Chemicon (Temecula, CA). Fluorescent secondary
antibodies were from Molecular Probes and Jackson ImmunoResearch
(West Grove, PA). Unless otherwise indicated, all other reagents were fromSigma Chemical Co. (St. Louis, MO).
Cell culture, transfection, and adenoviral infection. Normal human
skin fibroblasts (GM-5659D; Coriell Institute, Camden, NJ) were grown inEMEM with 10% fetal bovine serum (FBS); HeLa cells (American Type
Culture Collection, Rockville, MD) were grown in DMEM with 10% FBS.
Transfections were carried out by electroporation using a GenePulser Xcell
(Bio-Rad Laboratories, Hercules, CA) as described previously (25). Treatmentof cells with Cav1 siRNA was done using FuGene6 (Roche Diagnostics,
Indianapolis, IN). Cells were infected with Ad-KI-src, Ad-Dyn1K44A, or
‘‘empty virus’’ (Ad-empty) in culture medium for 4 hours followed by washing
with fresh medium and further incubation for 24 hours before use (25).Incubation with inhibitors. Inhibitors of endocytosis, src kinase, and
PKC were used as described (24–26). Briefly, cells were preincubated in
HEPES-buffered MEM (HMEM) containing inhibitors for 1 hour at 37jC;inhibitors were also present in all subsequent steps of the experiments. For
mhCD, cells were pretreated with 5 mmol/L drug for 30 minutes at 37jC.Incubation with fluorescent lipids and proteins. Incubations with
BODIPY-LacCer were done as described (25). Briefly, cells were incubatedfor 30 minutes at 10jC with 0.5 to 2.5 Amol/L BODIPY-LacCer/BSA, washed
twice with HMEM, and further incubated for 5 minutes at 37jC followed by
back exchange with 5% DF-BSA (6 � 10 minutes at 10jC) to remove any
fluorescent lipid remaining at the plasma membrane after endocytosis(30, 32). For labeled proteins, cells were preincubated with 5 Ag/mL AF594
transferrin or 50 Ag/mL AF594 albumin for 30 minutes at 10jC, furtherincubated for 5 minutes at 37jC, and acid stripped (26) to remove labeled
protein remaining at the cell surface.
Incubation with C8-lactosylceramide, cholesterol, and integrinantibody. Cells were incubated with 20 Amol/L C8-LacCer/BSA (30),
methyl-h-cyclodextrin (mhCD)/cholesterol complex (25), or anti-h1-
integrin antibody (2.5 Ag/mL) for 30 minutes at 10jC in HMEM. In
some experiments, samples were subsequently incubated for 5 minutes at37jC to allow endocytosis to occur.
Microscopy. Fluorescence microscopy was carried out using an
Olympus (Melville, NY) IX70 fluorescence microscope and the ‘‘Meta-
morph’’ image-processing program (Universal Imaging Corp., Downing-town, PA). Total internal reflection fluorescence (TIRF) microscopy was
carried out using an Olympus attachment for the IX70 microscope. In
any given experiment, all photomicrographs were exposed and processed
identically for a given fluorophore. Intracellular fluorescence wasquantified by analyzing images of z10 cells in at least three independent
experiments.
Miscellaneous procedures. SDS-PAGE and immunoblotting (27) andimmunofluorescence of formaldehyde-fixed cells (25) were done as
described previously. In vitro studies of src kinase activity were
determined in cell lysates using a kit from Upstate (Charlottesville, VA),
which measures phosphorylation of a model src peptide substrate.Statistical significance of quantified differences in cell fluorescence and
attachment was assessed by unpaired two-tailed t tests using the Prism4
program (GraphPad, San Diego, CA).
Results
Addition of C8-lactosylceramide or cholesterol inducesB1-integrin clustering into lipid domains. We first examined theeffect of C8-LacCer, a glycosphingolipid, or cholesterol/mhCDtreatments on the cell surface distribution of h1-integrin. Humanskin fibroblasts were treated with C8-LacCer or cholesterol for 30minutes at low temperature (10jC) to prevent endocytosis fromoccurring. The cells were then fixed without permeabilization andimmunostained to detect h1-integrin. Cells treated with 20 Amol/LC8-LacCer showed extensive clustering of h1-integrin comparedwith untreated cells (Fig. 1A, 1 versus 2). This clustering was similarto that observed when cells were incubated with h1-integrinantibody (Fig. 1A, 3). The clustering of h1-integrin induced byantibody was not sensitive to cholesterol depletion (Fig. 1A, 3 versus4). Similar results to those obtained with C8-LacCer (Fig. 1A, 2) werealso observed when cells were incubated with mhCD/cholesterolto increase plasma membrane cholesterol (data not shown).Previous studies from our laboratory have used fluorescent
(BODIPY-labeled) SL analogues to study lipid trafficking in cells(30, 32, 33). These analogues exhibit a shift in their fluorescenceemission from green to red wavelengths with increasingconcentrations in membranes (32, 33). We took advantage ofthis property to probe the domain organization of BODIPY-LacCer at the plasma membrane under different conditions. Inuntreated cells, the plasma membrane was uniformly labeled andemitted only green fluorescence (Fig. 1B, 1). In contrast, whencells were treated with nonfluorescent C8-LacCer or cholesterol,small ‘‘patches’’ of BODIPY-LacCer with increased red emission(shown as yellow/orange areas in green/red overlays) appeared atthe plasma membrane (Fig. 1B, 2 and 3), suggesting the formationof plasma membrane ‘‘domains’’ that were enriched inBODIPY-LacCer. Polyvalent antibody (IgG)–induced clustering ofh1-integrin also induced similar yellow/orange patches (Fig. 1B, 4);however, when monovalent anti-h1-integrin Fab fragments wereused, no clustering was detected (data not shown).Induction of yellow/orange plasma membrane microdomains by
antibody cross-linking of h1-integrin (Fig. 1B, 5) or C8-LacCer(data not shown) was inhibited by pretreatment of cells with
Cancer Research
Cancer Res 2005; 65: (18). September 15, 2005 8234 www.aacrjournals.org
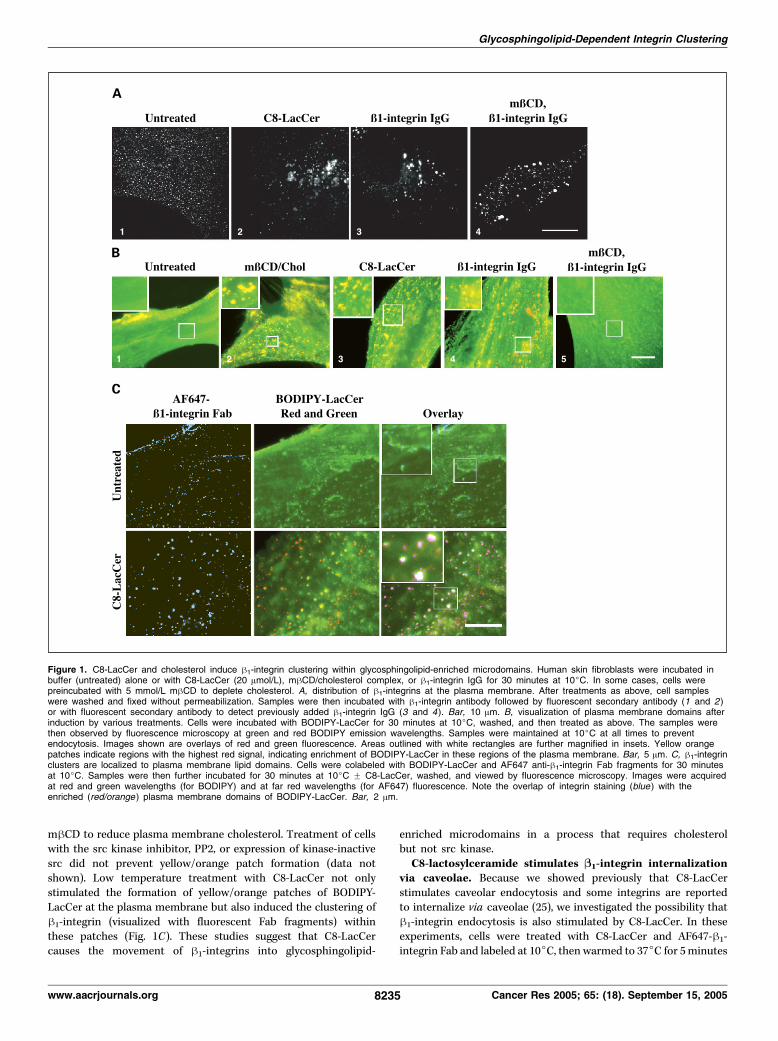
mhCD to reduce plasma membrane cholesterol. Treatment of cellswith the src kinase inhibitor, PP2, or expression of kinase-inactivesrc did not prevent yellow/orange patch formation (data notshown). Low temperature treatment with C8-LacCer not onlystimulated the formation of yellow/orange patches of BODIPY-LacCer at the plasma membrane but also induced the clustering ofh1-integrin (visualized with fluorescent Fab fragments) withinthese patches (Fig. 1C). These studies suggest that C8-LacCercauses the movement of h1-integrins into glycosphingolipid-
enriched microdomains in a process that requires cholesterolbut not src kinase.C8-lactosylceramide stimulates B1-integrin internalization
via caveolae. Because we showed previously that C8-LacCerstimulates caveolar endocytosis and some integrins are reportedto internalize via caveolae (25), we investigated the possibility thath1-integrin endocytosis is also stimulated by C8-LacCer. In theseexperiments, cells were treated with C8-LacCer and AF647-h1-integrin Fab and labeled at 10jC, then warmed to 37jC for 5minutes
Figure 1. C8-LacCer and cholesterol induce h1-integrin clustering within glycosphingolipid-enriched microdomains. Human skin fibroblasts were incubated inbuffer (untreated) alone or with C8-LacCer (20 Amol/L), mhCD/cholesterol complex, or h1-integrin IgG for 30 minutes at 10jC. In some cases, cells werepreincubated with 5 mmol/L mhCD to deplete cholesterol. A, distribution of h1-integrins at the plasma membrane. After treatments as above, cell sampleswere washed and fixed without permeabilization. Samples were then incubated with h1-integrin antibody followed by fluorescent secondary antibody (1 and 2 )or with fluorescent secondary antibody to detect previously added h1-integrin IgG (3 and 4). Bar, 10 Am. B, visualization of plasma membrane domains afterinduction by various treatments. Cells were incubated with BODIPY-LacCer for 30 minutes at 10jC, washed, and then treated as above. The samples werethen observed by fluorescence microscopy at green and red BODIPY emission wavelengths. Samples were maintained at 10jC at all times to preventendocytosis. Images shown are overlays of red and green fluorescence. Areas outlined with white rectangles are further magnified in insets. Yellow orangepatches indicate regions with the highest red signal, indicating enrichment of BODIPY-LacCer in these regions of the plasma membrane. Bar, 5 Am. C, h1-integrinclusters are localized to plasma membrane lipid domains. Cells were colabeled with BODIPY-LacCer and AF647 anti-h1-integrin Fab fragments for 30 minutesat 10jC. Samples were then further incubated for 30 minutes at 10jC F C8-LacCer, washed, and viewed by fluorescence microscopy. Images were acquiredat red and green wavelengths (for BODIPY) and at far red wavelengths (for AF647) fluorescence. Note the overlap of integrin staining (blue ) with theenriched (red/orange ) plasma membrane domains of BODIPY-LacCer. Bar, 2 Am.
Glycosphingolipid-Dependent Integrin Clustering
www.aacrjournals.org 8235 Cancer Res 2005; 65: (18). September 15, 2005
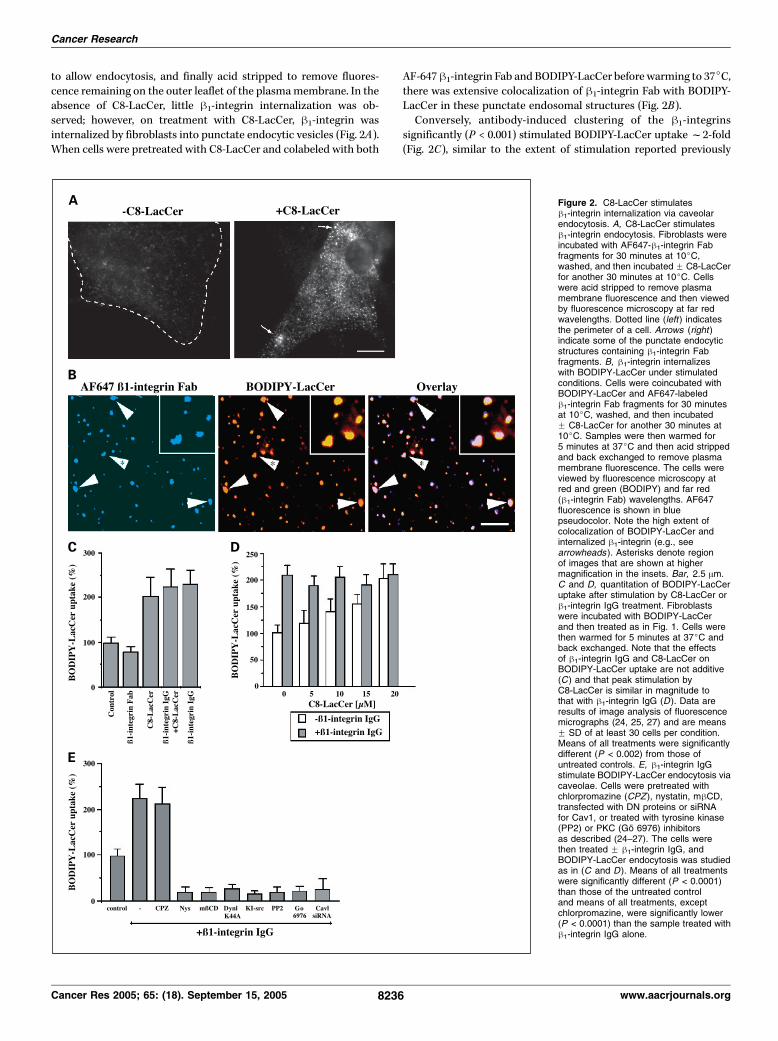
to allow endocytosis, and finally acid stripped to remove fluores-cence remaining on the outer leaflet of the plasmamembrane. In theabsence of C8-LacCer, little h1-integrin internalization was ob-served; however, on treatment with C8-LacCer, h1-integrin wasinternalized by fibroblasts into punctate endocytic vesicles (Fig. 2A).When cells were pretreated with C8-LacCer and colabeled with both
AF-647h1-integrin Fab and BODIPY-LacCer before warming to 37jC,there was extensive colocalization of h1-integrin Fab with BODIPY-LacCer in these punctate endosomal structures (Fig. 2B).Conversely, antibody-induced clustering of the h1-integrins
significantly (P < 0.001) stimulated BODIPY-LacCer uptakef2-fold(Fig. 2C), similar to the extent of stimulation reported previously
Figure 2. C8-LacCer stimulatesh1-integrin internalization via caveolarendocytosis. A, C8-LacCer stimulatesh1-integrin endocytosis. Fibroblasts wereincubated with AF647-h1-integrin Fabfragments for 30 minutes at 10jC,washed, and then incubated F C8-LacCerfor another 30 minutes at 10jC. Cellswere acid stripped to remove plasmamembrane fluorescence and then viewedby fluorescence microscopy at far redwavelengths. Dotted line (left ) indicatesthe perimeter of a cell. Arrows (right )indicate some of the punctate endocyticstructures containing h1-integrin Fabfragments. B, h1-integrin internalizeswith BODIPY-LacCer under stimulatedconditions. Cells were coincubated withBODIPY-LacCer and AF647-labeledh1-integrin Fab fragments for 30 minutesat 10jC, washed, and then incubatedF C8-LacCer for another 30 minutes at10jC. Samples were then warmed for5 minutes at 37jC and then acid strippedand back exchanged to remove plasmamembrane fluorescence. The cells wereviewed by fluorescence microscopy atred and green (BODIPY) and far red(h1-integrin Fab) wavelengths. AF647fluorescence is shown in bluepseudocolor. Note the high extent ofcolocalization of BODIPY-LacCer andinternalized h1-integrin (e.g., seearrowheads ). Asterisks denote regionof images that are shown at highermagnification in the insets. Bar, 2.5 Am.C and D, quantitation of BODIPY-LacCeruptake after stimulation by C8-LacCer orh1-integrin IgG treatment. Fibroblastswere incubated with BODIPY-LacCerand then treated as in Fig. 1. Cells werethen warmed for 5 minutes at 37jC andback exchanged. Note that the effectsof h1-integrin IgG and C8-LacCer onBODIPY-LacCer uptake are not additive(C ) and that peak stimulation byC8-LacCer is similar in magnitude tothat with h1-integrin IgG (D). Data areresults of image analysis of fluorescencemicrographs (24, 25, 27) and are meansF SD of at least 30 cells per condition.Means of all treatments were significantlydifferent (P < 0.002) from those ofuntreated controls. E, h1-integrin IgGstimulate BODIPY-LacCer endocytosis viacaveolae. Cells were pretreated withchlorpromazine (CPZ ), nystatin, mhCD,transfected with DN proteins or siRNAfor Cav1, or treated with tyrosine kinase(PP2) or PKC (Go 6976) inhibitorsas described (24–27). The cells werethen treated F h1-integrin IgG, andBODIPY-LacCer endocytosis was studiedas in (C and D). Means of all treatmentswere significantly different (P < 0.0001)than those of the untreated controland means of all treatments, exceptchlorpromazine, were significantly lower(P < 0.0001) than the sample treated withh1-integrin IgG alone.
Cancer Research
Cancer Res 2005; 65: (18). September 15, 2005 8236 www.aacrjournals.org

using C8-LacCer or cholesterol (25). Integrin cross-linking alsostimulated the endocytosis of fluorescent albumin, another markerfor caveolar uptake in fibroblasts (data not shown). Interestingly,the stimulation of BODIPY-LacCer uptake induced by incubationwith h1-integrin IgG clustering and C8-LacCer were not additive,suggesting that these treatmentsmay stimulate caveolar endocytosisby similar mechanisms (Fig. 2D). The stimulation of BODIPY-LacCeruptake by h1-integrin cross-linking was further characterized usinginhibitors of clathrin-mediated endocytosis (chloropromazine;DN Eps15; refs. 24, 34), inhibitors of caveolar uptake (mhCD;nystatin; ref. 24), Cav1 siRNA (35), and DN-Dyn1K44A, which blocksmultiple endocytic mechanisms (Fig. 2E). The inhibition profile wasconsistent with caveolar endocytosis. Moreover, the stimulateduptake of BODIPY-LacCer that was induced by h1-integrin cross-linking was also src and PKC dependent (Fig. 2E). Finally, we notethat transferrin uptake was not stimulated with h1-integrin cross-linking or by the addition of transferrin receptor antibody (data notshown). Addition of h1-integrin Fab fragments or aerolysin(to cluster glycophosphoinositol-anchored proteins at the plasmamembrane; ref. 36) also did not stimulate BODIPY-LacCer uptake(data not shown).C8-lactosylceramide promotes B1-integrin activation and
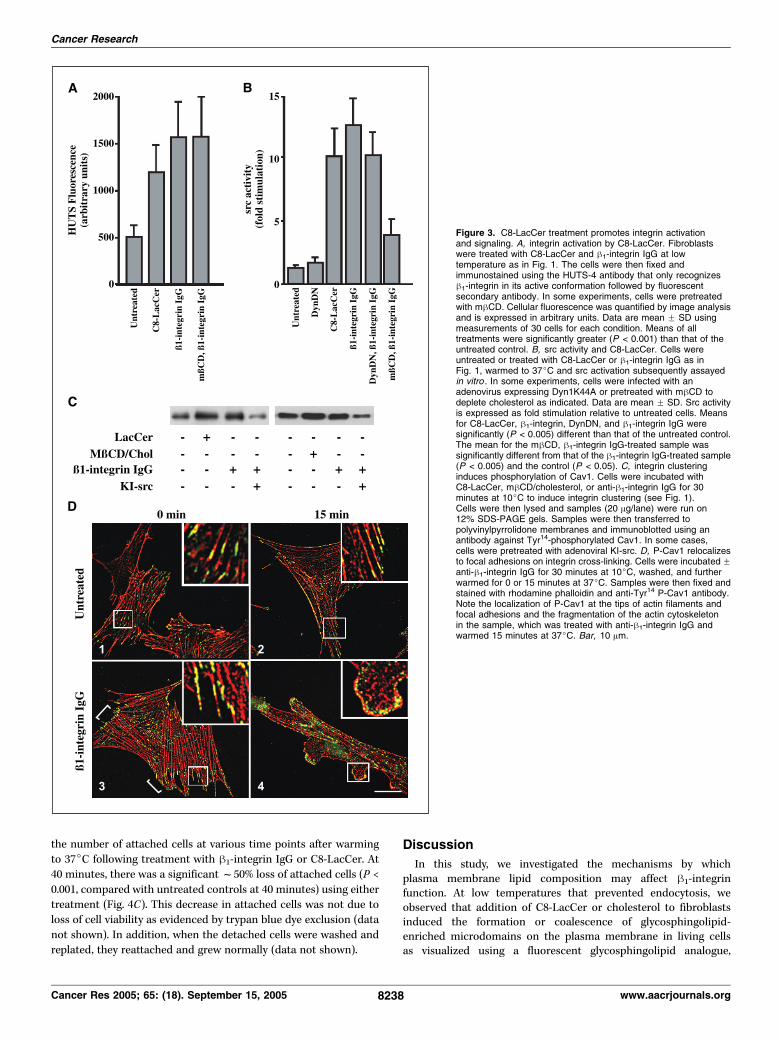
stimulates src activity. Integrin clustering is a required step in theinitiation of signaling by integrins. Thus, we investigated thepossibility that integrin clustering by glycosphingolipids stimulatesdownstream events associated with integrin signaling. First, wedetermined if glycosphingolipid treatment affected integrin activa-tion. C8-LacCer addition led to increased binding to h-integrin bythe HUTS-4 antibody (Fig. 3A), which only binds h1-integrins in theiractivated conformation (31), suggesting that h1-integrin wasactivated by these treatments. Because h1-integrin family membershave been shown to activate src kinases (37), we examined the effectof lipid-induced integrin clustering on src activation (Fig. 3B).Treatment with C8-LacCer at 10jC followed by a 5-minuteincubation at 37jC resulted in f10-fold activation of src (P <0.005), similar to the activation seen on h1-integrin cross-linkingwith an antibody (f12-fold activation). Similar src activation wasseen when cells were treated with mhCD/cholesterol (25). Acti-vation of src was not significantly reduced in cells infected with anadenovirus encoding DN-Dyn1K44A (Fig. 3B), which inhibits bothclathrin and caveolar endocytosis (25, 38, 39), indicating thatinternalization was not required for src activation by h1-integrin.However, treatmentwithmhCD to deplete cholesterol decreased srcactivation (P < 0.005) by integrin antibody by f60% (Fig. 3B),suggesting that integrin clustering into cholesterol-enriched plasmamembrane domains is required for src activation.Clustering of B1-integrin by C8-lactosylceramide causes
cytoskeletal alterations. Signaling via integrins is known toinitiate cytoskeletal remodeling (8, 17). Thus, we examined theeffects of C8-LacCer and cholesterol on the actin cytoskeleton. Oninternalization of BODIPY-LacCer via caveolae, we observedBODIPY-LacCer fluorescence in intracellular punctae thatappeared to be associated with actin filaments under both basalconditions and in cells stimulated with h1-integrin IgG and C8-LacCer (Supplementary Fig. S1). Previous studies have shown thatCav1 can be phosphorylated at Tyr14 by src family kinases inresponse to integrin activation (40). Further, on phosphorylation,Cav1 has been shown to localize at or near focal adhesions (40, 41).Thus, we investigated the possibility that C8-LacCer and choleste-rol treatment stimulated Cav1 phosphorylation. Treatment of cellswith C8-LacCer, mhCD/cholesterol, or h1-integrin antibody for
30 minutes at 10jC resulted in increased levels of phospho-Tyr14
Cav1 (P-Cav1) as assessed by immunoblotting (Fig. 3C). This phos-phorylation was significantly inhibited in cells that were infectedwith KI-src (Fig. 3C ; data not shown). This finding is in agreementwith previous studies showing that Tyr14 phosphorylation of Cav1occurs via the activity of a src kinase (41, 42). The increase inP-Cav1 levels in treated cells was even more pronounced (f65%)after a 5-minute incubation at 37jC (data not shown). In parallelexperiments, we found that total Cav1 levels were relativelyunchanged during each of these treatments (data not shown).We then examined the effects of integrin clustering on the
intracellular distribution of P-Cav1 using immunofluorescencemicroscopy. In control samples, P-Cav1 was localized along actinfilaments (Fig. 3D, 1) as reported previously (42). This distributionwas not significantly affected when cells were warmed for 15minutes at 37jC (Fig. 3D, 2). When cells were pretreated with h1-integrin IgG, P-Cav1 levels were enhanced at the tips of actinfilaments (i.e., focal adhesions; Fig. 3D, 3 ; e.g., at brackets). Onwarming of cells to 37jC, P-Cav1 became relocalized intonumerous puncta at the edges of the cells, whereas the actincytoskeleton in these regions began to depolymerize (Fig. 3D ,compare inset of 3 with inset of 4). Similar results to those shownin Fig. 3D using h1-integrin IgG were also obtained when cells werepretreated with C8-LacCer or mhCD/cholesterol (data not shown).Stimulation of caveolar endocytosis by integrin clustering
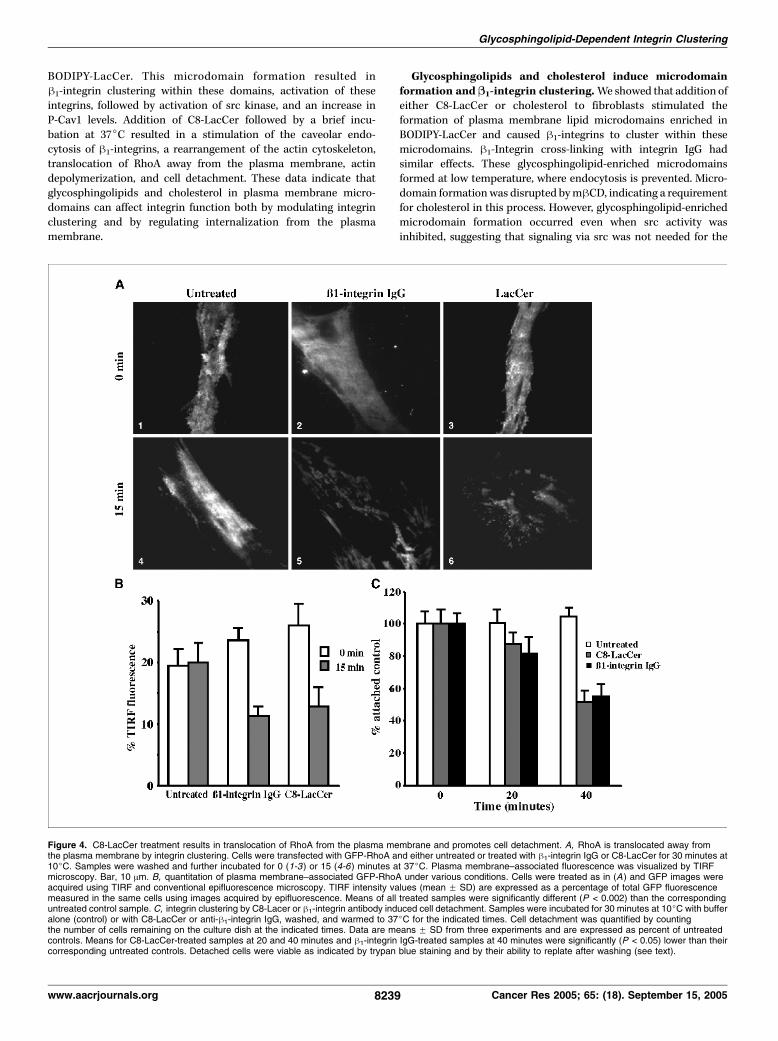
induces RhoA translocation from the plasma membrane andcell detachment. Integrins modulate the distribution and activityof Rho GTPases (43), and RhoA is also involved in stabilization ofcytoskeletal elements at focal adhesions (44, 45). In preliminarystudies, we found that incubations with C8-LacCer or cholesterolcaused cells to reorganize their actin cytoskeleton and retract,2
suggesting that Rho proteins might be affected by these treat-ments. We therefore next examined the effects of integrinclustering on the distribution of RhoA in live cells under variousconditions. Cells were transiently transfected with GFP-RhoA andthe extent of plasma membrane localization was determined usingTIRF microscopy. In control cells, the amount of GFP-RhoApresent at the plasma membrane was similar at 10jC (Fig. 4A, 1)or after a 15-minute incubation at 37jC (Fig. 4A, 4 , and B). Incontrast, when cells were treated with either h1-integrin antibodyor C8-LacCer at 10jC and subsequently shifted to 37jC, f50% ofthe plasma membrane–associated fluorescence were lost (Fig. 4A,2 versus 5 and 3 versus 6 and B). The translocation of GFP-RhoAfrom the plasma membrane was inhibited in cells expressingDyn1K44A (data not shown), suggesting that endocytosis isrequired for RhoA redistribution.Due to the known role of RhoA in regulation of actin organization
(43), and our observation that some actin depolymerizationoccurred on stimulation of caveolar endocytosis by h1-integrincross-linking (Fig. 3D, 4), we next examined the effect of integrinclustering on cell retraction and detachment. Cells were transfectedwith GFP-actin and incubated with h1-integrin IgG, and plasmamembrane–associated actin was visualized using TIRF microscopy.As seen in Supplementary Fig. S2 (and Supplemental Video), selec-ted regions of the cell in close proximity to the coverslip wereretracted on warming to 37jC. Similar results to those in Fig. S2were also seen using C8-LacCer or cholesterol treatments of cells(data not shown). Finally, wemonitored cell detachment by counting
2 Unpublished data.
Glycosphingolipid-Dependent Integrin Clustering
www.aacrjournals.org 8237 Cancer Res 2005; 65: (18). September 15, 2005
the number of attached cells at various time points after warming
to 37jC following treatment with h1-integrin IgG or C8-LacCer. At
40 minutes, there was a significantf50% loss of attached cells (P <
0.001, compared with untreated controls at 40 minutes) using either
treatment (Fig. 4C). This decrease in attached cells was not due to
loss of cell viability as evidenced by trypan blue dye exclusion (data
not shown). In addition, when the detached cells were washed and
replated, they reattached and grew normally (data not shown).
Discussion
In this study, we investigated the mechanisms by whichplasma membrane lipid composition may affect h1-integrinfunction. At low temperatures that prevented endocytosis, weobserved that addition of C8-LacCer or cholesterol to fibroblastsinduced the formation or coalescence of glycosphingolipid-enriched microdomains on the plasma membrane in living cellsas visualized using a fluorescent glycosphingolipid analogue,
Figure 3. C8-LacCer treatment promotes integrin activationand signaling. A, integrin activation by C8-LacCer. Fibroblastswere treated with C8-LacCer and h1-integrin IgG at lowtemperature as in Fig. 1. The cells were then fixed andimmunostained using the HUTS-4 antibody that only recognizesh1-integrin in its active conformation followed by fluorescentsecondary antibody. In some experiments, cells were pretreatedwith mhCD. Cellular fluorescence was quantified by image analysisand is expressed in arbitrary units. Data are mean F SD usingmeasurements of 30 cells for each condition. Means of alltreatments were significantly greater (P < 0.001) than that of theuntreated control. B, src activity and C8-LacCer. Cells wereuntreated or treated with C8-LacCer or h1-integrin IgG as inFig. 1, warmed to 37jC and src activation subsequently assayedin vitro . In some experiments, cells were infected with anadenovirus expressing Dyn1K44A or pretreated with mhCD todeplete cholesterol as indicated. Data are mean F SD. Src activityis expressed as fold stimulation relative to untreated cells. Meansfor C8-LacCer, h1-integrin, DynDN, and h1-integrin IgG weresignificantly (P < 0.005) different than that of the untreated control.The mean for the mhCD, h1-integrin IgG-treated sample wassignificantly different from that of the h1-integrin IgG-treated sample(P < 0.005) and the control (P < 0.05). C, integrin clusteringinduces phosphorylation of Cav1. Cells were incubated withC8-LacCer, mhCD/cholesterol, or anti-h1-integrin IgG for 30minutes at 10jC to induce integrin clustering (see Fig. 1).Cells were then lysed and samples (20 Ag/lane) were run on12% SDS-PAGE gels. Samples were then transferred topolyvinylpyrrolidone membranes and immunoblotted using anantibody against Tyr14-phosphorylated Cav1. In some cases,cells were pretreated with adenoviral KI-src. D, P-Cav1 relocalizesto focal adhesions on integrin cross-linking. Cells were incubated Fanti-h1-integrin IgG for 30 minutes at 10jC, washed, and furtherwarmed for 0 or 15 minutes at 37jC. Samples were then fixed andstained with rhodamine phalloidin and anti-Tyr14 P-Cav1 antibody.Note the localization of P-Cav1 at the tips of actin filaments andfocal adhesions and the fragmentation of the actin cytoskeletonin the sample, which was treated with anti-h1-integrin IgG andwarmed 15 minutes at 37jC. Bar, 10 Am.
Cancer Research
Cancer Res 2005; 65: (18). September 15, 2005 8238 www.aacrjournals.org
BODIPY-LacCer. This microdomain formation resulted inh1-integrin clustering within these domains, activation of theseintegrins, followed by activation of src kinase, and an increase inP-Cav1 levels. Addition of C8-LacCer followed by a brief incu-bation at 37jC resulted in a stimulation of the caveolar endo-cytosis of h1-integrins, a rearrangement of the actin cytoskeleton,translocation of RhoA away from the plasma membrane, actindepolymerization, and cell detachment. These data indicate thatglycosphingolipids and cholesterol in plasma membrane micro-domains can affect integrin function both by modulating integrinclustering and by regulating internalization from the plasmamembrane.
Glycosphingolipids and cholesterol induce microdomainformation and B1-integrin clustering.We showed that addition ofeither C8-LacCer or cholesterol to fibroblasts stimulated theformation of plasma membrane lipid microdomains enriched inBODIPY-LacCer and caused h1-integrins to cluster within thesemicrodomains. h1-Integrin cross-linking with integrin IgG hadsimilar effects. These glycosphingolipid-enriched microdomainsformed at low temperature, where endocytosis is prevented. Micro-domain formationwas disrupted bymhCD, indicating a requirementfor cholesterol in this process. However, glycosphingolipid-enrichedmicrodomain formation occurred even when src activity wasinhibited, suggesting that signaling via src was not needed for the
Figure 4. C8-LacCer treatment results in translocation of RhoA from the plasma membrane and promotes cell detachment. A, RhoA is translocated away fromthe plasma membrane by integrin clustering. Cells were transfected with GFP-RhoA and either untreated or treated with h1-integrin IgG or C8-LacCer for 30 minutes at10jC. Samples were washed and further incubated for 0 (1-3 ) or 15 (4-6) minutes at 37jC. Plasma membrane–associated fluorescence was visualized by TIRFmicroscopy. Bar, 10 Am. B, quantitation of plasma membrane–associated GFP-RhoA under various conditions. Cells were treated as in (A ) and GFP images wereacquired using TIRF and conventional epifluorescence microscopy. TIRF intensity values (mean F SD) are expressed as a percentage of total GFP fluorescencemeasured in the same cells using images acquired by epifluorescence. Means of all treated samples were significantly different (P < 0.002) than the correspondinguntreated control sample. C, integrin clustering by C8-Lacer or h1-integrin antibody induced cell detachment. Samples were incubated for 30 minutes at 10jC with bufferalone (control) or with C8-LacCer or anti-h1-integrin IgG, washed, and warmed to 37jC for the indicated times. Cell detachment was quantified by countingthe number of cells remaining on the culture dish at the indicated times. Data are means F SD from three experiments and are expressed as percent of untreatedcontrols. Means for C8-LacCer-treated samples at 20 and 40 minutes and h1-integrin IgG-treated samples at 40 minutes were significantly (P < 0.05) lower than theircorresponding untreated controls. Detached cells were viable as indicated by trypan blue staining and by their ability to replate after washing (see text).
Glycosphingolipid-Dependent Integrin Clustering
www.aacrjournals.org 8239 Cancer Res 2005; 65: (18). September 15, 2005

formation of these microdomains. The clustering of h1-integrin inthese domains seems to be the initial step in integrin activation andsignaling. This premise is supported by our demonstration thatC8-LacCer treatment caused a change in h1-integrin to its activeconformation (Fig. 3A). Further, both src activity and P-Cav1 levelsare increased on C8-LacCer addition. Our results are consistent withprevious studies showing that integrin activation by ligands or cross-linking antibodies can trigger the movement of integrins into lipidmicrodomains (18, 46). Several studies have shown that the cross-linking of glycosphingolipid-enriched microdomain constituents(e.g., glycophosphoinositol-linked proteins and GM1 ganglioside)can also stimulate integrin clustering (15, 16). However, to ourknowledge, our study is the first demonstration that increases ofplasmamembrane glycosphingolipid or cholesterol composition canrapidly promote integrin clustering within microdomains.Induction of glycosphingolipid-enriched microdomains
results in stimulated endocytosis via caveolae. The clusteringof glycosphingolipids into microdomains seems to be a prerequisitefor the stimulation of caveolar endocytosis observed in this study.Addition of C8-LacCer, cholesterol, and cross-linking of h1-integrinat low temperature each resulted in the clustering of BODIPY-LacCer and h1-integrins together in microdomains. Each of thesetreatments also elicited a significant stimulation of endocytosis viacaveolae when cells were warmed to 37jC. Stimulated uptake wasconsistent with endocytosis via caveolae based on its selectiveinhibition by several pharmacologic inhibitors and dominant-negative proteins (Fig. 2; ref. 25). C8-LacCer and h1-integrin cross-linking each stimulated caveolar endocytosis and src activity to asimilar degree and were not additive in their effects, suggestingthat both treatments stimulated endocytosis via similar mecha-nisms. The observations that C8-LacCer and cholesterol causeclustering of microdomains provides a partial mechanistic expla-nation for the selective stimulation of caveolar endocytosis that wereported previously (ref. 25; i.e., these treatments induce theselective clustering of glycosphingolipids and integrins into regionsof the plasma membrane, which then become sites for endocytosisvia caveolae). Importantly, our study suggests that glycosphingo-lipids and cholesterol at the plasma membrane may play importantroles in regulating the endocytic rate of integrins from the cellsurface.C8-lactosylceramide and cholesterol induce signaling via
B1-integrin and reorganization of the actin cytoskeleton.Treatment of fibroblasts with C8-LacCer or cholesterol initiated aseries of signaling events consistent with signaling via integrins. Onincubation of cells with C8-LacCer or cholesterol at 10jC,h1-integrins clustered in microdomains as stated above and werefound to be in an activated conformation as shown by binding withthe HUTS-4 antibody. Src was activated by treatment withC8-LacCer. This stimulation of src activity was not inhibited byDN-Dyn1, suggesting that src elevation precedes h1-integrinendocytosis. At 10jC, P-Cav1 levels were increased by C8-LacCeror cholesterol, similar to results seen with stimulation of cells withh1-integrin IgG. This increase in P-Cav1 levels was blocked byKI-src expression, consistent with the idea that stimulation of srcactivity precedes P-Cav1 elevation. On a brief (5-15 minutes) warm-up to 37jC, C8-LacCer caused a rearrangement of the actincytoskeleton, concomitant with translocation of RhoA away fromthe plasma membrane. By 40 minutes after treatment, f50% oftreated cells became detached from glass coverslips presumably asa result of integrin internalization and cytoskeletal changes.Interestingly, RhoA movement from the plasma membrane was
inhibited by DN-Dyn1 expression, suggesting that integrin inter-nalization (or stimulated caveolar endocytosis) is required for theobserved changes in RhoA distribution. This result is similar to areport by del Pozo et al. (47), which showed that the GTPase, rac1,is prevented from translocating away from the plasma membranewhen lipid microdomain endocytosis is inhibited in serum-stimulated 3T3 cells.Regulation of integrin functions by glycosphingolipids and
cholesterol. The results of these studies have elucidated twoimportant mechanisms by which plasma membrane glycosphingo-lipids can modulate h1-integrin function: enhancement of integrinclustering and stimulation of integrin removal from the plasmamembrane by endocytosis. Although these two mechanisms seemto modulate integrin function in opposite directions, both areconsistent with previous observations. First, because integrinclustering is known to increase the avidity of integrins for ECMand other ligands (48, 49), the observation that C8-LacCer andcholesterol promote the clustering of integrin at the plasmamembrane may be directly related to stimulatory effects of glyco-sphingolipids on integrin binding shown in some other studies.For example, the stimulation of integrin-mediated adhesion ofplatelets to collagen by gangliosides from neuroblastoma cells(mainly GD2 ganglioside) and atherosclerotic plaques (mainly GD3and GM3 gangliosides; refs. 9–11) could be due to effects onintegrin clustering. Similarly, studies in various cell types, whichhave shown a direct relationship between integrin-dependent celladhesion and glycosphingolipid levels (9, 13, 14), could reflect themodulation of integrin clustering by changes in glycosphingolipidcomposition.Although C8-LacCer addition initially causes a stimulation of
h1-integrin clustering and activation over time at 37jC, C8-LacCerpromotes the internalization of h1-integrin (Fig. 2), presumablyleading to reduced levels of integrin at the plasmamembrane. Loss ofintegrin at the cell surface may then lead to cell detachment(Fig. 4C). Inhibitory effects of gangliosides (GT1b and GD3) onintegrin-based cell adhesion and migration have been shown forkeratinocytes binding to fibronectin (50, 51). GD3 has also beenreported to inhibit the binding of neurepithelial cells to fibronectin(52). It remains to be determined if the observed inhibitory effects ofsome gangliosides on integrin function are due to stimulatedintegrin endocytosis and loss from the cell surface or other mecha-nisms (e.g., reducing integrin clustering by supplanting integrinassociation with other more favorable lipids). The net overall effectsof exogenous glycosphingolipids on integrin functionmay ultimatelydepend on the particular glycosphingolipids and integrin hetero-dimers present, the duration and temperature of treatment, and thepresence or absence of integrin ligands. Our studies thus far haveonly shown that C8-LacCer could affect integrin clustering andendocytosis. However, our previous study showing that, in additionto C8-LacCer, GM1 ganglioside could also stimulate caveolarendocytosis (25) suggests that C8-LacCer may not be unique amongglycosphingolipids in its ability to regulate h1-integrin function.Further study will be required to determine the specific effects ofindividual glycosphingolipid species on the clustering and endo-cytosis of other members of the integrin family.
Acknowledgments
Received 3/9/2005; revised 6/28/2005; accepted 7/8/2005.The costs of publication of this article were defrayed in part by the payment of page
charges. These articles must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
Cancer Research
Cancer Res 2005; 65: (18). September 15, 2005 8240 www.aacrjournals.org

References1. Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N.Carbohydrate-mediated cell adhesion in cancer metas-tasis and angiogenesis. Cancer Sci 2004;95:377–84.
2. Chatterjee S. Sphingolipids in atherosclerosis andvascular biology. Arterioscler Thromb Vasc Biol 1998;18:1523–33.
3. Hakomori SI. Inaugural article: the glycosynapse. ProcNatl Acad Sci U S A 2002;99:225–32.
4. Brown DA, London E. Structure and function ofsphingolipid- and cholesterol-rich membrane rafts.J Biol Chem 2000;275:17221–4.
5. Simons K, Toome D. Lipid rafts and signal transduc-tion. Nat Rev (Mol Cell Biol) 2000;1:31–9.
6. Marks DL, Pagano RE. Endocytosis and sorting ofglycosphingolipids in sphingolipid storage disease.Trends Cell Biol 2002;12:605–13.
7. Ffrench-Constant C, Colognato H. Integrins: versatileintegrators of extracellular signals. Trends Cell Biol 2004;14:678–86.
8. Hynes RO. Integrins: bidirectional, allosteric signalingmachines. Cell 2002;110:673–87.
9. Fang LH, Lucero M, Kazarian T, Wei Q, Luo FY,Valentino LA. Effects of neuroblastoma tumor ganglio-sides on platelet adhesion to collagen. Clin ExpMetastasis 1997;15:33–40.
10. Wen FQ, Jabbar AA, Patel DA, Kazarian T, ValentinoLA. Atherosclerotic aortic gangliosides enhance integrin-mediated platelet adhesion to collagen. ArteriosclerThromb Vasc Biol 1999;19:519–24.
11. Valentino LA, Ladisch S. Tumor gangliosides enhancea2h1 integrin-dependent platelet activation. BiochimBiophys Acta 1996;1316:19–28.
12. Burns GF, Lucas CM, Krissansen GW, et al. Synergismbetween membrane gangliosides and Arg-Gly-Asp-directed glycoprotein receptors in attachment to matrixproteins by melanoma cells. J Cell Biol 1988;107:1225–30.
13. Kazarian T, Jabbar AA, Wen FQ, Patel DA, ValentinoLA. Gangliosides regulate tumor cell adhesion tocollagen. Clin Exp Metastasis 2003;20:311–9.
14. Zheng M, Fang H, Tsuruoka T, Tsuji T, Sasaki T,Hakomori S. Regulatory role of GM3 ganglioside in a5h1
integrin receptor for fibronectin-mediated adhesion ofFUA169 cells. J Biol Chem 1993;268:2217–22.
15. Mitchell JS, Kanca O, McIntyre BW. Lipid micro-domain clustering induces a redistribution of antigenrecognition and adhesion molecules on human Tlymphocytes. J Immunol 2002;168:2737–44.
16. Krauss K, Altevogt P. Integrin leukocyte function-associated antigen-1-mediated cell binding can beactivated by clustering of membrane rafts. J Biol Chem1999;274:36921–7.
17. Hogg N, Laschinger M, Giles K, McDowall A. T-cellintegrins: more than just sticking points. J Cell Sci 2003;116:4695–705.
18. Upla P, Marjomaki V, Kankaanpaa P, et al. Clusteringinduces a lateral redistribution of a2h1 integrin frommembrane rafts to caveolae and subsequent proteinkinase C-dependent internalization. Mol Biol Cell 2004;15:625–36.
19. Marjomaki V, Pietiainen V, Matilainen H, et al.Internalization of echovirus 1 in caveolae. J Virol 2002;76:1856–65.
20. Minshall RD, Sessa WC, Stan RV, Anderson RG, MalikAB. Caveolin regulation of endothelial function. Am JPhysiol Lung Cell Mol Physiol 2003;285:L1179–83.
21. Cohen AW, Hnasko R, Schubert W, Lisanti MP. Roleof caveolae and caveolins in health and disease. PhysiolRev 2004;84:1341–79.
22. Mineo C, Anderson RG. Potocytosis. Robert FeulgenLecture. Histochem Cell Biol 2001;116:109–18.
23. Pelkmans L, Helenius A. Endocytosis via caveolae.Traffic 2002;3:311–20.
24. Puri V, Watanabe R, Singh RD, et al. Clathrin-dependent and -independent internalization of plasmamembrane sphingolipids initiates two Golgi targetingpathways. J Cell Biol 2001;154:535–47.
25. Sharma DK, Brown JC, Choudhury A, et al. Selectivestimulation of caveolar endocytosis by glycosphingoli-pids and cholesterol. Mol Biol Cell 2004;15:3114–22.
26. Sharma DK, Choudhury A, Singh RD, Wheatley CL,Marks DL, Pagano RE. Glycosphingolipids internalizedvia caveolar-related endocytosis rapidly merge withthe clathrin pathway in early endosomes and formmicrodomains for recycling. J Biol Chem 2003;278:7564–72.
27. Singh RD, Puri V, Valiyaveettil JT, Marks DL,Bittman R, Pagano RE. Selective caveolin-1-dependentendocytosis of glycosphingolipids. Mol Biol Cell 2003;14:3254–65.
28. Boyd ND, Chan BM, Petersen NO. h1 integrins aredistributed in adhesion structures with fibronectin andcaveolin and in coated pits. Biochem Cell Biol 2003;81:335–48.
29. Raghavan S, Vaezi A, Fuchs E. A role for ah1 integrinsin focal adhesion function and polarized cytoskeletaldynamics. Dev Cell 2003;5:415–27.
30. Martin OC, Pagano RE. Internalization and sorting ofa fluorescent analog of glucosylceramide to the Golgiapparatus of human skin fibroblasts: utilization ofendocytic and nonendocytic transport mechanisms.J Cell Biol 1994;125:769–81.
31. Luque A, Gomez M, Puzon W, Takada Y, Sanchez-Madrid F, Cabanas C. Activated conformations of verylate activation integrins detected by a group ofantibodies (HUTS) specific for a novel regulatory region(355-425) of the common h1 chain. J Biol Chem 1996;271:11067–75.
32. Chen CS, Martin OC, Pagano RE. Changes in thespectral properties of a plasma membrane lipid analogduring the first seconds of endocytosis in living cells.Biophys J 1997;72:37–50.
33. Pagano RE, Martin OC, Kang HC, Haugland RP. Anovel fluorescent ceramide analogue for studyingmembrane traffic in animal cells: accumulation at theGolgi apparatus results in altered spectral properties ofthe sphingolipid precursor. J Cell Biol 1991;113:1267–79.
34. Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly byan Eps15 mutant. J Cell Sci 1999;112:1303–11.
35. Cho KA, Ryu SJ, Oh YS, et al. Morphologicaladjustment of senescent cells by modulating caveolin-1 status. J Biol Chem 2004;279:42270–8.
36. Abrami L, Fivaz M, Glauser PE, Parton RG, van derGoot FG. A pore-forming toxin interacts with a GPI-anchored protein and causes vacuolation of theendoplasmic reticulum. J Cell Biol 1998;140:525–40.
37. Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS,Ginsberg MH, Shattil SJ. Src kinase activation bydirect interaction with the integrin b cytoplasmicdomain. Proc Natl Acad Sci U S A 2003;100:13298–302.
38. Damke H, Baba T, Warnock DE, Schmid SL.Induction of mutant dynamin specifically blocksendocytic coated vesicle formation. J Cell Biol 1994;127:915–34.
39. Henley JR, Krueger EW, Oswald BJ, McNiven MA.Dynamin-mediated internalization of caveolae. J CellBiol 1998;141:85–99.
40. Radel C, Rizzo V. Integrin mechanotransductionstimulates caveolin-1 phosphorylation and recruitmentof Csk to mediate actin reorganization. Am J PhysiolHeart Circ Physiol 2005;288:H936–45.
41. Volonte D, Galbiati F, Pestell RG, Lisanti MP. Cellularstress induces the tyrosine phosphorylation of caveolin-1 (Tyr(14)) via activation of p38 mitogen-activatedprotein kinase and c-Src kinase. Evidence for caveolae,the actin cytoskeleton, and focal adhesions as mechan-ical sensors of osmotic stress. J Biol Chem 2001;276:8094–103.
42. Lee H, Volonte D, Galbiati F, et al. Constitutive andgrowth factor-regulated phosphorylation of caveolin-1occurs at the same site (Tyr-14) in vivo : identification ofa c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol2000;14:1750–75.
43. Schwartz MA, Shattil SJ. Signaling networks linkingintegrins and Rho family GTPases. Trends Biochem Sci2000;25:388–91.
44. Arthur WT, Petch LA, Burridge K. Integrin engage-ment suppresses RhoA activity via a c-Src-dependentmechanism. Curr Biol 2000;10:719–22.
45. Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE,Gundersen GG. Localized stabilization of microtubulesby integrin- and FAK-facilitated Rho signaling. Science2004;303:836–9.
46. Leitinger B, Hogg N. The involvement of lipid rafts inthe regulation of integrin function. J Cell Sci 2002;115:963–72.
47. del Pozo MA, Alderson NB, Kiosses WB, Chiang HH,Anderson RG, Schwartz MA. Integrins regulate Ractargeting by internalization of membrane domains.Science 2004;303:839–42.
48. Brakebusch C, Bouvard D, Stanchi F, Sakai T, FasslerR. Integrins in invasive growth. J Clin Invest 2002;109:999–1006.
49. Decker L, Baron W, Ffrench-Constant C. Lipid rafts:microenvironments for integrin-growth factor interac-tions in neural development. Biochem Soc Trans 2004;32:426–30.
50. Sung CC, O’Toole EA, Lannutti BJ, et al. Integrin a5h1
expression is required for inhibition of keratinocytemigration by ganglioside GT1b. Exp Cell Res 1998;239:311–9.
51. Wang X, Sun P, Al-Qamari A, Tai T, Kawashima I,Paller AS. Carbohydrate-carbohydrate binding of gan-glioside to integrin a(5) modulates a(5)h(1) function.J Biol Chem 2001;276:8436–44.
52. Yanagisawa M, Nakamura K, Taga T. Roles of lipidrafts in integrin-dependent adhesion and gp130 signal-ling pathway in mouse embryonic neural precursorcells. Genes Cells 2004;9:801–9.
Glycosphingolipid-Dependent Integrin Clustering
www.aacrjournals.org 8241 Cancer Res 2005; 65: (18). September 15, 2005




















