
Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg 75 →Cys...
7
Hum Genet (1993) 92:499-505 human .. genet,cs Springer-Verlag 1993 Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75-->Cys mutation in the procollagen type II gene (COL2A1) Charlene J.Williams 1, Eileen L. Considine 1, Robert G. Knowlton 1, Antonio Reginato 2, Guillermo Neumann 3, David Harrison 1, Paul Buxton 1, Sergio Jimenez t, 4, Darwin J. Proekop 1, 4 1Department of Biochemistry and Molecular Biology, Thomas Jefferson University, 233 South Tenth Street, Philadelphia, PA 19107, USA 2Department of Medicine, Robert Wood Johnson Medical School, Camden, NJ 08103, USA 3National Institute of Health, Chiloe, Chile 4Department of Medicine, Thomas Jefferson University, 233 South Tenth Street, Philadelphia, PA 19107, USA Received: 25 February 1993 / Revised: 13 April 1993 Abstract. Direct sequencing of polymerase chain reac- tion (PCR)-amplified genomic DNA from a patient with spondyloepiphyseal dysplasia and precocious osteoarthri- tis revealed a single-base change in exon 11 of the type II procollagen gene (COL2A1), which produces an Arg---) Cys mutation in one allele. The proband is a member of a large Chilean kindred presenting with chondrodysplasia of the hips, knees, shoulders, elbows, and spine associated with severe, early-onset osteoarthritis. All affected indi- viduals exhibit mildly short stature; in addition, five out of seven affected family members display shortened metacarpals or metatarsals. DNA from affected and unaf- fected family members was PCR-amplified and analysis of restriction digests of the products determined that the mutation segregated with the disease with a lod score of 2.2 at zero recombination. The mutation, which resides in the triple-helical region of type II procollagen at amino acid position 75, is the second example of an Arg--+Cys mutation in the COL2A1 gene in heritable cartilaginous disease and is the first example of a point mutation in the amino terminal region of the otl(II) chain, that results in a spondyloepiphyseal dysplastic phenotype. Introduction The spondyloepiphyseal dysplasias (SEDs) constitute a heterogeneous group of disorders that are characterized by abnormal epiphyses and flattened vertebral bodies (Ri- moin and Lachman 1983). The epiphyseal abnormalities frequently result in precocious osteoarthritis of both weight-bearing and nonweight-bearing joints. Since type II collagen is a major component of affected tissues in SED, it has been hypothesized that structural defects in Correspondence to: C. J. Williams this matrix component may be responsible for some cases of SED. This hypothesis has been born out by the demon- stration of type II collagen abnormalities in cartilage tis- sue from SED patients and by reports of linkage to the type II procollagen gene (COL2A1) in some SED cases (Eyre et al. 1986; Godfrey and Hollister 1988; Horton et al. 1989; Murray et al. 1989; Anderson et al. 1990; Sher et al. 1991). Mutations in the COL2A1 gene have been con- firmed in four SED congenita cases (Lee et al. 1989; Tiller et al. 1990/1992; Chan and Cole 1991) and in sev- eral cases of achondrogenesis II/hypochondrogenesis (Vissing et al. 1989; Horton et al. 1992; Bogaert et al. 1992) During a research expedition to the Chiloe Islands, Chile, one of us (A.R.) discovered a sizable kindred with spondyloepiphyseal dysplasia, shortened metacarpals and metatarsals, precocious osteoarthritis and periarticular ap- atite like calcific deposits. Here we report the clinical and radiographic findings of this SED kindred. We also report molecular characterization of the disease, which was found to be associated with a mutation in the type II pro- collagen gene. A single-base substitution in one allele of the gene was detected, resulting in an Arg---~Cys mutation at amino acid position 75 of the triple helix. We present data to verify that the mutation segregates with the SED phenotype in this family. Materials and methods Clinical evaluation The proband is a 40-year-old woman who presented at birth with bilateral brachymetapody of her fourth and fifth metatarsals. Since age 12, she had experienced intermittent acute pain and swelling in her knees, ankles, and proximal interphalangeal joints. At age 21, she had persistent pain in her knees and hips and developed pro- gressive semiflexion contractures of her hips and fibrous ankylosis
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg 75 →Cys...

Hum Genet (1993) 92:499-505 human ..
genet,cs �9 Springer-Verlag 1993
Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75-->Cys mutation in the procollagen type II gene (COL2A1)
Charlene J.Will iams 1, Eileen L. Considine 1, Robert G. Knowlton 1, Antonio Reginato 2, Guil lermo Neumann 3, David Harrison 1, Paul Buxton 1, Sergio Jimenez t, 4, Darwin J. Proekop 1, 4
1 Department of Biochemistry and Molecular Biology, Thomas Jefferson University, 233 South Tenth Street, Philadelphia, PA 19107, USA 2 Department of Medicine, Robert Wood Johnson Medical School, Camden, NJ 08103, USA 3 National Institute of Health, Chiloe, Chile 4 Department of Medicine, Thomas Jefferson University, 233 South Tenth Street, Philadelphia, PA 19107, USA
Received: 25 February 1993 / Revised: 13 April 1993
Abstract. Direct sequencing of polymerase chain reac- tion (PCR)-amplified genomic DNA from a patient with spondyloepiphyseal dysplasia and precocious osteoarthri- tis revealed a single-base change in exon 11 of the type II procollagen gene (COL2A1), which produces an Arg---) Cys mutation in one allele. The proband is a member of a large Chilean kindred presenting with chondrodysplasia of the hips, knees, shoulders, elbows, and spine associated with severe, early-onset osteoarthritis. All affected indi- viduals exhibit mildly short stature; in addition, five out of seven affected family members display shortened metacarpals or metatarsals. DNA from affected and unaf- fected family members was PCR-amplified and analysis of restriction digests of the products determined that the mutation segregated with the disease with a lod score of 2.2 at zero recombination. The mutation, which resides in the triple-helical region of type II procollagen at amino acid position 75, is the second example of an Arg--+Cys mutation in the COL2A1 gene in heritable cartilaginous disease and is the first example of a point mutation in the amino terminal region of the otl(II) chain, that results in a spondyloepiphyseal dysplastic phenotype.
Introduction
The spondyloepiphyseal dysplasias (SEDs) constitute a heterogeneous group of disorders that are characterized by abnormal epiphyses and flattened vertebral bodies (Ri- moin and Lachman 1983). The epiphyseal abnormalities frequently result in precocious osteoarthritis of both weight-bearing and nonweight-bearing joints. Since type II collagen is a major component of affected tissues in SED, it has been hypothesized that structural defects in
Correspondence to: C. J. Williams
this matrix component may be responsible for some cases of SED. This hypothesis has been born out by the demon- stration of type II collagen abnormalities in cartilage tis- sue from SED patients and by reports of linkage to the type II procollagen gene (COL2A1) in some SED cases (Eyre et al. 1986; Godfrey and Hollister 1988; Horton et al. 1989; Murray et al. 1989; Anderson et al. 1990; Sher et al. 1991). Mutations in the COL2A1 gene have been con- firmed in four SED congenita cases (Lee et al. 1989; Tiller et al. 1990/1992; Chan and Cole 1991) and in sev- eral cases of achondrogenesis II/hypochondrogenesis (Vissing et al. 1989; Horton et al. 1992; Bogaert et al. 1992)
During a research expedition to the Chiloe Islands, Chile, one of us (A.R.) discovered a sizable kindred with spondyloepiphyseal dysplasia, shortened metacarpals and metatarsals, precocious osteoarthritis and periarticular ap- atite like calcific deposits. Here we report the clinical and radiographic findings of this SED kindred. We also report molecular characterization of the disease, which was found to be associated with a mutation in the type II pro- collagen gene. A single-base substitution in one allele of the gene was detected, resulting in an Arg---~Cys mutation at amino acid position 75 of the triple helix. We present data to verify that the mutation segregates with the SED phenotype in this family.
Materials and methods
Clinical evaluation
The proband is a 40-year-old woman who presented at birth with bilateral brachymetapody of her fourth and fifth metatarsals. Since age 12, she had experienced intermittent acute pain and swelling in her knees, ankles, and proximal interphalangeal joints. At age 21, she had persistent pain in her knees and hips and developed pro- gressive semiflexion contractures of her hips and fibrous ankylosis

500
of her knees. As a result of severe degenerative joint disease of her left hip, she underwent a total hip replacement at age 35, which was complicated by marked heterotopic periarticular calcification. She denied any low back pain. The proband's family history in- cludes multiple affected members: her mother, two of her four sib- lings, two of her children, and one niece are affected with identical precocious osteoarthritis. Her general physical examination was unremarkable; she ambulated with a walker due to severe degener- ative joint disease of the knees and left hip. Rotation of her tho- racic spine was limited and mobility of her lumbar spine was diffi- cult to assess due to the lack of hip motion and fixed extension of both knees. Radiological evaluation of her thoracic and lumbar spine clearly demonstrated flat vertebrae with anterior wedging, ir- regular vertebral endplates, and Schmorl's nodes. T12 was almost completely flat. Her right hip showed marked narrowing of the joint space with deformed femoral head and prominent soft tissue calcification. Both knees exhibited marked flattening of the inter- condylar notch and tibial plateau, joint space narrowing, and bone remodeling. Both shoulders demonstrated deformed humeral heads with bone remodeling and juxtaarticular calcific deposits. Hands and feet showed wide epiphyses, and brachymetapody of the fourth and fifth metatarsal bones, bilaterally, was evident. Sacroiliac joints were unremarkable. The findings were consistent with congenital brachymetapody, late-onset SED and precocious osteoarthritis. No ocular or oral manifestations were noted in the proband or other affected relatives. Detailed results of the family's history, physical examination, histological, biochemical, and sero- logical studies, as well as details of radiological examination of the proband and members of her family, have been described else- where (Reginato et al. 1990).
Linkage analysis
For restriction fragment length polymorphism (RFLP) analyses, digested genomic DNAs were electrophoretically resolved, blotted to nylon, and probed with the cosmid clone KFE1 (Sangiorgi et al. 1985; Knowlton et al. 1990). For analyses of the variable number tandem repeat (VNTR) located in the 3" untranslated region of COL2A 1, blots of restricted genomic DNAs were hybridized to a 4.5-kb EcoRI fragment of the clone HC2A (Nunez et al. 1985).
The probability of linkage between SED and the COL2A 1 poly- morphisms was estimated with the LIPED program (Ott 1974). For the calculations, the clinical syndrome was assumed to be domi- nantly inherited and fully penetrant in the individuals studied. The presence of dysplasia was verified by radiographic evidence. Fam- ily relationships were confirmed by analysis of several highly polymorphic DNA markers (Knowlton et al. 1986).
Genomic sequencing of COL2A1
The COL2A1 gene was directly sequenced from genomic DNA using primers for the polymerase chain reaction (PCR) amplifica- tion and sequencing of all 54 exons of COL2A1. The conditions for amplification and sequencing strategies with these primers have been described previously (Williams et al. 1992).
Genomic DNA was isolated by standard methods (Bell et al. 1981) and amplified by the PCR (Saiki et al. 1985) with an asym- metric ratio of primers to generate single-stranded templates for dideoxynucleotide sequencing (Sanger et al. 1977). Prior to se- quencing, PCR products were purified by precipitation with poly- ethylene glycol (Kusukawa et al. 1990), and the precipitate was re- suspended in deionized water.
Dideoxynucleotide sequencing was performed on the single- stranded templates using Sequenase version 2.0 (USB), the nu- cleotide analog 7-deaza-dGTP, and 35S-dATP as the labeled nu- cleotide (Sanger et al. 1977; Barnes et al. 1983). Sequencing reac- tions were analyzed on a 6% polyacrylamide/urea gel. The gel was then fixed, dried, and exposed to X-ray film overnight. Exon 11 was sequenced in three affected family members, three unaffected
members, and two unrelated individuals after the proband's se- quencing results demonstrated the mutation.
Restriction digest verification of the mutation
A subregion of exon 11 was PCR-amplified to produce a 286-bp fragment. The PCR reaction products were precipitated with poly- ethylene glycol and resuspended in distilled water. One-third of the purified PCR product was subjected to restriction digestion with BsiHKA1 as described by the manufacturer (New England Biolabs) to generate fragments of 158 bp, 96 bp, and 32 bp in nor- mal homozygotes and 190 bp, 158 bp, 96 bp, and 32 bp in het- erozygotes, containing one mutant allele. All available family members were tested.
R e s u l t s
The proband was first referred for examination, with a possible diagnosis of calcium pyrophosphate deposition disease, during a research trip by one of us (A.R.) to the Chiloe Islands. She has experienced jo in t pain since her second decade of life, and at age 38 she underwent a left total hip replacement, which was complicated by periar- ticular heterotopic calcification. By age 40, she was un- able to walk due to ankylosis of both hips as well as pro- gressive osteoarthritis of her knees. Figure 1 depicts the pedigree of the proband 's family. Three generations of the kindred were available for physical and radiological ex- aminations. Radiographs of hands, knees, and spine were performed for all studied members; radiographs of hips, shoulders, and elbows were also obtained for family members with precocious osteoarthritis. The results of the radiographic evaluations are reported in Table 1. At the t ime of the study, all affected members presented with bony enlargements of their wrists and metacarpopha- langeal joints. Five of the affected members exhibited brachymetapody with marked shortening of the meta- carpals or metatarsals, or both.
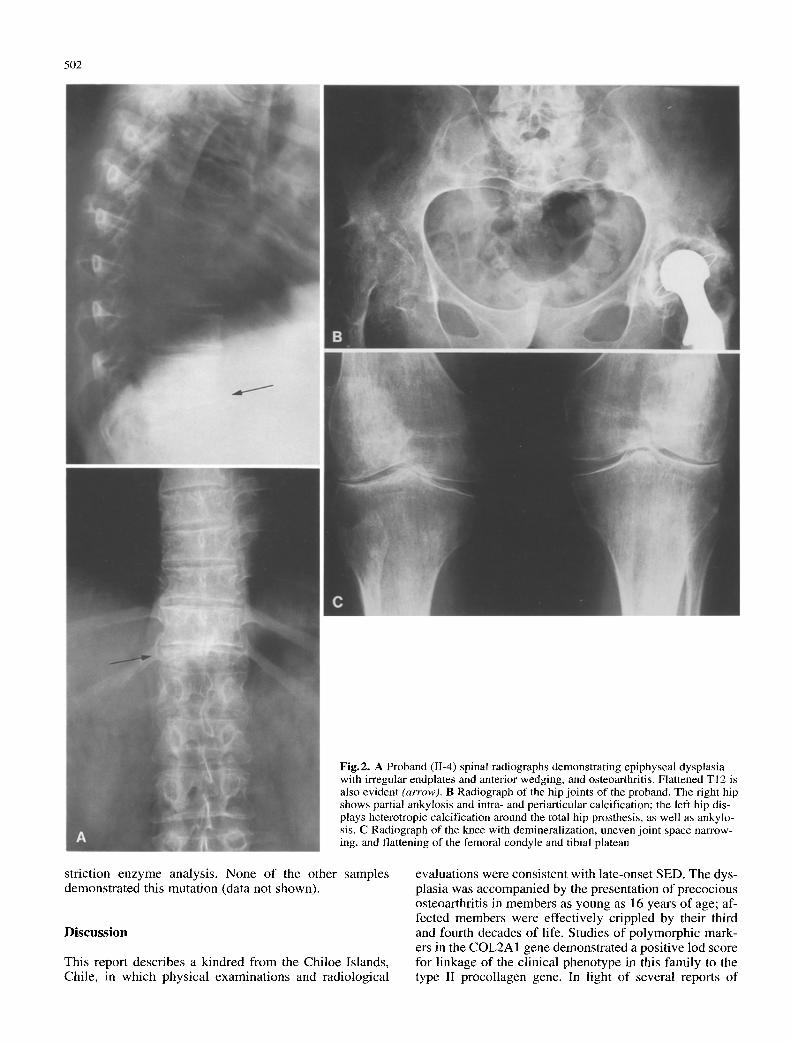
The radiographs in Fig. 2 A demonstrate the frontal and lateral view of the thoracic and lumbar spinal changes in the proband consistent with the diagnosis of SED. The vertebra plana of the proband 's spine at T12 is severe; se- vere platyspondylia, and even ankylosis, have been previ- ously reported in spondyloepiphyseal dysplasia (Deslan- dres and Menkes 1991; Barber 1960). Figure 2B shows radiographs of the proband 's knees and hips. Because of the presence of severe osteoarthritis, dysplastic changes in
I 2
3 + 6 1 II
1 2 3 4 5 6 7 8 9 ~O 11 12 13 14 15
Fig. 1. Family pedigree of SED/osteoarthritis kindred from Chiloe Islands, Chile. All affected family members (closed symbols) ex- hibited dysplastic changes of the spine and peripheral joints as well as precocious osteoarthritis. All affected individuals, except mem- bers IlI-2 and III-10, also displayed brachymetapody of the metatarsals or metacarpals, or both. Dashes in symbols indicate in- dividuals not studied; diagonal line, deceased; arrow, proband
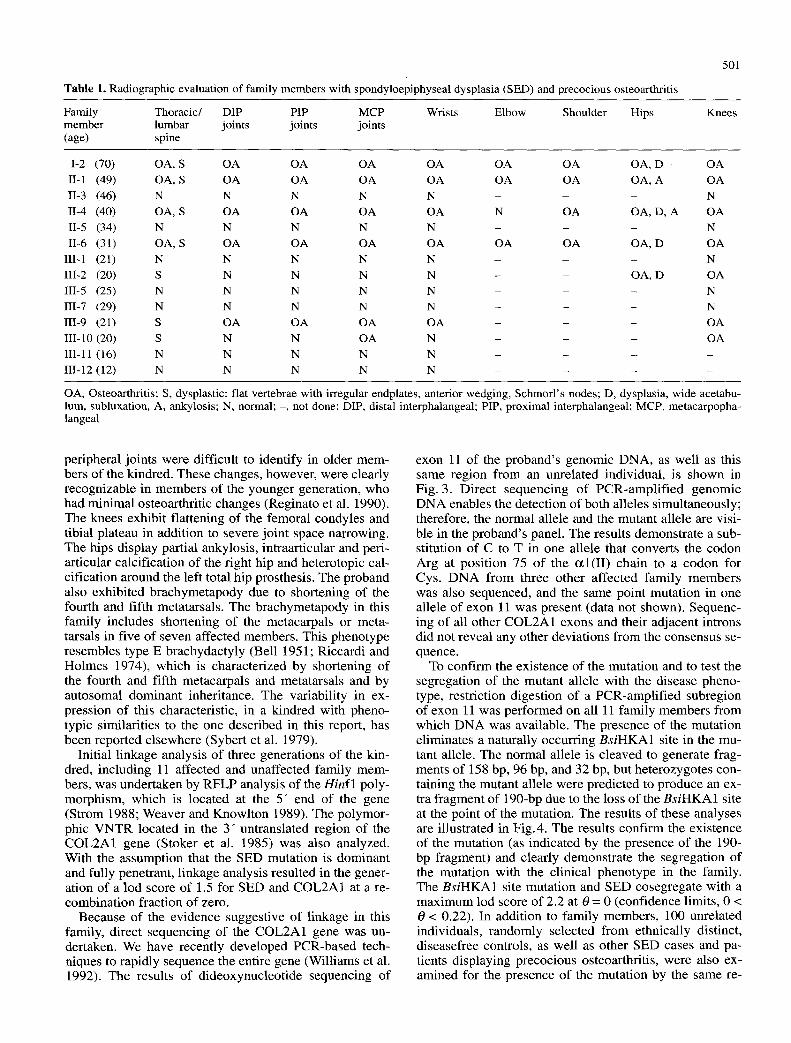
Table 1. Radiographic evaluation of family members with spondyloepiphyseal dysplasia (SED) and precocious osteoarthritis
501
Family Thoracic/ DIP PIP MCP member lumbar joints joints joints (age) spine
Wrists Elbow Shoulder Hips Knees
I-2 (70) OA, S OA OA OA OA OA OA OA, D OA II-1 (49) OA, S OA OA OA OA OA OA OA, A O A II-3 (46) N N N N N - - - N II-4 (40) OA, S OA OA OA OA N OA OA, D, A OA I1-5 (34) N N N N N - - - N II-6 (31) OA, S OA OA OA OA OA OA OA, D OA
III-1 (21) N N N N N - - - N III-2 (20) S N N N N - - OA, D OA III-5 (25) N N N N N - - - N III-7 (29) N N N N N - - - N III-9 (21) S OA OA OA OA - - - OA HI-t0 (20) S N N OA N - - - OA III-11 (16) N N N N N . . . . III-12 (12) N N N N N . . . .
OA, Osteoarthritis; S, dysplastic: flat vertebrae with irregular endplates, anterior wedging, Schmorl's nodes; D, dysplasia, wide acetabu- lure, subluxation, A, ankylosis; N, normal; -, not done; DIP, distal interphalangeal; PIP, proximal interphalangeal; MCP, metacarpopha- langeal
peripheral joints were difficult to identify in older mem- bers o f the kindred. These changes, however, were clearly recognizable in members of the younger generation, who had minimal osteoarthritic changes (Reginato et al. 1990). The knees exhibit flattening of the femoral condyles and tibial plateau in addition to severe joint space narrowing. The hips display partial ankylosis, intraarticular and peri- articular calcification o f the right hip and heterotopic cal- cification around the left total hip prosthesis. The proband also exhibited brachymetapody due to shortening of the fourth and fifth metatarsals. The brachymetapody in this family includes shortening of the metacarpals or meta- tarsals in five o f seven affected members. This phenotype resembles type E brachydactyly (Bell 1951; Riccardi and Holmes 1974), which is characterized by shortening of the fourth and fifth metacarpals and metatarsals and by autosomal dominant inheritance. The variability in ex- pression of this characteristic, in a kindred with pheno- typic similarities to the one described in this report, has been reported elsewhere (Sybert et al. 1979).
Initial l inkage analysis o f three generations o f the kin- dred, including 11 affected and unaffected family mem- bers, was undertaken by R F L P analysis o f the Hinf l poly- morphism, which is located at the 5" end of the gene (Strom 1988; Weaver and Knowlton 1989). The polymor- phic V N T R located in the 3" untranslated region of the C O L 2 A I gene (Stoker et al. 1985) was also analyzed. With the assumption that the SED mutation is dominant and fully penetrant, l inkage analysis resulted in the gener- ation of a aod score o f 1.5 for SED and COL2A1 at a re- combinat ion fraction o f zero.
Because o f the evidence suggestive of linkage in this family, direct sequencing of the COL2A1 gene was un- dertaken. We have recently developed PCR-based tech- niques to rapidly sequence the entire gene (Williams et al. 1992). The results of dideoxynucleot ide sequencing of
exon 11 of the proband 's genomic DNA, as well as this same region f rom an unrelated individual, is shown in Fig. 3. Direct sequencing of PCR-ampl i f i ed genomic D N A enables the detection of both alleles simultaneously; therefore, the normal allele and the mutant allele are visi- ble in the proband 's panel. The results demonstrate a sub- stitution of C to T in one allele that converts the codon Arg at posi t ion 75 o f the c~l(II) chain to a codon for Cys. D N A f rom three other affected family member s was also sequenced, and the same point mutat ion in one allele of exon 11 was present (data not shown). Sequenc- ing of all other COL2A1 exons and their adjacent introns did not reveal any other deviations f rom the consensus se- quence.
To confi rm the existence of the mutation and to test the segregation of the mutant allele with the disease pheno- type, restriction digestion o f a PCR-ampli f ied subregion of exon 11 was performed on all 11 family members f rom which D N A was available. The presence of the mutation eliminates a naturally occurring BsiHKA1 site in the mu- tant allele. The normal allele is cleaved to generate frag- ments of 158 bp, 96 bp, and 32 bp, but heterozygotes con- taining the mutant allele were predicted to produce an ex- tra f ragment of 190-bp due to the loss o f the BsiHKA1 site at the point o f the mutation. The results o f these analyses are illustrated in Fig. 4. The results conf i rm the existence of the mutation (as indicated by the presence o f the 190- bp fragment) and clearly demonstrate the segregation o f the mutation with the clinical phenotype in the family. The BsiHKA1 site mutation and SED cosegregate with a max imum lod score of 2.2 at 0 = 0 (confidence limits, 0 < 0 < 0.22). In addition to family members, 100 unrelated individuals, randomly selected f rom ethnically distinct, diseasefree controls, as well as other SED cases and pa- tients displaying precocious osteoarthritis, were also ex- amined for the presence o f the mutation by the same re-
502
Fig.2. A Proband (11-4) spinal radiographs demonstrating epiphyseal dysplasia with irregular endplates and anterior wedging, and osteoarthritis. Flattened TI2 is also evident (arrow). B Radiograph of the hip joints of the proband. The right hip shows partial ankylosis and intra- and periarticular calcification; the left hip dis- plays heterotropic calcification around the total hip prosthesis, as well as ankylo- sis. C Radiograph of the knee with demineralization, uneven joint space narrow- ing, and flattening of the femoral condyle and tibial plateau
striction enzyme analysis. None of the other samples demonstrated this mutation (data not shown).
Discuss ion
This report describes a kindred from the Chiloe Islands, Chile, in which physical examinations and radiological
evaluations were consistent with late-onset SED. The dys- plasia was accompanied by the presentation of precocious osteoarthritis in members as young as 16 years of age; af- fected members were effectively crippled by their third and fourth decades of life. Studies of polymorphic mark- ers in the COL2A1 gene demonstrated a positive lod score for linkage of the clinical phenotype in this family to the type II procollagen gene. In light of several reports of
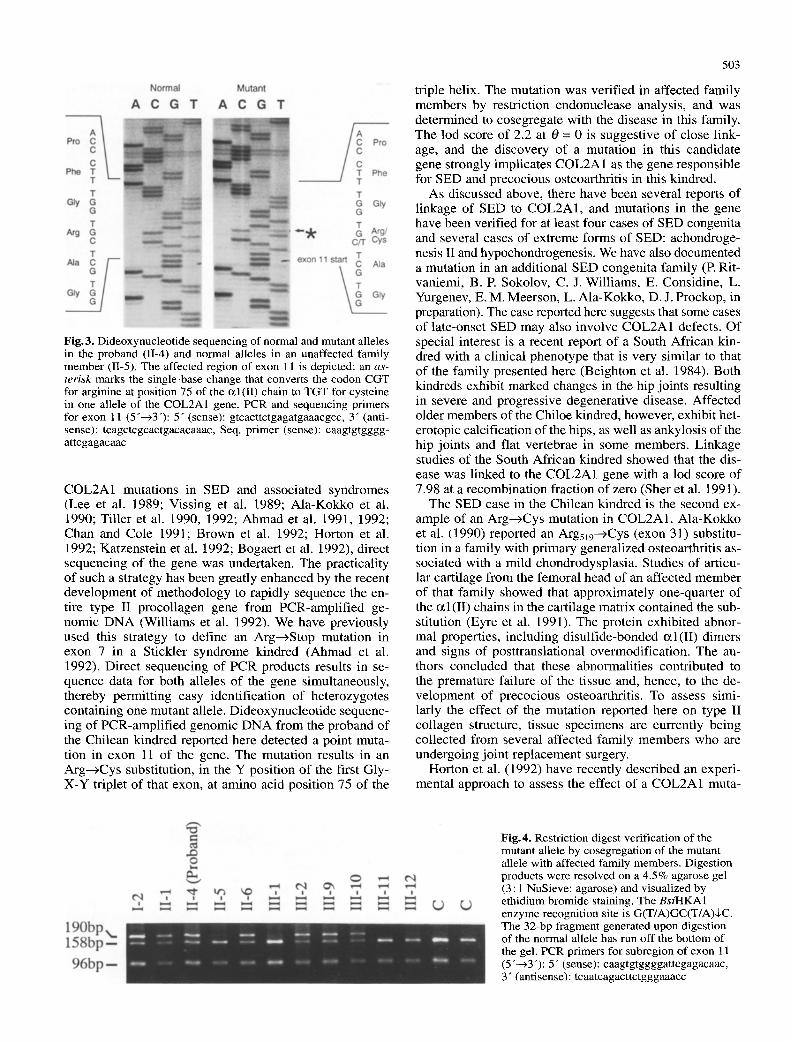
Fig.3. Dideoxynucleotide sequencing of normal and mutant alleles in the proband (1I-4) and normal alleles in an unaffected family member (11-5). The affected region of exon 11 is depicted; an as- terisk marks the single-base change that converts the codon CGT for arginine at position 75 of the c~l(II) chain to TGT for cysteine in one allele of the COL2A1 gene. PCR and sequencing primers for exon 11 (5"--~3"): 5" (sense): gtcacttctgagatgaaacgcc, 3" (anti- sense): tcagctcgcactgacacaaac, Seq. primer (sense): caagtgtgggg- attcgagacaac
COL2A1 mutations in SED and associated syndromes (Lee et al. 1989; Vissing et al. 1989; Ala-Kokko et al. 1990; Tiller et al. 1990, 1992; Ahmad et al. 1991, 1992; Chan and Cole 1991; Brown et al. 1992; Horton et al. 1992; Katzenstein et al. 1992; Bogaert et al. 1992), direct sequencing of the gene was undertaken. The practicality of such a strategy has been greatly enhanced by the recent development of methodology to rapidly sequence the en- tire type II procollagen gene from PCR-amplified ge- nomic DNA (Williams et al. 1992). We have previously used this strategy to define an Arg---)Stop mutation in exon 7 in a Stickler syndrome kindred (Ahmad et al. 1992). Direct sequencing of PCR products results in se- quence data for both alleles of the gene simultaneously, thereby permitting easy identification of heterozygotes containing one mutant allele. Dideoxynucleotide sequenc- ing of PCR-amplified genomic DNA from the proband of the Chilean kindred reported here detected a point muta- tion in exon 11 of the gene. The mutation results in an Arg---~Cys substitution, in the Y position of the first Gly- X-Y triplet of that exon, at amino acid position 75 of the
503
triple helix. The mutation was verified in affected family members by restriction endonuclease analysis, and was determined to cosegregate with the disease in this family. The lod score of 2.2 at 0 = 0 is suggestive of close link- age, and the discovery of a mutation in this candidate gene strongly implicates COL2A1 as the gene responsible for SED and precocious osteoarthritis in this kindred.
As discussed above, there have been several reports of linkage of SED to COL2A1, and mutations in the gene have been verified for at least four cases of SED congenita and several cases of extreme forms of SED: achondroge- nesis II and hypochondrogenesis. We have also documented a mutation in an additional SED congenita family (E Rit- vaniemi, B. P. Sokolov, C. J. Williams, E. Considine, L. Yurgenev, E. M. Meerson, L. Ala-Kokko, D. J. Prockop, in preparation). The case reported here suggests that some cases of late-onset SED may also involve COL2A1 defects. Of special interest is a recent report of a South African kin- dred with a clinical phenotype that is very similar to that of the family presented here (Beighton et al. 1984). Both kindreds exhibit marked changes in the hip joints resulting in severe and progressive degenerative disease. Affected older members of the Chiloe kindred, however, exhibit het- erotopic calcification of the hips, as well as ankylosis of the hip joints and flat vertebrae in some members. Linkage studies of the South African kindred showed that the dis- ease was linked to the COL2A1 gene with a lod score of 7.98 at a recombination fraction of zero (Sher et al. 1991).
The SED case in the Chilean kindred is the second ex- ample of an Arg---)Cys mutation in COL2A1. Ala-Kokko et al. (1990) reported an Arg519---~Cys (exon 31) substitu- tion in a family with primary generalized osteoarthritis as- sociated with a mild chondrodysplasia. Studies of articu- lar cartilage from the femoral head of an affected member of that family showed that approximately one-quarter of the ~ l ( I I ) chains in the cartilage matrix contained the sub- stitution (Eyre et al. 1991). The protein exhibited abnor- mal properties, including disulfide-bonded ~ l ( I I ) dimers and signs of posttranslational overmodification. The au- thors concluded that these abnormalities contributed to the premature failure of the tissue and, hence, to the de- velopment of precocious osteoarthritis. To assess simi- larly the effect of the mutation reported here on type II collagen structure, tissue specimens are currently being collected from several affected family members who are undergoing joint replacement surgery.
Horton et al. (1992) have recently described an experi- mental approach to assess the effect of a COL2A1 muta-
Fig. 4. Restriction digest verification of the mutant allele by cosegregation of the mutant allele with affected family members. Digestion products were resolved on a 4.5% agarose gel (3 : 1 NuSieve: agarose) and visualized by ethidium bromide staining. The B s i H K A I enzyme recognition site is G(T/A)GC(T/A)$C. The 32-bp fragment generated upon digestion of the normal allele has run off the bottom of the gel. PCR primers for subregion of exon 11 (5"---)3"): 5" (sense): caagtgtggggattcgagacaac, 3" (antisense): tcaatcagacttctgggaaacc

504
tion on chondrogenesis . Their studies utilized various cul- ture condit ions to expand the chondrocyte populat ion and to promote chondrogenesis in vitro. The chondrogenic condit ions permit ted an examinat ion of the morphology and biochemistry of the cartilagelike structures produced by chondrocytes isolated from an infant with hypochon- drogenesis. Ampl i f ica t ion of the cDNA from chondrocyte m R N A detected a Gly574-->Ser substi tution in COL2A1. The mutat ion apparently interfered with the intracellular transport and secretion of type lI procollagen and dis- rupted fibril assembly. Clearly, the abili ty to examine car- tilage tissue from patients with COL2A1 mutations, and to assess the effects of these mutat ions on chondrogenesis, provides powerful tools to delineate the consequence of COL2A1 mutat ions on collagen structure and cartilage matrix integrity. These approaches may be especially use- ful for determining posi t ion effects of similar mutat ions within the COL2A1 gene on proc~l (II) biosynthesis, post- translational processing, secretion, helix formation/stabil- ity, and interaction with other matrix constituents such as col lagen types 1X and XI. Ultimately, such studies may assist in establishing a relationship between the nature of COL2A1 mutat ions and the clinical expression of SED.
Acknowledgements. The authors express their gratitude to Drs. He- lena Kuivaniemi and Gerard Tromp for control DNA from unre- lated individuals. The authors are also grateful to Dr. Arnold S. Dion for restriction site searches and helpful discussions. This work was supported in part by NIH grant AR39740.
References
Ahmad NN, Ala-Kokko L, Knowlton RG, Jimenez SA, Weaver EJ, Maguire JI, Tasman W, Prockop DJ (1991) Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA 88 : 6624-6627
Ahmad NN, McDonald-McGinn DM, Zakai EH, Knowlton RG, LaRossa D, Dimascio J, Prockop DJ (1992) A second mutation in the type II procollagen gene (COL2A 1) causing Stickier syn- drome (arthro-ophthalmopathy) is also a premature termination codon. Am J Hum Genet 52:39-45
Ala-Kokko L, Baldwin CT, Moskowitz RW, Prockop DJ (1990) Single base mutation in the type II procollagen gene (COL2A1) as a cause of primary osteoarthritis associated with a mild chon- drodysplasia. Proc Natl Acad Sci USA 87:6565-6568
Anderson I J, Goldberg RB, Marion RW, Upholt WB, Tsipouras P (1990) Spondyloepiphyseal dysplasia congenita: genetic link- age to type II collagen (COL2AI). Am J Hum Genet 46: 896-9O 1
Barber HS (1960) An unusual form of familial osteodystrophy. Lancet II : 1220-1221
Barnes WM, Bevan M, Son PH (1983) Kilo-sequencing: creation of an ordered nest of asymmetric deletions across a large target sequence carried on phage M13. Methods Enzymol 101: 98-122
Beighton P, Christy G, Learmonth ID (1984) Namaqualand hip dysplasia: an autosomal dominant entity. Am J Med Genet 19: 161-169
Bell GLK, Karam JH, Rutter WJ (1981) Polymorphic DNA region adjacent to the 5" end of the human insulin gene. Proc Natl Acad Sci USA 78:5759-5763
Bell J (1951) On brachydactyly and symphalangism. In: Penrose EP (ed) Treasury of human inheritance. Cambridge University Press, Cambridge, pp 1-31
Bogaert R, Tiller GE, Weis MA, Gruber HE, Rimoin DL, Cohn DH, Eyre DR (1992) An amino acid substitution (Glya53---~Glu) in the collagen czl(II) chain produces hypochondrogenesis. J Biol Chem 267:22522-22526
Brown D, Nichols BE, Weingeist TA, Kimura AE, Scheffield VC, Stone EM (1992) Type II procollagen gene mutation in Stickler syndrome. Am J Hum Genet 51 [Suppl] :A301
Chan D, Cole WG (1991) Low basal transcription of genes for tis- sue-specific collagens by fibroblasts and lymphoblastoid cells: application to the characterization of a glycine 997 to serine substitution in c~l(II) collagen chains of a patient with spondy- loepiphyseal dysplasia. J Biol Chem 266:12487-12494
Deslandres C, Menkes CJ (1991) Spondyloepiphyseal dysplasia with ankylosing development: a case report. Rev Rhum Mal Os- teoartic 58:635-636
Eyre DR, Upton MP, Shapiro FD, Wilkinson RH, Vawter GW (1986) Nonexpression of cartilage type II collagen in a case of Langer-Saldino achondrogenesis. Am J Hum Genet 39:52- 67
Eyre DR, Weis MA, Moskowitz RW (1991) Cartilage expression of a type II collagen mutation in an inherited form of os- teoarthritis associated with a mild chondrodysplasia. J Clin In- vest 87:357-361
Godfrey M, Hollister DW (1988) Type II achondrogenesis/- hypochondrogenesis: identification of abnormal type II colla- gen. Am J Hum Genet 43:904-913
Horton WA, Campbell D, Machado MA, Chou J (1989) Type II collagen screening in the human chondrodysplasias. Am J Med Genet 34 : 579-583
Horton WA, Machado MA, Ellard J, Campbell D, Bartley J, Ramirez F, Vitale E, Lee B (1992) Characterization of a type II collagen gene (COL2AI) mutation identified in cultured chon- drocytes from human hypochondrogenesis. Proc Natl Acad Sci USA 89:4583-4587
Katzenstein PL, Campbell DF, Machado MA, Horton WA, Lee B, Ramirez F (1992) A type II collagen defect in a new family with SED tarda and early onset osteoarthritis (OA). 56th Ann Mtg Amer Coll Rheum, Atlanta, Ga, Abst No 41, $4l
Knowlton RG, Brown VA, Braman JC, Barker D, Schumm JW, Murray C, Takvorian T (1986) Use of highly polymorphic DNA probes for genotypic analysis following bone marrow transplan- tation. Blood 68 : 378-385
Knowlton RG, Katzenstein PL, Moskowitz RW, Weaver E J, Male- mud C J, Pathria MN, Jimenez SA, Prockop DJ (1990) Genetic linkage of a polymorphism in the type II procollagen gene (COL2AI) to primary osteoarthritis associated with a mild chondrodysplasia. N Engl J Med 322:526-530
Kusukawa N, Vemori T, Asada K, Kato I (1990) Rapid and reli- able protocol for direct sequencing of material amplified by the polymerase chain reaction. Biotechniques 9:66-72
Lee B, Vissing H, Ramirez F, Rogers D, Rimoin D (1989) Identi- fication of the molecular defect in a family with spondyloepi- physeal dysplasia. Science 244:978-980
Murray LW, Bautista J, James PL, Rimoin DL (1989) Type II col- lagen defects in the chondrodysplasias. I. Spondyloepiphyseal dysplasias. Am J Hum Genet 45:5-15
Nunez AM, Francomano C, Young MF, Martin GR, Yamada Y (1985) Isolation and partial characterization of genomic clones coding for a human proczl (II) collagen chain and demonstration of restriction fragment length polymorphism at the 3" end of the gene. Biochemistry 24 : 6343-6348
Ott J (1974) Estimation of the recombination fraction in human pedigrees: efficient computation of the likelihood for human linkage studies. Am J Hum Genet 26:588-597
Reginato A J, Knowlton RG, Diaz de Valdez M, Neumann A, Falasca GF, Jimenez SA, Prockop DJ (1990) Chondrodysplasia and precocious osteoarthritis in Chiloe Islanders. Arthritis Rheum 33 : $93
Riccardi VM, Holmes LB (1974) Brachydactyly type E: hereditary shortening of digits, metacarpals, metatarsals and long bones. J Pediatr 84: 251-254

505
Rimoin DL, Lachman RS (1983) The chondrodysplasias. In: Emery AEH, Rimoin DL (eds) Practice of medical genetics. Churchill Livingstone, New York, pp. 703-735
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymic application of [~-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350-1354
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74: 5463-5467
Sangiorgi FO, Benson-Chanda V, Wet WJ de, Sobel ME, Tsipouras P, Ramirez F (1985) Isolation and partial characteri- zation of the entire human procO(II) collagen gene. Nucleic Acids Res 13 : 2207-2225
Sher C, Ramesar R, Martell R. Learmonth I, Tsipouras P, Beighton P (1991) Mild spondyloepiphyseal dysplasia (Namaqualand type): genetic linkage to the type II collagen gene (COL2A1). Am J Hum Genet 48:518-524
Stoker NG, Cheah KSE, Griffin JR, Pope FM, Solomon E (1985) A highly polymorphic region 3" to the human type II collagen gene. Nucleic Acids Res 13:4613-4622
Strom CM (1988) A three allele restriction fragment length poly- morpbism within the human COL2A1 gene. Nucleic Acids Res 16 : 9077
Sybert VP, Byers PH, Hall JG (1979) Variable expression in a dominantly inherited skeletal dysplasia with similarities to
brachydactyly E and spondyloepiphyseal-spondyloperipheral dysplasia. Clin Genet 15:160-166
Tiller GE, Rimoin DL, Murray LW, Cohn DH (1990) Tandem du- plication within a type II collagen gene (COL2A1) exon in an individual with spondyloepiphyseal dysplasia. Proc Natl Acad Sci USA 87 : 3889-3893
Tiller GE, Weis MA, Eyre DR, Rimoin DL, Cohn DH (1992) RNA splicing mutation (G+5IVS20) in the gene for type II col- lagen (COL2AI) produces spondyloepiphyseal dysplasia con- genita. Am J Hum Genet 51 [Suppl] : A37
Vissing H, D'Alessio M, Lee B, Ramirez F, Godfrey M, Hollister DW (1989) Glycine to serine substitution in the triple helical domain of prowl (II) collagen results in a lethal perinatal form of short-limbed dwarfism. J Biol Chem 264:18 265-18 267
Weaver E J, Knowlton RG (1989) Characterization of a HinfI poly- morphism in the type II procollagen gene (COL2A1). Cyto- genet Cell Genet 54:1103
Williams CJ, Harrison DA, Hopkinson I, Baldwin CT, Ahmad NN, Ala-Kokko L, Korn RM, Buxton PG, Dimascio J, Consi- dine EL, Prockop DJ (1992) Detection of sequence variants in the gene for human type II procollagen (COL2AI) by direct se- quencing of polymerase chain reaction-amplified genomic DNA. Hum Mutat l :403-416




















