
Spectroscopic studies and quantum chemical investigations of (3,4-dimethoxybenzylidene)...
9
Spectroscopic studies and quantum chemical investigations of (3,4-dimethoxybenzylidene) propanedinitrile Ujval Gupta a , Vinay Kumar a , Vivek K. Singh a , Rajni Kant b , Yugal Khajuria a,⇑ a School of Physics, Shri Mata Vaishno Devi University, Kakryal, Katra 182320, Jammu & Kashmir, India b X-ray Crystallography Laboratory, Post-Graduate Department of Physics, University of Jammu, Jammu Tawi 180 006, India highlights FTIR, UV–Vis spectroscopy and TG analysis have been carried out. The molecular geometry and vibrational wave numbers have been studied. Mulliken charges, natural atomic charges and thermodynamic properties have been calculated. HOMO–LUMO analysis was performed. The natural bond orbital calculation has been carried out. graphical abstract article info Article history: Received 6 October 2014 Received in revised form 13 December 2014 Accepted 17 December 2014 Available online 27 December 2014 Keywords: FTIR UV–Vis TGA Density Functional Theory (DFT) VEDA NBO abstract The Fourier Transform Infrared (FTIR), Ultra-Violet Visible (UV–Vis) spectroscopy and Thermogravimetric (TG) analysis of (3,4-dimethoxybenzylidene) propanedinitrile have been carried out and investigated using quantum chemical calculations. The molecular geometry, harmonic vibrational frequencies, Mullik- en charges, natural atomic charges and thermodynamic properties in the ground state have been inves- tigated by using Hartree Fock Theory (HF) and Density Functional Theory (DFT) using B3LYP functional with 6-311G(d,p) basis set. Both HF and DFT methods yield good agreement with the experimental data. Vibrational modes are assigned with the help of Vibrational Energy Distribution Analysis (VEDA) pro- gram. UV–Visible spectrum was recorded in the spectral range of 190–800 nm and the results are com- pared with the calculated values using TD-DFT approach. Stability of the molecule arising from hyperconjugative interactions, charge delocalization have been analyzed using natural bond orbital (NBO) analysis. The results obtained from the studies of Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) are used to calculate molecular parameters like ioni- zation potential, electron affinity, global hardness, electron chemical potential and global electrophilicity. Ó 2014 Elsevier B.V. All rights reserved. Introduction Organic compounds containing functional group AC„N (nitriles) are commonly encountered in fruit pits, especially almonds, and during cooking of Brassica crops such as cabbage, brussel sprouts, and cauliflower. Nitrile derivatives are found in many useful compounds including methyl cyanoacrylate and are used in super glue, nitrile butadiene rubber which is a nitrile-con- taining polymer used in latex-free laboratory, and in medical gloves. Over 30 nitrile-containing pharmaceuticals are currently marketed for a diverse variety of medicinal indications with more than 20 additional nitrile-containing leads in clinical development. Propanedinitirile, a class of nitrile derivatives, and also known as Malononitrile, is reportedly used as intermediate for the synthesis http://dx.doi.org/10.1016/j.saa.2014.12.063 1386-1425/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author. E-mail address: [email protected] (Y. Khajuria). Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73 Contents lists available at ScienceDirect Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy journal homepage: www.elsevier.com/locate/saa
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Spectroscopic studies and quantum chemical investigations of (3,4-dimethoxybenzylidene)...

Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73
Contents lists available at ScienceDirect
Spectrochimica Acta Part A: Molecular andBiomolecular Spectroscopy
journal homepage: www.elsevier .com/locate /saa
Spectroscopic studies and quantum chemical investigationsof (3,4-dimethoxybenzylidene) propanedinitrile
http://dx.doi.org/10.1016/j.saa.2014.12.0631386-1425/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author.E-mail address: [email protected] (Y. Khajuria).
Ujval Gupta a, Vinay Kumar a, Vivek K. Singh a, Rajni Kant b, Yugal Khajuria a,⇑a School of Physics, Shri Mata Vaishno Devi University, Kakryal, Katra 182320, Jammu & Kashmir, Indiab X-ray Crystallography Laboratory, Post-Graduate Department of Physics, University of Jammu, Jammu Tawi 180 006, India
h i g h l i g h t s
� FTIR, UV–Vis spectroscopy and TGanalysis have been carried out.� The molecular geometry and
vibrational wave numbers have beenstudied.� Mulliken charges, natural atomic
charges and thermodynamicproperties have been calculated.� HOMO–LUMO analysis was
performed.� The natural bond orbital calculation
has been carried out.
g r a p h i c a l a b s t r a c t
a r t i c l e i n f o
Article history:Received 6 October 2014Received in revised form 13 December 2014Accepted 17 December 2014Available online 27 December 2014
Keywords:FTIRUV–VisTGADensity Functional Theory (DFT)VEDANBO
a b s t r a c t
The Fourier Transform Infrared (FTIR), Ultra-Violet Visible (UV–Vis) spectroscopy and Thermogravimetric(TG) analysis of (3,4-dimethoxybenzylidene) propanedinitrile have been carried out and investigatedusing quantum chemical calculations. The molecular geometry, harmonic vibrational frequencies, Mullik-en charges, natural atomic charges and thermodynamic properties in the ground state have been inves-tigated by using Hartree Fock Theory (HF) and Density Functional Theory (DFT) using B3LYP functionalwith 6-311G(d,p) basis set. Both HF and DFT methods yield good agreement with the experimental data.Vibrational modes are assigned with the help of Vibrational Energy Distribution Analysis (VEDA) pro-gram. UV–Visible spectrum was recorded in the spectral range of 190–800 nm and the results are com-pared with the calculated values using TD-DFT approach. Stability of the molecule arising fromhyperconjugative interactions, charge delocalization have been analyzed using natural bond orbital(NBO) analysis. The results obtained from the studies of Highest Occupied Molecular Orbital (HOMO)and Lowest Unoccupied Molecular Orbital (LUMO) are used to calculate molecular parameters like ioni-zation potential, electron affinity, global hardness, electron chemical potential and global electrophilicity.
� 2014 Elsevier B.V. All rights reserved.
Introduction
Organic compounds containing functional group AC„N(nitriles) are commonly encountered in fruit pits, especiallyalmonds, and during cooking of Brassica crops such as cabbage,
brussel sprouts, and cauliflower. Nitrile derivatives are found inmany useful compounds including methyl cyanoacrylate and areused in super glue, nitrile butadiene rubber which is a nitrile-con-taining polymer used in latex-free laboratory, and in medicalgloves. Over 30 nitrile-containing pharmaceuticals are currentlymarketed for a diverse variety of medicinal indications with morethan 20 additional nitrile-containing leads in clinical development.Propanedinitirile, a class of nitrile derivatives, and also known asMalononitrile, is reportedly used as intermediate for the synthesis
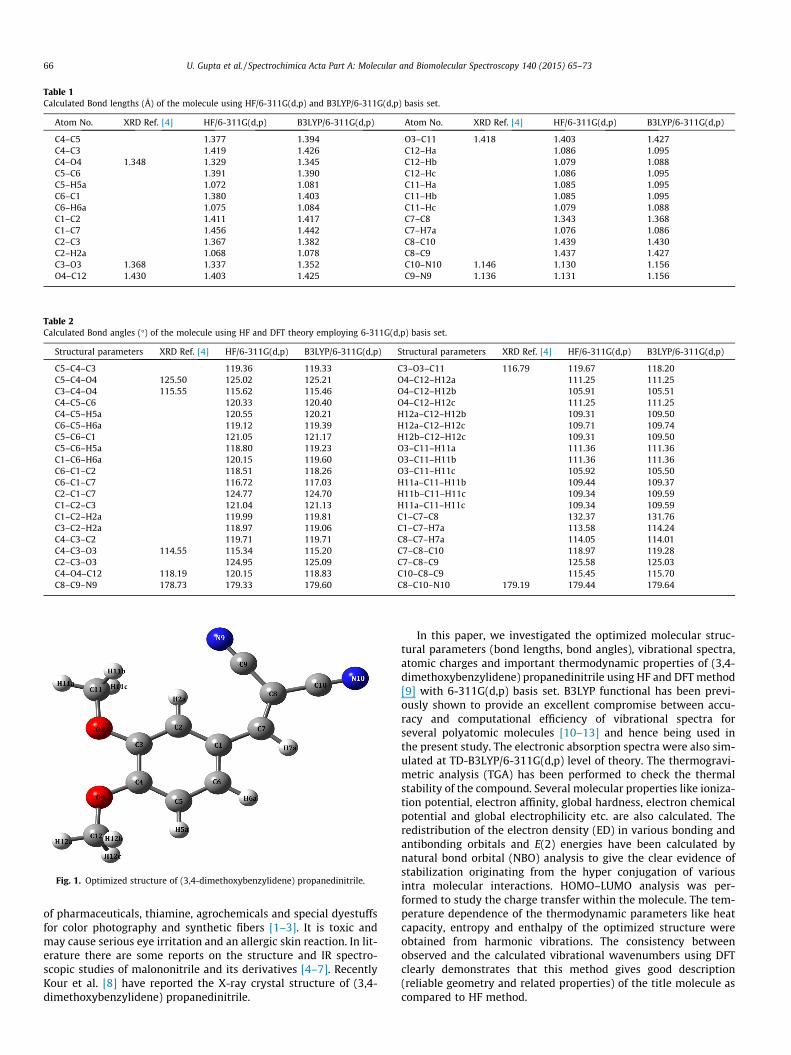
Table 1Calculated Bond lengths (Å) of the molecule using HF/6-311G(d,p) and B3LYP/6-311G(d,p) basis set.
Atom No. XRD Ref. [4] HF/6-311G(d,p) B3LYP/6-311G(d,p) Atom No. XRD Ref. [4] HF/6-311G(d,p) B3LYP/6-311G(d,p)
C4–C5 1.377 1.394 O3–C11 1.418 1.403 1.427C4–C3 1.419 1.426 C12–Ha 1.086 1.095C4–O4 1.348 1.329 1.345 C12–Hb 1.079 1.088C5–C6 1.391 1.390 C12–Hc 1.086 1.095C5–H5a 1.072 1.081 C11–Ha 1.085 1.095C6–C1 1.380 1.403 C11–Hb 1.085 1.095C6–H6a 1.075 1.084 C11–Hc 1.079 1.088C1–C2 1.411 1.417 C7–C8 1.343 1.368C1–C7 1.456 1.442 C7–H7a 1.076 1.086C2–C3 1.367 1.382 C8–C10 1.439 1.430C2–H2a 1.068 1.078 C8–C9 1.437 1.427C3–O3 1.368 1.337 1.352 C10–N10 1.146 1.130 1.156O4–C12 1.430 1.403 1.425 C9–N9 1.136 1.131 1.156
Table 2Calculated Bond angles (�) of the molecule using HF and DFT theory employing 6-311G(d,p) basis set.
Structural parameters XRD Ref. [4] HF/6-311G(d,p) B3LYP/6-311G(d,p) Structural parameters XRD Ref. [4] HF/6-311G(d,p) B3LYP/6-311G(d,p)
C5–C4–C3 119.36 119.33 C3–O3–C11 116.79 119.67 118.20C5–C4–O4 125.50 125.02 125.21 O4–C12–H12a 111.25 111.25C3–C4–O4 115.55 115.62 115.46 O4–C12–H12b 105.91 105.51C4–C5–C6 120.33 120.40 O4–C12–H12c 111.25 111.25C4–C5–H5a 120.55 120.21 H12a–C12–H12b 109.31 109.50C6–C5–H6a 119.12 119.39 H12a–C12–H12c 109.71 109.74C5–C6–C1 121.05 121.17 H12b–C12–H12c 109.31 109.50C5–C6–H5a 118.80 119.23 O3–C11–H11a 111.36 111.36C1–C6–H6a 120.15 119.60 O3–C11–H11b 111.36 111.36C6–C1–C2 118.51 118.26 O3–C11–H11c 105.92 105.50C6–C1–C7 116.72 117.03 H11a–C11–H11b 109.44 109.37C2–C1–C7 124.77 124.70 H11b–C11–H11c 109.34 109.59C1–C2–C3 121.04 121.13 H11a–C11–H11c 109.34 109.59C1–C2–H2a 119.99 119.81 C1–C7–C8 132.37 131.76C3–C2–H2a 118.97 119.06 C1–C7–H7a 113.58 114.24C4–C3–C2 119.71 119.71 C8–C7–H7a 114.05 114.01C4–C3–O3 114.55 115.34 115.20 C7–C8–C10 118.97 119.28C2–C3–O3 124.95 125.09 C7–C8–C9 125.58 125.03C4–O4–C12 118.19 120.15 118.83 C10–C8–C9 115.45 115.70C8–C9–N9 178.73 179.33 179.60 C8–C10–N10 179.19 179.44 179.64
Fig. 1. Optimized structure of (3,4-dimethoxybenzylidene) propanedinitrile.
66 U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73
of pharmaceuticals, thiamine, agrochemicals and special dyestuffsfor color photography and synthetic fibers [1–3]. It is toxic andmay cause serious eye irritation and an allergic skin reaction. In lit-erature there are some reports on the structure and IR spectro-scopic studies of malononitrile and its derivatives [4–7]. RecentlyKour et al. [8] have reported the X-ray crystal structure of (3,4-dimethoxybenzylidene) propanedinitrile.
In this paper, we investigated the optimized molecular struc-tural parameters (bond lengths, bond angles), vibrational spectra,atomic charges and important thermodynamic properties of (3,4-dimethoxybenzylidene) propanedinitrile using HF and DFT method[9] with 6-311G(d,p) basis set. B3LYP functional has been previ-ously shown to provide an excellent compromise between accu-racy and computational efficiency of vibrational spectra forseveral polyatomic molecules [10–13] and hence being used inthe present study. The electronic absorption spectra were also sim-ulated at TD-B3LYP/6-311G(d,p) level of theory. The thermogravi-metric analysis (TGA) has been performed to check the thermalstability of the compound. Several molecular properties like ioniza-tion potential, electron affinity, global hardness, electron chemicalpotential and global electrophilicity etc. are also calculated. Theredistribution of the electron density (ED) in various bonding andantibonding orbitals and E(2) energies have been calculated bynatural bond orbital (NBO) analysis to give the clear evidence ofstabilization originating from the hyper conjugation of variousintra molecular interactions. HOMO–LUMO analysis was per-formed to study the charge transfer within the molecule. The tem-perature dependence of the thermodynamic parameters like heatcapacity, entropy and enthalpy of the optimized structure wereobtained from harmonic vibrations. The consistency betweenobserved and the calculated vibrational wavenumbers using DFTclearly demonstrates that this method gives good description(reliable geometry and related properties) of the title molecule ascompared to HF method.
-0.4
-0.2
0.0
0.2
MullikenC12
H11bH11c
H12cH11a
H12b
H12aH5a H6a
H2a
O3 O4
N10N9
C11
C10
C3
C2
C7
C6
C8
C9
C4 HF DFT
Cha
rge/
eC1 C5
H7a
-0.6
-0.4
-0.2
0.0
0.2
0.4
NBOC1C2 C5
C6
C3C4
C7
C8
C9 C10
C11C12
N9 N10
O3 O4
H2a
H5aH6a H7a
H11a
H11bH11c
H12a
H12b
H12c
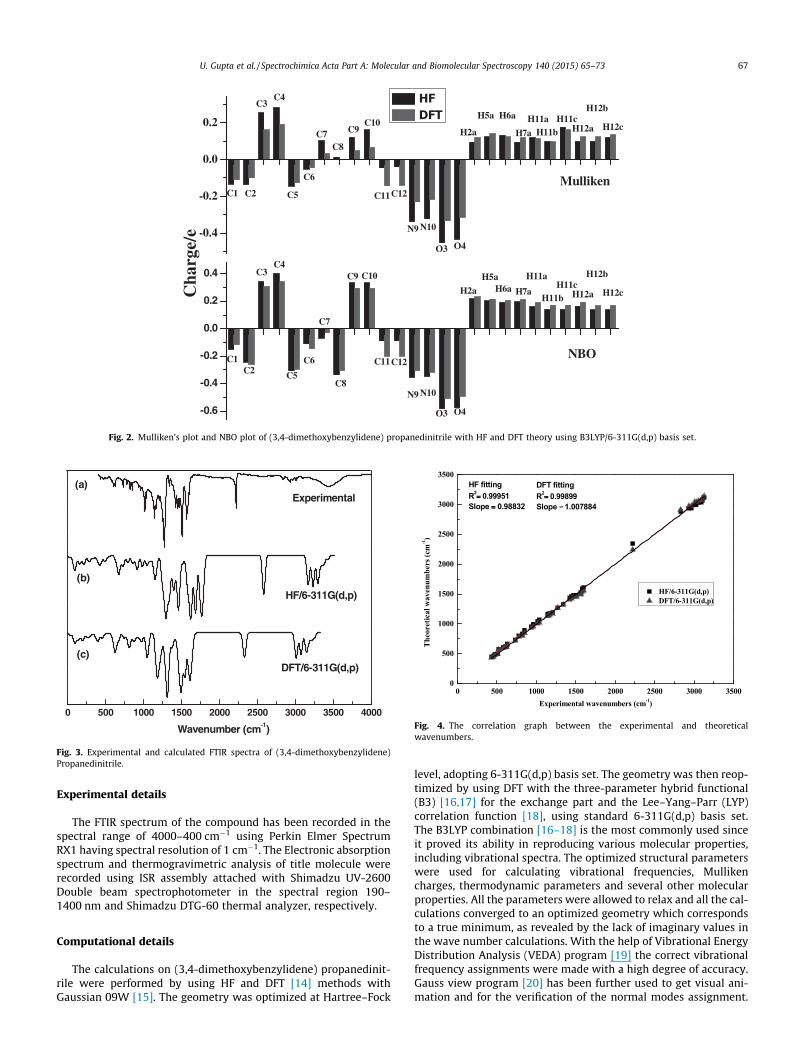
Fig. 2. Mulliken’s plot and NBO plot of (3,4-dimethoxybenzylidene) propanedinitrile with HF and DFT theory using B3LYP/6-311G(d,p) basis set.
0 500 1000 1500 2000 2500 3000 3500 4000
(b)
Experimental(a)
(c)
HF/6-311G(d,p)
DFT/6-311G(d,p)
Wavenumber (cm-1)
Fig. 3. Experimental and calculated FTIR spectra of (3,4-dimethoxybenzylidene)Propanedinitrile.
0 500 1000 1500 2000 2500 3000 35000
500
1000
1500
2000
2500
3000
3500DFT fittingR2= = == 0.99899Slope 1.007884
The
oret
ical
wav
enum
bers
(cm
-1)
HF/6-311G(d,p)DFT/6-311G(d,p)
Experimental wavenumbers (cm-1)
HF fittingR2= = == 0.99951Slope ==== 0.98832 =
Fig. 4. The correlation graph between the experimental and theoreticalwavenumbers.
U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73 67
Experimental details
The FTIR spectrum of the compound has been recorded in thespectral range of 4000–400 cm�1 using Perkin Elmer SpectrumRX1 having spectral resolution of 1 cm�1. The Electronic absorptionspectrum and thermogravimetric analysis of title molecule wererecorded using ISR assembly attached with Shimadzu UV-2600Double beam spectrophotometer in the spectral region 190–1400 nm and Shimadzu DTG-60 thermal analyzer, respectively.
Computational details
The calculations on (3,4-dimethoxybenzylidene) propanedinit-rile were performed by using HF and DFT [14] methods withGaussian 09W [15]. The geometry was optimized at Hartree–Fock
level, adopting 6-311G(d,p) basis set. The geometry was then reop-timized by using DFT with the three-parameter hybrid functional(B3) [16,17] for the exchange part and the Lee–Yang–Parr (LYP)correlation function [18], using standard 6-311G(d,p) basis set.The B3LYP combination [16–18] is the most commonly used sinceit proved its ability in reproducing various molecular properties,including vibrational spectra. The optimized structural parameterswere used for calculating vibrational frequencies, Mullikencharges, thermodynamic parameters and several other molecularproperties. All the parameters were allowed to relax and all the cal-culations converged to an optimized geometry which correspondsto a true minimum, as revealed by the lack of imaginary values inthe wave number calculations. With the help of Vibrational EnergyDistribution Analysis (VEDA) program [19] the correct vibrationalfrequency assignments were made with a high degree of accuracy.Gauss view program [20] has been further used to get visual ani-mation and for the verification of the normal modes assignment.
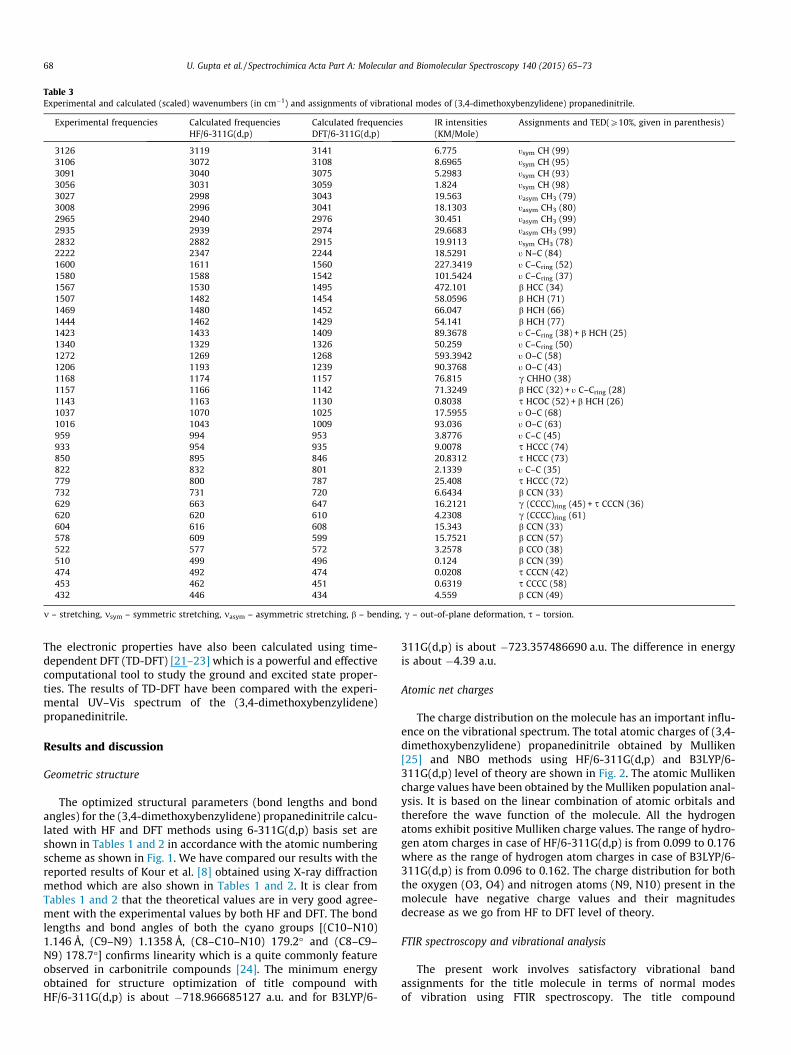
Table 3Experimental and calculated (scaled) wavenumbers (in cm�1) and assignments of vibrational modes of (3,4-dimethoxybenzylidene) propanedinitrile.
Experimental frequencies Calculated frequenciesHF/6-311G(d,p)
Calculated frequenciesDFT/6-311G(d,p)
IR intensities(KM/Mole)
Assignments and TED(P10%, given in parenthesis)
3126 3119 3141 6.775 ˆsym CH (99)3106 3072 3108 8.6965 ˆsym CH (95)3091 3040 3075 5.2983 ˆsym CH (93)3056 3031 3059 1.824 ˆsym CH (98)3027 2998 3043 19.563 ˆasym CH3 (79)3008 2996 3041 18.1303 ˆasym CH3 (80)2965 2940 2976 30.451 ˆasym CH3 (99)2935 2939 2974 29.6683 ˆasym CH3 (99)2832 2882 2915 19.9113 ˆsym CH3 (78)2222 2347 2244 18.5291 ˆ N–C (84)1600 1611 1560 227.3419 ˆ C–Cring (52)1580 1588 1542 101.5424 ˆ C–Cring (37)1567 1530 1495 472.101 b HCC (34)1507 1482 1454 58.0596 b HCH (71)1469 1480 1452 66.047 b HCH (66)1444 1462 1429 54.141 b HCH (77)1423 1433 1409 89.3678 ˆ C–Cring (38) + b HCH (25)1340 1329 1326 50.259 ˆ C–Cring (50)1272 1269 1268 593.3942 ˆ O–C (58)1206 1193 1239 90.3768 ˆ O–C (43)1168 1174 1157 76.815 c CHHO (38)1157 1166 1142 71.3249 b HCC (32) + ˆ C–Cring (28)1143 1163 1130 0.8038 s HCOC (52) + b HCH (26)1037 1070 1025 17.5955 ˆ O–C (68)1016 1043 1009 93.036 ˆ O–C (63)959 994 953 3.8776 ˆ C–C (45)933 954 935 9.0078 s HCCC (74)850 895 846 20.8312 s HCCC (73)822 832 801 2.1339 ˆ C–C (35)779 800 787 25.408 s HCCC (72)732 731 720 6.6434 b CCN (33)629 663 647 16.2121 c (CCCC)ring (45) + s CCCN (36)620 620 610 4.2308 c (CCCC)ring (61)604 616 608 15.343 b CCN (33)578 609 599 15.7521 b CCN (57)522 577 572 3.2578 b CCO (38)510 499 496 0.124 b CCN (39)474 492 474 0.0208 s CCCN (42)453 462 451 0.6319 s CCCC (58)432 446 434 4.559 b CCN (49)
m – stretching, msym – symmetric stretching, masym – asymmetric stretching, b – bending, c – out-of-plane deformation, s – torsion.
68 U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73
The electronic properties have also been calculated using time-dependent DFT (TD-DFT) [21–23] which is a powerful and effectivecomputational tool to study the ground and excited state proper-ties. The results of TD-DFT have been compared with the experi-mental UV–Vis spectrum of the (3,4-dimethoxybenzylidene)propanedinitrile.
Results and discussion
Geometric structure
The optimized structural parameters (bond lengths and bondangles) for the (3,4-dimethoxybenzylidene) propanedinitrile calcu-lated with HF and DFT methods using 6-311G(d,p) basis set areshown in Tables 1 and 2 in accordance with the atomic numberingscheme as shown in Fig. 1. We have compared our results with thereported results of Kour et al. [8] obtained using X-ray diffractionmethod which are also shown in Tables 1 and 2. It is clear fromTables 1 and 2 that the theoretical values are in very good agree-ment with the experimental values by both HF and DFT. The bondlengths and bond angles of both the cyano groups [(C10–N10)1.146 Å, (C9–N9) 1.1358 Å, (C8–C10–N10) 179.2� and (C8–C9–N9) 178.7�] confirms linearity which is a quite commonly featureobserved in carbonitrile compounds [24]. The minimum energyobtained for structure optimization of title compound withHF/6-311G(d,p) is about �718.966685127 a.u. and for B3LYP/6-
311G(d,p) is about �723.357486690 a.u. The difference in energyis about �4.39 a.u.
Atomic net charges
The charge distribution on the molecule has an important influ-ence on the vibrational spectrum. The total atomic charges of (3,4-dimethoxybenzylidene) propanedinitrile obtained by Mulliken[25] and NBO methods using HF/6-311G(d,p) and B3LYP/6-311G(d,p) level of theory are shown in Fig. 2. The atomic Mullikencharge values have been obtained by the Mulliken population anal-ysis. It is based on the linear combination of atomic orbitals andtherefore the wave function of the molecule. All the hydrogenatoms exhibit positive Mulliken charge values. The range of hydro-gen atom charges in case of HF/6-311G(d,p) is from 0.099 to 0.176where as the range of hydrogen atom charges in case of B3LYP/6-311G(d,p) is from 0.096 to 0.162. The charge distribution for boththe oxygen (O3, O4) and nitrogen atoms (N9, N10) present in themolecule have negative charge values and their magnitudesdecrease as we go from HF to DFT level of theory.
FTIR spectroscopy and vibrational analysis
The present work involves satisfactory vibrational bandassignments for the title molecule in terms of normal modesof vibration using FTIR spectroscopy. The title compound
Table 4Second-order perturbation theory analysis of Fock matrix in NBO basic corresponding to the intermolecular bonds of (3,4-dimethoxybenzylidene) propanedinitrile.
Donor (i) ED (i) (e) Acceptor (j) ED (j) (e) E(2) (kJ/mol)a E (j)–E (i) (a.u)b F(i,j) (a.u)c
p(C4–C5) 1.66204 p⁄(C6–C1) 0.40870 22.43 0.29 0.074p(C4–C3) 1.96590 p⁄(C2–C3) 0.02497 3.96 1.27 0.064r(C4–O4) 1.99095 r⁄(C2–C3) 0.02497 1.53 1.51 0.043r(C5–C6) 1.97411 r⁄(C4–O4) 0.02700 4.44 1.09 0.062r(C5–H7a) 1.97428 r⁄(C4–C3) 0.03670 4.26 1.03 0.059r(C5–H7a) 1.97428 r⁄(C6–C1) 0.02287 4.09 1.09 0.060p(C6–C1) 1.63475 p⁄(C2–C3) 0.32816 17.99 0.28 0.064p(C6–C1) 1.63475 p⁄(C7–C8) 0.24236 25.95 0.27 0.077r(C6–H8a) 1.97880 r⁄(C4–C5) 0.02698 4.21 1.08 0.060r(C6–H8a) 1.97880 r⁄(C1–C2) 0.02698 4.48 1.07 0.062r(C1–C2) 1.96760 r⁄(C6–C1) 0.02287 4.27 1.24 0.065r(C1–C2) 1.96760 r⁄(C3–O3) 0.02743 4.39 1.06 0.061p(C1–C7) 1.97300 p⁄(C7–C8) 0.02429 3.91 1.28 0.063p(C2–C3) 1.71353 p⁄(C4–C5) 0.37670 18.10 0.29 0.066r(O4–C12) 1.99105 r⁄(C4–C3) 0.03670 3.15 1.33 0.058r(O3–C11) 1.99085 r⁄(C4–C3) 0.03670 3.24 1.32 0.059r(C11–H16a) 1.99036 r⁄(C3–O3) 0.02743 4.20 0.90 0.055p(C7–C8) 1.96567 p⁄(C8–C10) 0.03575 4.72 1.25 0.068p(C7–C8) 1.81128 p⁄(C10–N10) 0.08879 20.92 0.40 0.084p(C7–C8) 1.81128 p⁄(C9–N9) 0.08768 20.47 20.39 0.082p(C10–N10) 1.99486 p⁄(C8–C10) 0.03575 8.18 1.53 0.101p(C9–N9) 1.99404 p⁄(C8–C9) 0.04050 8.04 1.55 0.100n2(O4) 1.80857 p⁄(C4–C5) 0.37670 34.75 0.34 0.102n2(O3) 1.82942 p⁄(C2–C3) 0.32816 33.14 0.34 0.098n1(N10) 1.96943 p⁄(C8–C10) 0.03575 12.22 0.99 0.098n1(N9) 1.96829 p⁄(C8–C9) 0.04050 12.03 1.00 0.098p(C4–C5) 0.37670 p⁄(C2–C3) 0.32816 267.38 0.01 0.081p⁄(C7–C8) 0.24236 p⁄(C9–N9) 0.08768 15.90 0.09 0.081
a E(2) means energy of hyperconjugative interactions.b Energy difference between donor and acceptor i and j NBO orbitals.c F(i, j) is the Fock matrix element between i and j NBO orbitals.
200 300 400 500 600 700 800
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.033
7 nm32
0 nm
427
nm
399
nm
Abs
orba
nce
(arb
.uni
ts)
Wavelength (nm)
290 nm
Fig. 5. Experimental UV–Vis spectrum of (3,4-dimethoxybenzylidene) propanedinitrile.
U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73 69
(3,4-dimethoxybenzylidene) propanedinitrile (C12H10N2O2) con-sists of 26 atoms and so it has (3N-6) 72 normal vibrational modes.All the vibrational frequency calculations were carried out afteroptimizing the structure of the molecule to its minimum energy.It is well known that the DFT method predicts vibrational calcula-tions with high accuracy which is applicable to large number ofcompounds [26] and in the present case DFT method also predictsvibrational frequencies with high accuracy (close to the experi-ment). The experimental FTIR spectrum is presented in Fig. 3 alongwith calculated FTIR spectra using HF and DFT method and the cor-relation graph between the experimental and corresponding calcu-lated wavenumbers is shown in Fig. 4. The experimental andcalculated wavenumbers and their assignments along with TED(total energy distribution) of title compound are given in Table 3.Only those calculated frequencies and assignments are given inthe Table 3 for which the corresponding experimental frequenciesare observed.
Electron correlations effects, insufficient basis sets to describethe molecular orbital exactly and anharmonicity are some of theimportant factors which results in higher values of calculatedwavenumbers than the corresponding experimental ones. Thesediscrepancies are removed either by computing anharmonic cor-rections explicitly or by introducing scalar field or even by directscaling of the calculated wavenumbers with a proper scaling factor[27,28]. The computed harmonic frequencies in the present studyhave been scaled by 0.967 in case of DFT/6-311G(d,p) and 0.909in case of HF/6-311G(d,p) for comparison. To assign bands in theexperimental vibrational spectra to molecular fragments one hasto recognize which atoms play a dominant role in the normalmodes corresponding to the bands in calculated spectra. With anincrease in the number of normal modes, the number of fragmentsthat may contribute to a mode increase quickly. PED (potentialenergy distribution) analysis using VEDA program is used to indi-cate in which mode a given fragment is participating. Most of the
fragments participate in several different modes. Such an analysisdetermines a potential energy connected with molecular frag-ments. The complete detail of the PED analyses can be found else-where [29,30]. The various vibrational modes of the title moleculeare discussed as under.
C–H vibrationsThe aromatic C–H stretching vibrations are normally occurs
above 3000 cm�1. In this region the bands are not affected by thenature of the substituents. In the present work, the C–H stretchingvibration of compound is observed in FTIR spectrum in the range2832–3126 cm�1 (Fig. 3) which is in good agreement with the
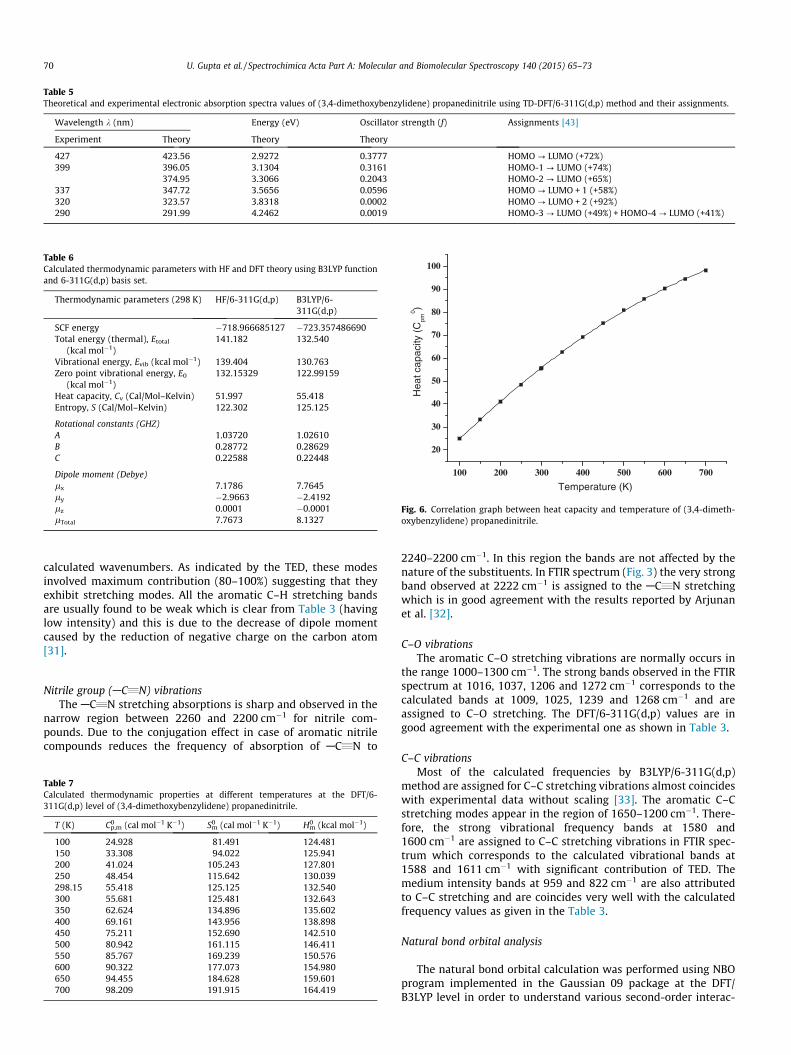
Table 5Theoretical and experimental electronic absorption spectra values of (3,4-dimethoxybenzylidene) propanedinitrile using TD-DFT/6-311G(d,p) method and their assignments.
Wavelength k (nm) Energy (eV) Oscillator strength (f) Assignments [43]
Experiment Theory Theory Theory
427 423.56 2.9272 0.3777 HOMO ? LUMO (+72%)399 396.05 3.1304 0.3161 HOMO-1 ? LUMO (+74%)
374.95 3.3066 0.2043 HOMO-2 ? LUMO (+65%)337 347.72 3.5656 0.0596 HOMO ? LUMO + 1 (+58%)320 323.57 3.8318 0.0002 HOMO ? LUMO + 2 (+92%)290 291.99 4.2462 0.0019 HOMO-3 ? LUMO (+49%) + HOMO-4 ? LUMO (+41%)
Table 6Calculated thermodynamic parameters with HF and DFT theory using B3LYP functionand 6-311G(d,p) basis set.
Thermodynamic parameters (298 K) HF/6-311G(d,p) B3LYP/6-311G(d,p)
SCF energy �718.966685127 �723.357486690Total energy (thermal), Etotal
(kcal mol�1)141.182 132.540
Vibrational energy, Evib (kcal mol�1) 139.404 130.763Zero point vibrational energy, E0
(kcal mol�1)132.15329 122.99159
Heat capacity, Cv (Cal/Mol–Kelvin) 51.997 55.418Entropy, S (Cal/Mol–Kelvin) 122.302 125.125
Rotational constants (GHZ)A 1.03720 1.02610B 0.28772 0.28629C 0.22588 0.22448
Dipole moment (Debye)lx 7.1786 7.7645ly �2.9663 �2.4192lz 0.0001 �0.0001lTotal 7.7673 8.1327
100 200 300 400 500 600 700
20
30
40
50
60
70
80
90
100
Temperature (K)
Hea
t cap
acity
(C
pm
0 )
Fig. 6. Correlation graph between heat capacity and temperature of (3,4-dimeth-oxybenzylidene) propanedinitrile.
70 U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73
calculated wavenumbers. As indicated by the TED, these modesinvolved maximum contribution (80–100%) suggesting that theyexhibit stretching modes. All the aromatic C–H stretching bandsare usually found to be weak which is clear from Table 3 (havinglow intensity) and this is due to the decrease of dipole momentcaused by the reduction of negative charge on the carbon atom[31].
Nitrile group (AC„N) vibrationsThe AC„N stretching absorptions is sharp and observed in the
narrow region between 2260 and 2200 cm�1 for nitrile com-pounds. Due to the conjugation effect in case of aromatic nitrilecompounds reduces the frequency of absorption of AC„N to
Table 7Calculated thermodynamic properties at different temperatures at the DFT/6-311G(d,p) level of (3,4-dimethoxybenzylidene) propanedinitrile.
T (K) C0p,m (cal mol�1 K�1) S0
m (cal mol�1 K�1) H0m (kcal mol�1)
100 24.928 81.491 124.481150 33.308 94.022 125.941200 41.024 105.243 127.801250 48.454 115.642 130.039298.15 55.418 125.125 132.540300 55.681 125.481 132.643350 62.624 134.896 135.602400 69.161 143.956 138.898450 75.211 152.690 142.510500 80.942 161.115 146.411550 85.767 169.239 150.576600 90.322 177.073 154.980650 94.455 184.628 159.601700 98.209 191.915 164.419
2240–2200 cm�1. In this region the bands are not affected by thenature of the substituents. In FTIR spectrum (Fig. 3) the very strongband observed at 2222 cm�1 is assigned to the AC„N stretchingwhich is in good agreement with the results reported by Arjunanet al. [32].
C–O vibrationsThe aromatic C–O stretching vibrations are normally occurs in
the range 1000–1300 cm�1. The strong bands observed in the FTIRspectrum at 1016, 1037, 1206 and 1272 cm�1 corresponds to thecalculated bands at 1009, 1025, 1239 and 1268 cm�1 and areassigned to C–O stretching. The DFT/6-311G(d,p) values are ingood agreement with the experimental one as shown in Table 3.
C–C vibrationsMost of the calculated frequencies by B3LYP/6-311G(d,p)
method are assigned for C–C stretching vibrations almost coincideswith experimental data without scaling [33]. The aromatic C–Cstretching modes appear in the region of 1650–1200 cm�1. There-fore, the strong vibrational frequency bands at 1580 and1600 cm�1 are assigned to C–C stretching vibrations in FTIR spec-trum which corresponds to the calculated vibrational bands at1588 and 1611 cm�1 with significant contribution of TED. Themedium intensity bands at 959 and 822 cm�1 are also attributedto C–C stretching and are coincides very well with the calculatedfrequency values as given in the Table 3.
Natural bond orbital analysis
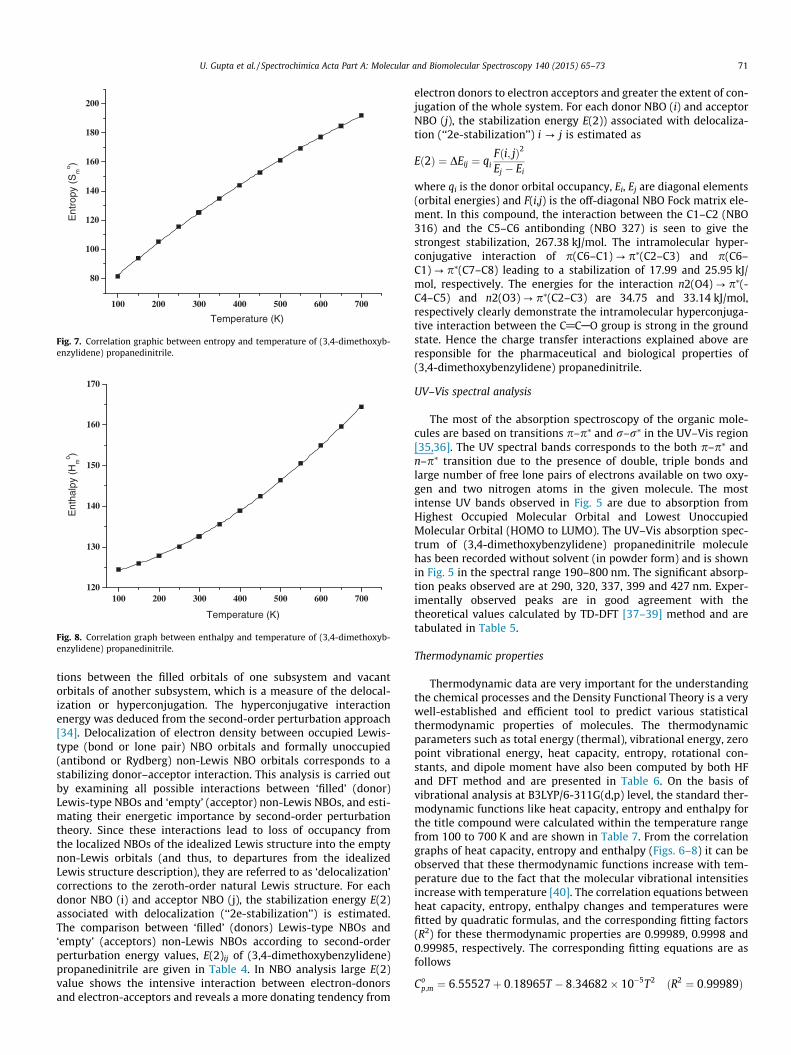
The natural bond orbital calculation was performed using NBOprogram implemented in the Gaussian 09 package at the DFT/B3LYP level in order to understand various second-order interac-
100 200 300 400 500 600 700
80
100
120
140
160
180
200
Ent
ropy
(S
m
o )
Temperature (K)
Fig. 7. Correlation graphic between entropy and temperature of (3,4-dimethoxyb-enzylidene) propanedinitrile.
100 200 300 400 500 600 700120
130
140
150
160
170
Ent
halp
y (H
m
0 )
Temperature (K)
Fig. 8. Correlation graph between enthalpy and temperature of (3,4-dimethoxyb-enzylidene) propanedinitrile.
U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73 71
tions between the filled orbitals of one subsystem and vacantorbitals of another subsystem, which is a measure of the delocal-ization or hyperconjugation. The hyperconjugative interactionenergy was deduced from the second-order perturbation approach[34]. Delocalization of electron density between occupied Lewis-type (bond or lone pair) NBO orbitals and formally unoccupied(antibond or Rydberg) non-Lewis NBO orbitals corresponds to astabilizing donor–acceptor interaction. This analysis is carried outby examining all possible interactions between ‘filled’ (donor)Lewis-type NBOs and ‘empty’ (acceptor) non-Lewis NBOs, and esti-mating their energetic importance by second-order perturbationtheory. Since these interactions lead to loss of occupancy fromthe localized NBOs of the idealized Lewis structure into the emptynon-Lewis orbitals (and thus, to departures from the idealizedLewis structure description), they are referred to as ‘delocalization’corrections to the zeroth-order natural Lewis structure. For eachdonor NBO (i) and acceptor NBO (j), the stabilization energy E(2)associated with delocalization (‘‘2e-stabilization’’) is estimated.The comparison between ‘filled’ (donors) Lewis-type NBOs and‘empty’ (acceptors) non-Lewis NBOs according to second-orderperturbation energy values, E(2)ij of (3,4-dimethoxybenzylidene)propanedinitrile are given in Table 4. In NBO analysis large E(2)value shows the intensive interaction between electron-donorsand electron-acceptors and reveals a more donating tendency from
electron donors to electron acceptors and greater the extent of con-jugation of the whole system. For each donor NBO (i) and acceptorNBO (j), the stabilization energy E(2)) associated with delocaliza-tion (‘‘2e-stabilization’’) i ? j is estimated as
Eð2Þ ¼ DEij ¼ qiFði; jÞ2
Ej � Ei
where qi is the donor orbital occupancy, Ei, Ej are diagonal elements(orbital energies) and F(i,j) is the off-diagonal NBO Fock matrix ele-ment. In this compound, the interaction between the C1–C2 (NBO316) and the C5–C6 antibonding (NBO 327) is seen to give thestrongest stabilization, 267.38 kJ/mol. The intramolecular hyper-conjugative interaction of p(C6–C1) ? p⁄(C2–C3) and p(C6–C1) ? p⁄(C7–C8) leading to a stabilization of 17.99 and 25.95 kJ/mol, respectively. The energies for the interaction n2(O4) ? p⁄(-C4–C5) and n2(O3) ? p⁄(C2–C3) are 34.75 and 33.14 kJ/mol,respectively clearly demonstrate the intramolecular hyperconjuga-tive interaction between the C@CAO group is strong in the groundstate. Hence the charge transfer interactions explained above areresponsible for the pharmaceutical and biological properties of(3,4-dimethoxybenzylidene) propanedinitrile.
UV–Vis spectral analysis
The most of the absorption spectroscopy of the organic mole-cules are based on transitions p–p⁄ and r–r⁄ in the UV–Vis region[35,36]. The UV spectral bands corresponds to the both p–p⁄ andn–p⁄ transition due to the presence of double, triple bonds andlarge number of free lone pairs of electrons available on two oxy-gen and two nitrogen atoms in the given molecule. The mostintense UV bands observed in Fig. 5 are due to absorption fromHighest Occupied Molecular Orbital and Lowest UnoccupiedMolecular Orbital (HOMO to LUMO). The UV–Vis absorption spec-trum of (3,4-dimethoxybenzylidene) propanedinitrile moleculehas been recorded without solvent (in powder form) and is shownin Fig. 5 in the spectral range 190–800 nm. The significant absorp-tion peaks observed are at 290, 320, 337, 399 and 427 nm. Exper-imentally observed peaks are in good agreement with thetheoretical values calculated by TD-DFT [37–39] method and aretabulated in Table 5.
Thermodynamic properties
Thermodynamic data are very important for the understandingthe chemical processes and the Density Functional Theory is a verywell-established and efficient tool to predict various statisticalthermodynamic properties of molecules. The thermodynamicparameters such as total energy (thermal), vibrational energy, zeropoint vibrational energy, heat capacity, entropy, rotational con-stants, and dipole moment have also been computed by both HFand DFT method and are presented in Table 6. On the basis ofvibrational analysis at B3LYP/6-311G(d,p) level, the standard ther-modynamic functions like heat capacity, entropy and enthalpy forthe title compound were calculated within the temperature rangefrom 100 to 700 K and are shown in Table 7. From the correlationgraphs of heat capacity, entropy and enthalpy (Figs. 6–8) it can beobserved that these thermodynamic functions increase with tem-perature due to the fact that the molecular vibrational intensitiesincrease with temperature [40]. The correlation equations betweenheat capacity, entropy, enthalpy changes and temperatures werefitted by quadratic formulas, and the corresponding fitting factors(R2) for these thermodynamic properties are 0.99989, 0.9998 and0.99985, respectively. The corresponding fitting equations are asfollows
Cop;m ¼ 6:55527þ 0:18965T � 8:34682� 10�5T2 ðR2 ¼ 0:99989Þ
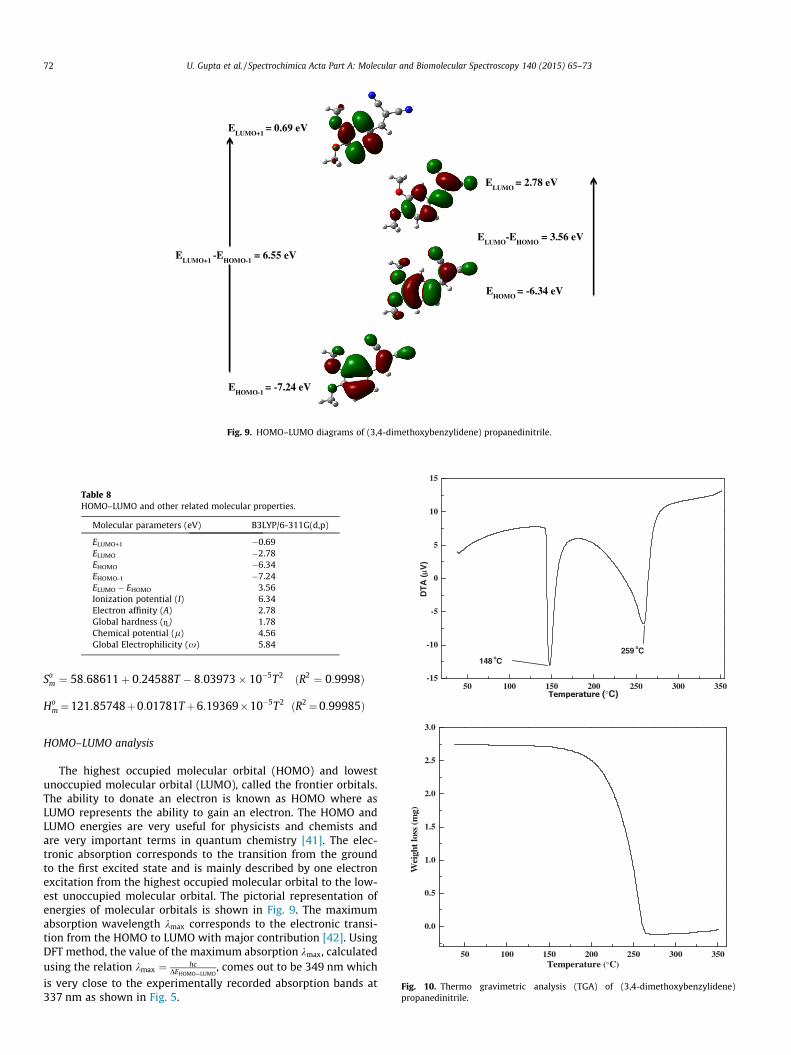
ELUMO+1
= 0.69 eV
ELUMO
= 2.78 eV
EHOMO
= -6.34 eV
EHOMO-1
= -7.24 eV
ELUMO+1
-EHOMO-1
= 6.55 eV
ELUMO
-EHOMO
= 3.56 eV
Fig. 9. HOMO–LUMO diagrams of (3,4-dimethoxybenzylidene) propanedinitrile.
Table 8HOMO–LUMO and other related molecular properties.
Molecular parameters (eV) B3LYP/6-311G(d,p)
ELUMO+1 �0.69ELUMO �2.78EHOMO �6.34EHOMO-1 �7.24ELUMO � EHOMO 3.56Ionization potential (I) 6.34Electron affinity (A) 2.78Global hardness (=) 1.78Chemical potential (l) 4.56Global Electrophilicity (x) 5.84
50 100 150 200 250 300 350-15
-10
-5
0
5
10
15
259 oC
DT
A (
µ V)
Temperature (°C)
148 oC
72 U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73
Som ¼ 58:68611þ 0:24588T � 8:03973� 10�5T2 ðR2 ¼ 0:9998Þ
Hom¼121:85748þ0:01781Tþ6:19369�10�5T2 ðR2¼0:99985Þ
50 100 150 200 250 300 350
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Wei
ght
loss
(m
g)
Temperature (°C)
Fig. 10. Thermo gravimetric analysis (TGA) of (3,4-dimethoxybenzylidene)propanedinitrile.
HOMO–LUMO analysis
The highest occupied molecular orbital (HOMO) and lowestunoccupied molecular orbital (LUMO), called the frontier orbitals.The ability to donate an electron is known as HOMO where asLUMO represents the ability to gain an electron. The HOMO andLUMO energies are very useful for physicists and chemists andare very important terms in quantum chemistry [41]. The elec-tronic absorption corresponds to the transition from the groundto the first excited state and is mainly described by one electronexcitation from the highest occupied molecular orbital to the low-est unoccupied molecular orbital. The pictorial representation ofenergies of molecular orbitals is shown in Fig. 9. The maximumabsorption wavelength kmax corresponds to the electronic transi-tion from the HOMO to LUMO with major contribution [42]. UsingDFT method, the value of the maximum absorption kmax, calculatedusing the relation kmax ¼ hc
DEHOMO—LUMO, comes out to be 349 nm which
is very close to the experimentally recorded absorption bands at337 nm as shown in Fig. 5.

U. Gupta et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 140 (2015) 65–73 73
The HOMO lying at �6.34 eV and is delocalized over entire mol-ecule whereas the LUMO located at �2.78 eV with large anti-bond-ing character which shows that the eventual charge transfer occurswithin the molecule, and the frontier orbital energy gap is �3.56eV. The lower energy gap means that the electrons are easilyexcited from ground to excited state. The larger the HOMO–LUMOenergy gap the harder the molecule and the smaller energy gapshows the high reactivity and low stability of the molecule. Theenergy gap explains the eventual charge transfer interaction withinthe molecule and in determining molecular electrical transportproperties. Both the HOMO and LUMO orbitals are the main orbitalthat take part in chemical stability. The ionization potentialI ¼ �EHOMO is directly proportional to the HOMO energy and theelectron affinity (A ¼ �ELUMO) is directly proportional to the LUMOenergy. The hardness corresponds to the gap between the HOMOand LUMO orbital energies. The hardness has been associated withthe stability of chemical system. The electron affinity can be usedin combination with ionization energy to give electronic chemicalpotential, l ¼ 1=2ðEHOMO þ ELUMOÞ. The global electrophilicityindex, x ¼ l2
2g is also calculated which measures the tendency ofchemical species to accept the electrons. All these molecular prop-erties above discussed are listed in Table 8.
Thermo gravimetric analysis
Thermal analysis is used to provide quantitative information onweight losses due to decomposition and/or evaporation of lowmolecular material as a function of time and temperature, thusgreatly facilitating the interpretation of thermal degradation pro-cesses. The sample weighing 2.74 mg was taken for the analysisand the thermogram is illustrated in Fig. 10. The thermal stabilityof (3,4-dimethoxy benzylidene) propanedinitrile was identified bythermo gravimetric (TG) and differential thermal analysis (DTA)were carried out simultaneously by using Shimadzu DTG-60 ther-mal analyser in a nitrogen free atmosphere from 38 to 350 �C at aheating rate of 15 �C/min. The molecule is stable up to 144 �C. Thethermogravimetry (TG) curve shows that the decomposition ofcomplex takes place. The weight loss starts at 177 �C and ends at267 �C, where total weight loss of compound takes place. TheDTA curve of the compound shows sharp endothermic peaks at148 and 259 �C (Fig. 10).
Conclusions
FT-IR, UV–Vis and TG measurements have been made for (3,4-dimethoxybenzylidene) propanedinitrile. The complete structural,vibrational, NBO, HOMO–LUMO analysis, and the thermodynamicproperties of the title molecule were performed on the basis ofHF and DFT calculations at B3LYP/6-311G(d,p) basis sets. The con-sistency between observed and the calculated vibrational wave-numbers using DFT clearly demonstrates that this method givesgood description (reliable geometry and related properties) of thetitle molecule as compared to HF method. TG analysis of the titlecompound shows that the molecule is stable up to 144 �C andshow two sharp endothermic peaks. The correlation graphsbetween the various thermodynamic parameters like heat capac-ity, enthalpy and entropy as a function of temperature in the range100–700 K are obtained. The correlation graphs show that the heatcapacities, entropies and enthalpies increase with increase in tem-perature because of the fact that the intensities of the molecularvibrations increase with increasing temperature.
Acknowledgement
Financial assistance from Science & Engineering Research Board(SERB), Department of Science and Technology, GOI (SR/S2/LOP-0020/2012) is gratefully acknowledged.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.saa.2014.12.063.
References
[1] F. Freeman, Chem. Rev. 69 (1969) 591–624.[2] A.J. Fatiadi, Synthesis (1978) 165–204.[3] S. Patai (Eds.), Chemistry of Triple Bonded Groups, The Chemistry of Functional
Groups, Suppl. C2, Wiley, New York, 1994.[4] Y.I. Binev, PhD Thesis, Bulgarian Academy of Sciences, Sofia, 2000.[5] F. Halverson, R.J. Francel, J. Chem. Phys. 17 (1949) 694–703.[6] L.L. Ames, D. White, D.E. Mann, J. Chem. Phys. 38 (1963) 910–917.[7] I.B. Yuri, A.T. Jordan, N.J. Ivan, G.B. Ivan, J. Mol. Struct. (Theochem.) 625 (2003)
207–214.[8] Dalbir Kour, D.R. Patil, D.R. Kumbhar, M.B. Deshmukh, Vivek K. Gupta, Rajni
Kant, Int. J. Chem. Appl. 6 (2014) 33–38.[9] R.G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules, Oxford
University Press, Oxford, 1989.[10] H.G. Korth, M.I. de Heer, P. Mulder, J. Phys. Chem. 106 (2002) 8779–8789.[11] P.K. Chowdhry, J. Phys. Chem. A 107 (2003) 5692–5696.[12] V. Chis, Chem. Phys. 300 (2004) 1–11.[13] A. Asensio, N. Kobko, J.J. Dannenberg, J. Phys. Chem. A 107 (2003) 6441–6443.[14] P. Hohenberg, W. Kohn, Phys. Rev. B 136 (1964) 864–871.[15] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M.Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.Honda, O. Kitao, H. Nakai, T. Vreven, Jr. J. A. Montgomery, J.E. Peralta, F. Ogliaro,M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J.Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M.Cossi, N. Rega, N.J. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J.Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C.Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G.Zakrzewski, G.A.Voth, P.Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman,J.V. Ortiz, J. Cioslowski, D. J. Fox Gaussian 09, Revision A.1, Gaussian Inc,Wallingford, CT, 2013.
[16] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.[17] A.D. Becke, Phys. Rev. A 38 (1988) 3098–3100.[18] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B37 (1988) 785–789.[19] M.H. Jamroz, Vibrational Energy Distribution Analysis VEDA 4, Warsaw, 2004.[20] A. Frisch, A.B. Neilson, A.J. Holder, GAUSSVIEW User Manual, Gaussian Inc.,
Pittsburgh, PA, 2000.[21] R.F.W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press,
Oxford (UK), 1990.[22] R. Bauernschmitt, R. Ahlrichs, Chem. Phys. Lett. 256 (1996) 454–464.[23] E. Kose, A. Atac, M. Karabacak, C. Karaca, M. Eskici, A. Karanfil, Spectrochim.
Acta A 97 (2012) 435–448.[24] M. Sekar, R. Velmurugan, A. Chandramohan, P. Rameshb, M.N. Ponnuswamy,
2-(6-Methyl-2, 3, 4, 9-tetrahydro-1H-carbazol-1ylidene) propanedinitrile,Acta Cryst. E67 (2011) 3270.
[25] R.S. Mulliken, J. Chem Phys. 23 (1955) 1833–1840.[26] Mehmet Karabacak, Leena Sinha, Onkar Prasad, Abdullah M. Asiri, Mehmet.
Cinar, Vikas K. Shukla, Spectrochim. Acta A 123 (2014) 352–362.[27] G. Keresztury, S. Holly, J. Varga, G. Besenyei, A.Y. Wang, J.R. Durig, Spectrochim.
Acta 49A (1993) 2007–2017.[28] G. Keresztury, Raman spectroscopy: theory, in: J.M. Chalmers, P.R. Griffith
(Eds.), Handbook of Vibrational spectroscopy, John Wiley & Sons, New York,2002.
[29] M.H. Jamroz, Jan Cz. Robert Brzozowski, J. Mol. Struct. 787 (2006) 172–183.[30] M.K. Jamroz, M.H. Jamroz, Jan Cz Dobrowolski, Jan A. Glinski, Matthew H.
Davey, Spectrochim. Acta Part A 78 (2011) 107–112.[31] H. Speeding, D.H. Wiffen, Proc. R. Soc. Lond. A 238 (1956) 245–255.[32] V. Arjunan, S. Thillai Govindaraja, A. Srilakshmi, C.V. Mythili, S. Mohan, Asian J.
Spectrosc. (2012) 65–80.[33] M. Kurt, P. Chinna Babu, N. Sundaraganesan, M. Cinar, M. Karabacak,
Spectrochim. Acta A 79 (2011) 1162–1170.[34] J. Choo, S. Kim, H. Joo, Y. Kwon, J. Mol. Struct. (Theochem.) 587 (2002) 1–8.[35] R.M. Silverstein, G.C. Bassler, T.C. Morrill, Spectrometric Identification of
Organic Compounds, John Wiley, Chichester, 1991.[36] F.A. Cotton, C.W. Wilkinson, Advanced Inorganic Chemistry, third ed.,
Interscience Publisher, New York, 1972.[37] M.E. Casida, C. Jamorski, K.C. Casida, D.R. Salahub, J. Chem. Phys. 108 (1998)
4439–4449.[38] E.K.U. Gross, W. Kohn, Adv. Quant. Chem. 21 (1990) 255–291.[39] O.J. Wacker, R. Kümmel, E.K.U. Gross, Phys. Rev. Lett. 73 (1994) 2915–2918.[40] J. Bevan Ott, J. Boerio-Goates, Chemical Thermodynamics: Principles and
Applications, Academic Press, San Diego, 2000.[41] K. Fukui, T. Yonezawa, H. Shingu, J. Chem. Phys 20 (1952) 722–725.[42] M. Karabacak, M. Cinar, Spectrochim. Acta A Mol. Biomol. Spectrosc. 86 (2012)
590–599.[43] S.I. Gorelsky, SWizard program, http://www.sg-chem.net/.







![Impedance spectroscopic investigations of ITO modified by new Azo-calix[4]arene immobilised into electroconducting polymer (MEHPPV)](https://static.fdokumen.com/doc/165x107/634532ab596bdb97a908d170/impedance-spectroscopic-investigations-of-ito-modified-by-new-azo-calix4arene.jpg)








![1-[2-(3,4-Dichlorobenzyloxy)-2-phenylethyl]-1 H -benzimidazole](https://static.fdokumen.com/doc/165x107/63152ee185333559270d05af/1-2-34-dichlorobenzyloxy-2-phenylethyl-1-h-benzimidazole.jpg)



