
Involvement of Nitric Oxide in Biofilm Dispersal of Pseudomonas aeruginosa

Bioorganic & Medicinal Chemistry 21 (2013) 7038–7046
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc
Spectroscopic identification and anti-biofilm properties of polarmetabolites from the medicinal plant Helichrysum italicum againstPseudomonas aeruginosa
0968-0896/$ - see front matter � 2013 Published by Elsevier Ltd.http://dx.doi.org/10.1016/j.bmc.2013.09.019
⇑ Corresponding author. Tel.: +39 0823 274564; fax: +39 0823 274571.E-mail address: [email protected] (B. D’Abrosca).
� These authors contributed equally to the work.
Brigida D’Abrosca a,⇑,�, Elisabetta Buommino b,�, Grazia D’Angelo a, Lorena Coretti b, Monica Scognamiglio a,Valeria Severino a, Severina Pacifico a, Giovanna Donnarumma b, Antonio Fiorentino a
a Department of Environmental, Biological and Pharmaceutical Sciences and Technologies, Second University of Naples, via Vivaldi 43, 81100 Caserta, Italyb Department of Experimental Medicine, Second University of Naples, via Costantinopoli 18, 80138 Naples, Italy
a r t i c l e i n f o a b s t r a c t
Article history:Received 17 June 2013Revised 5 September 2013Accepted 6 September 2013Available online 18 September 2013
Keywords:Helichrysum italicumStructural characterizationSpectroscopic analysisAnti-biofilm activityPseudomonas aeruginosa
Two new acylated styrylpyrones, one 5-methoxy-1(3H)-isobenzofuranone glucoside and a hydroxy-methyl-orcinol derivative, along with sixteen known aromatic metabolites, including lignans, quinic acidderivatives low-molecular weight phenol glucosides, have been isolated from the methanol extract ofHelichrysum italicum, a medicinal plant typical of the Mediterranean vegetation. The structures of thesecompounds have been elucidated on the basis of extensive 2D-NMR spectroscopic analyses, includingCOSY, TOCSY, HSQC, CIGAR-HMBC, H2BC and HSQC-TOCSY, along with Q-TOF HRMS2 analysis. Selectedcompounds were evaluated for their anti-biofilm properties against Pseudomonas aeruginosa.
� 2013 Published by Elsevier Ltd.
1. Introduction
Many pathogenic and commensal bacteria are capable of colo-nizing both the environment and the human host.1 These bacteriamust be able to adapt to sudden shifts in the availability of nutri-ents and to primary and secondary host immune defenses. A signif-icant factor contributing to the pathogenesis of a number ofmedically important bacterial strains is their ability to form a bio-film.2 Bacterial biofilms are highly organized surface-associatedcommunities of bacteria encased within a self produced extracellu-lar matrix, capable of growing in connection with different biolog-ical or inert surfaces such as artificial joints or catheters. Bacteriawithin a biofilm exhibit distinct phenotypes from planktonic cells,particularly with respect to growth and gene expression. Biofilmsare commonly associated with many health problems, such asendocarditis, otitis media, periodontitis, prostatitis, and urinarytract infections.2 Several bacteria, such as Escherichia coli, Staphylo-coccus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa,can form biofilms in the body tissues, leading to different infec-tions. In particular, chronic biofilm formation by P. aeruginosa incystic fibrosis lungs is a major cause of morbidity and mortality
for patients with cystic fibrosis.3 Thus, molecules that inhibit theformation of biofilm presumably may have therapeutic potential.In this context natural plant compounds useful for the new antimi-crobial and anti-biofilm drugs are of interest.4 Literature data evi-dence that a number of natural products, isolated from plants andmarine organisms possess the ability to inhibit or disperse bacte-rial biofilm.2 Halogenated furanones isolated from algae preventbiofilm formation in E. coli and Bacillus subtilis, while the diterpe-noids (agelasine class) isolated from the sponge genus Agelas,5
has been shown to have anti-biofilm capabilities without havinga bactericidal effect. Concerning plants, garlic extracts have shownthe ability to clear pulmonary P. areruginosa infection in mousemodel, ellagic acid derivatives from Rubus ulmifolius inhibit biofilmformation of a number of diverse S. aureus strains,6 while phloretininhibits E. coli O157:H7 biofilm formation.7 Recently, 4-epi-pimaricacid, isolated from Aralia cachemirica L., has been identified as a po-tent anti-biofilm agent for oral cavity pathogen.8
In the search for novel bioactive secondary metabolites fromnatural sources, the phytochemical study of plants from Mediterra-nean vegetation, one of the world biomes with the highest biodi-versity degree, is carried out.9 Helichrysum italicum G. Don is amedicinal plant from the Mediterranean area, widely used in folkmedicine as an anti-inflammatory and anti-infective plant. A previ-ous phytochemical investigation of H. italicum led to isolation ofheterodimeric phloroglucinyl pyrone, arzanol, reported as a potent

Table 1NMR data of compound 1 in CD3OD
Position dH dC DEPT HMBC(H ? C)
2 — 160.5 C —3 5.58 d (2.4) 89.0 CH 4, 5, 64 — 173.6 C —5 6.21 d (2.4) 102.2 CH 2, 3, 4, 76 — 166.7 C —7 6.76 d (15.9) 118.5 CH 2, 5, 10
8 7.39 d (15.9) 136.2 CH 2, 7, 20 , 60
10 — 130.9 C —20 7.55 d (8.7) 130.1 CH 8, 40
30 7.09 d (8.7) 118.1 CH 10 , 40
40 — 160.0 C —50 7.09 d (8.7) 118.1 CH 10 , 40
60 7.55 d (8.7) 130.1 CH 8, 40
1 4.95 d (7.5) 101.8 CH 40
200 3.48 ov 74.8 CH300 3.47 ov 77.4 CH400 3.42 ov 71.5 CH500 3.69 m 75.5 CH600 4.46 dd (12.3, 2.4)
4.20 dd (12.3, 6.3)64.5 CH2 1000
1000
1000 — 172.4 C —2000 2.62 d (14.1)
2.56 d (14.1)47.3 CH2 1000 , 3000 , 4000 , 6000Me
3000 — 70.9 C —4000 2.52 d (15.3)
2.34 d (15.3)47.8 CH2 2000 , 3000 , 5000 ,6000 Me
5000 — 178.0 C —6000 Me 1.29 s 27.9 CH3 2000 , 3000 , 4000
OMe 3.80 s 56.9 CH3 4
s = singlet, d = doublet, dd = doublet of doublet, m = multiplet, ov = overlapped; Jvalues (Hz) are reported in brackets.
B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046 7039
dual inhibitor of transcription factors (NF-jB) and inflammatoryenzymes [(mPGES)-1,5-LO].10 Recently, the antibacterial actionagainst multidrug-resistant S. aureus of heterodimeric phorogluci-nols from H. italicum was reported.11
In this Letter, the isolation and structural characterization of fournew metabolites–two styryl-pyrone derivatives, an acylated iso-benzofuranone and a hydroxymethyl-orcinol derivative–along withsixteen already known aromatic compounds, from methanolic ex-tract of H. italicum, is reported. The most representative compoundswere evaluated for anti-biofilm activity against P. aeruginosa.
2. Results and discussion
The molecular formula of compound 1 (Fig. 1) was calculated asC26H30O13 on the basis of 13C NMR data and of [M+H]+ and [M+Na]+
quasimolecular peaks appearing in the ESI Q-TOF HRMS at m/z551.1321 and at m/z 573.1178, respectively.
The 1H NMR spectrum (Table 1) revealed, in the downfield re-gion, the presence of two doublets at dH 5.58 and 6.21, and of anAA0BB0 spin system of a 1,4-disubstituted benzene ring as two dou-blets at dH 7.55 and 7.09. Furthermore, two olefinic protons of anAB spin system at dH 6.76 and 7.39, were evident; their couplingconstant value of 15.9 Hz, was consistent with the E-configurationfor the double bond. In the region of protons geminal to oxygen, ananomeric proton at dH 4.95, a diasterotopic methylene as two dou-blet of doublets at dH 4.46 and 4.20, besides four protons rangingfrom 3.30 to 3.70 ppm, were present, suggesting the presence ofa saccharide moiety in the molecule. A signal at dH 3.80, integratedfor three protons, suggested the presence of a methoxyl group inthe molecule. Finally, in the upfield region of the same spectrum,characteristic resonances of an aliphatic portion as two couplesof methylene signals at d 2.62/2.56, and 2.52/2.34, as well as a sin-glet methyl at d 1.29 were evident.
The 13C NMR spectrum (Table 1) showed 24 signals for 26 car-bons identified, on the basis of DEPT and HSQC experiments, as twomethyls, three methylenes, thirteen methines and eight quater-nary carbons, three of them attributable to carboxyl groups. Thesequence of the glycosidic moiety was established based on heter-onuclear two-bond correlations shown in the H2BC spectrum. Inparticular, correlations were observed between: the anomeric pro-ton (dH 4.95) and the C-200 carbon (dC 74.8); this carbon and the H-300 proton (dH 3.47); the latter proton and the methine carbon at dC
OHOH
HO
OR1OO
OR
5 R=OCH3 R1=malonyl6 R=OCH3 R1=H7 R=OH R1=H
O
ROR1O O
8 R=H R1=β-D-Glc9 R=β-D-Glc R1=H
O OOR
OHOH
HO
O
O
O
1 R=3-hydroxy-3-methyl)glutaryl2 R=malonyl3 R=H
OR
O
O
O
O
O
ORO
4 R=β-D-Glc
7
5
13
4
3a
1'
7a
1'
1''
2
4 76
4'
O
Figure 1. Styrylpyrones (1–4), isobenzofuranone (5–7), and 3-hydrox-ydihydrobenzofuran glycosides (8–9) from H. italicum.
71.5 (C-400); this carbon and the H-500 proton (dH 3.69); this protonand the methylene carbon at dC 64.5. These data, supported bythose shown in the HSQC-TOCSY experiment, suggested the pres-ence of a glucose moiety, whose identity was later confirmed byGC–MS analysis of the corresponding alditol acetate. The couplingconstant value of the anomeric proton (J = 7.5 Hz) allowed a b con-figuration for the C-10 carbon to be assigned. The absolute configu-ration of the sugar was determined by GC–MS analysis of theacylated thiazolidine derivatives, obtained from the reaction ofhydrolyzed 1 with L-cysteine methyl ester.12 The structure ofmetabolite 1 was deduced by HSQC and CIGAR–HMBC spectra asdescribed in Table 1.
All these data suggested the presence of desmethylyangonine,constituted by an a-pyrone core connected to a p-hydroxystyrylmoiety by a linkage between C-7 of the latter subunit and C-6 car-bon of c-pyrone. This hypothesis was confirmed based on correla-tions, shown in the HMBC experiment, from H-5 to C-7 (dC 118.5)and from H-8 to C-6.
Finally, the heterocorrelation between the anomeric doublet atdH 4.95 (bound to the carbon at dC 101.8) and the C-40 carbon ofthe aromatic ring at dC 160.2 allowed the glycosylation site tobe clearly defined. Moreover, further heterocorrelations (Table 1)suggested the presence of a 3-hydroxy-3-methylglutaryl moietyin the molecule.13 The downfield shifted values of the H-600 dou-blet of doublets of glucose and the correlations of these latterwith the carboxyl carbon at dC 172.4, allowed the localization ofthe acyl group at the C-6 carbon of the sugar. This structurewas confirmed by the ESI Q-TOF MS/MS analysis. The tandemMS analysis of the quasimolecular ion [M+Na]+ at m/z 551.1321produced the fragment ion at m/z 489.1537, arising from the con-temporary loss of CO2 and H2O, and the fragment ion at m/z245.0698, due to the loss of the hexose and acyl moiety. Thus, 1was identified as desmethylyangonine-40-O-[6-O00-(3-hydroxy-3-methylglutaryl)]-b-D-glucopyranoside.

7040 B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046
The ESI Q-TOF HRMS spectrum of 2 showed the quasimolecularpeaks [M+H]+ at m/z 493.1249 and [M+Na]+ at m/z 515.1052according to a molecular formula C23H24O12. The NMR data of com-pound 2 (Fig. 1), were very similar to those of the previous metab-olite. In fact, in the downfield region of 1H NMR spectrum (Table 2),the typical set of signals corresponding to the styrylpyrone skele-ton were shown. In the region of protons geminal to oxygen ananomeric proton at dH 4.90, a diasterotopic methylene as two dou-blets of doublets at dH 4.47 and 4.26, besides four protons rangingfrom 3.30 to 3.70 ppm, were evident, suggesting the presence of anacylated monosaccharide moiety in the molecule. In the same re-gion, a methoxyl group resonating at dH 3.80 was also evident.13C NMR, DEPT and HSQC spectra revealed signals for one methyl,thirteen methines (four of them attributed to C-20, C-30, C-50, and C-60 aromatic carbons) and seven quaternary carbons. The 2D NMRdata confirmed the presence of desmethylyangonine-400-O-b-D-glucopyranoside for 2.
The main difference was the lack of resonances attributable to3-hydroxy-3-methylglutaryl moiety. However, the downfield shiftof the values of the H-600 doublet of doublets, as well as their cor-relations with a carboxyl carbon at dC 171.0 suggested the pres-ence, in the compound 2, of a different acyl group at the C-600
carbon of the sugar. As no further signals were evident in the 1HNMR experiment in CD3OD, a spectrum of the compound was reg-istered in DMSO-d6; in these conditions, a methylene singlet wasevident at dH 2.92 (dC 45.7). This latter singlet heterocorrelatedwith both the carboxyl carbon at dC 173.1 and dC 171.0. These dataagreed with the presence of a malonic acid residue in the moleculeallowing to identify 2 as desmethylyangonine-40-O-(600-O-malo-nyl)-b-D-glucopyranoside. Compound 3 identified as desmethyly-angonine-40-O- b-D-glucopyranoside, was already isolated fromAcosmium panamense (Benth.) Yacovlev,14 while the styrylpyronedimer 4 was previously reported as constituent of the medicinalplant Achyrocline bogotensis.15
The molecular formula of compound 5 was determined asC18H20O12 on the basis of 13C NMR data and of the quasimolecularion [M+H]+ at m/z 429.0935 in the ESI Q-TOF HRMS.
Table 2NMR data of compound 2 in CD3OD
Position dH dC DEPT HMBC(H ? C)
2 — 160.7 C —3 5.58 d (2.4) 89.0 CH 4, 5,64 — 171.1 C —5 6.25 d (2.4) 102.1 CH 2, 3, 4, 76 — 166.8 C —7 6.85 d (15.9) 118.4 CH 2, 5, 10
8 7.39 d (15.9) 136.2 CH 2, 7, 20 , 60
10 131.0 C —20 7.57 d (8.4) 130.1 CH 8, 40
300 7.10 d (8.4) 118.0 CH 10 , 40
4000 160.0 C —50 7.10 d (8.4) 118.0 CH 10 , 40
60 7.57 d (8.4) 130.1 CH 8, 40
100 4.90 d (7.2) 101.8 CH 40
200 3.48 ov 74.8 CH300 3.48 ov 77.7 CH400 3.44 ov 71.3 CH500 3.70 m 75.5 CH600 4.47 dd (12.0, 2.1)
4.26 dd (12.0, 6.0)64.9 CH2
1000 — 171.0 C —2000 * * CH2
3000 — 173.1 C —OMe 3.80 s CH3 4
d = doublet, dd = doublet of doublet, m = multiplet, ov = overlapped, s = singlet, Jvalues (Hz) are reported in brackets.* Not detected in CD3OD.
The 1H NMR spectrum (Table 3) revealed, in the downfield re-gion, signals of two aromatic meta-coupled protons at dH 6.76and 6.71, and two protons as a singlet at dH 5.25. Moreover, onemethine as doublet at dH 5.07, two double doublets at dH 4.46and 4.26, as well as four protons ranging from 3.30 and3.80 ppm, due to a saccharide moiety, were also evident. Finally,a signal at dH 3.90 integrated for three protons indicated the pres-ence of a methoxyl group in the molecule. The 13C NMR spectrum(Table 3) showed 18 signals identified, on the basis of DEPT andHSQC experiments, as one methyls, three methylenes, sevenmethines and seven quaternary carbons, three of them attributableto carboxyl groups.
The sequence of NMR signals of the glycosidic moiety wasestablished on the basis of heteronuclear two-bond correlations,showed in the H2BC spectrum, suggesting the presence of a glu-cose moiety. The coupling constant value of the anomeric proton(J = 7.5 Hz) allowed a b configuration for the C100 carbon to beassigned.
The complete structural elucidation of compound 5 was per-formed by long range heterocorrelation experiments. The CI-GAR-HMBC experiment showed cross peaks between themethoxyl group at dH 3.90 and a quaternary carbon at dC 168.6,in turn correlated with the meta-coupled protons at dH 6.71 and6.76 (dc 103.9 and 101.5, respectively) and with the methylenesinglet a dH 5.25. This latter, as well as the aromatic proton atdH 6.71, heterocorrelated with the quaternary carbons at dC
107.3 and dC 158.0 and with the carboxyl carbon at dC 172.0. Allthese data suggested the presence of 5-methoxy-1-(3H)-iso-benzofuranone as algycon. The position of the sugar unit was indi-cated to be C-7 by CIGAR-HMBC correlations between the signalsat dH 5.07 and the carbon at dC 158.0 (C-7). The downfield shift ofthe values of the methylene diastereotopic protons of the sugarmoiety at dH 4.46 and dH 4.26 correlated in the CIGAR-HMBC withthe carboxyl carbon at dC 171.15, allowed us to hypothesize thepresence of an acyl group at the C-60 carbon of the sugar. Alsoin this case, the registration of the NMR data in DMSO-d6 alloweda malonic acid residue in the molecule to be identified. Therefore,5 was elucidated as 7-O-b-D-(6-O-malonyl)glucopyranosyl-5-hy-droxy-1-(3H)-isobenzofuranone.
Compounds 6 and 7 were previously isolated from flowers ofHelichrysum arenarium,16,17 while metabolites 8 and 9 were already
Table 3NMR data of compound 3 in CD3OD.
Position dH dC DEPT HMBC(H ? C)
1 — 172.0 C —3 5.25 s 70.7 CH2 1, 3a, 5, 7a,
10
3a — 153.6 C —4 6.76 d (1.8) 101.5 CH 3, 5, 6, 7, 7a5 — 168.6 C —6 6.71 d (1.8) 103.9 CH 4, 5, 7, 7a7 — 158.0 C —7a — 107.3 C —1’ 5.07 d (7.5) 101.1 CH 7, 30 , 50
2’ 3.57 dd (6.6, 2.4) 74.3 CH 5, 73’ 3.52 ov 77.2 CH4’ 3.45 ov 71.3 CH5’ 3.75 dt (6.3, 1.8) 75.7 CH6’a 4.46 dd (12.3, 1.8) 63.2 CH2 100 , 50 , 40
6’b 4.26 dd (12.3, 6.3) 100 , 50 ,100 — 171.1 C —200 * * CH2
*
300 — 173.0 C —OMe 3.90 s 56.8 CH3 5
s = singlet, d = doublet, dd = doublet of doublet, dt = doubleof triplet, ov = overlapped; Jvalues (Hz) are reported in brackets.* Not detected in CD3OD.
O
OH
O
HO
HO
OO
OHOH
HO
OHO
R
O
OH
O
HO
HO
OO
OHOH
HO
OHO
R
12 R=H15 R=OCH3
13 R=H14 R=OCH3
O
HOOH
OO
RO
10 R=β-D-Glc11 R=H
16 R=CH317 R=CH2OH
OHOH
HO
OHO
R
OHO
18
OHOH
HO
OHO O
OHO
OHOH
HO
OHO O
19
OOH
O
HOOC OH
OO
HOOH OH
OH
21
OOH
HO
HOOC OH
O
OHOH
20
13
5
1'
Figure 2. Neolignans (10–11), oxyneolignans (12–15) and low-molecular weight phenol glucosides (16–21) isolated from H. italicum.
B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046 7041
reported as constituent of Gnaphalium polycaulon.18 Neolignan 10was reported as constituent of leaves of Virburnum awabuki K.Koch,19 while metabolite 11, idenfied as dihydrodehydrodiconife-ryl alcohol, was isolated from Epimedium diphyllum.20 Compound12 was identified by comparing its NMR data with literaturedata.21 Metabolites 13 and 14, respectively named citrusin A andB, were already reported as constituent of Ligularia veitchiana.22
NMR data of neolignan glycoside 15 was in good accordance withTang et al.,23 while orcinol glucoside (16), was reported as a con-stituent of rhizomes of Curculigo breviscapa.24 The molecular for-mula of the new compound 17 (Fig. 2) was determined asC13H18O8 on the basis of 13C NMR and ESI Q-TOF HRMS data. The1H NMR spectrum of compound 17 showed in the downfield regionthree signals, each integrated for one proton, as two singlets at dH
6.58 and 6.47, and a multiplet at dH 6.45. These data were in goodaccordance with a 1,3,5-trisubstituted benzene ring. A methyleneas a singlet at dH 4.58, two doublets of doublets at dH 3.89 and3.69, besides four overlapped protons ranging from 3.40 to3.50 ppm, suggested the presence of a monosaccharide moiety inthe molecule identified as glucose on the basis of the 13C NMR data.The remaining seven carbon identified on the basis of an HSQC
0
25
50
1 2 3 4 6
Perc
enta
ge b
iofil
m in
hibi
tion
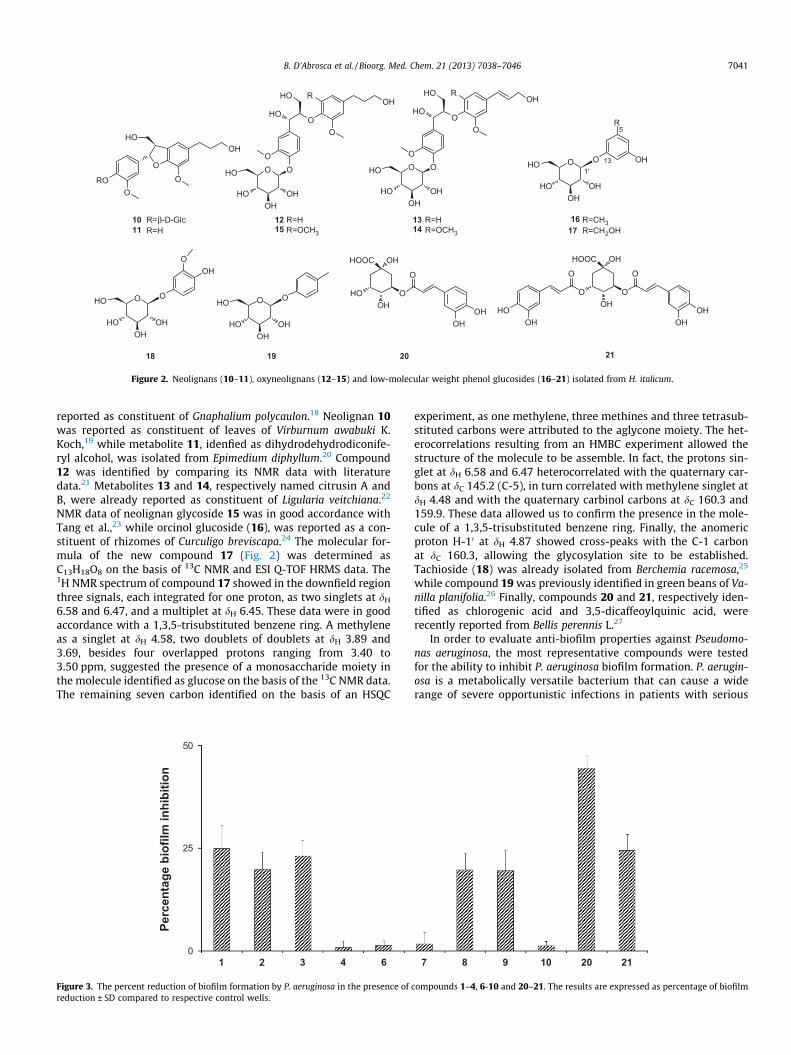
Figure 3. The percent reduction of biofilm formation by P. aeruginosa in the presence ofreduction ± SD compared to respective control wells.
experiment, as one methylene, three methines and three tetrasub-stituted carbons were attributed to the aglycone moiety. The het-erocorrelations resulting from an HMBC experiment allowed thestructure of the molecule to be assemble. In fact, the protons sin-glet at dH 6.58 and 6.47 heterocorrelated with the quaternary car-bons at dC 145.2 (C-5), in turn correlated with methylene singlet atdH 4.48 and with the quaternary carbinol carbons at dC 160.3 and159.9. These data allowed us to confirm the presence in the mole-cule of a 1,3,5-trisubstituted benzene ring. Finally, the anomericproton H-10 at dH 4.87 showed cross-peaks with the C-1 carbonat dC 160.3, allowing the glycosylation site to be established.Tachioside (18) was already isolated from Berchemia racemosa,25
while compound 19 was previously identified in green beans of Va-nilla planifolia.26 Finally, compounds 20 and 21, respectively iden-tified as chlorogenic acid and 3,5-dicaffeoylquinic acid, wererecently reported from Bellis perennis L.27
In order to evaluate anti-biofilm properties against Pseudomo-nas aeruginosa, the most representative compounds were testedfor the ability to inhibit P. aeruginosa biofilm formation. P. aerugin-osa is a metabolically versatile bacterium that can cause a widerange of severe opportunistic infections in patients with serious
7 8 9 10 20 21
compounds 1–4, 6-10 and 20–21. The results are expressed as percentage of biofilm
0
25
50
1 2 3 4 6 7 8 9 10 20 21
Perc
enta
g eb
iofil
m in
hibi
tion
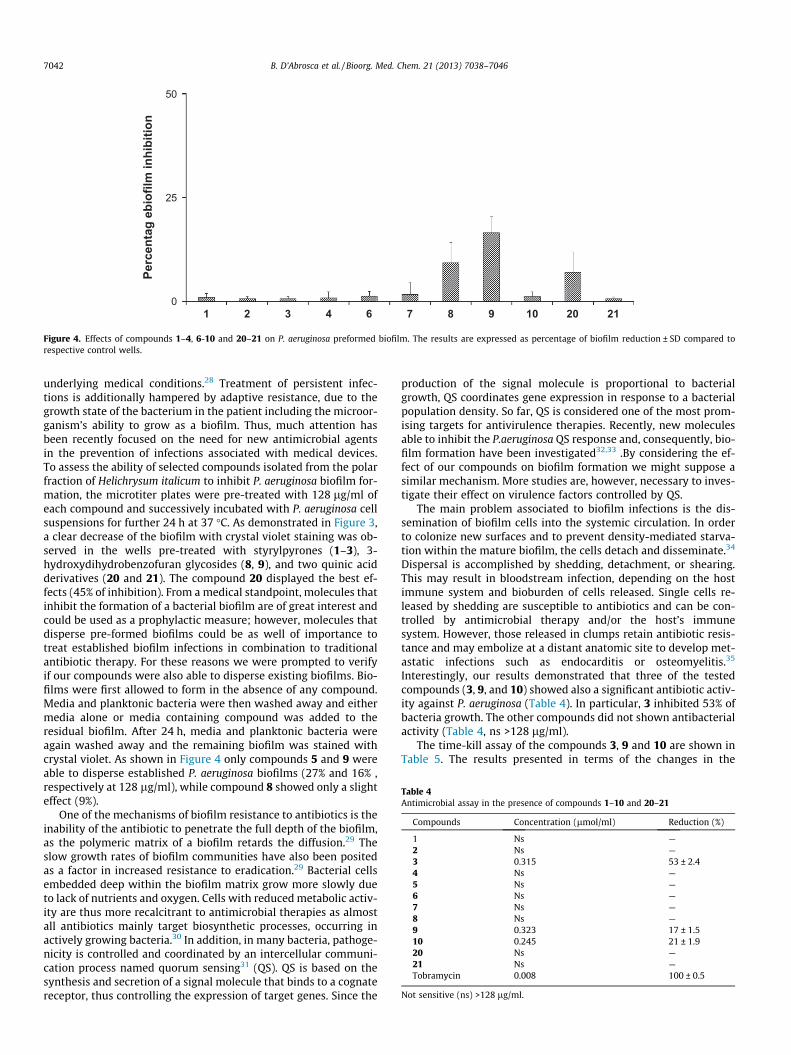
Figure 4. Effects of compounds 1–4, 6-10 and 20–21 on P. aeruginosa preformed biofilm. The results are expressed as percentage of biofilm reduction ± SD compared torespective control wells.
Table 4Antimicrobial assay in the presence of compounds 1–10 and 20–21
Compounds Concentration (lmol/ml) Reduction (%)
1 Ns —2 Ns —3 0.315 53 ± 2.44 Ns —5 Ns —6 Ns —7 Ns —8 Ns —9 0.323 17 ± 1.510 0.245 21 ± 1.920 Ns —21 Ns —Tobramycin 0.008 100 ± 0.5
Not sensitive (ns) >128 lg/ml.
7042 B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046
underlying medical conditions.28 Treatment of persistent infec-tions is additionally hampered by adaptive resistance, due to thegrowth state of the bacterium in the patient including the microor-ganism’s ability to grow as a biofilm. Thus, much attention hasbeen recently focused on the need for new antimicrobial agentsin the prevention of infections associated with medical devices.To assess the ability of selected compounds isolated from the polarfraction of Helichrysum italicum to inhibit P. aeruginosa biofilm for-mation, the microtiter plates were pre-treated with 128 lg/ml ofeach compound and successively incubated with P. aeruginosa cellsuspensions for further 24 h at 37 �C. As demonstrated in Figure 3,a clear decrease of the biofilm with crystal violet staining was ob-served in the wells pre-treated with styrylpyrones (1–3), 3-hydroxydihydrobenzofuran glycosides (8, 9), and two quinic acidderivatives (20 and 21). The compound 20 displayed the best ef-fects (45% of inhibition). From a medical standpoint, molecules thatinhibit the formation of a bacterial biofilm are of great interest andcould be used as a prophylactic measure; however, molecules thatdisperse pre-formed biofilms could be as well of importance totreat established biofilm infections in combination to traditionalantibiotic therapy. For these reasons we were prompted to verifyif our compounds were also able to disperse existing biofilms. Bio-films were first allowed to form in the absence of any compound.Media and planktonic bacteria were then washed away and eithermedia alone or media containing compound was added to theresidual biofilm. After 24 h, media and planktonic bacteria wereagain washed away and the remaining biofilm was stained withcrystal violet. As shown in Figure 4 only compounds 5 and 9 wereable to disperse established P. aeruginosa biofilms (27% and 16% ,respectively at 128 lg/ml), while compound 8 showed only a slighteffect (9%).
One of the mechanisms of biofilm resistance to antibiotics is theinability of the antibiotic to penetrate the full depth of the biofilm,as the polymeric matrix of a biofilm retards the diffusion.29 Theslow growth rates of biofilm communities have also been positedas a factor in increased resistance to eradication.29 Bacterial cellsembedded deep within the biofilm matrix grow more slowly dueto lack of nutrients and oxygen. Cells with reduced metabolic activ-ity are thus more recalcitrant to antimicrobial therapies as almostall antibiotics mainly target biosynthetic processes, occurring inactively growing bacteria.30 In addition, in many bacteria, pathoge-nicity is controlled and coordinated by an intercellular communi-cation process named quorum sensing31 (QS). QS is based on thesynthesis and secretion of a signal molecule that binds to a cognatereceptor, thus controlling the expression of target genes. Since the
production of the signal molecule is proportional to bacterialgrowth, QS coordinates gene expression in response to a bacterialpopulation density. So far, QS is considered one of the most prom-ising targets for antivirulence therapies. Recently, new moleculesable to inhibit the P.aeruginosa QS response and, consequently, bio-film formation have been investigated32,33 .By considering the ef-fect of our compounds on biofilm formation we might suppose asimilar mechanism. More studies are, however, necessary to inves-tigate their effect on virulence factors controlled by QS.
The main problem associated to biofilm infections is the dis-semination of biofilm cells into the systemic circulation. In orderto colonize new surfaces and to prevent density-mediated starva-tion within the mature biofilm, the cells detach and disseminate.34
Dispersal is accomplished by shedding, detachment, or shearing.This may result in bloodstream infection, depending on the hostimmune system and bioburden of cells released. Single cells re-leased by shedding are susceptible to antibiotics and can be con-trolled by antimicrobial therapy and/or the host’s immunesystem. However, those released in clumps retain antibiotic resis-tance and may embolize at a distant anatomic site to develop met-astatic infections such as endocarditis or osteomyelitis.35
Interestingly, our results demonstrated that three of the testedcompounds (3, 9, and 10) showed also a significant antibiotic activ-ity against P. aeruginosa (Table 4). In particular, 3 inhibited 53% ofbacteria growth. The other compounds did not shown antibacterialactivity (Table 4, ns >128 lg/ml).
The time-kill assay of the compounds 3, 9 and 10 are shown inTable 5. The results presented in terms of the changes in the

Table 5In vitro time-kill assessment of the compounds 3, 9 and 10
Compounds(128 lg/ml)
Log10 kill
0 h 4 h 8 h 16 h 24 h
3 2.322 1.157 1.103 1.055 1.0459 2.322 2.136 1.974 1.927 1.88110 2.322 2.043 1.858 1.811 1.765
B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046 7043
log10 CFU/mL of viable colonies indicated that the compound 3exhibited the best bactericidal activity already after 4 h of incuba-tion. Our results might, thus, suggest the ability of these moleculesto act both on preformed biofilm and on bacteria released by dis-persed biofilm, avoiding distant colonization.
3. Conclusion
Although antibiotic therapy generally reverses the symptomscaused by planktonic cells, bacteria within a biofilm can displayup to 1000-fold increased resistance to antibiotic or biocide treat-ment.36 For this reason biofilm infections typically show recurringsymptoms, even after cycles of antibiotic therapy. In this context,identification of natural compounds which can limit the formationof bacterial biofilms represent an important task for the research-ers working in this field.
Although polyphenols from natural source have widely been re-ported for their antimicrobial activities, few investigations on bac-terial biofilms have been carried out. Different classes of naturalphenols have been investigated against Pseudomonas aeruginosaand chlorogenic acid, vanillin and sinapic acid were found to exertsignificant anti-biofouling effect.37 Moreover Jagani et al38 re-ported that tannic acid, epigallocathechin and other phenols re-duced the formation of biofilm formation by P. aeruginosa.
In conclusion, the compounds 1–3, 8–9 and 20–21 identified inthis study showed significant antibiofilm activity against a strain ofP. aeruginosa forming biofilms. Even though this is a preliminaryscreening, the force of our study is the ability of natural productsto inhibit biofilm formation and to exhibit also a microbicidalactivity. However, further bioactive plant metabolites have to bescreened and synthetic analogue derivatives have to be projectedin the aim to discover new therapeutic agents useful to preventbiofilm formation of P. aeruginosa.
4. Experimental
4.1. General experiment procedures
Optical rotations were measured on a Perkin–Elmer 141 inMeOH solution. UV spectra were performed on UV-1700 Shimadzuspectrophotometer in MeOH. NMR spectra were recorded at300.03 MHz for 1H and 75.45 MHz for 13C on a Varian Mercury300 spectrometer Fourier transform NMR in CD3OD or DMSO-d6
solutions at 25 �C. Chemical shifts are reported in d (ppm) and ref-erenced to the residual solvent signal, J (coupling constant) are gi-ven in Hz. Standard pulse sequences and phase cycling from Varianlibrary were used for 1H,13C, DEPT, DQF-COSY, COSY, TOCSY, ,HSQC, H2BC, HMBC and CIGAR–HMBC experiments. 1H NMR spec-tra were acquired over a spectral window from 14 to 2 ppm, with1.0 s relaxation delay, 1.70 s acquisition time (AQ), 90� pulsewidth = 13.8 ls. The initial matrix was zero-filled to 64 K. 13C-NMR spectra were recorded in 1H broadband decoupling mode,over a spectral window from 235 to 15 ppm, 1.5 s relaxation delay,90� pulse width = 9.50 ls, AQ = 0.9 s. The number of scans for both1H and 13C-NMR experiments was chosen depending on the con-centration of the samples. Also for homonuclear and heteronuclear2D-NMR experiments, data points, number of scans and of incre-
ments were adjusted according to the sample concentrations. Cor-relation spectroscopy (COSY) and double quantum filtered COSY(DQF-COSY) spectra were recorded with gradient enhanced se-quence at spectral widths of 3000 Hz in both f2 and f1 domains;the relaxation delays were of 1.0 s. The total correlation spectros-copy (TOCSY) experiments were performed in the phase-sensitivemode with a mixing time of 90 ms. The spectral width was3000 Hz. Nuclear Overhauser effect spectroscopy (NOESY) experi-ments were performed in the phase-sensitive mode. The mixingtime was 500 ms and the spectral width was 3000 Hz. For all thehomonuclear experiments, the initial matrix of 512 � 512 datapoints was zero-filled to give a final matrix of 1 k � 1 k points. Pro-ton-detected heteronuclear correlations were measured. Hetero-nuclear single-quantum coherence (HSQC) experiments(optimized for 1J(H,C) = 140 Hz) were performed in the phase sensi-tive mode with field gradient; the spectral width was 12,000 Hz inf1 (13C) and 3000 Hz in f2 (1H) and 1.0 s of relaxation delay; thematrix of 1 k � 1 k data points was zero-filled to give a final matrixof 2 k � 2 k points. Heteronuclear 2 bond correlation (H2BC) spec-tra were obtained with T = 30.0 ms, and a relaxation delay of 1.0 s;the third-order low-pass filter was set for 130 < 1J(C,H) < 165 Hz.Heteronuclear multiple bond coherence (HMBC) experiment (opti-mized for nJ(H,C) = 8 Hz) was performed in the absolute value modewith field gradient; typically, 1H–13C gHMBC were acquired withspectral width of 18,000 Hz in f1 (13C) and 3000 Hz in f2 (1H)and 1.0 s of relaxation delay; the matrix of 1 k � 1 k data pointswas zero-filled to give a final matrix of 4 k � 4 k points. Constanttime inverse-detection gradient accordion rescaled heteronuclearmultiple bond correlation spectroscopy (CIGAR–HMBC) spectra(8 > nJ(H,C) > 5) were acquired with the same spectral width usedfor HMBC. Heteronuclear single-quantum coherence–total correla-tion spectroscopy (HSQC-TOCSY) experiments were optimize fornJ(H,C) = 8 Hz, with a mixing time of 90 ms. For accurate mass mea-surements the purified compounds were analyzed by electrosprayhybrid quadrupole orthogonal acceleration time-of-flight massspectrometer (Q-TOF) fitted with a Z-spray electrospray ion source(Waters S.p.A.). All analyses were performed in both positive andnegative ion mode. The capillary source voltage and the cone volt-age were set at 3500 V and 35 V, respectively. The source temper-ature was kept at 80 �C and nitrogen was used as a drying gas (flowrate about 50 L/h). The time-of-flight analyzer of the mass spec-trometer was externally calibrated with GFP from m/z 50 to1600. Accurate mass data were collected by directly infusing sam-ples (1.5 pmol/lL in CH3CN/H2O, 1:1) into the system at a flow rateof 15 lL/min. The acquisition and processing of data were per-formed with the MassLynx 4.1 software (Waters S.p.A., ManchesterUK). GC–MS qualitative analyses were carried out using a HP 6890GC instrument equipped with a 5975B VL MSD detector.
GC–MS analyses were carried out using a HP 6890 GC instru-ment (Zebron ZB-5MS column, He flow 1.0 ml/min) coupled witha 5973 N mass spectrometer, equipped with an electron ionizationsource (EIMS) operating with an electron energy of 70 eV. Full-scanmass spectra were collected between 0 and 600 amu at 2 scan/s.The MS was operated in the electron impact (EI) ionization modewith an electron energy of 70 eV. The ion source and quadrupoletemperatures were maintained at 230 and 150 �C, respectively.For the analyses of the acetylated alditols, the column head pres-sure was set at 7.41 p.s.i.
Analytical TLC was performed on Merck Kieselgel 60 F254 or RP-8 F254 plates with 0.2 mm layer thickness. Spots were visualized byUV light or by spraying with H2SO4/AcOH/H2O (1:20:4). The plateswere then heated for 5 min at 110 �C. Preparative TLC was per-formed on Merck Kieselgel 60 F254 plates, with 0.5 or 1.0 mm filmthickness. Column chromatography (CC) was performed on MerckKieselgel 60 (70–240 lm), Merck Kieselgel 60 (40–63 llm), Baker-bond C8 and C18 Sephadex LH-20, Amberlite XAD-4.

7044 B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046
4.2. Plant material
Plants of Helichrysum italicum (Roth) G. Don (Asteraceae) werecollected in May 2007, in the vegetative state, in Nature Reserveof Castel Volturno (Caserta, Italy), and identified by Dr. AssuntaEsposito of the Dept. of Environmental, Biological and Pharmaceu-tical Sciences and Technologies of Second University of Naples(SUN). A voucher specimen (CE0200) has been deposited at theHerbarium of the Department. Fresh plants harvested, separatedin leaves and stems were dried at 40 �C in a ventilated oven andstored in paper bags until the extraction process was carried out.
4.3. Extraction and isolation of compounds
Dried leaf material was powdered and extracted by ultrasoundassisted extraction (Elma� TransonicDigitals), one hour each, inmethanol. The extract was filtered on Whatman paper and concen-trated under vacuum. After removal of the solvent, a dried crudeextract was obtained (38.7 g) and stored at �80 �C until its purifi-cation. Analogously, a stem crude extract (9.5 g) was obtained. Themethanol extract was dissolved in distilled water (2.0 L) and sha-ken with EtOAc (3.0 L); the aqueous fraction was chromatographedon Amberlite XAD-4 and eluted with water first, to eliminate sug-ars, peptides, free amino acids and other primary metabolites, andthen with methanol.
The alcoholic eluate furnished 6.9 g of residual material whichwas chromatographed on Sephadex LH-20 eluting with MeOH/H2O polarity decreasing solutions and collecting five fractions(A–E). Fraction A, eluted with H2O, was chromatographed by SiO2
TLC (1.0 mm) eluting with the lower phase of the biphasic solutionCHCl3/MeOH/H2O (13:7:2) and gave one spot identified as com-pound 9 (39.0 mg). Fraction B, eluted with MeOH/H2O (1:3), waschromatographed by RP-18 CC and gave seven fractions: B1-B7.Fraction B1 eluted with MeCN/MeOH/H2O (1:1:18) was furtherpurified by SiO2 TLC (1.0 mm) eluting with the lower phase ofthe biphasic solution CHCl3/MeOH/H2O (13:7:2) and gave threespots. The first spot was identified as compound 20 (20.9 mg),the second as the metabolite 17 (3.0 mg) finally the third spotwas identified as compound 18 (4.2 mg). Fraction B2, eluted withMeCN/MeOH/H2O (1:1:8) was purified by SiO2 TLC (0.5 mm) elut-ing with the lower phase of the biphasic solution CHCl3/MeOH/H2O(13:7:2) to obtain pure compounds 6 (14.6 mg) and 2 (5.8 mg).Fraction B3 eluted with MeCN/MeOH/H2O (1:1:8) was rechromato-graphed by SiO2 TLC (1.0 mm) eluting with the organic phase of abiphasic solution CHCl3/MeOH/H2O (13:7:3) to obtain pure com-pound 1 (6.4 mg). Fraction B4 eluted with MeCN/MeOH/H2O(1:1:8) was purified by SiO2 TLC (0.5 mm) eluting with the lowerphase of the biphasic solution CHCl3/MeOH/H2O (13:7:3) and gavetwo spots. The first spot was identified as compound 12 (10.7 mg),while the second spot as the metabolite 9 (8.7 mg). Fraction B5,eluted with MeCN/MeOH/H2O (3:3:14), was rechromatographedby SiO2 TLC (0.5 mm) [eluent: lower phase of CHCl3/MeOH/H2O(13:7:2)] giving compounds 15 (3.1 mg) and 8 (6.7 mg). FractionB6, eluted with MeCN/MeOH/H2O (3:3:14), rechromatographedby SiO2 TLC (0.5 mm) [eluent: lower phase of CHCl3/MeOH/H2O(13:7:2)] furnished pure metabolite 4 (11.5 mg). Fraction B7 elutedwith MeCN/MeOH/H2O (1:1:8) was chromatographed by SiO2-flashCC eluting with the lower phase of the biphasic solution CHCl3/MeOH/H2O (13:7:4) and gave a fraction further purified by SiO2TLC (0.5 mm) [eluent: lower phase of CHCl3/MeOH/H2O(13:7:2)]; pure metabolites 7 (8.6 mg) and 19 (5.6 mg) were thusobtained.
Fraction C, eluted with MeOH/H2O (1:3), was chromatographedby RP-8 CC and gave two fraction. Fraction C1, eluted with MeCN/MeOH/H2O (1:1:8) was rechromatographed by SiO2 TLC (0.2 mm)eluting with the lower phase of the biphasic solution CHCl3/
MeOH/H2O (13:7:2) giving pure compound 11 (3.2 mg). FractionC2 eluted with MeCN/MeOH/H2O (3:3:14), after SiO2 TLC(0.5 mm) eluting with the lower phase of the biphasic solutionCHCl3/MeOH/H2O (13:7:3), furnished pure compound 10(18.6 mg).
Fraction D, eluted with MeOH/H2O (1:3), was purified by RP-18CC and gave five fractions: D1-D4. Fractions D1 and D2, both elutedwith MeCN/MeOH/H2O (1:1:8), were rechromatographed by SiO2
TLC (0.5 mm) [eluent: lower phase of CHCl3/MeOH/H2O (13:7:2)]giving the first compound 16 (4.8 mg) and the second the puremetabolite 13 (4.7 mg). Fraction D3, eluted with MeCN/MeOH/H2O (3:3:14), were rechromatographed by SiO2 TLC (0.5 mm) [elu-ent: lower phase of CHCl3/MeOH/H2O (13:7:2)] furnish compound14 (6.7 mg) while fraction D4, eluted with MeCN/MeOH/H2O(3:3:14), contained pure compound 4 (6.7 mg).
Fraction E, eluted with MeOH/H2O (1:3), was chromatographedby RP-18 CC and gave a fraction further purified by SiO2 TLC(1.0 mm) eluting with the lower phase of the biphasic solutionCHCl3/MeOH/H2O (13:7:2) to obtain pure metabolite 5 (20.7 mg).
Methanol extract from stems was dissolved in water and sha-ken with EtOAc to obtain organic (1.4 g) and aqueous fractions.The organic fraction was chromatographed on SiO2 to obtain twofractions A00–B0. Fraction A0, eluted with EtOAc, was chromato-graphed on Sephadex LH-20, eluting with hexane/CHCl3/MeOH(1:1:1) collecting fractions of 10 mL; fraction A10 was rechromato-graphed by SiO2 TLC (0.5 mm) eluting with the lower phase of thebiphasic solution CHCl3/MeOH/H2O (13:7:2) to obtain pure metab-olite 1 (17.9 mg). Fraction A200, instead, contained pure compound21 (207.1 mg). Fraction B0, eluted with MeOH, was purified by SiO2
TLC (1 mm) eluting with the lower phase of the biphasic solutionCHCl3/MeOH/H2O (13:7:2) to obtain three spots: the first wasidentified as the metabolite 2 (4.9 mg), the second as the com-pound 1 (12.5 mg) and finally the third as the pure compound 3(9.0 mg).
4.3.1. Desmethylyangonine-40-O-(600-O-(3-hydroxy-3-methyl)glutaryl)-b-D-glucopyranoside (1)
Light yellow oil, a25D �20.0 (c 0.32, MeOH); UV (MeOH) kmax
(loge) 353 (3.88), 281 (3.53) nm. 1H NMR (CD3OD, 300 MHz) data,see Table 1. 13C NMR (CD3OD, 75 MHz) data, see Table 1. Q-TOFHRMS (ESI+): m/z 589.1035 (calcd 589.1318 for C26H30KO12)[M+K]+; m/z 573.1178 (calcd 573.1579 for C26H30NaO13)[M+Na]+; m/z 551.1321 (calcd 551.1759 for C26H31O13) [M+H]+ ,489.1537 (calcd 489.1755 for C25H29O10) [M–CH2O3+H]+,245.0698 (calcd 245.0808 for C14H13O4) [M–C9H12O8+H]+.
4.3.2. Desmethylyangonine-40-O-(600-O-malonyl)-b-D-glucopyranoside (2)
light yellow oil, a25D =�7.14 (c 0.35, MeOH); UV (MeOH) kmax
(loge) 352 (2.96), 282 (2.95) nm. 1H NMR (CD3OD, 300 MHz) data,see Table 2. 13C NMR (CD3OD, 75 MHz) data, see Table 2. 1H NMR(DMSO, 300 MHz) dH: 7.58 (2H, d, J = 8.7 Hz , H-20/ H-60), 7.26 (1H,d, J = 15.9 Hz, H-8), 7.03 (2H, d, J = 8.7 Hz , H-30/ H-50), 6.85 (1H, d,J = 15.9 Hz, H-7), 6.25 (1H, d, J = 1.8 Hz, H-5), 5.58 (1H, d, J = 1.8 Hz,H-3), 4.90 (1H, d, J = 7.2 Hz, H-1000), 4.19 (1H, m, H-6a00), 4.04 (1H, m,H-6b00), 3.83 (3H, s, OCH3), 3.57 (1H, m, H-500), 3.33 (1H, m, H-300),3.26 (1H, m, H-200), 3.20 (1H, m, H-400), 2.92 (2H, s, H-200). 13CNMR (DMSO, 75 MHz) dC: 170.9 (C-4), 169.5 (C-1000), 167.9 (C-3000),162.7 (C-6), 158.6 (C-2), 158.2 (C-40), 133.8 (C-8), 129.0 (C-20/C-60), 128.8 (C-10), 117.7 (C-7), 116.5 (C-30/C-50), 100.7 (C-5), 99.9(C-100), 88.4 (C-3), 76.2 (C-500), 74.0 (C-300), 73.2 (C-200), 69.8 (C-400),63.1 (C-600), 56.4 (OMe), 45.7 (C-2000). Q-TOF HRMS (ESI+): m/z531.0781 (calcd. 531.0899 for C23H24KO12) [M+K]+; m/z 515.1052(calcd. 515.1165 for C23H24NaO12) [M+Na]+; m/z 493.1249 (calcd.493.1341 for C23H25O12) [M+H]+ , 245.0396 (calcd. 245.0808 forC14H13O4) [M–C9H12O8+H]+.

B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046 7045
4.3.3. 7-O-b-D-(60-O-malonyl)-glucopyranosyl-5-methoxy-1(3H)-isobenzofuranone (5)
Light yellow oil, a25D �27.3 (c 0.15, MeOH); UV (MeOH) kmax
(loge) 280 (3.14) nm. 1H NMR (CD3OD, 300 MHz) data, see Table 3.13C NMR (CD3OD, 75 MHz) data, see Table 3. 1H NMR (DMSO,300 MHz) dH: 6.75 (1H, s, H-4), 6.70 (1H, s, H-6), 5.13 (1H, d,J = 7.5 Hz, H-100), 4.20 (1H, m, H-6a00), 4.03 (1H, m, H-6b00), 3.83(3H, s, OCH3), 3.65 (1H, m, H-500), 3.38 (1H, m, H-300), 3.35 (1H, m,H-200), 3.28 (1H, m, H-400), 2.92 (2H, s, H-200). 13C NMR (DMSO,75 MHz) dC: 170.0 (C-3000), 169.5 (C-1000), 168.8 (C-1), 166.0 (C-5),156.8 (C-7), 151.6 (C-3a), 105.9 (C-7a), 101.5 (C-6), 100.1 (C-4),99.5 (C-100), 77.3 (C-500), 76.8 (C-300), 73.1 (C-2000), 69.6 (C-400), 63.0(C-600), 56.0 (OMe), 45.5 (C-2000). Q-TOF HRMS (ESI+): m/z429.0935 (calcd 429.1028 for C18H21O12) [M+H]+.
4.3.4. 1-O-b-D-glucopyranosyl-5-hydroxymethyl-orcinol (17)Light yellow oil, a25
D �43.08 (c 0.13, MeOH); UV (MeOH) kmax
(loge) 280 (3.47) nm. 1H NMR (CD3OD, 300 MHz) dH: 6.58 (1H, s,H-4), 6.47 (1H, s, H-6), 6.45 (1H, m, H-2), 4.87 (1H, s, H-10), 4.48(2H, s, H-7), 3.44 (1H, ov, H-20), 3.43 (2H, ov, H-30 and H-50), 3.41(1H, ov, H-40), 3.89 (1H, dd, J = 11.7, 1.8 Hz, H-60a), 3.69 (1H, dd,J = 11.7, 4.8 Hz, H-60b). 13C NMR (CD3OD, 75 MHz) dC: 160.3 (C-1),159.9 (C-3), 145.2 (C-5), 198.9 (C-6), 107.2 (C-4), 103.9 (C-1),102.3 (C-10), 78.1 (C-30), 78.0 (C-50), 74.9 (C-20), 71.3 (C-40), 62.5(C-60). Q-TOF HRMS (ESI+): m/z 303.1974 calcd 303.1974 forC13H19O8) [M+H]+.
5. Biological assay
All the compounds tested were dissolved in dimethylsulfoxide(DMSO, Sigma, Milan) and diluted in LB broth to give a stocksolution.
5.1. Bacterial strains and culture
Pseudomonas aeruginosa, a clinical mucoid strain (PaM02) wasrecovered from storage by subculture on Columbia Horse Bloodagar plates (Oxoid). For all experiments, cultures were preparedby inoculating a single colony into sterile Luria–Bertani (LB) brothand overnight incubation at 37 �C. The inoculum was collected anddiluted with normal saline.
5.2. Antimicrobial assay
The bacterial suspension to be used as the inoculum was dilutedto yield an optical density (OD) around 1 at 600 nm (correspondingto about 1 � 108 CFU/ml). Afterwards, the bacterial cell suspensionwas further diluted 100-folds to produce a bacterial cell suspensionof 10� 105 CFU/ml. Next, 100 lL of compounds (256 lg/ml) in LBbroth was added to 100 lL of the bacterial cell suspension in eachwell of the microplate to yield a final cell concentration of 5.0� 105
CFU/ml and a final compound concentration of 128 lg/ml. Somewells were reserved in each plate to test the sterility of the medium(no inoculum added) and inoculum viability (no compounds added).Control wells (negative control) were prepared growing P. aeruginosain LB broth plus the concentration of DMSO used to dilute each com-pounds. The turbidity of the medium was directly proportional to thegrowth of the bacteria, which was measured by a microtiter platereader (Tecan, Milan, Italy) at 600 nm absorbance. The antimicrobialagent tobramycin was used as positive control.
5.3. Determination of rate of kill
Assays for the rate of killing bacteria by the compounds 3, 9 and10 were carried out as previously reported with slight modifica-
tions.39 Compounds 3, 9 and 10 were incorporated into 10 mL LBbroth. Inoculums density, approximately 105 CFU/mL, was usedto inoculate 10 mL LB broth. The tubes were incubated at 37 �Con an orbital shaker at 120 rpm. Viability counts were performedat 0, 4, 8, 16 and 24 h of incubation at 37 �C by plating 0.1 mL undi-luted and 10-fold serial diluted samples onto LB plates in triplicate.After incubating at 37 �C for 24 h, emergent bacterial colonies werecounted, CFU/mL calculated, and compared with the count of theculture control without the extract (control).
5.4. Biofilm formation and effect of compounds on P.aeruginosa biofilm
Biofilm formation was assayed by measuring the ability ofcells to adhere to sterile 96-well polystyrene flat-bottom micro-titer plate (BD Falcon, Mississauga, Ontario Canada).33 Briefly, abacterial cell suspension to be used as the inoculum was ad-justed to 1.0 McFarland standard and was further diluted 10-folds to produce 1.0 � 107 CFU/ml. Next, 100 lL of the inoculumwere transferred to each well of a 96-well microplate, to allowbiofilm formation, and incubated at 37 �C for 24 h. After incuba-tion, the wells were rinsed with phosphate-buffered saline (PBS)and air dried at room temperature for 45 min. 200 lL of crystalviolet (1%) solution were added to each well, and the disheswere incubated for 30 min. Then, the wells were washed fourtimes with distilled water and immediately discolored with200 lL of 95% ethanol. Elapsed 45 min after the last procedure,100 lL of discolored solution was transferred to a well of anew plate and the crystal violet measured at 570 nm in an ELISAreader (MICROPLATES Reader, Biorad. Milan, Italy). To asses theactivity of the compounds against preformed P. aeruginosa bio-film, following 24 h of adhesion and biofilm formation, plank-tonic cells were removed from the wells and the plate wasrinsed with 100 lL of phosphate buffered saline (PBS). Cells ad-hered to the polystyrene microtiter plate were treated with100 lL of compounds diluted in LB broth to make the final con-centration of 128 lg/ml, and the plate was further incubated for24 h at 37 �C. Non-treated cells were incubated with 100 lL ofLB broth, which served as a negative control. Control wells wereprepared growing P. aeruginosa in LB broth plus the concentra-tion of DMSO used to dilute each compounds. To asses the abil-ity of compounds to prevent P. aeruginosa biofilm formation,100 lL of compounds, diluted in LB broth to make the final con-centration of 128 lg/ml, was added to polystyrene microtiterplate and incubated overnight at 4 �C. The compounds wereaspirated and the plates were washed once in sterile PBS. The96-well microtiter plates were then seeded with P. aeruginosafor biofilm formation, as above described, and the plate was fur-ther incubated for 24 h at 37 �C. Non-treated cells were pre-incu-bated with 100 lL of LB broth, which served as a negativecontrol.
At the end of each experiment crystal violet-staining has beenperformed to assess biofilm formation. The amount of biofilmformed was measured subtracting the absorbance from the absor-bance values of the control wells. The percentage of biofilm reduc-tion was calculated using the following formula: [(Ac�At)/Ac]� 100, were Ac is the OD570 for control well and At is OD570 forbiofilm in presence of compounds.
5.5. Statistical analysis
Each experiment was performed at least three times. The re-sults are expressed as mean ± standard deviation (SD). Student’s ttest was used to determine statistical differences between themeans, and p <0.05 was considered a significant difference.

7046 B. D’Abrosca et al. / Bioorg. Med. Chem. 21 (2013) 7038–7046
Acknowledgments
The author M. Scognamiglio aknowledges support by theL’Oréal UNESCO program ‘For Women in Science’ through the2012 National Fellowship ‘L’Oréal Italia Per le Donne e la Scienza’ .
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.bmc.2013.09.019.
References and notes
1. Johnjulio, W.; Fuge, L. H.; Kad, M.; Post, C. South. Med. J. 2012, 105, 24.2. Worthington, R. J.; Richards, J. J.; Melander, C. Org. Biomol. Chem. 2012, 10,
7457.3. Anderson, G. G.; Kenney, T. F.; Macleod, D. L.; Henig, N. R.; O’Toole, G. A. Pathog.
Dis. 2013, 67, 39.4. Kuzmaa, Ł.; Ró _zalski, M.; Walencka, E.; Ró _zalska, B.; Wysokinska, H.
Phytomedicine 2007, 14, 31.5. Stowe, S. D.; Richards, J. J.; Tucker, A. T.; Thompson, R.; Melander, C.; Cavanagh,
J. Mar. Drugs 2011, 9, 2010.6. Quave, C. L.; Estevez-Carmona, M.; Compadre, C. M.; Hobby, G.; Hendrickson,
H.; Beenken, K. E.; Smeltzer, M. S. PLoS One 2012, 7, e28737. http://dx.doi.org/10.1371/journal.pone.0028737.
7. Lee, J.-H.; Regmi, S. C.; Kim, J. A.; Cho, M. H.; Yun, H.; Lee, C.-S.; Lee, J. Infect.Immun. 2011, 79, 4819.
8. Ali, F.; Sangwan, P. L.; Koul, S.; Pandey, A.; Bani, S.; Abdullah, S. T.; Sharma, P. R.;Kitchlu, S.; Khan, I. A. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 149.
9. Fiorentino, A.; D’Abrosca, B.; Pacifico, S.; Scognamiglio, M.; D’Angelo, G.;Monaco, P. Bioorg. Med. Chem. 2010, 18, 8530.
10. Appendino, G.; Ottino, M.; Marquez, N.; Bianchi, F.; Giana, A.; Ballero, M.;Sterner, O.; Fiebich, B. L.; Munoz, E. J. Nat. Prod. 2007, 70, 608.
11. Taglialatela-Scafati, O.; Pollastro, F.; Chianese, G.; Minassi, A.; Gibbons, S.;Arunotayanun, W.; Mabebie, B.; Ballero, M.; Appendino, G. J. Nat. Prod. 2013,76, 346.
12. Scognamiglio, M.; D’Abrosca, B.; Fiumano, V.; Chambery, A.; Severino, V.;Tsafantakis, N.; Pacifico, S.; Esposito, A.; Fiorentino, A. Phytochemistry 2012, 84,125.
13. Jung, K. Y.; Do, J. C.; Son, K. H. Phytochemistry 1993, 34, 1196.14. Wiedenfeld, H.; Andrade-Cetto, A. Z. Naturforsch. 2003, 58c, 637.15. Sagawa, T.; Takaishi, Y.; Fujimoto, Y.; Duque, C.; Osorio, C.; Ramos, F.; Garzon,
C.; Sato, M.; Okamoto, Ma.; Oshikawa, T.; Ahmed, S. U. J. Nat. Prod. 2005, 68,502.
16. Morikawa, T.; Wang, L.-B.; Nakamura, S.; Ninomiya, K.; Yokoyama, E.; Matsuda,H.; Muraoka, O.; Wu, L.-J.; Yoshikawa, M. Chem. Pharm. Bull. 2009, 57, 853.
17. Morikawa, T.; Wang, L.-B.; Ninomiya, K.; Nakamura, S.; Matsuda, H.; Muraoka,O.; Wu, L.-J.; Yoshikawa, M. Chem. Pharm. Bull. 2009, 57, 361.
18. Sahakitpichan, P.; Disadee, W.; Ruchirawat, S.; Tripetch, K. Chem. Pharm. Bull.2011, 59, 1160.
19. Matsuda, N.; Sato, H.; Yaoita, Y.; Kikuchi, M. Chem. Pharm. Bull. 1996, 44, 1122.20. Miyase, T.; Ueno, A.; Nobuo, T.; Kobayashi, H.; Oguchi, H. Phytochemistry 1989,
28, 3483.21. Huo, C.; Liang, H.; Zhao, Y.; Wang, B.; Zhang, Q. Phytochemistry 2008, 69, 788.22. Zhu, H.; Tu, P. Z. Naturforsch. 2004, 59b, 1063.23. Tang, W.-Z.; Liu, Y.-B.; Yu, S.-S.; Qu, J.; Su, D.-M. Planta Med. 2007, 73, 484.24. Zhu, C. C.; Wang, K. J.; Wang, Z. Y.; Li, N. Bull. Korean Chem. Soc. 2010, 31, 224.25. Inoshiri, S.; Sasaki, M.; Kohda, H.; Otsuka, H.; Yamasaki, K. Phytochemistry
1987, 26, 2811.26. Dignum, M. J. W.; van der Heijden, R.; Kerler, J.; Winkel, C.; Verpoorte, R. Food
Chem. 2004, 85, 199.27. Scognamiglio, M.; Esposito, A.; D’Abrosca, B.; Pacifico, S.; Fiumano, V.;
Tsafantakis, N.; Monaco, P.; Fiorentino, A. Biochem. Syst. Ecol. 2012, 43, 108.28. Gellatly, S. L.; Hancock, R. E. Pathog. Dis. 2013, 67, 159.29. Mah, T. F.; O’Toole, G. A. Trends Microbiol. 2001, 9, 34.30. Mah, T. F.; Pitts, B.; Pellock, B.; Walker, G. C.; Stewart, P. S.; O’Toole, G. A. Nature
2003, 426, 306.31. Njoroge, J.; Sperandio, V. EMBO Mol. Med. 2009, 1, 201.32. Imperi, F.; Massai, F.; Pillai, C. R.; Longo, F.; Zennaro, E.; Rampioni, G.; Visca, P.;
Leoni, L. Antimicrob. Agents Ch. 2013, 57, 996.33. Nazzaro, F.; Fratianni, F.; Coppola, R. Int. J. Mol. Sci. 2013, 14, 12607.34. Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. J. Clin.
Invest. 2003, 112, 1466.35. Fux, C. A.; Wilson, S.; Stoodley, P. J. Bacteriol. 2004, 186, 4486.36. Costerton, J. W. Int. J. Antimicrob. Agents 1999, 11, 217.37. Plyuta, V.; Zaitseva, J.; Lobakova, E.; Zagoskina, N.; Kuznetsov, A.; Khmel, I. Acta
Path. Micro. Imm. 2013, 1.38. Jagani, S.; Chelikani, R.; Kim, D.-S. Biofuling 2009, 25, 321.39. Eliopoulos, G. M.; Eliopoulos, C. T. Clin. Microbiol. Rev. 1988, 1, 139.
Copyright © 2022 FDOKUMEN




















