
Sorsa et al Mol Cell Biochem 2004 266 87
21
Molecular and Cellular Biochemistry 266: 87–107, 2004. c 2004 Kluwer Academic Publishers. Printed in the Netherlands. The contractile apparatus as a target for drugs against heart failure: Interaction of levosimendan, a calcium sensitiser, with cardiac troponin c Tia Sorsa, 1 Piero Pollesello 1 and R. John Solaro 2 1 Orion Pharma, Cardiovascular Research, Espoo, Finland; 2 University of Illinois, Chicago, Department of Physiology and Biophysics, Chicago, IL, U.S.A. Received 1 March 2004; accepted 6 April 2004 Abstract Cardiac failure is one of the leading causes of mortality in developed countries. As life expectancies of the populations of these countries grow, the number of patients suffering from cardiac insufficiency also increases. Effective treatments are being sought and recently a new class of drugs, the calcium sensitisers, was developed. These drugs cause a positive inotropic effect on cardio-myocytes by interacting directly with the contractile apparatus. Their mechanism of action is not accompanied by an increase in intracellular calcium concentration at therapeutic doses, as seen for the older generation of positive inotropic drugs, and thus does not induce calcium-related deleterious effects such as arrhythmias or apoptosis. Levosimendan is a novel calcium sensitiser which has been discovered by using cardiac troponin C (cTnC) as target protein. This drug has been proved to be a well-tolerated and effective treatment for patients with severe decompensated heart failure. This review describes the basic principles of muscle contraction, the main components of the contractile apparatus and their roles in the heart contraction. The regulatory proteins troponin C (cTnC), troponin I (cTnI), troponin T (cTnT), and tropomyosin (Tm) and their interactions are discussed in details. The concept of calcium sensitisation is thereafter explained and a few examples of calcium sensitisers and their putative mechanisms are discussed. Finally, the binding of levosimendan to cTnC and its mechanism of action are described and the results discussed under the light of the action of this drug in vitro and in vivo. (Mol Cell Biochem 266: 87–107, 2004) Key words: cardiac troponin C, calcium sensitisation, drug interaction, regulation of muscle contraction, nuclear magnetic resonance, cardiovascular pharmacology Abbreviations: ATPase, adenosine triphosphatase; BSA, bovine serum albumin; Bis-Tris, Bis(2-hydroxyethyl)-imino- tris(hydroxymethyl)methane; cCTnC, C-domain of cardiac troponin C; cNTnC, N-domain of cardiac troponin C; cTn, het- erotrimeric cardiac troponin complex; cTnC, cardiac troponin C; CTnC, C-domain of troponin C; cTnC A-Cys , cardiac troponin C with mutations of C35S and C84S; cTnC CS , cardiac troponin C with mutation of C35S; cTnI, cardiac troponin I; cTnT, cardiac troponin T; DMSO, dimethyl sulfoxide; DTT, dithiolthreitol; FRET, fluoresence resonance energy transfer; HSQC, heteronu- clear single quantum correlation ; kDa, kilodalton; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; NMR, nuclear magnetic resonance; NOESY, nuclear Overhauser enhancement spectroscopy; NTnC, N-domain of troponin C; PAGE, polyacrylamide gel electrophoresis; PDB, Protein Data Bank; PKA, protein kinase A; PKC, protein kinase C; RDC, residual dipolar coupling; RP, reversed-phase chromatography; SAXS, small-angle X-ray scattering; SAR, structure-to-activity relationship; SDS, sodium dodecyl sulfate ; SR, sarcoplasmic reticulum; sTnC, skeletal troponin C; sTnI, skeletal troponin I; TFP, trifluoperazine; Tm, tropomyosin; Tn, heterotrimeric troponin complex; TnC, troponin C; TnI, troponin I; TnT, troponin T Address for offprints: P. Pollesello, Orion Pharma, P.O.Box 65 FIN-02101 Espoo, Finland (E-mail: [email protected])
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Sorsa et al Mol Cell Biochem 2004 266 87

Molecular and Cellular Biochemistry 266: 87–107, 2004.c© 2004 Kluwer Academic Publishers. Printed in the Netherlands.
The contractile apparatus as a targetfor drugs against heart failure: Interactionof levosimendan, a calcium sensitiser,with cardiac troponin c
Tia Sorsa,1 Piero Pollesello1 and R. John Solaro2
1Orion Pharma, Cardiovascular Research, Espoo, Finland; 2University of Illinois, Chicago, Department of Physiology andBiophysics, Chicago, IL, U.S.A.
Received 1 March 2004; accepted 6 April 2004
Abstract
Cardiac failure is one of the leading causes of mortality in developed countries. As life expectancies of the populations ofthese countries grow, the number of patients suffering from cardiac insufficiency also increases. Effective treatments are beingsought and recently a new class of drugs, the calcium sensitisers, was developed. These drugs cause a positive inotropic effecton cardio-myocytes by interacting directly with the contractile apparatus. Their mechanism of action is not accompanied by anincrease in intracellular calcium concentration at therapeutic doses, as seen for the older generation of positive inotropic drugs,and thus does not induce calcium-related deleterious effects such as arrhythmias or apoptosis.
Levosimendan is a novel calcium sensitiser which has been discovered by using cardiac troponin C (cTnC) as target protein.This drug has been proved to be a well-tolerated and effective treatment for patients with severe decompensated heart failure.
This review describes the basic principles of muscle contraction, the main components of the contractile apparatus and theirroles in the heart contraction. The regulatory proteins troponin C (cTnC), troponin I (cTnI), troponin T (cTnT), and tropomyosin(Tm) and their interactions are discussed in details. The concept of calcium sensitisation is thereafter explained and a fewexamples of calcium sensitisers and their putative mechanisms are discussed. Finally, the binding of levosimendan to cTnC andits mechanism of action are described and the results discussed under the light of the action of this drug in vitro and in vivo.(Mol Cell Biochem 266: 87–107, 2004)
Key words: cardiac troponin C, calcium sensitisation, drug interaction, regulation of muscle contraction, nuclear magneticresonance, cardiovascular pharmacology
Abbreviations: ATPase, adenosine triphosphatase; BSA, bovine serum albumin; Bis-Tris, Bis(2-hydroxyethyl)-imino-tris(hydroxymethyl)methane; cCTnC, C-domain of cardiac troponin C; cNTnC, N-domain of cardiac troponin C; cTn, het-erotrimeric cardiac troponin complex; cTnC, cardiac troponin C; CTnC, C-domain of troponin C; cTnCA-Cys, cardiac troponinC with mutations of C35S and C84S; cTnCCS, cardiac troponin C with mutation of C35S; cTnI, cardiac troponin I; cTnT, cardiactroponin T; DMSO, dimethyl sulfoxide; DTT, dithiolthreitol; FRET, fluoresence resonance energy transfer; HSQC, heteronu-clear single quantum correlation ; kDa, kilodalton; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight;NMR, nuclear magnetic resonance; NOESY, nuclear Overhauser enhancement spectroscopy; NTnC, N-domain of troponin C;PAGE, polyacrylamide gel electrophoresis; PDB, Protein Data Bank; PKA, protein kinase A; PKC, protein kinase C; RDC,residual dipolar coupling; RP, reversed-phase chromatography; SAXS, small-angle X-ray scattering; SAR, structure-to-activityrelationship; SDS, sodium dodecyl sulfate ; SR, sarcoplasmic reticulum; sTnC, skeletal troponin C; sTnI, skeletal troponin I;TFP, trifluoperazine; Tm, tropomyosin; Tn, heterotrimeric troponin complex; TnC, troponin C; TnI, troponin I; TnT, troponin T
Address for offprints: P. Pollesello, Orion Pharma, P.O.Box 65 FIN-02101 Espoo, Finland (E-mail: [email protected])

88
Structure, function and regulationof the cardiac contractile apparatus
Assembly of the contractile apparatus
The striated muscle cell contains myofibrils formed by repeat-ing units of sarcomeres, arranged in series, which are in turn iscomposed of parallel filaments of two species, commonly re-ferred as thin and thick because of their appearance under themicroscope (Fig. 1 A). Both in skeletal and cardiac muscle thesarcomere structure and the general mechanism of the musclecontraction are the same. Under the control of intracellularcalcium level, thin and thick filaments of the sarcomere slidepast each other and the sarcomere length shortens resultingin muscle contraction. Calcium triggers and regulates the se-
Fig. 1. Sketch of a sarcomere, thick filament and a thin filament structure. (A) Sarcomer is composed of parallel thin and thick filaments between Z discs.When muscle contracts the filaments slide past each other and the length of the sarcomere shortens. Strong cross-bridges form between thin and thick filaments.(B) Myosin is composed of coiled-coil tail and globular heads (S1) that include the binding sites for actin and ATP. A bipolar myosin polymer shows S1 headssticking outward at regular intervals for actin interaction. (C) Tropomyosin (Tm) is in the groove of the actin formed helical core of a thin filament and inregular intervals troponin complex (Tn) is attached on the thin filament.
ries of complex, interlinked protein – protein interactionsthat eventually result in the formation of the force generatingstrong crossbridges between the two filaments.
The thick filament of the sarcomere is a polymer com-posed of myosin molecules (Fig. 1 B). Myosin consists oftwo heavy and four light chains. A bundle of myosin heavychain coiled-coil tails forms the backbone of the thick fil-ament. The globular heads of the heavy chain N-terminusnamed as subfragment 1 (S1) point out from the thick fila-ment at regular intervals. These myosin heads interact withthe thin filaments to form strong crossbridges which even-tually result in the contraction of the sarcomere [1]. Minorscaffolding components of the thick filaments, C-, X- andH-proteins, keep myosin molecules together [2, 3]. Ad-ditional proteins are the M-line protein that connects thethick filaments together at the center of the sarcomere, and

89
titin which contributes to the elasticity and stability of thesarcomere [4].
The thin filament has a two-stranded helical structure(Fig. 1 C). The backbone of the thin filament is composedof polymerized globular actin monomers (G-actin). Actinmonomers consist of two equal-sized domains which areavailable for myosin interaction or interact with the corre-sponding sub-domains of the adjacent strand [5, 6]. On theactin scaffold, a series of regulatory proteins modulate theinteraction between the thick and thin filaments. The mostelongated of them, tropomyosin (Tm), is located in a grooveof the helical actin filament called F-actin (Fig. 1 C) [7]. Itis a long (∼40 nm) and flexible, largely α-helical coiled-coil dimer, which overlaps the neighboring tropomyosins ina head-to-tail configuration [8]. Each Tm spans over sevenactin monomers of the thin filament [9]. The overlapping re-gions of adjacent tropomyosins are mainly responsible forthe affinity of Tm for actin [8]. It binds to the actin filamentby electrostatic interaction [6, 10]. However, Tm on the thinfilament is not fixed in one position. It rolls over the surface ofthe thin filament depending on the phase of the contractioncycle. This movement is influenced by Ca2+ and it affectsmyosin S1 binding to actin (recent reviews by Gordon et al.[11, 12]).
Every tropomyosin is spatially coupled to a troponin com-plex (Tn), and the two form the calcium-dependent trigger ofthe contractile apparatus, i.e. a molecular switch on the thinfilament. One Tn complex interacts with one Tm moleculeat regular intervals and regulates the interaction of 12 to14 actin monomers with myosin [9, 13–15]. Tn is a het-erotrimeric protein assembly. It consists of a Tm binding unittroponin T (TnT), an actomyosin ATPase inhibitory unit tro-ponin I (TnI), and a calcium-binding unit troponin C (TnC)[16–18].
TnT is an asymmetric protein that attaches the Tn complexto a defined position on the thin filament. It is needed for full,Ca2+-dependent activity of the thin filament. TnT consists oftwo domains, an extended N-terminal domain T1 (residues1–158) and a globular T2 domain (residues 159–259). Theextended T1 domain interacts strongly with Tm [19]. It issuggested that this interaction mediates the activation of ac-tomyosin ATPase by stabilising the inactive state [20, 21].The C-terminus of T2 interacts with the N-terminus of TnI,CTnC, and the thin filament thus maintaining the cohesionof the complex [22–26]. The interaction of TnT with Tm iscalcium sensitive. Consequently, in the absence of calciumthe T2 region binds to Tm and blocks the ATPase activa-tion whereas in the presence of calcium the interaction withTnI/TnC is modified and it is detached from Tm [20, 27–30].
Calcium-binding to TnC initiates the cascade of eventsleading to muscle contraction. The interaction between TnCand TnI is essential for further transmission of the contractionsignal to the other components of the thin filament. Therefore,
their structures, roles and interactions are discussed in moredetail later in the text.
The skeletal and cardiac muscles differ in the thin filamentactivation properties, which is seen as a variation in thebalance between different conformational states of theregulatory proteins expressed in skeletal and cardiac muscle[31]. These tissue specific differences are explained bythe different physiological requirements.
Regulation of muscle contraction
Cardiac muscle contraction is defined as involuntary butskeletal muscle contracts voluntarily. In both cases the cal-cium ion initiates the contraction. The complex mechanismof calcium-directed regulation of striated muscle contractionhas been studied extensively over the years (for reviews seeLeavis and Gergely [32], Zot and Potter [33], da Silva andReinach [34], Grabarek et al. [35], Gergely et al. [36], Farahand Reinach [37], Tobacman [25], Lehrer and Geeves [38],Squire and Morris [39], Gordon et al. [11, 12]. The focus ofthis review is on the contraction of cardiac muscle, but oftena comparison between the tissues is needed because data arenot currently available about some of the events in cardiacmuscle contraction.
Several models for the regulation of the muscle contractionhave been proposed in the literature. Such models are basedon structural, biochemical and physiological data (for reviewsee Gordon et al. [11]). A three state model has been sug-gested by McKillop and Geeves [40]. In their model, aboutthree Tm positions on the actin filament and two steps ofmyosin binding to actin have been proposed. This agreeswell with current structural and biochemical data and hasbeen generally accepted as the model to describe the regula-tion of the thin filament activation [40–44]. The three statesof the thin filament are named blocked (B), closed (C) andopen (M). The two steps of myosin binding to actin initiallyinvolve a weak binding followed by a stronger binding. In theblocked state no myosin binding to actin occurs. In the closedstate, there is a weak interaction between myosin and actinthat becomes stronger binding in the open-force- generatingstate.
The regulation of the contraction cycle of both cardiac andskeletal muscles is calcium dependent (Fig. 2) [45]. Calciumrelease from the sarcoplasmic reticulum (SR) triggers a cas-cade of events that include changes in protein–protein interac-tions and structural changes in proteins leading to muscle con-traction (for recent reviews see Leavis and Gergely [32], Oht-suki et al. [17], Zot and Potter [33], Farah and Reinach .[37],and Solaro and Rarick [46]). In the resting muscle, the freeintracellular calcium concentration is low ([Ca2+]i ∼10−7
M) and TnI inhibits the ATPase activity of actomyosin bybinding with actin/Tm [47–49]. Tm occupies a position onthe thin filament where it is able to block (B) the interaction

90
Fig. 2. Calcium-dependent regulation of cardiac muscle contraction. In the relaxed state (diastole), there is no binding between actin and myosin. This is theblocked state (B). On calcium binding to cTnC, the inhibitory region of cTnI switches from actin to cTnC. This leads to the closed state (C) with weak bindingbetween actin and myosin. Tm is, in turn, moved further to the open state (M) allowing stronger interaction between actin and myosin and muscle contracts(systole). Black spheres indicate binding.
between actin and myosin hence the cross-bridge formationcan be inhibited. When calcium is released from the SR tocytosol ([Ca2+]i ∼10−5 M), it binds to TnC enabling TnI-binding. The ATPase inhibition ceases when the inhibitoryregion of TnI switches from actin to calcium-saturated TnC[50]. Moreover, Tm moves on the thin filament from blocked(B) to the closed state (C) [44, 51]. This movement allowsweak interactions between actin and myosin. Myosin andTm binding to actin induce a conformational change on actin[52, 53]. After which the connection between actin/Tm andTn breaks and Tm moves further to the open state (M) onthe surface of the thin filament [51]. Thus, myosin bindingsites are exposed to strong binding, and muscle contractionoccurs.
Troponin C
Striated-muscle troponin C is expressed in two isoforms invertebrates, in fast skeletal muscle skeletal troponin C (sTnC)and in slow skeletal and cardiac muscles cardiac troponin C(cTnC). Skeletal and cardiac TnC sequences are about 70% identical [54, 55]. The most significant sequential differ-ence between skeletal and cardiac TnC isomers is the inactivecalcium-binding site of the N-domain of cTnC. Mutations ofD29L and D31A and the insertion of V28 in the cardiac se-quence make the calcium-binding site I incapable of bindingcalcium [56].
Structure of troponin C
TnC is an 18 kDa acidic protein. High-resolution X-raydiffraction and nuclear magnetic resonance spectroscopy(NMR) structures of both skeletal [57–63] and cardiac TnC[64] show an elongated, largely α-helical, dumbbell-shaped
protein (Fig. 3). Both isomers consist of globular N- andC-domains that are connected by a linker helix, often re-ferred to as D/E linker. An N-terminal helix (N-helix) to-gether with A, B, C, and D helices compose the regulatoryor the N-domain of TnC (NTnC) and helices E, F, G, and Hform the structural or the C-domain (CTnC). The domains ofthe isolated TnC appear to be structurally and functionallyindependent from each other. The central D/E linker of iso-lated TnC between the domains is flexible and allows N- andC-domains to move independently of each other [65]. Thereappears to be a pH-dependent transition of the isolated TnC[66, 67]. The crystal structure determined at pH ∼5 revealsan elongated molecule with an extended D/E linker helix.Whereas closer to the physiological pH (pH ∼7), isolatedcTnC assumes a slightly more compact structure and the N-and C-domains are closer to each other [67]. Apart from thepH dependent transition, the differences in linker helix crystaland solution structures can also result from crystal packingforces that stabilise the more extended structure as is the casewith calmodulin [68]. The function of the linker is to keep thedomains apart though in optimal proximity to their target siteson troponin I [69–71]. TnI restricts the linker flexibility andthe positions of the domains are set so that the hydrophobicregions of each domain face each other [72].
Troponin C as calcium-binding protein
The calcium ion is a common factor in various cellularmechanisms and therefore there are numerous proteins thathave the ability to bind calcium. For example, Ca2+-bindingproteins function in cellular signal transmission, cytosolicCa2+ buffering, and also take part in Ca2+-dependent en-zymatic activities. Many of these calcium-binding proteinscontain an EF-hand calcium-binding motif(s) first described

91
Fig. 3. Ribbon presentation of calcium-saturated cTnC (Protein Data Bank entry 1AJ4 [64], www.rcsb.org/pdb). (A) Below the 3D-structure is shown thesecondary structure of cTnC. (B) Diagram showing cTnC-binding regions on cTnI sequence (Swiss-Prot ID P19429). Residues 1–32, the cardiac specificextension of cTnI contains two phosphorylation sites, S22 and S23, and has been proposed to interact with the N-domain of cTnC. Residues 33–70 interactwith the C-domain of cTnC anchoring TnC and TnI together. The inhibitory region, residues 128–147, interact with actin and cTnC according to the phase ofthe contraction cycle and is modulated by the calcium-dependent binding of the regulatory region cTnI148–166.
by Kretsinger and Nockolds [73]. In the EF-hand motif, twohelices, which are almost perpendicular to each other, areconnected by a loop comprising 12 amino acids. Certainside chains of the loop residues provide oxygen ligands forCa2+ chelation. calcium-binding to the EF-hand motif, whichis reversible, often results in significant structural changes.EF-hand motifs are usually arranged in pairs interacting viashort β-strands in the loops.
TnC belongs to the calmodulin group of EF-hand calcium-binding proteins. It functions as a calcium-dependent triggerfor the control of muscle contraction [74–76]. Binding of cal-cium ions to the regulatory domain of TnC alters the interac-tion between TnC and TnI and other thin filament proteins.Thus, the contraction signal is further transmitted to othersubunits of the contractile apparatus.
There are two helix–loop–helix EF-motifs in the regulatorydomain of TnC. The two calcium-binding sites of sNTnC and
one active site in cNTnC have a specific affinity for calcium(KCa 2×105 M−1) [74, 77]. These N-domain sites are referredto as regulatory sites because calcium-binding to them trig-gers the muscle contraction [74, 78, 79]. The paired calcium-binding sites of the N-domain are structurally and energet-ically coupled in the both TnC isoforms [80–82]. Thereare hydrogen bonds and van der Waals forces between theβ-strands of the EF-hands [59, 60, 83]. Calcium-binding tocNTnC causes perturbations throughout the N-domain se-quence, primarily in the defunct calcium-binding site I andin the active binding site II [81]. Calcium-binding to site II incTnC decreases the flexibility of the backbone and also thedefunct calcium-binding loop becomes more rigid [82].
In the C-domain of TnC, there are also two EF-hand motifcalcium-binding sites. These sites have a higher affinity tocalcium than the sites on the N-domain of TnC, KCa about2 × 107 M−1 and also a measurable affinity for magnesium

92
ions, KMg ∼5×103 M−1 [74, 84, 85]. The structure of the C-domain appears to be similar regardless of the bound metalion [86]. Ca2+/Mg2+-binding to the C-domain of TnC re-sults in two hydrogen bonds between the calcium-bindingloops and the conformation becomes more structured com-pared to the apo-form [87, 88]. It is generally believed thatthese C-domain sites are always occupied with either Mg2+orCa2+ under normal physiological conditions [35, 77]. There-fore, they are referred to as structural calcium-binding sites.In fact, calcium/magnesium binding to either site III or IVof TnC is enough for anchoring TnC to the thin filament[89–91].
Troponin C as trigger of muscle contraction
Binding of the calcium ion to TnC initiates the muscle con-traction process. Specifically, calcium ion binding to sites Iand II of sTnC and site II of the cTnC triggers the musclecontraction signal [91, 92]. The signal is further transmittedto other subunits of the contractile apparatus by structuralchanges and alterations in protein – protein interactions [93].The activation step induced by calcium-binding differs be-tween skeletal and cardiac isoforms. In the apo-form, bothisomers are energetically in the lowest state and the confor-mation of the N-domain of cTnC and sTnC stays closed. Thismeans that helices B and C are closer to the unit formed ofhelices N, A, and D (NAD), due to hydrophobic interactions[64, 94].
When the regulatory sites of sTnC are occupied by cal-cium ions, the domain structure is open and exposes a largehydrophobic surface [57, 83, 95–97]. Studies with mutatedsTnC, E41A-sTnC, which is incapable of binding calciumto site I, revealed that this site is the link between thecalcium-binding and the opening of the regulatory domain[98]. The binding of a second calcium ion releases sufficientenergy to overcome the energy barrier to open the structureof the sNTnC [80]. Helices B and C move relative to theNAD unit exposing a hydrophobic region where TnI binds[83, 97, 99, 100].
In the apo-form, cNTnC is more structured than its skele-tal counterpart. Short β-strands in calcium-binding loops areformed in the absence of calcium in cNTnC [101]. The activecalcium-binding site II in cNTnC alone is responsible for trig-gering cardiac muscle contraction [85, 102, 103]. Similarly,the binding of the Ca2+ to site II of sTnC does not induce largeconformational changes and the regulatory domain primarilystays closed [64, 94, 104]. It was proposed that in the equilib-rium between open-and closed-states of the calcium saturatedcNTnC the closed conformation dominates, which results inan average structure that resembles the closed conformationstate [104]. A similar state of open-and closed-forms in equi-librium has been demonstrated more clearly for a mutant(E104Q/E140Q) in the C-domain of calmodulin [105].
Despite the cardiac calcium-binding site I being inactive,its first 41 residues including the defunct site and the first N-terminal helix, are considered to be important for the normalfunction of cTnC. Calcium-binding primes the N-domain forcTnI binding even though not enough energy is released toopen the structure. The defunct site modifies the calcium-dependent contraction [103]. It forms a conformationally ac-tive region which modulates the overall conformational en-tropy and hence the populations of closed and open-states ofcNTnC [106]. The N-helix stabilises the N-domain of TnCand is involved in the fine-tuning of the calcium-binding affin-ity and the transmission of the signal in the activation of thethin filament [107–111].
Myocytes are sensitive to changes in pH. For example,a pH decrease in ischemic heart leads to a decrease in thecontraction force [112]. This results from protons competingwith calcium ions for binding to cTnC, which in turn reducesthe positive inotropic effect caused by calcium [113–115].Protons are also reported to alter TnC/TnI interactions thattransmit the calcium-binding signal [116].
Troponin I
TnI, the inhibitory subunit of the Tn complex, is responsi-ble for calcium-dependent inhibition of actomyosin ATPase[32, 93]. TnI is expressed in three isoforms in striated mus-cle: two isoforms are expressed in skeletal muscle and oneisoform in cardiac muscle [117]. It is a polar protein with alarge number of positively charged residues. TnI transmitsthe calcium-binding information to other units of the thin fil-ament by a large-scale conformational change and calcium-dependent interactions with other thin filament proteins [46,118, 119].
At present, the structure of TnI in isolation is poorly de-fined. It is proposed to have an open and extended confor-mation based on solvent accessibility [120]. However, low-and high-resolution structures of TnI with other componentsof the Tn complex are determined. Structural information onthe TnC/TnI complex based on SAXS, X-ray crystallographyand neutron scattering results are published in several arti-cles [119, 121–124]. In the absence of regulatory calcium,the TnC/TnI complex is rigid but becomes more flexible oncalcium-binding to the N-domain regulatory sites [125]. TnCremains in its extended dumbbell conformation even when itis complexed with TnI [65, 126]. The presence of TnI makesthe flexible linker of TnC more rigid and limits the indepen-dent tumbling of the N- and C-domains [127]. A recent studyof the cTnC/cTnI/cTnT198–298 complex showed that cTnC do-mains are closer to each other within the Tn complex thanin the crystal structure. However, they do not make physicalcontact with each other [128]. In contrast, the cTnI/cTnT inthis complex remains in a long, extended form.

93
Recently, crystal structures of the core domains of cardiacTn, cTnC1–161/cTnI31–163/cTnT183–288 and cTnC1–161/c TnI31–210/cTnT183–288, were published by Takeda et al. [129]. Theregions of cTnI, unbound to either cTnC or cTnT, are not welldefined or appear variable among the determined structures.This indicates flexibility in those parts of the polypeptidechain. The flexible nature of the troponin complex is sug-gested to be relevant to its physiological function [129].
The interactions between troponin C and troponin I
TnI makes several points of contact with TnC (Fig. 3). Theyinteract with each other in an antiparallel fashion, the N-terminus of TnI binds to the C-domain of TnC and vice versa[28, 130, 131]. The sequence of cTnI32–70 (sTnI1–40) bindsat the hydrophobic patch of cCTnC in a Ca2+/Mg2+ depen-dent manner and stays bound throughout the entire musclecontraction cycle [28, 86, 123, 131–137]. This interaction isresponsible for keeping the TnC/TnI complex together duringthe relaxed state. The N-terminal region of TnI also includesa TnT-binding region (residues 54–79), which is proposed tobe involved in the transmission of the Ca2+-binding signal tothe thin filament [129, 138].
The regulatory region of cTnI148–166 (sTnI116–131) interactswith the N-domain of TnC in a calcium-dependent manner.It modulates the interaction between the inhibitory regionof TnI, TnC, and actin filament during the contraction–relaxation cycle [99, 135, 139–142]. The regulatory regionof cTnI binds to cNTnC in the presence of calcium andpulls the inhibitory region away from the actin filament [46,143]. In the resting phase, the interaction between TnI andactin/Tm is stronger than between TnI and TnC. However,on calcium-binding to TnC, the interaction between TnI andTnC becomes stronger whereas the TnI-binding to actin/Tmbecomes weaker. The TnI oscilliates back and forth betweenthese two states in synchronisation with the contraction–relaxation cycle [93]. The binding of calcium to the skele-tal TnC is enough to induce the opening of the N-domain.Whereas, before the full N-domain opening can occur in thecardiac isoform, the binding of residues 150–166 of cTnI isrequired [141, 142]. This tissue-specific difference betweencardiac and skeletal thin filament activation is explained bydifferent thermodynamics and kinetics of the isoforms [31,144]. The difference could possibly be related to their adap-tation to different physiological requirements, i.e. control ofaction, the involuntary and voluntary action of contraction,and the speed of action.
The inhibitory region cTnI128–147 (sTnC96–115) switches be-tween actin and cTnC according to the phase of the musclecontraction and the relaxation cycle [28, 37, 145]. This regionalone is enough for the full inhibition of actomyosin ATPase[146]. In the absence of calcium, the inhibitory region of
TnI is bound to actin/Tm where it inhibits the ATPase activ-ity [16, 47]. The inhibition is removed on calcium-bindingwhen the inhibitory region is pulled away from actin [46, 50,147]. The exact binding site of the inhibitory peptide on TnChas been under debate. The binding site for the inhibitoryregion of cTnI has been proposed to be in the N-domain[99, 140, 148], in the C-domain [100, 139, 149–151], in bothdomains [28], or in the linker region between the domains[124, 142, 152, 153].
The controversy about the binding site for the inhibitory re-gion on TnC probably results from studies with short peptidesthat may show nonspecific binding. Furthermore, various iso-forms and in different lengths of peptides have been usedfor studying the role of various peptides in binding studies.Controversies may also result from different methods used.However, recent publications agree that the inhibitory regionof TnI binds to the C-domain of TnC [129, 154–157]. Inthese studies, the N-terminal portion of cTnI127–147 has beenthought to associate with the C-domain of cTnC, and theC-terminal part of the inhibitory region bridges the N- andC-domains of cTnC but do not have specific interaction withthe D/E linker [155, 156]. Moreover, the residues 131–133 ofTnI have been reported to bind to the hydrophobic pocket onthe C-domain [157]. It has also been shown that the inhibitoryregion could not replace cTnI33–80 in the C-domain of cTnC[152]. It was emphasised that cTnI33–80 has a structural roleand stays bound to the C-domain whereas cTnI128–147 has amore dynamic functional role [158].
On the basis of NMR and fluoresence resonance energytransfer (FRET) studies of the cardiac cTnC/cTnI complexa different structure of the inhibitory region to that of theskeletal isoform has been suggested [143, 156, 159]. Theseauthors suggest that the skeletal hair–pin loop does not exist inthe cardiac complex. Instead, they suggest that the inhibitoryregion is in an extended conformation in the presence of reg-ulatory calcium. The extended structure of cTnI138–148 of theinhibitory region was confirmed in the crystal structure of thecore complex [129].
The cardiac isoform of TnI has an N-terminal extensionof 32 residues, cTnI1–32 that interacts with the regulatory do-main of cTnC [160, 161]. Its function in the myofilament isnot entirely clear. The cardiac specific extension is highlyflexible in the presence of cTnC suggesting that it is eithernot bound to cTnC or binds only loosely [120]. It has alsobeen suggested that this region switches between the N- andC-domains of cTnC depending on the phosphorylation state[135]. It could therefore mediate signals from TnC domainsto each other. The unphosphorylated form interacts with theN-domain of cTnC and modulates the equilibrium betweenopen-and closed-states of Ca2+-cNTnC [127, 159–162]. Bis-phosporylation of cTnI disrupts this interaction and reducesthe calcium affinity of cTnC and thus the calcium sensitivityof the contraction [127, 159, 163, 164].

94
Fig. 4. Energy diagram for sTnC and cTnC in different states of the contraction cycle. On calcium binding to cNTnC an equilibrium between open and closedconformation exists in the cardiac isoform whereas the skeletal isoform is open showing no such conformational equilibrium [31, 165].
Calcium and cardiac troponin I binding induced changeson the regulatory domain of cardiac troponin C
In skeletal muscle, calcium-binding to sNTnC opens the do-main structure for sTnI binding, which eventually leads tomuscle contraction [36, 61, 97]. The activation mechanismof cardiac muscle differs from skeletal muscle. Calcium-binding to TnC is indeed the initiator in both muscle typesbut calcium-induced structural changes and energetics of theactivation differ. In a recent paper Paakkonen et al. [165]studied the activation sequence of cTnC on calcium and cTnIbinding by measuring NH residual dipolar couplings (RDCs).For this experiment, a dilute liquid crystalline medium isformed of filamentous Pf1 phages wherein protein moleculesaligned partially [166], which allows to measure RDCs andget angular information about structural motifs, especially α-helices and β-sheets [167–169]. Paakkonen et al. measuredthe NH RDCs in samples of uniformly 15N-labeled cNTnC inapo- and calcium-saturated forms and also in complex withcTnI. The experimental values were compared with RDC-values calculated from the respective PDB-structures. Themeasured values for NH RDCs correlated well with the cal-culated values of corresponding structures of TnC. In theapo-form (PDB entry 1SPY), the N-domain was closed andin the presence of calcium and cTnI (PDB entry 1MXL)the domain structure was fully open. However, the calciumform of cNTnC gave a poor quality factor and correlationbetween measured and calculated RDC-values. This couldbe explained by the conformational isomerism present in
the Ca2+-form of cTnC between open and closed confor-mations [104]. It has been shown that the closed state isenergetically more favored than the open state and there-fore it dominates [81, 142] (Fig. 4). calcium-binding primesthe N-domain for cTnI binding which brings in energy thatis sufficient to overcome the energy barrier and fully opensthe N-domain of cTnC [142]. In this open conformation, theskeletal and cardiac isoform structures closely resemble eachother. The populations of open and closed states in the liquidcrystalline phase are altered when compared to those of theliquid phase. The difference in populations can explain thepoor correlation between measured and calculated values forCa2+-cNTnC [165].
Phosphorylation of troponin I
The signaling process of myofilament activation can be mod-ulated not only with free intracellular [Ca2+] but also byphosphorylation of cTnI. This is characteristic of the car-diac isoform. Phosphorylation is known to affect cardiac con-tractility and different pathologies are known to affect cTnIphosphorylation. There are multiple protein kinase A (PKA)and protein kinase C (PKC) phosphorylation sites on cTnI.Ser22 and Ser23 can be phosphorylated by PKA in responseto β-adrenergic stimulation [170]. Phosphorylation of thesesites results in structural changes and affects their interactionswith TnC [164, 171–173]. The bisphosphorylated cardiacspecific extension interacts with cTnT and/or cTnI [174].Phosphorylation of Ser22 and Ser23 reduces the apparent

95
calcium sensitivity [163]. Ser42 and Ser44 in the part of cTnIthat binds to cCTnC and Thr142 in the inhibitory regionare all substrates for PKC [175]. Phosphorylation by PKCdecreases actomyosin ATPase activity. Phosphorylation ofSer42 and Ser44 influences the maximum tension of my-ofilaments whereas phosphorylation of Thr142 together withphosphorylation of Ser42 and Ser44 appears to regulate thinfilament sliding speed [176]. Recently it was reported thatthe phosphorylation of Thr142 of cardiac cTnI reduces theaffinity of the inhibitory peptide for calcium saturated cCTnC[177].
Calcium sensitisers
The binding of calcium ion to the N-domain of TnC initi-ates the contraction of the cardiac muscle and the impulse isfurther transmitted to other subunits of the contractile appa-ratus via TnC/TnI interaction. Therefore, the manipulation ofthis signaling pathway appears to be an effective and specifictreatment strategy for cardiac insufficiency that results fromdecreased ventricular function such as heart failure. Heartmuscle contraction can be enhanced by an increase in theintracellular calcium concentration. However, high levels ofintracellular calcium result in higher demands for oxygen.More energy in fact is needed for the handling of an increasedamount of calcium in the sarcoplasmic reticulum at every re-laxation. This further exacerbates the energy balance of analready weak muscle. In addition, arrhythmias may also resultfrom elevated intracellular calcium concentrations, which cansubsequently lead to death [178, 179]. An alternative idea wasintroduced over 20 years ago: calcium sensitiser [180, 181].Calcium sensitisers are a heterogeneous group of moleculesthat enhance the response of myofilaments to calcium with-out an increase in the intracellular calcium concentration andthus have a positive inotropic effect on cardiac contractility(recent reviews by Lee and Allen [182], Endoh [183, 184],and Arteaga et al. [185]).
A heart contracts when calcium binds to cNTnC. Con-versely, it relaxes when calcium dissociates from cNTnC.This feature makes the calcium saturated form of cTnC anideal target for calcium sensitisers [186]. Therefore, an addi-tional important feature of calcium sensitisers in order not toimpair the relaxation of cardiac muscle is that it dissociatesfrom cTnC when Ca2+ dissociates. It is essential that an ef-ficacious calcium sensitiser must not prohibit or inhibit anyprotein–protein interactions required for muscle contractionand relaxation. Rational drug design and understanding of theeffective mechanism of an efficacious drug candidate requiresknowledge of the relationship between structure and function(structure to activity relationship, SAR). SAR gives informa-tion about protein molecular structure and how it changes dur-ing the contraction–relaxation cycle. Moreover, it provides
data on how potential drugs affect the structure and interactwith other proteins in the contractile apparatus. Results fromSAR studies have been used to evaluate some potential candi-date calcium sensitising molecules including EMD57033, tri-fluoperazine (TFP), bepridil, and levosimendan. Before anyaccurate structural data from NMR or X-ray studies of cTnCwere available, the sTnC isoform was used as a template formodeling of drug-protein complexes. The binding site for hy-drophobic calcium sensitising compounds was thought to belocated on the hydrophobic patch that includes helices B, C,and D in the regulatory domain of TnC [187, 188].
The cTnC N-domain pathway
One of the possible mechanisms to enhance the apparentcalcium sensitivity of cardiac muscle involves the regula-tory domain of cTnC. Enhanced calcium sensitivity couldbe achieved by stabilising the calcium-dependent interactionbetween cNTnC and the regulatory region of cTnI, or byincreasing the calcium affinity of cNTnC. However, an in-crease in calcium-binding affinity would result in impairedrelaxation because Ca2+ would dissociate relatively slowlyfrom cNTnC (for review see Teramura and Yamakado [189]).A better alternative would be to modulate cTnC/cTnI inter-action prolonging the lifetime of the active form. Both do-mains of TnC have hydrophobic regions that are essentialfor cTnC/cTnI interaction; stabilisation of the complex andregulation of cardiac contraction. The hydrophobic region ofcTnC that includes Met81 is a binding site for cTnI and istherefore essential to the activity. This is not considered asan ideal binding site for calcium sensitisers [190]. Insteadit was suggested that another hydrophobic region of the N-domain including Met45, Met60, and Met80 of the regula-tory site II was a potential binding site for calcium sensi-tising ligands [188]. A pharmacological agent that binds tothis patch and does not interfere with the binding of cTnIcould enhance the apparent calcium sensitivity of the my-ofilaments. Agents that have been reported to have an effectby interacting with cTnC at this region are bepridil and TFP[188].
Bepridil (racemate of 1-isobutoxy-2-pyrrolidino-3-(N-benzylanilino) propane from Sigma, Fig. 5 A) has often beenused as a model molecule to study the mechanism of calciumsensitising of myofibrils even though it has been shown to beunsuitable for clinical use [191, 192]. Bepridil increases acto-myosin ATPase activity and affinity of Ca2+ for cTnC [193].Its calcium sensitising effect is thought to be derived from itscalcium-dependent binding to the regulatory domain of cTnCin the presence of cTnI and the stabilisation of the open con-formation [106, 194, 195]. The complex structure shows thatbepridil binding to cNTnC sterically hinders the closing ofthe conformation and enhances the affinity of TnC for cTnIand thus increases the activity of ATPase [195, 196]. Bepridil

96
Fig. 5. (A) Chemical structure of the bepridil molecule and a schematic presentation of a 3D-structure of the complex cNTnC/cTnI147–163 in the presence ofbepridil (PDB entry 1LXF) [196]. (B) Chemical structure of the EMD57033 molecule and a schematic presentation of a 3D-structure of cCTnC in the presenceof EMD57033 (PDB entry 1IH0) [202]. The asterisk marks the chiral carbon atom. The blue helix represents cTnI147–163.
binding to the N-domain of cTnC causes the N-terminus ofthe B helix to extend by forming a side-chain interaction be-tween Glu40 and Ser37 and thus stabilises the defunct site Iand enables the domain opening [195].
The cTnC C-domain pathway
A hydrophobic patch is located in the C-domain of cTnC.This is also reported to be the binding site for cTnI [131].Unlike the N-domain it is not considered to be a potentialbinding site for calcium sensitisers due to its structuralrole [188]. However, recently a modulatory role for theC-domain in thin filament activation has been proposed
[134]. EMD57033 (the R enantiomer of 5-[1-(3,4-dimethoxybenzoyl)-1,2,3,4-tetrahydro-6-quinolyl]-6-methyl-3,6-dihydro-2H-1,3,4-thiadiazin-2-one developed by Phar-maceutical Research, E. Merck, Darmstadt, Germany, Fig.5 B) has been reported to increase calcium sensitivityand force development in cardiac muscle [197–199]. Itinteracts stereoselectively and calcium-dependently withthe C-domain of cTnC in the absence or presence of cTnI[200–202]. These authors suggested that EMD57033 bindsto the structural domain of cTnC thus altering the interactionbetween cTnC and other thin filament proteins, especiallycTnI, thereby enhancing the apparent calcium sensitivity ofthe myocytes.

97
Levosimendan
Levosimendan (the R enantiomer of [4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl) phenyl] hydrazono} propan-edinitrile from Orion Pharma, Espoo, Finland, Fig. 6) is aninodilator designed for the treatment of acute cardiac fail-ure. It has been shown to act as a calcium sensitiser indemembranated cardiomyocytes [203], isolated intact car-diomyocytes [204], guinea pig cardiac muscle skinned fibers[205, 206], and in human heart failure skinned fibers [207].The positive inotropic effect of levosimendan is a result ofan increase in calcium sensitivity of the contractile proteinsand not an increase of the calcium influx at concentrationscomparable to the free plasma concentration reached in pa-tients during levosimendan treatment [203, 207]. Levosimen-dan induced tension increase is concentration-dependent andreversible [206, 208]. Levosimendan is accepted in clinicaluse as a calcium sensitiser in congestive heart failure and ithas been reported to be a specific and well-tolerated treatment[209].
Levosimendan has a calcium-dependent affinity for the car-diac Tn complex as well as for isolated cTnC. It is able tobind to both domains of cTnC [205]. Thus it has been sug-gested that the calcium sensitising effect of levosimendan ismediated by its calcium-dependent binding to cTnC [210].
The binding site for levosimendan on cTnC was hypoth-esised on the basis of the sTnC structure to locate at thehydrophobic pocket of the N-domain of cTnC near the linkerregion [187]. Preliminary NMR studies on the isolated N-terminal domain of cTnC revealed a spatial proximity of lev-osimendan and Met81, Met85 and Phe77 in the drug–proteincomplex [211]. Moreover, experimental data of several pointmutated cTnC samples support this hypothesis. The mutationof residues Cys84, Asp87, or Asp88 in cTnC has no effect onTnC’s calcium-binding properties but have an effect on thecalcium sensitisation of myocytes in the presence, or absenceof levosimendan [210]. However, conflicting results aboutthe affinity of levosimendan to cTnC have been presented.Kleerekoper and Putkey [212] studied several potentialcalcium sensitising agents and their affinities to cTnC. Theywere not able to detect any evidence of reversible levosimen-dan binding to calcium saturated cTnC and suggested the cal-
Fig. 6. Chemical structure of the levosimendan molecule. The asterisk marksthe chiral carbon atom on the levosimendan molecule.
cium sensitising action of levosimendan was derived from theinteraction with some other target protein than cTnC [212].
Binding of levosimendan to cardiactroponin C
Binding of levosimendan to isolated cardiac troponin C
The interaction of levosimendan with the isolated cTnCCS
was recently studied by following chemical shift changes inthe 1H-15N-HSQC spectrum on addition of the drug [213].This method is fast, has a high sensitivity and has been widelyused for detecting ligand—protein interactions. The majordrawback of the method is that it detects changes in thebackbone of the protein and most of the side chains can-not be followed. Based on chemical shift changes in the1H-15N-HSQC spectrum, interaction between levosimendanand cTnC was observed on isolated (Ca2+)3-cTnCCS (Fig. 7).It was known that levosimendan interacts with both domainsof the isolated cTnC as was previously shown by Haikalaet al. [205].
In the recent NMR study it was shown that levosimendaninduces chemical shift changes in a slow exchange process(kex < 50s−1) in the N-domain. In addition, small chemicalshift perturbations in fast time scale exchange are also in-duced by levosimendan in the same domain. There are twoobservable signals for many of the residues all over the reg-ulatory domain. The affinity of levosimendan to cNTnC, Kd
being about 10 µM, was estimated from the change in the in-tegrated volume of disappearing peaks and newly appearingpeaks along the levosimendan titration curve. No conclusionsregarding levosimendan binding site(s) in the N-domain werepossible. However, by comparing levosimendan-binding re-sults from various samples, an estimate for the binding regioncould be made. There were some clear differences betweenthe behavior of levosimendan binding to various cTnC sam-ples that pointed to a hypothesised binding site.
First, isolated Ca2+-cNTnC results did not reveal any evi-dence of a slow timescale conformational change caused bylevosimendan [213]. The second observation was that no slowtimescale exchange of the binding of levosimendan to the N-domain of the full-length (Ca2+)3-cTnCA-Cys occurred thoughthe C-domain interaction was clearly observable [213]. How-ever, the full-length (Ca2+)3-cTnCCS unambiguously showeda levosimendan interaction with the N-domain which oc-curred simultaneous with the C-domain binding [213, 214].It was clear from these results that levosimendan also bindsto the N-domain of cTnC. The binding region most probablylocated at the hydrophobic region of cTnC close to the D/Elinker. This has been modelled to be the interaction site forhydrophobic molecules [187]. Similarly, the prerequisite forfull-length cTnC including Cys84 for the interaction between

98
Fig. 7. Levosimendan induced chemical shift changes of cTnCCS followed by 1H-15 N HSQC spectrum at 800 MHz. Chemical shift changes larger than 4Hz (black line) and all the residues showing a slow exchange process are plotted on a schematic 3D structure of cTnC in red (PDB entry 1AJ4). Chemicalshift changes are also shown as a function of amino acid sequence. (A) Chemical shift perturbations in the fast exchange. (B) Chemical shift changes of slow
exchange timescale resulting in two sets of resonances. Chemical shift changes are shown as a distance in Hz from the original peak, �ν =√
�ν2H + �ν2
N .
levosimendan and the regulatory domain of cTnC to resultin the structural change as was also reported by Levijokiet al. [210]. Therefore, Cys84 is either essential for levosi-mendan binding, or its mutation to serine slightly alters theconformation of the binding region wherafter levosimendanbinding is no longer possible. This suggests that the bind-ing takes place close to the flexible linker. Consequently,small differences in this region due to the missing C-domainwere able to prevent levosimendan from binding to its targetprotein.
In a recent study, Sorsa et al. [213] used the small angleX-ray scattering technique to follow changes in the overallshape of cTnCCS on addition of levosimendan. Such methodallows to determine the distribution of distances within a pro-tein molecule. The data show that, despite a small change intothe overall protein shape induced by levosimendan, calcium-saturated cTnCCS remains in its elongated form in the pres-ence of the drug (Fig. 8). Levosimendan binding does notbring the domains in closer spatial proximity to each other ashas been shown to be the case for bepridil [195]. Interestingly,levosimendan binding increases the radius of gyration from20.2±0.5 to 21.7±0.6 A and slightly longer distances appearin the P(r) function in the presence of levosimendan. Sim-ilarly, a slight increase in the mean distance was observed
when cTnI binding to cTnC reduced the linker regionflexibility [141]. Thus these observations support the hy-pothesis that the levosimendan binding site to be locatedclose to the D/E linker making the flexible linker region
Fig. 8. P(r) function of cTnCCS with and without levosimendan. Drug bind-ing to calcium-saturated cTnCCS does not pull the domains together. Theradius of gyration is slightly larger for cTnCCS/levosimendan than cTnCalone and longer distances are also seen in the presence of levosimendanindicating drug binding close the linker region [213].

99
more rigid thus limiting the degrees of freedom of thedomains.
The interaction of bepridil with cTnC is shown to takeplace at the hydrophobic region of the N-domain and stabilisethe open conformation [106, 196]. A competitive bindingstudy between levosimendan and bepridil was attempted todetermine whether the binding site of both calcium sensitisersis the same. Unfortunately, there were several isoforms ofbepridil present under used conditions and no competitionexperiment could be completed.
The binding of levosimendan to the C-domain of cTnC,resulted in small-chemical shift changes assigning two po-tential binding sites in the structural domain. This interactionoccurred as a fast exchange process (kex > 104 s−1). TheseC-domain binding sites appear to be on opposite sides ofthe domain (Fig. 7). Levosimendan-induced chemical shiftchanges were compared with racemate EMD53998 inducedchanges and found to be very similar. However, EMD57033has been shown to have one binding site on the C-domain ofcTnC [201, 202]. The chemical shift changes were too smallto allow accurate determination of the affinity of levosimen-dan to cCTnC sites [213].
Binding of levosimendan to cardiac troponin C complexedwith cardiac troponin I
Levosimendan was shown to be able to interact with bothdomains of cTnC [205, 213], but the interaction site thatis responsible for the calcium-sensitising effect and themechanism were not known. Therefore, a more completebut still rather limited model of the contractile apparatuswas prepared to study the levosimendan interaction withcTnC. Two peptides of human cTnI, consisting of residues32–79 and 128–180, were used to make the complex with(Ca2+)3-cTnCCS. The cTnI32–79 comprises the C-domainbinding region. In contrast, the cTnI128–180 entity includesthe inhibitory region together with the regulatory regioncTnI147–166that interacts with the N-domain of cTnC. Thefull-length cTnI is rich in positively charged amino acidssuch as lysine and arginine, therefore nonspecific interac-tions between levosimendan and isolated cTnI would beexpected to take place to some extent. These peptides werechosen because they included all the essential cTnC bindingregions for the regulation of the cardiac contraction and tominimise nonspecific interactions (see review by Rosevearand Finley [215]).
Both cTnI peptides behaved as expected under the usedNMR conditions. A 1:1:1 complex of cTnCCS:cTnI32–79:cTnI128–180 was formed as indicated by the observed chem-ical shift changes in 1H-15N-HSQC spectra. The assign-ments obtained by Sorsa et al. (data not shown) agreewell with previously reported spectra and assignments of
cTnC/cTnI [159], thus indicating that the same structureof cTnC was formed with TnI peptides as when usingthe full-length cTnI. Therefore the complex used in thatstudy [214] is most likely to be in a functionally relevantconformation.
Levosimendan induced changes to the cTnCCS/cTnI32–79/cTnI128–180 complex were followed by 1H-15N-HSQC spectra.Binding of levosimendan to this complex resulted in chemicalshift changes and the significant chemical shift perturbationsare located only in the N-domain of cTnCCS (Fig. 9). Thisis an unambiguous indication of levosimendan bindingonly to the regulatory domain of cTnCCS in the presence ofcTnI. The C-domain binding sites detected with the isolatedcTnCCS are blocked by cTnI and thus are not a prerequisitefor the calcium sensitising effect of levosimendan to occur.Kleerekoper et al. [188] have suggested that the hydrophobicregion in the C-domain would not be an ideal binding site forcalcium sensitisers because cTnI may block the drug bindingsites.
Levosimendan binding in the presence of cTnI causesnumerous chemical shift changes in the 1H-15N-HSQCspectrum of cTnC, similar to those of the isolated cTnC.No exact binding site could be identified on the basis of thespectral changes. As the calcium-binding sites are structurallycoupled, Ca2+ or ligand binding to one part of the domainmay also cause observable changes in the other parts of thedomain [81]. A quite unexpected result was the significantchemical shift perturbations in the N-helix on levosimendanbinding. Gulati et al., have shown that the N-helix, togetherwith the inactive Ca2+ site I, is required for the full activity ofcTnC [107]. It has been suggested that the N-helix is impor-tant for the calcium-binding signal transmission within theTn complex to occur [109, 111]. Therefore, it is intuitivelyunderstandable that the binding of a calcium sensitiser willalso affect this helix even though its precise function andbinding sites are not known.
Fig. 9. Levosimendan-induced chemical shift changes on cTnCCS com-plexed with cTnI32–79 and cTnI128–180 determined by 1 H–15 N HSQCspectrum at 750 MHz. Chemical shift changes are shown as the distance
in Hz from the original peak, �ν =√
�ν2H + �ν2
N .
100
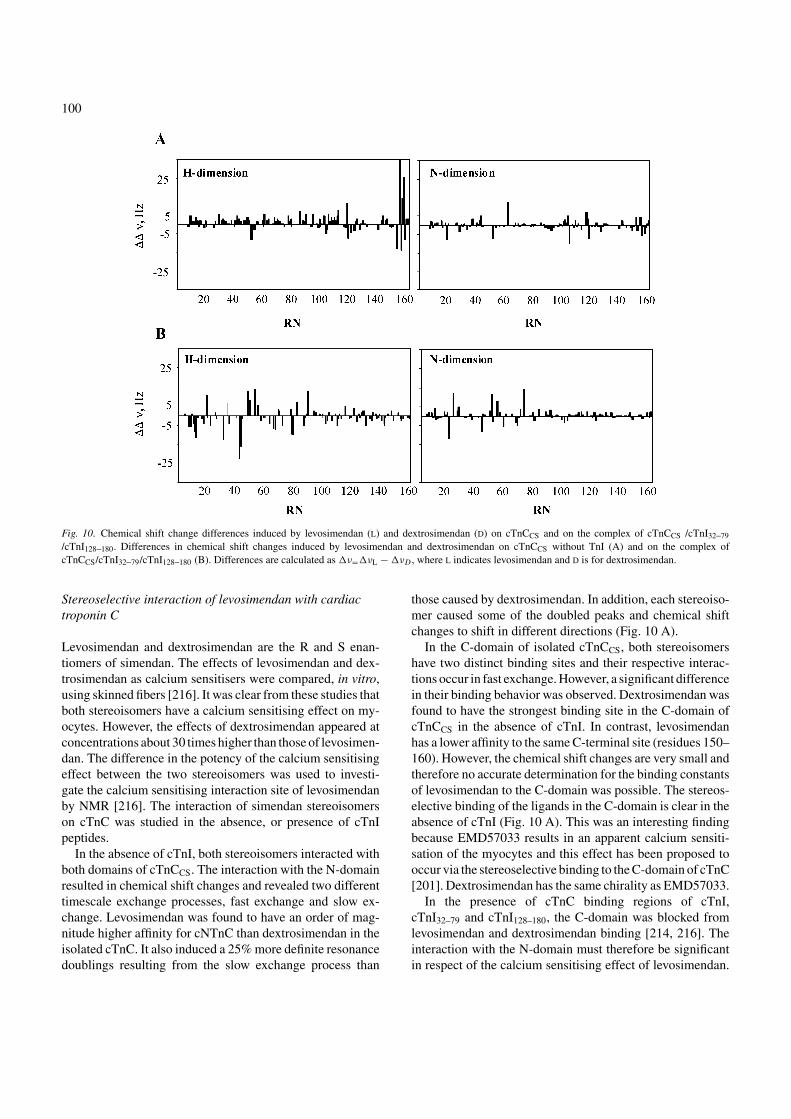
Fig. 10. Chemical shift change differences induced by levosimendan (L) and dextrosimendan (D) on cTnCCS and on the complex of cTnCCS /cTnI32–79
/cTnI128–180. Differences in chemical shift changes induced by levosimendan and dextrosimendan on cTnCCS without TnI (A) and on the complex ofcTnCCS/cTnI32–79/cTnI128–180 (B). Differences are calculated as �ν=�νL − �νD , where L indicates levosimendan and D is for dextrosimendan.
Stereoselective interaction of levosimendan with cardiactroponin C
Levosimendan and dextrosimendan are the R and S enan-tiomers of simendan. The effects of levosimendan and dex-trosimendan as calcium sensitisers were compared, in vitro,using skinned fibers [216]. It was clear from these studies thatboth stereoisomers have a calcium sensitising effect on my-ocytes. However, the effects of dextrosimendan appeared atconcentrations about 30 times higher than those of levosimen-dan. The difference in the potency of the calcium sensitisingeffect between the two stereoisomers was used to investi-gate the calcium sensitising interaction site of levosimendanby NMR [216]. The interaction of simendan stereoisomerson cTnC was studied in the absence, or presence of cTnIpeptides.
In the absence of cTnI, both stereoisomers interacted withboth domains of cTnCCS. The interaction with the N-domainresulted in chemical shift changes and revealed two differenttimescale exchange processes, fast exchange and slow ex-change. Levosimendan was found to have an order of mag-nitude higher affinity for cNTnC than dextrosimendan in theisolated cTnC. It also induced a 25% more definite resonancedoublings resulting from the slow exchange process than
those caused by dextrosimendan. In addition, each stereoiso-mer caused some of the doubled peaks and chemical shiftchanges to shift in different directions (Fig. 10 A).
In the C-domain of isolated cTnCCS, both stereoisomershave two distinct binding sites and their respective interac-tions occur in fast exchange. However, a significant differencein their binding behavior was observed. Dextrosimendan wasfound to have the strongest binding site in the C-domain ofcTnCCS in the absence of cTnI. In contrast, levosimendanhas a lower affinity to the same C-terminal site (residues 150–160). However, the chemical shift changes are very small andtherefore no accurate determination for the binding constantsof levosimendan to the C-domain was possible. The stereos-elective binding of the ligands in the C-domain is clear in theabsence of cTnI (Fig. 10 A). This was an interesting findingbecause EMD57033 results in an apparent calcium sensiti-sation of the myocytes and this effect has been proposed tooccur via the stereoselective binding to the C-domain of cTnC[201]. Dextrosimendan has the same chirality as EMD57033.
In the presence of cTnC binding regions of cTnI,cTnI32–79 and cTnI128–180, the C-domain was blocked fromlevosimendan and dextrosimendan binding [214, 216]. Theinteraction with the N-domain must therefore be significantin respect of the calcium sensitising effect of levosimendan.

101
The stereoisomers of simendan induced different chemicalshift changes in the NMR spectra of this protein model. InFig. 10 B, the differences between the stereoisomers-inducedchemical shift changes on cTnC are shown. No significantdifferences between the stereoisomers were detected in theC-domain interaction. On the other hand, the differencesbetween the stereoisomers on the N-domain were numerousand no exact binding site could be determined. It seemspossible that the suggested conformational isomerism ofcTnC masks the chemical shift changes resulting from thechange in chemical environment adjacent to the bound drug.However, it is possible that the chemical shift differencesobserved between the stereoisomers for the N-domainbinding were due to the chirality of this molecule and thusa stereospecific interaction.
Summary and conclusions
Structural information of protein complexes combined withthe knowledge of their functions elucidates the understand-ing of cellular mechanisms. Furthermore, knowledge abouta protein’s structure–activity relationship is especially usefulwhen new, effective treatments with specific mode of actionagainst different diseases are designed. Detailed informationof muscle contraction, roles of each protein of the contrac-tile apparatus and understanding of their interactions in vari-ous stages of the contraction cycle is essential for the designof the specific treatment for cardiac insufficiency. In recent
Fig. 11. Energy diagram of the effect of a potential calcium sensitiser. (A) Normal energy levels during excitation cycle in cardiac muscle [31, 165]. (B)Calcium sensitiser such as levosimendan possibly shifts the equilibrium from closed toward the open state and thus primes the N-domain for cTnI binding.Levosimendan in the complex of cTnC/cTnI may also prolong the lifetime of the complex and thus enhance the contraction.
years, cardiac troponin C has been in the focus of this re-search because of its central regulatory role in the contractionof cardiac muscle. Several molecules have been suggestedas candidates as calcium sensitisers because they modulatethe response of the contractile apparatus to the calcium sig-nal without an increase in intracellular calcium concentra-tion. However, many of them have unwanted side effects orare nonspecific and therefore they are unsuitable for clinicaluse.
On the other hand, levosimendan is used as a calcium sen-sitiser in congestive heart failure and it has been reported to bea specific and well-tolerated treatment. It undergoes stereos-elective interaction with the N-domain of calcium-saturatedcardiac troponin C in the presence of cardiac troponin I pep-tides of 32–79 and 128–180. On levosimendan binding tothe N-domain of cTnC, numerous chemical shift changesthroughout the entire N-domain were seen in the 1H-15N-HSQC spectrum. However, no NOEs could be fully assignedfor the short lifetime of the cTnC–levosimendan complex. Itmust be added that the stability and binding properties of lev-osimendan were adversely affected when DTT was present,and this reducing agent was observed to react with levosi-mendan. As a consequence, a previous report which failedto see an interaction of levosimendan on the isolated cNTnCdomain and Cys84Ser mutated cTnC in presence of DTT hasbeen the subject of controversy.
Meanwhile, several other hydrophobic calcium sensitisershave been reported to bind to the hydrophobic region in theN-domain of cTnC.

102
Indeed, in an earlier study, fluorescence spectroscopy ofdansylated N-terminal fraction of human recombinant cTnCwas used to show that amino acids near the D/E linker regionwere essential for levosimendan having a calcium sensitisingeffect on cTnC. In the same report some NOE cross signalsof levosimendan with amino acids of the hydrophobic pocketwere also shown and a hypothetic binding site was modeled.In more recent experiments no “exact” binding site for levosi-mendan on cTnC could be determined but, again, very strongindications were obtained that the D/E linker region is theprobable binding site of this calcium sensitiser. In fact, theconformation of the N-domain of cTnC in the proximity ofthe D/E linker region and the presence of some key aminoacids (e.g. Cys84) were found to be essential for the bindingof levosimendan. What is still missing is an exact 3D modelof the target-ligand complex.
It appears clear that a calcium sensitiser whose effects aremediated via cTnC should not prevent the interaction be-tween cTnI and cTnC. A potent calcium sensitiser will primethe cNTnC for cTnI binding calcium dependently. One of thelikely mechanisms to achieve this is to shift the equilibriumbetween open and closed states of the N-domain of cTnC,toward the open conformation (Fig. 11). The calcium sensi-tiser should enhance the cTnI binding to cTnC by loweringthe required energy level for the interaction to take place.This could result in the prolongation of the lifetime of thecTnC/cTnI complex, which is the active conformation trig-gering the contraction.
References
1. Haselgrove JC, Huxley HE: X-ray evidence for radial cross-bridgemovement and for the sliding filament model in actively contractingskeletal muscle. J Mol Biol 77(4): 549–568, 1973
2. Starr R, Almond R, Offer G: Location of C-protein, H-protein and X-protein in rabbit skeletal muscle fibre types. J Muscle Res Cell Motil6: 227–256, 1985
3. Seiler SH, Fischman DA, Leinwand LA: Modulation of myosin fil-ament organization by C-protein family members. Mol Biol Cell 7:113–127, 1996
4. Wang SM, Lo MC, Shang C, Kao SC, Tseng YZ: Role of M-line pro-teins in sarcomeric titin assembly during cardiac myofibrillogenesis. JCell Biochem 71: 82–95, 1998
5. Holmes KC, Popp D, Gebhard W, Kabsch W: Atomic model of theactin filament. Nature 347: 44–49, 1990
6. Lorenz M, Poole KJ, Popp D, Rosenbaum G, Holmes KC: Anatomic model of the unregulated thin filament obtained by X-ray fiberdiffraction on oriented actin–tropomyosin gels. J Mol Biol 246(1):108–119, 1995
7. Moore PB, Huxley HE, DeRosier DJ: Three-dimensional reconstruc-tion of F-actin, thin filaments and decorated thin filaments. J Mol Biol50: 279–295, 1970
8. Phillips GNJ, Fillers JP, Cohen C: Tropomyosin crystal structure andmuscle regulation. J Mol Biol 192(1): 111–131, 1986
9. Potter JD: The content of troponin, tropomyosin, actin, and myosinin rabbit skeletal muscle myofibrils. Arch Biochem Biophys 162(2):436–441, 1974
10. McLachlan AD, Stewart M: The 14-fold periodicity in alpha-tropomyosin and the interaction with actin. J Mol Biol 103(2):271–298, 1976
11. Gordon AM, Homsher E, Regnier M: Regulation of contraction instriated muscle. Rev Physiol Rev 80(2): 853–924, 2000
12. Gordon AM, Regnier M, Homsher E: Skeletal and cardiac musclecontractile activation: Tropomyosin “rocks and rolls”. Rev NewsPhysiol Sci 16: 49–55, 2001
13. Yates LD, Greaser ML: Troponin subunit stoichiometry and contentin rabbit skeletal muscle and myofibrils. J Biol Chem 258(9):5770–5774, 1983
14. Solaro RJ, Van Eyk J: Altered interactions among thin filament proteinmodulate cardiac function. J Mol Cell Cardiol 28: 217–230, 1996
15. Ohtsuki I, Shiraishi F: Periodic binding of troponin C×I and troponinI to tropomyosin–actin filaments. J Biochem 131: 739–743, 2002
16. Greaser ML, Gergely J: Reconstitution of troponin activity from threeprotein components. J Biol Chem 246(13): 4226–4233, 1971
17. Ohtsuki I, Maruyama K, Ebashi S: Regulatory and cytoskeletal proteinsof vertebrate skeletal muscle. Rev Adv Protein Chem 38: 1–67, 1986
18. Filatov VL, Katrukha AG, Bulargina TV, Gusev NB: Troponin:structure, properties, and mechanism of functioning. Rev Biochem(Mosc). 64(9): 969–985, 1999
19. Mak AS, Smillie LB: Non-polymerizable tropomyosin: Preparation,some properties and F-actin binding. Biochem Biophys Res Commun101: 208–214, 1981
20. Malnic B, Farah CS, Reinach FC: Regulatory properties of theNH2- and COOH-terminal domains of troponin T. ATPase activationand binding to troponin I and troponin C. J Biol Chem 273(17):10594–10601, 1998
21. Maytum R, Geeves MA, Lehrer SS: A modulatory role for thetroponin T tail domain in thin filament regulation. J Biol Chem 277:29774–29780, 2002
22. Pearlstone JR, Smillie LB: Binding of troponin-T fragments toseveral types of tropomyosin. Sensitivity to Ca2+ in the presence oftroponin-C. J Biol Chem 257(18): 10587–10592, 1982
23. Morris EP, Lehrer SS: Troponin-tropomyosin interactions. Fluores-cence studies of the binding of troponin, troponin T, and chymotryptictroponin T fragments to specifically labeled tropomyosin. Biochem-istry 23(10): 2214–2220, 1984
24. Schaertl S, Lehrer SS, Geeves MA: Separation and characterizationof the two functional regions of troponin involved in muscle thinfilament regulation. Biochemistry 34: 15890–15894, 1995
25. Tobacman LS: Thin filament-mediated regulation of cardiac contrac-tion. Rev Annu Rev Physiol 58: 447–481, 1996
26. Blumenschein TM, Tripet BP, Hodges RS, Sykes BD: Mapping theinteracting regions between troponins T and C. Binding of TnT andTnI peptides to TnC and NMR mapping of the TnT-binding site onTnC. J Biol Chem 276(39): 36606–36612, 2001
27. White SP, Cohen C, Phillips GNJ: Structure of co-crystals oftropomyosin and troponin. Nature 325: 826–828, 1987
28. Farah CS, Miyamoto CA, Ramos CH, Da S-AC, Quaggio RB,Fujimori K, Smillie LB, Reinach FC: Structural and regulatoryfunctions of the NH2- and COOH-terminal regions of skeletal muscletroponin I. J Biol Chem 269: 5230–5240, 1994
29. Potter JD, Sheng Z, Pan BS, Zhao J: A direct regulatory role fortroponin T and a dual role for troponin C in the Ca2+ regulation ofmuscle contraction. J Biol Chem 270(6): 2557–2562, 1995
30. Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R,Lehman W, Homsher E: The troponin tail domain promotes aconformational state of the thin filament that suppresses myosinactivity. J Biol Chem 277(31): 27636–27642, 2002
31. McKay RT, Saltibus LF, Li MX, Sykes BD: Energetics of the inducedstructural change in a Ca2+ regulatory protein: Ca2+ and troponin

103
I peptide binding to the E41A mutant of the N-domain of skeletaltroponin C. Biochemistry 39(41): 12731–12738, 2000
32. Leavis PC, Gergely J: Thin filament proteins and thin filament-linkedregulation of vertebrate muscle contraction. Revew. CRC Crit RevBiochem 16(3): 235–305, 1984
33. Zot AS, Potter JD: Structural aspects of troponin-tropomyosinregulation of skeletal muscle contraction. Review. Annu Rev BiophysBiophys Chem 16: 535–559, 1987
34. da Silva AC, Reinach FC: calcium-binding induces conformationalchanges in muscle regulatory proteins. Rev Trends Biochem Sci16(2): 53–57, 1991
35. Grabarek Z, Tao T, Gergely J: Molecular mechanism of troponin-Cfunction. Review. J Muscle Res Cell Motil 13(4): 383–393, 1992
36. Gergely J, Grabarek Z, Tao T: The molecular switch in troponin C.Rev. Adv Exp Med Biol. 332: 117–123, 1993
37. Farah CS, Reinach FC: The troponin complex and regulation ofmuscle contraction. Review. FASEB J 9: 755–767, 1995
38. Lehrer SS, Geeves MA: The muscle thin filament as a classical cooper-ative/allosteric regulatory system. J Mol Biol 277(5): 1081–1089, 1998
39. Squire JM, Morris EP: A new look at thin filament regulation invertebrate skeletal muscle. Review. FASEB J 12(10): 761–771, 1998
40. McKillop DF, Geeves MA: Regulation of the interaction betweenactin and myosin subfragment 1: Evidence for three states of the thinfilament. Biophys J 65(2): 693–701, 1993
41. Head JG, Ritchie MD, Geeves MA: Characterization of the equilib-rium between blocked and closed states of muscle thin filaments. EurJ Biochem 227(3): 694–699, 1995
42. Vibert P, Craig R, Lehman W: Steric-model for activation of musclethin filaments. J Mol Biol 266(1): 8–14, 1997
43. Maytum R, Lehrer SS, Geeves MA: Cooperativity and switchingwithin the three-state model of muscle regulation. Biochemistry38(3): 1102–1110, 1999
44. Xu C, Craig R, Tobacman L, Horowitz R, Lehman W: Tropomyosinpositions in regulated thin filaments revealed by cryoelectronmicroscopy. Biophys J 77(2): 985–992, 1999
45. Ebashi S, Endo M: Calcium ion and muscle contraction. Review. ProgBiophys Mol Biol 18: 123–183, 1968
46. Solaro RJ, Rarick HM: Troponin and tropomyosin: Proteins thatswitch on and tune in the activity of cardiac myofilaments. Rev CircRes 83(5): 471–480, 1998
47. Schaub MC, Perry SV: The relaxing protein system of striated muscle.Resolution of the troponin complex into inhibitory and calciumion-sensitizing factors and their relationship to tropomyosin. BiochemJ 115(5): 993–1004, 1969
48. Potter JD, Gergely J: Troponin, tropomyosin, and actin interactionsin the Ca2+ regulation of muscle contraction. Biochemistry 13:2697–2703, 1974
49. Hitchcock SE: Regulation of muscle contraction: Bindings of troponinand its components to actin and tropomyosin. Eur J Biochem 52(2):255–263, 1975
50. Weeks RA, Perry SV: Characterization of a region of the primarysequence of troponin C involved in calcium ion-dependent interactionwith troponin I. Biochem J 173(2): 449–457, 1978
51. Lehman W, Rosol M, Tobacman LS, Craig R: Troponin organizationon relaxed and activated thin filaments revealed by electron microscopyand three-dimensional reconstruction. J Mol Biol 307: 739–744, 2001
52. Rosol M, Lehman W, Craig R, Landis C, Butters C, Tobacman LS:Three-dimensional reconstruction of thin filaments containing mutanttropomyosin. Biophys J 78(2): 908–917, 2000
53. Tobacman LS, Butters CA: A new model of cooperative myosin-thinfilament binding. J Biol Chem 275(36): 27587–27593, 2000
54. Roher A, Lieska N, Spitz W: The amino acid sequence of humancardiac troponin C. Muscle Nerve 9: 73–77, 1986
55. Romero-Herrera AE, Castillo O, Lehmann H: Human skeletal muscleproteins. The primary structure of troponin C. J Mol Evol 8: 251–270,1976
56. van Eerd JP, Takahashi K: The amino acid sequence of bovine cardiactroponin-C. Comparison with rabbit skeletal troponin-C. BiochemBiophys Res Commun 64(1): 122–127, 1975
57. Herzberg O, James MN: Structure of the calcium regulatory muscleprotein troponin-C at 2.8 A resolution. Nature 313(6004): 653–659,1985
58. Sundaralingam M, Bergstrom R, Strasburg G, Rao ST, RoychowdhuryP, Greaser M, Wang BC: Molecular structure of troponin C fromchicken skeletal muscle at 3-angstrom resolution. Science 227(4689):945-948, 1985
59. Herzberg O, James MNG: Refined crystal structure of troponin Cfrom turkey skeletal muscle at 2.0 A resolution. J Mol Biol 203:761–779, 1988
60. Satyshur KA, Rao ST, Pyzalska D, Drendel W, Greaser M, Sundar-alingam M: Refined structure of chicken skeletal muscle troponinC in the two-calcium state at 2-A resolution. J Biol Chem 263(4):1628–1247, 1988
61. Slupsky CM, Sykes BD: NMR solution structure of calcium-saturatedskeletal muscle troponin C. Biochemistry 34: 15953–15964, 1995
62. Houdusse A, Love ML, Dominiguez R, Grabarek Z, Cohen C:Structures of four Ca2+-bound troponin C at 2.0 A resolution: Furtherinsights into the Ca2+-switch in the calmodulin superfamily. Structure5(12): 1695–1711, 1997
63. Soman J, Tao T, Phillips GNJ: Conformational variation of calcium-bound troponin C. Proteins: Struct Genet 37: 510–511, 1999
64. Sia SK, Li MX, Spyracopoulos L, Gagn, SM, Liu W, Putkey JA, SykesBD: Structure of cardiac muscle troponin C unexpectedly reveals aclosed regulatory domain. J Biol Chem 272(29): 18216–18221, 1997
65. Kleerekoper Q, Howarth JW, Guo X, Solaro RJ, Rosevear PR: Cardiactroponin I induced conformational changes in cardiac troponin Cas monitored by NMR using site-directed spin and isotope labeling.Biochemistry 34(41): 13343–13352, 1995
66. Wang CL, Zhan Q, Tao T, Gergely J: pH-dependent structural transitionin rabbit skeletal troponin C. J Biol Chem 262(20): 9636–9640, 1987
67. Wang CL, Leavis PC: Distance measurements in cardiac troponin C.Arch Biochem Biophys 276: 236–241, 1990
68. Heidorn DB, Trewhella J: Comparison of the crystal and solutionstructures of calmodulin and troponin C. Biochemistry 27(3):909–915, 1988
69. Sheng ZL, Francois JM, Hitchcock-DeGregori SE, Potter JD: Effectsof mutations in the central helix of troponin C on its biologicalactivity. J Biol Chem 266(9): 5711–5715, 1991
70. Babu A, Rao VG, Su H, Gulati J: Critical minimum length ofthe central helix in troponin C for the Ca2+ switch in muscularcontraction. J Biol Chem 268(26): 19232–19238, 1993
71. Ramakrishnan S, Hitchcock-DeGregori SE: Investigation of thestructural requirements of the troponin C central helix for function.Biochemistry 34(51): 16789–16796, 1995
72. Dvoretsky A, Abusamhadneh E, Howarth J, Rosevear PR: Solutionstructure of calcium-saturated cardiac troponin C bound to cardiactroponin I. J Biol Chem 277(41): 38565–38570, 2002
73. Kretsinger RH, Nockolds CE: Carp muscle calcium-binding protein.II. Structure determination and general description. J Biol Chemj248(9): 3313–3326, 1973
74. Potter JD, Gergely J: The calcium and magnesium binding sites ontroponin and their role in the regulation of myofibrillar adenosinetriphosphatase. J Biol Chem 250(12): 4628–4633, 1975
75. Babu A, Scordilis SP, Sonnenblick EH, Gulati J: The control ofmyocardial contraction with skeletal fast muscle troponin C. J BiolChem 262(12): 5815–5822, 1987

104
76. Babu A, Lehman W, Gulati J: Characterization of the Ca2+-switchin skeletal and cardiac muscles. FEBS Lett 251(1, 2): 177–182,1989
77. Zot HG, Potter JD: A Structural role for the Ca2+–Mg2+ sites ontroponin C in the regulation of muscle contraction. J Biol Chem 257:7678–7683, 1982
78. Sheng Z, Strauss WL, Francois JM, Potter JD: Evidence that bothCa2+-specific sites of skeletal muscle TnC are required for fullactivity. J Biol Chem 265: 21554–21560, 1990
79. Putkey JA, Liu W, Sweeney HL: Function of the N-terminal calcium-binding sites in cardiac/slow troponin C assessed in fast skeletalmuscle fibers. J Biol Chem 266: 14881–14884, 1991
80. Li MX, Gagne SM, Tsuda S, Kay CM, Smillie LB, Sykes BD: Calcium-binding to the regulatory N-domain of skeletal muscle troponin Coccurs in a stepwise manner. Biochemistry 34(26): 8330–8340, 1995
81. Li MX, Gagne SM, Spyracopoulos L, Kloks CP, Audette G, ChandraM, Solaro RJ, Smillie LB, Sykes BD: NMR studies of Ca2+ binding tothe regulatory domains of cardiac and E41A skeletal muscle troponinC reveal the importance of site I to energetics of the induced structuralchanges. Biochemistry 36(41): 12519–12525, 1997
82. Spyracopoulos L, Gagne SM, Li MX, Sykes BD: Dynamics andthermodynamics of the regulatory domain of human cardiac troponinC in the apo- and calcium-saturated states. Biochemistry 37(51):18032–18044, 1998
83. Strynadka NCJ, Chernia M, Sielecki AR, Li MX, Smillie LB, JamesMNG: Structural details of a calcium-induced molecular switch: X-raycrystallographic analysis of the calcium-saturated N-terminal domainof troponin C at 1.75 A resolution. J Mol Biol 273: 238–255, 1997
84. Leavis PC, Rosenfeld SS, Gergely J, Grabarek Z, Drabikowski W:Proteolytic fragments of troponin C. Localization of high and lowaffinity Ca2+ binding sites and interactions with troponin I andtroponin T. J Biol Chem 253(15): 5452–5459, 1978
85. Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD: Thecalcium and magnesium binding sites on cardiac troponin and theirrole in the regulation of myofibrillar adenosinetriphosphatase. J BiolChem 255(24): 11688–11693, 1980
86. Finley N, Dvoretsky A, Rosevear PR: Magnesium-calcium exchangein cardiac troponin C bound to cardiac troponin I. J Mol Cell Cardiol32: 1439–1446, 2000
87. Krudy GA, Brito RM, Putkey JA, Rosevear PR: Conformationalchanges in the metal-binding sites of cardiac troponin C induced bycalcium-binding. Biochemistry 31(6): 1595–1602, 1992
88. Lin X, Krudy GA, Howarth JW, Brito RM, Rosevear PR, PutkeyJA: Assignment and calcium dependence of methionyl epsilon C andepsilon H resonances in cardiac troponin C. Biochemistry 33(48):14434–14442, 1994
89. Negele JC, Dotson DG, Liu W, Sweeney HL, Putkey JA: Mutationof the high affinity calcium-binding sites in cardiac troponin C. J BiolChem 267(2): 825–831, 1992
90. Brito RM, Krudy GA, Negele JC, Putkey JA, Rosevear PR: Calciumplays distinctive structural roles in the N- and C-terminal domains ofcardiac troponin C. J Biol Chem 268(28): 20966–20973, 1993
91. Szczesna D, Guzman G, Miller T, Zhao J, Farokhi K, Ellemberger H,Potter JD: The role of the four Ca2+ binding sites of troponin C inthe regulation of skeletal muscle contraction. J Biol Chem 271(14):8381–8386, 1996
92. Grabarek Z, Tan RY, Wang J, Tao T, Gergely J: Inhibition of mutanttroponin C activity by an intra-domain disulphide bond. Nature345(6271): 132–135, 1990
93. Perry SV: Troponin I: inhibitor or facilitator. Rev Mol Cell Biochem190: 9–32, 1999
94. Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, SolaroRJ, Sykes BD: Calcium-induced structural transition in the regu-
latory domain of human cardiac troponin C. Biochemistry 36(40):12138–12146, 1997
95. Herzberg O, Moult J, James MN: A model for the Ca2+-induced con-formational transition of troponin C. A trigger for muscle contraction.J Biol Chem 261(6): 2638–2644, 1986
96. Herzberg O, Moult J, James MN: Molecular structure of troponin Cand its implications for the Ca2+ triggering of muscle contraction.Methods Enzymol 139: 610–632, 1987
97. Gagne SM, Tsuda S, Li MX, Smillie LB, Sykes BD: Structures ofthe troponin C regulatory domains in the apo and calcium-saturatedstates. Nat Struct Biol 2(9): 784–789, 1995
98. Gagne SM, Li MX, Sykes BD: Mechanism of direct coupling betweenbinding and induced structural changes in regulatory calcium-bindingproteins. Biochemistry 36(15): 4386–4392, 1997
99. McKay RT, Pearlstone JR, Corson DC, Gagne SM, Smillie LB,Sykes BD: Structure and interaction site of the regulatory domain oftroponin-C when complexed with the 96–148 region of troponin-I.Biochemistry 37(36): 12419–12430, 1998
100. McKay RT, Tripet BP, Pearlstone JR, Smillie LB, Sykes BD: Definingthe region of troponin-I that binds to troponin-C. Biochemistry 38(17):5478–5489, 1999
101. Brito RM, Putkey JA, Strynadka NC, James MN, Rosevear PR: Com-parative NMR studies on cardiac troponin C and a mutant incapable ofbinding calcium at site II. Biochemistry 30(42): 10236–10245, 1991
102. Johnson JD, Collins JH, Robertson SP, Potter JD: A fluorescent probestudy of Ca2+ binding to the Ca2+-specific sites of cardiac troponinand troponin C. J Biol Chem 255(20): 9635–9640, 1980
103. Putkey JA, Sweeney HL, Campbell AP: Site-directed mutation of thetrigger calcium-binding sites in cardiac troponin C. J Biol Chem 264:12370–12378, 1989
104. Paakkonen K, Annila A, Sorsa T, Pollesello P, Tilgmann C, KilpelainenI, Karisola P, Ulmanen I, Drakenberg T: Solution structure and mainchain dynamics of the regulatory domain (residues 1–91) of humancardiac troponin C. J Biol Chem 273(25): 15633–15638, 1998
105. Evenas J, Malmendal A, Thulin E, Carlstrom G, Forsen S: Ca2+binding and conformational changes in a calmodulin domain.Biochemistry 37: 13744–13754, 1998
106. Abusamhadneh E, Abbott MB, Dvoretsky A, Finley N, Sasi S, Rose-vear PR: Interaction of bepridil with the cardiac troponin C/troponinI complex. FEBS Lett 506: 51–54, 2001
107. Gulati J, Babu A, Su H: Functional delineation of the Ca2+-deficientEF-hand in cardiac muscle, with genetically engineered cardiac-skeletal chimeric troponin C. J Biol Chem 267: 25073–25077, 1992
108. Smith L, Greenfield NJ, Hitchcock-DeGregori SE: The effects ofdeletion of the amino-terminal helix on troponin C function andstability. J Biol Chem 269(13): 9857–9863, 1994
109. Fredricksen RS, Swenson CA: Relationship between stability andfunction for isolated domains of troponin C. Biochemistry 35(44):14012–14026, 1996
110. Chandra M, da Silva EF, Sorenson MM, Ferro JA, Pearlstone JR,Nash BE, Borgford T, Kay CM, Smillie LB: The effects of N helixdeletion and mutant F29W on the Ca2+ binding and functionalproperties of chicken skeletal muscle troponin. J Biol Chem 269(21):14988–14994, 1994
111. Smith L, Greenfield NJ, Hitchcock-DeGregori SE: Mutations in theN- and D-helices of the N-domain of troponin C affect the C-domainand regulatory function. Biophys J 76: 400–408, 1999
112. Fabiato A, Fabiato F: Effects of pH on the myofilaments and thesarcoplasmic reticulum of skinned cells from cardiac and skeletalmuscles. J Physiol 276: 233–255, 1978
113. Blanchard EM, Solaro RJ: Inhibition of the activation and troponincalcium-binding of dog cardiac myofibrils by acidic pH. Circ Res55(3): 382–391, 1984

105
114. Blanchard EM, Pan BS, Solaro RJ: The effect of acidic pH onthe ATPase activity and troponin Ca2+ binding of rabbit skeletalmyofilaments. J Biol Chem 259(5): 3181–3186, 1984
115. Parsons B, Szczesna D, Zhao J, Van Slooten G, Kerrick WG, PutkeyJA, Potter JD: The effect of pH on the Ca2+ affinity of the Ca2+regulatory sites of skeletal and cardiac troponin C in skinned musclefibres. J Muscle Res Cell Motil 18(5): 599–609, 1997
116. el-Saleh SC, Solaro RJ: Troponin I enhances acidic pH-induceddepression of Ca2+ binding to the regulatory sites in skeletal troponinC. J Biol Chem 263(7): 3274–3278, 1988
117. Dhoot GK, Gell PG, Perry SV: The localization of the different formsof troponin I in skeletal and cardiac muscle cells. Exp Cell Res 117(2):357–370, 1978
118. Van Eyk JE, Thomas LT, Tripet B, Wiesner RJ, Pearlstone JR, Farah CS,Reinach FC, Hodges RS: Distinct regions of troponin I regulate Ca2+-dependent activation and Ca2+ sensitivity of the acto-S1-TM ATPaseactivity of the thin filament. J Biol Chem 272: 10529–10537, 1997
119. Stone DB, Timmins PA, Schneider DK, Krylova I, Ramos CH,Reinach FC, Mendelson RA: The effect of regulatory Ca2+ on thein situ structures of troponin C and troponin I: A neutron scatteringstudy. J Mol Biol 281(4): 689–704, 1998
120. Dong WJ, Xing J, Chandra M, Solaro RJ, Cheung HC: Structuralmapping of single cystein mutants of cardiac troponin I. Proteins41(4): 438–447, 2000
121. Olah GA, Rokop SE, Wang CL, Blechner SL, Trewhella J: Troponin Iencompasses an extended troponin C in the Ca2+-bound complex: Asmall-angle X-ray and neutron scattering study. Biochemistry 33(27):8233–8239, 1994
122. Olah GA, Trewhella J: A model structure of the muscle protein complex4Ca2+ troponin C. Troponin I derived from small-angle scattering data:Implications for regulation. Biochemistry 33(43): 12800–12806, 1994
123. Vassylyev DG, Takeda S, Wakatsuki S, Maeda K, Maeda Y: Crystalstructure of troponin C in complex with troponin I fragment at 2.3 –A resolution. Proc Natl Acad Sci U S A 95(9): 4847–4852, 1998
124. Tung C-S, Wall ME, Gallagher SC, Trewhella J: A model of troponin-Iin complex with troponin-C using hybrid experimental data: Theinhibitory region is a beta-hairpin. Protein Sci 9: 1312–1326, 2000
125. Zhao X, Kobayashi T, Gryczynski Z, Gryczynski I, Lakowicz J, WadeR, Collins JH: Calcium-induced flexibility changes in the troponin C-troponin I complex. Biochim Biophys Acta 1479(1, 2): 247–254, 2000
126. Dong WJ, Robinson JM, Xing J, Umeda PK, Cheung HC: An inter-domain distance in cardiac troponin C determined by fluorescencespectroscopy. Protein Sci 9(2): 280–289, 2000
127. Abbott MB, Gaponenko V, Abusamhadneh E, Finley N, Li G, Dvoret-sky A, Rance M, Solaro RJ, Rosevear PR: Regulatory domain confor-mational exchange and linker region flexibility in cardiac troponin Cbound to cardiac troponin I. J Biol Chem 275(27): 20610–20617, 2000
128. Heller WT, Abusamhadneh E, Finley N, Rosevear PR, Trewhella J:The solution structure of a cardiac troponin C-troponin I-troponinT complex shows a somewhat compact troponin C interacting withan extended troponin I-troponin T component. Biochemistry 41(52):15654–15663, 2002
129. Takeda S, Yamashita A, K. M, Maeda Y: Structure of the coredomain of human cardiac troponin in the Ca2+-saturated form. Nature424(6944): 35–41, 2003
130. Sheng Z, Pan BS, Miller TE, Potter JD: Isolation, expression, andmutation of a rabbit skeletal muscle cDNA clone for troponin I. Therole of the NH2 terminus of fast skeletal muscle troponin I in itsbiological activity. J Biol Chem 267(35): 25407–25413, 1992
131. Krudy GA, Kleerekoper Q, Guo X, Howarth JW, Solaro RJ, RosevearPR: NMR studies delineating spatial relationship within the cardiactroponin I-troponin C complex. J Biol Chem 269: 23731–23735,1994
132. Pearlstone JR, Sykes BD, Smillie LB: Interactions of structural C andregulatory N domains of troponin C with repeated sequence motifs introponin I. Biochemistry 36(24): 7601–7606, 1997
133. Gasmi-Seabrook GM, Howarth JW, Finley N, Abusamhadneh E,Gaponenko V, Brito RM, Solaro RJ, Rosevear PR: Solution structuresof the C-terminal domain of cardiac troponin C free and bound tothe N-terminal domain of cardiac troponin I. Biochemistry 38(26):8313–8322, 1999
134. Calvert MJ, Ward DG, Trayer HR, Trayer IP: The importance of thecarboxyl-terminal domain of cardiac troponin C in a Ca2+ -sensitivemuscle regulation. J Biol Chem 275: 32508–32515, 2000
135. Ferrieres G, Pugniere M, Mani JC, Villard S, Laprade M, Doutre P,Pau B, Granier C: Systematic mapping of regions of human cardiactroponin I involved in binding to cardiac troponin C: N- and C-terminallow affinity contributing regions. FEBS Lett 479(3): 99–105, 2000
136. Mercier P, Spyracopoulos L, Sykes BD: Structure, dynamics, andthermodynamics of the structural domain of troponin C in complexwith the regulatory peptide 1-40 of troponin I. Biochemistry 40(34):10063–10077, 2001
137. Ngai SM, Pearlstone JR, Smillie LB, Hodges RS: Characterizationof the biologically important interaction between troponin C and theN-terminal region of troponin I. J Cell Biochem 83(1): 99–110, 2001
138. Rarick HM, Tang HP, Guo XD, Martin AF, Solaro RJ: Interac-tions at the NH2-terminal interface of cardiac troponin I modulatemyofilament activation. J Mol Cell Cardiol 31(2): 363–375, 1999
139. Tripet B, Van Eyk JE, Hodges RS: Mapping of a second actin-tropomyosin and a second troponin C binding site within the Cterminus of troponin I, and their importance in the Ca2+-dependentregulation of muscle contraction. J Mol Biol 271(5): 728–750, 1997
140. McKay RT, Tripet BP, Hodges RS, Sykes BD: Interaction of thesecond binding region of troponin I with the regulatory domain ofskeletal muscle troponin C as determined by NMR spectroscopy. JBiol Chem 272(45): 28494–28500, 1997
141. Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra M,Solaro RJ, Umeda PK, Cheung HC: Conformation of the regulatorydomain of cardiac muscle troponin C in its complex with cardiactroponin I. J Biol Chem 274(44): 31382–31390, 1999
142. Li MX, Spyracopoulos L, Sykes BD: Binding of cardiac troponin-I(147-163) induces a structural opening in human cardiac troponin-C.Biochemistry 38: 8289–8298, 1999
143. Dong WJ, Xing J, Robinson JM, Cheung HC: Ca2+ induces anextended conformation of the inhibitory region of troponin I in cardiacmuscle troponin. J Mol Biol 314(1): 51–61, 2001
144. Pearlstone JR, Chandra M, Sorenson MM, Smillie LB: Biologicalfunction and site II Ca2+-induced opening of the regulatory domain ofskeletal troponin C are impaired by invariant site I or II Glu mutations.J Biol Chem 275(45): 35106–35115, 2000
145. Kobayashi T, Kobayashi M, Gryczynski Z, Lakowicz JR, CollinsJH: Inhibitory region of troponin I: Ca2+-dependent structural andenvironmental changes in the troponin-tropomyosin complex and inreconstituted thin filaments. Biochemistry 39(1): 86–91, 2000
146. Syska H, Wilkinson JM, Grand RJ, Perry SV: The relationshipbetween biological activity and primary structure of troponin I fromwhite skeletal muscle of the rabbit. Biochem J 153(2): 375–387, 1976
147. Van Eyk JE, Strauss JD, Hodges RS, Ruegg JC: A syntheticpeptide mimics troponin I function in the calcium-dependentregulation of muscle contraction. FEBS Lett 323(3): 223–228,1993
148. Leszyk J, Grabarek Z, Gergely J, Collins JH: Characterization ofzero-length cross-links between rabbit skeletal muscle troponin Cand troponin I: Evidence for direct interaction between the inhibitoryregion of troponin I and the NH2-terminal, regulatory domain oftroponin C. Biochemistry 29(1): 299–304, 1990

106
149. Slupsky CM, Shaw GS, Campbell AP, Sykes BD: A 1H NMR studyof a ternary peptide complex that mimics the interaction betweentroponin C and troponin I. Protein Sci 1(12): 1595–1603, 1992
150. Howarth JW, Krudy GA, Lin X, Putkey JA, Rosevear PR: An NMRand spin label study of the effects of binding calcium and troponin I in-hibitory peptide to cardiac troponin C. Protein Sci 4(4): 671–680, 1995
151. Mercier P, Li MX, Sykes BD: Role of the structural domain oftroponin C in muscle regulation: NMR studies of Ca2+ binding andsubsequent interactions with regions 1-40 and 96-115 of troponin I.Biochemistry 39(11): 2902–2911, 2000
152. Abbott MB, Dvoretsky A, Gaponenko V, Rosevear PR: Cardiactroponin I inhibitory peptide: Location of interaction sites on troponinC. FEBS Lett 469(2, 3): 168–172, 2000
153. Luo Y, Wu JL, Li B, Langsetmo K, Gergely J, Tao T: Photocrosslinkingof benzophenone-labeled single cysteine troponin I mutants to otherthin filament proteins. J Mol Biol 296(3): 899–910, 2000
154. Tripet B, De Crescenzo G, Grothe S, O’Connor-McCourt M, HodgesRS: Kinetic analysis of the interactions between troponin C andthe C-terminal troponin I regulatory region and validation of a newpeptide delivery/capture system used for surface plasmon resonance.J Mol Biol 323(2): 345–362, 2002
155. Brown LJ, Sale KL, Hills R, Rouviere C, Song L, Zhang X, FajerPG: Structure of the inhibitory region of troponin by site directed spinlabeling electron paramagnetic resonance. Proc Natl Acad Sci USA99(20): 12765–12770, 2002
156. Dong WJ, Robinson JM, Stagg S, Xing J, Cheung HC: Ca2+-inducedconformational transition in the inhibitory and regulatory regions ofcardiac troponin I. J Biol Chem 278(10): 8686–8692, 2003
157. Lindhout DA, Sykes BD: Structure and dynamics of the C-domain ofhuman cardiac troponin C in complex with the inhibitory region ofhuman cardiac troponin I. J Biol Chem 278: 27024–27034, 2003
158. Li MX, Saude EJ, Wang X, Pearlstone JR, Smillie LB, Sykes BD:Kinetic studies of calcium and cardiac troponin I peptide binding tohuman cardiac troponin C using NMR spectroscopy. Eur Biophys J31: 245–256, 2002
159. Abbott MB, Dong WJ, Dvoretsky A, DaGue B, Caprioli RM, CheungHC, Rosevear PR: Modulation of cardiac troponin C-cardiac troponinI regulatory interactions by the amino-terminus of cardiac troponin I.Biochemistry 40(20): 5992–6001, 2001
160. Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W,Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, CheungHC, Rosevear PR: NMR analysis of cardiac troponin C-troponin Icomplexes: Effects of phosphorylation. FEBS Lett 453: 107–112, 1999
161. Gaponenko V, Abusamhadneh E, Abbott MB, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear PR: Effects of troponinI phosphorylation on conformational exchange in the regulatorydomain of cardiac troponin C. J Biol Chem 274: 16681–16684, 1999
162. Ward DG, Cornes MP, Trayer IP: Structural consequences of cardiactroponin I phosphorylation. J Biol Chem 277(44): 41795–41801, 2002
163. Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, So-laro RJ: The effect of troponin I phosphorylation on the Ca2+-bindingproperties of the Ca2+-regulatory site of bovine cardiac troponin. JBiol Chem 257(1): 260–263, 1982
164. Zhang R, Zhao J, Potter JD: Phosphorylation of both serine residues incardiac troponin I is required to decrease the Ca2+ affinity of cardiactroponin C. J Biol Chem 270(51): 30773–30780, 1995
165. Paakkonen K, Sorsa T, Drakenberg T, Pollesello P, Tilgmann C,Permi P, Heikkinen S, Kilpelainen I, Annila A: Conformations of theregulatory domain of cardiac troponin C examined by residual dipolarcouplings. Eur J Biochem 267: 6665–6672, 2000
166. Hansen MR, Mueller L, Pardi A: Tunable alignment of macro-molecules by filamentous phage yields dipolar coupling interactions.Nat Struct Biol 5(12): 1065–1074, 1998
167. Tjandra N, Bax A: Direct measurement of distances and angles inbiomolecules by NMR in a dilute liquid crystalline medium. Science278(5340): 1111–1114, 1997
168. Bax A, Tjandra N: High-resolution heteronuclear NMR of humanubiquitin in an aqueous liquid crystalline medium. J Biomol NMR10(3): 289–292, 1997
169. Tjandra N, Omichinski JG, Gronenborn AM, Clore GM, Bax A: Useof dipolar 1H-15N and 1H-13C couplings in the structure determinationof magnetically oriented macromolecules in solution. Nat Struct Biol4: 732–738, 1997
170. Moir AJ, Solaro RJ, Perry SV: The site of phosphorylation of troponinI in the perfused rabbit heart. The effect of adrenaline. Biochem J185(2): 505–513, 1980
171. Dong WJ, Chandra M, Xing J, She M, Solaro RJ, Cheung HC:Phosphorylation-induced distance change in a cardiac muscletroponin I mutant. Biochemistry 36(22): 6754–6761, 1997
172. Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ: Effects of pro-tein kinase A phosphorylation on signaling between cardiac troponinI and the N-terminal domain of cardiac troponin C. Biochemistry36(43): 13305–13311, 1997
173. Jaquet K, Lohmann K, Czisch M, Holak T, Gulati J, Jaquet R: A modelfor the function of the bisphosphorylated heart-specific troponin-IN-terminus. J Muscle Res Cell Motil 19(6): 647–659, 1998
174. Schmidtmann A, Lohmann K, Jaquet K: The interaction of thebisphosphorylated N-terminal arm of cardiac troponin I-A 31P-NMRstudy. FEBS Lett 513(2,3): 289–293, 2002
175. Noland TAJ, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V,Solaro RJ, Kuo JF: Cardiac troponin I mutants. Phosphorylation byprotein kinases C and A and regulation of Ca2+-stimulated MgATPaseof reconstituted actomyosin S-1. J Biol Chem 270(43): 25445–25454,1995
176. Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, HomsherE, Solaro RJ: Phosphorylation or glutamic acid substitution at proteinkinase C sites on cardiac troponin I differentially depress myofilamenttension and shortening velocity. J Biol Chem 278(13): 11265–11272,2003
177. Lindhout DA, Li MX, Schieve D, Sykes BD: Effects of T142 phos-phorylation and mutation R145G on the interaction of the inhibitoryregion of human cardiac troponin I with the C-domain of humancardiac troponin C. Biochemistry 41: 7267–7274, 2002
178. De Mello WC: Intercellular communication in cardiac muscle. RevCirc Res 51: 1–9, 1982
179. Kusuoka H, Koretsune Y, Chacko VP, Weisfeldt ML, Marban E:Excitation-contraction coupling in postischemic myocardium. Doesfailure of activator Ca2+ transients underlie stunning? Circ Res 66(5):1268–1276, 1990
180. Herzig JW, Feile K, Ihrig H, Ruegg JC: Inotropic intervention mayalter the Ca2+ sensitivity of the contractile structures of heart muscle.Pflugers Arch Eur J Physiol 384 (Suppl): R1, 1980
181. Solaro RJ, Ruegg JC: Stimulation of Ca2+ binding and ATPaseactivity of dog cardiac myofibrils by AR-L 115BS, a novel cardiotonicagent. Circ Res 51: 290–294, 1982
182. Lee JA, Allen DG: Calcium sensitisers: Mechanisms of action andpotential usefulness as inotropes. Cardiovasc Res 36: 10–20, 1997
183. Endoh M: Mechanism of action of Ca2+ sensitizers. Rev CardiovascDrugs Ther 15(5): 397–403, 2001
184. Endoh M: The therapeutic potential of novel cardiotonic agents. RevExpert Opin Investig Drugs 12(5): 735–750, 2003
185. Arteaga GM, Kobayashi T, Solaro RJ: Molecular actions of drugsthat sensitize cardiac myofilaments to Ca2+. Rev Ann Med 34(4):248–258, 2002
186. Haikala H, Linden I-B: Mechanisms of action of calcium-sensitizingdrugs. Review. J Cardiovasc Pharmacol 26(Suppl 1): S10–S19, 1995

107
187. Ovaska M, Taskinen J: A model for human cardiac troponin C and formodulation of its Ca2+ affinity by drugs. Proteins 11(2): 79–94, 1991
188. Kleerekoper Q, Liu W, Choi D, Putkey JA: Identification of bindingsites for bepridil and trifluoperazine on cardiac troponin C. J BiolChem 273(14): 8153–8160, 1998
189. Teramura S, Yamakado T: Calcium sensitizers in chronic heart failure:Inotropic interventions-reservation to preservation. Rev Cardiologia43(4): 375–385, 1998
190. Lin X, Dotson DG, Putkey JA: Covalent binding of peptides to theN-terminal hydrophobic region of cardiac troponin C has limitedeffects on function. J Biol Chem 271(1): 244–249, 1996
191. Kobayashi S, Reien Y, Ogura T, Saito T, Masuda Y, Nakaya H:Inhibitory effect of bepridil on hKv1.5 channel current: Comparisonwith amiodarone and E-4031. Eur J Pharmacol 430: 149–157, 2001
192. Stains JP, Gay CV: Inhibition of Na+/Ca2+ exchange with KB-R7943or bepridil diminished mineral deposition by osteoblasts. J BoneMiner Res 16: 1434–1443, 2001
193. Solaro RJ, Bousquet P, Johnson JD: Stimulation of cardiac myofila-ment force, ATPase activity and troponin C Ca2+ binding by bepridil.J Pharmacol Exp Ther 238(2): 502–507, 1986
194. MacLachlan LK, Reid DG, Mitchell RC, Salter CJ, Smith SJ:Binding of a calcium sensitizer, bepridil, to cardiac troponin C. Afluorescence stopped-flow kinetic, circular dichroism, and protonnuclear magnetic resonance study. J Biol Chem 265(17): 9764–9770,1990
195. Li Y, Love ML, Putkey JA, Cohen C: Bepridil opens the regulatoryN-terminal lobe of cardiac troponin C. Proc Natl Acad Sci U S A97(10): 5140–5145, 2000
196. Wang X, Li MX, Sykes BD: Structure of the regulatory N-domainof human cardiac troponin C in complex with human cardiactroponin I147–163 and bepridil. J Biol Chem 277: 31124–31133,2002
197. Beier N, Harting J, Jonas R, Klockow M, Lues I, Haeusler G: Thenovel cardiotonic agent EMD 53 998 is a potent “calcium sensitizer”.J Cardiovasc Pharmacol 18(1): 17–27, 1991
198. Lues I, Beier N, Jonas R, Klockow M, Haeusler G: The twomechanisms of action of racemic cardiotonic EMD 53998, calciumsensitization and phosphodiesterase inhibition, reside in differentenantiomers. J Cardiovasc Pharmacol 21(6): 883–892, 1993
199. Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, BeierN, Lakatta EG: Stereoselective actions of thiadiazinones on canine car-diac myocytes and myofilaments. Circ Res 73(6): 981–990, 1993
200. Pan B-S, Johnson RGJ: Interaction of cardiotonic thiadiazinonederivatives with cardiac troponin C. J Biol Chem 271(2): 817–823,1996
201. Li MX, Spyracopoulos L, Beier N, Putkey JA, Sykes BD: Inter-action of cardiac troponin C with Ca2+ sensitizer EMD57033 andcardiac troponin I inhibitory peptide. Biochemistry 39: 8782–8790,2000
202. Wang X, Li MX, Spyracopoulos L, Beier N, Chandra M, Solaro RJ,Sykes BD: Structure of the C-domain of human cardiac troponin C in
complex with the Ca2+ sensitizing drug EMD 57033. J Biol Chem276: 25456–25466, 2001
203. Szilagyi S, Pollesello P, Levijoki J, Kaheinen P, Haikala H, Edes I, PappZ: The mechanism of levosimendan and OR-1896-induced positive in-otropy in isolated guinea pig hearts. Eur J Pharm 486: 67–74, 2004
204. Lancaster MK, Cook SJ: The effects of levosimendan on [Ca2+]i
in guinea-pig isolated ventricular myocytes. Eur J Pharmacol 339:97–100, 1997
205. Haikala H, Kaivola J, Nissinen E, Wall P, Levijoki J, Linden I-B:Cardiac troponin C as a target protein for a novel calcium sensitizingdrug, levosimendan. J Mol Cell Cardiol 27: 1859–1866, 1995
206. Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG, SolaroRJ: Effects of Levosimendan, a cardiotonic agent targeted to troponinC, on cardiac function and on phosphorylation and Ca2+ sensitivityof cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart.Circ Res 77(1): 107–113, 1995
207. Hasenfuss G, Pieske B, Castell M, Kretschmann B, Maier LS, JustH: Influence of the novel inotropic agent levosimendan on isometrictension and calcium cycling in failing human myocardium. Circulation98: 2141–2147, 1998
208. Haikala H, Nissinen E, Etemadzadeh E, Levijoki J, Linden I-B:Troponin C-mediated calcium sensitization induced by levosimendandoes not impair relaxation. J Cardiovasc Pharmacol 25: 794–801, 1995
209. Kivikko M, Antila S, Eha J, Lehtonen L, Pentikainen PJ: Pharmaco-dynamics and safety of a new calcium sensitizer, levosimendan, andits metabolites during an extended infusion in patients with severeheart failure. J Clin Pharmacol 43: 43–51, 2002
210. Levijoki J, Pollesello P, Kaivola J, Tilgmann C, Sorsa T, Annila A,Kilpelainen I, Haikala H: Further evidence for the cardiac troponin Cmediated calcium sensitization by levosimendan: Structure-responseand binding analysis with analogs of levosimendan. J Mol Cell Cardiol32: 479–491, 2000
211. Pollesello P, Ovaska M, Kaivola J, Tilgmann C, Lundstrom K, Kalkki-nen N, Ulmanen I, Nissinen E, Taskinen J: Binding of a new Ca2+sensitizer, levosimendan, to recombinant human cardiac troponinC. A molecular modelling, fluorescence probe, and proton nuclearmagnetic resonance study. J Biol Chem 269: 28584–28590, 1994
212. Kleerekoper Q, Putkey JA: Drug binding to cardiac troponin C. J BiolChem 274(34): 23932–23939, 1999
213. Sorsa T, Heikkinen S, Abbott MB, Abusamhadneh E, Laakso T,Tilgmann C, Serimaa R, Annila A, Rosevear PR, Drakenberg T, Polle-sello P, Kilpelainen I: Binding of Levosimendan, a Calcium Sensitizer,to Cardiac Troponin C. J Biol Chem 276(12): 9337–9343, 2001
214. Sorsa T, Pollesello P, Permi P, Drakenberg T, Kilpelainen I: Interactionof levosimendan with cardiac troponin C in the presence of cardiactroponin I peptides. J Mol Cell Cardiol 35(9): 1055–1061, 2003
215. Rosevear PR, Finley N: Molecular mechanism of levosimendanaction: an update. J Mol Cell Cardiol 35: 1011–1015, 2003
216. Sorsa T, Pollesello P, Rosevear PR, Drakenberg T, Kilpelainen I:Stereoselective binding of levosimendan to cardiac troponin C causescalcium sensitization. Eur J Pharmacol 486: 1–8, 2004




















