
Slayt 1 - AVESİS
67
Lysosomes and lysosomal diseases The lysosome is the terminal degradative station of multiple trafficking routes, including endocytic and scavenging pathways. Prof. Dr. Nurten ÖZSOY
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Slayt 1 - AVESİS

Lysosomes and
lysosomal
diseases
The lysosome is the terminal
degradative station of multiple
trafficking routes, including
endocytic and scavenging
pathways.
Prof. Dr. Nurten ÖZSOY

Learning goals
• Describe the functions of lysosomes and
how vesicles are targeted to them.
• Understand the biological functions of
lysosomes
• Learn about the lysosomal storage
diseases

• "Lysosome" was the name given because of enzymes'ability to "lyse" the cell
• Initially referred to as "suicide bags" since one of thefunctions of lysosomes is to rupture when the cell dies
• Lysosomes were discovered by the Belgian cytologistChristian de Duve in 1949
• Rupturing releases hydrolytic enzymes that digest allcomponents of the cell, including proteins, DNA, RNA,carbohydrates, lipids and cellulose.
Lysosomes
Nobel-prize
1974
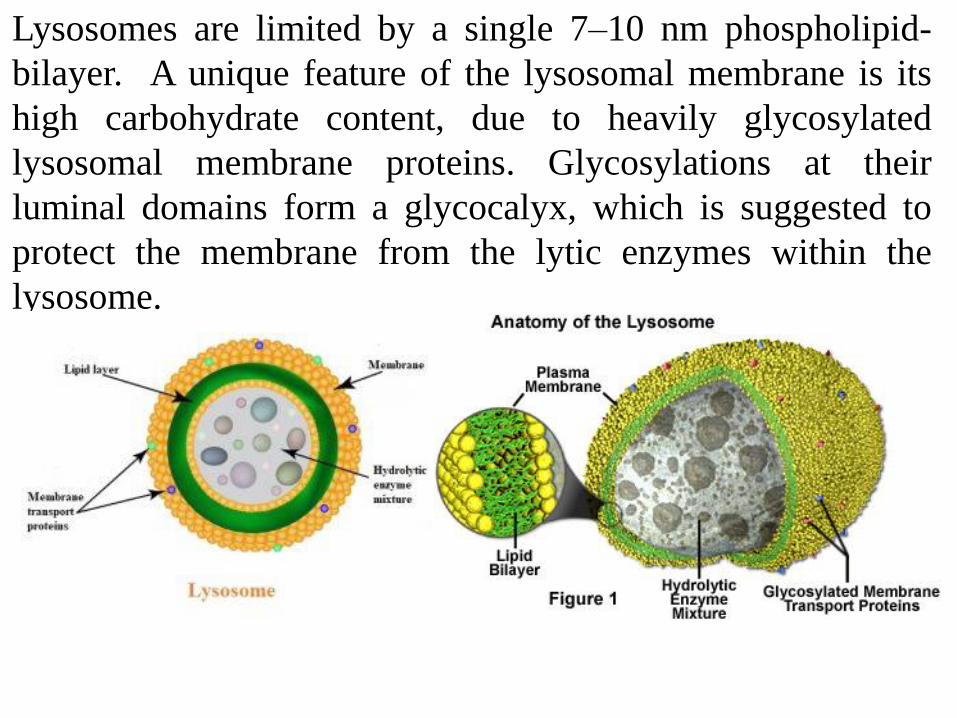
Lysosomes are limited by a single 7–10 nm phospholipid-
bilayer. A unique feature of the lysosomal membrane is its
high carbohydrate content, due to heavily glycosylated
lysosomal membrane proteins. Glycosylations at their
luminal domains form a glycocalyx, which is suggested to
protect the membrane from the lytic enzymes within the
lysosome.

Lysosome Types• Primary
• Newly formed without digestive substrate. Lysosomes are
formed by budding off of the Golgi apparatus, and the
hydrolytic enzymes within them are formed in the
endoplasmic reticulum. The enzymes are tagged with the
molecule mannose-6-phosphate, transported to the Golgi
apparatus in vesicles, and then packaged into the lysosomes.
They can be distinguished from endosomes by the lack of
mannose-6-phosphate receptors (MPRs).
• Can be secreted by exocytosis
• Secondary
• active form with enzyme + substrate
• formed by vesicle fusion event

Functions of lysosomes
Lysosomes function as the digestive system of the cell, serving both to degrade
material taken up from outside the cell and to digest obsolete components of the cell
itself.
1. Release enzymes outside of the cell (exocytosis) which may serve the purpose of
destroying materials around the cell.
2. Break-down 'digestion' of materials from inside the cell (autophagy) i.e. by fusing
with vacuoles from inside the cell.
This could include digesting worn-out organelles so that useful chemicals locked-
up in their structures can be re-used by the cell.
3. Break-down 'digestion' of materials from outside the cell (heterophagy)
i.e. by fusing with vacuoles from outside the cell.
This could include breaking-down material taken-in by phagocytes, which include
many types of white blood cells- also known as leucocytes. Specific mechanisms
of heterophagy can be:
phagocytic - by which cells engulf extracellular debris, bacteria or other
particles - only occurs in certain specialized cells
pinocytic - by which cells engulf extracellular fluid
endocytic - by which cells take-up particles such as molecules that have
become attached to the outer-surface of the cell membrane.

Deposit Degradation and Recycling
Normal Degradation
Missing Enzyme - Accumulation

Autophagy is a process of self-degradation of cellular
components.
-The products of these degradative reactions are made
available to the cell
-It is estimated that 1 to 1.5 percent of the proteins within a
healthy liver cell are degraded via autophagy per hour as part
of a normal process of cellular renovation.
Autophagy is upregulated in response to signals such as:
starvation
growth factor deprivation
ER stress
pathogen infection.

Types of autophagy
• Macro-autophagy
• Micro-autophagy
• Chaperone-mediated autophagy
While each is morphologically distinct, all three culminate in
the delivery of cargo to the lysosome for degradation and
recycling
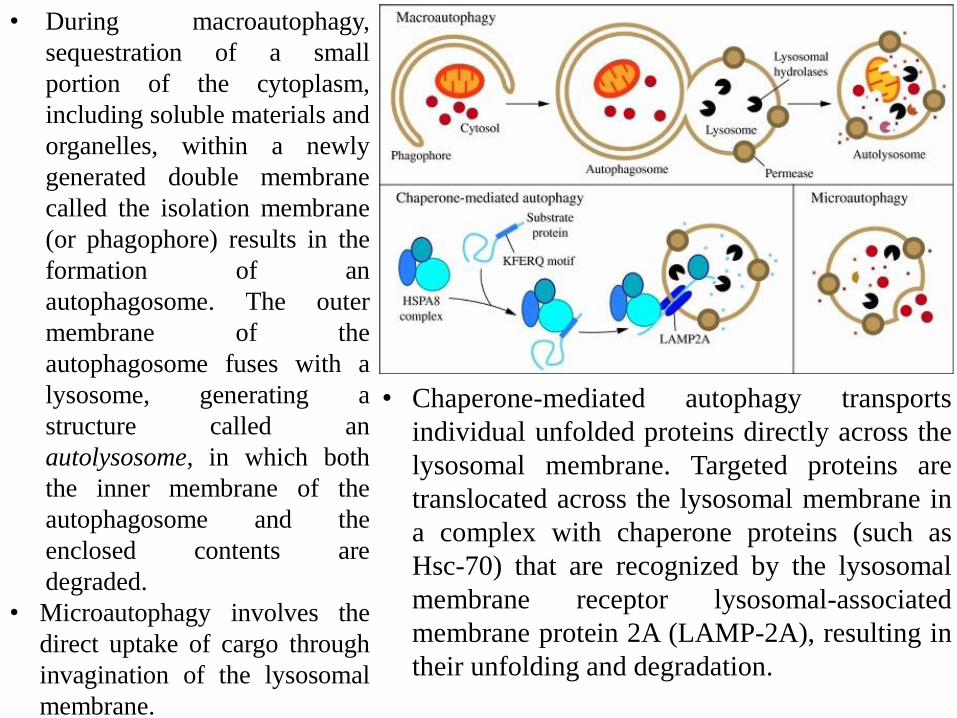
• During macroautophagy,
sequestration of a small
portion of the cytoplasm,
including soluble materials and
organelles, within a newly
generated double membrane
called the isolation membrane
(or phagophore) results in the
formation of an
autophagosome. The outer
membrane of the
autophagosome fuses with a
lysosome, generating a
structure called an
autolysosome, in which both
the inner membrane of the
autophagosome and the
enclosed contents are
degraded.
• Microautophagy involves the
direct uptake of cargo through
invagination of the lysosomal
membrane.
• Chaperone-mediated autophagy transports
individual unfolded proteins directly across the
lysosomal membrane. Targeted proteins are
translocated across the lysosomal membrane in
a complex with chaperone proteins (such as
Hsc-70) that are recognized by the lysosomal
membrane receptor lysosomal-associated
membrane protein 2A (LAMP-2A), resulting in
their unfolding and degradation.

In the cell there are two major degradation routes: the lysosomal
network and the ubiquitin-proteasome system. While the proteasome is
largely handling degradation of short-lived intracellular proteins,
lysosomes are degrading all kinds of macromolecules of intra- or
extracellular origin.

Lysosome Digestive Enzymes
•Acid hydrolases
• Enzymes are named on basis of substrate
• Protease - digests proteins
• Nuclease - digests nucleotides (DNA)
• Glycosidase - digests carbohydrates (sugars)
• Lipases - digests lipids (fats)
• Phospholipases - digests phospholipids
(membranes)
• Phosphatases - removes a phosphate group

• These enzymes work only at
low pH (highly acidic) levels.
The lysosome maintains this
pH differential by pumping
protons (H+ ions) from the
cytosol across the membrane
via proton pumps and
chloride ion channels.
• However because they can
only work at low pH levels
and the rest of the cell has a
neutral pH level, they can be
neutralized if they
accidentally escape from the
lysosome
•Acid Hydrolases
• Active at acid pH (5)
•Hydrogen ion pump in lysosomal membrane
• drives ions from cytoplasm into
lumenal space
• generates internal acidic environment

The lysosomal limiting membrane has multiple
functions including
• Acidification of the lysosomal matrix
• Sequestration of lysosomal enzymes
• Mediation of fusion events beween lysosomes and
other organelles
• Transport of degradation products to the
cytoplasm.

Lysosome-Associated Membrane Proteins
Lysosomal membrane proteins are usually highly
glycosylated proteins decorating the luminal surface of
lysosomal membranes. LAMP-1, LAMP-2 and Lysosomal
integral membrane protein 2 (LIMP-2) are the most
abundant components of this membrane.
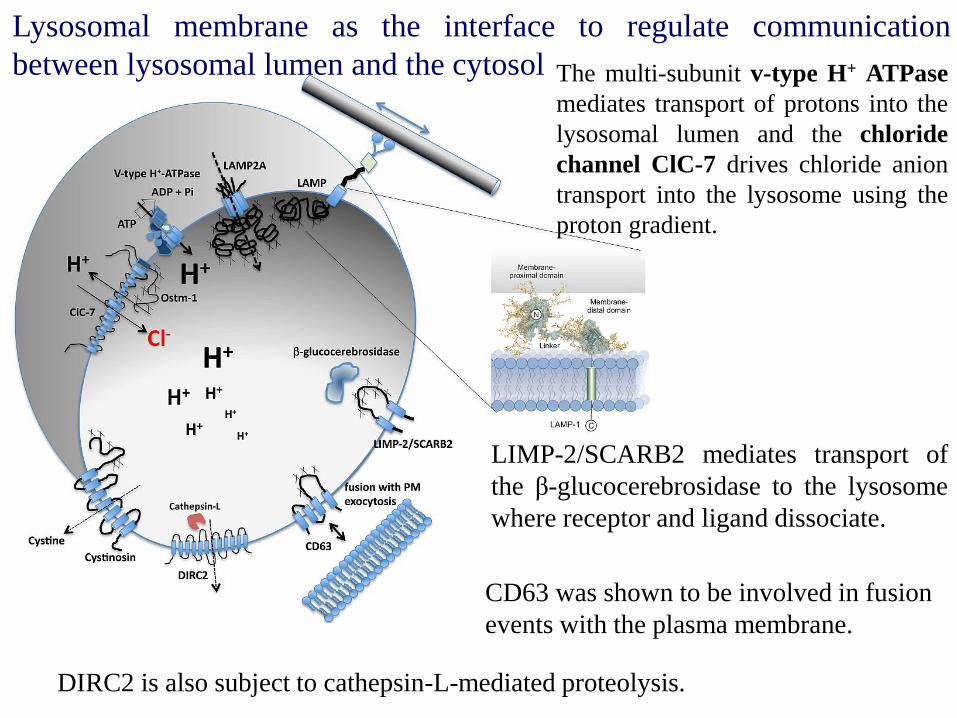
Lysosomal membrane as the interface to regulate communication
between lysosomal lumen and the cytosol
LIMP-2/SCARB2 mediates transport of
the β-glucocerebrosidase to the lysosome
where receptor and ligand dissociate.
The multi-subunit v-type H+ ATPase
mediates transport of protons into the
lysosomal lumen and the chloride
channel ClC-7 drives chloride anion
transport into the lysosome using the
proton gradient.
DIRC2 is also subject to cathepsin-L-mediated proteolysis.
CD63 was shown to be involved in fusion
events with the plasma membrane.

Lysosomal storage diseases
• Undegraded material accumulates within the
lysosomes of affected individuals
• Most of these diseases result from deficiencies
in single lysosomal enzymes.


LSD Sub-Categories
• When a lysosomal enzyme (or another proteinthat directs it) is deficient or malfunctioning,the substrate it targets accumulates, interferingwith normal cellular activity.
Healthy cell vs. LSD cell with accumulated substrate
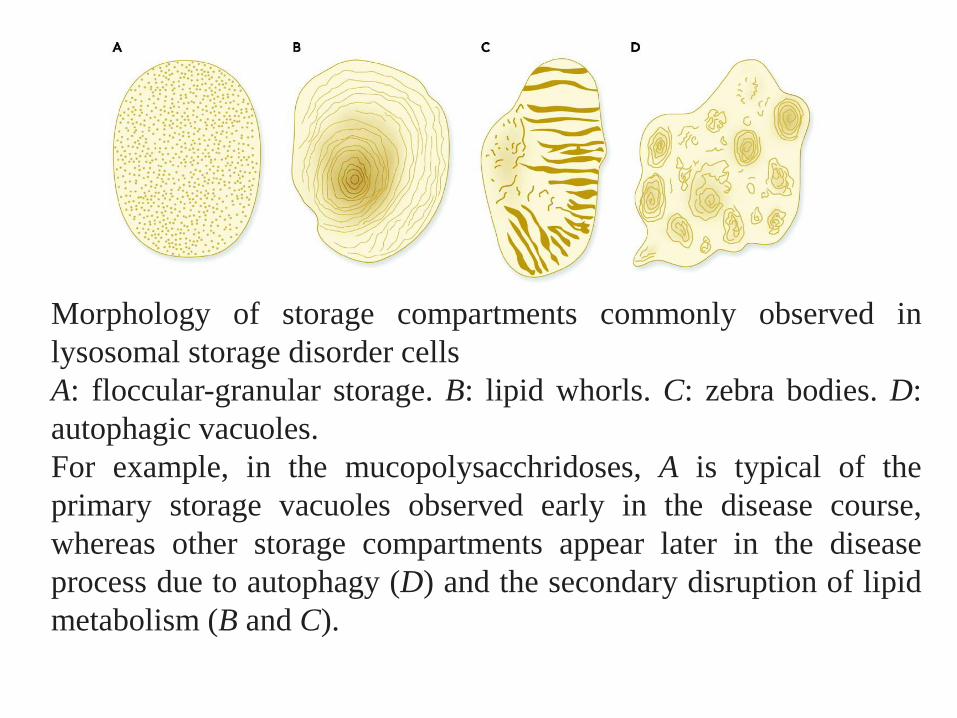
Morphology of storage compartments commonly observed in
lysosomal storage disorder cells
A: floccular-granular storage. B: lipid whorls. C: zebra bodies. D:
autophagic vacuoles.
For example, in the mucopolysacchridoses, A is typical of the
primary storage vacuoles observed early in the disease course,
whereas other storage compartments appear later in the disease
process due to autophagy (D) and the secondary disruption of lipid
metabolism (B and C).

Glycolipid structure — cerebrosides
• The carbohydrate component is linked by an O-
glycosidic bond to ceramide
• Cerebrosides contain a single sugar (Glu or Gal) or
few sugars; they are abundant in brain and myelin

Glycolipid structure — gangliosides
• Gangliosides are acidic glycosphingolipids
• They contain oligosaccharides with terminal, charged N-
acetyl neuraminic acids (NANA)
o Depending on the number of NANA sugars,
gangliosides are designated M, D, T, Q (e.g., GM).
o 'GM': a single (mono) sialic acid,
o GD, GT and GQ: two, three and four sialic acid residues
in the molecule, respectively
Ganglioside GM2
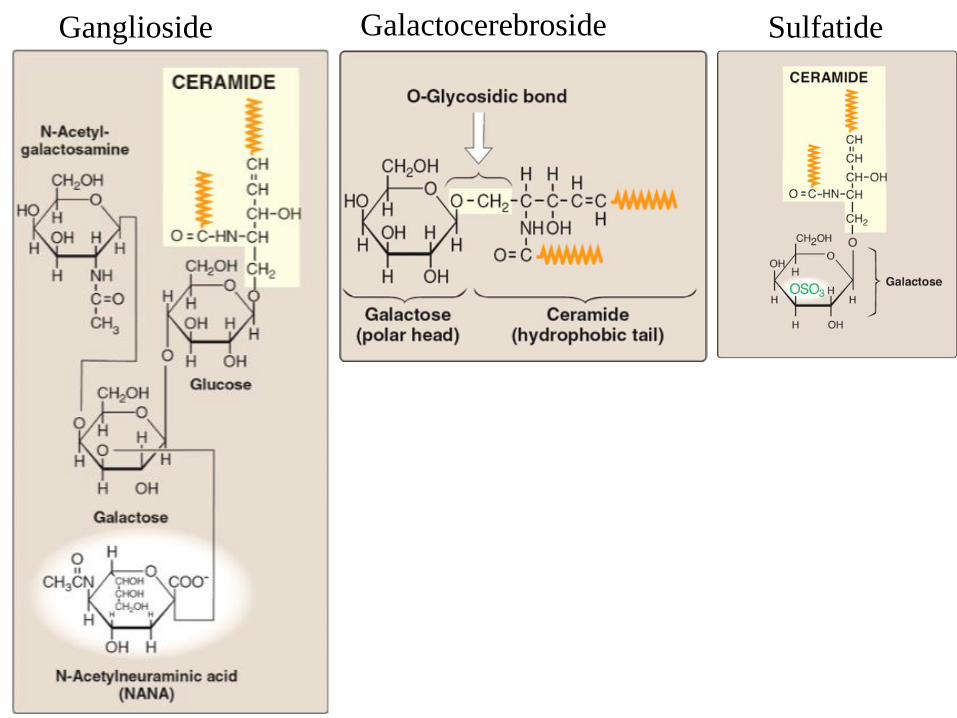
Ganglioside SulfatideGalactocerebroside

Biochemical and Cellular basis of
lysosomal storage disorders (LSD)

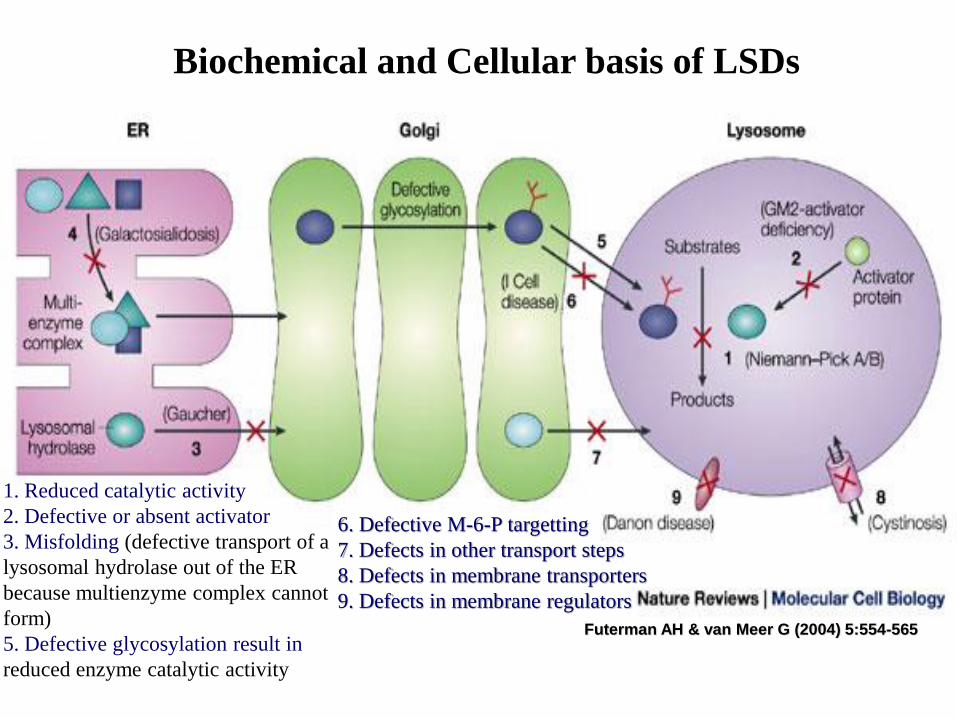
Biochemical and Cellular basis of LSDs
Futerman AH & van Meer G (2004) 5:554-565
1. Reduced catalytic activity
2. Defective or absent activator
3. Misfolding (defective transport of a
lysosomal hydrolase out of the ER
because multienzyme complex cannot
form)
5. Defective glycosylation result in
reduced enzyme catalytic activity
6. Defective M-6-P targetting
7. Defects in other transport steps
8. Defects in membrane transporters
9. Defects in membrane regulators

Biochemical and Cellular basis of lysosomal
storage disorders1. Most mutations result in the delivery of a defective enzyme with a
reduced catalytic activity to lysosomes
2. Another (activator) protein required for optimal hydrolase activity isdefective or absent
3. A mutation that causes misfolding results in defective transport of alysosomal hydrolase out of the endoplasmic reticulum
4. Alternatively, defective transport of a lysosomal hydrolase out of the ERoccurs because a multi-enzyme complex that is required for transportcannot form (Cathepsin A / sialidase / -galactosidase )
5. In the Golgi, defective glycosylation could result in an enzyme withreduced catalytic activity
6. Alternatively, defective glycosylation with mannose-6-phosphate in theGolgi could produce an enzyme that cannot reach lysosomes
7. Defects in other transport steps from the Golgi could also lead to an LSD
8. Defects in integral lysosomal membrane proteins with transporter roles
9. Defects in proteins that are involved in other vital regulatory events oflysosomal function (LAMP2, lysosomal associated membrane protein 2)

LSD: Common phenotypical features and affected organs
Central nervous system: neurodegeneration,
Spleen, liver : hepato and splenomegaly,
Skeleton: Facial dysmorphy,
Peripheral nervous system: peripheral neuropathy
Heart – cardiomyopathy, valve disease
Kidney : renal failure, nephrolithiasis (kidney stones)
Skin : agiokeratomas (vascular lesion characterized by epidermal
hyperplasia (hyperkeratosis)
Eye: cataracts, corneal clouding, cherryred spot, retinal degeneration
Ear: Sensorineural deafness
Bone marrow: anemia
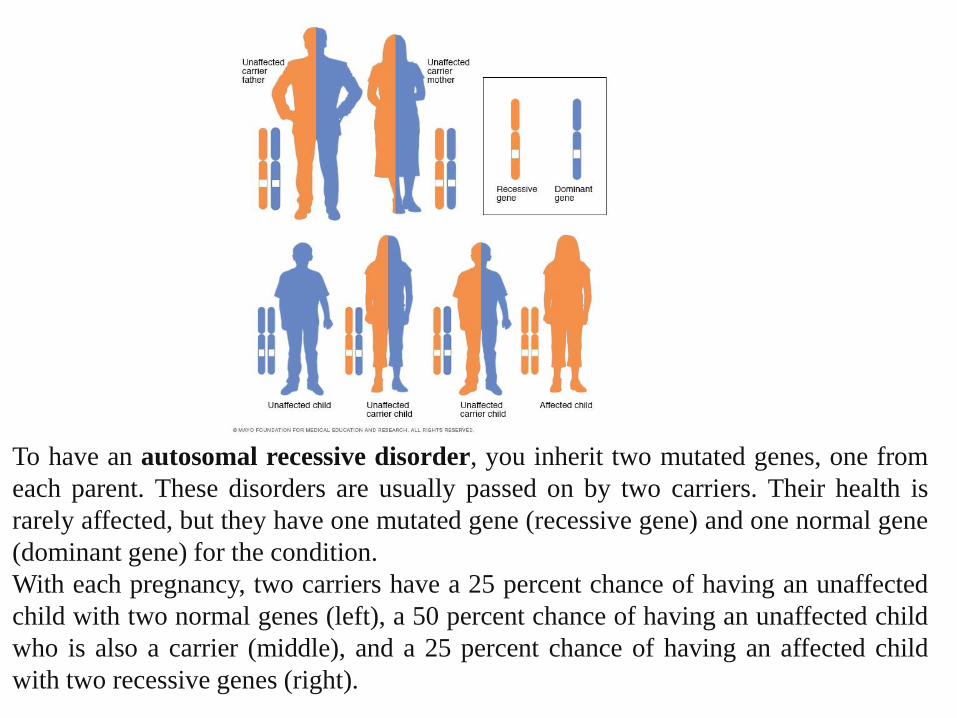
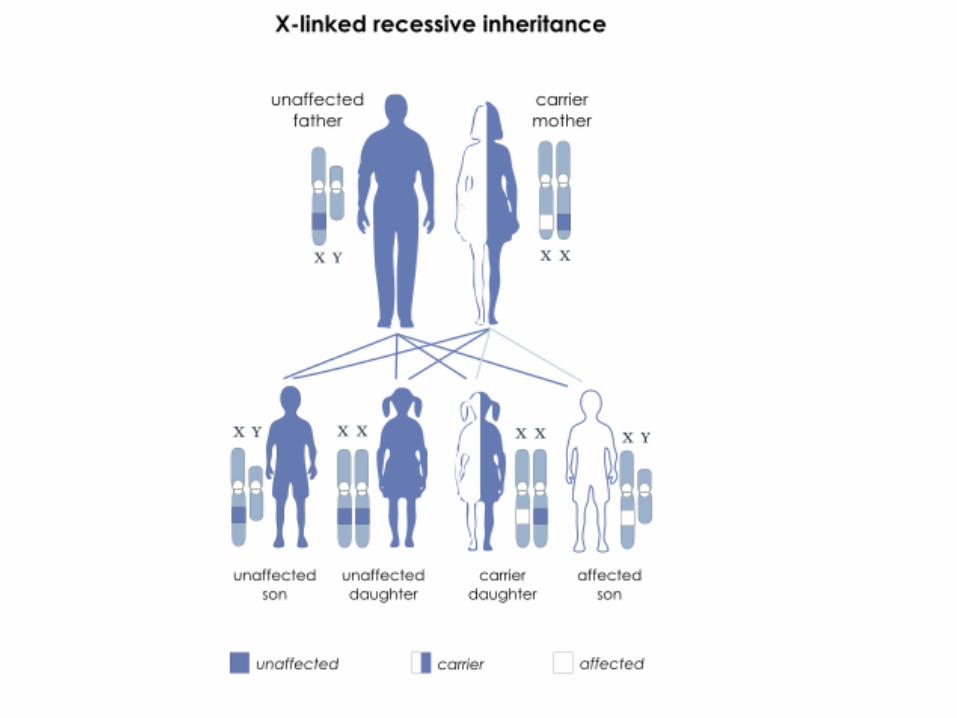
To have an autosomal recessive disorder, you inherit two mutated genes, one from
each parent. These disorders are usually passed on by two carriers. Their health is
rarely affected, but they have one mutated gene (recessive gene) and one normal gene
(dominant gene) for the condition.
With each pregnancy, two carriers have a 25 percent chance of having an unaffected
child with two normal genes (left), a 50 percent chance of having an unaffected child
who is also a carrier (middle), and a 25 percent chance of having an affected child
with two recessive genes (right).

• MPS I (Hurler) (iduronidase deficiency)
• MPS II (Hunter) (iduronate-2-sulfatase deficiency)
• MPS III (San filipo Types A, B, C and D) (heparan-N-sulfatase; N-acetyl-
glucosaminidase; acetyl Co-A glucosamine N-acetyl transferase; N-acetyl-
glucosamine-6-sulfatase deficiency)
• MPS IV (Morquio type A and B) (N-acetyl-galactosamine 6-sulfatase; -
galactosidase deficiency)
• MPS VI (Maroteaux-Lamy) (N-acetyl-galatosamine 4-sulfatase deficiency)
• MPS VII (Sly) (-glucuronidase deficiency)
• MPS IX (hyaluronidase deficiency)
• Multiple sulfatase deficiency
I - Defective metabolism of glycosaminoglycans
"the mucopolysaccharidoses "
Glycosaminoglycans: long unbranched polysaccharides containing a repeating disaccharide unit.

Typesof GAGs...
Due to the missing enzyme-missing degradation of different GAGs-
differnet type of MPS
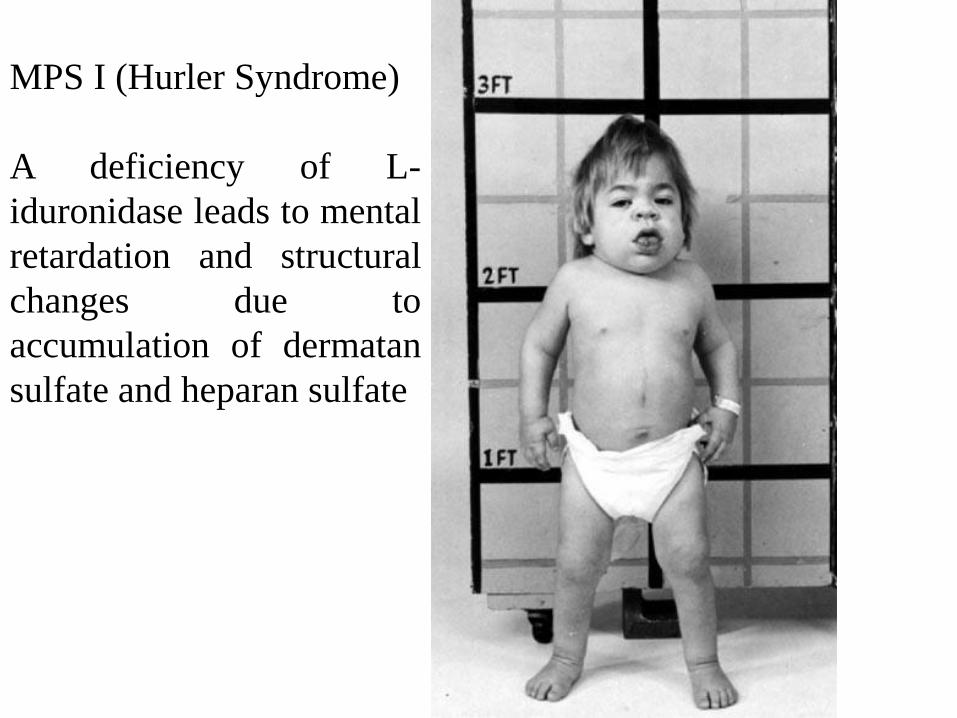
MPS I (Hurler Syndrome)
A deficiency of L-
iduronidase leads to mental
retardation and structural
changes due to
accumulation of dermatan
sulfate and heparan sulfate

MPS II (Hunter Syndrome)
X-linked disease due to a
deficiency of iduronate sulfatase
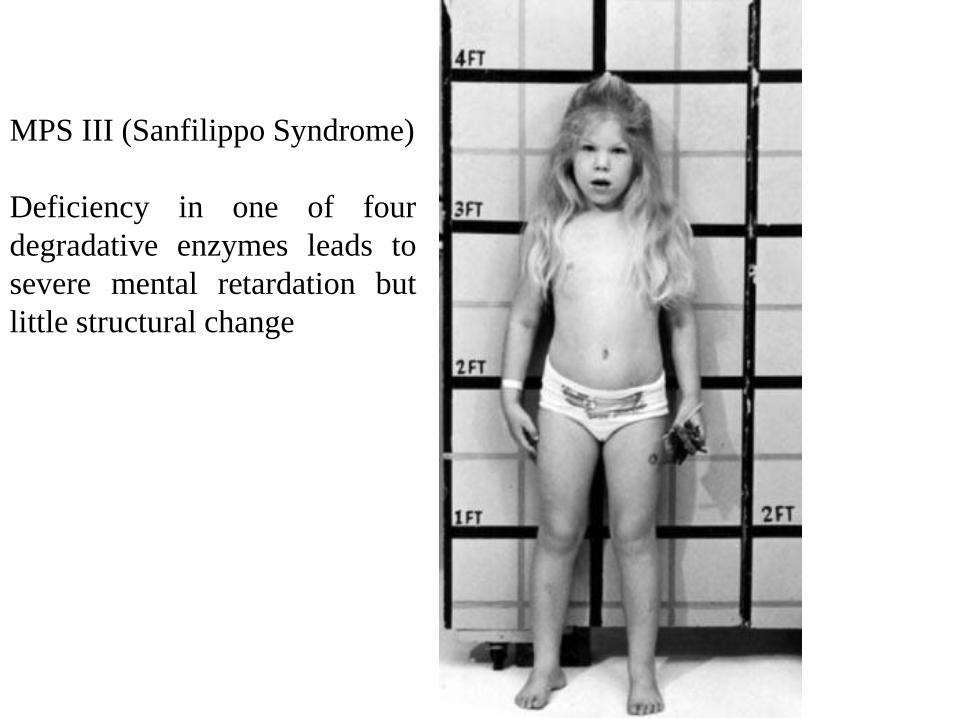
MPS III (Sanfilippo Syndrome)
Deficiency in one of four
degradative enzymes leads to
severe mental retardation but
little structural change

MPS IV (Morquio Syndrome)
Deficiency of a galactose-6-
sulfatase or a beta-
galactosidase leads to
accumulation of keratan
sulfate with normal
intelligence but severe
deformity

• Aspartylglucosaminuria
- deficient activity of the N-aspartyl-beta-glucosaminidase
• Fucosidosis, type I and II
- deficiency of the enzyme alpha-L-fucosidase
• Mannosidosis
- deficient activity of the enzyme alpha (beta)-D-mannosidase.
• Sialidosis, type I and II
- deficient activity of the enzyme alpha-N-acetyl neuraminidase
II - Defective degradation of glycan portion of
glycoproteins
III - Defective degradation of glycogen
• Pompe disease- deficiency of acid alpha-glucosidase

JC Pompe discovered the disease in 1932. The affected gene was identified in 1965 by
Henri G. Hers and he characterized the disease as resulting from the buildup of
glycogen in cells. Researchers realized that the deficiency of the enzyme alpha
glucosidase was the cause. Pompe
diseaseBundle of muscle fibers
Normal muscle fiber (cell)
Affected muscle
fiber (cell)
Sceletal
muscle
Lysosomes are compartments inside each
cell where glycogen is broken down
In Pompe disease, the buildup of
glycogen causes the lysosomes to
expand until they take up so much
space that the muscle cell is damaged
Glycogen begins to leak out of the
lysosomes and cause more damage to the
surrounding muscle cells. This leads to
muscle weakness that gets worse over time
Symptoms
• Early onset
• Cardiac and
respiratory
infections/failure
• Other difficulties:
Floppiness, inability
to gain weight
• Death (within 1 year)
• Late onset
• Difficulty breathing

• Niemann-Pick A & B
- acid sphingomyelinase deficiency
• Fabry disease
- deficient activity of lysosomal enzyme α -galactosidase A
• Farber disease
- ceramidase deficiency
• Gaucher disease, type I, II and III
- characterized by the deposition of glucocerebroside in cells of the
macrophage-monocyte system.
• GM1 gangliosidosis, type I, II and III
- deficiency of acid β–galactosidase, accumulation of GM1 ganglioside,
oligosaccharides, and the mucopolysaccharide keratan sulfate
• GM2 gangliosidosis (Tay-Sachs type I, II, III and Sandhoff)
- deficiency of hexosaminidase A results in accumulation of GM2 in the brain
• Krabbe disease
- deficiency of a galactosylceramide beta-galactosidase (GALC), which
degrades galactosylceramide, a major component of myelin
• Metachromatic leukodystrophy, type I, II and III
- the accumulation of sulfatides
IV- Defective degradation of sphingolipid components
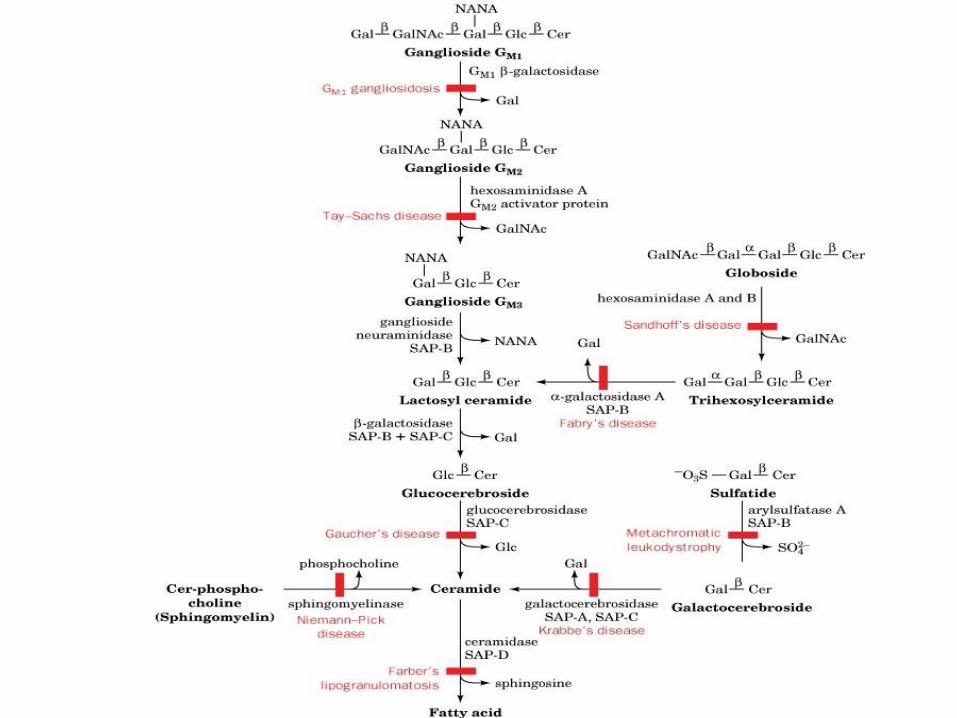
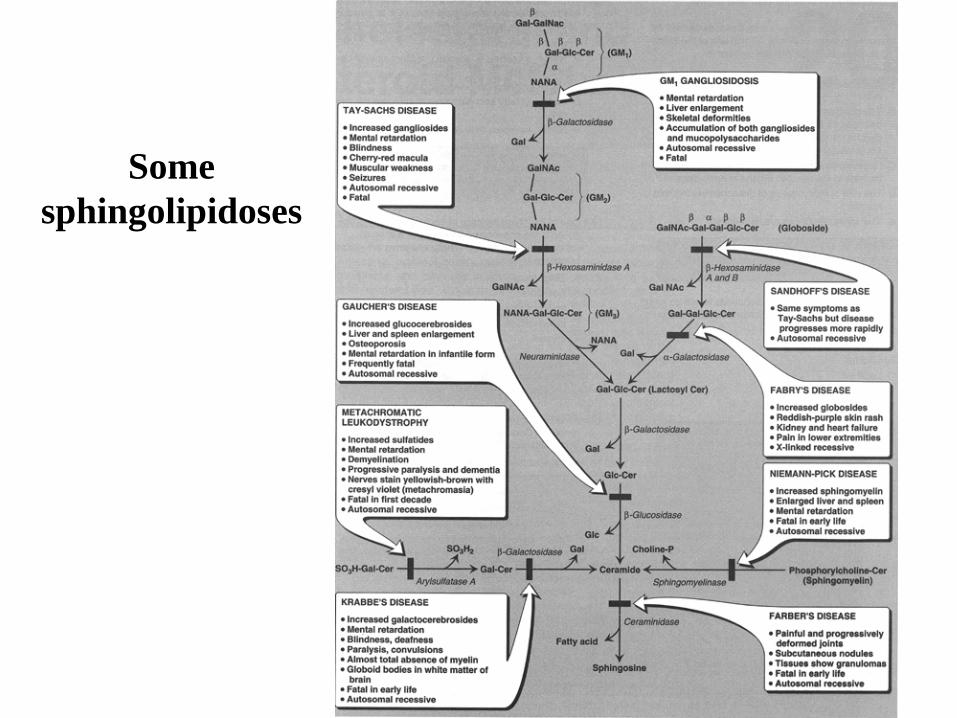
Some
sphingolipidoses
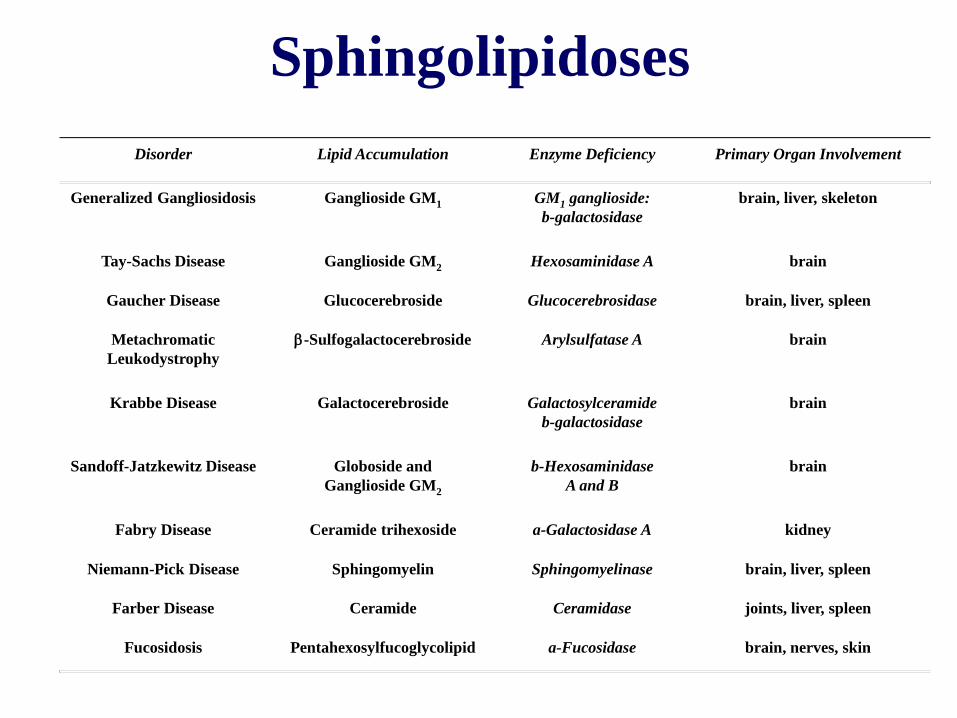
Disorder Lipid Accumulation Enzyme Deficiency Primary Organ Involvement
Generalized Gangliosidosis Ganglioside GM1 GM1 ganglioside:
b-galactosidase
brain, liver, skeleton
Tay-Sachs Disease Ganglioside GM2 Hexosaminidase A brain
Gaucher Disease Glucocerebroside Glucocerebrosidase brain, liver, spleen
Metachromatic
Leukodystrophy
-Sulfogalactocerebroside Arylsulfatase A brain
Krabbe Disease Galactocerebroside Galactosylceramide
b-galactosidase
brain
Sandoff-Jatzkewitz Disease Globoside and
Ganglioside GM2
b-Hexosaminidase
A and B
brain
Fabry Disease Ceramide trihexoside a-Galactosidase A kidney
Niemann-Pick Disease Sphingomyelin Sphingomyelinase brain, liver, spleen
Farber Disease Ceramide Ceramidase joints, liver, spleen
Fucosidosis Pentahexosylfucoglycolipid a-Fucosidase brain, nerves, skin
Sphingolipidoses

• Synthesis (Normal); Degradation (Defective).
• Substrate accumulates in organs.
• Progressive, early death.
• Phenotypic and genotypic variability.
• Autosomal recessive (mostly).
• Rare, Except in Ashkenazi Jewish.
• Usually only a single sphingolipid accumulates in the involved organs in
each disease
SPHINGOLIPIDOSES
• Diagnosis:
• Measure enzyme activity:
• Cultured fibroblasts or peripheral leukocytes.
• Cultured amniocytes or chorionic villi (prenatal).
• Histologic examination.
• DNA analysis.
• Treatment: e.g. for Gaucher disease:
• Replacement Therapy (e.g. recombinant human enzyme).
• Bone marrow transplantation.

Gaucher disease•Etiology: autosomal recessive inherited disease
•Epidemiology: most common lysosomal lipid storage disease
•Pathophysiology: deficiency of β-glucocerebrosidase → accumulation
of glucocerebroside in the brain, liver, spleen, bone marrow.
•Clinical features
• Vary according to the exact subtype of Gaucher disease
• Type I: non-neuronopathic Gaucher disease
• Type II: acute neuronopathic Gaucher disease
• Type III: chronic neuronopathic Gaucher disease
• Hepatosplenomegaly
• Bone: bone crises, osteoporosis, avascular necrosis of the femur
• Blood abnormalities: anemia, thrombocytopenia
• Pulmonary manifestations
• Growth delays
• Neurodegeneration
•Diagnosis:
• Reduced glucocerebrosidase activity in leukocytes or fibroblasts
• Accumulation of glucocerebrosidase in leukocytes or fibroblasts
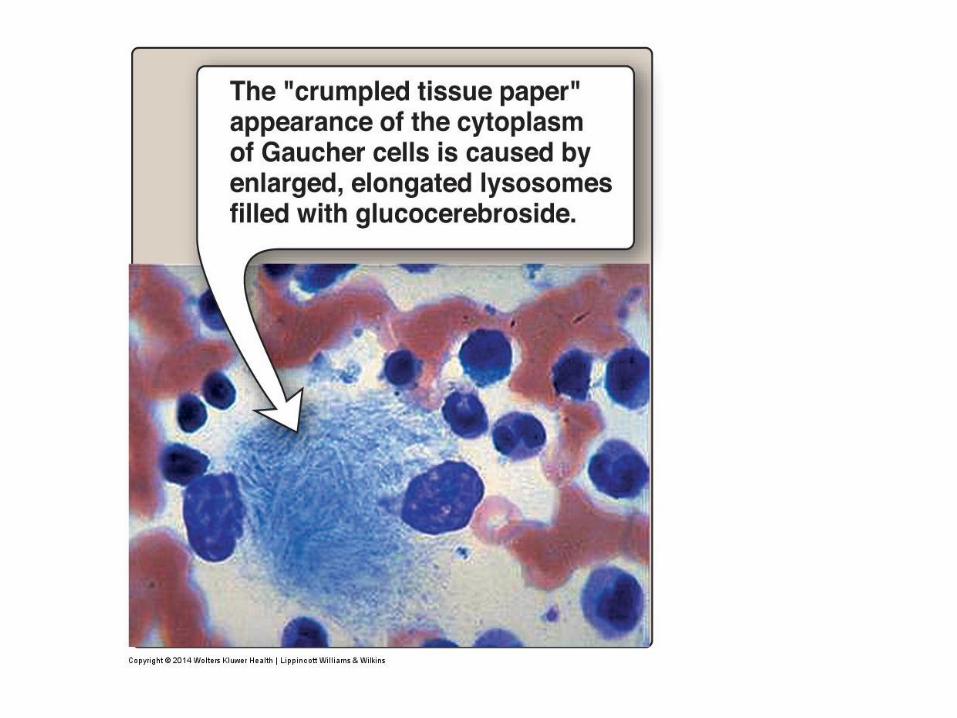
• Gaucher cell: Lipid-rich macrophages with an enlarged cytoplasm with
inclusions that resemble crumpled tissue paper on microscopy.
•Treatment: recombinant glucocerebrosidase
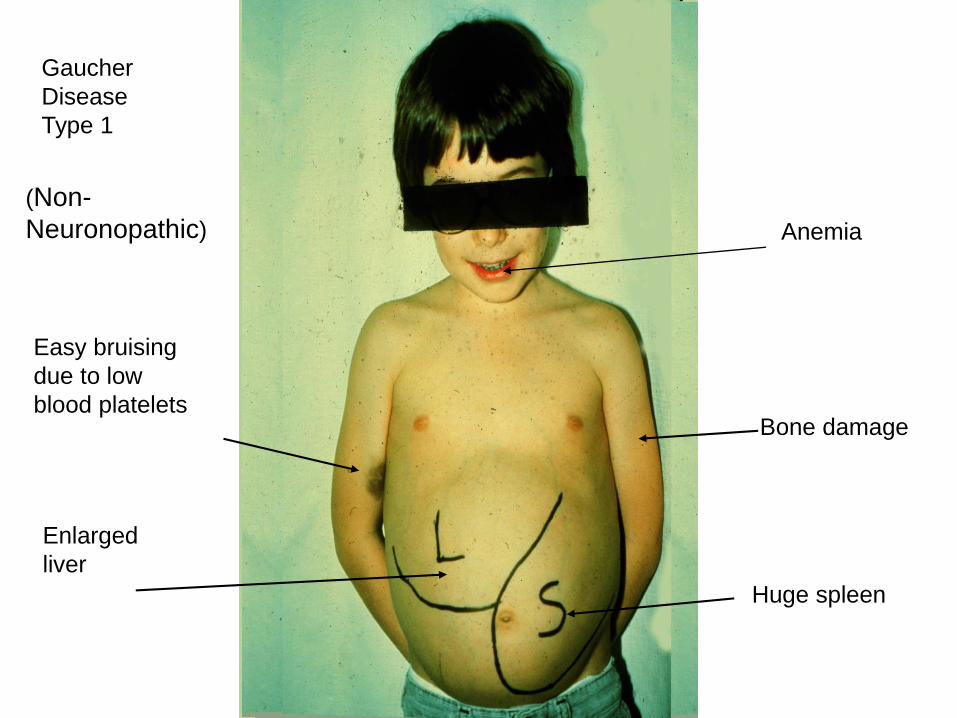
Gaucher
Disease
Type 1
(Non-
Neuronopathic)
Enlarged
liver
Huge spleen
Easy bruising
due to low
blood platelets
Anemia
Bone damage
Krabbe disease•Etiology: autosomal recessive inherited disease
•Pathophysiology: lack of or reduced activity of
the lysosomal enzyme galactocerebrosidase (GALC) → accumulation of
psychosine, galactocerebroside (toxic myelin degradation products) +
formation of globoid cells → demyelination + destruction
of oligodendrocytes up to decerebration
•Clinical features
• Major mental and motor deficits
• Developmental delay
• Peripheral neuropathy
• Limb stiffness
• Opisthotonic posture - causes the back to become extremely
rigid and curved or arched. The heels and head usually bend or
flex backward as far as possible in response.• Loss of vision (atrophy of the optical nerve)

• Diagnosis
• Globoid cell: A large multinucleated cell of mesodermal origin that is
found clustered in the brain tissue
•Treatment
• Before symptom onset, stem cell transplantation may improve outcome.
• Only supportive treatment is possible after symptom onset.

Tay-Sachs disease•Etiology: autosomal recessive inherited disease
•Epidemiology: more common in the Ashkenazi Jewish population
•Pathophysiology: hexosaminidase A deficiency → intracellular
accumulation of GM2 ganglioside → progressive neurodegeneration
•Clinical features
• Developmental delay
• Rapid reduction of physical and mental ability beginning at 6
months of age; patients typically die around the age of 4
• “Cherry-red” spot on macula
• Hypotonia
• Seizures
• Hearing impairment
• Note: no hepatomegaly (compared to Niemann-Pick disease)
•Diagnostics: lysosomes with “onion skin” appearance
•Treatment: supportive
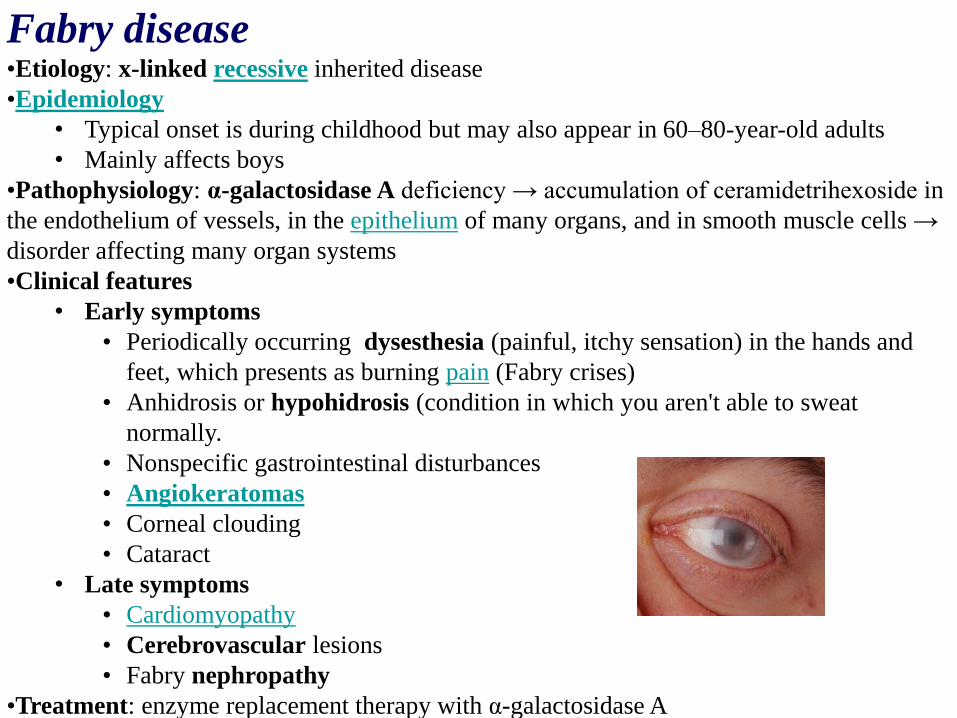
Fabry disease•Etiology: x-linked recessive inherited disease
•Epidemiology
• Typical onset is during childhood but may also appear in 60–80-year-old adults
• Mainly affects boys
•Pathophysiology: α-galactosidase A deficiency → accumulation of ceramidetrihexoside in
the endothelium of vessels, in the epithelium of many organs, and in smooth muscle cells →
disorder affecting many organ systems
•Clinical features
• Early symptoms
• Periodically occurring dysesthesia (painful, itchy sensation) in the hands and
feet, which presents as burning pain (Fabry crises)
• Anhidrosis or hypohidrosis (condition in which you aren't able to sweat
normally.
• Nonspecific gastrointestinal disturbances
• Angiokeratomas
• Corneal clouding
• Cataract
• Late symptoms
• Cardiomyopathy
• Cerebrovascular lesions
• Fabry nephropathy
•Treatment: enzyme replacement therapy with α-galactosidase A

Metachromatic leukodystrophy•Etiology: autosomal recessive inherited disease
•Pathophysiology: arylsulfatase A deficiency → cerebroside sulfate
accumulation in neural and non-neural tissue → central and peripheral
nervous system demyelination
•Clinical features:
• Infantile onset
• Motor regression and developmental delay
• Impaired memory
• Ataxia
• Hypotonia and hyporeflexia
• Optic nerve atrophy → loss of vision
• Flaccid paralysis (reduced muscle tone) followed by spastic
paralysis
• Childhood and juvenile onset
• Ataxia
• Dementia
• Peripheral neuropathy
•Treatment: supportive
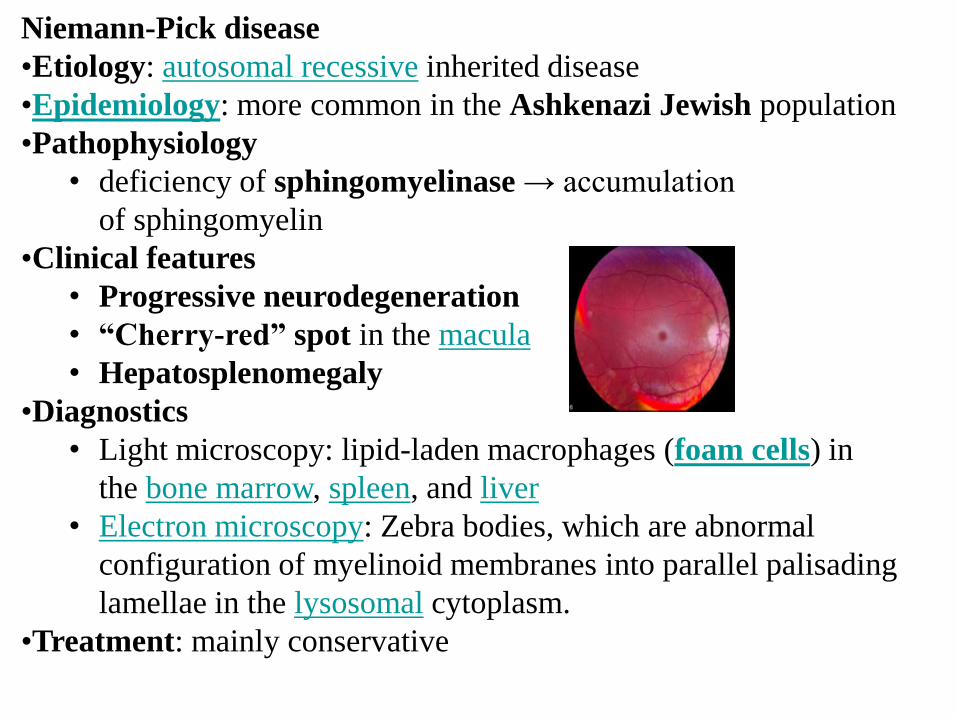
Niemann-Pick disease
•Etiology: autosomal recessive inherited disease
•Epidemiology: more common in the Ashkenazi Jewish population
•Pathophysiology
• deficiency of sphingomyelinase → accumulation
of sphingomyelin
•Clinical features
• Progressive neurodegeneration
• “Cherry-red” spot in the macula
• Hepatosplenomegaly
•Diagnostics
• Light microscopy: lipid-laden macrophages (foam cells) in
the bone marrow, spleen, and liver
• Electron microscopy: Zebra bodies, which are abnormal
configuration of myelinoid membranes into parallel palisading
lamellae in the lysosomal cytoplasm.
•Treatment: mainly conservative

• lipid-laden macrophages (foam cells) in the bone marrow, spleen,
and liver

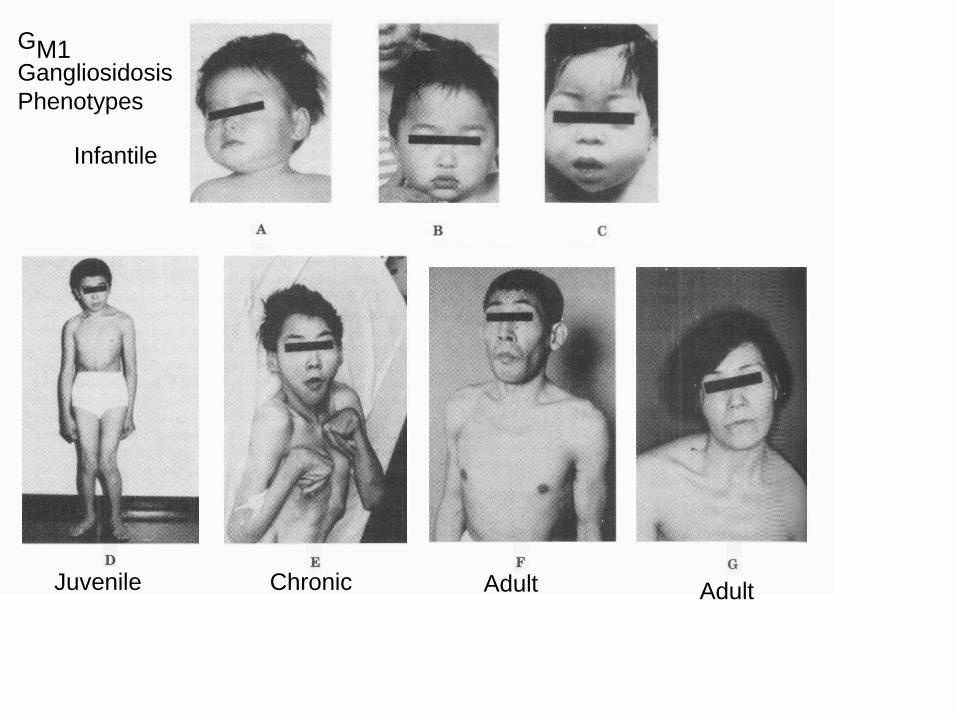
Juvenile
Infantile
Chronic Adult Adult
GM1Gangliosidosis
Phenotypes

Hypotonia
Hypotonia, commonly known as floppy baby syndrome, is a state of
low muscle tone often involving reduced muscle strength.

Coarse Facial Features
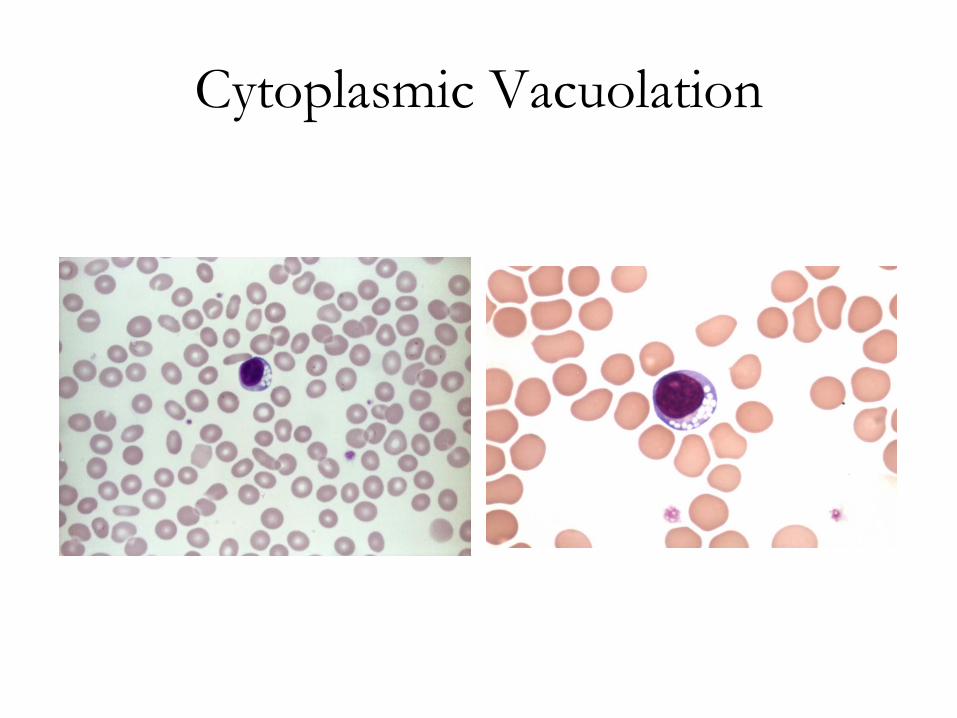
Cytoplasmic Vacuolation
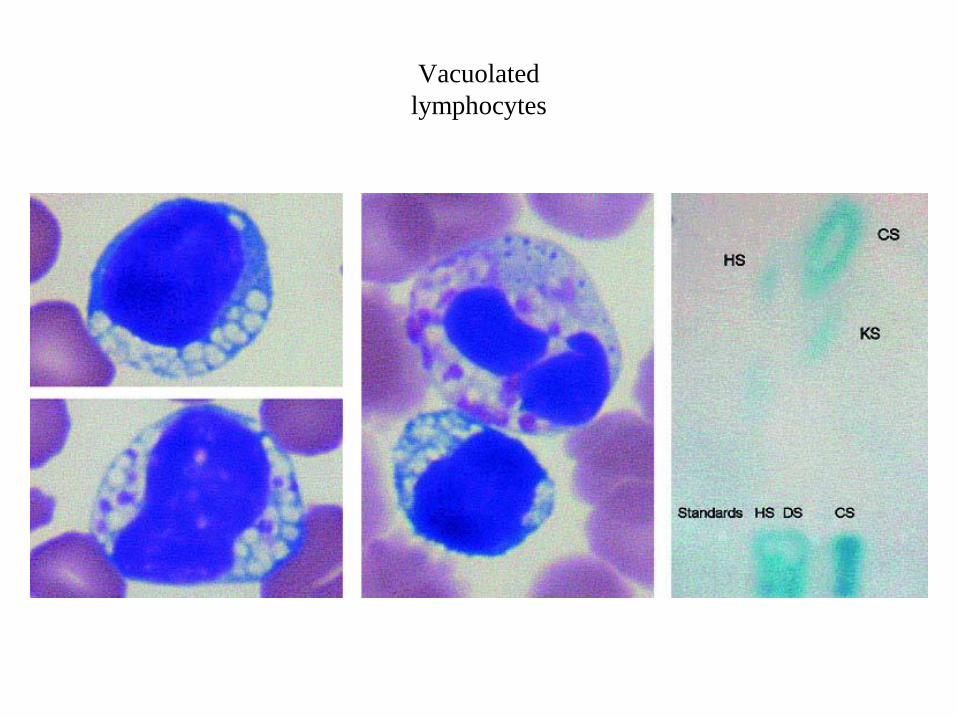
Vacuolated
lymphocytes
Lymphocytes with multiple medium-sized vacuoles (top left).
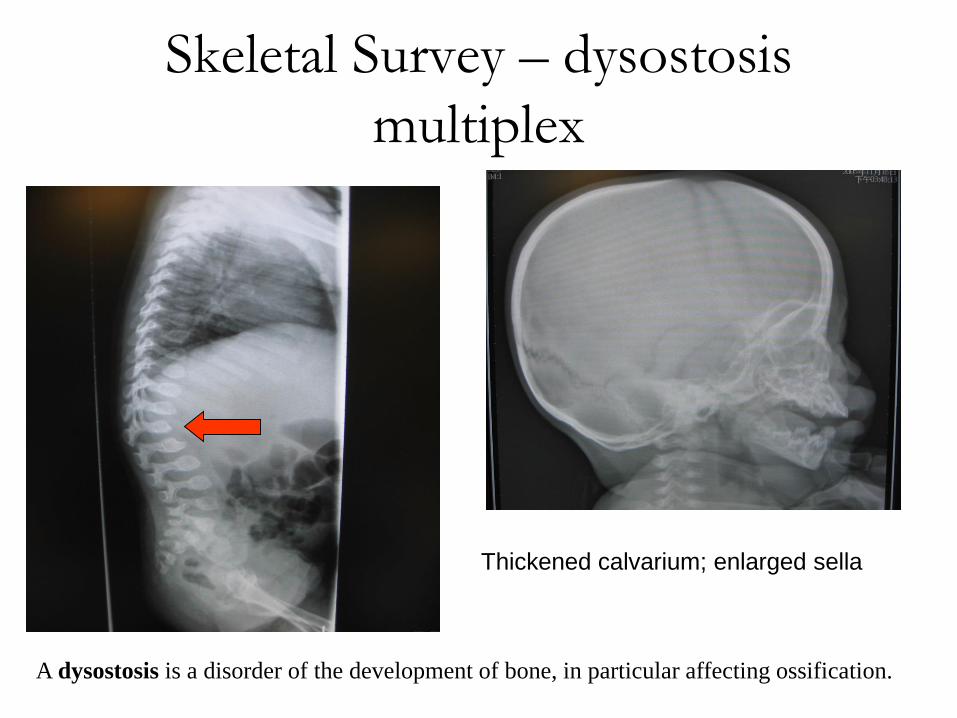
Skeletal Survey – dysostosis
multiplex
Thickened calvarium; enlarged sella
A dysostosis is a disorder of the development of bone, in particular affecting ossification.

V - Defective degradation of polypeptides
• Pycnodysostosis
- mutation in the gene that codes the enzyme cathepsin K
VI - Defective degradation or transport of
cholesterol, cholesterol esters, or other complex
lipids
• Neuronal ceroid lipofuscinosis, type I, II, III and IV
(Batten disease)
VII - Multiple deficiencies of lysosomal enzymes
• Galactosialidosis
• Mucolipidosis, type II and III
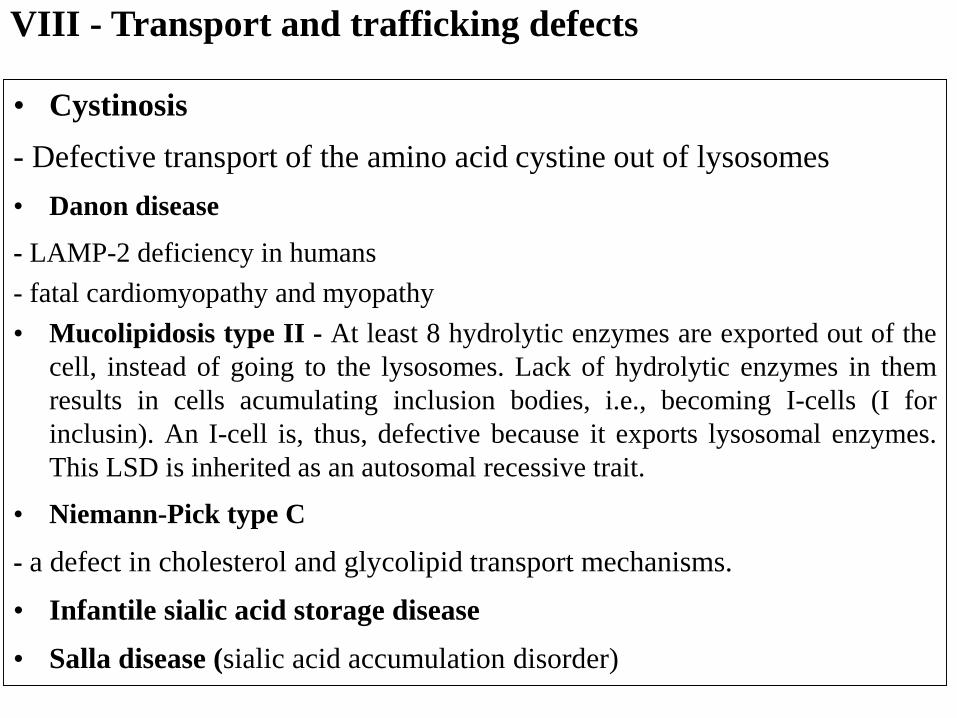
VIII - Transport and trafficking defects
• Cystinosis
- Defective transport of the amino acid cystine out of lysosomes
• Danon disease
- LAMP-2 deficiency in humans
- fatal cardiomyopathy and myopathy
• Mucolipidosis type II - At least 8 hydrolytic enzymes are exported out of the
cell, instead of going to the lysosomes. Lack of hydrolytic enzymes in them
results in cells acumulating inclusion bodies, i.e., becoming I-cells (I for
inclusin). An I-cell is, thus, defective because it exports lysosomal enzymes.
This LSD is inherited as an autosomal recessive trait.
• Niemann-Pick type C
- a defect in cholesterol and glycolipid transport mechanisms.
• Infantile sialic acid storage disease
• Salla disease (sialic acid accumulation disorder)

I-cell disease is a rare lysosomal storage disease in which the acid
hydrolases normally found in lysosomes are absent, resulting in an
accumulation of substrates normally degraded by these enzymes.
[Note: I-cell disease is so-named because of the large inclusion
bodies seen in cells of patients with this disease]. In addition, high
amounts of lysosomal enzymes are found in the patient’s plasma and
urine, indicating that the targeting process to lysosomes (rather than
the synthetic pathway of these enzymes) is deficient. Individuals
with I-cell disease are lacking the phosphotransferase needed to
phosphorylate the mannose residues of potential lysosomal
enzymes, causing the enzymes to be secreted (by default), rather
than being targeted to lysosomal vesicles. I-cell disease is
characterized by skeletal abnormalities, restricted joint movement,
coarse (dysmorphic) facial features, and severe psychomotor
impairment. Currently, there is no cure, and death from
cardiopulmonary complications usually occurs by age 10 years.

CURRENT AND FUTURE STRATEGIES FOR THE
TREATMENT OF METABOLIC STORAGE DISORDERS
1. BONE MARROW TRANSPLANTATION
2. ENZYME REPLACEMENT THERAPY
3. SUBSTRATE REDUCTION THERAPY
4. MOLECULAR CHAPERONE THERAPY
5. GENE THERAPY ?

How common are lysosomal diseases?
a. 1 in 5000 live births
b. 1 in 50,000 live births
c. 1 in 100,000 live births
Answer: a.

With what frequency are children born world-wide
with a lysosomal disease?
a. Every 30 minutes
b. Every 2 hours
c. Once a day
Answer: a.




![1 INCOTERMS 2010 (1)[1]](https://static.fdokumen.com/doc/165x107/631de3d1dc32ad07f3074e54/1-incoterms-2010-11.jpg)















