
sintesis conceptual 8 biología celular 2022
64
SINTESIS CONCEPTUAL 8 BIOLOGÍA CELULAR 2022 Distribución de las moléculas y organelas en células normales y en enfermedad Desde el punto de vista de la molécula y su señal se procederá a comprender los mecanismos de direccionamiento intracelular y los posibles defectos en condiciones patológicas.
-
Upload
khangminh22 -
Category
Documents
-
view
9 -
download
0
Transcript of sintesis conceptual 8 biología celular 2022

SINTESIS CONCEPTUAL 8BIOLOGÍA CELULAR 2022
Distribución de las moléculas y organelas en células normales y en enfermedad
Desde el punto de vista de la molécula y su señal se procederá a comprender los mecanismos de
direccionamiento intracelular y los posibles defectos en condiciones patológicas.

Objetivos básicos1) Describir los componentes de membrana de la célula y como están compuestas y localizadas las diferentes estructuras y organelas.2) Comprender las vías de síntesis de proteínas con localización citosólica, nuclear, de membrana o envueltas en membrana y de exportación extracelular.3) Comprender las señales direccionadoras de proteínas acopladas a los procesos de traducción y de plegamiento.4) Comprender las señales direccionadoras de proteínas dirigidas a vesículas y organelas acopladas a los procesos de maduración, transporte, degradación y secreción.4) Relacionar los conceptos del seminario asociados con enfermedades vinculadas a defectos de direccionamiento de proteínas o disfunciones del sistema de endomembranas.
Objetivos
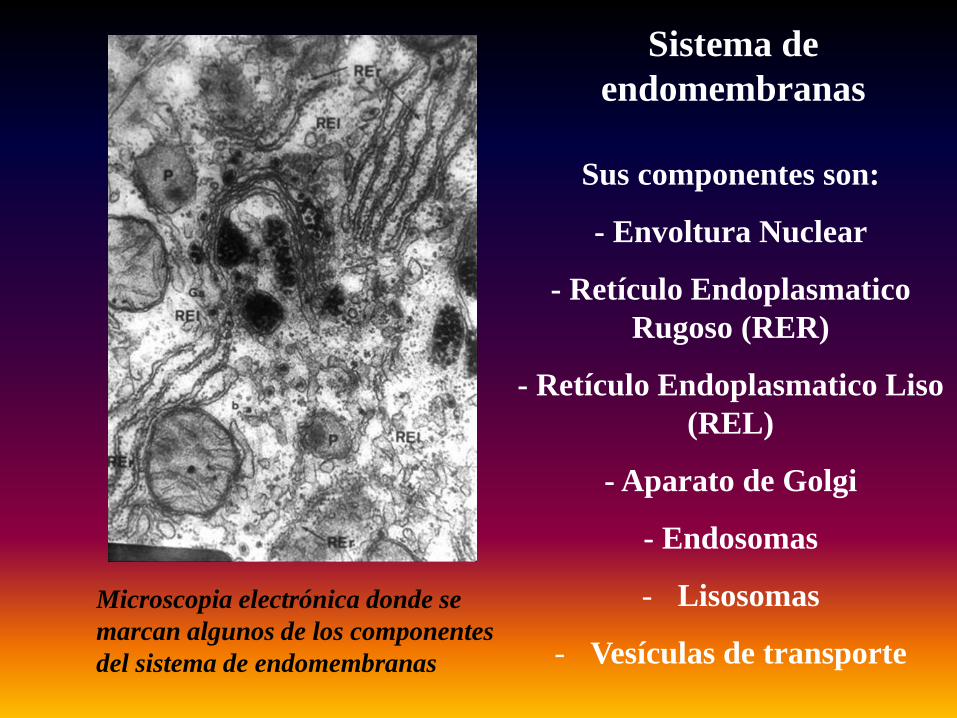
Sistema de endomembranas
Sus componentes son:
- Envoltura Nuclear
- Retículo EndoplasmaticoRugoso (RER)
- Retículo Endoplasmatico Liso (REL)
- Aparato de Golgi
- Endosomas
- Lisosomas
- Vesículas de transporte
Microscopia electrónica donde se marcan algunos de los componentes del sistema de endomembranas
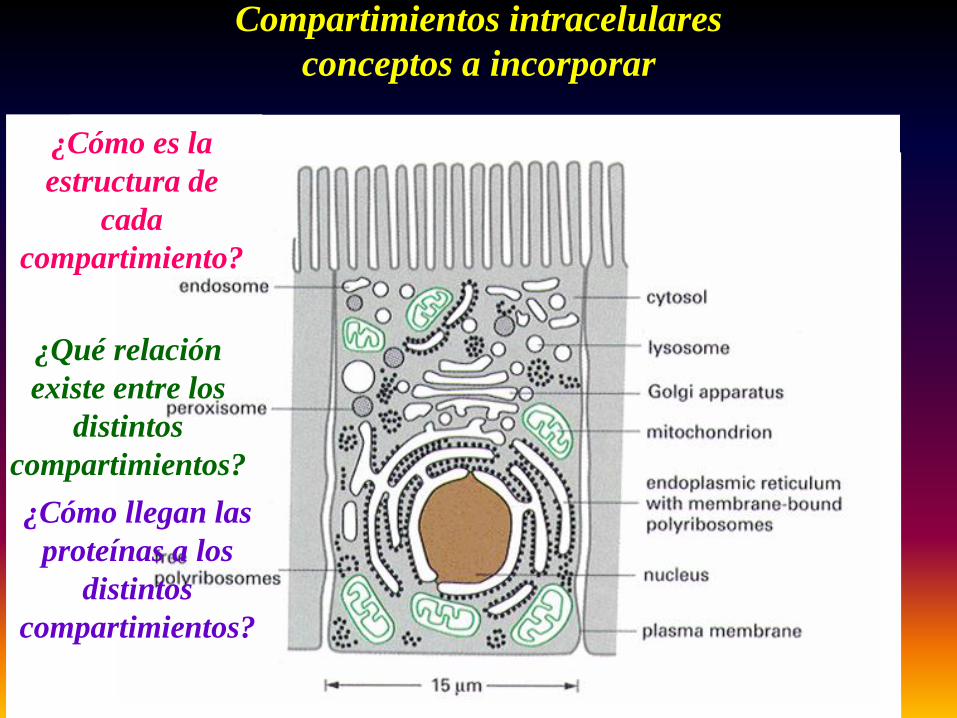
Compartimientos intracelularesconceptos a incorporar
¿Cómo llegan las proteínas a los
distintos compartimientos?
¿Qué relación existe entre los
distintos compartimientos?
¿Cómo es la estructura de
cada compartimiento?

COMO SE DETERMINA ADONDE DEBEN DIRIGIRSE
LAS PROTEINAS DENTRO DE LA CELULA?
POR MEDIO DE SEÑALES ENCRIPTADAS EN SU
SECUENCIA

Sintesis de Proteinas. Ribosomas libres y unidos a membranas
Todos las mensajeros inician su traducción en ribosomas libres en el citosol. Estos ribosomas según las señales de las proteínas
que se encuentran sintetizando son dirigidos al retículo endoplasmático rugoso o permanecen libres en el citosol
Ribosomas libres Ribosomas en RER

La envoltura nuclear
Propiedades y funciones
•La existencia de un núcleo es la principal característica que diferencia a una célula eucariota de una procariota.
•La presencia de una doble membrana delimitando al núcleo permite la aparición de mecanismos de regulación de la expresión génica que no ocurren en procariotas.
• Los ARNs eucariotas sufren un proceso de maduración en el núcleo antes de ser transportados al citosol para su traducción o funcion.
• La envoltura nuclear limita el acceso de factores transcripcionales desde el citosol al núcleo.
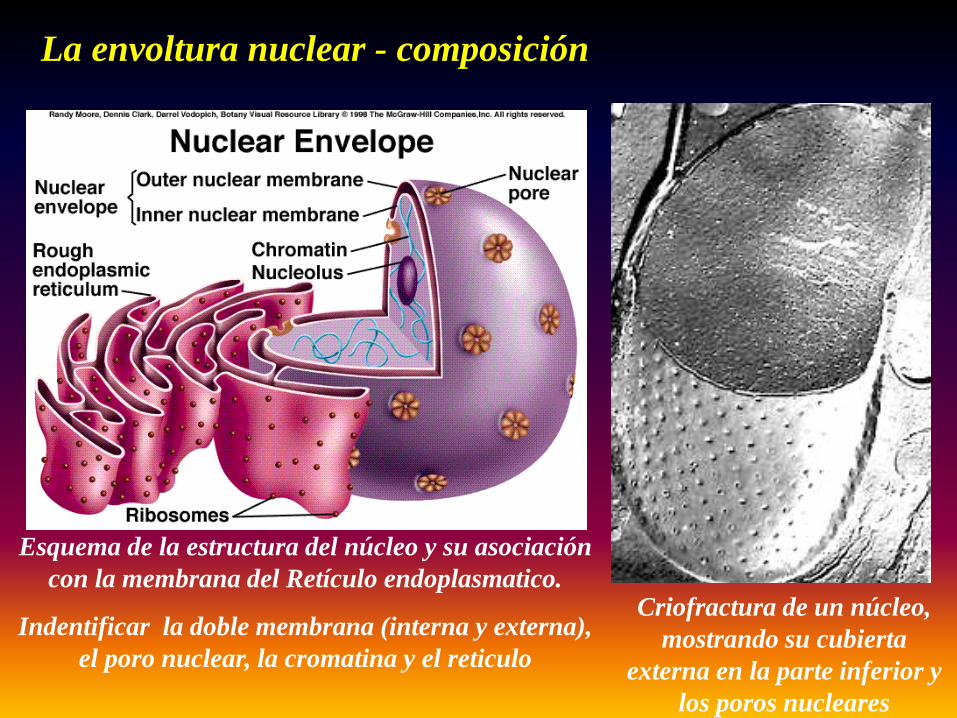
La envoltura nuclear - composición
Criofractura de un núcleo, mostrando su cubierta
externa en la parte inferior y los poros nucleares
Esquema de la estructura del núcleo y su asociación con la membrana del Retículo endoplasmatico.
Indentificar la doble membrana (interna y externa), el poro nuclear, la cromatina y el reticulo
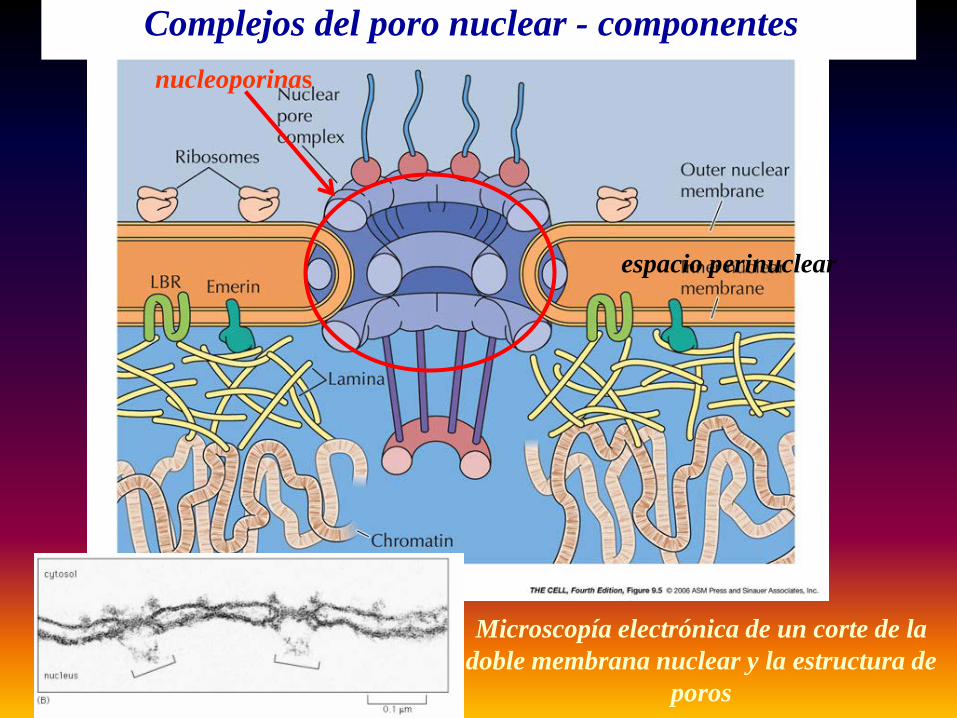
Complejos del poro nuclear - componentesnucleoporinas
espacio perinuclear
Microscopía electrónica de un corte de la doble membrana nuclear y la estructura de
poros

cara citoplasmática cara nucleoplasmáticacara nucleoplasmática
con lamina nuclear
Complejos del poro nuclear
Microscopía electrónica de barrido de ambas caras del complejo del poro nuclear
Reconocer componentes
Complejos del poro nuclear – sistema de entrada y salida de proteínas y mensajeros del núcleo
Estructura del Poro
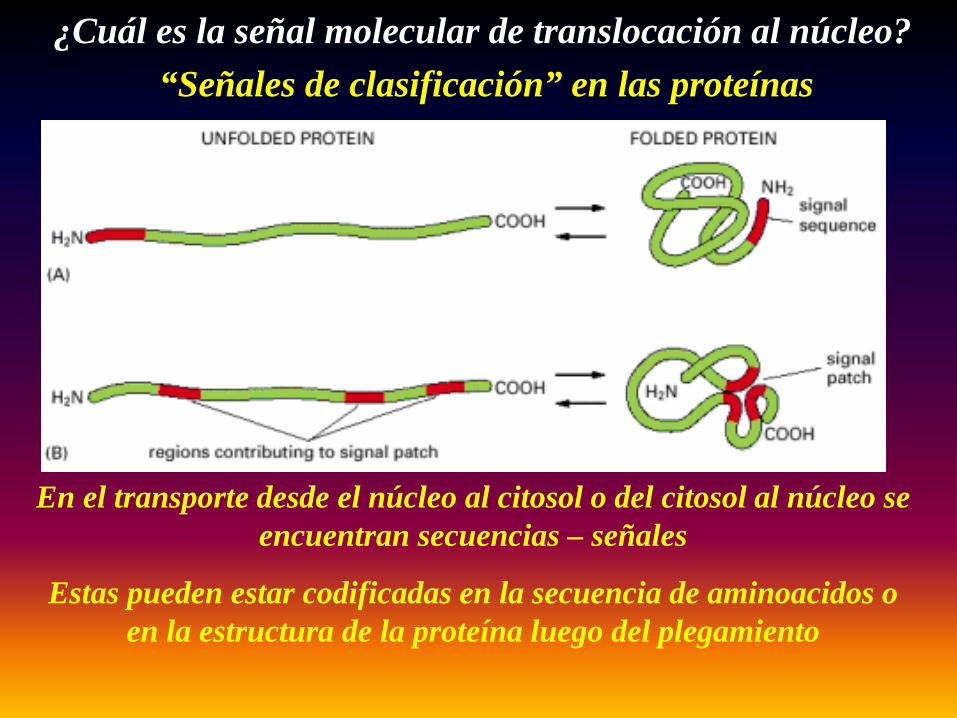
“Señales de clasificación” en las proteínas
En el transporte desde el núcleo al citosol o del citosol al núcleo se encuentran secuencias – señales
Estas pueden estar codificadas en la secuencia de aminoacidos o en la estructura de la proteína luego del plegamiento
¿Cuál es la señal molecular de translocación al núcleo?

NLS
(Nuclear Localization Signals o
Señales de Localización Nuclear)

Que pasa si se muta la codificación de la NLS? Se impide la traslocación al núcleo
Corroboraciones experimentales de la funcionalidad de la secuencia NLS
En el siguiente experimento se introdujo en las células una proteína fluorescente con distintas codificaciones de señales en su secuencia. Luego se observó donde se
dirigió la proteína.
Determine en cual de las dos imágenes la proteína esta en el núcleo y en cual en el citoplasma?

•La GTPasa llamada ‘Ran’ le da direccionalidad al transporte a través del poro nuclear. Ran se unea las proteínas que deben ser direccionadas hacia el núcleo o fuera de el.
•Si Ran esta unida a GDP en el citoplasma puede asociarse al poro nuclear y dirige a lasproteinas hacia el núcleo.
•Si Ran esta unida a GTP dentro del núcleo puede asociarse al poro y se dirige a las proteinashacia el citoplasma.
•Cuando Ran-GDP entra al núcleo, otra proteína, Ran-GEF (factor intercambiador de guanosina) lareconoce y le cambia el GDP por un GTP. Ahora Ran puede salir.
•Cuando Ran-GTP sale al citoplasma. Otra proteina Ran-GAP (activador de GTPasa) le saca unfosfato al GTP de Ran y lo transforma de nuevo en Ran-GDP, ahora puede entra de nuevo alnúcleo
Ciclo de entrada y salida a través del poro nuclear
Proteína activadora de la GTPasa
Factor intercambiador de guanosina
Es muy importante comprender como Ran
dirige el transporte
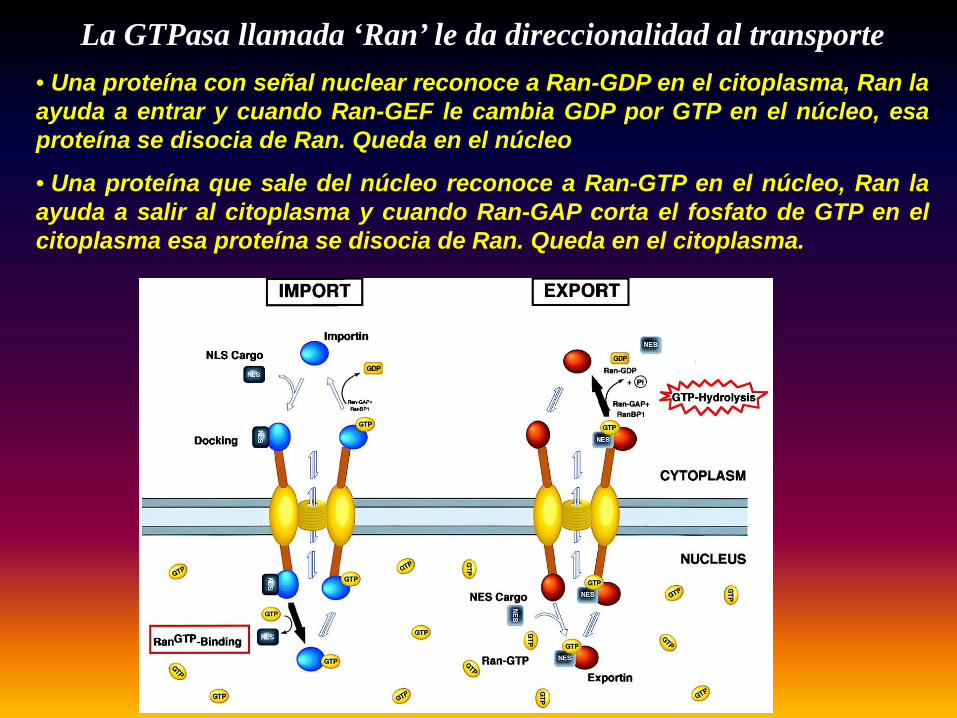
La GTPasa llamada ‘Ran’ le da direccionalidad al transporte• Una proteína con señal nuclear reconoce a Ran-GDP en el citoplasma, Ran laayuda a entrar y cuando Ran-GEF le cambia GDP por GTP en el núcleo, esaproteína se disocia de Ran. Queda en el núcleo
• Una proteína que sale del núcleo reconoce a Ran-GTP en el núcleo, Ran laayuda a salir al citoplasma y cuando Ran-GAP corta el fosfato de GTP en elcitoplasma esa proteína se disocia de Ran. Queda en el citoplasma.
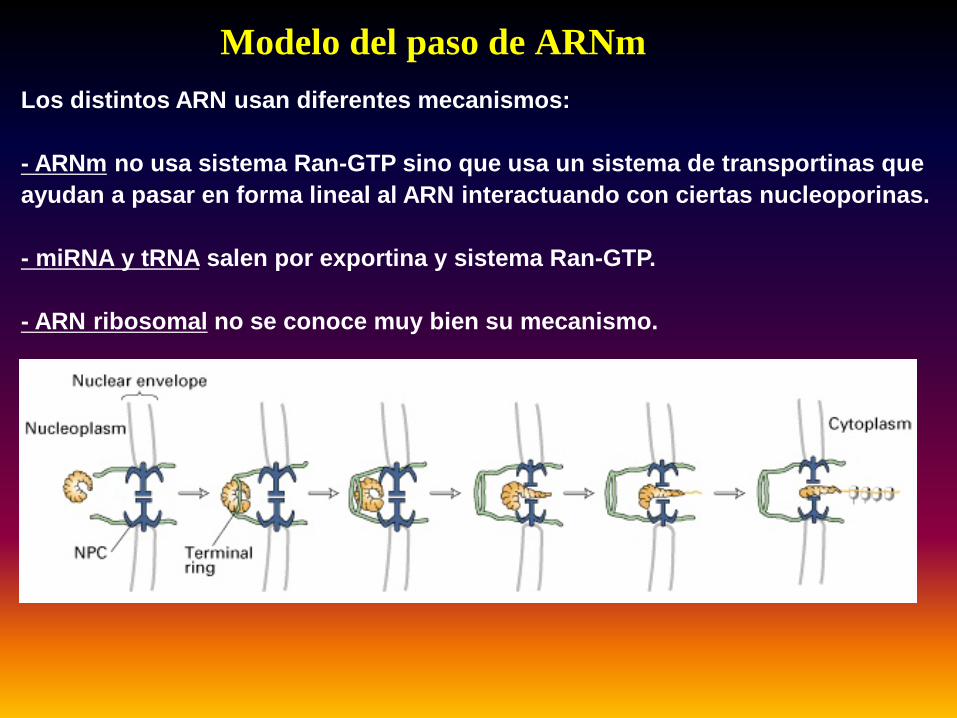
Modelo del paso de ARNmLos distintos ARN usan diferentes mecanismos:
- ARNm no usa sistema Ran-GTP sino que usa un sistema de transportinas que ayudan a pasar en forma lineal al ARN interactuando con ciertas nucleoporinas.
- miRNA y tRNA salen por exportina y sistema Ran-GTP.
- ARN ribosomal no se conoce muy bien su mecanismo.

Señales de Localización o importacion Mitocondrial
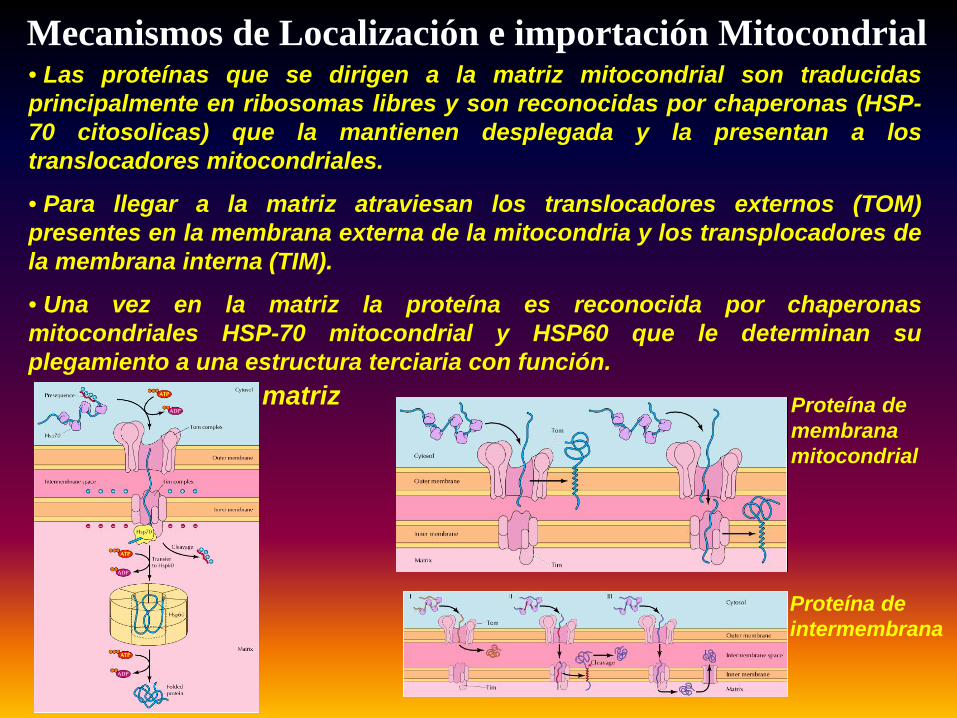
Mecanismos de Localización e importación Mitocondrial• Las proteínas que se dirigen a la matriz mitocondrial son traducidasprincipalmente en ribosomas libres y son reconocidas por chaperonas (HSP-70 citosolicas) que la mantienen desplegada y la presentan a lostranslocadores mitocondriales.
• Para llegar a la matriz atraviesan los translocadores externos (TOM)presentes en la membrana externa de la mitocondria y los transplocadores dela membrana interna (TIM).
• Una vez en la matriz la proteína es reconocida por chaperonasmitocondriales HSP-70 mitocondrial y HSP60 que le determinan suplegamiento a una estructura terciaria con función.
matriz Proteína de membrana mitocondrial
Proteína de intermembrana

Importación al RE
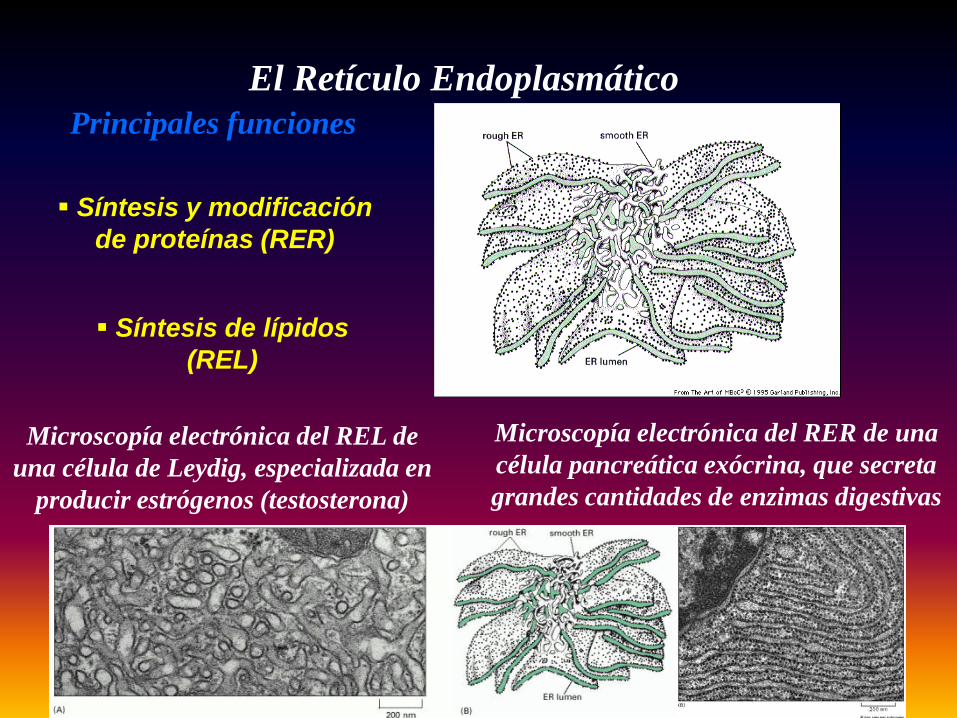
El Retículo EndoplasmáticoPrincipales funciones
Síntesis y modificación de proteínas (RER)
Síntesis de lípidos (REL)
Microscopía electrónica del RER de una célula pancreática exócrina, que secreta grandes cantidades de enzimas digestivas
Microscopía electrónica del REL de una célula de Leydig, especializada en
producir estrógenos (testosterona)

Mecanismo de importación al RE
Partícula de reconocimiento de la señal (PRS=SRP)
Ribosoma traduciendo una proteína en traslocación
• Las proteínas de membrana, las proteínas secretadas, o las proteínas de lossistemas de endomembranas (Ej: lisosomas) son inicialmente traducidas enribosomas libres. Sin embargo apenas aparece la codificación del péptidoseñal que las guía, la síntesis se detiene y el ribosoma es acoplado al RERgracias a la partícula de reconocimiento de señal (PRS).
• Luego la traducción continua en los ribosomas asociados al retículo y laproteína es translocada al lumen del retículo.

Modelo de translocación de una proteína soluble al lumen del RE
“peptidasa de la señal”
• Una proteína soluble que será secretada o parte del sistema de membranassiendo enviada al retículo. En rojo se encuentra el péptido señal que esclivado por la peptidasa presente en RER. Una vez cortada la señal la proteínapuede seguir la vía de retículo y adquirir modificaciones y plegamiento.
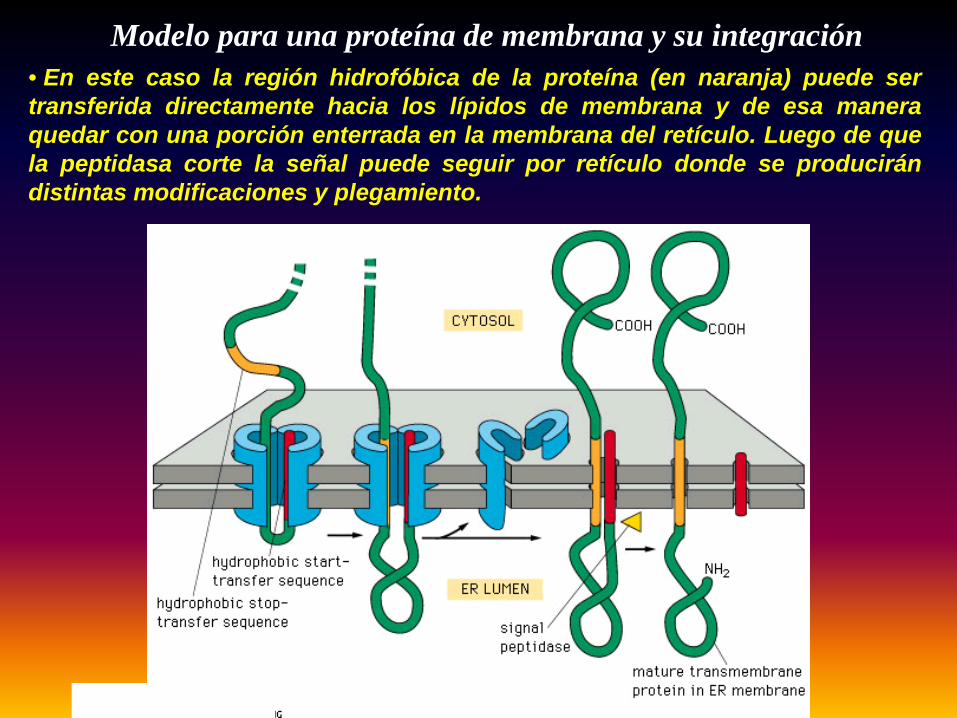
Modelo para una proteína de membrana y su integración• En este caso la región hidrofóbica de la proteína (en naranja) puede sertransferida directamente hacia los lípidos de membrana y de esa maneraquedar con una porción enterrada en la membrana del retículo. Luego de quela peptidasa corte la señal puede seguir por retículo donde se producirándistintas modificaciones y plegamiento.
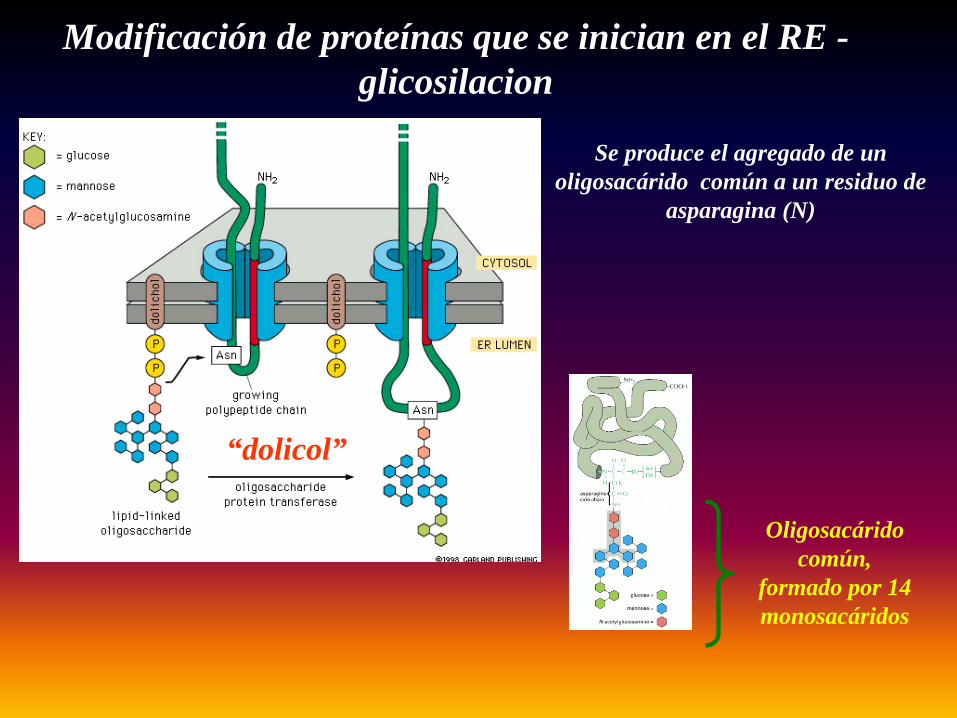
“dolicol”
Modificación de proteínas que se inician en el RE -glicosilacion
Oligosacárido común,
formado por 14 monosacáridos
Se produce el agregado de un oligosacárido común a un residuo de
asparagina (N)
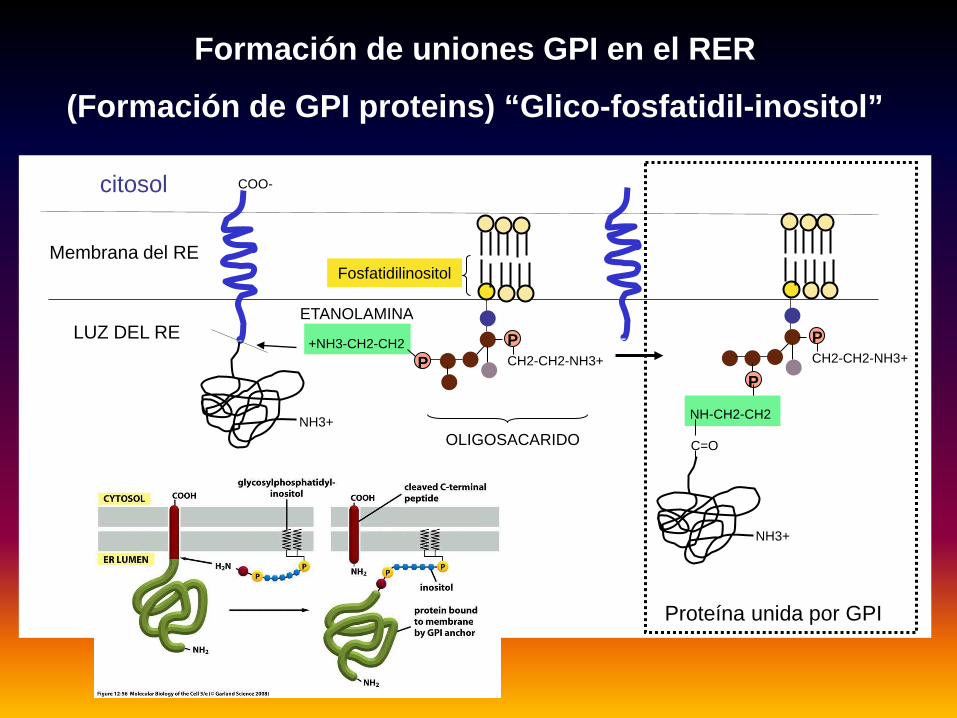
Formación de uniones GPI en el RER
(Formación de GPI proteins) “Glico-fosfatidil-inositol”
citosol
PPCH2-CH2-NH3+
+NH3-CH2-CH2
ETANOLAMINA
P
PCH2-CH2-NH3+
NH-CH2-CH2
C=OOLIGOSACARIDO
LUZ DEL RE
Membrana del RE
NH3+
NH3+
COO-
Proteína unida por GPI
Fosfatidilinositol
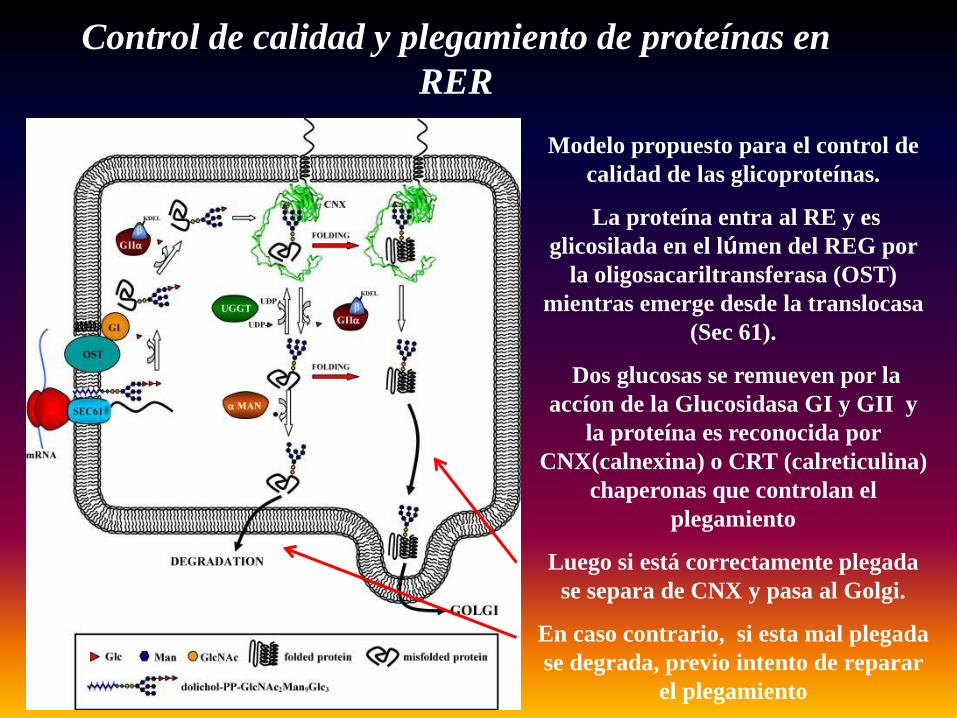
Plegamiento de la proteína en el REG
Modelo propuesto para el control de calidad de las glicoproteínas.
La proteína entra al RE y es glicosilada en el lúmen del REG por
la oligosacariltransferasa (OST) mientras emerge desde la translocasa
(Sec 61).
Dos glucosas se remueven por la accíon de la Glucosidasa GI y GII y
la proteína es reconocida por CNX(calnexina) o CRT (calreticulina)
chaperonas que controlan el plegamiento
Luego si está correctamente plegada se separa de CNX y pasa al Golgi.
En caso contrario, si esta mal plegada se degrada, previo intento de reparar
el plegamiento
Control de calidad y plegamiento de proteínas en RER

FUNCIONES DEL REG• Síntesis y modificaciones de proteínas de membrana,
lisosomales y de exportación (N-Glicosilación (Asp) y formación de proteínas GPI).
• Agregado a la N –Aspargina de la proteína un oligosacáridos formado por 14 monosacáridos a partir del Dolicol
• Participar en el plegamiento de la proteína
• Controlar el plegamiento protéico. Determinar si la proteína está correctamente plegada.
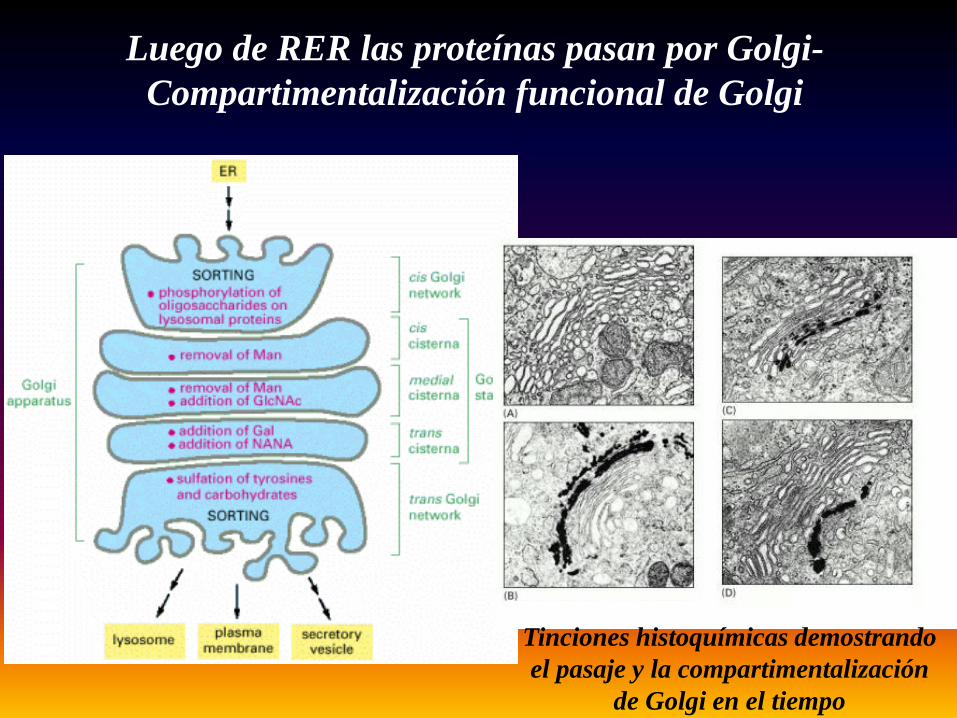
Luego de RER las proteínas pasan por Golgi-Compartimentalización funcional de Golgi
Tinciones histoquímicas demostrando el pasaje y la compartimentalización
de Golgi en el tiempo

Las cadenas de oligosacáridos iniciadas en RER terminan de procesarse y madurar en Golgi
• La incorporación de un oligosacarido en RER comienza a ser modificado.Esta modificación y procesamiento sigue en Golgi y puede adquirir diferentesmodificaciones. En el ejemplo la proteína glicosilada adquiere en Golgi dosmodificaciones finales. 1 es la manosa de 6 fosfatos que dirige esta proteínahacia el sistema de lisosomas. 2 es una formación compleja deoligosacaridos con altas cargas negativas que la llevan a ser posicionada enla membrana plasmática externa.

oligosacáridos complejos
oligosacáridos ricos
en manosa
Las cadenas de oligosacáridos se procesan en el Aparato de Golgi
En el Aparato de Golgi también se produce la O-glicosilación (en
residuos de Ser) y la glicosilación de lípidos a partir de ceramida.

FUNCIONES de GOLGI• Procesamiento y glicosilación de proteínas
(N- y O-glycosilation).• Distribución y exportación de proteínas (Ej.
Manosa-6-fosfato “Man-6P”).• Síntesis del fosfolípido esfingomielina y de
glicolípidos.
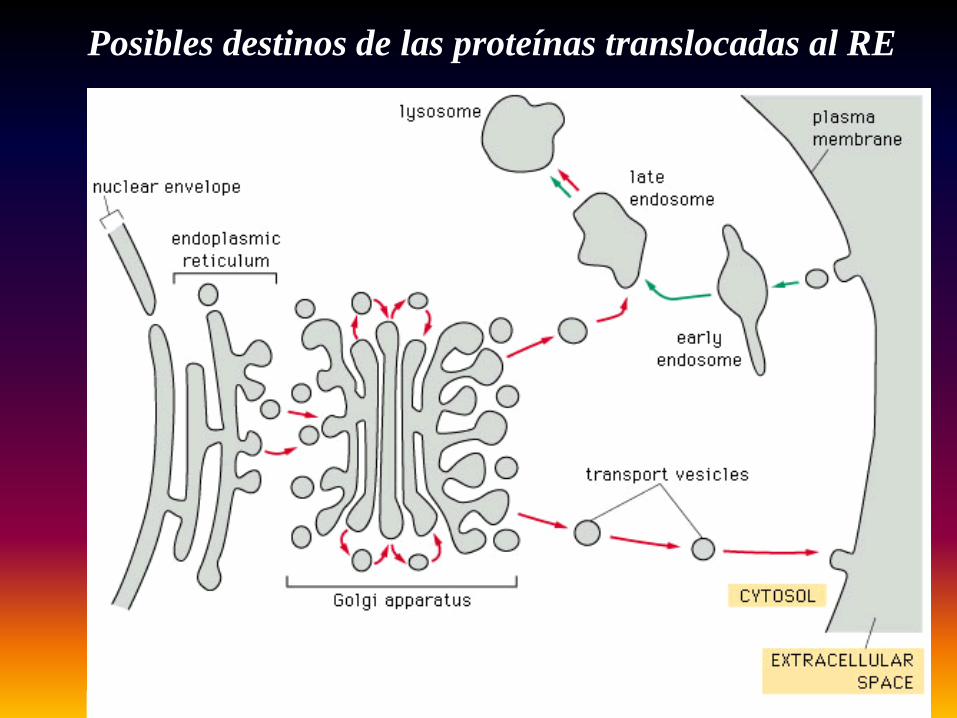
Posibles destinos de las proteínas translocadas al RE
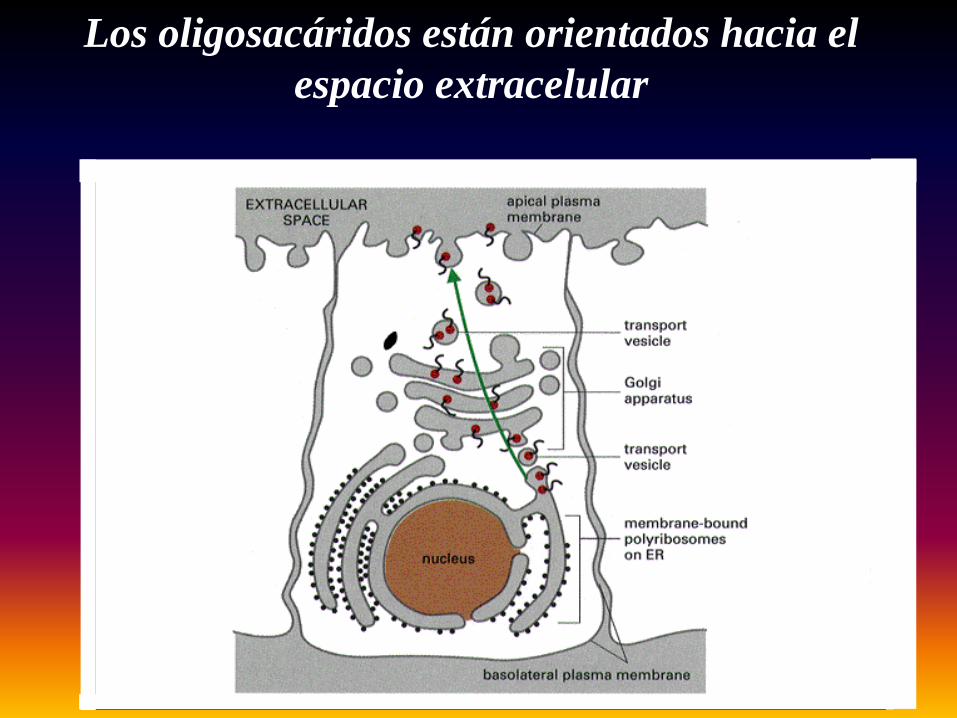
Los oligosacáridos están orientados hacia el espacio extracelular
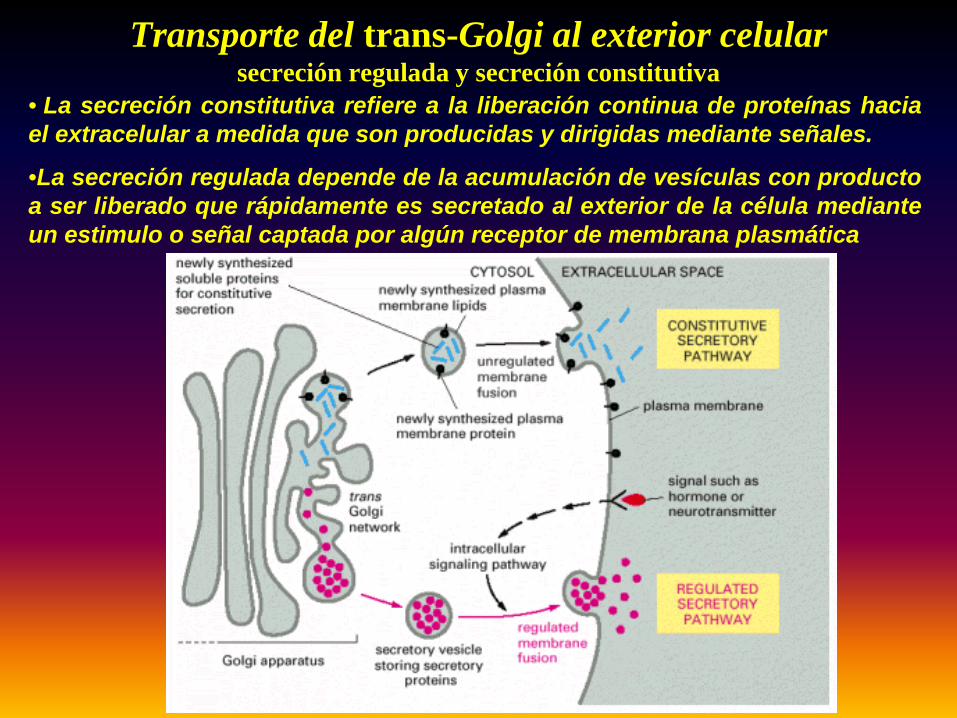
Transporte del trans-Golgi al exterior celularsecreción regulada y secreción constitutiva
• La secreción constitutiva refiere a la liberación continua de proteínas haciael extracelular a medida que son producidas y dirigidas mediante señales.
•La secreción regulada depende de la acumulación de vesículas con productoa ser liberado que rápidamente es secretado al exterior de la célula medianteun estimulo o señal captada por algún receptor de membrana plasmática
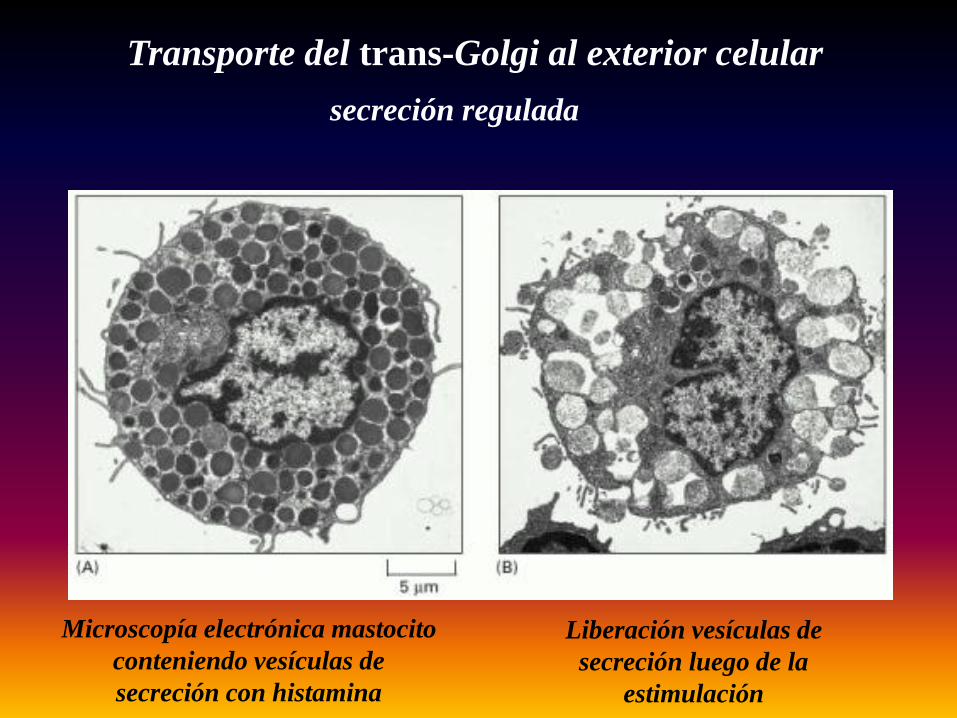
Transporte del trans-Golgi al exterior celularsecreción regulada
Microscopía electrónica mastocito conteniendo vesículas de secreción con histamina
Liberación vesículas de secreción luego de la
estimulación
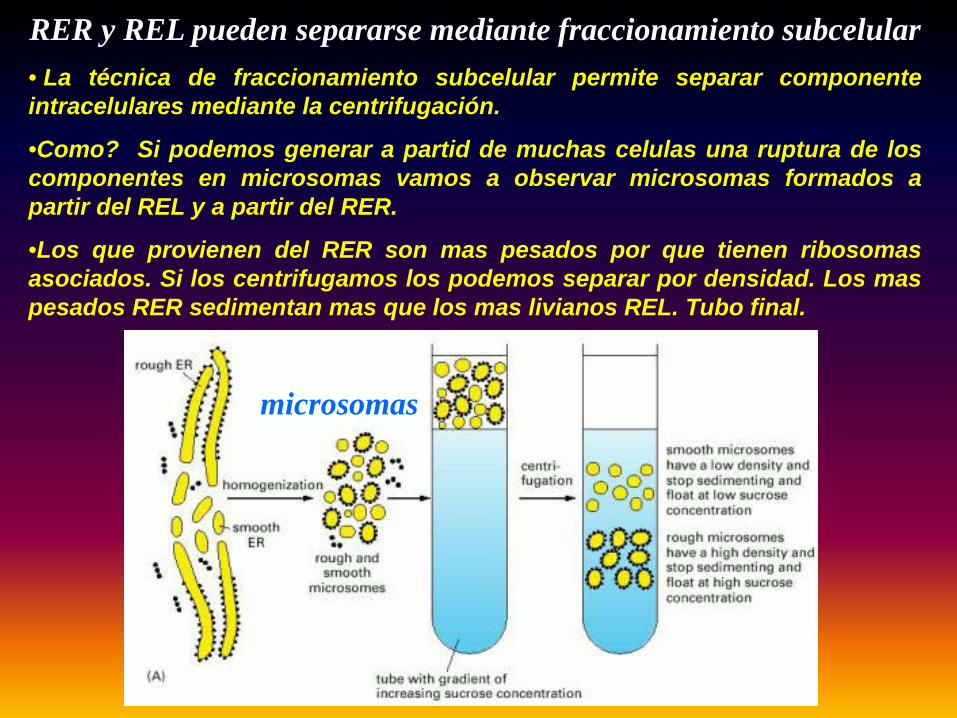
RER y REL pueden separarse mediante fraccionamiento subcelular
microsomas
• La técnica de fraccionamiento subcelular permite separar componenteintracelulares mediante la centrifugación.
•Como? Si podemos generar a partid de muchas celulas una ruptura de loscomponentes en microsomas vamos a observar microsomas formados apartir del REL y a partir del RER.
•Los que provienen del RER son mas pesados por que tienen ribosomasasociados. Si los centrifugamos los podemos separar por densidad. Los maspesados RER sedimentan mas que los mas livianos REL. Tubo final.
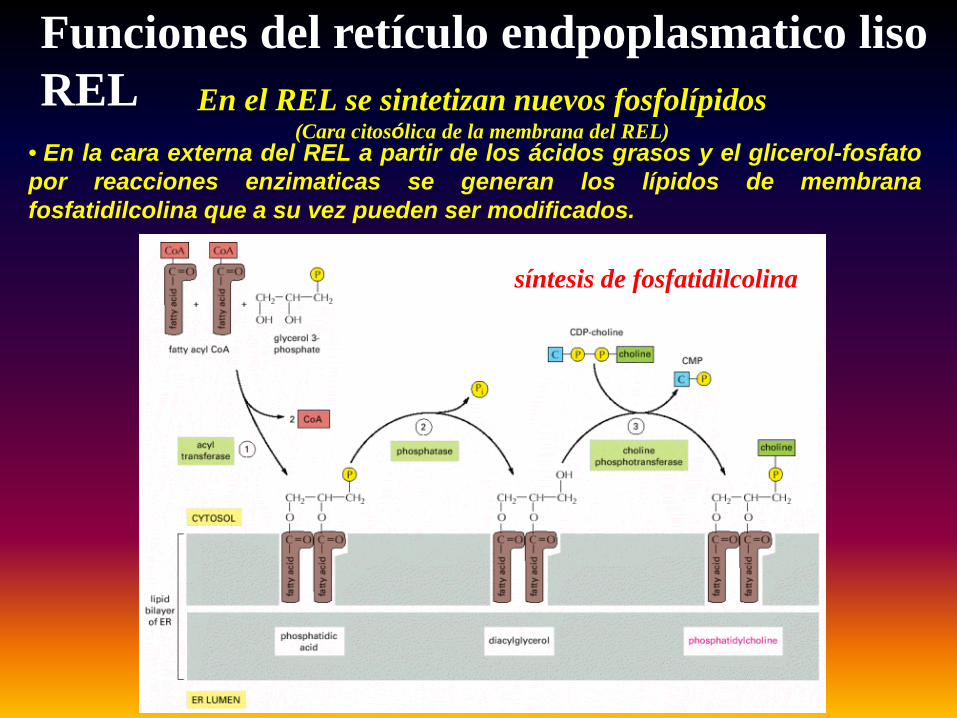
En el REL se sintetizan nuevos fosfolípidos(Cara citosólica de la membrana del REL)
síntesis de fosfatidilcolina
Funciones del retículo endpoplasmatico lisoREL
• En la cara externa del REL a partir de los ácidos grasos y el glicerol-fosfatopor reacciones enzimaticas se generan los lípidos de membranafosfatidilcolina que a su vez pueden ser modificados.
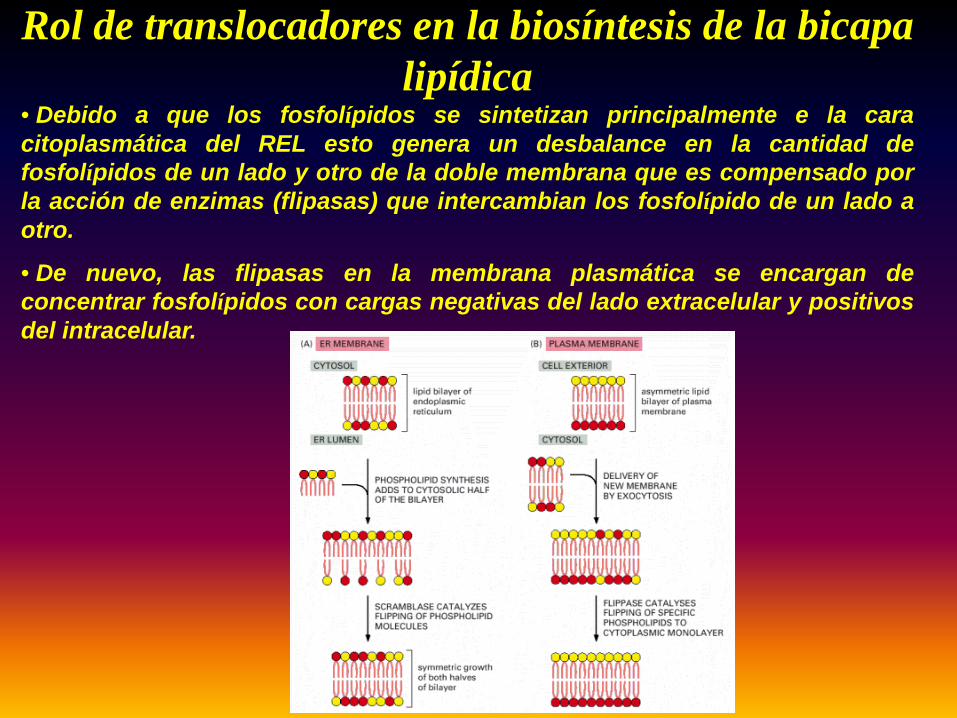
Rol de translocadores en la biosíntesis de la bicapa lipídica
• Debido a que los fosfolípidos se sintetizan principalmente e la caracitoplasmática del REL esto genera un desbalance en la cantidad defosfolípidos de un lado y otro de la doble membrana que es compensado porla acción de enzimas (flipasas) que intercambian los fosfolípido de un lado aotro.
• De nuevo, las flipasas en la membrana plasmática se encargan deconcentrar fosfolípidos con cargas negativas del lado extracelular y positivosdel intracelular.
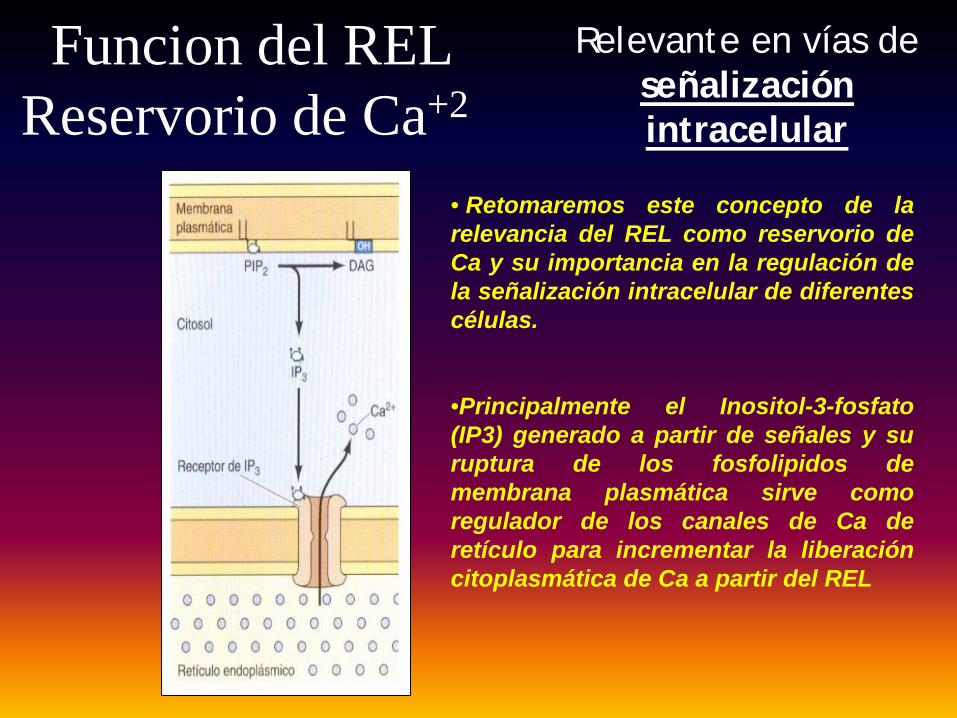
Funcion del REL Reservorio de Ca+2
Relevante en vías de señalización intracelular
• Retomaremos este concepto de larelevancia del REL como reservorio deCa y su importancia en la regulación dela señalización intracelular de diferentescélulas.
•Principalmente el Inositol-3-fosfato(IP3) generado a partir de señales y suruptura de los fosfolipidos demembrana plasmática sirve comoregulador de los canales de Ca deretículo para incrementar la liberacióncitoplasmática de Ca a partir del REL

Funciones del REL
• Glucogenólisis• Detoxificación de compuestos liposolubles.• Síntesis de Lípidos (fosfolípidos y colesterol) • Síntesis de hormonas esteroides (sintetizadas a
partir del colesterol)• Reservorio de Ca2+ intracelular

Fagocitosis: Entrada de partículas grandes , como bacterias a la célula
Pinocitosis: Entrada de fluídos o moléculas al interior de una célula
mediante vesículas pequeñas.
Entrada de material extracelular en vesículas formadas a partir de la membrana plasmática.ENDOCITOSIS
• Los endosomas son considerados parte del sistema deendomembranas. La endocitosis refiere al proceso de invaginación demembranas a partir de la membrana plasmática.
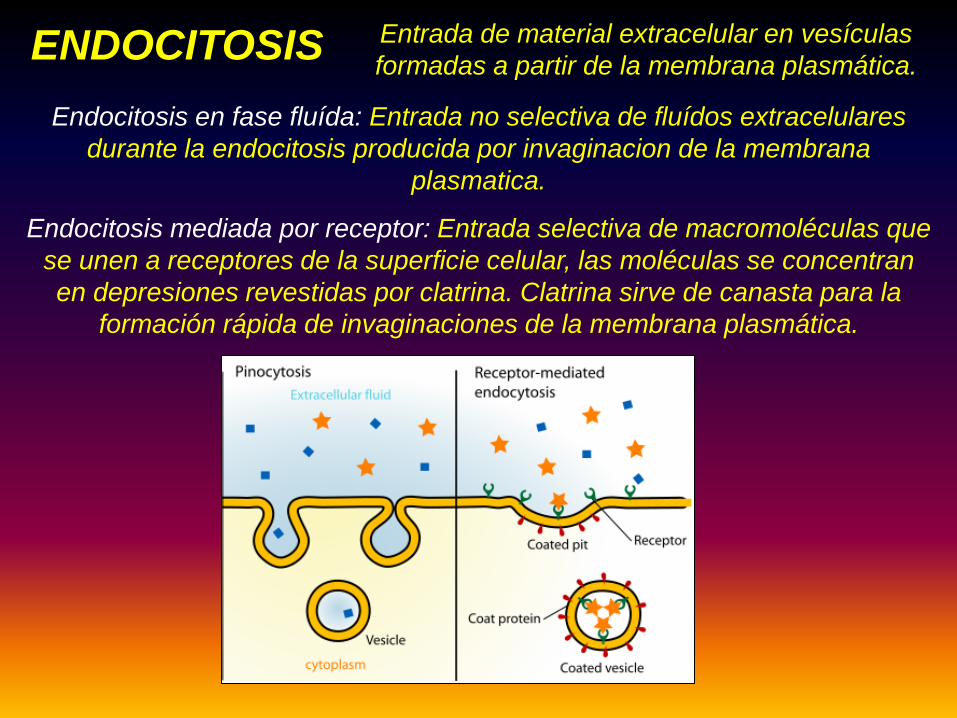
Endocitosis en fase fluída: Entrada no selectiva de fluídos extracelulares durante la endocitosis producida por invaginacion de la membrana
plasmatica.
Endocitosis mediada por receptor: Entrada selectiva de macromoléculas que se unen a receptores de la superficie celular, las moléculas se concentran en depresiones revestidas por clatrina. Clatrina sirve de canasta para la
formación rápida de invaginaciones de la membrana plasmática.
Entrada de material extracelular en vesículas formadas a partir de la membrana plasmática.ENDOCITOSIS
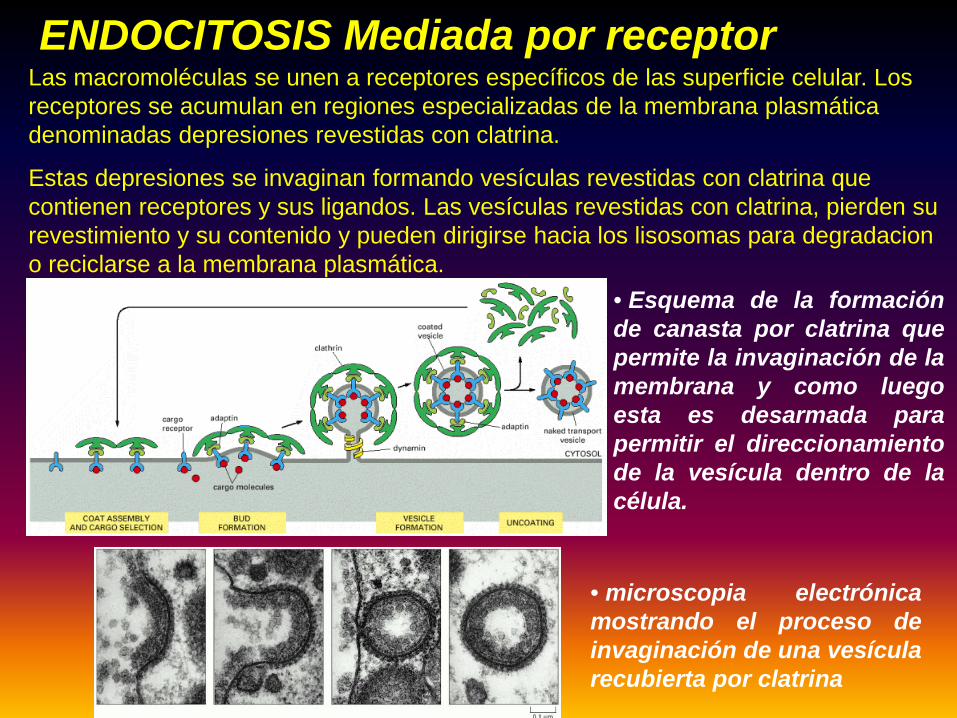
Las macromoléculas se unen a receptores específicos de las superficie celular. Los receptores se acumulan en regiones especializadas de la membrana plasmática denominadas depresiones revestidas con clatrina.
Estas depresiones se invaginan formando vesículas revestidas con clatrina que contienen receptores y sus ligandos. Las vesículas revestidas con clatrina, pierden su revestimiento y su contenido y pueden dirigirse hacia los lisosomas para degradacion o reciclarse a la membrana plasmática.
ENDOCITOSIS Mediada por receptor
• Esquema de la formaciónde canasta por clatrina quepermite la invaginación de lamembrana y como luegoesta es desarmada parapermitir el direccionamientode la vesícula dentro de lacélula.
• microscopia electrónicamostrando el proceso deinvaginación de una vesícularecubierta por clatrina
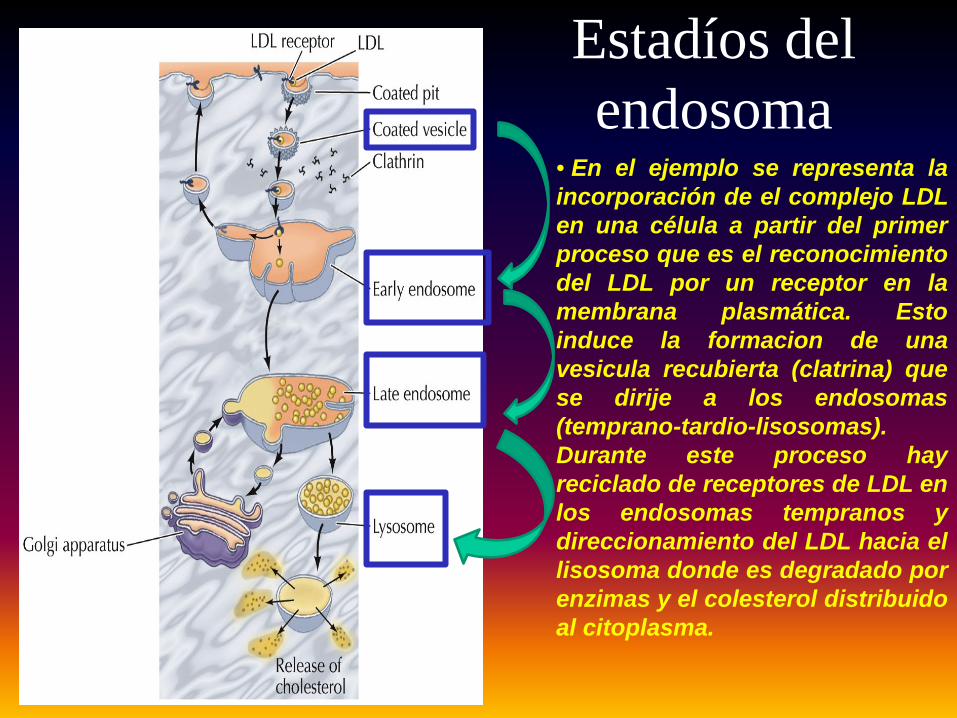
Estadíos del endosoma
• En el ejemplo se representa laincorporación de el complejo LDLen una célula a partir del primerproceso que es el reconocimientodel LDL por un receptor en lamembrana plasmática. Estoinduce la formacion de unavesicula recubierta (clatrina) quese dirije a los endosomas(temprano-tardio-lisosomas).Durante este proceso hayreciclado de receptores de LDL enlos endosomas tempranos ydireccionamiento del LDL hacia ellisosoma donde es degradado porenzimas y el colesterol distribuidoal citoplasma.

Endocitosis mediada por receptor independiente de Clatrina
Las células también poseen vías independientes de clatrina.
- Caveolas: pequeñas invaginaciones de la membrana plasmática (lipid raft especializados) que participan en transporte, transducción de señales en la via endocitica.
Se encuentran Organizadas por la acción de las caveolinas (Cav1,2,3) junto con el adaptador cavina.
Caveolina: Son una familia de proteínas que interactúan entre ellas para formar la estructura de las caveolas.
Las caveolas están implicadas en inflamación crónica y otras patologías como ateroesclerosis, distrofia muscular, dislipemias.

clatrina COP I COP II
Vesículas revestidas
Se conocen tres familias de proteínas de cubierta de vesículas de transporte
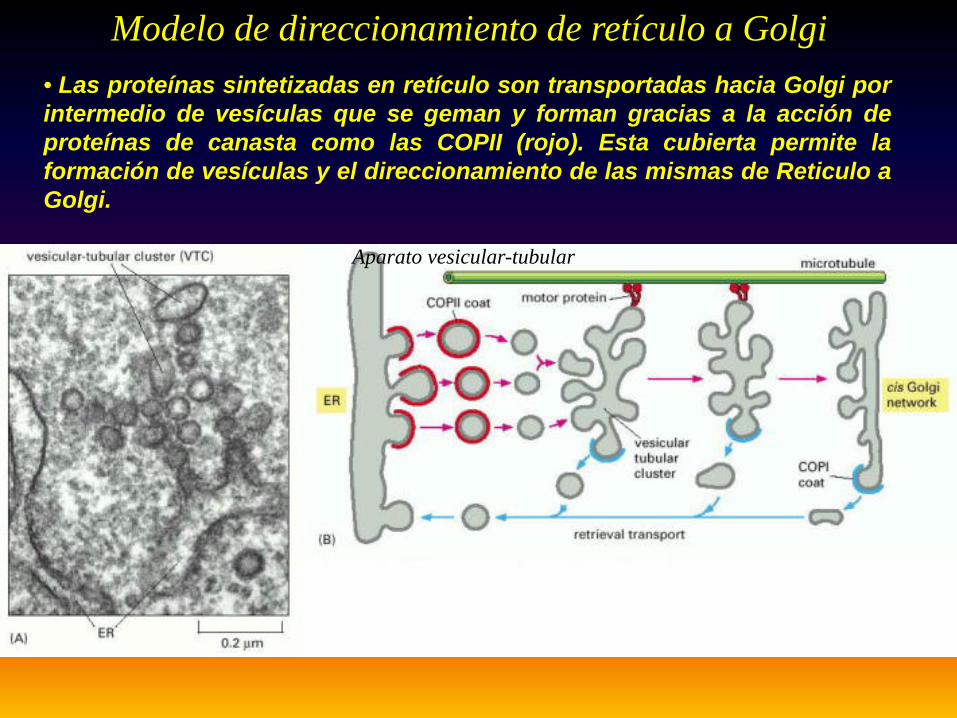
Aparato vesicular-tubular
Modelo de direccionamiento de retículo a Golgi• Las proteínas sintetizadas en retículo son transportadas hacia Golgi porintermedio de vesículas que se geman y forman gracias a la acción deproteínas de canasta como las COPII (rojo). Esta cubierta permite laformación de vesículas y el direccionamiento de las mismas de Reticulo aGolgi.
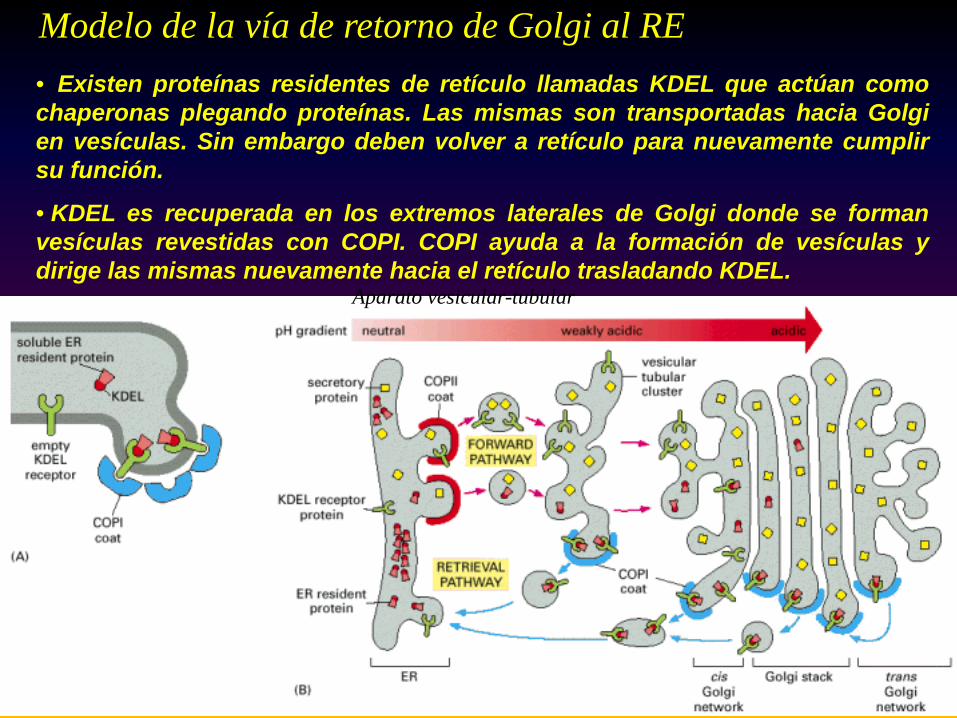
Aparato vesicular-tubular
Modelo de la vía de retorno de Golgi al RE • Existen proteínas residentes de retículo llamadas KDEL que actúan comochaperonas plegando proteínas. Las mismas son transportadas hacia Golgien vesículas. Sin embargo deben volver a retículo para nuevamente cumplirsu función.
• KDEL es recuperada en los extremos laterales de Golgi donde se formanvesículas revestidas con COPI. COPI ayuda a la formación de vesículas ydirige las mismas nuevamente hacia el retículo trasladando KDEL.
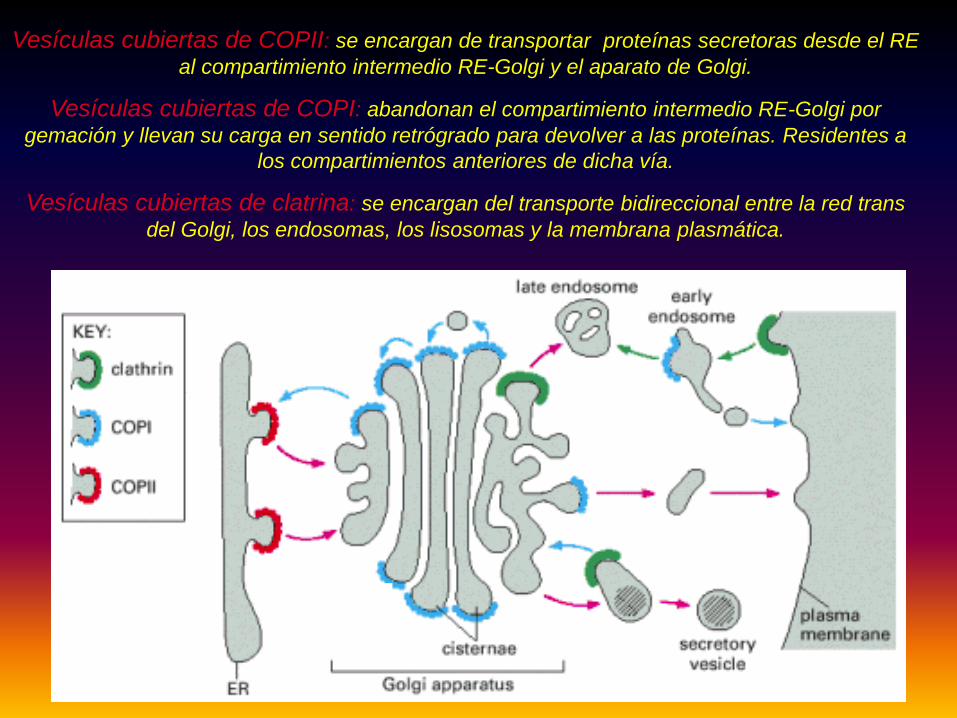
Vesículas cubiertas de COPI vs COPII vs Clatrina Vesículas cubiertas de COPII: se encargan de transportar proteínas secretoras desde el RE
al compartimiento intermedio RE-Golgi y el aparato de Golgi.
Vesículas cubiertas de COPI: abandonan el compartimiento intermedio RE-Golgi por gemación y llevan su carga en sentido retrógrado para devolver a las proteínas. Residentes a
los compartimientos anteriores de dicha vía.
Vesículas cubiertas de clatrina: se encargan del transporte bidireccional entre la red trans del Golgi, los endosomas, los lisosomas y la membrana plasmática.
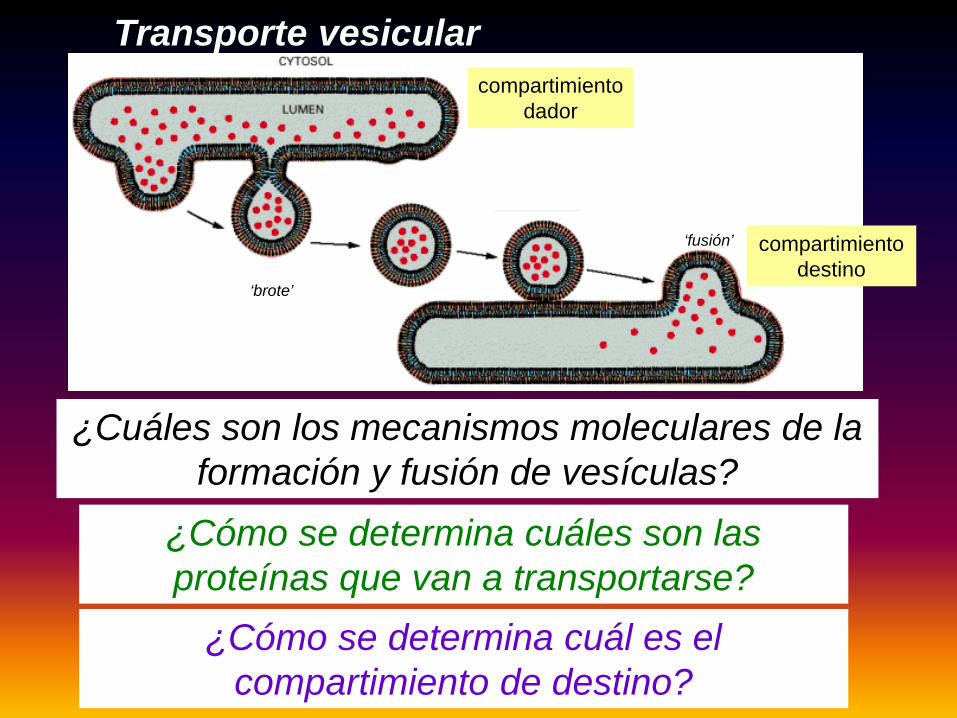
Transporte vesicular
¿Cuáles son los mecanismos moleculares de la formación y fusión de vesículas?
¿Cómo se determina cuáles son las proteínas que van a transportarse?
‘brote’
‘fusión’
¿Cómo se determina cuál es el compartimiento de destino?
compartimiento dador
compartimiento destino
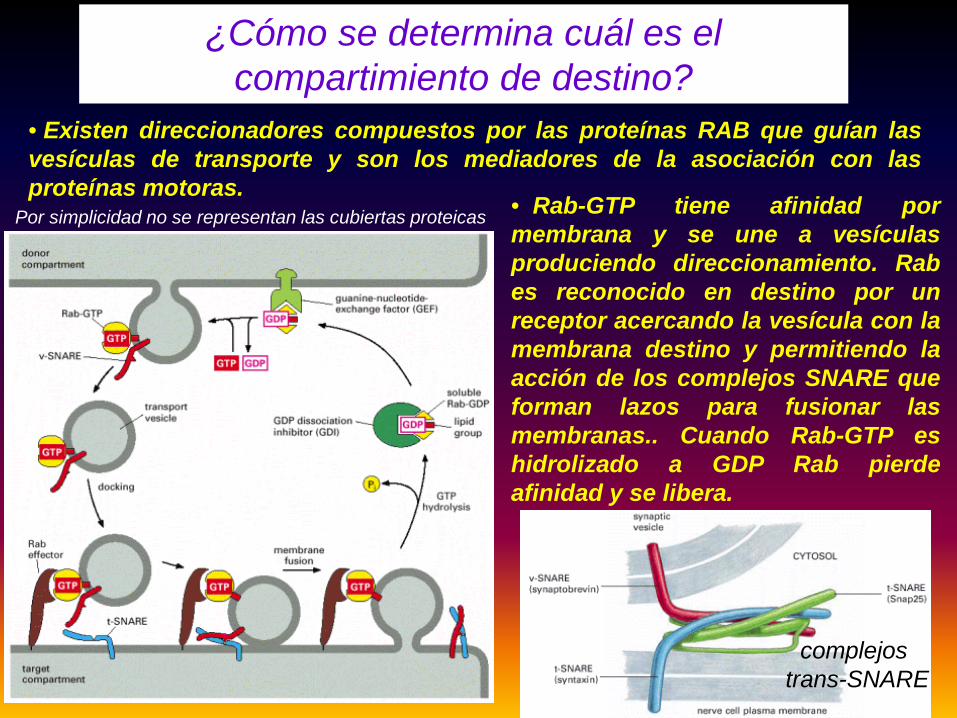
Por simplicidad no se representan las cubiertas proteicas
¿Cómo se determina cuál es el compartimiento de destino?
complejostrans-SNARE
• Existen direccionadores compuestos por las proteínas RAB que guían lasvesículas de transporte y son los mediadores de la asociación con lasproteínas motoras.
• Rab-GTP tiene afinidad pormembrana y se une a vesículasproduciendo direccionamiento. Rabes reconocido en destino por unreceptor acercando la vesícula con lamembrana destino y permitiendo laacción de los complejos SNARE queforman lazos para fusionar lasmembranas.. Cuando Rab-GTP eshidrolizado a GDP Rab pierdeafinidad y se libera.
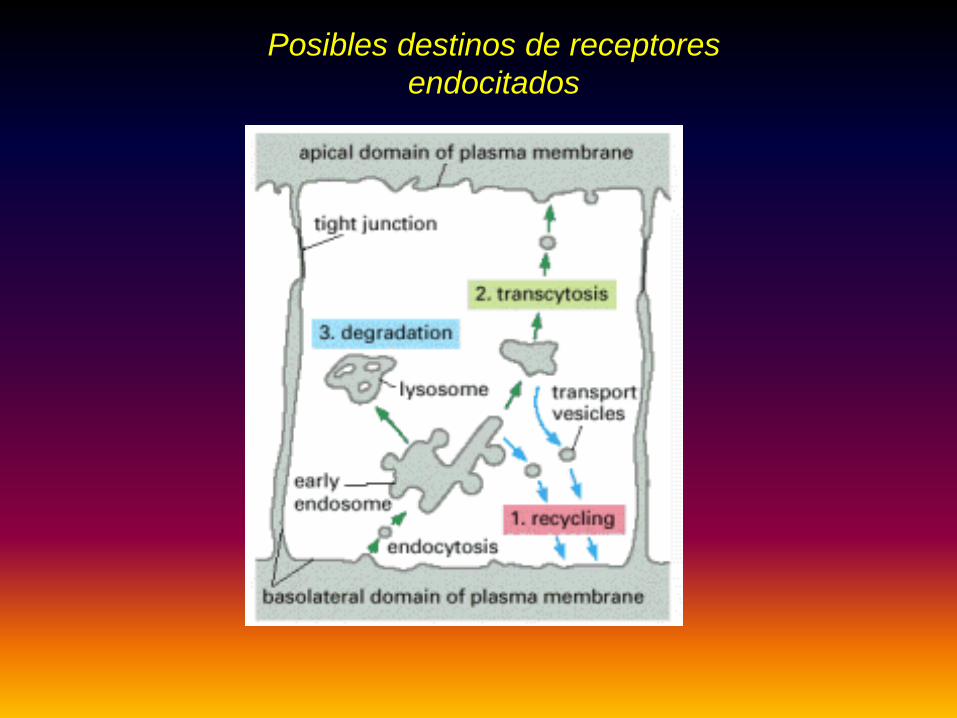
Posibles destinos de receptores endocitados

Son orgánelas rodeados de membrana que contienen una serie de enzimas que funcionan a un PH mas acido que el del citosol y son capaces de degradar todas las clases de polímeros biológicos: proteínas, ácidos nucleicos, carbohidratos y lípidos
Funcionan como el sistema digestivo de la célula, sirviendo tanto para degradar el material captado del exterior de la célula como para digerir los componentes obsoletos de la propia célula
Lisosomas
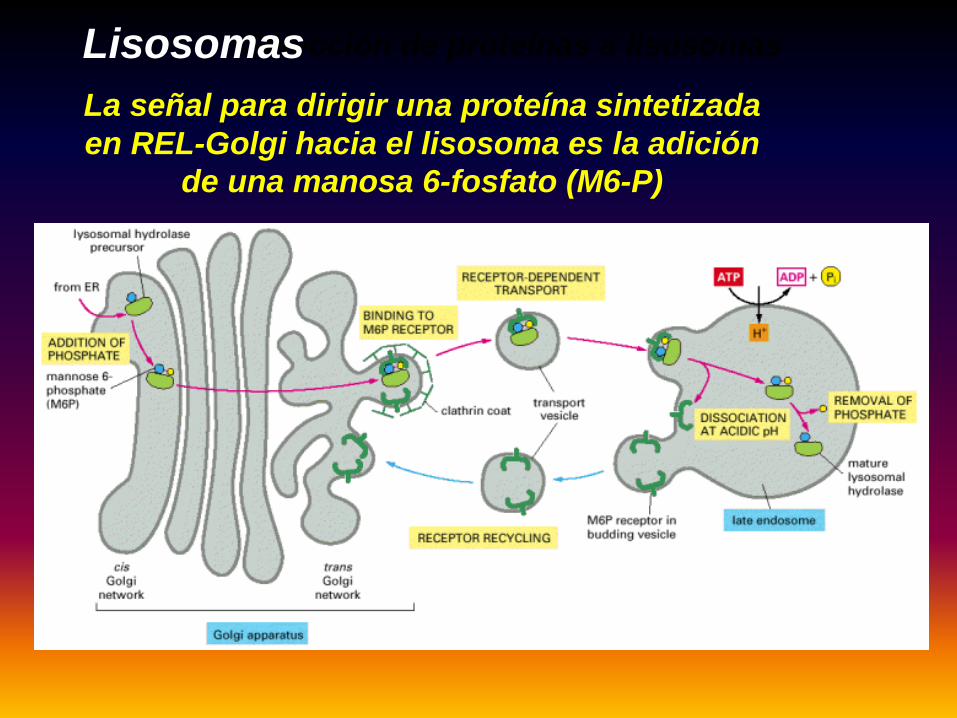
La señal para dirigir una proteína sintetizada en REL-Golgi hacia el lisosoma es la adición
de una manosa 6-fosfato (M6-P)
Selección de proteínas a lisosomasLisosomas

Algunas vías que llevan materiales a los lisosomas
Digestión Celular:
Fagocitosis
Endocitosis
Autofagia
Membranas que rodea al organoide que será degradado (Autofagia)
Además de degradar las moléculas englobadas por endocitosis, los
lisosomas digieren el material derivado de otras dos rutas:
Fagocitosis Autofagia
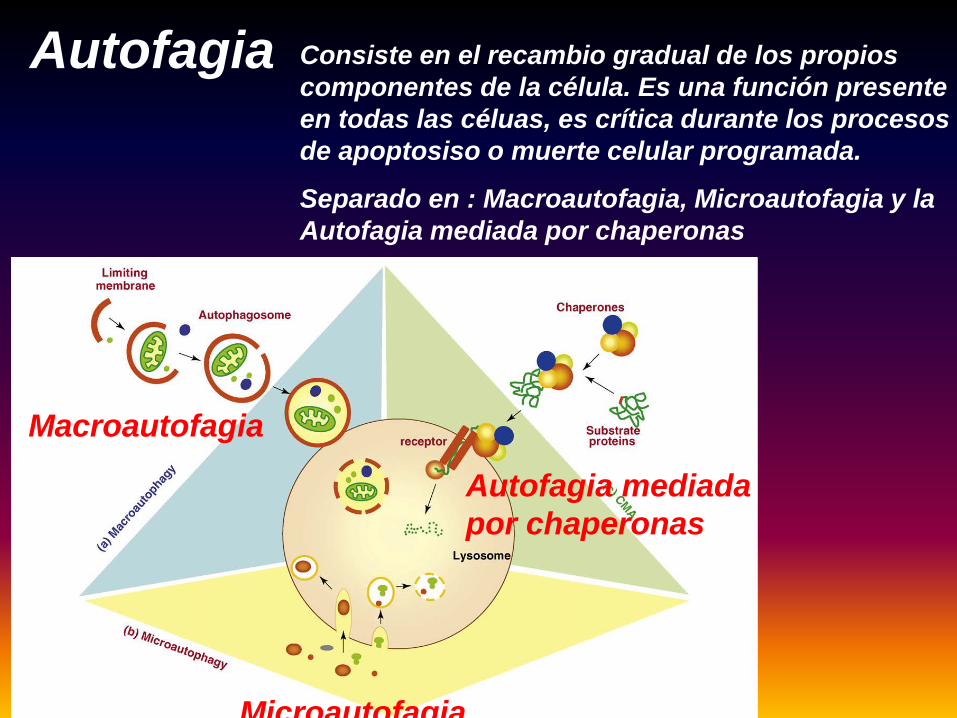
Autofagia Consiste en el recambio gradual de los propios componentes de la célula. Es una función presente en todas las céluas, es crítica durante los procesos de apoptosiso o muerte celular programada.
Separado en : Macroautofagia, Microautofagia y la Autofagia mediada por chaperonas
Macroautofagia
Microautofagia
Autofagia mediada por chaperonas
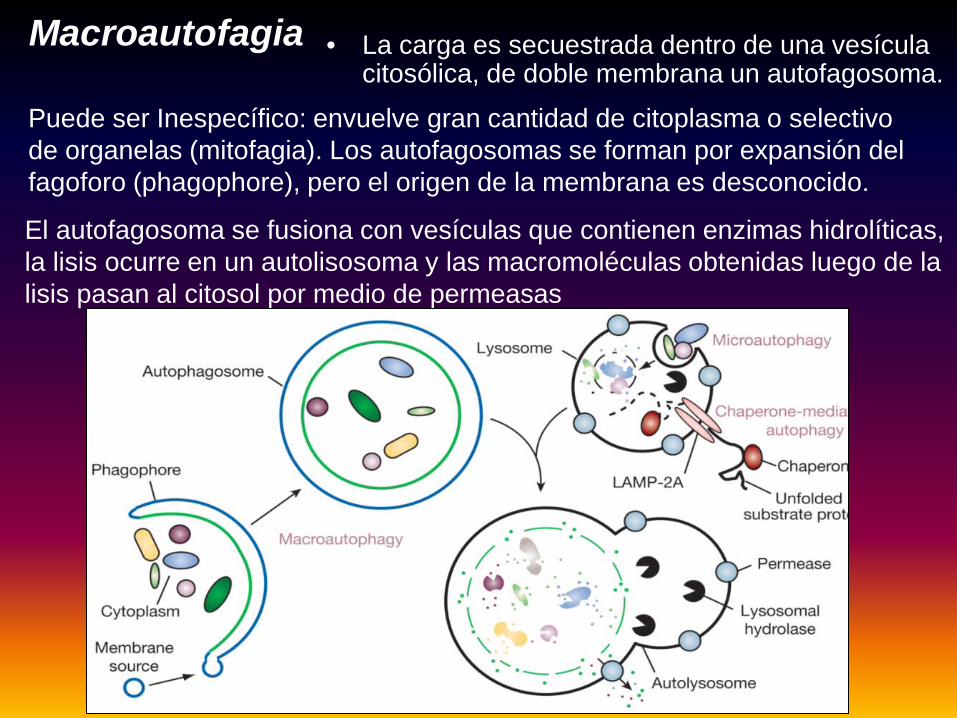
• La carga es secuestrada dentro de una vesícula citosólica, de doble membrana un autofagosoma.
Puede ser Inespecífico: envuelve gran cantidad de citoplasma o selectivo de organelas (mitofagia). Los autofagosomas se forman por expansión del fagoforo (phagophore), pero el origen de la membrana es desconocido.
Macroautofagia
El autofagosoma se fusiona con vesículas que contienen enzimas hidrolíticas, la lisis ocurre en un autolisosoma y las macromoléculas obtenidas luego de la lisis pasan al citosol por medio de permeasas

Comprende 4 estadíos: Nucleación, expansión, maduración y degradación.Nucleación: ocurre en respuesta a señales por stress celular, provoca el comienzo del crecimiento de membrana aislada (IM)= fagoforo. Expansión: IM se expande y secuestra proteínas citoplasmáticas, organelas o agregados proteicos. La expansión se completa cuando la membrana se cierra y forma una vesícula autofagosama de doble membrana.Maduración: Una vez cerrado, comienza la maduración fusionandose con compartimientos endocíticos, que incluyen endosomas tardíos y lisosomas-, esa fusión crea un anfisoma con contenido de autofagosoma y endosoma.Durante la maduración la luz del anfisoma se acidifica y la membrana adquiere enzimas hidrolíticas y lipasas, esto conduce a la formación de un autolisosoma.Degradación: Dentro del cual el contenido secuestrado se degrada y el reciclado vuelve al citosol
.
Macroautofagia

MicroautofagiaSecuestro de componentes citosólicos directamente por los lisosomas
por medio de invaginaciones de la membrana.
CMA: autofagia mediada por chaperonas: translocación de proteínas no plegadas a través de la membrana lisosomal por la acción de chaperonas
citosólicas y lisosomales Hsc 70 y el receptor integral de membrana
LAMP-2A (Lysosome-associated membrane protein type 2A).No hay formación de vesículas y fusión de membranas
Autofagia mediada por chaperonas (CMA)

A modo de ejemplo en estas ultimas diapositivas vamos a mencionar defectos en algunas señales de proteínas que generan casos de
enfermedad.

Podemos tener mutaciones que alteran la función de las proteínas.
Sin embargo, las proteínas pueden ser funcionales pero podemos tener alteraciones de direccionamiento de proteínas
que llevan a enfermedad.
Defectos en la degradación de proteínas y/o componentes de la célula en los lisosomas son responsables de más de 30 enfermedades congénitas humanas diferentes, que se
denominan enfermedades de depósito lisosómico.
La enfermedad se produce por acumulación del sustrato. El depósito de las moléculas no degradadas produce disfunción celular en los tejidos y órganos y viceromegalia..
La mayoría de estas enfermedades se deben a deficiencias en una única enzima de direccionamiento de proteínas hacia los lisosomas.
Enfermedades de depósito lisosómico
• ENFERMEDAD• TAY-SACHS• GAUCHER
• NIEMAN-PICK• HURLER
• ENZIMA• HEXOSAMINIDASA• GLUCOCEREBROSIDASA• ESFINGOMIELINASAS• IDURONIDASA
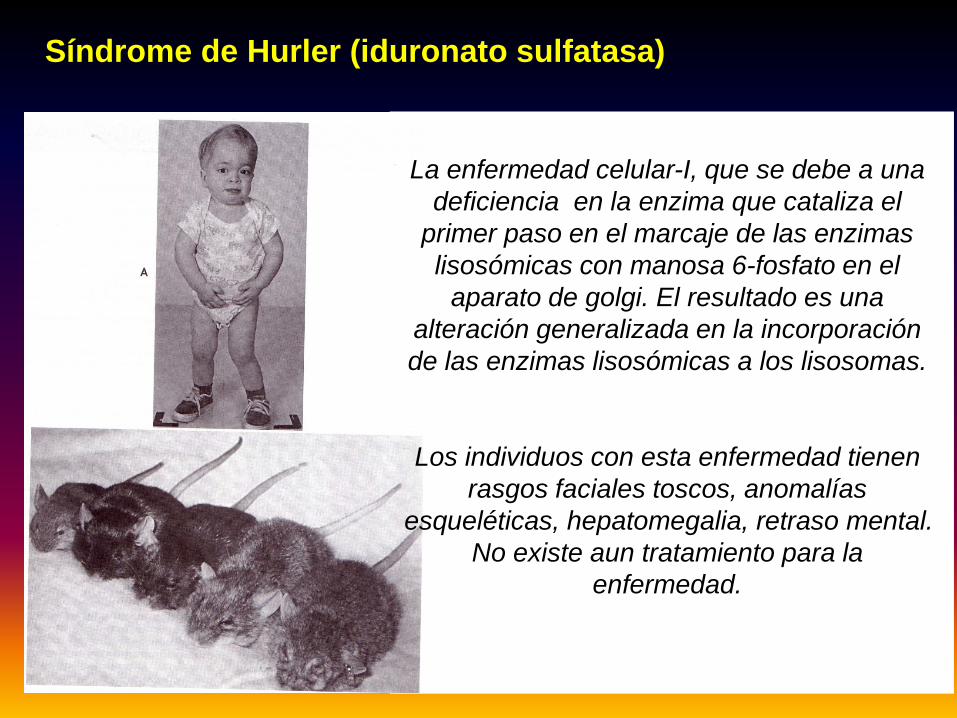
Síndrome de Hurler (iduronato sulfatasa)
La enfermedad celular-I, que se debe a una deficiencia en la enzima que cataliza el
primer paso en el marcaje de las enzimas lisosómicas con manosa 6-fosfato en el
aparato de golgi. El resultado es una alteración generalizada en la incorporación de las enzimas lisosómicas a los lisosomas.
Los individuos con esta enfermedad tienen rasgos faciales toscos, anomalías
esqueléticas, hepatomegalia, retraso mental. No existe aun tratamiento para la
enfermedad.
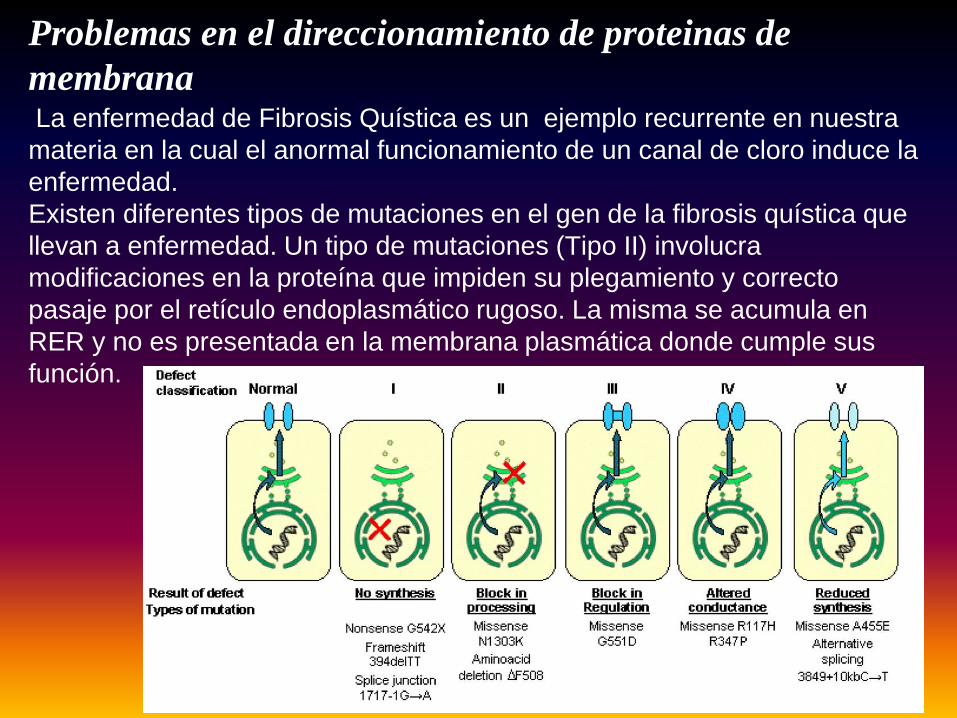
Problemas en el direccionamiento de proteinas de membranaLa enfermedad de Fibrosis Quística es un ejemplo recurrente en nuestra materia en la cual el anormal funcionamiento de un canal de cloro induce la enfermedad. Existen diferentes tipos de mutaciones en el gen de la fibrosis quística que llevan a enfermedad. Un tipo de mutaciones (Tipo II) involucra modificaciones en la proteína que impiden su plegamiento y correcto pasaje por el retículo endoplasmático rugoso. La misma se acumula en RER y no es presentada en la membrana plasmática donde cumple sus función.
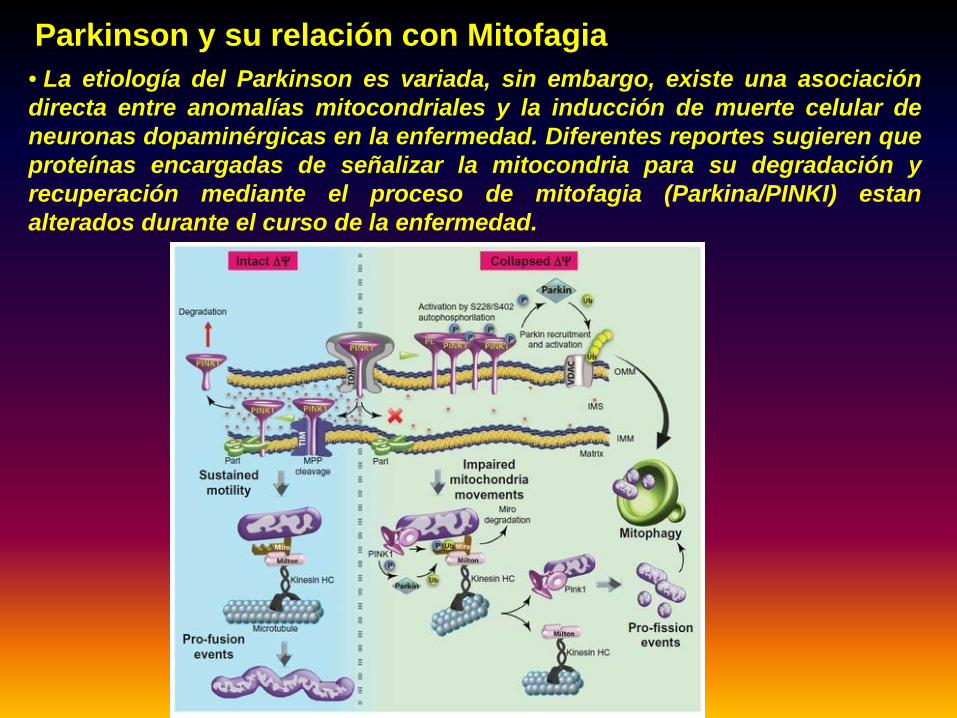
Parkinson y su relación con Mitofagia• La etiología del Parkinson es variada, sin embargo, existe una asociacióndirecta entre anomalías mitocondriales y la inducción de muerte celular deneuronas dopaminérgicas en la enfermedad. Diferentes reportes sugieren queproteínas encargadas de señalizar la mitocondria para su degradación yrecuperación mediante el proceso de mitofagia (Parkina/PINKI) estanalterados durante el curso de la enfermedad.




















