
Side chain liquid crystal polyurethanes with azobenzene mesogenic moieties: Influence of spacer...
12
Side Chain Liquid Crystal Polyurethanes with Azobenzene Mesogenic Moieties: Influence of Spacer Length on Hydrogen Bonding at Different Temperatures M. BRECL, M. ˇ ZIGON, T. MALAVA ˇ SI ˇ C National Institute of Chemistry, Hajdrihova 19, SLO-1001 Ljubljana, Slovenia Received 29 December 1997; accepted 18 March 1998 ABSTRACT: A series of side chain liquid crystal polyurethanes (CnCNPs), in which the spacer length was varied from 2 to 12 methylene units, were synthesized by the addition polymerization of a-[bis(2-hydroxyethyl)amino]-v-(4-cyanoazobenzene-49-oxy)- alkanes (CnCN-diols) with hexamethylene diisocyanate. The liquid crystalline proper- ties of CnCNPs were characterized by means of differential scanning calorimetry, polarizing optical microscopy, and X-ray diffraction. Polyurethanes with spacer length 4 or higher exhibited mesomophic properties. C4CNP and C5CNP exhibited an enan- tiotropic nematic mesophase, while C6-C12CNPs exhibited enantiotropic bilayer smec- tic mesophases. CnCNPs have a high tendency to crystallize; crystallization is kineti- cally controlled. Polyurethane’s backbone crystallization is closely related to hydrogen bonding. To establish the role of hydrogen bonding in mesophase formation as well as crystallization, Fourier transform infrared spectroscopy studies of CnCNPs were car- ried out at different temperatures focusing on H-bonds between the N—H and CAO groups of the urethane backbone. With increasing temperature, CAO and N—H stretching bands were evenly shifted to higher wavenumbers, with two exceptions (C4CNP and C5CNP) discussed in detail in the text. © 1998 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 36: 2135–2146, 1998 Keywords: SCLC polyurethanes; azobenzene mesogenic group; alkyl spacer; bilayer smectic phases; hydrogen bonding INTRODUCTION Side chain liquid crystal polymers (SCLCPs) have attracted attention due to their potential applica- tion in numerous areas, e.g. in nonlinear optics 1–4 and optical information storage devices. 5–7 Since they exhibited combined properties of polymers and of conventional monomer liquid crystals, they have been systematically studied since Finkel- mann first introduced polymers of this type. 8 A great deal of research has been focused on the relation between the structure of SCLCPs and their liquid crystalline behavior. The molecular structure of SCLCPs can be di- vided into three structural parts: the polymer chain, the flexible spacer, and the mesogenic unit. Ringsdorf et al. 9 have discovered that a flexible spacer should be inserted between the polymer backbone and the mesogenic side group to sepa- rate the motion of the polymer backbone, which tends to randomly coil up, from mesogenic groups which are ordered in the liquid crystalline state. This finding made it possible to synthesize SCLCPs systematically. Liquid crystalline properties (phase transitions and the nature of their mesophases) depend on the polymer chemical structure (the nature of the polymer backbone, the flexible spacer and its length, and the mesogenic unit), molar mass, polydispersity, and tacticity. Because of the com- Correspondence to: M. Brecl Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 36, 2135–2146 (1998) © 1998 John Wiley & Sons, Inc. CCC 0887-624X/98/122135-12 2135
Transcript of Side chain liquid crystal polyurethanes with azobenzene mesogenic moieties: Influence of spacer...

Side Chain Liquid Crystal Polyurethanes with AzobenzeneMesogenic Moieties: Influence of Spacer Length onHydrogen Bonding at Different Temperatures
M. BRECL, M. ZIGON, T. MALAVASIC
National Institute of Chemistry, Hajdrihova 19, SLO-1001 Ljubljana, Slovenia
Received 29 December 1997; accepted 18 March 1998
ABSTRACT: A series of side chain liquid crystal polyurethanes (CnCNPs), in which thespacer length was varied from 2 to 12 methylene units, were synthesized by theaddition polymerization of a-[bis(2-hydroxyethyl)amino]-v-(4-cyanoazobenzene-49-oxy)-alkanes (CnCN-diols) with hexamethylene diisocyanate. The liquid crystalline proper-ties of CnCNPs were characterized by means of differential scanning calorimetry,polarizing optical microscopy, and X-ray diffraction. Polyurethanes with spacer length4 or higher exhibited mesomophic properties. C4CNP and C5CNP exhibited an enan-tiotropic nematic mesophase, while C6-C12CNPs exhibited enantiotropic bilayer smec-tic mesophases. CnCNPs have a high tendency to crystallize; crystallization is kineti-cally controlled. Polyurethane’s backbone crystallization is closely related to hydrogenbonding. To establish the role of hydrogen bonding in mesophase formation as well ascrystallization, Fourier transform infrared spectroscopy studies of CnCNPs were car-ried out at different temperatures focusing on H-bonds between the N—H and CAOgroups of the urethane backbone. With increasing temperature, CAO and N—Hstretching bands were evenly shifted to higher wavenumbers, with two exceptions(C4CNP and C5CNP) discussed in detail in the text. © 1998 John Wiley & Sons, Inc. J PolymSci A: Polym Chem 36: 2135–2146, 1998Keywords: SCLC polyurethanes; azobenzene mesogenic group; alkyl spacer; bilayersmectic phases; hydrogen bonding
INTRODUCTION
Side chain liquid crystal polymers (SCLCPs) haveattracted attention due to their potential applica-tion in numerous areas, e.g. in nonlinear optics1–4
and optical information storage devices.5–7 Sincethey exhibited combined properties of polymersand of conventional monomer liquid crystals, theyhave been systematically studied since Finkel-mann first introduced polymers of this type.8 Agreat deal of research has been focused on therelation between the structure of SCLCPs andtheir liquid crystalline behavior.
The molecular structure of SCLCPs can be di-vided into three structural parts: the polymerchain, the flexible spacer, and the mesogenic unit.Ringsdorf et al.9 have discovered that a flexiblespacer should be inserted between the polymerbackbone and the mesogenic side group to sepa-rate the motion of the polymer backbone, whichtends to randomly coil up, from mesogenic groupswhich are ordered in the liquid crystalline state.This finding made it possible to synthesizeSCLCPs systematically.
Liquid crystalline properties (phase transitionsand the nature of their mesophases) depend onthe polymer chemical structure (the nature of thepolymer backbone, the flexible spacer and itslength, and the mesogenic unit), molar mass,polydispersity, and tacticity. Because of the com-
Correspondence to: M. BreclJournal of Polymer Science: Part A: Polymer Chemistry, Vol. 36, 2135–2146 (1998)© 1998 John Wiley & Sons, Inc. CCC 0887-624X/98/122135-12
2135

plexity of the system, it is difficult to determinethe effect of individual variables on liquid crystal-line behavior. Several systematic investigationsinto the correlation between the above-mentionedvariables and liquid crystalline behavior havebeen reported.10–18 Most of the research workrefers to SCLC polymers with an acrylic,methacrylic, or siloxane backbone9,10,19–24 andonly a limited number to the polyurethane-basedSCLC polymers.25–28 However, several of themain chain liquid crystal polyurethanes havebeen investigated.29–32
This article reports on the synthesis and char-acterization of a series of side chain liquid crystalpolyurethanes (CnCNPs) prepared by the addi-tion polymerization of a-[bis(2-hydroxyethyl)-amino]-v-(4-cyanoazobenzene-49-oxy)alkanes(CnCN-diols) with hexamethylene diisocyanate.To our knowledge, this is the second series of sidechain liquid crystalline polyurethanes for whichas many as 11 homologues have been synthe-sized.18
We investigated the influence of the spacerlength on the mesomorphic behavior of polyure-thane-based SCLCPs with an azobenzene meso-genic core and their thermal properties, as well asthe influence of the spacer length on hydrogenbonding between urethane groups at differenttemperatures. Namely, the polyurethane back-bone is different from most common polymers re-garding the possibility of H-bond formation. In
order to obtain a better understanding of hydro-gen bonding between the N—H and CAO groupsin CnCNP polyurethanes, FT-IR spectroscopystudies were carried out. The influence of theH-bonds of urethane groups on the mesophaseformation was of special interest.18
EXPERIMENTAL
The Synthesis of Monomers (CnCN-diol) andCorresponding Polyurethanes (CnCNP)
4-Hydroxy-4*-cyanoazobenzene (1)
The synthesis of 4-hydroxy-49-cyanoazobenzenehas been described elsewhere.33
a-Bromo-v-(4-cyanoazobenzene-4*-oxy)alkanes (2)
a-Bromo-v-(4-cyanoazobenzene-49-oxy)alkanes(2) were synthesized according to the proceduredescribed by Crivello.34
a-[Bis(2-hydroxyethyl)amino]-v-(4-cyanoazobenzene-4*-oxy)alkanes (CnCN-diols)
a-Bromo-v-(4-cyanoazobenzene-49-oxy)alkane (2)(0.01 mol) and diethanolamine (0.02 mol) weremixed in absolute ethanol (70 mL). The reactionmixture was heated under reflux for 20 h. Afterthe reaction was completed, the precipitate of thewarm reaction mixture was filtered off. The fil-
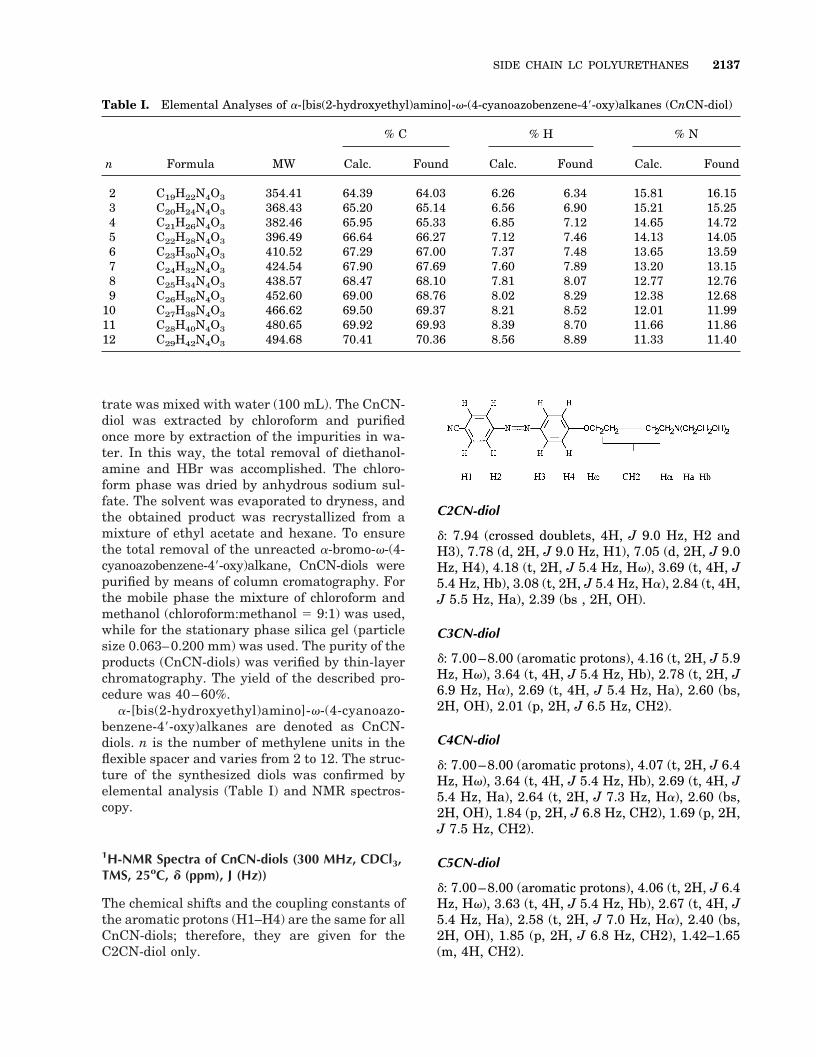
Scheme 1. Sequence of reactions leading to a series of SCLC polyurethanes (CnCNP).
2136 BRECL, ZIGON, AND MALAVASIC
trate was mixed with water (100 mL). The CnCN-diol was extracted by chloroform and purifiedonce more by extraction of the impurities in wa-ter. In this way, the total removal of diethanol-amine and HBr was accomplished. The chloro-form phase was dried by anhydrous sodium sul-fate. The solvent was evaporated to dryness, andthe obtained product was recrystallized from amixture of ethyl acetate and hexane. To ensurethe total removal of the unreacted a-bromo-v-(4-cyanoazobenzene-49-oxy)alkane, CnCN-diols werepurified by means of column cromatography. Forthe mobile phase the mixture of chloroform andmethanol (chloroform:methanol 5 9:1) was used,while for the stationary phase silica gel (particlesize 0.063–0.200 mm) was used. The purity of theproducts (CnCN-diols) was verified by thin-layerchromatography. The yield of the described pro-cedure was 40–60%.
a-[bis(2-hydroxyethyl)amino]-v-(4-cyanoazo-benzene-49-oxy)alkanes are denoted as CnCN-diols. n is the number of methylene units in theflexible spacer and varies from 2 to 12. The struc-ture of the synthesized diols was confirmed byelemental analysis (Table I) and NMR spectros-copy.
1H-NMR Spectra of CnCN-diols (300 MHz, CDCl3,TMS, 25oC, d (ppm), J (Hz))
The chemical shifts and the coupling constants ofthe aromatic protons (H1–H4) are the same for allCnCN-diols; therefore, they are given for theC2CN-diol only.
C2CN-diol
d: 7.94 (crossed doublets, 4H, J 9.0 Hz, H2 andH3), 7.78 (d, 2H, J 9.0 Hz, H1), 7.05 (d, 2H, J 9.0Hz, H4), 4.18 (t, 2H, J 5.4 Hz, Hv), 3.69 (t, 4H, J5.4 Hz, Hb), 3.08 (t, 2H, J 5.4 Hz, Ha), 2.84 (t, 4H,J 5.5 Hz, Ha), 2.39 (bs , 2H, OH).
C3CN-diol
d: 7.00–8.00 (aromatic protons), 4.16 (t, 2H, J 5.9Hz, Hv), 3.64 (t, 4H, J 5.4 Hz, Hb), 2.78 (t, 2H, J6.9 Hz, Ha), 2.69 (t, 4H, J 5.4 Hz, Ha), 2.60 (bs,2H, OH), 2.01 (p, 2H, J 6.5 Hz, CH2).
C4CN-diol
d: 7.00–8.00 (aromatic protons), 4.07 (t, 2H, J 6.4Hz, Hv), 3.64 (t, 4H, J 5.4 Hz, Hb), 2.69 (t, 4H, J5.4 Hz, Ha), 2.64 (t, 2H, J 7.3 Hz, Ha), 2.60 (bs,2H, OH), 1.84 (p, 2H, J 6.8 Hz, CH2), 1.69 (p, 2H,J 7.5 Hz, CH2).
C5CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.4Hz, Hv), 3.63 (t, 4H, J 5.4 Hz, Hb), 2.67 (t, 4H, J5.4 Hz, Ha), 2.58 (t, 2H, J 7.0 Hz, Ha), 2.40 (bs,2H, OH), 1.85 (p, 2H, J 6.8 Hz, CH2), 1.42–1.65(m, 4H, CH2).
Table I. Elemental Analyses of a-[bis(2-hydroxyethyl)amino]-v-(4-cyanoazobenzene-49-oxy)alkanes (CnCN-diol)
n Formula MW
% C % H % N
Calc. Found Calc. Found Calc. Found
2 C19H22N4O3 354.41 64.39 64.03 6.26 6.34 15.81 16.153 C20H24N4O3 368.43 65.20 65.14 6.56 6.90 15.21 15.254 C21H26N4O3 382.46 65.95 65.33 6.85 7.12 14.65 14.725 C22H28N4O3 396.49 66.64 66.27 7.12 7.46 14.13 14.056 C23H30N4O3 410.52 67.29 67.00 7.37 7.48 13.65 13.597 C24H32N4O3 424.54 67.90 67.69 7.60 7.89 13.20 13.158 C25H34N4O3 438.57 68.47 68.10 7.81 8.07 12.77 12.769 C26H36N4O3 452.60 69.00 68.76 8.02 8.29 12.38 12.68
10 C27H38N4O3 466.62 69.50 69.37 8.21 8.52 12.01 11.9911 C28H40N4O3 480.65 69.92 69.93 8.39 8.70 11.66 11.8612 C29H42N4O3 494.68 70.41 70.36 8.56 8.89 11.33 11.40
SIDE CHAIN LC POLYURETHANES 2137

C6CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.5Hz, Hv), 3.63 (t, 4H, J 5.4 Hz, Hb), 2.67 (t, 4H, J5.4 Hz, Ha), 2.56 (t, 2H, J 7.2 Hz, Ha), 2.38 (bs,2H, OH), 1.84 (p, 2H, J 7.4 Hz, CH2), 1.35–1.65(m, 6H, CH2).
C7CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.6Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H, J5.4 Hz, Ha), 2.54 (t, 2H, J 7.4 Hz, Ha), 2.38 (bs,2H, OH), 1.83 (p, 2H, J 6.9 Hz, CH2), 1.28–1.56(m, 8H, CH2).
C8CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.6Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H, J5.4 Hz, Ha), 2.53 (t, 2H, J 7.4 Hz, HA), 2.30 (bs,2H, OH), 1.83 (p, 2H, J 7.0 Hz, CH2), 1.20–1.56(m, 10H, CH2).
C9CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.6Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H, J5.4 Hz, Ha), 2.52 (t, 2H, J 7.4 Hz, Ha), 2.40 (bs,2H, OH), 1.83 (p, 2H, J 7.0 Hz, CH2), 1.20–1.56(m, 12H, CH2).
C10CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.6Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H, J5.4 Hz, Ha), 2.52 (t, 2H, J 7.4 Hz, HA), 2.42 (bs,
2H, OH), 1.83 (p, 2H, J 7.0 Hz, CH2), 1.20–1.56(m, 14H, CH2).
C11CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, J 6.6Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H, J5.4 Hz, Ha), 2.52 (t, 2H, J 7.4 Hz, Ha), 2.26 (bs,2H, OH), 1.83 (p, 2H, J 7.0 Hz, CH2), 1.20–1.56(m, 16H, CH2).
C12CN-diol
d: 7.00–8.00 (aromatic protons), 4.06 (t, 2H, a J6.6 Hz, Hv), 3.62 (t, 4H, J 5.4 Hz, Hb), 2.66 (t, 4H,J 5.4 Hz, Ha), 2.52 (t, 2H, J 7.4 Hz, Ha), 2.46 (bs,2H, OH), 1.83 (p, 2H, J 7.0 Hz, CH2), 1.20–1.56(m, 18H, CH2).
Polyurethanes (CnCNPs)
The CnCN-diols were dried in vacuo at 40oC for10 h. Dry CnCN-diol (0.002 mol), hexamethylenediisocyanate (0.002 mol), DMF (10 mL), and dibu-tyltin dilaurate (0.2% with respect to the diol) ascatalyst were placed into a 50 mL round-bottomedflask. All the reagents were weighed in a dry boxto avoid water contamination. The flask wasequipped with a drying tube. The reaction mix-ture was stirred by a magnetic stirrer at 70oCfor 3 h. Polymerization led to polyurethanes(CnCNPs). Polyurethanes were precipitated inethanol and purified by dissolution in DMF andreprecipitation in ethanol; after that they wereair-dried. Polyurethanes are denoted as CnCNP(Scheme 1), where n is the number of methylene
Table II. Elemental Analyses of Polyurethanes (CnCNP)
n Formula MW
% C % H % N1023 M# w
(g/mol) M# w/M# nCalc. Found Calc. Found Calc. Found
2 (C27H34N6O5)x (522.60)x 62.05 61.23 6.56 6.46 16.08 15.71 46 2.03 (C28H36N6O5)x (536.63)x 62.67 62.33 6.76 7.02 15.66 15.78 33 1.94 (C29H38N6O5)x (550.65)x 63.25 62.99 6.95 7.32 15.26 15.39 33 2.15 (C30H40N6O5)x (564.68)x 63.81 63.18 7.14 7.22 14.88 14.93 71 2.06 (C31H42N6O5)x (578.71)x 64.34 63.76 7.31 7.16 14.52 14.53 49 5.77 (C32H44N6O5)x (592.74)x 64.84 64.39 7.48 7.75 14.18 14.27 65 3.08 (C33H46N6O5)x (606.76)x 65.32 64.74 7.64 7.91 13.85 14.00 67 3.79 (C34H48N6O5)x (620.79)x 65.78 65.63 7.79 8.04 13.54 13.73 55 4.5
10 (C35H50N6O5)x (634.82)x 66.22 65.85 7.94 8.18 13.24 13.30 51 4.811 (C36H52N6O5)x (648.84)x 66.64 65.88 8.08 8.40 12.95 13.09 23 5.912 (C37H54N6O5)x (662.87)x 67.04 66.89 8.21 8.53 12.68 12.83 75 5.0
2138 BRECL, ZIGON, AND MALAVASIC

units in the spacer. Their composition was exam-ined by elemental analysis (Table II). Mass-aver-age molar masses (M# w) and polydispersities (M# w/M# n) as measured by SEC are given in Table II.I
Characterization Methods
NMR Spectroscopy
The 1H-NMR spectra were recorded at 25oC on aVarian VXR 300 spectrometer at 300 MHz usingTMS as an internal standard. CDCl3 was used asa solvent. Pulse length: 9.9 ms. Acquisition time:3.75 s. Delay time: 2.0 s. Number of scans: 32.
Elemental Analysis
Elemental analyses were done on a Perkin-Elmeranalyzer, model 240 C.
Size-Exclusion Chromatography (SEC)
Average molar masses and molar mass distribu-tions were determined by SEC on a modular Per-kin-Elmer liquid chromatograph equipped with aUV detector. A precolumn, a PL gel column Mixedc (30 cm x 7 mm, particle size 4–5 mm with alinear working range of molar masses between500 and 5 x 106 g/mol, Polymer Laboratories), andTHF as an eluent (1 mL/min) were used. Thecalibration was performed with polystyrene stan-dards (Toyo Soda Manufacturing) in the molarmass region of 500–3.7 3 106 g/mol.
Differential Scanning Calorimetry (DSC)
Thermal characterization was carried out by aPerkin-Elmer DSC 7 differential scanning calo-rimeter. The calibration was performed with in-dium. The given values of Tg, transition temper-atures, and corresponding enthalpies were aver-age values of at least two measurements taken atthe second heating and cooling cycle, respectively.All samples were treated in the same way. Eachsample was placed into a DSC heating cell at50oC, cooled to -50oC, maintained at -50oC for 3min, heated to 150oC, maintained at 150oC for 3min, cooled to -50oC, maintained at -50oC for 3min, and reheated once again. The scanning ratewas 10 K/min.
Polarizing Optical Microscopy (POM)
Optical textures were obtained by using a CarlZeiss polarizing optical microscope Stemi SV6equipped with a microscope camera MC 80 and a
Mettler Toledo FP82 hot stage. The sample waspressed between a glass slide and cover slip andobserved in the LC-temperature range.
Fourier Transform Infrared Spectroscopy (FTIR)
Infrared spectra at various temperatures wereobtained by a Perkin-Elmer FT-IR 1725 X spec-trophotometer equipped with a Spectra-Techheating cell. The resolution was 4 cm-1. The sam-ple was pressed between two NaCl crystal platesand heated to the desired temperature in theheating cell.
X-ray Diffraction
X-ray diffraction patterns were taken on a Sie-mens D-5000 diffractometer using Cu Ka radia-tion (l 5 1.54 Å) in 0.02o steps from 1 to 30o (in 2u)with 2 s per step. X-ray measurements were car-ried out on free-standing unoriented films approx-imately 0.5 mm thick prepared by moulding thepolymers at 150oC.
RESULTS AND DISCUSSIONS
The thermal properties of CnCNPs were investi-gated by polarized light microscopy, differentialscanning calorimetry, and infrared spectroscopy. Aswas expected, polyurethanes (CnCNPs) showed ahigh tendency to crystallize, which was the reasonwe were not able to obtain characteristic textures bypolarized light microscopy. Crystallization did notallow us to keep the sample in a liquid crystallinephase over longer periods in order to develop a clearoptical texture; furthermore, the isotropization tem-peratures of CnCNPs are low (from 34oC for C4CNPto 83oC for C12CNP) and, consequently, the viscos-ities of the samples are high. Figure 1 shows thefine grain texture of C12CNP obtained on coolingthe isotropic phase.
The thermal properties of the CnCNPs, partic-ularly crystallinity, strongly depend on the ther-mal history of the sample. A comparison of theresults obtained by differential scanning calorim-etry and infrared spectroscopy provides evidencethat backbone crystallization is closely related tothe hydrogen bonding of urethane groups (Fig.10). Side chain crystallization takes place as well.Both crystallizations are kinetically controlled,contrary to the transitions due to the mesophaseformation, which are not.
The second heating DSC scans of CnCNPs aregiven in Figure 2, and transition temperatures
SIDE CHAIN LC POLYURETHANES 2139
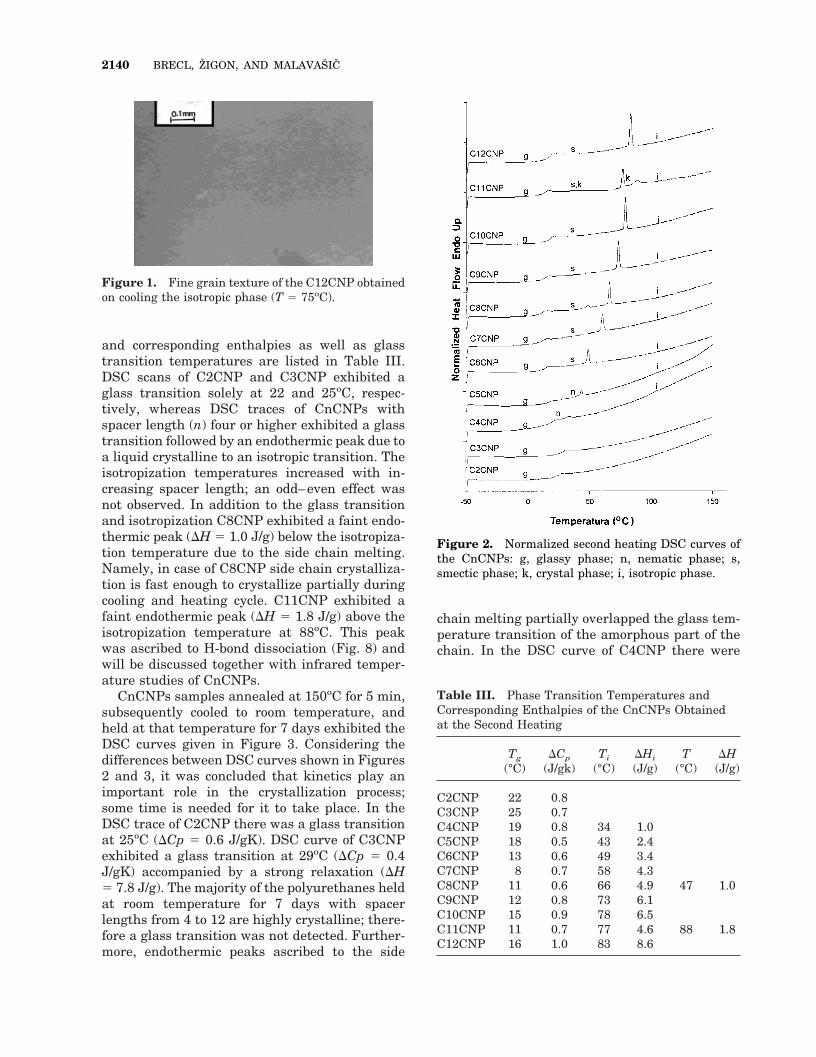
and corresponding enthalpies as well as glasstransition temperatures are listed in Table III.DSC scans of C2CNP and C3CNP exhibited aglass transition solely at 22 and 25oC, respec-tively, whereas DSC traces of CnCNPs withspacer length (n) four or higher exhibited a glasstransition followed by an endothermic peak due toa liquid crystalline to an isotropic transition. Theisotropization temperatures increased with in-creasing spacer length; an odd–even effect wasnot observed. In addition to the glass transitionand isotropization C8CNP exhibited a faint endo-thermic peak (DH 5 1.0 J/g) below the isotropiza-tion temperature due to the side chain melting.Namely, in case of C8CNP side chain crystalliza-tion is fast enough to crystallize partially duringcooling and heating cycle. C11CNP exhibited afaint endothermic peak (DH 5 1.8 J/g) above theisotropization temperature at 88oC. This peakwas ascribed to H-bond dissociation (Fig. 8) andwill be discussed together with infrared temper-ature studies of CnCNPs.
CnCNPs samples annealed at 150oC for 5 min,subsequently cooled to room temperature, andheld at that temperature for 7 days exhibited theDSC curves given in Figure 3. Considering thedifferences between DSC curves shown in Figures2 and 3, it was concluded that kinetics play animportant role in the crystallization process;some time is needed for it to take place. In theDSC trace of C2CNP there was a glass transitionat 25oC (DCp 5 0.6 J/gK). DSC curve of C3CNPexhibited a glass transition at 29oC (DCp 5 0.4J/gK) accompanied by a strong relaxation (DH5 7.8 J/g). The majority of the polyurethanes heldat room temperature for 7 days with spacerlengths from 4 to 12 are highly crystalline; there-fore a glass transition was not detected. Further-more, endothermic peaks ascribed to the side
chain melting partially overlapped the glass tem-perature transition of the amorphous part of thechain. In the DSC curve of C4CNP there were
Table III. Phase Transition Temperatures andCorresponding Enthalpies of the CnCNPs Obtainedat the Second Heating
Tg
(°C)DCp
(J/gk)Ti
(°C)DHi
(J/g)T
(°C)DH(J/g)
C2CNP 22 0.8C3CNP 25 0.7C4CNP 19 0.8 34 1.0C5CNP 18 0.5 43 2.4C6CNP 13 0.6 49 3.4C7CNP 8 0.7 58 4.3C8CNP 11 0.6 66 4.9 47 1.0C9CNP 12 0.8 73 6.1C10CNP 15 0.9 78 6.5C11CNP 11 0.7 77 4.6 88 1.8C12CNP 16 1.0 83 8.6
Figure 1. Fine grain texture of the C12CNP obtainedon cooling the isotropic phase (T 5 75oC).
Figure 2. Normalized second heating DSC curves ofthe CnCNPs: g, glassy phase; n, nematic phase; s,smectic phase; k, crystal phase; i, isotropic phase.
2140 BRECL, ZIGON, AND MALAVASIC
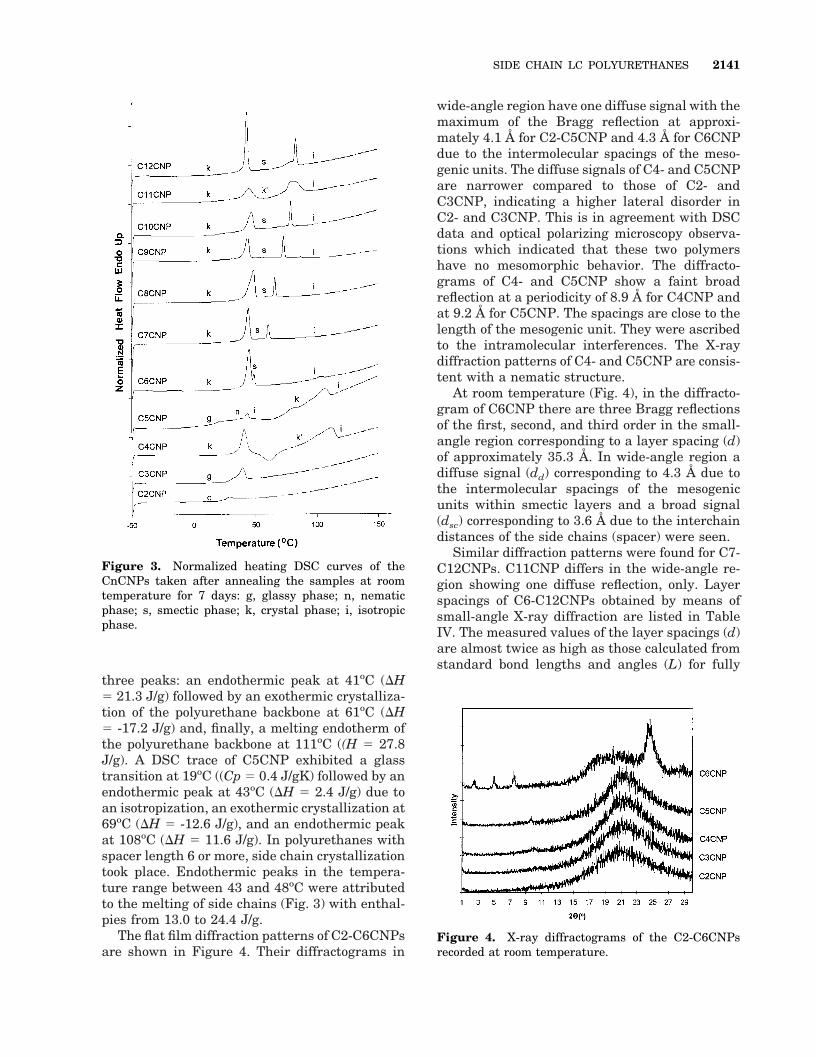
three peaks: an endothermic peak at 41oC (DH5 21.3 J/g) followed by an exothermic crystalliza-tion of the polyurethane backbone at 61oC (DH5 -17.2 J/g) and, finally, a melting endotherm ofthe polyurethane backbone at 111oC ((H 5 27.8J/g). A DSC trace of C5CNP exhibited a glasstransition at 19oC ((Cp 5 0.4 J/gK) followed by anendothermic peak at 43oC (DH 5 2.4 J/g) due toan isotropization, an exothermic crystallization at69oC (DH 5 -12.6 J/g), and an endothermic peakat 108oC (DH 5 11.6 J/g). In polyurethanes withspacer length 6 or more, side chain crystallizationtook place. Endothermic peaks in the tempera-ture range between 43 and 48oC were attributedto the melting of side chains (Fig. 3) with enthal-pies from 13.0 to 24.4 J/g.
The flat film diffraction patterns of C2-C6CNPsare shown in Figure 4. Their diffractograms in
wide-angle region have one diffuse signal with themaximum of the Bragg reflection at approxi-mately 4.1 Å for C2-C5CNP and 4.3 Å for C6CNPdue to the intermolecular spacings of the meso-genic units. The diffuse signals of C4- and C5CNPare narrower compared to those of C2- andC3CNP, indicating a higher lateral disorder inC2- and C3CNP. This is in agreement with DSCdata and optical polarizing microscopy observa-tions which indicated that these two polymershave no mesomorphic behavior. The diffracto-grams of C4- and C5CNP show a faint broadreflection at a periodicity of 8.9 Å for C4CNP andat 9.2 Å for C5CNP. The spacings are close to thelength of the mesogenic unit. They were ascribedto the intramolecular interferences. The X-raydiffraction patterns of C4- and C5CNP are consis-tent with a nematic structure.
At room temperature (Fig. 4), in the diffracto-gram of C6CNP there are three Bragg reflectionsof the first, second, and third order in the small-angle region corresponding to a layer spacing (d)of approximately 35.3 Å. In wide-angle region adiffuse signal (dd) corresponding to 4.3 Å due tothe intermolecular spacings of the mesogenicunits within smectic layers and a broad signal(dsc) corresponding to 3.6 Å due to the interchaindistances of the side chains (spacer) were seen.
Similar diffraction patterns were found for C7-C12CNPs. C11CNP differs in the wide-angle re-gion showing one diffuse reflection, only. Layerspacings of C6-C12CNPs obtained by means ofsmall-angle X-ray diffraction are listed in TableIV. The measured values of the layer spacings (d)are almost twice as high as those calculated fromstandard bond lengths and angles (L) for fully
Figure 4. X-ray diffractograms of the C2-C6CNPsrecorded at room temperature.
Figure 3. Normalized heating DSC curves of theCnCNPs taken after annealing the samples at roomtemperature for 7 days: g, glassy phase; n, nematicphase; s, smectic phase; k, crystal phase; i, isotropicphase.
SIDE CHAIN LC POLYURETHANES 2141

extended trans planar conformation of sidechains. Therefore, it could be concluded thatmesophases of C6-C12CNPs consist of bilayers(overlapping interdigitated structure). However,only the analyses of the X-ray diffraction patternsof oriented samples would enable an unambigu-ous assignment of the mesophases (SA or SC).
In order to obtain further information on thethermal behavior of CnCNP polyurethanes,FT-IR temperature studies focusing on the H-bonds between the N–H and the C5O groups ofthe urethane backbone were made. As knownfrom the literature,18,30,35–39 the N–H stretchingregion as well as the carbonyl stretching region ofpolyurethanes show at least three bands. Theband at the lowest wavenumber was attributed toordered hydrogen-bonded groups and at the high-est wavenumber to free (non-hydrogen-bonded)groups, respectively. The bands appearing be-tween these two bands originate from irregularlyH-bonded groups (disordered H-bonded groups).
Figure 5 shows IR spectra of C5CNP at differ-ent temperatures in the C5O and N–H stretchingregions together with the second derivative spec-tra indicating the individual components. Or-dered H-bonded carbonyl groups are distributedaround 1690 cm-1 and free C5O groups (non-H-bonded) around 1726 cm-1; between these fre-quency values irregularly bonded (disorderedH-bonded) groups were distributed around1707 cm-1.
It has been theoretically predicted and experi-mentally proven that the statistical random-coil
conformation of the polymer main chain is dis-torted in the liquid crystalline phase.40–43 Conse-quently, it can be supposed that mesophase for-mation should have influence on the strength ofthe hydrogen bonding in liquid crystal polyure-thanes. However, in the case of CnCNPs, the N–Hand C5O stretching regions in FT-IR spectra didnot change when the samples passed the isotro-pization temperature. On the other hand, it hasbeen found that spacer length has a great influ-ence on the strength of H-bonds in SCLC polyure-thanes18, which is also demonstrated for aCnCNP series. Recently, Koide et al. synthesizedside chain polyurethanes in which the hydrogenatoms of urethane groups were substituted formethylene groups.25,26 Surprisingly, no meso-morpnic properties were observed for such poly-urethanes, regardless of the spacer length.
The IR spectra of CnCNPs at different temper-atures differ in the N–H stretching (3200–3500
Figure 5. Infrared spectra of the C5CNP and corre-sponding second derivative spectra (2nd D) indicatingthe individual components.
Figure 6. FTIR spectra of the C6CNP at 30 and150oC.
Table IV. Layer Spacings (d) Obtained by Means ofSmall-Angle X-ray Diffraction and TheoreticallyCalculated Length (L) of Fully Extended MesogenicUnit with Spacer, Spacings of Diffuse Signals (dd)inthe Wide-Angle Region, and Interchain Distances ofthe Side Chains (dsc) for CnCNPs
L (Å) d (Å) dd (Å) dsc (Å)
C2CNP 15.6 4.1C3CNP 17.1 4.1C4CNP 17.8 4.1C5CNP 19.3 4.1C6CNP 20.1 35.3 4.3 3.6C7CNP 21.6 37.2 4.3 3.6C8CNP 22.5 39.9 4.3 3.6C9CNP 23.9 41.3 4.3 3.6C10CNP 24.9 44.0 4.3 3.6C11CNP 26.3 47.7 4.1C12CNP 27.3 53.8 4.3 3.6
2142 BRECL, ZIGON, AND MALAVASIC

cm-1) and C5O stretching (1600–1800 cm-1) re-gions, as well as in the amide II (1470–1570 cm-1)region, as is shown for C6CNP at 30 and 150oC inFigure 6. The maximum of the C5O band shiftedfrom 1702 cm-1 at 30oC to 1723 cm-1 at 150oC andof the N–H stretching band from 3335 cm-1 at30oC to 3363 cm-1 at 150oC. The changes can beseen in the amide II region as well. The maximumof the N–H bending band shifted from 1536 cm-1
at 30oC toward 1500 cm-1 with increasing temper-ature, due to H-bond dissociation.
The changes in frequency values for theband maxima of the C5O stretching vibration ofCnCNPs with increasing temperature are shownin Figures 7 and 8. Obviously, the strength ofhydrogen bonding depends not only on the tem-perature but also on the spacer length. For C2-
C3CNP, C6-C10CNP, and C12CNP, the fre-quency values evenly shifted toward 1724 cm-1,while during cooling they evenly decreased to ap-proximately 1700 cm-1 (depended on the spacerlength). As an example, the C5O and N–Hstretching regions of the infrared spectra ofC6CNP at different temperatures are shown inFigure 9.
As can be seen in Figures 7 and 10, for C4-C5CNP, the wavenumber of the C5O stretchingband did not change evenly with increasingtemperature. Infrared thermal analysis ofC4CNP in combination with DSC measurementrevealed that crystallization which took placeabove 40oC was related to hydrogen bonding.The band maximum of the C5O stretching vi-bration was at 1703 cm-1 at 40oC. With an in-crease in temperature to 60oC, the amount ofordered H-bonded carbonyl groups increased atthe expense of disordered H-bonded and non-H-
Figure 7. Shifts of the band maxima of the C2-C6CNPs for the C5O stretching vibrations as a func-tion of temperature.
Figure 8. Shifts of the band maxima of the C7-C12CNPs for the C5O stretching vibrations as a func-tion of temperature.
Figure 9. FTIR spectra of the C6CNP at differenttemperatures: (A) C5O stretching region; (B) N–Hstretching region. Spectra were recorded at a secondheating after the sample was annealed at 150oC for5 min.
SIDE CHAIN LC POLYURETHANES 2143
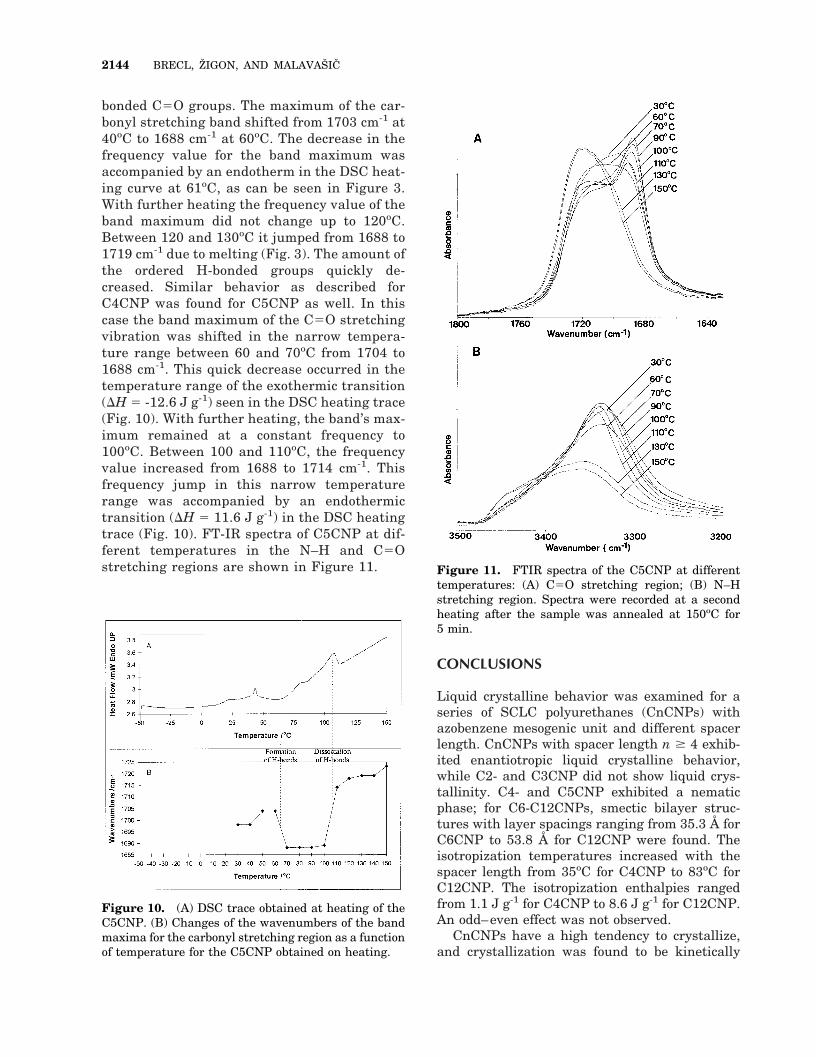
bonded C5O groups. The maximum of the car-bonyl stretching band shifted from 1703 cm-1 at40oC to 1688 cm-1 at 60oC. The decrease in thefrequency value for the band maximum wasaccompanied by an endotherm in the DSC heat-ing curve at 61oC, as can be seen in Figure 3.With further heating the frequency value of theband maximum did not change up to 120oC.Between 120 and 130oC it jumped from 1688 to1719 cm-1 due to melting (Fig. 3). The amount ofthe ordered H-bonded groups quickly de-creased. Similar behavior as described forC4CNP was found for C5CNP as well. In thiscase the band maximum of the C5O stretchingvibration was shifted in the narrow tempera-ture range between 60 and 70oC from 1704 to1688 cm-1. This quick decrease occurred in thetemperature range of the exothermic transition(DH 5 -12.6 J g-1) seen in the DSC heating trace(Fig. 10). With further heating, the band’s max-imum remained at a constant frequency to100oC. Between 100 and 110oC, the frequencyvalue increased from 1688 to 1714 cm-1. Thisfrequency jump in this narrow temperaturerange was accompanied by an endothermictransition (DH 5 11.6 J g-1) in the DSC heatingtrace (Fig. 10). FT-IR spectra of C5CNP at dif-ferent temperatures in the N–H and C5Ostretching regions are shown in Figure 11.
CONCLUSIONS
Liquid crystalline behavior was examined for aseries of SCLC polyurethanes (CnCNPs) withazobenzene mesogenic unit and different spacerlength. CnCNPs with spacer length n $ 4 exhib-ited enantiotropic liquid crystalline behavior,while C2- and C3CNP did not show liquid crys-tallinity. C4- and C5CNP exhibited a nematicphase; for C6-C12CNPs, smectic bilayer struc-tures with layer spacings ranging from 35.3 Å forC6CNP to 53.8 Å for C12CNP were found. Theisotropization temperatures increased with thespacer length from 35oC for C4CNP to 83oC forC12CNP. The isotropization enthalpies rangedfrom 1.1 J g-1 for C4CNP to 8.6 J g-1 for C12CNP.An odd–even effect was not observed.
CnCNPs have a high tendency to crystallize,and crystallization was found to be kinetically
Figure 10. (A) DSC trace obtained at heating of theC5CNP. (B) Changes of the wavenumbers of the bandmaxima for the carbonyl stretching region as a functionof temperature for the C5CNP obtained on heating.
Figure 11. FTIR spectra of the C5CNP at differenttemperatures: (A) C5O stretching region; (B) N–Hstretching region. Spectra were recorded at a secondheating after the sample was annealed at 150oC for5 min.
2144 BRECL, ZIGON, AND MALAVASIC

controlled. The isotropization temperatures werenot strongly affected by the thermal history of thesample. However, the degree of crystallizationwas highly sensitive to the annealing conditions.
In C6-C12CNPs, side chain crystallization wasobserved with melting temperatures from 44 to48oC and corresponding melting enthalpiesranged from 13 to 24 J g-1. Interchain distances(dsc) in crystal phase due to side chain crystalli-zaton obtained by means of wide-angle X-raydiffraction are 3.6 Å.
Though it was anticipated that the distortion ofthe statistical random-coil conformation duringthe mesophase formation would influence inter-molecular interactions and consequently hydro-gen bonding between urethane groups, the N–Hand C5O stretching regions of the FT-IR spectraof CnCNP did not change when the samplespassed the isotropization temperature. The car-bonyl stretching (1600–1800 cm-1) and N–Hstretching (3200–3500 cm-1) regions recorded as afunction of increasing and decreasing tempera-ture revealed not only the temperature depen-dence but also the influence of the spacer lengthon the formation of hydrogen bonding.
The frequency values of the band maxima(C5O stretching) changed with increasing tem-perature. In the IR spectra of C2-C3CNP andC6-C12CNPs, the frequency values evenly shiftedfrom approximately 1700 to 1724 cm-1 when sam-ples were heated. During cooling, the wavenum-bers evenly decreased. The exceptions were C4-and C5CNP. As temperature increased, a coldcrystallization of the main chain took place andthe band maxima of the C5O stretching vibra-tions shifted from 1704 to 1688 cm-1. With furtherheating the band’s maximum remained at a con-stant frequency (1688 cm-1) followed by a fre-quency jump to 1724 cm-1 in a narrow tempera-ture range which was accompanied by an endo-thermic transition (melting of the main chain) inthe DSC heating trace.
This work is a part of the project J2-7505. The authorswish to thank the Ministry of Science and Technologyof the Republic of Slovenia for financial support.
REFERENCES AND NOTES
1. M. Chen, L. Yu, L. R. Dalton, Y. Shi, and W. H.Steier, Macromolecules, 24, 5421 (1991).
2. M. Trollsas, F. Sahlen, U. W. Gedde, A. Hult, D.Hermann, P. Rudquist, L. Komitov, S. T. Lager-
wall, B. Stebler, J. Lindstrom, and O. Rydlund,Macromolecules, 29, 2590 (1996).
3. B. A. Reinhardt, TRIP, 1, 4 (1993).4. M. Eich and J. H. Wendorff , Makromol. Chem.,
186, 2639 (1985).5. M. S. Ho, A. Natansohn, and P. Rochon, Macromol-
ecules, 29, 44 (1996).6. X. Meng, A. Natansohn, C. Barrett, and P. Rochon,
Macromolecules, 29, 946 (1996).7. S. Hvilsted, F. Andruzzi, C. Kulinna, H. W. Siesler,
and P. S. Ramanujam, Macromolecules, 28, 2172(1995).
8. H. Finkelmann, M. Happ, M. Portugal, and H.Ringsdorf, Makromol. Chem., 179, 2541 (1978).
9. V. Percec and C. Pugh, in Side Chain Liquid Crys-tal Polymers, C. B. McArdle, Ed., Blackie, Glasgow,1989, Chap. 3, p. 30.
10. A. A. Craig and C. T. Imrie, Macromolecules, 28,3617 (1995).
11. H. Stevens, and G. Rehage, H. Finkelmann, Mac-romolecules, 17, 851 (1984).
12. C. T. Imrie, F. E. Karasz, and G. S. Attard, Macro-molecules, 26, 3803 (1993).
13. V. Percec, and A. Keller, Macromolecules, 23, 4347(1990).
14. T. Sagane and R. W. Lenz, Polym. J., 20, 923(1988).
15. Z. Komiya, C. Pugh, and R. R. Schrock, Macromol-ecules, 25, 3609 (1993).
16. T. Sagane and R. Lenz, Polymer, 30, 2269 (1989).17. V. Percec and D. Tomazos, in Comprehensive Poly-
mer Science, G. Allen, S. Aggarwal, and S. Russo,Eds., Pergamon Press, London, 1992, Chap. 14, p.299.
18. M. Brecl, and T. Malavasic, J. Polym. Sci.: Part A:Polym. Chem., 35, 2871 (1997).
19. D. J. Simmonds, in Liquid Crystal Polymers: FromStructures to Applications, A. A. Collyer, Ed.,Elsevier Applied Science, London and New York,1992, p. 349.
20. V. Percec and D. Tomazos, Macromolecules, 22,1989 (2062).
21. A. A. Craig and C. T. Imrie, J. Mater. Chem., 4,1705 (1994).
22. V. Percec and D. Tomazos, Adv. Mater., 4, 548(1992).
23. V. Percec and D. Tomazos, Polymer, 31, 1658(1990).
24. V. Percec and D. Tomazos, J. Polym. Sci.: Part A :Polym. Chem., 27, 999 (1989).
25. T. Mihara and N. Koide, Polym. J., 29, 134 (1997).26. T. Mihara and N. Koide, Polym. J., 29, 138 (1997).27. M. Tanaka and T. Nakaya, Makromol. Chem., 190,
3067 (1989).28. M. Tanaka and T. Nakaya, Makromol. Chem., 189,
771 (1988).29. F. Papadimitrakopoulos, E. Sawa, and W. J. Mac-
Knight, Macromolecules, 25, 4682 (1992).
SIDE CHAIN LC POLYURETHANES 2145

30. F. Papadimitrokopoulos, S. L. Hsu, and W. J.MacKnight, Macromolecules, 25, 4671 (1992).
31. J. B. Lee, T. Kato, T. Yoshida, and T. Uryu, Mac-romolecules, 26, 4989 (1993).
32. W. Mormann, and M. Brahm, Macromolecules, 24,1096 (1991).
33. Vogel’s Textbook of Practical Organic Chemistry,4th ed.; B. S. Furniss, A. J. Hannaford, V. Rog-ers, P. W. G. Smith, and A. R. Tatchell, Revisors,Longman Scientific and Technical, New York,1987.
34. J. V. Crivello, M. Deptolla, and H. Ringsdorf, Liq-uid Cryst., 3, 235 (1988).
35. M. M. Coleman, K. H. Lee, D. J. Skrovanek, andP. C. Painter, Macromolecules, 19, 2149 (1986).
36. S. K. Pollack, D. Y. Shen, S. L. Hsu, Q. Wang, andH. D. Stidham, Macromolecules, 22, 551 (1989).
37. J. B. Lee, T. Kato, S. Ujiie, K. Limura, and T. Uryu,Macromolecules, 28, 2165 (1995).
38. P. J. Stenhouse, E. M. Valles, S. W. Kantor, andW. J. MacKnight, Macromolecules, 22, 1467 (1989).
39. S. K. Pollack, G. Smyth, F. Papadimitrakopoulos,P. J. Stenhouse, S. L. Hsu, and W. J. MacKnight,Macromolecules, 25, 2381 (1992).
40. L. Noirez, J. P. Cotton, F. Hardouin, P. Keller, F.Moussa, G. Pepy, and C. Strazielle, Macromole-cules, 21, 2889 (1988).
41. P. Davidson, L. Noirez, J. P. Cotton, and P. Keller,Liq. Cryst., 10, 111 (1991).
42. V. Percec, B. Hahn, M. Ebert, and J. H. Wendorff,Macromolecules, 23, 2092 (1990).
43. F. Kuschel, A. Madicke, S. Diele, H. Utschik, B.Hisgen, and H. Ringsdorf, Polym. Bull., 23, 373(1990).
2146 BRECL, ZIGON, AND MALAVASIC


![Comparative studies of three‐ and four‐ring mesogenic esters containing p‐carborane, bicyclo[2.2.2]octane, cyclohexane, and benzene](https://static.fdokumen.com/doc/165x107/631ec0884c5c8fb3a00e583c/comparative-studies-of-three-and-fourring-mesogenic-esters-containing-pcarborane.jpg)

















