
Influence of UV irradiation on the blue and red light photoinduced processes in azobenzene...
10
Influence of UV irradiation on the blue and red light photoinduced processes in azobenzene polyesters F.J. Rodrı ´guez a , C. Sa ´nchez a , B. Villacampa a , R. Alcala ´ a, * , R. Cases a , M.V. Collados b , S. Hvilsted c , M. Strange d a Departamento de Fı ´sica de la Materia Condensada, Instituto de Ciencia de Materiales de Arago ´n, Universidad de Zaragoza, 50009 Zaragoza, Spain b Departamento de Fı ´sica Aplicada, Universidad de Zaragoza, 50009 Zaragoza, Spain c Danish Polymer Centre, Department of Chemical Engineering, Technical University of Denmark, DK-2800 Kgs Lyngby, Denmark d Risø National Laboratory, DK-4000 Roskilde, Denmark Received 7 January 2004; received in revised form 2 June 2004; accepted 15 June 2004 Abstract Birefringence induced in a series of liquid crystalline side-chain azobenzene polyesters with different substituent groups was investigated under irradiation with 488 and 633 nm linearly polarized lights. Two different initial conditions have been used: the effect of a previous irradiation with UV light that yields the films into the isotropic state at room temperature (RT) was compared with the quenching from temperatures above the isotropic transition temperature T i . UV–visible spectra of the thermally quenched films show the presence of aggregates when measured at RT. We have found that UV light irradiation creates a high concentration of cis isomers and breaks the aggregates, but they are formed again after a few days in dark at RT. Orientation of the chromophores perpendicular to the polarization of the 488 nm light and parallel to the polarization of the 633 nm light was confirmed by dichroism measurements. q 2004 Elsevier Ltd. All rights reserved. Keywords: Azobenzene; Polyesters; Photoinduced optical anisotropy 1. Introduction Photoinduced anisotropy in polymer films containing azobenzene units has been extensively studied [1–16]. Anisotropy is usually induced by illumination with linearly polarised blue or green light (488 or 514 nm) from an Ar C laser. Under this illumination, the orientation of the azo units changes through trans–cis–trans isomerization pro- cesses and a preferential orientation of the trans moieties, with its axis perpendicular to the polarisation direction of the exciting light, can be achieved. High and stable values of the optical anisotropy, which can be checked by dichroism and birefringence measurements, have been obtained, mainly in films of liquid crystalline polymers (LCP) with the azo units in the side chain. The strong molecular interactions among the azo moieties in LCP seems to enhance the photoinduced anisotropy and to improve its stability. Optical anisotropy has also been induced in some azo polymer films under illumination with linearly polarised red light from a He–Ne laser (633 nm), after pre-irradiation with either blue or ultraviolet (UV) light [17–19]. In this case, and depending on the irradiation conditions, the induced orientation of the azo moieties can either be parallel or perpendicular to the polarization direction of the red light. If the films are first irradiated with unpolarised UV light, a subsequent red light irradiation induces, in a first step, a preferential orientation of the trans moieties parallel to the red light polarization [18,20,21]. However, if the red light irradiation goes on for long times, a change in the preferential orientation direction from parallel to perpen- dicular has been found in some polymers [18]. That perpendicular orientation can also be induced in some azo-polymers under red light irradiation without a previous pre-irradiation with UV light [18,22]. 0032-3861/$ - see front matter q 2004 Elsevier Ltd. All rights reserved. doi:10.1016/j.polymer.2004.06.036 Polymer 45 (2004) 6003–6012 www.elsevier.com/locate/polymer * Corresponding author. Tel.: C34-976761333; fax: C34-976761229 E-mail address: [email protected] (R. Alcala ´).
Transcript of Influence of UV irradiation on the blue and red light photoinduced processes in azobenzene...

Influence of UV irradiation on the blue and red light photoinduced
processes in azobenzene polyesters
F.J. Rodrıgueza, C. Sancheza, B. Villacampaa, R. Alcalaa,*, R. Casesa, M.V. Colladosb,S. Hvilstedc, M. Stranged
aDepartamento de Fısica de la Materia Condensada, Instituto de Ciencia de Materiales de Aragon, Universidad de Zaragoza, 50009 Zaragoza, SpainbDepartamento de Fısica Aplicada, Universidad de Zaragoza, 50009 Zaragoza, Spain
cDanish Polymer Centre, Department of Chemical Engineering, Technical University of Denmark, DK-2800 Kgs Lyngby, DenmarkdRisø National Laboratory, DK-4000 Roskilde, Denmark
Received 7 January 2004; received in revised form 2 June 2004; accepted 15 June 2004
Abstract
Birefringence induced in a series of liquid crystalline side-chain azobenzene polyesters with different substituent groups was investigated
under irradiation with 488 and 633 nm linearly polarized lights. Two different initial conditions have been used: the effect of a previous
irradiation with UV light that yields the films into the isotropic state at room temperature (RT) was compared with the quenching from
temperatures above the isotropic transition temperature Ti. UV–visible spectra of the thermally quenched films show the presence of
aggregates when measured at RT. We have found that UV light irradiation creates a high concentration of cis isomers and breaks the
aggregates, but they are formed again after a few days in dark at RT. Orientation of the chromophores perpendicular to the polarization of the
488 nm light and parallel to the polarization of the 633 nm light was confirmed by dichroism measurements.
q 2004 Elsevier Ltd. All rights reserved.
Keywords: Azobenzene; Polyesters; Photoinduced optical anisotropy
1. Introduction
Photoinduced anisotropy in polymer films containing
azobenzene units has been extensively studied [1–16].
Anisotropy is usually induced by illumination with linearly
polarised blue or green light (488 or 514 nm) from an ArC
laser. Under this illumination, the orientation of the azo
units changes through trans–cis–trans isomerization pro-
cesses and a preferential orientation of the trans moieties,
with its axis perpendicular to the polarisation direction of
the exciting light, can be achieved. High and stable values of
the optical anisotropy, which can be checked by dichroism
and birefringence measurements, have been obtained,
mainly in films of liquid crystalline polymers (LCP) with
the azo units in the side chain. The strong molecular
interactions among the azo moieties in LCP seems to
0032-3861/$ - see front matter q 2004 Elsevier Ltd. All rights reserved.
doi:10.1016/j.polymer.2004.06.036
* Corresponding author. Tel.: C34-976761333; fax: C34-976761229
E-mail address: [email protected] (R. Alcala).
enhance the photoinduced anisotropy and to improve its
stability.
Optical anisotropy has also been induced in some azo
polymer films under illumination with linearly polarised red
light from a He–Ne laser (633 nm), after pre-irradiation with
either blue or ultraviolet (UV) light [17–19]. In this case,
and depending on the irradiation conditions, the induced
orientation of the azo moieties can either be parallel or
perpendicular to the polarization direction of the red light. If
the films are first irradiated with unpolarised UV light, a
subsequent red light irradiation induces, in a first step, a
preferential orientation of the trans moieties parallel to the
red light polarization [18,20,21]. However, if the red light
irradiation goes on for long times, a change in the
preferential orientation direction from parallel to perpen-
dicular has been found in some polymers [18]. That
perpendicular orientation can also be induced in some
azo-polymers under red light irradiation without a previous
pre-irradiation with UV light [18,22].
Polymer 45 (2004) 6003–6012
www.elsevier.com/locate/polymer

Fig. 1. (a) Chemical structure of the P6x4 polymers. (b) trans and cis
configurations of the azo moieties.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–60126004
Pre-irradiation with UV light produces a high concen-
tration of the cis isomer. Subsequent red light irradiation
induces the preferential back isomerization of the cis
moeities with its axis parallel to the red light polarisation
direction, to the trans form. But the presence of high cis
content in the films seems to produce some other changes in
the properties of the films. Thus, it has been reported that
UV irradiation can induce a liquid crystal (LC) to isotropic
phase transition at temperatures well below Ti (LC to
isotropic transition temperature without irradiation) [23].
This transition is usually accompanied by a strong decrease
in the light scattered by the film [24]. UV irradiation is also
able to break the aggregates of azobenzene moieties that
appear in many polymers and this can play an important role
in the photoinduced processes [25–27].
From all these results it is clear that the presence of the
cis isomer, induced by UV irradiation, strongly influences
the optical properties and the response of azobenzene
polymer films to light irradiation. In this paper we present a
study of the influence of UV pre-irradiation on the optical
properties of a series of side chain LC azobenzene
polyesters. The different polymers in this series were
chosen to have different terminal groups. In this way the
thermal properties as well as the aggregation trend are
modified along the series and the influence of those
properties on the photoinduced processes can be explored.
Optical absorption, dichroism and birefringence have been
studied using different irradiation conditions with either
blue light (488 nm) or red light (633 nm). The influence of
UV pre-irradiation on the photoinduced processes has been
analysed.
2. Experimental
The liquid crystalline polyesters used in this work were
prepared in a K2CO3 catalyzed transesterification of
equimolar amounts of diphenyl adipate [28] and the
appropriately 4-substituted (X) 2-[6-[4-[(4-Xphenyl)azo]-
phenoxy]hexyl]1,3-propanediol [29] in the melt under
vacuum at elevated temperature [30]. Further details are
provided elsewhere [31,32]. The chemical structure of the
investigated polyesters P6x4 is given in Fig. 1(a), while the
trans and cis configurations of the azo chromophore are
given in Fig. 1(b). The five different polyester substituents
Table 1
Transition temperatures and enthalpies and molecular mass of polyesters P6x4 m
x R Tg (8C) Ti (8C) D
c OCH3 22 63 1
e CF3 44 73 5
fa CH3 29 61 7
65 5
j Cl 9 54 2
k Br 24 81 3
a This polymer shows three phase transitions.
are shown in Table 1. Their glass transition temperatures
(Tg) and their isotropic transition temperatures (Ti) as well
as their molecular masses and transition enthalpies are also
collected in Table 1.
Films were produced by casting a solution of the
polymers in chloroform (0.5 mg in 200 ml) onto clean
glass substrates. Thickness was measured using a DEKTAK
profilometer. Thickness value ranged from 200 to 400 nm.
Before performing any experiment, all the films were heated
up to 20 8C above Ti for 10 min. Films were then fast
quenched to RT by putting them on a metallic plate.
A high pressure Hg lamp followed by a band pass UV
filter (maximum transmission at 350 nm) was used as the
UV light source. The measured light intensity was about
0.5 mW/cm2 in the film. The polymer films have been
easured heating up the polymers (third run) at 3 8C/min
H (cal/g) Mn Mw Mw/Mn
0.53 10,000 16,000 1.6
.81 27,000 42,000 1.6
.37 34,000 55,000 1.6
.36
.32 31,000 61,000 2.0
.99 35,000 62,000 1.8

F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–6012 6005
examined in the polarization microscope before and after
irradiation with UV light. Fast-quenched films show a
random distribution of micro-domains. However, we were
not able to identify the type of phase of those domains. In
films irradiated with UV light, the domain structure is barely
observed and the light scattering produced by the film is
drastically reduced. Vertically polarized light beams at 488
and 633 nm were provided by ArC and He–Ne lasers,
respectively. The intensity used was 75 mW/cm2 for the
488 nm light and 1 W/cm2 for the 633 nm light. Optical
absorption measurements have been performed in a Cary
500 Scan UV–Vis-NIR spectrophotometer. Linear (verti-
cal/horizontal) polarization of the measuring beams was
achieved using a Glan-Thomson prism. Birefringence
measurements were performed using the setup shown in
Fig. 2. The sample was placed between crossed polarizers
with their polarization directions at G458 with the vertical
axis. The light from a 780 nm diode laser transmitted
through the polarizer-sample-polarizer system was
measured with a Si detector as a probe of the photoinduced
anisotropy. The transmitted intensity I is given by the
equation:
I Z I0 sin2ðpjDnjd=lÞ
where I0 is the intensity transmitted by the as quenched films
between parallel polarizers, d the film thickness, Dn the
Fig. 2. Experimental set-up for bi
birefringence of the sample and l the wavelength of the
measuring light (780 nm). In isotropic samples (DnZ0), no
transmitted light is expected and thus, birefringence
measurements also enable us to detect the transition from
an anisotropic (Is0) to an isotropic (IZ0) state. An optical
pyrometer was used to check that the temperature of the
films did not increase during irradiation.
3. Experimental results and discussion
As we said above, the main goal of the present work is to
study the influence of UV light pre-irradiation on both the
optical properties and the photoinduced optical effects,
under several irradiation conditions, in films of side chain
liquid crystalline azo-polyesters with different end substi-
tutions. The polyesters generally have relatively high
molecular masses. As seen from Table 1 most of the
polyesters have Mn>27,000 stemming from DP’s >50 with
corresponding polydispersities (Mw/Mn) from 1.6 to 2.0.
P6c4 has a somehow lowerMn, however, this has previously
not caused significant changes in physical properties for this
type of polyesters [30]. Azobenzene units can appear in two
isomeric forms: trans and cis. Before any treatment, the azo
moieties are in the trans form that is the most stable one.
However, the azo moieties show a tendency to form
refringence measurements.

F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–60126006
different types of aggregates and the photoinduced effects
depend on the aggregation state. To check for the presence
of these aggregates in our films we have compared the
optical absorption spectrum of the different P6x4 polymer
measured at RT in a chloroform solution, with the spectra of
the polymer films measured at TZTiC10 8C and at RT
(after quenching the films from the isotropic phase to RT).
The results are given in Fig. 3. It can be seen that
independently of the end substitution, all the spectra
measured in solution show a main absorption band at
about 360 nm and a shoulder at about 450 nm that are
associated with the p–p* and n–p* transitions of the
azobenzene moieties, respectively. The spectra of the films
measured at TZTiC10 8C are similar (although slightly
broader) to those in solution but the ones at RT show new
bands that are due to the formation of different types of
azobenzene aggregates [16,26]. Besides, the strong decrease
Fig. 3. Optical absorption spectra of the different polyester measured:.. at RT in
from the isotropic phase to RT.
in the optical absorption when measured at RT can be
related with a tendency of the azobenzene units to be
oriented out of the plane of the film. This type of behaviour
has been previously reported by several authors [27,33,34].
In conclusion, the RT spectra of the different polymer films
show remarkable differences, indicating that the formation
of aggregates and their optical absorptions are strongly
influenced by the end substitution.
In a first step we have studied the time evolution of the
birefringence (Dn) induced with 350 nm linearly polarized
light in the polymer films quenched from the isotropic
phase. The evolution is similar in all the polymers. As an
example we give in Fig. 4 the evolution for P6c4. It can be
seen that birefringence increases with irradiation time,
reaches a maximum and then goes back to zero. The initial
growth is associated with the photoinduced trans–cis–trans
isomerization. Under irradiation with UV light in the
CHCl3 solution;.. at TZTiC10 8C in films; — at RT in films quenched
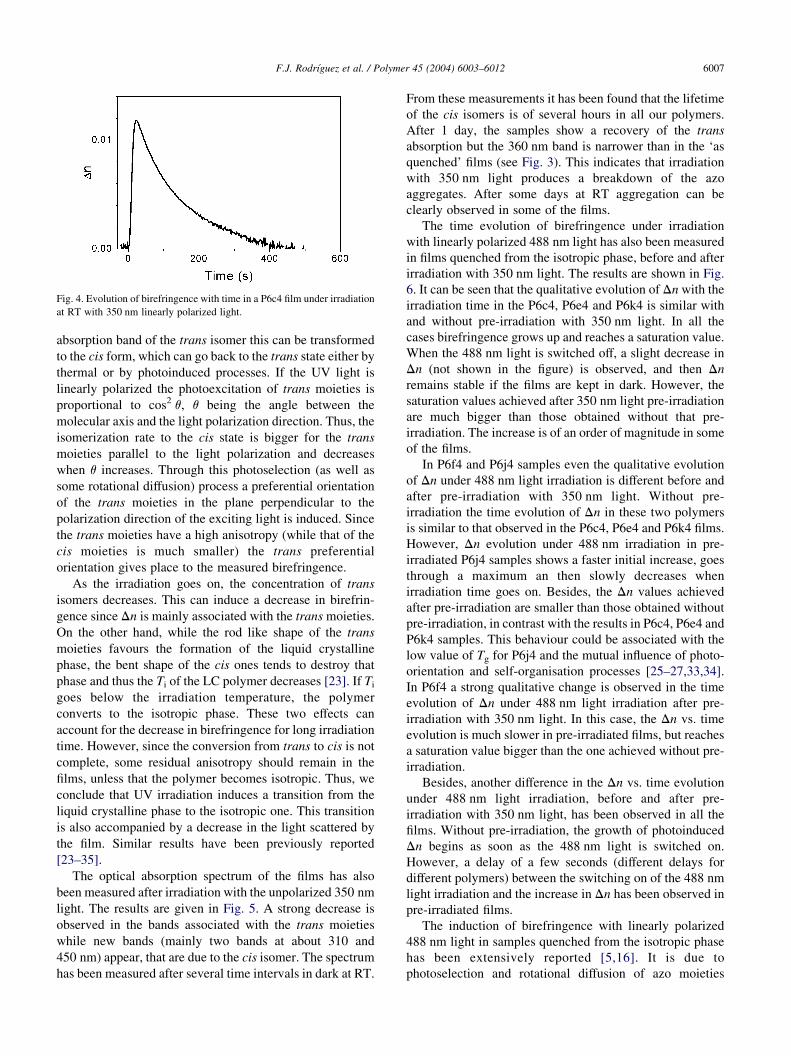
Fig. 4. Evolution of birefringence with time in a P6c4 film under irradiation
at RT with 350 nm linearly polarized light.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–6012 6007
absorption band of the trans isomer this can be transformed
to the cis form, which can go back to the trans state either by
thermal or by photoinduced processes. If the UV light is
linearly polarized the photoexcitation of trans moieties is
proportional to cos2 q, q being the angle between the
molecular axis and the light polarization direction. Thus, the
isomerization rate to the cis state is bigger for the trans
moieties parallel to the light polarization and decreases
when q increases. Through this photoselection (as well as
some rotational diffusion) process a preferential orientation
of the trans moieties in the plane perpendicular to the
polarization direction of the exciting light is induced. Since
the trans moieties have a high anisotropy (while that of the
cis moieties is much smaller) the trans preferential
orientation gives place to the measured birefringence.
As the irradiation goes on, the concentration of trans
isomers decreases. This can induce a decrease in birefrin-
gence since Dn is mainly associated with the transmoieties.
On the other hand, while the rod like shape of the trans
moieties favours the formation of the liquid crystalline
phase, the bent shape of the cis ones tends to destroy that
phase and thus the Ti of the LC polymer decreases [23]. If Tigoes below the irradiation temperature, the polymer
converts to the isotropic phase. These two effects can
account for the decrease in birefringence for long irradiation
time. However, since the conversion from trans to cis is not
complete, some residual anisotropy should remain in the
films, unless that the polymer becomes isotropic. Thus, we
conclude that UV irradiation induces a transition from the
liquid crystalline phase to the isotropic one. This transition
is also accompanied by a decrease in the light scattered by
the film. Similar results have been previously reported
[23–35].
The optical absorption spectrum of the films has also
been measured after irradiation with the unpolarized 350 nm
light. The results are given in Fig. 5. A strong decrease is
observed in the bands associated with the trans moieties
while new bands (mainly two bands at about 310 and
450 nm) appear, that are due to the cis isomer. The spectrum
has been measured after several time intervals in dark at RT.
From these measurements it has been found that the lifetime
of the cis isomers is of several hours in all our polymers.
After 1 day, the samples show a recovery of the trans
absorption but the 360 nm band is narrower than in the ‘as
quenched’ films (see Fig. 3). This indicates that irradiation
with 350 nm light produces a breakdown of the azo
aggregates. After some days at RT aggregation can be
clearly observed in some of the films.
The time evolution of birefringence under irradiation
with linearly polarized 488 nm light has also been measured
in films quenched from the isotropic phase, before and after
irradiation with 350 nm light. The results are shown in Fig.
6. It can be seen that the qualitative evolution of Dn with theirradiation time in the P6c4, P6e4 and P6k4 is similar with
and without pre-irradiation with 350 nm light. In all the
cases birefringence grows up and reaches a saturation value.
When the 488 nm light is switched off, a slight decrease in
Dn (not shown in the figure) is observed, and then Dnremains stable if the films are kept in dark. However, the
saturation values achieved after 350 nm light pre-irradiation
are much bigger than those obtained without that pre-
irradiation. The increase is of an order of magnitude in some
of the films.
In P6f4 and P6j4 samples even the qualitative evolution
of Dn under 488 nm light irradiation is different before and
after pre-irradiation with 350 nm light. Without pre-
irradiation the time evolution of Dn in these two polymers
is similar to that observed in the P6c4, P6e4 and P6k4 films.
However, Dn evolution under 488 nm irradiation in pre-
irradiated P6j4 samples shows a faster initial increase, goes
through a maximum an then slowly decreases when
irradiation time goes on. Besides, the Dn values achieved
after pre-irradiation are smaller than those obtained without
pre-irradiation, in contrast with the results in P6c4, P6e4 and
P6k4 samples. This behaviour could be associated with the
low value of Tg for P6j4 and the mutual influence of photo-
orientation and self-organisation processes [25–27,33,34].
In P6f4 a strong qualitative change is observed in the time
evolution of Dn under 488 nm light irradiation after pre-
irradiation with 350 nm light. In this case, the Dn vs. time
evolution is much slower in pre-irradiated films, but reaches
a saturation value bigger than the one achieved without pre-
irradiation.
Besides, another difference in the Dn vs. time evolution
under 488 nm light irradiation, before and after pre-
irradiation with 350 nm light, has been observed in all the
films. Without pre-irradiation, the growth of photoinduced
Dn begins as soon as the 488 nm light is switched on.
However, a delay of a few seconds (different delays for
different polymers) between the switching on of the 488 nm
light irradiation and the increase in Dn has been observed inpre-irradiated films.
The induction of birefringence with linearly polarized
488 nm light in samples quenched from the isotropic phase
has been extensively reported [5,16]. It is due to
photoselection and rotational diffusion of azo moieties
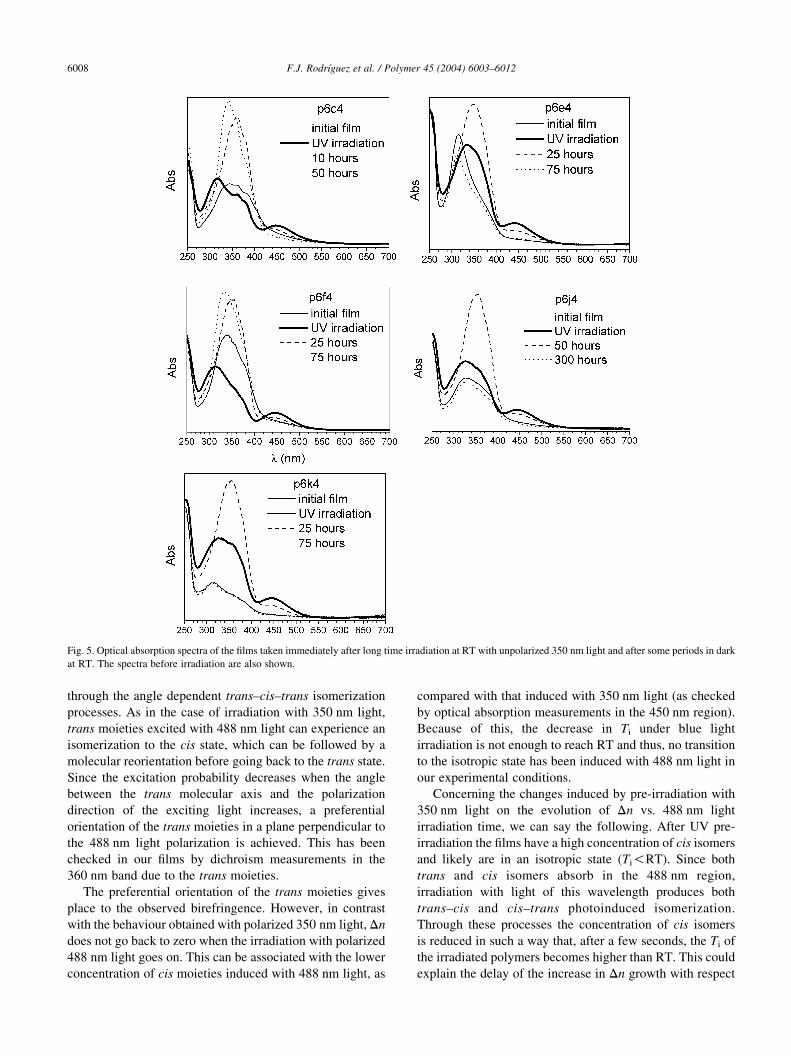
Fig. 5. Optical absorption spectra of the films taken immediately after long time irradiation at RT with unpolarized 350 nm light and after some periods in dark
at RT. The spectra before irradiation are also shown.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–60126008
through the angle dependent trans–cis–trans isomerization
processes. As in the case of irradiation with 350 nm light,
trans moieties excited with 488 nm light can experience an
isomerization to the cis state, which can be followed by a
molecular reorientation before going back to the trans state.
Since the excitation probability decreases when the angle
between the trans molecular axis and the polarization
direction of the exciting light increases, a preferential
orientation of the trans moieties in a plane perpendicular to
the 488 nm light polarization is achieved. This has been
checked in our films by dichroism measurements in the
360 nm band due to the trans moieties.
The preferential orientation of the trans moieties gives
place to the observed birefringence. However, in contrast
with the behaviour obtained with polarized 350 nm light, Dndoes not go back to zero when the irradiation with polarized
488 nm light goes on. This can be associated with the lower
concentration of cis moieties induced with 488 nm light, as
compared with that induced with 350 nm light (as checked
by optical absorption measurements in the 450 nm region).
Because of this, the decrease in Ti under blue light
irradiation is not enough to reach RT and thus, no transition
to the isotropic state has been induced with 488 nm light in
our experimental conditions.
Concerning the changes induced by pre-irradiation with
350 nm light on the evolution of Dn vs. 488 nm light
irradiation time, we can say the following. After UV pre-
irradiation the films have a high concentration of cis isomers
and likely are in an isotropic state (Ti!RT). Since both
trans and cis isomers absorb in the 488 nm region,
irradiation with light of this wavelength produces both
trans–cis and cis–trans photoinduced isomerization.
Through these processes the concentration of cis isomers
is reduced in such a way that, after a few seconds, the Ti of
the irradiated polymers becomes higher than RT. This could
explain the delay of the increase in Dn growth with respect

Fig. 6. Birefringence of the different films as a function of time under RT irradiation with linearly polarized 488 nm light. // after quenching from the
isotropic phase to RT; — after pre-irradiation with unpolarized 350 nm light.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–6012 6009
to the switching on of the 488 nm light irradiation. Then,
photo-orientation of trans moieties begins with a process
similar to that in the films that have not been pre-irradiated.
However, since the initial cis concentration is higher in the
pre-irradiated samples Ti is lower. Besides, the azo
aggregates that are present before UV irradiation are broken
after irradiation. These two facts, the decrease in Ti and the
breaking of aggregates, can produce and increase in the
mobility of the transmoieties similar to the one achieved by
increasing the temperature of the film. It has been reported
that the evolution of Dn with irradiation time is strongly
dependent on temperature [10,36]. The production effi-
ciency of Dn increases at low temperatures, goes through a
maximum and then decreases again to zero at high
temperatures. Thus, the effect of UV pre-irradiation could
be understood as equivalent to a heating up of the films and
this could account for the results shown in Fig. 6. A detailed
study of the temperature and light power dependence of all
these processes is under way in our laboratory.
Birefringence induced with linearly polarized 633 nm
light has also been measured before and after pre-irradiation
with UV light. It is known that red light can induce a
preferential orientation of the trans moieties that can either
be parallel or perpendicular to the polarization of the red
light, depending on the polymer and the irradiation
conditions. In our case, no birefringence has been induced
with 633 nm light in films quenched from the isotropic state.
After pre-irradiation with UV light some birefringence has
been observed. The results are given in Fig. 7. It can be seen
that in all the films, a photoinduced birefringence begins to
grow after a delay time. The growth of Dn continues, in
some of the films, for several hours, and then reaches a
saturation value. When the 633 nm light is switched off Dnremains stable if the samples are kept in dark. The saturation
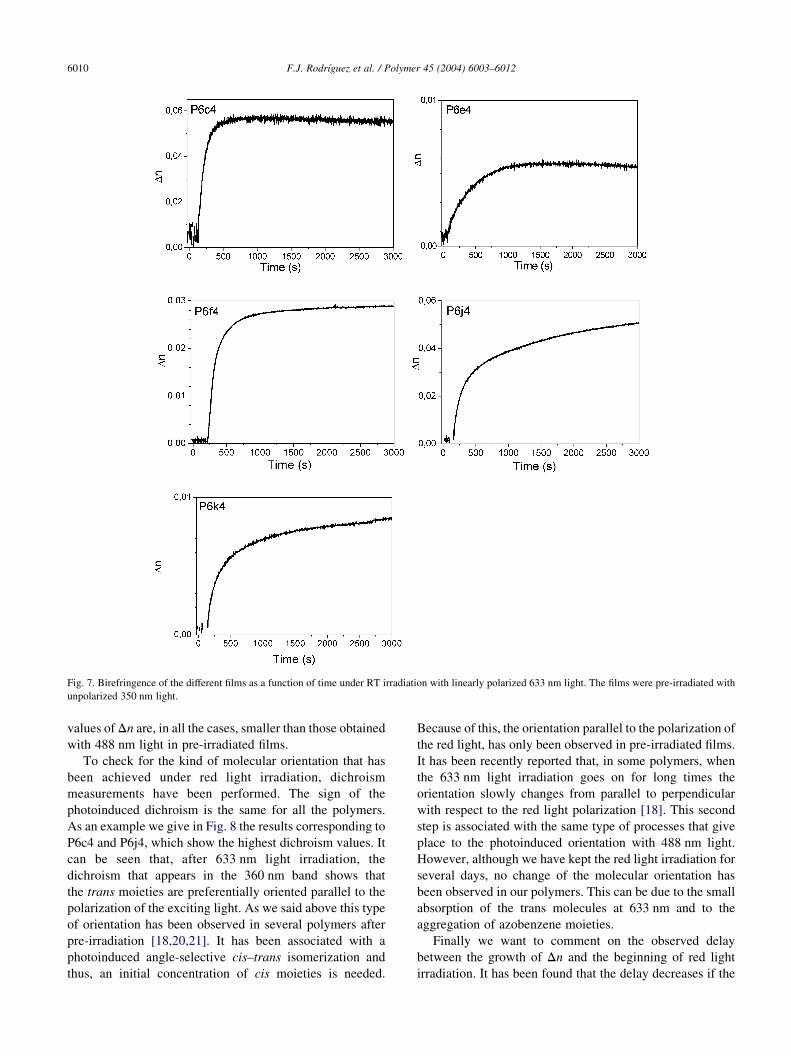
Fig. 7. Birefringence of the different films as a function of time under RT irradiation with linearly polarized 633 nm light. The films were pre-irradiated with
unpolarized 350 nm light.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–60126010
values of Dn are, in all the cases, smaller than those obtained
with 488 nm light in pre-irradiated films.
To check for the kind of molecular orientation that has
been achieved under red light irradiation, dichroism
measurements have been performed. The sign of the
photoinduced dichroism is the same for all the polymers.
As an example we give in Fig. 8 the results corresponding to
P6c4 and P6j4, which show the highest dichroism values. It
can be seen that, after 633 nm light irradiation, the
dichroism that appears in the 360 nm band shows that
the trans moieties are preferentially oriented parallel to the
polarization of the exciting light. As we said above this type
of orientation has been observed in several polymers after
pre-irradiation [18,20,21]. It has been associated with a
photoinduced angle-selective cis–trans isomerization and
thus, an initial concentration of cis moieties is needed.
Because of this, the orientation parallel to the polarization of
the red light, has only been observed in pre-irradiated films.
It has been recently reported that, in some polymers, when
the 633 nm light irradiation goes on for long times the
orientation slowly changes from parallel to perpendicular
with respect to the red light polarization [18]. This second
step is associated with the same type of processes that give
place to the photoinduced orientation with 488 nm light.
However, although we have kept the red light irradiation for
several days, no change of the molecular orientation has
been observed in our polymers. This can be due to the small
absorption of the trans molecules at 633 nm and to the
aggregation of azobenzene moieties.
Finally we want to comment on the observed delay
between the growth of Dn and the beginning of red light
irradiation. It has been found that the delay decreases if the
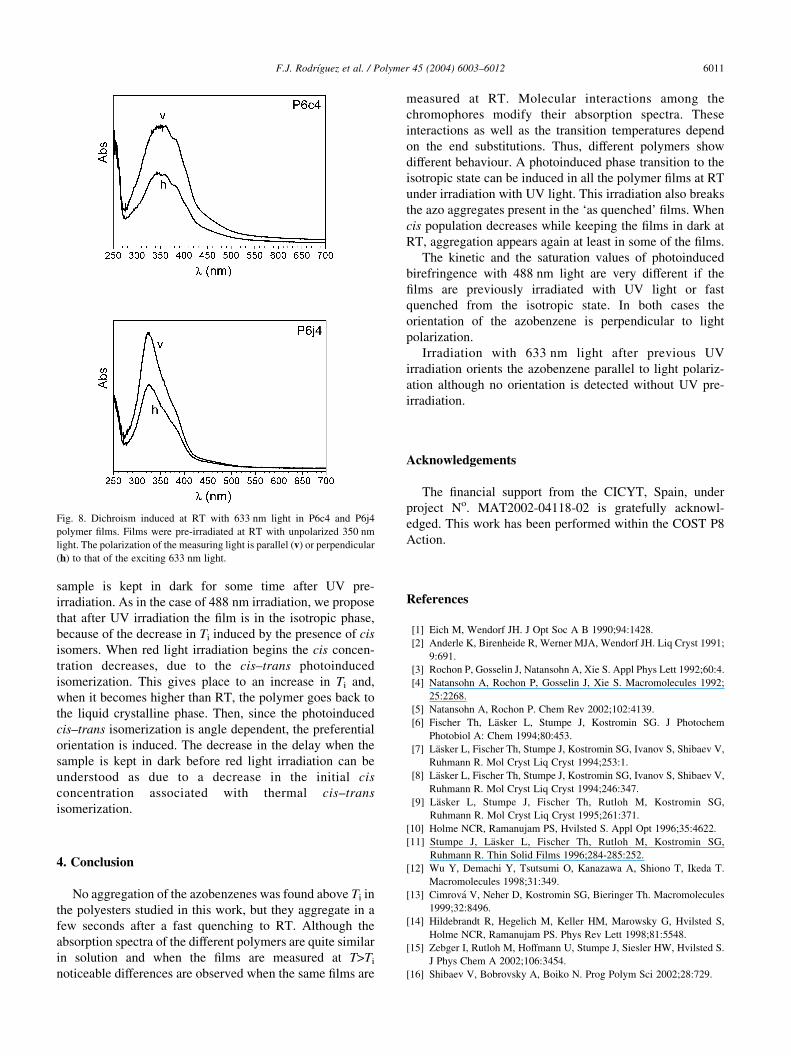
Fig. 8. Dichroism induced at RT with 633 nm light in P6c4 and P6j4
polymer films. Films were pre-irradiated at RT with unpolarized 350 nm
light. The polarization of the measuring light is parallel (v) or perpendicular
(h) to that of the exciting 633 nm light.
F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–6012 6011
sample is kept in dark for some time after UV pre-
irradiation. As in the case of 488 nm irradiation, we propose
that after UV irradiation the film is in the isotropic phase,
because of the decrease in Ti induced by the presence of cis
isomers. When red light irradiation begins the cis concen-
tration decreases, due to the cis–trans photoinduced
isomerization. This gives place to an increase in Ti and,
when it becomes higher than RT, the polymer goes back to
the liquid crystalline phase. Then, since the photoinduced
cis–trans isomerization is angle dependent, the preferential
orientation is induced. The decrease in the delay when the
sample is kept in dark before red light irradiation can be
understood as due to a decrease in the initial cis
concentration associated with thermal cis–trans
isomerization.
4. Conclusion
No aggregation of the azobenzenes was found above Ti in
the polyesters studied in this work, but they aggregate in a
few seconds after a fast quenching to RT. Although the
absorption spectra of the different polymers are quite similar
in solution and when the films are measured at T>Tinoticeable differences are observed when the same films are
measured at RT. Molecular interactions among the
chromophores modify their absorption spectra. These
interactions as well as the transition temperatures depend
on the end substitutions. Thus, different polymers show
different behaviour. A photoinduced phase transition to the
isotropic state can be induced in all the polymer films at RT
under irradiation with UV light. This irradiation also breaks
the azo aggregates present in the ‘as quenched’ films. When
cis population decreases while keeping the films in dark at
RT, aggregation appears again at least in some of the films.
The kinetic and the saturation values of photoinduced
birefringence with 488 nm light are very different if the
films are previously irradiated with UV light or fast
quenched from the isotropic state. In both cases the
orientation of the azobenzene is perpendicular to light
polarization.
Irradiation with 633 nm light after previous UV
irradiation orients the azobenzene parallel to light polariz-
ation although no orientation is detected without UV pre-
irradiation.
Acknowledgements
The financial support from the CICYT, Spain, under
project No. MAT2002-04118-02 is gratefully acknowl-
edged. This work has been performed within the COST P8
Action.
References
[1] Eich M, Wendorf JH. J Opt Soc A B 1990;94:1428.
[2] Anderle K, Birenheide R, Werner MJA, Wendorf JH. Liq Cryst 1991;
9:691.
[3] Rochon P, Gosselin J, Natansohn A, Xie S. Appl Phys Lett 1992;60:4.
[4] Natansohn A, Rochon P, Gosselin J, Xie S. Macromolecules 1992;
25:2268.
[5] Natansohn A, Rochon P. Chem Rev 2002;102:4139.
[6] Fischer Th, Lasker L, Stumpe J, Kostromin SG. J Photochem
Photobiol A: Chem 1994;80:453.
[7] Lasker L, Fischer Th, Stumpe J, Kostromin SG, Ivanov S, Shibaev V,
Ruhmann R. Mol Cryst Liq Cryst 1994;253:1.
[8] Lasker L, Fischer Th, Stumpe J, Kostromin SG, Ivanov S, Shibaev V,
Ruhmann R. Mol Cryst Liq Cryst 1994;246:347.
[9] Lasker L, Stumpe J, Fischer Th, Rutloh M, Kostromin SG,
Ruhmann R. Mol Cryst Liq Cryst 1995;261:371.
[10] Holme NCR, Ramanujam PS, Hvilsted S. Appl Opt 1996;35:4622.
[11] Stumpe J, Lasker L, Fischer Th, Rutloh M, Kostromin SG,
Ruhmann R. Thin Solid Films 1996;284-285:252.
[12] Wu Y, Demachi Y, Tsutsumi O, Kanazawa A, Shiono T, Ikeda T.
Macromolecules 1998;31:349.
[13] Cimrova V, Neher D, Kostromin SG, Bieringer Th. Macromolecules
1999;32:8496.
[14] Hildebrandt R, Hegelich M, Keller HM, Marowsky G, Hvilsted S,
Holme NCR, Ramanujam PS. Phys Rev Lett 1998;81:5548.
[15] Zebger I, Rutloh M, Hoffmann U, Stumpe J, Siesler HW, Hvilsted S.
J Phys Chem A 2002;106:3454.
[16] Shibaev V, Bobrovsky A, Boiko N. Prog Polym Sci 2002;28:729.

F.J. Rodrıguez et al. / Polymer 45 (2004) 6003–60126012
[17] Kulinna Ch, Zebger I, Hvilsted S, Ramanujam PS, Siesler HW.
Macromol Symp 1994;83:169.
[18] Kempe C, Rutloh M, Stumpe J. J Phys: Condens Matter 2003;15:813.
[19] Haosheng H, Wei Z, Wu P, Han L, Zhao Y, Che Y. Opt Lett 1994;
19:411.
[20] Sanchez C, Alcala R, Hvilsted S, Ramanujam PS. Appl Phys Lett
2000;77:1440.
[21] Sanchez C, Alcala R, Hvilsted S, Ramanujam PS. Appl Phys Lett
2001;78:3944.
[22] Wu Y, Mamiya JI, Tsutsumi O, Kanazawa A, Shiono T, Ikeda T. Liq
Cryst 2000;27:749.
[23] Ikeda T, Miyamoto T, Kurihara S, Tsukada M, Tazuke S. Mol Cryst
Liq Cryst 1990;182B:373.
[24] Lee HK, Kanazawa A, Shiono T, Ikeda T. ChemMater 1998;10:1402.
[25] Fischer Th, Lasker L, Rutloh M, Czapla S, Stumpe J. Mol Cryst Liq
Cryst 1997;299:293.
[26] Rutloh M, Stumpe J, Stachanov L, Kostromin SG, Shibaev V. Mol
Cryst Liq Cryst 2000;352:149.
[27] Meier JG, Ruhmann R, Stumpe J. Macromolecules 2000;(33):843.
[28] Hvilsted S, Andruzzi F, Cerrai P, Tricoli M. Polymer 1991;32:127.
[29] Hendann C, Siesler HW, Andruzzi F, Kulinna C, Hvilsted S. Mol
Cryst Liq Cryst 1998;319:207.
[30] Hvilsted S, Andruzzi F, Kulinna C, Siesler HW, Ramanujam PS.
Macromolecules 1995;28:2172.
[31] Pedersen M, Hvilsted S, Holme NCR, Ramanujam PS. Macromol
Symp 1999;137:115.
[32] Pedersen M. PhD Thesis New Azobenzene Side-Chain Polyesters for
Optical Information Storage, Technical University of Denmark; 1997.
[33] Fischer Th, Lasker L, Czapla S, Rubner J, Stumpe J. Mol Cryst Liq
Cryst 1997;298:213.
[34] Rosenhauer R, Fischer Th, Czapla S, Stumpe J, Vinuales A, Pinol M,
Serrano JL. Mol Cryst Liq Cryst 2001;364:295.
[35] Sanchez C, Alcala R, Hvilsted S, Ramanujam PS. J Appl Phys 2003;
93:4454.
[36] Song OK, Wang CH, Pauley MA. Macromolecules 1997;30:6913.




















