
Photoisomerization of amphiphilic azobenzene derivatives in Langmuir Blodgett films prepared as...
8
Photoisomerization of amphiphilic azobenzene derivatives in Langmuir Blodgett films prepared as polyion complexes, using ionic polymers Vishakha R. Shembekar a,1 , A.Q. Contractor a , S.S. Major b , S.S. Talwar b, * a Department of Chemistry, Indian Institute of Technology, Bombay, Mumbai-400 076, India b Department of Physics, Indian Institute of Technology, Bombay, Mumbai-400 076, India Abstract Polyion complexation in mixed Langmuir and Langmuir Blodgett (LB) films of photochromic amphiphilic azobenzene carboxylic acids, 11-[4- (4-hexylphenyl)azo] phenoxyundecanoic acid, 11-(4-phenylazo)phenoxyundecanoic acid, and diamine grafted poly(methylmethaacrylate) polymers has been studied. Monolayer behaviour of the pure components and mixed films was studied through pressure – area isotherms and LB films were characterized by spectroscopic, X-ray diffraction and Atomic force microscopy techniques. Aggregation (H-type), often observed in LB films of pure amphiphilic azo acids, was partly avoided in the mixed LB films as indicated by absorption spectral studies. Photoisomerization of the polyion complexed LB films was also studied. The results altogether demonstrate that amine grafted polymer enter into a polyion complexation with azo acid carboxylate group. LB films could be obtained by transfer of the composite monolayers and these LB films exhibited different levels of aggregation of the azo acids. Reversible photoisomerization was observed in LB films with unaggregated azo acid. Keywords: Polyion complexes; Azobenzenes; Langmuir-Blodgett films; Photoisomerization 1. Introduction Photochrome containing Langmuir Blodgett (LB) films have been explored as media for photochromic reactions aimed at constructing molecule based information storage or switch- ing devices [1–3]. In this context LB films of azobenzene derivatives have attracted increasing attention in recent years due to their diverse photofunctional applications [4–8]. These derivatives undergo facile cis – trans photoisomerization in solution, however isomerization is inhibited [9–12] in pure LB films due to insufficient Ffree volume_ in the higher order structure of LB films. Photoisomerization not only involves electronic changes but also involves changes in the molecular conformation and cross-sectional area due to difference in the shape of the cis and the trans isomers. Changes in the cross- sectional area and molecular conformation that are associated with azobenzene isomerization necessitate availability of adequate Ffree volume_ within the two dimensional LB film structure since the cross-sectional area of the cis isomer is larger than that of the trans isomer. Another factor limiting many potential applications of LB films is the poor chemical and thermal stability. Amongst various strategies [13–21] used to provide additional Ffree volume_, complexation of azobenzene deriva- tives with suitable polyions has received much attention partly due its versatility and convenience. This approach obviates the need for synthesis of specially designed complex amphiphilic molecules [22,23]. Polyion complex approach has been investigated extensively in last two decades for enhancing the stability of monolayers and influencing the orientation of the amphiphiles [24]. The polyion complex method is useful in stabilizing monolayers at the air–water interface by the electrostatic interaction between the monolayers and the water-soluble polyions [25–27]. This strategy has the advan- tage of providing azobenzene a favorable environment for the reversible cis – trans photoisomerization [25,26]. It has been reported by Nishiyama et al. [26] that polymers with ionic pendent groups are capable of forming complexes with carboxylic acid groups of acid at the air water interface. This
Transcript of Photoisomerization of amphiphilic azobenzene derivatives in Langmuir Blodgett films prepared as...

Photoisomerization of amphiphilic azobenzene derivatives in Langmuir
Blodgett films prepared as polyion complexes, using ionic polymers
Vishakha R. Shembekar a,1, A.Q. Contractor a, S.S. Major b, S.S. Talwar b,*
a Department of Chemistry, Indian Institute of Technology, Bombay, Mumbai-400 076, Indiab Department of Physics, Indian Institute of Technology, Bombay, Mumbai-400 076, India
Abstract
Polyion complexation in mixed Langmuir and Langmuir Blodgett (LB) films of photochromic amphiphilic azobenzene carboxylic acids, 11-[4-
(4-hexylphenyl)azo] phenoxyundecanoic acid, 11-(4-phenylazo)phenoxyundecanoic acid, and diamine grafted poly(methylmethaacrylate)
polymers has been studied. Monolayer behaviour of the pure components and mixed films was studied through pressure–area isotherms and
LB films were characterized by spectroscopic, X-ray diffraction and Atomic force microscopy techniques. Aggregation (H-type), often observed
in LB films of pure amphiphilic azo acids, was partly avoided in the mixed LB films as indicated by absorption spectral studies.
Photoisomerization of the polyion complexed LB films was also studied. The results altogether demonstrate that amine grafted polymer enter into
a polyion complexation with azo acid carboxylate group. LB films could be obtained by transfer of the composite monolayers and these LB films
exhibited different levels of aggregation of the azo acids. Reversible photoisomerization was observed in LB films with unaggregated azo acid.
Keywords: Polyion complexes; Azobenzenes; Langmuir-Blodgett films; Photoisomerization
1. Introduction
Photochrome containing Langmuir Blodgett (LB) films
have been explored as media for photochromic reactions aimed
at constructing molecule based information storage or switch-
ing devices [1–3]. In this context LB films of azobenzene
derivatives have attracted increasing attention in recent years
due to their diverse photofunctional applications [4–8]. These
derivatives undergo facile cis – trans photoisomerization in
solution, however isomerization is inhibited [9–12] in pure LB
films due to insufficient Ffree volume_ in the higher order
structure of LB films. Photoisomerization not only involves
electronic changes but also involves changes in the molecular
conformation and cross-sectional area due to difference in the
shape of the cis and the trans isomers. Changes in the cross-
sectional area and molecular conformation that are associated
with azobenzene isomerization necessitate availability of
adequate Ffree volume_ within the two dimensional LB film
structure since the cross-sectional area of the cis isomer is
larger than that of the trans isomer. Another factor limiting
many potential applications of LB films is the poor chemical
and thermal stability.
Amongst various strategies [13–21] used to provide
additional Ffree volume_, complexation of azobenzene deriva-
tives with suitable polyions has received much attention partly
due its versatility and convenience. This approach obviates the
need for synthesis of specially designed complex amphiphilic
molecules [22,23]. Polyion complex approach has been
investigated extensively in last two decades for enhancing the
stability of monolayers and influencing the orientation of the
amphiphiles [24]. The polyion complex method is useful in
stabilizing monolayers at the air–water interface by the
electrostatic interaction between the monolayers and the
water-soluble polyions [25–27]. This strategy has the advan-
tage of providing azobenzene a favorable environment for the
reversible cis – trans photoisomerization [25,26]. It has been
reported by Nishiyama et al. [26] that polymers with ionic
pendent groups are capable of forming complexes with
carboxylic acid groups of acid at the air water interface. This

298
phenomenon could be used for hooking the azo acids on
polymer backbone at a certain intervals thereby breaking the
aggregates of azo acid being formed in the monolayer [28]. LB
films of azobenzene containing polyion complexes have been
shown to exhibit reversible photoisomerization. The photo
stationary state cis isomer composition and the free volume
availability in such films depend on the nature of polyions and
the experimental conditions under which the film are prepared
[29,30].
In this paper we report investigations on monolayers and
transferred multilayers prepared as polyion complexes made
from amphiphilic azobenzene carboxylic acids, 11-[4-(4-
hexylphenyl)azo]phenoxyundecanoic acid(6A10) and 11-(4-
phenyl azo)phenoxyundecanoic acid(0A10), and diamine
random grafted poly(methylmetha acrylate) (PMMA) polymers
(GPn, 6–16% graft) by LB method and study of photoisome-
rization in LB films. The chemical structures of the azo acids
and the amine grafted polymers are shown in Fig. 1. The
protonated basic amino centers in the grafts are expected to
function as polyions for complexation with the ionized acid.
Monolayer behaviour has been examined by detailed pressure–
area isotherms (p–A) isotherm studies. p–A isotherm studies
of the monolayers essentially indicated absence of aggregation
in the monolayers. LB films were obtained by transfer of the
monolayers by vertical transfer methodology and characterized
by Fourier Transform Infrared spectroscpoy (FTIR), electronic
absorption spectroscopy (UV-Vis), X-ray diffraction (XRD)
and Atomic Force microscopy (AFM). The results demonstrate
that suitable pendent group grafts on polymers enter into
complexation with the carboxylate group on azo acid and can
serve as a basis for thin film media for reversible photoisome-
rization of the azo group.
2. Experimental details
2.1. Materials
11-[4-(4-hexylphenyl)azo]phenoxyundecanoic acid [31],
and 11-(4-phenylazo)phenoxyundecanoic acid [32], were
synthesized by diazotization of respective 4-substituted aniline
and coupling of the diazotized product with 11-bromoundeca-
nol followed by its oxidation using PDC ( 6A10, mp=106 -C,reported mp=106–108 -C, 0A10, mp=116–119 -C, reportedmp=116–118 -C) [31,32]. 4-substituted anilines, 11-bromoun-
Fig. 1. Structures of azo acids
decanol and dodecylbromide were bought from Aldrich.
The azo acids were repeatedly recrystallized to achieve a
purity of 99.9%. Purity of the azo acids was monitored using
high performance liquid chromatography (HPLC) (Shimadzu
LC-8A).
Grafted PMMA was prepared by refluxing 1, 2-diamino-
propane (10–50% of the PMMA) and PMMA in toluene for 8 h
in a Dean–Stark apparatus to remove methanol. Subsequently
grafted polymers were precipitated in hexane. The precipitated
polymers were purified and freed from unreacted diaminopro-
pane by repeated dissolution in toluene and precipitation in
hexane. The levels of grafting achieved were much lower than
the attempted graft level. Extent of grafting was determined by
amine value method. Grafting achieved and attempted in % for
different samples were GP1, 6.7(10); GP2, 8.7(20); GP3,
11(30); GP4, 13.1(40); GP5, 15.3(50), respectively. The
grafted PMMAwas further characterized by FTIR and Nuclear
Magnetic Resonance spectroscopy.
Measurements of surface pressure–area isotherms and the
automated deposition of the monolayers were carried out with
Langmuir trough (KSV 3000) equipped with an electronic
microbalance and a platinum Wilhelmy plate, kept in a clean
room, class 10,000. Deionized water with resistivity 18.2 MV
cm from Millipore system was employed for subphase
preparation. Pure azo acid monolayer behaviour and deposition
of LB films was studied with water subphase and subphase
containing 10�4 M CdCl2. Composite monolayers films of azo
acids and GPn polymers were made by spreading solutions of
the azo acid and polymer made by mixing parent solutions of
azo acid (0.56 mg ml�1) and GPn (0.62 mg ml�1) in HPLC
grade chloroform in 1 :1 M proportion of the presumed graft.
100 Al of the mixed solution of azo acid and GPn was spread
on the LB trough with water subphase at ambient temperature
and chloroform was allowed to evaporate. An equilibration
time of 40 min was allowed. The monolayers at the air–water
interface were compressed with the help of barriers (barrier
speed 3 mm/min). Formation of the condensed monolayer was
followed by p –A isotherms. Stability of the monolayers was
studied by noting the change in mean molecular area while
maintaining the monolayer at a specific pressure.
Monolayers were transferred onto quartz plates of dimen-
sions 1�1 in. (and on CaF2 plates of dimensions 1�0.5 in. for
FTIR) at a surface pressure of 15 mN/m with a dipping speed
of 3 mm/min. For all films either 11 or 22 monolayers were
and the grafted polymer.

299
transferred. Electronic absorption spectra of the films were
recorded using Shimadzu 260 UV-Vis spectrophotometer. A
clean identical quartz plate was used as reference. XRD studies
were carried out using a Philips X-ray Diffractometer (model
PW 1729) with Ka line from a copper target which produces
X-rays of wavelength 1.5406 A. The samples were scanned at
the rate of 2.4-/min in the 2h range of 4–20-. AFM studies
were done using Nanoscope III scanning probe microscope
with a microfabricated Si3N4 cantilever (force constant 0.5 N
m�1) stylus assembly. 5 nm to 2 Am images were obtained
under constant force conditions with a net tip force in the
region of 10–100 nN. The AFM measurements were
performed in the Fnon-contact_ mode to obtain molecular
resolution images.
3. Results and discussion
3.1. Monolayer studies
This section reports behaviour of composite monolayers of
azo acids and grafted PMMA. Monolayer behaviour of the pure
azo acid and pure grafted polymers was also examined for
comparison. Grafted PMMA monolayers were prepared by
spreading a chloroform solution of the polymer over water
subphase and composite monolayers of azo acids and polymers
GPn were prepared by spreading a chloroform solution of the
mixture of appropriate composition on the water subphase.
Surface pressure–area isotherms of diamine grafted PMMA
are shown in Fig. 2. Area per molecule axis is based on the
number of graft weighted PMMA monomer molecules.
Increased diamine grafting causes a shift of the p –A curve
to larger mean molecular area (Mma) values while retaining the
shape of the PMMA p–A curve. The curves exhibit two phase
transitions similar to PMMA. The first phase transition
probably involves folding of polymer chains on the water
surface and the second transition, which occurs at pressures
above ¨30 mN/m, probably involves an irreversible collapse.
The first phase transition in the grafts occurs around 15–
17 mN/m. The limiting mean molecular area (Lmma) obtained
from p–A isotherms of GPn by extrapolation of the steep part
of the curves before and after the first transition to zero surface
Fig. 2. p –A isotherms of the grafted polymers, GP1–GP5.
pressure, are plotted as function of graft composition in Fig. 3
(a) and (c), respectively. Collapse pressure for all the GPn
monolayers was ¨45 mN/m. In PMMA the ester groups
function as hydrophilic groups and it has been shown that in
the compressed monolayer the polymer backbone is stretched
out horizontally along the barrier at the interface [33]. The
gradual increase in the Lmma with the increase in the
percentage of amine graft as shown in Fig. 3(a) (except for
GP2 where the increase in Lmma is much larger) suggests that
grafting anchors grafted PMMA on water surface and polymer
chains in grafted polymers are probably more extended than in
PMMA. This may also result from increase in the area of the
grafted repeat unit and enhanced polarity due to presence of
free amino groups in the polymer. It is noted that the sharpness
of the first transition is less marked in the p–A curves as the
grafting level increases, however, the transition occurs at
almost the same surface pressure.
6A10–GPn (AGPn) Composite monolayers and LB films
were studied in greater detail as the amphiphilic azo acid 6A10
is well studied by the LB methodology. p –A isotherm of
composite 6A10–GP1 (AGP1) monolayer is shown in Fig.
4(a). The area per molecule axis is based on the total number of
GP1 and 6A10 molecules. Fig. 4(a) also shows the p–Aisotherms of pure GP1 and 6A10 under similar conditions.
Compression–decompression cycles up to pressure of 15 mN/
m did not show any hysteresis for the composite monolayer.
Also the monolayer was found to be quite stable at a pressure
of 15 mN/m. p –A isotherm of the composite monolayer shows
an expanded nature. The composite isotherm AGP1 is more
characteristic of the grafted polymer than that of the 6A10
isotherm. It showed two phase transitions similar to those
observed in pure grafted polymer monolayers. However, the
p–A isotherm curve shifts to larger mean molecular area
values compared to those for pure GP1 monolayer whereas the
collapse pressure was almost comparable to 6A10. Similar
monolayer behaviour was observed for composite films
containing polymers grafted to different extents Fig. 4(b)–
(e). Pre and post transition Lmma values obtained from the
composite curves AGPn are plotted as Fig. 3 (b) and (d),
respectively. Experimentally determined Lmma values from the
pre-transition region of the AGPn curves are smaller than that
expected for a simple mixture of components calculated using
the expression A (calculated)=Aava+Apvp where A is the area
of composite film and Aa, Ap areas and va, vp being the
respective mole fractions of 6A10 and the graft polymers GPn,
respectively (thus AGP1 Lmma is ¨18.5 A2 compared to
19.5 A2). This is suggestive of attractive interaction between
the azo acid and the polyion indicating polyion complex
formation and avoidance of aggregation of 6A10 molecules in
the closely packed composite monolayer at the air–water
interface [34]. Polyion complex formation is also supported by
the FTIR of the transferred films (vide infra). Comparison of
other isotherms of GPn and composite 6A10–GPn (1 :1),
respectively, also indicate similar behaviour. Lmma obtained
from the AGPn isotherms for the post transition region are
smaller for some of the cases than that obtained from the graft
polymer GPn isotherms as indicated in Fig. 3 (d) and (c),

Fig. 3. Variation of least mean molecular area for GPn and AGPn with graft
percent. Before transition (a) GPn and (b) AGPn; after transition (c) GPn and
(d) AGPn.
300
respectively. It may be noted that the phase obtained after the
first transition is less compressible in contrast with
corresponding GPn phase. Compressibility is least for AGP5
Fig. 4. (a) p –A isotherms of GP1, AGP1 and 6A10 on subphase water; (b)– (e)
p –A isotherms of AGPn and GPn (n =2–5).
which contains only 15% grafted polymer and thus may have
large amount of uncomplexed azo acid in the monolayer. The
similarity of the AGPn curves with the GPn curves may be a
reflection of the fact that the polymer is horizontally spread out
and makes large contribution to the mean molecular area in
contrast with azo acid which is oriented in the vertical direction.
Monolayer behaviour of some other amphiphilic azo acids
[35] and polymer GP2 composite systems were studied. GP2
was selected as a representative graft polymer for mixing with
the amphiphilic acids due to its potential for providing
relatively higher azo acid loading capacity, (GP3–GP5 have
higher loading capacities, but have shown greater extent of
aggregation of the azo acid in LB films ). Behaviour of the
composite monolayers was similar to that observed for AGP2
monolayer despite differences in the structure of the azo acids.
The p–A isotherm of composite 0A10–GP2 (AzGP2) mono-
layer is shown in Fig. 5 along with those of pure components.
Both, 0A10 and the composite p –A isotherms showed
considerably expanded nature and the p –A isotherm of
composite monolayer shows characteristics similar to that of
the graft polymer GP2 rather than that of the azo acid. The
composite monolayer was more stable in contrast with the
monolayer of the 0A10 with collapse pressure of 30 mNm�1.
No hysteresis was seen when the monolayer was compressed
and decompressed repeatedly to a surface pressure lower than
the collapse pressure. AzGP2 p –A isotherm curve shifted to
larger mean molecular area compared to pure GP2 monolayer.
Lmma observed for the composite film and FTIR of the of the
LB film of AZGP2 support the formation of polyion complex.
3.2. LB film studies
The composite monolayers of AGPn were transferred on to
the substrate at ¨15 mN m�1. This was done due to larger
mean molecular area prior to the first phase transition. All the
AGPn composite monolayers showed Y type transfer tending
towards Z type. The transfer ratios observed up to 22 layers are
given in Table 1. On deposition of still larger number of layers,
the transfer ratio decreased considerably with increasing
number of layers. The deposition ratios observed for AGPn
composites suggest the formation of poorly ordered and non-
uniform LB films.
Fig. 5. p –A isotherms of GP2, AzGP2 and 0A10 on subphase water.

Table 1
Transfer ratio (TR) obtained for lifting and dipping runs during transfer of
AGPn diamine grafted monolayer at air–water interface
Sr. no. 6A10–GPn TR (lifting) TR (dipping)
1 6A10–GP1 0.9T0.1 0.5T0.1
2 6A10–GP2 0.8T0.1 0.4T0.13 6A10–GP3 0.8T0.1 0.5T0.1
4 6A10–GP4 0.7T0.1 0.3T0.1
5 6A10–GP5 1.0T0.1 0.6T0.1
301
FTIR spectrum typical of AGPn films is shown in Fig. 6.
Absorption bands at 2924, 2855 cm�1 (CH2 mas and msstretching) and at 1586 cm�1 (C(O)O–mas stretching) establishthe presence of the azo acid component in the film and bands at
1728 cm�1 (ester carbonyl peak), ¨1660 cm�1 (amide
carbonyl) point to the presence of the grafted polymer in the
film. Polyion complex formation of 6A10 and the polymer in
the film is supported by the absence of 1710 cm�1 band for
carbonyl of the carboxylic acid and appearance of band at
1586 cm�1 for carboxylate anion and weak bands for
protonated amine at ¨3000–2800 cm�1 region. The carbonyl
stretching band at ¨1710 cm �1 present in the 6A10 azo acid
LB films is absent in the AGP1–AGP5 films despite the
expected presence of some azo acid in the uncomplexed form
in the composite films. The masking of this band by the large
ester carbonyl absorption is likely in films with excess acid.
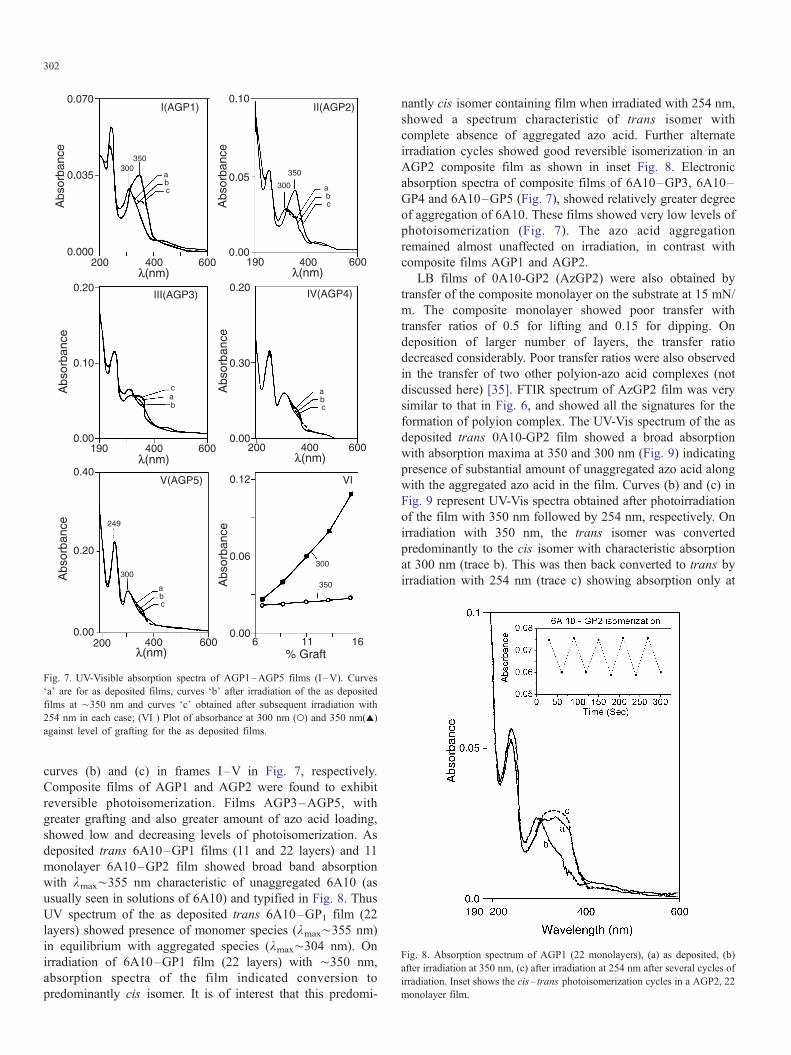
UV-Vis spectra of the as deposited trans 6A10–GP1-5
(AGP1–AGP5) films (22 layers) are shown in Fig. 7 (I–V).
Curves (a) in each frame represent the spectrum of the as
deposited film while the curves (b) and (c) are the spectra
obtained after photoirradiation of the films at 350 and 254 nm
sequentially for each of the films. In solution spectrum of 6A10
absorption bands are seen with maxima at ¨450 nm (assigned
as n–k* transition), ¨350, ¨238 nm (assigned to k–k*transitions) for azobenzene group and benzene ring, respec-
tively. A typical as deposited LB film of trans 6A10 exhibits
Fig. 6. FT-IR spectrum of AGP1 LB film on CaF2 (22
absorption bands with maxima at ¨300 and ¨250 nm and a
very weak absorption in the ¨400 nm region. These changes in
the LB film spectrum have been attributed to H-aggregation
with parallel stacking of the azobenzene chromophores of the
azo acid molecules in the LB film [36,37]. It is noteworthy that
as deposited AGP1, AGP2 LB films exhibit significant
absorbance in the 350 nm region besides absorption at
300 nm region suggesting presence of unaggregated azo acid.
Increased loading of the grafted polymer with the azo acid
AGP3–AGP5, Fig. 7 (III–V) curves (a), respectively, suggest
that only a small fraction of unaggregated acid is present in
AGP3–AGP5. The relative absorption intensities at 300 and
350 nm may be seen as an indication of the fraction of
molecules in the aggregated and unaggregated state, respec-
tively [29,38–40]. Fig. 7(VI) is a plot of absorbance of azo
acid in polyion composite films at 300 and 350 nm,
respectively, with the extent of graft. Examination of the
relative intensity at 300/350 nm indicates a gradual increase in
the fraction of aggregated 6A10 molecules in the films. Also,
there is finite and small increase in the fraction of complexed
azo acid molecules with increase in graft level although it is not
proportional to increase in the graft level. The increase in the
fraction of aggregated azo molecules from AGP1 to AGP5 is to
be expected in view of the excess of azo acid present above the
graft levels in the composite films AGP1 to AGP5, respec-
tively. Further, the number of monolayers deposited was also
found to affect the extent of the azo acid aggregation observed
in the film. An eleven layer LB film was found to show greater
fraction of the unaggregated acid in comparison with a 22 layer
film. This was noticed for LB films of AGP1, AGP2.
3.3. Photoisomerization in LB films
Photoisomerization of the composite LB films was exam-
ined by irradiation at 350 nm and subsequently at 254 nm and
the absorption spectrum after these irradiations are given by
layers) showing presence of both 6A10 and GP1.
Abs
orba
nce
Abs
orba
nce
Abs
orba
nce
Abs
orba
nce
Abs
orba
nce
Abs
orba
nce
λλ(nm) λ(nm)
λ(nm) λ(nm)
λ(nm) % Graft
0.070II(AGP2)I(AGP1)
III(AGP3) IV(AGP4)
V(AGP5) VI
0.035
0.000
0.10
0.05
0.00
0.20
0.10
0.00
0.40
0.20
0.00
0.12
0.06
0.00
0.20
0.30
0.00
200 400 600 190 400 600
190 400 600 200 400 600
200 400 600 6 11 16
350
350300
300abc a
bc
abc
abc
ab
c
300
249
300
350
Fig. 7. UV-Visible absorption spectra of AGP1–AGP5 films (I–V). Curves
Fa_ are for as deposited films, curves Fb_ after irradiation of the as deposited
films at ¨350 nm and curves Fc_ obtained after subsequent irradiation with
254 nm in each case; (VI ) Plot of absorbance at 300 nm (>) and 350 nm(r)
against level of grafting for the as deposited films.
Fig. 8. Absorption spectrum of AGP1 (22 monolayers), (a) as deposited, (b
after irradiation at 350 nm, (c) after irradiation at 254 nm after several cycles o
irradiation. Inset shows the cis– trans photoisomerization cycles in a AGP2, 22
monolayer film.
302
curves (b) and (c) in frames I–V in Fig. 7, respectively.
Composite films of AGP1 and AGP2 were found to exhibit
reversible photoisomerization. Films AGP3–AGP5, with
greater grafting and also greater amount of azo acid loading,
showed low and decreasing levels of photoisomerization. As
deposited trans 6A10–GP1 films (11 and 22 layers) and 11
monolayer 6A10–GP2 film showed broad band absorption
with kmax¨355 nm characteristic of unaggregated 6A10 (as
usually seen in solutions of 6A10) and typified in Fig. 8. Thus
UV spectrum of the as deposited trans 6A10–GP1 film (22
layers) showed presence of monomer species (kmax¨355 nm)
in equilibrium with aggregated species (kmax¨304 nm). On
irradiation of 6A10–GP1 film (22 layers) with ¨350 nm,
absorption spectra of the film indicated conversion to
predominantly cis isomer. It is of interest that this predomi-
nantly cis isomer containing film when irradiated with 254 nm,
showed a spectrum characteristic of trans isomer with
complete absence of aggregated azo acid. Further alternate
irradiation cycles showed good reversible isomerization in an
AGP2 composite film as shown in inset Fig. 8. Electronic
absorption spectra of composite films of 6A10–GP3, 6A10–
GP4 and 6A10–GP5 (Fig. 7), showed relatively greater degree
of aggregation of 6A10. These films showed very low levels of
photoisomerization (Fig. 7). The azo acid aggregation
remained almost unaffected on irradiation, in contrast with
composite films AGP1 and AGP2.
LB films of 0A10-GP2 (AzGP2) were also obtained by
transfer of the composite monolayer on the substrate at 15 mN/
m. The composite monolayer showed poor transfer with
transfer ratios of 0.5 for lifting and 0.15 for dipping. On
deposition of larger number of layers, the transfer ratio
decreased considerably. Poor transfer ratios were also observed
in the transfer of two other polyion-azo acid complexes (not
discussed here) [35]. FTIR spectrum of AzGP2 film was very
similar to that in Fig. 6, and showed all the signatures for the
formation of polyion complex. The UV-Vis spectrum of the as
deposited trans 0A10-GP2 film showed a broad absorption
with absorption maxima at 350 and 300 nm (Fig. 9) indicating
presence of substantial amount of unaggregated azo acid along
with the aggregated azo acid in the film. Curves (b) and (c) in
Fig. 9 represent UV-Vis spectra obtained after photoirradiation
of the film with 350 nm followed by 254 nm, respectively. On
irradiation with 350 nm, the trans isomer was converted
predominantly to the cis isomer with characteristic absorption
at 300 nm (trace b). This was then back converted to trans by
irradiation with 254 nm (trace c) showing absorption only at
)
f

0 2.5 5.0 nm
0
2.5
5.0 nm
0 nm
0.5 nm
1.0 nm
a
0 2.5 5.0 nm
0
2.5
5.0 nm
0 nm
0.9 nm
1.8 nm
b
Fig. 10. AFM of 17 layered LB film of (a) 6A10 and (b) AGP1 composite
deposited on quartz.
303
350 nm suggesting absence of aggregates of the azo acid.
Further irradiation of the film showed good reversible
isomerization. Storage of the film did not lead to reaggregation
of molecules.
In summary we note that low graft levels and low azo acid
loaded composite LB films exhibit presence of unaggregated
and aggregated azo molecules in proportions depending on the
graft level, the number of deposited layers and azo acid
loading. Composite films with low loading of azo acid exhibit
higher fraction of unaggregated azo molecules due to polyion
complexation and such films show facile reversible isomeriza-
tion. Further partial aggregation present in the as deposited
composite films was irreversibly destroyed in the isomerization
process. Films with higher loading of azo acid do not exhibit
facile reversible photoisomerization and do not indicate
significant effect on the aggregate proportion in the isomeri-
zation process. It may be speculated that even with higher graft
levels of polymers, higher loading of azo acid may not lead to
larger proportion of unaggregated azo acid although it requires
further experimental confirmation.
To understand the structural changes associated with
polyion complexation in the LB films, XRD and AFM of the
composite films were studied. XRD of the composite AGPn
and AzGP2 as deposited films did not show any diffraction
pattern pointing to absence of any layered order.
In-plane molecular packing of 6A10 and AGP1 composite
film was analyzed using atomic force microscopy. Scanning a
2�2 Am2 area showed the presence of 100 nm diameter
domains with internal structure (not shown). AFM results of
the fine structure within single domains for 6A10 and AGP1
composite film are shown in Fig. 10 (a) and (b). The AFM of
6A10 film shows an ordered molecular arrangement, Fig. 10
Fig. 9. UV-Visible spectrum of 0A10-GP2 (a) as deposited film with broad
absorption at 350 nm indicating presence of unaggregated species, (b) after
irradiation at 254 nm.
(a), the Fourier transform of which indicates a distorted
hexagonal structure with a =5.1T0.2 A and b =3.6T0.2 A.
The sharp and symmetric reflections seen in the Fourier
transform is an indicator of long range molecular order in the
film. The AFM image from the composite film, Fig. 10 (b)
shows a heavily deformed structure with lack of long range
molecular order. The presence of short range order in the
composite film clearly indicates that inclusion of randomly
grafted polymer into the LB film does not disrupt the molecular
order completely.
4. Conclusion
The above results demonstrate that amine containing
pendent group random PMMA grafts enter into complexation
with the carboxylate group of azo acids, to form stable and
transferable monolayers. LB films of the polyion complexes
obtained show partial deaggregation of amphiphilic azo acids;
the extent of aggregation depends on graft levels and the film
preparation conditions. Greater aggregation was noticed in LB
films with larger number of deposited monolayers. Although
no layered order was observed, AFM measurements indicated
that some degree of short range in plane order is observed in
the composite films. Unlike LB films of pure azo acids, LB
films of polyion complexes exhibited reversible photoisome-
rization. In some cases the photoisomerization process caused
enhanced irreversible disaggregation of the azo acid molecules.
Acknowledgements
Authors wish to thank Dr. M. Subramaniam for providing
1,2-diamino propane and valuable suggestions about grafting

304
of PMMA, Dr. P. Bhimalapuram for valuable discussions on
isotherm analysis, Prof. R.Srinivas and Professor S. Vitta for
discussions regarding AFM. SST thanks Sukhvinder Singh for
assistance in processing of figures.
References
[1] D.G. Whitten, Angew. Chem., Int. Ed. Engl. 18 (1979) 440.
[2] E. Ando, J. Miyazaki, K. Morimoto, K. Fukuda, Thin Solid Films 133
(1985) 21.
[3] T. Kawai, Thin Solid Films 301 (1997) 225.
[4] A. Yabe, Y. Kawabata, H. Niino, M. Matsumoto, A. Ouchi, H. Takahashi,
S. Tamura, W. Tagaki, H. Nakahara, K. Fukuda, Thin Solid Films 160
(1988) 33.
[5] H. Tachibana, T. Nakamura, M. Matsumoto, H. Komizu, E. Manda, H.
Niino, A. Yabe, Y. Kawabata, J. Am. Chem. Soc. 111 (1989) 3080.
[6] T. Seki, T. Tamaki, Y. Suzuki, Y. Kawanishi, K. Ichimura, Macromole-
cules 22 (1989) 3505.
[7] Z.F. Liu, K. Hashimoto, A. Fujishima, Nature 347 (1990) 658.
[8] J. Anzai, N. Sugaya, T. Osa, J. Chem. Soc., Perkin Trans. 2 (1994) 1897.
[9] D.G. Whitten, J. Am.Chem. Soc. 96 (1974) 594.
[10] L. Collins-Gold, D. Mobius, D.G. Whitten, Langmuir 2 (1986) 191.
[11] H. Nakahara, K. Fukuda, M. Shimomura, T. Kunitake, Nippon Kagaku
Kaishi (1998) 1001.
[12] W.F. Mooney, P.E. Brown, J.C. Russell, S.B. Costa, L.G. Pederson, D.G.
Whitten, J. Am. Chem. Soc. 106 (1984) 5659.
[13] O. Befort, D. Mobius, Thin Solid Films 243 (1994) 553.
[14] R.C. Ahuja, J. Maack, J. Phys. Chem. 99 (1995) 9221.
[15] K. Shirota, K. Kajikawa, H. Takezoe, A. Fukuda, Jpn. J. Appl. Phys. 29
(1990) 750.
[16] X. Xu, M. Era, T. Tsutsui, S. Saito, Thin Solid Films 178 (1989)
541.
[17] X. Xu, M. Era, T. Tsutsui, S. Saito, Thin Solid Films 178 (1989)
L138.
[18] M. Tanaka, Y. Ishizuka, M. Matsumoto, T. Nakamura, A. Yabe, H.
Nakanishi, Y. Kawabata, H. Takahashi, S. Tamura, W. Tagaki, H. Nakara,
K. Fukuda, Chem. Lett. (1987) 1307.
[19] Z.F. Liu, B.H. Loo, R. Baba, A. Fujishima, Chem. Lett. (1990) 1023.
[20] M. Matsumoto, D. Miyazaki, M. Tanaka, R. Azumi, E. Manda, Y. Kondo,
N. Yoshino, H. Tachibana, J. Am. Chem. Soc. 120 (1998) 1479.
[21] T. Kunitake, A. Tsuge, N. Nakashima, Chem. Lett. (1984) 1783.
[22] K. Nishiyama, M. Fujihira, Chem. Lett. (1988) 1257.
[23] O.N. Oliveira, Polymer 43 (2002) 3753.
[24] E.D. Goddard, Colloids Surf. 19 (1986) 301.
[25] R.A. Hall, M. Hara, W. Knoll, Langmuir 12 (1996) 2551.
[26] K. NIshiyama, M. Kurihara, M. Fujihira, Thin Solid Films 179 (1989)
477.
[27] M. Shimomura, T. Kunitake, Thin Solid Films 132 (1985) 243.
[28] H. Tachibana, R. Azumi, M. Tanaka, M. Matsumoto, S. Sako, H. Sakai,
M. Abe, Y. Kondo, N. Yoshino, Thin Solid Films 284–285 (1996) 73.
[29] N. Kimizuka, T. Kunitake, Chem. Lett. (1988) 827.
[30] R.A. Hall, M. Hara, W. Knoll, Langmuir 12 (1996) 2551.
[31] T. Seki, M. Sakuragi, Y. Kawanishi, Y. Suzuki, T. Tamaki, R. Fukuda,
K. Ichimura, Langmuir 9 (1993) 211.
[32] M. Era, T. Tsutsui, S. Saito, Langmuir 5 (1989) 1410.
[33] S.T. Kowel, G.G. Zhou, M.P. Srinivasan, P. Stoeve, B.P. Huggins, Thin
Solid Films 134 (1986) 209.
[34] M. Shimomura, R. Ando, T. Kunitake, Ber. Bunsenges. Phys. Chem. 87
(1983) 1134.
[35] Monolayer behaviour and LB film deposition of GP2 with 11-[4-
(-dodecyl phenyl)azo]phenoxyundecanoic acid (12A10), 4-[4-dodecy-
loxyphenyl)azo]benzoic acid (12A0) composites was also studied.
Vishakha Shembekar, Ph.D Thesis, IIT Bombay, 2000.
[36] W.F. Mooney, D.G. Whitten, J. Am. Chem. Soc. 108 (1986) 5712.
[37] J. Heesemann, J. Am. Chem. Soc. 102 (1980) 2167.
[38] D.L. Beveridge, H.H. Jaffe, J. Am. Chem. Soc. 88 (1966) 1948.
[39] X. Xu, M. Era, T. Tsutsui, S. Saito, Chem. Lett. (1988) 773.
[40] S. Dante, R. Advincula, C.W. Frank, P. Stroeve, Langmuir 15 (1999) 193.




















