
New mesogenic maleimide-styrene copolymers via N-oxyl mediated free radical copolymerization
14
Designed Monomers and Polymers , Vol. 6, No. 2, pp. 145– 158 (2003) Ó VSP 2003. Also available online - www.vsppub.com New mesogenic maleimide–styrene copolymers via N-oxyl mediated free radical copolymerization L. CIANGA 1;¤ , Y. YAGCI 1;† , E. G. FERNANDES 2 and G. GALLI 2 1 Istanbul Technical University, Department of Chemistry, Maslak, Istanbul 80626, Turkey 2 University of Pisa, Department of Chemistry and Industrial Chemistry, via Risorgimento 35, 56126 Pisa, Italy Abstract —New mesogenic N-substituted maleimide monomer was obtained starting 4-maleimido benzoic acid, hydroquinone and hexadecyloxy benzoic acid by successive Schotten–Bauman con- densations. The copolymerization of this monomer in 1 :1 ratio with styrene was performed both by stable free radical polymerization (SFRP) and conventional free radical polymerization (CFRP). High molecular weight polymeric material in high conversion and with low polydispersity was ob- tained under SFRP conditions. By using 1 H- and 13 C-NMR techniques, structural characteristics of the polymers were evidenced that could sustain the alternating character of the copolymers and the participationof the electrondonor–acceptorcomplex in the propagationstep. The copolymersformed a liquid crystal mesophase in the melt. This was ascribed to the strong tendency of the rod-like side groups on the stiff imide ring to order even in 1 : 1 dilute systems such as the alternating copolymers. Keywords: Liquid-crystalline polymers; stable free radical polymerization; alternating copolymers; mesogenic maleimide; styrene. 1. INTRODUCTION During the last two decades, side-group liquid-crystalline polymers (SG-LCPs) have received a great deal of attention, from both a scienti c point of view (for the study of the structure– properties relationships) and a commercial point of view (for the evaluation of possible applications). A considerable part of this interest has been directed towards the synthesis of new polymers (for a review, see Ref. [1]). One of the most fundamental questions still open regarding the structure – property relationships in SG-LCPs is the effect of molar mass and molar mass polydispersity on their mesophase behavior and properties. The development of polymerization ¤ Permanent address: ‘Petru Poni’ Institute of Macromolecular Chemistry, Iasi, Romania. † To whom correspondence should be addressed. E-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of New mesogenic maleimide-styrene copolymers via N-oxyl mediated free radical copolymerization

Designed Monomers and Polymers Vol 6 No 2 pp 145ndash158 (2003)Oacute VSP 2003Also available online - wwwvsppubcom
New mesogenic maleimidendashstyrene copolymers via N-oxylmediated free radical copolymerization
L CIANGA 1curren Y YAGCI 1dagger E G FERNANDES2 and G GALLI 2
1 Istanbul Technical University Department of Chemistry Maslak Istanbul 80626 Turkey2 University of Pisa Department of Chemistry and Industrial Chemistry via Risorgimento 35
56126 Pisa Italy
AbstractmdashNew mesogenic N-substituted maleimide monomer was obtained starting 4-maleimidobenzoic acid hydroquinone and hexadecyloxy benzoic acid by successive SchottenndashBauman con-densations The copolymerization of this monomer in 1 1 ratio with styrene was performed bothby stable free radical polymerization (SFRP) and conventional free radical polymerization (CFRP)High molecular weight polymeric material in high conversion and with low polydispersity was ob-tained under SFRP conditions By using 1H- and 13C-NMR techniques structural characteristics ofthe polymers were evidenced that could sustain the alternating character of the copolymers and theparticipationof the electrondonorndashacceptor complex in the propagationstep The copolymers formeda liquid crystal mesophase in the melt This was ascribed to the strong tendency of the rod-like sidegroups on the stiff imide ring to order even in 1 1 dilute systems such as the alternating copolymers
Keywords Liquid-crystalline polymers stable free radical polymerization alternating copolymersmesogenic maleimide styrene
1 INTRODUCTION
During the last two decades side-group liquid-crystalline polymers (SG-LCPs)have received a great deal of attention from both a scienti c point of view (forthe study of the structurendash properties relationships) and a commercial point of view(for the evaluation of possible applications) A considerable part of this interest hasbeen directed towards the synthesis of new polymers (for a review see Ref [1])
One of the most fundamental questions still open regarding the structurendash propertyrelationships in SG-LCPs is the effect of molar mass and molar mass polydispersityon their mesophase behavior and properties The development of polymerization
currenPermanent address lsquoPetru Ponirsquo Institute of Macromolecular Chemistry Iasi RomaniadaggerTo whom correspondence should be addressed E-mail yusufituedutr
146 L Cianga et al
methods such as living polymerization controlled polymerization and group-transfer polymerizations opens new avenues to the design and synthesis of liquid-crystalline polymer systems with well de ned structure controlled molar mass andnarrow molar mass distribution [1 2] Different types of liquid-crystalline polymerssynthesized by atom transfer radical polymerization (ATRP) [3 4] or stable freeradical polymerization (SFRP) [5 6] techniques were reported
Copolymerization of various combinations of mesogenicnonmesogenic mono-mers has proved to be a powerful tool to modulate the properties of different liquid-crystalline copolymer structures (for a review see Ref [1]) Surprisingly verylittle attention has been devoted to the synthesis of alternating copolymers (forexample see Refs [7 8] and references therein) In these systems one may controlthe monomer sequence distribution in semi-rigid copolymer chains and thus tailorthe properties of the polymer on a well-de ned structural basis [9]
N-Substituted polymaleimides belong to side-group polymers that have a ten-dency to order partially due to the rigid structure of the planar succinimide groupTo our knowledge there are few reports concerning preparation and characterizationof SG-LC polymaleimides Oishi et al reported about poly- and copolymaleimidesin which the cholesteryl mesogenic group was attached to the polymer backbonethrough a paraf nic [10 11] or an aromatic [12] spacer unit However the ther-motropic behavior of these polymers was not studied
Smectic ordering in poly(4-maleimidocholesteryl benzoate) poly(6-maleimido-cholesteryl hexanoate) and alternating copolymers with styrene reg and macr-methyl-styrene and p-ethoxystyrene was studied by Vukovic et al [13] Smectic ornematic phases were observed in a homologous series of SG-LCPs containing the4-cyanobiphenyl mesogenic group attached to the polymaleimide backbone throughparaf nic spacers [14] Smectic phases were also formed in polymaleimidesbased on phenyl benzoate mesogenic groups [15] The p-n-alkoxybenzoic acidsstarting with n-propyloxybenzoic acid exhibit liquid-crystalline behavior andtheir mesogenic potential increases with the length of the alkoxy group Phenylbenzoates as mesogenic groups may impart some special characteristics to theirSG-LCPs The primary signi cant characteristic of the phenyl benzoate moietiesmay be their strong interactions governed by electronic and geometrical factorsallowing the mesogenic group to achieve more ordered packing with respect to othermesogens [15]
In this paper we report the preparation of a new mesogenic N-substitutedmaleimide monomer (COBAMI) containing a ((hexadecyloxy)benzoyloxy)phenylmesogenic unit directly attached to the maleimide ring and its copolymerizationwith styrene (St) under stable free radical polymerization (SFRP) conditions andconventional free radical polymerization (CFRP) The aim of this work was thepreparation of alternating copolymers (Scheme 1) with controlled molecular struc-ture controlled molecular weight and narrow polydispersity that would exhibitliquid-crystalline behavior
Mesogenic maleimidendashstyrene copolymers 147
Scheme 1 Structure of the copolymers prepared and studied
2 EXPERIMENTAL
21 Materials
Maleic anhydride (Aldrich) was sublimated before use All solvents were puri edand dried by standard techniques before to use Acetic anhydride (Merck) p-aminobenzoic acid hydroquinone (Fluka) 4-hexadecyloxybenzoic acid (Across)(COBA) 2266-tetramethyl-piperidinyl-1-oxy (TEMPO) (Aldrich) and benzoylperoxide (BPO) (Fluka) were used without further puri cation Thionyl chlorideand styrene (Fluka) were distilled just before use
22 Preparation of intermediates and maleimide monomer
221 4-Maleimido benzoic acid (MBA) MBA was synthesized by the conden-sation reaction of maleic anhydride with p-aminobenzoic acid followed by cyclode-hydration using acetic anhydride and sodium acetate according to a procedure pub-lished elsewhere [16]
Yield 85 mp 234plusmnC (244plusmnC [16]) IR (cmiexcl1) 3460ndash 3200 (COOH) 3105ndash3100( CH) 1720 (O CNC O) 1610 (C C) 1217 (C O) 820 (phenyl) 720 (cisCH CH)
222 4-Maleimidobenzoic acid chloride (MBAC) MBAC was synthesizedaccording to a procedure described elsewhere [15]
Yield 73 mp 157ndash158plusmnC (168ndash169plusmnC [16]) IR (cmiexcl1) 1770 (COCl) 1715(O CNC O) 1600 (C C) 700 (cis CH CH)
223 N-[4-(40-hydroxyphenyloxycarbonyl) phenyl] maleimide (HPPMI)HPPMI was prepared as described previously [17]
Yield 79 mp 208plusmnC Anal calc for C17H11NO5 () C 6602 H 358 N453 Found C 6610 H 367 N 471
IR (cmiexcl1) 3450 (OH) 3105ndash3100 ( CH) 1765 1720 (imide I) 1705 (C Oester) 1600 (C C) 1380 (imide II) 1240 (C O C ester) 1190 (C (OH)phenolic) 1150 (imide III) 1070 (ester) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm in DMSO-d6) 953 (s 1H OH) 830ndash803 (q 2H Ar-H orthoto carbonyl group) 764ndash759 (q 2H Ar-H ortho to imide group) 725 (s 2H
148 L Cianga et al
HC CH) 71ndash706 (d 2H Ar-H ortho to O atom) 683ndash679 (d 2H Ar-H orthoto hydroxyl)
224 4-Hexadecyloxybenzoic acid chloride (COBAC) COBAC was preparedby dissolving 2175 g (006 mol) of acid in 150 ml of dry benzene To this solution2855 g (024 mol) of thionyl chloride was added dropwise and the resulting mixturewas re uxed for 24 h The solution was then cooled down to room temperatureand a solid precipitated by keeping the ask in the refrigerator overnight Theacid chloride was obtained as a white solid by vacuum ltration under nitrogenand subsequent drying in vacuum It was used in the next reaction without furtherpuri cation Yield 998
Anal calc for C23H37O2Cl () C 7250 H 979 Cl 931 Found C 7272 H980 Cl 943
IR (cmiexcl1) 2900 2860 (CH aliphatic) 1760 (COCl) 1600 (C C aromatic) 1460(CH3) 1280 (C O C) 1170 (CH2 chain) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm from TMS in CDCl3) 808ndash804 (d 2H aromatic) 696ndash692(d 2H aromatic) 406ndash401 (t 2H Ar O CH2 ) 183ndash178 (t 2H Ar OCH2 CH2 ) 17ndash125 (m 26H (CH213 ) 087ndash085 (t 3H CH3
225 40-[400-(Hexadecyloxy)benzoyl]oxyphenyl 4-maleimidobenzoate(COBAMI) (Scheme 2) A solution of 190 g (0005 mol) of COBAC in 20 mlof dry dichloromethane was added dropwise to a solution of 155 g (0005 mol) ofHPPMI in 200 ml dichloromethane containing 07 ml (0005 mol) of triethylamineThe reaction mixture was kept to re ux under nitrogen and with stirring for 96 hThe precipitate formed was ltered off and the solution was washed with 1 HCl(4 times) and then water (4 times) The organic layer was separated and dried overMgSO4 and after evaporation of dichloromethane the reaction product was washedthoroughly with hot hexane to remove unreacted traces of COBAC The pure prod-uct COBAMI (Scheme 3) was obtained by recrystallization from benzene
Yield 66 mp 156plusmnC Anal calc for C40H47NO7 () C 7348 H 725 N214 Found C 7357 H 732 N 215
IR (cmiexcl1) 3105ndash3050 (cis CH maleimide) 2920 2850 (aliphatic) 1770(imide I) 1708 (imide I C O ester) 1610 (C C aromatic and maleimide) 1420(CH3) 1400 (imide II) 1260 (C O C ether) 840 (p-disubstituted phenyl) 830(cis C H maleimide) 695 (imide IV carbonyl ring deformation)
1H-NMR (plusmn ppm from TMS in CDCl3) 832ndash829 (d 2H of C20 and C21)816ndash812 (d 2H of C7 and C9) 761ndash758 (d 2H of C6 and C10) 73ndash726(m 4H of C13 C14 C15 C17) 699ndash696 (d 2H of C22 and C24) 69 (s 2H ofC1 and C2)
13C-NMR (plusmn ppm in CDCl3) 16882 (C3 C4) 16477 (C11) 164208 (C18)163617 (C23) 148369 (C12 C16) 13612 (C5) 13446 (C1 C2) 13233(C7 C9) 13108 (C20 C21) 128318 (C8) 12525 (C6 C10) 12252 (C13 C14C15 C17) 12136 (C19) 114368 (C22 C24) 6842 (C25) 319 (C38)
Mesogenic maleimidendashstyrene copolymers 149
Scheme 2 Reaction pathway for the synthesis of monomer COBAMI
Scheme 3 Structure of COBAMI
2965ndash2940 (C28ndashC34) 2933 (C35 C36 C37) 29 (C26) 2596 (C27) 2268(C39) 1409 (C40)
23 General procedure for the synthesis of polymers
Styrene (St) and COBAMI were copolymerized using a comonomer feed ratio of1 1 under stable free radical polymerization (SFRP) or conventional free radicalpolymerization (CFRP) conditions
A round-bottom ask equipped with magnetic stirrer was vacuumed and back- lled with dry nitrogen several times Monomers initiators and solvent wereintroduced under inert atmosphere The ask was placed in a preheated oil bathAll the polymerizations were conducted at 125plusmnC for a given time (Table 1) Inthe copolymerizations under SFRP conditions rst the reaction mixture was kept at95plusmnC for 35 h to allow complete decomposition of BPO Then the mixtures werecooled and the resulting products were precipitated in methanol The polymer waspuri ed by repeated precipitations from chloroform solutions into methanol Theconversions determined gravimetrically and other characteristics of the resultingpolymers are reported in Table 1
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

146 L Cianga et al
methods such as living polymerization controlled polymerization and group-transfer polymerizations opens new avenues to the design and synthesis of liquid-crystalline polymer systems with well de ned structure controlled molar mass andnarrow molar mass distribution [1 2] Different types of liquid-crystalline polymerssynthesized by atom transfer radical polymerization (ATRP) [3 4] or stable freeradical polymerization (SFRP) [5 6] techniques were reported
Copolymerization of various combinations of mesogenicnonmesogenic mono-mers has proved to be a powerful tool to modulate the properties of different liquid-crystalline copolymer structures (for a review see Ref [1]) Surprisingly verylittle attention has been devoted to the synthesis of alternating copolymers (forexample see Refs [7 8] and references therein) In these systems one may controlthe monomer sequence distribution in semi-rigid copolymer chains and thus tailorthe properties of the polymer on a well-de ned structural basis [9]
N-Substituted polymaleimides belong to side-group polymers that have a ten-dency to order partially due to the rigid structure of the planar succinimide groupTo our knowledge there are few reports concerning preparation and characterizationof SG-LC polymaleimides Oishi et al reported about poly- and copolymaleimidesin which the cholesteryl mesogenic group was attached to the polymer backbonethrough a paraf nic [10 11] or an aromatic [12] spacer unit However the ther-motropic behavior of these polymers was not studied
Smectic ordering in poly(4-maleimidocholesteryl benzoate) poly(6-maleimido-cholesteryl hexanoate) and alternating copolymers with styrene reg and macr-methyl-styrene and p-ethoxystyrene was studied by Vukovic et al [13] Smectic ornematic phases were observed in a homologous series of SG-LCPs containing the4-cyanobiphenyl mesogenic group attached to the polymaleimide backbone throughparaf nic spacers [14] Smectic phases were also formed in polymaleimidesbased on phenyl benzoate mesogenic groups [15] The p-n-alkoxybenzoic acidsstarting with n-propyloxybenzoic acid exhibit liquid-crystalline behavior andtheir mesogenic potential increases with the length of the alkoxy group Phenylbenzoates as mesogenic groups may impart some special characteristics to theirSG-LCPs The primary signi cant characteristic of the phenyl benzoate moietiesmay be their strong interactions governed by electronic and geometrical factorsallowing the mesogenic group to achieve more ordered packing with respect to othermesogens [15]
In this paper we report the preparation of a new mesogenic N-substitutedmaleimide monomer (COBAMI) containing a ((hexadecyloxy)benzoyloxy)phenylmesogenic unit directly attached to the maleimide ring and its copolymerizationwith styrene (St) under stable free radical polymerization (SFRP) conditions andconventional free radical polymerization (CFRP) The aim of this work was thepreparation of alternating copolymers (Scheme 1) with controlled molecular struc-ture controlled molecular weight and narrow polydispersity that would exhibitliquid-crystalline behavior
Mesogenic maleimidendashstyrene copolymers 147
Scheme 1 Structure of the copolymers prepared and studied
2 EXPERIMENTAL
21 Materials
Maleic anhydride (Aldrich) was sublimated before use All solvents were puri edand dried by standard techniques before to use Acetic anhydride (Merck) p-aminobenzoic acid hydroquinone (Fluka) 4-hexadecyloxybenzoic acid (Across)(COBA) 2266-tetramethyl-piperidinyl-1-oxy (TEMPO) (Aldrich) and benzoylperoxide (BPO) (Fluka) were used without further puri cation Thionyl chlorideand styrene (Fluka) were distilled just before use
22 Preparation of intermediates and maleimide monomer
221 4-Maleimido benzoic acid (MBA) MBA was synthesized by the conden-sation reaction of maleic anhydride with p-aminobenzoic acid followed by cyclode-hydration using acetic anhydride and sodium acetate according to a procedure pub-lished elsewhere [16]
Yield 85 mp 234plusmnC (244plusmnC [16]) IR (cmiexcl1) 3460ndash 3200 (COOH) 3105ndash3100( CH) 1720 (O CNC O) 1610 (C C) 1217 (C O) 820 (phenyl) 720 (cisCH CH)
222 4-Maleimidobenzoic acid chloride (MBAC) MBAC was synthesizedaccording to a procedure described elsewhere [15]
Yield 73 mp 157ndash158plusmnC (168ndash169plusmnC [16]) IR (cmiexcl1) 1770 (COCl) 1715(O CNC O) 1600 (C C) 700 (cis CH CH)
223 N-[4-(40-hydroxyphenyloxycarbonyl) phenyl] maleimide (HPPMI)HPPMI was prepared as described previously [17]
Yield 79 mp 208plusmnC Anal calc for C17H11NO5 () C 6602 H 358 N453 Found C 6610 H 367 N 471
IR (cmiexcl1) 3450 (OH) 3105ndash3100 ( CH) 1765 1720 (imide I) 1705 (C Oester) 1600 (C C) 1380 (imide II) 1240 (C O C ester) 1190 (C (OH)phenolic) 1150 (imide III) 1070 (ester) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm in DMSO-d6) 953 (s 1H OH) 830ndash803 (q 2H Ar-H orthoto carbonyl group) 764ndash759 (q 2H Ar-H ortho to imide group) 725 (s 2H
148 L Cianga et al
HC CH) 71ndash706 (d 2H Ar-H ortho to O atom) 683ndash679 (d 2H Ar-H orthoto hydroxyl)
224 4-Hexadecyloxybenzoic acid chloride (COBAC) COBAC was preparedby dissolving 2175 g (006 mol) of acid in 150 ml of dry benzene To this solution2855 g (024 mol) of thionyl chloride was added dropwise and the resulting mixturewas re uxed for 24 h The solution was then cooled down to room temperatureand a solid precipitated by keeping the ask in the refrigerator overnight Theacid chloride was obtained as a white solid by vacuum ltration under nitrogenand subsequent drying in vacuum It was used in the next reaction without furtherpuri cation Yield 998
Anal calc for C23H37O2Cl () C 7250 H 979 Cl 931 Found C 7272 H980 Cl 943
IR (cmiexcl1) 2900 2860 (CH aliphatic) 1760 (COCl) 1600 (C C aromatic) 1460(CH3) 1280 (C O C) 1170 (CH2 chain) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm from TMS in CDCl3) 808ndash804 (d 2H aromatic) 696ndash692(d 2H aromatic) 406ndash401 (t 2H Ar O CH2 ) 183ndash178 (t 2H Ar OCH2 CH2 ) 17ndash125 (m 26H (CH213 ) 087ndash085 (t 3H CH3
225 40-[400-(Hexadecyloxy)benzoyl]oxyphenyl 4-maleimidobenzoate(COBAMI) (Scheme 2) A solution of 190 g (0005 mol) of COBAC in 20 mlof dry dichloromethane was added dropwise to a solution of 155 g (0005 mol) ofHPPMI in 200 ml dichloromethane containing 07 ml (0005 mol) of triethylamineThe reaction mixture was kept to re ux under nitrogen and with stirring for 96 hThe precipitate formed was ltered off and the solution was washed with 1 HCl(4 times) and then water (4 times) The organic layer was separated and dried overMgSO4 and after evaporation of dichloromethane the reaction product was washedthoroughly with hot hexane to remove unreacted traces of COBAC The pure prod-uct COBAMI (Scheme 3) was obtained by recrystallization from benzene
Yield 66 mp 156plusmnC Anal calc for C40H47NO7 () C 7348 H 725 N214 Found C 7357 H 732 N 215
IR (cmiexcl1) 3105ndash3050 (cis CH maleimide) 2920 2850 (aliphatic) 1770(imide I) 1708 (imide I C O ester) 1610 (C C aromatic and maleimide) 1420(CH3) 1400 (imide II) 1260 (C O C ether) 840 (p-disubstituted phenyl) 830(cis C H maleimide) 695 (imide IV carbonyl ring deformation)
1H-NMR (plusmn ppm from TMS in CDCl3) 832ndash829 (d 2H of C20 and C21)816ndash812 (d 2H of C7 and C9) 761ndash758 (d 2H of C6 and C10) 73ndash726(m 4H of C13 C14 C15 C17) 699ndash696 (d 2H of C22 and C24) 69 (s 2H ofC1 and C2)
13C-NMR (plusmn ppm in CDCl3) 16882 (C3 C4) 16477 (C11) 164208 (C18)163617 (C23) 148369 (C12 C16) 13612 (C5) 13446 (C1 C2) 13233(C7 C9) 13108 (C20 C21) 128318 (C8) 12525 (C6 C10) 12252 (C13 C14C15 C17) 12136 (C19) 114368 (C22 C24) 6842 (C25) 319 (C38)
Mesogenic maleimidendashstyrene copolymers 149
Scheme 2 Reaction pathway for the synthesis of monomer COBAMI
Scheme 3 Structure of COBAMI
2965ndash2940 (C28ndashC34) 2933 (C35 C36 C37) 29 (C26) 2596 (C27) 2268(C39) 1409 (C40)
23 General procedure for the synthesis of polymers
Styrene (St) and COBAMI were copolymerized using a comonomer feed ratio of1 1 under stable free radical polymerization (SFRP) or conventional free radicalpolymerization (CFRP) conditions
A round-bottom ask equipped with magnetic stirrer was vacuumed and back- lled with dry nitrogen several times Monomers initiators and solvent wereintroduced under inert atmosphere The ask was placed in a preheated oil bathAll the polymerizations were conducted at 125plusmnC for a given time (Table 1) Inthe copolymerizations under SFRP conditions rst the reaction mixture was kept at95plusmnC for 35 h to allow complete decomposition of BPO Then the mixtures werecooled and the resulting products were precipitated in methanol The polymer waspuri ed by repeated precipitations from chloroform solutions into methanol Theconversions determined gravimetrically and other characteristics of the resultingpolymers are reported in Table 1
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 147
Scheme 1 Structure of the copolymers prepared and studied
2 EXPERIMENTAL
21 Materials
Maleic anhydride (Aldrich) was sublimated before use All solvents were puri edand dried by standard techniques before to use Acetic anhydride (Merck) p-aminobenzoic acid hydroquinone (Fluka) 4-hexadecyloxybenzoic acid (Across)(COBA) 2266-tetramethyl-piperidinyl-1-oxy (TEMPO) (Aldrich) and benzoylperoxide (BPO) (Fluka) were used without further puri cation Thionyl chlorideand styrene (Fluka) were distilled just before use
22 Preparation of intermediates and maleimide monomer
221 4-Maleimido benzoic acid (MBA) MBA was synthesized by the conden-sation reaction of maleic anhydride with p-aminobenzoic acid followed by cyclode-hydration using acetic anhydride and sodium acetate according to a procedure pub-lished elsewhere [16]
Yield 85 mp 234plusmnC (244plusmnC [16]) IR (cmiexcl1) 3460ndash 3200 (COOH) 3105ndash3100( CH) 1720 (O CNC O) 1610 (C C) 1217 (C O) 820 (phenyl) 720 (cisCH CH)
222 4-Maleimidobenzoic acid chloride (MBAC) MBAC was synthesizedaccording to a procedure described elsewhere [15]
Yield 73 mp 157ndash158plusmnC (168ndash169plusmnC [16]) IR (cmiexcl1) 1770 (COCl) 1715(O CNC O) 1600 (C C) 700 (cis CH CH)
223 N-[4-(40-hydroxyphenyloxycarbonyl) phenyl] maleimide (HPPMI)HPPMI was prepared as described previously [17]
Yield 79 mp 208plusmnC Anal calc for C17H11NO5 () C 6602 H 358 N453 Found C 6610 H 367 N 471
IR (cmiexcl1) 3450 (OH) 3105ndash3100 ( CH) 1765 1720 (imide I) 1705 (C Oester) 1600 (C C) 1380 (imide II) 1240 (C O C ester) 1190 (C (OH)phenolic) 1150 (imide III) 1070 (ester) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm in DMSO-d6) 953 (s 1H OH) 830ndash803 (q 2H Ar-H orthoto carbonyl group) 764ndash759 (q 2H Ar-H ortho to imide group) 725 (s 2H
148 L Cianga et al
HC CH) 71ndash706 (d 2H Ar-H ortho to O atom) 683ndash679 (d 2H Ar-H orthoto hydroxyl)
224 4-Hexadecyloxybenzoic acid chloride (COBAC) COBAC was preparedby dissolving 2175 g (006 mol) of acid in 150 ml of dry benzene To this solution2855 g (024 mol) of thionyl chloride was added dropwise and the resulting mixturewas re uxed for 24 h The solution was then cooled down to room temperatureand a solid precipitated by keeping the ask in the refrigerator overnight Theacid chloride was obtained as a white solid by vacuum ltration under nitrogenand subsequent drying in vacuum It was used in the next reaction without furtherpuri cation Yield 998
Anal calc for C23H37O2Cl () C 7250 H 979 Cl 931 Found C 7272 H980 Cl 943
IR (cmiexcl1) 2900 2860 (CH aliphatic) 1760 (COCl) 1600 (C C aromatic) 1460(CH3) 1280 (C O C) 1170 (CH2 chain) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm from TMS in CDCl3) 808ndash804 (d 2H aromatic) 696ndash692(d 2H aromatic) 406ndash401 (t 2H Ar O CH2 ) 183ndash178 (t 2H Ar OCH2 CH2 ) 17ndash125 (m 26H (CH213 ) 087ndash085 (t 3H CH3
225 40-[400-(Hexadecyloxy)benzoyl]oxyphenyl 4-maleimidobenzoate(COBAMI) (Scheme 2) A solution of 190 g (0005 mol) of COBAC in 20 mlof dry dichloromethane was added dropwise to a solution of 155 g (0005 mol) ofHPPMI in 200 ml dichloromethane containing 07 ml (0005 mol) of triethylamineThe reaction mixture was kept to re ux under nitrogen and with stirring for 96 hThe precipitate formed was ltered off and the solution was washed with 1 HCl(4 times) and then water (4 times) The organic layer was separated and dried overMgSO4 and after evaporation of dichloromethane the reaction product was washedthoroughly with hot hexane to remove unreacted traces of COBAC The pure prod-uct COBAMI (Scheme 3) was obtained by recrystallization from benzene
Yield 66 mp 156plusmnC Anal calc for C40H47NO7 () C 7348 H 725 N214 Found C 7357 H 732 N 215
IR (cmiexcl1) 3105ndash3050 (cis CH maleimide) 2920 2850 (aliphatic) 1770(imide I) 1708 (imide I C O ester) 1610 (C C aromatic and maleimide) 1420(CH3) 1400 (imide II) 1260 (C O C ether) 840 (p-disubstituted phenyl) 830(cis C H maleimide) 695 (imide IV carbonyl ring deformation)
1H-NMR (plusmn ppm from TMS in CDCl3) 832ndash829 (d 2H of C20 and C21)816ndash812 (d 2H of C7 and C9) 761ndash758 (d 2H of C6 and C10) 73ndash726(m 4H of C13 C14 C15 C17) 699ndash696 (d 2H of C22 and C24) 69 (s 2H ofC1 and C2)
13C-NMR (plusmn ppm in CDCl3) 16882 (C3 C4) 16477 (C11) 164208 (C18)163617 (C23) 148369 (C12 C16) 13612 (C5) 13446 (C1 C2) 13233(C7 C9) 13108 (C20 C21) 128318 (C8) 12525 (C6 C10) 12252 (C13 C14C15 C17) 12136 (C19) 114368 (C22 C24) 6842 (C25) 319 (C38)
Mesogenic maleimidendashstyrene copolymers 149
Scheme 2 Reaction pathway for the synthesis of monomer COBAMI
Scheme 3 Structure of COBAMI
2965ndash2940 (C28ndashC34) 2933 (C35 C36 C37) 29 (C26) 2596 (C27) 2268(C39) 1409 (C40)
23 General procedure for the synthesis of polymers
Styrene (St) and COBAMI were copolymerized using a comonomer feed ratio of1 1 under stable free radical polymerization (SFRP) or conventional free radicalpolymerization (CFRP) conditions
A round-bottom ask equipped with magnetic stirrer was vacuumed and back- lled with dry nitrogen several times Monomers initiators and solvent wereintroduced under inert atmosphere The ask was placed in a preheated oil bathAll the polymerizations were conducted at 125plusmnC for a given time (Table 1) Inthe copolymerizations under SFRP conditions rst the reaction mixture was kept at95plusmnC for 35 h to allow complete decomposition of BPO Then the mixtures werecooled and the resulting products were precipitated in methanol The polymer waspuri ed by repeated precipitations from chloroform solutions into methanol Theconversions determined gravimetrically and other characteristics of the resultingpolymers are reported in Table 1
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

148 L Cianga et al
HC CH) 71ndash706 (d 2H Ar-H ortho to O atom) 683ndash679 (d 2H Ar-H orthoto hydroxyl)
224 4-Hexadecyloxybenzoic acid chloride (COBAC) COBAC was preparedby dissolving 2175 g (006 mol) of acid in 150 ml of dry benzene To this solution2855 g (024 mol) of thionyl chloride was added dropwise and the resulting mixturewas re uxed for 24 h The solution was then cooled down to room temperatureand a solid precipitated by keeping the ask in the refrigerator overnight Theacid chloride was obtained as a white solid by vacuum ltration under nitrogenand subsequent drying in vacuum It was used in the next reaction without furtherpuri cation Yield 998
Anal calc for C23H37O2Cl () C 7250 H 979 Cl 931 Found C 7272 H980 Cl 943
IR (cmiexcl1) 2900 2860 (CH aliphatic) 1760 (COCl) 1600 (C C aromatic) 1460(CH3) 1280 (C O C) 1170 (CH2 chain) 840 (p-disubstituted phenyl)
1H-NMR (plusmn ppm from TMS in CDCl3) 808ndash804 (d 2H aromatic) 696ndash692(d 2H aromatic) 406ndash401 (t 2H Ar O CH2 ) 183ndash178 (t 2H Ar OCH2 CH2 ) 17ndash125 (m 26H (CH213 ) 087ndash085 (t 3H CH3
225 40-[400-(Hexadecyloxy)benzoyl]oxyphenyl 4-maleimidobenzoate(COBAMI) (Scheme 2) A solution of 190 g (0005 mol) of COBAC in 20 mlof dry dichloromethane was added dropwise to a solution of 155 g (0005 mol) ofHPPMI in 200 ml dichloromethane containing 07 ml (0005 mol) of triethylamineThe reaction mixture was kept to re ux under nitrogen and with stirring for 96 hThe precipitate formed was ltered off and the solution was washed with 1 HCl(4 times) and then water (4 times) The organic layer was separated and dried overMgSO4 and after evaporation of dichloromethane the reaction product was washedthoroughly with hot hexane to remove unreacted traces of COBAC The pure prod-uct COBAMI (Scheme 3) was obtained by recrystallization from benzene
Yield 66 mp 156plusmnC Anal calc for C40H47NO7 () C 7348 H 725 N214 Found C 7357 H 732 N 215
IR (cmiexcl1) 3105ndash3050 (cis CH maleimide) 2920 2850 (aliphatic) 1770(imide I) 1708 (imide I C O ester) 1610 (C C aromatic and maleimide) 1420(CH3) 1400 (imide II) 1260 (C O C ether) 840 (p-disubstituted phenyl) 830(cis C H maleimide) 695 (imide IV carbonyl ring deformation)
1H-NMR (plusmn ppm from TMS in CDCl3) 832ndash829 (d 2H of C20 and C21)816ndash812 (d 2H of C7 and C9) 761ndash758 (d 2H of C6 and C10) 73ndash726(m 4H of C13 C14 C15 C17) 699ndash696 (d 2H of C22 and C24) 69 (s 2H ofC1 and C2)
13C-NMR (plusmn ppm in CDCl3) 16882 (C3 C4) 16477 (C11) 164208 (C18)163617 (C23) 148369 (C12 C16) 13612 (C5) 13446 (C1 C2) 13233(C7 C9) 13108 (C20 C21) 128318 (C8) 12525 (C6 C10) 12252 (C13 C14C15 C17) 12136 (C19) 114368 (C22 C24) 6842 (C25) 319 (C38)
Mesogenic maleimidendashstyrene copolymers 149
Scheme 2 Reaction pathway for the synthesis of monomer COBAMI
Scheme 3 Structure of COBAMI
2965ndash2940 (C28ndashC34) 2933 (C35 C36 C37) 29 (C26) 2596 (C27) 2268(C39) 1409 (C40)
23 General procedure for the synthesis of polymers
Styrene (St) and COBAMI were copolymerized using a comonomer feed ratio of1 1 under stable free radical polymerization (SFRP) or conventional free radicalpolymerization (CFRP) conditions
A round-bottom ask equipped with magnetic stirrer was vacuumed and back- lled with dry nitrogen several times Monomers initiators and solvent wereintroduced under inert atmosphere The ask was placed in a preheated oil bathAll the polymerizations were conducted at 125plusmnC for a given time (Table 1) Inthe copolymerizations under SFRP conditions rst the reaction mixture was kept at95plusmnC for 35 h to allow complete decomposition of BPO Then the mixtures werecooled and the resulting products were precipitated in methanol The polymer waspuri ed by repeated precipitations from chloroform solutions into methanol Theconversions determined gravimetrically and other characteristics of the resultingpolymers are reported in Table 1
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 149
Scheme 2 Reaction pathway for the synthesis of monomer COBAMI
Scheme 3 Structure of COBAMI
2965ndash2940 (C28ndashC34) 2933 (C35 C36 C37) 29 (C26) 2596 (C27) 2268(C39) 1409 (C40)
23 General procedure for the synthesis of polymers
Styrene (St) and COBAMI were copolymerized using a comonomer feed ratio of1 1 under stable free radical polymerization (SFRP) or conventional free radicalpolymerization (CFRP) conditions
A round-bottom ask equipped with magnetic stirrer was vacuumed and back- lled with dry nitrogen several times Monomers initiators and solvent wereintroduced under inert atmosphere The ask was placed in a preheated oil bathAll the polymerizations were conducted at 125plusmnC for a given time (Table 1) Inthe copolymerizations under SFRP conditions rst the reaction mixture was kept at95plusmnC for 35 h to allow complete decomposition of BPO Then the mixtures werecooled and the resulting products were precipitated in methanol The polymer waspuri ed by repeated precipitations from chloroform solutions into methanol Theconversions determined gravimetrically and other characteristics of the resultingpolymers are reported in Table 1
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)
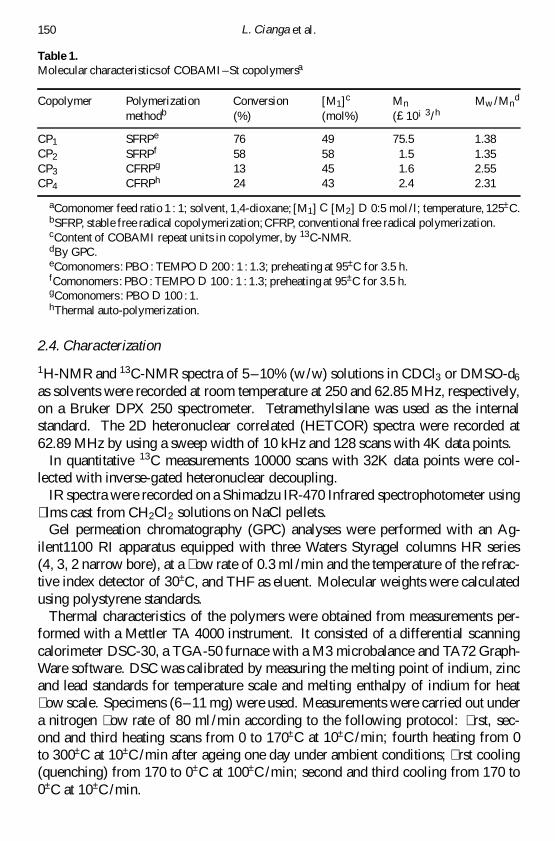
150 L Cianga et al
Table 1Molecular characteristicsof COBAMIndashSt copolymersa
Copolymer Polymerization Conversion [M1]c Mn Mw Mnd
methodb () (mol) (pound10iexcl3h
CP1 SFRPe 76 49 755 138CP2 SFRPf 58 58 15 135CP3 CFRPg 13 45 16 255CP4 CFRPh 24 43 24 231
aComonomer feed ratio 1 1 solvent 14-dioxane [M1] C [M2] D 05 mol l temperature 125plusmnCbSFRP stable free radical copolymerizationCFRP conventional free radical polymerizationcContent of COBAMI repeat units in copolymer by 13C-NMRdBy GPCeComonomers PBO TEMPO D 200 1 13 preheating at 95plusmnC for 35 hfComonomers PBO TEMPO D 100 1 13 preheating at 95plusmnC for 35 hgComonomers PBO D 100 1hThermal auto-polymerization
24 Characterization
1H-NMR and 13C-NMR spectra of 5ndash10 (ww) solutions in CDCl3 or DMSO-d6
as solvents were recorded at room temperature at 250 and 6285 MHz respectivelyon a Bruker DPX 250 spectrometer Tetramethylsilane was used as the internalstandard The 2D heteronuclear correlated (HETCOR) spectra were recorded at6289 MHz by using a sweep width of 10 kHz and 128 scans with 4K data points
In quantitative 13C measurements 10000 scans with 32K data points were col-lected with inverse-gated heteronuclear decoupling
IR spectra were recorded on a Shimadzu IR-470 Infrared spectrophotometer using lms cast from CH2Cl2 solutions on NaCl pellets
Gel permeation chromatography (GPC) analyses were performed with an Ag-ilent1100 RI apparatus equipped with three Waters Styragel columns HR series(4 3 2 narrow bore) at a ow rate of 03 mlmin and the temperature of the refrac-tive index detector of 30plusmnC and THF as eluent Molecular weights were calculatedusing polystyrene standards
Thermal characteristics of the polymers were obtained from measurements per-formed with a Mettler TA 4000 instrument It consisted of a differential scanningcalorimeter DSC-30 a TGA-50 furnace with a M3 microbalance and TA72 Graph-Ware software DSC was calibrated by measuring the melting point of indium zincand lead standards for temperature scale and melting enthalpy of indium for heat ow scale Specimens (6ndash11 mg) were used Measurements were carried out undera nitrogen ow rate of 80 mlmin according to the following protocol rst sec-ond and third heating scans from 0 to 170plusmnC at 10plusmnCmin fourth heating from 0to 300plusmnC at 10plusmnCmin after ageing one day under ambient conditions rst cooling(quenching) from 170 to 0plusmnC at 100plusmnCmin second and third cooling from 170 to0plusmnC at 10plusmnCmin
Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 151
The formation of liquid-crystalline phases was con rmed by cross-polarizedoptical microscopy observations with a Reichert Polyvar microscope equipped witha programmable Mettler FP52 heating stage
3 RESULTS AND DISCUSSION
31 Synthesis of N-phenylmaleimide monomer COBAMI
4-Maleimidobenzoic acid a very versatile reagent was often used in the last decadeto obtain different N-phenyl maleimide [12 13 16ndash18] or bismaleimide monomers[18 19]
As was previously reported [17] we used the same reagent for the synthesis ofthe HPPMI intermediate (Scheme 2) Then by a classical Schottenndash Baumanncondensation with 4-hexadecyloxybenzoic acid chloride (COBAC) the maleimidemonomer COBAMI was obtained
The structure of this new monomer was con rmed by infrared 13C- and 1H-NMRspectroscopies as well as by elemental analysis (see Experimental) In particularthe 2D HETCOR spectrum of COBAMI (Fig 1) shows cross signals between thepeaks at 13446 13233 13108 12525 12252 114368 6841 2965ndash2933 292596 2268 and 1409 ppm in the 13C-NMR spectrum and those at about 83 76728 699 and 69 in the 1H-NMR spectrum The latter signals were attributedrespectively to protons or carbons C1 C2 C7 C9 C20 C21 C6 C10 C13 C14C15 C17 C22 C24 C25 C28ndashC37 C26 C27 C39 and C40 and protons on themrespectively
Owing to its structure with both hydrophilic and hydrophobic groups the newmonomer is soluble in moderate concentrations in a large variety of solvents thebest solubility being qualitatively observed in chlorinated ones It is insoluble innon-polar solvents such as hexane heptane and cyclohexane
32 Copolymerization of maleimide monomer COBAMI with St
Copolymerizations of N-alkyl- or N-phenylmaleimides with St in bulk or solutionby lsquocontrolledrsquo living radical polymerization methods like ATRP [20 21] or SFRP[22ndash26] revealed that alternating copolymers or block copolymers with controlledmolecular weight and low polydispersities can be prepared
To assess the potential for copolymerization of COBAMI with St an initial com-parative study was performed in this work Thus two sets of copolymerizationswere developed in solution under SFRP and CFRP conditions respectively (Ta-ble 1) All polymerizations were performed in 14-dioxane as solvent at 125plusmnC withthe monomer feed ratio being 1 1 The initial comonomer concentration in solu-tion was kept constant In the copolymerizations under SFRP conditions (CP1 andCP2) a bimolecular initiator system consisting of 1 13 molar mixture of BPO andTEMPO was found to be optimal with a preheating at 95plusmnC for 35 h It can be
152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

152 L Cianga et al
Figure 1 2D HETCOR NMR spectrum of COBAMI (in CDCl3
pointed out that copolymers with low polydispersities (135 or 138) were obtainedand that conversion and molecular weight both depended on the reaction time
In both SFRP experiments the increase in the molecular weight along withthe increasing conversion was considered as an evidence of the pseudo-livingcharacter of the polymerization process with the participation of TEMPO radicalswhich are reversibly bound to the growing polymer chains As is normallyobserved for BPOTEMPO-mediated polymerizations [23] a drastic increase inthe polymerization rate and a signi cant reduction of the induction period couldalso be considered in the case of the COBAMIndashSt system when the molecularweights of the copolymers CP1 CP3 and CP4 are compared The low molecularweights of the copolymers CP3 and CP4 obtained under conventional free radicalpolymerization (CFRP) conditions at the same reaction temperature and time asfor CP1 could be due to the long induction period This is a consequence of thestructural characteristics of COBAMI the electron-de cient monomer in the studiedsystem such as (1) the presence of the electron withdrawing polar groups (COO)in the para-position on the N-phenyl ring which would decrease the reactivity ofthe double maleimide bond and (2) the steric hindrance due to the bulkiness of this
Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 153
p-substitutent For CP3 and CP4 high values for the polydispersities were observedas well
The composition of the copolymers was evaluated using quantitative 13C-NMRmeasurements In the 1H-NMR spectra the characteristic peaks of St merged to-gether with those of COBAMI and did not allow reliable evaluations of composi-tion It can be seen from Table 1 that the copolymer composition was nearly 1 1 inany copolymer prepared Therefore essentially alternating structures were obtaineddue to the formation of an electron donorndashacceptor complex
The structural characteristics of the copolymers were clari ed from their 13C- and1H-NMR spectra These spectra were as illustrated for CP1 in Figs 2A and 2Bbecause quite similar features were observed for all the synthesized copolymersThe peaks characteristic to all aliphatic and aromatic carbon atoms of CP1 arepresent for the COBAMI repeat units almost at the same positions as in the relevantmonomer except the signals of the carbonyls in the new formed succinimide ringsthat appeared as two small multiplets at 1767 ppm and 17517 ppm respectively(signal b in Fig 2B) and as a singlet at 16892 ppm (signal a in Fig 2B)The multiplets were attributed to the succinimide carbonyls near to the CH2
group in the St repeat units and the singlet was attributed to the carbonyls nearto CH group in the St repeat units The CH aromatic carbons in the St unitsappeared at 12634 12903 and 13449 ppm respectively The signal for thequaternary St carbon was broadened and up eld shifted (138ndash1415 ppm) due toboth long-range shielding and electron density perturbation by the proximity of theimide group containing bulky substitutent A similar behavior was reported forStndashbutylmaleimide copolymers [24]
The intensity ratio of the a-type and b-type carbon atoms in Fig 2B was 55 45This result suggests that the enchainment of styrene units is nearly ordered and thatthe bulky side chain sterically controls the addition of St monomer to the growingchain end in the propagation step Furthermore the coexistence of succinimide ringunits in trans and cis forms in the copolymer chains was proven by the presenceof signals at about 35 ppm and 37 ppm in the 1H-NMR spectra (Fig 2A) and at17517 ppm and 1767 ppm in the 13C-NMR spectra (Fig 2B) respectively Thesignals at 35 ppm and at 17517 ppm were attributed to the cis succinimide formwhile those at 37 ppm and at 1767 ppm to the trans form
It has been claimed [27] that the presence of cis succinimide units in Stndashmaleimidecopolymers should be considered as a structural evidence for the participation of theelectron donorndash acceptor complex in the propagation step Taking into account theresults of Mormann and Schmaltz [28] we conclude that the copolymers preparedpossess an alternating structure with a preference for the cis con guration over thetrans in a ratio of 7 4 as was calculated from the corresponding 1H-NMR integrals
33 Thermal properties of COBAMIndashSt copolymers
Prior to investigation of the liquid crystalline properties of the samples they wereanalyzed by thermogravimetric analysis (TGA) (Fig 3) N-substituted maleimide
154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

154 L Cianga et al
Figure 2 1H-NMR and 13C-NMR spectra of copolymer CP1 (in CDCl3
Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 155
Figure 3 Experimental and calculated TGA traces for copolymer CP1 and relevant model com-pounds
polymers are generally known to be thermally stable (see Refs [29ndash31] andreferences therein) with the nature of the N-substituent playing a role in affectingthe thermal degradation process (see Ref [31] and references therein)
Monomer COBAMI decomposed in nitrogen atmosphere in two overlapping stepsof weight loss leaving approx 21 wt residue at 680plusmnC The rst step of weightloss was approx 3 wt at an onset temperature (Ton of about 269plusmnC de ned at aslope of iexcl0048K and the last was at 346plusmnC at iexcl0053K On the other handpolystyrene tended to depolymerize in one step at Ton of about 322plusmnC de ned at aslope of iexcl0052K The TGA traces of the monomer COBAMI were similar tothose of the copolymers CP which suggests that the thermal decomposition of boththe imide monomer and comonomer unit in the copolymers can occur with a samemechanism Therefore we used COBAMI and poly(styrene) as a simple modelcompounds for the TGA analysis of the copolymers
By assuming that copolymer decomposition obeys an additivity rule [32] thesimulated and experimental TGA traces of the copolymers were compared withthose of the corresponding models Apparently the copolymers decomposed in asingle step leaving a residue comparable to that of COBAMI monomer Besidesthe Ton values of copolymers at about 309plusmnC were intermediate between those ofthe model compounds Nevertheless the early stage of the thermal decompositionof COBAMIndashSt copolymers seemed to occur at a higher temperature than thatexpected from the stability of its precursors This suggests that the more stablestyrene comonomer performs a protective effect on the less stable COBAMIcomonomer Furthermore above about 380plusmnC subsequent thermal decomposition
156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

156 L Cianga et al
Table 2Transition temperatures (in plusmnC) of COBAMIndashSt copolymers (by DSC at 10plusmnCmin)
Copolymer Tg Tcc Tm Ti
CP1 37 95 185 262CP2 52 84 158 ndCP3 45 104 130 ndCP4 46 126 166 nd
Glass cold crystallization melting and isotropization temperatures nd not detectable because ofpartial degradation Tg of CP2 and CP3 was determined with endotherm relaxation
occurred at a lower temperature than that indicated by the simulated trace untilapprox 470plusmnC when it began to overlap the COBAMI trace This last temperatureat which there was an apparent inversion of decomposition reaction corresponds tothe end of poly(styrene) homopolymer depolymerization
The additivity rule supposes that each counit and their decomposition productsdo not modify the decomposition reaction of the other However two concurrentprocesses can be postulated from the present results On the one hand the initialldquosynergisticrdquo effect implies a diffusion control of the degradation products of theCOBAMI counit from the more stable St co-unit On the other hand reactive speciesfrom COBAMI counit would be able to modify the degradation mechanism of Stcounit which acceleration began to be detected at about 380plusmnC A greater residuethan that expected from simulation can also result from the polymer chain endscontaining TEMPO [25]
The phase transition behavior of the copolymers was relatively complex and issummarized in Table 2 Each of them underwent cold-crystallization on heatingabove its glass transition temperature which resulted in a partial crystallinity ofthe samples (Fig 4) Above the melting temperature the copolymers formed amesophase but the mesophase-isotropic liquid transition or isotropization temper-ature was clearly detected in CP1 only (Ti D 262plusmnC) Isotropization of the othersamples was not unequivocally veri ed because of the concomitance of side reac-tions eg crosslinking (Fig 4) In this particular case the copolymers may not bestrictly classi ed as thermotropic liquid crystals The phase transitions were com-pletely reversible on cooling provided the sample was not taken above its degrada-tion temperature Observations by polarized light microscopy of schlieren texturessuggested that the mesophase was nematic Evidently the rigid rod-like side groupsubstituent on the stiff imide ring repeat unit can be packed in a crystalline struc-ture that is partially disrupted on heating to form a mesophase persistent over abroad existence range up to high temperatures The strong tendency to order of thepresent mesogenic side groups is not prevented in such diluted systems as the 1 1copolymers
While all the copolymers exhibited the same overall behavior their individualtransition temperatures depended on the molecular weight This is best illustratedby comparison of the high molecular weight CP1 with the oligomeric CP2 with the
Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

Mesogenic maleimidendashstyrene copolymers 157
Figure 4 DSC heating curves (10plusmnCmin) of representative CP copolymers
mesophase being stabilized by the increased molecular weight in the former sampleThe thermal transitions could also be affected although conceivably not in a signi -cant way by the chemical composition of the copolymers that slightly differed fromthe exactly alternating structure Further studies are necessary to clarify the nedetails of the mesophase structure and transitions of the COBAMIndashSt copolymers
Acknowledgements
The authors thank the Turkish TUumlBITAK the Italian CNR and Romanian CNCSISfor partial nancial support of the work L C is also thankful to TUumlBITAK for aNATO post-doctoral fellowship
REFERENCES
1 G Galli in Advanced Functional Molecules and Polymers Vol 1 H S Nalwa (Ed) p 271Overseas Publishers Association Singapore (2001)
2 A Omenat and J Lub Chem Mater 10 518 (1998)3 C Pugh C Chang and A M Kasko Polym Prepr 40 108 (1999)4 S M Ruder A Datta and S D Allen Polym Prepr 41 1241 (2000)5 C A Barbosa and A S Gomes Polym Bull 41 15 (1998)6 P Gopalan S Pragliola C K Ober P T Malher and H G Jeon Polym Prepr 40 372 (1999)7 M Laus M C Bignozzi A S Angeloni and E Chiellini Macromolecules 26 3999 (1993)
158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)

158 L Cianga et al
8 R P Nieuwhof A Koudijs A T M Marcelis and E J R Sudholter Macromolecules 32 6499(1999)
9 J Economy Angew Chem Int Ed Engl 29 1256 (1990)10 T Oishi Y Otsubo and M Fujimoto Polym J 24 527 (1992)11 T Oishi H Morikawa K Matsusaki and M Fujimoto Polym J 26 1331 (1994)12 T Oishi Y Otsubo K Matsusaki and M Fujimoto Polymer 34 1504 (1993)13 R Vukovic D Fles I Smith F E Karasz and A Waddon Polym Bull 38 347 (1997)14 I Gangadhara C Noel M Thomas and D Reyx J Polym Sci Part A Polym Chem 36 2531
(1998)15 F R Colomer J M M Duenas J L G Ribelles J M Barralas-Rienda and J M B de Ojeda
Macromolecules 26 155 (1993)16 T Oishi and M Fujimoto J Polym Sci Part A Polym Chem 30 1821 (1992)17 L Cianga and Y Yagci J Polym Sci Part A Polym Chem 40 995 (2002)18 L Cianga and N Olaru Design Monom Polym 3 209 (2000)19 F Sun Y Yang and R M Ottenbrite Polym Prepr 32 188 (1991)20 G Q Chen Z Q Wu J R Wu Z C Li and F M Li Macromolecules 33 232 (2000)21 X Jiang P Xia W Liu and D Yan J Polym Sci Part A Polym Chem 38 1203 (2000)22 G Schmidt-Naake and S Butz Macromol Rapid Commun 17 661 (1996)23 S Butz H Baethge and G Schmidt-Naake Macromol Chem Phys 201 2143 (2000)24 J Lokaj P Vlcek and J Kriz J Appl Polym Sci 74 2378 (1999)25 J Lokaj P Holler and J Kriz J Appl Polym Sci 76 1093 (2000)26 T E Chang K D Ahn and C G Cho Polym Prepr 41 1285 (2000)27 X Hao and K Fujimori Polym Prepr 40 476 (1999)28 W Mormann and K Schmaltz Macromolecules 27 7115 (1994)29 H Aida M Urushizaki H Maegawa and S Okazaki Kobunshi Ronbunshu 45 333 (1988)30 A Matsumoto T Kubota and T Otsu Macromolecules 23 4508 (1990)31 W Liu Y Liu C Chen Y Chen and F Xi Polym Degr Stabil 61 21 (1998)32 E G Fernandes I Giolito and E Chiellini Thermochim Acta 235 67 (1994)









![Comparative studies of three‐ and four‐ring mesogenic esters containing p‐carborane, bicyclo[2.2.2]octane, cyclohexane, and benzene](https://static.fdokumen.com/doc/165x107/631ec0884c5c8fb3a00e583c/comparative-studies-of-three-and-fourring-mesogenic-esters-containing-pcarborane.jpg)





![Ethene/norbornene copolymerization by [Me 2 Si(3- tert BuCp)(N tert Bu)]TiCl 2 /MAO-catalyst](https://static.fdokumen.com/doc/165x107/6312231a48b4e11f7d08cd0e/ethenenorbornene-copolymerization-by-me-2-si3-tert-bucpn-tert-buticl-2-mao-catalyst.jpg)




