
Short-term hypoxic vasodilation in vivo is mediated by bioactive nitric oxide metabolites, rather...
15
J Physiol 592.5 (2014) pp 1061–1075 1061 The Journal of Physiology Short-term hypoxic vasodilation in vivo is mediated by bioactive nitric oxide metabolites, rather than free nitric oxide derived from haemoglobin-mediated nitrite reduction Michele Umbrello 1,2 , Alex Dyson 1 , Bernardo Bollen Pinto 1 , Bernadette O. Fernandez 3 , Verena Simon 1,4 , Martin Feelisch 3 and Mervyn Singer 1 1 Bloomsbury Institute of Intensive Care Medicine, Division of Medicine, University College London, UK 2 Department of Pathophysiology & Transplantation, Universit` a degli Studi di Milano, Italy 3 University of Southampton School of Medicine, Southampton General Hospital, Tremona Road, Southampton, UK 4 Department of Anaesthesiology and Intensive Care, RWTH Aachen University, Aachen, Germany Key points Hypoxia increases blood flow through vasodilation, an adaptive response that increases oxygen availability to tissues. This response is probably mediated by nitric oxide (NO); different mechanisms have been postulated (increased de novo synthesis, release from preformed stores, reduced inactivation), but precise mechanisms remain controversial. In a short-term rodent model, hypoxaemia was associated with hypotension and lower plasma nitrite levels suggestive of nitrite bioactivation; this was only partially reversed by NO synthase inhibition. Administration of sodium nitrite produced marked vasodilation and increased nitrosylation. Scavenging of NO had little effect. Early hypoxic vasodilation is mediated by a complex interaction of multiple NO-related species, rather than by NO from haemoglobin-mediated reduction of nitrite per se. NO and its metabolites play a pivotal role in the body’s initial adaptation to an acute decrease in oxygen supply, probably offering protection against a supply–demand mismatch. Abstract Local increases in blood flow – ‘hypoxic vasodilation’ – confer cellular protection in the face of reduced oxygen delivery. The physiological relevance of this response is well established, yet ongoing controversy surrounds its underlying mechanisms. We sought to confirm that early hypoxic vasodilation is a nitric oxide (NO)-mediated phenomenon and to study putative pathways for increased levels of NO, namely production from NO synthases, intravascular nitrite reduction, release from preformed stores and reduced deactivation by cytochrome c oxidase. Experiments were performed on spontaneously breathing, anaesthetized, male Wistar rats under- going short-term systemic hypoxaemia, who received pharmacological inhibitors and activators of the various NO pathways. Arterial blood pressure, cardiac output, tissue oxygen tension and the circulating pool of NO metabolites (oxidation, nitrosation and nitrosylation products) were measured in plasma and erythrocytes. Hypoxaemia caused a rapid and sustained vasodilation, which was only partially reversed by non-selective NO synthase inhibition. This was associated with significantly lower plasma nitrite, and marginally elevated nitrate levels, suggestive of nitrite bioinactivation. Administration of sodium nitrite had little effect in normoxia, but produced significant vasodilation and increased nitrosylation during hypoxaemia that could not be reversed M. Umbrello and A. Dyson have contributed equally to this work. C 2014 The Authors. The Journal of Physiology C 2014 The Physiological Society DOI: 10.1113/jphysiol.2013.255687 ) at UCL Library Services on April 1, 2014 jp.physoc.org Downloaded from J Physiol (
Transcript of Short-term hypoxic vasodilation in vivo is mediated by bioactive nitric oxide metabolites, rather...

J Physiol 592.5 (2014) pp 1061–1075 1061
The
Jou
rnal
of
Phys
iolo
gy
Short-term hypoxic vasodilation in vivo is mediated bybioactive nitric oxide metabolites, rather than free nitricoxide derived from haemoglobin-mediated nitritereduction
Michele Umbrello1,2, Alex Dyson1, Bernardo Bollen Pinto1, Bernadette O. Fernandez3, Verena Simon1,4,Martin Feelisch3 and Mervyn Singer1
1Bloomsbury Institute of Intensive Care Medicine, Division of Medicine, University College London, UK2Department of Pathophysiology & Transplantation, Universita degli Studi di Milano, Italy3University of Southampton School of Medicine, Southampton General Hospital, Tremona Road, Southampton, UK4Department of Anaesthesiology and Intensive Care, RWTH Aachen University, Aachen, Germany
Key points
� Hypoxia increases blood flow through vasodilation, an adaptive response that increases oxygenavailability to tissues.
� This response is probably mediated by nitric oxide (NO); different mechanisms have beenpostulated (increased de novo synthesis, release from preformed stores, reduced inactivation),but precise mechanisms remain controversial.
� In a short-term rodent model, hypoxaemia was associated with hypotension and lower plasmanitrite levels suggestive of nitrite bioactivation; this was only partially reversed by NO synthaseinhibition. Administration of sodium nitrite produced marked vasodilation and increasednitrosylation. Scavenging of NO had little effect.
� Early hypoxic vasodilation is mediated by a complex interaction of multiple NO-related species,rather than by NO from haemoglobin-mediated reduction of nitrite per se.
� NO and its metabolites play a pivotal role in the body’s initial adaptation to an acute decreasein oxygen supply, probably offering protection against a supply–demand mismatch.
Abstract Local increases in blood flow – ‘hypoxic vasodilation’ – confer cellular protectionin the face of reduced oxygen delivery. The physiological relevance of this response is wellestablished, yet ongoing controversy surrounds its underlying mechanisms. We sought to confirmthat early hypoxic vasodilation is a nitric oxide (NO)-mediated phenomenon and to study putativepathways for increased levels of NO, namely production from NO synthases, intravascular nitritereduction, release from preformed stores and reduced deactivation by cytochrome c oxidase.Experiments were performed on spontaneously breathing, anaesthetized, male Wistar rats under-going short-term systemic hypoxaemia, who received pharmacological inhibitors and activatorsof the various NO pathways. Arterial blood pressure, cardiac output, tissue oxygen tension andthe circulating pool of NO metabolites (oxidation, nitrosation and nitrosylation products) weremeasured in plasma and erythrocytes. Hypoxaemia caused a rapid and sustained vasodilation,which was only partially reversed by non-selective NO synthase inhibition. This was associatedwith significantly lower plasma nitrite, and marginally elevated nitrate levels, suggestive of nitritebioinactivation. Administration of sodium nitrite had little effect in normoxia, but producedsignificant vasodilation and increased nitrosylation during hypoxaemia that could not be reversed
M. Umbrello and A. Dyson have contributed equally to this work.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society DOI: 10.1113/jphysiol.2013.255687
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1062 M. Umbrello and others J Physiol 592.5
by NO scavenging. Methodological issues prevented assessment of the contribution, if any, ofreduced deactivation of NO by cytochrome c oxidase. In conclusion, acute hypoxic vasodilationis an adaptive NO-mediated response conferred through bioactive metabolites rather than freeNO from haemoglobin-mediated reduction of nitrite.
(Received 24 March 2013; accepted after revision 29 December 2013; first published online 6 January 2014)Corresponding author M. Singer: Bloomsbury Institute of Intensive Care Medicine, University College London,Cruciform Building, Gower Street, London WC1E 6BT, UK. Email: [email protected]
Abbreviation ABP, arterial blood pressure; C-PTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide;NOS, nitric oxide synthase; POE-Hb, pyridoxylated haemoglobin poly-oxoethylene conjugate; RNNO, N-nitrosamines;RSNO, S-nitrosothiols; RXNO, sum of nitrosation products; SEITU, S-ethyl-isothiourea; sGC, soluble guanylatecyclase; SVR, systemic vascular resistance
Introduction
Mammals crucially require oxygen (O2) as the terminalelectron acceptor in the mitochondrial respiratory chainto provide sufficient energy for cell metabolism, organfunction and survival (Brand, 2005). Any seriousimbalance between O2 delivery and consumption can leadto a state of energetic crisis (Taylor & Pouyssegur, 2007).This is a critical factor underlying cell death (Brunelle &Chandel, 2002) and organ failure (Rixen & Siegel, 2005).
Several complementary mechanisms work in concert toprotect the tissues in the face of reduced O2 delivery. Theseinclude measures to facilitate O2 availability such as localincreases in blood flow – ‘hypoxic vasodilation’ (Roy &Brown, 1880; Dreyer, 1926; Hackel et al. 1954; Heistad& Wheeler, 1970; Rowell & Blackmon, 1987, 1989),with flow redistribution to ‘vital’ organs and increasedrecruitment of perfused microvessels (Tsai et al. 2003). Inparallel, additional strategies are employed that includedecreasing metabolic requirements to match energysupply and demand, upregulating glycolytic generation ofATP and modifying substrate availability and utilization,for example through the hormonal response to acute stress(Semenza, 2007).
Nitric oxide (NO) is a key determinant of myocardialfunction and vascular tone. It is essential for both globalregulation and regional distribution of blood flow andpressure (Welch & Loscalzo, 1994). NO also plays acrucial role in energy regulation and metabolism throughits modulatory effects on mitochondrial activity, andon protein function through nitrosylation, nitrosationor nitration (Clementi et al. 1999; Jobgen et al. 2006;Cooper & Giulivi, 2007). NO levels in blood or tissuesrepresent a balance between production/release andmetabolism/bioinactivation.
NO probably plays a key role in the body’s earlyadaptation to an acute energy supply–demand mismatch.However, considerable controversy persists with respectto its sources and mechanisms of action (Umbrello etal. 2013). Using a short-term rodent model of systemichypoxaemia, we sought to confirm that acute hypoxic
vasodilation is a NO-mediated phenomenon, and to studythe contribution of putative pathways known or suspectedto enhance NO bioactivity, namely increased productionfrom NO synthases (NOS), increased reduction of nitriteby red blood cells, increased release from preformedstorage forms of NO and reduced deactivation bymitochondrial cytochrome c oxidase.
Methods
Male Wistar rats of approximately 300 g body weight wereused in all experiments. Animals were purchased fromCharles River (Margate, Kent, UK) and housed in cagesof four on an alternating 12 h light–dark cycle, with freeaccess to chow (2018 rodent diet; Harlan Teklad, Madison,WI, USA) and tap water. Before instrumentation, animalswere allowed to acclimatize to the local environmentfor at least 1 week. All experiments were performedaccording to Home Office (UK) guidelines under the 1986Animal (Scientific Procedures) Act with University CollegeLondon Ethics Committee approval.
Instrumentation and monitoring
Spontaneously breathing animals were placed into a trans-parent plastic container and anaesthetized by 5% iso-flurane (Baxter Healthcare, Thetford, Norfolk, UK) inroom air. Following abolition of the righting reflex,animals were placed in the supine position on to a heatedmat to maintain rectal temperature at 37°C. Under 2–2.5%isoflurane, the left common carotid artery and right inter-nal jugular vein were located, isolated and cannulatedusing 0.96 mm outside diameter PVC tubing catheter(Biocorp Ltd, Huntingdale, WA, Australia). The arterialline was connected to a pressure transducer (Powerlab;AD Instruments, Chalgrove, Oxon, UK) for continuousmonitoring of arterial blood pressure (ABP). The venousline was used for subsequent administration of fluids anddrugs. A tracheostomy was sited using 2.08 mm externaldiameter polyethylene tubing (Portex Ltd, Hythe, Hants,
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1063
UK) to secure and suction the airways. This was connectedto a T-piece to maintain anaesthesia and to vary thefraction of inspired oxygen.
Continuous monitoring of muscle tissue PO2 (tPO2)levels was obtained by the use of Large Area Surface(LASTM) oxygen sensors (0.7 mm diameter) connected tothe OxyliteTM tissue monitoring system (Oxford Optronix,Abingdon, Oxon, UK), as described in detail elsewhere(Dyson et al. 2007). Briefly, the sensor sends shortpulses of light (475 nm) along a fibreoptic cable to aplatinum–complex fluorophore situated at the probe tip.This provides an 8 mm2 surface area in contact withthe tissue for tPO2 measurement. Upon interaction withoxygen, the fluorophore emits light (600 nm) back tothe detection unit, the lifetime of which is inverselyproportional to the local PO2 concentration withinthe tissue of interest. Unlike polarographic techniques,oxygen is not consumed during the measurement process;moreover, as the fluorescence decay is longer at lowervalues of PO2, highly accurate measurements can bemade in a hypoxic environment. A small incision wasmade at mid-thigh level, and the sensor inserted intothe vastus lateralis muscle to a depth of 10 mm usingan 18-gauge cannula for guidance (VenflonTM; BectonDickenson, Franklin Lakes, NJ, USA). The sensor was thenwithdrawn by 2 mm to prevent erroneous measurementsresulting from local haematoma. This was verified byseeing a rapid response in tPO2 following a change ininspired O2. ABP and muscle tPO2 measurements werecontinuously monitored and recorded on to a computerusing a Powerlab system and Chart 5.0 acquisition software(AD Instruments).
Isoflurane anaesthesia was reduced to 1.2% for theremainder of the experiment. Post-instrumentation, intra-vascular volume optimization was achieved by repeated0.5 ml intravenous challenges of 0.9% saline givenover 10 s every 5 min until ABP failed to increase>10%. A continuous infusion of 0.9% saline was thenadministered at a rate of 10 ml kg−1 h−1 for the durationof the experiment to ensure adequate filling conditionsthroughout. All animals were allowed to stabilize for atleast 30 min to achieve constant baseline physiologicalvariables before the beginning of the experiment.
At predefined time points, arterial blood samples(0.2 ml) were collected into heparinized capillary tubesfor blood gas analysis (ABL-625 analyser; Radiometer,Copenhagen, Denmark). Blood (0.5 ml) was also collectedinto tubes containing N-ethylmaleimide (10 mM) andEDTA (2.5 mM) to prevent artificial thiol nitrosation andto minimize transition metal-catalysed S-nitrosothiols(RSNO) degradation and trans-nitrosation reactions(Marley et al. 2000). Plasma and erythrocytes were thenobtained by centrifugation (800 g at 4°C for 10 min). Bothplasma and erythrocytes were immediately frozen in liquidnitrogen and stored at −80°C for subsequent analysis.
Transthoracic echocardiography was performed using aVivid 7 DimensionTM machine (GE Healthcare, Bedford,UK) with a 14 MHz probe recording at a depth of0–2 cm. Aortic blood flow velocities were determinedimmediately before the right carotid artery bifurcationusing pulsed-wave Doppler; the direction of blood flowwas confirmed by colour Doppler imaging. Stroke volumewas determined as the product of the velocity time integraland vessel cross-sectional area (� × [0.5 × diameter]2).Rats of this size have an aortic diameter of 0.26 cm (Slamaet al. 2003), thus a cross-sectional area of (0.13)2 × �was assumed for all animals studied. Heart rate (HR) wasdetermined by measuring the time between six consecutivecycles from the start of each Doppler trace to accountfor variation with respiration. Cardiac output (CO) wasdetermined as the product of stroke volume and HR.Systemic vascular resistance (SVR) was calculated as meanblood pressure divided by CO. The O2 content in thearterial blood and global O2 delivery were calculated usingstandard formulae.
Experimental protocols
Modulation of the various NO pathways was achievedthrough separate studies (Fig. 1). For each experimentalcondition, after recording of baseline haemodynamicvariables and blood sampling for NO metabolitedetermination (BL), animals were randomized to eitherbreathing room air (N) or a hypoxic mixture (H) at aninspired oxygen concentration (FiO2) of 0.125. They werethen allowed to stabilize for 10 min. Animals (six hypoxicand six normoxic per group) were then randomized intoone of the following groups:
i. sham, which only received the background infusion of0.9% saline;
ii. systemic vasoconstriction, using an intravenousinfusion of noradrenaline at a rate of 1.5 μg kg−1 min−1;
iii. NOS inhibition, where animals received a1.5 mg kg−1 min−1 infusion of the potent, non-selectiveNOS inhibitor S-ethyl-isothiourea (SEITU) (Southanet al. 1995);
iv. nitrite (NO2−) administration, where animals received
a 250 μg kg−1 min−1 infusion of sodium nitrite(NaNO2) (Badejo et al. 2010);
v. a combination of NOS inhibition with SEITU, andNaNO2 administration (doses as above);
vi. non-haem NO scavenging, using an infusion of thescavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (C-PTIO) (Yoshida et al.1994) running at 0.17 mg kg−1 min−1;
vii. haem-based NO scavenging, where animals received acontinuous infusion (50 mg kg−1 h−1) of pyridoxylatedhaemoglobin poly-oxoethylene (POE-Hb) (Kida et al.1995).
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1064 M. Umbrello and others J Physiol 592.5
To avoid any possible confounding effect, drugconcentrations were calculated so that all animals receivedat total of 10 ml kg−1 h−1 of intravenous fluid. At the endof each experiment (30 min), another aliquot of blood wasdrawn for the determination of NO metabolites. All drugsused were purchased from Sigma-Aldrich (Gillingham,
Dorset, UK) with the exception of noradrenaline (AbbottLabs, Maidenhead, Berks, UK). POE-Hb was kindlydonated by Apex Bioscience (Chapel Hill, NC, USA), anddissolved in 0.9% saline.
Drug doses were selected after extensive exploratorydose–response experiments (supporting information
Figure 1. Sites of intervention and study protocolA, pathways of NO production with identification of sites of action of inhibitors and activators used in thestudy. For details about the pathways, please refer to the text. The bolt denotes increase in the targetconcentration; the dashed line denotes a decrease, through enzymatic inhibition (SEITU) or conversion/directscavenging (C-PTIO and POE-Hb). B, study design showing doses and timings of pharmacological inhibitors andactivators. ABP, arterial blood pressure; BGA, arterial blood gas analysis; BL, baseline measurement; C-PTIO, organicNO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide; ECHO, echocardiography;L-NAME, non-selective NOS inhibitor N(G)-nitro-L-arginine methyl ester; N1/H1, measurement made 10 min afterrandomization to either hypoxia or normoxia; N2/H2, measurement made 20 min after the commencementof pharmacological inhibitors or activators; NaNO2, sodium nitrite; NO, nitric oxide; NO2
−, nitrite anion; NOS,nitric oxide synthase; POE-Hb, haem-based NO scavenger pyridoxylated haemoglobin poly-oxoethylene; RSNO,products of the nitrosation of critical thiol residues; SEITU, non-selective NOS inhibitor S-ethyl-isothiourea; sGC,soluble guanylate cyclase; tPO2, tissue (muscle) PO2.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1065
Fig. S1). We chose a dose of SEITU for subsequentstudy that gave an approximate 40 mmHg rise in ABPin normoxic rats. Notably, no further increases in bloodpressure were observed above this dose in either normoxicor hypoxic animals. We also attempted to modulate NOmetabolism by altering the redox state of cytochromec oxidase through administration of either hydrogensulphide (given as NaHS) or sodium azide (both 10−8
to 10−2 g kg−1 min−1). Unfortunately, major side effects(severe haemodynamic instability leading to prematuredeath) precluded in vivo study of this pathway.
Figure 1B depicts the experimental schedule. Bloodpressure and muscle tissue oxygen tension were monitoredcontinuously. Echocardiography and blood gas analysiswas performed at baseline, 10 and 30 min withsampling for plasma and erythrocyte levels of NO-relatedcompounds (see below) at baseline and 30 min. In selectedexperiments, additional echocardiography and blood gasanalysis was performed at 20 min before administrationof NaNO2.
After analysis of the results, we performed two setsof additional experiments to understand better themechanisms underlying our findings. The hypotheses weset out to test were: (i) whether the vasodilation seenunder hypoxia was caused by increased levels of NO perse or if a NO-related compound could play a role, and(ii) similarly, whether the vasodilating effect of nitritein hypoxia was caused by increased levels of NO perse or if a NO-related compound could play a role. Wethus treated two groups of hypoxic animals (n = 6 each)with a combination of (i) NOS inhibition followed by acontinuous infusion of POE-Hb, and (ii) NOS inhibitionand NaNO2 administration followed by a continuousinfusion of POE-Hb; all drugs were used at the same dosesas previously described. To avoid POE-Hb oxidation, thelatter experiment required separate venous access for theadministration of both drugs: in this group, SEITU andNaNO2 were administered via the jugular line. The leftfemoral vein was also cannulated for administration ofPOE-Hb.
Biochemical analyses
Frozen sample aliquots were thawed just before use.Plasma was used undiluted while the erythrocytepellet was subjected to hypotonic lysis in coldaqueous N-ethylmaleimide/EDTA (10/2.5 mM; 1:4 v/v)immediately before analysis. The concentration of nitrosocompounds was assessed using group-specific reductivedenitrosation by iodine-iodide in glacial acetic acid,with subsequent detection of liberated NO into thegas phase by its chemiluminescent reaction with ozone(Feelisch et al. 2002). ‘RSNO’ signifies mercury-labileS-nitrosated species (including nitrosothiols), whereas
‘RNNO’ signifies mercury-resistant nitroso adducts andmay include N-nitrosamines and metal nitrosyls otherthan NO-haem species. ‘RXNO’ represents the totalpool of nitrosated species (i.e. the sum of RSNOs andRNNOs). NO-haem species in erythrocytes (nitrosylhaemoglobin) were determined by parallel injection ofreplicate aliquots of blood homogenates into a solutionof 0.05 M ferricyanide in phosphate-buffered saline at pH7.5 and 37°C (Bryan et al. 2004). This method employsone-electron oxidation rather than reduction to achievedenitrosation, with the liberated NO being quantified bygas-phase chemiluminescence (CLD 77 AM; Eco Physics,Ann Arbor, MI, USA). Nitrate and nitrite were quantifiedby ion chromatography with on-line reduction of nitrateto nitrite and post-column Griess diazotization (ENO20Analyser; Eicom, Kyoto, Japan) (Rassaf et al. 2002).
Statistical analysis
All data are presented as means ± S.E.M. (n = 6 per group)unless otherwise specified. Statistics were performed onraw data using a repeated-measures two-way ANOVAfollowed by Tukey’s post hoc test (SigmaStat 11.0; SystatSoftware Inc., San Jose, CA, USA) to compare multiplegroups at multiple time points or unpaired Student’s t testto compare two groups. For all comparisons, P < 0.05 wasconsidered statistically significant.
Results
Model characterization and effects of hypoxia
Hypoxia (12.5% O2) significantly reduced arterial PO2
and haemoglobin saturation (Table 1). Muscle tPO2 levelshalved. Despite an unchanged respiratory rate, hypoxiawas associated with reduced PaCO2, but similar arterialpH. Though tidal volumes were not formally measured,there was an obvious increase in chest wall excursion.
Blood pressure fell significantly during hypoxia, and thiswas maintained over time despite no significant change ineither stroke volume or HR (Fig. 2; Fig. S1). Consequently,SVR was significantly lower in the hypoxic group. Globaloxygen delivery was reduced by half, and blood lactatelevels increased significantly (Table 1).
Noradrenaline, titrated to increase mean ABP by40 mmHg above normoxic levels, failed to reach thistarget under hypoxia (Fig. 2 and Table 1). Whereas thisagent increased SVR in the normoxic group, it had littleeffect during hypoxia. HR was significantly increased bynoradrenaline in both groups to the same extent. Oxygendelivery increased in both groups, though failed to reachbaseline levels in the hypoxia group. Lactate levels werelower than during hypoxia alone, and similar to thatmeasured in normoxic sham animals.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1066 M. Umbrello and others J Physiol 592.5
Tab
le1.
Hae
mo
dyn
amic
,res
pir
ato
ryan
dm
etab
olic
dat
a
SHA
MN
EPI
SEIT
UN
aNO
2SE
ITU
+N
aNO
2C
-PTI
OPO
E-H
b
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
a
sAB
P(m
mH
g)
117
±6
79±
9∗17
1±
11¶
104
±12
∗‡16
9±
11†
145
±13
∗‡99
±7†
64±
15∗
140
±23
88±
28∗
137
±28
92±
8∗‡
119
±14
101
±13
‡
dA
BP
(mm
Hg
)67
±1
42±
4∗11
5±
17†
59±
16∗‡
122
±9†
108
±19
‡63
±12
43±
11∗
109
±17
†63
±23
∗‡10
1±
15†
63±
11∗‡
96±
12†
71±
13∗‡
HR
(1m
in–1
)39
6±
3935
8±
4545
9±
9†45
8±
27‡
370
±39
335
±39
397
±48
350
±29
373
±50
361
±24
474
±47
†37
7±
100
431
±23
388
±45
VPe
ak(m
s–1)
0.94
±0.
070.
89±
0.05
0.72
±0.
02†
0.89
±0.
120.
31±
0.01
†0.
46±
0.09
∗‡0.
83±
0.11
†1
±0.
07∗
0.34
±0.
03†
0.43
±0.
09‡
0.93
±0.
171.
07±
0.08
†0.
83±
0.16
0.74
±0.
08
SV(m
l)0.
26±
0.02
0.29
±0.
030.
3±
0.05
0.23
±0.
02∗‡
0.1
±0.
01†
0.16
±0.
02∗‡
0.25
±0.
020.
32±
0.02
∗0.
1±
0.01
†0.
16±
0.01
∗‡0.
25±
0.05
0.32
±0.
020.
23±
0.04
0.22
±0.
03
PaO
2(k
Pa)
10.8
±0.
65.
6±
0.7∗
9.6
±1
4.5
±0.
3∗‡
11.4
±2.
46.
5±
1.6∗
11.4
±0.
36.
1±
0.8∗
12.8
±2.
35.
6±
0.8∗
10.4
±0.
74.
5±
0.6∗
‡10
.1±
0.3
5.2
±0.
5∗
PaC
O2
(kPa
)5.
5±
0.7
4.2
±0.
4∗4.
8±
0.5
3.5
±0.
4∗‡
4.7
±1.
24.
6±
0.4
5.3
±0.
64.
2±
0.8∗
3.6
±1†
3.9
±0.
54.
7±
0.4
4.7
±0.
84.
6±
0.4
4.4
±0.
8
pH
7.43
±0.
057.
47±
0.04
7.47
±0.
047.
58±
0.04
∗‡7.
46±
0.8
7.45
±0.
057.
44±
0.05
7.47
±0.
077.
53±
0.09
†7.
49±
0.05
7.47
±0.
027.
47±
0.8
7.48
±0.
017.
49±
0.08
SaO
2(%
)94
.7±
1.9
66.4
±9.
7∗94
.6±
4.6
62.5
±2.
8∗94
.2±
4.4
68.9
±19
.5∗
97.1
±2.
370
.6±
11.6
∗97
.1±
2.2
67.8
±14
.0∗
96.3
±0.
852
.4±
14.2
∗95
.1±
1.8
63.2
±5.
5∗
Hb
(g10
0m
l–1)
12.9
±1.
112
.1±
0.4
15.0
±1.
0†15
.0±
1.1‡
13.8
±0.
814
.1±
1.2‡
12.5
±1.
011
.8±
0.4
13.1
±1.
213
.4±
1.3‡
13.1
±0.
912
.8±
0.6
13.4
±0.
413
.5±
0.7‡
RR
(1m
in–1
)78
±8
66±
1087
±15
86±
1672
±25
68±
2080
±3
59±
9∗11
6±
2966
±13
∗65
±14
68±
1789
±1
76±
31
tPO
2(k
Pa)
5.8
±0.
73.
3±
0.5∗
6±
1.4
2.1
±0.
2∗‡
5.8
±1.
42.
7±
0.9∗
6±
0.6
3.3
±0.
6∗4.
9±
1.9
2.4
±0.
7∗‡
7.4
±1.
4†2.
9±
0.8∗
6.9
±0.
5†3
±0.
9∗
CaO
2(m
l100
ml–1
)17
.2±
1.4
11.3
±1.
5∗19
.7±
1.4†
13.1
±0.
9∗18
.4±
1.7
13.6
±4.
3∗17
.1±
1.3
11.7
±1.
7∗18
±1.
712
.7±
2.9∗
17.8
±1
9.4
±2.
2∗18
±0.
712
±0.
5∗
DO
2(m
lmin
–1)
16.9
±2.
49.
8±
1.6∗
22.4
±8.
613
.5±
2.5∗
‡6.
5±
0.8†
6.2
±3.
116
.6±
2.6
13.3
±2.
47.
6±
1†7.
8±
1.6
20±
29.
5±
2∗17
.1±
3.7
9.5
±0.
9∗
Lac
(mM
)1.
7±
0.5
2.9
±0.
8∗1.
2±
0.3
1.8
±0.
3‡2
±0.
84.
2±
1∗‡
1.3
±0.
33.
7±
1.8∗
2.5
±1.
14
±2.
22
±0.
83.
9±
0.5
1.6
±0.
42.
3±
0.7
CaO
2,a
rter
ialO
2co
nte
nt;
C-P
TIO
,NO
scav
eng
er2-
(4-c
arb
oxy
ph
enyl
)-4,
4,5,
5-te
tram
eth
yl-i
mid
azo
line-
1-o
xyl-
3-o
xid
e;d
AB
P,d
iast
olic
AB
P;D
O2,O
2d
eliv
ery
toti
ssu
es;H
b,h
aem
og
lob
in;H
R,h
eart
rate
;Lac
,lac
tate
;NaN
O2,s
od
ium
nit
rite
;
NEP
I,n
ora
dre
nal
ine;
NO
,n
itri
co
xid
e;Pa
CO
2,
arte
rial
par
tial
pre
ssu
reo
fC
O2;
PaO
2,
arte
rial
par
tial
pre
ssu
reo
fO
2;
POE-
Hb
,h
aem
NO
scav
eng
erp
yrid
oxy
late
dh
aem
og
lob
inp
oly
-oxo
eth
ylen
e;R
R,
resp
irat
ory
rate
;sA
BP,
syst
olic
AB
P;
SaO
2,a
rter
ialh
aem
og
lob
insa
tura
tio
n;S
EITU
,no
n-s
elec
tive
NO
syn
thas
ein
hib
ito
rS-
eth
yl-i
soth
iou
rea;
SV,s
tro
kevo
lum
e;tP
O2,t
issu
e(m
usc
le)
par
tial
pre
ssu
reo
fO
2;V
Peak
,blo
od
flo
wp
eak
velo
city
.∗P
<0.
05vs
.no
rmo
xia;
† P<
0.05
vs.
no
rmo
xia
sham
;‡P
<0.
05vs
.hyp
oxi
ash
am.n
=6
anim
als
per
gro
up
.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (
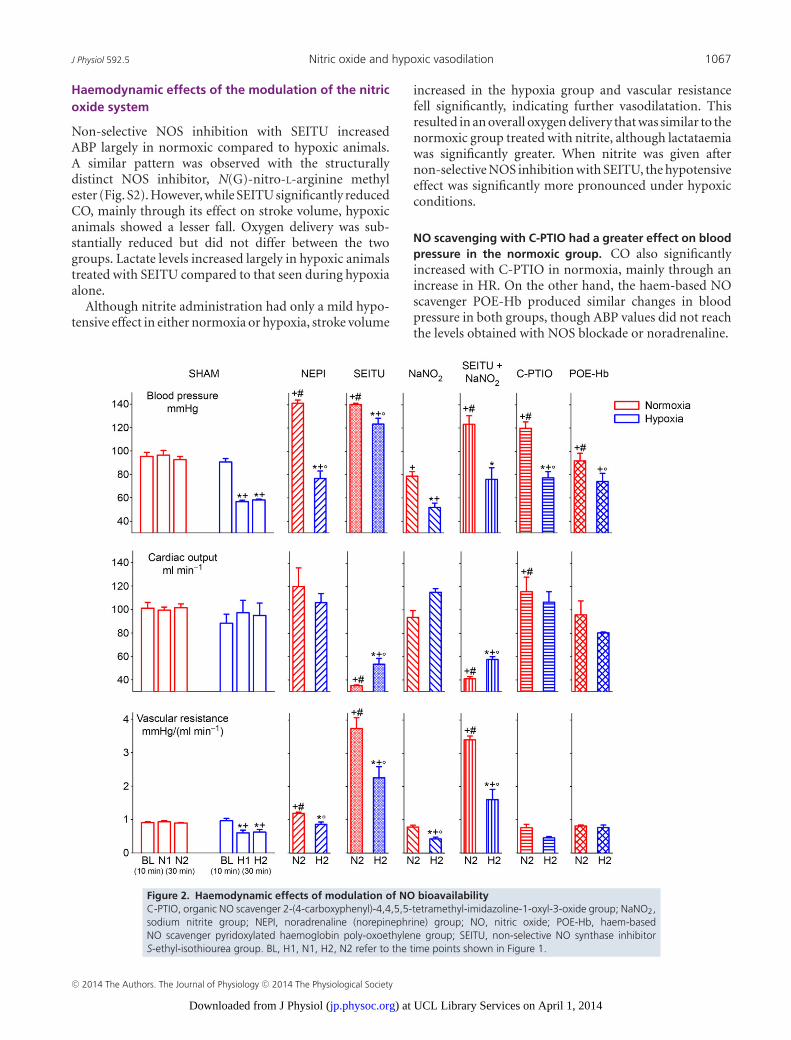
J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1067
Haemodynamic effects of the modulation of the nitricoxide system
Non-selective NOS inhibition with SEITU increasedABP largely in normoxic compared to hypoxic animals.A similar pattern was observed with the structurallydistinct NOS inhibitor, N(G)-nitro-L-arginine methylester (Fig. S2). However, while SEITU significantly reducedCO, mainly through its effect on stroke volume, hypoxicanimals showed a lesser fall. Oxygen delivery was sub-stantially reduced but did not differ between the twogroups. Lactate levels increased largely in hypoxic animalstreated with SEITU compared to that seen during hypoxiaalone.
Although nitrite administration had only a mild hypo-tensive effect in either normoxia or hypoxia, stroke volume
increased in the hypoxia group and vascular resistancefell significantly, indicating further vasodilatation. Thisresulted in an overall oxygen delivery that was similar to thenormoxic group treated with nitrite, although lactataemiawas significantly greater. When nitrite was given afternon-selective NOS inhibition with SEITU, the hypotensiveeffect was significantly more pronounced under hypoxicconditions.
NO scavenging with C-PTIO had a greater effect on bloodpressure in the normoxic group. CO also significantlyincreased with C-PTIO in normoxia, mainly through anincrease in HR. On the other hand, the haem-based NOscavenger POE-Hb produced similar changes in bloodpressure in both groups, though ABP values did not reachthe levels obtained with NOS blockade or noradrenaline.
Figure 2. Haemodynamic effects of modulation of NO bioavailabilityC-PTIO, organic NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide group; NaNO2,sodium nitrite group; NEPI, noradrenaline (norepinephrine) group; NO, nitric oxide; POE-Hb, haem-basedNO scavenger pyridoxylated haemoglobin poly-oxoethylene group; SEITU, non-selective NO synthase inhibitorS-ethyl-isothiourea group. BL, H1, N1, H2, N2 refer to the time points shown in Figure 1.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (
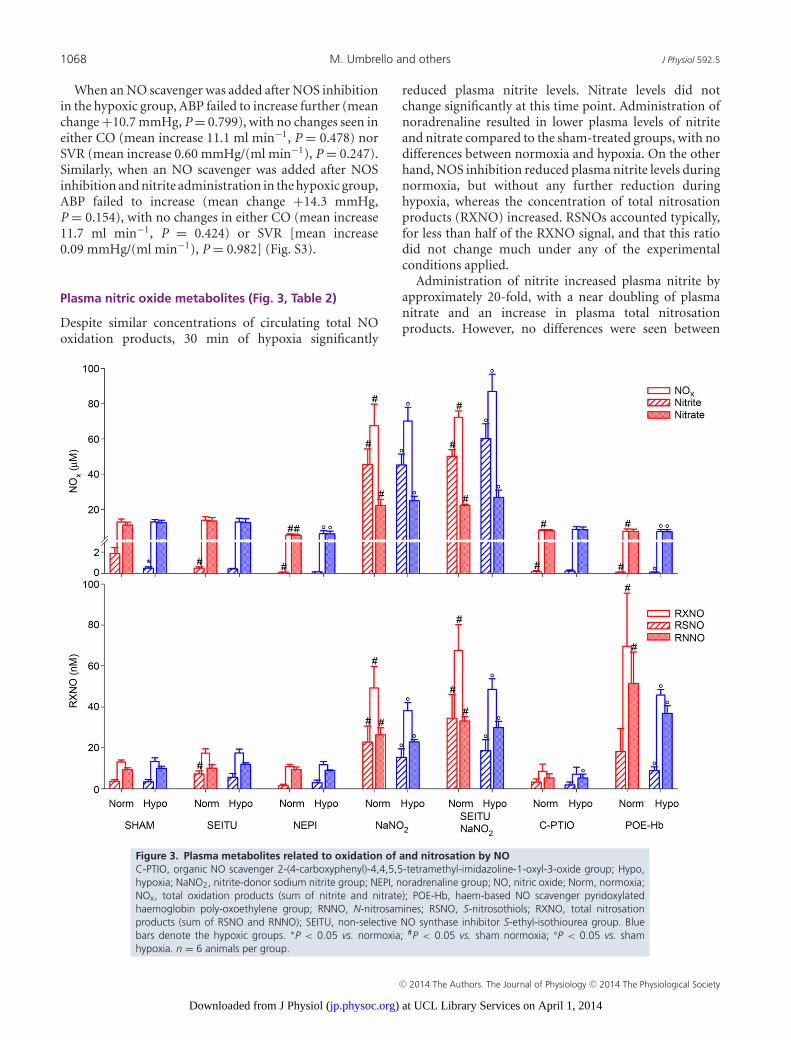
1068 M. Umbrello and others J Physiol 592.5
When an NO scavenger was added after NOS inhibitionin the hypoxic group, ABP failed to increase further (meanchange +10.7 mmHg, P = 0.799), with no changes seen ineither CO (mean increase 11.1 ml min−1, P = 0.478) norSVR (mean increase 0.60 mmHg/(ml min−1), P = 0.247).Similarly, when an NO scavenger was added after NOSinhibition and nitrite administration in the hypoxic group,ABP failed to increase (mean change +14.3 mmHg,P = 0.154), with no changes in either CO (mean increase11.7 ml min−1, P = 0.424) or SVR [mean increase0.09 mmHg/(ml min−1), P = 0.982] (Fig. S3).
Plasma nitric oxide metabolites (Fig. 3, Table 2)
Despite similar concentrations of circulating total NOoxidation products, 30 min of hypoxia significantly
reduced plasma nitrite levels. Nitrate levels did notchange significantly at this time point. Administration ofnoradrenaline resulted in lower plasma levels of nitriteand nitrate compared to the sham-treated groups, with nodifferences between normoxia and hypoxia. On the otherhand, NOS inhibition reduced plasma nitrite levels duringnormoxia, but without any further reduction duringhypoxia, whereas the concentration of total nitrosationproducts (RXNO) increased. RSNOs accounted typically,for less than half of the RXNO signal, and that this ratiodid not change much under any of the experimentalconditions applied.
Administration of nitrite increased plasma nitrite byapproximately 20-fold, with a near doubling of plasmanitrate and an increase in plasma total nitrosationproducts. However, no differences were seen between
Figure 3. Plasma metabolites related to oxidation of and nitrosation by NOC-PTIO, organic NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide group; Hypo,hypoxia; NaNO2, nitrite-donor sodium nitrite group; NEPI, noradrenaline group; NO, nitric oxide; Norm, normoxia;NOx, total oxidation products (sum of nitrite and nitrate); POE-Hb, haem-based NO scavenger pyridoxylatedhaemoglobin poly-oxoethylene group; RNNO, N-nitrosamines; RSNO, S-nitrosothiols; RXNO, total nitrosationproducts (sum of RSNO and RNNO); SEITU, non-selective NO synthase inhibitor S-ethyl-isothiourea group. Bluebars denote the hypoxic groups. ∗P < 0.05 vs. normoxia; #P < 0.05 vs. sham normoxia; °P < 0.05 vs. shamhypoxia. n = 6 animals per group.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1069
Tab
le2.
Rel
ativ
eco
mp
osi
tio
no
fo
xid
ativ
ean
dn
itro
sati
veN
Om
etab
olit
esin
pla
sma
and
eryt
hro
cyte
s
SHA
MN
EPI
SEIT
UN
aNO
2SE
ITU
+N
aNO
2C
-PTI
OPO
E-H
b
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
aN
orm
oxi
aH
ypo
xia
No
rmo
xia
Hyp
oxi
a
PLA
SMA
NO
2–/N
Ox
0.15
±0.
100.
05±
0.04
∗0.
02±
0.01
†0.
03±
0.02
0.04
±0.
02†
0.03
±0.
010.
66±
0.04
†0.
63±
0.06
‡0.
69±
0.05
†0.
67±
0.08
‡0.
02±
0.01
†0.
02±
0.02
0.02
±0.
01†
0.02
±0.
01
RSN
O/R
XN
O0.
28±
0.10
0.22
±0.
140.
14±
0.15
0.22
±0.
150.
41±
0.15
0.29
±0.
170.
40±
0.17
0.37
±0.
160.
44±
0.22
0.36
±0.
170.
34±
0.18
0.18
±0.
120.
22±
0.14
0.20
±0.
09
ERY
THR
OC
YTE
S
NO
2–/N
Ox
0.05
±0.
030.
07±
0.03
0.02
±0.
020.
01±
0.01
‡0.
05±
0.04
0.04
±0.
05‡
0.02
±0.
010.
02±
0.02
‡0.
03±
0.02
0.02
±0.
01‡
0.03
±0.
020.
03±
0.03
0.01
±0.
010.
02±
0.01
‡
RSN
O/R
SNO
0.15
±0.
100.
24±
0.10
0.22
±0.
160.
38±
0.14
0.40
±0.
25†
0.30
±0.
100.
27±
0.28
0.23
±0.
150.
38±
0.23
†0.
23±
0.16
0.31
±0.
230.
25±
0.10
0.30
±0.
09†
0.32
±0.
29
C-P
TIO
,N
Osc
aven
ger
2-(4
-car
bo
xyp
hen
yl)-
4,4,
5,5-
tetr
amet
hyl
-im
idaz
olin
e-1-
oxy
l-3-
oxi
de;
NaN
O2,
sod
ium
nit
rite
;N
EPI,
no
rad
ren
alin
e;N
O,
nit
ric
oxi
de;
NO
2–,
nit
rite
;N
Ox,
tota
lo
xid
atio
n(n
itri
te+
nit
rate
)sp
ecie
s;PO
E-H
b,
hae
mN
O
scav
eng
erp
yrid
oxy
late
dh
aem
og
lob
inp
oly
-oxo
eth
ylen
e;R
SNO
,S-n
itro
soth
iols
;R
XN
O,t
ota
ln
itro
sati
on
spec
ies
(S-n
itro
soth
iols
+N
-nit
rosa
min
es);
SEIT
U,n
on
-sel
ecti
veN
Osy
nth
ase
inh
ibit
or
S-et
hyl
-iso
thio
ure
a.∗ P
<0.
05vs
.n
orm
oxi
a;† P
<0.
05vs
.no
rmo
xia
sham
;‡P
<0.
05vs
.hyp
oxi
ash
am.n
=6
anim
als
per
gro
up
.
hypoxic and normoxic groups. Addition of NOS inhibitionto nitrite did not change the plasma NO metabolite profile.
Scavenging NO with C-PTIO reduced plasma nitritelevels to virtually zero, whereas nitrate levels wereconserved. Plasma nitrosation products were also reduced,though no differences were seen between hypoxia andnormoxia. Utilization of POE-Hb as the NO scavengerwas associated with a similar profile of plasma oxidationproducts. However, in stark contrast to C-PTIO, plasmanitrosation products increased compared to sham in bothnormoxic and hypoxic animals. Table 2 shows the relativecontribution of nitrite to total oxidation products, andthat of RSNO to total nitrosation products.
Erythrocyte nitric oxide metabolites (Fig. 4, Table 2)and nitrosyl haemoglobin (Fig. 5)
In sham animals, erythrocyte nitrite and nitrate levelswere conserved during hypoxia, with no differences seenin nitrosation products. Noradrenaline reduced intra-cellular nitrite levels in normoxic animals but RSNO andRNNO remained largely unchanged. NOS inhibition bySEITU had no impact upon erythrocyte NO oxidationand nitrosation products, and no differences were seenbetween hypoxia and normoxia states.
Notably, nitrite administration did not modify intra-erythrocytic nitrite levels. However, nitrate concentrationswere higher than in sham animals, particularly underhypoxic conditions. Nitrosyl haemoglobin levels werealso higher during hypoxia. Similar results were seenwhen nitrite was co-administered with the NOS inhibitor,SEITU. NO scavenging was associated with lower intra-cellular nitrite levels, yet most other NO metabolites wereunchanged. Of note, the NO metabolite profile observedin the presence of C-PTIO was different from that of atypical NO scavenger; besides the expected reductions inblood nitrite concentrations under normoxic and hypo-xic conditions due to oxygen-independent conversion ofNO to NO2, hypoxia was associated with increases innitrosation products other than RSNOs and increasesin nitrosyl haemoglobin. This suggests preferentialformation of N-nitrosated species in red blood cells thatact as potential NO donors with this particular compound.Thus, care should be taken when C-PTIO is used in in vivostudies, emphasizing the need to compare mechanisticallydistinct NO scavengers before reaching conclusions aboutthe possible involvement of NO in a given physiologicalprocess.
Discussion
Several complementary mechanisms protect the body’stissues in the face of a reduced O2 delivery. These includeredistribution of blood flow to ‘vital’ organs, and increased
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1070 M. Umbrello and others J Physiol 592.5
recruitment of perfused microvessels to facilitate O2
availability (Tsai et al. 2003). At the systemic level, the mostevident adaptation to hypoxaemia involves an increase inblood flow – ‘hypoxic vasodilation’ (Roy & Brown, 1880;Dreyer, 1926; Hackel et al. 1954; Heistad & Wheeler, 1970;Rowell & Blackmon, 1987, 1989). This aims to compensatefor the reduction in arterial O2 content by attempting tomaintain an acceptable O2 supply–demand balance. Thesympathetic nervous system is activated with increasedcatecholamine levels, albeit with a reduced response toexogenous vasopressors (vascular hyporeactivity).
Although the physiological relevance of hypoxic vaso-dilation is well established, ongoing controversy surroundsits underlying mechanisms. An increase in NO is stronglyimplicated. This key signalling and effector moleculeis a major determinant of vascular tone, an important
modulator of myocardial function, and essential for bothglobal regulation and regional distribution of blood flowand pressure (Welch & Loscalzo, 1994).
NO levels in blood and tissues represent abalance between production/release and metabolism/bioinactivation. Its best-recognized biosynthetic route isthe L-arginine–NO pathway (Moncada & Higgs, 1993;Boucher et al. 1999). However, alternative pathways areproposed, involving (i) nitrite and nitrate, oxidative endproducts traditionally considered to be inert (Modin etal. 2001), and (ii) RSNO, the reaction products betweenreactive NO species and thiol residues (Diesen et al. 2008).
Particularly under conditions of hypoxia, proteins fromthe haem-globin family (Gladwin & Kim-Shapiro, 2008)or from pteryn-based molybdenum enzymes (Li et al.2008) may catalyse the reduction of nitrite to NO, beyond
Figure 4. Erythrocytic metabolites related to oxidation of and nitrosation by NOC-PTIO, organic NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide group; Hypo,hypoxia; NaNO2, nitrite-donor sodium nitrite group; NEPI, noradrenaline group; NO, nitric oxide; Norm, normoxia;NOx, total oxidation products (sum of nitrite and nitrate); POE-Hb, haem-based NO scavenger pyridoxylatedhaemoglobin poly-oxoethylene group; RNNO, N-nitrosamines; RSNO, S-nitrosothiols; RXNO, total nitrosationproducts (sum of RSNO and RNNO); SEITU, non-selective NO synthase inhibitor S-ethyl-isothiourea group. Dashedbars denote the hypoxic groups. ∗P < 0.05 vs. normoxia; #P < 0.05 vs. sham normoxia; °P < 0.05 vs. shamhypoxia. n = 6 animals per group
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1071
their conventional physiological functions. Release of NOfrom RSNO storage pools may also be enhanced underlow oxygen conditions (Jia et al. 1996). These pathwaysmay provide important alternative sources of NO, whereincreased blood flow may be beneficial, such as duringhypoxia, particularly recognizing the critical requirementfor O2 as a cofactor for NOS. However, to date, manyof these mechanisms have only been demonstrated innon-physiological conditions, either in vitro or ex vivo,or indirectly implicated by utilization of pharmacologicaltools. Thus, an open question remains as to their in vivo(patho)physiological relevance.
The in vivo fate of NO is also highly complex; severalcatabolic pathways exist with varying relevance in differentbody compartments. The reaction with haemoglobin toform NO3
− constitutes the major catabolic pathway.However, in plasma in the presence of O2, the principalreaction is the formation of NO2
− (Ignarro et al.1993) occurring through oxidation by mitochondrialcytochrome c oxidase (Shiva et al. 2001; Pearce et al.2002). An alternative theory for the hypoxia-inducedincrease in NO is reduced elimination. Using an invitro model, Moncada and colleagues demonstrated thatcytochrome c oxidase in its oxidized state constantlyinactivates NO, contributing to its intracellular regulation(Palacios-Callender et al. 2007; Unitt et al. 2010). However,in the reduced state typical of hypoxia, an impairedinactivation would account for the witnessed increase inNO.
NO acts at multiple sites within the body(Martinez-Ruiz et al. 2011). It promotes vasorelaxationeither via activation of soluble guanylate cyclase (sGC) andsubsequent generation of cyclic GMP (cGMP), or throughS-nitrosation of critical thiols sited on ion channels. NOalso inhibits the mitochondrial electron transport chainthrough direct competition with oxygen at Complex IV(cytochrome c oxidase), and via nitrosation/nitration of
other respiratory chain complexes (Frost et al. 2005),ultimately causing a decrease in O2 consumption.
As the vasodilating (Movahed et al. 2003) and metabolic(Frost et al. 2005) effects of NO are both enhanced duringhypoxia, it probably plays a major role in re-equilibratingany O2 supply–demand mismatch (Umbrello et al. 2013).Hypoxia-induced vasorelaxation may be reversed byscavenging NO (Pohl & Busse, 1989), or inhibiting sGCwith methylene blue (Iwamoto et al. 1992). Whereasincreased NO synthesis by NOS may be a potential causeof hypoxic vasodilation (Edmunds & Marshall, 2001;Hunter et al. 2003), others report an ineffectiveness ofNOS inhibition on decreased hindlimb resistance (Valletet al. 1994) and dilation of small coronary arteries (Liu &Flavahan, 1997) induced by hypoxia.
We set out to study mechanisms underlying hypoxicvasodilation in vivo using a well-established short-termrodent model of systemic hypoxia (Dyson et al. 2007,2009). We chose FiO2 at 0.125 as this concentration wasassociated with the largest fall in PaO2 that did not carryshort-term lethality (Dyson et al. 2007). Our findingssupport the role of NO as a key mediator of hypoxic vaso-dilation.
Hypoxaemia caused an immediate and sustainedhypotension associated with muscle tissue hypoxia andincreased glycolytic flux. Vasopressor hyporesponsivenesswas confirmed by the limited increase in ABP withnoradrenaline. Notably, hypoxia reduced ABP and SVRbut had little effect on CO: this may be explained bythe balanced effect of an increased perfusion due tovasodilation and some degree of hypoxic myocardialdepression, possibly NO-mediated (Brady et al. 1993;Mohan et al. 1996).
Non-selective NOS inhibition with SEITU reversedhypoxic vasodilation but failed to achieve ABP valuesobtained from normoxic, SEITU-treated animals; weobserved a similar pattern using the non-selective NOS
Figure 5. Haem nitrosylation as marker ofNO availability in erythrocytesC-PTIO, organic NO scavenger2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide group; NaNO2,nitrite-donor sodium nitrite group; NEPI,noradrenaline group; NO, nitric oxide; NO-Hb,nitrosyl haemoglobin; POE-Hb, haem-basedNO scavenger pyridoxylated haemoglobinpoly-oxoethylene group; SEITU, non-selectiveNO synthase inhibitor S-ethyl-isothioureagroup. ∗P < 0.05 vs. normoxia; #P < 0.05 vs.sham normoxia; °P < 0.05 vs. sham hypoxia.n = 6 animals per group
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1072 M. Umbrello and others J Physiol 592.5
inhibitor N(G)-nitro-L-arginine methyl ester and infer thedifference in blood pressure between normoxic and hypo-xic rats is due to NO not generated by NOS but derivedfrom other pathways (nitrite reduction or release frompreformed nitroso pools). Hypoxia was associated withsignificantly lower plasma levels of nitrite, supportingthe concept of increased NO production originatingfrom nitrite reduction. When NOS was blocked bySEITU, plasma nitrite was lowered even under normoxicconditions. Most probably, this change is unrelated tonitrite reduction but a direct consequence of reducedNO production from NOS. Administration of nitritecaused little hypotensive effect in normoxia, and onlysmall reductions of ABP under hypoxia. However, strokevolume and CO, and thus SVR, were significantlyhigher in the hypoxic group, suggesting a stronger vaso-dilating effect of nitrite administration under hypo-xia. The failure of blood pressure to fall any furthermay be explained by being already low due to hypo-xia alone. However, nitrite administration significantlyincreased nitrosyl haemoglobin generation during hypo-xia, consistent with higher levels of NO production. Whengiven after non-selective NOS blockade with SEITU, nitritecaused a significantly greater hypotensive effect underconditions of hypoxia, accompanied by a further increasein nitrosyl haemoglobin levels.
Experiments with NO scavengers could distinguishwhether the vasodilating factor acting in hypoxia was dueto intravascular generation of NO per se or one or moreNO-related compound(s). Both C-PTIO and POE-Hb arespecific extracellular NO scavengers and both increasedABP irrespective of the presence of hypoxia. A possibleexplanation is that NO produced by endothelial cellsexerts its vasorelaxing activity by a paracrine effect onsurrounding vascular smooth cells, with no transit intothe bloodstream. However, the changes in the circulatingpool of NO oxidation and nitration products during NOscavenging suggest that a portion of the NO generatedin vascular tissue is transferred to the bloodstream.Alternatively, NO does not exert its vasodilatory actiondirectly, but rather through the action of other down-stream mediators. A study performed under physiologicalconditions found that nitrite was not directly reducedto NO but instead modulated several different signallingpathways (Bryan et al. 2005). In addition to sGC activation,post-translational modifications typically associated withNO occur, such as the formation of nitrosated, nitrosylatedand nitrated species (Bryan et al. 2005; Perlman et al.2009). Thus, nitrite may exert its signalling functionsdirectly, without the need for intermediary formationof free NO. Hypoxia markedly potentiates tissue NOproduction from nitrite in a dose-dependent manner(Feelisch et al. 2008). This occurs particularly inheart, liver and vascular tissue, with multiple haem,iron–sulphur cluster and molybdenum-based reductases
distributed among distinct subcellular compartmentsacting in a multifactorial and cooperative mannerto catalyse the reaction. Acute hypoxia also reducednitrite concentrations yet simultaneously enhanced theformation of NO metabolites such as RSNOs andRNNOs in an NO-independent manner, again consistentwith a pathway that generates bioactive NO metaboliteswithout the intermediacy of free NO. In this paradigm,conversion of nitrite to NO and the storage of NObioactivity as RSNOs may both be constituents of a morecomplex regulatory mechanism of interaction of multipleNO-related species. To further test this hypothesis, and itsin vivo relevance, we designed experiments whereby NOwas scavenged after hypoxia and NOS inhibition ± nitriteadministration. Notably, in both cases, blood pressurefailed to increase upon NO scavenging, further supportingthe hypothesis that actions of NO in hypoxia are mediatedby NO metabolite signalling pathways rather than by NOdirectly.
Our study has several limitations: first, we focusedon the acute hypoxic response, which may presentdifferent mechanisms from the response to chronichypoxia. However, 10-fold higher levels of circulatingNO products were found in Tibetan plateau residentscompared to sea-level dwellers (Erzurum et al. 2007).This was associated with increased resting forearm bloodflow, suggesting a NO-mediated adaptive mechanism thatoffsets the O2 lack caused by high altitude and persistsover time. Our rats were anaesthetized and spontaneouslybreathing, and responded to hypoxia with an increasein their ventilatory drive. The subsequent reduction inPaCO2 (compared to group-matched normoxic rats) was,however, limited, and probably more representative of thesituation that occurs in vivo. The (patho)physiology ofvascular adaptation to hypoxia is indeed complex. Studieshave demonstrated that NO is a major, though not thesole, contributor to this response (Liu & Flavahan, 1997),or that it acts within a network of synergistic factors (vonBeckerath et al. 1991; Park et al. 1992). Indeed, some ofour results may be explained by concomitant activationof other signalling pathways. Another limitation of ourstudy is that changes in NO metabolite status have onlybeen assessed in blood and not in the vasculature itself.While a contribution by NO generated in the vascular wallcan thus not be entirely excluded, this is unlikely to haveoccurred under our experimental conditions as the highconcentrations of the two NO scavengers used shouldhave created a sink for NO that would be efficient enoughto show clear reversal of the effects of nitrite; however, thisis not what we observed. Regrettably, we were unable toexplore the possibility that NO levels may be increased inhypoxia because of a reduced metabolism/bioinactivation.Although we tried several strategies to modulate theredox state of cytochrome c oxidase (namely azide andhydrogen sulphide administration), unavoidable side
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1073
effects prevented the in vivo construction of such a model.Owing to these methodological constraints we cannotexclude that reduced inactivation of NO by cytochromec oxidase may be an additional contributory factor.
In conclusion, we demonstrated that short-term hypo-xic vasodilation in vivo is an adaptive response mediatedby NO. The mediator seems to act through bioactivemetabolites, in a complex regulatory network of inter-action of multiple NO-related species, rather than directlyby NO itself.
References
Badejo AM, Jr, Hodnette C, Dhaliwal JS, Casey DB, Pankey E,Murthy SN, Nossaman BD, Hyman AL & Kadowitz PJ(2010). Mitochondrial aldehyde dehydrogenase mediatesvasodilator responses of glyceryl trinitrate and sodiumnitrite in the pulmonary vascular bed of the rat. Am J PhysiolHeart Circ Physiol 299, H819–H826.
Boucher JL, Moali C & Tenu JP (1999). Nitric oxidebiosynthesis, nitric oxide synthase inhibitors and arginasecompetition for L-arginine utilization. Cell Mol Life Sci 55,1015–1028.
Brady AJ, Warren JB, Poole-Wilson PA, Williams TJ & HardingSE (1993). Nitric oxide attenuates cardiac myocytecontraction. Am J Physiol Heart Circ Physiol 265,H176–H182.
Brand MD (2005). The efficiency and plasticity ofmitochondrial energy transduction. Biochem Soc Trans 33,897–904.
Brunelle JK & Chandel NS (2002). Oxygen deprivation inducedcell death: an update. Apoptosis 7, 475–482.
Bryan N, Rassaf T, Maloney R, Rodriguez C, Saijo F, RodriguezJ & Feelisch M (2004). Cellular targets and mechanisms ofnitros(yl)ation: an insight into their nature and kinetics invivo. Proc Natl Acad Sci U S A 101, 4308–4313.
Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, MilsomAB, Rassaf T, Maloney RE, Bharti A, Rodriguez J & FeelischM (2005). Nitrite is a signaling molecule and regulator ofgene expression in mammalian tissues. Nat Chem Biol 1,290–297.
Clementi E, Brown GC, Foxwell N & Moncada S (1999). Onthe mechanism by which vascular endothelial cells regulatetheir oxygen consumption. Proc Natl Acad Sci U S A 96,1559–1562.
Cooper CE & Giulivi C (2007). Nitric oxide regulation ofmitochondrial oxygen consumption II: Molecularmechanism and tissue physiology. Am J Physiol Cell Physiol292, C1993–C2003.
Diesen DL, Hess DT & Stamler JS (2008). Hypoxic vasodilationby red blood cells: evidence for an S-nitrosothiol–basedsignal. Circ Res 103, 545–553.
Dreyer NB (1926). Some effects of anoxaemia on thecirculation. Can Med Assoc J 16, 26–29.
Dyson A, Stidwill R, Taylor V & Singer M (2007). Tissue oxygenmonitoring in rodent models of shock. Am J Physiol HeartCirc Physiol 293, H526–H533.
Dyson A, Stidwill R, Taylor V & Singer M (2009). The impact ofinspired oxygen concentration on tissue oxygenation duringprogressive haemorrhage. Intensive Care Med 35, 1783–1791.
Edmunds NJ & Marshall JM (2001). Vasodilatation, oxygendelivery and oxygen consumption in rat hindlimb duringsystemic hypoxia: roles of nitric oxide. J Physiol 532,251–259.
Erzurum SC, Ghosh S, Janocha AJ, Xu W, Bauer S, Bryan NS,Tejero J, Hemann C, Hille R, Stuehr DJ, Feelisch M & BeallCM (2007). Higher blood flow and circulating NO productsoffset high-altitude hypoxia among Tibetans. Proc Natl AcadSci U S A 104, 17593–17598.
Feelisch M, Rassaf T, Mnaimneh S, Singh N, Bryan NS,Jourd’Heuil D & Kelm M (2002). Concomitant S-, N-, andheme-nitros(yl)ation in biological tissues and fluids:implications for the fate of NO in vivo. FASEB J 16,1775–1785.
Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, BauerS, Whitlock DR, Ford PC, Janero DR, Rodriguez J &Ashrafian H (2008). Tissue processing of nitrite in hypoxia:an intricate interplay of nitric oxide-generating and-scavenging systems. J Biol Chem 283, 33927–33934.
Frost MT, Wang Q, Moncada S & Singer M (2005). Hypoxiaaccelerates nitric oxide-dependent inhibition ofmitochondrial complex I in activated macrophages. Am JPhysiol Regul Integr Comp Physiol 288, R394–R400.
Gladwin MT & Kim-Shapiro DB (2008). The functional nitritereductase activity of the heme-globins. Blood 112,2636–2647.
Hackel DB, Goodale WT & Kleinerman J (1954). Effects ofhypoxia on the myocardial metabolism of intact dogs. CircRes 2, 169–174.
Heistad DD & Wheeler RC (1970). Effect of acute hypoxia onvascular responsiveness in man. I. Responsiveness to lowerbody negative pressure and ice on the forehead. II. Responsesto norepinephrine and angiotensin. 3. Effect of hypoxia andhypocapnia. J Clin Invest 49, 1252–1265.
Hunter CJ, Blood AB, White CR, Pearce WJ & Power GG(2003). Role of nitric oxide in hypoxic cerebralvasodilatation in the ovine fetus. J Physiol 549, 625–633.
Ignarro LJ, Fukuto JM, Griscavage JM, Rogers NE & Byrns RE(1993). Oxidation of nitric oxide in aqueous solution tonitrite but not nitrate: comparison with enzymaticallyformed nitric oxide from L-arginine. Proc Natl Acad SciU S A 90, 8103–8107.
Iwamoto J, Yoshinaga M, Yang SP, Krasney E & Krasney J(1992). Methylene blue inhibits hypoxic cerebralvasodilation in awake sheep. J Appl Physiol 73, 2226–2232.
Jia L, Bonaventura C, Bonaventura J & Stamler JS (1996).S-nitrosohaemoglobin: a dynamic activity of blood involvedin vascular control. Nature 380, 221–226.
Jobgen WS, Fried SK, Fu WJ, Meininger CJ & Wu G (2006).Regulatory role for the arginine-nitric oxide pathway inmetabolism of energy substrates. J Nutr Biochem 17,571–588.
Kida Y, Maeda M, Iwata S, Iwashita Y, Goto K & Nishi K(1995). Effects of pyridoxalated hemoglobin polyoxyethyleneconjugate and other hemoglobin-related substances onarterial blood pressure in anesthetized and conscious rats.Artif Organs 19, 117–128.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

1074 M. Umbrello and others J Physiol 592.5
Li H, Cui H, Kundu TK, Alzawahra W & Zweier JL (2008).Nitric oxide production from nitrite occurs primarily intissues not in the blood: critical role of xanthineoxidase and aldehyde oxidase. J Biol Chem 283,17855–17863.
Liu Q & Flavahan NA (1997). Hypoxic dilatation of porcinesmall coronary arteries: role of endothelium andKATP-channels. Br J Pharmacol 120,728–734.
Marley R, Feelisch M, Holt S & Moore K (2000). Achemiluminescense-based assay for S-nitrosoalbuminand other plasma S-nitrosothiols. Free Radic Res 32,1–9.
Martinez-Ruiz A, Cadenas S & Lamas S (2011). Nitric oxidesignaling: classical, less classical, and nonclassicalmechanisms. Free Radic Biol Med 51, 17–29.
Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E &Lundberg JO (2001). Nitrite-derived nitric oxide: a possiblemediator of ‘acidic-metabolic’ vasodilation. Acta PhysiolScand 171, 9–16.
Mohan P, Brutsaert DL, Paulus WJ & Sys SU (1996).Myocardial contractile response to nitric oxide and cGMP.Circulation 93, 1223–1229.
Moncada S & Higgs A (1993). The L-arginine-nitric oxidepathway. N Engl J Med 329, 2002–2012.
Movahed P, Hogestatt ED & Petersson J (2003). Effect ofhypoxia on vasodilator responses toS-nitroso-N-acetylpenicillamine and levcromakalim inguinea pig basilar artery. Naunyn Schmiedebergs ArchPharmacol 367, 532–537.
Palacios-Callender M, Hollis V, Mitchison M, Frakich N, UnittD & Moncada S (2007). Cytochrome c oxidase regulatesendogenous nitric oxide availability in respiring cells: apossible explanation for hypoxic vasodilation. Proc NatlAcad Sci U S A 104, 18508–18513.
Park KH, Rubin LE, Gross SS & Levi R (1992). Nitric oxide is amediator of hypoxic coronary vasodilatation. Relation toadenosine and cyclooxygenase-derived metabolites. Circ Res71, 992–1001.
Pearce LL, Kanai AJ, Birder LA, Pitt BR & Peterson J (2002).The catabolic fate of nitric oxide: the nitric oxide oxidase andperoxynitrite reductase activities of cytochrome oxidase.J Biol Chem 277, 13556–13562.
Perlman DH, Bauer SM, Ashrafian H, Bryan NS, Garcia-SauraMF, Lim CC, Fernandez BO, Infusini G, McComb ME,Costello CE & Feelisch M (2009). Mechanistic insights intonitrite-induced cardioprotection using an integratedmetabolomic/proteomic approach. Circ Res 104,796–804.
Pohl U & Busse R (1989). Hypoxia stimulates release ofendothelium-derived relaxant factor. Am J Physiol Heart CircPhysiol 256, H1595–H1600.
Rassaf T, Bryan NS, Kelm M & Feelisch M (2002).Concomitant presence of N-nitroso and S-nitroso proteinsin human plasma. Free Radic Biol Med 33,1590–1596.
Rixen D & Siegel JH (2005). Bench-to-bedside review: oxygendebt and its metabolic correlates as quantifiers of the severityof hemorrhagic and post-traumatic shock. Crit Care 9,441–453.
Rowell LB & Blackmon JR (1987). Human cardiovascularadjustments to acute hypoxaemia. Clin Physiol 7,349–376.
Rowell LB & Blackmon JR (1989). Hypotension induced bycentral hypovolaemia and hypoxaemia. Clin Physiol 9,269–277.
Roy CS & Brown JG (1880). The blood-pressure and itsvariations in the arterioles, capillaries and smaller veins.J Physiol 2, 323–446.
Semenza GL (2007). Oxygen-dependent regulation ofmitochondrial respiration by hypoxia-inducible factor 1.Biochem J 405, 1–9.
Shiva S, Brookes PS, Patel RP, Anderson PG & Darley-UsmarVM (2001). Nitric oxide partitioning into mitochondrialmembranes and the control of respiration at cytochrome coxidase. Proc Natl Acad Sci U S A 98, 7212–7217.
Slama M, Susic D, Varagic J, Ahn J & Frohlich ED (2003).Echocardiographic measurement of cardiac output in rats.Am J Physiol Heart Circ Physiol 284, H691–H697.
Southan GJ, Szabo C & Thiemermann C (1995). Isothioureas:potent inhibitors of nitric oxide synthases with variableisoform selectivity. Br J Pharmacol 114, 510–516.
Taylor CT & Pouyssegur J (2007). Oxygen, hypoxia, and stress.Ann N Y Acad Sci 1113, 87–94.
Tsai AG, Johnson PC & Intaglietta M (2003). Oxygengradients in the microcirculation. Physiol Rev 83,933–963.
Umbrello M, Dyson A, Feelisch M & Singer M (2013). The keyrole of nitric oxide in hypoxia: hypoxic vasodilation andenergy supply-demand matching. Antioxid Redox Signal 19,1690–1710
Unitt DC, Hollis VS, Palacios-Callender M, Frakich N &Moncada S (2010). Inactivation of nitric oxide bycytochrome c oxidase under steady-state oxygen conditions.Biochim Biophys Acta 1797, 371–377.
Vallet B, Curtis SE, Winn MJ, King CE, Chapler CK & Cain SM(1994). Hypoxic vasodilation does not require nitric oxide(EDRF/NO) synthesis. J Appl Physiol 76,1256–1261.
von Beckerath N, Cyrys S, Dischner A & Daut J (1991).Hypoxic vasodilatation in isolated, perfused guinea-pigheart: an analysis of the underlying mechanisms. J Physiol442, 297–319.
Welch G & Loscalzo J (1994). Nitric oxide and thecardiovascular system. J Card Surg 9, 361–371.
Yoshida M, Akaike T, Wada Y, Sato K, Ikeda K, Ueda S &Maeda H (1994). Therapeutic effects of imidazolineoxylN-oxide against endotoxin shock through its direct nitricoxide-scavenging activity. Biochem Biophys Res Commun202, 923–930.
Additional information
Competing interests
The authors declare no competing financial interests.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (

J Physiol 592.5 Nitric oxide and hypoxic vasodilation 1075
Author contributions
Experiments were performed at University College London.M.U., A.D., M.F. and M.S. conceived the study and designedthe experiments; M.U., A.D., B.B.P. and V.S. performed theexperiments and collected the physiological data. M.U. and V.S.undertook the data analysis. B.O.F. performed the biochemicalmeasurements. M.U. and M.S. drafted the first version of themanuscript; A.D., B.B.P., B.F. and M.F. revised it critically. Allauthors approved the final version of the manuscript.
Funding
This work was undertaken at UCLH/UCL who received aproportion of funding from the Department of Health’sNIHR Biomedical Research Centres funding scheme. M.U. wassupported by a Physiology Award from the Accademia Nazionaledei Lincei (Roma)/Royal Society (London).
Acknowledgement
None.
C© 2014 The Authors. The Journal of Physiology C© 2014 The Physiological Society
) at UCL Library Services on April 1, 2014jp.physoc.orgDownloaded from J Physiol (




















