
Role of residues 121 to 124 of vesicular stomatitis virus matrix protein in virus assembly and...
11
JOURNAL OF VIROLOGY, Apr. 2006, p. 3701–3711 Vol. 80, No. 8 0022-538X/06/$08.000 doi:10.1128/JVI.80.8.3701–3711.2006 Copyright © 2006, American Society for Microbiology. All Rights Reserved. Role of Residues 121 to 124 of Vesicular Stomatitis Virus Matrix Protein in Virus Assembly and Virus-Host Interaction John H. Connor,* Margie O. McKenzie, and Douglas S. Lyles Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, North Carolina 27157 Received 7 October 2005/Accepted 21 January 2006 The recent solution of the crystal structure of a fragment of the vesicular stomatitis virus matrix (M) protein suggested that amino acids 121 to 124, located on a solvent-exposed loop of the protein, are important for M protein self-association and association with membranes. These residues were mutated from the hydrophobic AVLA sequence to the polar sequence DKQQ. Expression and purification of this mutant from bacteria showed that it was structurally stable and that the mutant M protein had self-association kinetics similar to those of the wild-type M protein. Analysis of the membrane association of M protein in the context of infection with isogenic recombinant viruses showed that both wild-type and mutant M proteins associated with membranes to the same extent. Virus expressing the mutant M protein did show an approximately threefold-lower binding affinity of M protein for nucleocapsid-M complexes. In contrast to the relatively minor effects of the M protein mutation on virus assembly, the mutant virus exhibited growth restriction in MDBK but not BHK cells, a slower induction of apoptosis, and lower viral-protein synthesis. Despite translating less viral protein, the mutant virus produced more viral mRNA, showing that the mutant virus could not effectively promote viral translation. These results demonstrate that the 121-to-124 region of the VSV M protein plays a minor role in virus assembly but is involved in virus-host interactions and VSV replication by augmenting viral-mRNA translation. Virus assembly is a complex process involving a number of important steps. For enveloped viruses, the viral genome must be packaged into a nucleoprotein (nucleocapsid), which must be brought to the host membrane. This nucleocapsid must acquire a lipid bilayer by budding from the host membrane (for a review, see reference 14). For negative-strand RNA viruses of the order Mononegavirales and for the families Orthomyxoviridae and Retroviridae, these assembly functions appear to be carried out largely by viral matrix, or M, proteins (28, 32). This multifunc- tional nature of the M protein makes it a critical player in virus assembly, which has led to a great deal of interest in determin- ing how M proteins carry out such different functions. M pro- teins from different viruses have very different sequences and structures (32), a fact that has led to few initial clues about how they carry out their multiple functions and has highlighted the importance of studying individual M proteins to shed light on the wider class of functional analogs. The M protein of vesicular stomatitis virus (VSV) is a par- ticularly well-studied example of this class of proteins. VSV M protein has been shown to play several roles in the virus as- sembly pathway. VSV M is responsible for condensing the viral nucleocapsid, causing the collapse of the loosely coiled, flexible nucleocapsid into the characteristic tightly coiled, bullet-like shape found in assembled virions (23, 25, 26). Mutagenesis and proteolysis studies have shown that the interaction of M pro- tein with nucleocapsid requires an N-terminal sequence of the protein (2, 17). The N-terminal sequence (amino acids 1 to 50) is protease accessible in assembled nucleocapsid-M (NCM) complexes (17) and, by using a photoactivatable membrane probe, has been shown to associate with cellular membranes (20). The sequence PPPY contained within the N-terminal sequence is involved in a late stage of budding, allowing the release of budded virions from the membrane (15). In addition to the N-terminal sequence, several different regions in the middle of the protein sequence have been im- plicated in virus assembly, but these studies have yet to be extended to an understanding of whether these sequences co- operate and how they contribute to viral assembly and budding in an intact virus. In particular, proteolysis studies have sug- gested the contribution of a 4-amino-acid (aa) region at aa 121 to 124 in membrane association (9). A detailed understanding of why these regions are important has been hampered to some extent by the lack of structural information to indicate which residues of the M protein are part of the interior of the folded protein and which residues are located in solvent-exposed regions that can participate in mem- brane association and/or protein-protein interactions. This problem has been significantly reduced with the recent solution of the crystal structure of an M protein fragment (10). The ability to obtain crystals of the M protein required proteolytic removal of both the N-terminal sequence (aa 1 to 47) and the hydrophobic sequence aa 121 to 124 to prevent M protein self-association, but the resulting crystal structure represents a large portion of the M protein sequence. The structure showed that the M protein fragments formed a compactly folded struc- ture with two alpha helices cradled in a beta sheet (Fig. 1A). The structure indicated that residues 121 to 124 lie on a sol- vent-exposed loop. Based on this crystallographic data and the properties of the cleaved M protein fragment, it was proposed that the aa 121-to-124 sequence played a role in both M pro- tein self-association and M protein association with mem- * Corresponding author. Mailing address: Department of Biochem- istry, Wake Forest University School of Medicine, Winston-Salem, NC 27157. Phone: (336) 716-2157. Fax: (336) 716-7671. E-mail: jconnor @wfubmc.edu. 3701
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Role of residues 121 to 124 of vesicular stomatitis virus matrix protein in virus assembly and...

JOURNAL OF VIROLOGY, Apr. 2006, p. 3701–3711 Vol. 80, No. 80022-538X/06/$08.00�0 doi:10.1128/JVI.80.8.3701–3711.2006Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Role of Residues 121 to 124 of Vesicular Stomatitis Virus MatrixProtein in Virus Assembly and Virus-Host Interaction
John H. Connor,* Margie O. McKenzie, and Douglas S. LylesDepartment of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, North Carolina 27157
Received 7 October 2005/Accepted 21 January 2006
The recent solution of the crystal structure of a fragment of the vesicular stomatitis virus matrix (M) proteinsuggested that amino acids 121 to 124, located on a solvent-exposed loop of the protein, are important for Mprotein self-association and association with membranes. These residues were mutated from the hydrophobicAVLA sequence to the polar sequence DKQQ. Expression and purification of this mutant from bacteria showedthat it was structurally stable and that the mutant M protein had self-association kinetics similar to those ofthe wild-type M protein. Analysis of the membrane association of M protein in the context of infection withisogenic recombinant viruses showed that both wild-type and mutant M proteins associated with membranesto the same extent. Virus expressing the mutant M protein did show an approximately threefold-lower bindingaffinity of M protein for nucleocapsid-M complexes. In contrast to the relatively minor effects of the M proteinmutation on virus assembly, the mutant virus exhibited growth restriction in MDBK but not BHK cells, aslower induction of apoptosis, and lower viral-protein synthesis. Despite translating less viral protein, themutant virus produced more viral mRNA, showing that the mutant virus could not effectively promote viraltranslation. These results demonstrate that the 121-to-124 region of the VSV M protein plays a minor role invirus assembly but is involved in virus-host interactions and VSV replication by augmenting viral-mRNAtranslation.
Virus assembly is a complex process involving a number ofimportant steps. For enveloped viruses, the viral genome must bepackaged into a nucleoprotein (nucleocapsid), which must bebrought to the host membrane. This nucleocapsid must acquire alipid bilayer by budding from the host membrane (for a review,see reference 14). For negative-strand RNA viruses of the orderMononegavirales and for the families Orthomyxoviridae andRetroviridae, these assembly functions appear to be carried outlargely by viral matrix, or M, proteins (28, 32). This multifunc-tional nature of the M protein makes it a critical player in virusassembly, which has led to a great deal of interest in determin-ing how M proteins carry out such different functions. M pro-teins from different viruses have very different sequences andstructures (32), a fact that has led to few initial clues about howthey carry out their multiple functions and has highlighted theimportance of studying individual M proteins to shed light onthe wider class of functional analogs.
The M protein of vesicular stomatitis virus (VSV) is a par-ticularly well-studied example of this class of proteins. VSV Mprotein has been shown to play several roles in the virus as-sembly pathway. VSV M is responsible for condensing the viralnucleocapsid, causing the collapse of the loosely coiled, flexiblenucleocapsid into the characteristic tightly coiled, bullet-likeshape found in assembled virions (23, 25, 26). Mutagenesis andproteolysis studies have shown that the interaction of M pro-tein with nucleocapsid requires an N-terminal sequence of theprotein (2, 17). The N-terminal sequence (amino acids 1 to 50)is protease accessible in assembled nucleocapsid-M (NCM)
complexes (17) and, by using a photoactivatable membraneprobe, has been shown to associate with cellular membranes(20). The sequence PPPY contained within the N-terminalsequence is involved in a late stage of budding, allowing therelease of budded virions from the membrane (15).
In addition to the N-terminal sequence, several differentregions in the middle of the protein sequence have been im-plicated in virus assembly, but these studies have yet to beextended to an understanding of whether these sequences co-operate and how they contribute to viral assembly and buddingin an intact virus. In particular, proteolysis studies have sug-gested the contribution of a 4-amino-acid (aa) region at aa 121to 124 in membrane association (9).
A detailed understanding of why these regions are importanthas been hampered to some extent by the lack of structuralinformation to indicate which residues of the M protein arepart of the interior of the folded protein and which residues arelocated in solvent-exposed regions that can participate in mem-brane association and/or protein-protein interactions. Thisproblem has been significantly reduced with the recent solutionof the crystal structure of an M protein fragment (10). Theability to obtain crystals of the M protein required proteolyticremoval of both the N-terminal sequence (aa 1 to 47) and thehydrophobic sequence aa 121 to 124 to prevent M proteinself-association, but the resulting crystal structure represents alarge portion of the M protein sequence. The structure showedthat the M protein fragments formed a compactly folded struc-ture with two alpha helices cradled in a beta sheet (Fig. 1A).The structure indicated that residues 121 to 124 lie on a sol-vent-exposed loop. Based on this crystallographic data and theproperties of the cleaved M protein fragment, it was proposedthat the aa 121-to-124 sequence played a role in both M pro-tein self-association and M protein association with mem-
* Corresponding author. Mailing address: Department of Biochem-istry, Wake Forest University School of Medicine, Winston-Salem, NC27157. Phone: (336) 716-2157. Fax: (336) 716-7671. E-mail: [email protected].
3701

branes (9, 10). To determine the importance of this region inthe context of a nonproteolyzed M protein and during VSVinfection, we mutated the region from a hydrophobic sequenceto a polar sequence and analyzed its association with NCMcomplexes and with membranes in both the presence and theabsence of other viral components. Our findings do not sup-port a strong role for this region in membrane association.Instead, we have found that mutating amino acids 121 to 124 ofVSV M alters the virus-host interaction so that viral mRNAsare translated less efficiently and the induction of apoptosis isdelayed. While a role for the M protein in regulating apoptosishas been reported previously (8, 19), this is the first report ofa role for the M protein in controlling the translation of viralmRNAs.
MATERIALS AND METHODS
Chemicals, unless otherwise stated, were purchased from Fisher Scientific.Thapsigargin was purchased from Calbiochem. Antibodies against eIF2� andphospho-eIF2�, were purchased from Cell Signaling Technologies. Okadaic acidand microcystin were purchased from Alexis Pharmaceuticals. Ni-nitrilotriaceticacid beads were obtained from QIAGEN, and phosphocellulose (P11) was ob-tained from Whatman. Swiss-Model (30) was used to generate the coordinates ofthe M protein fragment from aa 51 to 227. This model includes amino acids 122to 127, which are missing in the crystal structure of the matrix protein.
Mutagenesis and bacterial expression of M protein. The gene encoding the Mprotein from the Orsay strain of VSV (2) was subcloned into the pET21dbacterial expression vector (Novagen). The sequence encoding aa 121 to 124 wasmutagenized using a previously described protocol (29). BL21(DE3) pLYSs cellswere transformed with the expression vector, and 500 nM IPTG (isopropyl-�-D-thiogalactopyranoside) was added when the cells reached an optical density at600 nm of 0.6. Following expression at 37°C for 3 h, the cells were spun down,lysed in 1/100 volume of lysis buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 250 mMNaCL, 20 mM imidazol), and purified using Ni-nitrilotriacetic acid beads. Fol-lowing adsorption onto the beads, the M protein was eluted in Ni elution buffer(50 mM Tris, pH 7.5, 250 mM NaCl, 400 mM imidazole). The resulting elutedprotein was diluted and passed over a P11 column and then eluted off of the P11column using a high-salt buffer (24). The resulting purified protein was dialyzedagainst 10 mM Tris, pH 7.5, 250 mM NaCl.
Stopped-flow analysis of M self-association. Purified wt or mutant M proteinwas diluted to 170 �g/ml in 10 mM Tris, pH 7.5, 650 mM NaCl and placed in thesample syringe of an Applied Photophysics stopped-flow spectrophotometer(22). The NaCl concentration was shifted to 65 mM or 135 nM NaCl by 10-folddilution in the appropriate buffer, and light scattering was analyzed as a functionof time after dilution as described previously (22).
Recovery of recombinant viruses. M genes encoding wild-type (wt) M protein(Orsay strain) and 121-to-124 mutant M protein were subcloned into an infec-tious cDNA clone described previously (19). Recombinant viruses were recov-ered as described previously (19). For all experiments, BHK cells were culturedin Dulbecco’s modified Eagle medium (DMEM) (Gibco-BRL) containing 7%fetal bovine serum (FBS) and 2 mM glutamine. The cells were grown to 80 to90% confluence and were infected with virus in DMEM with 2% FBS at amultiplicity of infection (MOI) of 10 PFU/cell in a small volume (500 �l per well
FIG. 1. Mutation of aa 121 to 124 of VSV M protein does not affect self-association. (A) Image of theoretical model (based on M crystalstructure; see Materials and Methods) illustrating the location of the presumptive loop region. (B) Diagram of wt M protein (top) and a mutantused in this study illustrating placement and sequence of mutations. (C) Coomassie staining of purified recombinant M proteins. Lane 1 is purifiedvirions as a marker for the migration of wild-type M protein. The star denotes a proteolytic fragment. mut, mutant. (D) Stopped-flow analysis ofself-association of M. The graph shows changes in light-scattering intensity following a shift of the salt concentration from 250 mM to either 65or 130 mM NaCl. The traces are representative of four experiments each from two separate protein preparations. Wild-type traces are shown inblack, and hydrophobic-loop mutant traces are in gray. The inset shows the first 12 seconds of each run to illustrate the lag seen in 130 mM NaClsamples.
3702 CONNOR ET AL. J. VIROL.

for a six-well dish). At 1 h postinfection, the culture volume was doubled by theaddition of DMEM plus 2% FBS.
Assays for M protein exchange with NCM complexes. The ability of radiola-beled wt or mutant M protein to exchange with unlabeled M protein in virionNCM complexes was assayed as described previously (7). Briefly, in vitro tran-scription and translation (Promega TNT) was used to produce [35S]methionine-labeled M protein. Radiolabeled M protein was mixed with increasing concen-trations of nonradiolabeled purified virions, which served as a source ofnucleocapsid-M complexes. Triton X-100 was added to permeabilize the virionenvelope and, following a 5-min incubation time to allow equilibrium exchangeof M protein, the NCM complexes were pelleted by centrifugation. The super-natants and pellets were then analyzed by sodium dodecyl sulfate-polyacrylamidegel electrophoresis (SDS-PAGE) and phosphorimager analysis.
Membrane association assays. For analysis of M protein in the absence ofother viral components, cells were infected with vaccinia virus expressing T7RNA polymerase (VTF7-3) at an MOI of 20, and then 60 min postinfection, thecells were transfected with appropriate pET21d-M plasmids and allowed toexpress M protein for 6 h. For analysis of M protein in the presence of other viralcomponents, cells were infected with recombinant viruses expressing recombi-nant wt (rwt) or mutant M protein at an MOI of 10 for 4 h. For both conditions,the cells were then starved for methionine for 10 min and then incubated inmedia containing [35S]methionine (200 �Ci/ml) for 60 min. The cells were thenwashed and lifted from the dish by scraping it. The cells were resuspended inhomogenization buffer and lysed using a Dounce homogenizer (100 to 125strokes). The postnuclear lysates were then adjusted to 60% sucrose and werefractionated on a 60-40-10% sucrose gradient by centrifugation at 35,000 � g for16 h at 4°C. Fractions were then collected from the top, M protein was immu-noprecipitated using 23H12 antibody (19), and immunoprecipitates were sepa-rated on a 10% SDS-PAGE gel and exposed using a phosphorimager.
Pulse-chase analysis of virus assembly. Cells were infected with rwt or ahydrophobic-loop mutant at an MOI of 10; 4 h postinfection, the cells werestarved for methionine for 10 min and then incubated with media containing[35S]methionine for 60 min. The radioactive media were replaced with non-radioactive media. At 30, 60, and 90 min postlabeling, the media were removedand lysates of cells were made. Virions were purified through a 15% sucrosecushion, and then virion and cell lysate fractions were run on an SDS-PAGE geland analyzed by phosphorimager analysis. The total amounts of radioactivity ineach fraction were back-calculated based on the phosphorimager analysis.
Northern blotting. Blotting was performed as described previously (4). Briefly,infected or mock-infected cells were lysed in 60-mm dishes with Trizol (Invitro-gen); 5 �g of total RNA from each sample was glyoxylated and separated on a1% agarose gel. Following ethidium bromide staining to check for loading, RNAwas transferred onto nitrocellulose membranes (Genescreen Plus; NEN) andprobed using a 32P-labeled M gene cDNA probe, and the results were deter-mined by phosphorescence imaging and analyzed using Imagequant software.
Immmunoblotting. Infected or mock-infected cells were lysed in six-welldishes using 400 �l EBC buffer containing 1 mM PMSF (phenylmethylsulfonylfluoride), 1 mM benzamidine, 100 nM okadaic acid, and 100 nM microcystin (4).The lysates were spun at 10,000 � g for 8 min in a refrigerated centrifuge, and360 �l of the supernatant was added to 40 �l of 10� sample buffer for SDS-PAGE. Equal volumes of lysate were electrophoresed on 12% SDS-PAGE gels.Following electrophoresis, the gels were electroblotted onto nitrocellulose mem-branes and blocked in Tris-buffered saline (pH 7.5) plus 5% dry milk. Antibodieswere diluted as recommended by the manufacturers.
Metabolic labeling and determination of the rate of protein synthesis. BHKcells were mock infected or infected with VSV and then labeled with [35S]me-thionine for 10 min at various times postinfection, as described above. Followinglabeling, the cells were washed three times with phosphate-buffered saline and lysedin 400 �l RIPA buffer for 10 min at 4°C. The lysates were spun at 10,000 � g for 8min. Supernatant (360 �l) was added to 40 �l 10� SDS-PAGE sample buffer,and 10 �l was electrophoresed on a 12% SDS-PAGE gel. The gels were stainedwith Coomassie blue to determine their protein contents, and then they wereanalyzed by phosphorescence imaging (Molecular Dynamics, Inc.). The phos-phorescence intensities were analyzed using Imagequant software. The rate ofviral-protein synthesis was determined from the experiments by quantitation ofthe radioactivity in the viral N/P and M bands in each lane. The rate of hostprotein synthesis was determined by quantitation of the radioactivity between theL and G bands and the N/P and M bands and below the M band. For all typesof experiment, three separate experiments were analyzed in this manner todetermine an average.
Cell rounding/blebbing and caspase 3 assays. Cells were grown to approxi-mately 50% confluence in six-well dishes, infected with wt control virus orhydrophobic-mutant virus, and placed on a Zeiss inverted microscope with a
six-well stage. Images were taken every 15 min using OpenLab (Improvision).Movies were made from streamed images, and occurrences of cell rounding andblebbing were tabulated using the time stamp record. Caspase 3 assays were doneusing a caspase 3 activity kit from R&D. Analysis was done according to themanufacturer’s instructions.
RESULTS
Earlier studies suggested that amino acids 121 to 124 areimportant for the self-association of M protein and the asso-ciation of M protein with membranes (9, 10). In the wild-typeM protein, the aa 121-to-124 region sequence, AVLA, is hy-drophobic and is likely to be a loop region, based on the crystalstructure and a theoretical model (Fig. 1A). Because theseprevious experiments utilized proteolytic fragments of the Mprotein, we wanted to determine the importance of this regionfor self-aggregation, membrane association, and the associa-tion of M with nucleocapsid in the context of the full-lengthprotein. We mutated the AVLA sequence to DKQQ (Fig. 1B),reasoning that this would replace hydrophobic residues withhydrophilic residues and that the charged Asp and Lys residueswould mimic the positive N-terminal and negative C-terminalcharges that would be generated following proteolysis in theregion.
Hydrophobic-loop mutations do not effect M aggregation.We were able to bacterially express and purify milligramamounts of the M protein as a C-terminal His-tagged proteinusing a two-step column procedure that removed contaminat-ing bacterial proteins (Fig. 1C). These preparations containedminor amounts of a 21-kDa proteolytic fragment of M that iscommon in purified M protein preparations. The presence ofthis fragment does not affect the self-association of intact Mprotein (11). This purified M protein was stable for up to aweek in a high-salt (250 mM NaCl) buffer. We assessed theability of the purified M protein to self-associate under condi-tions of reduced salt concentration. A solution of wt or hydro-phobic-loop mutant M (� mutant) protein in high-salt bufferwas mixed with a low-salt buffer so that the final NaCl concen-tration was either 65 mM or 130 mM. Under these lower-saltconditions, M protein will spontaneously self-associate to formaggregates. We determined the time course of self-associationby stopped-flow analysis of light-scattering intensity. Stopped-flow analysis allowed us to observe the changes in light scat-tering caused by the formation of large M protein aggregateson a time scale of milliseconds. At 130 mM salt (Fig. 1D, lowertraces), there was a brief lag before significant aggregationoccurred (Fig. 1D, inset), and most of the aggregation hadoccurred within 40 seconds. At the lower salt concentration of65 mM, the lag phase was not evident, and aggregation oc-curred to a greater extent (Fig. 1D, upper traces). The wt andmutant M proteins showed virtually identical aggregation char-acteristics in this assay, demonstrating that in the context of thefull-length protein, this region does not dramatically affect theaggregation of the M protein.
Effect of the hydrophobic-loop mutations on M protein as-sociation with NCM complexes or cellular membranes. Usingan M protein exchange assay that measures the reversiblebinding of M protein to the NCM complexes at physiologicalionic strength, we also determined whether mutations in thehydrophobic loop, aa 121 to 124, altered the ability of the Mprotein to interact with NCM complexes. There are both re-
VOL. 80, 2006 ROLE OF THE VSV M PROTEIN HYDROPHOBIC LOOP 3703
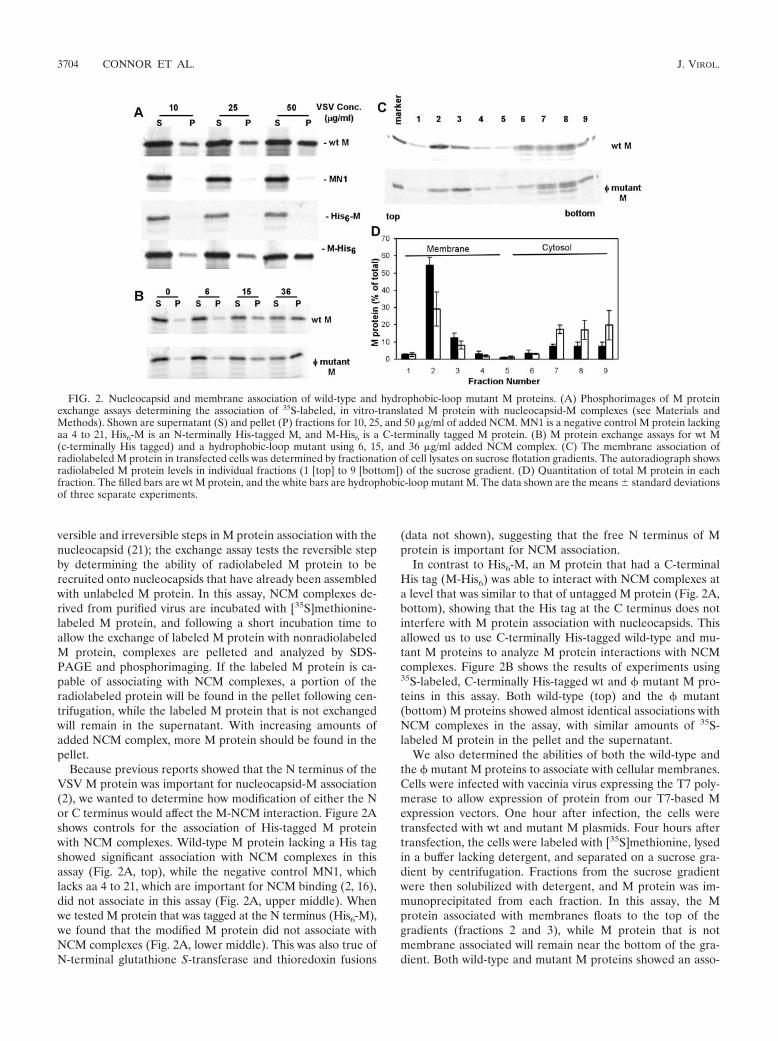
versible and irreversible steps in M protein association with thenucleocapsid (21); the exchange assay tests the reversible stepby determining the ability of radiolabeled M protein to berecruited onto nucleocapsids that have already been assembledwith unlabeled M protein. In this assay, NCM complexes de-rived from purified virus are incubated with [35S]methionine-labeled M protein, and following a short incubation time toallow the exchange of labeled M protein with nonradiolabeledM protein, complexes are pelleted and analyzed by SDS-PAGE and phosphorimaging. If the labeled M protein is ca-pable of associating with NCM complexes, a portion of theradiolabeled protein will be found in the pellet following cen-trifugation, while the labeled M protein that is not exchangedwill remain in the supernatant. With increasing amounts ofadded NCM complex, more M protein should be found in thepellet.
Because previous reports showed that the N terminus of theVSV M protein was important for nucleocapsid-M association(2), we wanted to determine how modification of either the Nor C terminus would affect the M-NCM interaction. Figure 2Ashows controls for the association of His-tagged M proteinwith NCM complexes. Wild-type M protein lacking a His tagshowed significant association with NCM complexes in thisassay (Fig. 2A, top), while the negative control MN1, whichlacks aa 4 to 21, which are important for NCM binding (2, 16),did not associate in this assay (Fig. 2A, upper middle). Whenwe tested M protein that was tagged at the N terminus (His6-M),we found that the modified M protein did not associate withNCM complexes (Fig. 2A, lower middle). This was also true ofN-terminal glutathione S-transferase and thioredoxin fusions
(data not shown), suggesting that the free N terminus of Mprotein is important for NCM association.
In contrast to His6-M, an M protein that had a C-terminalHis tag (M-His6) was able to interact with NCM complexes ata level that was similar to that of untagged M protein (Fig. 2A,bottom), showing that the His tag at the C terminus does notinterfere with M protein association with nucleocapsids. Thisallowed us to use C-terminally His-tagged wild-type and mu-tant M proteins to analyze M protein interactions with NCMcomplexes. Figure 2B shows the results of experiments using35S-labeled, C-terminally His-tagged wt and � mutant M pro-teins in this assay. Both wild-type (top) and the � mutant(bottom) M proteins showed almost identical associations withNCM complexes in the assay, with similar amounts of 35S-labeled M protein in the pellet and the supernatant.
We also determined the abilities of both the wild-type andthe � mutant M proteins to associate with cellular membranes.Cells were infected with vaccinia virus expressing the T7 poly-merase to allow expression of protein from our T7-based Mexpression vectors. One hour after infection, the cells weretransfected with wt and mutant M plasmids. Four hours aftertransfection, the cells were labeled with [35S]methionine, lysedin a buffer lacking detergent, and separated on a sucrose gra-dient by centrifugation. Fractions from the sucrose gradientwere then solubilized with detergent, and M protein was im-munoprecipitated from each fraction. In this assay, the Mprotein associated with membranes floats to the top of thegradients (fractions 2 and 3), while M protein that is notmembrane associated will remain near the bottom of the gra-dient. Both wild-type and mutant M proteins showed an asso-
FIG. 2. Nucleocapsid and membrane association of wild-type and hydrophobic-loop mutant M proteins. (A) Phosphorimages of M proteinexchange assays determining the association of 35S-labeled, in vitro-translated M protein with nucleocapsid-M complexes (see Materials andMethods). Shown are supernatant (S) and pellet (P) fractions for 10, 25, and 50 �g/ml of added NCM. MN1 is a negative control M protein lackingaa 4 to 21, His6-M is an N-terminally His-tagged M, and M-His6 is a C-terminally tagged M protein. (B) M protein exchange assays for wt M(c-terminally His tagged) and a hydrophobic-loop mutant using 6, 15, and 36 �g/ml added NCM complex. (C) The membrane association ofradiolabeled M protein in transfected cells was determined by fractionation of cell lysates on sucrose flotation gradients. The autoradiograph showsradiolabeled M protein levels in individual fractions (1 [top] to 9 [bottom]) of the sucrose gradient. (D) Quantitation of total M protein in eachfraction. The filled bars are wt M protein, and the white bars are hydrophobic-loop mutant M. The data shown are the means � standard deviationsof three separate experiments.
3704 CONNOR ET AL. J. VIROL.

ciation with membranes, indicated by M protein being found infractions 2 and 3 (Fig. 2C). Quantitation of the amount of Mpresent in each fraction as a percentage of the whole in threeseparate experiments is shown in Fig. 2D. A noticeable differ-ence between the wt and the hydrophobic mutant was evidentfrom this analysis. Mutant M protein was associated to a lesserextent than wt M protein. The total mutant M protein associ-ated with membrane fractions was 57% � 14% (mean � stan-dard deviation) that of wt M protein, and this difference wasstatistically significant (P � 0.05). These data suggest that, inthe absence of other viral components, the aa 121-to-124 re-gion of M protein plays a role in the association of M withmembranes.
Association of M protein with membranes during infectionwith recombinant mutant virus does not depend on the hydro-phobic-loop region. The finding that mutation of the hydro-phobic loop had an effect on the membrane association of Mprotein in transfection assays led us to determine the effect ofthis mutation in the context of infection with a recombinantmutant virus. We constructed infectious cDNAs encoding amutant M protein and an isogenic control that expressed thewild-type M protein. From these cDNAs, we rescued infectiousvirus expressing the wild-type M protein or the mutant Mprotein. Sequencing of reverse transcription-PCR productsproved that no additional mutations in the M gene had oc-curred during the recovery (data not shown). The associationwith cellular membranes of wt or mutant M protein expressedby the recombinant viruses was determined in experimentssimilar to those in Fig. 2D, except that virus-infected cells wereanalyzed rather than transfected cells. When the results of fiveindependent experiments were analyzed (Fig. 3A), they showeddifferences from the behavior of M protein expressed in theabsence of other viral components. First, it appeared that asmaller fraction of wt M associated with membranes duringvirus infection than with transfected cells. Second, the reducedassociation with the membrane that was previously seen inthe � mutant was not evident in the context of a virusinfection, leading to the conclusion that defects in mem-
FIG. 3. Characterization of recombinant M protein mutant virus. (A) Membrane association of M protein from wt VSV (black bars) or proteinfrom hydrophobic mutant VSV (white bars) was determined by fractionation of infected-cell lysates on sucrose flotation gradients. The error barsindicate standard deviations. (B) Association of wt and hydrophobic-mutant M proteins with NCM complexes from wild-type or mutant virions.The graph represents the quantitation of three separate experiments � standard deviations. The solid lines represent experiments in whichnucleocapsid-M complex was derived from wild-type control virus, and the dotted lines represent experiments in which NCM was derived frommutant virus. Wild-type control (■ ); hydrophobic mutant (E).
FIG. 4. (A) Pulse-chase analysis of M protein incorporated intovirions. The black bars are wild-type VSV, and the white bars are thehydrophobic-loop mutant. The bars represent the amount of radiola-beled M incorporated into virions over the amount of radiolabeled Mpresent in virions plus cells. (B) Virus titers of recombinant wild-typeVSV and recombinant VSV expressing the hydrophobic-loop mutant.Wild-type control (■ ); hydrophobic mutant (E). The inset shows anSDS-PAGE gel of purified virus from media of infected cells (24 hp.i.). All experiments are the averages of three separate experiments �standard deviations, except for B, which is five separate experiments.
VOL. 80, 2006 ROLE OF THE VSV M PROTEIN HYDROPHOBIC LOOP 3705

brane association are not apparent in the virus expressingthe mutant M protein.
We also used the recombinant mutant virus to test the effectof the hydrophobic-loop mutation on M protein binding tomutant NCM complexes. These experiments were similar tothose shown in Fig. 2B but included NCM complexes fromboth wt VSV and the � mutant virus. When NCM complexesderived from recombinant wt VSV were used, the wt andmutant M proteins showed equal abilities to associate withthese NCM complexes at all concentrations (Fig. 4B), similarto the results seen in Fig. 2B. When NCM complexes from therecombinant mutant virus were used in this assay, we found adifferent response. Both the wild-type and mutant M proteinsshowed significantly lower apparent affinities for mutant thanfor wild-type NCM complexes (Fig. 3B). These results showthat the association of M protein with NCM complexes isreduced when the hydrophobic loop is mutated; furthermore,it shows that this defect is not at the level of association ofindividual M protein molecules with the nucleocapsid, as themutant M protein associated well with NCM complexes fromwild-type virus. Rather, the defect was apparent only whenNCM complexes from the mutant virus were used, suggestingthat mutating the aa 121-to-124 region of the M protein affectseither the cooperativity of M protein association with the nu-cleocapsids or a higher order of binding, such as nucleocapsidcondensation.
To test whether this defect in NCM binding seen in the �mutant led to a defect in budding, cells were infected with wtor mutant virus and then labeled with [35S]methionine for 60min at 6 h postinfection (a time when only viral proteins aresynthesized) and then chased for 30, 60, or 90 min. Followingthe chase period, media and cell lysates were obtained, and theamounts of [35S]methionine-labeled M protein in budded viri-ons (released into the media) and in cell lysates (inside thecell) were determined. The amount of M protein in buddedvirions was then expressed as a fraction of the labeled M invirions plus the amount in cell lysates. The data from three ofthese experiments are depicted in Fig. 4A. For the wild-typevirus, the amount of M protein incorporated into virions in-creased steadily from just under 30% in the first half hour toalmost 50% at 90 min. The virus expressing the hydrophobic-loop mutant showed a similar steady increase in the amount ofM protein incorporated into virions over this time scale, but aslightly smaller fraction of the total M was incorporated (20 to38%). While this trend was seen at all times, it was not signif-icant within the margin of error of the assay.
Single-step virus growth experiments (Fig. 4B) showed thatthe virus containing the hydrophobic-loop mutations grew to aslightly lower titer (2 � 108 for the mutant virus versus 6 � 108
for the wt virus), but this was not statistically significant withinthe margin of error of the plaque assay. To further investigatethe small change in the virus titer, we purified virions from the
FIG. 5. Protein synthesis changes in cells infected with recombinant wt and hydrophobic-mutant viruses. (A) Cells were mock infected orinfected with recombinant wt control or hydrophobic (�M)-mutant VSV for the indicated times and labeled with 35S methionine for 10 min. Thecell lysates were electrophoresed on a 10% SDS-PAGE gel. A phosphorescence image of a representative gel is shown. (B) Quantification of hostprotein synthesis following virus infection. The error bars indicate standard deviations. (C) Quantification of viral-protein synthesis following virusinfection. (D) Cells were mock infected or infected with wt control, �M mutant, or a 2-amino-acid mutation (AVLA3AVQQ) for 8 h and thenlabeled and treated as described for panel A.
3706 CONNOR ET AL. J. VIROL.

media of rwt- and � mutant virus-infected cells, and the virionproteins were analyzed by SDS-PAGE and Coomassie bluestaining to determine whether there were differences in theamounts or the ratios of virus protein (Fig. 4C). Consistentwith the small drop in virus titer observed in the single-stepgrowth curve, we did see a difference in the total amount ofvirus proteins, with supernatants from the rwt virus-infectedcells showing approximately two to three times more viralprotein per ml of medium than the � mutant virus-infectedcells. There was no difference in the composition of the virusproteins between the mutant viruses, and the ratios of M pro-tein to G and N proteins were the same in both viruses (notshown).
Viruses with the � mutant M protein show defects in viraltranslation. Contrary to expectations based on previous data(9, 10), the recombinant M protein mutant virus showed onlyminor defects in M protein function related to virus assembly(Fig. 3C and 4). None of these had a large effect on virusgrowth (Fig. 3B). However, following the recovery of the re-combinant mutant virus, two dramatic phenotypes were noted:(i) the mutant virus showed defects in translation of viralmRNA and (ii) the virus showed a slower induction of apop-tosis in BHK cells. The defect in viral translation is illustratedin Fig. 5. Cells were infected at an MOI of 10 with recombinantwild-type VSV or with the mutant virus expressing the M proteinhydrophobic-loop M mutant. At increasing times postinfection,cells were pulse-labeled with [35S]methionine and analyzed bySDS-PAGE and phosphorimaging (Fig. 5A). The synthesis ofhost proteins is apparent as a ladder of dark bands in mock-infected cells and in lysates from early times postinfection,while viral proteins become apparent beginning at 4 h postin-fection (p.i.) and are largely the only proteins synthesized by8 h p.i. The recombinant wild-type virus and the virus express-ing the � mutant M protein showed similar abilities to suppresshost protein synthesis. Quantitation of the effect of virus in-fection on host protein synthesis (Fig. 5B) by analyzing threeseparate experiments showed that both viruses strongly in-hibited host gene expression, with equal kinetics. Strikingly,however, the hydrophobic-loop mutant appeared to synthesizeviral protein at a lower rate (Fig. 5A). Quantitation of viral-protein synthesis (Fig. 5C) showed that the rate of viral-proteinsynthesis was lower than that seen in the wild-type control at alltimes and was only 10 to 20% of the rate of the wild-type VSVat 8 h p.i., when viral-protein synthesis is near maximal in cellsinfected with wild-type virus (3). This was true both in BHKand in HeLa cells (data for BHK cells only are shown forsimplicity). To determine whether the change in promotingviral-protein synthesis could be caused by a subset of the mu-tations in the hydrophobic loop, we generated and recovered asecond mutant virus that contained only two amino acidchanges rather than four (AVLA3AVQQ). We then deter-mined the rate of protein synthesis of the rwt, � mutant, andAVQQ mutant at 8 h p.i. and found that the AVQQ mutantshowed strong viral-protein synthesis at that time, similar tothe wild-type virus (Fig. 4D).
One possible reason for lower viral-protein synthesis in the� mutant virus was a lower level of viral mRNA. The levels ofmRNA were determined by Northern blot analysis of M mRNAfrom mock-infected cells or cells infected with either wild-type ormutant virus for 6 or 8 h. These experiments (Fig. 6A) showed
that there was a greater accumulation of viral mRNA in cellsinfected with the recombinant mutant virus than in those in-fected with the wild-type control. Cells infected with the mu-tant virus accumulated at least twice as much mRNA as those
FIG. 6. Analysis of RNA accumulation, eIF2� phosphorylation,and mutation dominance in hydrophobic-mutant viruses. (A) TotalRNA was prepared from cells mock infected or infected with wild-typecontrol or hydrophobic-mutant VSV. The RNA was separated on a 1%agarose gel and transferred to a nitrocellulose membrane, and mRNAwas detected using a 32P-labeled probe against M protein mRNA.(B) Phosphorylation of eIF2� following virus infection. Extracts frommock- or virus-infected cells were resolved by SDS-PAGE, transferredto a nitrocellulose membrane, and probed for phospho-eIF2� (PhoseIF2�), total eIF2�, and actin. (C) Cells were infected with wild-typecontrol, hydrophobic-mutant virus, or both at the MOIs indicatedabove each lane; 8 h p.i., the cells were labeled with [35S]methionineand treated as described for panel A.
VOL. 80, 2006 ROLE OF THE VSV M PROTEIN HYDROPHOBIC LOOP 3707

infected with the wild-type virus, despite the fact that the rateof viral-protein synthesis was only half that of the wild-typevirus, demonstrating that mRNAs produced by this mutantvirus had a much lower translation efficiency than mRNAsproduced by wt virus.
A possible explanation for this reduced level of viral-proteinsynthesis in cells infected with the recombinant mutant virus isthe early stimulation of eIF2� phosphorylation as a host re-sponse to block viral-protein synthesis. Analysis of eIF2� phos-phorylation (Fig. 6B) in BHK cells following infection witheither wt or mutant virus showed that there was little if anydifference in the phosphorylation of this translation initiationfactor between the wild-type and mutant viruses. Both virusesinduced very little eIF2� phosphorylation at early times postin-fection, with increased phosphorylation becoming detectableabove the mock-infected control only at 8 h p.i., after thedefect in viral-protein synthesis was apparent.
An additional question about the decreased protein synthe-sis phenotype of the � mutant was whether the weak viral-protein translation represented a loss of function or whetherthe � mutation caused a dominant block of virus translationthrough an unknown mechanism. To test this, we performedcoinfections with the wild-type virus and the � mutant virus.Our expectations were that if the � mutant virus was causing a
dominant block of virus translation, the coinfection would dis-play the � mutant translation phenotype. If the � mutantmutation resulted in a loss of function, the coinfection wouldshow strong protein synthesis, like the wild-type virus. Figure6C shows an autoradiograph of [35S]methionine-labeled pro-tein extracts from wt VSV and � mutant VSV coinfectionslabeled at 8 h p.i. Coinfections were done using the � mutantat a constant MOI of 10, while the rwt virus used to coinfectwas varied from an MOI of 10 to 1. In cells that were infectedwith rwt virus alone, robust viral-protein synthesis was seen,and in viruses infected with the � mutant virus, reduced viral-protein synthesis was observed, as expected. In the cells thatwere coinfected with the wt and � mutant viruses, the strongprotein synthesis phenotype was always observed, demonstrat-ing that the protein synthesis defect appears to be a loss offunction in this M mutant virus that does not interfere with thestrong protein synthesis encouraged by the wild-type virus.
Virus with � mutant M protein shows slower induction ofCPE and cell death. A second unanticipated phenotype of the� mutant virus was that cells infected with the virus showeddelayed cytopathic effect (CPE) (cell rounding) following in-fection. This is illustrated in Fig. 7A, which shows images ofBHK cells infected with either the hydrophobic mutant virus orthe wild-type control virus at 10 h p.i. Almost all cells infected
FIG. 7. Apoptosis induction in cells infected with wt and hydrophobic-mutant recombinant viruses. (A) Phase-contrast images of BHK cellsinfected with hydrophobic-mutant virus (left) or wt control virus (right) at an MOI of 10 for 10 h. (B) Cells were infected with wild-type controlor hydrophobic-mutant virus and imaged by time-lapse microscopy (see Materials and Methods) for 48 h. Cell rounding is expressed as a fractionof the total number of cells in the field and as a function of time. (C) The cumulative fraction of cells entering apoptosis as determined bymembrane blebbing and cell lysis consistent with apoptosis plotted as a function of time. The data in panels B and C are averages of three separateexperiments with 150 cells per field. (D) Cells were mock infected or infected with wild-type control virus or hydrophobic-mutant virus for 8, 12,and 24 h. The cells were then lysed an analyzed for caspase 3-like activity using a fluorogenic substrate. The data are the averages of six separateexperiments � standard deviations.
3708 CONNOR ET AL. J. VIROL.

with the wild-type virus had rounded at this time, while amongthe cells infected by the � mutant virus there were still a largenumber of cells that had not yet become rounded. Immuno-fluorescent labeling of these cells for expression of VSV Gprotein showed that all cells were infected and expressing viralproteins (data not shown). We used time-lapse microscopy toimage and count the cells that were undergoing cell roundingand other signs of cell death. Quantitation of three experi-ments is shown in Fig. 7B for cell rounding and 7C for mem-brane blebbing and cell rupture. There was a small lag in cellrounding seen in cells infected with the hydrophobic-mutantvirus. While 50% of cells infected with wild-type virus showedcell rounding by 4 h p.i., a similar proportion of rounding incells infected with the hydrophobic-loop mutant did not occuruntil 15 to 20 h p.i. (Fig. 7B), a time at which 90% of wild-typecontrol-infected cells were rounded. The onset of membraneblebbing showed an even greater lag. In cells infected by wild-type virus, 50% of the cells showed signs of membrane bleb-bing and cell rupture by 35 h p.i. and all had undergone mem-brane blebbing by 48 h p.i. (Fig. 7C). In contrast, cells infectedwith virus expressing the � mutant M protein showed littlemembrane blebbing at 35 h p.i., and only 40% of these cellsblebbed by 48 h p.i. Taken together, these data suggestedthat while the � mutant M protein-expressing virus was stillcapable of inhibiting host cell gene expression, there was asignificant delay in the induction of other host responses,such as cell rounding and death. This was confirmed byanalysis of caspase 3 activity using a fluorogenic caspase 3substrate. These experiments (shown in Fig. 7D) showedthat there was less caspase 3 activity in the � mutant virus-infected cells at 8, 12, and 24 h p.i.
Virus with � mutant M protein shows decreased replicationin MDBK cells. The finding that the mutant VSV grew to titerssimilar to those of the wt in BHK cells but displayed decreasedprotein synthesis led us to characterize the growth of VSV inMDBK cells. Replication of VSV in BHK cells has been shownto be very robust, and mutants of VSV that synthesize less
protein than wild-type VSV often show little or no attenuationin these cells (12). Other cells, however, more effectively re-strict the replication of attenuated viruses (31). To determinewhether the � mutant virus displayed attenuation, we per-formed single-cycle and multiple-cycle growth experiments onthe wild-type and � mutant viruses in MDBK cell lines. Cellswere infected at an MOI of 10 for single-cycle experiments andat an MOI of 0.1 for multiple-cycle experiments. Plaque assaysof supernatants from either high or low MOI infection taken at24 h p.i. (Fig. 8A) showed that wt virus grew to titers of 3 �107, while the mutant virus grew to titers of 1 � 106 or 5 � 105,respectively, demonstrating that there was significant attenua-tion of virus growth in this mutant. Analysis of virus growth byWestern blotting of cell extracts at 24 h p.i. (data not shown)showed significant accumulation of VSV G protein in cellsinfected with wt VSV but almost no detectable G protein incells infected with the mutant virus. Consistent with this,MDBK cells infected with the wild-type virus at high and lowMOIs showed significant CPE at 24 h p.i. MDBK cells infectedwith the � mutant virus showed some CPE, but it was signifi-cantly lower than that seen with wild-type virus at both highand low MOIs (Fig. 8B).
DISCUSSION
The results presented here show that aa 121 to 124 of theVSV M protein play an important functional role in regulatingviral translation and the induction of apoptosis in the contextof a viral infection. The original aim of these studies—to testthe role of this region in controlling self-association and asso-ciation with cellular membranes—has led to the conclusionthat this domain plays a minimal role in both of these pro-cesses. By analyzing M protein association using stopped-flowassays, we were able to document the initial self-associationevents of purified M protein and to show that there was verylittle difference in the rate of association (Fig. 1D). This arguesthat the hydrophobic nature of the aa 121-to-124 loop does not
FIG. 8. Replication of wt and � mutant virus in MDBK cells. Cells were infected with VSV or the � mutant recombinant virus at an MOI of10 or 0.1. At 24 h p.i., the cells were analyzed as follows. (A) Virus titers were determined from cells infected at high or low MOI. Wild-type control(■ ); hydrophobic mutant (�). The titers shown are the means (plus standard deviations) of the results from three separate experiments usingmedia from cells at 24 h p.i. (B) Phase-contrast imaging showing rwt-infected cells in the top images and cells infected by mutant virus in the bottomimages.
VOL. 80, 2006 ROLE OF THE VSV M PROTEIN HYDROPHOBIC LOOP 3709

contribute significantly to the self-association of M protein. Itis still possible that the region plays a role in conformationalchanges or a similar structurally important event leading toself-association, but the hydrophobic nature of the loop is notcritical for this process.
Analyses of M protein association with membranes led totwo different conclusions, depending on the experimental de-sign. Wt or mutant M proteins expressed by recombinant vi-ruses did not behave differently in their association with mem-branes. This suggests that the hydrophobic loop plays a minorrole in interacting with the membrane during virus infection,contrary to previous hypotheses (10). However, expression ofM protein in the absence of other viral components did showthat the mutant M protein was reduced in membrane associ-ation, results which were similar to earlier reports (10). Wefound that analyzing M protein association with membranes inthe absence of other viral components can lead to higher levelsof association of wt M with the membrane, which may makeminor differences in the mutant M protein more apparent.Alternatively, factors beyond the M protein’s inherent affinityfor membranes control the level of M protein found associatedwith membranes during virus infection. This hypothesis is par-ticularly attractive, since previous experiments suggested thatthere is a regulation of M protein association with the nucleo-capsid at the membrane (7). This regulation could be at thelevel of interactions with viral or host components that controlmembrane association.
An interesting discovery with this virus was that the muta-tion of the aa 121-to-124 hydrophobic loop of M proteinchanged the behavior of NCM complexes derived from themutant virus. Both wild-type and mutant M proteins showed adecreased ability to associate with NCM complexes from mu-tant viruses. There was a fivefold decrease in the half-maximalbinding in this assay when mutant NCM complexes were used.This decrease was not enough to affect the assembly of thevirus, since there was little if any change in viral titers. This isconsistent with results with another mutant M protein with atwofold decrease in affinity for NCM complexes that had noeffect on assembly (21). These results suggest that regionsbeyond the N terminus of M are important for the assemblyand stability of the NCM complex. The mechanism of thischange in M protein exchange is not clear, as it could eitherreflect the fact that NCM complexes containing mutant Mprotein are in a different conformation or that the mutant Mprotein now lacks an important region for the cooperativeassociation of M protein with nucleocapsids. Further biochem-ical and biophysical studies will be required to determine theexact nature of this alteration.
Incorporating the aa 121-to-124 mutation into a recombi-nant virus uncovered a previously unrecognized function of Mprotein in controlling virus translation. As illustrated in Fig. 5,mutation of the hydrophobic loop caused the virus to increasemRNA production, but there was less viral-protein synthesis.Of particular interest is the fact that the mutation appears toresult in a loss of M function (illustrated by the coinfectionexperiments) (Fig. 6C) and of the kinetics of viral-proteinsynthesis in the mutant virus. In cells infected with the wild-type VSV, viral-protein synthesis increased even as host pro-tein synthesis was decreasing. In cells infected with the mutantvirus, translation of viral messages started out at lower levels
than that of the wild-type virus and decreased as host proteinsynthesis decreased (Fig. 5B and C), suggesting that in thewild-type virus M protein acts to maintain translation of viralmessages, while host messages are inhibited.
When these data are considered along with previous studiesthat have shown the M protein to be important for blockinghost protein synthesis, they suggest that there are two ways inwhich the M protein interacts with the host translation appa-ratus. The M protein is important for blocking host proteinsynthesis (1, 4), but it is also a translational activator for viralmRNA, allowing or facilitating high levels of viral-protein syn-thesis. The high levels of mRNA evident in the M mutant virus(Fig. 6A) are likely an adaptive response of the virus—an effortto produce more protein in response to low levels of transla-tion. The behavior of the � mutant virus is similar to that of thepreviously identified S2 VSV mutant. This virus also showed anincreased expression of viral mRNA and a decreased synthesisof viral protein (6), the first demonstration that a VSV factorwas important for allowing viral-mRNA translation. The mu-tations that caused the S2 phenotype have not been published,but our results suggest that they may reside near or within thehydrophobic loop of the M protein.
In its failure to activate translation of viral mRNA, the �mutant virus is analogous to protein synthesis-deficient mu-tants in another virus system, namely, adenovirus. Tempera-ture-sensitive mutants of the 100,000-molecular-weight (100K)protein in adenovirus show a loss of viral late-gene expression,even though high levels of mRNA are synthesized (13). Recentstudies have shown that the 100K protein becomes an integralpart of an adenovirus-specific eIF4F complex during late viralinfection (5) and link late viral mRNA to this translation ini-tiation complex and to driving its translation (33). Given thesimilarities in mutation effects (mutation leading to an increasein viral-mRNA accumulation but a decrease in the rate ofprotein synthesis) on the adenovirus 100K proteins and the�M mutation, it is attractive to speculate that M protein mayplay a role in facilitating VSV mRNA translation.
The �M mutant virus also showed a delayed induction ofapoptosis. This delay could be due to two phenomena. Earlierreports showed that the ability of M protein to induce apop-tosis was linked to its ability to inhibit host gene expression(19). The aa 121-to-124 M protein mutant retains this ability,so it is unlikely that changes in apoptosis are due to this aspectof M function. In the context of a virus infection of BHK cells,the inhibition of host gene expression by M protein leads toslower apoptosis (8, 18). It is possible that the slightly increasedinhibition of host gene expression seen at 8 h p.i. (Fig. 5A andB) further delays apoptosis in these cells. Additionally, it islikely that the lower levels of viral protein slow the induction ofapoptosis, since other viral components in addition to the Mprotein are involved in the induction of apoptosis in VSV-infected cells (19).
The deficiencies noted in the mutant virus likely contributeto the attenuated growth that we see in MDBK cells (Fig. 7).Cells infected with the � mutant virus showed less CPE, lowerlevels of protein accumulation, and a 1- to 2-log-unit loss inviral titer compared to wild-type recombinant VSV (Fig. 7B).The fact that this attenuation was noticeable in MDBK cellsbut less significant in BHK cells is similar to other VSV mu-tants. The S2 mutant discussed above and the host range-
3710 CONNOR ET AL. J. VIROL.

restricted mutants that are defective in mRNA methylation(12, 31) show similar phenotypes, growing well in BHK cellsbut poorly in other cell types. Both of these mutants havetranslational defects equal to or more severe than those seen inthe � mutant virus, suggesting that the translation defect isresponsible for this attenuation. Mutants in Thieler’s virus thathave translation defects also show a similar pattern of stronggrowth in BHK cells and restricted growth in other cell types(27), so cell-type differences may play a role in the abilities ofdifferent RNA virus mRNAs to gain access to the translationapparatus.
The findings presented here, combined with earlier work onthe importance of the M protein in apoptosis and inhibition ofhost gene expression, highlight the complex role of M in viralreplication and pathogenesis. Mutations like the one describedhere may have a primary effect (such as changing viral or hosttranslation) that leads to secondary effects that influence virus-host interactions, like apoptosis, or virus assembly events, likeM-nucleocapsid association. Understanding these roles will in-volve further efforts to delineate the specific functions andinteractions that M controls but will provide important insightinto how virus proteins directly and indirectly influence virus-host interactions and virus replication.
ACKNOWLEDGMENTS
We thank Griffith Parks for his critical reading of the manuscript.This work was supported by grants RO1 AI015892 and RO1
AI052304 to D.S.L.
REFERENCES
1. Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S.Lyles. 2003. Ability of the matrix protein of vesicular stomatitis virus tosuppress beta interferon gene expression is genetically correlated with theinhibition of host RNA and protein synthesis. J. Virol. 77:4646–4657.
2. Black, B. L., R. B. Rhodes, M. McKenzie, and D. S. Lyles. 1993. The role ofvesicular stomatitis virus matrix protein in inhibition of host-directed geneexpression is genetically separable from its function in virus assembly. J. Vi-rol. 67:4814–4821.
3. Connor, J. H., and D. S. Lyles. 2005. Inhibition of host and viral translationduring vesicular stomatitis virus infection. eIF2 is responsible for the inhi-bition of viral but not host translation. J. Biol. Chem. 280:13512–13519.
4. Connor, J. H., and D. S. Lyles. 2002. Vesicular stomatitis virus infectionalters the eIF4F translation initiation complex and causes dephosphorylationof the eIF4E binding protein 4E-BP1. J. Virol. 76:10177–10187.
5. Cuesta, R., Q. Xi, and R. J. Schneider. 2000. Adenovirus-specific translationby displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBOJ. 19:3465–3474.
6. Davis, N. L., and G. W. Wertz. 1980. A VSV mutant synthesizes a largeexcess of functional mRNA but produces less viral protein than its wild-typeparent. Virology 103:21–36.
7. Flood, E. A., M. O. McKenzie, and D. S. Lyles. 2000. Role of M proteinaggregation in defective assembly of temperature-sensitive M protein mu-tants of vesicular stomatitis virus. Virology 278:520–533.
8. Gaddy, D. F., and D. S. Lyles. 2005. Vesicular stomatitis viruses expressingwild-type or mutant M proteins activate apoptosis through distinct pathways.J. Virol. 79:4170–4179.
9. Gaudier, M., Y. Gaudin, and M. Knossow. 2001. Cleavage of vesicularstomatitis virus matrix protein prevents self-association and leads to crystal-lization. Virology 288:308–314.
10. Gaudier, M., Y. Gaudin, and M. Knossow. 2002. Crystal structure of vesic-ular stomatitis virus matrix protein. EMBO J. 21:2886–2892.
11. Gaudin, Y., A. Barge, C. Ebel, and R. W. Ruigrok. 1995. Aggregation of VSVM protein is reversible and mediated by nucleation sites: implications forviral assembly. Virology 206:28–37.
12. Grdzelishvili, V. Z., S. Smallwood, D. Tower, R. L. Hall, D. M. Hunt, andS. A. Moyer. 2005. A single amino acid change in the L-polymerase proteinof vesicular stomatitis virus completely abolishes viral mRNA cap methyl-ation. J. Virol. 79:7327–7337.
13. Hayes, B. W., G. C. Telling, M. M. Myat, J. F. Williams, and S. J. Flint. 1990.The adenovirus L4 100-kilodalton protein is necessary for efficient transla-tion of viral late mRNA species. J. Virol. 64:2732–2742.
14. Jayakar, H. R., E. Jeetendra, and M. A. Whitt. 2004. Rhabdovirus assemblyand budding. Virus Res. 106:117–132.
15. Jayakar, H. R., K. G. Murti, and M. A. Whitt. 2000. Mutations in the PPPYmotif of vesicular stomatitis virus matrix protein reduce virus budding byinhibiting a late step in virion release. J. Virol. 74:9818–9827.
16. Kaptur, P. E., M. O. McKenzie, G. W. Wertz, and D. S. Lyles. 1995. Assem-bly functions of vesicular stomatitis virus matrix protein are not disrupted bymutations at major sites of phosphorylation. Virology 206:894–903.
17. Kaptur, P. E., R. B. Rhodes, and D. S. Lyles. 1991. Sequences of the vesicularstomatitis virus matrix protein involved in binding to nucleocapsids. J. Virol.65:1057–1065.
18. Kopecky, S. A., and D. S. Lyles. 2003. Contrasting effects of matrix proteinon apoptosis in HeLa and BHK cells infected with vesicular stomatitis virusare due to inhibition of host gene expression. J. Virol. 77:4658–4669.
19. Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein andanother viral component contribute to induction of apoptosis in cells in-fected with vesicular stomatitis virus. J. Virol. 75:12169–12181.
20. Lenard, J., and R. Vanderoef. 1990. Localization of the membrane-associ-ated region of vesicular stomatitis virus M protein at the N terminus, usingthe hydrophobic, photoreactive probe 125I-TID. J. Virol. 64:3486–3491.
21. Lyles, D. S., and M. O. McKenzie. 1998. Reversible and irreversible steps inassembly and disassembly of vesicular stomatitis virus: equilibria and kineticsof dissociation of nucleocapsid-M protein complexes assembled in vivo.Biochemistry 37:439–450.
22. Lyles, D. S., M. O. McKenzie, and R. R. Hantgan. 1996. Stopped-flow,classical, and dynamic light scattering analysis of matrix protein binding tonucleocapsids of vesicular stomatitis virus. Biochemistry 35:6508–6518.
23. Lyles, D. S., M. O. McKenzie, P. E. Kaptur, K. W. Grant, and W. G. Jerome.1996. Complementation of M gene mutants of vesicular stomatitis virus byplasmid-derived M protein converts spherical extracellular particles intonative bullet shapes. Virology 217:76–87.
24. McCreedy, B. J., Jr., and D. S. Lyles. 1989. Distribution of M protein andnucleocapsid protein of vesicular stomatitis virus in infected cell plasmamembranes. Virus Res. 14:189–205.
25. Newcomb, W. W., and J. C. Brown. 1981. Role of the vesicular stomatitisvirus matrix protein in maintaining the viral nucleocapsid in the condensedform found in native virions. J. Virol. 39:295–299.
26. Newcomb, W. W., G. J. Tobin, J. J. McGowan, and J. C. Brown. 1982. In vitroreassembly of vesicular stomatitis virus skeletons. J. Virol. 41:1055–1062.
27. Pilipenko, E. V., A. P. Gmyl, S. V. Maslova, E. V. Khitrina, and V. I. Agol.1995. Attenuation of Theiler’s murine encephalomyelitis virus by modifica-tions of the oligopyrimidine/AUG tandem, a host-dependent translationalcis element. J. Virol. 69:864–870.
28. Pornillos, O., J. E. Garrus, and W. I. Sundquist. 2002. Mechanisms ofenveloped RNA virus budding. Trends Cell Biol. 12:569–579.
29. Sarkar, G., and S. S. Sommer. 1990. The “megaprimer” method of site-directed mutagenesis. BioTechniques 8:404–407.
30. Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: anautomated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385.
31. Simpson, R. W., J. F. Obijeski, and M. P. Morrongiello. 1979. Conditionallethal mutants of vesicular stomatitis virus. III. Host range properties, inter-fering capacity, and complementation patterns of specific hr mutants. Virol-ogy 93:493–505.
32. Timmins, J., R. W. Ruigrok, and W. Weissenhorn. 2004. Structural studieson the Ebola virus matrix protein VP40 indicate that matrix proteins ofenveloped RNA viruses are analogues but not homologues. FEMS Micro-biol. Lett. 233:179–186.
33. Xi, Q., R. Cuesta, and R. J. Schneider. 2004. Tethering of eIF4G to adeno-viral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev.18:1997–2009.
VOL. 80, 2006 ROLE OF THE VSV M PROTEIN HYDROPHOBIC LOOP 3711




















