
Role of Phosphatldylinositol 3-kinase as an effector of Ras
216
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Role of Phosphatldylinositol 3-kinase as an effector of Ras


Role of Phosphatldylinositol 3-kinase as an effector of Ras
Pablo Rodriguez-Viciana
A thesis submitted to University College London at the University of London for the degree of
Doctor of Philosophy
November 1997
Signal Transduction Laboratory Imperial Cancer Research Fund
London

ProQuest Number: U641929
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U641929
Published by ProQuest LLC(2015). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States Code.
Microform Edition © ProQuest LLC.
ProQuest LLC 789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106-1346

11
Abstract
Ras genes are found mutated in about 30% of all human malignancies, being one of the most frequent mutations in human cancer. They code for 21 kiloDalton GTPases that act as molecular switches at a critical crossroad in the control of proliferation, differentiation and many other cellular processes. When in its active, GTP-bound state, Ras can interact with its downstream targets or effectors to carry out its biological function. The best characterised Ras effector is the Raf serine/threonine kinase, through which Ras activates the MAP kinase cascade. The work presented in this thesis, describes the identification of Phosphoinositide 3-kinase (PI 3-kinase) as an additional Ras effector.
Ras, when bound to GTP, interacts through its effector domain with the pi 10 catalytic subunit of PI 3-kinase. This interaction results in stimulation of the lipid kinase activity of PI 3-kinase, both in intact cells and in an in vitro reconstitution system. Furthermore, expression of the dominant negative mutant N17 Ras, inhibits EGF and NGF-induced activation of PI 3-kinase in intact PC 12 cells.
In order to study the contribution of PI 3-kinase to Ras function, mutations in the effector domain of Ras have been generated, that selectively impair the ability of Ras to interact with different effectors. The Y40C mutation allows Ras to interact only with PI 3-kinase, T35S and D38E mutants interact only with Raf and the E37G mutant interacts only with RalGDS. These partial loss of function mutants, together with activated and dominant-negative versions of PI 3-kinase and other effectors, have been used to study the effects of the different Ras effector pathways in several cell systems.
It is shown that Ras needs to activate simultaneously several pathways in order to efficiently transform fibroblasts. Although no single pathway is sufficient, PI 3- kinase makes a critical contribution to Ras transforming potential: inhibition of PI 3- kinase function inhibits loss of contact inhibition, loss of anchorage-dependence for growth and morphological transformation induced by Ras. PI 3-kinase, through the activation of the Rac GTPase, is also a critical mediator of the effects of Ras on the actin cytoskeleton. In addition, evidence is presented that activation of PI 3-kinase may play a role in protecting tumour cells harbouring Ras mutations from programmed cell death. The results suggest that the PI 3-kinase pathway may be an attractive novel target for therapeutic intervention in the treatment of tumours where Ras is involved.

I l l
Acknowledgements
I wish to thank Carmela Cales for her support and directing me to London after my difficult first steps in science in Spain. I am grateful to Julian Downward for giving me the opportunity to work in his lab and for his constant support ever since. Thanks to past and present members of the lab, Tanya, Laszlo, Sean, Syd, Bengt, Liz, Barbara, Asim, Stefan, Hendrik, Petra, Sandra, Marjatta, Satu, and Norman, for helpful discussions and sharing the trials and tribulations of benchwork. Special thanks to Pat Warne whose help has been essential for a lot of the work in this thesis. Thanks to Karin, Ignacio and Bart for help with the figures. I would also like to express my gratitude to the Imperial Cancer Research Fund and to the people in charge of its excellent facilities, for providing such an exceptional environment to do a PhD.
Special thanks to Alison Lloyd for helpful and invigorating discussions throughout this work, for her critical reading of this thesis and for the last three wonderful years (and Gregory is included in the last).
Thanks to my family in Spain, Eduardo, my sisters Marta and Celia, and my mother Pilar for their love, support and friendship.
This thesis is dedicated to my mother.

IV
Contents
Title page........................................................................................................... .............1Abstract.................................................................................................................................ilAcknowledgements.............................................................................................................. illTable of contents.................................................................................................................. ivList of F igures............................................................................................................. xiList of Tables........................................................................................................................xiiiA bbrev iations................................................................................................................... xiv
CHAPTER 1Introduction.................................................................... i
1.1 Historical background...........................................................................................11.1.1 Cancer as a genetic disease........................................................................... 1
1.1.1.1 Acutely transforming retroviruses........................................ 11.1.1.2 Transformation assays and identification of cellular oncogenes................................................................................................. 2
1.1.2 Identification of ras genes.............................................................................21.1.3 Identification of point mutations in ras......................................................... 31.1.4 Activated ras genes in tumours.......................................................... 31.1.5 Ability of ras to transform cells in co-operation with other oncogenes..............................................................................................................61.1.6 The biological function of normal Ras......................................................... 7
1.2 Structure and biochemical properties of R as............................................... 81.2.1 Mammalian ras genes and Ras primary structure....................................... 81.2.2 Guanine nucleotide binding and GTPase activity........................................ 9
1.2.2.1 Biological significance of nucleotide binding...............................91.2.2.2 Nucleotide binding sequences...................................................... 101.2.2.3 Intrinsic nucleotide exchange........................................................101.2.2.4 Intrinsic GTPase activity...............................................................101.2.2.5 Effects of activating mutations.............................................11
1.2.3 Effector domain............................................................................................. 111.2.4 Three-dimensional structure of R as............................................................. 121.2.5 Post-translational processing of R as............................................................13

1.2.5.1 Prénylation...................................................................................... 131.2.5.2 Carboxy-terminal proteolysis and méthylation............................. 151.2.5.3 Palmitoylation.................................................................................151.2.5.4 Famesyl Transferase inhibitors as anti-Ras chemotherapeutic drugs............................................................................... 16
1.2.6 Ras in lower eukaryotes...............................................................................171.2.6.1 Ras in Saccharomyces cerevisiae.................................................. 181.2.6.2 Ras in Schizosaccharomyces pombe........................................... 191.2.6.3 Ras in Drosophila melanogaster and Caenorhabditiselegans.......................................................................................................... 19
1.3 Ras activation........................................................................................................... 201.3.1 G A Ps................................................................................ 21
1.3.1.1 pl20G A P........................................................................................221.3.1.1.1. Interacting proteins......................................................231.3.1.1.2 pl20GAP as an effector...............................................24
1.3.1.2 N eurofibrom in..........................................................................241.3.1.3 GAPl fam ily........................................................................... 251.3.1.4 Regulation of GAP activity............................................................ 26
1.3.1.4.1 Lipids may regulate GAP activity..............................271.3.2 Guanine nucleotide exchange factors......................................................27
1.3.2.1 SOS................................................................................................. 291.3.2.1.1 Sos phosphorylation...................................................... 30
1.3.2.2 RasGRF.......................................................................................... 31
1.4 Ras effectors................................................... .......................................................... 311.4.1 R af.............................................................................................................. 32
1.4.1.1 Structure of Raf proteins................................................................331.4.1.2 Activation of Raf by Ras................................................................331.4.1.3 Negative regulation of raf activation by cAMP/PKA................... 34
1.4.2 RalGEFs..........................................................................................................351.4.3 R IN ..............................................................................................................351.4.4 A F6..............................................................................................................36
1.5 Ras-related proteins................................................................................................361.5.1 The Ras family.............................................................................................. 37
1.5.1.1 R-Ras and TC21............................................................................. 371.5.1.2 Rap proteins................................................................................... 37
1.5.1.2.1 Activation........................................................................ 38

VI
1.5.1.2.2 Effectors........................................................................391.5.1.3 Ral proteins...................................................................................40
1.5.1.3.1 Activation........................................................................ 401.5.1.3.2 Effectors..........................................................................40
1.5.2 The Rho family...............................................................................................401.5.2.1 Exchange factors, GAPs an GDIs................................. 41
1.5.2.1.1 GEFs............................................................................... 411.5.2.1.2 G A Ps.................................... 421.5.2.1.3 GDIs................................................................................43
1.5.2.2. Effector function......................................................................... 431.5.2.2.1 ROS production.......................................................431.5.2.2.2 Gene regulation...............................................................441.5.2.2.3 Phospholipid metabolism............................................... 451.5.2.2.4 Protein Kinases...............................................................461.5.2.2.5 Effectors without enzymatic activity........................47
1.5.3 The Rah fam ily......................................................................................471.5.4 The ARP fam ily.................................................................................... 481.5.5 The Ran fam ily......................................................................................49
1.6 PI 3-K inase ............................................................................................................... 501.6.1 Classes of PI 3-kinase....................................................................................51
1.6.1.1 Class I PI 3-kinases.............................................................. 511.6.1.1.1 Class lA PI 3-kinases.....................................................511.6.1.1.2 Class IB PI 3-kinases.....................................................54
1.6.1.2 Class II PI 3-kinase........................................................................ 541.6.1.3 Class III PI 3-kinases............................................................541.6.1.4 PIK-related kinases.........................................................................54
1.6.2 Targets of PI 3-kinases.................................................................................. 551.6.2.1 Protein kinase B/Akt....................................................................... 55
1.6.2.1.1 Targets of PKB/Akt......................................................571.6.2.2 ARP exchange factors
Grpl, Cytohesin and ARNO.......................................................... 571.6.2.3 B tk ................................................................................................... 581.6.2.4 SH2..................................................................................................581.6.2.5 Protein kinase C .............................................................................. 58

VII
CHAPTER 2Materials and Methods..............................................................................6o
2.1 M a te ria ls .....................................................................................................................602.1.1 Reagents..........................................................................................................602.1..2 Antibodies......................................................................................................602.1.3 Constructs..................................................................................................... 60
2.2 M eth o d s..................................................................................................................... .642.2.1 Mutagenesis2.2.2 Purification of proteins.........................................................................64
2.2.2.1 Expression and Purification of GST-fusion proteins inbacteria..........................................................................................................642.2.2.2 Wild type and V12 c-Ha-Ras.........................................................642.2.2.3 A38 Ras and RalA ..........................................................................652.2.2.4 PI 3-kinase...................................................................................... 65
2.2.3 PI kinase assays............................................................................................. 652.2.4 Interaction of inmobilized Ras with soluble PI3K........................................ 662.2.5 Interaction of soluble Ras with inmobilized PI 3-kinase.............................. 662.2.6 GAP A ssays............................................................................................ 662.2.7 Scintillation Proximity Assay (SPA)............................................................. 672.2.8 Reconstitution of Ras into liposomes............................................................ 672.2.9 PI 3-kinase assays in liposomes.................................................................... 672.2.10 Transfections................................................................................................ 68
2.2.10.1 Electroporation............................................................................ 682.2.10 2 Lipofection with Tfx-50......................................................682.2.10.3 Lipofection with lipofectamine...................................................68
2.2.11 Western blots and inmunoprecipitations2.2.12 Focus formation assays............................................................................... 682.2.13 Growth in soft agar assays................................................................692.2.14 Generation of PC 12 clones expressing N 17 H-Ras2.2.15 Generation of retroviruses and infection of target cells.............................. 692.2.16 Measurement of 3' phosphoinositides in intact cells.................................. 692.2.18 Retroviral infections and PC 12 neurite outgrowth assays.................... 702.2.19 Microinjections and immunofluorescence................................................... 70

VIII
CHAPTER 3In vitro interaction between Ras and PI 3- kinase............................................................................... 72
3.1 Introduction..............................................................................................................72
3.2 Results........................................................................................................................723.2.1 Interaction of Ras and PI 3-kinase in vitro....................................................723.2.2 The interaction between Ras and PI 3-kinase is direct................................. 773.2.3 Ras interacts with the pi 10 subunit of PI 3-KINASE..........................773.2.4 Determination of the affinity of interaction between Ras andPI 3-kinase............................................................................................................... 793.2.5 Determination of the site on pi 10 that binds Ras..........................................803.2.6 Effect of a point mutation in the Ras binding site of pi 10a on the interaction with Ras......................................................................................... 80
CHAPTER 4Activation of PI 3-kinase by Ras............................83
4.1 Introduction ..............................................................................................................83
4.2 Results........................................................................................................................ 834.2.1 Dominant negative Ras inhibits elevation of PI 3' phosphorylatedlipid levels by growth factors in intact PC 12 cells.................................................. 834.2.2 Ras elevates PI 3' phosphorylated lipid levels in intact COS cells........... 864.2.3 Effect of interaction of pi 10 with p85 and with Ras on PI 3-kinase activity in cells.............................................................................................. 884.3.4 Effect of the K227E mutation in the Ras binding site of pi 10 on regulation of enzymatic activity............................................................................... 894.2.5 Activation of PI 3-kinase by Ras in a purified liposomereconstitution system................................................................................................90
4.3 Discussion to Chapters 3 and 4 ..........................................................................944.3.1 Regulation of PI 3-kinase activity by R as.....................................................954.3.2 Mechanism of activation................................................................................ 97

IX
CHAPTER 5Biological contribution of PI 3-kinase to Ras function I: effects on transformation.........................loo
5.1 Introduction..............................................................................................................100
5.2 R esults........................................................................................................................ 1005.2.1. In vitro interaction of mutant Ras proteins with effectors....................1005.2.2 Activation of effectors by Ras mutants in intact cells............................ 1065.2.3 Contribution of Ras effector pathways to transformation.......................1065.2.4 p85 expression inhibits focus formation by V12 Ras................................... 1105.2.5 p85 expression inhibits growth in soft agar of Ras-transformedcells............................................................................................................................ 112
CHAPTER 6 Biological contribution of PI 3-kinase to Ras function II: effects on PC12 differentiationand the actin cytoskeleton....................................................................115
6.1 Introduction ................................................................................................................115
6.2 R esults..........................................................................................................................1156.2.1 Role of Ras effector pathways in PCI2 cell differentiation.....................1156.2.2 Involvement of Ras effector pathways in cytoskeletalreorganisation............................................................................................................1176.2.3 Effect of LY294002 on PDGF and Ras induced cellular PIP3levels..........................................................................................................................1236.2.4 Effect of p85 expression on tumour cell lines with mutations inRas genes...................................................................................................................125
6.3 Discussion to Chapters 5 and 6 .......................................................................... 1276.3.1 Mutations in the effector domain discriminate between differentRas effectors........................................................................................................1276.3.2 Role of the different Ras-activated pathways in transformation...............1276.3.3. Inhibition of Ras transformation by p85 expression.................................1296.3.4 PC 12 differentiation........................................................................................130

6.3.5. Ras regulation of the actin cytoskeleton.............................................1316.3.6 Mechanism of p85 inhibition of PI 3-kinase function................................... 1356.3.7 Role of Ras in protection from apoptosis.......................................................135
CHAPTER 7General discussion........................................................ 137
7.1 Ras regulates several effector pathways........................................................137
7.2 Contribution of Ras to PI 3-kinase activation............................................. 137
7.3 Contribution of PI 3-kinase to Ras function................................................1387.3.1 Several pathways downstream of Ras are needed in combinationto transform cells .............................................................................................. 1387.3.2 PI 3-kinase as the connection between Ras and R ac.................................... 1397.3.3 PI 3-kinase activation of Rac could have pleitropic effects...........................1407.3.4 PI 3-kinase has several targets other than R ac.............................................. 141
7.4 From Ras function to cancer therapyPI 3-kinase as a target of pharmacological inhibition............................141
R e f e r e n c e s ................................................................143

X I
List of Figures
1.1. Post-translational modifications of Ras........................................................................14
1.2. Structure of mammalian GTPase activating proteins (GAPs) for Ras.......................22
1.3. Structure of mammalian exchange factors (GEFs) for Ras........................................ 29
1.4. Structure of known and putative mammalian Rho-family GEFs.............................. 42
1.5 . Phosphatldylinositol cycle......................................................................................... 50
1.6. Classification of PI 3-kinase family members.............................................................52
1.7. Adaptor subunits for class lA PI 3-kinases.................................................................53
3.1. Association of PI 3-Kinase activity with Ras in vitro.................................................73
3.2. Inhibition of the association of PI 3-Kinase activity with Ras by peptidesderived from the effector domain of Ras............................. 74
3.3. Inhibition of the association of PI 3-KINASE activity with Ras bymonoclonal antibodies..........................................................................................................75
3.4. Association of p85/pl 10 with Ras in vitro...................................................................76
3.5. The association between Ras and PI 3-kinase is direct...............................................78
3.6. Ras associates with the pi 10 catalytic subunit of PI 3-kinase....................................78
3.7. Affinity of Ras binding to PI 3-kinase......................................................................... 79
3.8. Determination of the binding site for Ras on the pi 10 catalytic domain.................. 81
3.9. Mutation in pi 10a that blocks binding to R as............................................................ 82
4.1. Dominant negative Ras inhibition of growth factor stimulation of 3' phosphorylated phosphoinositide levels in intact PCI2 cells............................................ 84
4.2. Dominant negative Ras does not inhibit all agonist stimulated responses................85
4.3. Elevation of 3' phosphorylated phosphoinositides levels in intact COScells by Ras............................................................................................................................87
4.4. Effect of K227E mutation on Ras elevation of PIP3 levels in intact Coscells.........................................................................................................................................90
4.5. Tyrosine phosphopeptide stimulation of kinase activity of p85ot/GST-pl 10a towards PI (4,5)P2 containing liposomes........................................... :...................92
4.6. Effect of Ras.GTP on PI 3-kinase activity in the liposome system........................... 93

X II
5.1. In vitro binding of effector mutants............................................................................. 101
5.2. Sensitivity of Ras effector mutants to Neurofibromin and pl20GAP........................103
5.3. Comparison of Ras mutant binding to Ral.GDS and Raf............................................104
5.4. Effects of Ras mutants on effector activity in COS cells............................................105
5.5. Transformation of NIH 3T3 by Ras mutants............................................................... 107
5.6. Transformation of NIH 3T3 cells by Ras effectors..................................................... 108
5.7. Transformation of NIH 3T3 by Ras mutants in combination with Raseffectors..................................................................................................................................109
5.8. Inhibition of Ras transformation of NIH 3T3 cells by p85 overexpression.............. 111
5.9. Effect of p85 overexpression on growth in soft agar by Ras and Srctransformed cells................................................................................................................... 113
5.10. Effect of p85 overexpression on morphological transformation of Rasand Src transformed cells......................................................................................................114
6.1. Induction of neurite outgrowth in PC 12 cells by Ras mutants andeffectors................................................................................................................................ 116
6.2. Effect of Ras mutants on actin rearrangement in Porcine AorticEndothelial cells................................................................................................................... 118
6.3. Effect of N17 Rac on Ras and PI 3-kinase induced actin rearrangement.................119
6.4. Effect of LY294002 on Ras and PI 3-kinase induced actinrearrangement........................................................................................................................ 120
6.5. Effect of p85 overexpression on Ras-induced ruffling................................................121
6.6. Effects of PKC inhibitors on Ras-induced ruffling......................................................122
6.7. Effect of LY294002 and p85 overexpression on Ras-induced cellular PIP3levels.......................................................................................................................................124
6.8. Effect of p85 overexpression on apoptosis induced by loss of anchorage in pancreatic tumour-derived cell lines.................................................................................... 126
6.9. A model of the function Ras mutants and effectors in cellular signalling pathways.................................................................................................................................134
7.1. Effector pathways activated by Ras............................................................................139

X l l l
List of Tables
1.1. Common elements across species in the signalling pathways from tyrosinekinase receptors to the MAP kinase cascade via activation of Ras................................... 20
1.2. Effector proteins for Rho-family GTPases.................................................................. 44
4.1. Levels of phosphatldylinositol (3,4) bisphosphate (PI(3,4)P2) and phosphatldylinositol (3,4,5) trisphosphate (PIP3) in transiently transfected 32P metabolically labelled COS cells......................................................................................... 89
4.2. Reconstitution of Ras into liposomes...........................................................................91

X IV
Abbreviations
ATP adenosine 5'-triphosphateBcr breakpoint cluster regionbp basepaircAMP adenosine 3':5'-cyclic monophosphateC. elegans Caenorhabditis eleganscDNA complementary DNACMV cytomegaloviruscpm counts per minuteCRIB Cdc42/Rac interactive bindingD. melanogaster Drosophila melanogasterDAG sn-1,2-diacylglycerolDMEM Dulbecco's modified Eagle's mediumDMSO dimethyl sulfoxideDMA deoxyribonucleic acidE. coli Escherichia coliECL enhanced chemiluminescenceEDTA ethylenediamine tetraacetic acidEGF epidermal growth factorEGTA [ethylene-bis(oxyethylenenitrilo)] tetra-acetic acidErk extracellular signal-regulated kinaseFACS fluorescence-activated cell sortingFak focal adhesion kinaseFCS fetal calf serumFrap FKBP12-rapamycin associated proteinG protein guanine nucleotide-binding proteinGAP GTPase activating proteinGDI guanine nucleotide dissociation inhibitorGDP guanosine diphosphateGEF guanine nucleotide exchange factorGDS guanine nucleotide dissociation stimulatorGrb2 growth factor receptor-bound protein 2GRD Gap related domainGST glutathione-S-transferaseGTP guanosine triphosphate

XV
HEPES N-2-hydroxyethylpiperazine-N'-2-ethanesulphonic acidHPLC high-pressure liquid chromatographyI(1,4,5)P3 inositol 1,4,5-trisphosphate
IFN interferon
Ig immunoglobulinIPTG isopropyl-2-D-thiogalactopyranosideJak Janus kinaseJnk Jun N-terminal kinasekDa kilodaltonLPA lysophosphatidic acidM molarm metrem micro (10"^)m milli(10-3)mAb monoclonal antibodyMap kinase mitogen-activated protein kinaseMBP myelin basic proteinMek Erk kinasemTor mammalian target of rapamycinMW molecular massN aminoNGF nerve growth factorp-NPP r-nitrophenol phosphatep70S6k p70 S6 kinasePAGE polyacrylamide gel electrophoresisPak p21-associated kinasePBSA phosphate-buffered saline APGR polymerase chain reactionPdBu phorbol 12,13-dibutyratePDGF platelet-derived growth factorPH domain pleckstrin homology domainPI 3-kinase phosphatidylinositol 3-kinasePI(3)P phosphatidylinositol 3-phosphatePI(3,4)P2 phosphatidylinositol 3,4-bisphosphate
PIP3 phosphatidylinositol 3,4,5-trisphosphate
PI(4,5)P2 phosphatidylinositol 4,5-bisphosphatePKB protein kinase B, also termed Akt or Rac protein kinasePKC protein kinase GPEG phospholipase G

X V I
PORI partner of Rac 1PTK protein tyrosine kinasePTP protein tyrosine phosphataseRTK receptor tyrosine kinaseS. cerevisiae Saccharomyces cerevisiaeS. pombe Schizosaccharomyces pombeSapk stress-activated protein kinaseSDS sodium dodecyl sulphateSH src homology domainShip SH2-containing inositol 5-phosphataseSHP SH2-containing protein tyrosine phosphataseSHP-1 previously known as: SHPTP-1, SHP, HCP, PTP 1CSHP-2 previously known as: SHPTP-2, SHPTP-3, Syp, PTP2C, PTP IDSLP-76 SH2 domain-containing leukocyte protein of 76 kDaSos son of sevenlessSRE serum response elementSRF serum rsponse factorSTAT signal transducer and activator of transcriptionTCP ternary complex factorTGF transforming growth factorTLC thin layer chromatographyTNF tumour necrosis factorTris tris(hydroxymethyl)aminomethaneUV ultravioletWASP Wiscott-Aldrich syndrome protein

CHAPTER 1 INTRODUCTION1.1 Historical background
1.1.1 Cancer as a genetic disease
The notion that cancer is a genetic disease arose from several lines of evidence. The realisation that the phenotype of neoplastic cells is heritable, passing to daughter cells like a stable genetic trait; the finding that most carcinogenic substances are also mutagenic together with the observation that cancer cells frequently harbour abnormal chromosomes and that some hereditary syndromes predispose to some forms of cancer, all suggested that genes lay at the heart of carcinogenesis.
However, the first direct evidence that genes could transform a normal cell into a cancerous one was provided by tumour viruses. In the mid-1960's, a conditional mutant of polyoma virus was described in which cellular transformation was temperature- sensitive and the transforming potential was shown to reside within the viral DNA. However, viral replication was also affected, so it was impossible to determine whether transformation was a primary or secondary consequence of viral gene expression. The genome and replication cycle of acutely transforming retroviruses proved more simple and manageable and the study of their mechanism of transformation has been instrumental in the understanding of the molecular biology of cancer.
1.1.1.1 Acutely transforming retrovirusesAn acutely transforming retrovirus was first identified by Rous in 1912 as a
filterable, transmissible agent ( later identified as a retrovirus and named Rous sarcoma virus or RSV) which could induce sarcomas in chickens (Rous, 1912). It was more than 50 years later, that the transforming principle of the RSV was mapped by genetic and biochemical means to a small region within the viral genome that was dispensable for the virus replication. This fragment of DNA was referred to as a viral oncogene and named v-src.(Hanafusa et al., 1963; Martin, 1970; Wang et al., 1976)
A major finding was made by Stehlin et al. (1976), using hybridisation techniques with the transforming gene of RSV as a probe, when it was found that the genome of vertebrate cells carry homologous sequences. This led to the proposition that viral oncogenes derive from cellular genes which have been inserted or "transduced" into the retroviral genome. Several dozens other transforming retroviruses have been isolated and for each of the viral oncogenes identified there is a corresponding cellular gene. Among these were the ras genes.

The realisation that viral oncogenes had a cellular origin led to suggestions that the cellular genes could be involved in human tumours. It was speculated that they could become activated by somatic mutations that would mimic the changes imposed upon them during retroviral transduction. The development of gene transfer technologies during the 1970s allowed the testing of this hypothesis.
1.1.1.2 Transformation assays and identification of cellular oncogenesThe observation that transfected RSV DNA could produce transformation of
recipient cells (Hill and Hillova, 1971) led many to attempt to repeat the experiments using DNA isolated from human tumours in the hope of isolating transforming cellular genes. The development of efficient DNA transfection methods and the use of NIH 3T3, an immortalised mouse embryo fibroblast cell line, as a recipient cell for neoplastic transformation, led to the development of a successful assay for cellular transforming genes (Cooper and Neiman, 1981; Shih et al., 1979; Weinberg, 1981). These gene transfer studies provided the first indication that dominant acting oncogenes play a role in human (Krontiris and Cooper, 1981; Perucho et al., 1981; Pulciani et al., 1982; Shih et al., 1981) and chemically-induced animal tumours (Balmain and Pragnell, 1983; Eva and Aaronson, 1983; Guerrero et al., 1984; Sukumar et al., 1983). Subsequent analysis revealed that many of the sequences responsible for transformation represented mutated alleles of cellular ras genes. Since then ras genes have been the focus of intense research.
1.1.2 Identification of ras genes
The ras genes were first identified as the transforming genes of the Harvey and Kirsten strains of rat sarcoma viruses (Harvey, 1964; Kirsten and Mayer, 1967) later shown to have derived from the cellular rat genes H-ras and K-ras respectively (DeFeo et al., 1981; Ellis et al., 1981).The H-ras gene has been transduced into the genome of two other strains of retroviruses, the BALB/c murine and Rasheed rat, sarcoma viruses (Andersen et al., 1981; Rasheed et al., 1983)
The ras genes were independently isolated with the development of transfection assays that established the presence of sequences in the DNA of tumours capable of transforming immortalised fibroblasts. Many of these transforming sequences turned out to be mutated versions of ras genes. In the case of the human bladder carcinoma lines EJ and T24 and several chemically induced mouse squamous carcinomas, cloning of the transforming gene revealed it to be an activated form of c-H-ras, the cellular homologue of the viral Harvey ras gene (Balmain and Pragnell, 1983; Parada et al., 1982; Santos et al., 1982; Sukumar et al., 1983). Activated forms of K-ras were also identified in a wide variety of tumour cell lines including colon, lung, pancreas and bladder (Der et al..

1982; Shimizu et al., 1983a) N-ras, a third ras gene with no viral counterpart, was detected as the transforming gene in several human sarcoma cell lines (Hall et al., 1983; Marshall et al., 1982) and a neuroblastoma line (Page et al., 1989; Shimizu et al., 1983a) and its proto-oncogenic form was subsequently isolated from normal human DNA (Brown et al., 1984)
1.1.3 Identification of point mutations in ras
The analysis of the transforming potential of chimaeric DNA molecules between the oncogenic ras gene found in the human bladder carcinoma cell lines EJ and T24 (later shown to be the same) and the normal cellular gene and comparison of the sequence in this region, revealed that a single nucleotide change (G to T in codon 12), causing a single amino acid substitution (valine for glycine) was sufficient for oncogenic conversion of the gene (Reddy et al., 1982; Tabin et al., 1982; Taparowsky et al., 1982)
Since this initial finding all three ras genes have been found to be activated in various tumours as a result of a single mutation in either codons 12, 13 or 61 (Bos, 1989). Recently a novel activating mutation, an insertion of 3 nucleotides between codons 10 and 11, resulting in the introduction of an additional arnino acid (glycine), has been detected in DNA from a patient with myeloid leukaemia (Bollag et al., 1996). Viral ras genes (with the exception of the BALB/c gene) in addition to mutations at position 12, have a second activating mutation at position 59 (Dhar et al., 1982; Rasheed et al., 1983; Reddy et al., 1985; Tsuchida et al., 1982).
In vitro mutagenesis studies have further shown that substitutions at position 59, 63, 116, 117, 119 and 146 can also confer transforming activity.(Fasano et al., 1984; Sigal et al., 1986a; Walter et al., 1986) although these mutations have not been found in human tumours. Further in vitro analysis has revealed that the substitution of glycine 12 for any other amino acid ,except proline (Seeburg et al., 1984) and the substitution of glutamine 61 for any other amino acid, but proline and glutamate (Der et al., 1986), leads to transforming activity.
Understanding the effects of these amino acid substitutions on the biological activity of the Ras protein and how this can ultimately lead to malignant cell growth, has become the subject of intense investigation in cancer research.
1.1.4 Activated ras genes in tumours
The gene transfer assays which revealed that cultured tumour cells carry activated ras genes were superseded by less laborious methods that also allowed the direct analysis of human tumours. Hybridisation with mutation-specific

oligonucleotides (Bos et al., 1984; Verlaan-de Vries et al., 1986), RNase mismatch assays (Sjolander and Lapetina, 1992; Winter et al., 1985) or direct sequence analysis (Bos, 1988)have been greatly aided by the development of the polymerase chain reaction (PCR) for amplifying DNA.
Ras mutations are found in 25-30% of all tumours examined and together with inactivation of the p53 and pl6 tumour supressor genes, are the most frequent mutations in human cancer. The incidence of ras activation varies greatly according to the type of tumour examined. It is extremely high in pancreatic carcinoma, one of the most aggressively malignant forms of human cancer (where 95% of tumours investigated harbour ras mutations), as well as thyroid tumours (60%) and adenocarcinoma of the colon (50%), whereas, ras mutations are rare in several tumour types such as the adenocarcinomas of breast and ovarian cancers (Bos, 1989; Rodenhuis, 1992)
There is some correlation between tumour type and mutations in specific ras genes. In particular, K-ras mutations predominate in adenocarcinomas of the lung (Rodenhuis et al., 1988), pancreas (Almoguera et al., 1988; Grünewald et al., 1989; S mit et al., 1988) and colon (Bos et al., 1987b; Forrester et al., 1987) and N-ras mutations are found preferentially in acute myelogenous leukaemias (Bos et al., 1987a; Cutler et al., 1993; Damen et al., 1993; Farr et al., 1988b; Lane et al., 1993; Needleman et al., 1986) However, certain tumours appear to lack such specificity, for example in thyroid tumours H-ras, N-ras and K-ras mutations occur with similar frequency (Lemoine et al., 1988; Suarez et al., 1988).
It is not yet clear why in some cases activation of specific ras genes associates with some types of cancer. Ras genes may have different functions in different cell types, although there is no evidence to support this possibility. Another possibility is that different ras genes are differentially susceptible to carcinogens or other factors contributing to specific tumours. For example chromatin structure could influence the accessibility of the three genes to mutagens in a tissue specific manner.
It is now well established that tumours arise from the sequential accumulation of several genetic changes. Epidemiological (Peto et ah, 1975) and genetic studies (Kinzleral and Vogelstein, 1996) have suggested that at least 5 to 7 genetic changes are required for progression to a fully malignant cancer. In agreement with this, mutation of ras genes have been shown to represent one in a series of steps involved in malignant transformation. There is evidence that ras activation can play a role in both early and late stages of tumour formation depending on the tumour type.
The identification of ras oncogenes in certain types of pre-neoplastic growths suggest that they may play a role in the early stages of tumour progression. An example of this is colon cancer, which develops from small benign polyps which are often found in the colons of healthy individuals, and must go through various stages of progression

before conversion to invasive carcinoma. K-ras mutations have been detected in these pre-malignant polyps in some cases, as well as in secondary stage adenomatous tissue in several tumours (Farr et ah, 1988a; Forrester et ah, 1987; Vogelstein et ah, 1988). This suggests that ras mutations occur before conversion to malignant carcinoma at an early (or intermediate) stage of tumour development. However, while mutant ras genes are found in the premalignant polyps, the mutation does not appear to be the initial event, as the smallest polyps do not contain ras mutations. Mutation of the adenomatous polyosis coli (APC) gene appears to be the initiating lesion in this type of tumours (Kinzleral and Vogelstein, 1996).
Chemically induced tumours in rodents also provide support for a role of ras activation as an early event in tumour progression. A single dose of N-nitroso-N- methylurea (NMU), a short lived carcinogen, results in mammary tumours in rats, most of which carry an activated H-ras gene and the type of mutation found is consistent with NMU having been the causative agent (Sukumar et al., 1983). In the mouse skin carcinoma model, premalignant papillomas also often contain ras mutations (Quintanilla et al., 1986)
Evidence of ras mutations playing a role in later stages of tumour progression comes from the study of acute myelogenous leukaemia (AML). Activated ras genes are also found in 25% of myelodisplastic syndromes (MDS), the pre-malignant precursor form of AML, suggesting that in this case, ras mutations may be involved in the late progression to fully malignant AML (Hirai et al., 1987; Liu et al., 1987; Lyons et al., 1988; Yunis et al., 1989)
Ras mutations have been found in self-regressing tumour tissue such as keratoacanthomas of the skin, which rarely progress to exhibit a fully malignant phenotype (Leon et al., 1988) illustrating again that ras gene mutation is not sufficient to induce tumour development.
The occurrence of ras mutations in some human tumours has been associated with exposure to particular carcinogens, shedding some light on the mechanism of ras activation during tumour formation. For example, a striking correlation has emerged in the case of lung adenocarcinoma where K-ras mutations are detected in 30% of smoking patients but less than 5% of non-smokers, suggesting that one of the carcinogenic agents present in cigarette smoke may induce this mutation (Slebos et al., 1991) In malignant melanoma, where ras mutations have been observed with an incidence of 20%, mainly in the N-ras gene, (Albino et al., 1984; Van't Veer et al., 1989) there is also a good correlation between the presence of ras mutations and exposure to sunlight. The type of DNA lesion observed in the N-ras genes is also consistent with ultra violet (UV) light causing the mutation (Van't Veer et al., 1989).
Studies of animal tumour systems have provided clear evidence for the idea that ras oncogenes can act as direct targets for specific carcinogens (Barbacid, 1990).

Mammary tumours induced in rats by treatment with NMU often have a G/A transition in codon 12 of H-ras, a mutation expected by the methylating activity of NMU. In addition, mouse skin carcinomas induced by the chemical dimethylbenzanthracene (DMBA) contained ATT transitions in codon 61 of H-ras (Balmain and Pragnell, 1983; Sukumar et al., 1983) The knowledge that particular ras mutations may be induced by different carcinogens should further our understanding of the role played by these agents in the pathogenesis of cancer.
1.1.5 Ability of ras to transform cells in co-operation with other oncogenes
A mutated ras gene is sufficient to transform many immortalised rodent cell lines. Expression of activated ras leads to changes in cell morphology and to the loss of contact inhibition for proliferation and movement. The cells also overcome the requirement of anchorage for growth, can proliferate in low serum conditions, and form tumours when injected into athymic mice (Barbacid, 1987), all of which are hallmarks of in vitro transformation in culture.
However, unlike immortalised cell lines, ras oncogenes are not sufficient to transform primary cells. Transformation of primary rat embryo fibroblasts (REFs) by ras oncogenes requires the co-operation of a second oncogene such as c-myc, N-myc , adenovirus ElA, SV40 T antigen, dominant negative p53, human papillomavirus E7, and HTLV-1 tax (Eliyahu et al., 1984; Land et al., 1983; Parada et al., 1984; Ruley, 1983; Ruley, 1990). More recently D-type cyclins, and Cdc25A and -B have been added to the list of genes co-operating with ras in this system (Galaktionov et al., 1995; Hinds et al., 1994; Lovec et al., 1994). In addition the availability of primary fibroblasts from gene knock-out mice has also identified loss of p53, pl6, p21 and IRF-1 as mutations that can co-operate with ras in transformation (Serrano et al., 1996). It was realised that ras oncogenes could not overcome senescence (in fact, it has recently been shown that oncogenic ras induces growth arrest and senescence in a variety of primary cells) and needed other genetic changes, mimicked by co-expression of "immortalising" nuclear oncogenes, or acquired during passage by established cell lines, to fully transform cells. The molecular mechanisms of oncogene co-operation are just beginning to come to light (Lloyd et al., 1997; Serrano et al., 1997).
The hypothesis that multiple, collaborating oncogenes, including ras, are involved in tumour formation is further supported by data from transgenic mice experiments. In one study, two strains of mice were generated carrying the ras and myc genes respectively, both driven by the mouse mammary tumour virus promoter. When these strains were interbred creating dual carriers expressing both oncogenes; tumour formation was much more rapid in these offspring than in either parent strain, indicating

that the two oncogenes are acting synergistically in vivo (Sinn et al., 1987). Additional evidence has come from studies where midgestation mouse embryos were infected in utero with retroviral vectors carrying a raslmyc double oncogene. This induced more rapid tumour formation than infection with either single oncogene virus, again indicating that ras co-operates with other oncogenes in tumourigenesis (Porfiri et al., 1994). However, in all these cases the clonal and stochastic nature of the tumours indicated the requirement for still more tumourigenic events.
It is clear that the biological properties acquired by mutant ras genes can play an important role in the process of multi-step carcinogenesis. The extensive studies of ras from the perspective of its role in tumour formation highlight its importance in cellular growth mechanisms.
1.1.6 The biological function of normal Ras
Ras proteins are now known to play a critical role in cell growth and differentiation. The first demonstration that endogenous Ras proteins are required for the proliferation of mammalian cells involved microinjection of a neutralising rat monoclonal antibody, Y 13-259 (Furth et al., 1982) that inactivates the three cellular Ras proteins. Injection of the antibody into cells transformed by the mutant v-H-ras induces their morphological reversion to a normal phenotype (Kung et al., 1986). Furthermore, injection of the antibody into quiescent NIK 3T3 cells inhibited mitogenesis in response to serum and a wide array of growth factors (Mulcahy et al., 1985) indicating a requirement for functional endogenous Ras during cell growth.
Y13-259 injection experiments also established that transformation of NIK 3T3 cells by oncogenes encoding growth factor receptors and other tyrosine kinase products such as src,fins, and/^.s was dependent on cellular Ras activity, whereas transformation induced by the cytoplasmic oncogenes raf and mos was independent of Ras (Smith et al., 1986). These seminal experiments suggested that Ras was likely to function as a critical mediator in the signalling pathways initiated after growth factor receptor activation at the plasma membrane, leading to cell proliferation. They also suggested that serine/treonine kinases like the raf gene product may lie downstream of Ras (or function in a separate pathway).
Amphibian oocyte microinjection studies have provided evidence that Ras acts as a signal transducer during the process of oocyte maturation. Microinjection of activated Ras protein induces maturation (the progression from prophase to metaphase of meiosis) of Xenopus laevis oocytes, which is characterised by germinal vesicle breakdown (GVBD) (Birchmeier et al., 1985). Insulin-induced oocyte maturation is inhibited by Y 13-259 antibody, suggesting that Ras is necessary to mediate this response and is positioned downstream of the insulin receptor (Kom et al., 1987).

8
In addition to their role in normal cell growth, Ras proteins are known to regulate differentiation of several cell types often having opposing effects depending of the cell type. Activated ras genes can induce the terminal differentiation of the rat pheochromocytoma cell line PC 12 mimicking the effect of nerve growth factor (NGF) (Bar-Sagi and Feramisco, 1985; Guerrero et ah, 1986; Noda et ah, 1985). Microinjection of the neutralising anti-Ras antibody Y 13-259 blocks neurite formation induced by NGF but has no effect on cAMP-induced differentiation (Hagag et ah, 1986) This provided a strong indication that Ras proteins are involved in the cell differentiation response promoted by nerve growth factor in PC 12 cells.
On the other hand, activated ras genes have been shown to block rather than promote differentiation of other cell types like skeletal myoblasts (Olsen et ah, 1987) and mouse basal kératinocytes (Yuspa et ah, 1985). This inhibition of differentiation has been mirrored by studies in transgenic mice, which have suggested that this property of ras could play a role in tumour formation. Expression of ras in the acinar cells of the foetal exocrine pancreas blocks differentiation of the cells leading to the development of mass hyperplasia (Quaife et ah, 1987).
A picture thus emerged that normal Ras proteins can exert different biological effects in different cell types. The Ras inactivation studies described provided the first convincing evidence that Ras participates in normal cell proliferation and differentiation, perhaps utilising common molecular mechanisms to couple growth factor signals to their intracellular responses. This has led to intense investigation to understand how Ras functions as a crucial regulator of cell growth and differentiation.
1.2 Structure and biochemical properties of Ras
1.2.1 Mammalian ras genes and Ras primary structure
Mammalian cells contain three functional ras genes, Yi-ras-, K-ras- and N-ra^ which code for 21 kiloDalton proteins of 189 amino acids. The genes share a common structure with four coding exons (Barbacid, 1987; Lowy and Willumsen, 1993). The K- ras gene contains two alternative fourth exons, 4A ( which contains one codon less) and 4B. In mammalian cells, splicing predominantly results in K-Ras4B proteins . However, viral K-Ras proteins code for K-Ras4A.
In addition to mammals, ras and mj-related genes have been identified in a wide variety of organisms including birds, insects, plants and yeast and their protein products are strongly conserved. Functional homology between the ras genes of

different species has been shown in several cases. A human ras gene can rescue a RAS- deficient yeast and a modified yeast ras gene can transform fibroblasts.
The three Ras proteins are remarkably similar, with the first 86 amino acids being identical and the next 78 amino acids showing 79% homology. However there is virtually no homology in the region between amino acids 165 and 185. This highly variable domain in each Ras protein is however highly conserved between human and mouse suggesting that it could have some functional role. Deletion of this region in an activated ras gene does not affect its transforming ability, suggesting that it is not required for effector function.
The hypervariable domain includes sites necessary for plasma membrane localisation. The last four C-terminal amino acids of Ras form a conserved CAAX motif (where C is cysteine, A an aliphatic residue and X, any residue) necessary for post- translational processing of Ras (see later).
Ras proteins are expressed in every cell type and tissue studied. It should be noted that ras expression is often high in non-proliferating, terminally differentiated tissues such as brain and heart suggesting that Ras proteins could be involved in processes other than proliferation and differentiation.Localization: Ras and caveolae
Ras proteins are localised at the inner leaflet of the plasma rriembrane and more recently they have been shown to be enriched in caveolae. Caveolae are specialised microdomains of the plasma membrane which appear as flask-shaped invaginations under the electron microscope (Parton, 1996). Ras has been shown to copurify with caveolin and it has been proposed that caveolin could act as a scaffolding protein that would localise Ras and other proteins to caveolae (Song et al., 1996). Based on the observation that a wide array of proteins involved in signal transduction, including Ras and other proteins in the Ras pathway (Lisanti et al., 1994; Mineo et al., 1996), are enriched in caveolae, it has been proposed that compartmentalisation to caveolae could facilitate coupling of activated receptors to their downstream effector pathways (Anderson, 1993; Lisanti et al., 1994).
1.2.2 Guanine nucleotide binding and GTPase activity
1.2.2.1 Biological significance of nucleotide bindingAll Ras proteins bind the nucleotides guanosine diphosphate (GDP) and
guanosine triphosphate (OTP) and possess an intrinsic GTPase activity (Barbacid, 1987; Ravichandran et al., 1995; Regnier et al., 1989; Sweet et al., 1984; Waters et al., 1995). Ras proteins are now known to be biologically active in the GTP-bound form and inactive when bound to GDP. This idea had been predicted by analogy to previously characterised guanine nucleotide-binding proteins and was subsequently confirmed by

1 0
assessing the ability of Ras bound with different guanine nucleotides to activate yeast adenylyl cyclase in vitro (known to be a RAS effector in yeast.) (Field et ah, 1987) Experiments to measure the biological activity of Ras bound to non-hydrolysable GTP analogues using the induction of NIH 3T3 transformation, Xenopus oocyte maturation and PC 12 neurite induction as quantitative assays also led to the same conclusion (Satoh et al., 1987; Sawasdikosol et al., 1995; Trahey and McCormick, 1987)
1.2.2.2 Nucleotide binding sequencesMutational analysis of the H-ras catalytic domain has defined several segments that are required for guanine nucleotide binding (Willumsen et al., 1986). These include several motifs identified in Ras that also appear in other purine nucleotide-binding proteins (Valencia et al., 1991; Wittinghofer and Pai, 1991). The first motif, GXXGXGKS, encompassing residues 10 to 17 of Ras, binds the a- and p- and y- phosphates. Within
the second conserved sequence, DXXG, at residues 57 to 60 in Ras, the aspartate binds a magnesium ion and the glycine interacts with the y-phosphate of GTP. Another motif
NKXD, found at residues 116 to 119 of Ras, is involved in binding to the guanine ring. Residues 144 to 146 of Ras also interact with the guanine nucleotide.
1.2.2.3 Intrinsic nucleotide exchangeGuanine nucleotides bind to Ras with high affinity and the dissociation constants for both GDP and GTP are about 2 x 10" 1 iM (Lowy and Willumsen, 1993) During nucleotide exchange, the rate-determining step is nucleotide release (Hall and Self, 1986). indeed, association rates are almost diffusion limited. The spontaneous rate of nucleotide release is dependent on the concentration of magnesium ions (Mg^+) present. In the presence of high, free Mg^+ (5mM), the half-life of the Ras-GDP complex is about 60 minutes at physiological temperature, whereas, in low Mg^+ (0.5|iM), this half-life is less than 30 seconds (Hall and Self, 1986). Within the cell,
millimolar magnesium concentrations exist, thus the basal nucleotide exchange rate on Ras is slow. Also, GTP is present in a large excess to GDP in vivo so Ras that does become nucleotide-free will rapidly bind GTP, resulting in active protein. Several cellular exchange factors which accelerate the rate of nucleotide exchange on Ras, causing its activation, have now been identified (see section 1.3).
1.2.2.4 Intrinsic GTPase activityRas proteins have an intrinsic GTPase activity that results in the hydrolysis of the y- phosphate of the bound GTP, leaving Ras in the GDP-bound state. This reaction is very slow and estimates of the half-life for hydrolysis of GTP by normal Ras lie between 30 minutes and 2 hours (Barbacid, 1987; Lowy and Willumsen, 1993).

1 1
The high resolution structures determined (to 1.35Â) by crystallography, revealed that a water molecule (water 175), hydrogen-bonded to the carbonyl of threonine 35, is positioned near the y-phosphate group and is likely to act as the attacking nucleophile during hydrolysis. The reaction is thought to result in an inversion of the y-phosphate
configuration, an event common in many phosphoryl-transfer reaction mechanisms (Wittinghofer and Pai, 1991). Lysine 16 and the magnesium ion also play a role in catalysis and are both bound to the p- and y-phosphates. These interactions are likely to
lower the activation state of the reaction and stabilise a transition state intermediate as hydrolysis proceeds. Hydrolysis is accelerated by GTPase activating proteins (GAPs) (see section 1.3.1), which may act by confining the GTP-binding pocket to the optimal conformation for hydrolysis out of the several conformations that are possible. The GAP proteins could possibly also contribute additional side-chains to the active site, encouraging catalysis.
1.2.2.5 Effects of activating mutationsActivating mutations, that result in the accumulation of Ras proteins trapped in their GTP-bound form, affect residues involved in nucleotide binding. Such mutations either impair GTPase activity or increase the rate of nucleotide exchange. For example, the mutations found in ras oncogenes in human tumours at positions 12, 13 or 61, which localise within the protein to sites of interaction with the P and y phosphates of the bound GTP, are associated with impaired GTPase activity. In vitro mutagenesis studies show that substitution of normal glycine 12 or glycine 61 by various amino acids reduces basal GTP hydrolysis on Ras and abolishes GTPase stimulation by GAPs (Adari et al., 1988; Martin et al., 1990; Trahey and McCormick, 1987). A substitution of alanine by threonine at position 59, as found in the viral H-ras and K-ras oncogenes, has been shown to have both reduced GTPase activity and increased exchange activity.(Velu et al., 1989). Activating mutations introduced in vitro at residues 116, 117, 119 or 146, all of which interact with the purine base of GTP, cause a dramatic increase in the nucleotide exchange rate of Ras (Barbacid, 1987; Lowy and Willumsen,1993)
1.2.3 Effector domain
Deletions or point mutations within residues 32 to 40 abolish the transforming ability of v-H-Ras without altering its membrane localisation, nucleotide-binding properties or expression level (Sigal et al., 1986b; Stone et al., 1988; Willumsen et al., 1986). It was hypothethised therefore, that this region of Ras could be involved in the interaction with its cellular targets and this region was designated an effector domain. The subsequent identification of Ras interacting proteins has shown that mutations in

1 2
this domain do indeed affect interaction with effectors (and with GAPs). In fact, it has proved an invaluable criteria for the identification of ras targets.
This region is shared with other members of the Ras family such as Rap-1, R-ras and TC21, which may have different functions, and it has been proposed that residues lying between positions 26 and 45 (that are not conserved between this different subfamilies), may be involved in conferring specificity to the effector interactions (Marshall et al., 1991; Nassar et al., 1996; Nur-E-Kamal et al., 1992; Zhang et al., 1990).Consistent with its role in effector function as defined by genetic means, data from Ras crystal analysis reveals that the conformation of residues 30 to 38 differs considerably between the GDP- and GTP-bound forms of the protein (Milburn et al., 1990; Schlichting et al., 1990).
1.2.4 Three-dimensional structure of Ras
The three-dimensional structures of both the GDP- and GTP-bound forms of Ras (Brunger et al., 1990; de Vos et al., 1988; Milburn et al., 1990; Pai et al., 1990) as well as certain mutants (Krengel et al., 1990; Tong et al., 1989) have been determined by X-ray crystallography.The secondary structure of H-Ras comprises five a-helices and a central six-stranded P-sheet (with five parallel strands and one strand running antiparallel to these). These p-strands and helices are connected by ten loops, five of which play a role in nucleotide binding (LI, L2, L4, L9 and LIO). Loops 1, 2 and 4 form the active site surrounding the y-phosphate group and loops 9 and 10 bind the guanine base (Valencia
et al., 1991; Wittinghofer and Pai, 1991)..The structural differences between the GTP- and GDP-bound forms of Ras are
limited to two regions of the protein that lie close together and contact the bound nucleotide. The conformational changes that occur upon GTP hydrolysis are initiated by the disruption of hydrogen bonds between the y-phosphate and residues of L2 and L4
(Wittinghofer and Pai, 1991). The first region affected, residues 30 to 38 (now called switch I), encompasses L2 and part of P-strand 2, and coincides with the effector site
defined by genetic means (see earlier). The second region identified (switch 2) spans residues 60 to 76 which includes L4 and a-helix 2 and is highly mobile existing in
multiple conformations (Pai et al., 1990). This region includes the epitope recognised by the neutralising antibody Y 13-259 (Lacal and Aaronson, 1986). Although early studies showed that deletions in this region did not severely affect the transforming ability of v-H-Ras (Willumsen et al., 1986), more recent analysis has identified mutations in this second switch that do compromise ras function and differentially affect

13
interaction with various effectors (Moodie et al., 1995). Residues in switch 2 have also been implicated in binding to nucleotide exchange factors (Quilliam et al., 1996).
1.2.5 Post-translational processing of Ras
Ras proteins are located at the inner face of the plasma membrane (Willingham et al., 1980) Initially, the primary translation product (pro-p21 Ras) is synthesised in the cytoplasm (Ulsh and Shih, 1984) This precursor subsequently undergoes a series of post-translational modifications at its C-terminus, increasing the hydrophobicity of the protein and permitting membrane localisation of the mature protein (Newman and Magee, 1993).
Mutational analysis established that the C-terminal sequences of Ras and its conserved residue cysteine 186 are required for both membrane association and the transforming ability of activated Ras (Willumsen et al., 1984; Willumsen et al., 1984). This cysteine residue forms part of a CAAX motif which is known to represent a consensus sequence for post-translational processing and is also found in several other proteins (Brown and Goldstein, 1993). The steps involved in the post-translational processing of Ras are now well characterised (see figure 2).
First, a prenyl group, the isoprenoid farnesyl (a product of the cholesterol synthesis pathway) is linked to the cysteine residue at position 186 (Hancock et al.,1989). After this, the three terminal amino acids (AAX) are removed by proteolytic cleavage leaving cysteine 186 at the C-terminus which is then further modified by carboxyl méthylation (Fujiyama and Tamanoi, 1990; Gutierrez et al., 1989; Hancock et al., 1991). Finally, in the case of H-Ras, N-Ras and K-Ras-4A, a second lipid modification step occurs. Cysteine residues lying within the hypervariable region, upstream of cysteine 186, undergo acétylation resulting in the reversible attachment of the fatty acid, palmitic acid by a thio-ester linkage (Hancock et al., 1989).
1.2.5.1 PrénylationThe first processing step, protein prénylation, is now known to be a widespread
mechanism which also affects many other proteins including Ras-related proteins, yeast mating factors, nuclear lamins, the y-subunit of transducin, rhodopsin kinase and the
peroxisomal protein PxF (Zhang and Casey, 1996)Mammalian farnesyl transferase (FTase), is the enzyme responsible for
prénylation of pro-p21, transferring a farnesyl group from farnesyldiphosphate (DPP) to the cystein residue of the CAAX motif (Casey and Seabra, 1996). It is an aP
heterodimer and is a zinc metalloenzyme which also requires magnesium for optimal activity (Reichman et al., 1992). The a subunit is also a component of geranylgeranyl
transferase type 1 (GGTasel), which catalyses the transfer of a geranyl group to the
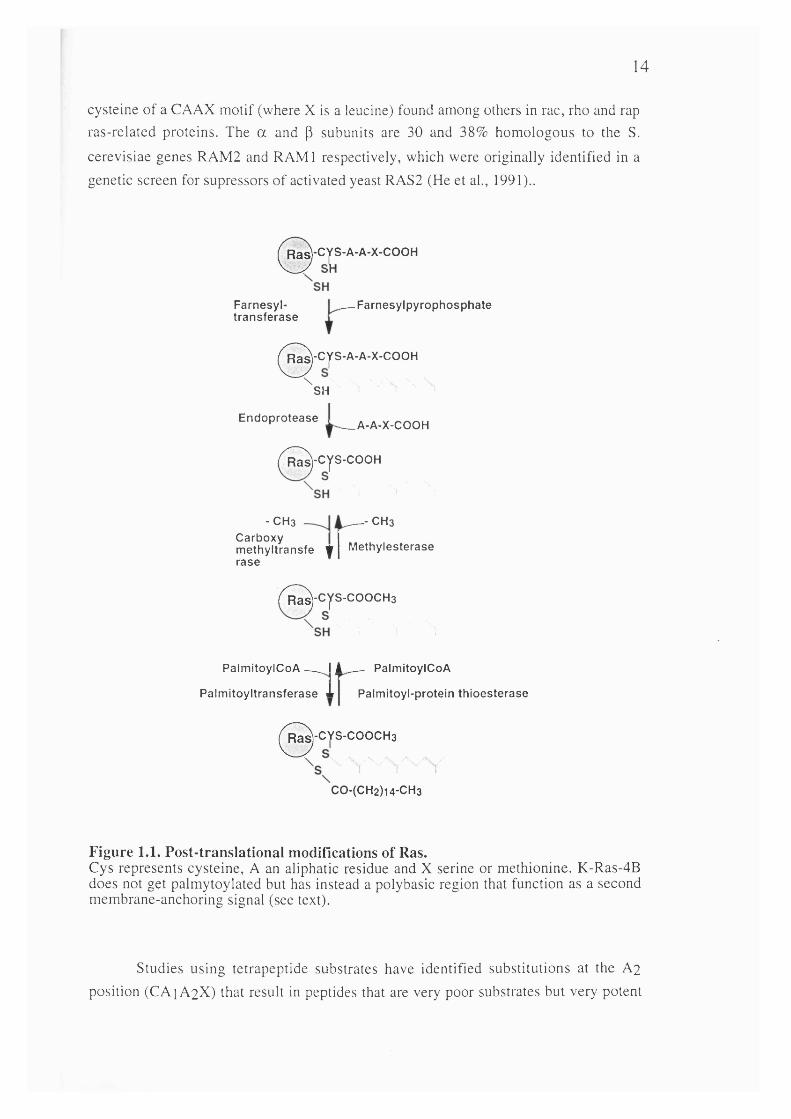
14
cysteine of a CAAX motif (where X is a leucine) found among others in rac, rho and rap ras-related proteins. The a and p subunits are 30 and 38% homologous to the S. cerevisiae genes RAM2 and RAMI respectively, which were originally identified in a genetic screen for supressors of activated yeast RAS2 (He et al., 1991)..
0 - c j sR^-Cys-A-A-X-COOH
Farnesyl- F arn esy ip y ro p h o sp h a tet r a n s fe ra se
^^^-CyS-A-A-X-COOH
^ S H ’ I
— A-A-X-CE n d o p ro te a se ^ a a v . c o O H
/^^-CyS-COOH
îfe Y
- CH3 - CH3Carboxy m ethyltransfe ra se
M ethylesterase
/"^-CyS-C00CH3
Palm itoy lC oA I A PalmitoylCoA
P alm itoy ltransfe rase X Palmitoyl-protein th io e s te ra se
^^^^-CYS-C00CH3
C0-(CH2)14-CH3
Figure 1.1. Post-translational modifications of Ras.Cys represents cysteine, A an aliphatic residue and X serine or methionine. K-Ras-4B does not get palmytoylated but has instead a polybasic region that function as a second membrane-anchoring signal (see text).
Studies using tetrapeptide substrates have identified substitutions at the A2 position (CA1A2X) that result in peptides that are very poor substrates but very potent

15
competitive inhibitors. One such peptide, CVFM, has served as the basis for the design of peptidomimetic inhibitors of FTase (see below).
In addition to promoting a correct subcellular localisation, prénylation may facilitate protein-protein interactions and regulate protein function (Casey and Seabra, 1996).. Consistent with this concept there is now evidence that farnesylation can affect the interaction of Ras proteins with both exchange factors and effectors (discussed later).
1.2.5.2 Carboxy-terminal proteolysis and méthylationProteolysis and carboxy-methylation steps contribute to efficient membrane
binding (Hancock et al., 1991). A proteolytic activity that cleaves the last three amino acids from farnesylated and geranylgeranylated peptides has been identified in microsomal membranes from mammalian cells but the gene has not been cloned as yet (Akopyan et al., 1994; Hancock et al., 1991). In S. cerevisiae two unrelated prenyl- proteases, named AFCl and RCEl, have been cloned. Deletion of the RCEl gene leads to a partial mislocalization of Ras2p within the cell and to a partial suppression of the heat-shock sensitivity conferred by activated RAS2 (Boyartchuk et al., 1997).
A membrane associated prenyl carboxyl metyltransferase activity has been detected in several mammalian tissues (Pillinger et al., 1994; Volker et al., 1991) It is enriched in the endoplasmic reticulum-microsome fraction and transfers the methyl group of S-adenosyl-L-methionine (AdoMet) to the cysteine exposed by the previous proteolytic step. Carboxy méthylation is a reversible step (Perez Sala et al., 1992) and has been proposed to play a role in signalling processes ((Leiser et al., 1995; Philips et al., 1993) although there is no evidence as yet for such a role in the case of the Ras proteins. Prenyl methylases have been cloned from S. cerevisiae (STE14), S.pombe (SpMam4) and Xenopus laevis (XMam4) (Imai et al., 1997), and shown to code for homologous proteins predicted to contain several membrane-spanning regions.
1.2.5.3 PalmitoylationH-ras that has undergone farnesylation, proteolysis and méthylation is still
mainly cytosolic and an additional modification step, which in the case of H- and N-Ras is palmitoylation, is required for Ras binding to the plasma membrane (Dudler and Gelb, 1996; Hancock et al., 1991). In H-Ras, Cys 181 and Cys 184 serve as palmitoylation sites (Hancock et al., 1989).K-Ras-4B does not get acetylated but has instead a stretch of six lysine residues (the polybasic domain) just upstream of the CAAX box, which work as an alternative

16
membrane association signal, presumably by favouring electrostatic interactions with negatively charged lipid head groups at the plasma membrane (Hancock et al., 1990).
Palmitoylation is achieved by estérification of cysteine thiol groups by palmitate. Unlike prénylation it is a reversible process and thus has the potential to be regulated (Mumby, 1997). Examples of proteins that exhibit agonist stimulated turnover of palmitate include the p-adrenergic receptor, Gs and endothelial nitric oxide synthase
(cNOS) but there is no evidence of such kind for Ras proteins as yet. Based on results obtained with eNOS (Garcia Gardena et al., 1996; Shaul et al., 1996) it has been proposed that palmitoylation could play a role in directing signalling proteins to caveolae (Mumby, 1997). Cycles of acylation/deacylation could regulate the lateral translocation of proteins, including Ras, between subdomains of the plasma membrane.
A membrane associated prenyl-palmytoyl transferase that acts on Ras has recently been purified (Liu et al., 1996) but its structure is not yet known. A protein with palmitoyl thiosterase activity that depalmitoylates Ras has also been identified and cloned (Camp et al., 1994) and shown to be a lysosomal protein (Verkrruyse and Hofmann, 1996).
It should be noted that the enzymatic steps discussed above, although responsible for membrane association, do not confer any specificity as to the type of membrane compartment to which the protein is targeted. For example, although Ras proteins are localised at the plasma membrane, another CAAX-containing protein, PxF, is found on peroxisomes (James et al., 1994). Most striking in this regard are the geranyl-geranylated Rab proteins, each of which is localised to distinct intracellular membranes (Novick and Brennwald, 1993). Additional factors must be involved in directing these proteins to specific membrane compartments. In the case of the Rab family, chimaeric analysis has revealed that the hypervariable C-terminal domain confers the specific membrane localization (Chavrier et al, 1991). Consistent with this possibility, mutations in Ras that drastically alter residues adjacent to the palmitoylated cysteins but do not abolish palmitoylation, result in proteins that accumulate in internal membranes (Willumsen et al., 1996).
Rab and Rho proteins are also known to interact with specific proteins or GDIs that regulate interaction with membranes (see later). No such proteins have yet been identified for the Ras family, but given the degree of conservation in the mechanisms by which the different GTPase families are regulated, it would not be surprising that similar proteins, as yet unidentified, may act on Ras.
1.2.5.4 Farnesyl Transferase inhibitors as anti-Ras chemotherapeutic drugsThe finding that membrane localization of oncogenic Ras is critical for its
transforming activity has led to a great interest in the development of inhibitors of the

17
enzymes involved in the post-translational processing of Ras for use in cancer chemotherapy. Most progess has been made with inhibitors of FTase but, no doubt, with the recent identification of the other enzymes involved in ras processing, they will also become the subject of extensive research.
Peptidomimetic inhibitors of FTase, based on modified CAAX peptides, inhibit Ras-dependent transformation of mammalian cells (James et al., 1993; Kohl et al.,1993) and slow tumour growth of Ras-transformed fibroblasts in nude mice at concentrations that are not toxic to the host (Gibbs et al., 1994; Hara et al., 1993; Kohl et al., 1994). A dramatic demonstration of the potential of FTase inhibitors in cancer chemotherapy has been shown in MMTV-H-ras transgenic mice which develop carcinomas of the mammary and salivary glands in a spontaneous and stochastic manner. Administration of the inhibitor (L-744,832) resulted in almost complete tumour regression without visible toxicity to the animal. Withdrawal of the inhibitor resulted in reappearance of the tumours which, in most cases, regressed again upon treatment (Kohl et al., 1995).
It is intriguing that the FTase inhibitors are not toxic to the cells at doses that block processing of H-ras and presumably other farnesylated proteins. Moreover, treatment of cells with the inhibitor under conditions that inhibit the processing of Ras did not affect the stimulation of MAP kinase by EOF in untransformed cells, a process known to be Ras-dependent (James et al., 1994). One possibility, is that geranylgeranylated relatives of Ras, such as R-Ras and TC21, may be able to substitute for Ras proteins in normal cell growth under certain circumstances (Graham et al., 1994), or that K-rasB, When FTase is inhibited, may serve as a substrate for GGTase-1 (James et al.,1995). K-RasB may in fact be resistant to FTase inhibitors (James et al., 1996; James et al., 1995; Lerner et al., 1995), a critical issue since K-RasB is the ras gene most commonly found mutated in human cancers. Another matter of concern is that at least some of the effects of FTase inhibitors may be the result of a block of the processing of farnesylated proteins other than Ras (like RhoB) (Lebowitz et al., 1995; Prendergast et al., 1994; Prendergast et al., 1995). Further evidence for this possibility comes from the observation that some cancer cells not harbouring oncogenic Ras can respond to FTase inhibitors (Nagasu et al., 1995; Sepp Lorenzino et al., 1995).
1.2.6 Ras in lower eukaryotes
In addition to mammalian cells, the regulation and function of Ras proteins has been studied in several yeast and invertebrate systems which are more amenable to genetic manipulation. Powerful genetic analysis techniques in these organisms have allowed the identification of genes involved in Ras-dependent pathways and yielded

18
much insight into the function of Ras. They have also highlighted the extraordinary degree of conservation in the signalling pathways regulated by Ras.In particular, the Ras signalling pathways of the yeast Saccharomyces cerevisiae and Schizosaccharomyces pombe, the fruit fly Drosophila melanogaster, and the nematode worm Caenorhabditis elegans and have undergone detailed investigation.
1.2.6.1 Ras in Saccharomyces cerevisiae
S. cerevisiae contain two Ras proteins, RAS 1 and RAS 2 which are essential for viability (disruption of both RAS 1 and RAS 2 genes is lethal) (Tatehell et al., 1984).In S. Cerevisiae, RAS proteins regulate adenylate cyclase (AC) which catalyses cyclic AMP production (Broach, 1991) cAMP plays an important role in the yeast response to nutrient deprivation and leads to a cAMP-dependent protein kinase cascade. When external nutrient supplies are limited, the cAMP concentration falls inside the cell leading to cell cycle arrest and, for diploid cells, sporulation. However, in strains containing an activated Val-19 RAS 2 mutant (equivalent to the mammalian Val-12 H-Ras), intracellular cAMP levels remain high despite starvation and the cells display abnormal phenotypes, including sensitivity to heat shock, loss of carbohydrate reserves, sensitivity to nutritional starvation and failure to sporulate (Broach, 1991).
RAS interacts with AC, the product of the CYRl gene, through a leucine rich repeat domain of AC (Colicelli et al., 1990; Minato et al., 1994; Suzuki et al., 1990) found in many other proteins (Kobe and Deisenhofer, 1994). Interaction of AC with a cyclase-associated protein (CAP), a protein identified in a genetic screen for suppression of the Vail9 RAS2 phenotype, is not required for binding of RAS to AC but greatly potentiates its activation (Shima et al., 1997).
Other yeast proteins identified that affect the RAS pathway include CDC25 which activates RAS and the IRA 1 and IRA 2 proteins which are GTPase activators all of which have mammalian equivalents (see section 1.3.1).
RAS has also been implicated in S. cerevisiae in regulating a MAP kinase module involved in pseudohyphal differentiation. Pseudohyphal differentiation, a morphogenetic pathway that involves Ste20 (a p65PAK homologue), S tell (MEKK homologue), Ste7 (MEK homologue) and the transcription factor Stel2 (Herskowitz,1995), is triggered by starvation of a/a diploid cells. Cells containing Vall9 RAS2 exhibit pseudohyphal development on rich medium (Gimeno et al., 1992), a process blocked by a dominant negative version of the yeast Cdc42 rho-family member (Mosch et al., 1996) suggesting the existence of a pathway analogous to that found in S. pombe (see below).

2 0
DROSOPHILA CAENORHABIDITIS MAMMALIAN
BOSS LIN-3 EGF
SEVENLESS LET-23 EGER
DRK SEM-5 GRB-2
CORKSCREW 7 SHP-2/PTP1D/SHPTP2? SLI-1 CBL
DOS 7 GAB-1
DSOS 7 SOS
GAPl 7 GAPs
DRASl LET-60 RAS
DKSR 7 KSR
DRAF LIN-45 RAF
DSORl MEK-2 MEK
ROLLED SUR-l/MPK-1 MAPK
TABLE 1.1. Common elements across species in the signalling pathways from tyrosine kinase receptors to the MAP kinase cascade via activation of Ras.
1.3 Ras activation
Ras proteins function as molecular switches cycling between an inactive GDP- bound and an active GTP-bound state. This transition is accompanied by a conformational change that allows the proteins to interact with their effectors and carry out their biological function. Ras proteins have very high affinity for guanine- nucleotides whilst having low intrinsic exchange and GTPase activities, and it became apparent long ago that by analogy with other GTP-binding proteins there had to be other cellular proteins regulating the activation of Ras in response to extracellular signals.
To date, two classes of proteins that jointly regulate the activation state of Ras (the GDP/GTP ratio) have been identified: Guanine-nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). GEFs promote dissociation of the bound nucleotide allowing binding of GTP (which in the cell is in excess over GDP) and

2 1
therefore lead to Ras activation. GAPs greatly stimulate the intrinsic GTPase activity of Ras and therefore lead to its inactivation.
1.3.1 GAPs
The existence of GAP proteins was first indicated by the observation that when Ras proteins were microinjected into Xenopus oocytes the rate of hydrolysis of GTP was much greater than the intrinsic GTPase rate of Ras measured in vitro (Trahey and McCormick, 1987). Subsequently, a GTPase stimulating activity was identified in extracts of Xenopus oocytes and mammalian cells. This protein was purified and cloned (Trahey et al., 1988; Vogel et al., 1988) and is now called pl20GAP. Two other types of RasGAPs are also known: Neurofibromin and three related molecules, GAPlm, GAplP^BP and GAPIII/R-RasGAP, referred to as the GAPl family. Apart from a conserved domain of around 300 amino acids (named GRD for GAP related domain) responsible for binding to and stimulating the GTPase activity of Ras, they are very different structurally, suggesting the existence of functional differences in their regulation and function.
Two other proteins, IQGAPl and 2, have a conserved GRD but they appear not to bind to or stimulate Ras GTPase activity, at least in vitro, (Brill et al., 1996) so the significance of the GRD domain is unclear and they will not be described here. Instead IQGAPs behave as effectors of Rho-family proteins, binding to the GTP-bound form (through a domain other than the GRD) of Rac and Cdc42 (see section 1.5.2.2).
Mutations that result in oncogenic activation of Ras (at positions 12, 13 and 61), result in proteins with decreased intrinsic GTPase activity. But in addition, they also render the protein insensitive to the GTPase stimulating effect of GAPs and it is now widely accepted that insensitivity to GAPs is the main mechanism by which activating mutations lead to the accumulation of Ras-GTP within the cell and therefore to the acquisition of transforming potential.
In addition to negatively regulating Ras, GAP proteins could also act as downstream targets of Ras signalling. All GAPs interact with the effector domain of Ras only when Ras is bound to GTP (Adari et al., 1988; Cales et al., 1988; Vogel et al., 1988) and activating mutations in Ras that render the protein insensitive to their GTPase stimulating effect can still interact with GAP proteins. Their in vitro behaviour is thus consistent with the properties expected of an effector protein. There is some evidence supporting this possibility, at least for pl20GAP, the best characterised of all Ras-GAPS (see below), but it cannot be excluded that a dual function in downregulating and mediating some aspect of Ras function could be a common property of all GAPs.

22
SH2 SH3 SH2 PH C2A GRD
pi 20 GAP
C2A G2B
(2GRD PH
> [ G A Plm
IRA GRD IRA
NF1
My NR IQ GRD
< > IQ-GAP
FIGURE 1.2. Structure of mammalian GTPase activating proteins (GAPs) for Ras. IQ-GAP, for which no RasGAP activity has been detected, contains a myosin-like region (My) and an aminoterminal region harbouring a unique repeated motif (NR). The other domains are described in the text.
1.3.1.1 pl20GAPp 120GAP was the first GAP protein to be identified and also the first protein
shown to directly interact with Ras. It binds to the effector domain of Ras and in
addition to the three Ras proteins it can also act as a GAP for the Ras-related GTPases
R-Ras and TC21. It also binds to Rapl with a 100-fold higher affinity than to Ras
although it cannot stimulate its GTPase activity.
p 120GAP has an amino-terminal hydrophobic domain, two SH2 domains
flanking an SH3 domain, a putative calcium-dependant phospholipid binding site (CalB
or C2A domain) and a PH domain. A 100 kDa isoform also exists that is only found in
the placenta and which lacks the amino-terminal hydrophobic domain but displays
similar catalytic proteins.
p l20G A P accelerates the rate of hydrolysis of GTP bound to Ras by up to
20.000-fold. However, the isolated catalytic domain can increase Ras GTPase activity
20-fold less efficiently than the full length molecule (Bollag and McCormick, 1992).
This suggests that the amino-terminal part of the molecule could positively regulate its
catalytic activity and that interaction with other molecules, perhaps through its SH2 and
SH3 domains (Bryant et al., 1996). Interestingly in several human basal cell carcinomas,
mutations in pl20GAP have been found in the C-terminal SH2 (Friedman et al., 1993)
although the biochemical and biological significance of these mutations is not known.

23
1.3.1.1.1. Interacting proteinsActivation of several tyrosine kinase growth factor receptors (e.g. PDGF-, EGF-,
CSF-1-receptors) leads to the tyrosine phosphorylation of pl20GAP (at position 457 in the case of the human protein) and in some cases to its association via its SH2 domains to the tyrosine phosphorylated receptor (e.g. PDGF- and CSF-1-receptors). Src-family proteins can also bind to and phosphorylate pl20GAP. Although tyrosine phosphorylation has not been shown to have any effect on pl20GAP catalytic activity in vitro, its interaction with membrane associated tyrosine kinases could result in its translocation from the cytosol to the membrane where it would have greater access to Ras.
In addition to tyrosine kinases, pl20GAP also interacts via its SH2 domains with two other proteins, p62 and p i90. p62 gets highly tyrosine phosphorylated in response to a wide array of extracellular signals as well as by expression of activated tyrosine kinases like v-Src, v-Abl, v-Fms. Interestingly, its tyrosine phosphorylation correlates with the transforming activity of the viral oncogene products as well as Bcr-Abl, a causative tyrosine kinase in human chronic myelogenous leukaemia. Conversely, p62 phosphorylation is decreased during integrin mediated cell substrate interaction (Sharma et al., 1996). The "real" p62 has been recently identified and named p62DOK (Carpino et al., 1997; Yamanashi and Baltimore, 1997). Apart from a putative PH domain it has no homology to known proteins and nothing is known about its function.
p i90 has at its amino-terminus several sequence motifs for guanine nucleotide binding and has indeed been shown to bind GTP. It also has a Bcr-domain that displays Gap activity towards the Rho GTPase (Settleman et al., 1992a; Settleman et al., 1992b) and could thus provide a point of cross talk between Ras and Rho GTPases. A 68 kDa protein, named G3BP, that interacts with the SH3 domain of pl20GAP only in growing but not quiescent cells has also been identified. This protein shares features in common with hnRNA-binding proteins (Parker et al., 1996).
pl20GAP has been shown to associate with several proteins through its CaLB/C2A domain (Davies et al., 1993). One of them, annexin VI, is a major calcium and phospholipid binding protein that translocates to the plasma membrane in response to increases in the levels of intracellular calcium. pl20GAP has also been shown to translocate to the plasma membrane in response to calcium elevation and this process was dependant on the CaLB domain (Gawler et al., 1995). It is therefore possible that the CaLB domain of pl20GAP could provide a point of cross talk between calcium- dependant and Ras signalling pathways.
The PH domain can bind in vitro to py subunits (Xu et al., 1996) suggesting the
possibility of regulation of pl20GAP by heterotrimeric G proteins. The PH and the CaLB domain have the potential to interact with lipids although this has not been yet

24
shown for pl20GAP. Such interactions could regulate their membrane localization and/or their activity (see below effect of lipids on GAP activity).
1.3.1.1.2 pl20GAP as an effectorSeveral lines of evidence, mostly indirect, suggest that pl20GAP, through its
non-catalytic segment, could mediate some effector aspect of Ras function. In atrial muscle cells pl20GAP is needed in combination with Ras to uncouple muscarinic receptors from the activation of potassium channels and a truncated molecule without the catalytic domain can bypass the need for Ras (Martin et al., 1992) . This suggests that interaction with Ras could induce a conformational change that would make the N- terminal segment available to interaction with other molecules. Although the biological significance of this finding to Ras function is not clear, it is noteworthy that the non- catalytic domain blocks fibroblast transformation by muscarinic receptors (Xu et al., 1994). This N-terminal region has also been implicated in co-operating with Ras in the activation of the fos promoter and the polyoma virus enhancer (Medema et al., 1992; Schweighoffer et al., 1992) and in the regulation of cell adhesion and the cytoskeleton (McGlade et al., 1993), with in the latter case, interaction through the SH2 domains with pl90RhoGAP being an attractive possible mechanism.
There is also evidence however, arguing against an important role of pl20GAP as an effector. There are, for example, effector mutants of Ras that fail to bind pl20GAP but are still transforming (Stone et al., 1993) and fibroblasts derived from pl20GAP-/- knock-out mice can still be transformed by Ras (van der Geer et al., 1997). Nevertheless, it is becoming increasingly apparent that Ras can simultaneously regulate multiple pathways with different phenotypes being regulated by different pathways (or combinations of pathways) in a cell type specific manner. A clarification of the role of pl20GAP in mediating some physiological function of Ras still awaits the better characterisation of the myriad of effects that Ras is known to elicit in different cell types.
1.3.1.2 NeurofibrominNeurofibromin, the product of the NFl gene also functions as a GAP for Ras.
NFl is the gene whose disruption is responsible for the human disorder neurofibromatosis type 1 (NFl) also known as von Recklinghausen's neurofibromatosis, one of the most common genetic diseases predisposing humans to cancer. It affects 1 in 3500 individuals and can result both from the inheritance of a defective NFl gene from an affected parent (30-40% of cases) but also as a result of sporadic spontaneous mutation in the gene (Bernards, 1995; McCormick, 1995). Clinical manifestations characteristic of this disease include cafe-au lait spots on the skin and Lisch nodules in

25
the eye (both due to abnormalities in melanocytes), benign neurofibromas and an increased risk of malignant tumours, primarily of neural crest origin.
Molecular cloning of the gene responsible for this disease revealed that it encodes a 250-320 kDa protein with a GAP-related domain, and was shown subsequently to stimulate the GTPase activity of Ras proteins. However, neurofibromin displays significant differences with pl20GAP in its biochemical properties. It has 20- fold higher affinity for Ras than pl20GAP but a 30-fold lower catalytic activity (Martin et al., 1990). There also is an alternative splice variant that contains an additional 21 amino acids in the GAP-related domain but has similar catalytic properties (Bollag and McCormick, 1992). Unlike the other Ras-GAPs, outside the GRD domain there are no other known functional domains, although it does share sequence similarity in regions surrounding the GRD to the IRAI and IRA2 yeast RAS-GAPs.
The finding that neurofibromin had Gap activity towards Ras led to suggestions that its deletion or inactivation in NFl tumours would result in the activation of Ras which would have a causal role in the disease. Indeed several cell lines derived from neurofibrosarcomas and schwammomas as well as leukaemic cells isolated from NFl patients displaying juvenile chronic myelogenous leukaemia (JCML) have constitutive high levels of Ras-GTP. Inactivation of Ras function (in the neurofibrosarcoma cell lines) either by microinjection of neutralising antibodies or by expression of the GRD domain of NFl leads to reversion of the transformed phenotype (Basu et al., 1992; DeClue et al., 1992), confirming the role of the Ras pathway in tumour formation in this disease. It should be noted however that in other tumour cell lines derived from neuroblastomas and melanomas from NFl patients, no increase in Ras-GTP levels have been detected (Johnson et al., 1993); although it cannot be ruled out that this negative result is related to the insensitivity of the assay or that Ras regulation is impaired in other ways (e.g. in response to specific growth factors (see later)).
It is not known whether, in addition to negatively regulating Ras, neurofibromin could also behave as an effector of Ras. Recent studies in Drosophila have suggested that neurofibromin could be involved in the regulation of a cAMP-dependant pathway in a manner independent of Ras (Guo et al., 1997; The et al., 1997). Interestingly neurofibromin displays most sequence similarity to IRAI and 2, which in yeast are necessary for the proper localization of yeast adenylate cyclase at the membrane. Whether these functions are conserved in mammalian cells and their relation to Ras signalling and the NFl syndrome will need further investigation.
1.3.1.3 GAPl family.Three other related proteins (around 70% identity), Gaplm (Maekawa et al.,
1994), GAPIP4BP (Cullen et al., 1995) and GAPIII/R-RasGAP (Baba et al., 1995; Yamamoto et al., 1995). display GAP activity for Ras. They are more closely related to

26
the Drosophila Gapl protein than to pl20GAP or Neurofibromin and are thus believed to be its mammalian counterpart. They all have C2A and C2B domains (which by analogy with synaptogamin2 are believed to be responsible for calcium-dependent and -independent phospholipid binding respectively) and a PH domain. Gaplm and Ga?IP4BP can bind inositol 1,3,4,5-tetrakisphosphate (IP4) with high affinity, with the PH domain being primarily responsible for this interaction. IP4 is thought to be generated by phosphorylation of inositol (1,4,5) triphosphate, one of the two products of phospholipase C (PLC) catalysed hydrolysis of PIP2, suggesting a possible connection between PLC/calcium signalling and Ras activation. In addition, based on the observation that other PH domains that bind IP4 can also bind PIP3 (e.g. the PH domain of Btk) it could be speculated that Gapl proteins could be regulated by the lipid products of PI 3-kinase. The observation that liposomes of a composition similar to that of the plasma membrane strongly inhibit GAP^P^BP and that this inhibition is reversed by IP4 suggests a possible mechanism of regulation inside the cell.
GAPlP^PP unlike all other Ras-GAPs can also stimulate Rapl GTPase activity. Interestingly its GAP activity on Rapl unlike that for Ras (see above) is not inhibited by phospholipid vesicles, suggesting that its GAP activity towards different Ras family members could be differentially regulated by lipid molecules (see also 1.3.1.4.1).
1.3.1.4 Regulation of GAP activity.Although increase in exchange factor activity seems to be mainly responsible for
Ras activation in response to most extracellular signals, regulation of GAP activity would also be expected to influence the activation state of Ras and there is clear evidence that GAP activity can be regulated in vivo in some instances. Phorbol ester stimulation of T-cells and erythropoietin stimulation of HEL cells leads to increases in Ras-GTP that correlate with an inhibition of GAP activity (Downward et al., 1990; Torti et al., 1992). Conversely, increased GAP activity has been observed in fibroblasts growing at high density (Hoshino et al., 1988). The fact that all three types of Ras-GAPs have very different structure apart from the GRD suggest that they could be differentially regulated. Some possible mechanisms have already been described and several other possibilities will be discussed below, although in all cases the in vivo significance needs further clarification.
Futher evidence of spatial and/or allosteric regulation of different GAP proteins comes from studies in cells in which the NFl or pl20GAP genes have been deleted, by spontaneous mutation in humans in the case of NFl or in knock-out mice of pl20GAP and NFl genes. As mentioned earlier some (but not all) cell lines derived from NFl human tumours have increased Ras-GTP levels although extracts from those same cells show normal GAP activity in vitro.

2 8
therefore Ras activation. GEFs proved difficult to purify and their eventual isolation relied on genetic studies of Ras function in lower eukaryotes.
In S. cerevisiae the CDC25 gene product was genetically mapped as being upstream of RAS and a 450 amino acid domain was mapped that displayed catalytic exchange activity in vitro. S. cerevisiae have a second protein , SCD25, with a region of strong homology to the catalytic domain of CDC25, that also acts as a RAS-GEF while the Ste6 gene product is a CDC25 homologue and Rasl-GEF in S. pombe.
Mammalian homologues of CDC25 were isolated using either a PGR based cloning strategy using primers to conserved regions of yeast CDC25 sequences or genetic complementation of CDC25-defective yeast strains. Strongly related genes named Ras-GRF or CDC25Mm (referred to henceforth as Ras-GRF) were thus isolated and shown to encode proteins with GEF activity for Ras (Chen et al., 1993; Martegani et al., 1992; Shou et al., 1992). These proteins are expressed exclusively in the brain suggesting the existence of other Ras-GEFs and genetic studies, this time in Drosophila, again proved instrumental for the identification of other mammalian Ras-GEFs.
In D. melanogaster, genetic studies of the Ras pathway in the development of the R7 photoreceptor cell identified the Son of Sevenless (SOS) gene product as a protein acting downstream of the Sevenless tyrosine kinase-receptor and upstream of Ras that had a CDC25 homology domain (Simon et al., 1991). Low stringency hybridisation allowed the cloning of two related but distinct mammalian homologues, Sosl and Sos2, (Bowtell et al., 1992; Chardin et al., 1993), that unlike Ras-GRF are ubiquitously expressed. Outside the catalytic CDC25 homology domain Sos and Ras- GRF differ considerably in their structure, suggesting differential regulatory mechanisms. Intriguingly both Sos and RasGRF contain Dbl-homology domains which in some cases have exchange activity for Rho/Rac GTPases. Although no such function has yet been found for these domains it raises the interesting possibility that RasGEFs may function as dual specificity exchange factors.
A third type of molecule, C3G, with a Ras-GEF homology domain was cloned as a protein binding to the SH3 domain of the adaptor molecule Crk (Tanaka et al.,1994). Although the C3G catalytic domain can complement loss of CDC25 function in yeast it is not yet clear whether C3G acts as a GEF for Ras or for other Ras-related proteins like Rapl (Gotoh et al., 1995).
Another unrelated molecule, SmgGDS, can catalyse nucleotide exchange on K- RasB but not H- or N-Ras. It shows only weak homology to the CDC25 catalytic domain and has instead 11 Armadillo repeats similar to those found in catenins and other proteins located at adherens junctions. In vivo SmgGDS can co-operate with wt- K-Ras to induce c-fos promoter expression and morphological transformation of NIH 3T3 cell (Fujioka et al., 1992). SmgGDS has however a broad specificity towards other Ras-related GTPases containing a polybasic domain like Rap 1, Rho A, Rac and Cdc42,

29
and in addition to exchange activity it inhibits the interaction of all these proteins with
membranes. Very little is known about SmgGDS and its in vivo contribution to the
activation of Ras although it could be speculated that it is involved in the co-ordinated
activation (and/or sub-cellular translocation) of several Ras-related GTPases in adherens
junctions.
PH CC IQ
DM PH Ras GEF proline
DM PH
SOS
Ras GEF
Ras GRF
proline Ras GEF
C3G
FIGURE 1.3. Structure of mammalian exchange factors (GEFs) for Ras.CC, coiled-coiled domain. DH, dbl-homology domain. The other domains are described in the text. It should be noted that no Ras-GEF activity has been detected in vitro for C3G.
1.3.2.1 SOSMammalian cells contain 2 related Sos genes, Sosl and 2, and Sosl has in
addition several isoforms. Sos genes code for 150 kDa proteins that contain a central
catalytic region with homology to CDC25, an N-terminal region with a Dbl-homology
domain and a PH domain, (which have been implicated in membrane localization and
regulation of activity in vivo), and a C-terminal proline rich tail that binds to SH3
domains of the Grb2, Grap, Nek and Crk adaptor proteins.
Genetic studies in Drosophila and C.elegans and biochemical studies in
mammalian cells have outlined a possible mechanism for the activation of Ras by
tyrosine-kinase receptors (Feig, 1993; Schlessinger, 1993) which involves in the most
basic model, the activated receptor, the adaptor molecule Grb2 and Sos.
Sos is normally found in the cytoplasm, bound through its C-terminal tail to the
SH3 domains of Grb2. Growth factor binding to its receptor (e.g. EGF-receptor) induces
dimerization and autophosphorylation which creates specific phosphotyrosine-
containing docking sites for SH2 domain containing proteins including Grb2. The
mechanism by which activated receptor recruitment of Grb2-Sos complexes catalyses

30
activation of Ras is still poorly understood but could involve both translocation of Sos from the cytosol to the membrane where Ras is located and/or allosteric regulation of Sos enzymatic activity upon complex formation (or by another signal at the plasma membrane). There are however numerous variations on this theme.
Although direct binding of Grb2 to the activated receptor can occur in some cases there are numerous alternative ways by which Grb2-Sos complexes can be recruited to receptor complexes through intermediate tyrosine phosphorylated adaptor molecules. Proteins like She, IRS-1, Gab-1, FRS2/SNT and the tyrosine phosphatase PTP1D/SHP-2/SHPTP2, can bind through SH2 or PTPB domains to the phosphorylated receptor, while at the same time provide binding sites for the SH2 domain of Grb2. In many cases, a single receptor can recruit Grb2-Sos through several mechanisms simultaneously.
Seven transmembrane receptors can also lead to Ras activation in some cell types through less well characterised mechanisms, that may however converge in some cases with those used by tyrosine kinase receptors. Thrombin, lysophosphatidic acid (LPA), m2 muscarinic and a-2 adrenergic agonists can activate Ras through a mechanism involving the Py subunits of pertussis toxin-sensitive heterotrimeric G- proteins (Bokoch, 1996). The mechanisms by which Py subunits lead to Ras activation are not well characterised although they again seem to require recruitment of Grb2-Sos complexes by, as is the case with tyrosine kinase receptors, several possible mechanisms depending on the agonist and cell type.
In some cases it may involve activation of Src-like tyrosine kinases that can bind to and phosphorylate She which can then recruit Grb2-Sos to specific membrane complexes. Src-like kinases could also contribute to Ras activation by transactivation of the EGF receptor (Daub et al., 1996). In a recent case it was reported that thrombin and LPA can activate Ras in a manner independent of Src and She that involves the binding of Grb2-SH3 domain to a 100 kDa tyrosine-phosphorylated protein that could be a SNT/FRS2 homologue (Kranenburg et al., 1997). Yet another mechanism could involve the calcium-dependant activation of the Pyk2 tyrosine kinase and subsequent tyrosine phosphorylation of She (Lev et al., 1995).
The many variations on Sos mediated Ras activation are likely to mirror the ability of Ras to respond to such a wide array of extracellular stimuli.
1.3.2.1.1 Sos phosphorvlationSos proteins can be phosphorylated on serine and threonine residues within the
proline-rich C-terminal domain by kinases acting downstream of Ras such as Erks and Rsk. Phosphorylation is accompanied by a shift in its molecular weight on SDS polyacrylamide gel electrophoresis and is believed to play a negative feedback role on Ras activation. While phosphorylation has not been shown to have any direct effect on

31
Sos catalytic activity it does modulate its interaction with other proteins in an agonist and cell-type specific manner. In some cases, such as in response to insulin or PDGF stimulation, Sos phosphorylation results in dissociation of the Sos-Grb2 complex. In other cases however, such as in response to EGF stimulation, Sos phosphorylation decreases the ability of the Sos-bound Grb2 to interact with tyrosine-phosphorylated proteins (Cherniack et al., 1995; Corbalan Garcia et al., 1996; Klarlund et al., 1995; Porfiri and McCormick, 1996; Rozakis-Adcock et al., 1995; Waters et al., 1995a; Waters et al., 1995b)..
1.3.2.2 RasGRFRasGRF contains 2 PH domains, a coiled-coil, an IQ (calcium-calmodulin binding) and a Dbl-homology domain N-terminal of a CDC25-catalytic domain. RasGRF lacks a proline-rich C-terminal tail that in Sos is responsible for Grb2 binding and coupling to tyrosine phosphorylated proteins, suggesting that RasGRF is not involved in tyrosine- kinase pathways leading to Ras activation. Instead, its brain restricted expression suggests that RasGRF may couple Ras to tissue-specific signals, perhaps involving G protein-coupled neurotransmitter receptors in neurons. Indeed, G protein (3y subunits have been shown to lead to RasGRF activation by a mechanism involving direct phosphorylation of RasGRF (Mattingly and Macara, 1996).RasGRF can also become activated in neurons by raising the calcium concentration, an effect that requires binding of calmodulin to the IQ-domain. An intact PH and coiled- coil domains appear to be important for targeting RasGRF to the membrane (Buchsbaum et al., 1996; Farnsworth and Feig, 1991; Farnsworth et al., 1995).
1.4 Ras effectors
It has long been known that introduction of activated Ras into a variety of immortalised cell lines could stimulate DNA synthesis and lead to their morphological transformation and that Ras could stimulate transcription from a variety of promoters including the c-fos, collagenase and the polyoma virus enhancer. Even before the biochemical pathways regulated by Ras were identified, it was known that Ras regulated more than one pathway: ground-breaking work from Lloyd and Marshall had shown that prolonged exposure to phorbol esters (a treatment known to downregulate protein kinase C) inhibited Ras induction of DNA synthesis but not c-myc expression and morphological transformation of Swiss 3T3 fibroblasts (Lloyd et al., 1989). It was therefore concluded that Ras must control at least two distinct signalling pathways: one leading to mitogenesis and another leading to morphological transformation.
However the “effector” molecules with which Ras interacted and the biochemical pathways regulated by Ras remained elusive for many years despite intense

32
research from many groups. It was not until 1993, with the advent of the yeast two hybrid screen to look for proteins that interacted with active, GTP-bound Ras, that Ras targets were finally found. The identification of Raf, a well known oncoprotein, as a direct Ras target seemed to explain a lot about Ras function. However other proteins with properties of Ras effectors were subsequently identified and it has now become apparent that Ras can interact with many different effector molecules and regulate many separate pathways, most of which are still very poorly characterised. More recently the use of mutations in the effector domain of Ras that selectively lose the interaction with some but not other effectors, has further confirmed that Ras can regulate diverse signalling pathways which have different phenotypes in different cell types.
1.4.1 Raf
A combination of genetic and biochemical experiments had shown that Raf functioned downstream of Ras regulating a kinase cascade, the Raf/Mek/ERK cascade, referred to as a MAPK cascade. The Raf kinase (a MAP kinase kinase kinase) phosphorylates and activates Mek, a dual-specificity MAP kinase kinase which in turn phosphorylates and activates the ERKs, members of the MAP kinase family. The MAP kinases (MAPK) form a distinctive class of proline-directed serine/threonine protein kinases that are activated by phosphorylation of a threonine and tyrosine residue separated by a single amino acid (T-X-Y motif). While very little is known about other Raf and Mek substrates, ERKs have a wide array of substrates which include other kinases (e.g. p90Rsk, MAPKAPK2) and transcription factors (e.g. Elk-1).
Microinjection of the Ras neutralising antibody Y 13-259 has been shown to revert transformation by Ras and tyrosine-kinase oncogenes but did not have any effect on transformation by Raf suggesting that Raf could lie in a linear pathway downstream of membrane-bound tyrosine kinases and Ras (Smith et al., 1986). Further support for this possibility came with the observation that dominant-negative mutants of Raf could block gene expression and transformation induced by Ras (Kolch et al., 1991). In addition, genetic analysis in C. elegans and D rosophila had again placed Raf homologues (lin45 and Draf respectively) in a signalling pathway downstream of Ras (Dickson et al., 1992; Han et al., 1993). Biochemical studies in mammalian cells also placed Ras upstream of Raf in the regulation of ERK activation in response to growth factors (de Vries Smits et al., 1992; Howe et al., 1992). However it was not until the advent of the two-hybrid system that it was shown that Raf is a direct target of Ras (Van Aelst et al., 1993; Vojtek et al., 1993; Wame et al., 1993; Zhang et al., 1993).

34
binding site (Rommel et al., 1996) although whether this second interaction helps to stabilise the Ras-Raf interaction or serves another purpose is not yet clear.
Raf translocation to the plasma membrane is thought to play a critical role in its activation by Ras. When Raf-1 is targeted to the membrane by addition of a CAAX motif, it is constitutively active. Interestingly, although it can be further activated by growth factors, this stimulation is independent of Ras, suggesting that Ras contribution to Raf-1 activation is to recruit it to the plasma membrane where other events then activate it (Leevers et al., 1994; Stokoe et al., 1994). The nature of the activating events at the plasma membrane are not yet fully understood, but phosphorylation of residues 340 and 341 by tyrosine-kinases including members of the Src and Jak family is thought to play a critical role (Marais et al., 1995; Xia et al., 1996).
It should be noted however that different Raf isoforms can be differentially regulated and in the case of B-Raf, Ras binding, in the presence of 14-3-3, is sufficient to activate its kinase activity in vitro arguing that in the case of B-Raf, Ras acts as an allosteric activator and that plasma membrane recruitment is not essential for B-Raf activation (Yamamori et al., 1995).
Other factors contributing to Raf activation could include serine/threonine phosphorylation (e.g. by Ksr or members of the PKC family), binding of an unknown lipid molecule (presumably through the CRD domain) and interacion with other proteins. Interaction with 14-3-3 for example could contribute to Raf activation by blocking phosphatase action (and hence inactivation) (Dent et al., 1995; Muslin et al.,1996). In addition, 14-3-3 proteins, due to their ability to homo/heterodimerize, could promote either dimerization of two Raf molecules, which has been shown to activate Raf-1 activity (Farrar et al., 1996; Luo et al., 1996) or could bring other signalling proteins (either activators or substrates) into the same protein complex (Braselmann and McCormick, 1995; Conklin et al., 1995).
1.4.1.3 Negative regulation of raf activation by cAMP/PKAElevation of the levels of cAMP and subsequent activation of PKA can be
growth stimulatory in some cell types (e.g. epithelial cells, Schwann cells, Swiss 3T3 fibroblasts) but it can suppress cell proliferation in others (e.g. NIH 3T3 and Rat-1 fibroblasts, adipocytes, arterial smooth muscle cells). In the latter case there is a correlation between growth inhibition and the ability of cAMP to block the activation of the Raf/ERK cascade in response to growth factor stimulation (Burgering et al., 1993; Cook and McCormick, 1993; Graves et ah, 1993; Sevetson et ah, 1993; Wu et ah, 1993). There is some evidence that this inhibition is mediated by phosphorylation of Raf by PKA: PKA can phosphorylate Raf-1 at several sites, predominantly serine 43, and this phosphorylation by PKA has been reported to inhibit the interaction with Ras (Wu

36
tgulation or function so its contribution to Ras function still awaits further tharacterisation.
U4AF6
|̂ F6isa 180 kDa protein highly homologous to the Drosophila protein Canoe, which is to function in the Notch developmental pathway. It has also been described as
[partner of ALL-1 in some acute lymphoblastic leukaemias (Prasad et al., 1993). F6 was identified independently as a Ras-interacting protein by the yeast two-hybrid
Aelst et al., 1994) and by affinity chromatography on a Ras-GTP column |Kuriyama et al., 1996) and its Ras-binding domain has been mapped to an N-terminal 4̂aa domain (residues 36-206). The biological relevance of this interaction remains to
tie elucidated.
liRas-related proteins
Ras belongs to a super-family of related low molecular weight GTPases which :|ludes over fifty members. Based on structural and functional similarities they have xen clustered into five sub-families: the Ras family, the Rho/Rac family, the Rah miy, the ARF (ADP-ribosylation factor) family and the Ran (Ras-related nuclear trotein) family (Hall, 1993). They regulate a wide variety of cellular processes Deluding growth, differentiation, cytoskeletal organisation, vesicle transport and püclear transport. Only recently has the high degree of interconnections between the iferent GTPase families become apparent.
Like Ras they function as molecular switches cycling between a GDP-and a ïïP-bound state that is regulated by specific GEFs, GAPs and in some cases GDIs, lowever the Rab and Ran families have important mechanistic differences with Ras *nd Rho/Rac families in the way that they couple their GTPase cycle to a biological espouse. Unlike Ras, in which only the GTP-bound form of the protein regulates ffector function, both the GDP- and GTP-bound forms are equally important for the I lab and Ran proteins to effect their biological responses. These two GTPase families * se the presence of GDP- and GTP-bound interacting proteins in different cellular ompartments to couple their GTPase cycle to the unidirectional transport of cellular omponents.

39
CSG, a molecule with a CDC25 homology domain that interacts with the SH3 of Crk has been reported to have Rapl exchange activity in vitro (Gotoh et al., 1995). Smg-GDS also displays GEF activity for Rapl although it also has a broad specificity towards other GTPases, making its contribution to in vivo activation difficult to interpret. Smg-GDS functions as an exchange factor for Rapl (a and b), Ki-Ras, RhoA, Rac(l and 2) and Cdc42 (Mizuno et al., 1991), all proteins with a polybasic domain. Smg-GDS binds to the prenylated forms of the GTPases and in addition to stimulate the exchange of nucleotide it inhibits their association with membranes in a manner analogous to Rho/Rab GDIs (see below) (Nakanishi et al., 1994).
Rapl can be activated in some cell types by a cAMP-dependant pathway and a possible mechanism could involve direct phosphorylation. Rapl is phosphorylated by PKA at serine 179, a residue that lies between the polybasic domain and the prenylated cystein (Altschuler and Lapetina, 1993; Sahyoun et al., 1991) and this phosphorylation is known to make the protein more sensitive to the action of Smg-GDS in vitro (Hata et al., 1991). A mutant in which serine 179 has been substituted for an aspartic acid to mimic the effect of phosphorylation behaves as an activated protein in inducing activation of MAP-kinase in PC12 cells (Vossler et al., 1997). Direct phosphorylation however, cannot account for Rapl activation in all cases as in platelets Rapl phosphorylation actually correlates with its inactivation suggesting that cAMP dependant pathways can have opposing effects on the activation of Rapl depending on the cell type.
Rapl is activated in platelets by thrombin and other activators of platelet function in a calcium dependant manner, whereas prostaglandin I2 (PGI2), which increases cAMP levels and results in Rapl phosphorylation, blocks agonist induced activation of Rapl and inhibits platelet activation (Franke et al., 1997).
1.5.1.2.2 EffectorsConsistent with the fact that Rap proteins have an identical effector domain as
Ras they have been shown to interact, at least in vitro, with all known Ras effectors. However as with the R-Ras/TC21 proteins interaction in vitro does not always correlate with activation in vivo, and the contribution of these interactions to Rap function (if any) is not yet clear.
pl20Ras-GAP binds with a 100-fold higher affinity to Rapl, than to Ras although it is unable to stimulate its GTPase activity (Freeh et al., 1990; Hata et al.,1990). Rap proteins also bind in vitro to RalGDS with a 100-fold higher affinity than Ras although (at least in COS cells) the interaction does not result in stimulation of RalGDS activity in vivo (Urano et al., 1996).

40
The situation with Raf proteins is more peculiar. Rap can bind to, but does not activate c-Raf-1. It can however activate B-Raf, both in vitro and in vivo (Ohtsuka et al., 1996; Vossler et al., 1997).
1.5.1.3 Ral proteinsThe Ral sub-family has two members, RalA and RalB, 85% identical between
them and 55% identical to Ras. However, unlike Rap and R-Ras/TC21 they differ from Ras at two important positions in the effector domain and consistent with this they do not interact with known Ras effectors.1.5.1.3.1 Activation.
Several Ral-GEFs/exchange factors are known, RalGDS, RGL, RLF and RSC (Albright et al., 1993; D'Adamo et al., 1997; Kikuchi et al., 1994; Wolthuis et al., 1996), which have been shown to behave as effectors of Ras and have been discussed in section 1.4.2. No Ral-GAPs have yet been cloned.1.5.1.3.2 Effectors.
Ral-GTP interacts with RalB P1/RIP 1/RLIPl, a protein that has GAP activity towards Rac and Cdc42 (Cantor et al., 1995; Jullien-Flores et al., 1995; Park and Weinberg, 1995) thus providing a point of cross talk between the Ral and Rac/Rho GTPase families. It is not known yet though, whether the interaction with Ral results in stimulation or inhibition of its Rac/Cdc42-GAP activity.
Another potential target molecule of Ral is phospholipase D (PLD) which hydrolyses phosphatidylcholine to generate phosphatidic acid (PA) and choline. Expression of activated and dominant-negative forms of Ral enhanced and suppressed respectively, activation of PLD activity by Ras and Src, but importantly, expression of activated Ral alone was not sufficient to stimulate PLD activity. In addition the association with PLD appears to be constitutive and not to take place through the effector domain as both Ral-GDP and Ral-GTP and an effector domain mutant associated with PLD whereas deletion of the first 11 amino acids abolished the interaction (Jiang et al., 1995). It is therefore possible that Ral could be playing a role in regulating PLD by localising it to specific signalling complexes where other proteins are involved in its activation (e.g. ARF and Rho, see later)
1.5.2 The Rho family
Members of the Rho family show about 30% amino acid homology to Ras. Currently, this group comprises twelve proteins in mammals: RhoA, B, C, RhoD, RhoE, RhoG, Racl and 2, Cdc42, G25K, TCIO and TTF. Early studies showed that the rho family played major roles in regulating the remodelling of the actin cytoskeleton

42
calcium binding EF-hand motif in Lbc; a PEST sequence in Tiam-1 and a serine/treonine kinase domain in Trio.
DH PHc-Dbl
Ost
PEST Tlam1 — @ —
Vav
Ect2
Lbc
Lfc
Dbs
FGD1
j Tim
Ber
Abr
Ras GRF
m Sos
PH
Ser/Thr Kinase
-C
PH IQ
proline
HZÏI
1-----------
1—
115 kDa
100 kDa
177 kDa1
Cys SH3 SH2 SH3
95 kDa
1-------- 84 kDa
h 47 kDa
h 69 kDa
SH3
1— CZH“ 129 kDa
Cys
H O 107 kDa
H 60 kDa
Rho GAP140 kDa
Rho GAP93 kDa
Ras GEF 5 ç ? j 140 kDa
Ras GEF Proline
150 kDa
FIGURE 1.4. Structure of known and putative mammalian Rho-family GEFs.DH, Dbl homology domain; EF, putative calcium binding EF-hand motif; PEST, protein instability domain; Cys, cysteine rich/ Zinc-butterfly motif providing a putative diacylglicerol-binding site; IQ, calmodulin-binding motif.
1.5.2.1.2 GAPs.There are at least 12 GAPs for the Rho-family proteins all of which share a 140
amino-acid, so called Bcr domain, responsible for the GTPase activating activity. Many of these GAPs have independent functional domains, suggesting that they may have additional roles, perhaps serving as links between different signalling pathways. The pl90 Rho-GAP, itself a GTP binding protein, associates with pl20 Ras-GAP (see later) providing a point of cross-talk between Ras and Rho pathways (Settleman et al., 1992).

44
Rac Cdc42 RhoACK - 4- -
PAK + 4- -
MLK2/3 -1- 4- -
WASP - 4- -
CITRON + / - - 4-
p67phox + - -
PORI + - -
IQGAP-1/2 4- + -
PIP5-kinase + *
ROCK/RHOK - - 4-
PKN/PRK - - 4-
Rhophilin - - 4-
Rhotekin - - 4-
MLC phosphatase - - 4-
pl40Dia - - 4-
PLA2 + * - -
PLD 4- 4- 4*
CIP4 - 4- -
TABLE 1.2. Effector proteins for Rho-family GTPases.*Rac and Rho have been shown to co-inmunoprecipitate with a PIP5-kinase activity, but the PIP5-kinase isoform involved or whether the interaction is direct is not known. Similarly, Rac has been shown to activate PLA] activity in vivo, but no direct interaction with any of the cloned PLA2 has been reported.
1.5.2.2.2 Gene regulation.Rho-family members have been shown to activate the NFkB transcription factor
by a mechanism that involves phosphorylation of iKBa (Perona et al., 1997) and at
least in the case of Rac, could be associated with their ability to generate ROS (Sulciner et ai., 1996).
Rho family proteins (at least when overexpressed) can activate Jun amino- terminal kinase (JNK)Zstress-activated protein kinase (SAPK) and p38/mpkl MAP kinase pathways (Coso et al., 1995; Minden et al., 1995; Olson et al., 1995; Teramoto et al., 1996). JNKs, upon activation, translocate to the nucleus where they can phosphorylate transcription factors such as c-jun, Elk-1 and ATF2 (Minden et al., 1995).

46
PIP5-KinaseRac and Rho have been implicated in the regulation of PIP5-Kinase in different cell types (Chong et ah, 1994; Hartwig et ah, 1995; Ren et ah, 1996; Tolias et ah, 1995). PIP5-Kinase associates with both the GDP- and GTP-bound forms of Rho and Rac, suggesting that other accessory molecules are required for its activation. PIP2, the lipid product of PIP5-Kinase can bind to, and affect the activities of, several actin-binding proteins (e.g. gelsolin, capZ, a-actinin and profilin) and so could be involved in
inducing actin polymerisation (e.g. by uncapping actin filament ends, allowing the addition of actin monomers and their elongation ) (Tapon and Hall, 1997; Zigmond,1996).
1.5.2.2.4 Protein Kinases.Rac/Cdc42 activated kinases: ACK and PAKs
Cdc42 binds to a non-receptor tyrosine kinase, p i20ACK, about which very little is known. Cdc42 and Rac bind to, and activate, the PAK serine/threonine kinase family which includes three members (PAK1,2 and 3) (Lim et ah, 1996; Sells et ah,1997). PAKs belong to a family of related kinases that includes the S. cerevisiae Ste20 kinases, that interacts with yeast Cdc42p and regulates a pheromone-responsive MAP kinase pathway. PAK can also bind to the SH3 domains of PLCy and Nek, suggesting the existence of additional regulatory mechanisms (Barodia et ah, 1995; Lu et ah, 1997).
PAK may be mediating Rac/Cdc42 induced activation of the JNK/p38 cascades (Bagrodia et ah, 1995; Brown et ah, 1996; Zhang et ah, 1995), although some Rac effector mutants exist that do not bind PAK but still activate JNK activity (Westwick et ah, 1997) suggesting that PAK independent mechanisms also exist.. Rac/Cdc42 could also lead to JNK activation through the mixed lineage kinases (MLKs), some of which contain CRIB (Cdc42/Rac interactive binding) motifs and can directly phosphorylate SEKl and MKK3/6, upstream regulators of JNK (Burbelo et ah, 1995; Rana et ah, 1996; Tibbies et ah, 1996).
PAKs have been shown to have some effects on the actin cytoskeleton (Manser et ah, 1997; Sells et ah, 1997) although rac effector mutants clearly show that filopodia and lamellipodia formation (and their associated focal complexes) are generated through a PAK-independent pathway (Joneson et ah, 1996; Lamarche et ah, 1996; Westwick et ah, 1997).
An exciting connection between PAKs and AIDS has recently emerged with the observation that the HIV-encoded nef protein associates with PAK-like kinases (Cullen, 1996; Lu et ah, 1996) suggesting that a PAK-dependant (and maybe Rac/Cdc42- dependant) pathway could be involved in the pathogenesis of AIDS.

47
Rho activated kinases: PKN and ROKsRho has been shown to bind to several serine/threonine kinases that could be
mediating some of Rho effects on the cytoskeleton These include PKN/PRK 1 and PRK2 (which also binds to Rac) (Amano et ah, 1996; Vincent and Settleman, 1997; Watanabe et al., 1996) whose kinase domain is related to that of PKC. Another kinase family regulated by Rho includes ROCK/ROKp, ROKa and Rho-kinase, with kinase
domains 70% homologous to that of myotonic dystrophy kinase. ROK can phosphorylate the myosin-binding subunit (MBS) of myosin light chain (MLC) phosphatase, which itself binds to Rho. This phosphorylation leads to a decrease in MLC-phosphatase activity with the subsequent accumulation of phosphorylated MLC which could then lead to increased binding of myosin to actin filaments and the formation of stress fibers (Kimura et al., 1996).
1.5.2.2.5 Effectors without enzvmatic activitv.Other molecules identified as effectors that have no enzymatic activity include
Rhophilin, Citron, WASP, PORI and IQGAPs.Cdc42 can bind to WASP, the product of the gene responsible for the Wiskott-Aldrich syndrome (WAS), a disease affecting platelets and lymphocytes (Aspenstrom et al., 1996; Symons et al., 1996). Overexpression of WASP causes a clustering of polymerised actin that is inhibited by co-expression of dominant-negative Cdc42. WASP shares some restricted homology with VASP, a protein known to interact with actin filaments, and can bind to the SH3 domains of Nek and Src-family kinases (Banin et al., 1996; Rivero et al., 1995).POR-1 binds to Rac and could be mediating rac effects on lamellipodia formation (Van et al., 1996). IQGAP (1 and 2) interact with both Rac and Cdc42 and contains domains responsible for calmodulin and SH3-domain binding . It also has a RasGAP domain although no RasGAP activity or binding to Ras has been detected (Brill et al., 1996; Hart et al., 1996).
1.5.3 The Rab family
The Rab family now consists of over thirty members, including the YPT land SEC 4 proteins of S. cerevisiae, which all have about 30% homology to Ras.
Rab family proteins regulate intracellular vesicle transport, with different family members, localised in distinct intracellular compartments, regulating discrete stages of membrane trafficking. They may function by allowing docking and/or fusion of vesicles to their target membrane after recognition between interacting proteins known as v- SNARES (on vesicle) and t-SNARES (on target membrane) has occurred. Defective prénylation of Rab due to disruption of geranylgeranyl-transferase, the enzyme that

48
catalyses their prénylation, has been associated with the human disorder choroideremia, an X-linked form of retinal degeneration, suggesting that some members of this family are essential for visual function .
The geranylgeranylated Rab proteins in their GDP-bound state are in the cytoplasm bound to GDIs. In response to an unknown signal, GDI is released from the complex resulting in the translocation of the protein to the target membrane, a process that is coupled with nucleotide exchange (Ullrich et ah, 1994). Following membrane fusion and inactivation by GAPs, GDIs extract the GDP-bound Rab from the membrane into the cytosol.
Known effectors of Rab proteins include Rabphilin-3A, a target for Rab3a and Rabaptin-5, an effector of Rab5 (Stenmark et al., 1995) that could be activated by translocation to membranes upon Rab activation in a manner analogous to the activation of Raf by Ras (Stahl et al., 1996).
1.5.4 The ARF family
ARF was first discovered as the factor essential for cholera toxin to ADP-ribosylate the G protein Gsa and later found to be a small GTP-binding protein distantly related to Ras. Like the Rab proteins, ARFs regulate intracellular vesicle transport and they have been implicated in the assembly, loading and targeting of vesicles to different membrane compartments (Rosenwald and Kahn, 1994).
ARFs are mainly cytosolic even though they are myristoylated. Upon activation they translocate to membranes and recruit coatamer proteins (COPs) to form coated vesicles. Several exchange factors for ARF have been recently identified, ARNO and Cytohesin-1 (Chardin et ah, 1996; Meacci et ah, 1997), both of which have a Sec7 homology domains (Sec7 is a yeast protein involved in secretion) responsible for ARF- GEF activity and a PH domains, which could play a role in their recruitment to membranes. The PH domain has recently been shown to bind to PIP3, the lipid product of PI 3-kinase, and their role as PI 3-KINASE targets will be discussed later.
ARFs can directly regulate PLD (Cockcroft et ah, 1994) which hydrolyses phosphatidylcholine to generate phosphatidic acid (PA). PA in turn can promote PIP2 synthesis, which can act as a cofactor for ARF-GAP (Randazzo and Kahn, 1994). The resulting nucleotide hydrolysis is required for the uncoating of vesicles before their fusion with their target membranes. PIP2 is also a cofactor for PLD activation and could provide a point of cross-talk with the Rac/Rho GTPases (see above Rac/Rho activation of PIP5-kinase and PLD).

49
1.5.5 The Ran family
The Ran proteins, Ran and TC4, are nuclear GTPases which participate in the transport of proteins and RNA through the nuclear pore (Melchior et ah, 1995). An exchange factor for Ran, RCCl, is located in the nucleus, associated to chromatin. Conversely, a Ran-GAP is located in the cytoplasm, suggesting that the nucleotide- bound state of Ran is involved in conferring direction of movement of Ran in and out of the nucleus.
According to a current model, nuclear transport works as follows (Gorlich and Mattaj, 1996). Importin-a and importin-(3 bind to a nuclear localization sequence (NLS)-containing protein in the cytoplasm via the NLS-binding domain of a-importin. The complex docks at the nuclear pore complex (NPC) via the interaction of p-importin
with components of the NPC (generically called nucleoporins) or fibers that extend from the NPC surface into the cytoplasm. Docking of the complex to the NPC requires Ran-GDP. Transport through the pore occurs through a poorly defined mechanism that requires GTP binding and hydrolysis by Ran (Gorlich and Mattaj, 1996). Once in the nucleoplasm, Ran-GTP, generated by the action of the nuclear exchange factor RCCl, binds to P-importin, displacing and releasing a-importin and its NLS-containing cargo in the nucleoplasm. It is not known how Ran-GTP returns to the cytoplasm and how that is coupled to RNA export, but once in the cytoplasm Ran-GAP would stimulate GTP hydrolysis and thus recycle the protein.
Ran has also been implicated in chromatin condensation and in regulating cell cycle progression at both Gl-S and S-G2 phases, but the mechanism by which it regulates these processes and whether these functions are related to its role in regulating nuclear transport or whether they are independently regulated processes is not yet known (Rush et al., 1996).
Several proteins that bind to Ran-GTP have been identified that share one or more copies of a conserved Ran binding motif (RanBD), including RanBPl which also binds and stimulates the activity of Ran-GAP and a nuclear pore protein, RanBP2/NUP358 (Coutavas et al., 1993; Yokoyama et al., 1995). There are also proteins that bind specifically to Ran-GDP like Ntf2p which like the other two is also essential for nuclear transport (Nehrbass and Blobel, 1996).

50
1.6 PI 3-Kinase
It has long been stablished that lipids not only have a structural role, but can also function as second messengers in signalling processes. The best known example of this is the hydrolysis of phosphatidylinositol PI(4,5)P2 by phospholipase C, that gives rise to diacylglycerol and inositol(l,4,5)P3, which lead to protein kinase C activation and intracellular Ca2+ release respectively. The lipids in the PI(4,5)P2 pathway are also substrates for PI 3-kinases.
PI 3-kinase activity was first detected associated with v-Ros and v-Src oncogene products (Macara et al., 1984; Sugimoto et al., 1984) and the enzyme responsible was later found to phosphorylate the D-3 position of the inositol ring (Whitman et al., 1988). At the same time PI(3,4,5)P3, a lipid not previously known to exist, was detected in activated neutrophiles (Traynor Kaplan et al., 1988) and in response to PDGF receptor stimulation (Auger et al., 1989). Interest in this enzyme was further stimulated by evidence that this enzyme was important for transformation by v-Src and the middle-T antigen of polyoma virus and for mitogenic stimulation by the PDGF receptor (Cantleyet al., 1991) and ever since PI 3-kinases have been the subject of intense research.
PI 4-kinase Pl(4)P 5-kinasePI -----------► PI(4)P ► PI(4,5)P2
PLCDAG
PI 3-kinase PI 3-kinaseclass 1, II, III class 1, II
PI 3-kinase class I
PI(3)P2 PI(3,4)P2
TPI(3,4,5)P3
I(1,4,5)P3
5-phosphatase
FIGURE 1.5 . Phosphatidylinositol cycle.Depicted are the phosphatidylinositol polyphosphates generated by distinct classes of the PI 3-kinase family (based on their in vitro substrate specificity). Note that not all possible reactions involved in PI metabolism are shown. For details see text
PI 3-kinases convert PI, PI(4)P and PI(4,5)P2 to PI(3)P, PI(3,4)P2 and PI(3,4,5)P3 (PIP3) respectively (figure 1.5). PI(3)P is constitutively present in

51
eukaryotic cells and its levels are largely unaltered upon cellular stimulation. In contrast PI(3,4)P2 and PIP3 are almost absent from resting cells but their intracellular concentration rises sharply upon stimulation with a wide variety of ligands, consistent with their role as second messengers (Carpenter and Cantley, 1996; Stephens et ah,1993). PI 3-kinase lipid products are not substrates of phospholipases. Instead a large family of recently identified phosphatases mediate their catabolism by removing the phosphate group at positions 3 or 5 of the inositol ring (Majerus, 1996).
1.6.1 Classes of PI 3-kinase
All PI 3-kinases have a catalytic domain and a PIK (Pl-kinase) domain, that is also found in PI4-kinases and is thought to play a role in substrate recognition/presentation. Based on other structural features and on their in vitro substrate specificity PI 3-kinases have been divided into three main classes (figure 1.6 ).
1.6.1.1 Class I PI 3-kinasesClass I PI 3-kinases can phosphorylate PI, PI(4)P and PI(4,5)P2 in vitro. In vivo,
however, their preferred substrate appears to be PI(4,5)P2. They all form heterodimeric complexes with adaptor proteins which link them to different upstream signalling events. According to the type of adaptor subunit with which they associate they can be subdivided into two subclasses, A and B.1.6.1.1.1 Class lA PI 3-kinases
Class lA PI 3-kinases contain 110-120kDa catalytic subunits, referred to as the pi 10 subunit, and an adaptor subunit that varies in size but always contain two SH2 domains. The SH2 domains bind to phosphorylated tyrosine residues (within a YXXM motif) and therefore link class lA PI 3-kinase catalytic subunits to tyrosine kinase signalling pathways. Three catalytic subunits, pi 10a, p and Ô, have been identified so
far in mammalian cells and homologous molecules are known in other species {C.elegans, Drosophila and Dyctiostelium) (figure 1.6). They contain several conserved domains including an N-terminal adaptor binding region, a PIK domain and a C- terminal kinase domain. The kinase domain retains some, but not all, of the sequence motifs present in the catalytic domains of protein kinases and indeed members of this PI 3-kinase class can phosphorylate a limited number of protein substrates which depending on the isoform can include the adaptor subunit or autophosphorylation of the catalytic subunit itself.

52
ClassLipid substra tes and
structural features of catalytic subunits
Subunits
Catalytic Adaptor
I
A
PI, PI(4)P, PI(4,5)P2
- ^ ^ -
pi 10a, p, 5(m) D pilo (Dm) AGE-1 (Ce) PIK1, PIK2 (Dd)
p85a,P (m) p55a,y(m) p50a (m) p60 (Dm)
B pllOy(m) PIK3 (Dd)
plOI (m)
II PI, PI(4)P PI3K-C2a/C pk-m /p170 (m)
PI3K-C2P (m)PI3K 68D/Cpk(Dm) PI3K-C2 (Ce)
?
III PI
--------- --Vps34p VpsISp (Sc)
pi 50 (m)
© adaptor-binding ^ Ras-binding C'_~> 02
^ 0 PIK kinase domainPI(3,4,5)P3 ^
3 e 5'©
FIGURE 1.6. Classification of PI 3-kinase family members.M, mammalian; Dm, drosophila melanogaster; Ce, Caenorhabditis elegans. Adapted from Vanhaesebroeck et al., 1997.
To date seven different adaptor subunits for this PI 3-kinase class lA have been
identified n mammals (encoded by 3 different genes) (figure 1.7). They all contain two
SH2 domains linked by an inter-SH2 region that contains the interaction site for the
pi 10 catalytic subunit.
The fist two identified adaptor subunits for class lA catalytic subunits have a Mr of approximately 85 kDa and were thus named p85s (p85a and p85(3).They are encoded
by different genes and contain in addition to their SH2 and inter-SH2 domains, an N-
terminal SH3 domain, and two proline-rich (SH3-binding) motifs flanking a Bcr-
homology (BH) domain. BH domains have been identified in a large number of
proteins, many of which posses GAP activity towards members of the Rho-family of
GTPases. However, no function or biochemical activity has yet been ascribed to the BH
domain of p85s.

53
SH3 BH N-SH2P1 P2 inter-SH2 §
U)
C-SH2
9-77 134-322 333-416 L-A 631-706+8 aa (p85ai)
p85a
+8 aa (p55al)
I
p 5 5 a /A S 5 3
p50a
p85(3
p 55y/p S sP 'K
FIG U RE 1.7. A daptor subunits for class lA PI 3-kinases.P, proline-rich region; BH, bcr homology domain. p50a and p55a (also known as AS53) are splice variants of p85a. whereas p85p and p55y (known also as p55PIK) are encoded by different genes. Triangles indicate further splice insertions in p85a and p55a (named p85ai and p55ai).
Five additional mammalian adaptor subunits have been identified more recently
(figure 1.7) (Antonetti et ah, 1996; Fruman et al., 1996; Inukai et al., 1996; Pons et al.,
1995). Several lack the SH3 domain, the first Pro-rich sequence and the BH domain and have instead a short unique N-terminal extension. One of these, p55y, is encoded by a
novel gene, whereas others are generated by alternative splicing of the p85a gene to
form transcripts encoding p50a or p55a subunits. Further alternative splicing of the
p55a and p85a transcripts can result in a 9 amino acid insertion within the inter-SH2
domain to generate p55ai and p85ai sububits.
The functional implications for this diversity in adaptor subunit is not yet clear,
but tissue-specific expression and the absence of functional domains in some of the
adaptors suggests possible differences in regulation and function. It is also noteworthy

54
that the insertions in the inter-SH2 in p55ai and p85ai occur in the vicinity of Serine 608, a target of regulatory phosphorylation by the pi 10a catalytic subunit (Dhand et al.,
1994) and could give rise to potential regulatory phosphorylation sites (Antonetti et al.,1996).1.6.1.1.2 Class IB PI 3-kinases
Class IB PI 3-kinases are stimulated by heterotrimeric Gpy subunits. There is only member to date, pllOy, that contains a PIK and catalytic domain (figure 1.6) but
does not interact with SH2 domain-containing adaptors. Instead it associates with a plOl subunit that has no known functional domain but that greatly potentiates activation by py subunits and G protein coupled receptors (Stephens et al., 1997).
1.6.1.2 Class II PI 3-kinaseClass II PI 3-kinases comprise several 200kDa enzymes which phosphorylate PI
and PI(4)P but not PI(4,5)P2. They all contain a C2B domain at their C-terminus (figure 1.6) that mediates interaction with phospholipids in a calcium-independent manner (MacDougall et al., 1995). The only identifiable motifs N-terminal to the PIK domain are proline-rich regions which presumably mediate interaction with SH3 domain- containing proteins. Very little is known about the regulation and function of this subfamily of PI 3-kinases.
1.6.1.3 Class III PI 3-kinasesClass III PI 3-kinases have a substrate specificity restricted to PI These PI 3-
kinases are homologous to Vps34p, the only PI 3-Kinase present in yeast, that is essential for the trafficking of newly formed proteins from the Golgi to the vacuole, the equivalent of the mammalian lysosome (Sheperd et al., 1996). Both yeast and human Vsp34 function as heterodimers complexed with a serine/treonine kinase, Vspl5 and p i50 respectively, which recruits the lipid kinase to the membrane.
Based on the established function of yeast Vps34 and on the observation of the relative invariant cellular levels of PI(3)P, it is hypothesised that class III PI 3-kinases and their PI(3)P lipid product play a housekeeping role in constitutive membrane trafficking and vesicle morphogenesis. It is worth mentioning here that class I PI 3- kinases can also play a role in vesicular trafficking and endocytosis (e.g. glucose transport) although in this case, in an agonist regulated manner.
1.6.1.4 PIK-related kinasesSeveral proteins have been recently identified which contain a region
homologous (20% identity) to the catalytic domain of PI 3-kinases but which seem to function as serine/treonine kinases rather than lipid kinases. They have been implicated in a diverse set of biological functions such as DNA repair, DNA recombination and

55
cell cycle control (Keith and Schreiber, 1995; Zakian, 1995). Members of this family include the DNA-dependent protein kinase (DNA-PK), the ataxia telangiectesia mutated (ATM) and related gene products and the rapamycin targets TOR/FRAP kinases.
1.6.2 Targets of PI 3-kinases
Although PI 3-kinases have been implicated in multiple biological responses such as control of the actin cytoskeleton, chemotaxis, mitogenesis, cell survival and differentiation, the molecules directly regulated by PI 3-kinases have proved more difficult to identify.
Several protein targets of PI 3-kinase funtion have been identified in the last few years based on several approaches. Chemical inhibitors of PI 3-Kinase, such as Wortmannin and LY294002, receptor mutants that no longer interact with PI 3-Kinase and the use of constitutively active and dominant negative PI 3-Kinase mutant subunits (Ap85, which lacks the binding site for the pi 10 catalytic subunit) have identified
pathways activated downstream of PI 3-kinases. But the identification of the direct targets of the PI 3-kinase lipid products has relied on the availability and use in biochemical studies of in vitro synthesised or chemical D3 phosphoinositides analogues.
Prominent among PI 3-kinases targets are pleckstrin-homology (PH) domain containing proteins. PH domains can mediate protein-protein interaction in some cases (e.g. with Py subunits of heterotrimeric G proteins and PKC) but are increasingly recognised as lipid binding domains, interacting in particular with phosphoinositides (and their soluble headgroups) and mediating recruitment of proteins to the membrane (Lemmon et al., 1997).
Different PH domains have specificity for distinct phophoinositides. Some PH domain-containing proteins including pleckstrin, phospholipase C-dl, dynamin, SOS and the B-adrenergic receptor kinase have been shown to bind PtdIns(4,5)P2 and/or Ins(l,4,5)P3. Yet others bind to D3 phosphoinositides. Among these the best characterised is the protein kinase PKB/Akt. Several other proteins are known to bind D3 phosphoinositides in vitro but the biological significance of these interactions still awaits further characterisation.
1.6.2.1 Protein kinase B/AktPKB/Akt was identified independently by three different groups in 1991. Two
groups cloned a 60kDa kinase based on its homology to both PKC (73% similarity to the kinase domain of PKCe) and PKA (68% similarity to the kinase domain of PKA);
thus the names PKB (Coffer and Woodgett, 1991) and RAC-PK (related to A and C kinases) (Jones et al., 1991). At the same time, this kinase was identified as the product of the oncogene v-akt of the acutely transforming retrovirus AKT8 found in a rodent T

56
cell lymphoma (Bellacosa et al., 1991). The viral oncogene encoded a fusion of the cellular Akt protein to the viral Gag structural protein.
In addition to a serine/treonine kinase domain, all three mammalian PKB/Akt molecules identified to date contain a PH domain (residues 1-106) that encompasses most of the N-terminal regulatory domain referred to as Akt homology (AH) domain (residues 1-147), a catalytic kinase domain and a small (70 amino acids) C-terminal extension.
PKB/Akt is activated in response to treatment of cells with a wide variety of growth stimuli, including PDGF, EGF, insulin, thrombin and NGF. Several findings have placed PKB/Akt downstream of a PI 3-kinase regulated pathway. Treatment with PI 3-kinase inhibitors or over-expression of dominant negative PI 3-kinase inhibit activation of PKB/Akt by many growth factors. In addition, PDGF receptor mutants that fail to bind (and activate) PI 3-kinase also fail to activate PKB/Akt (Burgering and Coffer, 1995; Franke et al., 1995; Kohn et al., 1995). Moreover, expression of constitutively active forms of PI 3-Kinase are sufficient to activate PKB/Akt in vivo (Franke et al., 1997; Klippel et al., 1996; Marte et al., 1997). It should be noted that PKB/Akt may also be regulated by PI 3-kinase independent pathways, as activation of PKB/Akt by heat shock and hyperosmolarity is insensitive to Wortmannin (Konishi et al., 1996).
The integrity of the PH domain has been found to be essential for activation of PKB/Akt in response to some (e.g. PDGF, EGF, serum) but not all (e.g. insulin) growth factors. Recent studies have shown that the lipid products of PI 3-Kinase PIP3 and PI(3,4)P2 can bind to the PH domain of PKB/Akt. Binding of PI(3,4)P2, but not PIP3, results in a modest increase in the activity of PKB/Akt in vitro (Franke et al., 1997; Freeh et al., 1997; Klippel et al., 1997). PI(3,4)P2 but not PIP3 also induces dimerization in vitro (Franke et al., 1997) a process that could play a role in the activation of the kinase activity (Datta et al., 1995). Binding of PI(3,4)P2 to the PH domain is most likely however not sufficient for full activation of PKB/Akt. Several lines of evidence have indicated that phosphorylation by upstream kinases is also involved.
Upon insulin or IGF-1 stimulation, PKB/Akt becomes activated independently of the PH domain (but in a wortmannin sensitive way) by a mechanism that involves phosphorylation at two sites, threonine 308 in the catalytic domain and serine 473 in the C-terminal tail (Alessi et al., 1996; Kohn et al., 1995). A kinase that phosphorylates threonine 308 and activates PKB/Akt has recently been purified and named PDKl (Alessi et al., 1997). This kinase is strongly activated by PIP3 and PI(3,4)P2 in vitro suggesting that it also is a direct target of PI 3-Kinase but a better understanding of its mechanism of activation will have to wait for the elucidation of its structure.

57
1.6.2.1.1 Targets of PKB/AktThe only direct PKB/Akt in vivo substrate identified so far is glycogen synthase
kinase 3 (GSK3). Phosphorylation of GSK3 by PKB/Akt results in its inactivation (Cross et al., 1995). GSK3 is involved in the regulation of several signalling pathways including glycogen synthesis, control of translation (eIF-2B) and transcription factors (API, CREB), the tumour supressor gene product APC and the Wingless developmental pathway in flies and dorsoventral patterning in frogs (Welsh et al., 1996).
Another target for PKB/Akt is the ribosomal protein S6 kinase, p70S6K, although there is no evidence that it is a direct substrate (Burgering and Coffer, 1995). p70S6K is involved in the regulation of translation of a subset of mRNAs in response to mitogenic stimulation of cells (Brown et al., 1996).
Recent studies have shown that PI 3-Kinase, through the activation of PKB/Akt, is involved in regulating cell survival (Franke et al., 1997) and this will be discussed later.
1.6.2.2 ARF exchange factors: Grpl, Cytohesin and ARNOThe GRPl (general receptor for phophoinositides) protein was identified using
an expression cloning approach to identify proteins that bind to PIP3 (Klarlund et al.,1997). GRPl is closely related to Cytohesin-1 and ARNO (Chardin et al., 1996; Kolanus et al., 1996). All three proteins contain a C-terminal PH domain while the remainder of the molecule shows homology with a portion of the yeast SEC7 gene (Sec7-homology domain). Cytohesin-1 was identified on the basis of its ability to bind to the cytoplasmic tail of the p2 integrin subunit and has been implicated in integrin inside-out signalling: overexpression of its Sec7-homology domain enhances P2-
integrin-dependant ICAM-1 binding by Jurkat T cells, whereas overexpression of the PH domain inhibited T-cell receptor stimulation of binding to ICAM-1.
The PH domains of Grpl and Cytohesin-1 bind with high affinity to PIP3 and Ins(l,3,4,5)P4 whereas the See? domain of ARNO and Cytohesin-1 (Meacci et al.,1997) stimulate nucleotide exchange activity on the ARF GTPases. The high homology between Grpl, ARNO and cytohesin 1 (90% identity in the PH domains) makes it likely that these proteins act as PIP3 regulated ARF exchange factors and further suggests a link, by way of ARF activation, between PI 3-Kinase and integrin signalling and possibly also protein sorting and vesicle trafficking (the latter two being known ARF regulated processes, see later). Interestingly, Centaurin-a, another protein that binds
PIP3 and Ins(l,3,4,5)P4 has homology to ARF-GAP (Hammonds Odie et al., 1996), further suggesting that PIP3 lipids could regulate ARF activation by several mechanisms.

58
1.6.2.3 BtkThe Bruton's tyrosine kinase (Btk), a cytoplasmic tyrosine kinase of the Tec
family, contains PH, Tec-homology, SH3 and SH2 domains N-terminal to the kinase domain. Mutations in its PH domain are responsible for X-linked immunodeficiency in mice (Xid) and X-linked agammaglobulinaemia (XLA) in humans. These diseases and the phenotype of Btk gene knockout mice indicate that Btk plays an essential role in the development and function of B lymphocytes.
The PH domain of Btk can bind with high affinity to PIP3 and Ins(l,3,4,5)P4 and with low affinity to PI(3,4)P2 and PI(4,5)P2 (Fukuda et al., 1996; Salim et al.,1996). The mutation in the PH domain found in Xid mice, reduces the affinity for PIP3.
Although PI 3-Kinase-dependant regulation of Btk has not yet been demonstrated in vivo, it can be speculated that binding to PIP3 could directly or indirectly activate Btk by localising it to the membrane, while Ins(l,3,4,5)P4 could act as a "switch off" signal, competing with PIP3 for binding to the PH domain and releasing the protein from the membrane. A similar mechanism could be speculated for other PIP3 and Ins(l,3,4,5)P4 binding proteins like the Grpl/ARNO/Cytohesin-1 family (see above).
1.6.2.4 SH2The SH2 domains of Src and the p85 subunit of PI 3-Kinase have been shown to
bind specifically to PIP3 (Rameh et al., 1995). This association could be competed with phosphotyrosine containing proteins, although mutational analysis showed that different residues are involved in the interaction with phosphotyrosine and phospholipid moieties. Treatment of cells with PI 3-Kinase inhibitors enhanced the association between the p85 subunit of PI 3-Kinase and IRS-1 upon insulin stimulation and it has been suggested that PIP3 binding to the SH2 domain of p85 could cause dissociation of PI 3-Kinase from tyrosine phosphorylated proteins and thus allow the recruitment of additional p85 molecules. It is also possible that PIP3 could directly recruit other SH2 containing proteins to the membrane.
1.6.2.5 Protein kinase CSeveral in vitro studies have shown that the PI 3-Kinase products PI(3,4)P2 and
PIP3 can activate novel and atypical PKC isoforms as well as the PKC-related kinase Prk/PKN (Nakanishi et al., 1993; Palmer et al., 1995; Toker et al., 1994). In several cases PI(4,5)P2 also stimulated kinase activity so the specificity and biological significance is not clear. In the case of PKCe and PKC-^, PDGF and EGF stimulated activation correlated with the ability of the PDGF receptor to associate with PI 3- KINASE and was blocked by wortmannin, supporting a role for these PKC isoforms

59
downstream of PI 3-Kinase (Akimoto et al., 1996; Moriya et al., 1996). Their contribution to PI 3-Kinase function remains however to be elucidated.

6 0
CHAPTER 2 Materials and Methods
2.1 Materials
2.1.1 Reagents
LY294002 was from Biomol; PD098059 was from New England Biolabs; foetal calf serum (PCS) was from GIBCO; soybean phosphatidylinositol and phosphatidylserine were from Sigma; [a-^^S]deoxy adenosine 5'a-thiotriphosphate (dATP) (1,000 Ci/mmol), [y-^^PJATP (5,000 Ci/mmol), [^^P]orthophosphate (10 mCi/ml) L-
[^^SJmethionine (1,000 Ci/mmol); [^H]phosphatidylinositol 4-phosphate (2-10 Ci/mmol), [^H]phosphatidylinositol 4,5-bisphosphate (2-10 Ci/mmol), and [^H]inositol 1,3,4,5-tetrakisphosphate (21 Ci/mmol) was from DuPont. CalphostinC and Bisidolylmaleimide were from Calbiochem. Unless otherwise stated all other reagents are from Sigma.
2.1..2 Antibodies
Monoclonal antibodies against p85a, p85|3, phosphotyrosine (4G10 and FB2), Ras (259
and 238), haemagglutinnin (12CA5) and a myc epitope (9E10) were supplied by the ICRF hybridoma Development Unit. Pan-Ras antibody was from Oncogene Science. Peroxidase-coupled seconday antibodies were from Sigma. Flurophore coupled secondary antibodies were from Dako.
2.1.3 Constructs
pcD N A 3-pll0a wt: pi 10a was digested out of pBS with Fspl, a BamHl linker added
and after digestion the resulting fragment was ligated into the BamHl site of pcDNA3 (Invitrogen).pcD N A 3-pll0a 3'myc:The sequence encoding the 9E10 epitope was inserted at the carboxi-terminus of pi lOa-wt by performing PCR of pcDNA3-pl 10a-wt with the oligos
TGGCTCAAAGACAAGAAC and GCCGGATCCTCATAGATCCTCCTCCGATATG AGTTTCTGTTCAGGGTTCAAAGCATGCTGCTT, digesting the resulting fragment with Clal and ligating it into pcDNA3-pl 10a digested with Clal and EcoRV. pM T2sm -pll0a 3'myc: p i 10a 3'myc was digested out of pcDNA3 with BamHl,
blunt-ended with klenow polymerase and ligated into the Smal site of pMT2sm

61
pcDNA3 p85BS-myc : Residues coding for the p85 binding site of p llO a (amino acids
1-108) were amplified and the sequence encoding the 9E10 epitope added to its amino- terminus by performing PCR with the oligos GCCGGATCCGCCACCATGGAACAG AAACTCATATCGGAGGAGGATCTACCTCCAAGACCATCATCATCAGGT and GCCGGATCCCTAACGGTTGCCTACTGGTTC and cloning the resulting fragment into the Baml site of pcDNA3.pMT2sm p85BS-myc: p85BS-myc generated as described aboved was digested out of pcDNAS as a KpnI-XhoI insert and cloned into the KpnI-XhoI sites of pMT2sm. pcDNA3 p85 and RasBS-myc: Residues coding for amino acids 1-314 of p i 10a,
comprising the p85-binding site and the Ras-binding domain, were amplified and the sequence encoding the 9E10 epitope inserted at its amino-terminus by performing PCR with the oligos GCCGGATCCGCCACCATGGAACAGAAACTCATATCGGA GAGGATCTACCTCCAAGACCATCATCATCAGGT and GCCGGATCCCTAAGC TGTGGAGATGCG and ligating the resulting fragment into the Baml site of pcDNAB. pcDN A 3-pll0a 5'myc: A 9E10 epitope was inserted at the amino-terminus of full length pi 10a by ligating the insert resulting from an EcoRl digestion of "pcDNAB p85 and RasBS-myc" to the vector-containing fragment of pcDNA3-pl 10a wt digested with
EcoRl.pcDNA3-p85a-interSH2-myc: The inter-SH2 domain of p85a (amino acids 358-617)
were amplified and a 9E10 epitope added by PCR with the oligos GCCGGATCCGCCACCATGGACGCATCTACTAAAATG and GCCGGATCCTCA TAGATCCTCCTCCGATATGAGTTTCTGTTCGGGCAAATCCTCATCATC and cloned into the Baml site of pcDNA3.pMT2-p85a-interSH2-myc: p85a-interSH2-myc generated as described aboved was
digested out of pcDNA3 as a KpnI-XhoI insert and cloned into the KpnI-XhoI sites of pMT2sm.pcDNA3 V12 H-ras: The V12 mutant H-ras cDNA was digested out of pExv3 with EcoRl and cloned into the EcoRl site of pcDNA3. pcDNA3 V12 Ras effector mutants.Mutageneis was performed on pcDNA3 V12 H-ras with oligos specific to each mutation and an oligo that destroyed the Xhol site of pcDNA3 and created an Hpal site.-Y32F oligo: created a Sail site CTGATCCAGAACCATTTTGTCGACGAATTTG ACCCCACTATAGAGGAT-K42A oligo: created a BssHII site. GAGGATTCCTACCGCGCGCAGGTGGTCATT GAT-E31K oligo: created a Sail site.CTGATCCAGAACCATTTTGTCGACAAATACGAC
CCCACTATAGAG-V45A oligo: created a Clal site. TACCGGAAGCAGGTGGAAATCGATGGGGAGA

62
CGTGC-G26J27 oligo: created a Sail site. ATCCAGCTGATCCAGGGCATTTTTGTCGACG AATACGACCCC-D38E oligo: destroyed a Sfcl site. GSCGAATACTACCCCACTATCGAGGAATCC TACCGGAAGCAGGTG-E37G oligo: destroyed a Sfcl site. GACGAATACTACCCCACTATCGGGGATTCC TACCGGAAGCAG-T35S oligo: destroyed a Sfcl site. GTGGACGAATACTACCCCAGTATAGAGGATT CCTACCGG-Y40C oligo: created a Pstl site. CTATAGAGGATTCCTGCAGGAAGCAGGTGGTC -Y64G oligo: created a Eco47III site. GCCGGCCAGGAGGAGGGCAGCGCTATG CGGGACCAGTACpSG5 Ras mutants: Ras mutants were digested out of pcDNA3 and cloned into the EcoRl site of pSG5 (Stratagene).pLXSN Ras mutants: Ras mutants were digested out of pcDNA3 and cloned into the EcoRl site of pLXSN (Miller et al., 1989).pcDNA3 p85aS608A. Serine 608 of p85a was mutated into alanine with an oligo,
AACACTGAAGACCAATATGCACTAGTAGAAGATGATGAGGAT, that created a Spel site, and an oligo that destroyed the Apal and Xbal sites of pcDNA3 and created a SacII site.pcDN A3-pll0a K227E: Lysine 227 of pllOa-wt was mutated into glutamic acid with the oligo GCTGAAGCAATCAGGGAAAAAACGCGTAGTATGTTGCTATCATCTG that also introduced a Mlul site by silent mutagenesis, and an oligo that destroyed the Xhol site of pcDNA3 and created an Hpal site.pcDN A 3-pll0a 5'myc-K227E: Created as for pcDNA3-pllOa K227E, but
performing the mutagenesis on pcDNA3-pl 10a 5'myc.pSG5 pllO s: pi lOwt, pi 10 5'-myc, pi 10 K227E and pi 10 5'-myc K227E were taken out of pcDNA3 as BamHl inserts and subcloned into the BamHl of pSG5. pLXSN pllOs: pi lOwt, pi 10 5'-myc, pi 10 K227E and pi 10 5'-myc K227E were taken out of pcDNA3 as BamHl inserts and subcloned into the BamHl of pLXSN. pGEX-2T V12 Ras mutants: A BamHl site was added in frame to the second codon of H-Ras by performing PCR on the corresponding pcDNA3 plasmids with the oligo CGCGGATCCATGACGGAATATAAGCTG and an Sp6 primer. The resulting fragments were ligated into pGEX-2T digested with BamHl and EcoRl. pRSETB V12 Ras mutants: Ras mutants on a V12 background were taken out as BamHl-EcoRl inserts from pGEX-2T and cloned, in frame with a six histide tag, into the Bglll-EcoRl sites of pRSETB vector (Invitrogen).pRSETB G12 Ras mutants: To have the effector mutants on a G 12 background the V12 mutation was mutated back to G 12 by performing PCR on the corresponding

63
pGEX-2T plasm ids. Reaction A inculded the m utagenic oligo 1 = GCTGGTGGTGGTGGGCGCCGGCGGTGTGGGCAAGAGTGCG and oligo 2= AACGCGCGAGGCAGATCG for pGEX-2T. Reaction B included the mutagenic oligo 3= CGCACTCTTGCCCACACCGCCGGCGCCCACCACCACCAGC and oligo 4= TTCTTAGCAAGCTACCTG for pGEX-2T. Both, reactions A and B, were subjected to 10 cycles of amplification, the resulting fragments isolated, pooled and subjected to a further 15 cycles of amplification with oligos 2 and 4. The resulting fragments were digested with BamHl and EcoRl and ligated into the Bglll-EcoRl sites of pRSETB. pGEX-2T RasBS of p llO a: Residues coding for amino acids 133 to 314 of p i 10a,
were amplified by PCR with the oligos GCGGATCCCCAGAAGTACAGGAC and GCGAATTCAAGCTGTGGAGATGCG and cloned into pGEX-2T digested with BamHl and EcoRl.pGEX-2T RasBS of p ll0a-K 227E : As above but the PCR was done using pcDNA3- pl 10a K227E as a template.pGEX-2T RasBS of pllOP: Residues coding for amino acids 133-314 of pllOp were
amplified by PCR with the oligos GCGGATCCAATGAATTTCGAAGA and GCGAATTCATCGATTTATGGCAGC and cloned into pGEX-2T digested with BamHl and EcoRl.GST-deletion mutants of p i 10Amino terminal deletion mutants of pi 10a were made by ExoIII deletion of bovine
pi 10, inserted in the pAcG3 vector [Davies, 1993 #954] .pBS 110a: Bovine pi 10a. Kind gift from M. WaterfieldpBS p85a: Bovine p85a. Kind gift from M. Waterfield
pExv3 wt H-Ras: Human H-Ras. Kind gift from C. CalespExv3 V12 H-Ras: Kind gift from C. CalespExv3 V12, S186 H-Ras: Kind gift from C. MarshallpExv3 V12 , S181,S184 H-Ras: Kind gift from C. MarshallpExv3 V12 K-Ras: Kind gift from C. MarshallpExv3 V12 K6Q K-Ras: Kind gift from C. MarshallpExv3 wt R acl: Kind gift from A. HallpExv3 V12 R acl: Kind gift from A. HallpExv3 N17 R acl: Kind gift from A. HallpExv3 wt RhoA: Kind gift from A. HallpExv3 V14 RhoA: Kind gift from A. HallpExv3 MEK-EE: Kind gift from C. MarshallpExv3 MEK-A221: Kind gift from C. MarshallpRSV N17 Ras: Kind gift from H. Bos

64
2.2 Methods
2.2.1 Mutagenesis:Point mutations were carried out, unless otherwise stated, using the In Vitro Mutagenesis Kit (Pharmacia) according to the manufacturers protocol. The oligos creating the point mutations were designed to either create a novel restriction site or destroy an existing one by silent mutagenesis.
2.2.2 Purification of proteins2.2.2.1 Expression and Purification of GST-fusion proteins in bacteriawt Racl, V12 Racl, N17 Racl wt Cdc42, V12 Cdc42, N17 Cdc42, V14 RhoA, C3 transferase, Raf-RBD, RalGDS-RBD and pllO-RBD were expressed in the pGEX-2T vectoras GST-fusion proteins.A 100ml overnight culture of bacteria carrying the corresponding pGEX expression plasmid were diluted 1:10 in L-broth/100 pg/ml ampicillin and grown for 1-2 hours. Expression of fusion protein was induced by incubation with 0.4 mM isopropyl p-D-
thiogalactopyranoside (Sigma) for an additional 2-4h at 37 ®C. E. coli were harvested by centrifugation at 4,000 rpm for 15 min at 4 °C, the pellet was resuspended in 15ml of ice-cold GST lysis bujfer (1% Triton X-100, 5mM MgCl2, 1 mM PMSF,lmM DTT in
PBSA).The bacteria were lysed by sonication on ice (4x1 min) and cell debris was removed by centrifugation at 10,000 rpm for 10 min at 4 °C. The supernatant was incubated with 1 ml of 50%(w/v) suspension of glutathione agarose beads (Sigma) which had been washed thoroughly in PBSA containing 2 mM EDTA. After 1 h to 2 h at 4 ®C, the suspension was centrifuged at room temperature in a bench top centrifuge and the beads were washed 2 times in GST lysis buffer and 3 times in PBSA 5mM MgCl2 to remove
unbound proteins. GST fusion proteins were eluted from the beads by competition with 25 mM reduced glutathione in 50 mM Tris pH8. Proteins were dialysed against PBSA, 5mM MgCl2, ImM DTT at 4 and the protein concentration was determined.
2.2.2.2 Wild tvpe and V I2 c-Ha-RasUnmodified wild type and V I2 c-Ha-Ras was purified from detergent free homogenate of recombinant baculovirus infected Sf9 cells as described in [Page, 1989 #592]. Post-translationally modified wild type Ras was purified from the particulate pellet from the above cells. The pellet was solubilised in the same homogenisation buffer to which 40mM n-octylglucoside had been added. After clarification by centrifugation at 350,000 X g for 10 minutes, the solubilised Ras was purified as for the unmodified protein but with all buffers including 40mM n-octylglucoside.

65
2.2.2.3 A38 Ras and RalAA38 Ras and RalA were purified from bacteria following the same protocol as for unmodified Ras.
2.2.2.4 PI 3-kinasePI 3-kinase was partially purified from Sf9 cells co-infected with baculovirus encoding p85 and baculovirus encoding pi 10. The cytosolic supernatant was loaded into a Superdex-200 column and fractions were analysed by lipid kinase assays on p85 immunoprecipitates. The peak of activity was pooled and used for further experiments. Alternatively cells were infected with a GST-pllO and p85 baculoviruses and the GST- pl 10/p85 complex was purified as for bacterial GST-fusion proteins.
2.2.3 PI kinase assaysThe immunoprecipitates were washed once in the appropriate lysis buffer, twice in washing buffer 1 (100 mM Tris pH7.5, 0.5 M LiCl, 100 )iM NagVO^) and twice in washing buffer 2 (50 mM Hepes pH7.4, lOOmM NaCl, 1 mM EDTA, 100 |iM NagVOzt). After last wash, beads were drained with a Hamilton syringe. The
immunoprecipitates were assayed for inositol lipid kinase activity using unilamellar liposomes composed of a 1:1 mixture of L-a-phosphatidylinositol (PI) and L-a-
phosphatidyl-L-serine (PS), which were prepared by sonication (30 s, 30 s break, 1 min, 30 s break, 30 s; 50% level) of the lipid mix (PI and PS, both at a concentration of 1 mg per ml) in 25 mM HEPES buffer, pH7.4 containing 1 mM EDTA on ice. The washed immunoprecipitates were resuspended in 10 pi lipid mixture and the reaction was initiated by the addition of 40 |il PI kinase reaction buffer (25 nM [y-^^P]ATP (5 |iCi), 125 pM ATP, 12.5 mM MgCl2, 125 mM NaCl, 25 mM Hepes pH7.4, 250 pM adenosine). The final concentrations in the assay were: 20 nM [y-^^PJATP (5 pCi), 100 pM ATP, 10 mM MgCl], 100 mM NaCl, 25 mM Hepes pH7.4, 200 pM EDTA, and 200 pM adenosine (to inhibit PI 4-kinase activity, [Whitman, 1988 #580]). After 15 min at 25 °C the reaction was terminated by the addition of 500 pi chloroform : methanol (1:2) in 1% conc. HCl plus 125 pi chloroform and 125 pi HCl (10 mM). The mixture was vigorously mixed and then centrifuged to separate the phases. 200 pi of the
organic (lower) layer was removed (with a chloroform sealed tip) and washed once with 400 pi methanol : 100 mM HCl plus 2 mM EDTA (1:1). The organic phase was extracted, dried in vacuo, and resuspended in 25 pi chloroform. The phospholipids were
separated by thin layer chromatography (TLC) on 1% potassium oxalate pre-treated Silica gel 60 plates (Whatman) in propanol-1 : 2 M acetic acid (65:35 (v/v)) developing solvents. Incorporation of ^^P into PI was quantified using a Phosphorlmager (Molecular Dynamics).

6 6
2.2.4 Interaction of inmobilized Ras with soluble PI3KRas and Ras-related proteins were coupled covalently to Affigel-10 beads (BioRad) following the manufacturers protocol.GTPases coupled to beads were bound to GDP or GTP as for soluble Ras (see below). PI3K was added in 200|il of PBS,5mM MgCl2,l%
Triton X 100 and the mixture was rotated at 4°C for 2 hours.When doing competition experiments the Ras beads were preincubated with peptides or proteins for 2h and then PI3K was added for a further 2h. Beads were washed 2x with PBS,1% Triton X 100,5mM MgCl2; 2x lOOmM Tris pH 7.5, 500mM LiCl, 5mM MgCl2 and 2x 50mM Tris pH7.5, lOOmM NaCl, 5mM MgCl2. After last wash beads were drained and used
for invitro lipid kinase assays or resuspended in SDS PAGE sample buffer and used for western blotting.
2.2.5 Interaction of soluble Ras with inmobilized PI 3-kinaseFifty pi binding buffer (50mM Hepes pH7.5, 50mM NaCl, 5mM MgCl2: 5mg/ml BSA) containing 0.5pM Ras and 200pM GDP or GTP were incubated at 37®C for 5minutes. Magnesium chloride was added to lOmM followed by 20pl of 1:1 suspension of
glutathione agarose beads bound to GST fusion protein.The volume was made up to 200plwith PBS,5mM MgCl2,l% Triton X 100 and the mixture was rotated at 4°C for 2 hours.The beads were then washed with 4xlml of cold 50mM Hepes pH 7.5, lOOmM NaCl, 0.1 mg/ml BSA, 5mM MgCl2 /0.1% Triton X-100 and finally resuspended in SDS PAGE sample buffer. Bound Ras was detected by immunoblotting using 259 monoclonal antibody or pan-Ras antibody (Oncogene Science).
2.2.6 GAP AssaysFifty pi binding buffer (see above) containing 2.4pM Ras and 3.5pCi y-32p qTP
(specific activity 5000Ci/mmol) were incubated at 37®C for 5 minutes. Magnesium chloride was then added to lOmM and the volume made up to 125pl with 50mM Hepes pH7.5: 5mM MgCl2:lmM GDP. 25pl were dispensed into 2 control tubes of 25pl buffer and 2 tubes of 25pl buffer containing GAP. All tubes were incubated at 30°C for 20 minutes and then had 1ml PBS: 5mM MgCl2 added. The contents of each tube were
filtered through a nitrocellulose disc which was washed twice with 5ml PBS:5mM MgCl2 and then counted.For neurofibromin activity a soluble fusion protein of GST and the catalytic domain of NFl was used.p l2 0 ^ ^ P was partially purified from Sf9 cells infected with bacolovirus expressing pl20GAP.

67
2.2.7 Scintillation Proximity Assay (SPA)Fifty \x\ binding buffer (see above) containing 7.5|iM Ras and 60}iCi ^H-GTP (specific
activity 7.8Ci/mmol) were incubated at 37°C for 5 minutes.Magnesium chloride was added to lOmM and the volume made up to 160jxl with 50mM Tris pH 7.5:5mM M gCl2
(T:M). Twenty |il of this and of a 1:5 dilution were added to 20pl T:M containing 2.5mg
GST or GST fusion protein. After 1 hour on ice lOOjil SPA Protein A beads
(Amersham) coated with 5jLtg rabbit anti GST immunoglobulins were added with a
further 60jil T:M. The mixture was rotated at 4°C for 2 hours and then counted in a P
scintillatoin counter set with an open window.
2.2.8 Reconstitution of Ras into liposomes.Liposomes were made by sonication of dried lipids in kinase buffer (40mM Hepes pH 7.5, 1 mg/ml fatty acid free BSA, 2mM EGTA, lOOmM NaCl, 5mM M gCl2 , 50|iM
ATP). The final lipid concentration was: 640|iM phosphatidylethanolamine, 600|iM
phosphatidylserine, 280pM phosphatidylcholine, bOpM sphingom yelin, 60pM
PI(4,5)P2 . Sonication was on ice for 4 x 2 0 " with 4mm probe at 15pm amplitude in an
MSB Soniprep 150. Modified Ras was added to the liposomes at a final concentration of l|xM and incubated on ice for 20 minutes. Liposomes were collected by centrifugation
at 350,000 X g for 20 minutes. The supernatant was removed and liposome pellet
resonicated in the same volume of kinase buffer. To load Ras with nucleotide, the mixture was made lOmM in EDTA, and then 20pM GTP or GDP added. After
incubation at 30°C for 10 minutes, 15 mM MgCl2 was added. When antibodies were
used to block Ras effects, 300pg/ml of purified monoclonal antibody was added to the
initial incubation of Ras with liposomes.
2.2.9 PI 3-kinase assays in liposomes.Liposome mixture was made as described above, either with or without Ras. Tyrosine phosphopeptides were added as indicated, along with lOng p85a/G ST-pl 10a and IpCi [y.32p]^TP pej. point. The total assay volume was 25pl, which represented a 50%
dilution of the liposome preparation. Assays were mixed continuously for 20 minutes at 20°C, then stopped by adding 400pl of chloroform:methanol (1:2) and lOOpl of 2.4M
HCl. After vortexing and centrifugation (15,000 x g, 3 minutes), the lower chloroform
layer was put into fresh tubes and dried down in a Speedvac. The residue was dissolved in 2 5 p l o f c h lo ro fo rm and run on a s i l ic a tic p la te in
Chloroform:Methanol:Acetone:Acetic acid:water (40:13:15:12:7). The PIP3 spot was
quantitated using a Molecular Dynamics Phosphorimager.

69
Tfx-50 lipofection protocol. For study of inhibitory effects on transformation, 10 ng of V12 Ras or 5 ng v-Src plasmid were co-transfected with l|ig of inhibitory construct.by
the lipofectamine-transfection protocol. 2 days after transfection (or whe confluent) cells were transferred to 10cm dishes.After reaching confluence cells were kept in DMEM containing 5% calf serum and the medium changed 3 times a week. After 2 weeks cells were stained with 0.5% crystal violet in 10% methanol, rinsed thorougly with distilled water and photographed.
2.2.13 Growth in soft agar assays1 or 5x10^ cells were mixed with 4ml of 0.5% low melting point agarose (PMC) agarose and overlayed onto a layer of 4ml of 0.9% agarose in 6cm dishes and allowed to set for Ih at RT. 0.5 mis of fresh medium was added twice a week. After 3 weeks, cells were stained by adding 1ml of 0.2% neutral red in PBS and the plates were photographed.
2.2.14 Generation of PC12 clones expressing N17 H-Ras:PC 12 cells were transfected by the calcium phosphate method with RSVneoNl? H-Ras and 0418 resistant clones were picked and analysed by western blot. Two clones, 7 and 8, expressing highest levels of N17-Ras were chosen for further experiments. All were insensitive to NGF induced differentiation.
2.2.15 Generation of retroviruses and infection of target cells.Retrovirus were generated by cloning inserts into the pLXSN vector and transfection into the GP+E 86 packaging cell line. Pools of G418 resistant colonies were selected and when confluent the medium was collected and frozen as aliquots in the presence of 8 P-g/ml polyberene. Virus aliquots were thawed immediately before addition to
subconfluent cells. After 3-16 hours incubation the medium was replaced and G418 was added when appropiate 24h later.
2.2.16 Measurement of 3’ phosphoinositides in intact cells.Cells, in 6cm dishes, were washed with phosphate-free DMEM and labelled in 2ml phosphate-free DMEM containing 1 mg/ml fatty acid free BSA, 20mM HEPES pH 7.2 for 2h with 400pCi or 0/N with 200p.Ci ^^P orthophosphate per dish. After washing
with cold PBSA cells were lysed by addition of 1ml cold 2.4M HCl, 5mM TBAS (tetrabutylammonium hydrogen sulphate. Sigma).Cells were scraped into a 15ml polypropylene falcon tube. 1.5 ml cold CHClgimethanol (1:2) containing 10 pg phosphoinositides (Sigma P6023) was added followed by 1ml CHCI3. Samples were vortexed and centrifuged for 3min at 3000 rpm to sepearate phases.The lower (CHCI3)

70
phase was transferred to a new 15 ml tube and the upper (aquaous) phase re-extracted twice with 1 ml CHCI3,.1/10 of the combined CHCI3 phase was run on an oxalated silica gel 60 tic plate, using a CHCl3:methanol:acetone:acetic acidiwater (40:13:15:12:7) system. The rest of the CHCI3 phase was dried in a Speedvac and deacylated by incubating 35 minutes at 50°C with methylamine. The dried pellet was resuspended in 500p,l of butanol and the polar glicerophophoinositols extracted twice with 500jil of water. Samples were loaded on a
Spherisorb HPLC column (Anachem) and eluted at 1ml min'^ with a gradient of Milli Q water (A) versus 2.5 M NaH2P04 pH3.8 (%B) with program 1: t=0, %B=0; t=5, %B=0;
t=65, %B=14; t=70, %B=20; t=75, %B=50, t=80, %B=50; t=81, %B=0 ; t=150, %B=0. Fractions were collected every 0.5 min and counted by Cerenkov emission.
2.2.17 Measurement of Raf activityRaf activity was assayed in the absence of serum by cotransfection of a myc-tagged c- Raf-1 construct and performing a coupled MEK/ERK2 kinase assay on 9E10 immunoprecipitates as described by [Leevers, 1992 #365]. For lipid analysis in NIH 3T3 cells, ras retrovirus-containing supernatant was added to subconfluent NIH 3T3 cells and replaced 4 hours later with fresh medium. 24 hours later the cells were labelled overnight in 0.2% dialysed calf serum and PIP3 levels were measured by the same method as for COS cells.
2.2.18 Retroviral infections and PC12 neurite outgrowth assaysRetroviruses were generated by cloning inserts into the pLXSN vector and transfection into the GP+E 86 ecotropic packaging cell line. Pools of 0418 resistant cells were selected and when confluent the medium was collected and frozen as aliquots in the presence of polybrene. 5x 10^ PCI2 cells were seeded on 6 cm dishes the day before and the retrovirus containing medium was added to the cells overnight. The following day fresh medium (DME plus 10% horse serum and 5% fetal calf serum) containing 0.5 mg/ml 0418 was added and photographs of the cells taken after three days. Expression of the desired proteins was checked in parallel infections by immunoblotting.
2.2.19 Microinjections and immunofluorescence1x10^ Porcine Aortic Endothelial (PAE) cells were seeded in 13mm glass coverslips two days before ,and serum starved overnight in 0.5% fetal calf serum before injections. When injecting proteins, V12 Rac, V12 Cdc42, N17 Rac and N17 Cdc42 were expressed as OST- fusion proteins, purified and cleaved as described in (Self and Hall,1995). V12 Ras proteins expressed as either OST- or as histidine-tagged fusion proteins were found to have very poor biological activity; the V12 Ras protein used for microinjection experiments was purified from the soluble fraction of Sf-9 cells infected

71
with a V12 Ras baculovirus. When microinjecting DNA, plasmids were injected into the nucleus at a concentration of lOOpg/ml and the cells fixed after 5 hours. Guinea pig IgG
at 2 mg/ml was co-injected in all cases to help identify injected cells. LY294002 was used at 20|iM. An Eppendorf microinjector (Model No. 5242) and micromanipulator
(Model No. 5170) was used. Cells were fixed and immunofluorescence carried out as described in [Ridley, 1995 #1039]. All micrographs shown are representative examples ofmicroinjected cells from at least four similar experiments.

72
CHAPTER 3 In vitro interaction between Ras and PI 3-kinase
3.1 Introduction
At the time this thesis was started virtually nothing was known about the cellular targets of mammalian Ras or about the biochemical pathways by which Ras carried out its known effects on gene expression, cellular proliferation and differentiation. The aim of my thesis was to try to identify proteins that interacted with Ras and to try to gain insight into the biochemical pathways regulated by Ras.There was a report in the literature of PI 3-kinase activity being immunoprecipitated with the anti-Ras antibody Y 13-259 upon overexpression of activated H-Ras in a rat liver epithelial cell line (Sjolander et al., 1991). With the cloning of the regulatory and catalytic subunits of PI 3-kinase (Escobedo et al., 1991; Hiles et al., 1992; Otsu et al., 1991; Skolnik et al., 1991) and the generation of recombinant baculoviruses expressing both PI 3-kinase subunits (Hiles et al., 1992; Otsu et al., 1991) it became possible to further analyse whether Ras interacted, directly or indirectly, with PI 3-kinase and if so, characterise this interaction.When this work was started only one pi 10 catalytic subunit was known (now referred to as pi 10a). As discussed in section 1.6.1 many PI 3-kinase family members are now
known, sharing a conserved catalytic domain but displaying considerable differences in their structure and regulation. For simplicity purposes and unless otherwise stated, in the results and discussion chapters ahead I will often use the term PI 3-kinase to refer to the complexes containing the pi 10a catalytic subunit.
3.2 Results
3.2.1 Interaction of Ras and PI 3-kinase in vitro.
Purified Ha-Ras proteins were directly coupled to activated agarose beads. The immobilised Ras proteins were loaded with either GTP or GDP and then incubated with purified PI 3-kinase (from an Sf9 cell co-infection with baculoviruses expressing both p85a and pi 10a subunits of the kinase). After washing, the amount of PI 3-kinase
activity bound to the Ras beads was measured. As shown in figure 3.1 A, beads carrying wild type Ras protein loaded with non-hydrolysable analogues of GTP bound high
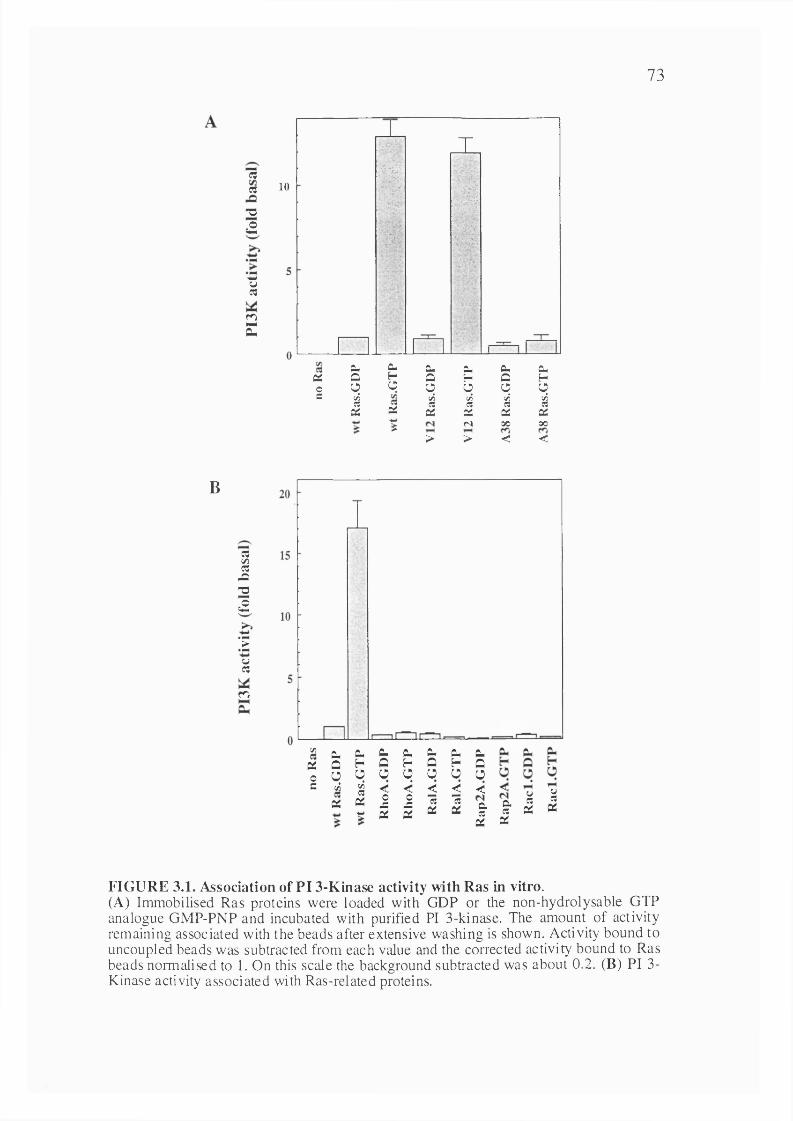
73
esw-oa
-wVCQ
m
E
10
T% * *
' ' A
% A' î y
A
» v w
CL. C. X X Cu CuQi Q H Q H O HO O O O Ü q OC t/5 c/5 c/5 c/5 c/5a « « « a
X X X X X XN 00 00>
> > < <
B
S«ÆT3a
*>
uC3
m
c/) Cu X X X X X XX Û h Q H 0 H 00 q q O q o 0 0c c/5 %
X< < < < <
X0JS
0Æ 15 "B
X a-w X X X «X
Q .«0:
I I
FIGURE 3.1. Association of PI 3-Kinase activity with Ras in vitro.(A) Immobilised Ras proteins were loaded with GDP or the non-hydrolysable GTP analogue GMP-PNP and incubated with purified PI 3-kinase. The amount of activity remaining associated with the beads after extensive washing is shown. Activity bound to uncoupled beads was subtracted from each value and the corrected activity bound to Ras beads normalised to 1. On this scale the background subtracted was about 0.2. (B) PI 3- Kinase activity associated with Ras-related proteins.
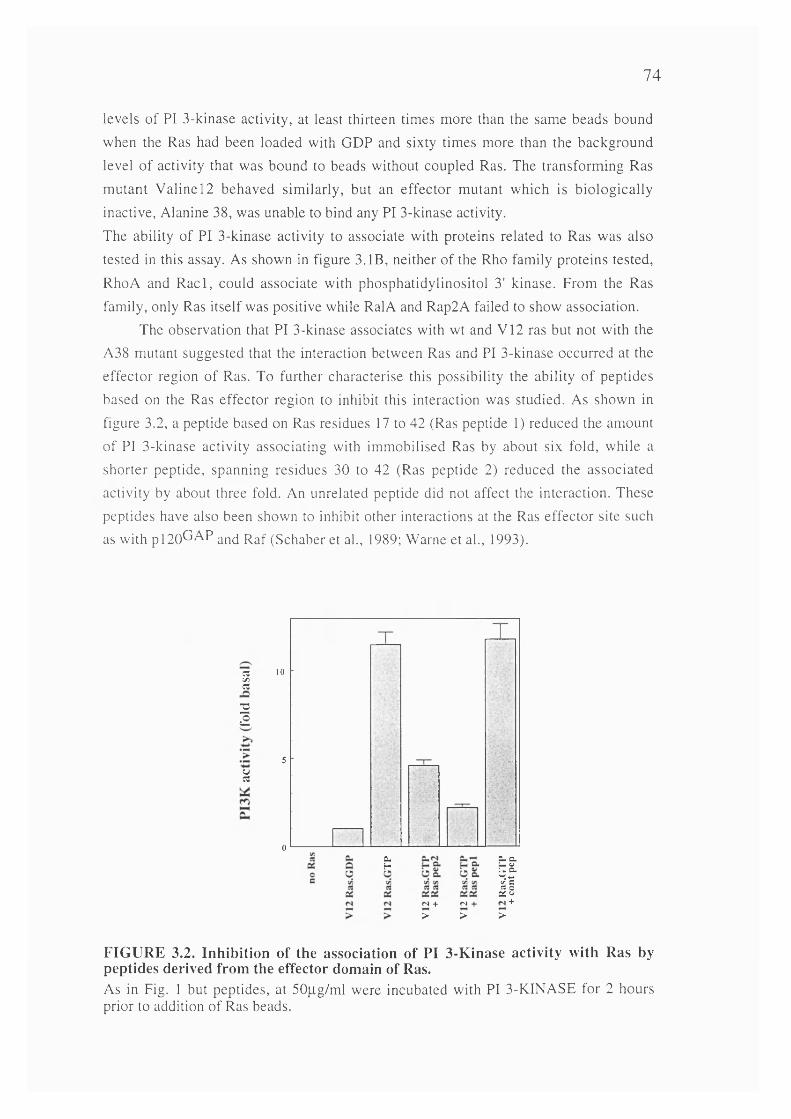
74
levels of PI 3-kinase activity, at least thirteen times more than the same beads bound
when the Ras had been loaded with GDP and sixty times more than the background
level of activity that was bound to beads without coupled Ras. The transforming Ras
mutant Valine 12 behaved similarly, but an effector mutant which is biologically
inactive. Alanine 38, was unable to bind any PI 3-kinase activity.
The ability of PI 3-kinase activity to associate with proteins related to Ras was also
tested in this assay. As shown in figure 3. IB, neither of the Rho family proteins tested,
RhoA and R a d , could associate with phosphatidylinositol 3' kinase. From the Ras
family, only Ras itself was positive while RalA and Rap2A failed to show association.
The observation that PI 3-kinase associates with wt and V 12 ras but not with the
A38 mutant suggested that the interaction between Ras and PI 3-kinase occurred at the
effector region of Ras. To further characterise this possibility the ability of peptides
based on the Ras effector region to inhibit this interaction was studied. As shown in
figure 3.2, a peptide based on Ras residues 17 to 42 (Ras peptide 1) reduced the amount
of PI 3-kinase activity associating with immobilised Ras by about six fold, while a
shorter peptide, spanning residues 30 to 42 (Ras peptide 2) reduced the associated
activity by about three fold. An unrelated peptide did not affect the interaction. These
peptides have also been shown to inhibit other interactions at the Ras effector site such
as with p l2 0 ^ '^ P and Raf (Schaber et al., 1989; Warne et al., 1993).
SC3
2a
uC3
10
5
0c.H »N +
>N + >
+>
FIGURE 3.2. Inhibition of the association of PI 3-Kinase activity with Ras by peptides derived from the effector domain of Ras.As in Fig. 1 but peptides, at 50pg/ml were incubated with PI 3 -KINASE for 2 hours prior to addition of Ras beads.

75
To further characterise the site on Ras that is responsible for this interaction, the
immobilised Ras was incubated with various monoclonal antibodies prior to interaction
w ith PI 3-kinase. As shown in figure 3.3, a control antibody (FB2, anti-
phosphotyrosine) and a non-neutralising anti-Ras antibody, Y 13-238, have no effect on
the amount of PI 3-kinase binding to Ras, whereas the neutralising anti-Ras antibody
Y 13-259 (Furth et ah, 1982) greatly inhibits the interaction.
«.Q13
â
uC3
Ow
15
10
5
0C l.Û
FIGURE 3.3. Inhibition of the association of PI 3-KINASE activity with Ras by monoclonal antibodies.Ras beads were incubated with the indicated antibodies for 2 hours prior to the addition of PI 3-KINASE.
In order to confirm the association of PI 3-kinase with Ras.GTP, the interaction of purified Ras with purified p85a/pl 10a was studied directly by Western blotting for the
p85a subunit. In these experiments, p i 10 was expressed in a baculovirus construct in
Sf9 cells as a glutathione S-transferase fusion protein, along with normal p85. The active p85a/G ST-pl 10a complex was then purified in a single step on glutathione
agarose. As shown in figure 3.4A, p85a/G ST-pl 10a complexes with wild type or
Valine 12 H-Ras when it is bound to GTP, but binds very much more weakly to the GDP
bound form. No interaction is seen between p85 and the effector mutant Alanine 38. When p85p/GST-pl 10a is used in these experiments, it is found also to bind to Ras in a
GTP-dependent manner. Similar results are found when p85a/p l 10 is used that is not
fused to GST (data not shown).
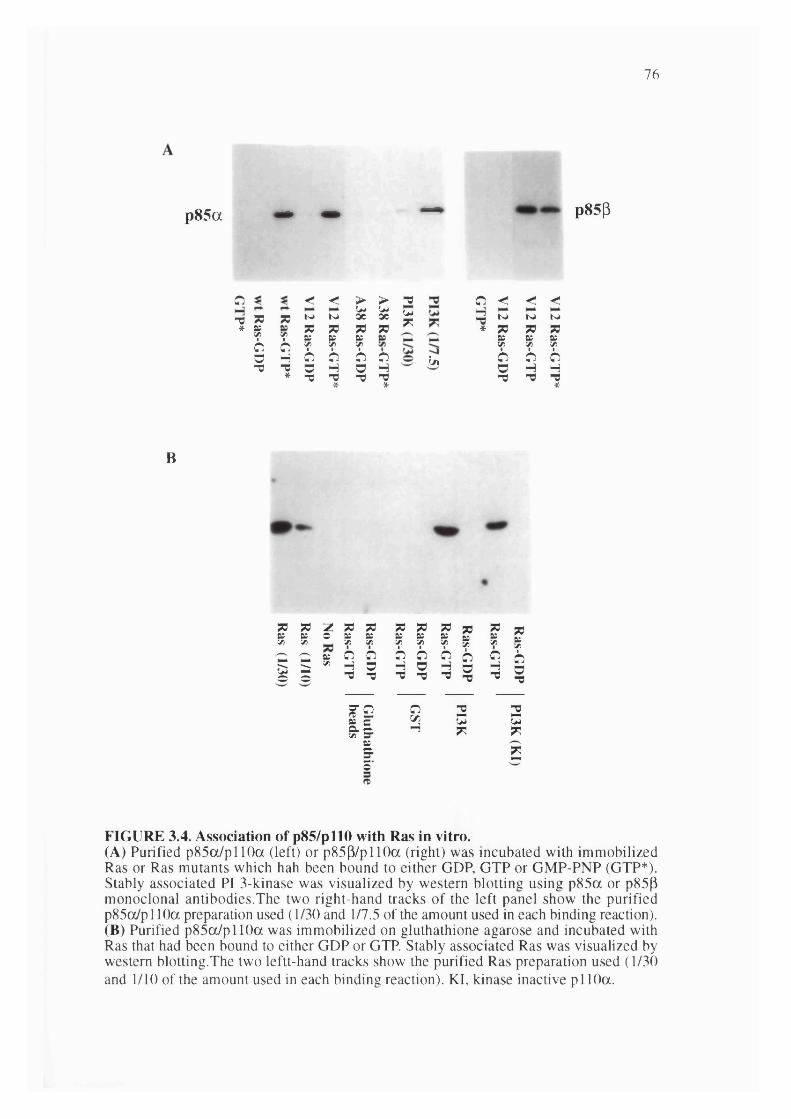
76
p85a — — — P85P
o < < > > XwX
w w-0 X X w w 3C ac*
? yoH
XK
XBS
Xit Xco
VI6
?o
?O c
-0 x o H o H* -0 X* -0 X*
In
O < < <□ K> K> K>* 73 7S 73» tS Ky y y
Ô O Oo H H-0 -0 - 0
*
B
X 73 -Z 73 73 X X X x ^ Xg s * s g ? ? I f * gI r ̂i 11 i I e i
I M I iI :
FIGURE 3.4. Association of p85/pll0 with Ras in vitro.(A) Purified p85ot/pllOa (left) or p85p/p llO a (right) was incubated with immobilized Ras or Ras mutants which hah been bound to either GDP, GTP or GMP-PNP (GTP*). Stably associated PI 3-kinase was visualized by western blotting using p85a or p85(3 monoclonal antibodies.The two right-hand tracks of the left panel show the purified p85o/pllO a preparation used (1/30 and 1/7.5 of the amount used in each binding reaction).(B) Purified p85cx/pllOa was immobilized on gluthathione agarose and incubated with Ras that had been bound to either GDP or GTP. Stably associated Ras was visualized by western blotting.The two leftt-hand tracks show the purified Ras preparation used (1/30 and 1/10 of the amount used in each binding reaction). KI, kinase inactive pi 10a.

77
Another way in which the interaction of Ras and PI 3-kinase can be demonstrated is by incubating soluble Ras, bound either to GTP or GDP, to p85a/GST-pl 10a that has
been immobilised on glutathione agarose. Bound Ras is visualised by Western blotting. As shown in figure 3.4B, Ras does not bind to control or GST agarose beads, but interacts in a GTP-dependent manner with p85a/GST-pllOa beads. In addition, Ras
binds to a point mutated PI 3-kinase whose pi 10 subunit lacks lipid kinase activity (R916P, (Dhand et al., 1994)), indicating that the enzymatic activity of the kinase is not required for its interaction with Ras.
3.2.2 The interaction between Ras and PI 3-kinase is directThe above data show that purified Ras interacts with purified PI 3-kinase in a
GTP-dependent manner, but does not rule out the possibility that this interaction occurs through a contaminating protein that copurifies with PI 3-KINASE purified from insect cells. To address this, purified p85a/GST-pllOa, which was apparently homogenous,
was bound to purified Ras.GTP immobilised on agarose beads, washed, and the bound proteins analysed by SDS-polyacrylamide gel electrophoresis followed by staining with Coomassie Brilliant Blue. As shown in figure 3.5, the p85 and GST-pl 10a subunits can
be seen binding to the Ras beads. No other proteins from the PI 3-kinase preparation are accumulated on the Ras beads, indicating that it is not a subfraction of p85/pl 10 that is bound to a contaminating protein that is being retained on Ras. The only other proteins present are found in the Ras agarose bead preparation: they are bovine serum albumin at 68 kDa, which comes from prewashing the beads with buffers containing BSA to reduce non-specific binding, and very broad bands below 45 kDa which are recognised by Ras- specific antibodies and are probably oligomerised Ras fragments (data not shown). It is therefore likely that Ras interacts directly with PI 3-kinase.
3.2.3 Ras interacts with the p i 10 subunit of PI 3-KINASEIn order to determine whether the interaction with PI 3-kinase is through the
catalytic pi 10 subunit or the regulatory p85 subunit, Ras bound either to GTP or GDP was incubated with glutathione agarose immobilised GST-pl 10a, GST-p85a or GST- pl 10a/p85a. As shown in figure 3.6, Ras-GTP bound to pi 10a and p85/pl 10 but not to p85a alone. The interaction of Ras with PI 3-kinase is therefore mediated through the
pi 10 catalytic subunit. Addition of increasing amounts of monomeric p85 does not inhibit the binding of monomeric pi 10a to Ras.GTP in vitro, even though the
stoichiometric formation of p85/pllO heterodimers can be demonstrated (data not shown).
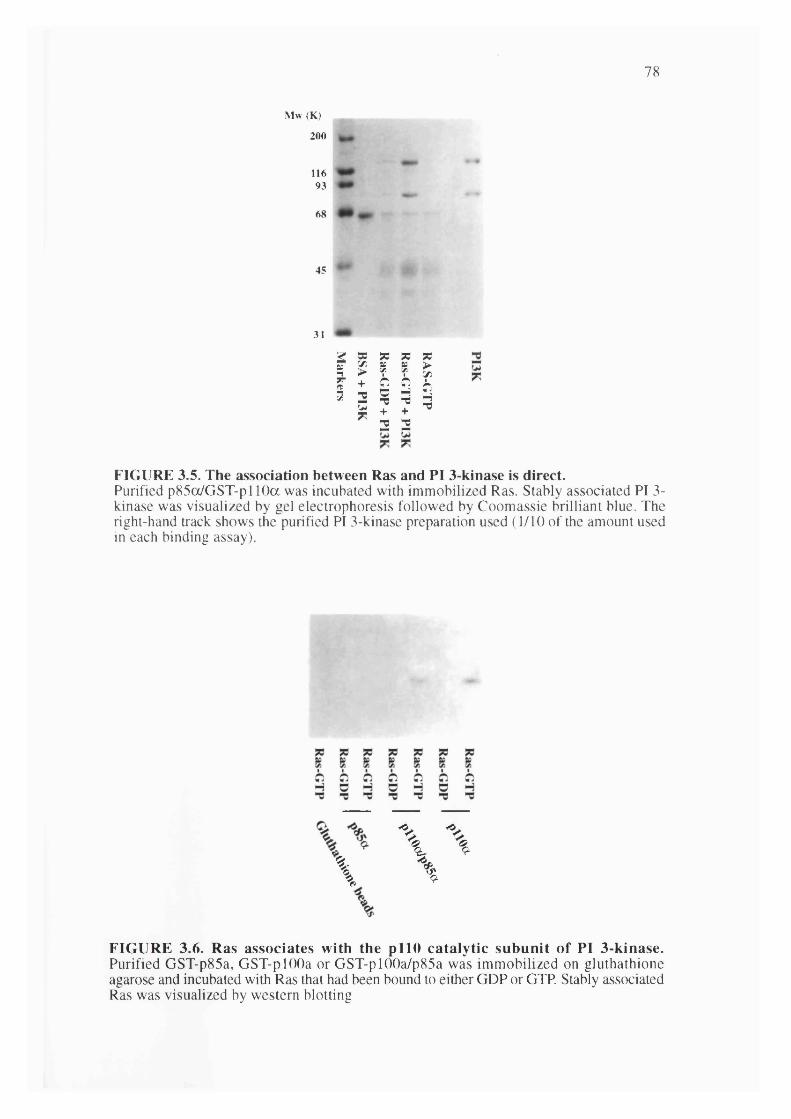
78
M w ( K )
200
11693
68
45
31
m2 2
FIGURE 3.5. The association between Ras and PI 3-kinase is direct.Purified p85o/GST-pl 10a was incubated with immobilized Ras. Stably associated PI 3- kinase was visualized by gel electrophoresis followed by Coomassie brilliant blue. The right-hand track shows the purified PI 3-kinase preparation used (1/10 of the amount used in each binding assay).
\ % \ %
FIGURE 3.6. Ras associates with the pllO catalytic subunit of PI 3-kinase.Purified GST-p85a, GST-pl00a or GST-plOOa/p85a was immobilized on gluthathione agarose and incubated with Ras that had been bound to either GDP or GTP. Stably associated Ras was visualized by western blotting

79
3.2.4 Determination of the affinity of interaction between Ras and PI 3-kinaseIn order to estimate the strength of the interaction between GTP-bound Ras and
PI 3-kinase, purified baculovirus expressed H-Ras protein was loaded with [a-^^P]GTP
and then increasing amounts allowed to bind to purified baculovirus expressed p85/GST-pllO in solution in the presence of excess unlabelled guanine nucleotide. The PI 3-kinase fusion protein was recovered using glutathione agarose, washed, and the amount of [a-^^P]GTP labelled Ras specifically bound determined by Cherenkov
counting. As shown in figure 3.7, the amount of Ras binding to PI 3-kinase saturates at about 300nM Ras. Half maximal binding is achieved at approximately 150nM Ras, giving a rough estimate of the binding affinity of Ras for PI 3-kinase. A slightly weaker binding was seen using the region of pi 10a that interacts with Ras (see below)
expressed as a bacterial GST fusion protein. Figure 3.7B shows the binding of this PI 3- kinase fragment, immobilised on glutathione agarose, to soluble Ras protein loaded with [a-^^P]GTP. The binding assays shown were carried out at close to physiological ionic
strength. At low ionic strength (with omission of the added lOOmM NaCl), the binding of Ras to p85/GST-pl 10a was about two fold stronger (data not shown).
4000 -O
3000
2000
1000
o 6 -0 200 800400 600 1000
5000
4000
3000
2000
1000
o 6 -0 1000800400 600200
[ R a s .G T P ] (n M ) [ R a s .G T P ] (n M )
FIGURE 3.7. Affinity of Ras binding to PI 3-kinase.A, purified baculovirus expressed soluble V12 Ras was labelled with y32-GTP and then varyingamounts mixed with soluble purified baculovirus expressed p85a/GST-pl 10a. After 1 hour, the PI 3-kinase was captured on gluthathione agarose, washed and the amount of labelled Ras bound was measured by Cherenkov counting. The background obtained when GST was usedinstead of p85a/GST-pllOa was subtracted for each point. Data shown is the average of triplicates with standard error shown. B, as above, but bacterially expressed pi 10 fragment, GST-pl 10 133-314, was used.

80
3.2.5 Determination of the site on p i 10 that binds RasIn order to determine where on the p llO a catalytic subunit Ras.GTP interacts with
PI 3-kinase, a number of deletion mutants were expressed in baculovirus. One mutant, A3-125, lacked the minimal p85 binding site (Dhand et al., 1994) while others lacked
larger stretches from the amino terminus. The interaction of these purified mutant proteins with immobilised Ras was examined: as shown in figure 3.8, only A3-125 interacted with Ras, while other mutants, including A1-395, failed to bind. This
suggested that the interaction site may lie between amino acid residues 125 and 395 of pi 10a. A GST fusion protein containing most of this area (amino acids 133-314) was
therefore expressed in bacteria and was found to be able to interact with Ras in a GTP- dependent manner, thus defining amino acid residues 133 to 314 of p i 10a as a sufficient binding site for Ras.GTP. A similar region of pi lOp was also expressed as a
GST fusion protein in bacteria and found to interact with Ras in a GTP-dependent manner; pi lOP is therefore also likely to be a target for Ras.
3.2.6 Effect of a point mutation in the Ras binding site of p i 10a on the interaction with Ras
The sequence of the three cloned forms of pi 10 in this region is compared in figure 3.9A. Conserved residues are boxed and a consensus sequence is written below. The homology over this region is 25% identity between pi 10a and p, 18% between pi 10a and y and 20% between pi lOp and y. This compares with overall homologies between the entire catalytic subunits of 42% identity between pi 10a and p, 36% between pi 10a and y and 34% between pllOP and y (Hiles et al., 1992; Hu et al., 1993;
Stoyanov et al., 1995).In order to investigate the significance of the Ras/pl 10 interaction in the activation
of PI 3-kinase in cells, a number of point mutations were made in p i 10. One of these, the substitution of the conserved lysine residue 227 of pi 10a by glutamic acid, resulted
in generation of a protein that was unable to interact with Ras. When expressed in an in vitro transcription/translation system, pi 10a K227E displayed PI 3-kinase activity,
suggesting that the overall folding of the protein was not disrupted (data not shown). However, this mutation completely blocked the ability of a GST fusion protein containing pi 10a amino acids 133-314 to bind to Ras.GTP (figure 3.9B).

81
I I I I I I I I I I 11% % % % \
% % %% \
B
>p85 >Ras Kinase
1# ■ ■■ ■ ■—
1068-----------------------------------#
126# -
pllO interaction with Ras.GTP
+
+396
108-#
133
422#-
314
FIGURE 3.8. Determination of the binding site for Ras on the pllO catalytic domain.(A) Purified deletion mutants of pi 10a expressed as GST fusion proteins in baculoviruses, or bacterially expressed GST fusion proteins of short fragments of pi00a or p, were mixed with soluble Ras bound to either GTP or GDP. The GST fusion proteins were recovered on glutathione-agarose, washed and probed for the presence of Ras by Western blotting using anti-Ras antibodies. (B) Summary of the Ras binding behaviour or the pi 10 mutants shown in (A).

82
141 pllOa 150 pllOp 143 pllOyConsensusi
RNILNVCKEA VDLRDLNSPH SRAMYVYPPN VESSPELPKH lYNKLDKGQI RKMRKFSEEK ILSLVGLSWM DWLKQTYPPE HEPS..IPEN LEDKLYGGKL RGLVTPRMAE VASRDPKLYA MHPWVTSKP LPEY LWKKIANNCIR------- e- v-srd--s-- typP- -e-s--lPe- l--Kl--g-
191 pllOa IWIWVIVSP NNDKQKYTLK INHDCVPEQV lAEAIRKKTR SMLLSSEQLK197 pllOp . . . .IVAVHF ENCQDVFSFQ VSPNMNPIKV NELAIQKR...... LTIHGKE180 pllOy FIVI HRSTTSQTIK VSPDDTPGAI LQSFFTKMAK KKSLM....DConsensus v-v-- -n----- t-k vspd--P--v---ai-K------- L-----
241 pllOa LCVLEYQGKY ILKVCGCDEY FLEKYPLSQY KYIRSCIMLG RMPN..LMLM241 pllOp DEVSPY..DY VLQVSGRVEY VFGDHPLIQF QYIRNCVMNR ALPHFILVEC22 6 pllOy IPESQSEQDF VLRVCGRDEY LVGETPIKNF QWVRHCLKNG EEIHWLDTPConsensus --vs-y--dy vL-VcGrdEY --g--Pl-qf qyiR-C-mng --ph--L--
289 pllOa A K E S L Y S Q L P M D C F T M P S Y S289 pllO(3 C K IK K M Y E Q E M I A I E A A I N R
B
Oo•V
CH"t
% 'Q-4 ? ,
% %
FIGURE 3.9. Mutation in pllOa that blocks binding to Ras.(A) Alignment of sequences from the Ras binding site of the p 110 subunit of PI 3-kinases isoforms. The sequences of pi 10a, plOOp and pllOy are shown. Conserved residues are in bold. A consensus sequence is written below: residues conserved in all three sequences are in upper case, those conserved in two out of three are in a lower case. Lysine 227 in pi 10a is marked with an asterisk. (B) Binding to Ras of GST fusion proteins containing residues 133-314 of pi 10a: effect of the K227E mutation. Performed as in figure 3.8.

83
CHAPTER 4 Activation of PI 3-kinase by Ras4.1 Introduction
In the previous chapter PI 3-kinase has been shown to directly interact with Ras in vitro in the manner expected of an effector protein. In this chapter the biological significance of this interaction is analysed by studying the effects of activated Ras on the activity of PI 3-kinase, both in vivo and in vitro, as well as investigating the contribution of endogenous cellular Ras to PI 3-kinase activation by extracellular signals.
4.2 Results
4.2.1 Dominant negative Ras inhibits elevation of PI 3' phosphorylated lipid levels by growth factors in intact PC12 cells
To investigate the possible physiological significance of the ability of GTP-bound Ras to interact with PI 3-kinase, the effect of expressing a dominant negative mutant of Ras, Asnl7, on growth factor stimulation of PI 3' phosphorylated lipid levels in intact cells was studied. Since most cell types cannot proliferate while expressing Asnl7 Ras, the rat pheochromocytoma cell line PC 12 in which Ras regulates differentiation rather than proliferation was chosen. PCI2 cells were transfected with an RSV Asnl7 Ras construct and clones of cells selected. The ability of nerve growth factor and epidermal growth factor to elevate PI 3' phosphorylated lipid levels in these cells was compared to the parental cells. Cells were metabolically labelled with ^^P-orthophosphate and levels of the 3' phosphorylated phosphoinositides determined following NGF or EGF stimulation. As shown in figures 4.1 A and 4 .IB, the amounts of PI(3,4)P2 and PIP3 in
wild type PC 12 cells increases by between ten and twenty fold in response to EGF or NGF. By contrast, in the Asnl7 Ras expressing PCI2 cells, the growth factor stimulation of PI(3,4)P2 levels is attenuated by about five fold. The response of the PIP3
lipid pool to growth factors is attenuated somewhat less, by about three fold, due to the expression of Asnl7 Ras in PC 12 cells. The incomplete inhibition probably reflects the fact that several mechanisms exist whereby growth factors can control phosphatidylinositol 3' kinase activity, only one of which involves Ras. The time courses of the responses are similar in both wild type and Asnl7 Ras expressing PC 12 cells.
In order to be certain that the inhibition of the growth factor induced elevation of PI(3,4)P2 and PIP3 in dominant negative Ras expressing cells is specific to this pathway
and not a general phenomenon affecting other signalling pathways, a number of other
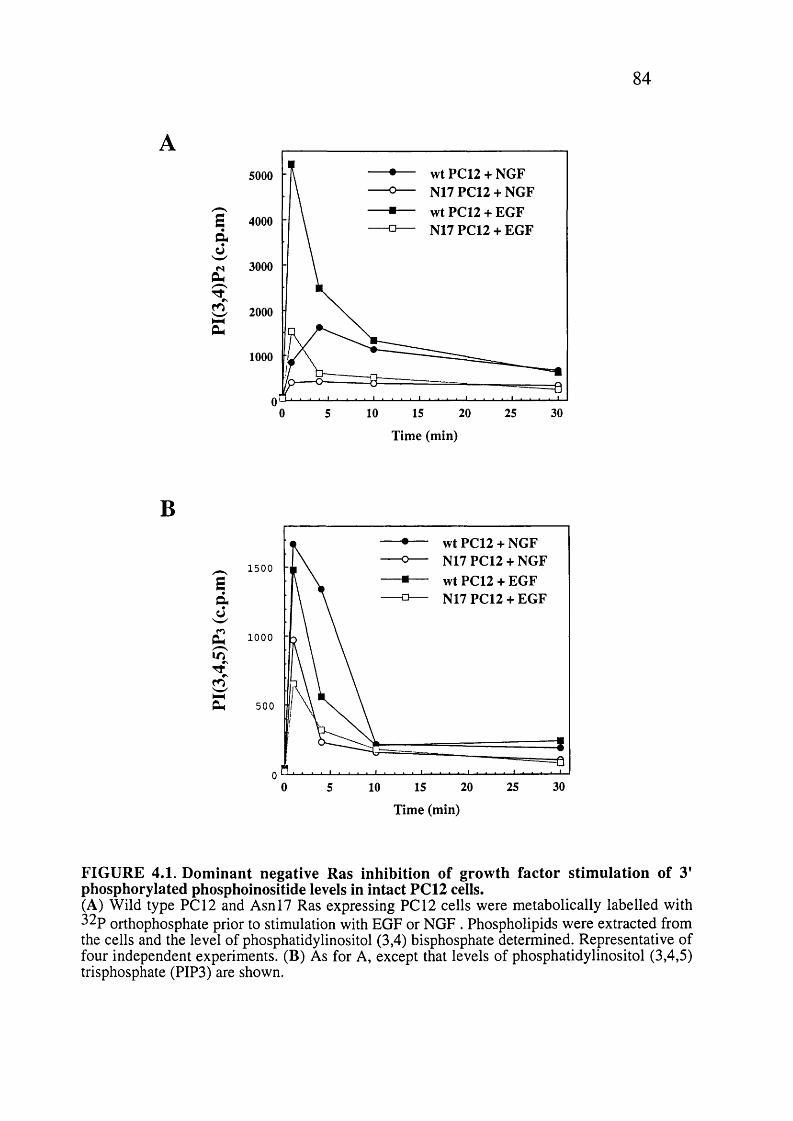
84
wt PC12 + NGF N17 PC12 + NGF wt PC12 + EGF N17 PC12 + EGF
5000
£ 4000d
3000
2000
1000
0 10 20 25 30155
B
Sd
d
%
Time (min)
wt PC12 + NGF N17 PC12 + NGF wt PC12 + EGF N17 PC12 + EGF
1500
1000
500
03020 250 10 155
Time (min)
FIGURE 4.1. Dominant negative Ras inhibition of growth factor stimulation of 3' phosphorylated phosphoinositide levels in intact PC12 cells.(A) Wild type PC 12 and Asnl7 Ras expressing PCI2 cells were metabolically labelled with ^^P orthophosphate prior to stimulation with EGF or NGF . Phospholipids were extracted from the cells and the level of phosphatidylinositol (3,4) bisphosphate determined. Representative of four independent experiments. (B) As for A, except that levels of phosphatidylinositol (3,4,5) trisphosphate (PIP3) are shown.

85
B
EcB
4
3
2
0
M w (KDa)
200 -
97 -
68 -
45 -
2 2 2 z z zc Q 0 3 :: :3W W W
+ +
FIGURE 4.2. Dominant negative Ras does not inhibit all agonist stimulated signals.(A) Antiphosphotyrosine immunoprecipitates (PY IPs) were made of PC 12 or N17 Ras- expressing PCI2 cells treated wth EGF or NGF for 1 and 4 min respectively. These immunoprecipitates were assayed for PI 3-kinase activity in vitro. Activity is expressed as multiples of the amount in similar IPs from unstimulated cells. (B) Wild-type and N17 Ras expressing PC 12 cells were metabolically labelled with 3H- inositol before stimulation with EGF or NGF. Soluble inositol polyphosphates were extracted and the levels of Ins(l,4)P2 and Ins(l,4,5)P3 were determined. Elevation above the levels in u n s tim u la ted c e lls is show n. (C) Antiphosphotyrosine western blots of whole cell lysates from (A).

8 6
growth factor stimulated events were studied. Firstly, the amount of PI 3-kinase activity found in anti-phosphotyrosine immunoprecipitates from stimulated cells was measured in an in vitro assay. As shown in figure 4.2A, EGF and NGF treatment of PC12 cells lead to an approximately four and three fold stimulation respectively of the amount of PI 3-kinase activity found in anti-phosphotyrosine immunoprecipitates. Expression of Asnl7 Ras in the PCI2 cells does not reduce this activity, indicating that growth factor control of PI 3-kinase through association with phosphotyrosine containing proteins is normal even in the presence of dominant negative mutant Ras protein.
In addition, the production of inositol phosphates (inositol (1,4,5) trisphosphate and inositol (1,4) bisphosphate) by phosphatidylinositol-specific phospholipase C in response to EGF and NGF is also unaltered by Asnl7 Ras expression (figure 4.2A). Furthermore, expression of Asnl7 Ras in PCI2 cells does not obviously alter the ability of EGF and NGF to induce tyrosine phosphorylation of total cellular proteins (figure 4.2C). It is therefore likely that the expression and function of growth factor receptor tyrosine kinases and the regulation of PI 3-kinase by tyrosine phosphoprotein binding are not being directly altered by the presence of Asnl7 Ras in these PC 12 cells.
4.2.2 Ras elevates PI 3’ phosphorylated lipid levels in intact COS cellsFrom the ability of Ras and PI 3-kinase to interact in vitro and the inhibitory effect
of Asnl7 Ras on PI 3' phosphorylated lipid accumulation in vivo, it appears likely that Ras is regulating the function of PI 3-kinase in intact cells. To explore this possibility further, the effect of over-expressing Ras transiently on 3' phosphorylated phosphoinositides in COS cells was investigated.
As shown in figure 4.3A, when a vector expressing activated mutant V ail2 Ras was transfected into COS cells, some elevation was found in the levels of PI(3,4)P2 and PIP3 in the intact cells. Expression of the p85a subunit plus the pi 10a subunit of PI 3-
kinase together had a similar effect on these lipids. However, when all three proteins were expressed together they co-operated to give a very large increase in the level of 3' phosphorylated phosphoinositides. In figure 4.3B it is shown that wild type Ras is almost as potent as V ail2 Ras in elevating these lipids, presumably reflecting the very high levels of Ras protein expression achieved in this system, while the effector mutant Vall2Ala38 Ras is inactive.
V-Src also caused a similar level of activation. A number of other Ras-related proteins were studied in this assay, including RhoA and Racl. None of these have any effect on the 3' phosphorylated phosphoinositide levels. The ability of v-Src to elevate the levels of these lipids was partially blocked by the dominant negative mutant of Ras, Asnl7, consistent with the hypothesis that v-Src can activate PI 3-kinase through both a Ras-dependent and a Ras-independent pathway, the latter presumably involving tyrosine phosphoprotein binding to p85.
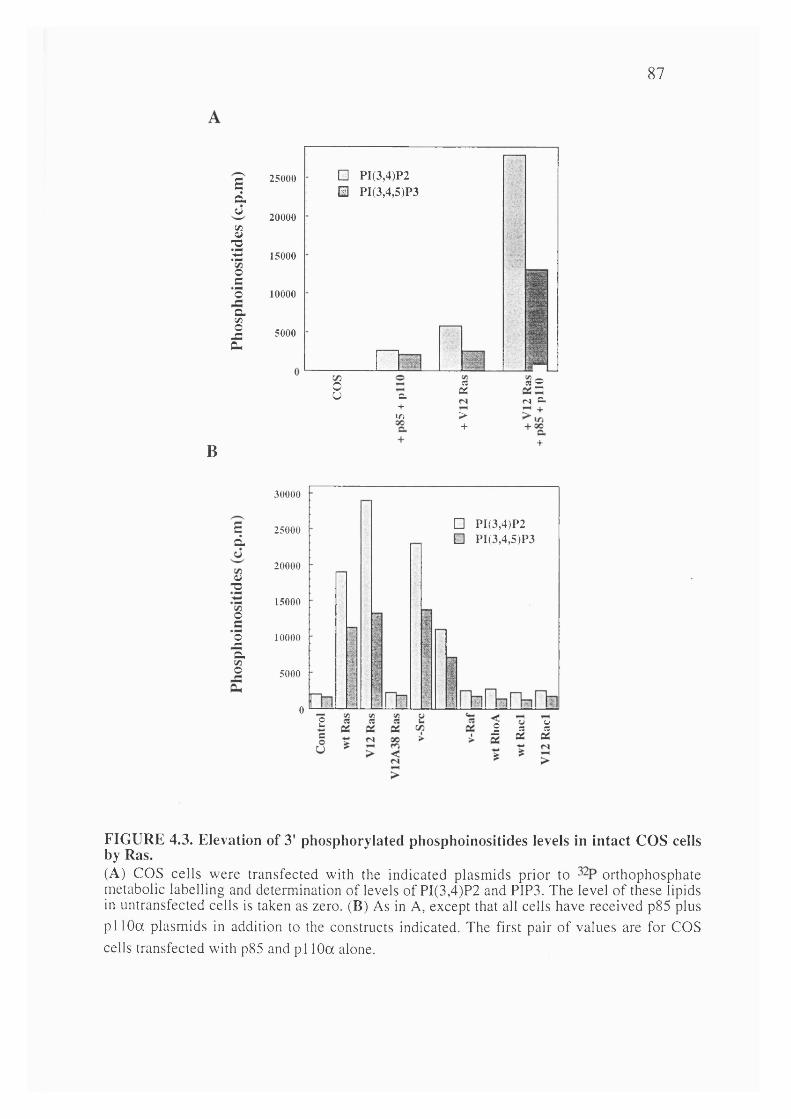
87
25000
20000
BdVs
13. t i 15000©coaViOÆeu
10000
5000
□ PI(3,4)P2 B PI(3,4,5)P3
Ou c.+if)
B
« «2a (S o.+>+
+
cd
©■o
o’©U3O.O
30000
□ PI(3,4)P2 0 PI(3,4,5)P3
25000
20000
15000
10000
5000
0
i C32 un
FIGURE 4.3. Elevation of 3’ phosphorylated phosphoinositides levels in intact COS cells by Ras.(A) COS cells were transfected with the indicated plasmids prior to orthophosphate metabolic labelling and determination of levels of PI(3,4)P2 and PIP3. The level of these lipids in untransfected cells is taken as zero. (B) As in A, except that all cells have received p85 plusp i 10a plasmids in addition to the constructs indicated. The first pair of values are for COScells transfected with p85 and pi 10a alone.

8 8
The v-Raf serine/threonine kinase, or an amino terminally truncated, activated version of c-Raf (ARaf), which act immediately downstream of Ras to activate the MAP
kinase pathway, do not cause elevation of 3' phosphorylated phosphoinositides, so it is unlikely that the effects seen with Ras are indirect through secondary events such as
autocrine production of growth factors (see also discussion). As also reported previously by others (Howe et al., 1992; Kyriakis et al., 1992) in this system transfection of either
activated Ras or Raf could be shown to activate MAP kinase (data not shown).
4.2.3 Effect of interaction of p i 10 with p85 and with Ras on PI 3-kinase activity in cells
The Cos cell transient cotransfection assay was further used to explore aspects of the regulation of PI 3-kinase activity. As shown in Table 4.1, the p85 regulatory subunit had minimal effect on the PI(3,4)P2 and PIP3 levels in cells transfected with the pi 10 catalytic subunit or pi 10 plus activated Ras. Immunoprécipitation of pi 10 through a carboxy terminal myc epitope tag followed by immunoblotting for p85 revealed that in the absence of cotransfected p85, pi 10 was greater than 95% monomeric, or at least not bound to p85 a or p (data not shown). This suggests that the stimulation of PI 3-kinase activity of pi 10a by Ras seen in this system does not rely on signalling through p85,
such as phosphotyrosine/SH2 domain interactions.Cotransfection of the inter SH2 domain of p85 (the binding site for p i 10) with
p i 10 leads to a relatively weak elevation of PI(3,4)P2 and PI(3,4,5)P3 levels, as suggested by previous reports (Hu et al., 1995). However, inclusion of the p85 binding site of p i 10 (amino acids 1-125) as a separate fragment, which might be expected to sequester endogenous p85, did not alter the ability of pi 10 to elevate PI(3,4)P2 and PIP3 levels either in the presence or absence of Ras. Immunoprécipitation of this
fragment using an epitope tag showed that it was capable of binding to endogenous or cotransfected p85 (data not shown). Furthermore, cotransfection of a S608A mutant of p85, which is resistant to phosphorylation by pi 10 (Dhand et al., 1994), did not greatly affect the activity of PI 3-kinase in this system.
Cos cell transient transfection was also used to study whether post-translational modification and membrane localisation of Ras was required in order for it to activate PI 3-kinase. Farnesylation of Ras at C l86 is absolutely required for it to influence the activity of PI 3-kinase: very similar results have been reported for Ras activation of Raf and MAP kinase (Leevers and Marshall, 1992) . By contrast, removal of the second membrane localisation signal, either palmitoylation at C181 and C l84 for H- and N-Ras (Hancock et al., 1989) or a polybasic region near the carboxy terminus for K-Ras (Hancock et al., 1990), only slightly reduces Ras activation of PI 3-kinase. Although these mutants are not predominantly associated with the plasma membrane (Hancock et al., 1989; Hancock et al., 1990), they do retain a significant amount of transforming

89
activity and are still able to activate Raf and MAP kinase quite efficiently (Cadwallader et al., 1994). It is possible that there is a large excess of Ras expressed in these assays, such that only a minor fraction at the membrane is required for activation of PI 3-kinase: however, the absolute requirement for farnesylation is clear.
Plasmids transfected PI(3,4)P2 PI(3,4,5)P3(% of level with V12 Ras + p85 4- p i 10)
None 0 0pi 10a 1 2 . 7 5 . 1pllO +V12H-Ras 106 122pi 10 + p85a 1 0 . 6 6 . 1pi 10 + p85 + V12 Ras 100 100p85 inter SH2 + pi 10 2 3 . 3 1 1 . 4p85 inter SH2 + pi 10 + V12 Ras 106 123pllO (l-125)-kpllO 1 4 . 8 5 . 5p ll0 (l-1 2 5 ) + p l l0 + V12 Ras 108 124pllO + p85 S608A 1 8 . 6 9 . 9pi 10 + p85 S608A + V12 Ras 112 5 7 . 2pllO +V12RasS181/184 7 7 . 0 5 4 . 9pllO +V12RasS186 7 . 7 2 . 9pllO +V12K-Ras 102 9 8 . 2pllO +V12K-Ras K6Q 108 8 4 . 3
TABLE 4.1. Levels of phosphatidylinositol (3,4) bisphosphate (PI(3,4)P2) and phosphatidylinositol (3,4,5) trisphosphate (PIP3) in transiently transfected ^^P metabolically labelled COS cells.Phosphoinositides were analysed by HPLC (200,000 c.p.m. of PI(3,4)P2). Phospholipid
levels are expressed as a percentage of the level seen in cells transfected with V12 H- Ras plus p85 and pi 10 plasmids. Background corresponding to that seen in the same fractions from untransfected cells was subtracted. The background subtracted was no more than 3% (800 cpm for PIP3) and 600 cpm for PI(3,4)P2).
4.3.4 Effect of the K227E mutation in the Ras binding site of p i 10 on regulation of enzymatic activity
The effect of the K227E mutation in the Ras binding site of pi 10 on the ability of Ras to activate PI 3-kinase in intact cells was also investigated. As shown in figure 4.4, transient transfection of either V12 Ras or p85/pl 10 into Cos cells causes modest elevation of the cellular levels of PIP3. Transfection of both activated Ras and p85/pl 10 together gives a much greater than additive elevation of PIP3 levels, suggesting that Ras
is directly activating PI 3-kinase. Use of the K227E mutant pi 10 in these transfections
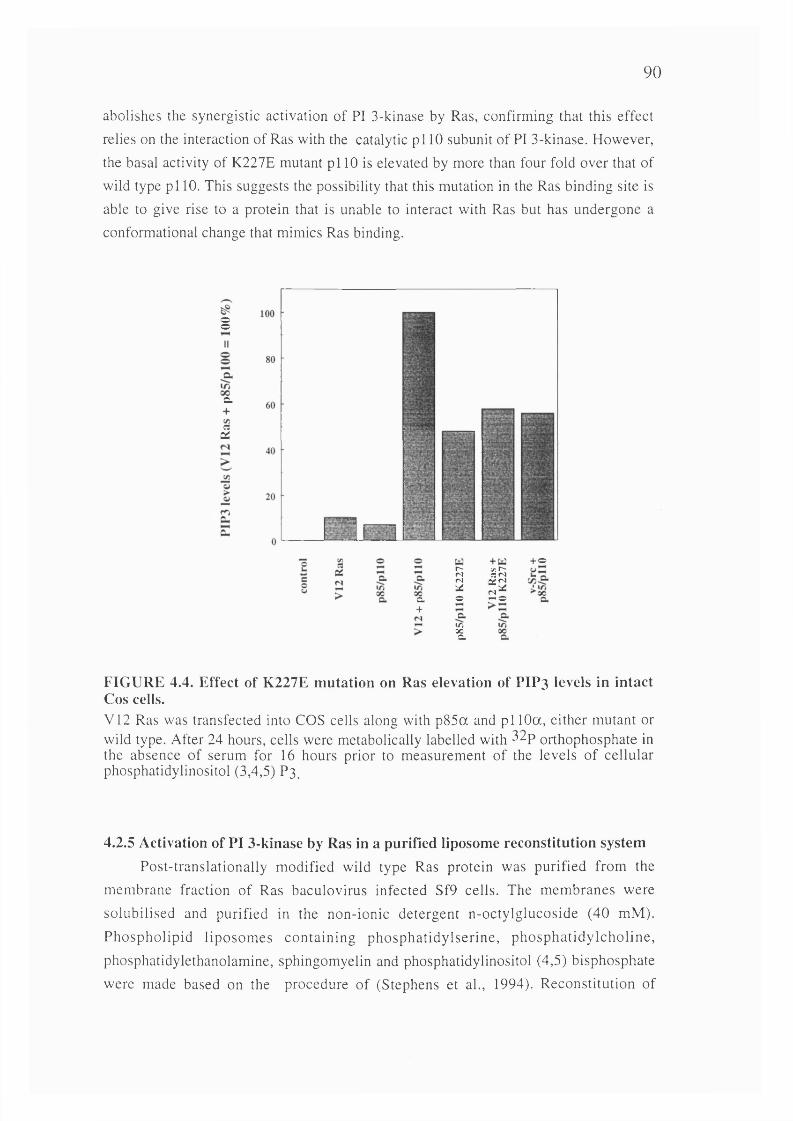
90
abolishes the synergistic activation of PI 3-kinase by Ras, confirming that this effect
relies on the interaction of Ras with the catalytic p i 10 subunit of PI 3-kinase. However,
the basal activity of K227E mutant p i 10 is elevated by more than four fold over that of
wild type pi 10. This suggests the possibility that this mutation in the Ras binding site is
able to give rise to a protein that is unable to interact with Ras but has undergone a
conformational change that mimics Ras binding.
§
c.
C3(Z
a.+
Io
II
+ o
FIGURE 4.4. Effect of K227E mutation on Ras elevation of PIP3 levels in intact Cos cells.V 12 Ras was transfected into COS cells along with p85a and p i 10a, either mutant or wild type. After 24 hours, cells were metabolically labelled with ^^P orthophosphate in the absence of serum for 16 hours prior to measurement of the levels of cellular phosphatidylinositol (3,4,5) P 3 .
4.2.5 Activation of PI 3-kinase by Ras in a purified liposome reconstitution systemPost-translationally modified wild type Ras protein was purified from the
membrane fraction of Ras baculovirus infected Sf9 cells. The membranes were
solubilised and purified in the non-ionic detergent n-octylglucoside (40 mM).
Phospholipid liposom es containing phosphatidylserine, phosphatidylcholine,
phosphatidylethanolamine, sphingomyelin and phosphatidylinositol (4,5) bisphosphate
were made based on the procedure of (Stephens et ah, 1994). Reconstitution of

91
modified Ras into the liposomes was achieved by adding a concentrated stock solution of Ras to the liposomes in the absence of additional detergent, such that the final detergent concentration in the mixture was only 1 mM n-octylglucoside, roughly 5% of the critical micellar concentration. The total lipid:detergent ratio was 1.6:1. Following mixing and incubation on ice for 10 minutes, the liposomes were collected by ultracentrifugation. As shown in Table 4.2, about 35% of the modified Ras could be recovered stably associated with the liposomes, as judged from the inclusion of a prelabelled Ras.[a-^^P]GTP tracer. Unmodified Ras was unable to associate with
liposomes. When the liposome pellet was resuspended using sonication, roughly 80% of the associated Ras was accessible to the medium as judged by the ability of EDTA to exchange labelled for unlabelled GTP.
Modified Ras Unmodified Ras No Ras(% of total input) (% of total input)
Total R as.[a-^^P]G TP added 100 100R as.[a-32p]G TP associated
with liposomes 3 4 . 8 ± 0 . 2 0 . 4 ± O. l[a-^2p]G T P dissociating fromliposomes in presence of EDTA 2 8 . 3 ± 0 . 3 0 . 2 ± 0 . 0 4
Total [a-^^P]G TP addedwithout Ras 100
[a-^^P]G TP associatedwith liposomes 0 . 0 9
[a-^^P]G TP dissociating from Ras in liposomesin presence of EDTA 0 .0 2
TABLE 4.2. Reconstitution of Ras into liposomes.Post-translationally modified Ras, or unmodified Ras, was labelled with [a-^^P]GTP and reconstituted into liposomes as described in "Methods". Ras associating with liposomes was estimated by counting anti-Ras monoclonal antibody Y13-259 immunoprecipitates of solubilised washed liposomes. Total Ras input was also measured by immunoprécipitation. Label released with EDTA and the "No Ras" controls were measured by direct counting of liposome or supernatant fractions.
The ability of purified baculovirus expressed PI 3-kinase (p85a/GST-pl 10a) to phosphorylate the PI(4,5)P2 in these liposomes was tested. As shown in figure 4.5, PI 3- kinase efficiently phosphorylated PI(4,5)P2 to give PIP3. In absence of Ras, the activity
of the lipid kinase could be stimulated about three fold by the addition of a tyrosine phosphopeptide based on the autophosphorylation site Y751 of the human PDGF
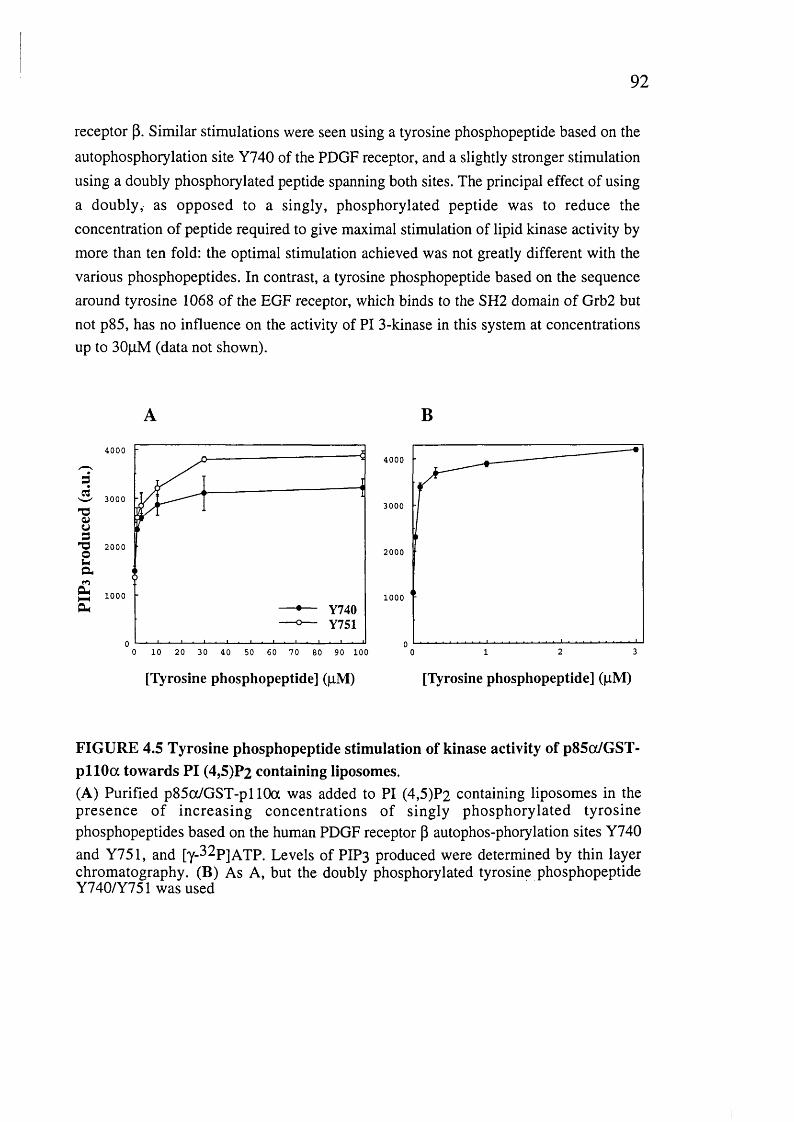
92
receptor p. Similar stimulations were seen using a tyrosine phosphopeptide based on the
autophosphorylation site Y740 of the PDGF receptor, and a slightly stronger stimulation using a doubly phosphorylated peptide spanning both sites. The principal effect of using a doubly, as opposed to a singly, phosphorylated peptide was to reduce the concentration of peptide required to give maximal stimulation of lipid kinase activity by more than ten fold: the optimal stimulation achieved was not greatly different with the various phosphopeptides. In contrast, a tyrosine phosphopeptide based on the sequence around tyrosine 1068 of the EGF receptor, which binds to the SH2 domain of Grb2 but not p85, has no influence on the activity of PI 3-kinase in this system at concentrations up to BOpM (data not shown).
B
40004000
Seg
w 3000
■gusT 3 2 0 0 0
2 a£
3000
2000
1000 1000Y740Y751
10 20 30 40 60 70 90 100
[Tyrosine phosphopeptide] (pM) [Tyrosine phosphopeptide] (pM)
FIGURE 4.5 Tyrosine phosphopeptide stimulation of kinase activity of p85a/GST- p llO a towards PI (4,5)P2 containing liposomes.(A) Purified p85cx/GST-pllOa was added to PI (4,5)P2 containing liposomes in the presence of increasing concentrations of singly phosphorylated tyrosinephosphopeptides based on the human PDGF receptor P autophos-phorylation sites Y740and Y751, and [y-^^P]ATP. Levels of PIP3 produced were determined by thin layer chromatography. (B) As A, but the doubly phosphorylated tyrosine phosphopeptide Y740/Y751 was used

93
6000
5000Sd
GDPGTERas.GDPRasGTP
4000
3000ka
g 2000
1000
10 20 300
[Tyrosine phosphopeptide Y740] (|iM)
B3
éT3o3
2a£
6000 Ras.GDPRas.GTPRas.GTP,259Ras.GTP,238
4000
2000
03010 200
[Tyrosine phosphopeptide Y751] (|iM)
FIGURE 4.6. Effect of Ras.GTP on PI 3-kinase activity in the liposome system.(A) The kinase activity of p85a/GST-pl 10a towards PI (4,5)?2 was measured in liposomes into which had been reconstituted post-translationally modified Ras that wasbound to GTP or GDP. The influence of increasing concentrations of PDGF receptor p Y740 phosphopeptide was determined. The effect of GTP or GDP alone in the absence of Ras was also measured. Average of triplicates with standard error shown. Representative of five experiments. (B) The influence of anti-Ras monoclonal antibodies Y13-259 and Y13-238 on the stimulation of PI 3-kinase activity by Ras.GTP was measured as in A, except that tyrosine phosphopeptide Y751 was used. Average of duplicates with standard error shown.
In order to study the effect of Ras on PI 3-kinase activity, Ras reconstituted into liposomes was loaded with either GTP or GDP in the absence of magnesium. PI 3- kinase phosphorylation of PI(4,5)P2 to give PIP3 was then measured. As shown in

94
figure 4.6, GDP bound Ras did not significantly affect the activity of PI 3-kinase in this system. GDP or GTP alone were also without effect in the absence of Ras. However, GTP bound Ras caused a significant increase in the activity of PI 3-kinase. The ras.GTP induced stimulation observed in the absence of phosphopeptide Y740 was about 2.5 fold and was similar in the presence of saturating concentrations of peptide. However, the activation of PI 3-kinase activity by Ras.GTP was most marked at low concentrations of tyrosine phosphopeptide. For example, at 1 |iM Y740 phosphopeptide there was only a
small stimulation of PI 3-kinase activity in the absence of Ras.GTP, but maximal stimulation in the presence of Ras.GTP, a stimulation by Ras.GTP of more than four fold. Similar results are seen for the Y751 phosphopeptide (figure 4.6B): in the absence of peptide the Ras induced stimulation was three fold, while in the presence of saturating peptide Ras.GTP stimulated 3.5 fold. At IpM Y751, Ras.GTP induced a five
fold activation of PI 3-kinase. Comparable results were found for the doubly phosphorylated peptide, including the leftward shift of the dose response curve induced by Ras.GTP (data not shown).
To be certain that this effect was due to Ras and not contaminants in the Ras preparation, the neutralising Ras monoclonal antibody Y13-259 was added to the Ras preparation prior to reconstitution. Excess antibody was removed during the preparation of the Ras liposomes. As shown in figure 4.6B, treatment of Ras in this way with Y13- 259, but not the non-neutralising Y13-238 antibody, blocked the ability of Ras.GTP to stimulate PI 3-kinase activity. These data indicate that GTP bound Ras can activate the enzymatic activity of PI 3-kinase in a defined system, and that the interaction is synergistic with tyrosine phosphopeptides that bind to the SH2 domains of p85.
4.3 Discussion to Chapters 3 and 4
The regulation of PI 3-kinase in response to growth factor treatment of cells could involve a number of mechanisms: p85 is phosphorylated on tyrosine residues in response to treatment of cells with growth factors such as PDGF and it binds through its SH2 domains to autophosphorylation sites on receptor tyrosine kinases and related sites on other molecules, such as IRS-1, CD 19 and CD28 (Pages et al., 1994; Stephens et al., 1993; Tuveson et al., 1993). Tyrosine phosphorylation of p85 has not been shown to change its catalytic activity, but binding to the tyrosine phosphorylated insulin receptor substrate, IRS-1, and to tyrosine phosphorylated peptides, particularly those based on the sequences of the p85 binding sites in the PDGF receptor and IRS-1, does lead to a relatively small increase in the catalytic activity of PI 3-kinase (Backer et al., 1992; Carpenter et al., 1993; Myers et al., 1992). It is possible that in the intact cell much stronger stimulations result from such interactions due to translocation of PI 3-kinase

95
from the cytosol to the plasma membrane where its substrate lipids are located. In addition, another mechanism of regulation could involve proline-rich sequences in p85 binding to SH3 domains of Src-family kinases such as Lck (Prasad et ah, 1993; Vogel and Fujita, 1993).
Most mitogenic stimuli activate both Ras and PI 3-kinase (Lowy and Willumsen, 1993; Stephens et ah, 1993). Here evidence is presented that interaction with Ras-GTP provides another possible mechanism of regulation of PI 3-kinase. The co- inmunoprecipitation of PI 3-kinase activity with Ras was previously reported by Lapetina et al. (Sjôlander et ah, 1991). Here I have extended those observations to show that the interaction of Ras and PI 3-kinase is direct, requires Ras to be in the active, GTP-bound conformation and occurs through the effector site on Ras. Ras can therefore interact with PI 3-kinase in vitro in the manner expected for a true effector interaction. The original observation of PI 3-kinase activity in Y13-259 immunoprecipitates of Ras (Sjôlander et ah, 1991) indicated that it was unlikely that the interaction was through the effector site on Ras since this antibody blocks its biological activity. However, I find that Y 13-259 strongly inhibits the interaction of Ras and PI 3-kinase : it is possible that differences exist between various preparations of this antibody, or that it does not completely inhibit the interaction.
4.3.1 Regulation of PI 3-kinase activity by RasThe question of whether Ras is involved in the regulation of PI 3-kinase within
intact cells is critical. The demonstration here that a dominant negative Ras mutant severely inhibits the ability of NGF and EGF to elevate PI 3' phosphorylated lipid levels in PCI2 cells in vivo suggests that endogenous Ras is necessary for at least some extracellular signals to activate PI 3-kinase (this point will be discussed again in the final discussion). Similar inhibition of PI 3-kinase activation by PDGF upon expression of N17 Ras has been observed more recently (Klinghofer et al., 1996).
Further in vivo evidence implicating Ras in the regulation of PI 3-kinase comes from the transient transfections in COS cells: the elevation of cellular 3' phosphorylated phosphoinositides caused by Ras when co-expressed with p85 and pi 10 indicates that Ras is capable of activating PI 3-kinase. It is possible that expression of activated Ras could lead to the autocrine production of growth factors that could cause activation of PI 3-kinase through a tyrosine kinase mediated pathway. While the experiments have been designed to minimise the impact of autocrine growth factor production, a more compelling argument for a direct effect of Ras is that activated Raf does not have the same stimulatory effect as Ras on 3' phosphorylated phosphoinositide levels. This is despite the fact that Raf causes production of a similar set of autocrine growth factors as does Ras.

96
The behaviour of the K227E mutant of pi 10 further establishes, not only that this residue is essential for interaction with Ras, but also that the direct interaction of Ras with PI 3-kinase is necessary for the elevation of PIP3 levels in Cos cells transfected
with activated Ras and p85/pllO. This indicates that Ras-induced production of autocrine growth factors is not sufficient by itself to cause the increase in PIP3 seen and
that direct interaction with Ras is required.The data from the COS cells indicate that there exists a point of divergence in the
signalling pathway downstream of Ras and upstream of the Raf kinase which could be accounted for by a direct activation of PI 3-kinase by Ras.
The site of the interaction of Ras with PI 3-kinase is shown to be between residues 133 and 314 on the catalytic p i 10 subunit, with lysine residue 227 being essential for the interaction. This is a region immediately carboxy-terminal to the site of interaction between pi 10 and the regulatory subunit p85. This suggests a model in which the lipid kinase receives regulatory signals through two domains in its amino-terminal region: one signal comes from tyrosine phosphoproteins, and possibly from SH3 domains of Src family kinases, through p85 and its interaction with the first 150 amino acids of pi 10. The other signal comes from the direct interaction of Ras.GTP with the neighbouring region of p i 10, roughly spanning the next 150 amino acid residues. It is likely that optimal activation of PI 3-kinase requires input from both signalling pathways.
Evidence for this model comes from in vitro activation studies where Ras synergizes with tyrosine phosphopeptides interacting with the SH2 domains of the p85 subunit to activate PI 3-kinase activity in a liposome reconstitution system (see below). In addition, the observation that N17 Ras expression inhibits the increase in PI 3' phosphorylated lipids upon NGF and EGF stimulation of PCI2 cells without affecting PI 3-kinase interaction with tyrosine phosphorylated proteins further argues that both Ras-GTP binding to the catalytic pi 10 subunit and tyrosine-phosphorylated proteins (and possibly other molecules) binding to the regulatory p85 subunit cooperate in activating PI 3-kinase under normal conditions. Another related case is that of polyoma virus middle-T antigen, which is phosphorylated at multiple tyrosine residues by p60Src. A non-transforming mutant of middle-T exists that does not bind to Shc/Grb2 and therefore does not activate Ras (Campbell et al., 1994; Dilworth et al., 1994). This mutant still binds normally to PI 3-kinase through its tyrosine phosphorylated p85 binding site but does not cause elevation of 3' phosphorylated lipids (Ling et al., 1992).
Since growth factors such as PDGF stimulate both tyrosine phosphorylation of p85 binding sites and activation of Ras, both these pathways will be activated in response to the growth factor, allowing potentiation of the activation signals being transmitted to PI 3-kinase. This is analogous to the stimulation of Raf-1 by growth

97
factors: multiple signalling pathways feeding into Raf-1 activation are triggered by a single growth factor, including Ras activation, Src-family tyrosine-kinase activation and protein kinase C activation (see section 1.4.1) Ras contributes to Raf-1 activation by translocating it to the plasma membrane(Leevers et al., 1994; Stokoe et al., 1994) while Src-type kinases directly phosphorylate and further stimulate Raf-1 activity. It is possible that this growth factor induced activation of multiple signalling pathways that then synergise together to activate Ras effectors such as Raf and PI 3-kinase allows the generation of strong downstream signals from activation of small numbers of receptors. It may also allow for the greatest flexibility in the range of responses generated by different growth factors that activate seemingly similar sets of signalling pathways.
At the time this studies were started pi 10a was the only PI 3-kinase catalytic subunit known. It is now clear that pi 10a is just one member of an still expanding
family of PI 3-kinase isoforms which based on structural and functional similarities has been divided into several sub-families (see section 1.6). The sequence homology in the region that interacts with Ras suggests that regulation by Ras is a common feature of class I PI 3-kinases.It is shown here that pi 10(3 behaves like pi 10a with regard to its ability to interact with Ras-GTP. p i 106, another recently identified class IA PI 3-kinase member has also been shown to interact with Ras-GTP (Vanhaesebroeck et al., 1997). Interestingly, pllOy, a member of the class IB PI 3-kinase sub-family that is activated by (3y subunits of
heterotrimeric G proteins also interacts in vitro with Ras-GTP (Rubio et al., 1997). It is therefore possible that stimuli that activate heterotrimeric G proteins could synergistically activate class IB PI 3-kinases through both (3y subunits and Ras in a
manner analogous to the activation described for class lA PI 3-kinases by tyrosine- kinase pathways and Ras.
4.3.2 Mechanism of activationThe in vitro reconstitution system described here clearly shows that Ras
interaction with PI 3-kinase can lead to activation of its lipid kinase activity. This appears to occur synergistically with the previously described activation of PI 3-kinase by tyrosine phosphopeptide interaction with the SH2 domains of the p85 subunit. Although the strongest effects are seen at suboptimal tyrosine phosphopeptide concentrations, Ras stimulation of PI 3-kinase activity is seen both in the absence of phosphopeptide and in the presence of saturating amounts of peptide. The exact mechanism by which this stimulation is achieved is not clear: one possibility is a simple translocation of PI 3-kinase to the liposome where both Ras and its substrate PI(4,5)P2
are located. In addition, interaction with Ras-GTP could result in a conformational change on PI 3-kinase and allosteric activation.

98
The observation that the presence of Ras.GTP alters the concentration dependence of the stimulation of PI 3-kinase activity by tyrosine phosphopeptides in vitro suggests that there may indeed be an allosteric effect on PI 3-kinase. The Ras-induced change in the concentration dependence for tyrosine phosphopeptide could reflect an increase in the affinity of the SH2 domains of p85 for the tyrosine phosphopeptide induced by Ras.GTP. Since Ras binds to p i 10 at the site next to that where pi 10 binds to p85, this is perhaps not unlikely.
The data with the K227E mutation of p i 10 is also consistent with a conformational change taking place upon binding to Ras. The fact that the K227E pi 10 is no longer able to interact with Ras but has high basal activity when expressed in Cos cells suggests that this reversal of charged residues at an essential site for the Ras-pllO interaction may cause a conformational change that in some ways mimics that occurring when Ras binds.
The stimulation is dependent on the post-translational modification of Ras: this is not needed for Ras binding to p i 10 but is essential for Ras to associate with the liposomes. Bacterially expressed Ras will bind to PI 3-kinase but will not stimulate its activity in the liposome system. This is consistent with post-translationally modified Ras being able to bind to liposomes and therefore bring PI 3-kinase closer to its lipid substrates. However a direct allosteric effect of post-translational modification (and famesylation in particular) on the activation of PI 3-kinase cannot be ruled out.
In the Cos system famesylation but not palmitoylation (of H-Ras) or the polybasic C-terminal region (of K-Ras) is critical for Ras activation of PI 3-kinase activity . Ras mutants that are only farnesylated are mainly cytosolic (Hancock et al., 1990; Hancock et al., 1991) arguing that famesylation rather than membrane localization is important for stimulation of PI 3-kinase. However, it is difficult to exclude the possibility that under conditions of overexpression there is a small fraction of the protein that weakly associates with the plasma membrane and is responsible for the activation of PI 3- kinase.
It should be noted however that there are precedents for Ras famesylation being required for allosteric activation of effector proteins. Famesylation of yeast RAS2 (and Ras) greatly stimulates activation of yeast Adenylate Cyclase in a membrane free in vitro system, without having any effect on the affinity of the interaction (Horiuchi et al., 1992; Kuroda et al., 1993; Shima et al., 1997). A similar effect has been seen with Ras in vitro activation of B-Raf (Okada et al., 1996; Yamamori et al., 1995). Post- translational modification is also required for Sos stimulation of exchange activity on Ras (Porfiri et al., 1994). It is therefore possible that post-translational modification, in addition to facilitating recruitment of PI 3-kinase (and other effectors) to membrane

99
compartments, is also contributing to an allosteric activation of PI 3-kinase (and other effectors).
The data presented here provide evidence for regulation of PI 3-kinase by Ras both in vitro and in vivo. A number of mechanisms for regulating PI 3-kinase therefore exist. Such redundancy in signalling pathways is a common feature in many systems. It is likely that these pathways can synergise in some cases to cause strong activation of the lipid kinase. Multiple pathways co-operating to activate PI 3-kinase would also allow for flexibility in the response to different growth factors. It could be speculated for example that the magnitude or time course of the activation of PI 3-kinase in response to different agonists could vary depending on the combination of regulatory pathways activated simultaneously (e.g. interaction with tyrosine phosphoproteins and Ras-GTP).

100
CHAPTER 5 Biological contribution of PI 3-kinase to Ras function I: effects on transformation
5.1 Introduction
Ras has been shown to interact with and activate PI 3-kinase, both in vitro and in vivo. A crucial issue arising from these results was whether PI 3-kinase was a physiologically relevant Ras effector and what aspect of Ras function PI 3-kinase was responsible for. One of the approaches that was pursued in order to address the contribution of PI 3-kinase to Ras function was to generate partial loss of function mutations in the effector domain of Ras.
Many mutations in this effector region (residues 32 to 40) are known to inhibit the biological function of Ras and to block the interaction of Ras with target proteins. Based on the observation that there is apparent homology between the Ras-binding domain (RED) of Raf and that of PI 3-kinase, it was rationalised that mutations in this effector domain could be generated which would cause Ras to lose the interaction with some effectors, while still retaining the ability to interact with others. Whilst in the process of generating and screening mutants, there were two reports in the literature describing three such partial loss of function effector mutations (Joneson et al., 1996; White et al., 1995) although in both cases they were identified as mutants that did or did not interact with Raf or some other unidentified molecule.
This chapter describes the identification of Ras effector mutants that selectively lose the ability to interact with PI 3-kinase and other effectors. These partial loss of function mutants, together with activated and dominant-negative versions of PI 3-kinase and other Ras effectors have been used to study the contribution of PI 3-kinase to transformation by Ras.
5.2 Results
5.2.1. In vitro interaction of mutant Ras proteins with effectorsA large array of mutations in the effector domain of Ras were generated by in
vitro mutagenesis and after expression as recombinant proteins the Ras mutants were screened for their ability to interact in vitro with the effectors c-Raf-1, Ral.GDS and PI 3-kinase (figure 5.1). Purified full length baculovirus expressed pi 10a fused to GST
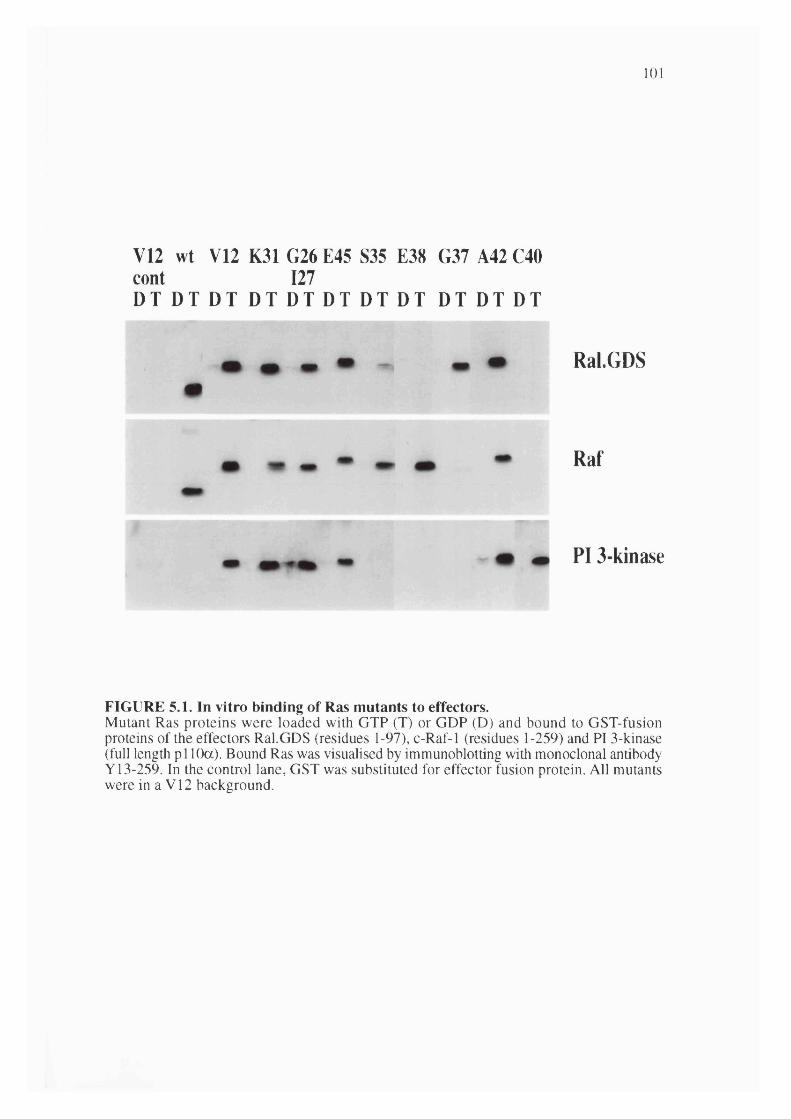
101
V12 wt V12 K31 G26 E45 S35 E38 G37 A42 C40 cont 127D T D T D T D T D T D T D T D T D T D T D T
Ral.GDS
Raf
PI 3-kinase
FIGURE 5.1. In vitro binding of Ras mutants to effectors.Mutant Ras proteins were loaded with GTP (T) or GDP (D) and bound to GST-fusion proteins of the effectors Ral.GDS (residues 1-97), c-Raf-1 (residues 1-259) and PI 3-kinase (full length pi 10a). Bound Ras was visualised by immunoblotting with monoclonal antibody Y13-259. In the control lane, GST was substituted for effector fusion protein. All mutants were in a V12 background.

102
and bound to its regulatory p85 subunit was used while Raf-1 and Ral.GDS were used as bacterially expressed GST fusion proteins containing the Ras binding site alone (Spaargaren and Bischoff, 1994; Wame et ah, 1993).
It can be seen in figure 5.1 that in most cases, Ras mutants show similar ability, or lack of ability, to bind to these three effectors. A number, however, are capable of preferentially binding to one effector while failing to bind to the other two. In particular, E38 and S35 are able to bind to Raf-1 but not significantly to Ral.GDS or p i 10a, while G37 will bind to Ral.GDS but not Raf-1 or pi 10a, and C40 will bind to p i 10a but not
Raf-1 or Ral.GDS. The use of only the Ras binding site of Raf and Ral.GDS in these binding assays may have some limitations, particularly if additional sites of interaction with Ras exist: this approach was used because of difficulties in obtaining active forms of the full length proteins. In the case of pi 10a, both full length protein and the Ras
binding site alone have been used to study Ras interactions with broadly similar results.The ability of these mutant Ras proteins to interact productively with GAPs was
investigated using an enzymatic assay: the ability of purified baculovirus expressed full length p l 20GAP or purified bacterially expressed GAP-related domain of neurofibromin (Xu et al., 1990) to induce hydrolysis of [y-^^pjQTP bound to the G12
version of the various effector mutants was measured. As shown in figure 5.2, each of the Ras mutants G37, E38 and C40 fail to show stimulation of GTPase activity in response to p l2 0 ^ ^ P or neurofibromin, while S35 shows marginal sensitivity to p l20^^P . It should be noted however that this assay measures GAP sensitivity but not interaction. In the case of the V12 mutation for example, GAPs fail to stimulate GTP hydrolysis despite normal binding. It cannot be excluded therefore that the effector mutants described here form non-productive complexes with pl20GAP or neurofibromin.
The ability of these four Ras effector mutants, plus activated V12 Ras, to interact with the various Ras binding proteins was investigated in more detail. A scintillation proximity assay (Skinner et al., 1994) was used to estimate the relative binding affinities of these Ras mutants for Raf-1 and Ral.GDS. As shown in figure 5.3, V12 Ras interacted strongly with Raf-1 in this assay: the affinity of this interaction has been reported to be about 40nM. No interaction could be seen with G37 and C40. E38 interacted with Raf-1 with about a five fold reduced affinity relative to V I2, while S35 bound somewhat more weakly. In the case of Ral.GDS, E38 and C40 showed no detectable binding, while the affinity of G37 binding to Ral.GDS was reduced by about three fold relative to V I2, while that of S35 was reduced by more than ten fold. The scintillation proximity assay proved to be unsuitable for studying the interaction of Ras with either PI 3-kinase or neurofibromin, possibly reflecting the relatively low affinities of these interactions, both around 200nM. Previous use of this assay to study Ras

103
interaction with neurofibromin relied on the use of L61 Ras which has a higher affinity for GAPs (Skinner et al., 1994).
125
*o O
g §
IIII
100 -
75 -
50 -
25 -
1 -4
sLT) CO r -m m mCT) W Ü < 5
150
: = V C
£C O
*S*3cu
100 -
nâ 3
FIGURE 5.2. Sensitivity of Ras effector mutants to Neurofibromin and pl20GAP.Mutant Ras proteins were loaded with [y-32p]GTP were loaded and incubated with the purified GAP-related domain of neurofibromin (GST fusion) or purified full-length pl20GAP. GTP remaining bound at end of assay is shown as a percentage of the amount of V12 Ras incubated with Neurofibromin. All other mutants were in a G 12 background.

104
15000
B&'w 'twCQ
£10000
c.2 5000V
IGNH
100 200 300 400
[Ras] (nM)
500
V12S35G37E38C40
£ac/] 15000ûqa
J=
s 5000
10000
200 300 400
[Ras] (nM)
V12S35G37E38C40
FIGURE 5.3. Comparison of Ras mutant binding to Ral.GDS and Raf.Ras mutant interaction with effectors was measured by scintillation proximity assay. All mutants were His tagged in a V12 background. GST fusion proteins spanning the Ras binding site in Ral.GDS and Raf were used.
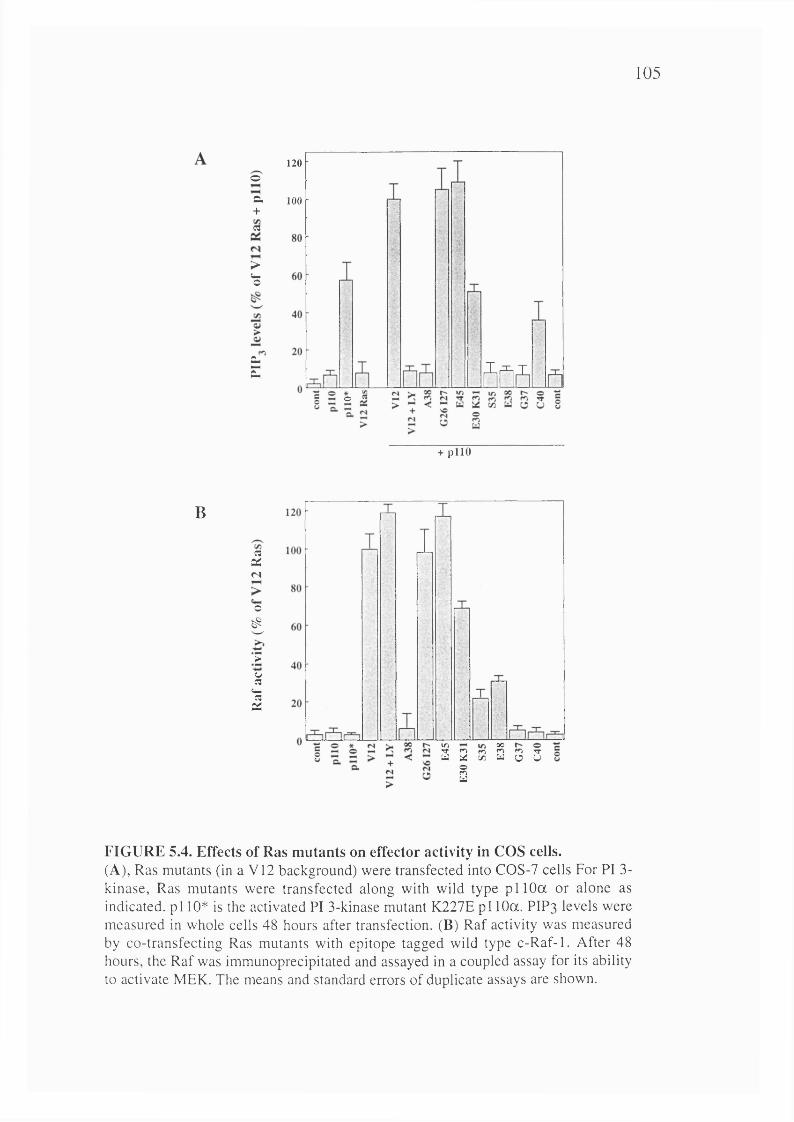
105
120©
Q.+«
>Cmo
100
CLZ 5ffi
©
+ p l l O
B
csXn
o
6
*>uR
Cmega:
i S 8 2 3 3 2
FIGURE 5.4. Effects of Ras mutants on effector activity in COS cells.(A), Ras mutants (in a V12 background) were transfected into COS-7 cells For PI 3- kinase, Ras mutants were transfected along with wild type p i 10a or alone as indicated, p i 10* is the activated PI 3-kinase mutant K227E p i 10a. PIP3 levels were measured in whole cells 48 hours after transfection. (B) Raf activity was measured by co-transfecting Ras mutants with epitope tagged wild type c-Raf-1. After 48 hours, the Raf was immunoprecipitated and assayed in a coupled assay for its ability to activate MEK. The means and standard errors of duplicate assays are shown.

106
5.2.2 Activation of effectors by Ras mutants in intact cellsThe ability of the Ras mutants described above to activate effector pathways was
investigated in intact cells. PI 3-kinase activation was investigated by transfecting Ras effector mutants, in a V12 activated background, into COS cells along with the pi 10a
catalytic subunit of PI 3-kinase. As mentioned earlier V12 Ras synergises strongly with PI 3-kinase expression to give greatly elevated levels of the lipid product phosphatidylinositol (3,4,5) trisphosphate (PIP3). As shown in figure 5.4, S35, G37 and E38 Ras all failed to elevate PIP3 levels in this assay, while C40 stimulated to about
35% of the level achieved with V12 Ras. The in vivo activity of these mutants thus mirrored their in vitro binding affinity for PI 3-kinase.
Raf activation by Ras mutants was also determined in COS cells by cotransfection of Ras mutants (in a V12 background) with tagged Raf (Leevers and Marshall, 1992). V12 Ras strongly stimulates Raf, while G37 and C40 are entirely inactive. S35 and E38 Ras both activate Raf, but about four fold less strongly than V12. Again, the ability of Ras mutants to activate Raf in this cell based assay correlates well with their in vitro binding to Raf.
5.2.3 Contribution of Ras effector pathways to transformationIn order to assess the importance of the various Ras effector pathways in
fibroblast transformation, the ability of the Ras effector mutants described above, and also of the activated effectors themselves, to transform NIH 3T3 cells was studied by focus formation assays. As shown in figure 5.5, none of the three Ras effector mutants G37, S35 and C40 in a V12 background were able to induce transformation by themselves. However, as has been reported for G37 and S35 (White et al., 1995), each pairwise combination of G37, S35 and C40 was able to induce transformation, albeit considerably less strongly than V12 Ras itself (in figure 5.5, 20ng of V12 Ras plasmid was used, but 1.5|ig of every other construct). It is therefore likely that each of the three
mutants is capable of activating a separate effector pathway, each of which can contribute to transformation. In these assays we found that E38 behaved very similarly to S35 (data not shown).
The ability of downstream effectors of Ras to transform cells was also determined (figure 5.6). Raf activated by deletion of the amino-terminal regulatory region is a weaker oncogene by itself than Ras, but it synergises strongly with activated mutants of PI 3-kinase, Rac and Rho, none of which are transforming by themselves. Interestingly, PI 3-kinase will not synergise with Rac, suggesting that they function on the same pathway, but PI 3-kinase will synergise with Rho. Rac and Rho also synergise
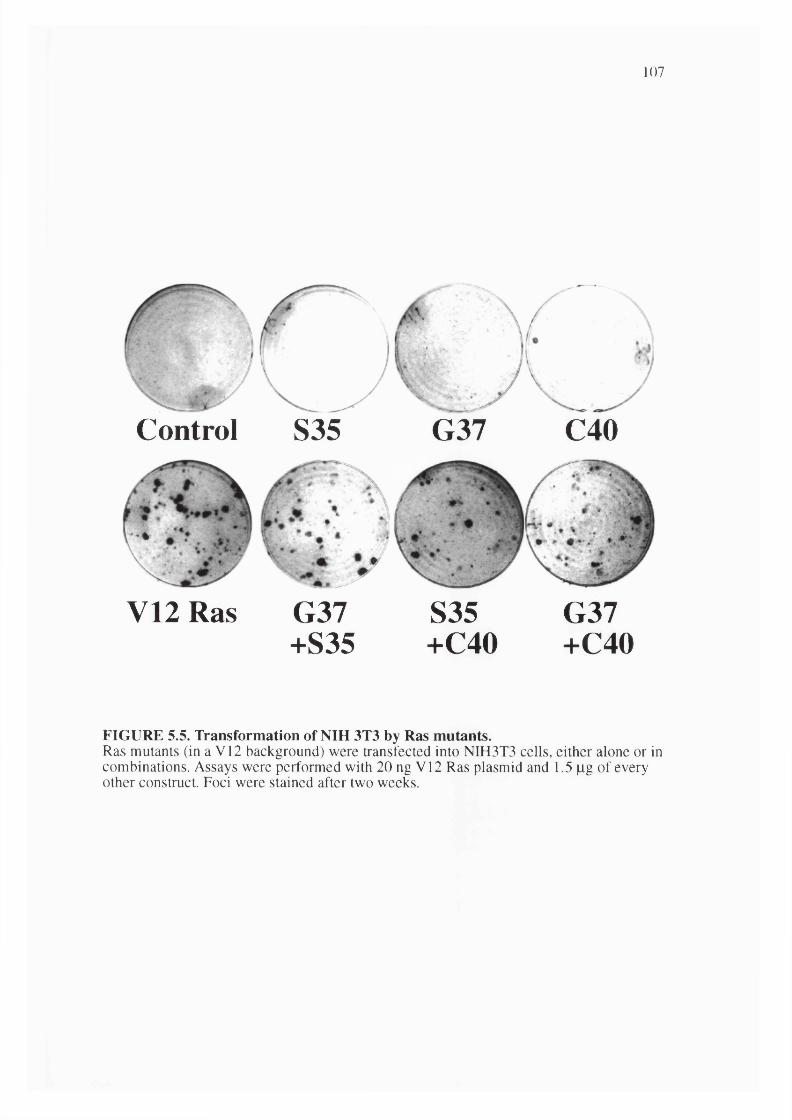
107
V12 Ras
Control S35
• %
G37+S35
G37 C40
S35+C40
G37+C40
FIGURE 5.5. Transformation of NIH 3T3 by Ras mutants.Ras mutants (in a V12 background) were transfected into NIH3T3 cells, either alone or in combinations. Assays were performed with 20 ng V12 Ras plasmid and 1.5 pg of every other construct. Foci were stained after two weeks.
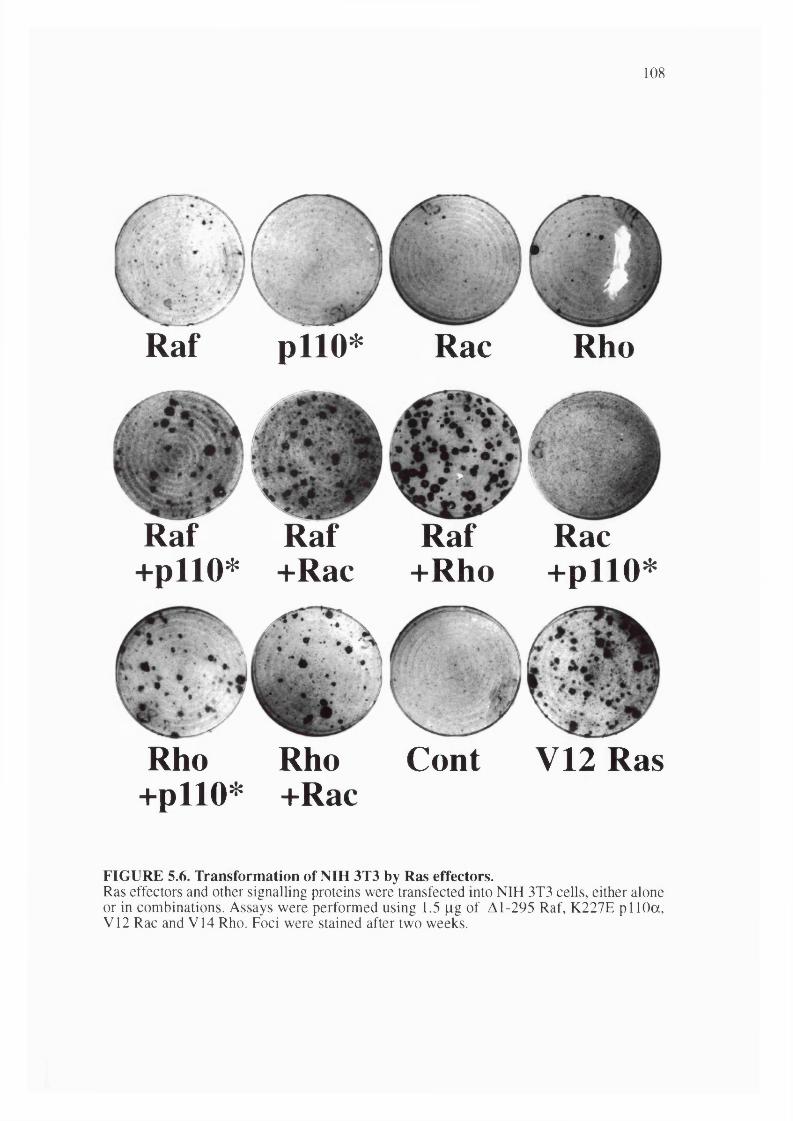
108
/
Raf pllO* Rac Rho
Raf Raf +pllO* +Rac
Raf Rac +Rho +pllO*
/
Rho Rho Cont +pllO* +Rac
V12 Ras
FIGURE 5.6. Transformation of NIH 3T3 by Ras effectors.Ras effectors and other signalling proteins were transfected into NIH 3T3 cells, either alone or in combinations. Assays were performed using 1.5 fig of A1-295 Raf, K227E pi 10a, V12 Rac and V14 Rho. Foci were stained after two weeks.

109
S35 plus: Raf
G37 plus: Raf Rac Rho
pllO*
pllO*
C40 plus: Raf Rho pllO*
FIGURE 5.7. Transformation of NIH 3T3 by Ras mutants in combination with Ras effectors.Ras effectors and other signalling proteins were transfected into NIH 3T3 cells, either alone or in combinations. Assays were performed using 1.5 |ag of the Ras mutants and Al-295 Raf, K227E pi 10a,V12 Rac and V14 Rho. Foci were stained after two weeks.

110
in the transformation assay, suggesting that they are not acting in the same pathway. We were unable to see any effects of activated L72 Ral in these assays (data not shown).
Co-operation of Ras effector mutants with effectors themselves was also tested (figure 5.7). S35 Ras synergised with PI 3-kinase, Rac and Rho, but not Raf. C40 Ras synergised with Raf and Rho but not Rac or PI 3-kinase. G37 Ras synergised with Raf, Rac and PI 3-kinase, and more weakly with Rho. From these data it can be concluded that at least the Raf/MAP kinase pathway (activated by S35) and the PI 3-kinase/Rac pathway (activated by C40) act downstream of Ras to induce transformation synergistically. The role of Ral.GDS in transformation is less clear: we were unable to see any effects of activated Ral, but synergy between Ral and Raf has been reported recently (Urano et al., 1996). It is possible that Ral.GDS activates other targets in addition to Ral, and also that G37 might interact with other unknown effectors in addition to Ral.GDS.
5.2.4 p85 expression inhibits focus formation by V12 RasA direct way of testing the hypothesis that the PI 3-kinase pathway is important
in the transformation of cells by Ras is to determine the effect of inhibition of this enzyme on V12 Ras transformation of NIH 3T3 cells. The long term nature of transformation assays means that the use of chemical inhibitors of PI 3-kinase, particularly unstable ones such as wortmannin, is not ideal. We therefore used the well characterised dominant negative mutant of the PI 3-kinase regulatory subunit, p85, which lacks the binding site for the catalytic subunit, pi 10 (Dhand et al., 1994). This dominant negative PI 3-kinase ("p85 AiSH2-N", also widely referred to elsewhere as "Ap85") strongly inhibits focus formation by V12 Ras, but not by v-Src (figure 5.8).
The expression of this mutant, or any other form of p85, does not reduce the efficiency of formation and growth of drug resistant colonies of these cells, either in the presence or absence of activated Ras (data not shown): the reduction in transformed foci is therefore not due to a general toxicity resulting from inhibition of PI 3-kinase, but appears to be a specific effect on the ability of Ras, but not Src, to transform these cells. By contrast, the strong inhibition of transformation by both Ras and Src seen on coexpression of dominant negative N17 Rac, is accompanied by a very marked reduction, greater than 80%, in drug resistant colony formation, suggesting that expression of N17 Rac is inhibitory for normal as well as transformed cell growth (data not shown). As would be expected, dominant negative MEK, an inhibitor of the Raf/MAP kinase pathway, significantly blocks Ras transformation, showing that the Raf pathway is also critical for Ras transformation.
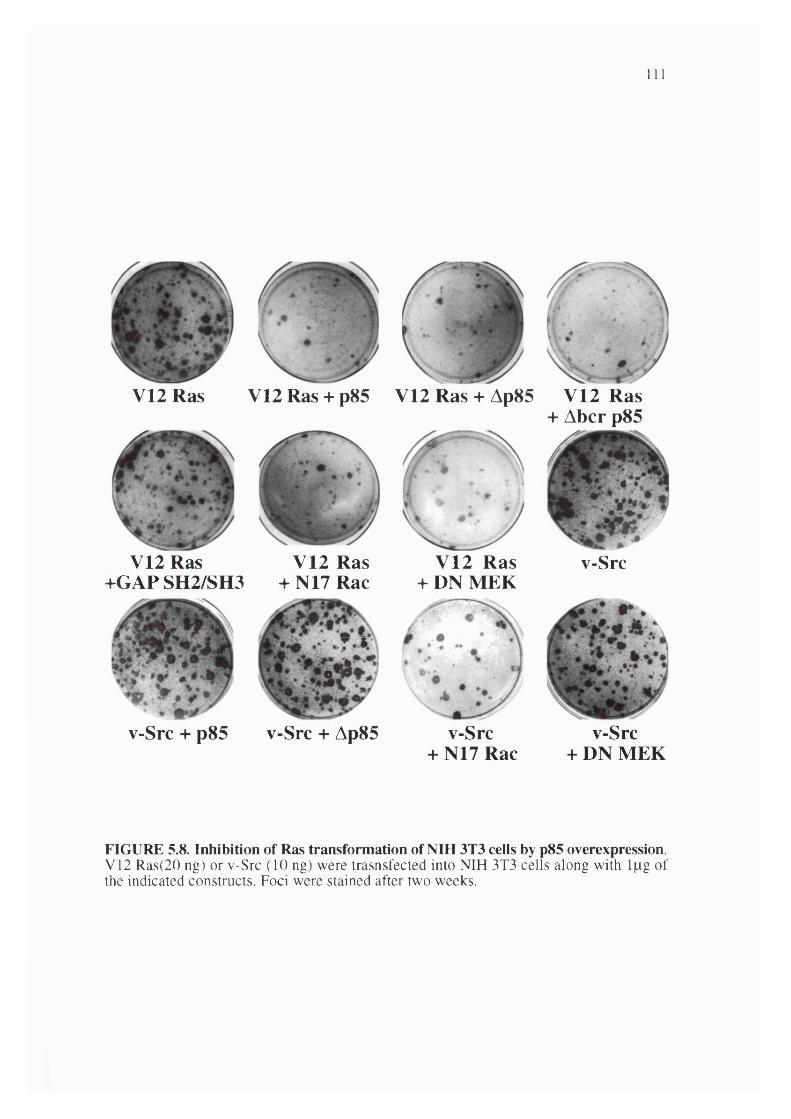
I l l
V12 Ras V12 Ras + p85 V 1 2 R a s + Ap85 V 12 R as + Abcr p85
V12 Ras + G A P SH 2/SH 3
v-Src
v-Src + p85
V 12 Ras + N17 Rac
T A
V 12 R as + DN M EK
v-Src + Ap85 v-Src+ N17 Rac
y .
v-Src + DN M EK
FIGURE 5.8. Inhibition of Ras transformation of NIH 3T3 cells by p85 overexpression.V12 Ras(20 ng) or v-Src (10 ng) were trasnsfected into NIH 3T3 cells along with Ipg of the indicated constructs. Foci were stained after two weeks.

112
In addition to p85 AiSH2-N, the inhibitory ability of full length p85 and also mutants lacking the bcr-related Rho GAP-like region (p85 Abcr) or a region in the inter- SH2 domain C-terminal to the pi 10 interaction site (p85 AiSH2-C) were also tested. All
three of these inhibited Ras, but not Src, transformation. Overexpression of full length p85 inhibits PI 3-kinase signalling probably by titering out stimulatory molecules that bind to the regulatory subunit in much the same way that p85 AiSH2-N does.
Presumably some productive complexes with pllO will form, but at high levels of p85 expression these will be effectively removed from upstream signalling by competition from free p85. The bcr-related region of p85 has been reported to interact with the Rho family proteins Cdc42 and Rac (Zheng et al., 1994); the fact that p85 Abcr has identical inhibitory activity to p85 and p85 AiSH2-N in this assay suggests that direct interaction
of p85 with Rac or Cdc42 is not likely to be a critical part of its mechanism of inhibition of Ras transformation. The expression of the SH2 and SH3 domains of p l2 0 ^ ^ P does not inhibit Ras transformation, suggesting that the inhibitory effect of p85 is not non-specific due to over-expression of any SH2 and SH3 domains.
5.2.5 p85 expression inhibits growth in soft agar of Ras-transformed cellsIn order to further characterize the inhibitory effect of blocking PI 3-kinase
function the effect of expression of p85 subunits on anchorage-independent growth of Ras-transformed cells was analyzed in soft agar assays. To avoid the problem of clonal variability, pools of NIH 3T3 cells expressing V12 Ras or v-Src were infected with retroviruses expressing p85 subunits and drug resistant pools were isolated.
As shown in 5.9, expression of full length p85a substantially inhibited colony formation of Ras transformed cells in soft agar. Interestingly the expression of the p85P
isoform had a more dramatic effect ( 100% inhibition) on anchorage-independent growth of Ras-transformed cells. Expression of these constructs had no significant effect on the anchorage-independent growth of v-Src transformed cells.
p85 expression was not toxic to growth of normal cells on the dish nor did it have any significant effect on the anchorage-dependant growth of Ras-transformed cells under subconfluent conditions (data not shown). However, as shown in figure 5.10, expression of p85P (but not p85a ) did cause a striking reversion of the morphological
transformation of Ras whereas it did not have any apparent effect on the morphology of v-Src expressing cells.

113
Ras + empty Ras + p85a Ras + p85p
1 .4 .
Src + empty Src + p85a Src + p85(3
FIGURE 5.9. Expression of p85 inhibits growth in soft agar of Ras, but not Src, transformed cells.V12 Ras or v-Src expressing-NIH 3T3 cells were infected with a control retrovirus (empty) or retroviruses expressing p85a or p85p. Pools of drug resistant cells were isolated and soft agar assays performed with 500cells/plate. Colonies were stained after 3 weeks.
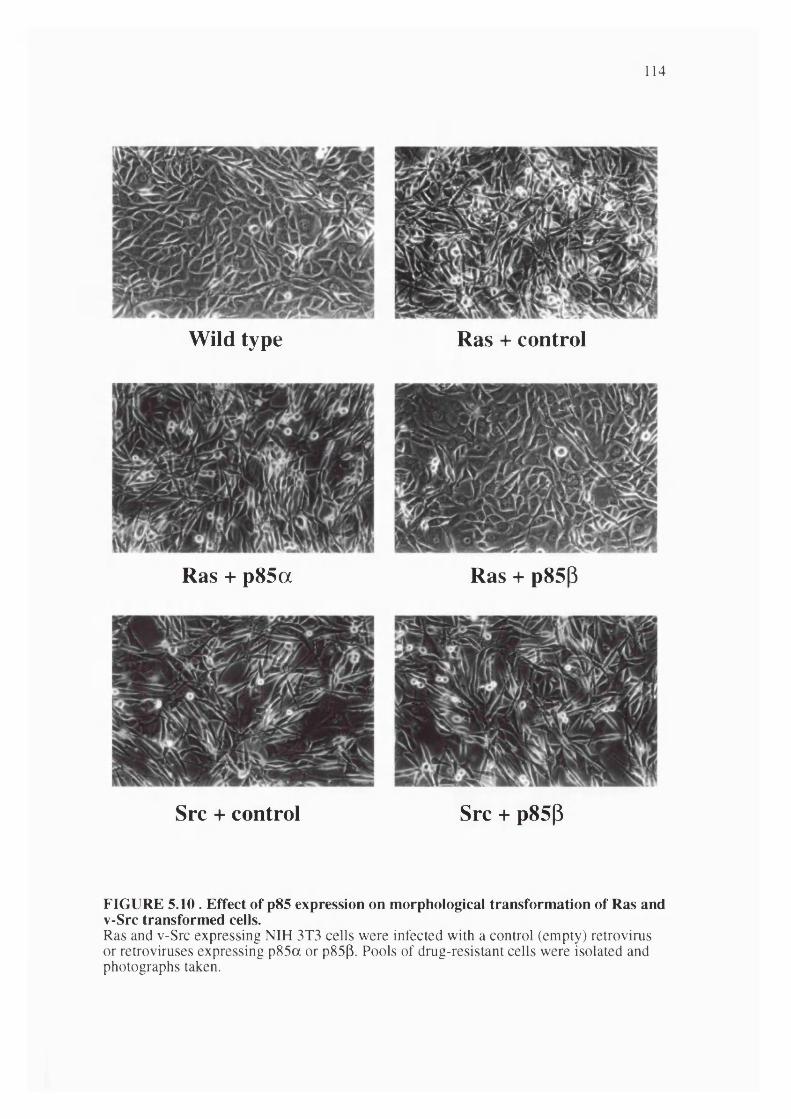
114
Wild type Ras + control
Ras + p85a Ras + p85p
Src + control Src + p85p
FIGURE 5.10 . Effect of p85 expression on morphological transformation of Ras and v-Src transformed cells.Ras and v-Src expressing NIH 3T3 cells were infected with a control (empty) retrovirus or retroviruses expressing p85a or p85p. Pools of drug-resistant cells were isolated and photographs taken.

115
CHAPTER 6 Biological contribution of PI 3-kinase to Ras function II: effects on PC12 differentiation and the actin cytoskeleton
6.1 Introduction
In addition to their well characterised roles as inducers of cellular proliferation, Ras proteins are known to have profound and rapid effects on the actin cytoskeleton. When microinjected into fibroblasts, activated Ras protein induces membrane ruffling due to cortical actin rearrangement within a few minutes (Bar Sagi and Feramisco, 1986). Membrane ruffling or lamellipodia formation is also induced by a number of growth factors and by microinjection of activated Rac protein, (Ridley et al., 1992). Since dominant negative Rac blocks induction of ruffling by Ras and by growth factors, it is likely that Rac functions downstream of Ras and growth factor receptors in the pathway leading to actin rearrangement. In Swiss 3T3 cells, Ras microinjection will also lead to actin stress fibre formation through a Rho mediated pathway that lies downstream of Rac (Ridley and Hall, 1992). These observations suggested that Ras is able to control the activation state of Rac and hence regulate the actin cytoskeleton. On the other hand, inhibition of PI 3-kinase had been reported to block growth factor- induced membrane ruffling in several cell types (Kotani et al., 1994; Wennstrôm et al., 1994). These results suggested that Ras activation of PI 3-kinase could be mediating some of Ras effects on the actin cytoskeleton. In this chapter, I have used the Ras partial loss of function mutants together with activated and dominant-negative forms of PI 3- kinase and other effectors to study the pathway by which Ras controls the actin cytoskeleton leading to the induction of membrane ruffles. In addition, effector mutants and activated effectors have also been used to study the effects of the different Ras effector pathways on the neuronal differentiation of the rat pheochromocytoma cell line PC 12.
6.2 Results
6.1.1 Role of Ras effector pathways in PC12 cell differentiationActivated Ras is known to be able to induce neurite outgrowth in PC 12 cells.
The effector pathways used by Ras in this system were studied by infecting PC 12 cells with retroviruses expressing effector mutants of activated Ras, or activated downstream
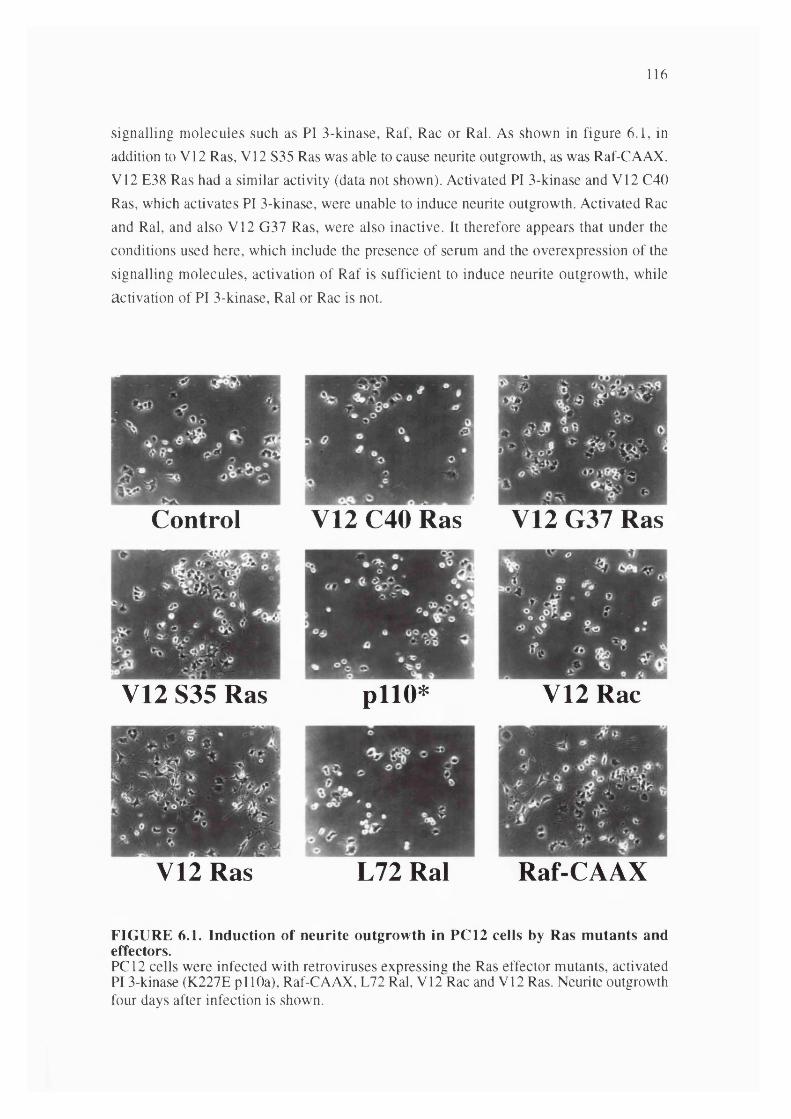
116
signalling molecules such as PI 3-kinase, Raf, Rac or Ral. As shown in figure 6.1, in addition to V12 Ras, VI2 S35 Ras was able to cause neurite outgrowth, as was Raf-CAAX. V12 E38 Ras had a similar activity (data not shown). Activated PI 3-kinase and V12 C40 Ras, which activates PI 3-kinase, were unable to induce neurite outgrowth. Activated Rac and Ral, and also V12 G37 Ras, were also inactive. It therefore appears that under the conditions used here, which include the presence of serum and the overexpression of the signalling molecules, activation of Raf is sufficient to induce neurite outgrowth, while activation of PI 3-kinase, Ral or Rac is not.
Control V12 C40 Ras V12 G37 Ras
V12 S35 Ras pllO* V12 Rac
V12 Ras L72 Ral Raf-CAAX
FIGURE 6.1. Induction of neurite outgrowth in PC12 cells by Ras mutants and effectors.PCI2 cells were infected with retroviruses expressing the Ras effector mutants, activated PI 3-kinase (K227E pi 10a), Raf-CAAX, L72 Ral, VI2 Rac and V12 Ras. Neurite outgrowth four days after infection is shown.

117
1.6.2 Involvement of Ras effector pathways in cytoskeletal reorganisationMicroinjection of activated Ras protein or cDNA into porcine aortic endothelial
(PAE) cells induces rapid cortical actin rearrangement and membrane ruffling. Expression vectors for the various Ras effector mutants, and the activated effectors themselves, were injected into PAE cells and the cells subsequently stained with phalloidin-rhodamine to visualise F actin. As shown in figure 6.2, while activated V12 Ras gave strong membrane ruffling, S35 and G37 Ras in V12 backgrounds failed to induce actin rearrangements. V I2 C40 Ras induced membrane ruffling, although more weakly than V12 Ras. It therefore appears likely that PI 3-kinase is a downstream effector involved in Ras induced ruffling. An activated mutant of PI 3-kinase was sufficient to cause membrane ruffling. Activated Rac gave strong ruffling, while activated Raf-CAAX and L72 Ral did not (figure 6.2).
PI 3-kinase induced ruffling was inhibited by co-expression of dominant negative N17 Rac, but not dominant negative N17 Ras or N17 Cdc42, while V12 Ras induced ruffling was sensitive to dominant negative Rac but not Cdc42 (figure 6.3). This NI7 Cdc42 construct was able to inhibit filopodia formation induced by CSF-1 in macrophages (data not shown).
In order to investigate whether PI 3-kinase activity was responsible for the Ras induced membrane ruffling, PAE cells microinjected with V12 Ras, V12 C40 Ras, activated PI 3-kinase or V12 Rac were treated with the PI 3-kinase inhibitor LY294002. This inhibitor completely blocked the ability of V12 C40 Ras and activated PI 3-kinase to induce membrane ruffling (figure 6.4). In addition this drug also blocked PDGF induced ruffling in these cells (data not shown and (Wennstrom et al., 1994)). However, LY294002 had absolutely no effect on the activation of the MAP kinase ERK2 in response to PDGF or phorbol ester in these cells (data not shown). Ruffling induced by V12 Ras was not fully blocked by LY294002, although some clear diminution of the extent of ruffling was reproducibly noticed. V12 Rac induced ruffling was unaffected by LY294002. For all these experiments, similar effects to LY294002 were also seen with wortmannin, an unrelated inhibitor of PI 3-kinase activity (data not shown). It therefore appears that PI 3-kinase, and not Raf or Ral.GDS, is one of the Ras effectors involved in the induction of cortical actin rearrangement, membrane ruffling and lamellipodia formation. However, it is possible that another ruffling pathway also exists which is activated by V12 Ras but not V12 C40 Ras and is insensitive to chemical inhibitors of PI 3-kinase.
In order to further investigate this PI 3-kinase inhibitor resistant pathway linking Ras to Rac and membrane ruffling, the effects of a number of other inhibitors on Ras induced membrane ruffling were studied. The p85 derived dominant negative PI 3- kinase constructs were co-expressed in PAE cells with activated Ras or Rac.
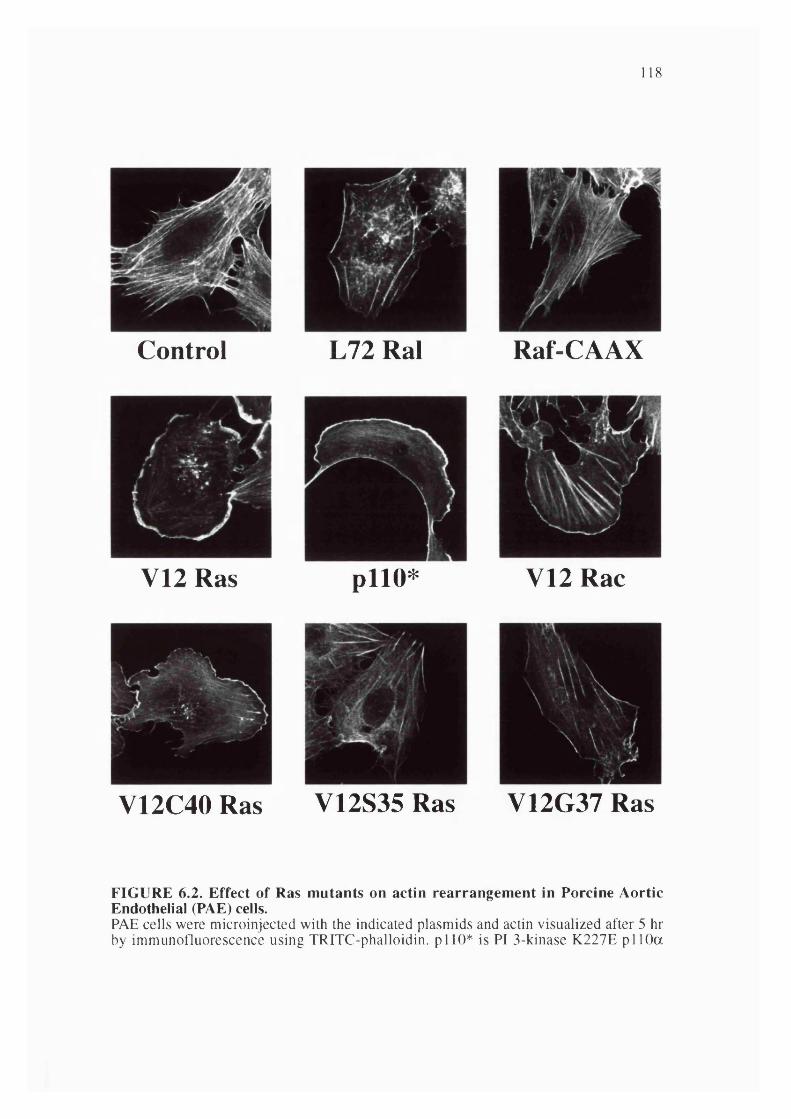
118
Control
V12 Ras
L72 Ral Raf-CAAX
pllO* V12 Rac
V12C40 Ras V12S35 Ras V12G37 Ras
FIGURE 6.2. Effect of Ras mutants on actin rearrangement in Porcine Aortic Endothelial (PAE) cells.PAE cells were microinjected with the indicated plasmids and actin visualized after 5 hr by immunofluorescence using TRITC-phalloidin. pi 10* is PI 3-kinase K227E pi 10a

119
Control
V12 Ras
V 12R as + N 17R ac
pllO *
pllO * + N17 Ras
pllO * + N17 Rac
V12 Ras + N17 Cdc42 pllO * + N17 Cdc42
FIGURE 6.3. Effect of N17 Rac on Ras and PI 3-kinase induced actin rearrangement.PAE cells were microinjected with V12 Ras protein or K227E pi 10a plasmid, along with dominant negative Ras, Rac, or Cdc42. Cells were fixed after 5 hr.
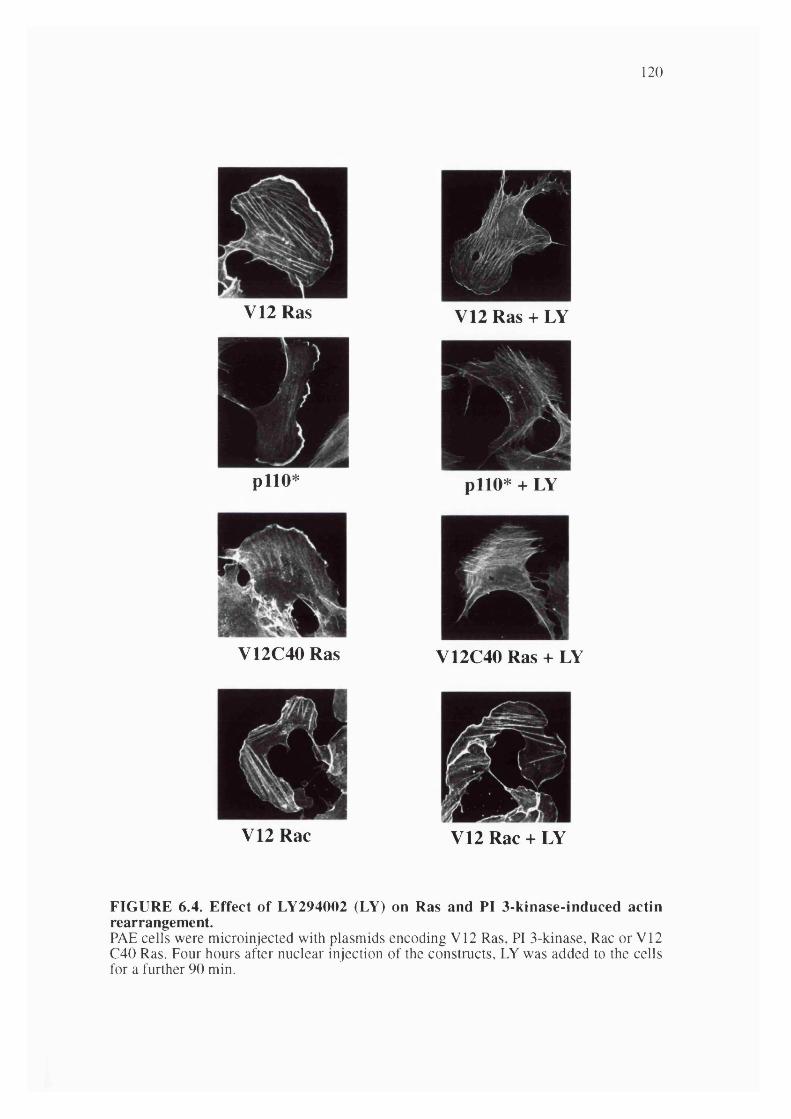
120
V12 Ras
pllO*
V12 Ras + LY
pllO* + LY
V12C40 Ras V12C40 Ras + LY
V12 Rac V12 Rac + LY
FIGURE 6.4. Effect of LY294002 (LY) on Ras and PI 3-kinase-induced actin rearrangement.PAE cells were microinjected with plasmids encoding VI2 Ras, PI 3-kinase, Rac or V12 C40 Ras. Four hours after nuclear injection of the constructs, LY was added to the cells for a further 90 min.
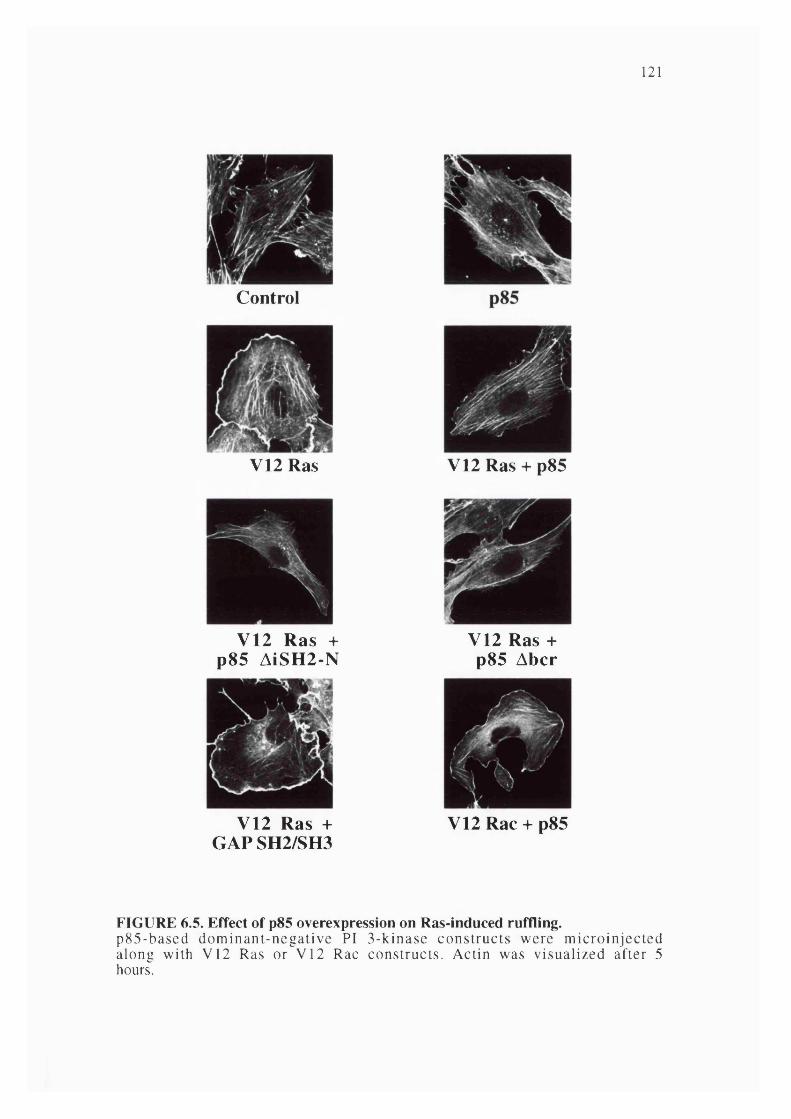
121
Control
V12 Ras V12 Ras + p85
V12 Ras + p85 AISH2-N
V12 Ras + p85 Abcr
V12 Ras + GAPSH2/SH3
V12 Rac + p85
FIGURE 6.5. Effect of p85 overexpression on Ras-induced ruffling.p85-based dominant-negative PI 3-kinase constructs were m icroinjected along with V 12 Ras or V12 Rac constructs. Actin was visualized after 5 hours.
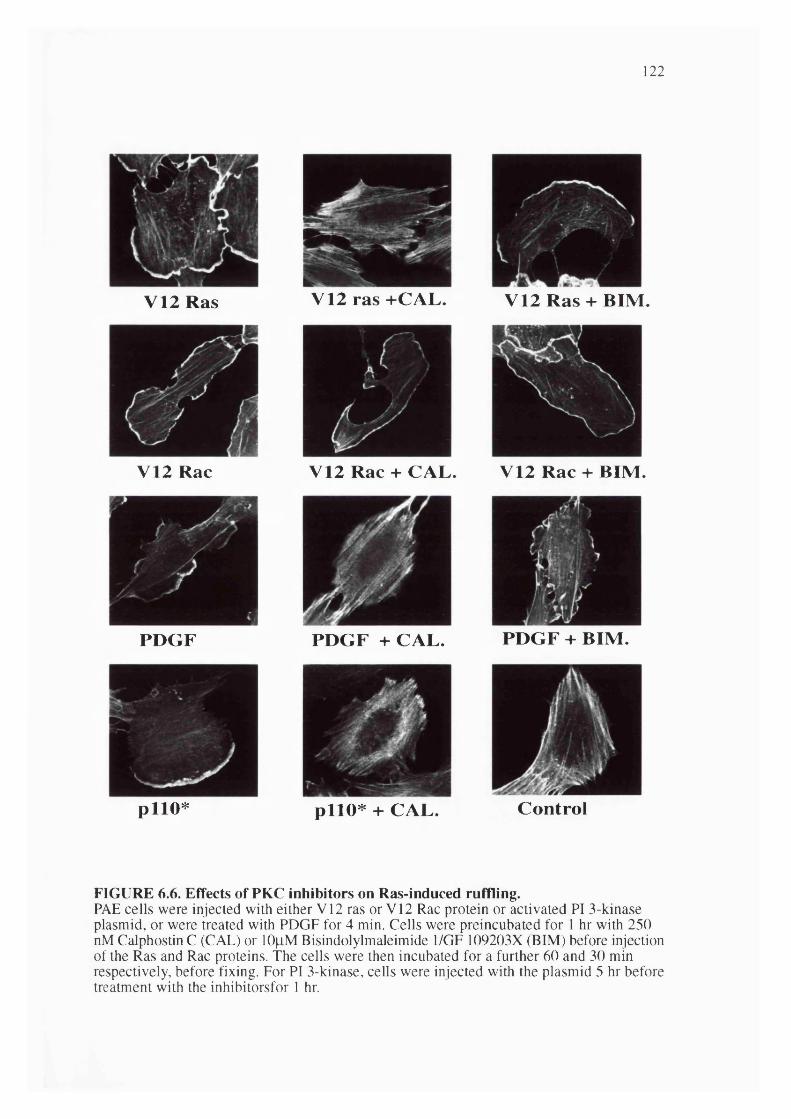
122
V 12 Ras V 12 ras +C A L. V 12 R as + BIM .
V 12 Rac V 12 Rac + C A L. V12 R ac + BIM .
PD G F P D G F + CAL. P D G F + BIM .
p l io * p l io * + CAL. C ontrol
FIGURE 6.6. Effects of PKC inhibitors on Ras-induced ruffling.PAE cells were injected with either V12 ras or V12 Rac protein or activated PI 3-kinase plasmid, or were treated with PDGF for 4 min. Cells were preincubated for 1 hr with 250 nM Calphostin C (CAL) or lOfiM Bisindolylmaleimide 1/GF 109203X (BIM) before injection of the Ras and Rac proteins. The cells were then incubated for a further 60 and 30 min respectively, before fixing. For PI 3-kinase, cells were injected with the plasmid 5 hr before treatment with the inhibitorsfor 1 hr.

123
As shown in figure 6.5, expression of p85 totally and reproducibly blocked V12 Ras induced membrane ruffling. In addition, p85 AiSH2-N, p85 Abcr and p85 AiSH2-C all
completely inhibited V12 Ras induced ruffling. These constructs also block PDGF induced ruffling ((Wennstrom et al., 1994) and data not shown). The p l20GAP gH2 and SH3 construct was without effect. The effect of these p85 based dominant negative PI 3-kinase constructs appeared to be specific for Ras and growth factor induced ruffling since they had absolutely no effect on ruffling induced by V12 Rac. These data indicate that the mechanism by which Ras controls the actin cytoskeleton through Rac is entirely dependent on normal PI 3-kinase function acting upstream of Rac. The inability of chemical inhibitors of PI 3-kinase to inhibit fully Ras induced actin rearrangement may suggest that drug resistant forms of PI 3-kinase are involved.
As shown in figure 6.6, the protein kinase C inhibitor calphostin C was a very good inhibitor of ruffling induced both by Ras and by PDGF in PAE cells. Calphostin C was also able to fully inhibit activated PI 3-kinase induced ruffling. However, another protein kinase C inhibitor, the bisindoylmaleimide GF 109203X, was not effective. The inhibitor GF 109203X was used under conditions where it inhibited phorbol ester activation of ERK2 (data not shown). Calphostin C was unable to inhibit ruffling induced by activated Rac, placing the target for this drug downstream of Ras and PI 3- kinase and upstream of Rac. Since calphostin C acts on diacylglycerol binding zinc fingers, while bisindoylmaleimide acts on the kinase domain, it is possible that a diacyl glycerol binding protein other than a protein kinase C family member is important in the control of membrane ruffling: possible candidates include the Rho family exchange factors Vav, Vav2, FGDl and Lfc and the Rac-GAP chimaerin.
6.2.3 Effect of LY294002 on PDGF and Ras induced cellular PIP3 levelsThe data in figure 6.4 and 6.5 raises the possibility that Ras may be capable of
affecting the activity of PI 3-kinases that are not fully sensitive to chemical inhibitors such as LY294002 and wortmannin. In order to explore this possibility further, the effect of LY294002 on the levels of PI 3-kinase produced lipids was investigated in NIH 3T3 cells (figure 6.7A). PDGF induced PIP3 elevation is entirely reversed by treatment of cells with 20jiM LY294002, a dosage which should entirely inhibit the activity of pi 10a (Vlahos et al., 1994). When V12 Ras was expressed in these cells, the levels of PIP3 were constitutively elevated to about half the peak level achieved with PDGF. This elevation of PIP3 was, however, substantially resistant to the effects of the
drug; even extended treatment with very high concentrations of LY294002 left about half this level of PIP3. It is therefore likely that Ras couples to forms of PI 3-kinase that
are resistant to this drug, as well as activating conventional drug-sensitive isoforms.

124
B
cTiTTTfCu(mo
o>><u3
,/Ax
_ESSE3.
-
u.OCcu
nofN5
=L®
5+k.O
nfS>
zLOfS
«aM>
OS
C5a:ri>
o®
5na:
>
Oa.
«cafS>
in m% ao
c . U
+ <</5a
C S+
t/>
=L0(S
(S aa : +
> V )( S «
a:> n
>
FIGURE 6.7. Effect of LY294002 and p85 overexpression on Ras-induced cellular PIP3 levels.(A) PIP3 levels were measured in metabolically labelled, serum-starved NIH 3T3 cells. Cells were treated with LY294002 for 90 min before lysis. PDGF (50 ng/ml) treatment was for 3 min before lysis. V12 Ras was expressed by retroviral infection for 48 hr before labelling. (B) V12 Ras and PI 3-kinase constructs (wild-type p i 10), were transfected into COS-7 cells. PIP3 levels were measured 48 hr after transfection.
In order to confirm that expression of the regulatory p85 subunit of PI 3-kinase can block the ability of V12 Ras to induce elevated levels of PIP3 in intact cells, even
though LY294002 fails to do so, COS cells were transfected with V12 Ras in the presence or absence of p85 or Ap85. The cells were then metabolically labelled with
orthophosphate, and PIP3 levels determined. As shown in figure 6.7B, both p85 or
Ap85 were able to almost completely reverse V12 Ras induced elevation of PI 3-kinase
lipid products. These constructs did not influence Ras expression levels (data not shown).

125
6.2.4 Effect of p85 expression on tumour cell lines with mutations in Ras genes.It has been shown that expression of p85 (in particular p85p) has a very potent
effect on the reversion of the transformed phenotype of Ras-expressing NIH 3T3 cells. This effect appears to be non-toxic and specific to Ras (but not v-Src) transformation. It was important to determine whether the inhibitory effect of p85 could also be seen in cells derived from human tumours containing activated Ras.To this effect, several cell lines derived from pancreatic tumours (kindly provided by Nick Lemoine), both with wt Ras genes and with mutations in K-Ras genes, were infected with retroviruses expressing p85a or p85P and the effect of p85 expression on
their tumorigenic potential was studied. These experiments are still ongoing at the time of writing of this thesis and only some preliminary results can be presented on the apoptotic behaviour of various cell lines.
Work from this lab (Kwaja et al., 1997) has shown that in epithelial MDCK cells expression of activated Ras, through the activation of PI 3-kinase, protects cells from apoptosis induced by loss of anchorage to the extracellular matrix. It is likely that this anti-apoptotic property of Ras is of great relevance to its role in human tumorigenesis. To address this possibility and study the effect of inhibiting PI 3-kinase function on protection from loss of anchorage-induced apoptosis, the apoptotic behaviour of the tumour cell lines was studied when the cells were put in suspension. An ELISA based method that detects chromatin degradation as measured by the appearance in the cytoplasm of histone-associated DNA fragments (nucleosomes) was used to compare attached cells (a), to cells that had been left in suspension overnight (s).
Figure 67 shows that BxPc3 cells, that contain wt Ras genes, undergo significant apoptosis when in suspension and expression of either p85a nor p85(3 had no significant
effect on this behaviour. However, in the case of the ASPC-1, Colo357 and HPAF cell lines, all of which contain activating mutations in K-ras genes, two observations can be made. All three control-infected cell lines appear more resistant to loss of anchorage- induced apoptosis. In addition, expression of both p85 isoforms significantly stimulates the cells to undergo apoptosis only when in suspension. These results are consistent with the notion that Ras activation in human tumours, through the activation of PI 3- kinase, may be involved in protecting carcinoma cells from apoptosis induced by loss of anchorage, a process most likely of great relevance to their malignant/metastatic potential. Furthermore, these results suggest that interfering with the PI 3-kinase pathway represents an attractive target for pharmacological inhibition in the treatment of human cancers where Ras activation is involved.

126
1.5
BxPc3 Aspcl1.5
E *IOO
0.5 -
1 -
0.5 -
a s a s a sempty p85a p85p
a s a s a sempty p85a p85P
1.5
EcssQO
1 -
0.5 -
0 -
HPAF Colo3571.5 -
I -
0.5 -
empty p85a p85p empty p85a p85p
FIGURE 6.8. Effect of p85 overexpression on apoptosis induced by loss of anchorage in pancreatic tumour-derived cell lines.BxPc3 cells (containing wt Ras genes) and ASPC-1, HPAF and Colo357 cells (with mutations in K-ras) were kept on the dish (a, attached) or detached and kept in suspension (s) on polyHema coated dishes for 16 hours. DNA fragmentation was quantified by ELISA with the Cell Death Detection kit (Boehringer Mannheim).

127
6.3 Discussion to Chapters 5 and 6
6.3.1 Mutations in the effector domain discriminate between different Ras effectorsMany mutations in the effector region of Ras, residues 32 to 40, are known to
inhibit the biological function of Ras and to block the interaction of Ras with target proteins. Relatively subtle mutations in this region have been generated that lead to partial loss of function mutants in which interaction with some effectors is maintained but with others is lost. Some of the mutants described here had been previously identified by other groups. For example. White et al. have shown that S35 Ras will interact with Raf, but not Byr2, a MAP kinase kinase kinase from Ss. pombe, while G37 interacts with Byr2 but not Raf (White et al., 1995). Both mutants are unable to transform mammalian cells, but will co-operate together to give transformation; the normal mammalian target for G37 Ras was not identified. Similarly, Joneson et al. have reported that S35 Ras is able to induce MAP kinase activation, but not membrane ruffling, when microinjected into fibroblasts, while C40 Ras induces membrane ruffling but not MAP kinase activation (Joneson et al., 1996).The two mutants acting together will induce DNA synthesis, but either one alone is ineffective. Again, the target of C40 Ras was not known.
A series of mutants have been generated and screened for interaction with PI 3- kinase and other known Ras effectors, namely Raf and RalGDS. It is shown here that C40 Ras interacts only with PI 3-kinase, G37 Ras interacts only with Ral.GDS and S35 and E38 Ras interact only with Raf. None of these mutants interact productively with p l20^ ^ P or neurofibromin.
It should be noted that Ras is known to interact through its effector domain with other proteins in addition to the 3 effectors studied here. Indeed G37, C40 and S35 have all been shown to interact with AF6 (Khosravi-Far et al., 1996), a putative effector about which very little is known. It is therefore possible that some of these other proteins (or other effectors yet to be identified) may be contributing to some of the effects seen upon expression of the Ras mutants. However, in the studies presented here the behaviour of the Ras effector mutants, both on transformation assays and actin cytoskeleton rearrangement, was mimicked by activated versions of their known interacting effectors.
6.3.2 Role of the different Ras-activated pathways in transformationAs a way to assess the contribution of the different effector pathways to the
transforming potential of Ras the behaviour of the different effector mutants and activated versions of the effectors themselves has been studied in focus formation assays in NIH 3T3 fibroblasts, an assay that measures loss of contact inhibition. It is

128
shown that no single effector mutant is sufficient to transform cells but each mutant synergyzes with each of the other two to recover some transforming potential. Similar results have been obtained by Khosravi-Far et al. (Khosravi-Far et al., 1996).
Since Ras mutants that activate only Raf, such as E38 and S35, are very poorly transforming, it would appear that activation of endogenous Raf protein alone is not sufficient to transform murine fibroblasts. This is despite the fact that overexpression of strongly activated Raf is transforming by itself, possibly through a mechanism involving secretion of autocrine growth factors (McCarthy et al., 1995). In the work presented here, both a cytosolic Raf mutant activated by deletion of its amino terminal regulatory domain and RafCAAX, which is constitutively localized to the plasma membrane, behaved similarly and were consistently found to be at least 100 fold weaker in their focus forming ability as compared to V12 Ras. It is also clear from the cooperation seen by the G37 and C40 mutants that Ras may transform cells through Raf- independent pathways.Reasonable transformation is achieved by the Ras mutants that activate only Raf in synergy with either Ras mutants that activate Ral.GDS or PI 3-kinase, or with activated PI 3-kinase, Rac or Rho. Similarly, the Ras mutant that activates only PI 3-kinase synergises with those activating other pathways, including the one that activates only Ral.GDS while activated PI 3-kinase synergises with Rho and Raf, but not Rac.
The synergy on transformation seen with G37 was not mimicked by activated Ral: possibly RalGDS may activate other targets in addition to Ral, or Ral.GDS may not be the only effector binding to G37 Ras. There is recent evidence supporting both possibilities. RalGDS, but not activated Ral, has been shown to cooperate with activated Raf to transform cells (White et al., 1996) and to stimulate c-fos promoter activity (Okazaki et al., 1997) arguing that RalGDS may have other functions in addition to activation of Ral. The lack of effect of constitutively active Ral could also be explained if Ral needs nucleotide turnover in order to carry out its function. On the other hand, RIN1, another putative Ras target, has been shown to still interact with the G37 mutant (Han et al., 1997), suggesting that effectors other than RalGDS may be mediating some of the function retained by G37 Ras.
Looking further down the effector pathways, Raf can synergise with Rac or Rho, as shown previously by others (Khosravi-Far et al., 1995; Qiu et al., 1995a; Qiu et al., 1995b), and PI 3-kinase can synergise with Rho, but not Rac. This suggests that Rac and PI 3-kinase lie on the same pathway, most likely with PI 3-kinase acting upstream of Rac (Hawkins et al., 1995), but that Rho lies on a different pathway; this ties in with the ability of Rac and Rho to synergise with each other in causing transformation. Placing Rac and Rho on different pathways differs from the model derived from actin cytoskeleton rearrangements in Swiss 3T3 cells in which Rac acts upstream of Rho

129
(Ridley and Hall, 1992; Ridley et al., 1992). This apparent inconsistency may reflect differences in cell types or, perhaps more likely, differences in the nature of the biological assay: it is possible that distinct pools of Rho and Rac GTPases exist, with those proteins involved in actin rearrangement being differently regulated from those involved in, for example, transcriptional regulation. Transformation is likely to reflect all aspects of Rac and Rho signalling, not just cytoskeletal effects.
It appears that at least two effector pathways need to be activated simultaneously by Ras in order for efficient transformation to result, at least as measured by focus forming ability/loss of contact inhibition. These pathways can be Raf plus PI 3-kinase, Raf plus RalGDS or PI 3-kinase plus RalGDS, or another effector capable of binding to G37 Ras. Even so, the synergistic transformation seen with two different Ras mutants in figures 5.5 falls well short of the efficiency of transformation seen with V I2 Ras alone. This may reflect the fact that the partial loss of function effector mutants of Ras are significantly compromised in their ability to activate even the effectors that they still interact with (see figures 5.3 and 5.4). Alternatively, more than two effector pathways may need to be activated for truly high efficiency transformation. In support of the latter possibility is the observation that the transformation efficiencies obtained with combinations of activated downstream effectors (e.g. RafCAAX and P110K227E) is still well short of that seen with Ras (and similar to that seen with pairs of effector mutants). This is despite the fact that activation of downstream components (e.g. activation of MAP kinase and PIP3 elevation seen with RafCAAX and P1I0K227E respectively) , at least in transient transfection assays, is as strong (or stronger) as that seen with Ras (figure 5.4B and data not shown).
6.3.3. Inhibition of Ras transformation by p85 expressionEven though the PI 3-kinase pathway is not sufficient to transform cells, the fact
that dominant negative forms of PI 3-kinase based on the p85 regulatory subunit are able to strongly inhibit V12 Ras transformation indicates that this pathway is one of those normally used by Ras oncogenes to establish the transformed cell phenotype.
Over-expression of different mutants of p85a strongly inhibit Ras focus-forming
ability. This effect, unlike that seen with N17 Rac, does not seem to reflect a general toxic effect : N17 Rac, but not p85a, strongly inhibit the number of drug-resistant
colonies arising from control transfections. This is more clearly illustrated when p85 is introduced by retroviral infection into either control NIH 3T3 or stably expressing Ras- transformed cells: p85 over-expression has very little effect on the growth rate of control cells. In the case of Ras-transformed cells expression of either p85a or p85p has
no effect on the growth of cells under low density conditions (subconfuent). Only under

130
high density conditions (when the cells reach confluence) does p85 over-expressing markedly inhibit cell growth.
When anchorage-independent growth was analyzed in soft agar assays the effects of p85 expression, particularly p85p, were even more dramatic: p85a results in about a 60% inhibition in the number of colonies, whereas p85P expressing cells were completely unable to grow in soft agar. p85p, but not p85a, also caused a striking
reversion to the morphological transformation of Ras-expressing cells.The inhibitory effect of p85 expression was not only non-toxic to normal cells, it seems also specific for Ras transformation. Transformation by v-Src appears to be much less sensitive to inhibition of PI 3-kinase signalling: neither p85a nor p had any detectable
effect on focus formation, anchorage-independent growth or morphological transformation of v-Src expressing cells. This probably reflects the fact that v-Src uses several signalling pathways to transform cells, only some of which involve Ras (see for example Aftab et al., 1997). Alternatively it could be speculated that p85 expression selectively disrupts PI 3-kinase activation by Ras: Src may also be activating PI 3- kinase by a Ras-independant pathway that is less sensitive to disruption by p85 overexpression.
Whatever the mechanisms (see later) it is clear that there must be significant differences between the p85a and P isoforms that may account for the stronger effect of p8SP in their inhibition of Ras transformation. This could reflect the fact that p85p is
involved in differential interactions or has higher affinity for binding to some unknown molecule required for the activation or function of PI 3-kinase. It should be noted that there are already examples in the literature of differences between p85a and p in their potential regulation and/or function: upon activation of T-cells, p85a and p are
differentially phosphorylated (Reif et al., 1993) and Cbl interacts preferentially with p85p (Hartley et al., 1995).
6.3.4 PC12 differentiationRas has long been known to induce neuronal differentiation of the rat
pheocromocytoma derived PC 12 cell line. It is shown here that the ability of Ras mutants to induce neurite outgrowth of PC 12 cells correlates well with the activation of Raf (figure 6.1). In addition expression of activated Raf, but not PI 3-kinase or Ral, is sufficient to give neurites. This is consistent with previous results: activated MEK induces neurite outgrowth (Cowley et al., 1994) and inhibition of Raf/MAP kinase pathway function inhibits NGF induced neurite outgrowth (Cowley et al., 1994; Pang et al., 1995). The Raf pathway therefore seems the main pathway by which Ras induces neurite outgrowth of PC 12 cells.

131
However, the PI 3-kinase inhibitor wortmannin has been found to cause collapse of preexisting neurites and inhibition of their formation in response to NGF (Jackson et ah, 1996; Kimura et ah, 1994). It seems likely therefore that PI 3-kinase activity, although not sufficient, is required for neurite outgrowth. Under the conditions used to induce neurite outgrowth with activated MEK (Cowley et al., 1994), and in the experiments presented here, there may be a significant basal PI 3-kinase activity provided by serum factors or by adhesion to extracellular matrix.It is also worth noting that neurite extension is just one, easy to score morphological marker of neuronal differentiation, a process that also includes expression of several neuronal specific proteins. It is therefore possible that Ras-activated pathways other than the Raf pathway may be involved in other aspects of PCI2 differentiation. For example, Ras but not Raf, induces expression of SCO 10 mRNA (D'Arcangelo and Halegoua, 1993)
6.3.5. Ras regulation of the actin cytoskeletonIn addition to their well characterised roles as inducers of cellular proliferation,
Ras proteins are known to have profound and rapid effects on the actin cytoskeleton. When microinjected into fibroblasts, activated Ras protein induces membrane ruffling due to cortical actin rearrangement within a few minutes (Bar Sagi and Feramisco, 1986). Membrane ruffling or lamellipodia formation is also induced by a number of growth factors and by microinjection of activated Rac protein, (Ridley et al., 1992). Since dominant negative Rac blocks induction of ruffling by Ras and by growth factors, it is likely that Rac functions downstream of Ras and growth factor receptors in the pathway leading to actin rearrangement. In Swiss 3T3 cells, Ras microinjection will also lead to actin stress fibre formation through a Rho mediated pathway that lies downstream of Rac (Ridley and Hall, 1992). These observations suggested that Ras is able to control the activation state of Rac and hence regulate the actin cytoskeleton. It is shown here that Ras controls Rac activation and induction of membrane ruffling through activation of PI 3-kinase.
Activated PI 3-kinase is sufficient to induce membrane ruffling, as is the case for Ras, through a Rac mediated mechanism. The only Ras partial loss of function mutant to cause ruffling is C40: this, together with the inability of Raf or Ral to cause ruffling, rules out Raf and Ral.GDS as effectors on the ruffling pathway. Since C40 Ras stimulates PI 3-kinase and causes membrane ruffling in an LY294002 and wortmannin sensitive manner in PAE cells, it is very likely that this mutant is causing ruffling through interaction with a drug sensitive PI 3-kinase.
In contrast to C40 Ras induced ruffling, V12 Ras induced ruffling is not fully sensitive to PI 3-kinase inhibitors in PAE cells (figure 6.4) or in Swiss 3T3 cells (Nobes et al., 1995). This has previously been interpreted as evidence that Ras controls Rac-

132
dependent ruffling by a PI 3-kinase independent mechanism. However, the use of a different means of inhibiting PI 3-kinase function, expression of dominant negative constructs based on the p85 regulatory subunit, shows that the V 12 Ras induced ruffling is entirely sensitive to this form of disruption of PI 3-kinase signalling. Similarly to the inhibitory effect seen on transformation, only the SH2 and SH3 domains of p85 are required for this inhibitory effect on ruffling, while other SH2 and SH3 domain containing constructs such as one derived from p l2 0 ^ ^ ^ are inactive. Since the bcr domain of p85 is not required for inhibition of Ras induced ruffling, and p85 constructs do not inhibit Rac induced ruffling, it is unlikely that direct interaction of p85 with GTP-bound Rac or Cdc42 is responsible for this inhibition (Zheng et al., 1994).
The fact that V12 Ras induced ruffling is completely blocked by p85 overexpression but only partially sensitive to LY294002 or wortmannin treatment suggests that Ras could activate an isoform of PI 3-kinase that is partially resistant to these inhibitors. It is already clear that Ras will interact with at least four different PI 3- kinase family members (pi 10a and P as shown here, pi 105 (Vanhaesebroeck et al., 1997), pllOy (Rubio et al., 1997), although none of these are known to be resistant to these drugs. Measurement of PIP3 levels in V12 Ras expressing NIH 3T3 cells indeed shows that the Ras, but not PDGF, induced elevation of this lipid is partially resistant to treatment of cells with LY294002 (figure 6.7 A) . A class II (C2 domain containing) PI 3-kinase isoform has recently been reported that is about fifty times less sensitive than pi 10a to wortmannin (Virbasius et al., 1996). Interestingly, a very closely related
protein in Drosophila is wortmannin sensitive (MacDougall et al., 1995; Molz et al., 1996), suggesting that minor differences in sequence may have a large effect on drug sensitivity of these lipid kinases. There is no evidence however, that Ras is regulating class II PI 3-kinases (S. Watton personal communication). Recently Inukai et al. (Inukai et al., 1997) found that the p50a regulatory subunit associates with a pi 10 isoform that
is not labelled by tritiated wortmannin, suggesting the existence of such inhibitor- resistant class lA pi 10 isoforms that are excellent candidates to behave as effectors of Ras responsible for the resistance to inhibition of the Ras-induced ruffling by LY294002 and wortmannin observed in some cell types
The protein kinase C inhibitor calphostin C, which binds to the cysteine-rich zinc finger domains of diacylglicerol-binding PKCs, blocks PI 3-kinase and Ras induced ruffling, but not ruffling induced by activated Rac. On the other hand, bisindoylmaleimide, a PKC inhibitor that binds to the catalytic domain of most PKC isoforms, fails to block Ras-induced ruffling. This suggests that calphostin C is inhibiting a molecule, downstream of PI 3-kinase and upstream of Rac, other than PKCs. Several other proteins in addition to PKCs are known to contain cysteine-rich

133
zinc finger domains, including several molecules involved in Rac activation. Both Rac- GAPs like chimaerins (Diekman et al., 1991) and Rho/Rac family exchange factors such as Vav, Vav2, Lfc and FGDl contain cysteine-rich zinc finger domains and are thus good candidates to be mediating the inhibitory effect of calphostin C on Ras- induced ruffling. It is not yet clear whether calphostin C affects the function of Rac GAPs or GEFs, but it is known to be able to inhibit at least one non-PKC zinc finger protein, Unc-13 in C. elegans (Kazanietz et al., 1995). It is worth noting that Vav, Vav2, Lfc and FGDl (and all other putative Rac-GBFs) contain PH domains, which as discussed in section 1.6.2 can act in some cases as binding domains for the lipid products of PI 3-kinase, further strengthening the possibility that they may be PI 3-kinase targets. A model summarising the data presented on the actin cytoskeleton is shown in figure 6.9 .
Since many growth factors rapidly induce both PI 3-kinase inhibitor sensitive membrane ruffling and Ras activation, it is possible that Ras function is required for the growth factor receptors to induce PI 3-kinase activity and hence to cause actin rearrangement. In the case of Swiss 3T3 cells, PDGF induced ruffling does not appear to be inhibited by neutralising Ras antibodies (Ridley et al., 1992); however, in MDCK cells, hepatocyte growth factor induced ruffling is entirely sensitive to these Ras antibodies (Ridley et al., 1995). In both cases, Rac function is required for ruffling to occur. We have found that the ability of N17 Ras to inhibit ruffling and other PI 3- kinase dependant responses (e.g. Akt activation) vary greatly with cell type and the nature of the external stimulus. It is clear that there are multiple pathways leading to PI 3-kinase activation: in some cases Ras is necessary, in other cases it is not. A very similar situation has been described for growth factor induction of ERK2 MAP kinase in fibroblasts, where Ras is required for PDGF and insulin induced, but not EGF induced, activation in Rat-1 and NIH 3T3 fibroblasts, but both PDGF and EGF induced ERK2 activation are insensitive to inhibition of Ras in Swiss 3T3 fibroblasts, while insulin requires Ras function (Burgering et al., 1993).
The fact that PDGF induced ruffling in PAE cells, unlike that induced by Ras, does not show a significant degree of resistance to LY294002 may suggest that the contribution of endogenous Ras to PDGF induced PI 3-kinase activation is relatively small in this case. Alternatively, Ras may participate in the activation of multiple PI 3- kinase isoforms and the specificity as to which agonist activates which PI 3-kinase isoform is provided by other growth-factor receptor activated signals. In this model the comparatively stronger and constitutive signal provided by V12 Ras expression (as opposed to the small transient activation of endogenous Ras by growth factors) would be sufficient to activate several PI 3-kinase isoforms simultaneously without the need for co-operating signals.

134
Ras
Raf pllOa otherPI3Ks Ral.GDS
LY 294002
MEK
ERK
> 7 (RacGEF)
calph. C I
RacII
Ruffling
Ral
Figure 6.9. A model of the function Ras mutants and effectors in cellular signalling pathways.The Ras mutants S35 and E38 activate only Raf, C40 Ras activates only PI 3-kinase and G37 activates only Ral.GDS. Ras induced ruffling in porcine aortic endothelial cells proceeds through a PI 3-kinase pathway, involving isoforms that are sensitive to LY 294002, but also through a inhibitor resistant isoform(s). Whatever the isoform involved there is a calphostin C sensitive step downstream of PI -3 kinase and upstream of Rac for which Rac GEFs (e.g.. Vav, Vav2, Lfc, FGDl) are very good candidates. The Raf pathway is necessary for neurite outgrowth in PC 12 cells, while at least two effector pathways are needed for efficient transformation of NIH 3T3 cells. See discussion for details.

135
6.3.6 Mechanism of p85 inhibition of PI 3-kinase functionThe exact mechanism by which overexpression of p85 mutants interferes with
PI 3-kinase has not been defined, but is likely to involve titering out of upstream signalling molecules which are needed for activation of PI 3-kinase both by receptor tyrosine kinases and by Ras. As shown in figure 6.7B, when pi 10 is not co-expressed, over-expression of p85 or Ap85 does indeed inhibit the ability of V12 Ras to activate PI 3-kinase in whole cells as determined by measuring PIP3 levels. Although Ras directly
interacts with and moderately activates pi 10 in vitro, it is likely that in order to achieve efficient activation of PI 3-kinase by V12 Ras in whole cells at least basal levels of other signals are also required, including one mediated by p85. Endogenous pi 10 is likely to be effectively uncoupled from stimulatory signals feeding into p85 by massive over-expression of this protein. A similar scenario has been described for PI 3-kinase y, where even though py subunits interact with the pi 10 catalytic subunit and stimulate its
activity two-fold, the presence of the p 101 regulatory subunit amplifies the activation by Py more than 100-fold (Stephens et al., 1997). Over-expression of Ras and of p85
have also been shown to exert opposing influences on mammalian PI 3-kinase activity in a model system in Ss. pombe (Kodaki et al., 1994).
It cannot be ruled out that a function of PI 3-kinase in addition to its lipid kinase activity is important in Ras induced membrane ruffling. Over-expression of p85 does not inhibit the lipid kinase activity of cotransfected pi 10a (Table 4.1) but does block membrane ruffling induced by activated pi 10a in PAE cells (data not shown). It is
therefore possible that over-expression of p85 is inhibiting PI 3-kinase function by other mechanisms in addition to inhibiting its lipid kinase activity. For example, a model could be proposed where p85 could interact with molecules with downstream effector function (e.g. a Rac-GEF) and both this interaction and lipid binding to a PH domain would be required in combination for activation of this putative effector. Under conditions of p85 over-expression, an excess of free p85 would sequester the molecule away from the p85/pllO heterodimer and uncouple regulation by p85 from lipid binding. Another mechanism by which overexpression of p85 could block PI 3-kinase could be related to the observation that the SH2 domains of P85 can bind PIP3 (Rameh et al., 1995). Under this scenario overexpression of p85 would sequester PI 3-kinase lipid products.
6.3.7 Role of Ras in protection from apoptosisIn the last few years it has been appreciated that deregulation of programmed
cell death (or apoptosis) is involved in the pathogenesis of many diseases, including

136
cancer. Apoptosis induced by loss of anchorage to the extracellular matrix probably represents a critical defence mechanism that a tumour cell has to overcome, in the process of malignant transformation, in order to acquire metastatic potential. It is also becoming apparent that activation of anti-apoptotic pathways is likely to be involved in resistance to chemotherapy and radiotherapy, which in the vast majority of cases kill tumour cells by inducing apoptosis.
It has become clear in the last year that PI 3-kinase, through the activation of PKB/Akt, is involved in the protection of different cell types from cell death in response to various apoptotic signals (Franke et al., 1997). Expression of activated forms of PI 3- kinase or PKB/Akt has been shown to protect Cos? epithelial cells from UV irradiation induced apoptosis (Kulik et al., 1997), neuronal cells from apoptosis induced by withdrawal of the survival factor IGF-1 (Dudek et al., 1997) and Rat-1 fibroblasts from apoptosis induced by c-myc expression in the absence of serum factors (Kauffman-Zeh et al., 1997). Activated PI 3-kinase and PKB/Akt also protect MDCK epithelial cells from apoptosis induced by detachment of adherent cells from their extracellular matrix (Khwaja et al., 1997).
Ras is known to protect epithelial cells from apoptosis induced by loss of anchorage (Frisch and Francis, 1994; Rak et al., 1995) and this survival effect has recently been shown to result from the activation of PI 3-kinase (Khwaja et al., 1997). In addition, there are numerous examples in the literature of Ras oncogene expression protecting different cell types from drug and radiation induced apoptosis (Basu and Cline, 1995; Fernandes et al., 1996; Jansen et al., 1997). All these studies indicate that the PI 3-kinase mediated anti-apoptotic effect could be playing a major role in Ras oncogenic potential in human cancers. The data presented here on the effects of p85 expression on the apoptotic behaviour of tumour-derived cell lines further highlights this possibility.
Cell lines derived from pancreatic tumours with mutations in Ras genes seem more resistant than cells with wt Ras to loss of anchorage-induced apoptosis, consistent with the idea that activated Ras is capable of conferring protection from apoptosis, which is likely to be contributing to the tumorigenic phenotype in vivo. Importantly, over-expression of p85, selectively makes cells with activated Ras undergo apoptosis upon of loss of anchorage. These results suggest that PI 3-kinase may represent an ideal target for pharmacological inhibition in the treatment of human tumours in which Ras is involved.

137
CHAPTER 7 General discussion
7.1 Ras regulates several effector pathways
At the time this thesis was started very little was known about the cellular targets of mammalian Ras or about the biochemical pathways by which it carried out its known effects on gene expression, cellular proliferation and differentiation. Little was also known about the mechanisms by which Ras proteins were activated in response to extracellular signals. In the last five years staggering progress has been made on both aspects of Ras biology, its regulation and function.
A major step in the elucidation of Ras function was taken with the finding that the Raf serine/threonine kinases interacted directly with active Ras (Moodie et al., 1993; Van Aelst et al., 1993; Vojtek et al., 1993; Wame et al., 1993; Zhang et al., 1993), thus providing a well characterized connection to the MAP kinase pathway and its pleitropic effects on gene expression, mitogenesis and differentiation. It seemed that after many years of arduous research, Ras biochemical function had been finally deciphered (Hall, 1993). However it soon became apparent that there were other molecules in addition to Raf that, at least in vitro, behaved as potential effectors of Ras.
In this thesis PI 3-kinase has been identified as an additional direct target of Ras. Ras, when in its active GTP-bound conformation, can interact through its effector domain with the pi 10 catalytic subunit of PI 3-kinase and importantly, this interaction leads to stimulation of its lipid kinase activity both in vitro and in vivo.
The ability of Ras to regulate several effector pathways seems to have emerged early in evolution. In S.Pombe, Ras can control both a MAP-kinase cascade through direct interaction with Byr2 and a morphogenic pathway through direct interaction with Scdl, a GEF for a Rho-family GTPase. Moreover, interaction with multiple effectors seems to be a common feature of all members of the Ras superfamily of GTPases: if the still-increasing number of proteins interacting with the Rho-family GTPases is anything to go by, there are still many Ras effectors yet to be identified.
7.2 Contribution of Ras to PI 3-kinase activation.
A major issue in understanding the physiological significance of the interaction between Ras and PI 3-kinase, seen in vitro or when one or both of the components are overexpressed exogenously within the cell, is to determine the contribution of endogenous Ras to PI 3-kinase activation by extracellular signals. It has been shown

138
here that inhibition of Ras activation by overexpression of N17 Ras inhibits PI 3-kinase activation by NGF and EGF in PCI2 cells. Similar results have been obtained after stimulation of HepG2 with PDGF cells transfected with the PDGF receptor (Klinghoffer et al., 1996). In both cases the inhibition is not complete (60-80% inhibition) suggesting that interaction with Ras is just one of a number of pathways by which one single agonist can regulate PI 3-kinase activation. The ability of an enzyme to integrate regulatory signals coming from multiple independent pathways is a common feature in many systems. It is likely that combinations of these pathways will cooperate to modulate the magnitude and/or the kinetics of the lipid kinase activity. It is also likely that in some cases these pathways may be redundant and Ras-independant pathways may be sufficient for full activation of PI 3-kinase activity. The contribution of Ras to PI 3-kinase activation by extracellular signals will thus vary in each case depending on the cell type and the nature of the stimulus.
7.3 Contribution of PI 3-kinase to Ras function.
Another crucial issue for understanding the in vivo significance of the interaction between Ras and PI 3-kinase is to determine the contribution of PI 3-kinase to Ras biological function. To this effect the identification of mutations in the effector domain of Ras that lead to the selective loss of interaction with some, but not all, effectors has provided useful tools to dissect Ras regulated pathways in vivo. These partial loss of function Ras mutants, together with constitutively active and dominant negative forms of PI 3-kinase and other Ras effectors, have allowed the study of the contribution of PI 3-kinase to Ras function in different cell systems.
7.3.1 Several pathways downstream of Ras are needed in combination to transform cells
Ras needs to activate simultaneously several effector pathways in order for efficient transformation to result. The use of partial loss of function mutants of Ras (and of activated versions of effectors) in focus formation assays in NIH 3T3 fibroblasts, described in this work and by other groups (White et al., 1995; Khosravi-Far et al., 1996), show that activation of any single pathway downstream of Ras is not sufficient for transformation. However, PI 3-kinase plays a critical contribution to Ras transforming potential: inhibition of PI 3-kinase function dramatically inhibits the loss of contact inhibition, loss of anchorage-dependence for growth and morphological transformation induced by Ras.

139
MAPKs
\MEK
RINPKCs RAF
PDK AF6
RASAKT/PKB pl20GAPPI3Ks
ARE GEFs ARF GAPs
pl90RhoGAP
RAL GDS(RAC GEFs) (RAC GAPs)
RAL
ARFs RAL-BPlPLD
CDC42 RHORAC
FIGURE 7.1. Effector pathways activated by Ras.Ras and Ras-related GTPases are circled. All components and connections are explained in the text. Ral-BPl has been shown to display GAP activity towards Rac and Cdc42. It should be noted however, that it is not known how Ral interaction with Ral-BPl or pl20GAP interaction with pl90RhoGAP regulate their respective GAP activities.
7.3.2 PI 3-kinase as the connection between Ras and RacA recurrent theme that is emerging from knowledge of Ras effector pathways is
the number of interconnections between GTPases families. Figure 7.1 shows Ras at the top of a GTPase cascade and particularly noteworthy are the various pathways by which Ras seems to be controlling the Rho-family GTPases.
Work in this thesis and by other groups (Hawkins et al., 1995; Reif et al., 1996; Wennstrom et al., 1994) places PI 3-kinase downstream of growth factor receptors and Ras and upstream of Rac in the regulation of membrane ruffling. The exact mechanism by which PI 3-kinase leads to the activation of Rac still remains to be elucidated but as

140
discussed earlier Rac-GEFs, all of which contain PH domains (and can potentially bind 3’ phosphoinositides) and in particular GEFs containing cysteine rich domains (that would account for the inhibitory effect of calphostin C), are good candidates to be direct PI 3-kinase targets.
The biological significance of membrane ruffling is not clear but its occurrence generally correlates with cell motility and in particular with directed migration or chemotaxis (Ridley, 1994). Interestingly both Ras and PI 3-kinase have been implicated in the chemotactic response to specific agonists in various cell types. In MDCK cells both the inhibition of Ras and PI 3-kinase activation block the scattering response induced by HGF/SF (Hartmann et al., 1994; Ridley et al., 1995; Royal and Park, 1995). In addition, dominant negative Ras blocks PDGF-, but not fibronectin-induce chemotaxis in NIH 3T3 cells (Kundra et al., 1995). N17 Ras expression or 259 antibody microinjection also block the motile response induced by bFGF and mechanical wounding in bovine endothelial cells (Fox et al., 1994; Sosnowski et al., 1993). Similarly, inhibition of PI 3-kinase blocks the chemotactic response (as well as membrane ruffling) induced by PDGF in porcine aortic endothelial cells (Wenstromm et al., 1994). Furthermore, cells expressing PDGF receptor mutants that no longer bind to and activate PI 3-kinase can no longer migrate towards PDGF while cells expressing a PDGF receptor mutant that no longer binds pl20GAP migrate better than wild type cells. This suggests that GAP, via downregulation of Ras, negatively regulates chemotaxis (Kundra et al., 1994). Interestingly, it has been shown recently that the PDGF receptor mutant defective for pl20GAP binding does indeed lead to a stronger activation of Ras which correlates with a stronger activation of PI 3-kinase (Klinghoffer et al., 1996). All these results are consistent with Ras-induced activation of PI 3-kinase being an important component of the chemotactic response to certain growth factors. Furthermore, the ability of Ras to regulate cell motility is likely to play a role in the invasive and metastatic potential of tumour cells in which Ras is activated.
7.3.3 PI 3-kinase activation of Rac could have pleitropic effectsRegardless of its biological function, membrane ruffling provides a "read-out"
for Rac activation and as discussed in section 1.5.2.2, activated Rac is known to have several other functions in addition to the regulation of the actin cytoskeleton. It can therefore be predicted that, in addition to membrane ruffling, Ras could regulate several other Rac-dependant functions through the activation of PI 3-kinase. For example, Rac is known to be involved in the generation of reactive oxygen species (ROS) by phagocytic and non-phagocytc cells (see section 1.5.2.2.1) and at least in neutrophils, agonist stimulated ROS production is sensitive to PI 3-kinase inhibitors (Arcaro and Wymann, 1993; Vlahos et al., 1995). It has recently been shown that Ras can induce ROS generation through a Rac-dependant pathway (Irani et al., 1997; Sundaresan et al..

141
1996) and treatment of Ras-transformed NIH 3T3 cells with chemical anti-oxidants inhibits mitogenesis in these cells (Irani et al., 1997). It is therefore possible that ROS generation by Ras could be an important function of PI 3-kinase mediated activation of Rac. Additionally, both Ras and Rac are known to activate the NFkB and AP-1 transcription factors and future studies should determine the contribution of the PI 3- kinase pathway to Ras effects on gene expression.
7.3.4 PI 3-kinase has several targets other than RacEvidence has been presented that Ras activation of PI 3-kinase, through an as yet
unidentified intermediate, leads to the activation of Rac. As described in section 1.6.2 however, several known targets of PI 3-kinase are known (and more no doubt remain to be identified), all of which could potentially be contributing to Ras function. The best characterized direct target of PI 3-kinase is Akt/PKB and Ras has indeed been shown to activate Akt/PKB in a PI 3-kinase-dependent manner (Klippet et al., 1996; Kaufmann- Zeh et al., 1996; Marte et al., 1997; Kwaja et al., 1997). Furthermore, Akt/PKB appears to be a critical mediator of the survival pathways protecting cells from apoptosis, a process most likely of the utmost relevance for Ras oncogenic potential. Other recently identified targets of PI 3-kinase include GEFs for the ARF GTPases (section 1.6.2.2) suggesting that PI 3-kinase could be involved in the regulation of ARF activation in a manner analogous to that proposed for Rac activation. ARFs are known to activate PLD and have been implicated in protein sorting and vesicle trafficking. Future experiments should determine whether Ras can indeed regulate ARF activation and if so what the contribution of ARFs to Ras function is.
7.4 From Ras function to cancer therapy: PI 3-kinase as a target of pharmacological inhibition.
Ras genes are found mutated in 25-30% of human tumours. In addition activation of Ras by an indirect mechanism, is probably involved in an even larger number of human tumours. For example, by loss of GAP activity in Neurofibromatosis type I, by activation or overexpression of receptors known to activate Ras as is the case with the EGF receptor and ErbB2 oncogene in many breasts tumours or by bcr-abl in Philadelphia chromosome positive chronic myelogenous leukaemia. Ras proteins thus represent a very good example of how knowledge of the biochemical and biological properties of a protein can be translated into rational drug design for use in cancer treatment. Farnesyltransferase inhibitors provide a good example of such a strategy.
A better understanding of the different effector pathways activated by Ras and their different effects in the context of different cell types could potentially be of use in the rational design of drugs that will inhibit a relevant pathway for a specific effect in a

142
particular cell type. This would be more specific and less toxic than completely blocking Ras function, as is the case with the farnesyltransferase inhibitors.
In the work discussed in this thesis it has been shown that inhibition of PI 3- kinase function selectively inhibits malignant transformation by Ras in immortalised fibroblasts. More importantly, in cell lines derived from human tumours harbouring Ras mutations, aberrant activation of the PI 3-kinase pathway seems to be playing a role in protecting tumour cells from apoptosis. These results suggests that PI 3-kinase represents an ideal target for therapeutic intervention in the treatment of tumours where Ras activation is playing a role.
A lot more work and time are needed to translate knowledge of Ras function into viable therapies for the treatment of human cancer but PI 3-kinase, no doubt, will be at the heart of several new anti-tumour strategies.

143
ReferencesAdari, H., Lowy, D. R., Willumsen, B. M., Der, C. J., and McCormick, F. (1988). Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 240, 518-21.
Ahmed, S., Lee, J., Kozma, R., Best, A., Monfries, C., and Lim, L. (1993). A novel functional target for tumor-promoting phorbol esters and lysophosphatidic acid. The p21rac-GTPase activating protein n-chimaerin. J Biol Chem 268, 10709-10712.
Akimoto, K., Takahashi, R., Moriya, S., Nishioka, N., Takayanagi, J., Kimura, K., Fukui, Y., Osada, S., Mizuno, K., Hirai, S., Kazlauskas, A., and Ohno, S. (1996). EGFor PDGF receptors activate atypical PKCX through phosphatidylinositol 3-kinase. EMBO J. 15, 788-798.
Akopyan, T. N., Couedel, Y., Oriowski, M., Fournie Zaluski, M. C., and Roques, B. P. (1994). Proteolytic processing of farnesylated peptides: assay and partial purification from pig brain membranes of an endopeptidase which has the characteristics of E.G. 3.4.24.15. Biochem-Biophys-Res-Commun 198, 787-94.
Albino, A. P., Le Strange, R., Oliff, A. L., Furth, M. E., and Old, L. J. (1984). Transforming ras genes from human melanoma: a manifestation of tumour heterogeneity? Nature 308, 69-72.
Albright, C. F., Giddings, B. W., Liu, J., Vito, M., and Weinberg, R. A. (1993). Characterization of a guanine nucleotide dissociation stimulator for a ras-related GTPase. EMBO-J 12, 339-47.
Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P., and Hemmings, B. A. (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 75, 6541 -6551.
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P., Reece, C., and Cohen, P. (1997). Purification and characterisation of a phosphatidylinositol 3,4,5 trisphosphate dependent protein kinase (PDKl) that phosphorylates and activatesprotein kinase Ba. Curr. Biol. 7, 261-269.
Almoguera, C., Shibata, D., Forrester, K., Martin, J., Arnheim, N., and Perucho, M. (1988). Most human carcinomas of the exocrine pancreas contain mutant c-K-ra5 genes. Cell 53, 549-554.
Altschuler, D., and Lapetina, E. G. (1993). Mutational analysis of the cAMP-dependent protein kinase-mediated phosphorylation site of Rap lb. J Biol Chem 268, 7527-31.
Altschuler, D. L., Peterson, S. N., Ostrowski, M. C., and Lapetina, E. G. (1995). Cyclic AMP-dependent activation of Rap lb. J BIOL CHEM. Journal of Biological Chemistry 270, 10373-10376.
Amano, M., Mukai, H., Ono, Y., Chihara, K., Matsui, T., Hamajima, Y., Okawa, K., Iwamatsu, A., and Kaibuchi, K. (1996). Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271, 648-50.
Andersen, P. R., Devare, S. G., Tronick, S. R., Ellis, R. W., Aaronson, S. A., and Scolnick, E. M. (1981). Generation of BALB-MuSV and Ha-MuSV by Type C Virus

144
Transduction of Homologous Transforming Genes from Different Species. Cell 26, 129- 134.
Anderson, R. G. (1993). Caveolae: where incoming and outgoing messengers meet. Proc-Natl-Acad-Sci-U-S-A 90, 10909-13.
Antonetti, D. A., Algenstaedt, P., and Kahn, C. R. (1996). Insulin receptor substrate 1 binds two novel splice variants of the regulatory subunit of phosphatidylinositol 3- kinase in muscle and brain. Mol-Cell-Biol 16, 2195-203.
Arcaro, A., and Wymann, M. P. (1993). Wortmannin is a potent phosphatidylinositol 3- kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochemical Journal.
Aspenstrom, P., Lindberg, U., and Hall, A. (1996). Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Current Biology 6, 70-5.
Auger, K. R., Serunian, L. A., Soltoff, S. P., Libby, P., and Cantley, L. C. (1989). PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57, 167-75.
Baba, H., Fuss, B., Urano, J., Poullet, P., Watson, J. B., Tamanoi, F., and Macklin, W.B. (1995). GapIII, a new brain-enriched member of the GTPase-activating protein family. Journal of Neuroscience Research 41, 846-58.
Backer, J. M., Myers, M. G., Shoelson, S. E., Chin, D. J., Sun, X.-J., Miralpeix, M., Hu, P., Margolis, B., Skolnik, Y., Schlessinger, J., and White, M. J. (1992). (IRS-1 tyrosine phosphopeptide activation of Phosphotidyl inositol 3'kinase). EMBO J. 11, 3469-3479.
Bagrodia, S., Derijard, B., Davis, R. J., and Cerione, R. A. (1995). Cdc42 and PAK- mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. Journal of Biological Chemistry 270, 27995-8.
Balmain, A., and Pragnell, I. B. (1983). Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-raj oncogene. Nature 303, 72-74.
Banin, S., Truong, O., Katz, D. R., Waterfield, M. D., Brickell, P. M., and Gout, I. (1996). Wiskott-Aldrich syndrome protein (WASp) is a binding partner for c-Src family protein-tyrosine kinases. Current Biology 6, 981-8.
Bar Sagi, D., and Feramisco, J. R. (1986). Induction of membrane ruffling and fluid- phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233, 1061-1068.
Bar-Sagi, D., and Feramisco, J. R. (1985). Microinjection of the ras Oncogene Protein into PC 12 cells Induces Morphological Differentiation. Cell 42, 841-848.
Barbacid, M. (1987). ras genes. Annu Rev Biochem 56, 779-827.
Barbacid, M. (1990). ras oncogenes: their role in neoplasia. Eur. J. Clin. Invest. 20, 225-35.
Basu, A., and Cline, J. S. (1995). Oncogenic transformation alters cisplatin-induced apoptosis in rat embryo fibroblasts. Int-J-Cancer 63, 597-603.
Basu, T., Gutmann, D. H., Fletcher, J. A., Glover, T. W., Collins, F. S., and Downward, J. (1992). Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356, 713-715.

145
Bellacosa, A., Testa, J. R., Staal, S. P., and Tsichlis, P. N. (1991). A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 254, 274-277.
Bernards, A. (1995). Neurofibromatosis type 1 and Ras-mediated signaling: filling in the GAPs. Biochim-Biophys-Acta 1242,43-59.
Birchmeier, C., Broek, D., and Wigler, M. (1985). Ras proteins can induce meiosis in xenopus oocytes. Cell 43, 615-621.
Bokoch, G. M. (1996). Interplay between Ras-related and heterotrimeric GTP binding proteins: lifestyles of the BIG and little. FASEB-J 10, 1290-5.
Bokoch, G. M., Quilliam, L. A., Bohl, B. P., Jesaitis, A. J., and Quinn, M. T. (1991). Inhibition of rap la binding to cytochrome b558 of nadph oxidase by phosphorylation of rap la. Science 254, 1794-6.
Bollag, G., Adler, P., elMasry, N., and Shannon, K. (1996). Biochemical characterization of a novel KRAS insertion mutation from a human leukemia. Journal of Biological Chemistry 271, 32491-32494.
Bollag, G., Clapp, D. W., Shih, S., Adler, P., Zhang, Y. Y., Thompson, P., Lange, B. J., Freedman, M. H., McCormick, P., Jacks, T., and Shannon, K. (1996). Loss of NPl results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat-Genet 12, 144-8.
Bollag, G., and McCormick, P. (1991). Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 351, 576-579.
Bollag, G., and McCormick, P. (1992). GTPase activating proteins. Semin-Cancer-Biol 3, 199-208.
Bollag, G., and McCormick, P. (1992). NP is enough of GAP. Nature 356, 663-664.
Bos, J. L. (1988). The ras gene family and human carcinogenesis. Mutat. Res. 195, 255- 271.
Bos, J. L. (1989). ras oncogenes in human cancer: a review. Cancer Res. 49, 4682-4689.
Bos, J. L., Pearon, E. R., Hamilton, S. R., Verlaan-de Vries, M., Van Boom, J. H., Van der Eb, A. J., and Vogelstein, B. (1987b). Prevalence of ras mutations in human colorectal cancers. Nature 327, 293-297.
Bos, J. L., Verlaan-de Vries, M., Jansen, A. M., Veeneman, G. H., Van Boom, J. H., and A.J., V. d. E. (1984). Three different mutations in codon 61 of the human N-ras gene detected by synthetic oligonucleotide hybridization. Nucleic Acids Res. 12, 9155- 9163.
Bos, J. L., Verlaan-de Vries, M., Van der Eb, A. J., Jansenn, J. W. G., Delwel, R., Lowenberg, B., and Colly, L. P. (1987a). Mutations in N-ras predominate in acute myeloid leukemia. Blood 69, 1237-1241.
Bowtell, D., Pu, P., Simon, M., and Senior, P. (1992). Identification of murine homologues of the Drosophila son of sevenless gene: potential activators of ras. Proc. Natl. Acad. Sci. USA 89, 6511-6515.

146
Boyartchuk, V. L., Ashby, M. N., and Rine, J. (1997). Modulation of Ras and a-factor function by carboxyl-terminal proteolysis [see comments]. Science 275, 1796-800.
Braselmann, S., and McCormick, F. (1995). Bcr and Raf form a complex in vivo via 14- 3-3 proteins. EMBO-J 14,4839-48.
Brill, S., Li, S., Lyman, C. W., Church, D. M., Wasmuth, J. J., Weissbach, L., Bernards, A., and Snijders, A. J. (1996). The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol-Cell-Biol 16,4869-78.
Broach, J. R. (1991). RAS genes in Saccharomyces cerevisiae: signal transduction in search of a pathway. Trends Genet. 7, 28-33.
Brown, J. L., Stowers, L., Baer, M., Trejo, J., Coughlin, S., and Chant, J. (1996). Human Ste20 homologue hPAKl links GTPases to the JNK MAP kinase pathway. Current Biology 6, 598-605.
Brown, M. S., and Goldstein, J. L. (1993). Protein prénylation. Mad bet for Rab [news; comment]. Nature 366, 14-5.
Brown, R., Marshall, C. J., Pennie, S. G., and Hall, A. (1984). Mechanism of activation of an N-ra5 gene in the human fibrosarcoma cell line HT 1080. EMBO J. 3, 1321-1326.
Brtva, T. R., Drugan, J. K., Ghosh, S., Terrell, R. S., Campbell-Burk, S., Bell, R. M., and Der, C. J. (1995). Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem. 270, 9809-9812.
Brunger, A. T., Milburn, M. V., Tong, L., deVos, A. M., Jancarik, J., Yamaizumi, Z., Nishimura, S., Ohtsuka, E., and Kim, S. H. (1990). Crystal structure of an active form of RAS protein, a complex of a GTP analog and the HRAS p21 catalytic domain. Proc. Natl. Acad. Sci. U S A 57, 4849-4853.
Buchsbaum, R., Telliez, J. B., Goonesekera, S., and Feig, L. A. (1996). The N-terminal pleckstrin, coiled-coil, and IQ domains of the exchange factor Ras-GRF act cooperatively to facilitate activation by calcium. Mol-Cell-Biol 16, 4888-96.
Burgering, B. M. T., and Coffer, P. J. (1995). Protein kinase B (c-akt) in phosphatidylinositol 3-kinase signal transduction. Nature 376, 599-602.
Burgering, B. M. T., de Vries-Smits, A. M. M., Medema, R. H., van Weeren, P. C., Tertoolen, L. G. J., and Bos, J. L. (1993). Epidermal growth factor induces phosphorylation of extracellular signal-regulated kinase 2 via multiple pathways. Mol. Cell. Biol. 13, 7248-7256.
Burgering, B. M. T., Pronk, G. J., van Weeren, P. C., Chardin, P., and Bos, J. L. (1993). cAMP antagonizes p21ras directed activation of extracellular signal-regulated kinase 2 and phosphorylation of mSos nucleotide exchange factor. EMBO J. 12,4211-4220.
Cadwallader, K. A., Paterson, H., MacDonald, S. G., and Hancock, J. F. (1994). N- terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localisation. Mol. Cell. Biol. 14,4722-4730.
Cales, C., Hancock, J. F., Marshall, C. J., and Hall, A. (1988). The cytoplasmic protein GAP is implicated as the target for regulation by the ras gene product. Nature 332, 548- 51.

147
Camp, L. A., Verkruyse, L. A., Afendis, S. J., Slaughter, C. A., and Hofmann, S. L.(1994). Molecular cloning and expression of palmitoyl-protein thioesterase. J-Biol- Chem 269, 23212-9.
Campbell, K. S., Ogris, E., Burke, B., Su, W., Auger, K. R., Druker, B. J., Schaffhausen, B. S., Roberts, T. M., and Pallas, D. C. (19^94). Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc. Natl. Acad. Sci. USA 91, 6344-6348.
Cantley, L. C., Auger, K. R., Carpenter, C., Duckworth, B., Graziani, A., Kapeller, R., and Soltoff, S. (1991). Oncogenes and signal transduction. Cell 64, 281-302.
Cantor, S. B., Urano, T., and Feig, L. A. (1995). Identification and characterization of Ral-binding protein-1, a potential downstream target of Ral GTPases. Mol. Cell. Biol. 15, 4578-4584.
Carpenter, C. L., Auger, K. R., Chaudhuri, M., Yoakin, M., Schaffhausen, B., Shoelson,S., and Cantley, L. C. (1993). (PDGF receptor tyrosine phoshphopeptide stimulation of phosphatidyl inositol 3'kinase activity). J. Biol. Chem. 268, 9478-9483.
Carpenter, C. L., and Cantley, L. C. (1996). Phosphoinositide kinases. Curr-Opin-Cell- Biol 8, 153-8.
Carpino, N., Wisniewski, D., Strife, A., Marshak, D., Kobayashi, R., Stillman, B., and Clarkson, B. (1997). p62(dok): a constitutively tyrosine-phosphorylated, GAP- associated protein in chronic myelogenous leukemia progenitor cells. Cell 88, 197-204.
Casey, P. J., and Seabra, M. C. (1996). Protein prenyltransferases. J-Biol-Chem 271, 5289-92.
Cerione, R. A., and Zheng, Y. (1996). The Dbl family of oncogenes. Current Opinion in Cell Biology 5,216-222.
Chan, A. M. L., Miki, T., Meyers, K. A., and Aaronson, S. A. (1994). Human oncogene of the ras superfamily unmasked by expression cDNA cloning. Proc. Natl. Acad. Sci. USA 91, 7558-7562.
Chang, E. C., Barr, M., Wang, Y., Jung, V., Xu, H. P., and Wigler, M. H. (1994). Cooperative interaction of S. pombe proteins required for mating and morphogenesis. Cell 79, 131-141.
Chardin, P., Camonis, J. H., Gale, N. W., Van Aelst, L., Schlessinger, J., Wigler, M., and Bar-Sagi, D. (1993). Human Sosl: a guanine nucleotide exchange factor for Ras that binds to Grb2. Science 260, 1338-1343.
Chardin, P., Paris, S., Antonny, B., Robineau, S., Beraud Dufour, S., Jackson, C. L., and Chabre, M. (1996). A human exchange factor for ARE contains Sec7- and pleckstrin- homology domains. Nature 384, 481-4.
Chen, L., Zhang, L. J., Greer, P., Tung, P. S., and Moran, M. F. (1993). A murine CDC25/ras-GRF-related protein implicated in Ras regulation. Dev. Genet. 14, 339-346.
Cherniack, A. D., Klarlund, J. K., Conway, B. R., and Czech, M. P. (1995). Disassembly of Son of sevenless proteins from Grb2 during p21ras desensitization by insulin. J. Biol. Chem 270, 1485-1488.

148
Chong, L. D., Traynor-Kaplan, A., Bokoch, G. M., and Schwartz, M. A. (1994). The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 79, 507-513.
Clark, G. J., Kinch, M. S., Gilmer, T. M., Burridge, K., and Der, C. J. (1996). Overexpression of the Ras-related TC21/R-Ras2 protein may contribute to the development of human breast cancers. Oncogene. 72, 169-176.
Cockcroft, S., Thomas, G. M., Fensome, A., Geny, B., Cunningham, E., Gout, I., Hiles,I., Totty, N. F., Truong, O., and Hsuan, J. J. (1994). Phospholipase D: a downstream effector of ARF in granulocytes. Science 263, 523-6.
Coffer, P. J., and Woodgett, J. R. (1991). Molecular cloning and characterisation of a novel protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur. J. Biochem. 201,475-481.
Colicelli, J., Field, J., Ballester, R., Chester, N., Young, D., and Wigler, M. (1990). Mutational mapping of RAS-responsive domains of the Saccharomyces cerevisiae adenylyl cyclase. Mol-Cell-Biol 10, 2539-43.
Conklin, D. S., Galaktionov, K., and Beach, D. (1995). 14-3-3 proteins associate with cdc25 phosphatases. Proc-Natl-Acad-Sci-U-S-A 92, 7892-6.
Cook, S. J., and McCormick, F. (1993). Inhibition by cAMP of Ras-dependent activation of Raf [see comments]. Science 262, 1069-72.
Cooper, G. M., and Neiman, P. E. (1981). Two distinct candidate transforming genes of lymphoid leukosis virus-induced neoplasms. Nature 292, 857-8.
Corbalan Garcia, S., Degenhardt, K. R., and Bar Sagi, D. (1996). Insulin-induced dissociation of Sos from Grb2 does not contribute to the down regulation of Ras activation. Oncogene 12, 1063-8.
Coso, O. A., Chiariello, M., Yu, J. C., Teramoto, H., Crespo, P., Xu, N., Miki, T., and Gutkind, J. S. (1995). The small GTP-binding proteins R ad and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81, 1137-1146.
Coutavas, E., Ren, M., Oppenheim, J. D., D'Eustachio, P., and Rush, M. G. (1993). Characterization of proteins that interact with the cell-cycle regulatory protein Ran/TC4. Nature 366, 585-7.
Cowley, S., Paterson, H., Kemp, P., and C.J., M. (1994). Activation of map kinase kinase is necessary and sufficient for pci2 differentiation and for transformation of NIH 3T3 cells. Cell 77, 841-852.
Cox, A. D., Brtva, T. R., Lowe, D. G., and Der, C. J. (1994). R-Ras induces malignant, but not morphologic, transformation of NIH3T3 cells. Oncogene 9, 3281-3288.
Cross, D. A. E., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A.(1995). Inhibition of glycogen synthase kinase-3 by insulin mediated protein kinase B. Nature 378, 785-789.
Cullen, B. R. (1996). HIV-1: is Nef a PAK animal? Current Biology 6, 1557-9.
Cullen, P. J., Hsuan, J. J., Truong, O., Letcher, A. J., Jackson, T. R., Dawson, A. P., and Irvine, R. F. (1995). Identification of a specific Ins(l,3,4,5)P4-binding protein as a member of the GAPl family. Nature 376, 527-530.

149
Cutler, R. L., Liu, L., Damen, J. E., and Krystal, G. (1993). Multiple cytokines induce the tyrosine phosphorylation of She and its association with Grb2 in hematopoietic cells. J. Biol. Chem. 268, 21463-21465.
D'Adamo, D. R., Novick, S., Kahn, J. M., Leonardi, P., and Pellicer, A. (1997). rsc: A novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene. 14, 1295-1305.
D'Arcangelo, G., and Halegoua, S. (1993). A branched signaling pathway for nerve growth factor is revealed by Src-, Ras-, and Raf-mediated gene inductions. Mol-Cell- Biol 73,3146-55.
Damen, J. E., Liu, L., Cutler, R. L., and Krystal, G. (1993). Erythropoietin stimulates the tyrosine phosphorylation of She and its association with Grb2 and a 145 kD tyrosine phosphorylated protein. Blood 82, 2296-2303.
Datta, K., Franke, T. P., Chan, T. O., Makris, A., Yang, S.-I., Kaplan, D. R., Morrison, D. K., Golemis, E. A., and Tsichlis, P. N. (1995). AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol. Cell. Biol. 15, 2304-2310.
Daub, H,, Weiss, F. U., Wallasch, C., and Ullrich, A. (1996). Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557-60.
Davies, A. H., Jowett, J. B. M., and Jones, I. A. (1993). Creation of Exo III deletion mutants. Bio/Technology 11, 933-936.
de Vries Smits, A. M., Burgering, B. M., Leevers, S. J., Marshall, C. J., and Bos, J. L. (1992). Involvement of p21ras in activation of extracellular signal-regulated kinase 2 [see comments]. Nature 357, 602-4.
DeClue, J. E., Papageorge, A. G., Fletcher, J. A., Diehl, S. R., Ratner, N., Vass, W. C., and Lowy, D. R. (1992). Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69, 265-273.
DePeo, D., Gonda, M. A., Young, H. A., Chang, E. H., Lowy, D. R., Scolnick, E. M., and Ellis, R. W. (1981). Analysis of two divergent rat genomic clones homologous to the transforming gene of Harvey murine sarcoma virus. Proc. Natl. Acad. Sci. USA 78, 3328-3332.
Dent, P., Reardon, D. B., Morrison, D. K., and Sturgill, T. W. (1995). Regulation of Raf-1 and Raf-1 mutants by Ras-dependent and Ras-independent mechanisms in vitro [published erratum appears in Mol Cell Biol 1995 Sep;15(9):5203]. Mol-Cell-Biol 15, 4125-35.
Der, C. J., Krontiris, T. G., and Cooper, G. M. (1982). Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc. Natl. Acad. Sci. USA 79, 3637-3640.
Der, C. J., Pan, B. T., and Cooper, G. M. (1986). rasH mutants deficient in GTP binding. Mol Cell Biol 6, 3291-4.
de Vos, A. M., Tong, L., Milburn, M. V., Matias, P. M., Jancarik, J., Noguchi, S., Nishimura, S., Miura, K., Ohtsuka, P., and Kim, S. H. (1988). Three-dimensional Structure of an Oncogene Protein: Catalytic Domain of Human c-W-ras p21. Science 239, 888-893.

150
Dhand, R., Hara, K., Hiles, L, Bax, B., Gout, L, Panayotou, G., Fry, M. J., Yonezawa, K., Kasuga, M., and Waterfield, M. D. (1994). PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO. J 13, 511-21.
Dhand, R., Hiles, I., Panayotou, G., Roche, S., Fry, M. J., Gout, I., Totty, N. F., Truong, O., Vicendo, P., Yonezawa, K., Kasuga, M., and Waterfield, M. D. (1994). PI 3-kinase is a dual-specificity enzyme: autoregulation by an intrinsic protein-serine kinase- activity. EMBO J. 13, 522-533.
Dhar, R., Ellis, R. W., Shih, T. Y., Oroszlan, S., Shapiro, B., Maizel, J., Lowy, D., and Scolnick, E. M. (1982). Nucleotide sequence of the p21 Transforming Protein of Harvey Murine Sarcoma Virus. Science 217, 934-937.
Dickson, B., and Hafen, E. (1994). Genetics of signal transduction in invertebrates. Curr-Opin-Genet-Dev 4, 64-70.
Dickson, B., Sprenger, F., Morrison, D., and Hafen, E. (1992). Raf functions downstream of Rasl in the Sevenless signal transduction pathway. Nature 360, 600-603.
Diekman, D., Brill, S., Garrett, M., Totty, N., Hsuan, J., Monfries, C., Hall, C., Lim, L., and Hall, A. (1991). Bcr encodes a GTPase activating protein for p21rac. Nature 351, 400-402.
Diekmann, D., Abo, A., Johnston, C., Segal, A. W., and Hall, A. (1994). Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science 265, 531-3.
Dilworth, S. M., Brewster, C. E., Jones, M. D., Lanfrancone, L., Pelicci, G., and Pelicci, P. G. (1994). Transformation by polyoma virus middle T-antigen involves the binding and tyrosine phosphorylation of She. Nature 367, 87-90.
Downward, J., Graves, J. D., Warne, P. H., Rayter, S., and Cantrell, D. A. (1990). Stimulation of p2H^^ upon T-cell activation. Nature 346, 719-723.
Drugan, J. K., Khosravi Far, R., White, M. A., Der, C. J., Sung, Y. J., Hwang, Y. W., and Campbell, S. L. (1996). Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J-Biol-Chem 271, 233-7.
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R. J., Cooper, G. M., Segal, R. A., Kaplan, D. R., and Greenberg, M. E. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661-665.
Dudler, T., and Gelb, M. H. (1996). Palmitoylation of Ha-Ras facilitates membrane binding, activation of downstream effectors, and meiotic maturation in Xenopus oocytes. J-Biol-Chem 271, 11541-7.
Eliyahu, D., Raz, A., Gruss, P., Givol, D., and Oren, M. (1984). Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312, 646- 649.
Ellis, R. W., DeFeo, D., Shih, T. Y., Gonda, M. A., Young, H. A., Tsuchida, N., Lowy, D. R., and Scolnick, E. M. (1981). The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 292, 506-511.

151
Escobedo, J. A., Navankasattusas, S., Kavanaugh, W. M., Milfay, D., Fried, V. A., and Williams, L. T. (1991). cDNA cloning of a novel 85 kd protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF beta-receptor. Cell 65, 75-82.
Eva, A., and Aaronson, S. A. (1983). Frequent Activation of c-kis as a Transforming Gene in Fibrosarcomas Induced by Methlycholanthrene. Science 220, 955-956.
Farnsworth, C. L., and Feig, L. A. (1991). Dominant Inhibitory mutations in the Mg binding site of Ras-H prevent its activation by GTP. Mol. Cell. Biol. 11,4822-4829.
Farnsworth, C. L., Freshney, N. W., Rosen, L. B., Ghosh, A., Greenberg, M. E., and Feig, L. A. (1995). Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376, 524-527.
Farr, C. J., Marshall, C. J., Easty, D. J., Wright, N. A., Powell, S. C., and Paraskeva, C. A. (1988a). A study of ras mutations in colonic adenomas from familial polyposis coli patients. Oncogene 3, 673-678.
Farr, C. J., Saiki, R. K., Erlich, H. A., McCormick, F., and Marshall, C. J. (1988b). Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc. Natl. Acad. Sci. USA 85, 1629-1633.
Farrar, M. A., Alberol, I., and Perlmutter, R. M. (1996). Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization [see comments]. Nature 383, 178-81.
Fasano, O., Aldrich, T., Tamanoi, F., Taparowsky, E., Furth, M., and Wigler, M. (1984). Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. USA 81, 4008-4012.
Feig, L. A. (1993). The many roads that lead to Ras. Science 260, 767-768.
Fernandes, R. S., McGowan, A. J., and Cotter, T. G. (1996). Mutant H-ras overexpression inhibits drug and U.V. induced apoptosis. Anticancer-Res 16, 1691-705.
Field, J., Broek, D., Kataoka, T., and Wigler, M. (1987). Guanine nucleotide activation of, and competition between, RAS proteins from Saccharomyces cerevisiae. Mol Cell Biol 7,2128-33.
Forrester, K., Almoguera, C., Han, K., Grizzle, W. E., and Perucho, M. (1987). Detection of high incidence of Y^-ras oncogenes during human carcinogenesis. Nature 327, 298-303.
Fox, P. L., Sa, G., Dobrowolski, S. F., and Stacey, D. W. (1994). The regulation of endothelial cell motility by p21 ras. Oncogene 9, 3519-26.
Franke, B., Akkerman, J. W., and Bos, J. L. (1997). Rapid Ca2+-mediated activation of Rapl in human platelets. Embo Journal 16, 252-9.
Franke, T. F., Kaplan, D. R., and Cantley, L. C. (1997). PI3K: downstream AKTion blocks apoptosis. Cell 88 ,435-7.
Franke, T. F., Kaplan, D. R., Cantley, L. C., and Toker, A. (1997). Direct regulation of the akt protooncogene product by phosphatidylinositol -3,4- bisphosphate. Science 275, 665-668.
Franke, T. F., Yang, S.-I., Chan, T. O., Datta, K., Kazlauskas, A., Morrison, D. K., Kaplan, D. R., and Tsichlis, P. N. (1995). The protein kinase encoded by the Akt proto

152
oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727- 736.
Freeh, M., Andjelkovic, M., Ingley, E., Reddy, K. K., Falck, J. R., and Hemmings, B. A. (1997). High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of of RAC/Protein kinase B and their influence on the kinase activity. J. Biol. Chem. 272, 8474-8481.
Freeh, M., John, J., Pizon, V., Chardin, P., Tavitian, A., Clark, R., McCormick, F., and Wittinghofer, A. (1990). Inhibition of GTPase activating protein stimulation of ras-p21 GTPase by the Krev-1 gene product. Science 249, 169-171.
Friedman, E., Gejman, P. V., Martin, G. A., and McCormick, F. (1993). Nonsense mutations in the C-terminal SH2 region of the GTPase activating protein (GAP) gene in human tumours. Nat-Genet 5, 242-7.
Frisch, S. M., and Francis, H. (1994). Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619-626.
Fruman, D. A., Cantley, L. C., and Carpenter, C. L. (1996). Structural organization and alternative splicing of the murine phosphoinositide 3-kinase p85 alpha gene. Genomics 37, 113-21.
Fujioka, H., Kaibuchi, K., Kishi, K., Yamamoto, T., Kawamura, M., Sakoda, T., Mizuno, T., and Takai, Y. (1992). Transforming and c-fos promoter/enhancer- stimulating activities of a stimulatory GDP/GTP exchange protein for small GTP- binding proteins. J. Biol. Chem. 267, 926-930.
Fujiyama, A., and Tamanoi, F. (1990). RAS2 protein of Saccharomyces cerevisiae undergoes removal of methionine at N terminus and removal of three amino acids at C terminus. J-Biol-Chem 265, 3362-8.
Fukuda, M., Kojima, T., Kabayama, H., and Mikoshiba, K. (1996). Mutation of the pleckstrin homology domain of Bruton's tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5-tetrakisphosphate binding capacity. J-Biol-Chem 271, 30303-6.
Furth, M. E., Davis, L. J., Fleurdelys, B., and Scolnick, E. M. (1982). Monoclonal antibodies to the p21 products of the transforming gene of Harvey murine sarcoma virus and of the cellular ras gene family. J Virol 43, 294-304.
Gabig, T. G., Crean, C. D., Mantel, P. L., and Rosli, R. (1995). Function of wild-type or mutant Rac2 and Rap la GTPases in differentiated HL60 cell NADPH oxidase activation. Blood. 85, 804-811.
Galaktionov, K., Lee, A. K., Eckstein, J., Draetta, G., Meckler, J., Loda, M., and Beach, D. (1995). CDC25 phosphatases as potential human oncogenes. Science 269, 1575-7.
Garcia Gardena, G., Oh, P., Liu, J., Schnitzer, J. E., and Sessa, W. C. (1996). Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc-Natl-Acad-Sci-U-S-A 93, 6448-53
Gawler, D. J., Zhang, L. J., and Moran, M. F. (1995). Mutation-deletion analysis of a Ca(2-f)-dependent phospholipid binding (CaLB) domain within p i20 GAP, a GTPase- activating protein for p21 ras. Biochem-J 307,487-91.
Ghosh, S., Xie, W. Q., Quest, A. F., Mabrouk, G. M., Strum, J. C., and Bell, R. M. (1994). The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J-Biol-Chem 269, 10000-7.

153
Gibbs, J. B., Oliff, A., and Kohl, N. E. (1994). Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 77, 175-8.
Gimeno, C. J., Ljungdahl, P. O., Styles, C. A., and Fink, G. R. (1992). Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell 68, 1077-90.
Golubic, M., Tanaka, K., Dobrowolski, S., Wood, D., Tsai, M. H., Marshall, M., Tamanoi, P., and Stacey, D. W. (1991). The GTPase stimulatory activities of the neurofibromatosis type 1 and the yeast IRA2 proteins are inhibited by arachidonic acid. EMBO J. 10, 2897-2903.
Gorlich, D., and Mattaj, I. W. (1996). Nucleocytoplasmic transport. [Review] [77 refs]. Science 271, 1513-8.
Gotoh, T.,Hattori, S, Nakamura, S., Kitayama, H., Noda, M., Takai, Y., Kaibuchi, K., Matsui, H., Hatase, O., Takahashi, H. et al. (1995). Identification of Rapl as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol-Cell-Biol 15, 6746-53.
Gotoh, T; Niino, Y; Tokuda, M; Hatase, O., Nakamura, S., Nakamura,S; Matsuda, M and Hattori S. (1997). Activation of R-Ras by Ras-Guanine nucleotide-releasing Factor. J. Biol. Chem. 272, 18602-07.
Graham, S. M., Cox, A. D., Drivas, G., Rush, M. G., Deustachio, P., and Der, C. J. (1994). Aberrant function of the ras-related protein tc21/r-ras2 triggers malignant transformation. Mol. Cell. Biol. 14,4108-4115.
Graham, S. M., Vojtek, A. B., Huff, S. Y., Cox, A. D., Clark, G. J., Cooper, J. A., and Der, C. J. (1996). TC21 causes transformation by Raf-independent signaling pathways. Mol Cell Biol 76,6132-40.
Graves, L. M., Bornfeldt, K. E., Raines, E. W., Potts, B. C., Macdonald, S. G., Ross, R., and Krebs, E. G. (1993). Protein kinase A antagonizes platelet-derived growth factor- induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proc-Natl-Acad-Sci-U-S-A 90, 10300-4.
Grünewald, K., Lyons, J., Frohlich, A., Feichtinger, H., Weger, R. A., Schwab, G., Jansenn, J. W. G,, and Bartram, C. R. (1989). High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int. J. Cancer 43.
Guerrero, I., Calzada, P., Mayer, A., and Pellicer, A. (1984). A molecular approach to leukemogenesis: Mouse lymphomas contain an activated c-ras oncogene. Proc. Natl. Acad. Sci. USA 81, 202-205.
Guerrero, I., Wong, H., Pellicer, A., and Burstein, D. E. (1986). Activated N-r<3J Induces Neuronal Differentiation of PC 12 Rat Pheochromocytoma Cells. J. Cell. Physiol. 729,71-76.
Guo, H. P., The, I., Hannan, P., Bernards, A., and Zhong, Y. (1997). Requirement of Drosophila NPl for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276, 795-8.
Gutierrez, L., Magee, A. I,, Marshall, C. J., and Hancock, J. P. (1989). Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy- terminal proteolysis. EMBO-J 8, 1093-8.

154
Habets, G. G. M., Scholtes, E. H. M., Zuydgeest, D., Van der Kammen, R. A., Stam, J.C., Berns, A., and Collard, J. G. (1994). Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell 77, 537-549.
Hafner, S., Adler, H. S., Mischak, H., Janosch, P., Heidecker, G., Wolfman, A., Pippig,S., Lohse, M., Ueffing, M., and Kolch, W. (1994). Mechanism of inhibition of Raf-1 by protein kinase A. Mol-Cell-Biol 14, 6696-703.
Hagag, N., Halegoua, S., and Viola, M. (1986). Inhibition of growth factor-induced differentiation of PC 12 cells by microinjection of antibody to ras p21. Nature 319, 680- 682.
Hall, A. (1993). Ras-related proteins. Curr. Op. Cell. Biol. 5, 265-268.
Hall, A., Marshall, C. J., Spurr, N. K., and Weiss, R. A. (1983). Identification of transforming gene in human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature 303, 396-400.
Hall, A., and Self, A. J. (1986). The Effect of Mg2+ on the Guanine Nucleotide Exchange Rate of p21N-ras. J. Biol. Chem. 261, 10963-10965.
Hammond, S. M., Jenco, J. M., Nakashima, S., Cadwallader, K., Gu, Q., Cook, S., Nozawa, Y., Prestwich, G. D., Frohman, M. A., and Morris, A. J. (1997). Characterization of two alternately spliced forms of phospholipase D l. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. Journal of Biological Chemistry 272, 3860-8.
Hammonds Odie, L. P., Jackson, T. R., Profit, A. A., Blader, I. J., Turck, C. W., Prestwich, G. D., and Theibert, A. B. (1996). Identification and cloning of centaurin- alpha. A novel phosphatidylinositol 3,4,5-trisphosphate-binding protein from rat brain. J-Biol-Chem 277, 18859-68.
Han, J. W., McCormick, P., and Macara, I. G. (1991). Regulation of ras-GAP and the Neurofibromatosis -1 gene product by eicosanoids. Science 252, 576-579.
Han, L., and Colicelli, J. (1995). A human protein selected for interference with Ras function interacts directly with Ras and competes with Rafl. Mol-Cell-Biol 15, 1318- 23.
Han, 1., Wong, D., Dhaka, A., Afar, D., Witte, O., and Collicelli, J. (1997). Protein binding and signalling properties of RINl suggest a unique effector function. Proc, Natl. Sci. 94, 4954-4959.
Han, M., Golden, A., Han, Y., and Sternberg, P. W. (1993). C.elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 363, 133-140.
Hancock, J. P., Cadwallader, K., and Marshall, C. J. (1991). Méthylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO-J 10, 641-6.
Hancock, J. P., Cadwallader, K., Paterson, H., and Marshall, C. J. (1991). A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO-J 10,4033-9.
Hancock, J. P., Magee, A. I., Childs, J. E., and Marshall, C. J. (1989). All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167-1177.

155
Hancock, J. F., Paterson, H., and Marshall, C. J. (1990). A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63, 133-139.
Hara, M,, Akasaka, K., Akinaga, S., Okabe, M., Nakano, H., Gomez, R., Wood, D., Uh, M., and Tamanoi, F. (1993). Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc-Natl-Acad-Sci-U-S-A 90, 2281-5.
Hart, M. J., Sharma, S., elMasry, N., Qiu, R. G., McCabe, P., Polakis, P., and Bollag, G.(1996). Identification of a novel guanine nucleotide exchange factor for the Rho GTPase. J-Biol-Chem 277, 25452-8.
Hartley, D., Meisner, H., and Corvera, S. (1995). Specific association of the beta- isoform of the p85 subunit of phosphatidylinositol-3 kinase with the protooncogene c- cbl. J. Biol. Chem. 270, 18260-18263.
Hartmann, G., Weidner, K. M., Schwarz, H., and Birchmeier, W. (1994). The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase Met requires intracellular action of Ras. J. Biol. Chem. 269, 21936-21939.
Hartwig, J. H., Bokoch, G. M., Carpenter, C. L., Janmey, P. A., Taylor, L. A., Toker, A., and Stossel, T. P. (1995). Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 82, 643-53.
Harvey, J. J. (1964). An Unidentified Virus which causes the Rapid Production of Tumours in Mice. Nature 204, 1104-1105.
Hata, Y., Kaibuchi, K., Kawamura, S., Hiroyoshi, M., Shirataki, H., and Takai, Y.(1991). Enhancement of the actions of smg p21 GDP/GTP exchange protein by the protein kinase A-catalysed phosphorylation of smg p21. J. biol. Chem. 266, 6571-6577.
Hata, Y., Kikuchi, A., Sasaki, T., Schaber, M. D., Gibbs, J. B., and Takai, Y. (1990). Inhibition of the ras p21 GTPase-activating protein-stimulated GTPase activity of c-Ha- ras p21 by smg p21 having the same putative effector domain as ras p21s. J. Biol. Chem. 265,7104-7107.
Hawkins, P. T., Eguinoa, A., Qiu, R. G., Stokoe, D., Cooke, F. T., Walters, R., Wennstrom, S., Claesson-Welsh, L., Evans, T., Symons, M., and Stephens, L. (1995). PDGF stimulates an increase in GTP-rac via activation of phosphoinositide 3-kinase. Current Biology 5, 393-403.
He, B., Chen, P., Chen, S. Y., Vancura, K. L., Michaelis, S., and Powers, S. (1991). RAM2, an essential gene of yeast, and RAMI encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc-Natl-Acad-Sci- U-S-A 88, 11373-7.
Herrmann, C., Martin, G. A., and Wittinghofer, A. (1995). Quantitative analysis of the complex between p21(ras) and the Ras-binding domain of the human Raf-1 protein kinase. J. Biol. Chem. 270, 2901-2905.
Herskowitz, I. (1995). MAP kinase pathways in yeast: for mating and more. Cell 80, 187-97.
Hiles, I. D., Otsu, M., Volinia, S., Fry, M. J., Gout, I., Dhand, R., Panayotou, G., Ruiz- Larrea, F., Thompson, A., Totty, N. F., and Waterfield, M. D. (1992).

156
Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell 70, 419-429.
Hill, C. S., Wynne, J., and Treisman, R. (1995). The Rho family GTPases RhoA, Racl, and CDC42Hs regulate transcriptional activation by SRF. Cell 57, 1159-1170.
Hill, M., and Hillova, J. (1971). Recombinational events between exogenous mouse DNA and newly synthesized DNA strands of chicken cells in culture. Nat-New-Biol 257,261-5.
Hinds, P. W., Dowdy, S. F., Ng Eaton, E., Arnold, A., and Weinberg, R. A. (1994). Function of a human cyclin gene as an oncogene. Proc. Natl. Acad. Sci. USA 97, 709- 713.
Hirai, H., Kobayashi, Y., Mano, H., Hagiwara, K., Maru, Y., Omine, M., Mizoguchi, H., Nishida, J., and Takayu, F. (1987). A point mutation at codon 13 of the N-ras oncogene in myelodysplastic syndrome. Nature 327,430-432.
Hofer, F., Fields, S., Schneider, C., and Martin, G. S. (1994).Activated ras interacts with the ral guanine-nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 97, 11089-11093.
Horiuchi, H., Kaibuchi, K., Kawamura, M., Matsuura, Y., Suzuki, N., Kuroda, Y., Kataoka, T., and Takai, Y. (1992). The posttranslational processing of ras p21 is critical for its stimulation of yeast adenylate cyclase. Molecular & Cellular Biology 72, 4515- 20.
Hoshino, M., Kawakita, M., and Hattori, S. (1988). Characterization of a factor that stimulates hydrolysis of GTP bound to ras gene product p21 (GTPase-activating protein) and correlation of its activity to cell density. Mol-Cell-Biol 8, 4169-73 issn: 0270-7306.
Howe, L. R., Leevers, S. J., Gomez, N., Nakielny, S., Cohen, P., and Marshall, C. J.(1992). Activation of the MAP kinase pathway by the protein kinase raf. Cell 77, 335- 342.
Hu, C. D., Kariya, K., Tamada, M., Akasaka, K., Shirouzu, M., Yokoyama, S., and Kataoka, T. (1995). Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J-Biol-Chem 270, 30274-7.
Hu, P., Mondino, A., Skolnik, E. Y., and Schlessinger, J. (1993). Cloning of a novel ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol. Cell. Biol. 13, 7677-7688.
Hu, Q., Klippel, A., Muslin, A. J., Fantl, W. J., and Williams, L. T. (1995). Ras- dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 268, 100-102.
Huang, Y., Saez, R., Chao, L., Santos, E., Aaronson, S. A., and Chan, A. L. (1995). A novel insertional mutation in the TC21 gene activates its transforming activity in a human leiomyosarcoma cell line. Oncogene. 77, 1255-1260.
Hughes, D. A., Fukui, Y., and Yamamoto, M. (1990). Homologous activators of ras in fission and budding yeast. Nature 344, 355-357.
Imai, Y., Davey, J., Kawagishi Kobayashi, M., and Yamamoto, M. (1997). Genes encoding farnesyl cysteine carboxyl methyltransferase in Schizosaccharomyces pombe and Xenopus laevis. Mol-Cell-Biol 77, 1543-51.

157
Inukai, K., Anai, M., Van Breda, E., Hosaka, T., Katagiri, H., Funaki, M., Fukushima, Y., Ogihara, T., Yazaki, Y., Kikuchi, Oka, Y., and Asano, T. (1996). A novel 55-kDa regulatory subunit for phosphatidylinositol 3-kinase structurally similar to p55PIK Is generated by alternative splicing of the p85alpha gene. J-Biol-Chem 277, 531%20.
Inukai, K., Funaki, M., Ogihara, T., Oka, Y., and Asano, T. (1997). p85alpha gene generates three isoforms of regulatory subunit forphosphatidylinositol 3-kinase (PI 3- Kinase), p50alpha, p55alpha, and p85alpha, with different PI 3-kinase activity elevating responses to insulin. Journal of Biological Chemistry. 272(12), 7873-82,.
Irani, K., Xia, Y., Zweier, J. L., Sollott, S. J., Der, C. J., Fearon, E. R., Sundaresan, M., Finkel, T., and Goldschmidt Clermont, P. J. (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. SCIENCE. Science. 275, 1649-16^52.
Jackson, T. R., Blader, I. J., Hammonds-Odie, L. P., Burga, C. R., Cooke, F., Hawkins, P. T., Wolf, A. G., Heldman, K. A., and Theibert, A. B. (1996). Initiation and maintenance of NGF stimulated neurite outgrowth requires activation of a phosphoinositide 3-kinase. J. Cell Sci. 109, 289-300.
James, G., Goldstein, J. L., and Brown, M. S. (1996). Resistance of K-RasBV12 proteins to farnesyltransferase inhibitors in Rati cells. Proc-Natl-Acad-Sci-U-S-A 93, 4454-8.
James, G. L., Goldstein, J. L., and Brown, M. S. (1995). Polylysine and CVIM sequences of K-RasB dictate specificity of prénylation and confer resistance to benzodiazepine peptidomimetic in vitro. J-Biol-Chem 270, 6221-6.
James, G. L., Goldstein, J. L., Brown, M. S., Rawson, T. E., Somers, T. C., McDowell, R. S., Crowley, C. W., Lucas, B. K., Levinson, A. D., and Marsters, J. C., Jr. (1993). Benzodiazepine peptidomimetics: potent inhibitors of Ras famesylation in animal cells [see comments]. Science 260, 1937-42.
James, G. L., Goldstein, J. L., Pathak, R. K., Anderson, R. G., and Brown, M. S. (1994). PxF, a prenylated protein of peroxisomes. J-Biol-Chem 269, 14182-90.
Jansen, B., Schlagbauer Wadi, H., Eichler, H. G., Wolff, K., van Elsas, A., Schrier, P. I., and Pehamberger, H. (1997). Activated N-ras contributes to the chemoresistance of human melanoma in severe combined immunodeficiency (SCID) mice by blocking apoptosis. Cancer-Res 57, 362-5.
Jelinek, T., Dent, P., Sturgill, T. W., and Weber, M. J. (1996). Ras-induced activation of Raf-1 is dependent on tyrosine phosphorylation. Mol-Cell-Biol 16, 1027-34.
Jiang, H., Luo, J. Q., Urano, T., Frankel, P., Lu, Z. M., Foster, D. A., and Feig, L. A.(1995). Involvement of Ral GTPase in v-src induced phospholipase-D activation. Nature 378,409-412.
Johnson, M. R., Look, A. T., DeClue, J. E., Valentine, M. B., and Lowy, D. R. (1993). Inactivation of the NFl gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP.Ras. Proc-Natl-Acad-Sci-U-S-A 90, 5539-43.
Jones, P. F., Jakubowicz, T., Pitossi, F. J., Maurer, F., and Hemmings, B. A. (1991). Molecular cloning and identification of a serine/threonine protein kinase of the second- messenger subfamily. Proc. Natl. Acad. Sci. USA 88, 4171-4175.

158
Joneson, T., McDonough, M., Bar-Sagi, D., and Van Aelst, L. (1996). RAC regulation of actin polymerization and proliferation by a pathway distinct from Jun kinase. Science 274, 1374-6.
Joneson, T., White, M. A., Wigler, M. H., and Bar-Sagi, D. (1996). Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science 277, 810-812.
Jullien-Flores, V., Dorseuil, O., Romero, F., Letoumeur, F., Saragosti, S., Berger, R., Tavitian, A., Gacon, G., and Camonis, J. H. (1995). Bridging ral GTPase to rho pathways: rlip76, a ral effector with cdc42/rac GTPase-activating protein activity. J. Biol. Chem. 270, 22473-22477.
Kauffman-Zeh, A., Rodriguez-Viciana, P., Ulrich, E., Gilbert, C., Coffer, P., Downward, J., and Evan, G. (1997). Suppression of c-myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature 3S5, 544-548.
Kazanietz, M. G., Lewin, N. E., Bruns, J. D., and Blumberg, P. M. (1995). Characterization of the cysteine rich region of the Caenorhabditis elegans protein Unc- 13 as a high affinity phorbol ester receptor. Analysis of ligand binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J. Biol. Chem. 270, 10777-10783.
Keith, C. T., and Schreiber, S. L. (1995). PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 270, 50-1.
Khosravi-Far, R., Solski, P. A., Clark, G. J., Kinch, M. S., and Der, C. J. (1995). Activation of Racl, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 75, 6443-6453.
Khosravi-Far, R., White, M. A., Westwick, J. K., Solski, P. A., Chrzanowska- Wodnicka, M., Van Aelst, L., Wigler, M. H., and Der, C. J. (1996). Oncogenic Ras activation of Raf/MAP kinase independent pathways is sufficient to cause tumorigenic transformation. Mol. Cell. Biol. 16, 3923-3933.
Khwaja, A., Rodriguez-Viciana, P., Wennstrom, S., Warne, P. H., and Downward, J. (1997). Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783-2793.
Kikuchi, A., Demo, S. D., Ye, Z. H., Chen, Y. W., and Williams, L. T. (1994). Ral-gds family members interact with the effector loop of ras p21. Mol. Cell. Biol. 14, 7483- 7491.
Kimura, K., Hattori, S., Kabuyama, Y., Shizawa, Y., Takayanagi, J., Nakamura, S., Toki, S., Matsuda, Y., Onodera, K., and Fukui, Y. (1994). Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 269, 18961-18967.
Kimura, K., Ito, M., Amano, M., Chihara, K., Fukata, Y., Nakafuku, M., Yamamori, B., Feng, J., Nakano, T., Okawa, K., Iwamatsu, A., and Kaibuchi, K. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) [see comments]. Science 273, 245-8.
Kirsten, W. W., and Mayer, L. A. (1967). Morphologic Responses to a Murine Erythroblastosis Virus. J.Natl. Cancer Inst. 39, 311-335.
Klarlund, J. K., Cherniack, A. D., and Czech, M. P. (1995). Divergent mechanisms for homologous desensitization of p21ras by insulin and growth factors. J. Biol. Chem. 270, 23421-23428.

159
Klarlund, J. K., Guilherme, A., Holik, J. J., Virbasius, J. V., Chawla, A., and Czech, M. P. (1997). Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains [see comments]. Science 275, 1927- 30.
Klinghofer, R. A., Duckworth, B., Valius, M., Cantley, L., and Kazlauskas, A. (1996). PDGF-dependent activation of PI3 kinase is regulated by receptor binding of SH2 domain-containing proteins which influence Ras activity. Mol. Cell. Biol. 16, 5905- 5914.
Klippel, A., Kavanaugh, W. M., Pot, D., and Williams, L. T. (1997). A specific product of phosphatidylinositol 3-kinase directly activates the protein-kinase akt through its pleckstrin homology domain. Mol. Cell. Biol. 17,338-344.
Klippel, A., Reinhard, C., Kavanaugh, W. M., Apell, G., Escobedo, M. A., and Williams, L. T. (1996). Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal transducing kinase pathways. Mol. Cell. Biol. 16, 4117-4127.
Kobe, B., and Deisenhofer, J. (1994). The leucine-rich repeat: a versatile binding motif. Trends-Biochem-Sci 19, 415-21.
Kodaki, T., Woscholski, R., Hallberg, B., Rodriguez-Viciana, P., Downward, J., and Parker, P. J. (1994). The activation of phosphatidylinositol 3-kinase by Ras. Current Biology 4, 798-806.
Kohl, N. E., Mosser, S. D., deSolms, S. J., Giuliani, E. A., Pompliano, D. L., Graham, S. L., Smith, R, L., Scolnick, E. M., Oliff, A., and Gibbs, J. B. (1993). Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor [see comments]. Science 260, 1934-7.
Kohl, N. E., Omer, C. A., Conner, M. W., Anthony, N. J., Davide, J. P., deSolms, S. J., Giuliani, E. A., Gomez, R. P., Graham, S. L., Hamilton, K., and et al. (1995). Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice [see comments]. Nat-Med 1, 792-7.
Kohl, N. E., Wilson, F. R., Mosser, S. D., Giuliani, E., deSolms, S. J., Conner, M. W., Anthony, N. J., Holtz, W. J., Gomez, R. P., Lee, T. J., and et al. (1994). Protein farnesyltransferase inhibitors block the growth of ras-dependent tumors in nude mice. Proc-Natl-Acad-Sci-U-S-A 91, 9141-5.
Kohn, A. D., Kovacina, K. S., and Roth, R. A. (1995). Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J. 14, 4288-4295.
Kolanus, W., Nagel, W., Schiller, B., Zeitlmann, L., Godar, S., Stockinger, H., and Seed, B. (1996). Alpha L beta 2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell 86, 233-42.
Kolch, W., Heidecker, G., Lloyd, P., and Rapp, U. R. (1991). Raf-1 protein kinase is required for growth of induced NIH 3T3 cells. Nature 349, 426-428.
Konishi, H., Matsuzaki, H., Tanaka, M., Ono, Y., Tokunaga, C., Kuroda, S., and Kikkawa, U. (1996). Activation of RAC-protein k in ase by h eat sh o ck and hyperosm olarity stress through a pathway independent of phosphatidylinositol 3- kinase. Proc. Natl. Acad. Sci. USA 93,7639-7643.

160
Korn, L. J., Siebel, C. W., McCormick, F., and Roth, R. A. (1987). Ras p21 as a potential mediator of insulin action in Xenopus oocytes. Science 236, 840-843.
Kornfeld, K. (1997). Vulval development in Caenorhabditis elegans. Trends-Genet 13, 55-61.
Kotani, K., Yonezawa, K., Hara, K., Ueda, H., Kitamura, Y., Sakaue, H., Ando, A., Chavaneau, A., Calas, B., Grigorescu, P., Nishiyama, M., Waterfield, M. D., and Kasuga, M. (1994). Involvement of phosphoinositide 3-kinase in insulin or IGF-1 induced membrane ruffling. EMBO J. 13, 2313-2321.
Kozma, R., Ahmed, S., Best, A., and Lim, L. (1996). The GTPase-activating protein n- chimaerin cooperates with Racl and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol Cell Biol 16, 5069-80.
Kozma, R., Sarner, S., Ahmed, S., and Lim, L. (1997). Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, R acl, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Molecular and Cellular Biology 17, 1201-1211.
Krengel, U., Schlichting, L., Scherer, A., Schumann, R., Freeh, M., John, J., Kabsch, W., Pai, E. F., and Wittinghofer, A. (1990). Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 62, 539-548.
Krieger Brauer, H. I., and Kather, H. (1995). The stimulus-sensitive H202-generating system present in human fat-cell plasma membranes is multireceptor-linked and under antagonistic control by hormones and cytokines. Biochem J 307, 543-8.
Krontiris, T. G., and Cooper, G. M. (1981). Transforming activity of human tumor DNAs. Proc. Natl. Acad. Sci. USA 78, 1181-1184.
Kulik, G., Klippel, A., and Weber, M. J. (1997). Anti-apoptotic signalling by the IGF-I receptor, PI 3-kinase and Akt. Mol. Cell. Biol. 17, 1595-1606.
Kundra, V., Escobedo, J. A., Kazlauskas, A., Kim, H. K., Rhee, S. G., Williams, L. T., and Zetter, B. R. (1994). Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature 367,474-6.
Kung, H. F., Smith, M. R., Bekesi, E., Manne, V., and Stacey, D. W. (1986). Reversal of transformed phenotype by monoclonal antibodies against Ha-ras p21 proteins. Exp Cell Res 762,363-71.
Kuriyama, M., Harada, N., Kuroda, S., Yamamoto, T., Nakafuku, M., Iwamatsu, A., Yamamoto, D., Prasad, R., Croce, C., Canaani, E., and Kaibuchi, K. (1996). Identification of AF-6 and canoe as putative targets for Ras. J-Biol-Chem 277, 607-10.
Kuroda, Y., Suzuki, N., and Kataoka, T. (1993). The effect of posttranslational modifications on the interaction of Ras2 with adenylyl cyclase. Science 259, 683-6.
Kwak, J. Y., Lopez, I., Uhlinger, D. J., Ryu, S. H., and Lambeth, J. D. (1995). RhoA and a cytosolic 50-kDa factor reconstitute GTP gamma S-dependent phospholipase D activity in human neutrophil subcellular fractions. Journal of Biological Chemistry 270, 27093-8.
Kyriakis, J. M., App, H., Zhang, X.-F., Banerjee, P., Brautigan, D. L., Rapp, U. R., and Avruch, J. (1992). Raf-1 activates MAP kinase-kinase. Nature 358,417-421.

161
Lacal, J. C., and Aaronson, S. A. (1986). Monoclonal antibody Y 13-259 recognizes an epitope of the p21 ras molecule not directly involved in the GTP-binding activity of the protein. Mol Cell Biol 6, 1002-9.
Lamarche, N., Tapon, N., Stowers, L., Burbelo, P. D., Aspenstrom, P., Bridges, T., Chant, J., and Hall, A. (1996). Rac and Cdc42 induce actin polymerization and 01 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell 57,519-529.
Land, H., Parada, L. F., and Weinberg, R. A. (1983). Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304, 596-602.
Lane, H. A., Fernandez, A., Lamb, N. J. C., and Thomas, G. (1993). p70S6k function is essential for G1 progression. Nature 363, 170-172.
Largaespada, D. A., Brannan, C. I., Jenkins, N. A., and Copeland, N. G. (1996). Nfl deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat-Genet 12, 137-43.
Lebowitz, P. F., Davide, J. P., and Prendergast, G. C. (1995). Evidence that farnesyltransferase inhibitors suppress Ras transformation by interfering with Rho activity. Mol-Cell-Biol 15, 6613-22.
Leevers, S. J., and Marshall, C. J. (1992). Activation of extracellular signal-regulated kinase ERK2 by p21ras oncoprotein. EMBO J. 11, 569-574.
Leevers, S. J., Paterson, H. F., and C.J., M. (1994). Requirement for ras in raf activation is overcome by targeting raf to the plasma-membrane. Nature 369,411-414.
Leiser, M., Efrat, S., and Fleischer, N. (1995). Evidence that Rapl carboxylmethylation is involved in regulated insulin secretion. Endocrinology 136, 2521-30.
Lemoine, N. R., Mayall, E. S., Wyllie, F. W., Farr, C. J., Hughes, D., Padua, R. A., Thurston, V., Williams, E. D., and Wynford-Thomas, D. (1988). Activated ras oncogenes in human thyroid cancers. Cancer Res. 48 ,4459-4463.
Leon, J., Kamino, H., Steinberg, J. J., and Pellicer, A. (1988). H-ra^ activation in benign and self-regressing skin tumors (keratoacanthomas) in both humans and an animal model system. Mol. Cell. Biol. 8, 786-793.
Lerner, E. C., Qian, Y., Hamilton, A. D., and Sebti, S. M. (1995). Disruption of oncogenic K-Ras4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J-Biol-Chem 270, 26770-3.
Lev, S., Moreno, H., Martinez, R., Canoll, P., Peles, E., Musacchio, J. M., Plowman, G. D., Rudy, B., and Schlessinger, J. (1995). Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions [see comments]. Nature 376, 737-45.
Lim, L., Manser, E., Leung, T., and Hall, C. (1996). Regulation of phosphorylation pathways by p21 GTPases. The p21 Ras-related Rho subfamily and its role in phosphorylation signalling pathways. [Review] [123 refs]. European Journal of Biochemistry 242, 171-85.
Ling, L. E., Druker, B. J., Cantley, L. C., and Roberts, T. M. (1992). Transformation- defective mutants of polyomavirus middle T antigen associate with phosphatidylinositol

162
3-kinase (PI 3-kinase) but are unable to maintain wild-type levels of PI 3-kinase products in intact cells. J-Virol 66, 1702-8.
Lisanti, M. P., Scherer, P. E., Vidugiriene, J., Tang, Z., Hermanowski Vosatka, A., Tu, Y. H., Cook, R. F., and Sargiacomo, M. (1994). Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J-Cell-Biol 126, 111-26.
Liu, L., Dudler, T., and Gelb, M. H. (1996). Purification of a protein palmitoyltransferase that acts on H-Ras protein and on a C-terminal N-Ras peptide. J- Biol-Chem 277, 23269-76.
Liu, R., Hjelle, B., Morgan, R., Hecht, P., and Bishop, J. M. (1987). Mutations of the Kirsten-mj proto-oncogene in human preleukemia. Nature 330, 186-188.
Lloyd, A. C., Obermuller, P., Staddon, S., Barth, C. P., McMahon, M., and Land, H. (1997). Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes-Dev 11, 663-77.
Lloyd, A. C., Paterson, H. P., Morris, J. D., Hall, A., and Marshall, C. J. (1989). p21H- ras-induced morphological transformation and increases in c-myc expression are independent of functional protein kinase C. EMBO J 8, 1099-1104.
Lo, Y. Y. C., and Cruz, T. P. (1995). Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J BIOL CHEM. Journal of Biological Chemistry 270, 11727-11730.
Lopez Barahona, M., Bustelo, X. R., and Barbacid, M. (1996). The TC21 oncoprotein interacts with the Ral guanosine nucleotide dissociation factor. Oncogene. Oncogene. 72, 463-470.
Lovec, H., Sewing, A., Lucibello, P. C., Müller, R., and Moroy, T. (1994). Oncogenic activity of cyclin Dl revealed through cooperation with Ha-ras: a link between cell cycle control and malignant transformation. Oncogene 9, 323-326.
Lowe, D. G., Capon, D. J., Delwart, E., Sakaguchi, A. Y., Naylor, S. L., and Goeddel,D. V. (1987). Structure of the human and murine R-ras genes, novel genes closely related to ras proto-oncogenes. Cell 48, 137-146.
Lowy, D. R., and Willumsen, B. M. (1993). Function and Regulation of RAS. Annu. Rev. Biochem. 62, 851-891.
Lu, X., Wu, X., Plemenitas, A., Yu, H., Sawai, E. T., Abo, A., and Peterlin, B. M.(1996). CDC42 and Racl are implicated in the activation of the Nef-associated kinase and replication of HIV-1. Current Biology 6, 1677-84.
Luo, Z., Tzivion, G., Belshaw, P. J., Va was, D., Marshall, M., and Avruch, J. (1996). Oligomerization activates c-Raf-1 through a Ras-dependent mechanism [see comments]. Nature 383, 181-5.
Lyons, J., Jansenn, J. W. G., Bartram, C., Layton, M., and Mufti, G. J. (1988). Mutation of Ki-mj oncogenes in myelodysplastic syndromes. Blood 71, 1707-1712.
Macara, I. G., Marinetti, G. V., and Balduzzi, P. C. (1984). Transforming protein of avian sarcoma virus UR2 is associated with phosphatidylinositol kinase activity: possible role in tumorigenesis. Proc-Natl-Acad-Sci-U-S-A 81, 2728-32.

163
MacDougall, L. K., Domin, J., and Waterfield, M. D. (1995). A family of phosphoinositide 3-kinases in Drosophila identifies a new mediator of signal transduction. Current Biology 5, 1404-1415.
Maekawa, M., Li, S., Iwamatsu, A., Morishita, T., Yokota, K., Imai, Y., Kohsaka, S., Nakamura, S., and Hattori, S. (1994). A novel mammalian Ras GTPase-activating protein which has phospholipid-binding and Btk homology regions. Molecular & Cellular Biology 14, 6879-85.
Majerus, P. W. (1996). Inositols do it all. Genes-Dev 10, 1051-3.
Malcolm, K. C., Elliott, C. M., and Exton, J. H. (1996). Evidence for Rho-mediated agonist stimulation of phospholipase D in rati fibroblasts. Effects of Clostridium botulinum C3 exoenzyme. Journal of Biological Chemistry 271, 13135-9.
Malcolm, K. C., Ross, A. H., Qiu, R. G., Symons, M., and Exton, J. H. (1994). Activation of rat liver phospholipase D by the small GTP-binding protein RhoA. Journal of Biological Chemistry 269, 25951-4.
Manser, E., Huang, H. Y., Loo, T. H., Chen, X. Q., Dong, J. M., Leung, T., and Lim, L.(1997). Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Molecular & Cellular Biology 77, 1129-43.
Marais, R., Light, Y., Paterson, H. P., and Marshall, C. J. (1995). Ras recruits Raf-1 to the plasma-membrane for activation by tyrosine phosphorylation. EMBO J. 14, 3136- 3145.
Maridonneau Parini, I., and De Gunzburg, J. (1992). Association of rapl and rap2 proteins with the specific granules of human neutrophils. Translocation to the plasma membrane during cell activation. J Biol Chem 267, 6396-6402.
Marshall, C. J., Hall, A., and Weiss, R. A. (1982). A transforming gene present in human sarcoma cell lines. Nature 299, 171-173.
Marshall, M. S., Davis, L. J., Keys, R. D., Mosser, S. D., Hill, W. S., Scolnick, E. M., and Gibbs, J. B. (1991). Identification of amino acid residues required for Ras p21 function. Mol. Cell. Biol. 11, 3997-4004.
Marte, B. M., and Downward, J. (1997). PKB/Akt: connecting PI 3-kinase to cell survival and beyond. Trends Biochem. Sci. in press.
Marte, B. M., Rodriguez-Viciana, P., Wennstrom, S., Warne, P. H., and Downward, J.(1997). PI 3-kinase and PKB/Akt act as an effector pathway for R-Ras. Current Biology 7, 63-70.
Martegani, E., Vanoni, M., Zippel, R., Coccetti, P., Brambilla, R., Ferrari, C., Sturani,E., and Alberghina, L. (1992). Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a saccharomyces cerevisiae ras activator. EMBO J. 6,2151-2157.
Martin, G. A., Viskochil, D., Bollag, G., McCabe, P. C., Crosier, W., Haubruck, H., Conroy, L., Clark, R., O'Connell, P., Cawthon, R. M., Innis, M. A., and McCormick, F. (1990). The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63, 843-849.
Martin, G. A., Yatani, A., Clark, R., Conroy, L., Polakis, P., Brown, A. M., and McCormick, F. (1992). GAP domains responsible for ras p21-dependent inhibition of muscarinic atrial K-f- channel currents [see comments]. Science 255, 192-4.

164
Martin, G. S. (1970). Rous sarcoma virus: a function required for the maintenance of the transformed state. Nature 227, 1021-3.
Mattingly, R. R., and Macara, I. 0 . (1996). Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein beta gamma subunits. Nature 382, 268-72.
McCarthy, S. A., Samuels, M. L., Pritchard, C. A., Abraham, J. A., and McMahon, M.(1995). Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev. 9, 1953-1964.
McCormick, F. (1995). Ras signaling and NFl. Curr-Opin-Genet-Dev 5, 51-5.
McGlade, J., Brunkhorst, B., Anderson, D. M., G., Settleman, J., Dedhar, S., Rozakis- Adcock, M., Chen, L. B., and Pawson, T. (1993). The N-terminal region of GAP regulates cytoskeletal structure and cell adhesion. EMBO J. 72, 3073-3081.
Meacci, E., Tsai, S. C., Adamik, R., Moss, J., and Vaughan, M. (1997). Cytohesin-1, a cytosolic guanine nucleotide-exchange protein for ADP-ribosylation factor. Proc-Natl- Acad-Sci-U-S-A 94, 1745-8.
Medema, R. H., de Laat, W. L., Martin, G. A., McCormick, F., and Bos, J. L. (1992). GTPase activating protein SH2-SH3 domains induce gene expression in a ras-dependent fashion. Mol. Cell. Biol. 72, 3425-3430.
Meier, B., Radeke, H. H., Selle, S., Younes, M., Sies, H., Resch, K., and Habermehl, G. G. (1989). Human fibroblasts release reactive oxygen species in response to interleukin- 1 or tumour necrosis factor-alpha. Biochem J 263, 539-45.
Michaud, N. R., Fabian, J. R., Mathes, K. D., and Morrison, D. K. (1995). 14-3-3 is not essential for Raf-1 function: identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-independent manner. Mol-Cell-Biol 75, 3390-7.
Milburn, M. V., Tong, L., deVos, A. M., Brunger, A., Yamaizumi, Z., Nishimura, S., and Kim, S.-H. (1990). Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939- 945.
Miller, M. J., Prigent, S., Kupperman, E., Rioux, L., Park, S. H., Feramisco, J. R., White, M. A., Rutkowski, J. L., and Meinkoth, J. L. (1997). RalGDS functions in Ras- and cAMP-mediated growth stimulation. J-Biol-Chem 272, 5600-5.
Minato, T., Wang, J., Akasaka, K., Okada, T., Suzuki, N., and Kataoka, T. (1994). Quantitative analysis of mutually competitive binding of human Raf-1 and yeast adenylyl cyclase to Ras proteins. J-Biol-Chem 269, 20845-51.
Minden, A., Lin, A., Claret, F. X., Abo, A., and Karin, M. (1995). Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPses Rac and Cdc42Hs. CELL. Cell. 81, 1147-1157.
Mineo, C., James, G. L., Smart, E. J., and Anderson, R. G. (1996). Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J- Biol-Chem277, 11930-5.
Mizuno, T., Kaibuchi, K., Yamamoto, T., Kawamura, M., Sakodo, T., Fujioka, H., Matsuura, Y., and Takai, Y. (1991). A stimulatory GDP/GTP exchange protein for smg

165
p21 is active on the post-translationally processed from of c-Ki-ras p21 and rho A p21. Proc. Natl. Acad. Sci. USA 88, 6442-6446.
Molz, L., Chen, Y.-W., Hirano, M., and Williams, L. T. (1996). Cpk is a novel class of Drosophila Ptdlns 3-kinase containing a C2 domain. J. Biol. Chem. 271, 13892-13899.
Moriya, S., Kazlauskas, A., Akimoto, K., Hirai, S., Mizuno, K., Takenawa, T., Fukui, Y., Watanabe, Y., Ozaki, S., and Ohno, S. (1996). Platelet-derived growth factor activates protein kinase C epsilon through redundant and independent signaling pathways involving phospholipase C gamma or phosphatidylinositol 3-kinase. Proc- Natl-Acad-Sci-U-S-A 93, 151-5.
Mosch, H. U., Roberts, R. L., and Fink, G. R. (1996). Ras2 signals via the Cdc42/Ste20/mitogen-activated protein kinase module to induce filamentous growth in Saccharomyces cerevisiae. Proc-Natl-Acad-Sci-U-S- A 93, 5352-6.
Mulcahy, L. S., Smith, M. R., and Stacey, D. W. (1985). Requirement for ras protooncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 313, 241- 243.
Mumby, S. M. (1997). Reversible palmitoylation of signaling proteins. Curr-Opin-Cell- Biol 9, 148-54.
Muslin, A. J., Tanner, J. W., Allen, P. M., and Shaw, A. S. (1996). Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889- 97.
Myers, M. G., Backer, J. M., Sun, X. J., Shoelson, S., Hu, P., Schlessinger, J., Yoakim, M., Schaffhausen, B., and White, M. F. (1992). IRS-1 activates phosphatidylinositol 3'- kinase by associating with src homology 2 domains of p85. Proc. Natl. Acad. Sci. USA 89, 10350-10354.
Nagasu, T., Yoshimatsu, K., Rowell, C., Lewis, M. D., and Garcia, A. M. (1995). Inhibition of human tumor xenograft growth by treatment with the farnesyl transferase inhibitor B956. Cancer-Res 55, 5310-4.
Nakanishi, H., Brewer, K. M., and Exton, J. H. (1993). Activation of the Ç isozyme of protein kinase C by phosphatidylinositol (3,4,5) trisphosphate. J. Biol. Chem. 268, 13- lb.
Nassar, N., Horn, G., Herrmann, C., Block, C., Janknecht, R., and Wittinghofer, A.(1996). Ras/Rap effector specificity determined by charge reversal [see comments]. Nat-Struct-Biol 3, 723-9.
Nassar, N., Horn, G., Herrmann, C., Scherer, A., McCormick, F., and Wittinghofer, A. (1995). The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Rafl in complex with Rapl A and a GTP analogue. Nature 375, 554-60.
Needleman, S. W., Kraus, M. H., Srivastava, S. K., Levine, P. H., and Aaronson, S. A. (1986). High frequency of N-ras activation in acute myelogenous leukemia. Blood 67, 753-757.
Nehrbass, U., and Blobel, G. (1996). Role of the nuclear transport factor plO in nuclear import. Science 272, 120-2.
Newman, C. M., and Magee, A. I. (1993). Posttranslational processing of the ras superfamily of small GTP-binding proteins. Biochim-Biophys-Acta 1155, 79-96.

166
Nobes, C. D., and Hall, A. (1995). Rho, Rac and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell 81, 53-62.
Nobes, C. D., Hawkins, P., Stephens, L., and Hall, A. (1995). Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J. Cell Sci. 108, 225-233.
Noda, M. (1993). Structures and functions of the K rev-1 transformation suppressor gene and its relatives. Biochim. Biophys. Acta 1155, 97-109.
Noda, M., Ko, M., Ogura, A., Liu, D., Amano, T., Takano, T., and Ikawa, Y. (1985). Sarcoma viruses carrying ras oncogenes induce differentiation properties in a neuronal cell line. Nature 318, 73-75.
Nonaka, H., Tanaka, K., Hirano, H., Fujiwara, T., Kohno, H., Umikawa, M., Mino, A., and Takai, Y. (1995). A downstream target of RHOl small GTP-binding protein is PKCl, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. Embo Journal 14, 5931-8.
Novick, P., and Brennwald, P. (1993). Friends and family: the role of the Rab GTPases in vesicular traffic. Cell 75, 597-601.
Nur-E-Kamal, M. S. A., Sizeland, A., D'Abaco, G., and Maruta, H. (1992). Asparagine 26, glutamic acid 31, valine 45, and tyrosine 64 of Ras proteins are required for their oncogenicity. J. Biol. Chem. 267, 1415-1418.
Ohtsuka, T., Shimizu, K., Yamamori, B., Kuroda, S., and Takai, Y. (1996). Activation of brain B-Raf protein kinase by Rap IB small GTP-binding protein. J Biol Chem 271, 1258-61.
Okada, T., Masuda, T., Shinkai, M., Kariya, K., and Kataoka, T. (1996). Posttranslational modification of H-Ras is required for activation of, but not for association with, B-Raf. Journal of Biological Chemistry 271,4671-8.
Okazaki, M., Kishida, S., Hinoi, T., Hasegawa, T., Tamada, M., Kataoka, T., and Kikuchi, A. (1997). Synergistic activation of c-fos promoter activity by Raf and Ral GDP dissociation stimulator. Oncogene 14, 515-21.
Olson, M. F., Ashworth, A., and Hall, A. (1995). An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through Gl. Science 269, 1270-2.
Otsu, M., Hiles, I., Gout, I., Fry, M. J., Ruiz-Larrea, F., Panayotou, G., Thompson, A., Dhand, R., Hsuan, J., Totty, N., and Waterfield, M. D. (1991). Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell 65, 91-104.
Page, M. J., Hall, A., Rhodes, S., Skinner, R. H., Murphy, V., Sydenham, M., and Lowe, P. N. (1989). Expression and characterization of the Ha-ras p21 protein produced at high levels in the insect/baculovirus system. J. Biol. Chem. 264, 19147-19154.
Pages, F., Ragueneau, M., Rottapel, R., Truneh, A., Nunes, J., Imbert, J., and Olive, D.(1994). Binding of phosphatidylinositol 3-OH kinase to CD28 is required for T cell signalling. Nature 369, 327-329.
Pai, E. F., Krengel, U., Petsko, G. A., Goody, R. S., Kabsch, W., and Wittinghofer, A.(1990). Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35

167
 resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 9, 2351- 2359.
Palmer, R. H., Dekker, L. V., Woscholski, R., Le Good, J. A., Gigg, R., and Parker, P. J.(1995). Activation of PRKl by phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate. A comparison with protein kinase C isotypes. J-Biol-Chem 270, 22412-6.
Pang, L., Sawada, T., Decker, S. J., and Saltiel, A. R. (1995). Inhibition of MAP kinase kinase blocks the differentiation of PC12 cells induced by nerve growth factor. J. Biol. Chem. 270, 13585-13588.
Parada, L. F., Land, H., Weinberg, R. A., Wolf, D., and Rotter, V. (1984). Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312, 649-651.
Parada, L. P., Tabin, C. J., Shih, C., and Weinberg, R. A. (1982). Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 297, 474- 478.
Park, S. H., and Weinberg, R. A. (1995). A putative effector of ral has homology to rho/rac GTPase-activating proteins. Oncogene I I , 2349-2355.
Parker, P., Maurier, P., Delumeau, I., Duchesne, M., Faucher, D., Debussche, L., Dugue, A., Schweighoffer, P., and Tocque, B. (1996). A Ras-GTPase-activating protein SH3-domain-binding protein. Mol-Cell-Biol 16, 2561-9.
Parton, R. G. (1996). Caveolae and caveolins. Curr-Opin-Cell-Biol 8, 542-8.
Peppelenbosch, M. P., Qiu, R. G., de, V., Smits, A. M., Tertoolen, L. G., de, L. S., McCormick, P., Hall, A., Symons, M. H., and Bos, J. L. (1995). Rac mediates growth factor-induced arachidonic acid release. Cell 81, 849-56.
Perez Sala, D., Gilbert, B. A., Tan, E. W., and Rando, R. R. (1992). Prenylated protein methyltransferases do not distinguish between farnesylated and geranylgeranylated substrates. Biochem-J 284, 835-40.
Perona, R., Montaner, S., Saniger, L., Sanchez Perez, I., Bravo, R., and Lacal, J. C.(1997). Activation of the nuclear factor-kappa B by Rho, CDC42, and Rac-1 proteins. GENES DEV. Genes and Development 11, 463-475.
Perucho, M., Goldfarb, M., Shimizu, K., Lama, C., Pogh, J., and Wigler, M. (1981). Human-Tumor-Derived Cell Lines Contain Common and Different Transforming Genes. Cell 27,467-476.
Peto, R., Roe, P. J., Lee, P. N., Levy, L., and Clack, J. (1975). Cancer and ageing in mice and men. Br-J-Cancer 32 ,411-26.
Philips, M. R., Pillinger, M. H., Stand, R., Volker, C., Rosenfeld, M. G., Weissmann, G., and Stock, J. B. (1993). Carboxyl méthylation of Ras-related proteins during signal transduction in neutrophils. Science 259, 977-80.
Pillinger, M. H., Volker, C., Stock, J. B., Weissmann, G., and Philips, M. R. (1994). Characterization of a plasma membrane-associated prenylcysteine-directed alpha carboxyl methyltransferase in human neutrophils. J-Biol-Chem 269, 1486-92.

168
Pons, S., Asano, T., Glasheen, E., Miralpeix, M., Zhang, Y., Fisher, T. L., Myers, M. G., Jr., Sun, X. J., and White, M. F. (1995). The structure and function of p55PIK reveal a new regulatory subunit for phosphatidylinositol 3-kinase. Mol-Cell-Biol 75, 4453-65.
Porfiri, F., Evans, T., Chardin, P., and Hancock, J. F. (1994). Prénylation of Ras proteins is required for efficient hSosl-promoted guanine nucleotide exchange. J. Biol. Chem. 2(59, 22672-22677.
Porfiri, F., and McCormick, F. (1996). Regulation of epidermal growth factor receptor signaling by phosphorylation of the ras exchange factor hSOSl. J-Biol-Chem 277, 5871-7.
Prasad, K. V. S., Kapeller, R., Janssen, O., Repke, H., Duke-Cohan, J. S., Cantley, L.C., and Rudd, C. F. (1993). Phosphatidylinositol (PI) 3-kinase and PI 4-kinase bindingto the CD4-p56^^^ complex: the p56^^^ SH3 binds to PI 3-kinase but not PI 4-kinase. Mol. Cell. Biol. 75, 7708-7717.
Prasad, R., Gu, Y., Alder, H., Nakamura, T., Canaani, O., Saito, H., Huebner, K., Gale, R. P., Nowell, P. C., Kuriyama, K., and et al. (1993). Cloning of the ALL-1 fusion partner, the AF-6 gene, involved in acute myeloid leukemias with the t(6; l l ) chromosome translocation. Cancer-Res 53, 5624-8.
Prendergast, G. C., Davide, J. P., deSolms, S. J., Giuliani, F. A., Graham, S. L., Gibbs, J. B., Cliff, A., and Kohl, N. F. (1994). Farnesyltransferase inhibition causes morphological reversion of ras-transformed cells by a complex mechanism that involves regulation of the actin cytoskeleton. Mol-Cell-Biol 14, 4193-202.
Prendergast, G. C., Khosravi-Far, R., Solski, P. A., Kurzawa, H., Lebowitz, P. F., and Der, C. J. (1995). Critical role of Rho in cell transformation by oncogenic Ras. Oncogene 10, 2289-2296.
Pritchard, C. A., Samuels, M. L., Bosch, F., and McMahon, M. (1995). Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol-Cell-Biol 75, 6430-42.
Pulciani, S., Santos, F., Lauver, A. V., Long, L. K., Robbins, K. C., and Barbacid, M. (1982). Oncogenes in human tumor cell lines: Molecular cloning of a transforming gene from human bladder carcinoma cells. Proc. Natl. Acad. Sci. USA 70, 2845-2849.
Qiu, R. G., Chen, J., Kirn, D., McCormick, F., and Symons, M. (1995a). An essential role for rac in ras transformation. Nature 374,457-459.
Qiu, R. G., Chen, J., McCormick, F., and Symons, M. (1995b). A role for rho in ras transformation. Proc. Natl. Acad. Sci. USA 92, 11781-11785.
Quaife, C. J., Pinkert, C. A., Omitz, D. M., Palmiter, R. D., and Brinster, R. L. (1987). Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell 48, 1023-34.
Quilliam, L. A., Hisaka, M. M., Zhong, S., Lowry, A., Mosteller, R. D., Han, J., Drugan, J. K., Broek, D., Campbell, S. L., and Der, C. J. (1996). Involvement of the switch 2 domain of Ras in its interaction with guanine nucleotide exchange factors. J- Biol-Chem277, 11076-82.
Quinn, M. T., Parkos, C. A., Walker, L., Orkin, S., Dinauer, M. C., and Jesaitis, A. J. (1989). Association of a ras-related protein with cytochrome b of human neutrophils. Nature 342, 198-200.

169
Rak, J., Mitsuhashi, Y., Erdos, V., Huang, S. N., Filmus, J., and Kerbel, R. S. (1995). Massive programmed cell death in intestinal epithelial cells induced by three- dimensional growth conditions: suppression by mutant c-H-ras oncogene expression. J- Cell-Biol 131, 1587-98.
Rameh, L. E., Chen, C. S., and Cantley, L. C. (1995). Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine- phosphorylated proteins. Cell 83, 821-30.
Randazzo, P. A., and Kahn, R. A. (1994). GTP hydrolysis by ADP-ribosylation factor is dependent on both an ADP-ribosylation factor GTPase-activating protein and acid phospholipids [published erratum appears in J Biol Chem 1994 Jun 10;269(23):16519]. Journal of Biological Chemistry 269, 10758-63.
Rasheed, S., Norman, G. L., and Heidecker, G. (1983). Nucleotide Sequence of the Rasheed Rat Sarcoma Virus Oncogene: New Mutations. Science 221, 155-157.
Ravichandran, K. S., Lorenz, U., Shoelson, S. E., and Burakoff, S. J. (1995). Interaction of She with Grb2 regulates association of Grb2 with mSos. Mol. Cell. Biol. 15, 593-600.
Reddy, E. P., Lipman, D., Andersen, P. R., Tronick, S. R., and Aaronson, S. A. (1985). Nucleotide Sequence Analysis of the BALB/c Murine Sarcoma Virus Transforming Gene. J. Virol. 53, 984-987.
Reddy, E. P., Reynolds, R. K., Santos, E., and Barbacid, M. (1982). A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149-52.
Regnier, D. C., Kozak, C. A., Kingsley, D. M., Jenkins, N. A., Copeland, N. G., Langdon, W. Y., and Morse, H. C. d. (1989). Identification of two murine loci homologous to the v-cbl oncogene. J. Virol. 63, 3678-3682.
Reichman, C. T., Mayer, B. J., Keshav, S., and Hanafusa, H. (1992). The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell-Growth-Differ 3, 451-60.
Reif, K., Gout, I., Waterfield, M. D., and Cantrell, D. A. (1993). Divergent regulation ofphosphatidylinositol 3-kinase p85a and p85(3 isoforms upon T cell activation. J. Biol. Chem. 268, 10780-10788.
Reif, K., Nobes, C. D., Thomas, G., Hall, A., and Cantrell, D. A. (1996). Phosphatidylinositol 3-kinase signals activate a selective subset of Rac/Rho-dependent effector pathways. Curr. Biol. 6, 1445-1455.
Ren, X. D., Bokoch, G. M., Tray nor Kaplan, A., Jenkins, G. H., Anderson, R. A., and Schwartz, M. A. (1996). Physical association of the small GTPase Rho with a 68-kDa phosphatidylinositol 4-phosphate 5-kinase in Swiss 3T3 cells. Mol-Biol-Cell 7, 435-42.
Rey, I., Taylor-Harris, P., van Erp, H., and Hall, A. (1994). R-ras interacts with rasGAP, neurofibromin and c-raf but does not regulate cell growth or differentiation. Oncogene 9, 685-692.
Ridley, A., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389-399.

170
Ridley, A., Paterson, H. F., Johnston, C. L., Diekmann, D., and Hall, A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401-410.
Ridley, A. J. (1995). Microinjection of Rho and Rac into quiescent Swiss 3T3 cells. Methods Enzymol. 256B, 313-319.
Ridley, A. J. (1994). Membrane ruffling and signal transduction. Bioessays 16, 321-7.
Ridley, A. J., Comoglio, P. M., and Hall, A. (1995). Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol. Cell. Biol. 15, 1110-1122.
Rivero, L. O., Marcilla, A., Sameshima, J. H., and Robbins, K. C. (1995). Wiskott- Aldrich syndrome protein physically associates with Nek through Src homology 3 domains. Molecular & Cellular Biology 15, 5725-31.
Rodenhuis, S. (1992). ras and human tumors. Semin. Cancer Biol. 3, 241-47.
Rodenhuis, S., Slebos, R. J. C., Boot, A. J. M., Evers, S. G., Mooi, W. J., Wagenaar, S.S., Van Bodegom, P. C., and Bos, J. L. (1988). K-ras oncogene activation in adenocarcinoma of the lung: incidence and possible clinical significance. Cancer Res. 48, 5738-5741.
Rommel, C., Radziwill, G., Lovric, J., Noeldeke, J., Heinicke, T., Jones, D., Aitken, A., and Moelling, K. (1996). Activated Ras displaces 14-3-3 protein from the amino terminus of c-Raf-1. Oncogene 12, 609-19.
Rozakis-Adcock, M., van der Geer, P., Mbamalu, G., and Pawson, T. (1995). MAP kinase phosphorylation of mSosl promotes dissociation of mSosl-Shc and mSosl-EGF receptor complexes. Oncogene 11, 1417-1426.
Royal, I., and Park, M. (1995). Hepatocyte growth factor-induced scatter of MDCK cells requires phosphatidylinositol 3-kinase. J. Biol. Chem. 270, 27780-27787.
Rubinfeld, B., Munemitsu, S., Clark, R., Conroy, L., Watt, K., Crosier, W. J., McCormick, F., and Polakis, P. (1991). Molecular cloning of a GTPase activating protein specific for the Krev-1 protein p21(rapl). Cell 65, 1033-1042.
Rubio, I., Rodriguez-Viciana, P., Downward, J., and Wetzker, R. (1997). Ras Interaction with Phosphoinositide 3-kinase gamma. Biochemical Journal.
Ruley, H. E. (1983). Adenovirus early region lA enables viral and cellular transforming genes to transform primary cells in culture. Nature 304, 602-606.
Ruley, H. E. (1990). Transforming collaborations between ras and nuclear oncogenes. Cancer-Cells 2, 258-68.
Sahyoun, N., McDonald, O. B., Farrell, F., and Lapetina, E. G. (1991). Phosphorylation of a Ras-related GTP-binding protein. Rap-lb, by a neuronal Ca24-/calmodulin- dependent protein kinase, CaM kinase Gr. Proc Natl Acad Sci U S A 88, 2643-7.
Salim, K., Bottomley, M. J., Querfurth, E., Zvelebil, M. J., Gout, I., Scaife, R., Margolis, R. L., Gigg, R., Smith, C. I., Driscoll, P. C., Waterfield, M. D., and Panayotou, G. (1996). Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton's tyrosine kinase. EMBO-J 15, 6241-50.

171
Santos, E., Tronick, S. R., Aaronson, S. A., Pulciani, S., and Barbacid, M. (1982). T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALE- and Harvey-MSV transforming genes. Nature 298, 343-347.
Satoh, T., Nakamura, S., and Kaziro, Y. (1987). Induction of neurite formation in PC 12 cells by microinjection of proto-oncogenic Ha-ras protein preincubated with guanosine- 5'-0-(3-thiotriphosphate). Mol Cell Biol 7, 4553-6.
Sawasdikosol, S., Ravichandran, K. S., Lee, K. K., Chang, J. H., and Burakoff, S. J.(1995). Crk interacts with tyrosine-phosphorylated pi 16 upon T cell activation. J. Biol. Chem 270, 2893-2896.
Schaber, M. D., Garsky, V. M., Boylan, D., Hill, W. S., Scolnick, E. M., Marshall, M.S., Sigal, I. S., and Gibbs, J. B. (1989). Ras interaction with the GTPase-activating protein (GAP). Proteins: Structure, Function and Genetics 6, 306-315.
Schlessinger, J. (1993). How receptor tyrosine kinases activate Ras. Trends-Biochem- Sci 18, 273-5.
Schlichting, I., Almo, S. C., Rapp, G., Wilson, K., Petratos, K., Lentfer, A., Wittinghofer, A., Kabsch, W., Pai, E. P., Petsko, G. A., and Goody, R. S. (1990). Time- resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature 345, 309-315.
Schweighoffer, P., Barlat, I., Chevalier-Multon, M. C., and Tocque, B. (1992). Implication of GAP in Ras-dependent transactivation of a polyoma enhancer sequence. Science 256, 825-827.
Seeburg, P. H., Colby, W. W., Capon, D. J., Goeddel, D. V., and Levinson, A. D. (1984). Biological properties of human c-Ha-mj 1 genes mutated at codon 12. Nature 372,71-75.
Sells, M. A., Knaus, U. G., Bagrodia, S., Ambrose, D. M., Bokoch, G. M., and Chernoff, J. (1997). Human p21-activated kinase (Pakl) regulates actin organization in mammalian cells. Current Biology 7, 202-10.
Sepp Lorenzino, L., Ma, Z., Rands, E., Kohl, N. E., Gibbs, J. B., Oliff, A., and Rosen, N. (1995). A peptidomimetic inhibitor of farnesyhprotein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer-Res 55, 5302-9.
Serrano, M., Lee, H., Chin, L., Cordon Cardo, C., Beach, D., and DePinho, R. A.(1996). Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27-37.
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and pl6INK4a. Cell 88, 593-602.
Settleman, J., Albright, C. P., Poster, L. C., and Weinberg, R. A. (1992). Association between GTPase activators for Rho and Ras families. Nature 359, 153-4.
Settleman, J., Narasimhan, V., Poster, L. C., and Weinberg, R. A. (1992). Molecular cloning of cDNAs encoding the GAP-associated protein p i90: implications for a signalling pathway from ras to the nucleus. Cell 69, 539-549.
Sevetson, B. R., Kong, X., and Lawrence, J. C., Jr. (1993). Increasing cAMP attenuates activation of mitogen-activated protein kinase. Proc-Natl-Acad-Sci-U-S-A 90, 10305-9.

172
Shaul, P. W., Smart, E. J., robinson, L., German, Z., Yuhanna, I. S., Ying, Y., Anderson, R. G., and Michel, T. (1996). Acylation targets emdotheil nitric-oxide synthase to plasmalemmal caveolae. J-Biol-Chem 271, 6518 -̂22.
Shih, C., Padhy, L. C., Murray, M., and Weinberg, R. A. (1981). Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 29^9, 261- 264.
Shih, C., Shilo, B. Z., Goldfarb, M. P., Dannenberg, A., and Weinberg, R. A. (1979). Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc-Natl-Acad-Sci-U-S-A 76, 5714-8.
Shima, F., Yamawaki Kataoka, Y., Yanagihara, C., Tamada, M., Okada, T., Kariya, K., and Kataoka, T. (1997). Effect of association with adenylyl cyclase-associated protein on the interaction of yeast adenylyl cyclase with Ras protein. Mol-Cell-Biol 17, 1057- 64.
Shimizu, K., Goldfarb, M., Suard, Y., Perucho, M., Li, Y., Kamata, T., Feramisco, J., Stavnezer, E., Fogh, J., and Wigler, M. (1983a). Three human transforming genes are related to the viral ras oncogenes. Proc. Natl. Acad. Sci. USA 80, 2112-2116.
Shou, C., Farnsworth, C. L., Neel, B. G., and Feig, L. A. (1992). Molecular cloning of cDNAs encoding a guanine-nucleotide releasing factor for ras-p21. Nature 358, 351- 354.
Sigal, I. S., Gibbs, J. B., DAlonzo, J. S., and Scolnick, E. M. (1986b). Identification of effector residues and a neutralizing epitope of Ha-ra^-encoded p21. Proc. Natl. Acad. Sci. USA 83 ,4725-4729.
Sigal, I. S., Gibbs, J. B., DAlonzo, J. S., Temeles, G. L., Wolanski, B. S., Socher, S. H., and Scolnick, E. M. (1986a). Mutant raj-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc. Natl. Acad. Sci. USA 83, 952-956.
Simon, M. A., Bowtell, D. D. L., Dodson, G. S., Laverty, T. R., and Rubin, G. M.(1991). Rasl and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701-716.
Sinn, E., Muller, W., Pattengale, P., Tepler, I., Wallace, R., and Leder, P. (1987). Coexpression of MMTV/v-Ha-raj and MMTV/c-myc Genes in Transgenic Mice: Synergistic Action of Oncogenes In Vivo. Cell 49 ,465-475.
Sjolander, A., and Lapetina, E. (1992). Agonist-induced association of the p21ras GTPase-activating protein with phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 189, 1503-1508.
Sjolander, A., Yamamoto, K., Huber, B. E., and Lapetina, E. C. (1991). Association of p2 U^s phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 88, 7908-7912.
Skinner, R. H., Picardo, M., Gane, N. M., Cook, N. D., Morgan, L., Rowedder, J., and Lowe, P. N. (1994). Direct measurement of the binding of ras to neurofibromin using a scintillation proximity assay. Anal. Biochem. 1994, 259-265.
Skolnik, E. Y., Margolis, B., Mohammadi, M., Lowenstein, E., Fischer, R., Drepps, A., Ullrich, A., and Schlessinger, J. (1991). Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell 65, 83-90.

173
Slebos, R. J. C., Hruban, R. N., Dalesio, O., Mooi, W. J., Gérard, J., Offerhaus, G. J., and Rodenhuis, S. (1991). K-ras activation and smoking in adenocarcinoma of the human lung. J. Natl. Cancer Inst. 83, 1024-1027.
Smit, V. T. H. B. M., Boot, A. J. M., Smits, A. M. M., Fleuren, G. J., Comelisse, C. J., and Bos, J. L. (1988). K-ras codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 16, 7773-7782.
Smith, M. R., DeGudicibus, S. J., and Stacey, D. W. (1986). Requirement for c-ras proteins during viral oncogene transformation. Nature 320, 540-3.
Song, S. K., Li, S., Okamoto, T., Quilliam, L. A., Sargiacomo, M., and Lisanti, M. P.(1996). Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J-Biol-Chem 271, 9690-7.
Sosnowski, R. G., Feldman, S., and Feramisco, J. R. (1993). Interference with endogenous ras function inhibits cellular responses to wounding. Journal of Cell Biology 121, 113-9.
Spaargaren, M., and Bischoff, J. R. (1994). Identification of the guanine-nucleotide dissociation stimulator for ral as a putative effector molecule of r-ras, h-ras, k-ras, and rap. Proc. Natl. Acad. Sci. USA 91, 12609-12613.
Stenmark, H., Vitale, G., Ullrich, O., and Zerial, M. (1995). Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell 83, 423-32.
Stephens, L., Smrcka, A., Cooke, F. T., Jackson, T. R., Sternweis, P. C., and Hawkins, P. T. (1994). A novel phosphoinositide 3 kinase activity in myeloid-derived cells isactivated by G protein (3y subunits. Cell 77, 83-93.
Stephens, L. R., Eguinoa, A., Erdjument, B. H., Lui, M., Cooke, F., Coadwell, J., Smrcka, A. S., Thelen, M., Cadwallader, K., Tempst, P., and Hawkins, P. T. (1997). The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, pl01.C ell59, 105-14.
Stephens, L. R., Jackson, T. R., and Hawkins, P. T. (1993). Agonist-stimulated synthesis of phosphatidylinositol(3,4,5)-trisphosphate: a new intracellular signalling system. Biochim. Biophys. Acta 1179, 27-75.
Stokoe, D., MacDonald, S. G., Cadwallader, K., Symons, M., and Hancock, J. F. (1994). Activation of raf as a result of recruitment to the plasma-membrane. Science 264, 1463-1467.
Stone, J. C., Colleton, M., and Bottorff, D. (1993). Effector domain mutations dissociate p21ras effector function and GTPase activating protein interaction. Mol. Cell. Biol. 13, 7311-7320.
Stone, J. C., Vass, W. C., Willumsen, B. M., and Lowy, D. R. (1988). ^2\-ras Effector Domain Mutants Constructed by "Cassette" Mutagenesis. Mol. Cell. Biol. 8, 3565-3569.
Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoyanova, S., Vanhaesebroeck, B., Dhand, R., Numberg, B., Gierschik, P., Seedorf, K., Hsuan, J. J., Waterfield, M. D., and Wetzker, R. (1995). Cloning and characterization of a G protein activated human phosphoinositide-3 Idnase. Science 269, 690-693.

174
Suarez, H. G., Du Villard, J. A., Caillou, B., Schlumberger, M., Tubiana, M., Parmentier, C., and Monier, R. (1988). Detection of activated ras oncogenes in human thyroid carcinomas. Oncogene 2.
Sugimoto, Y., Whitman, M., Cantley, L. C., and Erikson, R. L. (1984). Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proc-Natl-Acad-Sci-U-S-A 81, 2117-21.
Sukumar, S., Notario, V., Martin-Zanca, D., and Barbacid, M. (1983). Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H- mj-1 locus by single point mutations. Nature 306, 658-61.
Sulciner, D. J., Irani, K., Yu, Z. X., Ferrans, V. J., Goldschmidt Clermont, P., and Finkel, T. (1996). r a d regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappa B activation. MOL CELL BIOL. Molecular and Cellular Biology 16, 7115-7121.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995). Requirement for generation of H202 for platelet-derived growth factor signal transduction. Science 270, 296-9.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Sulciner, D. J., Gutkind, J. S., Irani, K., Goldschmidt Clermont, P. J., and Finkel, T. (1996). Regulation of reactive-oxygen- species generation in fibroblasts by Racl. BIOCHEM J. Biochemical Journal 318, 379- 382.
Suzuki, N., Choe, H. R., Nishida, Y., Yamawaki Kataoka, Y., Ohnishi, S., Tamaoki, T., and Kataoka, T. (1990). Leucine-rich repeats and carboxyl terminus are required for interaction of yeast adenylate cyclase with RAS proteins. Proc-Natl-Acad-Sci-U-S-A 57,8711-5.
Sweet, R. W., Yokoyama, S., Kamata, T., Feramisco, J. R., Rosenberg, M., and Gross, M. (1984). The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 311, 273-5.
Symons, M., Derry, J. M., Karlak, B., Jiang, S., Lemahieu, V., Mccormick, F., Francke, U., and Abo, A. (1996). Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84, 723-34.
Tabin, C. J., Bradley, S. M., Bargmann, C. I., Weinberg, R. A., Papageorge, A. G., Dhar, R., Lowy, D. R., and Chang, E. H. (1982). Mechanism of activation of a human oncogene. Nature 300, 143-149.
Tanaka, S., Morishita, T., Hashimoto, Y., Hattori, S., Nakamura, S., Shibuya, M., Matuoka, K., Takenawa, T., Kurata, T., and Nagashima, K. (1994). C3g, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the src homology-3 domains of crk and grb2 ash proteins. Proc. Natl. Acad. Sci. USA 91, 3443-3447.
Taparowsky, E., Suard, Y., Fasano, O., Shimizu, K., Goldfarb, M., and Wigler, M. (1982). Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature 300, 762-765.
Tapon, N., and Hall, A. (1997). Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. [Review] [44 refs]. Current Opinion in Cell Biology 9, 86-92.
Tatchell, K., Chaleff, D. T., DeFeo Jones, D., and Scolnick, E. M. (1984). Requirement of either of a pair of ras-related genes of Saccharomyces cerevisiae for spore viability. Nature 309, 523-7.

175
Teramoto, H., Coso, O. A., Miyata, H., Igishi, T., Miki, T., and Gutkind, J. S. (1996). Signaling from the small GTP-binding proteins Racl and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. Journal of Biological Chemistry 277, 27225-8.
The, I., Hannigan, G. E., Cowley, G. S., Reginald, S., Zhong, Y., Gusella, J. F., Hariharan, I. K., and Bernards, A. (1997). Rescue of a Drosophila NFl mutant phenotype by protein kinase A. Science 27(5, 791-4.
Toker, A., Meyer, M., Reddy, K. K., Falck, J. R., Aneja, R., Aneja, S., Parra, A., Burns,D. J., Balias, L. M., and Cantley, L. C. (1994). Activation of protein kinase C family members by the novel phosphoinositides PI-3,4-P2 and PI-3,4,5-P3. J. Biol. Chem. 2(59, 32358-32367.
Tolias, K. F., Cantley, L. C., and Carpenter, C. L. (1995). Rho family GTPases bind to phosphoinositide kinases. Journal of Biological Chemistry 270, 17656-9.
Tong, L. A., A.M., d., Milburn, M. V., Jancarik, J., Noguchi, S., Nishimura, S., Miura, K., Ohtsuka, E., and Kim, S. H. (1989). Structural differences between a ras oncogene protein and the normal protein. Nature 337, 90-93.
Torti, M., Marti, K. B., Altschuler, D., Yamamoto, E. G., and Lapetina, E. (1992). Erythropoietin induces p21ras activation and pl20GAP tyrosine phosphorylation in human erythroleukemia cells. J. Biol. Chem. 267, 8293-8298.
Trahey, M., and McCormick, F. (1987). A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542-5.
Trahey, M., Wong, G., Halenbeck, R., Rubinfeld, B., Martin, G. A., Ladner, M., Long, C. M., Crosier, W. J., Watt, K., Koths, K., and et, a. 1. (1988). Molecular cloning of two types of GAP complementary DNA from human placenta. Science 242, 1697-700.
Traynor Kaplan, A. E., Harris, A. L., Thompson, B. L., Taylor, P., and Sklar, L. A. (1988). An inositol tetrakisphosphate-containing phospholipid in activated neutrophils. Nature 334, 353-6.
Tsai, M.-H., Roudebush, M., Dobrowolski, S., Yu, C.-L., Gibbs, J. B., and Stacey, D. W. (1991). Ras GTPase activating protein physically associates with mitogenically active phospholipids. Mol. Cell. Biol. I I , 2785-2793.
Tsai, M.-H., Yu, C.-L., Wei, F.-S., and Stacey, D. W. (1989). The effect of GTPase activating protein upon ras is inhibited by mitogenically responsive lipids. Science 243, 522-526.
Tsuchida, M., Ohtsubo, E., and Ryder, T. (1982). Nucleotide Sequence of the Oncogene Encoding the p21 Transforming Protein of Kirsten Murine Sarcoma Virus. Science 277, 937-939.
Tuveson, D. A., Carter, R. H., Soltoff, S. P., and Fearon, D. T. (1993). CD19 of B cells as a surrogate kinase insert region to bind phosphatidylinositol 3-kinase. Science 260, 986-989.
Ullrich, O., Horiuchi, H., Bucci, C., and Zerial, M. (1994). Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange [see comments]. Nature 368, 157-60.

176
Ulsh, L. S., and Shih, T. Y. (1984). Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol-Cell-Biol 4, 1647-52.
Urano, T., Emkey, R., and Feig, L. A. (1996). Ral-GTPases mediate a distinct downstream signaling pathway from ras that facilitates cellular-transformation. EMBO J. 75,810-816.
Valencia, A., Chardin, P., Wittinghofer, A., and Sander, C. (1991). The ras Protein Family: Evolutionary Tree and Role of Conserved Amino Acids. Biochemistry 30, 4637-4648.
Van Aelst, L., Barr, M., Marcus, S., Polverino, A., and Wigler, M. (1993). Complex formation between Ras and Raf and other protein kinases. Proc. Natl. Acad. Sci. USA 90, 6213-6217.
Van Aelst, L., White, M. A., and Wigler, M. H. (1994). Ras partners. Cold Spring Harb Symp Quant Biol 59, 181-6.
Van, A. L., Joneson, T., and Bar, S. D. (1996). Identification of a novel Racl- interacting protein involved in membrane ruffling. Embo Journal 15, 3778-86.
van der Geer, P., Henkemeyer, M., Jacks, T., and Pawson, T. (1997). Aberrant Ras regulation and reduced pi 90 tyrosine phosphorylation in cells lacking pi 20-Gap. Mol- Cell-Biol 17, 1840-7.
Van't Veer, L. J., Burgering, B. M. T., Versteeg, R., Boot, A. J. M., Ruiter, D. J., Osanto, S., Schrier, P. L., and Bos, J. L. (1989). N-ras mutations in human cutaneous melanoma correlated with sun exposure. Mol. Cell. Biol. 9, 3114-3116.
Vanhaesebroeck, B., Welham, M., Kotani, K., Stein, R., and Waterfield, M., D. (1997). p i 10 delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. 94, 4330-4335.
Velu, T. J., Vass, W. C., Lowy, D. R., and Tambourin, P. E. (1989). Harvey murine sarcoma virus: influences of coding and noncoding sequences on cell transformation in vitro and oncogenicity in vivo. J. Virol. 63, 1384-1392.
Verlaan-de Vries, M., Bogaard, M. E., Van den Elst, H., Van Boom, J. H., Van der Eb, A. J., and Bos, J. L. (1986). A dot-blot screening procedure for mutated ras oncogenes using sythetic oligonucleotides. Gene 50, 313-320.
Vincent, S., and Settleman, J. (1997). The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Molecular & Cellular Biology 77, 2247-56.
Virbasius, J. V., Guilherme, A., and Czech, M. P. (1996). Mouse pl70 is a novel phosphatidylinositol 3-kinase containing a c2 domain. J. Biol. Chem. 277, 13304- 13307.
Vlahos, C. J., Matter, W. P., Hui, K. Y., and Brown, R. F. (1994). A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl) -8-phenyl-4H-1 -benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241-5248.
Vlahos, C. J., Matter, W. P., Brown, R. P., Traynor, K. A., Heyworth, P. G., Prossnitz,E. R., Ye, R. D., Marder, P., Schelm, J. A., Rothfuss, K. J., and et, a. 1. (1995). Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. Journal of Immunology 154, 2413-22.

177
Vogel, L. B., and Fujita, D. J. (1993). The SH3 domain of is involved in binding to phosphatidylinositol 3'-kinase from T lymphocytes. Mol. Cell. Biol. 13, 7408-7417.
Vogel, U. S., Dixon, R. A., Schaber, M. D., Diehl, R. E., Marshall, M. S., Scolnick, E. M., Sigal, I. S., and Gibbs, J. B. (1988). Cloning of bovine GAP and its interaction with oncogenic ras p21. Nature 335, 90-93.
Vogelstein, B., Fearon, E. R., S.R., H., Kern, S. E., Preisinger, A. C., Leppert, M., Nakamura, Y., Whyte, R., Smits, A. M. M., and Bos, J. L. (1988). Genetic alterations during clolrectal tumor development. N. Engl. J. Med. 319, 525-532.
Vojtek, A. B., Hollenberg, S. M., and Cooper, J. A. (1993). Mammalian Ras interacts directly with the Serine/Threonine kinase Raf. Cell 74, 205-214.
Volker, C., Lane, P., Kwee, C., Johnson, M., and Stock, J. (1991). A single activity carboxyl methylates both farnesyl and geranylgeranyl cysteine residues. FEBS-Lett 295, 189-94.
Vossler, M. R., Yao, H., York, R. D., Pan, M. G., Rim, C. S., and Stork, P. J. S. (1997). cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap 1-dependent pathway. CELL. Cell. 89, 73-82.
Walter, M., Clark, S. G., and Levinson, A. D. (1986). The Oncogenic Activation of Human p21™-' by a Novel Mechanism Science 233,649-652.
Wang, L., Galehouse, D., Mellon, P., Duesberg, P., Mason, W. S., and Vogt, P. K. (1976). Mapping oligonucleotides of Rous sarcoma virus RNA that segregate with polymerase and group-specific antigen markers in recombinants. Proc-Natl-Acad-Sci- U-S-A 73, 3952-6.
Warne, P. H., Vicinia, P. R., and Downward, J. (1993). Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364, 352-355.
Watanabe, G., Saito, Y., Madaule, P., Ishizaki, T., Fujisawa, K., Morii, N., Mukai, H., Ono, Y., Kakizuka, A., and Narumiya, S. (1996). Protein kinase N (PKN) and PKN- related protein rhophilin as targets of small GTPase Rho. Science 271, 645-8.
Waters, S. B., Holt, K. H., Ross, S. E., Syu, L. J., Guan, K. L., Saltiel, A. R., Koretzky, G. A., and Pessin, J. E. (1995). Desensitization of Ras activation by a feedback disassociation of the S0S-Grb2 complex. J-Biol-Chem 270, 20883-6.
Waters, S. B., Yamauchi, K., and Pessin, J. E. (1995). Insulin-stimulated disassociation of the Sos-Grb2 complex. Mol. Cell. Biol. 15, 2791-2799.
Weinberg, R. A. (1981). Use of transfection to analyze genetic information and malignant transformation. Biochim-Biophys-Acta 651, 25-35.
Welsh, G. I., Wilson, C., and Proud, C. G. (1996). Glycogen Synthase Kinase 3. Trends Cell Biol. 6, 274-278.
Wennstrom, S., Hawkins, P., Cooke, F., Hara, K., Yonezawa, K., Kasuga, M., Jackson, T., Claesson-Welsh, L., and Stephens, L. (1994). Activation of phosphoinositide 3- kinase is required for PDGF-stimulated membrane ruffling. Current Biology 4, 385-393.
Wennstrom, S., Siegbahn, A., Yokote, K., Arvidsson, A.-K., Heldin, C.-H., Mori, S., and Claesson-Welsh, L. (1994). Membrane ruffling and chemotaxis transduced by the

178
PDGF receptor require the binding site for phosphatidylinositol 3'kinase. Oncogene 9, 651-660.
Westwick, J. K., Lambert, Q. T., Clark, G. J., Symons, M., Van, A. L., Pestell, R. G., and Der, C. J. (1997). Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Molecular & Cellular Biology 77, 1324-35.
White, M. A., Nicolette, C., Minden, A., Polverino, A., Vanaelst, L., Karin, M., and Wigler, M. H. (1995). Multiple ras functions can contribute to mammalian-cell transformation. Cell 80, 533-541.
White, M. A., Vale, T., Camonis, J. H., Schaefer, E., and Wigler, M. H. (1996). A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem. 277, 16439-16442.
Whitehead, I. P., Campbell, S., Rossman, K. L., and Der, C. J. (1997). Dbl family proteins. Biochimica et Biophysica Acta Reviews on Cancer 1332, Fl-f23.
Whitman, M., Downes, C. P., Keeler, M., Keller, T., and Cantley, L. (1988). Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol- 3-phosphate. Nature 332, 644-6.
Willumsen, B. M., Christensen, A., Hubbert, N. L., Papageorge, A. G., and Lowy, D. R. (1984). The p21 ras C-terminus is required for transformation and membrane association. Nature 310, 583-6.
Willumsen, B. M., Cox, A. D., Solski, P. A., Der, C. J., and Buss, J. E. (1996). Novel determinants of H-Ras plasma membrane localization and transformation. Oncogene 13, 1901-9.
Willumsen, B. M., Norris, K., Papageorge, A. G., Hubbert, N. L., and Lowy, D. R. (1984). Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO-J 3, 2581-5.
Willumsen, B. M., Papageorge, A. G., Kung, H. F., Bekesi, E., Robins, T., Johnsen, M., Vass, W. C., and Lowy, D. R. (1986). Mutational analysis of a ras catalytic domain. Mol Cell Biol 6, 2646-54.
Winter, E., Yamamoto, F., Almoguera, C., and Perucho, M. (1985). A method to detect and characterize point mutations in transcribed genes: amplification and overexpression of the mutant c-Ki-mj allele in human tumour cells. Proc. Natl. Acad. Sci. USA 82, 7575-7579.
Wittinghofer, A., and Pai, E. F. (1991). The structure of Ras protein: a model for a universal molecular switch. Trends Biochem. Sci. 16, 382-387.
Wolthuis, R. M., Bauer, B., van't Veer, L. J., de Vries Smits, A. M., Cool, R. H., Spaargaren, M., Wittinghofer, A., Burgering, B. M., and Bos, J. L. (1996). RalGDS-like factor (Rif) is a novel Ras and Rap 1 A-associating protein. Oncogene 13, 353-62.
Wu, J., Dent, P., Jelinek, T., Wolfman, A., Weber, M. J., and Sturgill, T. W. (1993). Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3',5'- monophosphate [see comments]. Science 262, 1065-9.
Xia, K., Mukhopadhyay, N. K., Inhorn, R. C., Barber, D. L., Rose, P. E., Lee, R. S., Narsimhan, R. P., D'Andrea, A. D., Griffin, J. D., and Roberts, T. M. (1996). The

179
cytokine-activated tyrosine kinase JAK2 activates Raf-1 in a p21 ras-dependent manner. Proc-Natl-Acad-Sci-U-S-A 93, 11681-6.
Xiao, G. H., Shoarinejad, F., Jin, P., Golemis, E. A., and Yeung, R. S. (1997). The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. Journal of Biological Chemistry 272, 6097- 100.
Xu, G., Lin, B., Tanaka, K., Dunn, D., Wood, D., Gesteland, R., White, R., Weiss, R., and Tamanoi, F. (1990). The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S.cerevisiae. Cell 63, 835-841.
Xu, N., Coso, O., Mahadevan, D., De Blasi, A., Goldsmith, P. K., Simonds, W. P., and Gutkind, J. S. (1996). The PH domain of Ras-GAP is sufficient for in vitro binding to beta gamma subunits of heterotrimeric G proteins. Cell-Mol-Neurobiol 16, 51-9.
Xu, N., McCormick, P., and Gutkind, J. S. (1994). The non-catalytic domain of rasGAP inhibits transformation induced by G protein coupled receptors. Oncogene 9, 597-601.
Yamamori, B., Kuroda, S., Shimizu, K., Pukui, K., Ohtsuka, T., and Takai, Y. (1995). Purification of a Ras-dependent mitogen-activated protein kinase kinase kinase from bovine brain cytosol and its identification as a complex of B-Raf and 14-3-3 proteins. J- Biol-Chem 270, 11723-6.
Yamamoto, T., Matsui, T., Nakafuku, M., Iwamatsu, A., and Kaibuchi, K. (1995). A novel GTPase-activating protein for R-Ras. J-Biol-Chem 270, 30557-61.
Yamanashi, Y., and Baltimore, D. (1997). Identification of the Abl- and rasGAP- associated 62 kDa protein as a docking protein, Dok. Cell 88, 205-11.
Yokoyama, N., Hayashi, N., Seki, T., Pante, N., Ohba, T., Nishii, K., Kuma, K., Hayashida, T., Miyata, T., Aebi, U., and et, a. 1. (1995). A giant nucleopore protein that binds Ran/TC4. Nature 376, 184-8.
Yoshida, Y., Kawata, M., Miura, Y., Musha, T., Sasaki, T., Kikuchi, A., and Takai, Y.(1992). Microinjection of smg/rapl/krev-1 p21 into swiss 3t3 cells induces dna synthesis and morphological changes. Mol Cell Biol 12, 3407-14.
Yu, C. L., Tsai, M. H., and Stacey, D. W. 0990). Serum stimulation of NIH 3T3 cells induces the production of lipids able to inhibit GTPase activating protein activity. Mol. Cell. Biol. 10, 6683-6689.
Yunis, J. J., Boot, A. J. M., Mayer, M. G., and Bos, J. L. (1989). Mechanisms of ras mutation in myelodysplastic syndrome. Oncogene 4, 609-614.
Yuspa, S. H., Kilkenny, A. E., Stanley, J., and Lichti, U. (1985). Kératinocytes blocked in phorbol ester-responsive early stage of terminal differentiation by sarcoma viruses. Nature 314,459-62.
Zakian, V. A. (1995). ATM-related genes: what do they tell us about functions of the human gene? Cell 82, 685-7.
Zhang, P. L., and Casey, P. J. (1996). Protein prénylation: molecular mechanisms and functional consequences. Annu Rev Biochem 65, 241-69.

180
Zhang, K., Noda, M., Vass, W. C., Papageorge, A. G., and Lowy, D. R. (1990). Identification of small clusters of divergent amino acids that mediate the opposing effects of ras and Krev-1 . Science 249, 162-165.
Zhang, S., Han, J., Sells, M. A., Chernoff, J., Knaus, U. G., Ulevitch, R. J., and Bokoch, G. M. (1995). Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pakl. Journal of Biological Chemistry 270, 23934-6.
Zhang, X.-F., Settleman, J., Kyriakis, J. M., Takeuchi-Suzuki, E., Elledge, S. J., Marshall, M. S., Bruder, J. T., Rapp, U. R., and Avruch, J. (1993). Normal and Oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364, 308-313.
Zhang, Z., Yuori, K., Wang, H., Reed, J. C., and Ruoslahti, E. (1996). Integrin activation by R-ras. Cell 85, 61-69.
Zheng, Y., Bagrodia, S., and Cerione, R. A. (1994). Activation of phosphoinositide 3- kinase activity by Cdc42Hs binding to p85. J. Biol. Chem 269, 18727-18730.
Zigmond, S. H. (1996). Signal transduction and actin filament organization. [Review] [90 refs]. Current Opinion in Cell Biology 8, 66-73.

ARTICLES
52. Church, S. E.. LeHuray. A. P.. Grant. A. R.. Delevaux. M H & Gray. J. E Geochim. cosmochim. Acta SO, 317-328 (1986).
53. Ohmoto. H. & Rye. R. 0. in Geochemistry of Hydrothermal Ore Deposits 2nd edn (ed. Barnes. H. L) 50»-567 fWiley. New York. 1979).
54. Rye. R. 0. Econ. Geo/. M , 733-753 (1993).55. Stoftregen. R. E. Econ. Geo/. 82, 1575-1591 (1987).56. Sénger-von Oepen. P.. Friedrich. G. A Kisters. A. Miner. Deposlta 25, 536-541 (1990).57. Heald. P.. Foley. N. K. & Haytw. D O. Econ. Geo/. 82, 1-26 (1987).58. Hedenquist J. W. & Henley. R. W. Econ. Geo/. 80, 1379-1406 (1985).59. White. N. C. & Hedenquist, J. W. J. Geochem. Exptor. 38, 445-474 (1990).60. Simmons. S. F.. Gemmell. J. B. & Sawkins. F. J. Econ. Geo/. 83, 1619-1641 (1988),61. O'Neil, J. R. & Silberman. M. L Econ. Geo/. 88, 902-909 (1974).62. Criss. R. E.. Fleck. R. J. & Taylor, H. P. i t J. geophys. Res. #8 ,13335-13356 (1991).63. Doe. B. R. & Zartman. R. E. in Geochemistry of Hydrothermal Ore Deposits 2nd edn (ed.
Barnes. H. L) 22-70 (Wiley. New York, 1979).64. Norman. D. I. & Musgrave. J. A. Geochim. cosmochim. Acta 88, 1119-1131 (1994).65. Giggenbach. W. F. Econ. Geo/. 87 ,1927-1944 (1992).66. Brown. K. L Econ. Geo/. 81, 979-1983 (1986).67. Reyes. A. G.. Giggenbach, W. F., Saleras. J. R. M., Salonga. N. D. & Vergara. M. C. Geotherm-
k s 22, 479-519 (1993).68. Hedenquist, J. W.. Aoki. M. & Shlnohara. H. Geology 22, 585-588 (1994).69. Symonds, R. B.. Reed. M. H. & Rose. W. I. Geochim. cosmochim. Acta 88, 633-657 (1992).70. Reed. M. H. in Magrrtatk ContrltHJtions to Hydrothermal Systems (ed. Hedenquist, J. W.)
135-140 (Rep. No. 279. Geol. Surv. Japan. Tsukuba. 1992).71. Hemley. J. J.. Cygan. G L. Fein. J. S.. Robinson. G. R. & d Angelo. W. M. Econ. Geol. 87,
1-22 (1992).72. Richards. J. P. Geology 20, 547-550 (1992).73. van leeuwen. Th. M.. Leach, T.. Hawke. A. & Hawke. M. M. J. Geochem. Explor. 35, 1-61
(1990).
74. Torgerson. T. EOS 71, 1. 5. 13 (1990)75. Crowe. D. E.. Valley. J w & Baker. K. L. Geochim. cosmochim. Acta 54, 2075-2092
(1990).76. Heinrich. C. A.. Ryan. C. G . Mernagh. T P & Eadington. P. j Econ. Geol 87, 1566-1583
(1992).77. Lepel. E. A.. Stefansson. K. M. i Zoller. W. H. J. geophys. Res. 83, 6213-6220 (1978).78. Buat-Menard. P. 4 Arnold. M. Geophys. Res. Lett. 8, 245-248 (1978).79. Andres. R. J.. Kyle. P. R. 4 Chuan. R. L. Geol. Rdsch. 82, 687-695 (1993).80. Krauskopf. K. B Introduction to Geochemistry (McGraw-Hill. New York. 1979).81. Gilmour. P. in Advances in Geology of the Porphyry Copper Deposits (ed. Tit ley. S. R.) 7-
35 (Univ. Arizona Press. Tucson. 1982)82. Prtzer. K. S. 4 Pabalan. R. T. Geochim. cosmochim Acta 80, 1445-1454 (1986).83. Hedenquist, J. W.. Simmons. S. F.. Giggenbach. W. F. 4 EkJridge. C. S. Geology 21, 731-
734 (1993184. Craig. H. in Nuclear Geology in Geothermal Area (ed. Tongiorgi. E.) 17-53 (Consiglio Nazion
ale delle Richerche Laboratorio di Geo/ogia Nucleare. Pisa. 1963).85. Sheppard. S. M. F. in Stab/e Isotopes in High Temperature Geological Processes (eds Valley.
J. W.. Taylor. H. P. Jr 4 O'Neil. J. R.) 165-183 (Miner. Soc. Am.. Washington DC. 1986).86. Hedenquist J W. in 4th Orcum-Padfk Energy and Mineral Resources Conf. Trans, (ed.
Horn. M. K.) 513-524 (Am. Ass, Petrol Geol.. Tulsa. 1987).
ACKNOWLEDGEMENTS. We thank Y. Matsuhisa and members of the Japan-US Seminar on Magmatic Contributions to HydrotJiermal Systems tor providing the impetus to write this paper: P. Allard and 8. W. C2iristenson allowed use of tTieir unpublished data; E Lougee helped draft the figures and E. Roedder contributed the photographs; W. F. Giggenbach. R. W. Henley. H. Shinohara and R. H. Sillitoe shared their insight and A. T, Anderson Jr. A. Arribas Jr. R. 0. Fournier, J. J. Rytuba. R. H. Sillitoe. B. E. Taylor and N. C. White provided abundant cntical comments on the manuscript
ARTICLES
Phosphatidylinositol-3-OH kinase as a direct target of RasPablo Rodriguez-Viciana , Patricia H. Warne , Ritu Dhand , Bart Vanhaesebroeck , Ivan G out\ Michael J. Fry\ Michael D. Waterfield^^ & Julian Downward^• Imperial Cancer Research Fund, 44 Lincoln’s Inn Fields, London WC2A 3PX, UKt Ludwig Institute for Cancer Research, 91 Riding House S tre e t London W IP 8BT, UKÎ Department of Biochemistry and Molecular Biology, University College, Gower S tre e t London WCIE 6BT, UK
Ras (p21'”*) interacts directly with the catalytic subunit of phosphatidyiinositoi-3-OH kinase in a GTP-dependent manner through the Ras effector site. In vivo, dominant negative Ras mutant N17 inhibits growth factor Induced production of 3' phosphoryiated phosphoinositides in PC12 ceils, and transfection of Ras, but not Raf, into COS ceils results In a large elevation in the level of these lipids. Therefore Ras can probably regulate phosphatidyiinositoi-3-OH kinase, providing a point of divergence In signalling pathways downstream of Ras.
T h e Ras proteins are key regulators o f cell growth and other functions' ; the elucidation o f the identities o f their critical cellular targets, or ‘effectors’ through which they exert their biological effects has long been a major goal o f research into the regulation of eukaryotic cellular proliferation. The interaction o f Ras proteins with their effectors occurs through a stretch o f amino acids (32-40), generally known as the effector region, which assumes a different conformation in GTP-bound (active) Ras compared to GDP-bound (inactive) Ras, An effector o f Ras would be expected to possess, or control, some sort o f enzymatic activity that is regulated by its interaction with Ras GTP, and which is responsible for at least some of the effects o f Ras on the cell. It is possible, indeed likely, that Ras has multiple effectors.
The first protein for which evidence for a possible Ras effector role was found was pl20°'^'‘ (ref, 2). This protein interacts only with GTP-bound Ras and fails to interact with most effector region mutants. This is also true for the GAP-related protein neurofibromin, the product o f the N F / gene\ Because both these
§ To whom correspondence should be addressed.
n ature • VOL 370 • 18 AUGUST 1994
proteins strongly stimulate the GTPase activity o f Ras they are both negative regulators o f Ras: this activity could be incorporated in an effector as a negative feedback mechanism. Some reports indicate that p i20°' ̂ can have an influence downstream o f Ras in various signalling pathways^’’, but it seems unlikely that it can account for all the signals emerging from Ras, The effectors discovered so far that are most likely to mediate the growth stimulatory signals from Ras are the cytoplasmic serine/ threonine Raf kinases. These function downstream o f Ras and activate the MAP kinase, and possibly other, pathways*. It is not clear whether Raf can activate all the signalling pathways that Ras can, but it can act as a very potent oncogene, unlike GAP. Recently several groups have reported that Ras directly interacts in a GTP-dependent manner with the amino-terminal domain o f Raf through its effector region’ ' I t remains to be determined whether interaction o f Raf with Ras GTP can result in an increase o f its kinase activity,
R af kinases may not be the sole mediators o f the effects of Ras on cellular behaviour. Phosphatidylinositol-3-OH kinase (PI(3)K ) coimmunoprecipitates with Ras"^^, This suggests that
527
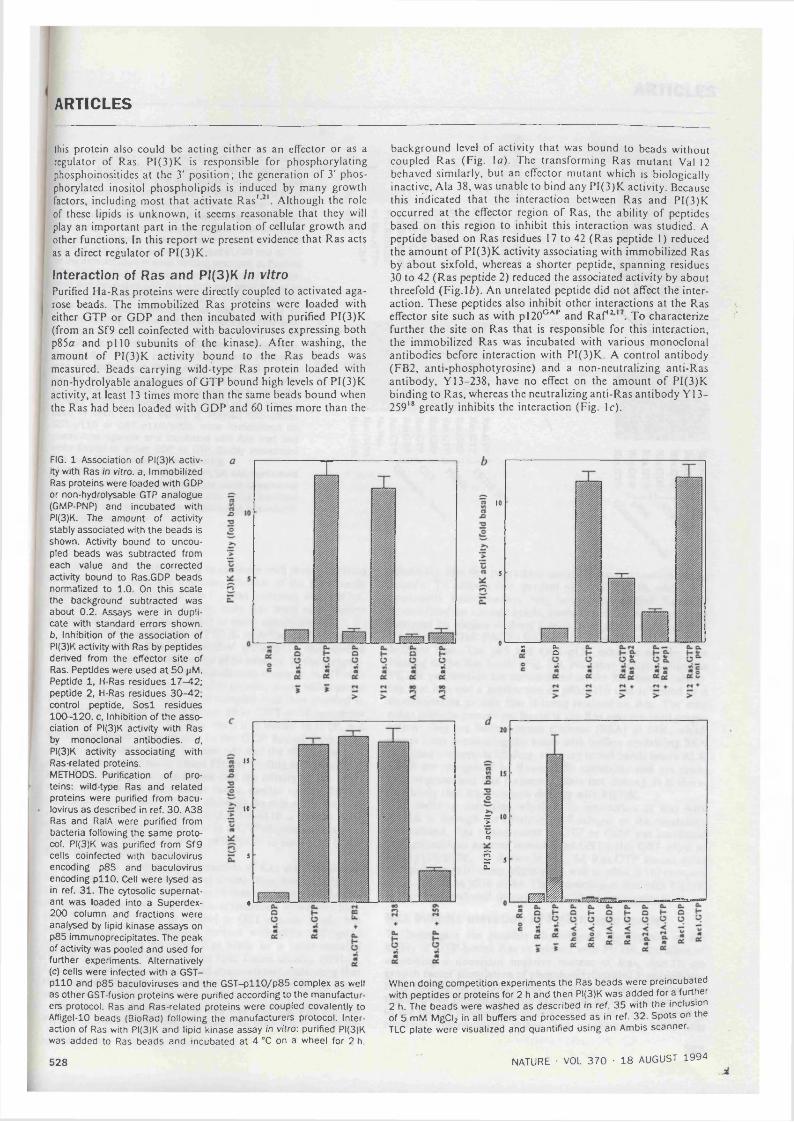
ARTICLES
this protein also could be acting cither as an effector or as a regulator of Ras. PI(3)K is responsible for phosphorylating phosphoinositides at the 3' position; the generation o f 3' phosphoryiated inositol phospholipids is induced by many growth factors, including most that activate Ras' Although the role of these lipids is unknown, it seems reasonable that they will play an important part in the regulation of cellular growth and other functions. In this report we present evidence that Ras acts as a direct regulator o f PI(3)K.
Interaction of Ras and PI(3)K In vitroPurified Ha-Ras proteins were directly coupled to activated agarose beads. The immobilized Ras proteins were loaded with either GTP or GDP and then incubated with purified P1(3)K (from an Sf9 cell coinfected with baculoviruses expressing both p85a and pi 10 subunits o f the kinase). After washing, the amount o f P1(3)K activity bound to the Ras beads was measured. Beads carrying wild-type Ras protein loaded with non-hydrolyable analogues o f GTP bound high levels o f P1(3)K activity, at least 13 times more than the same beads bound when the Ras had been loaded with GDP and 60 times more than the
background level o f activity that was bound to beads without coupled Ras (Fig. la). The transforming Ras mutant Val 12 behaved similarly, but an effector mutant which is biologically inactive, Ala 38, was unable to bind any P1(3)K activity. Because this iridicated that the interaction between Ras and P1(3)K occurred at the effector region o f Ras, the ability o f peptides based on this region to inhibit this interaction was studied. A peptide based on Ras residues 17 to 42 (Ras peptide 1) reduced the amount o f P1(3)K activity associating with immobilized Ras by about sixfold, whereas a shorter peptide, spanning residues 30 to 42 (Ras peptide 2) reduced the associated activity by about threefold (Fig. 16). An unrelated peptide did not affect the interaction. These peptides also inhibit other interactions at the Ras effector site such as with pl20°'^'’ and Raf'^ To characterize further the site on Ras that is responsible for this interaction, the immobilized Ras was incubated with various monoclonal antibodies before interaction with P1(3)K. A control antibody (FB2, anti-phosphotyrosine) and a non-neutralizing anti-Ras antibody, Y13-238, have no effect on the amount o f P1(3)K binding to Ras, whereas the neutralizing anti-Ras antibody Y 13- 259'* greatly inhibits the interaction (Fig. Ic).
FIG. 1 Association of Pi(3)K activity with Ras in vitro, a, Immobilized Ras proteins were loaded with GDP or non-hydrolysable GTP analogue (GMP-PNP) and incubated with PI(3)K. The amount of activity stably associated with the beads is shown. Activity bound to uncoupled beads was subtracted from each value and the corrected activity bound to Ras.GDP beads normalized to 1.0. On this scale the background subtracted was about 0.2. Assays were in duplicate with standard errors shown, b. Inhibition of the association of PI(3)K activity with Ras by peptides denved from the effector site of Ras. Peptides were used a t 50 pM.Peptide 1. H-Ras residues 17-42; peptide 2. H-Ras residues 30-42; control peptide, S o s l residues 100-120. c. Inhibition of the association of PI(3)K activity with Ras by monoclonal antibodies, d,PI(3)K activity associating with Ras-related proteins.METHODS. Purification of proteins: wild-type Ras and related proteins were purified from bacu- lovirus as described in ref. 30. A38 Ras and RalA were purified from bacteria following the sam e protocol. PI(3)K was purified from Sf9 cells coinfected with baculovirus encoding p85 and baculovirus encoding p llO . Cell were lysed as in ref. 31. The cytosolic supernatant was loaded into a Superdex- 200 column and fractions were analysed by lipid kinase assays on p85 immunoprecipitates. The peak of activity was pooled and used for further experiments. Alternatively (c) cells were infected with a GST- p llO and p85 baculoviruses and the G ST-pllO /p85 complex as well as other GST-fusion proteins were purified according to the manufacturers protocol. Ras and Ras-related proteins were coupled covalently to Affigel-10 beads (BioRad) following the manufacturers protocol. Interaction of Ras with PI(3)K and lipid kinase assay in vitro: purified PI(3)K was added to Ras beads and incubated at 4 °C on a wheel for 2 h.
528
15
to
5
0
10
5
0 o.oS
I%
§ sCL.
J
When doing competition experiments the Ras beads were preincubated with peptides or proteins for 2 h and then PI(3)K was added for a further 2 h. The beads were washed as described in ref. 35 with the inclusion of 5 mM MgClz in all buffers and processed as in ref. 32. Spots on the TLC plate were visualized and quantified using an Ambis scanner.
NATURE ■ VOL 370 • 18 AUGUST 1994

ARTICLES
FIG. 2 Association of p 8 5 /p l l0 with Ras in vitro, a. Purified p85a/G ST -pllO (left) or p85/3/G ST-pllO (right) was incubated with immobilized Ras or m utants of Ras which had been bound to either GDP, GTP or a non-hydrolysable analogue of GTP. GMP-PNP (GTP*). Stably associated PI(3)K was visualized by western blotting using anti-p58a or anti-p85)3 monoclonal antibody. The two right-hand tracks of the left panel show the purified p 8 5 a /p l l0 preparation used (1 /30 and 1 /7 .5 of the amount used in each binding reaction), b. Purified p85a/G ST -pllO was immobilized on glutathione agarose and incubated with Ras which had been bound to either GDP or the non-hydrolysable analogue of GTP. GMP-PNP. Stably associated Ras was visualized by western blotting. The two left-hand tracks show the purified Ras preparation used (1 /30 and 1 /1 0 0 of the amount used in each binding reaction. Kl. kinase inactive p llO . c. Purified p85o/G ST -pllO was incubated with immobilized Ras. Stably associated PI(3)K was visualized by gel electrophoresis followed by staining with Coomassie brilliant blue. The right-hand track shows the purified PI(3)K preparation used (1 /10 of the amount used in each binding assay), d. Purified GST-p85a. GST-pllO or G ST-pllO /p85o were immobilized on glutathione agarose and incubated with Ras that had been bound to either GDP or GTP. Stably associated Ras was visualized by western blotting.METHODS. Interaction of Ras and PI(3)K was performed as in Fig. 1. Western blots were done using monoclonal antibody against p85a. p 8 5 P or Ras using standard protocols^^ with development by enhanced chemilumi- nescence (Amersham).
pH5(*
73 t:
m < < H ^
T|
p85(5
Ras
c o c rH C H O -S -5 "3 -=
o n o cH C H C -3 "3 -5 13
% %%
AMK)211(1 W
11693
68
^ 7^% :45
31S 53 »
He
73 T : 7373 73 73 73
o o o o o o aH o H a"9 13 "C "9
H O H 13 13 13
J I I
V
The ability of PI(3)K activity to associate with proteins related to Ras was also tested in this assay. Neither o f the Rho family proteins tested, RhoA and R acl, could associate with PI(3)K (Fig. \d ). From the Ras family, only Ras itself was positive whereas R alA and Rap2A failed to show association.
To confirm the association o f PI(3)K with Ras.GTP, the interaction of purified Ras with purified p85a/pI10 was studied directly by western blotting for the p85a subunit. In these experiments, pi 10 was expressed in a baculovirus construct in Sf9 cells as a glutathione S transferase fusion protein, along with normal p85. The active p85a/G ST-pl 10 complex was then purified in a single step on glutathione agarose. p85a/G ST -pl 10 complexes with wild-type or Val 12 H-Ras when it is bound to GTP, but binds very much more weakly to the GDP bound form (Fig. 2a). No interaction is seen between p85 and the effector mutant Ala 38. The proportion o f added PI(3)K binding to Ras in these and similar experiments indicates that the affinity o f the interaction is in the low micromolar range, similar to that for the interaction o f Ras and pl20°'''", although the rate o f dissociation of the complex is slow. When p85/?/GST-pl 10 is used in these experiments, it also binds to Ras in a GTP-dependent manner. Similar results are found when p85a/p llO is used that is not fused to GST (data not shown).
Another way in which the interaction o f Ras and PI(3)K can be demonstrated is by incubating soluble Ras, bound either to GTP or GDP, with p85a/G ST-pl 10 that has been immobilized on glutathione agarose. Bound Ras is visualized by western blotting. Ras does not bind to control or GST agarose beads, but interacts in a GTP-dependent manner with p85a/G ST -pl 10 beads (Fig. 2b). In addition, Ras binds to a point-mutated PI(3)K whose pi 10 subunit lacks lipid kinase activity (R916P,I. Hiles and M.D.W., unpublished observations), indicating that the enzymatic activity o f the kinase is not required for its interaction with Ras.
The above data show that purified Ras interacts with purified P1(3)K in a GTP-dependent manner, but does not rule out the
possibility that this interaction occurs through a contaminating protein. To address this, purified p85a/G ST -pl 10, which was apparently homogeneous, was bound to purified Ras GTP immobilized on agarose beads, washed, and the bound proteins analysed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAG E) followed by staining with Coomassie brilliant blue. The p85 and GST-pllO subunits can be seen binding to the Ras beads (Fig. 2c). N o other proteins from the PI(3)K preparation are accumulated on the Ras beads, indicating that it is not a subfraction of p85/p l 10 that is bound to a contaminating protein that is being retained on Ras. The only other proteins present are found in the Ras agarose bead preparation: they are bovine serum albumin (ESA) at 68K, which comes from prewashing the beads with buffers containing ESA to reduce nonspecific binding, and very broad bands below 45 K which are recognized by Ras-specific antibodies and are probably oligomerized Ras fragments (data not shown). It is therefore likely that Ras interacts directly with PI(3)K.
In order to determine whether the interaction o f Ras with PI(3)K is through the catalytic pi 10 subunit or the regulatory p85 subunit, Ras bound either to GTP or GDP was incubated with glutathione agarose immobilized GST-pllO, GST-p85a or GST-pl 10/p85a. As shown in Fig. 2d, Ras GTP bound most efficiently to pi 10 alone, slightly less well to p 85a /p l 10 complex and not at all to p85a alone. The interaction o f Ras with PI(3)K is therefore mediated through the pi 10 catalytic subunit.
Ras inhibits eievation of Pi(3)K iipid ieveisTo investigate the possible physiological significance o f the ability o f GTP-bound Ras to interact with PI(3)K, the effect o f expressing a dominant negative mutant o f Ras, Asn 17, on growth factor stimulation o f phosphatidylinositol 3' phosphoryiated lipid levels in intact cells was studied. Because most cell types cannot proliferate while expressing Asn 17 Ras, we chose to use the rat phaeochromocytoma cell line PC 12 in which Ras regulates differentiation rather than proliferation. PC 12 cells
NATURE • VOL 370 • 18 AUGUST 1994 5 2 9

ARTICLES
were transfected with a Rous sarcoma virus (RSV) Asn 17 Ras construct and clones o f cells selected. The ability o f nerve growth factor and epidermal growth factor to elevate phosphatidylinositol 3' phosphoryiated lipid levels in these cells was compared to the parental cells. Cells were metabolically labelled with '̂P- orthophosphate and levels o f the 3' phosphoryiated phosphoinositides determined after nerve growth factor (N G F ) or epidermal growth factor (EOF) stimulation. The amounts o f phosphatidylinositol (3,4) bisphosphate (PtdIns(3 ,4 )P2) and phosphatidylinositol (3,4,5) trisphosphate (PtdIns(3,4,5)Pj) in wild-type ?C \2 cells increases by between 10- and 20-fold in response to EG F or NGF (Fig. 3a, b). By contrast, in the Asn 17 Ras-expressing PC 12 cells, the growth factor stimulation of PtdIns(3 ,4 )P2 levels is attenuated by about fivefold. The response o f PtdIns(3,4,5)P, lipid pool to growth factors is attenuated somewhat less, by about threefold, because o f the expression o f Asn 17 Ras in PC 12 cells. The incomplete inhibition probably reflects the fact that several mechanisms exist whereby growth factors can control PI(3)K activity, only one o f which involves Ras. The time courses o f the responses are similar in both wild-type and As.n- 17 Ras expressing PCI 2 cells. Although the simplest explanation for these data, given our in vitro observations reported above, is that Ras is involved in the activation PI(3)K, it cannot be ruled out that Ras is acting at the level o f lipid breakdown. Active Ras could cause inhibition of phosphoinositide 3' phosphatases.
To be certain that the inhibition of the growth factor induced elevation of PtdIns(3,4 )P2 and PtdIns(3 ,4 ,5 )P3 in dominant negative Ras-expressing cells is specific to this pathway and not a general phenomenon affecting other signalling pathways, a number o f other growth factor-stimulated events were studied. The amount of PI(3)K activity found in anti-phosphotyrosine immunoprecipitates from stimulated cells was measured in an in vitro assay. EGF and NG F treatment o f PC 12 cells lead to a roughly four- and threefold stimulation, respectively, o f the amount o f PI(3)K activity found in anti-phosphotyrosine immunoprecipitates (Fig. 3c). Expression o f Asn 17 Ras in the PC 12 cells does not reduce this activity, indicating that growth factor control of PI(3)K through association with phosphotyro- sine-containing proteins is normal even in the presence o f dominant negative mutant Ras protein. In addition, the production of inositol phosphates (inositol(l,4,5)trisphosphate and inositol(l,4)bisphosphate) by phosphatidylinositol-specific pho- spholipase C in response to EGF and NG F is also unaltered by Asn 17 Ras expression (see Fig. 3d). Furthermore, expression of Asn 17 Ras in PC 12 cells does not obviously alter the ability of EGF and NGF to induce tyrosine phosphorylation o f total cellular proteins (see Fig. 3c). It is therefore likely that the expression and function o f growth factor receptor tyrosine kinases and the regulation o f PI(3)K by tyrosine phosphoprotein binding are not being directly altered by the presence o f Asn 17 Ras in these PC 12 cells.
Ras elevates phosphoryiated lipid ieveisFrom the ability of Ras and PI(3)K to interact in vitro and the inhibitory effect o f Asn 17 Ras on phosphatidylinositol 3' phosphoryiated lipid accumulation in vivo, it seems likely that Ras is regulating the function o f PI(3)K in intact cells. To explore this possibility further, the effect o f overexpressing Ras transiently on 3' phosphoryiated phosphoinositides in COS cells was investigated. When a vector expressing activated mutant Val 12 Ras was transfected into COS cells, some elevation was found in the levels o f PtdIns(3 ,4 )P2 and PtdIns(3,4,5)Pj in the intact cells (Fig. 4a). Expression of the p85 a-subunit plus the pi 10 subunit of PI(3)K together had a similar effect on these lipids. However, when all three proteins were expressed together they cooperated to give a very large increase in the level o f 3' phosphoryiated phosphoinositides. Wild-type Ras is almost as potent as Val 12 Ras in elevating these lipids (Fig. 46) presumably reflecting the very high levels o f Ras protein expression
5 3 0
achieved in this system, whereas the effector mutant Val 12-* Ala 38 Ras is inactive. u-Src also caused a similar level o f activation. A number of other Ras-related proteins were studied in this assay, including RhoA and Racl. None of these have any effect on the 3' phosphoryiated phosphoinositide levels. The ability o f v-Src to elevate the levels of these lipids was partially blocked by the dominant negative mutant of Ras, Asn 17, consistent with the hypothesis that v-Src can activate P1(3)K through both a Ras-dependent and a Ras-independent pathway, the latter presumably involving tyrosine phosphoprotein binding to p85. The v-Raf serine/threonine kinase, or an amino-termin- ally truncated, activated version o f c-Raf (ARaf), which act immediately downstream o f Ras to activate the MAP kinase pathway, do not cause elevation o f 3' phosphoryiated phosphoinositides, so it is unlikely that the effects seen with Ras are indirect through secondary events such as autocrine production o f growth factors (see also discussion). As also reported previously by others” -̂ ®, in this system transfection o f either activated Ras or Raf could be shown to activate MAP kinase (data not shown). As in Fig. 3, we cannot at this stage rule out a role for Ras in the control o f the rate o f breakdown o f 3' phosphoryiated phosphoinositides, but feel the simplest explanation is that Ras directly contributes to the activation o f PI(3)K.
DiscussionThe regulation o f PI(3)K in response to growth factor treatment of cells could involve a number o f mechanisms: p85 is phosphoryiated on tyrosine residues in response to treatment o f cells with growth factors such as PDGF and it binds through its SH2 domains to autophosphorylation sites on receptor tyrosine kinases and related sites on other molecules, such as IRS-1, CD19 and C D 28^'^\ Tyrosine phosphorylation of p85 has not been shown to change its catalytic activity, but binding to the tyrosine phosphoryiated insulin receptor substrate, IRS-1, and to tyrosine phosphoryiated peptides, particularly those based on the sequences o f the p85 binding sites in the PDGF receptor and IRS-1, does lead to a relatively small increase in the catalytic activity o f PI(3)K^'* *̂. It is possible that in the intact cell, much stronger stimulations result from such interactions because of translocation o f PI(3)K from the cytosol to the plasma membrane where its substrate lipids are located. In addition, another mechanism of regulation could involve proline-rich sequences in p85 binding to SH3 domains o f Src-family kinases such as L c k '" \
Most mitogenic stimuli activate both Ras and PI(3)K.'-^'. This could reflect the fact that there are common components, such as tyrosine kinases, in the upstream regulatory pathways for both proteins. It could also be the result o f a system in which Ras is capable, directly or indirectly, o f regulating PI(3)K or vice versa. There has been a report that phosphorylation o f PDGF receptor at sites involved in the binding o f p85 is required for the activation o f Ras by PDGF^’. However, this appears to occur only in certain cell types^*. Furthermore, another adaptor protein. Nek, is now known also to bind to one o f these sites^’ . It therefore seems unlikely that PI(3)K acts upstream of Ras.
The alternative possibility, that Ras acts to control PI(3)K, receives support from the data that are presented here. The association o f PI(3)K activity with Ras was previously reported by Lapetina” , who also showed that p85 could be detected in association with Ras by immunoblotting” . Here we have extended these observations to show that the interaction o f Ras and PI(3)K is direct, requires Ras to be in the active, GTP-bound conformation and occurs through the effector site on Ras. Ras can therefore interact with PI(3)K in vitro in the manner expected for a true effector interaction. We have not yet been able to reproducibly demonstrate that this interaction results in an increase in the lipid kinase activity. The original observation o f phosphatidylinositol activity in Y 13-259 immunoprecipitates of Ras^ ̂ indicated that it was unlikely that the interaction was through the effector site on Ras because this antibody blocks
NATURE ■ VOL 370 • 18 AUGUST 1994
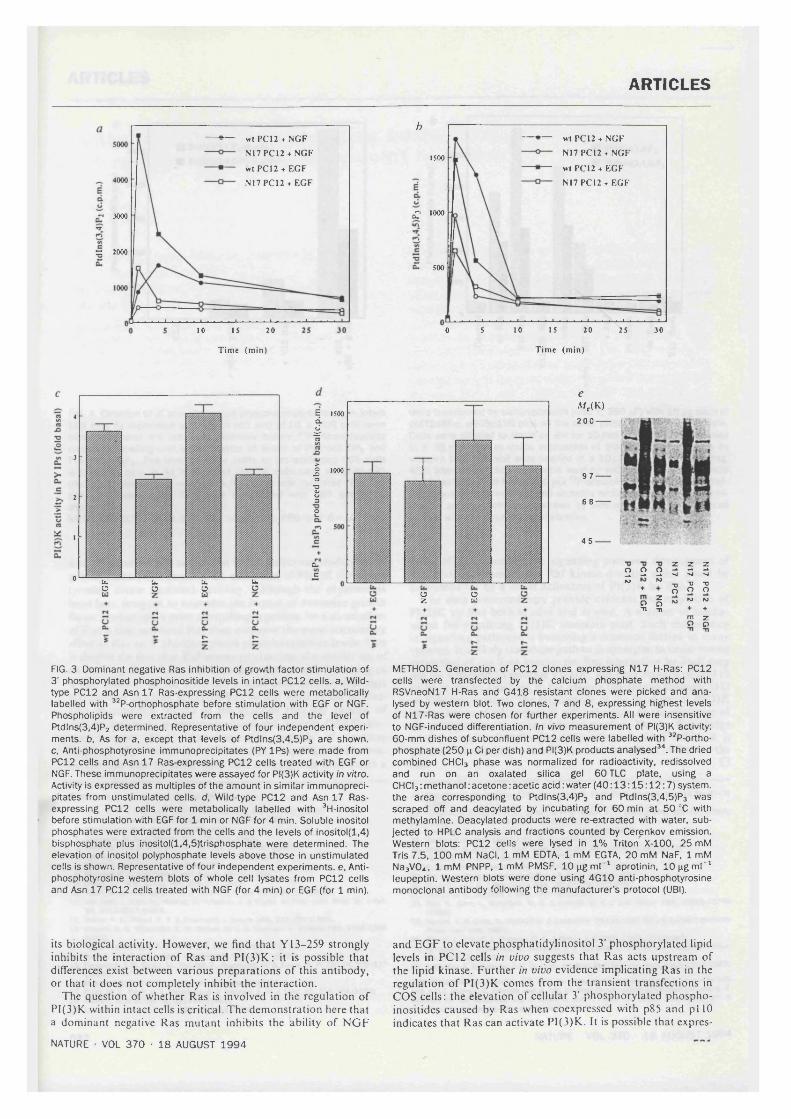
ARTICLES
wt PC12 + NGF N17PC12 + NGF
wt PCI2 + EGF N17PC12 + EGF
r* 3000
= 2000
10 15 20
Time (min)
hwt PCI2 + NGF N17PCI2 + NGF
wt PC12 + EGF N17 PC12 + EGF
1500
1000ô?
500
5 10 15 20 25 300
Time (min)
4
3
2
I
0 U .ob.O h.Ob.ü
E 1500
I
I 1000
12
Cl.
C
uZ s
eA/r(K)200--- Iff
9 7 -
6 8 -
45
2 2 2 m“ • “ S S S
S I “ 7 rm z o o
FIG. 3 Dominant negative Ras inhibition of growth factor stimulation of 3' phosphoryiated phosphoinositide levels in intact PC12 cells, a. Wild- type PC12 and Asn 17 Ras-expressing PC12 cells were metabolically labelled with ^^P-orthophosphate before stimulation with EGF or NGF. Phospholipids were extracted from the cells and the level of Ptdlns(3 ,4 )P2 determined. Representative of four independent experiments. b. As for a, except that levels of Ptdlns(3 ,4 ,5 )P3 are shown, c, Anti-phosphotyrosine immunoprecipitates (PY IPs) were made from PC12 cells and Asn 17 Ras-expressing PC12 cells treated with EGF or NGF. These immunoprecipitates were assayed for PI(3)K activity In vitro. Activity is expressed as multiples of the amount in similar immunoprecipitates from unstimulated cells, d, Wild-type PC12 and Asn 17 Ras- expressing PC12 cells were metabolically labelled with ^H-inositol before stimulation with EGF for 1 min or NGF for 4 min. Soluble inositol phosphates were extracted from the cells and the levels of inositol(l,4) bisphosphate plus inositol(l,4,5)trisphosphate were determined. The elevation of inositol polyphosphate levels above those in unstimulated cells is shown. Representative of four independent experiments, e. Anti- phosphotyrosine western blots of whole cell lysates from PCI2 cells and Asn 17 PCI2 cells treated with NGF (for 4 min) or EGF (for 1 min).
METHODS. Generation of PC12 clones expressing N17 H-Ras; PC12 cells were transfected by the calcium phosphate method with RSVneoN17 H-Ras and G418 resistant clones were picked and analysed by western blot. Two clones, 7 and 8 , expressing highest levels of N17-Ras were chosen for further experiments. All were insensitive to NGF-induced differentiation. In vivo m easurem ent of PI(3)K activity. 60-mm dishes of subconfluent PC12 cells were labelled with ^^P-ortho- phosphate (250 p Ci per dish) and PI(3)K products analysed^\ The dried combined CHCI3 phase was normalized for radioactivity, redissolved and run on an oxalated silica gel 60 TLC plate, using a CHCI3 ; methanol : acetone : acetic acid ; water (4 0 :1 3 :1 5 :1 2 :7 ) system, the area corresponding to Ptdlns(3,4)Pz and Ptdlns(3 ,4 ,5 )P3 was scraped off and deacylated by incubating for 60 min a t 50 °C with methylamine. Deacylated products were re-extracted with water, subjected to HPLC analysis and fractions counted by Cerenkov emission. Western blots: PC12 cells were lysed in 1% Triton X-100, 25 mM Tris 7.5, 100 mM NaCI, 1 mM EDTA, 1 mM EGTA. 20 mM NaF, 1 mM Na3V0 4 . Im M PNPP, 1 mM PMSF, lO p g m P " aprotinin, lO p g m l'" leupeptin. Western blots were done using 4G10 anti-phosphotyrosine monoclonal antibody following the manufacturer’s protocol (UBI).
its biological activity. However, we find that Y 13-259 strongly inhibits the interaction o f Ras and PI(3)K; it is possible that differences exist between various preparations o f this antibody, or that it does not completely inhibit the interaction.
The question o f whether Ras is involved in the regulation of PI(3)K within intact cells is critical. The demonstration here that a dominant negative Ras mutant inhibits the ability o f NG F
and EGF to elevate phosphatidylinositol 3' phosphoryiated lipid levels in PC 12 cells in vivo suggests that Ras acts upstream of the lipid kinase. Further in vivo evidence implicating Ras in the regulation o f PI(3)K comes from the transient transfections in COS cells: the elevation o f cellular 3' phosphoryiated phosphoinositides caused by Ras when coexpresscd with p85 and pi 10 indicates that Ras can activate PI(3)K. It is possible that expres-
NATURE • VOL 370 • 18 AUGUST 1994
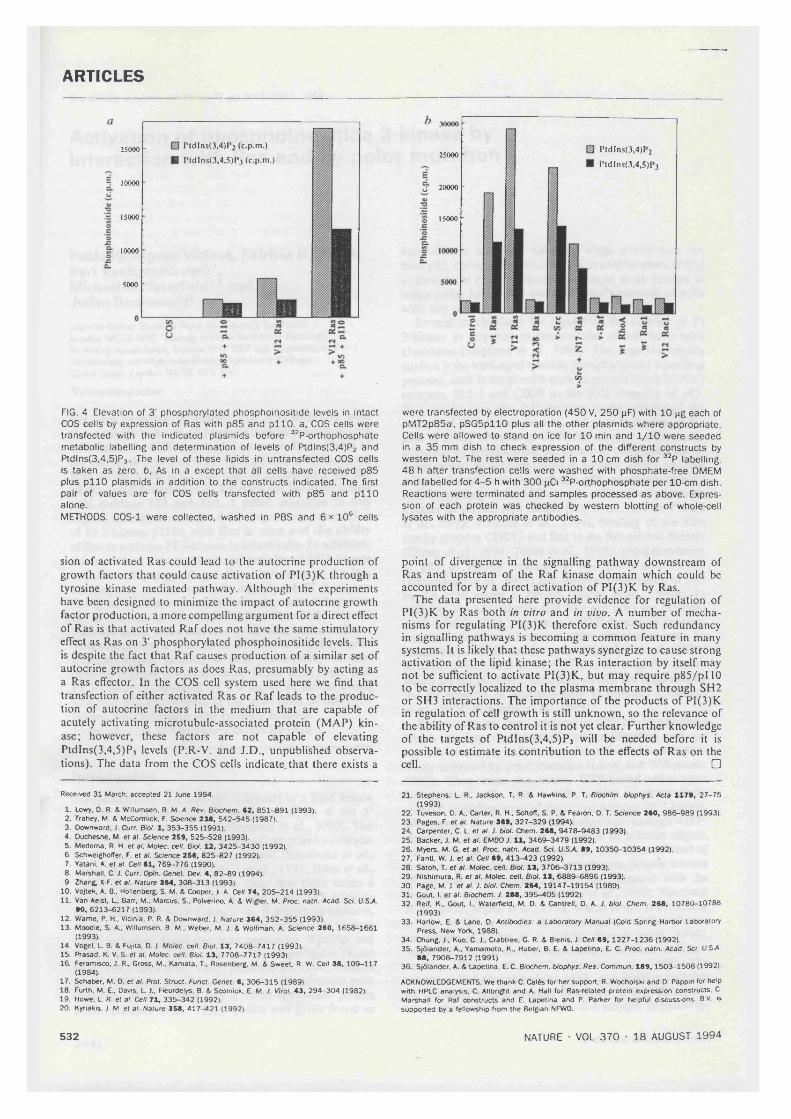
ARTICLES
25000
20000
- I5000
10000
5000
PldIns(3,4)P2 (c.p.m.) PtdIns(3,4,5)P3 (c.p.m.)
P td ln s(3 ,4 )l 2
PtdIns(3.4,S)I 325000
U 20000
15000
FIG. 4 Elevation of 3' phosphoryiated phosphoinositide levels in intact COS cells by expression of Ras with p85 and p llO . a. COS cells were transfected with the indicated plasmids before ^^P-orthophosphate metabolic labelling and determination of levels of Ptdlns(3,4)P2 and Ptdlns(3,4.5)P3. The level of these lipids in untransfected COS cells is taken as zero. b. As in a except that all cells have received p85 plus p llO plasmids in addition to the constructs indicated. The first pair of values are for COS cells transfected with p85 and p llO alone.METHODS. COS-1 were collected, washed in PBS and 6 x lO® cells
were transfected by electroporation (450 V, 250 pF) with 10 pg each of pMT2p85a, pSG 5pllO plus all the other plasmids where appropriate. Cells were allowed to stand on ice for 10 min and 1 /1 0 were seeded in a 35 mm dish to check expression of the different constructs by western blot. The rest were seeded in a 10 cm dish for ^^P labelling. 48 h after transfection cells were washed with phosphate-free DMEM and labelled for 4 -5 h with 300 pCi ^^P-orthophosphate per 10-cm dish. Reactions were terminated and sam ples processed a s above. Expression of each protein was checked by western blotting of whole-cell lysates with the appropriate antibodies.
sion of activated Ras could lead to the autocrine production o f growth factors that could cause activation o f PI(3)K through a tyrosine kinase mediated pathway. Although the experiments have been designed to minimize the impact o f autocrine growth factor production, a more compelling argument for a direct effect o f Ras is that activated Raf does not have the same stimulatory effect as Ras on 3' phosphoryiated phosphoinositide levels. This is despite the fact that Raf causes production o f a similar set o f autocrine growth factors as does Ras, presumably by acting as a Ras effector. In the COS cell system used here we find that transfection of either activated Ras or Raf leads to the production o f autocrine factors in the medium that are capable o f acutely activating microtubule-associated protein (MAP) kinase; however, these factors are not capable o f elevating PtdIns(3 ,4 ,5)P3 levels (P.R-V. and J.D., unpublished observations). The data from the COS cells indicate, that there exists a
point o f divergence in the signalling pathway downstream of Ras and upstream o f the Raf kinase domain which could be accounted for by a direct activation o f PI(3)K by Ras.
The data presented here provide evidence for regulation o f PI(3)K by Ras both in vitro and in vivo. A number o f mechanisms for regulating PI(3)K therefore exist. Such redundancy in signalling pathways is becoming a common feature in many systems. It is likely that these pathways synergize to cause strong activation o f the lipid kinase; the Ras interaction by itself may not be sufficient to activate PI(3)K, but may require p85/pl 10 to be correctly localized to the plasma membrane through SH2 or SH3 interactions. The importance o f the products o f PI(3)K in regulation o f cell growth is still unknown, so the relevance of the ability o f Ras to control it is not yet clear. Further knowledge o f the targets o f PtdIns(3 ,4 ,5)P3 will be needed before it is possible to estimate its contribution to the effects o f Ras on the cell. □
Received 31 March; accepted 21 June 1994.
1. Lowy. D. R. & Willumsen. B. M. A. Rev. Biochem. 62, 851-891 (1993).2. Trahey. M, & McCormick. F. Science 236, 542-545 (1987t.3. Downward. J. Curr. Biol. 1, 353-355 (1991).4. Duchesne. M. ef al. Science 259, 525-528 (1993).5. Medema, R. H. et al. Molec. cell. Bid. 12, 3425-3430 (1992).6. SchweigholTer. F. et al. Science 256, 825-827 (1992).7. Yatani, A. et al. Cell 61, 769-776 (1990).8. Marshall. C. J. Curr. Oprn. Genet. Dev. 4, 82-89 (1994).9. Zhang, X-F. et al. Nature 364, 308-313 (1993).
10. Vojtek. A. B.. Hollenberg, S. M. & Cooper. J. A. Cell 74, 205-214 (1993).11. Van AelsL L, Ban, M.. Marcus. S.. Polverino. A. & Wigler. M. Proc. natn. Acad. Sci. U.SA.
90, 6213-6217 (1993).12. Warne. P. H.. Vicinia, P. R. & Downward. J. Nature 364, 352-355 (1993).13. Moodie. S. A.. Willumsen. B. M.. Weber. M. J. & Wolfman. A. Science 260, 1658-1661
(1993).14. Vogel. L B. & Fujita. D. J. Molec. cell. Biol. 13, 7408-7417 (1993).15. Prasad. K. V. S. et al. Molec. cell. Biol. 13, 7708-7717 (1993).16. Feramisco. J. R.. Gross, M., Kamata. T.. Rosenberg. M. & Sweet. R. W. Cell 38, 109-117
(1984).17. Schaber. M. D. et al. Prot. Struct. Funct. Genet. 6, 306-315 (1989).18. Furth. M. E.. Davis. L J.. Fleurdelys. B. & Scolnick, E. M. J. Virol. 43, 294-304 (1982).19. Howe. L. R. et al. Cell 71, 335-342 (1992).20. Kyriakis. J, M. et al. Nature 358, 417-421 (1992).
21. Stephens. L R., Jackson. I. R. & Hawkins. P. T. Biochim. biophys. Acta 1179, 27-75 (1993).
22. Tuveson. D. A.. Carter, R. H.. Soltoff, S. P. & Fearon. D. T. Science 260, 986-989 (1993).23. Pages. F. et al. Nature 16», 327-329 (1994).24. Carpenter. C. L. et al. J. bid. Chem. 268, 9478-9483 (1993).25. Backer. J. M. et al. EMBO J. 11, 3469-3479 (1992).26. Myers, M. G. et al. Proc. natn. Acad. Sci. U S A . 89, 10350-10354 (1992).27. Fantl. W. J. et al. Cell 6», 413-423 (1992).28. Satoh. T. et al. Molec. cell. Bid. 13, 3706-3713 (1993).29. Nishimura. R. et al. Molec. cell. Bid. 13, 6889-6896 (1993).30. Page. M. J. et al. J. bid. Chem. 264 ,19147-19154 (1989).31. Gout. I. et al. Biochem. J. 288, 395-405 (1992).32. Reif. K.. Gout I.. Waterfield. M. D. & Cantrell. D. A. J. blol. Chem. 268, 10780-10788
(1993).33. Harlow. E. & Lane. D. Antibodies: a Laboratory Manual (Cold Spring Harbor Latx>ratory
Press. New York. 1988).34. Chung, J.. Kuo. C. J.. Crabtree. G. R. & Blenis, J. Cell 69, 1227-1236 (1992).35. Sjolander. A.. Yamamoto. K.. Hutjer. B. E. & Lapetina. E. C. Proc. natn. Acad. Sci. USA.
88, 7908-7912 (1991).36. Sjôlander. A. & Lapetina. E. C. Biochem. biophys. Res. Commun. 189 ,1503-1508 (1992).
ACKNOWLEDGEMENTS. We thank C. Calés for her support R. Wocholski and D. Pappin for help with HPLC analysis. C. Allbright and A. Hall for Ras related protein expression constructs. C. Marstrall for Raf constructs and E. Lapetina and P. Parker for helpful discussions. B.V. is supported by a fellowship from the Belgian NFWO.
5 3 2 NATURE ■ VOL 370 ■ 18 AUGUST 1994

The EMBO Jo u rn a l v o l.15 n o .10 p p .2442-2451, 1996
Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation
Pablo Rodriguez-Viciana, Patricia H.Warne, Bart Vanhaesebroeck”', Michael D.Waterfield^^ and Julian Downward^Imperial Cancer Research Fund. 44 Lincoln's Inn Fields.London WC2A 3PX. 'Ludwig Institute for Cancer Research.91 Riding House Street. London W IP 8BT and ^Department of Biochemistry and Molecular Biology. University College.Gower Street. London W C IE 6BT. UK
^Corresponding author
We have reported previously that Ras interacts with the catalytic subunit of phosphoinositide 3-kinase (PI 3-kinase) in a GTP-dependent manner. The affinity of the interaction of Ras GTP with p85(x/pll0a is shown here to be -150 nM. The site of interaction on the pllOa and p isoforms of PI 3-kinase lies between amino acid residues 133 and 314. A point mutation in this region, K227E, blocks the GTP-dependent interaction of PI 3-kinase pllOa with Ras in vitro and the ability of Ras to activate PI 3-kinase in intact cells. In addition, this mutation elevates the basal activity of PI 3-kinase in intact cells, suggesting a direct influence of the Ras binding site on the catalytic activity of PI 3-kinase. Using an in vitro reconstitution assay, it is shown that the interaction of Ras*GTP, but not Ras*GDP, with PI 3-kinase leads to an increase in its enzymatic activity. This stimulation is synergistic with the effect of tyrosine phosphopeptide binding to p85, particularly at sub- optimal peptide concentrations. These data show that PI 3-kinase is regulated by a number of mechanisms, and that Ras contributes to the activation of this lipid kinase synergistically with tyrosine kinases.Keywords: phosphoinositide 3-kinase/Raf/Ras
IntroductionPhosphoinositide 3-kinase (PI 3-kinase) is a lipid kinase capable of phosphorylating phosphoinositides at the 3' position of the inositol ring (Stephens et a i, 1993). The first characterized form of PI 3-kinase comprised a catalytic (pi 10) and a regulatory (p85) subunit (Escobedo et a i, 1991; Otsu et a i, 1991; Skolnik et a i, 1991; Hiles et a i,1992). Recently, it has become clear that there exists a family of PI 3-kinases with at least three p85 subunits and at least three p i 1 0 subunits (a , P and y); in addition, a number o f less closely related lipid and protein kinases exist including Saccharomyces cerevisiae Vps34p and TOR 1 and 2 and their mammalian homologues (Kunz et a i, 1993; Schu et a i, 1993; Brown et a i, 1994; Sabatini et a i, 1994), the product o f the ataxia telangiectasia gene (Savitsky et a i, 1995) and a DNA-dependent protein kinase (Hartley et a!., 1995). pi 10a and P are found as
heterodimers with p85 subunits, while p llO y does not bind p85. Phosphatidylinositol (3,4,5) trisphosphate (PIP3 ) is thought to be the primary product o f these kinases in intact cells: its level is elevated upon treatment o f cells with any o f a wide variety o f agonists.
Several mechanisms for regulating the activity o f PI 3-kinase in response to extracellular stimuli have been elucidated (Stephens et a i, 1993). The most thoroughly studied is the binding o f tyrosine phosphoryiated signalling proteins, such as the platelet-derived growth factor (PDGF) receptor, lRS-1 and CD28 to the SH2 domains o f p85. This results in a few fold increase in the activity o f the a form o f PI 3-kinase in vitro, and in cells it may, in the case o f PDGF receptor or CD28 for example, additionally stimulate by translocating the kinase to the plasma membrane where its substrate lipid is located. Other possible means o f regulation through the p85 regulatory subunit include binding o f the SH3 domain o f Src family tyrosine kinases to proline-rich motifs (Liu et a i, 1993b; Prasad et a i, 1993; Pleiman et a i, 1994), binding o f the Rho family proteins CDC42 and Rac to the Bcr-related domain (Zheng et a i, 1994; Tolias et a i, 1995), phosphorylation o f p85 at Tyr580 (Hayashi et a i, 1993) and Tyr508 (Kavanaugh et a i, 1994) and autophosphorylation o f p85 by the p llO kinase at Ser608 (Dhand et a i, 1994b). In addition, we have shown that Ras can bind the catalytic p i 10 subunit o f PI 3-kinase in a GTP-dependent manner, and have found evidence that this interaction may be stimulatory within intact cells (Kodaki et a i, 1994; Rodriguez-Viciana et a i, 1994). Other forms of PI 3-kinase which do not bind p85, including pllOy, are regulated by the Py subunits o f heterotrimeric G proteins (Stephens et a i, 1994; Stoyanov et a i, 1995).
Ras proteins are small monomeric GTP binding proteins which are key regulators o f cell proliferation and are capable o f causing cellular transformation when constitu- tively activated by point mutation (Lowy and Willumsen,1993). They enter the active GTP-bound state upon treatment o f cells with a wide variety o f extracellular stimuli, including most o f those that activate PI 3-kinase. Ras exerts its effect on cell behaviour through GTP- dependent interaction with a number o f cellular targets, commonly known as effectors. The best characterized o f these are the serine/threonine cytoplasmic protein kinases o f the Raf family (Rapp, 1991). Ras interacts with the regulatory region o f Raf (Moodie et a i, 1993; Van Aelst et a i, 1993; Vojtek et a i, 1993; Warne et a i, 1993; Zhang et a i, 1993) to cause its translocation to the plasma membrane (Leevers et a i, 1994; Stokoe et a i, 1994), where further unknown events occur to activate its kinase activity (Dent et a i, 1995; Marais et a i, 1995). The activated Raf phosphorylates MEK and thereby switches on the MAP Idnase cascade (Marshall, 1994). This pathway, which results in activation o f a number o f transcrip-
2442 Oxford U niversity P ress

R as-PI 3 -k in a se i n t e r a c t i o n s
lion factors and other enzymes, is capable o f stimulating proliferation of some cell types, at least when constilutively activated. There is good evidence, however, that other pathways are triggered by Ras that are important in transformation: Ras proteins containing certain mutations in the effector interaction site can synergize with other Ras mutants in this region, suggesting that multiple effector molecules are interacting with Ras at a similar site (White et a i, 1995). Transformation by Ras may involve both MAP kinase and Rac pathways (Qiu et a i, 1995). Genetic evidence for multiple effector pathways for Ras in the yeasts S.cerevisiae and Schizosaccharomyces pombe has been known for some time (Toda et a i, 1985; Wang et a i, 1991; Chang et a i, 1994).
A number of possible alternative effectors for mammalian Ras are known in the handful o f proteins that are capable of interacting with the Ras effector site in a GTP-dependent manner. The GTPase-activating proteins p l 2 oGAP neurofibromin and Gap-1 are all negative regulators o f Ras which stimulate its conversion from active GTP-bound form to inactive GDP-bound form (Lowy and Willumsen, 1993). In the case o f pl20 '̂^**, there is some evidence for downstream function, although its possible targets are not known. The other GAPs appear to be unlikely to have effector function. Ral GDS and a related protein have been found to interact with the effector site of Ras in the yeast two-hybrid system (Hofer et a i, 1994; Kikuchi et a i, 1994; Spaargaren and Bischoff, 1994). These proteins may be capable o f inducing activation o f the Ras family member Ral in response to activated Ras; at present the function o f Ral is unknown, so the significance o f the Ral GDS pathway is unclear. A recent report has also indicated that protein kinase is able to interact with Ras GTP in vitro in a manner that suggests it might be an effector (Diaz-Meco et a i, 1994). In addition, it has been reported that Ras can interact with MEKK, an activator o f the Jun kinase pathway, in a GTP- dependent manner (Russell et a i, 1995).
We have shown previously that Ras can interact with the catalytic p i 10a subunit o f PI 3-kinase in a GTP- dependent manner (Rodriguez-Viciana et a i, 1994). This interaction occurs through the effector region o f Ras. In intact cells there is evidence that Ras can stimulate PI 3-kinase activity and is required for its optimal activation in response to growth factors (Kodaki et a i, 1994; Rodriguez-Viciana et a i, 1994). It is possible that the Ras GTP and the tyrosine phosphoprotein interactions with p 85 /p ll0 synergize to give full activation o f PI 3-kinase. The ability o f Ras to contribute to PI 3-kinase activation may be an important part o f its downstream signalling, with major implications for cellular transformation. Here we provide further details o f the nature o f the interaction between Ras and PI 3-kinase. The Ras binding site o f p i 1 0 is identified and a mutation in this region is shown to block Ras-induced activation o f PI 3-kinase, and also to elevate basal lipid kinase activity. Data are presented from an in vitro reconstitution system showing that Ras GTP can activate the lipid kinase activity o f PI 3-kinase directly, and that this effect is synergistic with tyrosine phosphopeptide binding to p85. These data support the idea that multiple cellular signalling pathways are activated by Ras, including Raf and PI 3-kinase.
4000 -oI
3000
20002II
1000
06̂0 200 400 600 800 1000
[Ras.CTP] (nM)
soooEs-
4000
3000
20002
1000
0&0 200 400 600 800 1000
(Ras.GTPJ (nM)
Fig. 1. Affinity o f Ras binding to PI 3-kinase. (A) Purified baculovirus-expressed soluble V I2 H-Ras was labelled with [y-^^PlGTP and then varying amounts were mixed with soluble purified baculovirus-expressed p85a /G S T -p !I0 a . After 1 h. the PI 3-kinase was captured on glutathione-agarose, washed and the amount o f labelled Ras bound was measured by Cerenkov counting. The background obtained when GST was used instead of p85a/G ST- p l lO a was subtracted for each point. Data shown are the average o f triplicates with standard error shown. (6 ) As above, but bacterially expressed p i 10 fragment. G ST-pIIO 133-314. was used.
ResultsInteraction o f Ras-GTP with Pi 3-kinase In order to estimate the strength o f the interaction between GTP-bound Ras and PI 3-kinase, purified baculovirus- expressed H-Ras protein was loaded with [a-^^PJGTP and then increasing amounts were allowed to bind to purified baculovirus-expressed p85/G ST-pllO in solution in the presence o f excess unlabelled guanine nucleotide. The PI 3-kinase fusion protein was recovered using glutathione-agarose, washed and the amount o f [a-^^P]GTP- labelled Ras specifically bound determined by Cerenkov counting. As shown in Figure lA , the amount o f Ras binding to PI 3-kinase saturates at ~300 nM Ras. Half- maximal binding is achieved at ~150 nM Ras, giving a rough estimate o f the binding affinity o f Ras for PI 3-kinase. A slightly weaker binding was seen using the region o f p i 10 that interacts with Ras (see below) expressed as a bacterial GST fusion protein. Figure IB shows the binding o f this PI 3-kinase fragment, inunobil-
2 4 4 3

P.Rodriguez-Vic iana et a i
OL %A
p2 1
o o o o o o o o o o o o o oO H O H O H O H O H O H O H"3 T "cn3"g"g"g"C"QH •^ io 'ü 'o
Q- O’ Q-V
Vd x?o : V)V
B>p85 I >Ras
1068
126
396
422
108
133 314
Fig. 2. Determination of the binding site for Ras on the catalytic domain of pi 10. (A) Purified deletions mutants of pi I Oct. expressed as GST fusion proteins in baculovirus, or bacterially expressed GST fusion proteins of short fragments of p llO a or P, were mixed with soluble Ras bound to either GTP or GDP. The GST fusion proteins were recovered on glutathione-agarose. washed and probed for the presence of Ras by Western blotting using anti-Ras antibodies. (B) Summary of the Ras binding behaviour of the pi 10 mutants shown in (A).
ized on glutathione-agarose, to soluble Ras protein loaded with [a-^^P]GTR The binding assays shown were carried out at close to physiological ionic strength. At low ionic strength (with omission o f the added 100 mM NaCl), the binding of Ras to p85/G ST -plI0 was ~3-fold stronger (data not shown).
Determination o f the site on p llO that binds Ras PI 3-kinase interacts with Ras through its catalytic p llO subunit: p llO will bind to Ras GTP in the absence o f the regulatory p85 subunit (Rodriguez-Viciana et a i, 1994). Addition of increasing amounts o f monomeric p85 does not inhibit the binding o f monomeric p i 10 to Ras-GTP in vitro, even though the stoichiometric formation o f p85/ pi 1 0 heterodimers can be demonstrated (data not shown). In order to determine where on the p 110a catalytic subunit Ras GTP interacts with PI 3-kinase, a number o f deletion mutants were expressed in baculovirus. One mutant, A 3- 125, lacked the minimal p85 binding site (Dhand et a i, 1994a), while others lacked larger stretches from the amino-terminus. The interaction o f these purified mutant proteins with immobilized Ras was examined: as shown in Figure 2, only A3-125 interacted with Ras, while other mutants, including A 1-395, failed to bind. This suggested that the interaction site may lie between amino acid residues 125 and 395 o f p i 10. A GST fusion protein containing most o f this area (amino acids 133-314) was therefore expressed in bacteria and was found to be able to interact with Ras in a GTP-dependent manner, thus defining amino acid residues 133-314 o f p i 10a as a sufficient binding site for Ras-GTP. A similar region o f pllOP was also expressed as a GST fusion protein in bacteria and found to interact with Ras in a GTP-dependent manner; p llo p is therefore also likely to be a target for Ras.
Effect o f a point mutation in the Ras binding site of p llO on regulation o f enzym atic activity The sequence o f the three cloned forms o f p i 10 in this region is compared in Figure 3A. Conserved residues are boxed and a consensus sequence is written below. The homology over this region is 25% identity between pi 10a and p, .18% between pi 10a and y and 20% between p llo p and y. This compares with overall homologies between the entire catalytic subunits o f 42% identity between
pi 10a and p, 36% between p i 10a and y and 34% between pi lOP and y (Hiles et a i, 1992; Hu et a i, 1993; Stoyanov et al., 1995). In order to investigate the significance of the R as-pllO interaction in the activation o f PI 3-kinase in cells, a number o f point mutations were made in p i 1 0 . One o f these, the substitution o f the conserved lysine residue 227 o f p i 10a by glutamic acid, resulted in generation o f a protein that was unable to interact with Ras. When expressed in an in vitro transcription-translation system, p i 10a K227E displayed phosphoinositide 3-OH kinase activity, suggesting that the overall folding o f the protein was not disrupted (data not shown). However, this mutation completely blocked the ability o f a GST fusion protein containing p i 10a amino acids 133-314 to bind to Ras-GTP (Figure 3B). The effect o f this mutation on the ability o f Ras to activate PI 3-kinase in intact cells was also investigated. As shown in Figure 3C, transient transfection o f either V12 Ras or p85/pllO into COS cells causes modest elevation o f the cellular levels o f PIP3 . Transfection o f both activated Ras and p85/pllO together gives a much greater than additive elevation o f PIP3 levels, suggesting that Ras is activating PI 3-kinase directly. Use o f the K227E mutant p i 10 in these transfections abolishes the synergistic activation o f PI 3-kinase by Ras, confirming that this effect relies on the interaction o f Ras with the catalytic p i 10 subunit o f PI 3-kinase. However, the basal activity o f K227E mutant p i 10 is elevated by >4-fold over that o f wild-type pi 10. This suggests the possibility that this mutation in the Ras binding site is able to give rise to a protein that is unable to interact with Ras but has undergone a conformational change that mimics Ras binding.
Effect o f interaction o f p i t 0 w ith p85 and with Ras on PI 3-kinase activity in cellsThe COS cell transient co-transfection assay was used further to explore aspects o f the regulation o f PI 3-kinase activity. As shown in Table I, the p85 regulatory subunit had minimal effect on the phosphatidylinositol (3,4) bisphosphate [PI(3,4)P;] and PIP3 levels in cells transfected with the p i 1 0 catalytic subunit or p i 1 0 plus activated Ras. Immunoprécipitation o f p i 10 through a carboxy-terminal myc epitope tag followed by immuno- blotting for p85 revealed that, in the absence o f co-
2444

Ras-PI 3 -k inase in te ra c t io n s
A14 1 pilOfr ISO pllO[5 14 3 pllOyC o n s e n s u s
RNILNVCKEA VDLRDLNSPH SRAMYVYPPN VESSPELPKH lYNKLDKGQI RKMRKFSEEK ILSLVGLSWM DWLKQTYPPE HEPS..IPEN LEDKEYGGKL RGLVTPRMAE VASRDPKLYA MHPWVTSKP........ LPEY LWKKIANNCIR ------- e- v-srd--s-- CypP- -e-s--lPe- l--Kl--g-i
191 pi 10a IWIWVIVSP NNDKQKYTLK INHDCVPEQV lAEAIRKKTR SMLLSSEQLK197 plloP . . . .IVAVHF ENCQDVFSFQ VSPNMNPIKV NELAIQKR...... LTIHGKE180 pllOy FIVI HRSTTSQTIK VSPDDTPGAI LQSFFTKMAK KKSLM....DConsensus v-v— -n t-k vspd— P — v ai-K L ------
B
Ras
241 pllOa LCVLEYQGKY ILKVCGCDEY FLEKYPLSQY KYIRSCIMLG RMPN..LMLM241 pllOP DEVSPY..DY VLQVSGRVEY VFGDHPLIQF QYIRNCVMNR ALPHFILVEC226 pllOy IPESQSEQDF VLRVCGRDEY LVGETPIKNF QWVRHCLKNG EEIHWLDTPConsensus --vs-y--dy vL-VcGrdEY --g--Pl-qf qyiR-C-mng --ph--L---
289 pllOa AKESLYSQLP MDCFTMPSYS289 pllOp CKIKKMYEQE MIAIEAAINR276 pllOy PDPALDEVRK EEWPLVDDCTConsensus -k--l-
/>
%%
%
1 b3 U
+ 5: i>4^ 02 =
it>
1
Fig. 3. Mutation in pIlO a that blocks binding to Ras. (A) Alignment of sequences from the Ras binding site of the catalytic pi 10 subunit of PI 3-kinase isoforms. The sequences of pi lOo, pi lOP and pIlOy are shown. Conserved residues are in bold. A consensus sequence is written below; residues conserved in all three sequences are in upper case, those conserved in two out of three arc in lower case. Lysine residue 227 in pi 10a is marked with an asterisk. (B) Binding to Ras o f GST fusion proteins containing residues 133-314 of pi 10a; effect of the mutation K227E. Performed as in Figure 2. (C) Effect of the K227E mutation on Ras elevation of PIP3 levels in intact COS cells. V I2 Ras was transfected into COS cells along with p85a and pi 10a. either mutant or wild-type. After 24 h, cells were metabolically labelled with p^PJorthophosphate in the absence of serum for 16 h prior to measurement of the levels o f cellular PIP3.
transfected p85, p i 10 was >95% m onom eric, or at least not bound to p 85a or (3 (data not shown). This suggests that the stimulation o f PI 3-kinase activity o f p i 10 by Ras seen in this system does not rely on signalling through p85, such as phosphotyrosine-SH 2 domain interactions. Co-transfection o f the inter-SH2 domain o f p85 (the binding site for p i 10) with p i 10 leads to a relatively weak elevation o f PI(3 ,4 )P2 and PIP3 levels, as suggested by previous reports (Hu et al., 1995). However, inclusion o f the p85 binding site o f p i 10 (amino acids 1-125) as a separate fragment, which might be expected to sequester endogenous p85, did not alter the ability o f p i 10 to elevate P1(3 ,4 )P2 and PIP3 levels either in the presence or absence o f Ras. Immunoprécipitation o f this fragment using an epitope tag showed that it was capable o f binding to endogenous or co transfected p85 (data not shown).
Furthermore, co-transfection o f a S608A mutant o f p85, which is resistant to phosphorylation by p i 10 (Dhand et a i , 1994b), did not greatly affect the activity o f PI 3-kinase in this system.
COS cell transient transfection was also used to study whether membrane localization o f Ras was required in order for it to activate PI 3-kinase. Famesylation o f Ras at C l 86 is absolutely required for it to influence the activity o f PI 3-kinase: very similar results have been reported for Ras activation o f Raf and M AP kinase (Leevers and Marshall, 1992). By contrast, removal o f the second membrane localization signal, either palmitoylation at C181 and C184 for H- and N-Ras (Hancock et a i , 1989) or a polybasic region near the carboxy-terminus for K-Ras (Hancock et a i , 1990), only slightly reduces Ras activation o f PI 3-kinase. Although these mutants are
2445

P.Rodriguez-Viciana e t al.
not associated predominantly with the plasma membrane (Hancock el a i, 1989, 1990), they do retain a significant amount of transforming activity and are still able to activate Raf and MAP kinase quite efficiently (Cadwallader el a i, 1994). It is possible that there is a large excess of Ras expressed in these assays, such that only a minor fraction at the membrane is required for activation o f PI 3-kinase: however, the absolute requirement for famesylation is clear.
Activation o f PI 3-kinase b y Ras in a purified liposome reconstitution systemPost-translationally modified wild-type Ras protein was purified from the membrane fraction o f Ras baculovirus- infected Sf9 cells. The membranes were solubilized and purified in the non-ionic detergent n-octylglucoside (40 mM). Phospholipid liposomes containing phos- phatidylserine, phosphatidylcholine, phosphatidylethanol- amine, sphingomyelin and phosphatidylinositol (4,5) bisphosphate [PI(4 ,5 )P2 ] were made based on the proced-
Table I. The levels o f F1(3.4 )P2 and PIP3 in transiently transfected ^^P metabolically labelled COS cells
Plasmids transfected Percentage o f level with V I2 Ras + p85 + p llO
PI(3,4)P2 PIP]
None 0 0p IlO a 12.7 5.1p i 10 + V I2 H-Ras 106 122pllO + p85a 10.6 6.1p i 10 + p85 -t- V I2 Ras 100 100p85 inter-SH2 + p llO 23.3 11.4p85 inter-SH2 p i 10 V I2 Ras 106 123p llO (1-125) + pllO 14.8 5.5p llO (1-125) + p llO + V I2 Ras 108 124p llO + p85 S608A 18.6 9.9p llO + p85 S608A + V I2 Ras 112 57.2p llO + V I2 Ras S 18I/I84 77.0 54.9p llO + V12 Ras S I86 7.7 2.9p llO + V I2 K-Ras 102 98.2p i 10 + V I2 K-Ras K6Q 108 84.3
Phosphoinositides were analysed by HPLC [200 000 c.p.m. of PI(4j 5)P2|. Phospholipid levels arc expressed as a percentage o f the level seen in cells transfected with V12 H-Ras plus p85 and p i 10 plasmids. Background corresponding to that seen in the same fractions from untransfected cells was subtracted. The results shown are representative o f at least three different experiments. The background subtracted was no more than 3% [800 c.p.m. for PIP3 and 600 c.p.m. for PI(3.4)P2).
urc o f Stephens el a i (1994). Reconstitution of modified Ras into the liposomes was achieved by adding a concentrated stock solution o f Ras to the liposomes in the absence o f additional detergent, such that the final detergent concentration in the mixture was only 1 mM n-octylglucoside, -5% o f the critical micellar concentration. The total lipid:detergent ratio was 1.6:1. Following mixing and incubation on ice for 1 0 min, the liposomes were collected by ultracentrifugation. As shown in Table II, -35% of the modified Ras could be recovered stably associated with the liposomes, as judged from the inclusion o f a prelabelled Ras (a-^^P]GTP tracer. Unmodified Ras was unable to associate with liposomes. When the liposome pellet was resuspended using sonication, -80% o f the associated Ras was accessible to the medium, as judged by the ability o f EDTA to exchange labelled for unlabelled GTP.
The ability o f purified baculovirus-expressed PI 3-kinase (p85ct/G ST-pII0a) to phosphorylate the PI(4,5)P2 in these liposomes was tested. As shown in Figure 4, PI 3-kinase efficiently phosphoryiated PI(4 ,5 )P2 to give PIP3 . In the absence o f Ras, the activity o f the lipid kinase could be stimulated -3 -fo ld by the addition o f a tyrosine phosphopeptide based on the autophosphorylation site Y 751 o f the human PDGF receptor p. Similar stimulations were seen using a tyrosine phosphopeptide based on the autophosphorylation site Y740 o f the PDGF receptor, and a slightly stronger stimulation using a doubly phosphoryiated peptide spanning both sites. The principal effect o f using a doubly, as opposed to a singly, phosphoryiated peptide was to reduce the concentration o f peptide required to give maximal stimulation o f lipid kinase activity by > lO-foId: the optimal stimulation achieved was not greatly different with the various phosphopeptides. In contrast, a tyrosine phosphopeptide based on the sequence around Tyrl068 o f the epidermal growth factor (EGF) receptor, which binds to the SH2 domain o f Grb2 but not p85, has no influence on the activity o f PI 3-kinase in this system at concentrations up to 30 pM (data not shown).
In order to study the effect o f Ras on PI 3-kinase activity, Ras reconstituted into liposomes was loaded with either GTP or GDP in the absence o f magnesium. PI 3-kinase phosphorylation o f PI(4 ,5 )P2 to give PIP3 was then measured. As shown in Figure 5A, GDP-bound Ras did not significantly affect the activity o f PI 3-kinase in this system. GDP or GTP alone were also without effect in the absence o f Ras. However, GTP-bound Ras caused
Table II. Reconstitution o f Ras into liposomes
Percentage o f total input
Modified Ras Unmodified Ras No Ras
Total Ras [ot-^^PjGTP addedRas [a-^^P]GTP associated with liposomes(a-^^PJGTP dissociating from liposomes in the presence o f EDTATotal [a-^^P]GTP added without Ras[a-^^P]GTP associated with liposomes[a-^^P]GTP dissociating from Ras in liposomes in the presence o f EDTA
100 100 34.8 ± 0.6 0.4 ± 0.1 28.3 ± 0.3 0.2 ± 0.04
1000.090.02
Post-translationally modified Ras. or unmodified Ras, was labelled with (a-^^PlG TP and reconstituted into liposomes as described in Materials and methods. Ras associating with liposomes was estimated by counting anti-Ras monoclonal antibody Y 13-259 immunoprecipitates o f solubilized washed liposomes. Total Ras input was also measured by immunoprécipitation. L.abel released with EDTA and the ‘No Ras’ controls were measured by direct counting o f liposome or supernatant fractions.
2446

R a s PI 3 -k in a se i n te r a c t io n s
A
■ctICL
2000
Y740Y75I
0 10 2 0 3 0 4 0 SO 6 0 7 0 8 0 9 0 100
5000
1
!Ou
3000
2000
z1000,
(Tyrosine phosphopeptide Y740] (pM)
GDPOTPRas.GDPRas.GTP
(Tyrosine phosphopeptide] (pM )
4000
6es
3000
II 2000
D.2CL 1000
(Tyrosine phosphopeptide 740/751] (pM )
Fig. 4. Tyrosine phosphopeptide stimulation o f kinase activity o f p85o/G ST-pl lOa towards PI(4.5)P2-containing liposomes.(A) Purified p85(X/GST-pllOa was added to PI(4 .5)P2-containing liposomes in the presence of increasing concentrations o f singly phosphorylated tyrosine phosphopeptides. based on the human PDGF receptor P autophosphorylation sites Y740 and Y751, and (y-^’PJATP. Levels o f PIP3 produced were determined by TLC. (B) As (A), but the doubly phosphorylated tyrosine phosphopeptide Y740/Y751 was used. Average o f duplicates with standard errors shown. Representative o f four experiments.
a significant increase in the activity o f PI 3-kinase. The Ras GTP-induced stimulation observed in the absence o f phosphopeptide Y740 was ~2.5-foId and was similar in the presence o f saturating concentrations o f peptide. However, the activation o f PI 3-kinase activity by Ras-GTP was most marked at low concentrations o f tyrosine phosphopeptide. For example, at 1 (iM Y740 phosphopeptide there was only a small stimulation o f PI 3-kinase activity in the absence o f Ras-GTP, but maximal stimulation in the presence o f Ras-GTP, a stimulation by Ras-GTP of >4-fold. Similar results are seen for the Y751 phosphopeptide (Figure 5B): in the absence o f peptide the Ras- induced stimulation was 3-fold, while in the presence o f saturating peptide Ras-GTP stimulated 3.5-fold. At 1 p,M Y 751, Ras-GTP induced a 5-fold activation o f PI 3-kinase, Comparable results were found for the doubly phosphorylated peptide, including the leftward shift o f the d ose- response curve induced by Ras-GTP (data not shown). To be certain that this effect was due to Ras and not contaminants in the Ras preparation, the neutralizing Ras monoclonal antibody Y 13-259 was added to the Ras preparation prior to reconstitution. Excess antibody was removed during the preparation o f the Ras liposomes. As shown in Figure 5B, treatment o f Ras in this way with
(000
4000
2Z
3 0
[Tyrosine phosphopeptide Y751] (pM)
Ras.GDPRas.GTPRas.GTP,259Ras.GTP,238
Fig. 5. Effect o f Ras-GTP on PI 3-kinase activity in the liposome system. (A) The kinase activity o f p 8 5 a /G S T -p llO a towards P1(4 ,5)P2 was measured in liposomes into which had been reconstituted post-translationally modified Ras that was bound to GTP or GDP. The influence o f increasing concentrations o f PDGF receptor p Y740 phosphopeptide was determined. The effect o f GTP or GDP alone in the absence o f Ras was also measured. Average o f triplicates with standard error shown. Representative o f five experiments. (B) The influence o f anti-Ras monoclonal antibodies Y 13-259 and Y 13-238 on the stimulation o f PI 3-kinase activity by Ras-GTP was measured as in (A), except that tyrosine phosphopeptide Y751 was used. Average of duplicates with standard error shown. Representative o f three experiments.
Y 13-259, but not the non-neutralizing Y 13-238 antibody, blocked the ability o f Ras-GTP to stimulate PI 3-kinase activity. These data indicate that GTP-bound Ras can activate the enzymatic activity o f PI 3-kinase in a defined system, and that the interaction is synergistic with tyrosine phosphopeptides that bind to the SH2 domains o f p85.
DiscussionThe data presented here provide new evidence for the importance o f the interaction between Ras and PI 3-kinase in the control o f cell growth. We have established that the interaction with full-length p85/pllO occurs with a relatively high binding affinity o f ~ 150 nM for V 12 H-Ras. This compares with ~50 nM for the interaction o f this transforming Ras mutant with the amino-terminal part o f c-R afl (Herrmann et a i, 1995), which is thought to be stronger than the interaction with full-length Raf. Given the established physiological importance o f the Ras-Raf interaction, the strength o f the interaction between Ras and PI 3-kinase suggests that it is at least a good candidate for a biologically significant effector pathway. It should be noted, however, that biologically significant interactions
2447

P.Rodriguez-Viciana et al.
need not be o f high affinity: Ras-GTP binds to p i20^^’’ with an affinity o f ~2 p.M (Vogel et al., 1988).
The site of the interaction o f Ras with PI 3-kinase is shown here to be between residues 133 and 314 on the catalytic pi 10 subunit, with lysine residue 227 being essential for the interaction. This is a region immediately carboxy-terminal to the site o f interaction between p i 1 0
and the regulatory subunit p85. This suggests a model in which the lipid kinase receives regulatory signals through two domains in its amino-terminal region. One signal comes from tyrosine phosphoproteins, and possibly from SH3 domains o f Src family kinases and from Rho family proteins, through p85 and its interaction with the first 150 amino acids o f p i 10. The other signal comes from the direct interaction of Ras-GTP with the neighbouring region of p i 10, roughly spanning the next 150 amino acid residues. It is likely that optimal activation o f PI 3-kinase requires input from both signalling pathways. Since growth factors such as PDGF stimulate both tyrosine phosphorylation o f p85 binding sites and activation o f Ras, both these pathways will be activated in response to the growth factor, allowing potentiation o f the activation signals being transmitted to PI 3-kinase. This is analogous to the stimulation o f MAP kinase by growth factors: multiple signalling pathways feeding into MAP kinase activation are triggered by a single growth factor, including Ras activation, protein kinase C activation and intracellular calcium elevation (Burgering et a i, 1993). Ras contributes to Raf activation by translocating it to the plasma membrane (Leevers et a i, 1994; Stokoe et a i, 1994), while protein kinase C can phosphorylate and activate Raf directly (Sozeri et a i, 1992; Kolch et a i, 1993). It is possible that this growth factor-induced activation o f multiple signalling pathways, that then synergize together to activate Ras effectors such as Raf and PI 3-kinase, allows the generation o f strong downstream signals from activation o f small numbers o f receptors. It may also allow for the greatest flexibility in the range o f responses generated by different growth factors that activate seem ingly similar sets o f signalling pathways.
It is now clear that a number o f different forms o f growth factor-regulated PI 3-kinases exist, including pi 10a, P and y. We have shown here that both p i 10a and P interact with Ras in a GTP-dependent manner. The sequence similarity o f p llO y to p i 10a and P in the Ras binding site indicates that it too is likely to interact with Ras. In this case, stimuli that activate heterotrimeric G proteins could activate p llO y synergistically both through direct interaction and through stimulation o f Ras (Koch et a i, 1994), in a situation analogous to that described above for tyrosine kinase pathways.
The behaviour o f the K227E mutant o f p i 10 establishes not only that this residue is essential for interaction with Ras, although not for in vitro basal PI 3-kinase activity of the catalytic subunit by itself, but also that the direct interaction o f Ras with PI 3-kinase is necessary for the elevation o f PIP3 levels in COS cells transfected with activated Ras and p85/pllO. This indicates that Ras- induced production o f autocrine growth factors is not sufficient by itself to cause the increase in PIP3 seen, but that direct interaction with Ras is also required, supporting the model in which multiple signals are required for PI 3-kinase activation, including direct interaction o f Ras with
pi 1 0 and interaction of p85 with tyrosine phosphoproteins. The fact that the basal activity of K227E pi 10 is high in Cos cells suggests that this reversal o f charged residues at an essential site for the Ras-pl 10 interaction may cause a conformational change that in some ways mimics that occurring when Ras binds. We currently are exploring whether less radical amino acid changes result in a PI 3-kinase that does not bind Ras but also does not have elevated basal activity.
Use o f the in vitro reconstitution system described here clearly shows that Ras interaction with PI 3-kinase can lead to activation o f its lipid kinase activity. This appears to occur synergistically with the previously described activation o f PI 3-kinase by tyrosine phosphopeptide interaction with the SH2 domains o f the p85 subunit, with the strongest effects being seen at suboptimal phosphopeptide concentrations, although Ras stimulation o f PI 3 -^nase activity is seen both in the absence o f tyrosine phosphopeptide and in the presence o f saturating amounts of peptide. The exact mechanism by which this stimulation is achieved is not clear: one possibility is a simple translocation o f PI 3-kinase to the liposome where both Ras and its substrate P1(4,5)P% are located. The stimulation is dependent on the post-translational modification o f Ras: this is not needed for Ras binding to p i 10 but is essential for Ras to associate with the liposomes. Bacterially expressed Ras will bind to PI 3-kinase but w ill not stimulate its activity in the liposome system. However, there is precedent for Ras famesylation being required for a productive interaction with another protein, Sos (Porfiri et a i, 1994), although it is not absolutely required for binding (Liu et a i, 1993a). In addition, the activation of the kinase activity o f p l 10 by the mutation K227E suggests that a conformational change might also be involved, as does the fact that the presence o f Ras-GTP alters the concentration dependence o f the stimulation o f the kinase by tyrosine phosphopeptides. The Ras-induced change in the concentration dependence for tyrosine phosphopeptide could reflect an increase in the affinity o f the SH2 domains o f p85 for the tyrosine phosphopeptide induced by Ras-GTP. Since Ras binds to p llO at the site next to that where pllO binds to p85, this is perhaps not unlikely. Whatever the mechanism, the stimulation o f PI 3-kinase activity by Ras reported here is the first time that the enzymatic activity o f a mammalian effector o f Ras has been shown to be controlled by Ras in a completely defined system. No regulation o f the protein kinase activity o f purified Raf by interaction with Ras-GTP has yet been reported.
It has been suggested that the Rho family protein Rac lies on a signalling pathway downstream o f PI 3 -kinase which controls, amongst other things, the polymerization of actin beneath the plasma membrane to create membrane ruffles (Ridley et a i, 1992; Kotani et a i, 1994; Wennstrôm et a i, 1994a,b). Activated Ras can induce activation of Rac, at least as judged by downstream morphological events (Ridley et a i, 1992). PIP3 may be able to control the activation state o f Rac through decreasing Rac- GAP activity or increasing Rac exchange factor activity (Hawkins et a i, 1995). A recent report has shown that an activated mutant o f Rac has some transforming activity, which can synergize strongly with activated Raf, but not Ras (Qiu et a i, 1995), suggesting that both Rac and the
2448

R s s—PI 3-k in ase in te r a c t io n s
MAP kinase pathway are separate effector mechanisms used by Ras. Taken together, these reports plus the data presented here suggest that Ras activates the MAP kinase pathway through interaction with Raf and the Rac pathway through interaction with PI 3-kinase, and that the function o f both of these pathways is required for efficient transformation o f fibroblasts by Ras. For the activation o f both Raf and PI 3-kinase pathways in Ras-transformed cells, it is likely that direct Ras-effector interaction is potentiated by the autocrine production o f growth factors leading to receptor tyrosine kinase activation. In support o f the possibility that PI 3-kinase is required for cell proliferation and could be playing an essential role downstream of Ras, it has also been reported that microinjection o f neutralizing antibodies against PI 3-kinase blocks the ability o f a number o f growth factors to induce DNA synthesis in fibroblasts (Roche et a i, 1994).
Altogether, the data presented here provide strong evidence for a physiological role for PI 3-kinase in addition to Raf in signalling downstream o f Ras. At least five different effector systems have now been identified that might operate downstream o f Ras, raising the possibility that a very complex set o f signals emerges from Ras which could co-operate in a various ways to cause cellular transformation. At present, it is not clear how important other putative effectors o f Ras are in its control o f cell growth, but it is clearly possible that GAPs, RalGDSs, MEKKs or PKCÇ could also be contributing to the overall phenotype o f Ras transformation. When strongly activated mutants o f Ras are highly expressed as in a transformation assay, it is possible that just one potently activated effector pathway is required for measurable loss o f growth control. This may be very different from the situation in normal cells stimulated with physiological levels o f growth factors where the expression level o f Ras is low and its activation is moderate and transient: under these circumstances, function o f several signalling pathways downstream of Ras may be required in order for cell proliferation to result.
Materials and methodsExpression and purification o f proteinsUnmodified wild-type and V12 c-Ha-Ras was purified from a detergent- free homogenate o f recombinant baculovirus-infected S(9 cells as described in Page et al. (1989). Post-translationally modified wild-type Ras was purified from the particulate pellet from the above cells. The pellet was solubilized in the same homogenization buffer to which 40 mM n-octylglucoside had been added. After clarification by centrifugation at 350 000 g for 10 min, the solubilized Ras was purified as in Page et al. (1989) but with all buffers including 40 mM n-octylglucoside. To prepare PI 3-kinase, Sf9 cells were co-infected with G S T -p llO a and p85a bacuioviruses and the G ST-pllO /p85 complex purified according to the manufacturer's protocol.
Amino-terminal deletion mutants o f p l 10a were made by Exolll deletion o f bovine p llO , inserted in the pAcG3 vector (Davies et al., 1993).
plIO constructs, pl 10 in pBluescript was digested with F sp l, a BamHl linker ligated and, after digestion with BamHl, the resulting fragment ligated into the BamHl site o f pcDNA3 (Invitrogen).The 9E I0 epitope was inserted at the amino-terminus o f p lIO by PCR. Point mutations in pl 10 and p85 were done using the U.S.E. in vitro mutagenesis kit (Pharmacia) according to the manufacturer's protocol. For COS transfections. all the p l 10 variants were subcloned into the BamHl site o f pSGS (Stratagene).
The p85 binding domain o f p llO (amino acids 1-108) and the inter- SH2 domain o f p85 (amino acids 358-617) were amplified and a 9E I0
epitope inserted by PCR. and cloned into the BamHl site o f pcDNA.V For transfections they were subcloned as Kpnl-Xhal fragments into pMT2.
Interaction o f Ras w ith Pl 3-kinase in vitro Either baculovirus-expressed p85o/G ST-pl lO a or bacterially expressed G S T -p l 10 133-314 was used in solution at 50 nM. The binding buffer used was 50 mM HEPES pH 7.5, 100 mM NaCI. 0.1 mg/ml bovine serum albumin (BSA), 5 mM MgClj. To determine the affinity o f the R as-PI 3-kinase interaction, unmodified Ras was loaded with (yA^P)GTP (36 pCi per set o f 30 assays, diluted with 3 pmol o f cold GTP). Varying concentrations o f Ras were incubated with Pl 3-kinase in 50 pl assays for I h at 4*C. Five pl o f glutathione-agarose was then added per assay for 30 min at 4"C with shaking, and then the beads were washed with 4X 1 ml o f cold 50 mM HEPES pH 7.5, 100 mM NaCI, 0.1 mg/ml BSA, 5 mM MgCiyO. I % Triton X-KX). and the pellets counted. Backgrounds without Pl 3-kinase were subtracted.
For detection o f Ras binding to PI 3-kinase by immunoblotting, assays were performed as above, using 1 pM Ras which was bound to either unlabelled GTP or GDP. Ras stably associated with PI 3-kinase was detected by immunoblotting using pan-Ras antibody (Oncogene Science).
M easurem ent o f PIP3 in COS-7 cellsCells were harvested, washed and resuspended at 5X10* in HeBS and transfected by electroporation (250 V, 125 pF) with the indicated plasmids and 50 pg o f sonicated herring sperm DNA. The amount of plasmid in different transfections was made equal with empty vector. Cells were allowed to stand at room temperature for 10 min and seeded in a 60 mm dish. At 40-48 h after transfection, cells were washed with phosphate-free DMEM and labelled overnight with 200 pCi [^^PJorthophosphate per dish. Reactions were terminated and samples analysed as described in Chung et al. (1992). One-tenth o f the combined CHCI5 phase was run on an oxalated silica gel 60 TLC plate, using a CHClj:methanol:acetone:acetic acid:water (40:13:15:12:7) system. The rest o f the CHCI3 phase was dried in a Speedvac and deacylated by incubating for 60 min at 5 0 'C with methylamine. The dried pellet was resuspended in 500 pl o f butanol and then re-extracted twice with 500 pl o f water. Samples were run on a Spherisorb HPLC column (Anachem) and fractions counted by Cerenkov emission. Expression o f each protein was checked by Western blotting o f whole cell lysates with the appropriate antibodies.
Reconstitution o f Ras into liposom esLiposomes were made by sonication of dried lipids in kinase buffer (40 mM HEPES pH 7.5, 1 mg/ml fatty acid-free BSA. 2 mM EGTA. 100 mM NaCI, 5 mM MgCl%, 50 pM ATP). The final lipid concentration was: 640 pM phosphatidylethanolamine, 600 pM phosphatidylserine, 280 pM phosphatidylcholine. 60 pM sphingomyelin, 60 pM PI(4 .5)P2. Sonication was on ice for 4X 20 min with a 4 mm probe at 15 pm amplitude in an MSE Soniprep 150. Modified Ras was added to the liposomes at a final concentration o f 1 pM and incubated on ice for 20 min. Liposomes were collected by centrifugation at 350 000 g for 20 min. The supernatant was removed and liposome pellet resonicated in the same volume o f kinase buffer. To load Ras with nucleotide, the mixture was made 10 mM in EDTA, and then 20 pM GTP or GDP added. After incubation at 30°C for 10 min, 15 mM M gC l; was added. W hen antibodies were used to block Ras effects, 300 pg/m l o f purified monoclonal antibody was added to the initial incubation o f Ras with liposomes.
Pi 3-kinase assays in liposom esLiposome mixture was made as described above, either with or without Ras. Tyrosine phosphopeptides were added as indicated, along with 10 ng o f p85a/G S T -p ll()a and I pCi (y-^^P]ATP per point. The total assay volume was 25 p l, which represented a 50% dilution o f the liposome preparation. Assays were mixed continuously for 20 min at 20*C, then stopped by adding 400 p l o f chloroform:methanol ( 1:2) and 100 p l o f 2.4 M HCl. After vortexing and centrifugation (15 000 g, 3 m in). the lower chloroform layer was put into fresh tubes and dried down in a Speedvac. The residue was dissolved in 25 p l o f chloroform and run on a silica TLC plate in chloroform:methanol:acetone:acctic acid:water (40:13:15:12:7). The P lP j spot was quantitated using a Molecular Dynamics Phosphorlmager.
AcknowledgementsChristophe Pierreux is thanked for his help in the construction of the p i 10 ExolII deletion mutants. Thanks to Anne Vojtek and Jonathan
2449

P .Rodriguez-V ic iana e t al.
Cooper for sharing unpublished data. B.V. is supported in part by the Belgian National Fund for Scientific Research.
ReferencesBrown.E.J.. Albers.M.W.. Shin.T.B.. Ichikawa.K.. Keith.C.T., Lane.W.S.
and Screiber.S.L. (1994) _A mammalian protein targeted by G I- arresting rapamycin receptor complex. Nature, 369. 756-758.
Burgering.B.M.T., de Vrics-Smits.A.M.M.. Mcdema,R.H.. van Weeren. PC.. Tcrtoolen.L.G.J. and Bos.J.L. (1993) Epidermal growth factor induces phosphorylation o f extracellular signal-regulated kinase 2 via multiple pathways. Mol. Cell. Biol., 13. 7248-7256.
Cadwallader.K.A.. Paterson.H.. MacDonald.S.G. and HancockJ.F.(1994) N-terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localisation. Mol. Cell. Biol., 14. 4722^730 .
Chang.E C.. Barr.M.. Wang.Y.. Jung.V., Xu.H.P. and Wigler.M.H. (1994) Cooperative interaction o f S.pombe proteins required for mating and morphogenesis. Cell, 79, 131-141.
Chung J . . Kuo.CJ.. Crabtree.G.R. and B lenisJ. (1992) Rapamycin-FKBP specifically blocks growth-dependent activation o f and signalling by the 70 kDa S6 protein kinases. Cell, 69. 1227-1236.
Davies.A.H.. Jow ettJ.B .M . and Jones.I.A. (1993) Creation o f ExoIIl deletion mutants. Bio/Tecfmology, 11. 933-936.
Dent.P.. Jelinek.T.. Morrison.D.K.. W ebcr.MJ. and Stui^gill.T.W. (1995) Reversal o f raf-1 activation by purified and membrane associated protein phosphatases. Science, 268. 1902-1906.
Dhand.R.. Hara.K.. Hiles.I., Bax.B.. Gout.1.. Fanayotou.G.. Fry.M.).. Yonezawa.K.. Kasuga.M. and Waterfield.M.D. (1994a) PI 3-kinase: structural and functional analysis o f intersubunit interactions. EMBOJ., 13.511-521.
Dhand.R.. Hiles.I.. Fanayotou.G., Roche.S.. Fry.M J.. Gout.I.. Totty.N.F., Truong.O., Vicendo.P., Yonezawa.K., Kasuga,M. and Waterfield.M.D. (1994b) PI 3-kinase is a dual-specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. EMBO J., 13. 522-533.
Diaz-Meco.M.T.. Lozano J . . Municio.M.M.. Bena.E.. Frutos.S., Sanz,L. and M oscatJ. (1994) Evidence for the in vitro and in vivo interaction o f ras with protein kinase C zeta. J. Biol. Chem., 269. 31706-31710.
EscobedoJ.A.. Navankasattusas.S.. Kavanaugh.W.M., Milfay.D.. Fried. V A. and Williams.L.T. (1991) cDNA cloning of a novel 85 kDa protein that has SH2 domains and regulates binding o f PI3-kinase to the PDGF beta-receptor. Cell, 65. 75-82.
HancockJ.F.. Magee.A.I., ChildsJ.E . and M arshall.CJ. (1989) All ras proteins arc polyisoprenylated but only some are palmitoylated. Cell, 57. 1167-1177.
Hancock J.F.. Paterson.H. and M arshall.CJ. (1990) A polybasic domain or palmitoylation is required in addition to the CAAX m otif to localize p21 ras to the plasma membrane. Cell, 63, 133-139.
Hanley.K.O., Gell,D., Smith,G.C.M., Zhang.H., Divecha,N., Connelly, M.A., Admon,A., Leesmiller,S.P., Anderson.C.W. and Jackson,S.P.(1995) DNA-dcpendent protein-kinase catalytic subunit— a relative o f phosphatidyl!nositol 3-kinase and the ataxia-telangiectasia gene product. Cell, 82, 849-856.
Hawkins.P.T., Eguinoa.A., Qiu,R.G., Stokoc.D., Cooke,F.T., Walters,R., Wennstrom.S., Claesson-Welsh,L., Evans.T., Symons.M . and Stephens.L. (1995) PDGF stimulates an increase in GTF-rac via activation of phosphoinositide 3-kinase. Curr. Biol., 5, 393-403.
Hayashi,H., Nishioka,Y, Kamohara,S., Kanai.F., Ishii.K., Fukui.Y., Shibasaki,F., Takenawa,T., Kido.H., Katsunuma,N. and Ebina,Y. (1993) The alpha-type 85-kDa subunit o f phosphatidylinositol 3- kinase is phosphorylated at tyrosine 368, tyrosine 580. and tyrosine 607 by the insulin receptor. J. Biol. Chem., 268. 7107-7117.
Herrmann.C., Martin.G.A. and Wittinghofer.A. (1995) Quantitative- analysis o f the complex between p2 l(ras) and the ras-binding domain o f the human raf-1 protein-kinase. J. Biol. Chem., 270. 2901-2905.
Hiles.LD.. Otsu.M.. Volinia.S., Fry.M J., Gout.I.. Dhand.R.. Fanayotou.G.. Ruiz-Larrea,F., Thompson.A., Totty.N.F. and Waterfield.M.D. (1992) Phosphatidylinositol 3-kinase: structure and expression o f the 110 kDa catalytic subunit. Cell, 70. 419-429.
Hofcr.F.. Ficlds.S.. Schneider.C. and Martin,G.S. (1994) Activated ras interacts with the ral guanine-nucleotide dissociation stimulator. Proc. Natl Acad. Sci. USA, 91. 11089-11093.
Hu.P. Mondino.A.. Skolnik.E.Y. and SchlessingerJ. (1993) Cloning of a novel ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol. Cell. Biol., 13. 7677-7688.
Hu.Q.. Klippci.A.. Musiin.A J-. F-anti.W J. and Williams.L.T. (1995) Ras dependent induction of cellular responses by constitutively active phosphalidylinositol-3 kinase. Science. 268, I (XT 102.
Kavanaugh.W.M.. Turck.C.W.. KlippeI.A. and Williams.L.T. (1994) Tyrosinc-508 of the 85-kilodalton subunit o f phosphatidylinositol kinase is phosphorylated by the platelet-derived growth-factor receptor. Biochemistry. 33. 110 4 6 -11050.
Kikuchi.A.. Demo.S.D.. Yc.Z.H., Chcn.Y.W. and W illiams.L.T (1994) Ral-gds family members interact with the effector loop of ras p2 l. Mol. Cell. Biol., 14. 7483-7491.
K och.W J.. Hawes.B.E.. A llen.LF. and Lefkowitz.R.J. (1994) Direct evidence that g(i)-coupled receptor stimulation o f mitogen activated protein kinase is mediated by g beta gamma activation o f p2 l(ras). Proc. Natl Acad. Sci. USA, 91. 12706-12710.
Kodaki.T.. Wosholski.R.. Hallberg.B.. Rodriguez-Viciana.P. Down ward J . and Parker.PJ. (1994) The activation o f phosphatidylinositol 3-kinase by Ras. Curr. Biol., 4. 798-806.
Kolch.W.. Heidecker.G.. Kochs.G.. HummeI.R.. Vahidi.H.. Mischak.H.. Finkenzeller.G., Marme.D. and Rapp.U. (1993) Protein kinase C activates Raf-1 by direct phosphorylation. Nature, 364, 249-252.
Kotani,K., Yonezawa.K.. Hara.K.. Ueda.H.. Kitamura.Y.. Sakaue.H.. Ando.A.. Chavaneau.A.. Calas.B.. Grigorescu.F., Nishiyama.M.. Waterfield.M.D. and Kasuga.M. (1994) Involvement o f phosphoinositide 3-kinase in insulin or IGF-1 induced membrane ruffling. EMBOJ., 13. 2313-2321.
K unzJ.. Henriquez.R., Schneider.U.. Deuter-Reinhard.M.. Movva.N.R. and Hall.M.N. (1993) Target o f rapamycin in yeast. TOR2. is an essential phosphatidylinositol kinase homolog required for G I progression. Cell, 73. 585-596.
Lee vers,S. J. and M arshall,CJ. (1992) Activation o f extracellular signal- regulated kinase ERK2 by p21 ras oncoprotein. EMBOJ., 11,569-574.
Leevers,S.J., Paterson,H.F. and M arshall.CJ. (1994) Requirement for ras in raf activation is overcome by targeting raf to the plasma- membrane. Nature, 369. 411-414.
Liu.B.X., Wei.W. and Broek.D. (1993a) The catalytic domain o f the mouse sosi gene product activates Ras proteins in vivo and in vitro. Oncogene, 8 . 3081-3084.
Liu.X.. Marengere.L.E.M.. Koch.C.A. and Pawson.T. (1993b) The v-src SH3 domain binds phosphatidylinositol 3 '-kinase. Mol. Cell. Biol., 13. 5225-232.
Lowy.D.R. and Willumscn.B.M. (1993) Function and regulation o f RAS. Annu. Rev. Biochem., 62. 851-891.
Marais.R.. L ight,Y, Paterson.H.F. and M arshall.CJ. (1995) Ras recruits raf-1 to the plasma-membrane for activation by tyrosine phosphorylation. EMBO J., 14. 3136-3145.
M arshall.C J. (1994) MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev., 4, 82-89.
Moodie,S.A., Willumsen,B.M., W ebcr,M J. and Wolfman.A. (1993) Complexes o f Ras-GTP with Raf-1 and mitogen activated protein kinase kinase. Science, 260, 1658-1661.
Otsu,M ., Hiles,!., G outJ., Fry,M J., Ruiz-Larrea.F., Fanayotou.G., Thompson,A., Dhand.R., H suanJ., Totty.N. and Waterfield.M.D. (1991) Characterization o f two 85 kDa proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3- kinase. Cell, 65, 91-104.
Page,M J., Hall,A., Rhodes,S., Skinner.R H., M urphy,V, Sydenham,M. and Lowe.P.N. (1989) Expression and characterization o f the Ha ras p2 I protein produced at high levels in the insect/baculovirus system. J. Biol. Chem., 264, I9 I4 7 -I9 I5 4 .
Pleiman.C.M ., Hertz,W.M. and Cam bierJ.C. (1994) Activation o f phosphatidylinositol 3-kinase by src family kinase SH3 binding to the p85 subunit Science, 263, 1609-1612.
Porfiri.E., Evans,T , Chardin,?, and Hancock J.F . (1994) Prénylation o f Ras proteins is required for efficient hSos I-promoted guanine nucleotide exchange. J. Biol. Chem., 269, 22672-22677.
Prasad.K .VS., Janssen.O., Kapeller.R.. Cantley.L C. and Rudd.C.E.(1993) The p59fyn SH3 domain mediates binding to phosphatidylinositol 3-kinase in T cells. Proc. Natl Acad. Sci. USA, 90, 7366-7370.
Qiu.R.G., C henJ., Kim,D., McCormick.F. and Symons.M. (1995) An essential role for rac in ras transformation. Nature, 374, 457-459.
Rapp.U.R. (1991) The Raf serine/threonine kinases. Oncogene, 6 , 495-500.
Ridley. A., Paterson.H.F.. Johnston.C.L.. Dickmann.D. and Hall.A. ( 1992) The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell, 70. 401-410.
2450

R as-PI 3 -k in a se in te ra c t io n s
Kothe.S.. KocgI.M. and Counncidgc.S.A. ( ly y j) The phosphatidyl- inosiioi .Vkinasc ci is required for DNA synthesis induced hy some, hut not all. growth factors. Proc. Nutl Acad. Sci. USA. 91. 9IK 5-9I89.
Rodriguez-Viciana.P.. Warnc.P.H., Dhand.R.. Vanhaescbroeck.B., Gout.I.. Fry.M.J.. Waterfield.M.D. and Downward.]. (1994) Phosphatidylinositol-3-OH kinase as a direct target o f Ras. Nature. 370. 527-532.
Russell.M.. Lange-Carlcr.C. and Johnson.G.L. (1995) Direct interaction between ras and the kinase domain o f mitogen activated protein kinase kinase kinase (m ekki). J. Biol. Chem.. 270. 11757-11760.
Sabatini.D.M.. Erdjument-Bromagc.H.. Lui.M.. Tempst.P. and Snydcr.S.H. (1994) RAFTI: a mammalian protein that binds to FKBPI2 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 78. 35-43.
Savitsky.K.. Barshira.A.. Gilad.S.. Rotman.G.. Ziv.Y.. Vanagaite.L.. Tagle.D.A.. Smith.S., UzieI.T., Sfez.S. and Shiloh.Y. (1995) A single ataxia-telangiectasia gene with a product similar to PI-3 kinase. Science. 268. 1749-1753.
Schu.P.V.. Takegawa,K.. Fry.M J., S tackJ.H .. Waterfield.M.D. and Emr.S.D. (1993) Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 260. 88-91.
Skolnik.E.Y.. MargoIis.B.. Mohammadi,M., Lowenstein.E.. Fischer.R.. Drepps.A.. Ullrich,A. and SchlessingerJ. (1991) Cloning o f PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell. 65. 83-90.
Sozeri.O.. VoIlmer.K., Liyanage.M., Frith.D.. Kour.G.. Mark.G.E. and StabeI.S. (1992) Activation of the c-Raf protein kinase by protein kinase C phosphorylation. Oncogene. 7. 2259-2262.
Spaargaren.M. and BischoffJ.R. (1994) Identification o f the guanine- nucleotide dissociation stimulator for ral as a putative effector molecule o f r-ras. h-ras. k-ras. and rap. Proc. Natl Acad. Sci. USA. 91.12609-12613.
Stephens.L.R.. Jackson.T.R. and Hawkins.P.T. (1993) Agonist-stimulated synthesis o f phosphatidyIinositol(3.4.5)-trisphosphate: a newintracellular signalling system. Biochim. Biophys. Acta. 1179. 27-75.
Stephens.L.. Smrcka.A., Cooke.F.T, Jackson.T.R.. Stemweis.P.C. and Hawkins.P.T. (1994) A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein Py subunits. Cell. 77. 83-93.
Stokoe.D.. MacDonaId.S.G.. Cadwallader.K.. Symons.M. and Hancock J.F . (1994) Activation o f raf as a result o f recruitment to the plasma-membrane. Science. 264. 14 6 3 -1467.
Stoyanov.B., Volinia.S., Hanck.T. Rubio.L. Loubtchenkov.M.. Malek.D.. Stoyanova.S.. Vanhaesebroeck.B.. Dhand.R.. Numbcrg.B.. Gierschik.P. Scedorf.K., H suanJJ.. Waterfield.M.D. and Wetzker.R.(1995) Cloning and characterization o f a G protein activated human phosphoinositide-3 kinase. Science. 269. 6 9 0 ^ 9 3 .
Toda.T. Uno.I.. lshikaw a.T. Powers.S.. K ataoka.T . Broek.D.. Cameron.S., Broach J . . Matsumoto.K. and Wigler.M. (1985) In yeast Ras proteins are controlling elements o f adenylate cyclase. Cell. 40. 27-36.
Tolias.K.F., Cantley.L.C. and Carpcnter.C.L. (1995) Rho-family GTPases bind to phosphoinositide kinases. J. Biol. Chem.. 270. I7656-I7659.
Van Aelst.L.. Barr.M.. Marcus.S.. Polverino.A. and Wigler.M. (1993) Complex formation between Ras and Raf and other protein kinases. Proc. Natl Acad. Sci. USA. 90, 6213-6217.
VogeI.U.S.. Dixon.R.A.. Schaber,M.D.. Diehl.R.E., Marshall.M.S.. Scolnick.E.M.. Sigal.I.S. and GibbsJ.B. (1988) Cloning o f bovine GAP and its interaction with oncogenic ras p21. Nature. 335. 90-93.
Vojtek.A.B.. Hollenberg.S.M. and CooperJ.A. (1993) Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 74. 205-214.
W ang.Y. Xu.H.-P, Riggs.M., Rodgers.L. and Wigler.M. (1991) byr2. a Schizosaccharomyces pombe gene encoding a protein kinase capable o f partial suppression o f the ras I mutant phenotype. Mol. Cell. Biol..11. 3554-3563.
Wame.PH.. Vicinia.P.R. and DownwardJ. (1993) Direct interaction o f Ras and the amino-terminal region of Raf-1 in vitro. Nature. 364. 352-355.
Wennstrom.S.. Hawkins.P, Cookc.F.. Hara.K.. Yonezawa.K.. Kasuga.M.. Jackson.T. Claesson-Welsh.L. and Stephens.L. (1994a) Activation o f phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr. Biol.. 4. 385-393.
Wennstrom.S.. Siegbahn.A.. Yokote.K.. Arvidsson.A.-K.. Heldin.C.-H.. Mori.S. and Claesson-Welsh.L. (1994b) Membrane ruffling and chemotaxis transduced by the PDGF receptor require the binding site for phosphatidylinositol 3 ' kinase. Oncogene. 9. 651-660.
Whiic.M.A.. Nicolclic.C.. Mindcn.A.. Polverino.A.. Vanaclst.L.. Karin.M. and Wigler.M.H. (1995) Multiple ras functions can contribute to mammalian-cell transformation. Cell. 80. 533-541.
Zhang.X.-F.. Settlem anJ.. KyriakisJ.M .. Takeuchi-Suzuki.E.. El ledge. S J .. Marshall.M.S.. B rudcrJ.T . Rapp.U.R. and AvruchJ. (1993) Normal and oncogenic p21 ras proteins bind to the amiho-tcrminal regulatory domain of c-Raf-1. Nature. 364. 308-313.
Zheng. Y . Bagrodia.S. and Cerionc.R.A. (1994) Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J. Biol. Chem. 269. 18727-18730.
Received on October 27. 1995: revised on January 5. 1996
2451

Cell, Vol. 89, 457-467, May 2, 1997, Copyright ©1997 by Cell Press
Role of Phosphoinositide 3-OH Kinase in Cell Transformation and Control of the Actin Cytoskeleton by RasPablo Rodriguez-Viciana,* Patricia H. Warne,* Asim Khwaja,* Barbara M. Marte,* Darryl Pappin,* Pam ela D as,t Michael D. W aterfield,#Anne Ridley,t and Julian Downward**lmperial Cancer Research Fund London WC2A 3PX United Kingdomt Ludwig Institute for Cancer ResearchLondon W1P 8BTUnited Kingdomt Department of BiochemistryUniversity College LondonLondon WC1E 6BTUnited Kingdom
Summary
The pathw ays by which mammalian Ras proteins induce cortical actin rearrangem ent and cause cellular transform ation are investigated using partial loss of function m utants of Ras and activated and inhibitory form s of various postu lated ta rg e t enzym es for Ras. Efficient transform ation by Ras requires activation of o ther d irect effectors in addition to the MAP kinase kinase kinase Raf and is inhibited by inactivation of the PI 3-kinase pathway. Actin rearrangem ent correla tes with the ability of R as m utants to activate PI 3-kinase. Inhibition of PI 3-kinase activity blocks Ras induction of m em brane ruffling, while activated PI 3-kinase is sufficient to induce m em brane ruffling, acting through Rac. The ability of activated Ras to stim ulate PI 3-kinase in addition to Raf is therefore im portant in Ras transform ation of mammalian cells and essential in Ras-induced cytoskeletal reorganization.
Introduction
In addition to their well characterized roles a s inducers of cellular proliferation, Ras proteins are known to have profound and rapid effects on the actin cytoskeleton; both pathways are likely to be important in establishing and maintaining the transformed phenotype. When microinjected into fibroblasts, activated mutant Ras protein induces membrane ruffling due to cortical actin rearrangem ent within a few minutes (Bar Sagi and Fera- misco, 1986). Membrane ruffling or lamellipodia formation is also induced by a number of growth factors and by microinjection of activated mutant Rac protein, a m ember of the Rho subfamily of Ras-related small GTPases (Ridley et al., 1992). Since dominant-negative mutant Rac blocks induction of ruffling by Ras and by growth factors, it is likely that Rac functions downstream of Ras and growth factor receptors in the pathway leading to actin rearrangement. In Swiss 3T3 cells, Ras microinjection also leads to actin stress fiber formation through a Rho-mediated pathway that lies downstream of Rac (Ridley and Hall, 1992). These observations provide compelling, though indirect, evidence that Ras is
able to control the activation sta te of Rac and, hence, regulate the actin cytoskeleton.
At present, there is no consensus as to how Ras is able to control Rac proteins and actin polymerization. Several proteins have been identified that interact directly with active, GTP-bound Ras, but not with inactive, GDP-bound Ras: these are all potential effectors, or downstream targets, of Ras. The best characterized of these is the Raf family of cytoplasmic serine/threonine kinases (Marshall, 1996). Ras interaction with Raf leads to activation of the MAP kinase pathway with subsequent regulation of transcription factors; this pathway has been shown to be essential for fibroblast proliferation, but there is evidence that it is not directly involved in short-term control of the cytoskeleton (Joneson et al., 1996).
A number of reports support the existence of other effector pathways operating downstream of Ras in addition to the Raf/MAP kinase pathway (White et al., 1995; Joneson et al., 1996). Other putative mammalian Ras effectors include the family of Ras GTPase-activating proteins, among them pi 20*̂ '*, neurofibromin, and Gapi (Marshall, 1996), which are negative regulators of Ras but could possibly influence Rho family proteins through the pi 20®*'’-associated protein pi 90, a Rho family GAP. Another family of putative Ras effectors, Ral.GDS, and two related proteins activate the Ras family member Ral in response to activation of Ras (Urano e t al., 1996). At present, the function of Ral is unknown, although a role in the regulation of phospholipase D has been suggested. A protein that interacts with activated Ral has recently been identified (Cantor et al., 1995) that contains a Rac GAP domain, establishing another effector pathway by which Ras might perhaps be able to influence the activity of Rho family proteins.
In fission yeast, Ras has been shown to directly regulate two effectors. One is a MAP kinase kinase kinase, Byr2, the other is S c d l, a guanine nucleotide exchange factor for the Rho family protein Cdc42, which is involved in regulation of cell morphology (Chang et al., 1994). There is therefore a precedent for Ras directly controlling a Rho family exchange factor, although no such connection has yet em erged in mammalian systems; yeast does not always accurately mirror mammalian pathways, with adenylyl cyclase being a Ras effector in S. cerevisiae, but not in other species.
One other mammalian Ras effector has been described that might be capable of mediating Ras control of the cytoskeleton. Phosphoinositide 3-OH kinase (PI 3-kinase) interacts with Ras.GTP but not with Ras.GDP and is activated both in vitro and in vivo a s a result of this interaction (Kodak! et al., 1994; Rodriguez-Viciana et al., 1994, 1996). PI 3-kinase has been implicated in the regulation of the actin cytoskeleton by growth factors such as PDGF and insulin (Kotani et al., 1994; Wennstrôm et al., 1994; Nobes e t al., 1995).
Many mutations in the effector region of Ras, residues 32-40, are known to inhibit the biological function of Ras and to block the interaction of Ras with target proteins. Recently, it has becom e clear that relatively subtle mutations in this region might lead to partial loss of function

Cell458
V12 wt V12 K31 G26 E45 S35 E38 G37 A42 €40 cont 127D T D T D T D T D T D T D T D T D T D T D T
Sims
R al.G D S
R af
PI 3-k in ase
B
II|R b | InMI
I'&1Ï 0 3 7
FJSCM
Figure 1. In Vitro Binding of Ras Mutants to Effectors
(A) Mutant Ras proteins were loaded with GTP (I) or GDP (D) and bound to GST-fusion proteins of the effectors Ral.GDS (residues 1-97), c-Raf-1 (residues 1-259), and PI 3-kinase (full-length pi 10). Bound Ras was visualized by immunoblotting with monoclonal antibody Y13-259. In the control lane (cont), GST was substituted for effector fusion protein. All mutants were in a V I2 background.(B) Ras mutant interaction with effectors was measured by scintillation proximity assay. GST-fusion proteins spanning the Ras-binding site in Ral.GDS and Raf were used. The means and standard errors of triplicate assays are shown.
mutants in which interaction with some effectors is maintained but with others is lost (Joneson et al., 1996; White et al., 1995) (see Discussion). In this paper, we establish which mammalian effectors interact with the Ras partial loss of function mutants C40, G37, S35, and E38. We use these mutants, plus activated versions of Ras effector pathways including Raf and phosphoinositide 3-kinase (PI 3-kinase), to establish that at least two of these pathways need to be activated for efficient transformation of murine fibroblasts. Ras transformation is strongly inhibited by blocking PI 3-kinase activation. The mutants are also used to investigate the means by which Ras controls the actin cytoskeleton in mammalian cells. PI 3-kinase is shown to be the effector by which Ras induces membrane ruffling, since Ras-induced ruffling is blocked by inhibition of PI 3-kinase function, and PI 3-kinase activity is demonstrated to be sufficient to cause membrane ruffling, in a manner involving Rac and a target of calphostin C. Activation of Raf or Ral.GDS is not sufficient to cause membrane ruffling.
Results
In Vitro Interaction of Mutant Ras Proteins with EffectorsIn order to gain further insight into the relative importance of the various different effectors of Ras, we have screened a large array of Ras mutants for their ability to interact in vitro with the proven and suspected effectors c-Raf-1, Ral.GDS, and PI 3-kinase pi 10a (Figure 1 A). The PI 3-kinase used was full-length p85/GST- p llO a purified from a baculovirus expression system (Rodriguez-Viciana et al., 1996), while the Raf-1 and Ral.GDS used were bacterially expressed GST-fusion proteins containing the Ras-binding site alone (Warne
et al., 1993; Spaargaren and Bischoff, 1994). It is apparent that in most cases, Ras mutants show similar ability, or lack of ability, to bind to these three effectors. A number, however, are capable of preferentially binding to one effector while failing to bind to the other two. In particular, E38 and S35 are able to bind to Raf-1 but not significantly to Ral.GDS or pi 10a, while G37 will bind to Ral.GDS but not to Raf-1 or pi 1 Da, and 040 will bind to pi 10a but not to Raf-1 or Ral.GDS. Each of the Ras mutants G37, E38, and 040 fail to show stimulation of GTPase activity in response to the GTPase-activating proteins p120'^^ and neurofibromin, while S35 shows marginal sensitivity to pi 20'^'’ (data not shown). The use of only the Ras-binding site of Raf and Ral.GDS in these binding assays may have some limitations, particularly if additional sites of interaction with Ras exist: this ap proach was used because of difficulties in obtaining active forms of the full-length proteins. In the case of pi 10a, both full-length protein and the Ras-binding site alone (Rodriguez-Viciana et al., 1996) have been used to study Ras interactions with broadly similar results.
The ability of these four Ras effector mutants, plus activated VI2 Ras, to interact with the various Ras- binding proteins was investigated in more detail. A scintillation proximity assay (Skinner et al., 1994) was used to estimate the relative binding affinities of these Ras mutants for Raf-1 and Ral.GDS. As shown in Figure 1B, VI2 Ras interacted strongly with Raf-1 in this assay: the affinity of this interaction has been reported to be about 40 nM. No interaction could be seen with G37 and 040. E38 interacted with Raf-1 with about a 5-fold-reduced affinity relative to VI2, while S35 bound somewhat more weakly. In the case of Ral.GDS, E38 and 040 showed no detectable binding, while the affinity of G37 binding to Ral.GDS was reduced by about 3-fold relative to VI2,
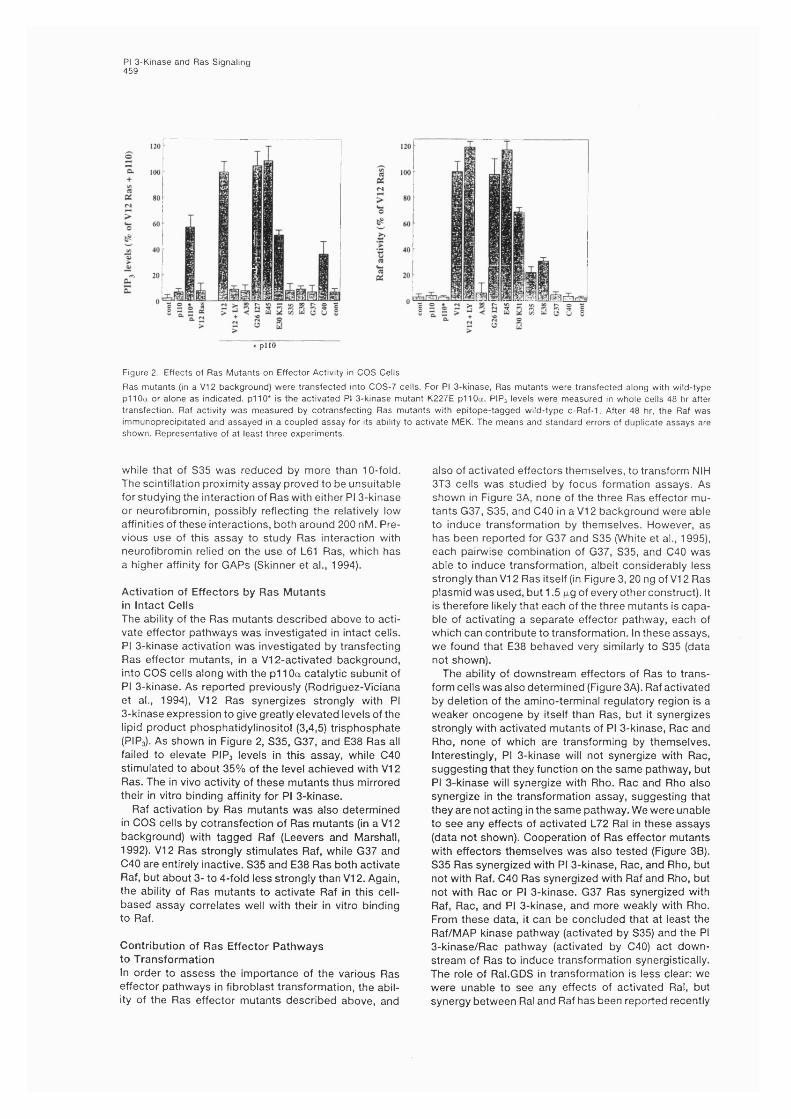
PI 3-Kinase and Ras Signaling459
+ p lio
Figure 2. Effects of Ras Mutants on Effector Activity in COS Cells
Ras mutants (in a V I2 background) were transfected into COS-7 cells. For PI 3-kinase, Ras mutants were transfected along with wild-type pi 10a or alone as indicated, p i 10* is the activated Pl 3-kinase mutant K227E pi 10a. PIP3 levels were measured in whole cells 48 hr after transfection. Raf activity was measured by cotransfecting Ras mutants with epitope-tagged wild-type c-Raf-1. After 48 hr. the Raf was immunoprecipitated and assayed in a coupled assay for its ability to activate MEK. The means and standard errors of duplicate assays are shown. Representative of at least three experiments.
while that of S35 was reduced by more than 10-fold. The scintillation proximity assay proved to be unsuitable for studying the interaction of Ras with either PI 3-kinase or neurofibromin, possibly reflecting the relatively low affinities of these interactions, both around 200 nM. Previous use of this assay to study Ras interaction with neurofibromin relied on the use of L61 Ras, which has a higher affinity for GAPs (Skinner et al., 1994).
Activation of Effectors by Ras Mutants In Intact CellsThe ability of the Ras mutants described above to activate effector pathways was investigated in intact cells. PI 3-kinase activation was investigated by transfecting Ras effector mutants, in a VI 2-activated background, into COS cells along with the pi 10a catalytic subunit of PI 3-kinase. As reported previously (Rodriguez-Viciana et al., 1994), VI2 Ras synergizes strongly with PI 3-kinase expression to give greatly elevated levels of the lipid product phosphatidylinositol (3,4,5) trisphosphate (PIP3). As shown in Figure 2, S35, G37, and E38 Ras all failed to elevate PIP3 levels in this assay, while C40 stimulated to about 35% of the level achieved with VI2 Ras. The in vivo activity of these mutants thus mirrored their in vitro binding affinity for PI 3-kinase.
Raf activation by Ras mutants was also determined in COS cells by cotransfection of Ras mutants (in a VI2 background) with tagged Raf (Leevers and Marshall, 1992). VI2 Ras strongly stimulates Raf, while G37 and 040 are entirely inactive. 835 and E38 Ras both activate Raf, but about 3- to 4-fold less strongly than VI2. Again, the ability of Ras mutants to activate Raf in this cell- based assay correlates well with their in vitro binding to Raf.
Contribution of Ras Effector Pathways to TransformationIn order to assess the importance of the various Ras effector pathways in fibroblast transformation, the ability of the Ras effector mutants described above, and
also of activated effectors themselves, to transform NIH 3T3 cells was studied by focus formation assays. As shown in Figure 3A, none of the three Ras effector mutants G37, 835, and C40 in a VI2 background were able to induce transformation by themselves. However, as has been reported for G37 and 835 (White et al., 1995), each pairwise combination of G37, 835, and C40 was able to induce transformation, albeit considerably less strongly than VI2 Ras itself (in Figure 3, 20 ng of VI2 Ras plasmid was used, but 1.5 fig of every other construct). It is therefore likely that each of the three mutants is capable of activating a separate effector pathway, each of which can contribute to transformation. In these assays, we found that E38 behaved very similarly to 835 (data not shown).
The ability of downstream effectors of Ras to transform cells was also determined (Figure 3A). Raf activated by deletion of the amino-terminal regulatory region is a weaker oncogene by itself than Ras, but it synergizes strongly with activated mutants of PI 3-kinase, Rac and Rho, none of which are transforming by themselves. Interestingly, PI 3-kinase will not synergize with Rac, suggesting that they function on the same pathway, but PI 3-kinase will synergize with Rho. Rac and Rho also synergize in the transformation assay, suggesting that they are not acting in the same pathway. We were unable to see any effects of activated L72 Ral in these assays (data not shown). Cooperation of Ras effector mutants with effectors themselves was also tested (Figure 3B). 835 Ras synergized with PI 3-kinase, Rac, and Rho, but not with Raf, 040 Ras synergized with Raf and Rho, but not with Rac or PI 3-kinase. G37 Ras synergized with Raf, Rac, and PI 3-kinase, and more weakly with Rho. From these data, it can be concluded that at least the Raf/MAP kinase pathway (activated by 835) and the PI 3-kinase/Rac pathway (activated by 040) act downstream of Ras to induce transformation synergistically. The role of Ral.GD8 in transformation is less clear: we were unable to see any effects of activated Ral, but synergy between Ral and Raf has been reported recently
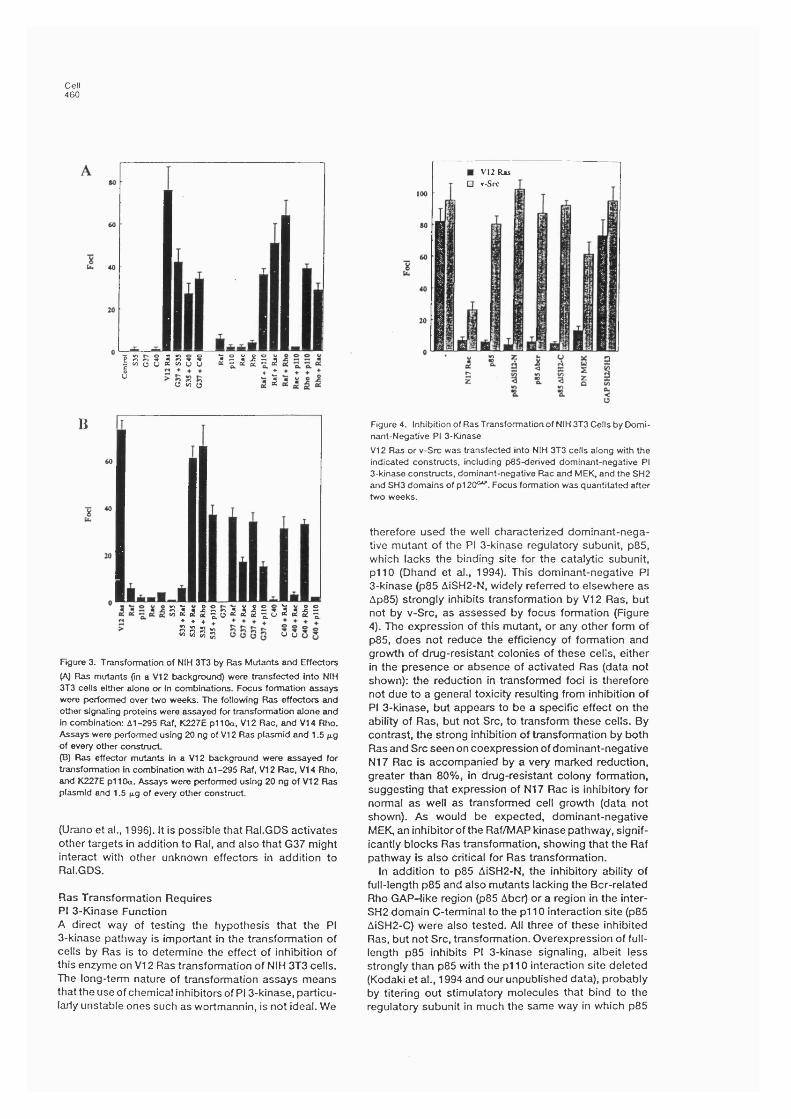
Cell460
P s S i S ? ? 2 I I I ya a iU W o
B
S 2 5 | | g2 ^ I | s 2 ^ I |: : : :BBBg o u
Figure 3. Transformation of NIH 3T3 by Ras Mutants and Effectors
(A) Ras mutants (in a V I2 background) were transfected into NIH 3T3 cells either alone or in combinations. Focus formation assays were performed over two weeks. The following Ras effectors and other signaling proteins were assayed for transformation alone and in combination: 61-295 Raf. K227E p i 10a, V I2 Rac, and V I4 Rho. Assays were performed using 20 ng of V I2 Ras plasmid and 1.5 jig of every other construct(B) Ras effector mutants in a V I2 background were assayed for transformation in combination with 61-295 Raf, V I2 Rac, V I4 Rho, and K227E p i 10a. Assays were performed using 20 ng of V I2 Ftas plasmid and 1.5 jig of every other construct.
(Urano et a!., 1996). It Is possible that Ral.GDS activates other targets in addition to Ral, and also that G37 might interact with other unknown effectors in addition to Ral.GDS.
Ras Transformation Requires PI 3-Kinase FunctionA direct way of testing the hypothesis that the PI 3-kinase pathway is important in the transformation of cells by Ras is to determine the effect of inhibition of this enzyme on VI2 Ras transformation of NIH 3T3 cells. The long-term nature of transformation assays means that the use of chemical inhibitors of PI 3-kinase, particularly unstable ones such as wortmannin, is not ideal. We
V12 Ras □ v-Six
Figure 4. Inhibition of Ras Transformation of NIH 3T3 Cells by Dominant-Negative PI 3-Kinase
V I2 Ras or v-Src was transfected Into NIH 3T3 cells along with the indicated constructs, including p85-derived dominant-negative PI 3-kinase constructs, dominant-negative Rac and MEK, and the SH2 and SH3 domains of p i 20°"’. Focus formation was quantitated after two weeks.
therefore used the well characterized dominant-negative mutant of the PI 3-kinase regulatory subunit, p85, which lacks the binding site for the catalytic subunit, p l io (Dhand et al., 1994). This dominant-negative PI 3-kinase (p85 AiSH2-N, widely referred to elsewhere as Ap85) strongly inhibits transformation by VI2 Ras, but not by v-Src, as assessed by focus formation (Figure 4). The expression of this mutant, or any other form of p85, does not reduce the efficiency of formation and growth of drug-resistant colonies of these cells, either in the presence or absence of activated Ras (data not shown): the reduction in transformed foci is therefore not due to a general toxicity resulting from inhibition of PI 3-kinase, but appears to be a specific effect on the ability of Ras, but not Src, to transform these cells. By contrast, the strong inhibition of transformation by both Ras and Src seen on coexpression of dominant-negative N17 Rac is accompanied by a very marked reduction, greater than 80%, in drug-resistant colony formation, suggesting that expression of N17 Rac is inhibitory for normal as well as transformed cell growth (data not shown). As would be expected, dominant-negative MEK, an inhibitor of the Raf/MAP kinase pathway, significantly blocks Ras transformation, showing that the Raf pathway is also critical for Ras transformation.
In addition to p85 AiSH2-N, the inhibitory ability of full-length p85 and also mutants lacking the Bcr-related Rho GAP-like region (p85 Abcr) or a region in the inter- SH2 domain C-terminal to the pi 10 interaction site (p85 AiSH2-C) were also tested. All three of these inhibited Ras, but not Src, transformation. Overexpression of full- length p85 inhibits PI 3-kinase signaling, albeit less strongly than p85 with the pi 10 interaction site deleted (Kodaki et al., 1994 and our unpublished data), probably by titering out stimulatory molecules that bind to the regulatory subunit in much the same way in which p85

PI 3-Kinase and Ras Signaling461
Table 1. Inhibition of Growth in Soft Agar of Ras-Transformed NIH 3T3 Cell Lines by Expression of p85 Constructs
Vector VI2 Ras Clone 1 VI2 Ras Clone 2 v-Src
Control 272 ± 37 218 ± 45 326 + 82p85a 42 ± 5 48 ± 12 2 9 1 + 6 8p85p 5 i 1 6 + 2 278 + 89Ap85a 22 ± 3 28 + 7 310 + 45
The numbers of colonies visible by the naked eye are listed. Clone 1 expresses low and Clone 2, high levels of VI2 Ras. Representative of two separa te experiments.
AÎSH2-N does. Presumably, som e productive com plexes with p l io will form, but a t high levels of p85 expression, these will be effectively removed from upstream signaling by competition from free p85. The Bcr- related region of p85 has been reported to Interact with the Rho family proteins Cdc42 and Rac (Zheng e t al.,1994); the fact that p85 Abcr has identical activity to p85 and p85 AISH2-N in this assay suggests that direct interaction of p85 with Rac or Cdc42 is not likely to be a critical part of the mechanism of Ras transformation. The expression of the SH2 and SH3 domains of p i 20®"’ does not inhibit Ras transformation, suggesting that the inhibitory effect of p85 is not nonspecific due to overexpression of any SH2 and SH3 domains.
To confirm the inhibitory effect of blocking PI 3-kinase function on the ability of oncogenic Ras to transform cells, NIH 3T3 cell lines stably transformed by VI2 Ras were infected with retroviruses expressing dominant- negative p85 constructs. Drug-resistant pools of infected cells were assayed for their ability to grow in suspension in soft agar. As shown in Table 1, expression of either full-length p85a or p8Sp, or Ap85a, all substantially inhibited colony formation of Ras-transformed cells in soft agar. Expression of these constructs had no significant effects on v-Src-induced anchorage-independent cell growth.
Involvement of Ras Effector Pathw ays in Cytoskeletal Reorganization Microinjection of activated Ras protein or cDNA into porcine aortic endothelial (PAE) cells induces rapid cortical actin rearrangement and membrane ruffling. Expression vectors for the various Ras effector mutants, and the activated effectors them selves, were injected into PAE cells and the cells subsequently stained with phal- loidin-rhodamine to visualize F actin. As shown in Figure 5A, while activated VI2 Ras gave strong mernbrane ruffling, S35 and G37 Ras in VI2 backgrounds failed to induce actin rearrangements. VI2 040 Ras induced membrane ruffling, although more weakly than VI2 Ras. It therefore appears likely that PI 3-kinase is a downstream effector involved in Ras-induced ruffling. An activated mutant of PI 3-kinase was sufficient to cause membrane ruffling. Activated Rac gave strong ruffling, while activated Raf-CAAX and L72 Ral did not. PI 3-kinase-induced ruffling was inhibited by coexpression of dominant-negative N17 Rac, but not dom inant-negative N17 Ras or N17 Cdc42, while VI2 R asnnduced ruffling was sensitive to dominant-negative Rac but not Cdc42 (Figure 58). This N17 Cdc42 construct w as able
to inhibit filopodia formation induced by extracellular stimuli in another system (A. R., unpublished data).
To investigate whether PI 3-kinase activity was responsible for the Ras-induced membrane ruffling, PAE cells microinjected with VI2 Ras, VI2 C40 Ras, activated PI 3-kinase, or VI2 Rac were treated with the PI 3-kinase inhibitor LY294002. This inhibitor completely blocked the ability of VI2 C40 Ras and activated PI 3-kinase to induce membrane ruffling (Figure 6A). In addition, this drug blocked PDGF-induced ruffling in these cells (data not shown and Wennstrôm et al., 1994). However, LY294002 had absolutely no effect on the activation of the MAP kinase ERK2 in response to PDGF or phorbol ester in these cells (data not shown). Ruffling induced by VI2 Ras was not fully blocked by LY294002, although som e clear diminution of the extent of ruffling was reproducibly noticed. VI2 Rac-induced ruffling was unaffected by LY294002. For all of these experiments, similar effects to LY294002 were also seen with wortmannin, an unrelated inhibitor of PI 3-kinase activity (data not shown). It therefore appears that PI 3-kinase, and not Raf or Ral.GDS, is one of the Ras effectors involved in the induction of cortical actin rearrangem ent, m em brane ruffling, and lamellipodia formation. However, it is possible that another ruffling pathway also exists that is activated by VI2 Ras but not VI2 040 Ras and is insensitive to chemical inhibitors of PI 3-kinase.
To further investigate this PI 3-kinase inhibitor- resistant pathway linking Ras to Rac and m em brane ruffling, the effects of a number of other inhibitors on Ras-induced membrane ruffling were studied. The p85- derived dominant-negative PI 3-kinase constructs were coexpressed in PAE cells with activated Ras or Rac. As shown in Figure 68, expression of p85 totally and reproducibly blocked VI2 R asnnduced m em brane ruffling. In addition, p85 AiSH2-N, p85 Abcr, and p85 AiSH2-C all completely inhibited VI2 R as-induced ruffling. These constructs also block PDGF-induced ruffling (Wennstrôm et al., 1994 and da ta not shown). The p i 20®"* SH2 and SH3 construct w as without effect. The effect of these p85-based dom inant-negative PI 3-kinase constructs appeared to be specific for Ras- and growth factor-induced ruffling, since they had abso lutely no effect on ruffling induced by VI2 Rac (Figure 68 and da ta not shown). These da ta indicate that the mechanism by which Ras controls the actin cytoskeleton through Rac is entirely dependent on normal PI 3-kinase function acting upstream of Rac. The inability of chemical inhibitors of PI 3-kinase to inhibit fully Ras- induced actin rearrangement may suggest that drug- resistant forms of PI 3-kinase couple to Ras.
As shown in Figure 60, the protein kinase 0 inhibitor calphostin C was a very good inhibitor of ruffling induced both by Ras and by PDGF in PAE cells. Calphostin C w as also able to fully inhibit activated PI 3-kinase-induced ruffling. However, another protein kinase C inhibitor, the bisindoylmaleimide G F 109203X, w as not effective. The inhibitor GF 109203X w as used under conditions where it inhibited phorbol ester activation of ERK2 (data not shown). Since calphostin 0 ac ts on diacyl glycerol- binding zinc fingers, while bisindoylmaleimide ac ts on the kinase domain, it is possible that a diacyl glycerol- binding protein other than a protein kinase C family

Cell462
B
C ontro l L72 Ral Raf-CAAX
w
V 1 2 R a s plio* V 1 2 R a c
C on tro l
V 1 2 R a s p lio * + N17 Ras
V I2 C 4 0 R a s V12S35 Ras V12G37 R as V12 R as + N17 R ac p llO * + N 1 7 R ac
V12 R as + N17 Cdc42 p llO * + N17 Cdc42
Figure 5. Effect of Ras Mutants and Effectors on Actin Rearrangement in Porcine Aortic Endottielial Cells
(A) PAE cells were microinjected with the indicated plasmids (pi 10* is the activated PI 3-kinase K227E pi 10a) and actin visualized after 5 hr by Immunofluorescence using TRfTC-phalloidin.(B) Cells were microinjected with V I2 Ras protein or K227E pi 10a plasmid, along with dominant-negative Ras, Rac, or Cdc42 protein or plasmid, respectively.
member is important in the control of membrane ruffling: possible candidates include the Rho family exchange factors Vav, Vav2, FGD1, and Lfc. Calphostin 0 was unable to inhibit ruffling induced by activated Rac, placing the target for this drug downstream of Ras and Pl 3-kinase and upstream of Rac.
Effect of LY294002 on PDGF- and Ras-induced Cellular PIP, LevelsThe data in Figure 6B raises the possibility that Ras may be capable of affecting the activity of PI 3-kinases that are not fully sensitive to chemical inhibitors such as LY294002 and wortmannin. To further explore this possibility, the effect of LY294002 on the levels of PI 3-kinase- produced lipids was investigated in NIH 3T3 cells (Figure 7A). PDGF-induced PIP, elevation is entirely reversed by treatment of cells with 20 jiM LY294002, a dosage that should entirely inhibit the activity of pi 10a (Vlahos et al., 1994). When VI2 Ras was expressed in these cells, the levels of PIP, were constitutively elevated to about half the peak level achieved with PDGF. This elevation of PIP, was, however, substantially resistant to the effects of the drug; even extended treatment with very high concentrations of LY294002 left about half this level of PIP,. It is therefore likely that Ras couples to forms of PI 3-kinase that are resistant to this drug, and also activates conventional drug-sensitive isoforms.
To confirm that expression of the regulatory p85 subunit of PI 3-kinase can block the ability of VI2 Ras to induce elevated levels of PIP, in intact cells, even though
LY294002 fails to do so, COS cells were transfected with VI2 Ras in the presence or absence of p85 or Ap85. The cells were then metabolically labeled with “ P-orthophosphate, and PIP, levels were determined. As shown in Figure 7B, both p85 and Ap85 were able to almost completely reverse VI2 Ras-induced elevation of PI 3-kinase lipid products. These constructs did not influence Ras expression levels (data not shown).
Discussion
The various partial loss of function mutants of Ras used in this paper help to define the effector pathways acting downstream of Ras. It is clear that Ras can trigger multiple signaling pathways in addition to the well characterized Raf/MAP kinase cascade (White et al., 1995; Joneson et al., 1996; Marshall, 1996). We describe here the ability of four different effector site mutants of Ras to stimulate known effectors: 040 Ras interacts with PI 3-kinase, G37 Ras interacts with Ral.GDS, and 835 and E38 Ras interact with Raf. None of these mutants interact productively with pi 2 0 ^ or neurofibromin. The ability of these mutants to interact with other potential mammalian effectors of Ras is not known. 835 Ras has previously been reported to interact selectively with Raf, but not with the unrelated MAP kinase kinase kinase, Byr2, from Schizosaccharomyces pombe, while G37 Ras interacts with Byr2 but not with Raf (White et al.,1995): together, the two synergize strongly in transformation of NIH 3T3 cells, as also shown here. The mutant
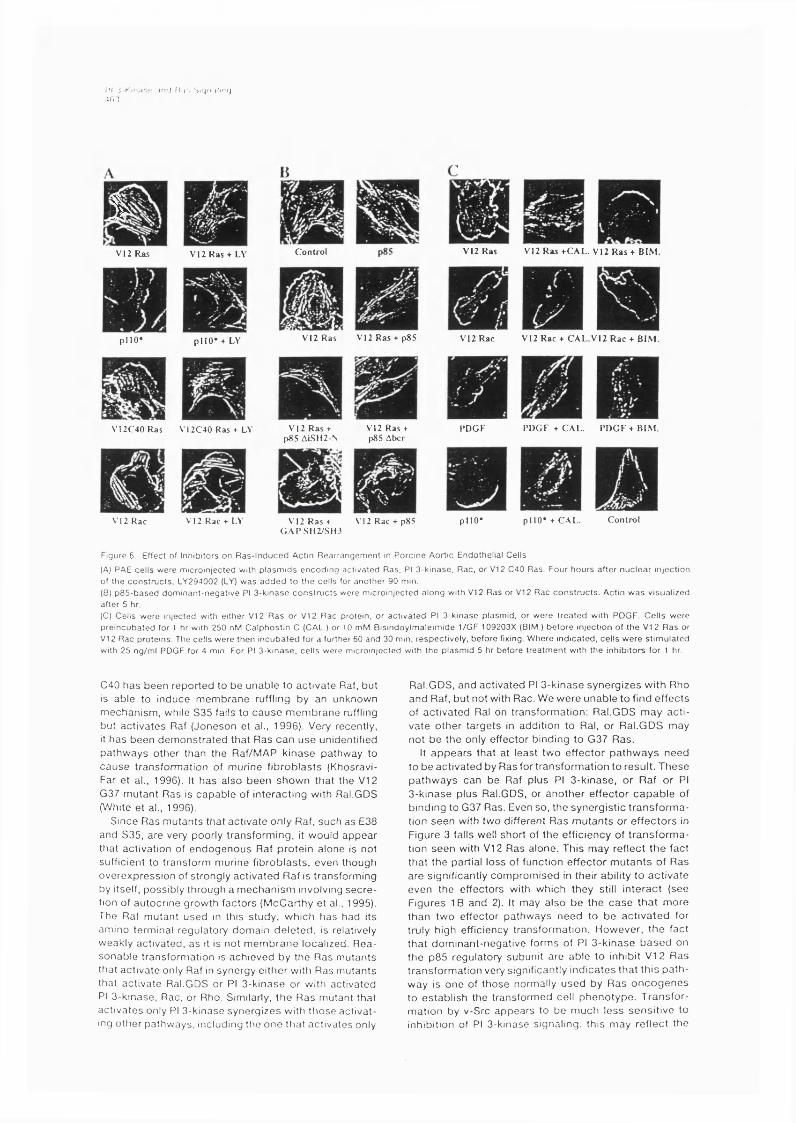
f'l ) Kin.i'.c , \ n( l K . f . Siqn-ilif'fjir.i
iV12 Ras V12 R as + L\
p l i o * p l l O* + LY
V 12C40 Ras V 12C 40 Ras + L \
V I 2 Rac V I 2 Rac + I.V
C o n t ro l V12 R as + C A L . V12 R a s + B IMV12 Ras
V I2 R as V I 2 R a s + p85 V I 2 Rac V12 Rac + C A L .V 1 2 R a c + B IM .
V I 2 R as + p85 ALSH2-N
V12 Ras + p85 Abcr
P D G F P D G F t G AL . P D G F + B IM
V12 Ras + G A P S H 2 / S H 3
V I 2 Rac + p85 p l i o * p l i o * + C A L . C o n t ro l
Figure 6. Effect of infiibitors on R a s - in d u c e d Actin R ea r ra n g em e n t in P o rc in e Aortic Endottielial Cells
(A) PAE cells w ere m icro in |e c ted witfi p l a s m id s e n c o d in g ac t iva ted Ras, PI 3 -k inase , Rac , or V I 2 C40 Ras. Four tiour s af ter n u c l e a r in ject ion of ttie c o n s t r u c t s , LY294002 (LY) w a s a d d e d to ttie ce lls for anot tie r 90 min.(B) p 8 5 - b a s e d d o m in an t -n e g a t iv e PI 3 -k in a se c o n s t r u c t s were micro in jec ted a lo ng witti V I 2 R as or V I 2 R ac c o n s t r u c t s . Actin w a s vi sual ized after 5 tir.(C) Cells w ere in jec ted witti eitfier V I 2 R a s o r VI 2 Rac protein, or ac t iv a t ed PI 3 -k in a se pla sm id , or w e re t r e a t e d witfi PDGF. C e l ls w ere p r e i n c u b a te d for 1 tir witfi 250 nfVI Calp tios tin C (CAL.) or 10 mM Bis in doy lmale im id e 1/GF 109203X (BIM.) b e fo re in ject ion of ttie VI 2 R a s or VI 2 Rac p ro te in s . The ce lls w ere th en i n c u b a te d for a further 60 an d 30 min, re spec t ive ly , b e fo re fixing. W h ere ind ica ted , ce ll s w e r e s t im u la t ed with 25 ng /m l PDGF for 4 min. For PI 3 -k in ase , ce ll s were m icro in jec te d with th e p la sm id 5 hr be fo re t r e a tm e n t with th e inhib itors for 1 hr
C40 h a s b een rep o rted to be u nab le to ac tiv a te Raf, but is ab le to induce m em b ran e ruffling by an unknown m ech an ism , while S35 fails to c a u s e m em b ran e ruffling but ac tiv a te s Raf (Jo n eso n et al., 1996). Very recently. I t h as b e en d e m o n s tra ted th at R as c a n u se unidentified p a th w ay s o th er than the Raf/MAP k in ase pathw ay to c a u s e transform ation of m urine f ib ro b la s ts (Khosravi- Far e t al., 1996). It h a s a lso b een sh o w n th a t the VI 2 G37 m utan t R as is c ap a b le of in te rac tin g with Ral.GDS (White e t al., 1996).
S ince R as m u tan ts that activ a te only Raf, su ch a s E38 and S35, a re very poorly transform ing , it would a p p ea r that activation of e n d o g e n o u s Raf p ro te in alone is not sufficient to transform m urine f ib ro b la sts , even though o v erex p ressio n of strongly ac tiv a ted R af is transform ing by itself, possib ly th rough a m ech an ism involving s e c re tion of au tocrine grow th fac to rs (M cC arthy e t al., 1995). The Raf m utan t u sed in th is study , w hich h as had its am m o term inal regulatory dom ain d e le te d , is relatively w eakly activ ated , a s it is not m em b ran e localized. R e a so n ab le transform ation is ach iev ed by the R as m utan ts that a c tiv a te only Raf in synergy e ith e r with R as m utan ts that a c tiv a te Ral.GDS or PI 3 -k in ase or with activated PI 3 -k inase, Rac, or Rho. Similarly, th e R as m utant that a c tiv a te s only PI 3 -k inase sy n e rg ize s witti ttio se ac tiv a ting o th e r pa thw ays, including the o n e th a t a c tiv a tes only
Ral.GDS, and ac tiv a ted PI 3 -k in ase sy n e rg ize s w ith Rho an d Raf, bu t not with Rac. W e w ere u n ab le to find e ffe c ts of ac tiv a ted Ral on transfo rm ation : Ral.GDS m ay a c t iv a te o th e r ta rg e ts in addition to Ral, o r Ral.G D S m ay no t b e th e only e ffecto r binding to G37 R as.
It a p p e a rs th at a t least tw o e ffec to r p a th w a y s n eed to b e a c tiv a ted by R as for tran sfo rm atio n to resu lt. T h ese p a th w ay s can b e Raf p lus PI 3 -k in ase , or R af or PI 3 -k in ase p lus Ral.GDS, or a n o th e r e ffec to r c a p a b le of b inding to G37 Ras. Even so , the sy n e rg istic t ra n s fo rm a tion se e n with two different R as m u ta n ts o r e ffe c to rs in F igure 3 falls well sh o rt of th e efficiency of tra n s fo rm a tion se e n with V I2 R as a lone. This m ay reflec t th e fact th a t th e partial lo ss of function e ffe c to r m u ta n ts of R as a re significantly co m p ro m ised in their ability to a c tiv a te ev en the e ffec to rs with w hich they still in te ra c t (see F igu res IB an d 2). It m ay a lso b e the c a s e th a t m ore th an tw o effec to r p a th w ay s n e ed to b e a c tiv a te d for truly high efficiency transfo rm ation . H ow ever, th e fact th a t dom in an t-n eg a tiv e form s of PI 3 -k in ase b a s e d on the p85 regulatory subunit a re ab le to inhibit VI 2 R as tran sfo rm atio n very significantly in d ic a te s th a t th is p a th w ay is o n e of th o se norm ally u se d by R as o n c o g e n e s to e s tab lish the tran sfo m ied cell p h e n o ty p e . T ran s fo rm ation by v-Src a p p ea rs to be m uch le s s se n s itiv e to inhibition of PI 3 -k inase signaling: th is m ay re flec t the

Cell464
B
Figure 7. Effect of LY294002 and p85 Overexpression on Ras- induced Cellular PIP, Levels
(A) PIP, levels were measured In metabolically labeled, serum- starved NIH 3T3 cells. Cells were treated with LY294002 for 90 min tiefore lysis. PDGF (50 ng/ml) treatment was for 3 min before lysis. V I2 Ras was expressed by retroviral infection for 48 hr before labeling.(B) V I2 Ftas and PI 3-kinase constructs, including wild-type p i 10, were transfected into COS-7 cells. PIP, levels were measured in whole cells 48 hr after transfection.
fact that v-Src uses several signaling pathways to transform cells, only some of which involve Ras, Src may be also able to control PI 3-kinase in a Ras-independent manner.
Looking further down the effector pathways, Raf can synergize with Rac or Rho, as shown previously by others (Khosravi-Far et al., 1995; Qiu et al., 1995a, 1995b), and PI 3-kinase can synergize with Rho, but not with Rac. This suggests that Rac and PI 3-kinase lie on the same pathway, most likely with PI 3-kinase acting upstream of Rac (Hawkins et al., 1995), but that Rho lies on a different pathway; this ties in with the ability of Rac and Rho to synergize with each other in causing transformation. Placing Rac and Rho on different pathways differs from the model derived from actin cytoskeleton rearrangements in Swiss 3T3 cells in which Rac
acts upstream of Rho (Ridley et al., 1992; Ridley and Hall, 1992). This apparent inconsistency may reflect differences in cell types or, perhaps more likely, differences in the nature of the biological assay: it is possible that distinct pools of Rho and Rac GTPases exist, with those proteins involved in actin rearrangement being differently regulated from those involved in, for example, transcriptional regulation. Transformation is likely to reflect all aspects of Rac and Rho signaling, not just cytoskeletal effects.
The definition of a set of effector mutants of Ras that each engage only one effector pathway allows one to dem onstrate that multiple pathways are required for efficient transformation of fibroblasts. However, are these mutants able to provide information about the m echanism by which Ras acts to give a specific short-term biological response? In the case of Ras induction of membrane ruffling and lamellipodia, it has been shown that activated Ras induces very strong ruffling through a Rac-mediated pathway (Ridley et al., 1992). We have shown here that activated PI 3-kinase also induces membrane ruffling, again through a Rac-mediated mechanism. The only Ras partial loss of function mutant to cause ruffling is 040: this, together with the inability of Raf or Ral to cause ruffling, rules out Raf and Ral.GDS as effectors on the ruffling pathway. Since 040 Ras stimulates PI 3-kinase and causes membrane ruffling in an LY294002- and wortmannin-sensitive manner in PAE cells, it is very likely that this mutant is causing ruffling through interaction with a drug-sensitive Pl 3-kinase.
In contrast to 040 Ras-induced ruffling, VI2 Ras- induced ruffling is not fully sensitive to PI 3-kinase inhibitors in porcine aortic endothelial cells (Figure 6A) or in Swiss 3T3 cells (0. D. Nobes and A. Hall, personal communication). This has previously been interpreted as evidence that Ras controls Rac-dependent ruffling by a PI 3-kinase-independent mechanism. However, the use of a different means of inhibiting PI 3-kinase function, expression of dominant-negative constructs based on the p85 regulatory subunit, shows that the VI2 R as- induced ruffling is entirely sensitive to this form of disruption of PI 3-kinase signaling. Other SH2 and SH3 domain-containing constructs, such as one derived from pi 20"^, are inactive. Since the Bcr domain of p85 is not required for inhibition of Ras-induced ruffling, and p85 constructs do not inhibit Rac-induced ruffling, it is unlikely that direct interaction of p85 with GTP-bound Rac or Cdc42 is responsible for this inhibition (Zheng et al., 1994). The exact mechanism by which overexpression of p85 mutants interferes with PI 3-kinase has not been defined, but is likely to involve titering out upstream signaling molecules that are needed for activation of Pl 3-kinase both by receptor tyrosine kinases and by Ras. As shown in Figure 78, overexpression of p85 or Ap85 does indeed inhibit the ability of VI2 Ras to activate PI 3-kinase in whole cells, as determined by measuring PIP] levels. Although Ras directly interacts with and moderately activates pi 10 in vitro, it is likely that in order to achieve efficient activation of PI 3-kinase by VI2 Ras in whole cells, at least basal levels of other signals are also required, including one mediated by p85. Endogenous p l io is likely to be effectively uncoupled from stimulatory signals feeding into p85 by massive overexpression of this protein. Overexpression of Ras and of

PI 3-Kinase and Ras Signaling465
p85 have previously been shown to exert opposing influences on mammalian PI 3-kinase activity in a model system in S. pom be (Kodaki e t al., 1994), suggesting that in the absence of upstream positive signals, p85 has a negative influence on the enzymatic activity of p l io within a cellular context. It cannot be ruled out that a function of PI 3-kinase, in addition to its lipid kinase activity, is important in Ras-induced membrane ruffling.
The fact that VI2 Rasnrtduced ruffling is sensitive to mutant p85 overexpression but not to LY294002 or wortmannin treatm ent suggests that Ras could activate an isoform of PI 3-kinase that is profoundly resistant to these inhibitors: it is already clear that Ras will interact with at least four different PI 3-kinase family members (P. R. V., P. H. W., M. D. W., and J . D., unpublished data), although none of these are known to be resistant to these drugs. M easurement of PIP, levels in VI2 Ras expressing NIH 3T3 cells shows tha t the Ras-induced, but not PDGF-induced, elevation of this lipid is partially resistant to treatment of cells with LY294002 (Figure 7). A novel C2 domain containing PI 3-kinase isoform has recently been reported that is about fifty tim es less sensitive than p i 10a to wortmannin (Virbasius e t al. 1996).
Since many growth factors rapidly induce both PI 3-kinase inhibitor-sensitive m em brane ruffling and Ras activation, it is possible that Ras function is required for the growth factor receptors to induce PI 3-kinase activity and, hence, to cause actin rearrangement. Indeed, it has been recently reported that in NIH 3T3 cells, Ras function is required for efficient activation of Pi 3-kinase in response to PDGF (Klinghofer e t al., 1996). In the ca se of Swiss 3T3 cells, PDGF-induced ruffling does not appear to be inhibited by neutralizing Ras antibodies (Ridley e t al., 1992); however, in MDCK cells, hepatocyte growth factor-induced ruffling is entirely sensitive to these Ras antibodies (Ridley et al., 1995). In both cases, Rac function is required for ruffling to occur. We have found that the ability of anti-Ras antibodies or N17 Ras to inhibit ruffling varies greatly with cell type and the nature of the external stimulus. It is clear that there are multiple pathways leading to Pi 3-kinase activation: in som e cases, Ras is necessary; in other cases, it is not. A very similar situation has been described for growth factor induction of ERK2 MAP kinase in fibroblasts, in which Ras is required for PDGF- and insulin-induced, but not EGF-induced, activation in Rat-1 and NiH 3T3 fibroblasts, but both PDGF- and EGF-induced ERK2 activation is insensitive to inhibition of Ras in Swiss 3T3 fibroblasts, while insulin requires Ras function (Burgering et al., 1993). The fact that PDGF-induced ruffling in PAE cells does not show a significant degree of resistance to LY294002 suggests that the contribution of endogenous Ras to PDGF-induced Pi 3-kinase activation is relatively small in this case, at least assum ing that endogenous Ras interacts with the sam e PI 3-kinase isoforms as does VI2 Ras.
The possibility that endogenous Ras normally activates multiple effector pathways is supported by the observation that, in a number of mammalian system s, Ras function is required for growth factor activation of PI 3-kinase (Rodriguez-Viciana e t al,, 1994; Klinghofer et al., 1996) as well as Raf (Marshall, 1996). Probably the m ost compelling evidence to date that Ras normally
controls more than one pathway com es from a com bination of genetic and two-hybrid analysis in yeast, particularly in S. pombe, where Ras control of Byr2 and S cdl is well established. The data presented here indicate that activated Ras is capable of performing multiple roles in mammalian cells. Activation of both the Raf and PI 3-kinase pathways are needed for efficient transformation by Ras oncogenes, while activated Ras-induced cortical actin rearrangement operates through a PI 3-kinase, and not a Raf or Ral.GDS, pathway.
Experimental Procedures
Plasm ids and ProteinsThe VI2 mutant H-ras cDNA w as cloned Into the pcDNA3 vector, and point m utations were carried out using the In Vitro M utagenesis Kit (Pharmacia). For bacterial expression, a BamHl site w as added in-frame to the second codon by PCR and the Inserts cloned Into the pGEX (Pharmacia) and pRSET (Invltrogen) vectors for expression as GST- and histidine-tagged fusion proteins, respectively. To have the effector m utants on a G12 background for GAP assays, the VI2 mutation w as m utated back to G12 by PCR and the Inserts cloned into the pRSET vector. Fusion proteins were purified according to the m anufacturer's protocols. The p i 10K227E m utant has t>een d e scribed elsewhere (Rodriguez-Viciana e t al., 1996). pSSawt, p85 AISH2-N (deletion of amirx) acids 478-513). p85 AISH2-C (deletion of amino a d d s 494-690), and p85Abcr (deletion of amino acids 134-302) were subcloned into the EcoRI site of pSG5. Full-length Ral.GDS expression construct w as a gift from Robert Weinberg; Ral.GDS fusion protein, from Johannes Bos; the N-tenminal (residues 1-666) p120®*^ SH2/SH3 construct, from Channing Der; the dom inant-negative MEK construct (A221), from Chris Marshall; and the dominant-negative Rac (N17 rac), from Alan Hall. Retroviruses were generated by cloning Inserts Into the pLXSN vector and transfecting them Into the G P+E86 packaging cell line. Pools of G418-resistant colonies were selected and, when confluent, the medium w as collected and frozen a s aliquots in the p resence of polytxene.
Ras/Effector In Vitro Binding A ssayRas-t>inding assays were carried out a s descrit)ed In Warne e t al.(1993) and Rodriguez-Vidana et al. (1994). Ras (0.5 pM) and effector- GST (0.5 pg) were used In a 200 pl assay . ^ In tillation Proximity Assay w as performed a s described In Skinner e t al. (1994).
M easurem ent of Raf and PI 3-Klnase Activity in COS-7 CellsConstructs were electroporated Into COS-7 cells a s described previously (Rodriguez-Viciana e t al., 1994). After 48 hr, PI 3-klnase activity w as assayed In the absence of serum by m easuring 3 ' p hosphorylated phosphdnosltide lipids, a s described by Carter and Downes (1992). Raf activity w as assayed In tfie absen ce of serum by cotransfecting a Myc-tagged c-Raf-1 construct and by performing a coupled MEK/ERK2 kinase assay on 9E10 Im m unopredpitates a s described by Leevers and Marshall (1992). For lipid analysis In NIH 3T3 cells, Ras retrovirus-containing supernatant w as added to sub confluent NIH 3T3 cells and replaced 4 hr later with fresh medium. After 24 hr incutiation to to allow expression of Ras protein, the cells were labeled for 16 hr with "P -orthophosphate In phosphate- free medium containing 0.2% dialysed calf serum , and PIP, levels were m easured by the sam e method a s described above for COS cells.
Transform ation A ssays In NIH 3T3 CellsFor the data In Figures 3 and 4, low passag e NIH 3T3 cells were se eded a t 1.25 x 10* cells per well In 6-well d ishes the day t>efore lipofection with 1.5 ng of each plasmid, apart from VI2 R as plasmid, in which 20 ng w as used, using the Tfx-50 reagent (Promega) a c cording to the manufacturer's protocols. Two days later, the cells were transferred to 10 cm plates. After reaching confluence, they were kept for two vreeks In DME medium containing 5% calf serum, after which they were stained with 0.5% crystal vio let For study of Inhibitory effects on transformation, 40 ng of V12 Ras o r 20 ng

Cell466
v-Src plasmid was cotransfected with 2 jig of inhibitory construct. Quantitation plotted is the m eans and standard errors of duplicate assays and is representative of a t least three experiments. For the da ta in Table 1, stably transformed NIH 3T3 cells expressing low (Clone 1, about equal level of VI2 Ras to endogenous Ras) or high levels (Clone 2, about 8-fold more VI2 Ras than endogenous Ras) of VI2 Ras with a puromycin resistance marker were Infected with retroviruses expressing p85a, p8Sp, or ApBSa (p85 AISH2-N) with a neo resistance marker. Cells were se lected with G418 for 5 days, during which time all control uninfected cells died, then soft agar colony formation assays were se t up with 1000 cells in the p resence of G418. Colonies visible by the naked eye were scored in duplicates a t 3 weeks.
M icroinjections and Im m unofluorescence Porcine aortic endothelial 1 x 1 0 * cells were seeded in 13 mm glass coverslips two days before and serum starved overnight In 0.5% fetal calf serum before injections. When Injecting proteins, VI2 Rac, VI2 Cdc42, N17 Rac, and N17 Cdc42 were expressed a s GST-fusion proteins, purified, and cleaved a s described in Self and Hall (1995). VI2 Ras proteins expressed either a s GST- or a s histidine-tagged fusion proteins were found to have very poor biological activity; the VI2 Ras protein used for microinjection experim ents w as purified from the soluble fraction of Sf-9 cells Infected with a VI2 Ras t>acu- lovirus. When microinjecting DNA. plasmids were injected into the nucleus at a concentration of 100 (jig/ml, and the cells fixed after 5 hr. Guinea pig IgG a t 2 mg/ml w as coinjected in all c a se s to help identify injected cells. LY294002 w as used a t 20 p.M. An Eppendorf microinjector (Model No. 5242) and micromanipulator (Model No. 5170) were used. Cells were fixed, and Immunofluorescence w as carried out a s described in Ridley (1995). All m icrographs shown are representative examples of microinjected cells from a t least four similar experiments.
Acknow ledgm ents
Correspondence regarding this paper should be add ressed to J . D. ([email protected]). We extend our thanks to Chris Gilbert, Ritu Garg, Rosa Guasch, and Helen Flinn for help with microinjection; Chris Marshall, Alan Hall, Robert Weinberg, and Hans Bos for supplying constructs; Kate Nobes for helpful discussions; Bart Vanhaesebroeck for PI 3-kinase constructs and advice; and Peter Jordan and Andrew Edwards for help with confocal microscopy.
Received June 19,1996; revised February 13,1997.
R eferences
Bar Sagi, D., and Feramisco, J.R. (1986). Induction of m em brane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233,1061-1068.Burgering, B.M.T., de Vries-Smtts, A.M.M., Medema, R.H., van W eeren, P.C., Tertoolen, LG.J., and Bos, J .L (1993). Epidermal growth factor induces phosphorylation of extracellular signal- regulated kinase 2 via multiple pathways. Mol. Cell. Biol. 73, 7248- 7256.
Cantor, S B., Urano, T., and Feig, LA. (1995). Identification and ctraracterization of Ral-binding proteIn-1, a potential downstream target of Ral GTPases. Mol. Cell. Biol. 75, 4578-4584.Carter, A.N., and Downes, C.P. (1992). Phosphatidylinositol 3-kinase is activated by nerve growth factor and epidermal growth factor in PCI 2 cells. J . Biol. Chem. 267,14563-14567,Chang, EC., Barr, M., Wang, Y., Jung, V., Xu, H P., and Wigler, M.H.(1994). Cooperative interaction of S. pom be proteins required for mating and morphogenesis. Cell 79, 131-141.Dhand, R., Hara, K., Hiles, I., Bax, B., Gout, I., Panayotou, G., Fry, M.J., Yonezawa, K., Kasuga, M., and Waterfield, M.D. (1994). PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 73, 511-521.
Hawkins, P.T., Eguinoa, A., Qiu, R.G., Stokoe, D., Cooke, F.T., Walters, R., Wennstrôm, S., Claesson-Welsh, L , Evans, T., Symons, M.,
and S tephens, L (1995). PDGF stimulates an increase in GTP-rac via activation of phosphoinositide 3-kinase. Curr. Biol. 5, 393-403. Joneson, T., White, M.A., Wigler, M.H., and Bar-Sagi, D. (1996). Stimulation of m em txane ruffling and MAP kinase activation by d istinct effectors of Ras. Science 277, 810-812.Khosravi-Far, R., Solski, P.A., Clark, G J . , Kinch, M.S., and Der, C.J.(1995). Activation of R a d , Rho A, and mitogen-activated protein kinases Is required for Ras transformation. Mol. Cell. Biol. 75,6443- 6453.Khosravi-Far, R., White, M.A., Westwick, J.K., Solski, P.A., Chrza- nowska-Wodnicka, M., Van Aelst, L , Wigler, M.H., and Der, C J .(1996). Oncogenic Ras activation of Raf/MAP kinase Independent pathways Is sufficient to cause tumorigenic transformation. Mol. Cell. Biol. 76, 3923-3933.Klinghofer, R.A., Duckworth, B., Valius, M., Cantley, L , and Kaz- lauskas, A. (1996). PDGF-dependent activation of PI3 kinase is regulated by receptor binding of SH2 domain-containing proteins which influence Ras activity. Mol. Cell. Biol. 76, 5905-5914.Kodaki, T., Woscholskl, R., Hallberg, B., Rodriguez-Viciana, P., Downward, J ., and Parker, P J . (1994). The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 4, 798-806.Kotani, K., Yonezawa, K., Hara, K., Ueda, H., Kitamura, Y., Sakaue, H., Ando, A., Chavaneau, A., Calas, B., Grigorescu, F., et al. (1994). Involvement of phosphoinositide 3-kinase In Insulin or lGF-1 induced m em brane ruffling. EMBO J. 73, 2313-2321.Leevers, S J . , and Marshall, C J . (1992). Activation of extracellular signal-regulated kinase ERK2 by p21 ras oncoprotein. EMBO J . 77, 569-574.Marshall, C J . (1996). Ras effectors. Curr. Opin. Cell Biol. 8 ,197-204. McCarthy, S.A., Samuels, M L , Pritchard, CA., Abraham, J.A., and McMahon, M. (1995). Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev. 9,1953-1964.Nobes, C D., Hawkins, P., Stephens, L , and Hall, A. (1995). Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J . Cell Sci. 706, 225-233.Oiu, R.G., Chen, J., Kim, D., McCormick, F., and Symons, M. (1995a). An essential role for rac in ras transformation. Nature 374,457-459. Oiu, R.G., Chen, J., McCormick, F., and Symons, M. (1995b). A role for rtw in ras transformation. Proc, Natl. Acad. Sci. USA 92, 11781-11785.Ridley, A., and Hall, A. (1992). The small GTP-binding protein rho regulates the assem bly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389-399.Ridley, A., Paterson, H.F., Johnston, C .L , Diekmann, D., and Hall,A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401-410.Ridley, A.J. (1995). Microinjection of Rho and Rac into quiescent Swiss 3T3 cells. Meth. Enzymol. 256B, 313-319.Ridley, A.J., Comoglio, P.M., and Hall, A. (1995). Regulation of sc a tter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol. Cell. Biol. 75,1110-1122- Rodriguez-Viciana, P., Warne, P H., Dhand, R., Vanhaesebroeck,B., Gout, I., Fry, M J., Waterfield, M.D., and Downward, J . (1994). Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370, 527-532.Rodriguez-Viciana, P., Wame, P.H., Vanhaesebroeck, B., Waterfield, M.D., and Downward, J. (1996). Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J . 75, 2442-2451.Self, A.J., and Hall, A. (1995). Purification of recom binant Rho/Rac/ G25K from Escherichia coli. Meth. Enzymol. 256B, 3-10.Skinner, R.H., Picardo, M., Gane, N.M., Cook, N.D., Morgan, L , Rowedder, J., and Lowe, P.N. (1994). Direct m easurem ent of the binding of ras to neurofibromin using a scintillation proximity assay . Anal. Biochem. 7994, 259-265.Spaargaren, M., and Bischoff, J.R. (1994). Identification of the g uanine-nucleotide dissociation stimulator for ral a s a putative effector

PI 3-Kinase and Ras Signaling467
molecule of r-ras, h-ras, k-ras, and rap. Proc. Natl. Acad. Sci. USA 91. 12609-12613.Urano, T., Emkey, R., and Feig, LA. (1996). Ral-GTPases m ediate a distinct downstream signaling pathway from ras that facilitates cellular-transformation. EMBO J . 15, 810-816.Virbasius, J.V., Guilherme, A., and Czech, M.P. (1996). Mouse p i 70 is a novel phosphatidylinositol 3-kinase containing a c2 domairu J. Biol. Chem. 271,13304-13307.Vlahos, C J ., Matter, W.F., Hui, K.Y., and Brown, R.F. (1994). A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyQ-8- phenyl-4H-1 -benzopyran-4-one (LY294002). J . Biol. Chem. 269, 5241-5248.Wame, P.H., Vicinia, P.R., and Downward, J . (1993). Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364, 352-355.W ennstrôm, S., Hawkins, P., Cooke, F., Hara, K., Yonezawa, K., Kasuga, M., Jackson, T., Claesson-W elsh, L , and S tephens, L(1994). Activation of phosphoinositide 3-kinase Is required for PDGF- stimulated membrane ruffling. Curr. Biol. 4, 385-393.White, M.A., Nicolette, C., Minden, A., Pofverino, A., Vanaelst, L , Karin, M., and Wigler, M.H. (1995). Multiple Ras functions can contribute to mammalian cell transformation. Cell 80, 533-541.White, M.A., Vale, T., Camonis, J.H., Schaefer, E., and Wigler, M.H.(1996). A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J . Biol. Chem. 271,16439- 16442.Zheng, Y., Bagrodia, S., and Cerione, R.A. (1994). Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J . Biol. Chem, 269,18727-18730.




















