
The uridine in “U-turn”: Contributions to tRNA-ribosomal binding

Molecular Cell
Article
Ribosomal Protein L3: Gatekeeper to the A SiteArturas Meskauskas1,* and Jonathan D. Dinman1,*1Department of Cell Biology and Molecular Genetics, University of Maryland, Microbiology Building, Room 2135, College Park,
MD 20742, USA
*Correspondence: [email protected] (A.M.), [email protected] (J.D.D.)DOI 10.1016/j.molcel.2007.02.015
SUMMARY
Ribosomal protein L3 (L3) is an essential and in-dispensable component for formation of thepeptidyltransferase center. Atomic resolutionribosome structures reveal two extensions ofL3 protruding deep into the core of the largesubunit. The central extension of L3 in Saccha-romyces cerevisiae was investigated usinga combination of molecular genetic, biochemi-cal, chemical probing, and molecular modelingmethods. A reciprocal relationship betweenribosomal affinity for eEF-1A stimulated bindingof aa-tRNA and for eEF2 suggests that the cen-tral extension of L3 may function as an allostericswitch in coordinating binding of the elongationfactors. Opening of the aa-tRNA accommo-dation corridor promoted resistance to the Asite-specific translational inhibitor anisomycin,suggesting a competitive model for anisomycinresistance. These changes were also found toinhibit peptidyltransferase activity, stimulatingprogrammed �1 ribosomal frameshifting andpromoting virus propagation defects. Thesestudies provide a basis for deeper insight intorational design of small molecule antiviraltherapeutics.
INTRODUCTION
Multicomponent nanoscale biomachines must be able to
coordinate orderly series of functions. For example, the
elongation phase of protein synthesis requires that the ri-
bosome perform at least nine sequentially ordered steps.
The emerging view is one in which each event triggers al-
losteric rearrangements of specific segments of ribosomal
RNAs (rRNAs) and ribosomal proteins, thus enabling infor-
mation transfer among and coordination between the var-
ious functional centers of the ribosome (reviewed in Noller
[2007]). Although rRNA carries out the catalytic activity
and comprises the bulk of the ribosome, many ribosomal
proteins are also required for proper function. Ribosomal
protein L3 (L3) in particular appears to be extremely im-
portant: its sequence and structure are highly conserved
across all three kingdoms (reviewed in Brodersen and
Molec
Nissen [2005]); it is one of the few ribosomal proteins
required for peptidyltransferase activity (Schulze and
Nierhaus, 1982); and it is one of only two proteins capable
of initiating in vitro assembly of E. coli large ribosomal sub-
units (Nowotny and Nierhaus, 1982). Like many ribosomal
proteins, L3 contains a globular domain on the cytoplas-
mic face of the large subunit and has two long tentacle-
like structures that extend deep into the mostly rRNA
core. L3’s central extension reaches all the way to the A
site side of the peptidyltransferase center (PTC), where
a tryptophan at position 255 (in the S. cerevisiae L3 pro-
tein), which is located at its tip, makes the closest ap-
proach of any amino acid to the peptidyltransferase center
active site (Klein et al., 2004; Meskauskas et al., 2005). Re-
gions of two important rRNA helical structures are also
anchored on this central extension (also called the ‘‘W fin-
ger’’ after the tryptophan residue): helix 95, and the struc-
ture formed by helices 90–92. Helix 95 contains the sarcin/
ricin loop (SRL), one of the key structures recognized by
the translation elongation factors, and the target of ribo-
some-inactivating proteins such as ricin (reviewed in Irvin
and Uckun [1992]). The helix 90–92 structure is important
because it forms one side of the corridor along which the
30 ends of aa-tRNAs slide during the process of accom-
modation (Sanbonmatsu et al., 2005). The N-terminal
extension of L3 may also interact with this structure.
Molecular genetic and biochemical studies have shown
that L3 plays an important role in aa-tRNA binding, pepti-
dyltransferase activity, drug resistance, translational
frame maintenance, and virus replication, and that it also
acts as a binding site for a ribosome inhibitory protein
(Wickner et al., 1982; Meskauskas et al., 2003, 2005; Peltz
et al., 1999; Petrov et al., 2004; Bosling et al., 2003; Fried
and Warner, 1981; Hudak et al., 1999; Jimenez et al.,
1975; Pringle et al., 2004; Schindler et al., 1974). Satura-
tion mutagenesis suggested that L3 may function to
transmit information between the SRL and the PTC
(Meskauskas et al., 2005).
The current study employed a combination of targeted
mutagenesis, structural, biochemical, and genetic
approaches to examine the function of the W finger.
Mutagenesis experiments suggest that the tip of this
structure interacts with a nearby base or bases in 25S
rRNA, presumably A2940 (E. coli A2572). The W finger ap-
pears to be intrinsically flexible, but its range of movement
is strictly limited in one direction. Changes in the confor-
mation of the W finger resulting from mutation of W255
to cysteine (W255C) induce significant conformational
ular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 877

Molecular Cell
L3: The A Site Gatekeeper
Figure 1. Putative Interaction between
the Tip of the L3 W Finger and A2940 of
25S rRNA Is Critical for Cell Viability
(A) Predicted interactions between amino acid
substitutions at position 255 with large subunit
rRNA. Images were modeled using data for
yeast proteins obtained from cryo-EM studies
threaded into the H. marismortui X-ray crystal
structures (Spahn et al., 2004). U2607 of the
H. marismortui 23S rRNA (shown here) is the
structural equivalent of E. coli A2572, and of
A2940 in the S. cerevisiae 25S rRNA.
(B) Viability of substitution mutants at position
255 of L3 as monitored by 10-fold dilution
spot assays on �Trp 5-FOA plates. Apparently
viable W255E and W255A colonies were
proved to be revertants to tryptophan by DNA
sequence analysis.
rearrangements of the A site side of the PTC, the helix
90–92 structure, helix 95, and the SRL. These are accom-
panied by resistance to anisomycin, increased affinity for
aa-tRNA, decreased affinity for eEF2, and decreased
rates of peptidyltransfer. Other mutants of nearby histi-
dines had the opposite effects on elongation factor and
aa-tRNA binding to ribosomes. These findings suggest
a model in which L3 may play an important role in synchro-
nizing the processes of aa-tRNA accommodation and
translocation by functioning as sensor of the tRNA occu-
pancy status of the A site region of the PTC.
RESULTS
Targeted Substitutions for Tryptophan at Position
255 of L3 Point to a Crucial Interaction with A2940
in the Yeast Ribosome
Previous studies showed that substitution of W255 with
cysteine (W255C) conferred resistance to anisomycin, in-
creased rates of programmed �1 ribosomal frameshifting
(�1 PRF), inability to maintain the killer virus (the Mak�
phenotype), increased affinity for aa-tRNA, and de-
creased peptidyltransferase activity (see Meskauskas
et al. [2003]). This broad spectrum of phenotypes sug-
gested a crucial role for W255 in ribosome function, raising
questions regarding its nature. Molecular modeling using
numerous X-ray crystal structures from H. marismortui
(Ban et al., 2000), E. coli (Schuwirth et al., 2005), and
T. thermophilus (Selmer et al., 2006) reveals that the struc-
ture of this core region of the large subunit is highly
conserved and that W255 is positioned at the tip of the
W finger (data not shown). In the current study, molecular
modeling was based on the H. marismortui structures
because (1) they provided the highest available levels of
resolution, and (2) its amino acid sequence in the central
extension of L3 most closely resembles that of the yeast
protein. This analysis revealed a potential base stacking
interaction along the A site-proximal side of the PTC be-
tween W255 of L3 and A2940 in helix 90 of 25S rRNA
(A2572 in E. coli and U2607 in H. marismortui 23S rRNA,
Figure 1A). The ‘‘plasmid shuffle’’ method (Rose et al.,
878 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevie
1990) was used to determine the viability of a set of amino
acid substitutions for W255 as the only forms of L3. Amino
acid substitutions were chosen to reflect the full range of
biochemical properties of their side chains. In agreement
with the proposed aromatic stacking, substitution of other
aromatic amino acids for tryptophan did not affect cell
growth (Figure 1B, Table 1). Arginine, with its long, basic
side chain, was also viable, but basic substitutions
showed decreased viability in proportion to reductions in
side-chain lengths, indicating that electrostatic interac-
tions may to some extent complement the requirement
for aromatic side chains. The acidic and small amino
acid substitutions, predicted to be unable to interact
with nearby bases (Figure 1A), all resulted in lethal pheno-
types (Figure 1B), as did deletion of W255 (data not
shown). DNA sequence analyses of clones rescued from
apparently viable W255E and W255A colonies revealed
that all were revertants to tryptophan. The set of viable
substitutions clearly indicates the requirement of aromatic
or positively charged amino acids at position 255 for cell
viability, the only exception being the original W255C mu-
tant (see next paragraph addressing this issue). These
findings also point to the importance of putative interac-
tions between the tip of the L3 W finger and rRNA bases,
the most likely candidate being A2940.
Intrinsic but Directionally Constrained Flexibility
of the W Finger
Given that small amino acids at position 255 were lethal,
how could substitution with cysteine be viable? Examina-
tion of nearby amino acids revealed a second cysteine,
C251, located four residues in the N-terminal direction.
We hypothesized that formation of a disulfide bond be-
tween C251 and C255 may promote structural rearrange-
ments of W finger, repositioning nearby aromatic or basic
residues to establish new contacts with A2940 (E. coli
A2572) or other nearby bases. If true, then disruption of
the disulfide bond should prevent such repositioning,
resulting in lethality. Two complementary experiments
were designed to test this hypothesis. Figure 2A shows
that while the W255C and C251S single mutants were
r Inc.
Molecular Cell
L3: The A Site Gatekeeper
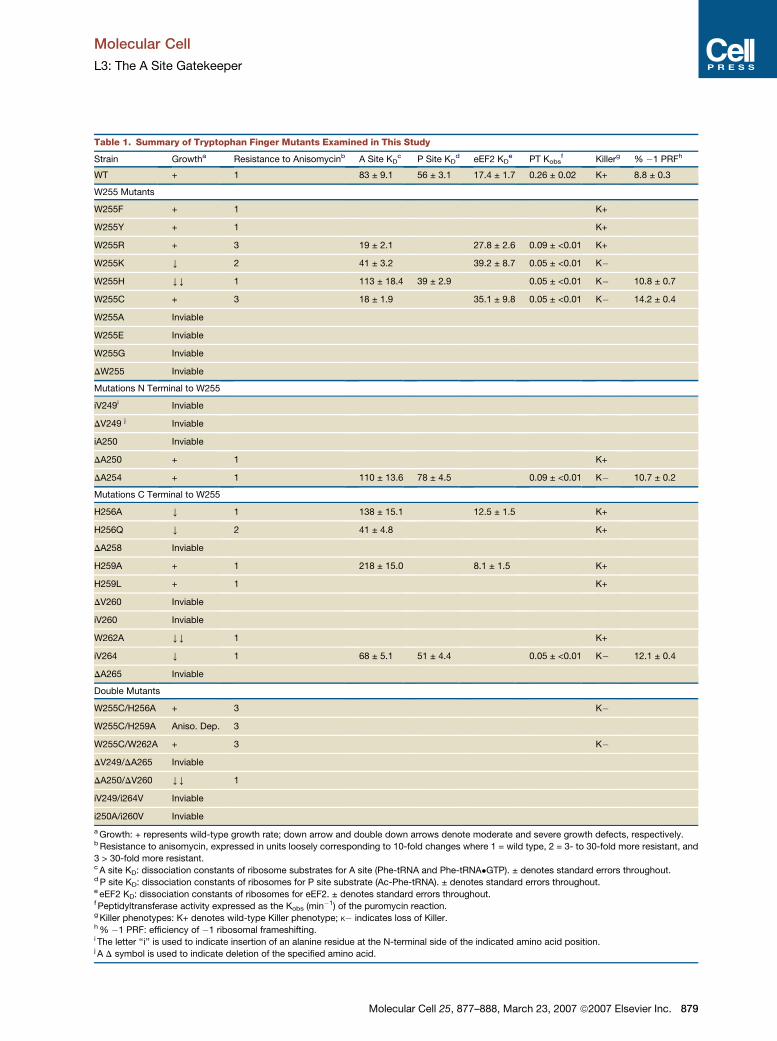
Table 1. Summary of Tryptophan Finger Mutants Examined in This Study
Strain Growtha Resistance to Anisomycinb A Site KDc P Site KD
d eEF2 KDe PT Kobs
f Killerg % �1 PRFh
WT + 1 83 ± 9.1 56 ± 3.1 17.4 ± 1.7 0.26 ± 0.02 K+ 8.8 ± 0.3
W255 Mutants
W255F + 1 K+
W255Y + 1 K+
W255R + 3 19 ± 2.1 27.8 ± 2.6 0.09 ± <0.01 K+
W255K Y 2 41 ± 3.2 39.2 ± 8.7 0.05 ± <0.01 K�
W255H YY 1 113 ± 18.4 39 ± 2.9 0.05 ± <0.01 K� 10.8 ± 0.7
W255C + 3 18 ± 1.9 35.1 ± 9.8 0.05 ± <0.01 K� 14.2 ± 0.4
W255A Inviable
W255E Inviable
W255G Inviable
DW255 Inviable
Mutations N Terminal to W255
iV249i Inviable
DV249 j Inviable
iA250 Inviable
DA250 + 1 K+
DA254 + 1 110 ± 13.6 78 ± 4.5 0.09 ± <0.01 K� 10.7 ± 0.2
Mutations C Terminal to W255
H256A Y 1 138 ± 15.1 12.5 ± 1.5 K+
H256Q Y 2 41 ± 4.8 K+
DA258 Inviable
H259A + 1 218 ± 15.0 8.1 ± 1.5 K+
H259L + 1 K+
DV260 Inviable
iV260 Inviable
W262A YY 1 K+
iV264 Y 1 68 ± 5.1 51 ± 4.4 0.05 ± <0.01 K� 12.1 ± 0.4
DA265 Inviable
Double Mutants
W255C/H256A + 3 K�
W255C/H259A Aniso. Dep. 3
W255C/W262A + 3 K�
DV249/DA265 Inviable
DA250/DV260 YY 1
iV249/i264V Inviable
i250A/i260V Inviable
a Growth: + represents wild-type growth rate; down arrow and double down arrows denote moderate and severe growth defects, respectively.b Resistance to anisomycin, expressed in units loosely corresponding to 10-fold changes where 1 = wild type, 2 = 3- to 30-fold more resistant, and
3 > 30-fold more resistant.c A site KD: dissociation constants of ribosome substrates for A site (Phe-tRNA and Phe-tRNA�GTP). ± denotes standard errors throughout.d P site KD: dissociation constants of ribosomes for P site substrate (Ac-Phe-tRNA). ± denotes standard errors throughout.e eEF2 KD: dissociation constants of ribosomes for eEF2. ± denotes standard errors throughout.f Peptidyltransferase activity expressed as the Kobs (min�1) of the puromycin reaction.g Killer phenotypes: K+ denotes wild-type Killer phenotype; K� indicates loss of Killer.h % �1 PRF: efficiency of �1 ribosomal frameshifting.i The letter ‘‘i’’ is used to indicate insertion of an alanine residue at the N-terminal side of the indicated amino acid position.j A D symbol is used to indicate deletion of the specified amino acid.
Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 879

Molecular Cell
L3: The A Site Gatekeeper
Figure 2. Intrinsic but Limited Flexibility
of the L3 W Finger
(A) Wild-type L3, W255C, and C251S are viable
as the sole forms of L3, but the W255C/C251S
double mutant is not.
(B) The W255C mutant is hypersensitive to the
reducing agent dithiothreitol.
(C) H259 is absolutely required for viability in
combination with W255C, and H256 is partially
required, suggesting that the putative inter-
actions normally made between W255 and
A2940 (E. coli A2572) are compensated for by
a combination of H259 and H256 in the
W255C mutant.
(D) Insertions or deletions in the W finger,
resulting in shifting of W255 toward the
N-terminal side of the central extension (green
up arrow) are mostly viable (except iV260 and
DV249), while those that shift W255 toward
the C-terminal side (red down arrow) are lethal.
(E) Model describing these observations.
Modeling was based on the cryo-EM map of
the yeast 80S ribosome threaded onto the
X-ray crystal structure of H. marismortui (Spahn
et al., 2004). (Top panel) Mutated cysteine at
position 255 presumably forms a disulfide
bridge with C251 (red dots), repositioning
H256 and H259 to interact with A2940 (shown
as H. marismortui U2607). (Bottom panel) In
general, mutants that allow movement of the
tip of the W finger toward the N-terminal side
of the L3 W finger (green arrow) reposition his-
tidines on the C-terminal side to interact with
A2940, thus helping to restabilize the A site re-
gion of the PTC. In contrast, mutations that
move the tip in the other direction (red arrow)
are lethal because there are no basic or aro-
matic amino acid residues on the N-terminal
side of W255 to interact with A2940.
viable, the W255C/C251S double mutant was lethal.
Figure 2B shows that 4 mM DTT severely inhibited growth
of the W255C mutant, but not of wild-type cells. These re-
sults support the existence of a disulfide bond between
C255-C251 in the W255C mutant.
880 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevie
Formation of a disulfide bond between C255 and C251
would shift the tip of central extension toward the N termi-
nus, potentially moving basic or aromatic residues on the
C-terminal side of C255 into position to interact with
A2940. Examination of the proximal amino acids on the
r Inc.

Molecular Cell
L3: The A Site Gatekeeper
C-terminal side of position 255 revealed three candidates:
H256, H259, and W262. To test this hypothesis, these res-
idues were changed to alanine either individually or in
combination with the W255C mutation (Figure 2C). Among
the single mutants, H256A and H259A did not affect
cell growth, while W262A was viable, albeit it negatively
impacted growth rates. Among the double mutants, the
W255C/H256A allele was viable but conferred reduced
growth, the W255C/H259A double mutant was inviable,
and growth of the W255C/W262A mutant was indistin-
guishable from wild-type cells. These data suggest that
in the W255C mutant, H259 (likely in combination with
H256) is repositioned to interact with A2940, although
the possibility that other bases may be involved cannot
be excluded. A cartoon of this model is shown in the
upper panel of Figure 2E.
These results indicate that the requirement for aromatic
or positively charged residues at the tip of the L3 W finger
can be met by adjacent positive residues, provided that
they are moved into correct position subsequent to for-
mation of the putative disulfide bond between C251 and
C255 in the W255C mutant. Although the data do not allow
determination of the exact conformation of the W finger
in the W255C mutant, they suggest that a certain degree
of flexibility of the W finger in the direction of the N termi-
nus can be tolerated and that this flexibility may be an
inherent feature of L3. The next issue was to determine
the extent and limitations of the W finger’s flexibility. The
amino acid sequence surrounding the tip of the W finger
is N-RKVACIGA(W255)HPAHVMW-C. On the N-terminal
side, the amino acids proximal to W255 are all aliphatic,
and the nearest best candidates for interactions with
rRNA are seven and eight residues away (arginine and
lysine). As discussed above, the C-terminal side contains
the two nearby histidines and a tryptophan. Insertional
mutagenesis of amino acids on the N-terminal side of
W255 would move adjacent N-terminal residues toward
the position occupied by W255, and these mutants should
be inviable. Deletions of residues C terminal to W255
should have similar effects. In contrast, deletions on the
N-terminal side and insertions on the C-terminal side of
W255 were predicted to be viable because they should
bend the tip of the W finger toward the N terminus, moving
H256 into position where it could interact with A2940. To
test this model, L3 mutants containing amino acid dele-
tions or alanine insertions on either side of the W finger
were generated so as to either push or pull the W finger
in the N- or C-terminal directions as described above.
The results of this experiment are shown in Figure 2D.
As predicted, changes which should bend the W finger to-
ward its C-terminal side (indicated by the red down arrow),
e.g., insertions of alanine after V249 and A250, or deleting
A258, V260, and A265, were all lethal. Conversely, three
of the changes predicted to bend the W finger toward its
N-terminal side (indicated by the green arrow), i.e., inser-
tion of alanine after V264, and deleting A250 or A254,
were viable. The lethality due to insertion of alanine after
V260 or by deletion of V249 is likely to have resulted
Mole
from complex and unpredictable changes, either in the lo-
cal ribosome structure or in L3 itself. The effects of sym-
metrical double insertions and deletion mutations in the
W finger, predicted to move the tip of the W finger further
into or away from the PTC, respectively, were also as-
sayed. As shown in Table 1, the symmetrical double inser-
tion mutants (iV249/iV264, iV260/iA250) and one of the
double deletion mutants (DV249/DA265) were lethal. The
DV260/DA250 double deletion mutation was viable but
conferred a very severe growth defect on cells (Table 1).
These findings suggest that the W finger cannot move in
toward or away from the PTC without affecting cell viabil-
ity. In summary, three different series of targeted muta-
tions to the W finger of L3 are consistent with the notion
that the W finger is flexible but that its range of motion is
very tightly constrained within the densely crowded inner
core of the large subunit. Specifically, the data suggest
that its flexibility appears to be limited toward the N-termi-
nal side of W255 in the direction of the extended helix
formed by helices 90–92. A cartoon of this model is shown
in the lower panel of Figure 2E.
Mutations in the W Finger Affect 25S rRNA
Structure in the Sarcin/Ricin Loop, along the
Pathway Taken by Accommodating aa-tRNA,
and the A Site-Proximal Side of the PTC
The data presented above suggested that the W finger
mutants may affect local ribosome structure. To examine
this, ribosomes purified from wild-type and mutant cells
were probed for structural changes using three base-
specific solvent-accessible reagents: dimethylsulfate
(DMS), kethoxal, and carbodiimide metho-p-toluenesulfo-
nate (CMCT). rRNAs were extracted, and modified bases
were identified by primer extension using reverse tran-
scriptase to detect methylation at the N3 position of
uridines and the N1 position of guanosines (CMCT), at
the N1 and N2 positions of guanosines (kethoxal), and at
the N1 position of adenosines and N3 position of cytidines
(DMS) (Inoue et al., 1985). The primers used (see Experi-
mental Procedures) were designed to probe functional
regions of domain V (including the A site and P site loops,
the PTC, and the helices adjacent to these structures, i.e.,
helices 73, 74, 89–93), and helices 94–96.
The results of these experiments are shown in Figures
3A–3C and are summarized in Figures 3D and 3E. The
W255C mutant significantly altered the structure of the
PTC, especially in the A site loop and along helices
90–92 (Figure 3A). In the helix 90–92 structure, A2889,
C2898, G2913, C2924, A2929, A2932, A2935, A2940,
and C2941 were significantly deprotected from chemical
modification. In helix 89, U2861 showed decreased reac-
tivity to CMCT. Similar, albeit weaker deprotection pat-
terns were noted in the W255R, W255H, W255K,
H256Q, and DA254 mutants (data not shown). The stron-
ger effects due to the W255C mutation may reflect the
large conformational changes of the W finger due to the
putative C251-C255 disulfide bond described above.
cular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 881

Molecular Cell
L3: The A Site Gatekeeper
Figure 3. Structure Probing of Wild-Type and Mutant Ribosomes
(A–C) Autoradiograms of reverse transcriptase primer extension reactions spanning sequence in helices 89–93 (A), helix 73 (B), and the helix 94–95
regions (C). Sequencing reactions (left sides of panels) are labeled corresponding to the rRNA sense strand. Mutants are indicated at top, and bases
with altered chemical protection patterns are indicated to the left. Below each panel, ‘‘U’’ stands for untreated, ‘‘D’’ is DMS, ‘‘C’’ denotes CMCT, and
‘‘K’’ indicates kethoxal.
882 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc.

Molecular Cell
L3: The A Site Gatekeeper
L3 also makes contacts with bases in helix 73 at the A
site-proximal side of the PTC (Klein et al., 2004). Chemical
protection experiments revealed increased modification
of two bases, A2398 and A2400, in the W255R and
W255K mutants (Figure 3B). The region comprising the
SRL, which forms part of the elongation factor-binding
site, was also probed for structural changes (Figure 3C).
A3020 and A3023, both located in the SRL, and A3010
(base of helix 95) were deprotected in the W255C mutant.
A few additional changes were also noted in the vicinity
of the SRL in the W255R (deprotection of C3003, G3014,
and G3014), H256Q (deprotection of A3023), and DA245
(enhanced protection of A3020) mutants. The major
changes in protection patterns in the vicinity of the PTC
and SRL are summarized on a two-dimensional rendering
of the yeast 25S rRNA (Figure 3D). Molecular modeling of
these changes to the three-dimensional structure of the
H. marismortui large ribosomal subunit (Figure 3E) reveals
that the W finger mutants affect the conformation of re-
gions of 25S rRNA that are involved in interactions with
the aa-tRNA from where it first encounters the ribosome
in complex with eEF-1A, along the pathway taken by
accommodating aa-tRNA (indicated by the orange arrow)
(Sanbonmatsu et al., 2005), and into the A site region of
the PTC in the core of the large subunit. Taken together,
the rRNA chemical protection studies indicate that this
group of W finger mutants promoted a more accessible or
‘‘open’’ conformation of these regions of the large subunit.
An Inverse Relationship between Ribosomal
Affinity for aa-tRNA and eEF2
We previously showed that the W255C mutant promoted
increased affinity for aa-tRNA (Meskauskas et al., 2005).
This analysis was extended in the current study in which
aa-tRNA binding properties were compared between
wild-type and nine mutants. Preparations of non-salt-
washed ribosomes containing soluble factors were used
to monitor aa-tRNA/ribosome interactions because bind-
ing of aa-tRNA to the ribosomal A site requires elongation
factor 1 (Schilling-Bartetzko et al., 1992). The saturation
curves of [14C]Phe-tRNA binding shown in Figure 4A
were analyzed using GraphPad Prism software fitted to a
nonlinear regression one-site binding curve [Y = Bmax*X /
(KD + X)], where Y denotes bound [14C]Phe-tRNA values,
X is input [14C]Phe-tRNA, and Bmax is maximal ligand bind-
ing. As summarized in Table 1, KD values for aa-tRNA var-
ied over an�10-fold range, from a low of�20 nM (W255C
and W255R) to >200 nM (H259A).
Coordination of the translation elongation cycle requires
that binding of the eEF-1A�aa-tRNA�GTP ternary com-
Molec
plex and eEF2 to the elongation factor-binding region be
mutually exclusive. To examine eEF2/ribosome interac-
tions, affinity-purified 63 His-tagged eEF2 was incubated
with GDPNP and salt-washed ribosomes isolated from
cells expressing wild-type L3 as well as six mutant forms
of L3. Ribosomes were harvested by centrifugation, the
pellets were treated with EDTA, and cofractionating
eEF2 was ADP ribosylated using diphtheria toxin and
[14C]NAD+. Ribosome-associated eEF2 was then quanti-
fied by liquid scintillation. Analyses of the saturation
curves (Figure 4B) revealed notable differences in eEF2
binding among the different ribosomes. As summarized
in Table 1, KD values for eEF2 varied over a 5-fold range,
from a low of�8 nM (H259A) to �40 nM (W255K). Inspec-
tion of these data clearly shows a reciprocal relationship
between eEF-1A stimulated binding of aa-tRNA and bind-
ing of eEF2.
Ribosomes isolated from wild-type cells and from three
mutants (DA254, W255H, and iV264) were also assayed
with respect to their affinities for Ac[14C]Phe-tRNA. As
summarized in Table 1 (data from saturation curves shown
in Figure 4C), only small changes in P site binding were ob-
served for the W255H and DA254 mutants. These findings
are consistent with previous data showing that the W255C
mutation did not affect Ac-Phe-tRNA binding to the P site
(Meskauskas et al., 2005).
Anisomycin Resistance Correlates with Increased
Affinity for aa-tRNA
As shown in Figure 3E, anisomycin competes with the 30
end of aa-tRNA for the same binding site in the A site of
the peptidyltransferase center (Hansen et al., 2003). Prior
examination of the randomly generated series of L3 mu-
tants demonstrated a correlation between anisomycin re-
sistance and increased ribosomal affinity for aa-tRNA
(Meskauskas et al., 2005). To further examine this relation-
ship, 10-fold dilution spot assays were performed on rich
media alone or on media containing 50 mg/ml of anisomy-
cin, and growth of cells in the presence of drug relative to
no drug was used to score for anisomycin resistance. The
results of these experiments are summarized in Table 1. At
position 255, the aromatic substitutions (W255F and
W255Y) did not affect anisomycin sensitivity. The longer
basic substitutions (W255R and W255K) showed aniso-
mycin resistance. Changing H256 to alanine (H256A) did
not promote resistance to anisomycin, but substitution
with glutamine (H256Q) promoted drug resistance. Nei-
ther the H259L nor H259A mutants were anisomycin resis-
tant, but these mutations in combination with W255C
(H256A/W255C, W255C/H259A, and W262A/W255C)
(D) Localization of bases in yeast 25S rRNA whose modification patterns were affected by the L3 mutants. Protected and deprotected bases are in-
dicated by open and filled arrows, respectively; bases of 25S rRNA located within 4 A of W255 are boxed in gray.
(E) rRNA protection data mapped onto the H. marismortui large subunit crystal structure. Base numbering follows the S. cerevisiae sequence shown in
(D), and E. coli numbering is shown in parentheses. The mutants described above result in conformational changes of helices 90–92 (orange and
yellow), helix 95 (light blue), and helix 73 (gray). Nucleotides with altered chemical protection patterns are shown in pale blue. Bases forming the
two A site gates are in yellow. Bases interacting with anisomycin are in salmon. A2819 (E. coli A2451) is shown in red. The path taken by the 30
end of the aa-tRNA from the SRL to the PTC (Sanbonmatsu et al., 2005) is shown as an orange arrow.
ular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 883

Molecular Cell
L3: The A Site Gatekeeper
Figure 4. Biochemical Characterization of Ribosomes Extracted from Cells Expressing L3 W Finger Mutants
(A) Single site isotherms of eEF-1A stimulated binding of [14C]Phe-tRNA to ribosomal A sites.
(B) eEF2 binding isotherms for wild-type and mutant ribosomes.
(C) Binding isotherms of Ac-[14C]Phe-tRNA to ribosomal P sites.
(D) Characterization of peptidyltransferase activities of wild-type and mutant ribosomes by first order time plots of Ac-[14C]Phe-puromycin formation.
Error bars denote standard deviations.
were all drug resistant. In fact, W255C/H259A, which was
inviable on drug-free media, grew well in the presence of
anisomycin, i.e., it was anisomycin dependent. None of
the viable insertion or deletion mutants affected growth
in the presence of anisomycin. Examination of Table 1
shows that decreasing dissociation constants for
[14C]Phe-tRNA correlated with the degree of anisomycin
resistance. Approximately 4-fold decreases in KD values
correlated with >30-fold increases, and �2-fold de-
creases correlated with 3- to 30-fold increases in aniso-
mycin resistance, respectively.
Increased Rates of�1 PRF and Inability to Propagate
the Killer Virus Correlate with Decreased
Peptidyltransferase Activity
Maintenance of the yeast killer virus is exquisitely sensitive
to subtle defects in the translational apparatus (reviewed
in Wickner [1996]), providing a rapid and sensitive assay
to monitor the effects of the W finger mutants on ribosome
function. The killer virus maintenance phenotypes of the
mutants generated in this study are summarized in Table
1. At position 255, the healthy substitutions (W255F,
W255Y, and W255R) were able to maintain the killer virus
(Mak+ phenotype), but the less viable W255K and W255H
were not (Mak�). In the C-terminal direction, changes at
position 256 (H256A and H256Q) and 259 (H259L and
H259A) did not affect killer virus maintenance. However,
884 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier
the double mutants of this series in combination with
W255C revealed interesting trends: W255C/H256A and
W262A/W255C were Mak�, indicating that the W255C
mutation is dominant in this aspect. Of the viable insertion
and deletion mutants, two promoted the Mak� phenotype
(DA254 and iV264), and one maintained the killer virus
(DA250). The L-A viral replicase is synthesized by a �1
PRF event; viral particle assembly and propagation of
the killer virus depends on proper rates of�1 PRF (Dinman
and Wickner, 1992). PRF efficiency was monitored for
wild-type cells and four of the Mak� strains (W255H,
W255C, DA254, and iV264). Consistent with previous
studies, there was a strong correlation between increased
levels of �1 PRF and the Mak� phenotype (see Table 1).
Prior studies suggested a correlation between de-
creased peptidyltransferase activity and increased �1
PRF and the Mak� phenotype (Meskauskas et al., 2003),
but a later investigation suggested that other factors
may influence the relationships between these parame-
ters (Meskauskas et al., 2005). Peptidyltransferase activi-
ties were significantly decreased in all six mutants tested
(DA254, W255C, W255H, W255K, W255R, and iV264,
see Figure 4D and Table 1), and in general, this cor-
responded with the inability of cells to maintain the killer
virus (see Table 1). The W255R mutant was the exception:
although peptidyltransferase activity in this mutant was in-
hibited to the same extent as, for example, DA254 (which
Inc.

Molecular Cell
L3: The A Site Gatekeeper
had elevated levels of�1 PRF and was Mak�), this mutant
nonetheless stably maintained the Killer+ phenotype.
DISCUSSION
The elongation cycle requires coordination of an ordered
series of events. The elongation factor 1�aa-tRNA�GTP
ternary complex must first interact with the ribosomes’
factor-binding site. At the decoding center in the small
subunit, the presence of cognate codon:anticodon
base-pairing is transduced up the body of the aa-tRNA,
activating GTP hydrolysis by elongation factor 1 (Valle
et al., 2002; Cochella and Green, 2005). This initiates ac-
commodation of the aa-tRNA into the PTC. Successful ac-
commodation has been hypothesized to be accompanied
by rRNA structural changes that both close the aa-tRNA
accommodation corridor (Sanbonmatsu et al., 2005),
and activate the PTC (Schmeing et al., 2005). Concur-
rently, the structure of the factor-binding site must also
change to favor elongation factor 2 binding, setting the
stage for translocation (Sergiev et al., 2005). The rRNA
structure probing and ribosome binding data presented
in the current study suggest a model describing how L3
may help to coordinate these ribosome-associated func-
tions by acting as a ‘‘gatekeeper’’ to the A site.
Computer modeling studies suggest that during ac-
commodation, the aa-tRNA 30 end moves through a ‘‘cor-
ridor’’ formed by helix 89 on one side and the helix 90–92
structure on the other (Sanbonmatsu et al., 2005). The
chemical protection data clearly show that specific bases
in the helix 90–92 side of this pathway are generally depro-
tected in the W255C mutant as compared to wild-type ri-
bosomes. Thus, the increased accessibility of bases in
this structure to small chemicals suggests that it is in a rel-
atively open conformation (Figure 3A). The computational
simulations of aa-tRNA accommodation also suggest that
the aa-tRNA 30 end passes through two rRNA-based
structures or ‘‘gates.’’ The first gate encountered by aa-
tRNA (gate 1) is formed by U2491 and C2556 (yeast
U2860 and C2924), while the second (gate 2) is formed
by C2573 (yeast C2941) (see Figures 3D and 3E). Notably,
the gate bases in the helix 90–92 structure (C2924 and
C2941) were deprotected, consistent with an open con-
formation. Interestingly, U2861 was protected from chem-
ical attack. It is possible that reorientation of the helix 89
gate 1 base U2860 had a shielding effect on U2861, con-
sistent with the idea that the W255C mutant promotes
opening of the accommodation corridor. Additionally,
the chemical protection studies suggest that these con-
formational changes in this and other mutants are promul-
gated along the entire pathway traveled by the aa-tRNA 30
end, from the SRL down to the PTC (Figures 3B and 3C).
Opening of the aa-tRNA accommodation corridor
should affect the biochemical properties of the ribosome.
Specifically, such changes should promote increased af-
finity for aa-tRNA, which is supported by the observation
of decreased KD values for aa-tRNA by the W255C,
W255R, W255K, and H256Q mutants. The increased
Molec
eEF2 KD values for these mutants are also consistent
with the notion that ternary complex and eEF2 binding
should be mutually exclusive. This is further supported
by the H256A and H259A mutants, which promoted in-
creased affinity for eEF2 at the expense of their ability to
interact with aa-tRNA. These biochemical observations
suggest that two classes of mutants were generated:
one promoting ribosomal conformations more favorable
to eEF2 binding, the other favoring binding of the eEF-
1A�aa-tRNA�GTP ternary complex. These data suggest
that in the L3 mutants, bases in the PTC and along the
aa-tRNA accommodation pathway have been artificially
shifted into conformations that are normally transitory dur-
ing translation in wild-type cells. Specifically, we propose
that some W finger mutants (e.g., W255C, W255R,
W255K, and perhaps H256Q) favor positioning of the
SRL into the eEF-1A�aa-tRNA�GTP binding state, and
the accommodation corridor into the open conformation,
while others (H256A and H269A) favor the closure of the
corridor and eEF2 binding.
These findings suggest a model wherein the W finger
may normally assume different positions within the ribo-
some during these two factor-binding states, possibly ac-
companied by different patterns of putative interactions
between amino acids in the W finger and 25S rRNA bases.
This is cartooned in Figure 5. By this model, movement of
the L3 W finger may participate, either actively or pas-
sively, in an allosteric process to coordinate these events.
Specifically, positioning of the W finger toward its N-termi-
nal side, i.e., directed away from the PTC and toward the
factor-binding site, may help both to open the helix 90–92
face of the accommodation corridor and to position the
SRL so as to favor binding of the eEF-1A�aa-tRNA�GTP
ternary complex. We suggest that, while in this conforma-
tion, A2940 of 25S rRNA may interact with H259 of L3. The
model proposes that, during accommodation, the W fin-
ger swings toward its C-terminal side (toward the PTC)
and that the A2940-W255 interaction is established upon
completion of aa-tRNA accommodation into the A site.
Additionally, because the aa-tRNA and eEF2 KD values
for the H256A mutant were between those observed for
the W255 mutants (W255C, W255R, and W255K) and
H259A, it is also possible that interaction between
A2940 and H256 could constitute a transitional inter-
mediate between the two states. We propose that, during
rearrangement of the W finger, the helix 90–92 structure
moves to close the accommodation gates. Concurrently,
the SRL atop helix 95 is repositioned so as to form the
eEF2 competent binding site. This sequence of events
would allow the occupancy status of the A site to be com-
municated to the translation factor-binding region, pre-
venting entry of new aa-tRNA into the A site during trans-
location. It must be noted that our results have only
revealed amino acid residues in the W finger that may be
involved in these conformational events, not their rRNA
counterpart bases. Although the chemical properties of
the substituted amino acids suggest their involvement
in interactions with nucleotides, the actual rRNA base
ular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 885

Molecular Cell
L3: The A Site Gatekeeper
Figure 5. Model for the Role of L3 in
Coordinating Functions of the Elongation
Factor-Binding Site: L3 as the Gate-
keeper to the A Site
(Left) Positioning of the L3 W finger (green) away
from the PTC (red oval) positions the helix 90–
92 side of the aa-tRNA accommodation corri-
dor so as to open the accommodation gates
(red circles), allowing accommodation (dotted
gray line) of the 30 end of aa-tRNA (aa). ‘‘pep’’
denotes the 30 end of the peptidyl-tRNA. This
W finger conformation also positions helix 95
to form the eEF-1A�aa-tRNA�GTP ternary
complex binding site and promotes interaction between L3 H259 and 25S rRNA A2940, denoted by ‘‘A’’ (E. coli A2572). (Right) During/after accom-
modation, repositioning of the W finger toward the PTC closes the gates, repositions helices 95 and 90–92 to form the eEF2 binding site, and
promotes the W255-A2940 stacking interaction.
involved was not experimentally determined, but A2940
represents the most likely candidate based on the crystal
structures.
Resistance of the W255C mutant to anisomycin has
been known for many years (Fried and Warner, 1981),
but the underlying mechanism has been unclear. Early
studies showed that the W255C mutation did not affect
anisomycin binding (Jimenez and Vazquez, 1975), sug-
gesting that structural and/or conformational changes in
resistant ribosomes may reduce the ability of anisomycin
to compete with aa-tRNA for entry into the A site. The
chemical protection data presented in the current study
show structural changes in the large subunit consistent
with a more open conformation in the vicinity of the A
site of the PTC and in the aa-tRNA accommodation corri-
dor in the anisomycin-resistant mutants. Opening of the
accommodation corridor should facilitate aa-tRNA pas-
sage into the A site, consistent with the decreased KD
values for aa-tRNAs. In contrast, because of its small mo-
lecular radius relative to aa-tRNA, access to the PTC by
anisomycin should not be as intrinsically limited by the
two gates, and hence should not be stimulated as much
by gate opening. Therefore, it is possible that these muta-
tions lower the barriers encountered by aa-tRNA relative
to anisomycin during accommodation, thus altering the ki-
netic partitioning ratios for these two molecules, resulting
in anisomycin resistance. An alternate, nonexclusive ex-
planation may be that changes in the interactions between
anisomycin and bases in the PTC (see Figure 3E) conse-
quent to these mutations may contribute to anisomycin re-
sistance. Interestingly, the �OH group of the N-terminal
serine of L3 (S2) is predicted to interact with C2924, the
helix 90 ‘‘Gate 1’’ residue (see Figure 3E). The S2T mutant
was also strongly anisomycin resistant (Meskauskas et al.,
2005), and experiments are ongoing to characterize
a bank of S2 mutants.
An ‘‘entropic catalysis’’ model of peptidyltransfer posits
that precise positioning of substrates and water reorgani-
zation in the PTC allows formation of a highly ordered
electrostatic network that stabilizes reaction intermedi-
ates, providing the proper environment for the ribosome
to accelerate peptide bond formation (reviewed in Rod-
nina et al. [2007]). The rRNA structure-probing experi-
886 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevie
ments in the current study are consistent with this model
insofar as distortion of the topology of the A site side of
the PTC by the W finger mutants could affect positioning
of the aa-tRNA 30 end, promoting the observed peptidyl-
transfer defects. It has also been proposed that the PTC
is activated by an ‘‘induced fit’’ mechanism, wherein ac-
commodation of the aa-tRNA acceptor stem into the A
site reorganizes the geometry of the PTC, making the ester
group of the peptidyl-tRNA accessible for nucleophilic at-
tack (Schmeing et al., 2005). The involvement of the L3 W
finger in this process remains an intriguing possibility.
It is remarkable how atomic scale allosteric events can
propagate themselves all the way to the biological level.
Translating our understanding of molecular structure and
biochemical properties of molecules may lead to concrete
medical benefits. For example, understanding how these
mutants affect the interactions between ribosomes and
trans-acting factors and how this leads to resistance to
A site-specific antibiotics can provide the foundation for
the design of new antibacterials to counter resistance of
pathogenic bacteria to drugs such as tiamulin (Pringle
et al., 2004). This research may also aid in the develop-
ment of new antivirals insofar as it furthers our under-
standing of how changes at the atomic level can affect
peptidyltransfer, and hence �1 PRF and virus propaga-
tion. It is hoped that atomic resolution structures of yeast
ribosomes will be solved in the near future and that
achievement of this milestone will enable the models pro-
posed herein to be directly visualized.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, Genetic Manipulation, and Media
E. coli DH5a was used to amplify plasmid DNA. Transformation of
E. coli and yeast, and preparation of yeast growth media (YPAD,
synthetic drop out medium, and 4.7 MB plates for testing the Killer
phenotype), were as previously reported (Dinman and Wickner,
1992). Restriction enzymes were obtained from MBI Fermentas
(Vilnius, Lithuania). The QuikChange XL II Site-Directed Specific Muta-
genesis Kit was obtained from Stratagene (La Jolla, CA). Macrogen
(Seoul, South Korea) performed DNA sequence analysis. Oligonucleo-
tide primers were purchased from IDT (Coralville, IA). The yeast strains
used in this study were all derived from the rpl3-gene disruption (rpl3D)
strain JD1090 (MATa ura3-52 lys2-801 trp1d leu2- his3 RPL3::HIS3
pRPL3-URA3-CEN6 [L-A HN M1]) (Meskauskas et al., 2003). Mutants
r Inc.

Molecular Cell
L3: The A Site Gatekeeper
of rpl3 were generated using the wild-type RPL3 gene in pJD225
(Meskauskas et al., 2003), synthetic oligonucleotides, and the Quik-
Change XL II Kit. The pYDL dual luciferase reporter series of plasmids
for monitoring programmed ribosomal frameshifting (PRF) were used
as previously described (Harger and Dinman, 2003). Ten-fold dilution
spot assays were performed on rich media alone and on media con-
taining 50 mg/ml of anisomycin, and growth of cells in the presence
of drug relative to no drug was used to score for anisomycin
resistance.
Characterization of Peptidyltransferase Activity, tRNA,
and eEF2 Binding Studies
Yeast phenylalanyl-tRNAs were aminoacylated with [14C]Phe-tRNA to
monitor A site binding, Ac-[14C]Phe-tRNA was generated to monitor P
site binding, these were purified by HPLC, and equilibrium binding
studies were performed as previously described (Meskauskas et al.,
2005). Peptidyltransfer assays were performed essentially as previ-
ously described (Dresios et al., 2001; Meskauskas et al., 2005). 63
His-tagged eEF2 was purified from TKY675 yeast cells (kindly pro-
vided by Dr. T. Kinzy) as previously described (Ortiz et al., 2006). For
eEF2 binding experiments, reaction mixes containing 25 pmol of
salt-washed 80S ribosomes and various concentrations of 63 His-
tagged eEF2 (5–40 pmol) in binding buffer (50 mM Tris-HCl [pH 7.5],
50 mM ammonium acetate, 10 mM magnesium acetate, 2 mM DTT,
and 100 mM GDPNP) were incubated for 5 min at room temperature,
layered on top of the 300 ml sucrose cushion (10% sucrose in binding
buffer), and centrifuged at 50,000 rpm for 20 min at 4�C in a MLS50
(Beckman) rotor. Pellets (bound fraction) were washed and resus-
pended in 50 ml of DT buffer (50 mM Tris-HCl [pH 7.5], 20 mM DTT,
and 1 mM EDTA). Ribosome-bound eEF2 was ADP ribosylated for
20 min at 37�C by adding 100 pmol of [14C]NAD+ and 0.2 mg of diph-
theria toxin. Reaction mixes were precipitated with TCA, and amounts
of [14C]ADP-ribosylated eEF2 were determined by liquid scintillation
counting. Control values (lacking eEF2, not exceeding 10% of the low-
est count) were subtracted. KD values were determined assuming sin-
gle binding sites using GraphPad Prism software.
Isolation and Chemical Probing of Mutant Ribosomes
Ribosomes were isolated as described (Meskauskas et al., 2005) and
were synchronized by puromycin treatment in buffer C (50 mM Tris-
HCl [pH 7.5], 5 mM magnesium acetate, 50 mM ammonium chloride,
0.1 mM PMSF, 0.1 mM DTE, and 25% glycerol) containing 1 mM
GTP, 1 mM puromycin, and 500 pmol of ribosomes. After incubation
for 30 min at 30�C, ribosomes were sedimented through a cushion
of buffer B (20 mM Tris-HCl [pH 7.5], 10 mM magnesium acetate,
0.1 mM PMSF, 0.1 mM DTE, 0.5 M KCl, and 25% glycerol) and
suspended in buffer C at 10 pmol/ml. Chemical probing with DMS,
kethoxal, and CMCT, followed by RT primer extension analysis of
modified RNAs, was performed as described (Stern et al., 1988).
Primers (numbered after the first transcribed base of yeast 25S
rRNA) employed for these analyses were 2957 (50-AACCTGTCTCAC
GACGG-30), 3043 (50-CCTGATCAGACAGCCGC-30), 2877 (50-GGTAT
GATAGGAAGAGC-30), 2435 (50-CCTCTATGTCTCTTCAC-30 ), and 2675
(50-GTTCTACTGGAGATTTCTG-30).
Computational Analysis of Ribosome Structure
The X-ray crystal structure of the H. marismortui 50S ribosomal subunit
(Ban et al., 2000) and the cryoelectron microscopy (cryo-EM) recon-
struction of Saccharomyces cerevisiae ribosomal proteins threaded
onto the X-ray crystal structure of the H. marismortui 50S ribosomal
subunit (Spahn et al., 2004) were visualized using PyMOL (DeLano
Scientific LLC).
ACKNOWLEDGMENTS
We would like to thank the members of our laboratory, with special
thanks to Jennifer Baxter-Roshek and Alexey Petrov for stimulating
Mole
discussions, and to Rasa Rakauskaite and Johnathan Russ for techni-
cal assistance. This work was supported by grants from the NIH to
J.D.D. (GM58859) and from the American Heart Association to A.M.
(AHA 0630163N).
Received: November 27, 2006
Revised: January 22, 2007
Accepted: February 14, 2007
Published: March 22, 2007
REFERENCES
Ban, N., Nissen, P., Hansen, J., Moore, P.B., and Steitz, T.A. (2000).
The complete atomic structure of the large ribosomal subunit at
2.4 A resolution. Science 289, 905–920.
Bosling, J., Poulsen, S.M., Vester, B., and Long, K.S. (2003). Resis-
tance to the peptidyl transferase inhibitor tiamulin caused by mutation
of ribosomal protein l3. Antimicrob. Agents Chemother. 47, 2892–
2896.
Brodersen, D.E., and Nissen, P. (2005). The social life of ribosomal pro-
teins. FEBS J. 272, 2098–2108.
Cochella, L., and Green, R. (2005). An active role for tRNA in decoding
beyond codon:anticodon pairing. Science 308, 1178–1180.
Dinman, J.D., and Wickner, R.B. (1992). Ribosomal frameshifting effi-
ciency and Gag/Gag-pol ratio are critical for yeast M1 double-stranded
RNA virus propagation. J. Virol. 66, 3669–3676.
Dresios, J., Panopoulos, P., Frantziou, C.P., and Synetos, D. (2001).
Yeast ribosomal protein deletion mutants possess altered peptidyl-
transferase activity and different sensitivity to cycloheximide. Bio-
chemistry 40, 8101–8108.
Fried, H.M., and Warner, J.R. (1981). Cloning of yeast gene for tricho-
dermin resistance and ribosomal protein L3. Proc. Natl. Acad. Sci.
USA 78, 238–242.
Hansen, J.L., Moore, P.B., and Steitz, T.A. (2003). Structures of five an-
tibiotics bound at the peptidyl transferase center of the large ribosomal
subunit. J. Mol. Biol. 330, 1061–1075.
Harger, J.W., and Dinman, J.D. (2003). An in vivo dual-luciferase assay
system for studying translational recoding in the yeast Saccharomyces
cerevisiae. RNA 9, 1019–1024.
Hudak, K.A., Dinman, J.D., and Tumer, N.E. (1999). Pokeweed antiviral
protein accesses ribosomes by binding to L3. J. Biol. Chem. 274,
3859–3864.
Inoue, T., Sullivan, F.X., and Cech, T.R. (1985). Intermolecular exon li-
gation of the rRNA precursor of Tetrahymena: oligonucleotides can
function as 50 exons. Cell 43, 431–437.
Irvin, J.D., and Uckun, F.M. (1992). Pokeweed antiviral protein: ribo-
some inactivation and therapeutic applications. Pharmacol. Ther. 55,
279–302.
Jimenez, A., and Vazquez, D. (1975). Quantitative binding of antibiotics
to ribosomes from a yeast mutant altered on the peptidyl-transferase
center. Eur. J. Biochem. 54, 483–492.
Jimenez, A., Sanchez, L., and Vazquez, D. (1975). Simultaneous ribo-
somal resistance to trichodermin and anisomycin in Saccharomyces
cerevisiae mutants. Biochim. Biophys. Acta 383, 427–434.
Klein, D.J., Moore, P.B., and Steitz, T.A. (2004). The roles of ribosomal
proteins in the structure assembly, and evolution of the large ribosomal
subunit. J. Mol. Biol. 340, 141–177.
Meskauskas, A., Harger, J.W., Jacobs, K.L.M., and Dinman, J.D.
(2003). Decreased peptidyltransferase activity correlates with in-
creased programmed -1 ribosomal frameshifting and viral mainte-
nance defects in the yeast Saccharomyces cerevisiae. RNA 9, 982–
992.
cular Cell 25, 877–888, March 23, 2007 ª2007 Elsevier Inc. 887

Molecular Cell
L3: The A Site Gatekeeper
Meskauskas, A., Petrov, A.N., and Dinman, J.D. (2005). Identification
of functionally important amino acids of ribosomal protein L3 by satu-
ration mutagenesis. Mol. Cell. Biol. 25, 10863–10874.
Noller, H.F. (2007). Structure of the bacterial ribosome and some impli-
cations for translational regulation. In Translational Control in Biology
and Medicine, M.B. Mathews, N. Sonenberg, and J.W.B. Hershey,
eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press),
pp. 41–58.
Nowotny, V., and Nierhaus, K.H. (1982). Initiator proteins for the as-
sembly of the 50S subunit from Escherichia coli ribosomes. Proc.
Natl. Acad. Sci. USA 79, 7238–7242.
Ortiz, P.A., Ulloque, R., Kihara, G.K., Zheng, H., and Kinzy, T.G. (2006).
Translation elongation factor 2 anticodon mimicry domain mutants af-
fect fidelity and diphtheria toxin resistance. J. Biol. Chem. 281, 32639–
32648.
Peltz, S.W., Hammell, A.B., Cui, Y., Yasenchak, J., Puljanowski, L., and
Dinman, J.D. (1999). Ribosomal protein L3 mutants alter translational
fidelity and promote rapid loss of the yeast killer virus. Mol. Cell.
Biol. 19, 384–391.
Petrov, A., Meskauskas, A., and Dinman, J.D. (2004). Ribosomal
protein L3: influence on ribosome structure and function. RNA Biol.
1, 59–65.
Pringle, M., Poehlsgaard, J., Vester, B., and Long, K.S. (2004). Muta-
tions in ribosomal protein L3 and 23S ribosomal RNA at the peptidyl
transferase centre are associated with reduced susceptibility to tiamu-
lin in Brachyspira spp. isolates. Mol. Microbiol. 54, 1295–1306.
Rodnina, M.V., Beringer, M., and Wintermeyer, W. (2007). How ribo-
somes make peptide bonds. Trends Biochem. Sci. 32, 20–26.
Rose, M.D., Winston, F., and Hieter, P. (1990). Methods in Yeast
Genetics (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory
Press).
Sanbonmatsu, K.Y., Joseph, S., and Tung, C.S. (2005). Simulating
movement of tRNA into the ribosome during decoding. Proc. Natl.
Acad. Sci. USA 102, 15854–15859.
Schilling-Bartetzko, S., Franceschi, F., Sternbach, H., and Nierhaus,
K.H. (1992). Apparent association constants of transfer-RNAs for the
ribosomal A-site, P-site, and E-site. J. Biol. Chem. 267, 4693–4702.
888 Molecular Cell 25, 877–888, March 23, 2007 ª2007 Elsevie
Schindler, D., Grant, P., and Davies, J. (1974). Trichodermin resis-
tance—mutation affecting eukaryotic ribosomes. Nature 248, 535–
536.
Schmeing, T.M., Huang, K.S., Strobel, S.A., and Steitz, T.A. (2005). An
induced-fit mechanism to promote peptide bond formation and ex-
clude hydrolysis of peptidyl-tRNA. Nature 438, 520–524.
Schulze, H., and Nierhaus, K.H. (1982). Minimal set of ribosomal com-
ponents for reconstitution of the peptidyltransferase activity. EMBO J.
1, 609–613.
Schuwirth, B.S., Borovinskaya, M.A., Hau, C.W., Zhang, W., Vila-San-
jurjo, A., Holton, J.M., and Cate, J.H. (2005). Structures of the bacterial
ribosome at 3.5 A resolution. Science 310, 827–834.
Selmer, M., Dunham, C.M., Murphy, F.V., Weixlbaumer, A., Petry, S.,
Kelley, A.C., Weir, J.R., and Ramakrishnan, V. (2006). Structure of
the 70S ribosome complexed with mRNA and tRNA. Science 313,
1935–1942.
Sergiev, P.V., Bogdanov, A.A., and Dontsova, O.A. (2005). How can
elongation factors EF-G and EF-Tu discriminate the functional state
of the ribosome using the same binding site? FEBS Lett. 579, 5439–
5442.
Spahn, C.M., Gomez-Lorenzo, M.G., Grassucci, R.A., Jorgensen, R.,
Andersen, G.R., Beckmann, R., Penczek, P.A., Ballesta, J.P., and
Frank, J. (2004). Domain movements of elongation factor eEF2 and
the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J.
23, 1008–1019.
Stern, S., Moazed, D., and Noller, H.F. (1988). Structural analysis of
RNA using chemical and enzymatic probing monitored by primer ex-
tension. Methods Enzymol. 164, 481–489.
Valle, M., Sengupta, J., Swami, N.K., Grassucci, R.A., Burkhardt, N.,
Nierhaus, K.H., Agrawal, R.K., and Frank, J. (2002). Cryo-EM reveals
an active role for aminoacyl-tRNA in the accommodation process.
EMBO J. 21, 3557–3567.
Wickner, R.B. (1996). Double-stranded RNA viruses of Saccharomy-
ces cerevisiae. Microbiol. Rev. 60, 250–265.
Wickner, R.B., Porter-Ridley, S., Fried, H.M., and Ball, S.G. (1982).
Ribosomal protein L3 is involved in replication or maintenance of the
killer double-stranded RNA genome of Saccharomyces cerevisiae.
Proc. Natl. Acad. Sci. USA 79, 4706–4708.
r Inc.
Copyright © 2022 FDOKUMEN




















