
Rh-catalyzed hydrogenation of amino acids to bio - Lirias
40
1 Rh-catalyzed hydrogenation of amino acids to bio- based amino alcohols: tackling challenging substrates and application to protein hydrolysates Annelies Vandekerkhove, Laurens Claes, Free De Schouwer, Cédric Van Goethem, Ivo F. J. Vankelecom, Bert Lagrain and Dirk E. De Vos* Department of Microbial and Molecular Systems, Centre for Surface Chemistry and Catalysis KU Leuven Celestijnenlaan 200F, post box 2461 3001 Heverlee, Belgium KEYWORDS. Hydrogenation – amino acids – amino alcohols – biomass – rhodium ABSTRACT. While protein-rich biomass waste is nowadays mainly used for animal feed, conversion of its amino acid constituents to nitrogenous chemicals is a potential higher value route. To that end, the hydrogenation of amino acids to amino alcohols was studied in this work. Using a bimetallic Rh-MoOx/SiO2 catalyst, glutamic acid was for the first time hydrogenated to the aminodiol in high yield. By minimizing partial reduction and consecutive hydrogenolysis, and by suppressing the competitive cyclization to pyroglutamic acid (and derivatives thereof), glutamidiol was obtained in 77% yield at 70 bar H2 and 80 °C. High yields (typically > 80%) and selectivities were also achieved for most other natural amino acids, except for the S-containing amino acids
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Rh-catalyzed hydrogenation of amino acids to bio - Lirias

1
Rh-catalyzed hydrogenation of amino acids to bio-
based amino alcohols: tackling challenging
substrates and application to protein hydrolysates
Annelies Vandekerkhove, Laurens Claes, Free De Schouwer, Cédric Van Goethem, Ivo F. J.
Vankelecom, Bert Lagrain and Dirk E. De Vos*
Department of Microbial and Molecular Systems, Centre for Surface Chemistry and Catalysis
KU Leuven
Celestijnenlaan 200F, post box 2461
3001 Heverlee, Belgium
KEYWORDS. Hydrogenation – amino acids – amino alcohols – biomass – rhodium
ABSTRACT. While protein-rich biomass waste is nowadays mainly used for animal feed,
conversion of its amino acid constituents to nitrogenous chemicals is a potential higher value route.
To that end, the hydrogenation of amino acids to amino alcohols was studied in this work. Using
a bimetallic Rh-MoOx/SiO2 catalyst, glutamic acid was for the first time hydrogenated to the
aminodiol in high yield. By minimizing partial reduction and consecutive hydrogenolysis, and by
suppressing the competitive cyclization to pyroglutamic acid (and derivatives thereof), glutamidiol
was obtained in 77% yield at 70 bar H2 and 80 °C. High yields (typically > 80%) and selectivities
were also achieved for most other natural amino acids, except for the S-containing amino acids

2
cysteine and methionine, which act as catalyst poisons. This limitation was overcome by applying
a simple oxidation step with performic acid prior to the hydrogenation. The system was applied
successfully to a mixture of amino acids obtained by hydrolysis of pre-oxidized bovine serum
albumin. Amino alcohols were produced with high overall conversion (> 90%) and selectivity
(88%) without the need for an intermediate, expensive and difficult separation step. The reaction
proceeds with very high atom economies for both carbon and nitrogen, and generates only water
as a by-product.
Introduction
β-Amino alcohols are an interesting class of compounds with a broad range of applications, for
instance as intermediates in the production of pharmaceuticals (e.g. nelfinavir, ethambutol),1,2 as
chiral auxiliaries (e.g. pseudoephedrine)3 and as polymer precursors (e.g. lysinol).4 Classical
synthetic approaches often start from fossil hydrocarbon resources and consist of multiple steps
involving the use of homogeneous catalysts, toxic reagents and hazardous solvents.2,5–9
Alternatively, the one-step reduction of natural α-amino acids provides a bio-based route towards
β-amino alcohols, with excellent atom economy for both carbon and nitrogen. Moreover, many
amino acids are inexpensive and safe reagents, which are nowadays produced from renewable
resources by large-scale fermentation processes1,10,11 or can be recovered from cheap agricultural
protein-rich waste, such as animal slaughter waste, algae, sugar beet vinasses etc.12 This reaction
may therefore be particularly attractive regarding the chemocatalytic upgrading of protein-rich
biomass into nitrogenous chemicals, being highly complementary to recently developed strategies
for selective defunctionalization of amino acids.13-28

3
The reduction of carboxylic acids is not straightforward. Transition-metal catalyzed
hydrogenation, where nucleophilic hydride species are produced in situ by the dissociation of
molecular hydrogen (H2), is the most attractive approach in terms of green chemistry because only
water is generated as a co-product. Besides homogeneous catalysts also supported monometallic
(e.g. Re/TiO2)29 and bimetallic (e.g. Pd-Re, Ru-Re, Pt-Mo etc.) hydrogenation catalysts have been
reported.30 The latter are much more effective for this purpose, because the more oxophilic metal
activates the carbonyl carbon for a hydride attack.31–34 However, α-amino acids and ester
derivatives thereof are even more challenging substrates. The reduction can be performed either
with metal hydride reagents (e.g. LiAlH4, NaBH4), or by homogeneous and heterogeneous
transition metal-catalyzed hydrogenation. The first approach typically proceeds under mild
conditions (< 80 °C and atmospheric pressure), but involves the use of hazardous reactants and
solvents (e.g. THF).35 Moreover, the hydride reagents are used in excess, with concomitant salt
waste generation.1,36–40 Amino acid esters have been hydrogenated using homogeneous Ru and
heterogeneous Cu catalysts.41–43 Although amino alcohols were obtained in good to excellent
yields, the methods proceed in organic solvents under 40-50 bar H2 and eventually require a
protecting group on the amine moiety in the substrate. The first attempts towards the hydrogenation
of free amino acids in aqueous media with heterogeneous Ru and Ni catalysts required very high
pressures (200 bar at 100 °C) and long reaction times (up to 30 h); however, amino alcohols were
only obtained in moderate yields.44 Bimetallic Ru-Re catalysts, either unsupported45 or
immobilized on carbon46, operate under slightly milder conditions (150-200 bar H2 at 80-120 °C)
and amino alcohols have been produced in higher yields (31-80%); however, the product obtained
from glutamic acid, the most abundant constituent in plant-based proteins, was not unambiguously
characterized. Moreover, sintering is a major disadvantage under these conditions.47 The supported

4
Ru/C catalyst is more resistant to sintering and facilitates the hydrogenation at substantially lower
pressures. The product selectivity is strongly influenced by the reaction conditions: amino alcohols
have been obtained in high yields (> 92%) at 70 bar H2 and 100 °C,48 whereas the hydrogenation-
decarbonylation to primary amines occurs as a competitive side reaction at 40 bar H2 and 150 °C.22
However, in both cases highly acidic conditions are required to protonate the carboxylate group in
amino acids (pKa ≈ 2-3)49 in order to increase the electrophilic character of the carbonyl group for
a hydride attack. Nevertheless, the highest activity in amino acid hydrogenation has been observed
for a bimetallic Rh-MoOx/SiO2 catalyst.50,51 In this material, metallic Rh and MoOx species are
expected to be in close proximity and the adsorption of amino acids on the catalyst surface is
facilitated by stabilizing the adsorbed substrate via hydrogen bonding between MoOx species and
the carboxylic acid group. In this way, amino alcohols have been obtained in > 90% yield under
very mild conditions (80 bar H2 at 50 °C); primary amines have been observed as minor side
products as a result of the consecutive C-O hydrogenolysis of amino alcohols.50,51 Remarkably,
these bimetallic Rh-based catalysts show also excellent performance in C-O hydrogenolysis
reactions and have been applied successfully in the valorization of biomass-derived (poly)alcohols
and ethers.52–58
Despite the development of several performant catalytic systems for amino acid hydrogenation,
the substrate scope remains an important challenge. Up to now only a few simple amino acids (viz.
glycine, alanine, valine, leucine, isoleucine, proline, serine, threonine and lysine) or ester
derivatives thereof have been modified successfully to the corresponding amino
alcohols.3,4,42,45,46,48,50,51,59,60 The hydrogenation of other amino acids is more challenging due to
the presence of multiple and potentially interfering functional groups in the side chain. For
instance, the aromatic moieties in phenylalanine, tyrosine and tryptophan are preferentially

5
reduced compared to the carboxylic acid61 and glutamic acid is converted readily to pyroglutamic
acid under typical reaction conditions.59,62 Moreover, the hydrogenation of mixtures is currently
limited to synthetic mixtures of three amino acids with an aliphatic side chain.60
In this work, we optimized a chemocatalytic system for the hydrogenation of many natural amino
acids, even the challenging ones, under mild conditions. An oxidative pre-treatment of sulfur-
containing amino acids has been developed to avoid catalyst poisoning. The successful application
to both pure amino acids and protein hydrolysates demonstrates the potential of this system for the
valorization of biomass waste. Indeed, a mixture of amino alcohols provides potential applications
as precursors for epoxy resins. These polymers are obtained upon reaction with polyoxiranes under
basic conditions and have applications in e.g. coatings, adhesives, electrical insulators.4,63
Experimental section
Catalyst synthesis
A Rh-MoOx/SiO2 catalyst (4 wt% Rh, Mo/Rh molar ratio = 1:8) was synthesized by two
impregnation steps according to a procedure reported by Tomishige and coworkers.50 First, an
aqueous solution of RhCl3.3H2O (25 mM, 20 ml) was added to Aerosil 380 silica powder (1 g).
This suspension was stirred at ambient temperature to evaporate water and afterwards the pre-
catalyst was dried overnight in an oven at 60 °C. In the second step, the pre-catalyst was contacted
with an aqueous solution of (NH4)6Mo7O24.4H2O (0.36 mM, 20 ml) and dried in a similar manner.
Finally, the material was granulated (250-500 µm), calcined at 500 °C (2.5 °C min-1,
100 ml min-1 O2, 3 h) and reduced at 500 °C (2.5 °C min-1, 100 ml min-1 H2, 3 h) in a quartz U-
tube. A Ru-Re black catalyst was prepared in situ by treating a suspension of Re2O7 (0.0074 g)
and RuO2 (0.0077 g) in water (10 ml) at 80 bar H2 and 120 °C for 1 h.45 The synthesis was

6
performed in a 50 ml high-pressure Parr batch reactor. The pre-reduced catalyst was used as such
for the hydrogenation of amino acids (vide infra). Procedures for the synthesis of other supported
Rh- and Pt-based catalysts studied in this work are provided in the supplementary information.
Catalyst characterization
The Rh-MoOx/SiO2 catalyst was characterized by (scanning) transmission electron microscopy
((S)TEM) and energy-dispersive X-ray spectroscopy (EDX). TEM specimens were prepared by
depositing catalyst particles on a Lacey carbon-coated TEM grid (300 mesh Cu grid, Pacific Grid
Tech, USA). TEM and STEM images as well as EDX elemental maps were collected on a JEOL
ARM200F TEM instrument operated at 200 kV and equipped with a cold field emission gun and
a probe aberration corrector.
Hydrogenation of amino acids
Reactions were performed in a 50 ml high-pressure Parr batch reactor equipped with a Teflon
liner. In a typical reaction, the Teflon liner was charged with the amino acid(s) (0.11 M), the Rh-
MoOx/SiO2 catalyst (4 wt% Rh, Mo/Rh = 1:8, Rh/amino acid(s) = 3.5 mol%) and water (15 ml).
A highly acidic medium (pH 1.8) was obtained by the addition of phosphoric acid (0.3 M) to the
aqueous suspension. Experiments with the Ru-Re black catalyst were performed by the addition
of the amino acid (0.11 M) and phosphoric acid (0.3 M) to the aqueous solution containing the pre-
reduced catalyst (vide supra, Ru/amino acid = 3.5 mol%). After insertion of the Teflon liner, the
reactor was sealed, purged 3 times with N2, 3 times with H2 and finally pressurized with 65 bar H2.
The reactor was heated to 80 °C while stirring mechanically (350 rpm). During the heating step,
the hydrogen pressure increased to 70 bar. After the applied reaction time, the reactor was allowed
to cool down in an ice bath. The gas was released and the solid catalyst was removed by
centrifugation. Finally, the crude reaction mixture was analyzed by nuclear magnetic resonance

7
(NMR) spectroscopy and high-pressure liquid chromatography (HPLC) to determine both the
amino acid conversion and the product selectivity.
Oxidation of sulfur-containing amino acids
Amino acids with thiol and thioether moieties were oxidized prior to hydrogenation. First,
performic acid was produced in situ by reacting hydrogen peroxide (29-32 wt% aqueous solution,
1 ml) and formic acid (9 ml) at ambient temperature for 1 h, and then this mixture was cooled to
0 °C.64 The substrate (cysteine, methionine or a protein) was added to the cold oxidant solution
(molar ratio performic acid/sulfur = 3:1) and the reaction proceeded for 4 h in an ice bath. Finally,
the excess of formic acid and water were evaporated by heating under vacuum and the solid residue
was dried further overnight in a vacuum oven at 100 °C.
Protein hydrolysis
A standard procedure for protein hydrolysis was followed.65 To that end, a sample of bovine
serum albumin (1 g; either as such, or pre-treated with performic acid) was dissolved in aqueous
hydrochloric acid (6 M, 100 ml). The sample was incubated under nitrogen to prevent amino acid
oxidation by flushing the headspace with N2 for 60 s and subsequently the sample was heated to
110 °C for 24 h. Afterwards, the hydrolysates were evaporated to dryness at 110 °C and the residue
was suspended in water (5 ml, ± 1.5 M) and filtered to remove any remaining solid particles. Amino
acid levels were determined by high-performance liquid chromatography (HPLC) after 2- to 10-
fold dilution of the samples, in order to have an excess of the derivatisation agent (vide infra).
Product analysis and identification
Samples for NMR spectroscopy were prepared by mixing 300 µl of the crude reaction mixture
with 300 µl of deuterium oxide. The spectra were recorded on a Bruker Ascend 400 MHz NMR
spectrometer equipped with a BBO 5 mm atma probe and a sample case. The broad water signal

8
at δ = 4.7 ppm was suppressed by applying a zgpr pulse program: p1 8 μs, ns 32, d1 5 s, aq 2.55 s,
plw1 15 W, plw9 5.7·10-5 W, o1P on the resonance signal of water, determined and selected
automatically. In addition, three types of 2D NMR experiments were performed to identify
products derived from glutamic acid, tyrosine and arginine. 1H-1H NMR (COSY) according to the
following pulse program: p0 9.75 µs, p1 9.75 µs, ns 32, d1 1.89 s, plw1 15 W; 1H-13C NMR over
1 bond (HSQC) according to the pulse program: p1 9.75 µs, p2 19.50 µs, p3 7.50 µs, p4 15 µs, ns
24, d1 1.44 s, plw1 15 W, plw2 120 W, plw12 1.05 W; and 1H-13C NMR over 2 or 3 bonds
(HMBC) according to the pulse program: p1 9.75 µs, p2 19.50 µs, p3 7.50 µs, ns 40, d1 1.39 s,
plw1 15 W, plw2 120 W.
ICP-OES analyses were used to determine the Rh and Mo content of the reaction mixture of
leucine hydrogenation (6 h, 80 °C, 70 bar H2) using a Varian 720-ES equipped with a double-pass
glass cyclonic spray chamber, a Sea Spray concentric glass nebulizer and a high solids torch.
HPLC was used for both qualitative and quantitative analysis of protein hydrolysates and crude
reaction mixtures after hydrogenation. Measurements were performed on an Agilent 1200 Series
SL binary system equipped with a G1322A degasser, a G1312B binary pump, a G1367A
automated sample injector, a G1316A thermostatted column compartment and a G1314A variable
wavelength detector (VWD). Data were processed with Agilent Chemstation software version
B.04.03. The method was adapted from an Agilent Application Note.66,67 An automated pre-
column derivatization procedure was applied to convert the amine groups in amino acids, amino
alcohols and amines into chromophoric moieties, in order to increase the sensitivity for UV
detection. Primary amines were modified with o-phthalaldehyde (OPA) and 3-mercaptopropionic
acid, whereas secondary amines were treated with 9-fluorenylmethyl chloroformate (FMOC).
These chromophores were detected at 338 nm and 262 nm, respectively. Pyroglutamic acid and

9
pyroglutaminol, which contain a lactam moiety, were detected at 212 nm without any
derivatisation. Separation of these compounds was achieved on an Agilent ZORBAX Eclipse Plus
C18 reversed phase column (250 mm x 4.6 mm i.d., 5.0 µm particles), maintained at 40 °C. The
aqueous mobile phase (A) consisted of a 40 mM solution of NaH2PO4.H2O in Milli-Q water at pH
7.8 and the organic mobile phase (B) consisted of a mixture of methanol, acetonitrile and Milli-Q
water (45:45:10 v/v/v). Elution conditions: a flow rate of 1.5 mL min-1 and a mixture of 98% A
and 2% B was used as eluent from 0 to 0.84 min. Then a linear gradient was applied from 2% to
20% B in 0.84 to 10 min, 20% to 50% B in 10 to 45 min, 50% to 100% B in 45 to 54 min, 100%
B in 54 to 56.6 min, and 100% to 2% B in 56.6 to 57 min. The injection volume was 1.0 µl. After
each analysis, the column was purged with the initial solvent mixture (98% A, 2% B) for 5 min.
Results and discussion
Hydrogenation of glutamic acid: catalyst screening and optimization of reaction
parameters
The hydrogenation of glutamic acid (1a) was studied as a model reaction, thereby aiming at the
selective production of glutamidiol (1d), a useful polymer building block e.g. for the production
of epoxy resins. The product mixture was expected to be complex (Scheme 1). Glutamic acid
contains two distinct carboxylic acid groups with different reactivity (pKa,α-COOH = 2.2 and pKa,γ-
COOH = 4.3), which both can be reduced to an alcohol under highly acidic conditions; hence besides
glutamidiol also the monohydrogenated intermediates 4-amino-5-hydroxy-pentanoic acid (1b) and
5-hydroxy-norvaline (1c) may be present. Moreover, according to earlier observations, either
alcohol group in glutamidiol is susceptible to C-O hydrogenolysis as well,50,51 thereby generating

10
2-amino-1-pentanol (1e) and 4-amino-1-pentanol (1f) and eventually also 2-pentanamine (1g) as
side products. The selectivity can also be affected by the intramolecular condensation of glutamic
acid to pyroglutamic acid (2a), which occurs in parallel to the hydrogenation and proceeds easily
upon heating the reaction mixture.23,68 Finally, pyroglutamic acid can be reduced to
pyroglutaminol (2b) and further to prolinol (2c).59
Scheme 1. Possible reaction pathways in the hydrogenation of glutamic acid (1a): reduction to 4-
amino-5-hydroxy-pentanoic acid (1b), 5-hydroxy-norvaline (1c) and glutamidiol (1d);
consecutive C-O hydrogenolysis of 1d to 2-amino-1-pentanol (1e), 4-amino-1-pentanol (1f) and
2-pentanamine (1g). Pyroglutamic acid (2a) is obtained by thermal lactamisation of 1a and can be
reduced to pyroglutaminol (2b) and further to prolinol (2c).
Several Pt-, Ru- and Rh-based catalysts were evaluated on their performance in the model
reaction under selected conditions (Table 1 and ESI, Table S1). The hydrogenation did not proceed
in the absence of a catalyst; in this case however glutamic acid was converted selectively to
pyroglutamic acid (Table S1, entry 1). Among the known catalysts for amino acid hydrogenation,
only the bimetallic Rh-MoOx/SiO2 catalyst showed a reasonable activity, and the desired

11
glutamidiol was obtained in 34% yield at near-complete conversion (Table 1, entry 3). In contrast,
Ru-Re black and monometallic Ru-, Rh- and Pt-based catalysts were almost inactive: the reaction
mixtures consisted mainly of pyroglutamic acid, and eventually also traces of the
monohydrogenated products 1b and 1c were present (Table 1, entries 1, 2 and 4; Table S1, entries
9-12). The selectivity to 1b was always much higher than to 1c, as the carboxylic acid at the α-
carbon is more reactive due to the close presence of a positively charged, electron withdrawing
amine, rendering the carbonyl carbon more susceptible to a hydride attack. Substitution of Rh by
Pt in the bimetallic catalyst was also not beneficial (Table S1, entries 2-3). The bimetallic Rh-
MoOx catalyst was therefore selected for further optimization (Table S1, entries 4-8). However,
the catalytic performance could not be improved upon variation of the metal loading or by using
other support materials. In the latter case, the activity of the Rh-MoOx catalysts generally decreased
with the surface area (SiO2 > zeolite beta >> TiO2, ZrO2) and hence with the dispersion of the
active sites on the support. The best performance was achieved by immobilizing the Rh-MoOx
species on a SiO2 support and by applying a Rh loading of 4 wt%, which is in good agreement
with the observations of Tomishige and coworkers.50,51
Table 1. Catalytic hydrogenation of glutamic acid (1a): catalyst screening.[a]
Entry Catalyst X1a (%) S1b-1c (%) S1d-1f (%) S1g (%) S2a-2c (%)
1 Ru/C (5 wt% Ru) 61 9 < 1 < 1 91
2[b] Ru-Re black 37 < 1 < 1 < 1 37
3 Rh-MoOx/SiO2 (4 wt% Rh) 98 28 46 2 25
4 Rh/SiO2 (5 wt% Rh) 60 15 < 1 < 1 85
[a] Conditions: 1a (0.11 M), catalyst (Ru/1a or Rh/1a = 1.7 mol%), H3PO4 (0.3 M), water (15 ml),
H2 (70 bar), 100 °C, 6 h. Conversion (X) and selectivity (S) were determined by 1H NMR
spectroscopy and HPLC. [b] Reaction at Ru/1a = 3.5 mol%, 80 °C, 17 h.

12
The bimetallic Rh-MoOx/SiO2 catalyst has already been characterized thoroughly before51 and
therefore only the spatial distribution of Rh and MoOx species was determined by STEM-EDX
elemental mapping. Both metals were well dispersed over the SiO2 support, rather than being
present as large particles (Figure 1, a). Moreover, Rh and Mo were in close proximity (Figure 1, b
and c), which provides evidence for the proposed mechanism of amino acid hydrogenation (vide
supra).
(a)
(b)
(c)
Figure 1. TEM micrographs of the bimetallic Rh-MoOx/SiO2 catalyst: (a) bright field STEM, (b)
STEM-EDX elemental mapping of Rh, and (c) STEM-EDX elemental mapping of Mo.
Next, the catalytic system for the hydrogenation of glutamic acid was optimized in terms of
temperature, H2 pressure, acidity, time and catalyst loading in order to increase the yield of
glutamidiol (Table 2 and Figures S1-S4). The conversion of glutamic acid increased at higher
temperatures, but side reactions such as the lactamisation to pyroglutamic acid and C-O
hydrogenolysis of glutamidiol occurred to a higher extent as well; an optimum was reached at
80 °C (Figure S1). For instance, the selectivity to cyclic products 2a-2c could be diminished from
38% to 18% by decreasing the temperature from 140 °C to 70 °C, albeit that the conversion after
6 h dropped from > 99% to nearly 40% (Table 2, entries 1-3). The influence of other reaction
parameters was therefore studied at 70 °C. The hydrogenation proceeded to a larger extent in

13
highly acidic media and at higher H2 pressures, thereby reaching a plateau at 70 bar H2 (Figures
S2-S3). Mass transfer limitations as a result of the low solubility of H2 in aqueous media can be
avoided under these conditions; otherwise the selectivity to pyroglutamic acid and reduced
derivatives will be more pronounced. Substantial improvements to the yield of glutamidiol were
achieved by extending the reaction time from 6 h to 48 h, and even more by a 10-fold increase in
catalyst loading to 17.7 mol% Rh (Table 2, entries 4-5 and Figure S4). Finally, further optimization
based on these insights allowed to obtain glutamidiol (1d) in 77% yield under mild conditions
(Table 2, entries 6-8), which is the best result that has been reported for this challenging amino
acid so far.
Table 2. Rh-catalyzed hydrogenation of glutamic acid (1a): optimization of reaction parameters.[a]
Entry Time
(h)
Temperature
(°C)
Rh/1a
(mol%)
X1a
(%)
S1b
(%)
S1c
(%)
S1d
(%)
S1e
(%)
S1f
(%)
S1g
(%)
S2a-2c
(%)
1 6 100 1.7 98 24 4 35 5 6 2 24
2 6 70 1.7 38 64 6 11 < 1 < 1 < 1 18
3 6 140 1.7 > 99 4 1 22 8 19 8 38
4 48 70 1.7 99 25 3 45 4 5 1 18
5[b] 6 70 17.7 > 99 < 1 < 1 75 9 12 4 < 1
6 17 90 2.5 > 99 1 < 1 54 7 12 3 23
7 17 80 2.5 > 99 6 1 54 6 8 2 24
8 17 80 3.5 > 99 1 < 1 77 11 8 2 17
[a] Conditions: 1a (0.11 M), Rh-MoOx/SiO2 (4 wt% Rh, Mo/Rh = 1:8), H3PO4 (0.3 M), water
(15 ml), H2 (70 bar). Conversion (X) and selectivity (S) were determined by 1H NMR spectroscopy
and HPLC. [b] 1a (0.022 M).
Hydrogenation of amino acids: substrate scope
The hydrogenation of other natural amino acids was evaluated under the optimized conditions, viz.
3.5 mol% Rh, 0.3 M H3PO4, 70 bar H2, 80 °C (Table 3). The aliphatic amino acids glycine (3a),

14
alanine (4a), valine (5a), leucine (6a), isoleucine (7a) and proline (8a) were easily reduced to the
corresponding amino alcohols: high conversions and very good selectivities were already obtained
after 6 h. However, under these conditions C-O hydrogenolysis of amino alcohols was already
quite pronounced, in particular for alaninol (4b). The reactivity of valine (5a) and isoleucine (7a)
was diminished by the steric hindrance at the β-position; for isoleucine longer reaction times were
required to achieve complete conversion. In contrast to earlier reports on amino acid
hydrogenation,50,51 the chirality present in the amino acid substrate could not be retained under
these conditions, e.g. the enantiomeric excess of leucinol (6b) was only 64%. Serine (9a) and
threonine (10a), which contain an alcohol group in the side chain, were successfully hydrogenated
as well. Note that C-O hydrogenolysis of the aminodiols serinol (9b) and threoninol (10b) still
provides amino alcohols, respectively 2-amino-1-propanol (4b) and 3-amino-2-butanol; the former
is also the hydrogenation product of alanine (4a). Consecutive C-O hydrogenolysis of 9b and 10b
to amines was however not observed after 6 h. The basic amino acids lysine (11a), ornithine (12a),
arginine (13a) and histidine (14a) were less reactive compared to the aliphatic amino acids, which
is reflected in the longer reaction times required to achieve high conversions; however, even after
17 h the conversion of histidine (14a) was limited to about 70%. Moreover, in the case of arginine
(13a) besides C-O hydrogenolysis of the amino alcohol also hydrolysis of the guanidine moiety
was observed, thereby generating ornithinol (12b) as an additional side product. Nevertheless, the
overall selectivity towards both argininol (13b) and ornithinol (12b) amounts up to 90%. The
imidazole group in the side chain of histidine (14a) remained intact. The hydrogenation of aspartic
acid (15a) proceeded more easily than that of glutamic acid (1a), because the lactamisation of the
former is thermodynamically unfavorable under these conditions. Accordingly, the selectivity to
aspartidiol (15b) was considerably higher than for glutamidiol (1d), respectively 88% versus 77%.

15
Glutamine (16a) and asparagine (17a) behaved similarly to glutamic acid (1a) and aspartic acid
(15a), because the amide groups in the side chains of these amino acids were easily hydrolyzed in
the highly acidic medium. In this way, aspartidiol (15b) was obtained in 94% yield from asparagine
(17a). However, the selectivity to glutamidiol (1d) from glutamine (16a) was only 32%, which is
substantially lower compared to the glutamic acid case (1a), where glutamidiol was obtained in
77% yield. The lactamisation to pyroglutamic acid (2a) is probably facilitated upon hydrolysis of
the amide group. Next, the hydrogenation of phenylalanine (18a) and tyrosine (19a) proceeded
with full conversion. However, besides the carboxylic acid also the aromatic moieties in these
substrates were completely transformed into saturated cyclohexyl and 4-hydroxycyclohexyl
groups (18b, 19b). Moreover, two different amino alcohols were obtained from tyrosine because
the 4-hydroxycyclohexyl group in 19b was susceptible to C-O hydrogenolysis as well; the overall
selectivity to amino alcohols was 78%. Tryptophan (20a) could not be hydrogenated, because the
heterocyclic indole group was entirely degraded under acidic conditions; this observation is well-
known from protein hydrolysis.65 Finally, the hydrogenation of the sulfur-containing amino acids
cysteine (21a) and methionine (22a) was unsuccessful, probably as a result of catalyst
poisoning.69,70 A similar result was obtained for cystine (23a), which is the dimer form of cysteine.
This observation, however, poses an important challenge regarding the hydrogenation of amino
acids in peptide and protein hydrolysates, because cysteine and methionine are always present in
these mixtures, albeit in low concentrations.

16
Table 3. Rh-catalyzed hydrogenation of amino acids to amino alcohols.[a]
Entry Amino acid Products
1
X3a > 99% S3b = 88% S3c = 12%
2
X4a > 99% S4b = 75% S4c = 25%
3
X5a = 94% S5b = 86% S5c = 14%
4
X6a > 99% S6b = 85% S6c = 15%
5[b]
X7a > 99% S7b = 86% S7c = 14%
6
X8a > 99% S2c = 84% S8c = 16%
7
X9a > 99% S9b = 88% S4b = 12%
8
X10a > 99% S10b = 90% S10c = 10%

17
Table 3. Rh-catalyzed hydrogenation of amino acids to amino alcohols.[a] (continued)
Entry Amino acid Products
9[b]
X11a = 96% S11b = 87% S11c = 13%
10[b]
X12a = 92% S12b = 83% S12c = 17%
11[b]
S13b = 75% S13c = 9%
X13a > 99%
S12b = 15% S12c = 1%
12[b]
X14a = 71% S14b = 90% S14c = 10%
13[b]
X15a > 99% S15b = 88% S15c = 12%
14[b]
S1d = 32% S1e = 2%
X16a > 99%
S2b = 46% S2c = 20%

18
Table 3. Rh-catalyzed hydrogenation of amino acids to amino alcohols (all reaction products are
shown).[a] (continued)
Entry Amino acid Products
15[b]
X17a > 99% S15b = 94% S15c = 6%
16
X18a > 99% S18b = 75% S18c = 25%
17[b]
S19b = 31% S19c = 18%
X19a > 99%
S18b = 47% S18c = 4%
[a] Conditions: amino acid (0.11 M), Rh-MoOx/SiO2 (4 wt% Rh, Mo/Rh = 1:8, Rh/amino acid=
3.5 mol%), H3PO4 (0.3 M), water (15 ml), H2 (70 bar), 80 °C, 6 h. Conversion (X) and selectivity
(S) were determined by 1H NMR spectroscopy and HPLC. [b] Reaction for 17 h.

19
Catalyst recycling
The stability of the Rh-MoOx/SiO2 catalyst under typical reaction conditions was demonstrated
by recycling the material in the hydrogenation of isoleucine (7a) for three times (ESI, Figure S5).
After each run the catalyst was recovered by centrifugation, washed with deionized water and dried
at 60 °C. Elemental analysis of the reaction medium obtained after the first run revealed that Rh
leaching was negligible (0.01%), whereas Mo leaching was more pronounced (8.2%).
Nevertheless, recycling was successful since both the conversion of 7a and the selectivity to
isoleucinol (7b) remained constant throughout the four runs.
Influence of sulfur-containing compounds on amino acid hydrogenation
Platinum group metal catalysts are known to be inhibited by irreversible coordination of sulfur-
containing groups with lone pairs to the metal surface.69–71 Freifelder suggested that catalyst
poisoning can be avoided by oxidation of thiols and thioethers to sulfonic acids and sulfones,
respectively.71 This hypothesis was verified by applying the hydrogenation procedure on mixtures
of threonine (10a) and various sulfur-containing additives. Threoninol (10b) was obtained in 89%
yield in the presence of sulfolane and methanesulfonic acid (Table 4, entries 3,4); these results are
nearly identical to the yield in the absence of any additive (entry 1). However, the reaction did not
proceed at all in the presence of dimethyl sulfoxide, which still contains a lone pair (entry 2). These
experiments clearly demonstrate that complete oxidation of the sulfur-containing moieties in
cysteine and methionine is the key to prevent deactivation of the hydrogenation catalyst.

20
Table 4. Rh-catalyzed hydrogenation of threonine (10a) in the presence of several sulfur-
containing additives.[a]
Entry Additive X10a (%) S10b (%)
1 - > 99 90
2 Dimethyl sulfoxide < 1 /
3 Sulfolane > 99 89
4 Methanesulfonic acid > 99 89
[a] Conditions: 10a (0.11 M), Rh-MoOx/SiO2 (4 wt% Rh, Mo/Rh = 1:8, Rh/10a = 3.5 mol%),
additive (5 mol%), H3PO4 (0.3 M), water (15 ml), H2 (70 bar), 80 °C, 6 h. Conversion (X) and
selectivity (S) were determined by 1H NMR spectroscopy.
Oxidation and subsequent hydrogenation of cysteine and methionine
The oxidation of sulfur-containing compounds has already been studied extensively.
Nevertheless, the method of choice for the oxidation of cysteine and methionine should be highly
efficient, allow to modify thiols and thioethers simultaneously, proceed in aqueous conditions and
preferably use an environmentally benign oxidant, such as hydrogen peroxide, or even oxygen.
Catalytic oxidation of thioethers is often limited to the sulfoxide stage and this approach is
therefore considered to be less useful for our purpose.72–76 Inspired by the analytical determination
of sulfur-containing amino acids in proteins, oxidation with performic acid is expected to be more
promising. This unstable oxidant is produced in situ from hydrogen peroxide and formic acid, and
is able to transform cysteine and methionine selectively into cysteic acid and methionine sulfone,
respectively. In this way even cystine can be converted to cysteic acid. The excess of formic acid
can be easily removed afterwards via evaporation under vacuum.
First, the hydrogenation of cysteic acid (24a) and methionine sulfone (25a), which were prepared
by oxidation with performic acid for 4 h, was studied with Ru- and Rh-based catalysts. Again, Rh-
MoOx/SiO2 outperformed Ru/C and after 16 h the amino alcohols derived from cysteic acid (24a)

21
and methionine sulfone (25a) were obtained in about 90% yield at complete conversion (Table 5).
These experiments demonstrate that the noble metal catalysts are not poisoned by the sulfonic acid
or the sulfone moieties present in the substrates.
Table 5. Catalytic hydrogenation of cysteic acid (24a) and methionine sulfone (25a).[a]
Amino acid Amino alcohol Catalyst Time
(h)
X
(%)
S
(%)
24a
24b
Rh-MoOx/SiO2 6 59 88
16 > 99 87
Ru/C 6 13 96
16 78 94
25a
25b
Rh-MoOx/SiO2 6 57 90
16 > 99 90
Ru/C 6 18 96
16 48 95
[a] Conditions: pre-oxidized amino acid (0.11 M), catalyst (3.5 mol% Rh or Ru), H3PO4 (0.3 M),
water (15 ml), H2 (70 bar), 80 °C. Conversion (X) and selectivity (S) were determined by 1H NMR
spectroscopy.
The duration of the oxidative pre-treatment has a large impact on the outcome of the
hydrogenation (Figure 2). Indeed, by extending the oxidation time to 6 h, the conversions of
cysteic acid and methionine sulfone in the hydrogenation already decreased to respectively 91%
and 22%, whereas the catalytic activity was completely inhibited after 16 h of oxidation. The
decrease in catalytic activity can probably be attributed to overoxidation at longer pre-treatment
times, which has been suggested earlier for the performic acid-mediated oxidation of cysteine and
methionine.64,77 Additional signals were observed in the 1H NMR spectra of the oxidized amino
acids (Figures S6-S7), but the identity of the degradation products remains unclear. We suspect
that the amino acid moiety is susceptible to oxidation, possibly concurring with decarboxylation

22
as was also observed in previous work by our research group.14 Consequently, the oxidative pre-
treatment step should be limited to 4 h in order to maximize amino acid conversions.
Figure 2. Effect of the pre-treatment time during performic acid mediated oxidation of cysteine
and methionine on the conversion (X) in the Rh-catalyzed hydrogenation of cysteic acid (blue) and
methionine sulfone (orange). Conditions: pre-oxidized amino acid (0.11 M), Rh-MoOx/SiO2
(3.5 mol% Rh), H3PO4 (0.3 M), water (15 ml), H2 (70 bar), 80 °C, 6 h.
Hydrogenation of a protein hydrolysate
Finally, the hydrogenation procedure was applied to mixtures of amino acids obtained by acid-
mediated protein hydrolysis, in order to demonstrate the potential for biomass valorization. Bovine
serum albumin (BSA), which is present in cow’s blood plasma and has a well-known amino acid
composition,78–83 was selected as a model protein for animal slaughter waste. The hydrolysis was
preceded by an oxidative treatment with performic acid in order to eliminate potential catalyst
poisons in proteins, in particular the sulfur-containing amino acid residues. The composition of
the BSA hydrolysate and the product mixtures obtained after hydrogenation was determined by
reversed phase HPLC (Figure 3, Table 6 and ESI, Tables S2-S4). The mixtures contained about
60 compounds, which were separated based on differences in polarity, with the more polar amino
acids eluting first (4.3-34.4 min), followed by amino alcohols (11.4-50.8 min) and the least polar
0
20
40
60
80
100
4 6 16
X(%
)
Pretreatment time (h)

23
amines (17.3-52.4 min). The chromatograms obtained after several reaction times demonstrate that
the amino acids were progressively converted into a mixture of amino alcohols and amines, hence
that the catalyst was not poisoned (Figure S9). However, when the oxidative pre-treatment was
omitted, the hydrogenation did not proceed at all (Figure S8).
Figure 3. Reversed phase HPLC analysis of amino acids after oxidation and hydrolysis of BSA
(A), and subsequent hydrogenation for 48 h (B). The signals and elution ranges of amino acids
(▲, red), amino alcohols (●, green) and amines (♦, blue) are highlighted in and below the
chromatograms. Gradient elution was performed by combining an aqueous mobile phase (A;
40 mM NaH2PO4 in Milli-Q water at pH 7.8) and an organic mobile phase (B; methanol,
acetonitrile and Milli-Q water (45:45:10 v/v/v)). Conditions: 1.5 mL min-1; 98% A and 2% B from
0 to 0.84 min, from 2% to 20% B in 0.84 to 10 min, 20% to 50% B in 10 to 45 min, 50% to 100%
B in 45 to 54 min, 100% B in 54 to 56.6 min, and 100% to 2% B in 56.6 to 57 min.

24
Several side products of reactions that coincide with the hydrogenation products were taken into
account when calculating the amino acid conversions and the amino alcohol selectivities.
Hydrogenation of some natural amino acids may generate other non-natural amino acids, which
were observed as reaction intermediates: e.g. 3-amino-4-hydroxybutanoic acid and 4-amino-5-
hydroxy-pentanoic acid by reduction of the carboxylic acid at the α-carbon of aspartic acid and
glutamic acid, but also α-amino-cyclohexanepropanoic acid and α-amino-4-hydroxy-
cyclohexanepropanoic acid by hydrogenation of the aromatic moieties in phenylalanine and
tyrosine. Similarly, C-O hydrogenolysis of aminodiols may produce other amino alcohols: e.g. 3-
amino-1-butanol and 2-amino-1-butanol from aspartidiol (15b), 4-amino-1-pentanol and 2-amino-
1-pentanol from glutamidiol (1d), 2-amino-1-propanol from serinol (9b), 3-amino-2-butanol from
threoninol (10b), and 4-(2-aminopropyl)-cyclohexanol from β-amino-4-hydroxy-
cyclohexanepropanol (19b). Some amino alcohols may even originate from different amino acids:
2-amino-1-propanol can be generated by reduction of alanine (4a) or by C-O hydrogenolysis of
serinol (9b), whereas prolinol (2c) can be obtained by reduction of either proline (8a) or
pyroglutaminol (2b). Since both pathways cannot be distinguished in these mixtures, 2-amino-1-
propanol was entirely assigned to alanine, and prolinol to proline.
The mixture of amino acids was hydrogenated more slowly compared to single-compound
experiments (Table 6). Trace amounts of residual formic acid after oxidation could slow down the
hydrogenation as was observed in the discrepancy between the hydrogenation rates of cysteic acid
and methionine sulfone obtained either commercially or after oxidation of cysteine and methionine
(ESI Table S5). Also, amino acid hydrogenation can be affected by degradation products of
tryptophan. For instance, a mixture of leucine, isoleucine, valine, glycine and tryptophan was
hydrogenated more slowly compared to the same mixture in which tryptophan replaced by
25
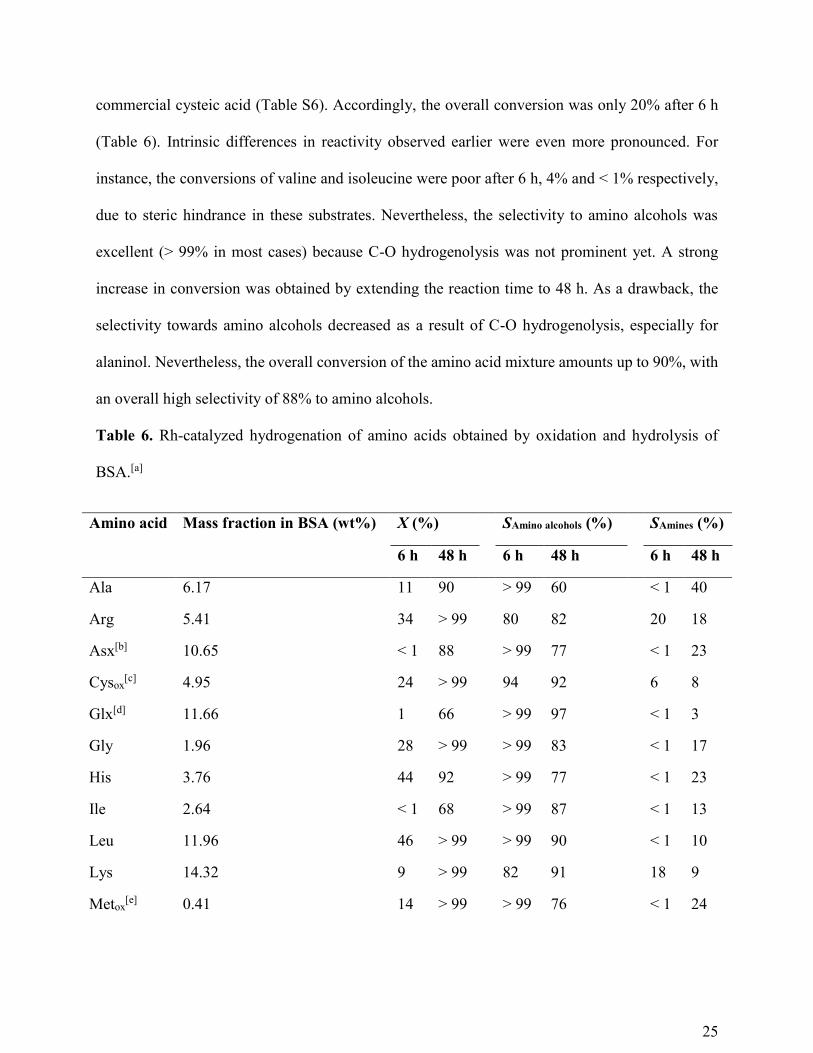
commercial cysteic acid (Table S6). Accordingly, the overall conversion was only 20% after 6 h
(Table 6). Intrinsic differences in reactivity observed earlier were even more pronounced. For
instance, the conversions of valine and isoleucine were poor after 6 h, 4% and < 1% respectively,
due to steric hindrance in these substrates. Nevertheless, the selectivity to amino alcohols was
excellent (> 99% in most cases) because C-O hydrogenolysis was not prominent yet. A strong
increase in conversion was obtained by extending the reaction time to 48 h. As a drawback, the
selectivity towards amino alcohols decreased as a result of C-O hydrogenolysis, especially for
alaninol. Nevertheless, the overall conversion of the amino acid mixture amounts up to 90%, with
an overall high selectivity of 88% to amino alcohols.
Table 6. Rh-catalyzed hydrogenation of amino acids obtained by oxidation and hydrolysis of
BSA.[a]
Amino acid Mass fraction in BSA (wt%) X (%) SAmino alcohols (%) SAmines (%)
6 h 48 h 6 h 48 h 6 h 48 h
Ala 6.17 11 90 > 99 60 < 1 40
Arg 5.41 34 > 99 80 82 20 18
Asx[b] 10.65 < 1 88 > 99 77 < 1 23
Cysox[c] 4.95 24 > 99 94 92 6 8
Glx[d] 11.66 1 66 > 99 97 < 1 3
Gly 1.96 28 > 99 > 99 83 < 1 17
His 3.76 44 92 > 99 77 < 1 23
Ile 2.64 < 1 68 > 99 87 < 1 13
Leu 11.96 46 > 99 > 99 90 < 1 10
Lys 14.32 9 > 99 82 91 18 9
Metox[e] 0.41 14 > 99 > 99 76 < 1 24

26
Phe 6.41 26 87 > 99 91 < 1 9
Pro 6.29 47 > 99 > 99 > 99 < 1 < 1
Ser 2.01 10 96 > 99 > 99 < 1 < 1
Thr 3.86 33 95 > 99 > 99 < 1 < 1
Tyr 1.75 44 > 99 > 99 > 99 < 1 < 1
Val 5.79 4 67 > 99 88 < 1 12
Overall 20 90 96 88 4 12
[a] Conditions: amino acids (0.11 M), Rh-MoOx/SiO2 (4 wt% Rh, Mo/Rh = 1:8, Rh/amino acids
= 3.5 mol%), H3PO4 (0.3 M), water (15 ml), H2 (70 bar), 80 °C. Conversion (X) and selectivity (S)
determined by HPLC. [b] Asx = aspartic acid + asparagine. [c] Cysox = cysteic acid. [d] Glx =
glutamic acid + glutamine. [e] Metox = methionine sulfone.
In conclusion, a Rh-based chemocatalytic system for the hydrogenation of amino acids was
optimized successfully for the selective production of amino alcohols under mild conditions. When
the reaction was performed in an acidic medium with a bimetallic Rh-MoOx/SiO2 catalyst at 80 °C
and 70 bar H2, glutamic acid was transformed in 77% yield to glutamidiol, which is an interesting
polymer precursor. Deactivation of the Rh catalyst can be avoided by applying a straightforward
oxidation procedure with performic acid, which allows to convert the sulfur-containing groups in
cysteine and methionine into inert sulfonic acid and sulfone moieties. As a proof of concept a
mixture of amino acids, obtained by hydrolysis of a pre-oxidized protein, was hydrogenated to a
mixture of predominantly amino alcohols with high overall conversion and selectivity. The
approach is characterized by excellent atom economies for both carbon and nitrogen and provides
opportunities for the valorization of protein-rich biomass waste.
ASSOCIATED CONTENT

27
Experimental details, product identification, additional experimental results (catalyst screening,
optimization reaction parameters, catalyst recycling, oxidation of cysteine and methionine,
protein hydrolysis, hydrogenation of a protein hydrolysate).
AUTHOR INFORMATION
Corresponding Author
* E-mail: [email protected] *
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval
to the final version of the manuscript.
Funding Sources
Agency for Innovation by Science and Technology (IWT) and Flanders Innovation &
Entrepreneurship (VLAIO) for (post-)doctoral fellowships.
Flemish government for long-term structural funding (Methusalem grant CASAS).
Belgian Federal Government (Belspo, IAP-PAI 7/05) for research project funding.
Hercules project AKUL/13/19.
ACKNOWLEDGEMENTS
A.V., L.C., F.D.S. and C.V.G. and are grateful to the Agency for Innovation by Science and
Technology (IWT) and Flanders Innovation & Entrepreneurship (VLAIO) for (post-)doctoral
fellowships (grants 141449 (A.V.), 131344 (F.D.S.), 141697 (C.V.G.) and HBC.2017.0366
(L.C.)). D.E.D.V. acknowledges the Flemish government for long-term structural funding
(Methusalem grant CASAS) FWO-FNRS for support in the EoS BioFACT project, and FWO for

28
research project funding. The authors are grateful for Hercules project AKUL/13/19 funding,
which financed the TEM equipment and also to prof. Jin Won Seo for help with the TEM
measurements. The authors thank Birgit Claes, Carlos Marquez and Karel Duerinckx for their
assistance with HPLC, ICP and NMR measurements.

29
REFERENCES
(1) Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T.
Industrial Methods for the Production of Optically Active Intermediates. Angew. Chemie -
Int. Ed. 2004, 43 (7), 788–824.
(2) Shivani, P. B.; Asit, K. C. Zinc(II) Perchlorate Hexahydrate Catalyzed Opening of Epoxide
Ring by Amines: Applications to Synthesis of (RS)/(R)-Propranolols and (RS)/(R)/(S)-
Naftopidils. J. Org. Chem. 2007, 72 (10), 3713–3722.
(3) Ager, D. J.; Prakash, I.; Schaad, D. R. 1,2-Amino Alcohols and Their Heterocyclic
Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96, 835–876.
(4) Metkar, P. S.; Scialdone, M. A.; Moloy, K. G. Lysinol: A Renewably Resourced Alternative
to Petrochemical Polyamines and Aminoalcohols. Green Chem. 2014, 16 (10), 4575–4586.
(5) Au, C. W. G.; Pyne, S. G. Asymmetric Synthesis of Anti-1,2-Amino Alcohols via the
Borono-Mannich Reaction: A Formal Synthesis of (-)-Swainsonine. J. Org. Chem. 2006,
71 (18), 7097–7099.
(6) Barbazanges, M.; Meyer, C.; Cossy, J.; Turner, P. Synthesis of 1,2-Amino Alcohols by
Sigmatropic Rearrangements of 3-(N-Tosylamino)Allylic Alcohol Derivatives. Chem. - A
Eur. J. 2011, 17 (16), 4480–4495.
(7) Hu, Y.; Wu, W.; Dong, X.-Q.; Zhang, X. Efficient Access to Chiral 1,2-Amino Alcohols
via Ir/f-Amphox-Catalyzed Asymmetric Hydrogenation of α-Amino Ketones. Org. Chem.
Front. 2017, 4 (8), 1499–1502.
(8) Mou, X.-Q.; Chen, X.-Y.; Chen, G.; He, G. Radical-Mediated Intramolecular β-C(Sp3)−H

30
Amidation of Alkylimidates: Facile Synthesis of 1,2-Amino Alcohols. Chem. Commun.
2018, 54, 515–518.
(9) Wang, Z.; Cui, Y. T.; Xu, Z. B.; Qu, J. Hot Water-Promoted Ring-Opening of Epoxides and
Aziridines by Water and Other Nucleopliles. J. Org. Chem. 2008, 73 (6), 2270–2274.
(10) Anastassiadis, S. L-Lysine Fermentation. Recent Pat. Biotechnol. 2007, 1 (1), 11–24.
(11) Sano, C. History of Glutamate Production. Am. J. Clin. Nutr. 2009, 90 (3), 728–732.
(12) Lammens, T. M.; Franssen, M. C. R.; Scott, E. L.; Sanders, J. P. M. Availability of Protein-
Derived Amino Acids as Feedstock for the Production of Bio-Based Chemicals. Biomass
and Bioenergy 2012, 44, 168–181.
(13) Claes, L.; Matthessen, R.; Rombouts, I.; Stassen, I.; De Baerdemaeker, T.; Depla, D.;
Delcour, J. A.; Lagrain, B.; De Vos, D. E. Bio-Based Nitriles from the Heterogeneously
Catalyzed Oxidative Decarboxylation of Amino Acids. ChemSusChem 2015, 8 (2), 345–
352.
(14) Claes, L.; Verduyckt, J.; Stassen, I.; Lagrain, B.; De Vos, D. E. Ruthenium-Catalyzed
Aerobic Oxidative Decarboxylation of Amino Acids: A Green, Zero-Waste Route to
Biobased Nitriles. Chem. Commun. 2015, 51 (30), 6528–6531.
(15) Lammens, T. M.; De Biase, D.; Franssen, M. C. R.; Scott, E. L.; Sanders, J. P. M. The
Application of Glutamic Acid α-Decarboxylase for the Valorization of Glutamic Acid.
Green Chem. 2009, 11 (10), 1562–1567.
(16) Teng, Y.; Scott, E. L.; van Zeeland, A. N. T.; Sanders, J. P. M. The Use of L-Lysine
Decarboxylase as a Means to Separate Amino Acids by Electrodialysis. Green Chem. 2011,

31
13, 624–630.
(17) Teng, Y.; Scott, E. L.; Witte-van Dijk, S. C. M.; Sanders, J. P. M. Simultaneous and
Selective Decarboxylation of L-Serine and Deamination of l-Phenylalanine in an Amino
Acid Mixture - a Means of Separating Amino Acids for Synthesizing Biobased Chemicals.
N. Biotechnol. 2016, 33 (1), 171–178.
(18) Le Nôtre, J.; Scott, E. L.; Franssen, M. C. R.; Sanders, J. P. M. Biobased Synthesis of
Acrylonitrile from Glutamic Acid. Green Chem. 2011, 13, 807–809.
(19) But, A.; Le Nôtre, J.; Scott, E. L.; Wever, R.; Sanders, J. P. M. Selective Oxidative
Decarboxylation of Amino Acids to Produce Industrially Relevant Nitriles by Vanadium
Chloroperoxidase. ChemSusChem 2012, 5 (7), 1199–1202.
(20) Dawes, G. J. S.; Scott, E. L.; Le Nôtre, J.; Sanders, J. P. M.; Bitter, J. H. Deoxygenation of
Biobased Molecules by Decarboxylation and Decarbonylation – a Review on the Role of
Heterogeneous, Homogeneous and Bio-Catalysis. Green Chem. 2015, 17 (6), 3231–3250.
(21) Verduyckt, J.; Van Hoof, M.; De Schouwer, F.; Wolberg, M.; Kurttepeli, M.; Eloy, P.;
Gaigneaux, E. M.; Bals, S.; Kirschhock, C. E. A.; De Vos, D. E. PdPb-Catalyzed
Decarboxylation of Proline to Pyrrolidine: Highly Selective Formation of a Biobased Amine
in Water. ACS Catal. 2016, 6 (11), 7303–7310.
(22) Verduyckt, J.; Coeck, R.; De Vos, D. E. Ru-Catalyzed Hydrogenation-Decarbonylation of
Amino Acids to Bio-Based Primary Amines. ACS Sustain. Chem. Eng. 2017, 5 (4), 3290–
3295.
(23) De Schouwer, F.; Claes, L.; Claes, N.; Bals, S.; Degrève, J.; De Vos, D. E. Pd-Catalyzed

32
Decarboxylation of Glutamic Acid and Pyroglutamic Acid to Bio-Based 2-Pyrrolidone.
Green Chem. 2015, 17, 2263–2270.
(24) De Schouwer, F.; Cuypers, T.; Claes, L.; De Vos, D. E. Metal-Catalyzed Reductive
Deamination of Glutamic Acid to Bio-Based Dimethyl Glutarate and Methylamines. Green
Chem. 2017, 19 (8), 1866–1876.
(25) De Schouwer, F.; Adriaansen, S.; Claes, L.; De Vos, D. E. Bio-Based N-Alkyl-2-
Pyrrolidones by Pd-Catalyzed Reductive N-Alkylation and Decarboxylation of Glutamic
Acid. Green Chem. 2017, 19, 4919–4929.
(26) Matthessen, R.; Claes, L.; Fransaer, J.; Binnemans, K.; De Vos, D. E. Decarboxylation of a
Wide Range of Amino Acids with Electrogenerated Hypobromite. European J. Org. Chem.
2014, No. 30, 6649–6652.
(27) Könst, P. M.; Franssen, M. C. R.; Scott, E. L.; Sanders, J. P. M. A Study on the Applicability
of L-Aspartate α-Decarboxylase in the Biobased Production of Nitrogen Containing
Chemicals. Green Chem. 2009, 11 (10), 1646–1652.
(28) Könst, P. M.; Franssen, M. C. R.; Scott, E. L.; Sanders, J. P. M. Stabilization and
Immobilization of Trypanosoma Brucei Ornithine Decarboxylase for the Biobased
Production of 1,4-Diaminobutane. Green Chem. 2011, 13 (5), 1167.
(29) Toyao, T.; Siddiki, S. M. A. H.; Touchy, A. S.; Onodera, W.; Kon, K.; Morita, Y.; Kamachi,
T.; Yoshizawa, K.; Shimizu, K. TiO2-Supported Re as a General and Chemoselective
Heterogeneous Catalyst for Hydrogenation of Carboxylic Acids to Alcohols. Chem. - A Eur.
J. 2017, 23 (5), 1001–1006.

33
(30) Pritchard, J.; Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. A. Heterogeneous
and Homogeneous Catalysis for the Hydrogenation of Carboxylic Acid Derivatives:
History, Advances and Future Directions. Chem. Soc. Rev. 2015, 44 (11), 3808–3833.
(31) Ullrich, J.; Breit, B. Selective Hydrogenation of Carboxylic Acids to Alcohols or Alkanes
Employing a Heterogeneous Catalyst. ACS Catal. 2018, 8 (2), 785–789.
(32) Toyao, T.; Siddiki, S. M. A. H.; Morita, Y.; Kamachi, T.; Touchy, A. S.; Onodera, W.; Kon,
K.; Furukawa, S.; Ariga, H.; Asakura, K.; et al. Rhenium-Loaded TiO2: A Highly Versatile
and Chemoselective Catalyst for the Hydrogenation of Carboxylic Acid Derivatives and the
N-Methylation of Amines Using H2 and CO2. Chem. - A Eur. J. 2017, 23 (59), 14848–
14859.
(33) Kang, K. H.; Hong, U. G.; Bang, Y.; Choi, J. H.; Kim, J. K.; Lee, J. K.; Han, S. J.; Song, I.
K. Hydrogenation of Succinic Acid to 1,4-Butanediol over Re-Ru Bimetallic Catalysts
Supported on Mesoporous Carbon. Appl. Catal. A Gen. 2015, 490, 153–162.
(34) Mizugaki, T.; Nagatsu, Y.; Togo, K.; Maeno, Z.; Mitsudome, T.; Jitsukawa, K.; Kaneda, K.
Selective Hydrogenation of Levulinic Acid to 1,4-Pentanediol in Water Using a
Hydroxyapatite-Supported Pt–Mo Bimetallic Catalyst. Green Chem. 2015, 17 (12), 5136–
5139.
(35) Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C. R.; Abou-Shehada, S.; Dunn, P.
J. CHEM21 Selection Guide of Classical- and Less Classical-Solvents. Green Chem. 2016,
18 (1), 288–296.
(36) Abiko, A.; Masamune, S. An Improved, Convenient Procedure for Reduction of Amino

34
Acids to Aminoalcohols: Use of NaBH4-H2SO4. Tetrahedron Lett. 1992, 33 (38), 5517–
5518.
(37) Dickman, D. A.; Meyers, A. I.; Smith, G. A.; Gawley, R. E. Reduction of A-Amino Acids:
L-Valinol. Org. Synth. 1990, 7, 530.
(38) Hedeo Seki, Kenji Koga, Hisayuki Matsuo, Sadao Ohki, Ichiro Matsuo, S. Y. Studies on
Optically Active Amino Acids. V. Synthesis of Optically Active α-Aminoalcohols by the
Reduction of α-Amino Acid Esters with Sodium Borohydride. Chem. Pharm. Bull. 1965,
13 (8), 995–1000.
(39) McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. A Convenient Reduction of
Amino Acids and Their Derivatives. J. Org. Chem. 1993, 58 (13), 3568–3571.
(40) Poindexter, G. S.; Meyers, A. I. Reduction of Amino Acids to Amino Alcohols. A
Comparison of Various Methods with Regard to Potential Racemization. Tetrahedron Lett.
1977, 18 (40), 3527–3528.
(41) Kuriyama, W.; Ino, Y.; Ogata, O.; Sayo, N.; Saito, T. A Homogeneous Catalyst for
Reduction of Optically Active Esters to the Corresponding Chiral Alcohols without Loss of
Optical Purities. Adv. Synth. Catal. 2010, 352 (1), 92–96.
(42) Gao, C.; Xiao, X.; Mao, D.; Lu, G. Preparation of L-Phenylalaninol with High Ee
Selectivity by Catalytic Hydrogenation of L-Phenylalaninate over Cu/ZnO/Al2O3 Catalyst.
Catal. Sci. Technol. 2013, 3 (4), 1056.
(43) Zhang, S.; Yu, J.; Li, H.; Mao, D.; Lu, G. High-Effective Approach from Amino Acid Esters
to Chiral Amino Alcohols over Cu/ZnO/Al2O3 Catalyst and Its Catalytic Reaction

35
Mechanism. Sci. Rep. 2016, 6, 1–15.
(44) Antons, S.; Beitzke, B. Process for Preparing Optically Active Amino Alcohols. US
5536879, 1996.
(45) Antons, S.; Tilling, A. S.; Wolters, E. Method for Producing Optically Active Amino
Alcohols. US 6310254, 2001.
(46) Mägerlein, W.; Dreisbach, C.; Hugl, H.; Tse, M. K.; Klawonn, M.; Bhor, S.; Beller, M.
Homogeneous and Heterogeneous Ruthenium Catalysts in the Synthesis of Fine Chemicals.
Catal. Today 2007, 121, 140–150.
(47) Besson, M.; Gallezot, P. Deactivation of Metal Catalysts in Liquid Phase Organic
Reactions. Catal. Today 2003, 81 (4), 547–559.
(48) Jere, F. T.; Miller, D. J.; Jackson, J. E. Stereoretentive C-H Bond Activation in the Aqueous
Phase Catalytic Hydrogenation of Amino Acids to Amino Alcohols. Org. Lett. 2003, 5,
527–530.
(49) Dawson, R. M. C.; Elliot, D. C.; Elliot, W. H.; Jones, K. M. Data for Biochemical Research;
Oxford, Clarendon Press, 1959.
(50) Tamura, M.; Tamura, R.; Takeda, Y.; Nakagawa, Y.; Tomishige, K. Catalytic
Hydrogenation of Amino Acids to Amino Alcohols with Complete Retention of
Configuration. Chem. Commun. 2014, 50, 6656–6659.
(51) Tamura, M.; Tamura, R.; Takeda, Y.; Nakagawa, Y.; Tomishige, K. Insight into the
Mechanism of Hydrogenation of Amino Acids to Amino Alcohols Catalyzed by a
Heterogeneous MoOx-Modified Rh Catalyst. Chem. - A Eur. J. 2015, 21, 3097–3107.

36
(52) Arai, T.; Tamura, M.; Nakagawa, Y.; Tomishige, K. Synthesis of 2-Butanol by Selective
Hydrogenolysis of 1,4-Anhydroerythritol over Molybdenum Oxide-Modified Rhodium-
Supported Silica. ChemSusChem 2016, 9 (13), 1680–1688.
(53) Koso, S.; Ueda, N.; Shinmi, Y.; Okumura, K.; Kizuka, T.; Tomishige, K. Promoting Effect
of Mo on the Hydrogenolysis of Tetrahydrofurfuryl Alcohol to 1,5-Pentanediol over
Rh/SiO2. J. Catal. 2009, 267 (1), 89–92.
(54) Koso, S.; Watanabe, H.; Okumura, K.; Nakagawa, Y.; Tomishige, K. Comparative Study
of Rh-MoOx and Rh-ReOx Supported on SiO2 for the Hydrogenolysis of Ethers and
Polyols. Appl. Catal. B Environ. 2012, 111–112, 27–37.
(55) Shimao, A.; Koso, S.; Ueda, N.; Shinmi, Y.; Furikado, I.; Tomishige, K. Promoting Effect
of Re Addition to Rh/SiO2 on Glycerol Hydrogenolysis. Chem. Lett. 2009, 38 (6), 540–541.
(56) Shinmi, Y.; Koso, S.; Kubota, T.; Nakagawa, Y.; Tomishige, K. Modification of Rh/SiO2
Catalyst for the Hydrogenolysis of Glycerol in Water. Appl. Catal. B Environ. 2010, 94 (3–
4), 318–326.
(57) Tomishige, K.; Tamura, M.; Nakagawa, Y. Role of Re Species and Acid Cocatalyst on Ir-
ReOx/SiO2 in the C-O Hydrogenolysis of Biomass-Derived Substrates. Chem. Rec. 2014,
14 (6), 1041–1054.
(58) Tomishige, K.; Nakagawa, Y.; Tamura, M. Selective Hydrogenolysis and Hydrogenation
Using Metal Catalysts Directly Modified with Metal Oxide Species. Green Chem. 2017, 19
(13), 2876–2924.
(59) Holladay, J. E.; Werpy, T. A.; Muzatko, D. S. Catalytic Hydrogenation of Glutamic Acid.

37
Appl. Biochem. Biotechnol. 2004, 113, 857–869.
(60) Pimparkar, K. P.; Miller, D. J.; Jackson, J. E. Hydrogenation of Amino Acid Mixtures to
Amino Alcohols. Ind. Eng. Chem. Res. 2008, 47, 7648–7653.
(61) Sun, B.; Süss-Fink, G. Ruthenium-Catalyzed Hydrogenation of Aromatic Amino Acids in
Aqueous Solution. J. Organomet. Chem. 2016, 812, 81–86.
(62) Bhandare, S. G.; Vaidya, P. D. Kinetics of Hydrogenation of Serine and Glutamic Acid in
Aqueous Solution over a Ru/C Catalyst. Ind. Eng. Chem. Res. 2017, 56 (14), 3797–3803.
(63) Auvergne, R.; Caillol, S.; David, G.; Boutevin, B.; Pascault, J.-P. Biobased Thermosetting
Epoxy: Present and Future. Chem. Rev. 2014, 114, 1082–1115.
(64) Moore, S. On the Determination of Cystine as Cysteic. J. Biol. Chem. 1963, 238, 235–237.
(65) Rombouts, I.; Lamberts, L.; Celus, I.; Lagrain, B.; Brijs, K.; Delcour, J. A. Wheat Gluten
Amino Acid Composition Analysis by High-Performance Anion-Exchange
Chromatography with Integrated Pulsed Amperometric Detection. J. Chromatogr. A 2009,
1216 (29), 5557–5562.
(66) Henderson, J. W.; Brooks, A. Improved Amino Acid Methods Using Agilent ZORBAX
Eclipse Plus C18 Columns for a Variety of Agilent LC Instrumentation and Separation
Goals; Tokyo, Agilent Technologies, 2010.
(67) Matsushita, K. Automatic Precolumn Derivatization of Amino Acids and Analysis by Fast
LC Using the Agilent 1290 Infinity LC; Tokyo, Agilent Technologies, 2010.
(68) Teng, Y.; Scott, E. L.; Sanders, J. P. M. Separation of L-Aspartic Acid and L-Glutamic Acid

38
Mixtures for Use in the Production of Bio-Based Chemicals. J. Chem. Technol. Biotechnol.
2012, 87, 1458–1465.
(69) Bartholomew, C. H.; Agrawal, P. K.; Katzer, J. R. Sulfur Poisoning of Metals. Adv. Catal.
1982, 31, 135–242.
(70) Maxted, E. B. The Poisoning of Metallic Catalysts. Adv. Catal. 1951, 3, 129–178.
(71) Freifelder, M. Practical Catalytic Hydrogenation - Techniques and Applications; John
Wiley & Sons, Inc.: New York, 1971.
(72) Hajjami, M.; Kolivand, S. New Metal Complexes Supported on Fe3O4 Magnetic
Nanoparticles as Recoverable Catalysts for Selective Oxidation of Sulfides to Sulfoxides.
Appl. Organomet. Chem. 2016, 30 (5), 282–288.
(73) Muñoz, M.; Gallo, M.; Gutiérrez-Alejandre, A.; Gazzoli, D. Molybdenum-Containing
Systems Based on Natural Kaolinite as Catalysts for Selective Oxidation of Aromatic
Sulfides. Appl. Catal. B, Environ. 2017, 219, 683–692.
(74) Nikoorazm, M.; Jabbari, A. Synthesis and Characterization of M-5NSA-MCM-41, (M = Cr,
Fe) as Reusable Catalysts for the Selective Oxidation of Sulfides to Sulfoxides and
Oxidative Coupling of Thiols into Disulfides in the Presence of H2O2. J. Porous Mater.
2017, 24 (2), 477–486.
(75) Rostamnia, S.; Gholipour, B.; Liu, X.; Wang, Y.; Arandiyan, H. NH2-Coordinately
Immobilized Tris(8-Quinolinolato)Iron onto the Silica Coated Magnetite Nanoparticle:
Fe3O4@SiO2-FeQ3 as a Selective Fenton-like Catalyst for Clean Oxidation of Sulfides. J.
Colloid Interface Sci. 2018, 551, 447–455.

39
(76) Veisi, H.; Eshbala, F. H.; Hemmati, S.; Baghayeri, M. Selective Hydrogen Peroxide
Oxidation of Sulfides to Sulfones with Carboxylated Multi-Walled Carbon Nano Tubes
(MWCNTs-COOH) as Heterogeneous and Recyclable Nanocatalysts under Organic
Solvent-Free Conditions. RSC Adv. 2015, 5 (14), 10152–10158.
(77) Schram, E.; Moore, S.; Bigwood, E. J. Chromatographic Determination of Cystine as
Cysteic Acid. Biochem. J. 1954, 57 (1), 33–37.
(78) Brand, E.; Kassell, B.; Saidel, L. J. Chemical, Clinical and Immunological Studies on the
Products of Human Plasma Fractionation. III. Amino Acid Composition of Plasma Proteins.
J. Clin. Invest. 1944, 23, 437–444.
(79) Henderson, L. M.; Snell, E. E. A Uniform Medium for Determination of Amino Acids with
Various Microorganisms. J. Biol. Chem. 1948, 175, 15–29.
(80) Hier, S. W.; Graham, C. E.; Ruth, F.; Klein, D. The Microbiological Determination of
Amino Acids in Animal Proteins. J. Biol. Chem. 1945, 161, 705–716.
(81) Shemin, D. Amino Acid Determinations on Crystalline Bovine and Human Serum Albumin
by the Isotope Dilution Method. J. Biol. Chem. 1945, 159, 439–443.
(82) Stein, W. H.; Moore, S. Amino Acid Composition of B-Lactoglobulin and Bovine Serum
Albumin. J. Biol. Chem. 1949, 178, 79–91.
(83) Velick, S. F.; E., R. The Amino Acid Composition of Aldolase and D-Glyceraldehyde
Phosphate Dehydrogenase. J. Biol. Chem. 1948, 173, 627–639.

40
SYNOPSIS
A broad range of amino acids and a protein hydrolysate were successfully hydrogenated to
amino alcohols, providing opportunities for the valorization of protein-rich waste streams.




















