
Rgs16 and Rgs8 in embryonic endocrine pancreas and mouse models of diabetes
14
INTRODUCTION Diabetes affects over 246 million people worldwide and accounts for about 6% of annual global mortality (www.idf.org). This disease is characterized by defective glucose metabolism and hyperglycemia resulting from the destruction of insulin-producing -cells within the pancreas (type 1), or defects in insulin signaling (type 2). Diabetes has no cure, although there are palliative treatments to control its symptoms. There is a great need to understand the cellular and molecular basis for islet cell proliferation and differentiation in an effort to generate -cell regenerative therapies for diabetic patients. Although groundbreaking work has advanced our ability to drive stem cells towards the pancreatic endocrine cell fate in culture (D’Amour et al., 2005; D’Amour et al., 2006; Kroon et al., 2008), much remains unknown about the molecular pathways regulating the differentiation of islet cell lineages (Lammert et al., 2001; Cleaver and Melton, 2003; Lammert et al., 2003; Collombat et al., 2006; Oliver-Krasinski and Stoffers, 2008) and the mechanisms underlying islet regeneration (Dor et al., 2004; Bonner- Weir et al., 2008; Xu et al., 2008). New tools required for the development of diabetes therapies can be fashioned using embryonic genes that are expressed during pancreas development and later reactivated during pancreatic - cell regeneration in models of diabetes (Inada et al., 2008; Xu et al., 2008). The earliest candidate genes are expressed in pancreatic ‘progenitor cells’ within the pre-pancreatic endoderm at around embryonic day (E)8.75-9.0 (Golosow and Grobstein, 1962; Gittes and Rutter, 1992; Kim and MacDonald, 2002; Yoshitomi and Zaret, 2004). By E12.5-14.5, endocrine progenitor cells proliferate, delaminate and begin coalescing into small islet-like clusters. During postnatal development, these clusters acquire recognizable islet anatomy; in mice, this consists of a core of -cells (that produce insulin) surrounded by a mantle of mostly -cells (that produce glucagon), but also -cells (somatostatin), -cells (ghrelin) and PP (pancreatic polypeptide) cells (Kim and MacDonald, 2002; Cleaver and Melton, 2003; Collombat et al., 2006). In adulthood, there is little endocrine cell proliferation unless animals experience metabolic stresses that challenge their glucose homeostasis. The cellular origin of the new endocrine cells remains controversial. Studies from Melton and others demonstrate that new -cells derive from replication of pre-existing -cells rather than through proliferation of endogenous specialized progenitors (Dor et al., 2004; Teta et al., 2007). Work from Bonner-Weir, by contrast, supports the existence of ‘foci of regeneration’ or pools of endocrine progenitors within the pancreatic ducts (Bonner-Weir et al., 2004). Recent work by Heimberg and colleagues has shown that the adult pancreatic ducts have the ability to generate new - cell formation in response to extreme pancreatic injury (Gradwohl et al., 2000; Xu et al., 2008). It is therefore plausible that both mechanisms occur, but depend on unspecified signals within the microenvironment. New biomarkers are therefore needed to further identify and examine expanding islets in different injury or disease models. These biomarkers will provide direct and rapid in vivo validation of conditions that stimulate -cell replication and expansion. G protein-coupled receptor (GPCR) signaling pathways have been associated with -cell neogenesis. Glucagon-like peptide 1 (Glp-1) and exendin-4 (Byetta) are GPCR agonists that stimulate -cell replication and neogenesis and improve glucose tolerance in mouse models of type 1 diabetes (Xu et al., 1999; Tourrel et al., RESEARCH ARTICLE Disease Models & Mechanisms 567 Disease Models & Mechanisms 3, 567-580 (2010) doi:10.1242/dmm.003210 © 2010. Published by The Company of Biologists Ltd 1 Department of Molecular Biology and 5 Department of Cell Biology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-9148, USA 2 Department of Internal Medicine and Touchstone Diabetes Center, University of Texas Southwestern Medical Center, 75390 TX, USA 3 Hamon Center for Therapeutic Oncology Research, University of Texas Southwestern Medical Center, 75390 TX, USA 4 Department of Pharmacology, University of Texas Southwestern Medical Center, 6001 Forest Park Rd, Dallas, TX 75390-9041, USA *These authors contributed equally to this work ‡ Authors for correspondence ([email protected]; [email protected]) SUMMARY Diabetes is characterized by the loss, or gradual dysfunction, of insulin-producing pancreatic -cells. Although -cells can replicate in younger adults, the available diabetes therapies do not specifically target -cell regeneration. Novel approaches are needed to discover new therapeutics and to understand the contributions of endocrine progenitors and -cell regeneration during islet expansion. Here, we show that the regulators of G protein signaling Rgs16 and Rgs8 are expressed in pancreatic progenitor and endocrine cells during development, then extinguished in adults, but reactivated in models of both type 1 and type 2 diabetes. Exendin-4, a glucagon-like peptide 1 (Glp-1)/incretin mimetic that stimulates -cell expansion, insulin secretion and normalization of blood glucose levels in diabetics, also promoted re-expression of Rgs16::GFP within a few days in pancreatic ductal- associated cells and islet -cells. These findings show that Rgs16::GFP and Rgs8::GFP are novel and early reporters of G protein-coupled receptor (GPCR)-stimulated -cell expansion after therapeutic treatment and in diabetes models. Rgs16 and Rgs8 are likely to control aspects of islet progenitor cell activation, differentiation and -cell expansion in embryos and metabolically stressed adults. Rgs16 and Rgs8 in embryonic endocrine pancreas and mouse models of diabetes Alethia Villasenor 1 , Zhao V. Wang 2 , Lee B. Rivera 3 , Ozhan Ocal 4 , Ingrid Wernstedt Asterholm 2 , Philipp E. Scherer 2,5 , Rolf A. Brekken 3,4 , Ondine Cleaver 1, * ,‡ and Thomas M. Wilkie 4, * ,‡ Disease Models & Mechanisms DMM
Transcript of Rgs16 and Rgs8 in embryonic endocrine pancreas and mouse models of diabetes

INTRODUCTIONDiabetes affects over 246 million people worldwide and accountsfor about 6% of annual global mortality (www.idf.org). This diseaseis characterized by defective glucose metabolism and hyperglycemiaresulting from the destruction of insulin-producing -cells withinthe pancreas (type 1), or defects in insulin signaling (type 2).Diabetes has no cure, although there are palliative treatments tocontrol its symptoms. There is a great need to understand thecellular and molecular basis for islet cell proliferation anddifferentiation in an effort to generate -cell regenerative therapiesfor diabetic patients. Although groundbreaking work has advancedour ability to drive stem cells towards the pancreatic endocrine cellfate in culture (D’Amour et al., 2005; D’Amour et al., 2006; Kroonet al., 2008), much remains unknown about the molecular pathwaysregulating the differentiation of islet cell lineages (Lammert et al.,2001; Cleaver and Melton, 2003; Lammert et al., 2003; Collombatet al., 2006; Oliver-Krasinski and Stoffers, 2008) and themechanisms underlying islet regeneration (Dor et al., 2004; Bonner-Weir et al., 2008; Xu et al., 2008).
New tools required for the development of diabetes therapiescan be fashioned using embryonic genes that are expressed duringpancreas development and later reactivated during pancreatic -cell regeneration in models of diabetes (Inada et al., 2008; Xu et al.,2008). The earliest candidate genes are expressed in pancreatic
‘progenitor cells’ within the pre-pancreatic endoderm at aroundembryonic day (E)8.75-9.0 (Golosow and Grobstein, 1962; Gittesand Rutter, 1992; Kim and MacDonald, 2002; Yoshitomi and Zaret,2004). By E12.5-14.5, endocrine progenitor cells proliferate,delaminate and begin coalescing into small islet-like clusters.During postnatal development, these clusters acquire recognizableislet anatomy; in mice, this consists of a core of -cells (that produceinsulin) surrounded by a mantle of mostly -cells (that produceglucagon), but also -cells (somatostatin), -cells (ghrelin) and PP(pancreatic polypeptide) cells (Kim and MacDonald, 2002; Cleaverand Melton, 2003; Collombat et al., 2006). In adulthood, there islittle endocrine cell proliferation unless animals experiencemetabolic stresses that challenge their glucose homeostasis.
The cellular origin of the new endocrine cells remainscontroversial. Studies from Melton and others demonstrate thatnew -cells derive from replication of pre-existing -cells ratherthan through proliferation of endogenous specialized progenitors(Dor et al., 2004; Teta et al., 2007). Work from Bonner-Weir, bycontrast, supports the existence of ‘foci of regeneration’ or poolsof endocrine progenitors within the pancreatic ducts (Bonner-Weiret al., 2004). Recent work by Heimberg and colleagues has shownthat the adult pancreatic ducts have the ability to generate new -cell formation in response to extreme pancreatic injury (Gradwohlet al., 2000; Xu et al., 2008). It is therefore plausible that bothmechanisms occur, but depend on unspecified signals within themicroenvironment. New biomarkers are therefore needed tofurther identify and examine expanding islets in different injury ordisease models. These biomarkers will provide direct and rapid invivo validation of conditions that stimulate -cell replication andexpansion.
G protein-coupled receptor (GPCR) signaling pathways havebeen associated with -cell neogenesis. Glucagon-like peptide 1(Glp-1) and exendin-4 (Byetta) are GPCR agonists that stimulate-cell replication and neogenesis and improve glucose tolerance inmouse models of type 1 diabetes (Xu et al., 1999; Tourrel et al.,
RESEARCH ARTICLE
Disease Models & Mechanisms 567
Disease Models & Mechanisms 3, 567-580 (2010) doi:10.1242/dmm.003210© 2010. Published by The Company of Biologists Ltd
1Department of Molecular Biology and 5Department of Cell Biology, University ofTexas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-9148,USA2Department of Internal Medicine and Touchstone Diabetes Center, University ofTexas Southwestern Medical Center, 75390 TX, USA3Hamon Center for Therapeutic Oncology Research, University of TexasSouthwestern Medical Center, 75390 TX, USA4Department of Pharmacology, University of Texas Southwestern Medical Center,6001 Forest Park Rd, Dallas, TX 75390-9041, USA*These authors contributed equally to this work‡Authors for correspondence ([email protected];[email protected])
SUMMARY
Diabetes is characterized by the loss, or gradual dysfunction, of insulin-producing pancreatic -cells. Although -cells can replicate in younger adults,the available diabetes therapies do not specifically target -cell regeneration. Novel approaches are needed to discover new therapeutics and tounderstand the contributions of endocrine progenitors and -cell regeneration during islet expansion. Here, we show that the regulators of G proteinsignaling Rgs16 and Rgs8 are expressed in pancreatic progenitor and endocrine cells during development, then extinguished in adults, but reactivatedin models of both type 1 and type 2 diabetes. Exendin-4, a glucagon-like peptide 1 (Glp-1)/incretin mimetic that stimulates -cell expansion, insulinsecretion and normalization of blood glucose levels in diabetics, also promoted re-expression of Rgs16::GFP within a few days in pancreatic ductal-associated cells and islet -cells. These findings show that Rgs16::GFP and Rgs8::GFP are novel and early reporters of G protein-coupled receptor(GPCR)-stimulated -cell expansion after therapeutic treatment and in diabetes models. Rgs16 and Rgs8 are likely to control aspects of islet progenitorcell activation, differentiation and -cell expansion in embryos and metabolically stressed adults.
Rgs16 and Rgs8 in embryonic endocrine pancreas andmouse models of diabetesAlethia Villasenor1, Zhao V. Wang2, Lee B. Rivera3, Ozhan Ocal4, Ingrid Wernstedt Asterholm2, Philipp E. Scherer2,5,Rolf A. Brekken3,4, Ondine Cleaver1,*,‡ and Thomas M. Wilkie4,*,‡
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

2001; Kodama et al., 2005; Chu et al., 2007; Sherry et al., 2007; Wanget al., 2008). Glucose homeostasis is also improved in human type2 diabetics (Fineman et al., 2003; Kendall et al., 2005). GPCRsactivate G protein pathways, such as Gs- and Gq/11-stimulatedcAMP and calcium signaling, respectively (Gilman, 1987; Simonet al., 1991). The regulators of G protein signaling (RGS) proteinsare GTPase activating proteins (GAPs) for Gq/11 and Gi/o class subunits, and thereby regulate the frequency and duration of GPCRsignaling (Berman et al., 1996; Koelle and Horvitz, 1996; Ross andWilkie, 2000). Interestingly, the expression of some Rgs genes isinduced during GPCR signaling and can thereby serve as anindicator of active GPCR signaling (Dohlman et al., 1996).Furthermore, Rgs genes can be co-incidence detectors, induced bycross-talk pathways, to regulate Gi/q signaling in the same cell.Thus, Rgs gene expression can be used to identify where and whenGPCR signaling is active during development and in tissues ofhealthy or diseased adults.
The Rgs genes were first implicated in glucose homeostasis whenwe discovered that hepatic glucose production elevates theexpression of Rgs16 in hepatocytes during fasting (Huang et al.,2006). To identify Rgs8/16 in other tissues that regulate glucosehomeostasis, we used two lines of bacterial artificial chromosome(BAC) transgenic mice that express either Rgs16::GFP or Rgs8::GFP.These reporter genes faithfully reproduce endogenous Rgs16 andRgs8 mRNA expression in mice (Gong et al., 2003; Su et al., 2004;Huang et al., 2006; Morales and Hatten, 2006). Identifying theligands, conditions and cell types that induce the expression of theseRgs genes is likely to further the development of novel therapiesfor metabolic diseases.
Here, we show that Rgs16::GFP and Rgs8::GFP are expressedduring pancreatic endocrine cell development and proliferation.Pancreatic progenitor cells and differentiating -cells express Rgs16and Rgs8, beginning in the dorsal pancreatic anlagen and continuingthroughout embryogenesis. In the perinatal pancreas, Rgs-expressing cells aggregate into islets in tight association withpancreatic blood vessels. In adults, pancreatic expression of Rgs16and Rgs8 becomes quiescent. However, under conditions of chronicglucose stress, Rgs16 and Rgs8 are re-expressed in adult pancreaticislets. Here, we demonstrate Rgs16 expression in four differentmodels of adult -cell expansion, including in (1) PANIC-ATTACmice, a model of type 1 diabetes (Wang et al., 2008); and, togetherwith Rgs8 expression, in (2) obese, hyperglycemic ob/ob mice, amodel of type 2 diabetes (Chua et al., 2002; Prentki and Nolan,2006); (3) midgestation pregnant females; and (4) followingtreatment with the GPCR agonist exendin-4. Rgs16 and Rgs8 aresensitive reporters of conditions that stimulate the early stages ofislet progenitor cell differentiation and -cell expansion indevelopment and disease.
RESULTSRgs8/16 gene expression in endocrine progenitors duringdevelopmentTwo separate lines of GFP-expressing BAC transgenic mice (Fig.1A,B) (Gong et al., 2003) were used to determine the expressionof the tandemly duplicated Rgs16 and Rgs8 genes during pancreaticdevelopment. Both Rgs16::GFP and Rgs8::GFP were expressedthroughout embryonic and neonatal pancreas development (Fig.1C,D; Fig. 2 and Table 1).
Rgs16::GFP was expressed throughout the early gut tubeendoderm (E8.5) from the foregut to the tip of the open hindgut(Fig. 2A and data not shown) and became restricted to the earlyliver and dorsal pancreatic bud epithelium after embryonic turning(E9.5-10.5) (Fig. 2B,C). During dorsal bud outgrowth, Rgs16 wasexpressed in a punctate pattern within the pancreatic epitheliumin a subpopulation of cells known to contain mostly epithelial andendocrine cell types (Fig. 2C-E). In postnatal stages, Rgs16expression became restricted to aggregates of endocrine cellsforming the islets of Langerhans (Fig. 2F).
In contrast to the broad distribution of early Rgs16::GFPexpression in the endoderm, Rgs8::GFP expression was initiallylocalized to a distinct dorsal patch in a region fated to give rise tothe pancreatic endoderm (E8.5) (Fig. 2G) (Wells and Melton, 1999).This striking expression was initiated prior to any cellular ormolecular evidence of pancreas specification, such as expressionof Pdx1/Ipf1 or Ngn3 (Villasenor et al., 2008). By E9.5, Rgs8expression, like Rgs16, became largely restricted to the formingpancreatic bud (Fig. 2H,I). Later, Rgs8 was expressed in a patternthat was almost identical to that of Rgs16, in scattered clusters ofcells in the central region of the developing bud (Fig. 2J), but wasalso weakly present in the exocrine pancreas (Fig. 2K,L). Overall,both patterns of expression were consistent with endocrine celldistribution until postnatal stages (Fig. 2K,L).
Because the spatiotemporal distribution of Rgs16- and Rgs8-expressing cells was reminiscent of endocrine precursors, theirexpression was compared with that of Ngn3, a well-characterizedendocrine progenitor marker (Gradwohl et al., 2000). Rgs16 and
dmm.biologists.org568
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLE
Fig. 1. Rgs8::GFP and Rgs16::GFP are expressed in the pancreatic bud.Rgs8 and Rgs16 are tandemly duplicated genes that are separated by42,682 bp (43 kb) of intragenic DNA (Sierra et al., 2002). (A,B)Transgenic micecontaining a BAC with enhanced GFP (eGFP) inserted at the translationinitiation site of either Rgs8::GFP (GENSAT ID: BX478, constructed from RP23-184B11) (A) or Rgs16::GFP (GENSAT ID: BX843, constructed from RP23-101N8)(B) (Gong et al., 2003). (C,D)Expression of Rgs8::GFP (C) and Rgs16::GFP (D) inthe dorsal pancreas during initial budding (arrows). Embryos were collected atthe indicated stages. dp, dorsal pancreatic bud; nt, neural tube. Bar, 250mm.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Rgs8 were expressed in a very similar pattern to Ngn3 (Fig. 2)throughout development and until approximately 2 weekspostnatal, when all three genes were extinguished in islets. However,the Rgs::GFP genes were expressed earlier than Ngn3::GFP (Fig.2A,G,M) (see also Villasenor et al., 2008). Outside the pancreaticbud, endodermal Rgs8/16 and Ngn3 expression diverged, withRgs16::GFP in the budding liver (Fig. 2B), Rgs8::GFP in the adjacentdorsal endoderm (Fig. 2H) and Ngn3::GFP faint in the posteriorduodenal endoderm (Fig. 2N,O).
In contrast to Rgs8::GFP and Ngn3::GFP, both of which wereextinguished shortly after birth, Rgs16::GFP remained strong inscattered cells along veins, ducts and arteries for the first 3-4 weeksof postnatal development (see below). Rgs16::GFP expression wasstronger than Rgs8::GFP throughout embryonic pancreaticdevelopment (the photographic exposure time of Rgs8::GFP was
on average about twofold longer than that of Rgs16::GFP).Endogenous Rgs16 mRNA was also about twofold more abundantthan Rgs8 mRNA in E14.5 pancreas [quantitative PCR (qPCR); datanot shown]. Therefore, further investigation was focused onRgs16::GFP expression during development and in adult modelsof islet regeneration/expansion.
Rgs16::GFP in embryonic pancreatic endocrine cellsTo determine whether endocrine cells within the pancreasexpressed Rgs16, the co-expression of Rgs16::GFP and endocrinecell markers was analyzed (Fig. 3). We examined Rgs16 expressionat E8.75 through to E10.5, as the bud first evaginates and the firsttransition endocrine cells emerge, and at E15.5 during the rapidwave of endocrine expansion and differentiation called the‘secondary transition’. Rgs16::GFP was expressed initially in most
Disease Models & Mechanisms 569
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLE
Fig. 2. Rgs16::GFP, Rgs8::GFP and Ngn3::GFP expression in the developing pancreatic bud. (A)Rgs16::GFP is broadly expressed in the endoderm at E8.5 butresolves (B) to the dorsal pancreas and liver bud by E9.5. (C)E10.5, expression in pancreatic endoderm, but not in mesoderm. (D,E)Expression in groups of cellsalong the central axis of the pancreatic tree, as branching elaborates. (F)In the postnatal pancreas, expression becomes restricted to forming islets (arrows). Bycontrast, (G) Rgs8::GFP is first expressed in a more localized domain within the foregut (arrowhead). (H-L)Similar to Rgs16, Rgs8 expression continues in scatteredcells within the pancreatic endoderm between E9.5-10.5 (H,I), in the central region of the branching pancreatic bud between E12.5-E16.5 (J,K), and strongly informing islets after birth (L). (G,M)Note that Rgs8::GFP is expressed in the dorsal pancreas prior to Ngn3::GFP (arrowheads), which is first detected in the dorsalpancreas at about E9.5 (compare G,H with M,N). (C-F,I-L,O-R) All three genes are expressed in the endocrine pancreas from E9.5 in a very similar pattern.(F,L,R) Arrows mark coalescing islets during postnatal development. The expression of Ngn3-eGFP was lower, but detectable, up to a few weeks post-birth.(D,E,J,K,P,Q) Dotted lines define the limit of the developing pancreas at each stage. Top row: schematics of the embryonic structures displayed in the rows below.aip, anterior intestinal portal; d, duodenum; dp, dorsal pancreatic bud; gte, gut tube endoderm; h, heart; l, liver; m, mesoderm; nf, neural folds; nt, neural tube;s, somite; vp, ventral pancreatic bud; ys, yolk sac. Bars, 100mm (A-E,G-K,M-Q); 50mm (F,L,R).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

cells of the pancreatic bud epithelium and overlapped with thepancreatic progenitor markers Sox9 and Pdx1 (Fig. 3A,B;supplementary material Fig. S1A,B and Table S1). At about E9.5,Rgs16::GFP expression overlapped with differentiating glucagon-expressing cells and began to be excluded from Sox9-expressingcells (supplementary material Fig. S1C and Table S1). Shortlythereafter, Rgs16::GFP expression became largely restricted todifferentiating glucagon-expressing endocrine cells and to scatteredE-cadherin-positive epithelial cells that expressed low levels ofPdx1 but not Sox9 (Fig. 3C-E and supplementary material Fig.S1D-F).
Following the secondary transition at E15.5, Rgs16::GFPexpression was largely excluded from the tubular epithelium.Most Rgs16::GFP-positive (Rgs16::GFP+) cells co-expressedsynaptophysin and islet hormones in delaminating endocrine cells(Fig. 3K-M). Only a small number of Rgs16::GFP+ cells co-expressedeither E-cadherin (3.5%) or Sox9 (5%) in the epithelium (Fig. 3F,G;supplementary material Fig. S2A,B and Table S1 and data notshown). Interestingly, Rgs16::GFP was never co-expressed withNgn3 (Fig. 3H; supplementary material Fig. S2C and Table S1).
In contrast to the lack of overlap with Ngn3, we found that 53%of Rgs16::GFP co-localized with Pdx1 (Fig. 3I; supplementarymaterial Fig. S2H and Table S1), a pancreatic progenitor markerthat also partially overlaps with insulin expression at E15.5 (Offieldet al., 1996). Rgs16::GFP co-localized with other markers ofendocrine cell fate, such as 52% with Nkx6.1 and 97% withsynaptophysin (Fig. 3J,K; supplementary material Fig. S2D,I andTable S1). Rgs16::GFP was co-expressed with terminal endocrinedifferentiation markers such as glucagon and insulin (Fig. 3L,M andsupplementary material Fig. S2E-G), indicating that Rgs16 isexpressed in islet lineages. Consistent with these expression
patterns, greater than 98% of Rgs16::GFP+ cells were found to beKi67 negative at E15.5, indicating that, at this stage, the vast majorityof Rgs16::GFP-expressing cells were non-replicating (only 1.45%were Rgs16+/Ki67+) (supplementary material Fig. S3 and Table S1,and data not shown). As expected, Rgs16 expression did not co-localize with amylase-positive cells in acinar tissue (Fig. 3N).Rgs16-expressing cells were near blood vessels [marked by plateletendothelial cell adhesion molecule (PECAM)] (Fig. 3O;supplementary material Fig. S2J and data not shown), reflectingthe known close association of endocrine and vascular tissues. Atmidgestation, Rgs16::GFP is predominately expressed in transient,post-delaminating endocrine cells that are aggregating into pre-isletclusters. Together, these co-labeling data establish Rgs16 as amarker for both early pancreatic progenitors and laterdifferentiating cells of the endocrine lineage.
Rgs16::GFP in postnatal pancreasFrom E18.5 until a few weeks after birth, endocrine cells aggregatealong the axes of pancreatic branches to form islets (Cleaver, 2004).During the early stages of this process, strong Rgs16 expressioncould be detected within the forming islets (Fig. 4A) and always inclose association with blood vessels (Fig. 4B). Expression in isletendocrine cells declined rapidly after postnatal day (P)11 and wasabsent by P15 (compare Fig. 4A,B with 4C-E; supplementarymaterial Fig. S9). By contrast, Rgs16::GFP remained in discrete cellsand small cell clusters (two to ten cells) located along the centralaxis of distinct pancreatic branches (Fig. 4D-F). We termed thesevessel- and ductal-associated Rgs16::GFP-positive cells (VDACs)(Fig. 4H). VDACs were frequently clustered around triad branchpoints, where vessels and ducts branch coordinately (Fig. 4F).Relatively strong VDAC expression could be observed throughoutearly postnatal stages, up to and during the first 10 days followingweaning (pups were weaned at P16) (Table 1). VDACs were notobserved by 12 days post-weaning (Fig. 4G) or in the normalglycemic adult pancreas (data not shown).
To identify the types of vessels associated with VDACs,endothelial-lined blood vessels were distinguished frompancreatic ducts by immunofluorescence (Fig. 5A,B). Rgs16::GFP+
VDACs were located along ducts (DBA) (Fig. 5B) closelyassociated with blood vessels (PECAM) (Fig. 5A) and lymphatics(LYVE-1) (Fig. 5E). In addition, VDACs were often in closeproximity to developing islets expressing synaptophysin (Fig. 5D)and, later, differentiation markers such as insulin (Fig. 5E). Indeed,Rgs16::GFP+ cells were often observed around the periphery ofislets or along duct/vessels triads in proximity to islets(supplementary material Fig. S10).
To elucidate the identity of Rgs16-expressing VDACs, a rangeof endocrine markers of early versus late endocrine differentiationwere examined. A small proportion of VDACs (~5%) expressed theearliest endocrine progenitor marker Sox9 (Fig. 5F), but noneexpressed later markers of endocrine differentiation such assynaptophysin, insulin, glucagon, Glut2, somatostatin, PP or ghrelin(supplementary material Fig. S4 and data not shown). Furthermore,VDACs did not express markers of vascular endothelial, smoothmuscle or macrophage lineages (supplementary material Fig. S4).Some Rgs16+ VDACs are therefore similar to early progenitor cellsin the epithelium in their expression of Sox9 and all are distinctfrom delaminating embryonic Rgs16+ endocrine cells (Fig. 3C-F).
dmm.biologists.org570
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLE
Table 1. Expression of Rgs16::GFP in embryonic and postnatal
pancreas
Stage
Islet (Rgs16::GFP
expression)
VDACs (Rgs16::GFP
expression)
E8.5 +/– NA
E8.75-9.0 ++ NA
E9.5-16.5 +++ NA
P0 +++ NA
P1 +++ NA
P3 +++ NA
P7 +++ NA
P10 +++ +
P11 + ++
P14 – ++
P15 – ++
P16-30 – +
Adult (2 months) – –
Adult (3 months) – –
Arbitrary expression levels were assigned by independent visual analysis of relativeintensity of fluorescence (two observers). Live dissected pancreatic tissue wasexamined in PBS, under fluorescence microscopy. P, pancreas-stage postnatal day. Expression could be detected either in forming islets (middle column) or within scattered cells along ducts and blood vessels (VDACs, right column). NA, not applicable because strong expression in islets at these stages masks potential VDAC expression; (–) absent; (+) present; (++) medium; (+++) strong.
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Rgs16::GFP re-activated in the pancreas of pregnant femalesTo determine whether Rgs16-expressing cells re-emerge in thepancreas of metabolically stressed mice, the pancreata of pregnantfemales were examined at different times during gestation. Isletmass in females expands during pregnancy (Van Assche, 1978).Although Rgs16::GFP expression was never observed in normaladult pancreas, either within islets or as VDACs, Rgs16 was re-expressed along vessel and duct tracts in mid- to late-gestationfemale pancreas (supplementary material Fig. S5). Indeed, startingat approximately 8 days of gestation, rare GFP+ cells could be readilyobserved along the central axes of lateral pancreatic branches(supplementary material Fig. S5A), and increasing numbers ofVDACs were present between E10.5-15.5 along many differentbranches (supplementary material Fig. S5B-E). Rgs16::GFPexpression was coincident with the initiation of a known phase of-cell expansion in pregnant females (Gupta et al., 2007), but then
faded between E16.5 and E18.5 (supplementary material Fig. S5F;data not shown). Between these stages, all pregnant femalesexpressed Rgs16::GFP in VDACs but only one expressedRgs16::GFP in the islets. By contrast, lactating females did notexpress Rgs16::GFP in either VDACs or islets (data not shown).Rgs16::GFP expression along the ducts in pregnant females suggeststhat increased maternal metabolic demands might be conveyed byGPCR signaling and regulated by RGS proteins.
Rgs16::GFP re-activated in regenerating -cells of type 1 diabeticmiceGiven the correlation of Rgs16 expression with -cell proliferation,both in the embryo and pregnant adults, we asked whetherRgs16::GFP might also be re-expressed in islets during -cellregeneration. Recently, islet regeneration was shown to occurfollowing ablation in PANIC-ATTAC (pancreatic islet -cell
Disease Models & Mechanisms 571
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLE
Fig. 3. Rgs16::GFP is expressed in early pancreatic progenitors and in delaminated cells of the endocrine lineage at E15.5. Rgs16::GFP is initially expressedin a subset of cells within the early pancreatic epithelium, at E8.75, and is co-expressed with Sox9 (A) and Pdx1(B). (C)A subset of Rgs16::GFP cells can be seenwithin the pancreatic bud epithelium, as shown by E-cadherin staining, at E10.0. (D)Later, Rgs16::GFP expression becomes mutually exclusive with Sox9 and (E)overlaps with the endocrine marker glucagon. By E15.5, Rgs16::GFP is primarily not co-expressed with E-cadherin (F) or Sox9 (G) in the pre-ductal epithelium, orwith Ngn3 in endocrine cells prior to their delamination (H) (note that, in panel F, E-cadherin is expressed at low levels in budding endocrine cells that expresshigh levels of Rgs16::GFP, green arrow). A few (2%) Rgs16::GFP-expressing cells, however, still reside in the epithelium (see inset in F). At E15.5, Rgs16::GFP is co-expressed (white arrows) with markers of differentiating endocrine cells: Pdx1(I), nkx6.1(J), synaptophysin (K), insulin (L) and glucagon (M). It is almost completelyco-expressed with pooled antibodies to all differentiated endocrine cells (anti-ins, -gluc, -somatostatin, -ghrelin). Rgs16::GFP is not co-expressed with amylase (N)in exocrine cells or with PECAM (O) in vascular cells. (G-O)Nuclei are identified by DAPI staining of DNA (blue). The insets in F-M display either co-expression (F,I-M) or lack of co-expression (G,H) of markers, as indicated. Confocal micrographs, 50� (A,B); 40� (C-O).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

apoptosis through targeted activation of caspase 8) mice (Wang etal., 2008). In this model of type 1 diabetes, pancreatic -cells weretargeted for cell death by the regulated expression of an FKBP-caspase 8 fusion protein, resulting in hyperglycemia.
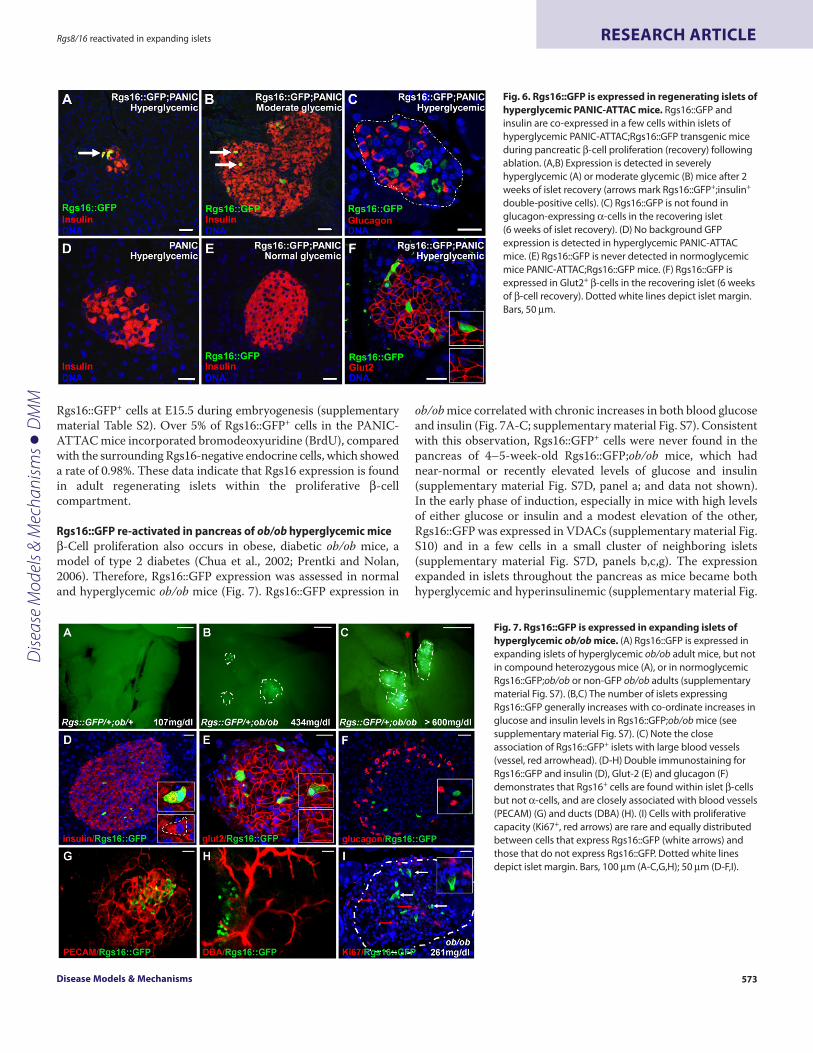
To assess the expression of Rgs16 during islet regeneration, theRgs16::GFP transgene was crossed into the background of thePANIC-ATTAC mice. Following -cell apoptosis, Rgs16::GFP wasco-expressed with insulin in a subset of pancreatic -cells during thefirst two weeks of islet regeneration (Fig. 6). The percentage ofRgs16::GFP+ cells was higher in hyperglycemic mice with more severehyperglycemia and islet destruction than in mice with moderateglucose levels (Fig. 6A,B; supplementary material Fig. S6). Rgs16 wasspecifically expressed in -cells, as assessed by either insulin (Fig.6A,B) or Glut2 co-staining (Fig. 6F), and was not observed in otherendocrine cell types, such as -cells (Fig. 6C). Rgs16::GFP was alsonot expressed in the pancreas of either parental PANIC-ATTAC (Fig.6D) or normoglycemic Rgs16::GFP;PANIC-ATTAC mice (Fig. 6E;supplementary material Fig. S6A,B,E).
A time course study showed that hyperglycemia (>300 mg/dl)appeared in about half the males during the first three to five daysof FKBP-ligand injection (supplementary material Fig. S6A,B).Rgs16::GFP was induced by chronic, but not acute, hyperglycemia.For example, Rgs16::GFP was not visible in day 5 hyperglycemicmice (n2) and only first appeared by day 7 in VDACs and a fewcells within one or a small cluster of neighboring islets(supplementary material Fig. S6E, panel a). Rgs16::GFP expressionexpanded to more cells in more islets throughout the pancreas overa 50-day interval of -cell regeneration (supplementary materialFig. S6D,E). Of note, if the blood insulin was at least 0.5 ng/ml, theday 5 blood glucose levels were good predictors of later Rgs16::GFPexpression (supplementary material Fig. S6B-D).
Interestingly, the Rgs16::GFP+ cells in this model of -cellregeneration displayed a higher proliferative capacity than the
dmm.biologists.org572
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLE
Fig. 4. Rgs16::GFP+ cells are associated with blood vessels during neonatal islet formation. Neonatal Rgs16::GFP expression is observed in coalescing isletsthroughout the postnatal pancreas (at postnatal day indicated). (A)Rgs expression is evident in endocrine cells as they aggregate into clusters along blood vessels atP0. (B)A forming islet (white arrow) can be seen overlying a blood vessel (white arrowhead). (C)Expression continues in islets until approximately P11 (short redarrows), but expression remains in scattered cells, or VDACs, along the axes of lateral branches (short white arrows). (D-F)At weaning (P16), the expression ofRgs16::GFP is no longer detectable in islets (D); however, expression continues in VDACs lining axial blood vessels at the center of lateral pancreatic branches (shortwhite arrows) (E,F). (F)GFP+ cells (arrow) are tightly associated with tracts of blood vessels and associated pancreatic ducts, and are especially enriched at vascularbranch points (red arrowhead; the red dotted line delineates a mature islet). (G)Expression vanishes by P27 (12 days past weaning). (C-E)The dotted white linesdepict margins of lateral pancreatic branches in postnatal pancreas. (H)Schematic of VDACs location along the ‘triad’ composed of artery (red), vein (blue) and duct(yellow). The cross section shows the approximate location of VDACs (green) at the interface between ducts and blood vessels. Scale bars, 100mm.
Fig. 5. Rgs16::GFP+ cells are associated with ducts and blood vesselsduring postnatal islet formation. (A-E)Whole-mount staining for GFP in P15BAC transgenic neonates demonstrates the close association of Rgs16-expressing VDACs (arrows) with blood vessels (PECAM) (A), ducts (DBA lectin)(B), endocrine cells (synaptophysin) (C), late endocrine cells (insulin) (D) andlymphatic vessels (Lyve-1) (E). Note the rare Rgs16::GFP+ cells observed withinislets (yellow arrow in D). At higher resolution, a portion of VDACs (5%) can beseen to express Sox9 (F). (A-E)Confocal micrographs of whole-mountpreparations, 30� (A-E). Confocal micrographs of sectioned tissue, 100� (F).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM
Rgs16::GFP+ cells at E15.5 during embryogenesis (supplementarymaterial Table S2). Over 5% of Rgs16::GFP+ cells in the PANIC-ATTAC mice incorporated bromodeoxyuridine (BrdU), comparedwith the surrounding Rgs16-negative endocrine cells, which showeda rate of 0.98%. These data indicate that Rgs16 expression is foundin adult regenerating islets within the proliferative -cellcompartment.
Rgs16::GFP re-activated in pancreas of ob/ob hyperglycemic mice-Cell proliferation also occurs in obese, diabetic ob/ob mice, amodel of type 2 diabetes (Chua et al., 2002; Prentki and Nolan,2006). Therefore, Rgs16::GFP expression was assessed in normaland hyperglycemic ob/ob mice (Fig. 7). Rgs16::GFP expression in
ob/ob mice correlated with chronic increases in both blood glucoseand insulin (Fig. 7A-C; supplementary material Fig. S7). Consistentwith this observation, Rgs16::GFP+ cells were never found in thepancreas of 4–5-week-old Rgs16::GFP;ob/ob mice, which hadnear-normal or recently elevated levels of glucose and insulin(supplementary material Fig. S7D, panel a; and data not shown).In the early phase of induction, especially in mice with high levelsof either glucose or insulin and a modest elevation of the other,Rgs16::GFP was expressed in VDACs (supplementary material Fig.S10) and in a few cells in a small cluster of neighboring islets(supplementary material Fig. S7D, panels b,c,g). The expressionexpanded in islets throughout the pancreas as mice became bothhyperglycemic and hyperinsulinemic (supplementary material Fig.
Disease Models & Mechanisms 573
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLE
Fig. 6. Rgs16::GFP is expressed in regenerating islets ofhyperglycemic PANIC-ATTAC mice. Rgs16::GFP andinsulin are co-expressed in a few cells within islets ofhyperglycemic PANIC-ATTAC;Rgs16::GFP transgenic miceduring pancreatic -cell proliferation (recovery) followingablation. (A,B)Expression is detected in severelyhyperglycemic (A) or moderate glycemic (B) mice after 2weeks of islet recovery (arrows mark Rgs16::GFP+;insulin+
double-positive cells). (C)Rgs16::GFP is not found inglucagon-expressing -cells in the recovering islet(6 weeks of islet recovery). (D)No background GFPexpression is detected in hyperglycemic PANIC-ATTACmice. (E)Rgs16::GFP is never detected in normoglycemicmice PANIC-ATTAC;Rgs16::GFP mice. (F)Rgs16::GFP isexpressed in Glut2+ -cells in the recovering islet (6 weeksof -cell recovery). Dotted white lines depict islet margin.Bars, 50mm.
Fig. 7. Rgs16::GFP is expressed in expanding islets ofhyperglycemic ob/ob mice. (A)Rgs16::GFP is expressed inexpanding islets of hyperglycemic ob/ob adult mice, but notin compound heterozygous mice (A), or in normoglycemicRgs16::GFP;ob/ob or non-GFP ob/ob adults (supplementarymaterial Fig. S7). (B,C)The number of islets expressingRgs16::GFP generally increases with co-ordinate increases inglucose and insulin levels in Rgs16::GFP;ob/ob mice (seesupplementary material Fig. S7). (C)Note the closeassociation of Rgs16::GFP+ islets with large blood vessels(vessel, red arrowhead). (D-H)Double immunostaining forRgs16::GFP and insulin (D), Glut-2 (E) and glucagon (F)demonstrates that Rgs16+ cells are found within islet -cellsbut not -cells, and are closely associated with blood vessels(PECAM) (G) and ducts (DBA) (H). (I)Cells with proliferativecapacity (Ki67+, red arrows) are rare and equally distributedbetween cells that express Rgs16::GFP (white arrows) andthose that do not express Rgs16::GFP. Dotted white linesdepict islet margin. Bars, 100mm (A-C,G,H); 50mm (D-F,I).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

S7D, panels d-f,j,k). Rgs16 expression occurred in -cells, but not-cells of the ob/ob islets, as Rgs16::GFP+ cells expressed insulinand Glut2 (Fig. 7D,E) but not glucagon (Fig. 7F). This -cellspecificity within islets and gradual expansion of Rgs16 expression
was similar to that observed in the islets of Rgs16::GFP;PANIC-ATTAC mice. As anticipated, Rgs16::GFP+ islet -cells were foundin close association with islet capillaries (Fig. 7G) and ducts (Fig.7H). Additionally, Rgs8::GFP was similarly induced specifically inthe islets of hyperglycemic and hyperinsulinemic ob/ob mice(supplementary material Fig. S8).
In older ob/ob mice with high glucose and insulin levels (as inFig. 7C and supplementary material Fig. S7C,D, panel k),Rgs16::GFP expression was observed in up to half of the pancreaticislets (supplementary material Fig. S9), each containing 1-5%Rgs16::GFP+ -cells (in the three animals counted, 78% of ob/obislets expressed Rgs16 in 0.5-5% of total endocrine cells, 17%expressed Rgs16 in 6-20% of total cells and 5% expressed Rgs16 in21-50% of total cells). The majority of Rgs16::GFP+ islets were foundalong the central region of the main axis of the pancreas (Fig. 7C)but Rgs16::GFP expression appeared in islets of all sizes throughoutthe pancreas (supplementary material Fig. S7D).
Although -cells in islets of ob/ob mice are known to proliferate(Bock et al., 2003), relatively modest numbers of Ki67+ cells wereobserved in hyperglycemic animals (36% of the ob/ob islets had0.5-1% Ki67+ cells, 59% had 1-3% Ki67+ cells and about 5% had 3-5% Ki67+ cells). In addition, Rgs16::GFP+ -cells in the islets of olderob/ob mice did not display a more highly proliferative state thanRgs16::GFP– -cells (supplementary material Table S3). Indeed,Rgs16-expressing cells co-stained for Ki67+ at the same rate as thesurrounding -cells, at slightly over 1% (supplementary materialTable S3). The total number of -cells expressing Rgs16::GFPdeclined in the pancreas of ob/ob mice as glycemia was loweredfollowing initial -cell expansion, then increased withhyperinsulinemia in older ob/ob mice (supplementary material Fig.S7).
Exendin-4 induces Rgs16::GFP in adult pancreasFinally, to assess Rgs16::GFP expression using another model of -cell proliferation, we stimulated -cell neogenesis and isletexpansion by injection of Rgs16::GFP transgenic mice with exendin-4, a known GPCR agonist (Tourrel et al., 2001). Within 3 days oftwice-daily injections of exendin-4 plus glucose, Rgs16 expressionwas induced in some VDACs but rarely, if at all, in islets (Fig. 8A,B).Later, by the sixth day of injection, Rgs16::GFP expression wasobserved in more VDACs and also in -cells within a few islets(Fig. 8C,D,H). Rgs16::GFP+ islets were also observed in miceinjected daily with exendin-4 alone (three of five mice). As seenpreviously in PANIC and ob/ob mice, Rgs16 expression firstappeared in a small cluster of neighboring islets (Fig. 8E,F). Bycontrast, expression was not observed in islets in mice injected withglucose alone or PBS (Fig. 8G and supplementary material Fig. S11).These results confirmed that Rgs16::GFP can be used as a reporterof GPCR-stimulated -cell neogenesis in the pancreas.
DISCUSSIONIn this study, we introduce regulators of G protein signaling asgenetic beacons of -cell expansion during development anddisease. Rgs16 and Rgs8 are expressed in pancreatic progenitor andendocrine cells during development, then extinguished in adults,but reactivated in models of both type 1 and type 2 diabetes.Furthermore, Rgs16 expression is activated in response to exendin-4, a GPCR agonist that is used to stimulate -cell expansion in type
dmm.biologists.org574
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLE
Fig. 8. Exendin-4 induces Rgs16::GFP expression in adult pancreas.Normoglycemic Rgs16::GFP BAC transgenic male mice (8-12 weeks) wereinjected (intraperitoneally) twice daily for 6 days with PBS, glucose, exendin-4plus glucose (Ex+Glc), or exendin-4 alone (Ex). (A,B)After 3 days, Rgs16::GFP+
VDACs were observed but only in Ex+Glu-treated mice; (B) bright field of panelA (n3/group). (C-F)After 6 days, Rgs16::GFP+ VDACs and/or islets wereobserved in Ex+Glu (C,D) or Ex-treated (E,F) mice; (D,F) bright field of panels Cand E. (G)An islet from a PBS-injected mouse at day 3; GFP expression was notobserved in any PBS-treated mice (n2). (H)An islet of an Ex+Glc-injectedmouse at day 5; GFP expression was observed in the islets of Ex+Glu-injectedmice (n2) (see supplementary material Fig. S11C). Dotted white lines depictislet margin. Bars, 100mm (A-F); 50mm (G-H).
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

2 diabetes (Xu et al., 1999; Tourrel et al., 2001; Fineman et al., 2003;Kendall et al., 2005; Kodama et al., 2005; Xu et al., 2006; Chu et al.,2007; Sherry et al., 2007; Wang et al., 2008). Based on theirendocrine expression and known biochemical activities, Rgs16 andRgs8 are likely to have redundant functions in the endocrinepancreas and may promote -cell differentiation and maturation,although definitive functional characterization awaits simultaneousdeletion of both genes in -cells. Rgs16::GFP and Rgs8::GFPtransgenic mice are unique tools for comparative analysis of theendocrine pancreas during -cell expansion/regeneration indevelopment, disease and response to therapy.
RGS regulation of GPCR signalingRGS proteins are essential regulators of GPCR signaling, controllingthe timing of both signal activation and termination in diverseprocesses such as chemotaxis, cell cycle exit or re-entry, and neuralprocessing (Berman et al., 1996; Doupnik et al., 1997; Zeng et al.,1998; Zerangue and Jan, 1998; Luo et al., 2001). An intriguing aspectof mammalian Rgs genes related to Rgs16, but first noted in yeastSst2/Rgs1, is that their expression is induced by ligands that arefeedback regulated by the RGS protein (Dohlman et al., 1996; Rossand Wilkie, 2000). Thus, the expression of Rgs16 and Rgs8 can beused to identify where and when GPCR signaling is active duringdevelopment and in adult tissues of healthy or diseased individuals.
Rgs8/16::GFP expression in embryonic pancreasUsing Rgs8/16 BAC transgenic reporter lines, we assessedexpression during key time points of pancreas development. Ourresults showed that, similar to Pdx1 and Ngn3 (Kim andMacDonald, 2002; Villasenor et al., 2008; Cleaver and MacDonald,2009), Rgs16::GFP and Rgs8::GFP are first observed throughout thepancreatic epithelium, but later become restricted to -cells. BothRgs16::GFP and Rgs8::GFP are first expressed in the ‘proto-differentiated’ early pancreatic epithelium at E8.75, when theessential progenitor cells are set aside for later pancreasdevelopment (Offield et al., 1996; Zhou et al., 2007; Cleaver andMacDonald, 2009). At this early developmental stage, Rgs16::GFPis co-expressed with the pancreatic progenitor marker Sox9 in mostmultipotent progenitors. Interestingly, Rgs16::GFP and Rgs8::GFPare induced in the pancreatic bud in this early epithelium prior toNgn3::GFP, a well-characterized marker of pancreatic endocrineprogenitors (Villasenor et al., 2008).
With the onset of endocrine differentiation, Rgs16::GFPexpression becomes restricted to endocrine cells. By E15.5,Rgs16::GFP expression is predominantly restricted to delaminatedand/or differentiated endocrine cells that co-express synaptophysin,but not Ngn3. These Rgs16::GFP+ endocrine cells are primarily non-replicating, as less than 1.5% of Rgs16::GFP+ cells are Ki67+. Giventhese data, Rgs16 may promote -cell differentiation and/or inhibitcell cycle re-entry in delaminating endocrine cells.
During development, Rgs16 expression identifies transient cellstates, an earlier progenitor state, and a later state that anticipatesand initiates hormone expression. The two ‘waves’ of Rgs16expression, in the early pancreatic epithelium and later endocrinelineages, are similar to the ‘biphasic’ expression of pancreatic genes,such as Pdx1, Hlxb9 and Ngn3, before and after the secondarytransition (Pictet and Rutter, 1972; Offield et al., 1996; Li andEdlund, 2001; Villasenor et al., 2008; Cleaver and MacDonald, 2009).
For example, Pdx1 expression is first observed throughout thepancreatic epithelium and its derivatives, but after birth it becomesrestricted to differentiating islets (Offield et al., 1996). Thequalitatively different expression domains for Rgs16 may signifytemporal regulation within a single cell lineage or expression intwo separate lineages, the first appearing in the early epitheliumof the pancreatic bud and the other following transient expressionof Ngn3.
Rgs8/16::GFP expression in models of diabetesDespite the strong expression of Rgs16 and Rgs8 throughoutembryonic and neonatal isletogenesis, their expression ceases after4 weeks of age in endocrine pancreas under normal conditions.However, both genes were re-expressed in models of -cellexpansion or regeneration, suggesting that these genes are likelyto play a role in either endocrine proliferation, differentiation, orin response to chronic changes in glucose metabolism. The absenceof RGS::GFP expression in normal adult pancreatic endocrine cellssuggests that, if Rgs16 and Rgs8 have a physiological role in adultislets, low basal levels are sufficient to regulate daily postprandialinsulin release and glucose homeostasis. Dietary approaches to alterblood glucose and metabolism, such as high-fat or high-disaccharide diets, or twice-daily intraperitoneal injections ofglucose, failed to induce Rgs8/16 expression in otherwise-normalpancreatic islets (data not shown). However, the pancreaticexpression of Rgs16 in pregnant females during midgestation, inexendin-4-treated mice, and in the islets of hyperglycemic type 1and type 2 diabetic mice suggests that Rgs16 is a biomarker forcells that have reactivated an aspect of the embryonic -cellprogram during adult -cell proliferation/regeneration in responseto glucose stress.
Rgs16::GFP becomes re-expressed in hyperglycemic PANIC-ATTAC and ob/ob mice during the early phase of -cellregeneration, indicating that Rgs16 may regulate -cell replicationduring compensatory islet expansion. In these two models, thepattern of expression is strikingly similar during disease progressionand recovery. Hyperglycemia precedes Rgs16 induction in bothPANIC and ob/ob mice (supplementary material Figs S6 and S7,respectively). This lag in expression suggests that Rgs16 is notdirectly induced in the pancreas by elevated blood glucose, but byphysiologic responses to chronic glucose stress. Elevated insulinmay contribute to increased Rgs16::GFP expression but is notsufficient, as some young hyperinsulinemic ob/ob mice express littleor no Rgs16::GFP (supplementary material Fig. S7). However,combined hyperglycemia and hyperinsulinemia (and severe obesity)in older ob/ob mice are strongly correlated with Rgs16::GFPexpression (supplementary material Fig. S7).
Another striking similarity during disease progression is Rgs16distribution. In both mouse models of diabetes, Rgs16 expressionis first observed in the -cells of one or a small cluster of neighboringislets. As the disease progresses, Rgs16::GFP expression expands tomore cells, within more islets, throughout the pancreas(supplementary material Figs S6, S7). Rgs16::GFP expression persistsin non-proliferative -cells in both models of diabetes(supplementary material Tables S2 and S3), reflecting expressionduring embryogenesis and neonatal isletogenesis (supplementarymaterial Table S1). This progressive and heterogeneous expressionof Rgs in islets during -cell expansion in PANIC and ob/ob mice
Disease Models & Mechanisms 575
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLED
iseas
e M
odel
s & M
echa
nism
s
DM
M

reflects the known heterogeneous response to hyperglycemia andearly diabetes (Atkinson and Gianani, 2009).
Rgs16 has been suggested to inhibit -cell expansion duringpostnatal development and in ob/ob mice (Poy et al., 2009). Indeed,Rgs16 and Rgs8 feedback may inhibit the GPCR signaling thatinitiates -cell expansion. Endogenous Rgs16 and Rgs8 genes aresimilarly induced in the islets of 10-week-old ob/ob mice in bothC57BL/6 and BTBR genetic backgrounds (Keller et al., 2008), where-cell expansion occurs in the C57BL/6 mice but not in BTBR mice.Blood insulin levels are elevated in both backgrounds of mice, butmodestly in ob/ob BTBR mice (to about 10 ng/ml) in a constrainedeffort to manage insulin resistance and hyperglycemia. Thisindicates that BTBR islets perceive chronic glucose stress andinduce Rgs16 and Rgs8, even though they fail to undergo -cellexpansion. These signals may be transmitted within the localenvironment of responding islets and augment cellular responsesto elevated glucose and insulin (Llona, 1995). Feedback inhibitionby Rgs16 (and Rgs8) could promote the cell cycle exit or preventthe cell cycle re-entry of maturing -cells that are still exposed tomitogens during diabetic islet expansion.
Rgs16::GFP a biomarker of GPCR stimulationExendin-4 is an incretin mimetic prescribed for type 2 diabeticsto improve glucose homeostasis by suppressing glucagonsecretion, stimulating insulin release and promoting -cellexpansion (Xu et al., 1999; Tourrel et al., 2001; Kodama et al.,2005; Bond, 2006; Lee et al., 2006; Sherry et al., 2007). Exendin-4 is a protease-resistant homolog of mammalian Glp-1, and bothare agonists of the Glp-1 receptor (Glp-1R). Agonist bindingevokes Gs-mediated increases in intracellular cAMP levels,promotes mitogenic and anti-apoptotic signals, and activates c-fos and other early response genes (Lee and Nielsen, 2009).However, RGS proteins are not Gs-GAPs (Berman1996).Therefore, Glp-1R either activates Gi or Gq proteins as well, orstimulates the synthesis or release of other agonists of Gi/q-coupled GPCRs in the pancreas, which would be regulated byRgs16 and Rgs8 (Wang et al., 2006).
Daily injection of exendin-4 induced Rgs16::GFP expression inthe pancreas. Rgs16::GFP expression was initiated in VDACs,whereas -cell expression was observed in islets by day 6.Interestingly, Glp-1R is specifically expressed in pancreatic ductsand islet -cells, and exendin-4 was reported to promote theemergence of insulin-positive cells in cultured pancreatic ducts (Xuet al., 2006; Tornehave et al., 2008). The pattern of Rgs16::GFPinduction by exendin-4 is strikingly similar to the early phase inPANIC and ob/ob mice, in which small numbers of VDACs appeartransiently, and islet expression is first seen in one or a few closelyclustered islets (supplementary material Fig. S10). Althoughspeculative, the location of VDACs along pancreatic ducts, and theirappearance early in the time course of the hyperglycemic responsein mouse models of diabetes or in exendin-4 treatment, raises thealluring possibility that these represent an endocrine progenitorcell type. The contribution of endocrine progenitors within adultducts to -cell regeneration is controversial. Future lineage-tracingexperiments will be required to conclusively determine theendocrine potential of Rgs16+ VDACs.
The induction of Rgs genes in VDACs and -cells impliesregulation of endogenous GPCRs that are important in the process
of -cell differentiation during development and -cell regenerationin models of metabolic stress and disease. Many GPCRs areexpressed in mouse islets but their functions are unknown (Regardet al., 2007). Furthermore, GPCRs that are expressed in embryonicendocrine progenitors, such as the Gq-coupled receptor Gpr56 (Guet al., 2004) (www.genepaint.org), may be induced during -cellexpansion in diabetes. A subset of these GPCRs may be regulatedby Rgs16 and/or Rgs8 during pancreas development and isletregeneration. A challenge is to identify additional mitogens thatstimulate -cell expansion. A practical approach would be toidentify molecules that induce Rgs16 expression in surrogates ofpancreatic ducts and/or endocrine progenitors, such as primarycultured cells from pancreatic ductal adenocarcinoma (Aguirre etal., 2003), ‘endocrine-differentiated’ embryonic stem (ES) cells(Kroon et al., 2008; Serafimidis et al., 2008), or induced pluripotentstem (iPS) cells (Takahashi and Yamanaka, 2006; Nakamura et al.,2009). Superlative candidate molecules could be identified by invivo validation in Rgs16::GFP transgenic mice, as demonstratedhere for exendin-4.
We propose that Rgs16 and Rgs8 are fetal pancreatic endocrinegenes that have redundant functions during embryonic pancreasand -cell development and, later in adults, during islet regenerationin type 1 diabetes (PANIC-ATTAC) and islet expansion in type 2diabetes (ob/ob) (Herrera et al., 1994; Nir et al., 2007; Cano et al.,2008; Wang et al., 2008). Future work will identify the ligands andreceptor-signaling pathways that are regulated by Rgs16 and Rgs8in hyperglycemic type 1 and type 2 diabetic mice. These ligands,GPCRs and regulatory RGS proteins are likely to be importantsignaling molecules in the progression towards diabetes, and keytargets for diabetes therapy.
METHODSGreen fluorescent protein (GFP) visualization in the pancreas fromembryos, pups and adultsRgs16::GFP and Rgs8::GFP BAC transgenic mice were generatedby the Gene Expression Nervous System Atlas (GENSAT) project(Gong et al., 2003; Morales and Hatten, 2006). Pancreata werecollected from Rgs16::GFP and Rgs8::GFP embryos (E8.5 throughto E16.5) and from postnatal stages P0 (birth) to P28. Pups wereweaned at P16 for time course studies of Rgs16::eGFP expressionpost-weaning. Tissues were dissected and transferred into ice-cold1� PBS buffer. GFP visualization was accomplished by removingthe embryonic gut tube and isolating the midgut, including thepancreas, stomach and spleen. Tissue fragments were equilibratedin 40% glycerol for viewing. The pancreas was visualized using aZeiss NeoLumar fluorescent microscope and photographed usingan Olympus DP70 camera. Ngn3::eGFP embryos were generatedby mating Ngn3::eGFP heterozygous males (generously providedby Klaus Kaestner) with CD1 females. Embryos were dissected atdifferent developmental stages, and pancreata were dissected andvisualized as described above. Pancreata from adult pregnantfemales and ob/ob;Rgs16::eGFP transgenic mice were treatedsimilarly to those from embryos, as described above.
Immunofluorescence staining of frozen sectionsE15.5 dorsal pancreata were dissected from Rgs16::eGFP pregnantfemales and were fixed overnight in 4% PFA in 1� PBS at 4°C. Thenext day the tissues were washed several times with 1� PBS,
dmm.biologists.org576
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLED
iseas
e M
odel
s & M
echa
nism
s
DM
M

equilibrated in 30% sucrose overnight and embedded in optimalcutting temperature (OCT) medium (Tissue-Tek). Cryosections(10 mm) of complete pancreata were mounted on SuperfrostPlusslides (Fisher) and immunofluorescence was carried out using:chicken anti-GFP (1:500; Aves Labs), guinea pig anti-insulin (1:300;DakoCy), rabbit anti-glucagon (1:1000; LINCO), rat anti-CD31(1:300; BD Pharmingen), rabbit anti-synaptophysin (1:200;DakoCy), 5 mg/ml DBA (1:200; Vector Labs), rabbit anti-Pdx1 (1:600)and mouse anti-ngn3 (1:4000) (Beta Cell Biology Consortium,kindly provided by Dr Raymond MacDonald). TRITC secondaryantibodies were from Jackson ImmunoResearch Laboratories andanti-chicken Alexa 488 was from Invitrogen. Slides werecounterstained with DAPI and mounted with ProLong Goldantifade reagent (Invitrogen). Images were acquired on a LSM 510META Zeiss confocal microscope. For PANIC-ATTAC samples,histology and immunofluorescence were performed as describedpreviously (Wang et al., 2008). Briefly, the pancreas was fixed in10% buffered formalin overnight. Paraffin sections (5 mm) wereincubated with guinea pig anti-insulin (1:500; DakoCy) and rabbitanti-GFP (1:100; Invitrogen). The secondary antibodies used weredonkey anti-guinea pig FITC (1:250; Jackson ImmunoResearch) anddonkey anti-rabbit Cy3 (1:500; Jackson ImmunoResearch). Imageswere taken on a Leica TCS SP5 confocal microscope (Leica).
Immunofluorescence on paraffin sectionsE15.5 dorsal pancreata (Rgs16::eGFP) or adult pancreata fromob/ob;Rgs16::eGFP or PANIC-ATTAC;Rgs16::eGFP mice weredissected and fixed overnight in 4% PFA in 1� PBS at 4°C. Thenext day, the tissues were washed several times with 1� PBS,dehydrated and paraffin embedded in Paraplast Plus tissueembedding medium (McCormick). Sections (10 mm) of completepancreata were mounted on SuperfrostPlus slides. Sections werede-waxed in xylene; rehydrated through an ethanol series; washedseveral times in 1� PBS; treated with R-Buffer A or R-Buffer B(Electron Microscopy Sciences) in the 2100 Retriever; blocked for1-2 hours with CAS-Block (Invitrogen); and incubated with chickenanti-GFP (1:200), rabbit anti-sox9 (1:400; Chemicon), mouse anti-e-cadherin (1:200; Invitrogen), rabbit anti-glut2 (1:100; Abcam) andthe four-hormones cocktail: guinea pig anti-glucagon (1:600; kindlyprovided by Raymond MacDonald), guinea pig anti-insulin (1:600;DakoCy), goat anti-ghrelin (1:300; Beta Cell Consortium) and goatanti-somatostatin (1:300; Santa Cruz). TRITC secondary antibodieswere from Jackson ImmunoResearch and anti-chicken Alexa 488was from Invitrogen. Slides were counterstained with DAPI andmounted with ProLong Gold antifade reagent (Invitrogen). Imageswere acquired on a LSM 510 META Zeiss confocal microscope.
Quantification of rates of replicationImmunofluorescence in paraffin sections of E15.5 dorsal pancreata(Rgs16::eGFP) or adult pancreata from ob/ob;Rgs16::GFP andPANIC-ATTAC;Rgs16::GFP transgenic mice were performedutilizing chicken anti-GFP (1:200) and either rabbit anti-Ki67(1:200; Invitrogen) or rat anti-Brdu (1:50; Serotec). A total of eightfields of three different E15.5 embryonic pancreata, 70 islets of threedifferent ob/ob;Rgs16::GFP pancreata, and 13 islets for PANIC-ATTAC;Rgs16::GFP transgenic pancreata were studied. The totalnumber of endocrine cells per islet/field, the total number of Ki67+
cells, the total number of Rgs16+ cells, and the total overlap between
Ki67 and Rgs16 were all determined, and rates of replication werecalculated.
Immunofluorescence staining of frozen sections for VDACsP15 pancreata from Rgs16::GFP mice were harvested andimmediately embedded in OCT medium and frozen. Sections(10 mm) were cut and allowed to dry overnight at room temperature.Sections were then fixed for 5 minutes in ice-cold acetone andrinsed several times in 1� TBST (1� TBS supplemented with 0.2%Tween). Sections were then blocked in 20% Aquablock in 1� TBST,and stained using chicken anti-GFP (1:100) and one of the followingprimary antibodies: rabbit anti-sox9 (1:1000; Chemicon), guineapig anti-insulin (1:200; DakoCy), rabbit anti-glucagon (1:300;LINCO), goat anti-somatostatin (1:300; Santa Cruz), rabbit anti-synaptophysin (1:300; DakoCy), rabbit anti-glut2 (1:200; Millipore),25 mg/ml rat anti-MECA32 (Rolf Brekken), 20 mg/ml rabbit anti-NG2 (Millipore), 20 mg/ml rat-anti Mac1(AbD Serotec). Sectionswere stained overnight at 4°C then washed three times in 1� TBST.Bound primary antibody was visualized using appropriate Cy3- orFITC-conjugated secondary antibodies. Nuclei were visualized withDAPI. Images were taken using a Nikon Eclipse E600 microscope.
Whole-mount immunofluorescencePancreata were fixed for 1 hour in 4% PFA in PBS, washed anddehydrated to 70% ethanol. Embryos were then washed in 50%methanol for 1 hour and rinsed twice in 1� PBS. The tissue waspermeabilized for 1 hour in 1% TritonX 100 in 1� PBS, blockedin Cas-Block (Zymed), and immunofluorescence was then carriedout using: chicken anti-GFP (1:250; Aves Labs), guinea-pig anti-insulin (1:200; DakoCy), rabbit anti-glucagon (LINCO), rat anti-CD31 (1:200; BD Pharmingen), rabbit anti-synaptophysin (1:200;DakoCy), 5 mg/ml DBA (1:200; Vector Labs), and rabbit anti-LYVE(1:1000; Ambion). TRITC secondary antibodies were from JacksonImmunoResearch and anti-chicken Alexa 488 was from Invitrogen.Tissues were dehydrated, cleared with BABB (1:2 mix of benzylalcohol:benzyl benzoate), and visualized using a Zeiss NeoLumarfluorescent microscope and photographed using an Olympus DP70camera or LSM 510 META Zeiss confocal microscope.
ob/ob;Rgs16::eGFP and ob/ob;Rgs8::eGFP miceob/+ breeder mice were obtained from Jackson Labs. Doubleheterozygous Rgs16::eGFP;ob/+ mice were intercrossed to obtainob/ob, ob/ob;Rgs16::eGFP and ob/+;Rgs16::eGFP mice. Animalswere fed a standard rodent chow diet (Teklad) ad libitum. Bloodwas acquired through tail clipping. Glucose levels were measuredby using a glucometer (AscensiaElite), and insulin levels weremeasured by using a rat/mouse insulin ELISA kit (Millipore) andread with a Sunrise microplater reader (Tecan).
PANIC-ATTAC;Rgs16::eGFP transgenic micePANIC-ATTAC transgenic mice were generated as describedpreviously (Wang et al., 2008). Briefly, the rat insulin promoter wasused to drive the expression of an FKBP-caspase 8 fusion protein.Homozygous PANIC-ATTAC animals were crossed to Rgs16::eGFPtransgenic mice. All progeny were hemizygous for the PANIC-ATTAC transgene. At about 12 weeks of age, animals were groupedinto hemizygous PANIC-ATTAC;Rgs16::GFP transgenic mice(n2), hemizygous PANIC-ATTAC;Rgs16::eGFP-negative mice
Disease Models & Mechanisms 577
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLED
iseas
e M
odel
s & M
echa
nism
s
DM
M

(n4) and FVB control mice (n3). The dimerizer AP20187 wasadministered to animals according to the manufacturer’srecommendations (Ariad Pharmaceuticals). For hemizygousPANIC-ATTAC mice, dimerizer (0.2 mg/g body weight) wasinjected either twice per day, every other day for eight days, or onceper day at 12.00 h for five consecutive days. Fed glucose levels weremonitored using a glucometer and strips (Abbott Diabetes Care).After five days treatment, hemizygous PANIC-ATTAC malesshowed either moderate (glucose <300 mg/dl) or severe (glucose>300 mg/dl) hyperglycemia. Animals were sacrificed two weekslater, with the pancreas processed for immunofluorescent staining(Fig. 6), or at the times indicated for analysis of GFP expression(supplementary material Fig. S6). Insulin levels were assessed asper ob/ob mice.
Exendin-4+/–, glucose or PBS treatmentMale mice aged 8-12 weeks (30-40 g) were injected,intraperitoneally, twice daily at zeitgeber time ZT6 (6 hours afterlights on) and ZT11. Exendin-4 was injected at a concentration of200 ng/mouse time point in a volume of 100 ml, glucose at 4.5 g/kg,and PBS at a volume of 100 ml.
GFP quantification of exendin-4/glucose imagesOriginal JPEG RGB images, at a resolution of 1360�1024 pixels,of mouse pancreas taken with a 48� objective magnification wereconverted into 8-bit gray-scale format without rescaling in ImageJ(NIH). Background levels with a rolling ball radius of 50 pixels weresubtracted from images. Varying lower threshold adjustmentswere selected based on the contours of GFP+ regions. Integrateddensities as the sum of 8-bit gray values, on a 0-255 scale per pixel,were obtained from each particle with a size of three pixels orgreater. The sum of analyzed integrated densities as a total GFPvalue was calculated using MS Excel. Three-dimensional bar graphscorrelating the levels of blood glucose, insulin, Rgs16::GFPexpression in islets, and the age of ob/ob mice or the time aftertreatment in PANIC mice, were created by Matlab.ACKNOWLEDGEMENTSWe thank M. E. Hatten (Rockefeller University) for kindly providing the Rgs16::GFPand Rgs8::GFP BAC transgenic mice; Klaus Kaestner (University of PennsylvaniaSchool of Medicine) for providing the Ngn3-eGFP mice; Diana Chong for technicalassistance; Raymond MacDonald for providing antibodies; Galvin Swift for E14.5mRNA; Ariad Pharmaceuticals for providing the dimerizer for the PANIC-ATTACmice; Victor Pashkov for genotyping, qPCR and discussions; Alex Artyukhin forassistance with image analysis; and Raymond MacDonald, Galvin Swift, NilsHalberg, Michael Dellinger and Deborah Clegg for helpful discussions and/orreading the manuscript. This work was supported by NIH R0161395,P50MH066172, Welch I-1382 (to T.M.W.); NIH R01CA118240 (to R.A.B.); JDRF 1-2008-16 (to P.E.S.); and NIH DK079862 and JDRF 99-2007-472 (to O.C.). Depositedin PMC for release after 12 months.
COMPETING INTERESTSThe authors declare no competing financial interests.
AUTHOR CONTRIBUTIONSO.C. and T.M.W. conceived the project and, with A.V., Z.V.W., R.A.B. and P.E.S.,designed the experiments. A.V., Z.V.W., L.B.R., O.O., I.W.A., O.C. and T.M.W.performed the experiments. All authors analyzed data and edited the manuscript.A.V., O.C. and T.M.W. wrote the manuscript.
SUPPLEMENTARY MATERIALSupplementary material for this article is available athttp://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003210/-/DC1
Received 17 March 2009; Accepted 26 March 2010.
REFERENCESAguirre, A. J., Bardeesy, N., Sinha, M., Lopez, L., Tuveson, D. A., Horner, J.,
Redston, M. S. and DePinho, R. A. (2003). Activated Kras and Ink4a/Arf deficiencycooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17,3112-3126.
Atkinson, M. A. and Gianani, R. (2009). The pancreas in human type 1 diabetes:providing new answers to age-old questions. Curr. Opin. Endocrinol. Diabetes Obes.16, 279-285.
Berman, D. M., Wilkie, T. M. and Gilman, A. G. (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445-452.
Bock, T., Pakkenberg, B. and Buschard, K. (2003). Increased islet volume butunchanged islet number in ob/ob mice. Diabetes 52, 1716-1722.
Bond, A. (2006). Exenatide (Byetta) as a novel treatment option for type 2 diabetesmellitus. Proc. (Bayl. Univ. Med. Cent.) 19, 281-284.
Bonner-Weir, S., Toschi, E., Inada, A., Reitz, P., Fonseca, S. Y., Aye, T. and Sharma, A.(2004). The pancreatic ductal epithelium serves as a potential pool of progenitorcells. Pediatr. Diabetes 5 Suppl. 2, 16-22.
Bonner-Weir, S., Inada, A., Yatoh, S., Li, W. C., Aye, T., Toschi, E. and Sharma, A.(2008). Transdifferentiation of pancreatic ductal cells to endocrine -cells. Biochem.Soc. Trans. 36, 353-356.
Cano, D. A., Rulifson, I. C., Heiser, P. W., Swigart, L. B., Pelengaris, S., German, M.,Evan, G. I., Bluestone, J. A. and Hebrok, M. (2008). Regulated -cell regenerationin the adult mouse pancreas. Diabetes 57, 958-966.
Chu, Z. L., Jones, R. M., He, H., Carroll, C., Gutierrez, V., Lucman, A., Moloney, M.,Gao, H., Mondala, H., Bagnol, D. et al. (2007). A role for -cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulinrelease. Endocrinology 148, 2601-2609.
Chua, S., Jr, Liu, S. M., Li, Q., Yang, L., Thassanapaff, V. T. and Fisher, P. (2002).Differential beta cell responses to hyperglycaemia and insulin resistance in twonovel congenic strains of diabetes (FVB-Lepr (db)) and obese (DBA-Lep (ob)) mice.Diabetologia 45, 976-990.
Cleaver, O. (2004). Blood vessel signals during development and beyond. Curr. Top.Dev. Biol. 62, 1-36.
dmm.biologists.org578
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLE
TRANSLATIONAL IMPACT
Clinical issueDiabetes exacts one of the highest annual costs for treatment of any disease inthe world and is the seventh leading cause of death in the USA. Most cases ofboth type 1 and type 2 diabetes involve the actual or functional loss of insulin-producing pancreatic -cells. Although -cells can replicate, little is knownabout how the process is regulated and available therapies for diabetes cannotspecifically stimulate -cell regeneration. There is hope that understanding thesignals that control islet cell proliferation and differentiation will help generatetargeted -cell regenerative therapies for diabetic patients.
ResultsThis study shows that two members of the gene family regulators of G proteinsignaling (RGS), Rgs8 and Rgs16, coordinate -cell expansion with metabolicneed. Rgs8 and Rgs16 are expressed during islet development and in mousemodels of type 1 and type 2 diabetes. Exendin-4, a Glp-1/incretin mimetic thatstimulates -cell expansion in diabetics, also promotes the expression ofRgs16::GFP in pancreatic ductal-associated cells and islet -cells in mice. As thename implies, RGS members regulate the frequency and duration of G protein-coupled receptor (GPCR) signaling. Since GPCR pathways are also associatedwith -cell expansion, this suggests that RGS proteins serve as sensitive andearly beacons of G protein signaling in -cell progenitor expansion andregeneration during development and in metabolically stressed adults.
Implications and future directionsThis study indicates that the RGS proteins Rgs8 and Rgs16 coordinate GPCRsignaling with islet development and, later in life, with metabolic stress. Theearly response of these proteins to metabolic changes suggests that they maybe useful screening indicators. Furthermore, understanding the mechanismsthat regulate GPCR induction of pancreatic -cell proliferation may provide acrucial inroad to diabetes therapy. Future work should determine the potentialof RGS proteins to promote -cell development and islet regeneration.
doi:10.1242/dmm.005850
Dise
ase
Mod
els &
Mec
hani
sms
D
MM

Cleaver, O. and Melton, D. A. (2003). Endothelial signaling during development. Nat.
Med. 9, 661-668.Cleaver, O. and MacDonald, R. J. (2009). Developmental molecular biology of the
pancreas. In Handbook of Pancreatic Cancer (ed. J. A. J. Neoptolemos, M. Buchler andR. Urrutia), pp. 71-118. New York, NY: Springer.
Collombat, P., Hecksher-Sorensen, J., Serup, P. and Mansouri, A. (2006). Specifyingpancreatic endocrine cell fates. Mech. Dev. 123, 501-512.
D’Amour, K. A., Agulnick, A. D., Eliazer, S., Kelly, O. G., Kroon, E. and Baetge, E. E.(2005). Efficient differentiation of human embryonic stem cells to definitiveendoderm. Nat. Biotechnol. 23, 1534-1541.
D’Amour, K. A., Bang, A. G., Eliazer, S., Kelly, O. G., Agulnick, A. D., Smart, N. G.,Moorman, M. A., Kroon, E., Carpenter, M. K. and Baetge, E. E. (2006). Productionof pancreatic hormone-expressing endocrine cells from human embryonic stemcells. Nat. Biotechnol. 24, 1392-1401.
Dohlman, H. G., Song, J., Ma, D., Courchesne, W. E. and Thorner, J. (1996). Sst2, anegative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae:expression, localization, and genetic interaction and physical association with Gpa1(the G-protein alpha subunit). Mol. Cell. Biol. 16, 5194-5209.
Dor, Y., Brown, J., Martinez, O. I. and Melton, D. A. (2004). Adult pancreatic -cellsare formed by self-duplication rather than stem-cell differentiation. Nature 429, 41-46.
Doupnik, C. A., Davidson, N., Lester, H. A. and Kofuji, P. (1997). RGS proteinsreconstitute the rapid gating kinetics of G-activated inwardly rectifying K+channels. Proc. Natl. Acad. Sci. USA 94, 10461-10466.
Fineman, M. S., Bicsak, T. A., Shen, L. Z., Taylor, K., Gaines, E., Varns, A., Kim, D.and Baron, A. D. (2003). Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type2 diabetes. Diabetes Care 26, 2370-2377.
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev.
Biochem. 56, 615-649.Gittes, G. K. and Rutter, W. J. (1992). Onset of cell-specific gene expression in the
developing mouse pancreas. Proc. Natl. Acad. Sci. USA 89, 1128-1132.Golosow, N. and Grobstein, C. (1962). Epitheliomesenchymal interaction in pancreatic
morphogenesis. Dev. Biol. 4, 242-255.Gong, S., Zheng, C., Doughty, M. L., Losos, K., Didkovsky, N., Schambra, U. B.,
Nowak, N. J., Joyner, A., Leblanc, G., Hatten, M. E. et al. (2003). A gene expressionatlas of the central nervous system based on bacterial artificial chromosomes. Nature
425, 917-925.Gradwohl, G., Dierich, A., LeMeur, M. and Guillemot, F. (2000). neurogenin3 is
required for the development of the four endocrine cell lineages of the pancreas.Proc. Natl. Acad. Sci. USA 97, 1607-1611.
Gu, G., Wells, J. M., Dombkowski, D., Preffer, F., Aronow, B. and Melton, D. A.(2004). Global expression analysis of gene regulatory pathways during endocrinepancreatic development. Development 131, 165-179.
Gupta, R. K., Gao, N., Gorski, R. K., White, P., Hardy, O. T., Rafiq, K., Brestelli, J. E.,Chen, G., Stoeckert, C. J., Jr and Kaestner, K. H. (2007). Expansion of adult -cellmass in response to increased metabolic demand is dependent on HNF-4. Genes
Dev. 21, 756-769.Herrera, P. L., Huarte, J., Zufferey, R., Nichols, A., Mermillod, B., Philippe, J.,
Muniesa, P., Sanvito, F., Orci, L. and Vassalli, J. D. (1994). Ablation of isletendocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc.
Natl. Acad. Sci. USA 91, 12999-13003.Huang, J., Pashkov, V., Kurrasch, D. M., Yu, K., Gold, S. J. and Wilkie, T. M. (2006).
Feeding and fasting controls liver expression of a regulator of G protein signaling(Rgs16) in periportal hepatocytes. Comp. Hepatol. 5, 8.
Inada, A., Nienaber, C., Katsuta, H., Fujitani, Y., Levine, J., Morita, R., Sharma, A.and Bonner-Weir, S. (2008). Carbonic anhydrase II-positive pancreatic cells areprogenitors for both endocrine and exocrine pancreas after birth. Proc. Natl. Acad.
Sci. USA 105, 19915-19919.Keller, M. P., Choi, Y., Wang, P., Davis, D. B., Rabaglia, M. E., Oler, A. T., Stapleton, D.
S., Argmann, C., Schueler, K. L., Edwards, S. et al. (2008). A gene expressionnetwork model of type 2 diabetes links cell cycle regulation in islets with diabetessusceptibility. Genome Res. 18, 706-716.
Kendall, D. M., Riddle, M. C., Rosenstock, J., Zhuang, D., Kim, D. D., Fineman, M. S.and Baron, A. D. (2005). Effects of exenatide (exendin-4) on glycemic control over30 weeks in patients with type 2 diabetes treated with metformin and asulfonylurea. Diabetes Care 28, 1083-1091.
Kim, S. K. and MacDonald, R. J. (2002). Signaling and transcriptional control ofpancreatic organogenesis. Curr. Opin. Genet. Dev. 12, 540-547.
Kodama, S., Toyonaga, T., Kondo, T., Matsumoto, K., Tsuruzoe, K., Kawashima, J.,Goto, H., Kume, K., Kume, S., Sakakida, M. et al. (2005). Enhanced expression ofPDX-1 and Ngn3 by exendin-4 during beta cell regeneration in STZ-treated mice.Biochem. Biophys. Res. Commun. 327, 1170-1178.
Koelle, M. R. and Horvitz, H. R. (1996). EGL-10 regulates G protein signaling in the C.elegans nervous system and shares a conserved domain with many mammalianproteins. Cell 84, 115-125.
Kroon, E., Martinson, L. A., Kadoya, K., Bang, A. G., Kelly, O. G., Eliazer, S., Young,H., Richardson, M., Smart, N. G., Cunningham, J. et al. (2008). Pancreaticendoderm derived from human embryonic stem cells generates glucose-responsiveinsulin-secreting cells in vivo. Nat. Biotechnol. 26, 443-452.
Lammert, E., Cleaver, O. and Melton, D. (2001). Induction of pancreaticdifferentiation by signals from blood vessels. Science 294, 564-567.
Lammert, E., Cleaver, O. and Melton, D. (2003). Role of endothelial cells in earlypancreas and liver development. Mech. Dev. 120, 59-64.
Lee, C. S., De Leon, D. D., Kaestner, K. H. and Stoffers, D. A. (2006). Regeneration ofpancreatic islets after partial pancreatectomy in mice does not involve thereactivation of neurogenin-3. Diabetes 55, 269-272.
Lee, Y. C. and Nielsen, J. H. (2009). Regulation of beta cell replication. Mol. Cell
Endocrinol. 297, 18-27.Li, H. and Edlund, H. (2001). Persistent expression of Hlxb9 in the pancreatic
epithelium impairs pancreatic development. Dev. Biol. 240, 247-253.Llona, I. (1995). Synaptic like microvesicles: do they participate in regulated
exocytosis? Neurochem. Int. 27, 219-226.Luo, X., Popov, S., Bera, A. K., Wilkie, T. M. and Muallem, S. (2001). RGS proteins
provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol. Cell 7, 651-660.
Morales, D. and Hatten, M. E. (2006). Molecular markers of neuronal progenitors inthe embryonic cerebellar anlage. J. Neurosci. 26, 12226-12236.
Nakamura, K., Salomonis, N., Tomoda, K., Yamanaka, S. and Conklin, B. R. (2009).G(i)-coupled GPCR signaling controls the formation and organization of humanpluripotent colonies. PLoS One 4, e7780.
Nir, T., Melton, D. A. and Dor, Y. (2007). Recovery from diabetes in mice by beta cellregeneration. J. Clin. Invest. 117, 2553-2561.
Offield, M. F., Jetton, T. L., Labosky, P. A., Ray, M., Stein, R. W., Magnuson, M. A.,Hogan, B. L. and Wright, C. V. (1996). PDX-1 is required for pancreatic outgrowthand differentiation of the rostral duodenum. Development 122, 983-995.
Oliver-Krasinski, J. M. and Stoffers, D. A. (2008). On the origin of the beta cell. Genes
Dev. 22, 1998-2021.Pictet, R. L. and Rutter, W. J. (1972). Development of the embryonic pancreas. In
Endocrine Pancreas: Handbook of Physiology (ed. D. F. Steiner and N. Frienkel), pp. 25-66. Baltimore: Williams and Wilkins.
Poy, M. N., Hausser, J., Trajkovski, M., Braun, M., Collins, S., Rorsman, P., Zavolan,M. and Stoffel, M. (2009). miR-375 maintains normal pancreatic - and -cell mass.Proc. Natl. Acad. Sci. USA 106, 5813-5818.
Prentki, M. and Nolan, C. J. (2006). Islet beta cell failure in type 2 diabetes. J. Clin.
Invest. 116, 1802-1812.Regard, J. B., Kataoka, H., Cano, D. A., Camerer, E., Yin, L., Zheng, Y. W., Scanlan, T.
S., Hebrok, M. and Coughlin, S. R. (2007). Probing cell type-specific functions of Giin vivo identifies GPCR regulators of insulin secretion. J. Clin. Invest. 117, 4034-4043.
Ross, E. M. and Wilkie, T. M. (2000). GTPase-activating proteins for heterotrimeric Gproteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev.
Biochem. 69, 795-827.Serafimidis, I., Rakatzi, I., Episkopou, V., Gouti, M. and Gavalas, A. (2008). Novel
effectors of directed and Ngn3-mediated differentiation of mouse embryonic stemcells into endocrine pancreas progenitors. Stem Cells 26, 3-16.
Sherry, N. A., Chen, W., Kushner, J. A., Glandt, M., Tang, Q., Tsai, S., Santamaria, P.,Bluestone, J. A., Brillantes, A. M. and Herold, K. C. (2007). Exendin-4 improvesreversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody byenhancing recovery of -cells. Endocrinology 148, 5136-5144.
Sierra, D. A., Gilbert, D. J., Householder, D., Grishin, N. V., Yu, K., Wensel, T. G.,Otero, G., Copeland, N. G., Jenkins, N. A., Wilkie, T. M. et al. (2002). Evolution ofthe regulators of G-protein signaling multigene family in mouse and human.Genomics 79, 177-185.
Simon, M. I., Strathmann, M. P. and Gautam, N. (1991). Diversity of G proteins insignal transduction. Science 252, 802-808.
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., Zhang, J., Soden,R., Hayakawa, M., Kreiman, G. et al. (2004). A gene atlas of the mouse and humanprotein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101, 6062-6067.
Takahashi, K. and Yamanaka, S. (2006). Induction of pluripotent stem cells frommouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663-676.
Teta, M., Rankin, M. M., Long, S. Y., Stein, G. M. and Kushner, J. A. (2007). Growthand regeneration of adult beta cells does not involve specialized progenitors. Dev.
Cell 12, 817-826.Tornehave, D., Kristensen, P., Romer, J., Knudsen, L. B. and Heller, R. S. (2008).
Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J. Histochem.
Cytochem. 56, 841-851.
Disease Models & Mechanisms 579
Rgs8/16 reactivated in expanding islets RESEARCH ARTICLED
iseas
e M
odel
s & M
echa
nism
s
DM
M

Tourrel, C., Bailbe, D., Meile, M. J., Kergoat, M. and Portha, B. (2001). Glucagon-likepeptide-1 and exendin-4 stimulate -cell neogenesis in streptozotocin-treatednewborn rats resulting in persistently improved glucose homeostasis at adult age.Diabetes 50, 1562-1570.
Van Assche, F. A., Aerts, L. and De Prins, F. (1978). A morphological study of theendocrine pancreas in human pregnancy. Br. J. Obstet. Gynaecol. 85, 818-820.
Villasenor, A., Chong, D. C. and Cleaver, O. (2008). Biphasic Ngn3 expression in thedeveloping pancreas. Dev. Dyn. 237, 3270-3279.
Wang, C., Kerckhofs, K., Van de Casteele, M., Smolders, I., Pipeleers, D. and Ling, Z.(2006). Glucose inhibits GABA release by pancreatic -cells through an increase inGABA shunt activity. Am J. Physiol. Endocrinol. Metab. 290, E494-E499.
Wang, Z. V., Mu, J., Schraw, T. D., Gautron, L., Elmquist, J. K., Zhang, B. B.,Brownlee, M. and Scherer, P. E. (2008). PANIC-ATTAC, a mouse model for inducibleand reversible -cell ablation. Diabetes 57, 2137-2148.
Wells, J. M. and Melton, D. A. (1999). Vertebrate endoderm development. Annu. Rev.Cell Dev. Biol. 15, 393-410.
Xu, G., Stoffers, D. A., Habener, J. F. and Bonner-Weir, S. (1999). Exendin-4 stimulatesboth -cell replication and neogenesis, resulting in increased -cell mass andimproved glucose tolerance in diabetic rats. Diabetes 48, 2270-2276.
Xu, G., Kaneto, H., Lopez-Avalos, M. D., Weir, G. C. and Bonner-Weir, S. (2006). GLP-1/exendin-4 facilitates -cell neogenesis in rat and human pancreatic ducts. DiabetesRes. Clin. Pract. 73, 107-110.
Xu, X., D’Hoker, J., Stange, G., Bonne, S., De Leu, N., Xiao, X., Van de Casteele,M., Mellitzer, G., Ling, Z., Pipeleers, D. et al. (2008). Beta cells can begenerated from endogenous progenitors in injured adult mouse pancreas. Cell132, 197-207.
Yoshitomi, H. and Zaret, K. S. (2004). Endothelial cell interactions initiate dorsalpancreas development by selectively inducing the transcription factor Ptf1a.Development 131, 807-817.
Zeng, W., Xu, X., Popov, S., Mukhopadhyay, S., Chidiac, P., Yagaloff, K. A., Fisher, S.L., Ross, E. M., Muallem, S., Wilkie, T. M. et al. (1998). The N-terminal domain ofRGS4 confers receptor-selective inhibition of G protein signaling. J. Biol. Chem. 273,34687-34690.
Zerangue, N. and Jan, L. Y. (1998). G-protein signaling: fine-tuning signaling kinetics.Curr. Biol. 8, R313-R316.
Zhou, Q., Law, A. C., Rajagopal, J., Anderson, W. J., Gray, P. A. and Melton, D. A.(2007). A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell13, 103-114.
dmm.biologists.org580
Rgs8/16 reactivated in expanding isletsRESEARCH ARTICLED
iseas
e M
odel
s & M
echa
nism
s
DM
M




















