
Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24...
11
Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters Qinbo Zhou a , Rachel Gallagher a , Rafael Ufret-Vincenty a , Xinyu Li a , Eric N. Olson b,1 , and Shusheng Wang a,c,1 Departments of a Ophthalmology, b Molecular Biology, and c Pharmacology, University of Texas Southwestern Medical Center, Dallas, TX 75390 Contributed by Eric N. Olson, April 6, 2011 (sent for review March 2, 2011) MicroRNAs (miRNAs) modulate complex physiological and patholog- ical processes by repressing expression of multiple components of cellular regulatory networks. Here we demonstrate that miRNAs encoded by the miR-23∼27∼24 gene clusters are enriched in endo- thelial cells and highly vascularized tissues. Inhibition of miR-23 and miR-27 function by locked nucleic acid-modified anti-miRNAs represses angiogenesis in vitro and postnatal retinal vascular devel- opment in vivo. Moreover, miR-23 and miR-27 are required for path- ological angiogenesis in a laser-induced choroidal neovascularization mouse model. MiR-23 and miR-27 enhance angiogenesis by promot- ing angiogenic signaling through targeting Sprouty2 and Sema6A proteins, which exert antiangiogenic activity. Manipulating miR-23/ 27 levels may have important therapeutic implications in neovascular age-related macular degeneration and other vascular disorders. blindness | MAP kinase signaling | semaphorins | Akt | proangiogenic T he growth of blood vessels through angiogenesis is a delicately controlled process that involves endothelial cell (EC) activa- tion, proliferation, migration, and maturation (1). Physiological angiogenesis is required for normal vascular development as well as vascular homeostasis during adulthood. Pathological angio- genesis, commonly induced by tissue ischemia or inflammation, underlies numerous vascular disorders, such as age-related mac- ular degeneration (AMD), a leading cause of blindness in the elderly (2). Choroidal neovascularization (CNV), which involves abnormal growth of blood vessels in the back of the eye, is a hallmark of neovascular AMD (3). Although the pathogenic mechanisms underlying AMD are still largely unknown, vascular endothelial growth factor (VEGF) has been shown to play a causal role in the development of CNV (4). Anti-VEGF agents have demonstrated efficacy in treating CNV in neovascular AMD (5, 6) but have limited efficacy and potential side effects (7, 8). Recent studies have revealed important roles for microRNAs (miRNAs) in cardiovascular diseases and other disorders (9). miRNAs are small noncoding RNAs that negatively regulate gene expression by inducing mRNA degradation or inhibiting trans- lation through binding to the 3′ untranslated region (3′UTR) of target mRNAs (10). Often, miRNAs modulate broad collections of mRNAs encoding multiple components of complex biological pathways. Several miRNAs have been implicated in angiogenesis (11, 12). A group of miRNAs has also been shown to be sub- stantially decreased in a laser-induced CNV model (13). The miR-23∼27∼24 clusters are highly expressed in ECs (14– 17). Two miR-23∼27∼24 clusters exist in the vertebrate genome: an intergenic miR-23a∼27a∼24–2 cluster and an intronic miR- 23b∼27b∼24–1 cluster. Members of these clusters are involved in cell cycle control, proliferation, and differentiation of various cell types (18). Here, we show that inhibition of miR-23/27 impairs angiogenesis in vitro and postnatal retinal vascular development in vivo. Moreover, silencing of miR-23/27 suppresses laser-in- duced CNV in mice. The proangiogenic functions of miR-23/27 correlate with the repression of Sprouty2 and Sema6A, which negatively regulate angiogenic signaling. Results Structure and Expression Pattern of miR-23∼27∼24 Cluster Members. The mouse miR-23a∼27a∼24–2 cluster is intergenic on chro- mosome 8, and the miR-23b∼27b∼24–1 cluster is located in in- tron 4 of an alanine aminopeptidase gene on chromosome 13 (Fig. 1A). The miR-23a∼27a∼24–2 cluster encodes a primary miRNA (pri-miRNA) transcript composed of three miRNAs: miR-23a, miR-27a, and miR-24–2, and the miR-23b∼27b∼24–1 cluster encodes a pri-miRNA transcript containing miR-23b, miR-27b, and miR-24–1(Fig. S1A). The mature miRNA se- quences of miR-23a/b, miR-27a/b, and miR-24 are conserved among vertebrate species (Fig. S1B). miR-23a and miR-27a differ by only one nucleotide near their 3′ ends compared with their paralogs miR-23b and miR-27b, whereas the sequence of miR-24–1 and miR-24–2 is the same (Fig. S1C). miR-23a and miR-23b share most, if not all, predicted target genes by Tar- getScan and DIANA Lab, as do miR-27a and miR-27b (19, 20). Northern-blot analyses revealed that miR-23a/b, miR-27a/b, and miR-24 are expressed at the highest levels in the lung and heart, which are highly vascularized tissues (Fig. 1B). Their ex- pression is also detectable in other organs, including the eye. Real- time PCR confirmed the enrichment of the mature miRNAs of these two clusters in the lung and heart (Fig. S1D). Real-time PCR was further performed to determine the expression of miR- 23∼27∼24 cluster members in different cell types. Our results showed that the expression of all miR-23∼27∼24 cluster members is enriched in ECs compared to the other cell types tested (Fig. 1C), consistent with previous reports (14–21) and a recent report that expression of the miR-23b∼27b∼24-1 host gene is enriched in ECs in vivo (21). Taken together, our results indicate that miR-23∼27∼24 cluster members are enriched in ECs and highly vascularized tissues, suggesting a potential role in EC function. Modulation of Sprouting Angiogenesis by miR-23 and miR-27 in Vitro. To study the EC function of miR-23∼27∼24 cluster members in vitro, human umbilical vein EC line (HUVECs) were transfected with locked nucleic acid (LNA)-modified anti-miRs of miR-23a/b, miR-27a/b, or a scramble control and tested for EC network for- mation on Matrigel. More than 90% knockdown of miR-23a/b or miR-27a/b expression was achieved by LNA-anti-miR-23a/b or anti-miR-27a/b transfection, respectively, indicating the efficiency and specificity of miRNA knockdown by LNA anti-miRs (Fig. 2A). Hereafter, miR-23a/b and miR-27a/b will be referred to as miR-23 and miR-27. Of note, there was compensatory up-regulation of miR-23 or miR-27 when miR-27 or miR-23 were knocked down. When cultured on Matrigel, ECs form a primary vascular network. Knockdown of miR-27, and to a lesser extent miR-23, impaired the formation of capillary-like structures in HUVECs cultured on Matrigel, as quantified by the reduced branching points upon miR-27 or miR-23 inhibition (Fig. 2 B and C). These results suggest that miR-27 and miR-23 are required for proper capillary tube formation in vitro. Author contributions: E.N.O. and S.W. designed research; Q.Z., R.G., R.U.-V., and X.L. performed research; and E.N.O. and S.W. wrote the paper. The authors declare no conflict of interest. 1 To whom correspondence may be addressed. E-mail: [email protected] or [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1105254108/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1105254108 PNAS | May 17, 2011 | vol. 108 | no. 20 | 8287–8292 CELL BIOLOGY
Transcript of Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24...

Regulation of angiogenesis and choroidalneovascularization by members ofmicroRNA-23∼27∼24 clustersQinbo Zhoua, Rachel Gallaghera, Rafael Ufret-Vincentya, Xinyu Lia, Eric N. Olsonb,1, and Shusheng Wanga,c,1
Departments of aOphthalmology, bMolecular Biology, and cPharmacology, University of Texas Southwestern Medical Center, Dallas, TX 75390
Contributed by Eric N. Olson, April 6, 2011 (sent for review March 2, 2011)
MicroRNAs (miRNAs) modulate complex physiological and patholog-ical processes by repressing expression of multiple components ofcellular regulatory networks. Here we demonstrate that miRNAsencoded by the miR-23∼27∼24 gene clusters are enriched in endo-thelial cells and highly vascularized tissues. Inhibition of miR-23and miR-27 function by locked nucleic acid-modified anti-miRNAsrepresses angiogenesis in vitro and postnatal retinal vascular devel-opment in vivo. Moreover, miR-23 and miR-27 are required for path-ological angiogenesis in a laser-induced choroidal neovascularizationmouse model. MiR-23 and miR-27 enhance angiogenesis by promot-ing angiogenic signaling through targeting Sprouty2 and Sema6Aproteins, which exert antiangiogenic activity. Manipulating miR-23/27 levelsmay have important therapeutic implications in neovascularage-related macular degeneration and other vascular disorders.
blindness | MAP kinase signaling | semaphorins | Akt | proangiogenic
The growth of blood vessels through angiogenesis is a delicatelycontrolled process that involves endothelial cell (EC) activa-
tion, proliferation, migration, and maturation (1). Physiologicalangiogenesis is required for normal vascular development as wellas vascular homeostasis during adulthood. Pathological angio-genesis, commonly induced by tissue ischemia or inflammation,underlies numerous vascular disorders, such as age-related mac-ular degeneration (AMD), a leading cause of blindness in theelderly (2). Choroidal neovascularization (CNV), which involvesabnormal growth of blood vessels in the back of the eye, is ahallmark of neovascular AMD (3). Although the pathogenicmechanisms underlying AMD are still largely unknown, vascularendothelial growth factor (VEGF) has been shown to play a causalrole in the development of CNV (4). Anti-VEGF agents havedemonstrated efficacy in treating CNV in neovascular AMD (5, 6)but have limited efficacy and potential side effects (7, 8).Recent studies have revealed important roles for microRNAs
(miRNAs) in cardiovascular diseases and other disorders (9).miRNAs are small noncoding RNAs that negatively regulate geneexpression by inducing mRNA degradation or inhibiting trans-lation through binding to the 3′ untranslated region (3′UTR) oftarget mRNAs (10). Often, miRNAs modulate broad collectionsof mRNAs encoding multiple components of complex biologicalpathways. Several miRNAs have been implicated in angiogenesis(11, 12). A group of miRNAs has also been shown to be sub-stantially decreased in a laser-induced CNV model (13).The miR-23∼27∼24 clusters are highly expressed in ECs (14–
17). Two miR-23∼27∼24 clusters exist in the vertebrate genome:an intergenic miR-23a∼27a∼24–2 cluster and an intronic miR-23b∼27b∼24–1 cluster. Members of these clusters are involved incell cycle control, proliferation, and differentiation of various celltypes (18). Here, we show that inhibition of miR-23/27 impairsangiogenesis in vitro and postnatal retinal vascular developmentin vivo. Moreover, silencing of miR-23/27 suppresses laser-in-duced CNV in mice. The proangiogenic functions of miR-23/27correlate with the repression of Sprouty2 and Sema6A, whichnegatively regulate angiogenic signaling.
ResultsStructure and Expression Pattern of miR-23∼27∼24 Cluster Members.The mouse miR-23a∼27a∼24–2 cluster is intergenic on chro-
mosome 8, and the miR-23b∼27b∼24–1 cluster is located in in-tron 4 of an alanine aminopeptidase gene on chromosome 13(Fig. 1A). The miR-23a∼27a∼24–2 cluster encodes a primarymiRNA (pri-miRNA) transcript composed of three miRNAs:miR-23a, miR-27a, and miR-24–2, and the miR-23b∼27b∼24–1cluster encodes a pri-miRNA transcript containing miR-23b,miR-27b, and miR-24–1 (Fig. S1A). The mature miRNA se-quences of miR-23a/b, miR-27a/b, and miR-24 are conservedamong vertebrate species (Fig. S1B). miR-23a and miR-27adiffer by only one nucleotide near their 3′ ends compared withtheir paralogs miR-23b and miR-27b, whereas the sequence ofmiR-24–1 and miR-24–2 is the same (Fig. S1C). miR-23a andmiR-23b share most, if not all, predicted target genes by Tar-getScan and DIANA Lab, as do miR-27a and miR-27b (19, 20).Northern-blot analyses revealed that miR-23a/b, miR-27a/b,
and miR-24 are expressed at the highest levels in the lung andheart, which are highly vascularized tissues (Fig. 1B). Their ex-pression is also detectable in other organs, including the eye. Real-time PCR confirmed the enrichment of the mature miRNAs ofthese two clusters in the lung and heart (Fig. S1D). Real-timePCR was further performed to determine the expression of miR-23∼27∼24 cluster members in different cell types. Our resultsshowed that the expression of all miR-23∼27∼24 cluster membersis enriched in ECs compared to the other cell types tested (Fig.1C), consistent with previous reports (14–21) and a recent reportthat expression of the miR-23b∼27b∼24-1 host gene is enrichedin ECs in vivo (21). Taken together, our results indicate thatmiR-23∼27∼24 cluster members are enriched in ECs and highlyvascularized tissues, suggesting a potential role in EC function.
Modulation of Sprouting Angiogenesis by miR-23 and miR-27 in Vitro.To study the EC function of miR-23∼27∼24 cluster members invitro, human umbilical vein EC line (HUVECs) were transfectedwith locked nucleic acid (LNA)-modified anti-miRs of miR-23a/b,miR-27a/b, or a scramble control and tested for EC network for-mation on Matrigel. More than 90% knockdown of miR-23a/b ormiR-27a/b expression was achieved by LNA-anti-miR-23a/b oranti-miR-27a/b transfection, respectively, indicating the efficiencyand specificity of miRNAknockdown by LNAanti-miRs (Fig. 2A).Hereafter, miR-23a/b andmiR-27a/b will be referred to as miR-23and miR-27. Of note, there was compensatory up-regulation ofmiR-23 or miR-27 when miR-27 or miR-23 were knocked down.When cultured onMatrigel, ECs form a primary vascular network.Knockdown of miR-27, and to a lesser extent miR-23, impairedthe formation of capillary-like structures in HUVECs cultured onMatrigel, as quantified by the reduced branching points uponmiR-27 or miR-23 inhibition (Fig. 2 B and C). These resultssuggest that miR-27 and miR-23 are required for proper capillarytube formation in vitro.
Author contributions: E.N.O. and S.W. designed research; Q.Z., R.G., R.U.-V., and X.L.performed research; and E.N.O. and S.W. wrote the paper.
The authors declare no conflict of interest.1To whom correspondence may be addressed. E-mail: [email protected] [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1105254108/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1105254108 PNAS | May 17, 2011 | vol. 108 | no. 20 | 8287–8292
CELL
BIOLO
GY
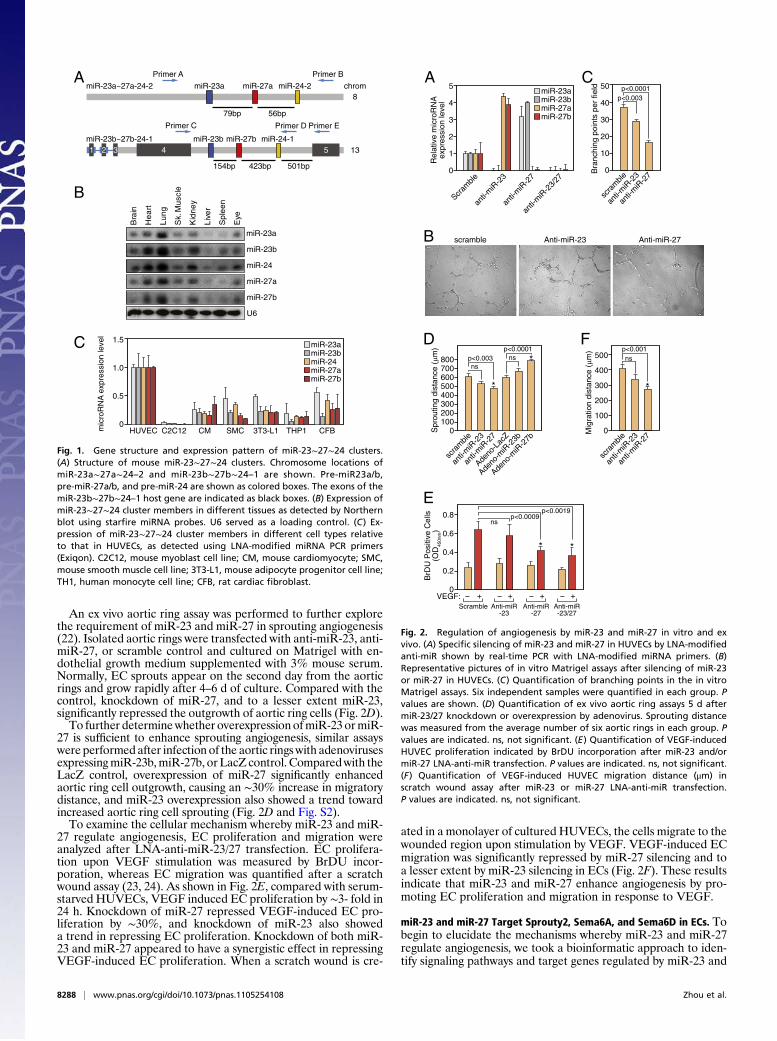
An ex vivo aortic ring assay was performed to further explorethe requirement of miR-23 and miR-27 in sprouting angiogenesis(22). Isolated aortic rings were transfected with anti-miR-23, anti-miR-27, or scramble control and cultured on Matrigel with en-dothelial growth medium supplemented with 3% mouse serum.Normally, EC sprouts appear on the second day from the aorticrings and grow rapidly after 4–6 d of culture. Compared with thecontrol, knockdown of miR-27, and to a lesser extent miR-23,significantly repressed the outgrowth of aortic ring cells (Fig. 2D).To further determinewhether overexpression ofmiR-23 ormiR-
27 is sufficient to enhance sprouting angiogenesis, similar assayswere performed after infection of the aortic rings with adenovirusesexpressingmiR-23b,miR-27b, or LacZ control. Comparedwith theLacZ control, overexpression of miR-27 significantly enhancedaortic ring cell outgrowth, causing an ∼30% increase in migratorydistance, and miR-23 overexpression also showed a trend towardincreased aortic ring cell sprouting (Fig. 2D and Fig. S2).To examine the cellular mechanism whereby miR-23 and miR-
27 regulate angiogenesis, EC proliferation and migration wereanalyzed after LNA-anti-miR-23/27 transfection. EC prolifera-tion upon VEGF stimulation was measured by BrDU incor-poration, whereas EC migration was quantified after a scratchwound assay (23, 24). As shown in Fig. 2E, compared with serum-starved HUVECs, VEGF induced EC proliferation by ∼3- fold in24 h. Knockdown of miR-27 repressed VEGF-induced EC pro-liferation by ∼30%, and knockdown of miR-23 also showeda trend in repressing EC proliferation. Knockdown of both miR-23 and miR-27 appeared to have a synergistic effect in repressingVEGF-induced EC proliferation. When a scratch wound is cre-
ated in a monolayer of cultured HUVECs, the cells migrate to thewounded region upon stimulation by VEGF. VEGF-induced ECmigration was significantly repressed by miR-27 silencing and toa lesser extent by miR-23 silencing in ECs (Fig. 2F). These resultsindicate that miR-23 and miR-27 enhance angiogenesis by pro-moting EC proliferation and migration in response to VEGF.
miR-23 and miR-27 Target Sprouty2, Sema6A, and Sema6D in ECs. Tobegin to elucidate the mechanisms whereby miR-23 and miR-27regulate angiogenesis, we took a bioinformatic approach to iden-tify signaling pathways and target genes regulated by miR-23 and
AmiR-27amiR-23a
79bp 56bp
miR-24-2
Primer A Primer B
4miR-27bmiR-23b
154bp
miR-24-1
423bp 501bp
Primer C Primer D Primer E
5
miR-23a~27a-24-2
miR-23b~27b-24-113
chrom8
1 2 3
B
Bra
in
Hea
rt
Lung
Eye
Sk.
Mus
cle
Live
r
Kid
ney
Spl
een
miR-23b
miR-24
miR-27b
U6
miR-27a
miR-23a
C 1.5
0HUVEC 3T3-L1 CFBCM SMCC2C12 THP1
0.5
1.0
mic
roR
NA
exp
ress
ion
leve
l
miR-23amiR-23bmiR-24miR-27amiR-27b
Fig. 1. Gene structure and expression pattern of miR-23∼27∼24 clusters.(A) Structure of mouse miR-23∼27∼24 clusters. Chromosome locations ofmiR-23a∼27a∼24–2 and miR-23b∼27b∼24–1 are shown. Pre-miR23a/b,pre-miR-27a/b, and pre-miR-24 are shown as colored boxes. The exons of themiR-23b∼27b∼24–1 host gene are indicated as black boxes. (B) Expression ofmiR-23∼27∼24 cluster members in different tissues as detected by Northernblot using starfire miRNA probes. U6 served as a loading control. (C) Ex-pression of miR-23∼27∼24 cluster members in different cell types relativeto that in HUVECs, as detected using LNA-modified miRNA PCR primers(Exiqon). C2C12, mouse myoblast cell line; CM, mouse cardiomyocyte; SMC,mouse smooth muscle cell line; 3T3-L1, mouse adipocyte progenitor cell line;TH1, human monocyte cell line; CFB, rat cardiac fibroblast.
A 5
0
Scram
ble
anti-
miR
-27
anti-
miR
-23/
27
anti-
miR
-23
4
3
2
1
Rel
ativ
e m
icro
RN
Aex
pres
sion
leve
l
miR-23amiR-23bmiR-27amiR-27b
scramble Anti-miR-23 Anti-miR-27B
C50
0
scra
mble
anti-
miR
-27
anti-
miR
-23
p<0.0001p<0.003
Bra
nchi
ng p
oint
s pe
r fie
ld
30
20
10
40
D800
0Spr
outin
g di
stan
ce (µm
)
500600700
400300200100
scra
mble
anti-
miR
-27
anti-
miR
-23
Adeno
-Lac
Z
p<0.003ns
p<0.0001ns
Adeno
-miR
-23b
Adeno
-miR
-27b
*
*
E0.8
0VEGF: +
p<0.0009ns
Scramble
+Anti-miR
-23
+Anti-miR
-27
+Anti-miR-23/27
p<0.0019
BrD
U P
ositi
ve C
ells
(OD
450n
m) 0.6
0.4
0.2
* *
F500
0
scra
mble
anti-
miR
-27
anti-
miR
-23
p<0.001ns
Mig
ratio
n di
stan
ce (µm
)
300
200
100
400
*
− − − −
Fig. 2. Regulation of angiogenesis by miR-23 and miR-27 in vitro and exvivo. (A) Specific silencing of miR-23 and miR-27 in HUVECs by LNA-modifiedanti-miR shown by real-time PCR with LNA-modified miRNA primers. (B)Representative pictures of in vitro Matrigel assays after silencing of miR-23or miR-27 in HUVECs. (C) Quantification of branching points in the in vitroMatrigel assays. Six independent samples were quantified in each group. Pvalues are shown. (D) Quantification of ex vivo aortic ring assays 5 d aftermiR-23/27 knockdown or overexpression by adenovirus. Sprouting distancewas measured from the average number of six aortic rings in each group. Pvalues are indicated. ns, not significant. (E) Quantification of VEGF-inducedHUVEC proliferation indicated by BrDU incorporation after miR-23 and/ormiR-27 LNA-anti-miR transfection. P values are indicated. ns, not significant.(F) Quantification of VEGF-induced HUVEC migration distance (μm) inscratch wound assay after miR-23 or miR-27 LNA-anti-miR transfection.P values are indicated. ns, not significant.
8288 | www.pnas.org/cgi/doi/10.1073/pnas.1105254108 Zhou et al.
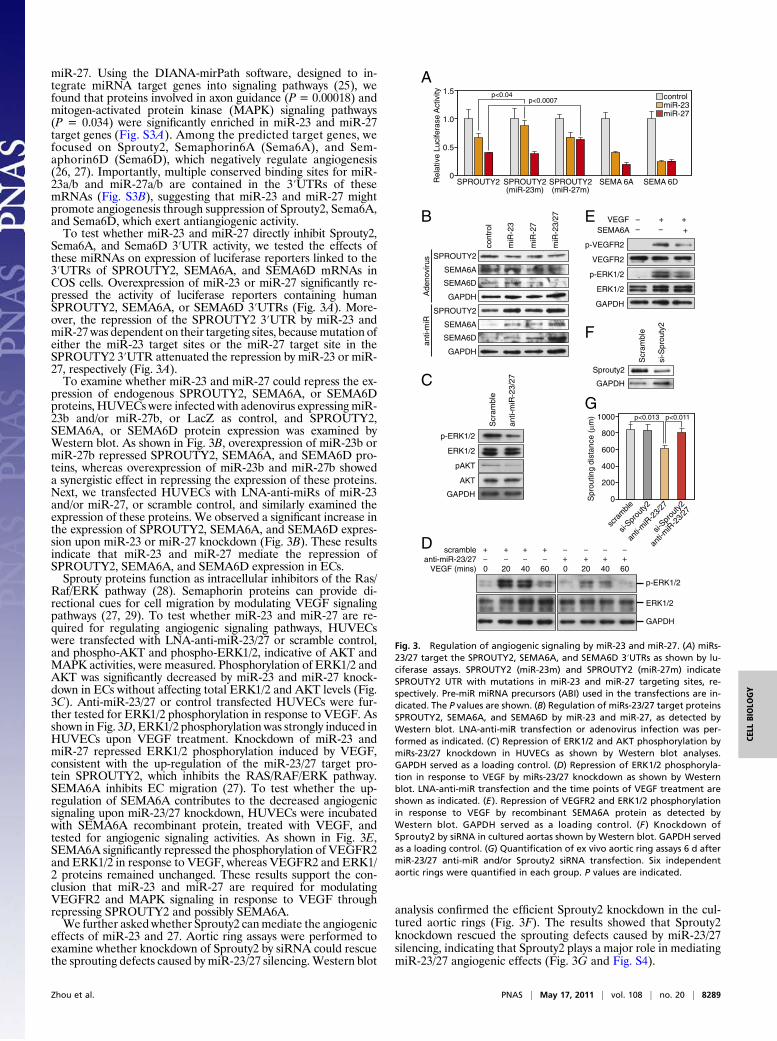
miR-27. Using the DIANA-mirPath software, designed to in-tegrate miRNA target genes into signaling pathways (25), wefound that proteins involved in axon guidance (P = 0.00018) andmitogen-activated protein kinase (MAPK) signaling pathways(P = 0.034) were significantly enriched in miR-23 and miR-27target genes (Fig. S3A). Among the predicted target genes, wefocused on Sprouty2, Semaphorin6A (Sema6A), and Sem-aphorin6D (Sema6D), which negatively regulate angiogenesis(26, 27). Importantly, multiple conserved binding sites for miR-23a/b and miR-27a/b are contained in the 3′UTRs of thesemRNAs (Fig. S3B), suggesting that miR-23 and miR-27 mightpromote angiogenesis through suppression of Sprouty2, Sema6A,and Sema6D, which exert antiangiogenic activity.To test whether miR-23 and miR-27 directly inhibit Sprouty2,
Sema6A, and Sema6D 3′UTR activity, we tested the effects ofthese miRNAs on expression of luciferase reporters linked to the3′UTRs of SPROUTY2, SEMA6A, and SEMA6D mRNAs inCOS cells. Overexpression of miR-23 or miR-27 significantly re-pressed the activity of luciferase reporters containing humanSPROUTY2, SEMA6A, or SEMA6D 3′UTRs (Fig. 3A). More-over, the repression of the SPROUTY2 3′UTR by miR-23 andmiR-27 was dependent on their targeting sites, becausemutation ofeither the miR-23 target sites or the miR-27 target site in theSPROUTY2 3′UTR attenuated the repression by miR-23 or miR-27, respectively (Fig. 3A).To examine whether miR-23 and miR-27 could repress the ex-
pression of endogenous SPROUTY2, SEMA6A, or SEMA6Dproteins, HUVECs were infected with adenovirus expressing miR-23b and/or miR-27b, or LacZ as control, and SPROUTY2,SEMA6A, or SEMA6D protein expression was examined byWestern blot. As shown in Fig. 3B, overexpression of miR-23b ormiR-27b repressed SPROUTY2, SEMA6A, and SEMA6D pro-teins, whereas overexpression of miR-23b and miR-27b showeda synergistic effect in repressing the expression of these proteins.Next, we transfected HUVECs with LNA-anti-miRs of miR-23and/or miR-27, or scramble control, and similarly examined theexpression of these proteins. We observed a significant increase inthe expression of SPROUTY2, SEMA6A, and SEMA6D expres-sion upon miR-23 or miR-27 knockdown (Fig. 3B). These resultsindicate that miR-23 and miR-27 mediate the repression ofSPROUTY2, SEMA6A, and SEMA6D expression in ECs.Sprouty proteins function as intracellular inhibitors of the Ras/
Raf/ERK pathway (28). Semaphorin proteins can provide di-rectional cues for cell migration by modulating VEGF signalingpathways (27, 29). To test whether miR-23 and miR-27 are re-quired for regulating angiogenic signaling pathways, HUVECswere transfected with LNA-anti-miR-23/27 or scramble control,and phospho-AKT and phospho-ERK1/2, indicative of AKT andMAPK activities, were measured. Phosphorylation of ERK1/2 andAKT was significantly decreased by miR-23 and miR-27 knock-down in ECs without affecting total ERK1/2 and AKT levels (Fig.3C). Anti-miR-23/27 or control transfected HUVECs were fur-ther tested for ERK1/2 phosphorylation in response to VEGF. Asshown in Fig. 3D, ERK1/2 phosphorylation was strongly induced inHUVECs upon VEGF treatment. Knockdown of miR-23 andmiR-27 repressed ERK1/2 phosphorylation induced by VEGF,consistent with the up-regulation of the miR-23/27 target pro-tein SPROUTY2, which inhibits the RAS/RAF/ERK pathway.SEMA6A inhibits EC migration (27). To test whether the up-regulation of SEMA6A contributes to the decreased angiogenicsignaling upon miR-23/27 knockdown, HUVECs were incubatedwith SEMA6A recombinant protein, treated with VEGF, andtested for angiogenic signaling activities. As shown in Fig. 3E,SEMA6A significantly repressed the phosphorylation of VEGFR2and ERK1/2 in response to VEGF, whereas VEGFR2 and ERK1/2 proteins remained unchanged. These results support the con-clusion that miR-23 and miR-27 are required for modulatingVEGFR2 and MAPK signaling in response to VEGF throughrepressing SPROUTY2 and possibly SEMA6A.We further asked whether Sprouty2 canmediate the angiogenic
effects of miR-23 and 27. Aortic ring assays were performed toexamine whether knockdown of Sprouty2 by siRNA could rescuethe sprouting defects caused by miR-23/27 silencing.Western blot
analysis confirmed the efficient Sprouty2 knockdown in the cul-tured aortic rings (Fig. 3F). The results showed that Sprouty2knockdown rescued the sprouting defects caused by miR-23/27silencing, indicating that Sprouty2 plays a major role in mediatingmiR-23/27 angiogenic effects (Fig. 3G and Fig. S4).
G1000
0
anti-
miR
-23/
27
si-Spr
outy2
anti-
miR
-23/
27
si-Spr
outy2
scra
mble
200
600
400
800
Spr
outin
g di
stan
ce (µm
) p<0.013 p<0.011
F
GAPDH
Sprouty2
elbmarc
S
2yt uor pS-i s
GAPDH
ERK1/2
p-ERK1/2
VEGFR2
p-VEGFR2
VEGF + +SEMA6A +
E
D
ERK1/2
GAPDH
p-ERK1/2
VEGF (mins) 0 20 40 60anti-miR-23/27
scramble + + + +
0 20 40 60+ ++ +
C
GAPDH
AKT
pAKT
ERK1/2
p-ERK1/2
elbmarc
S
72/ 32-Ri
m-it na
B
Ade
novi
rus
cont
rol
miR
-23
miR
-27
miR
-23/
27
GAPDH
SEMA6D
SEMA6A
SPROUTY2
GAPDH
SEMA6D
SEMA6A
SPROUTY2
anti-
miR
A1.5
0
0.5
1.0
Rel
ativ
e Lu
cife
rase
Act
ivity
SPROUTY2(miR-23m)
SPROUTY2(miR-27m)
SEMA 6A SEMA 6DSPROUTY2
controlmiR-23miR-27
p<0.04p<0.0007
−− −
− − − −− − − −
Fig. 3. Regulation of angiogenic signaling by miR-23 and miR-27. (A) miRs-23/27 target the SPROUTY2, SEMA6A, and SEMA6D 3′UTRs as shown by lu-ciferase assays. SPROUTY2 (miR-23m) and SPROUTY2 (miR-27m) indicateSPROUTY2 UTR with mutations in miR-23 and miR-27 targeting sites, re-spectively. Pre-miR miRNA precursors (ABI) used in the transfections are in-dicated. The P values are shown. (B) Regulation of miRs-23/27 target proteinsSPROUTY2, SEMA6A, and SEMA6D by miR-23 and miR-27, as detected byWestern blot. LNA-anti-miR transfection or adenovirus infection was per-formed as indicated. (C) Repression of ERK1/2 and AKT phosphorylation bymiRs-23/27 knockdown in HUVECs as shown by Western blot analyses.GAPDH served as a loading control. (D) Repression of ERK1/2 phosphoryla-tion in response to VEGF by miRs-23/27 knockdown as shown by Westernblot. LNA-anti-miR transfection and the time points of VEGF treatment areshown as indicated. (E). Repression of VEGFR2 and ERK1/2 phosphorylationin response to VEGF by recombinant SEMA6A protein as detected byWestern blot. GAPDH served as a loading control. (F ) Knockdown ofSprouty2 by siRNA in cultured aortas shown by Western blot. GAPDH servedas a loading control. (G) Quantification of ex vivo aortic ring assays 6 d aftermiR-23/27 anti-miR and/or Sprouty2 siRNA transfection. Six independentaortic rings were quantified in each group. P values are indicated.
Zhou et al. PNAS | May 17, 2011 | vol. 108 | no. 20 | 8289
CELL
BIOLO
GY

Regulation of Retinal Vascular Development by miR-23 and miR-27 inVivo. To further address the role of miR-23 and miR-27 in an-giogenesis in vivo, we performed miRNA loss-of-function studiesin neonatal mouse retinas, a well-established model of angio-genesis (30). Vascularization of the outer retina commences atpostnatal d 0 (P0) from the central retinal artery, and the ECssprout to reach the peripheral region at about P8. Anti-miRstargeting miR-23 and miR-27, or scramble control, were injectedintravitreously into the eye at P2, and retinas were isolated at P6for RNA, protein, and flat mount staining of the developingretinal vasculature (Fig. 4A). Subretinal injection of LNA-anti–miR-23/27 resulted in >90% reduction in retinal miR-23/27 levelscompared with the controls (Fig. 4B). Of note, compensatory up-regulation of miR-24 was observed (Fig. 4B). Thus, LNA-anti-miRs displayed high efficacy and specificity in the retina in vivo.To examine the effect of miR-23/27 knockdown on postnatal
retinal vascular development, P6 retinal flat mounts were stainedwith intercellular adhesion molecule-2 (ICAM-2) to visualize thevessels. Scramble LNAs did not affect retinal vascular de-velopment compared with the noninjection controls, based onICAM-2 staining. However, in anti-miR-23/27-injected retinas,there was an ∼10% decrease in sprouting distance and an ∼20%decrease in vascular coverage at 4 d after anti-miR-23/27 injectioncompared with the controls (Fig. 4 C and D). At higher magnifi-cation, fewer newly formed vessel sprouts were observed in theanti-miR-23/27-injected retinas compared with the controls (Fig.4C, c and d). During retinal vascular development, ECs sproutalong glial fibrillary acidic protein (GFAP)-positive astrocytes.GFAP staining indicated that astrocyte coverage was not affectedby miR-23/27 knockdown (Fig. 4C, e and f), suggesting that theretinal EC sprouting phenotype evoked by miR-23/27 knockdownis not secondary to potential astrocyte defects. Moreover, consis-tent with our in vitro results, miR-23 and miR-27 target proteins,Sprouty2, Sema6A, and Sema6D, were significantly up-regulatedupon miR-23/27 knockdown in the retinas (Fig. 4E). Taken to-gether, our results indicate that miR-23 and miR-27 modulateretinal vascular development in vivo.
Requirement of miR-23 and miR-27 in CNV in Vivo. The angiogenicdefects resulting from miR-23 and miR-27 knockdown in vitroand in vivo suggested that miR-23 and miR-27 might play animportant role in neovascularization in response to injury. Weadopted a laser-induced CNV mouse model, the most reliableCNV animal model, to test the role of miR-23 and miR-27 inCNV (31). We first examined the expression of miR-23∼27∼24clusters in the retina/choroid after laser injury. Adult C57BL/6mice were subjected to rupture of Bruch’s membrane in sixlocations by laser photocoagulation (140 mV, 100 ms, 100 μm)(32). As demonstrated by real-time PCR, miR-23a, miR-27a, andmiR-24 levels were significantly up-regulated, and miR-23b andmiR-27b expression was also increased in the retinal/choroidalregion at 1 wk after laser injury (Fig. 5A).To directly test the requirement of miR-23/27 in CNV, LNA-
anti-miRs targeting miR-23 and miR-27, or scramble control,were intravitreously injected to knockdown miR-23 and miR-27in the eye immediately following laser injury in three locations(Fig. 5B). A secondary injection was performed on the followingday to ensure efficient knockdown. Eyes were collected at 2 wkafter laser injury for ICAM-2 staining and confocal imaging (33).As shown in Fig. 5C, >90% knockdown efficiency of miR-23 andmiR-27 in the retina/choroid/sclera was achieved by anti-miR in-jection compared with the controls. Of note, we observed an up-regulation of miR-23∼27∼24 members after scramble control in-jection compared with noninjection controls, possibly reflectingthe increased inflammatory response due to injection. We testedanti-platelet/endothelial cell adhesion molecule (PECAM)-1 an-tibody, isolectin-B4, and FITC-dextran staining besides anti-ICAM-2 staining and found ICAM-2 gave the best staining forCNV, consistent with a recent report (33). Quantification of theCNV area, as shown by ICAM-2 staining, revealed that silencing ofmiR-23 and miR-27 repressed the CNV area by >50% comparedwith scramble control (Fig. 5 D and F). The repression of CNV bymiR-23/27 was also confirmed by ICAM-2 staining of the lesion
sections (Fig. 5E). Compared with the noninjection controls, weobserved a mild increase in CNV in scramble-injected samples,possibly due to increased CNV induced by miR∼23∼27∼24 up-regulation (Fig. S5B). Taken together, our data indicates that miR-23 and miR-27 are required for laser-induced CNV in vivo.
DiscussionThe findings of this study reveal an important role for miR-23 andmiR-27 in angiogenesis and CNV (Fig. 6). We provide evidencethat silencing of miR-23 and miR-27 represses sprouting angio-genesis in vitro and in vivo, as well as CNV in a laser-induced CNVmouse model. The proangiogenic actions of miR-23 and miR-27correlate with the repression of their target mRNAs encodingSprouty2 and Sema6A, which negatively regulate MAPK andVEGFR2 signaling in response to angiogenic factors. Thus, in theabsence of miR-23 and miR-27, Sprouty2 and Sema6A proteinsare up-regulated, with consequent dampening of MAPK andVEGFR2 signaling. Conversely, overexpression of miR-23 andmiR-27 represses these targets, relieving their repressive influenceon these pathways.
AP0 P2 P6
Injection RNA, Protein,Retinal Flatmount
B3
0anti-miR-23/27Scramble
1
2
mic
roR
NA
exp
ress
ion
leve
l
miR-23amiR-23bmiR-24miR-27amiR-27b
C a
fd
ec
b
Con
trol
anti-
miR
-23/
27
D1800
10001200
16001400
800
0200
600400
Spr
outin
g D
ista
nce
(µm
)
anti-
miR
-23/
27
Scram
ble
p<0.0019
**
30
0
20
10
% o
f vas
cula
r co
vera
gean
ti-m
iR-2
3/27
Scram
ble
p<0.0049
*
E
GAPDH
Sema6D
Sema6A
Sprouty2
elbmarc
S
72/ 32-Ri
m-it na
Fig. 4. Regulation of retinal vascular development by miR-23 and miR-27.(A) Experimental setup for retinal injections. LNA-anti-miRs were injected atP2 and retinal samples are collected at P6 for RNA, protein analyses, andflatmount staining. (B) Detection of miR-23∼27∼24 family members in theretina after LNA anti-miR treatment by real-time PCR with LNA-modifiedmiRNA primers. (C) Vasculature of the retina at P6 after LNA anti-miRtreatment at P2, as visualized by ICAM-2 (green) and GFAP (red) staining inflat mount preparation. Anti-scramble (Upper) and anti-miR-23/27 (Lower).Scale bar (Left): 100 μm. Scale bar (Middle and Right): 50 μm. Arrows point tothe new retinal vascular sprouts. (D) Quantification of sprouting distance(μm) and vascular coverage of the retinal vasculature from 12 miR-23/27 anti-miR treated retinas compared with 12 scramble controls. P values are in-dicated. (E) Western blot analyses showing the up-regulation of miRs-23/27target proteins Sprouty2, Sema6A, and Sema6D upon miRs-23/27 knock-down in P6 retinas.
8290 | www.pnas.org/cgi/doi/10.1073/pnas.1105254108 Zhou et al.
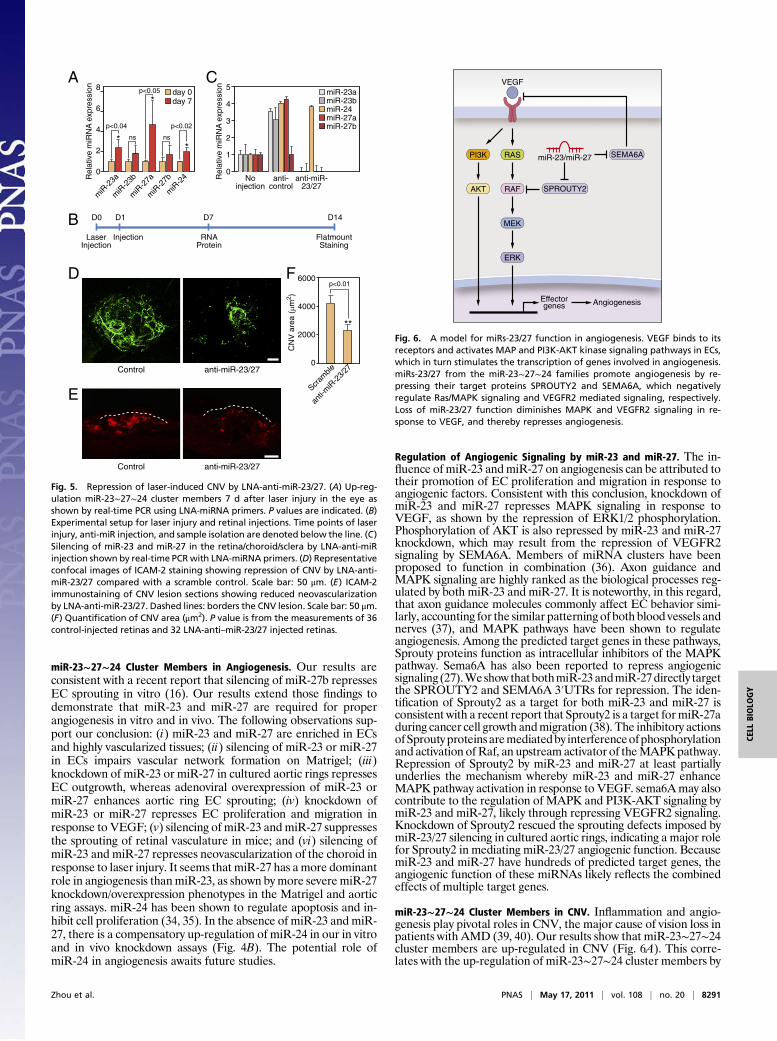
miR-23∼27∼24 Cluster Members in Angiogenesis. Our results areconsistent with a recent report that silencing of miR-27b repressesEC sprouting in vitro (16). Our results extend those findings todemonstrate that miR-23 and miR-27 are required for properangiogenesis in vitro and in vivo. The following observations sup-port our conclusion: (i) miR-23 and miR-27 are enriched in ECsand highly vascularized tissues; (ii) silencing of miR-23 or miR-27in ECs impairs vascular network formation on Matrigel; (iii)knockdown of miR-23 or miR-27 in cultured aortic rings repressesEC outgrowth, whereas adenoviral overexpression of miR-23 ormiR-27 enhances aortic ring EC sprouting; (iv) knockdown ofmiR-23 or miR-27 represses EC proliferation and migration inresponse to VEGF; (v) silencing of miR-23 and miR-27 suppressesthe sprouting of retinal vasculature in mice; and (vi) silencing ofmiR-23 and miR-27 represses neovascularization of the choroid inresponse to laser injury. It seems that miR-27 has a more dominantrole in angiogenesis thanmiR-23, as shown bymore severe miR-27knockdown/overexpression phenotypes in the Matrigel and aorticring assays. miR-24 has been shown to regulate apoptosis and in-hibit cell proliferation (34, 35). In the absence of miR-23 and miR-27, there is a compensatory up-regulation of miR-24 in our in vitroand in vivo knockdown assays (Fig. 4B). The potential role ofmiR-24 in angiogenesis awaits future studies.
Regulation of Angiogenic Signaling by miR-23 and miR-27. The in-fluence of miR-23 andmiR-27 on angiogenesis can be attributed totheir promotion of EC proliferation and migration in response toangiogenic factors. Consistent with this conclusion, knockdown ofmiR-23 and miR-27 represses MAPK signaling in response toVEGF, as shown by the repression of ERK1/2 phosphorylation.Phosphorylation of AKT is also repressed by miR-23 and miR-27knockdown, which may result from the repression of VEGFR2signaling by SEMA6A. Members of miRNA clusters have beenproposed to function in combination (36). Axon guidance andMAPK signaling are highly ranked as the biological processes reg-ulated by both miR-23 and miR-27. It is noteworthy, in this regard,that axon guidance molecules commonly affect EC behavior simi-larly, accounting for the similar patterning of both blood vessels andnerves (37), and MAPK pathways have been shown to regulateangiogenesis. Among the predicted target genes in these pathways,Sprouty proteins function as intracellular inhibitors of the MAPKpathway. Sema6A has also been reported to repress angiogenicsignaling (27).Weshow that bothmiR-23andmiR-27directly targetthe SPROUTY2 and SEMA6A 3′UTRs for repression. The iden-tification of Sprouty2 as a target for both miR-23 and miR-27 isconsistent with a recent report that Sprouty2 is a target formiR-27aduring cancer cell growth andmigration (38). The inhibitory actionsofSproutyproteinsaremediatedby interferenceofphosphorylationand activation ofRaf, an upstream activator of theMAPKpathway.Repression of Sprouty2 by miR-23 and miR-27 at least partiallyunderlies the mechanism whereby miR-23 and miR-27 enhanceMAPK pathway activation in response to VEGF. sema6Amay alsocontribute to the regulation of MAPK and PI3K-AKT signaling bymiR-23 and miR-27, likely through repressing VEGFR2 signaling.Knockdown of Sprouty2 rescued the sprouting defects imposed bymiR-23/27 silencing in cultured aortic rings, indicating a major rolefor Sprouty2 in mediating miR-23/27 angiogenic function. BecausemiR-23 and miR-27 have hundreds of predicted target genes, theangiogenic function of these miRNAs likely reflects the combinedeffects of multiple target genes.
miR-23∼27∼24 Cluster Members in CNV. Inflammation and angio-genesis play pivotal roles in CNV, the major cause of vision loss inpatients with AMD (39, 40). Our results show that miR-23∼27∼24cluster members are up-regulated in CNV (Fig. 6A). This corre-lates with the up-regulation of miR-23∼27∼24 cluster members by
A8
0
miR
-23a
miR
-27a
miR
-24
miR
-23b
miR
-27b
2
6
4
Rel
ativ
e m
iRN
A e
xpre
ssio
nday 0day 7
p<0.04
ns ns*p<0.02
*
*p<0.05
B D0 D1 D7 D14
LaserInjection
Injection RNAProtein
FlatmountStaining
C5
0No
injectionanti-miR-
23/27anti-
control
2
3
4
Rel
ativ
e m
iRN
A e
xpre
ssio
n
1
miR-23amiR-23bmiR-24miR-27amiR-27b
D
Control anti-miR-23/27
E
Control anti-miR-23/27
F 6000
0
4000
2000
CN
V a
rea
(µm
2 )
anti-
miR
-23/
27
Scram
ble
p<0.01
**
Fig. 5. Repression of laser-induced CNV by LNA-anti-miR-23/27. (A) Up-reg-ulation miR-23∼27∼24 cluster members 7 d after laser injury in the eye asshown by real-time PCR using LNA-miRNA primers. P values are indicated. (B)Experimental setup for laser injury and retinal injections. Time points of laserinjury, anti-miR injection, and sample isolation are denoted below the line. (C)Silencing of miR-23 and miR-27 in the retina/choroid/sclera by LNA-anti-miRinjection shown by real-time PCRwith LNA-miRNA primers. (D) Representativeconfocal images of ICAM-2 staining showing repression of CNV by LNA-anti-miR-23/27 compared with a scramble control. Scale bar: 50 μm. (E) ICAM-2immunostaining of CNV lesion sections showing reduced neovascularizationby LNA-anti-miR-23/27. Dashed lines: borders the CNV lesion. Scale bar: 50 μm.(F) Quantification of CNV area (μm2). P value is from the measurements of 36control-injected retinas and 32 LNA-anti–miR-23/27 injected retinas.
MEK
ERK
RAF SPROUTY2
SEMA6ARASPI3K
VEGF
AKT
miR-23/miR-27
Effectorgenes Angiogenesis
Fig. 6. A model for miRs-23/27 function in angiogenesis. VEGF binds to itsreceptors and activates MAP and PI3K-AKT kinase signaling pathways in ECs,which in turn stimulates the transcription of genes involved in angiogenesis.miRs-23/27 from the miR-23∼27∼24 families promote angiogenesis by re-pressing their target proteins SPROUTY2 and SEMA6A, which negativelyregulate Ras/MAPK signaling and VEGFR2 mediated signaling, respectively.Loss of miR-23/27 function diminishes MAPK and VEGFR2 signaling in re-sponse to VEGF, and thereby represses angiogenesis.
Zhou et al. PNAS | May 17, 2011 | vol. 108 | no. 20 | 8291
CELL
BIOLO
GY

proinflammatory stimuli (Fig. S5A) (41, 42), suggesting a role formiR-23∼27∼24 cluster members in linking inflammation to an-giogenesis in CNV. Indeed, miR-27b was recently shown to con-tribute to LPS-mediated inflammation by targeting PPAR-γ (41).Our finding that silencing of miR-23 and miR-27 represses laser-induced CNV indicates that miR-23∼27∼24 cluster members in-deed play a causative role in CNV.However, howmiR-23, -27, and-24 regulate inflammation in CNV is yet to be investigated. Be-cause miR-23∼27∼24 clusters are highly conserved from mice tohumans, therapeutic manipulation of miR-23/27 represents a po-tential strategy in treating CNV in patients with neovascular AMDand other vascular diseases. The identification of miR-23 andmiR-27 as important regulators of MAPK activation also suggestsroles for these miRNAs in cancer.
Materials and MethodsLNA-Anti-miRs, Pre-miR Precursors, siRNAs, and miRNA expressing adeno-viruses. LNA-anti-miRs for miR-23a/b, miR-27a/b, or scramble controls for invitro and in vivo studies were synthesized from Exiqon. SiRNAs for SPROUTY2were synthesized from Dharmacon. Sequences for SPROUTY2 siRNA, in vitroand in vivo LNA-anti-miRs, are described in SI Materials and Methods.Ambion pre-miR miRNA precursor of hsa-miR-23b, hsa-miR-27b, or negativecontrol was synthesized from Applied Biosystem. Adenovirus expressingmiR-23b, miR-27b, or lacZ was generated from mouse genomic DNAencoding miR-23b or miR-27 as described (24).
Cell Culture, Cell Proliferation, Scratch-Wound, In Vitro Matrigel, and AorticRing Sprouting Assay. HUVEC cell culture, cell proliferation, scratch-wound, in
vitro Matrigel assays, as well as aortic ring sprouting assays are described inSI Materials and Methods.
RNA, Western Blot Analysis, and Reporter Assay. RNA isolation, real-time RT-PCR, Western blot analysis, and reporter assays were performed according tostandard procedures. Detailed methods are outlined in SI Materials andMethods.
Neonatal Retinal Injection, Postnatal Retinal Angiogenesis, and Laser-InducedCNV. In vivo injection in the mouse retina was performed primarily as de-scribed (43). Laser photocoagulation was induced in 6- to 8-wk-old maleC57BL/6J mice as described (32). Detailed protocols are summarized in SIMaterials and Methods.
Institutional Compliance and Animal Care. All experiments using animals wereapproved by the Institutional Animal Care and Use Committee at Universityof Texas Southwestern Medical Center.
ACKNOWLEDGMENTS. We thank Jose Cabrera for graphics. We are grate-ful to C. Kuo, C. Lowenstein, and W. Sessa for comments on the manu-script. S.W. was supported by a Startup fund from the Department ofOphthalmology at UT Southwestern Medical Center, National Institutesof Health Grant EY020799, and an unrestricted grant from Researchto Prevent Blindness. E.N.O. was supported by grants from the NationalInstitutes of Health, the Donald W. Reynolds Center for Clinical Cardiovas-cular Research, The Robert A. Welch Foundation (Grant I-0025), theFoundation Leducq’s Transatlantic Network of Excellence in CardiovascularResearch Program, the American Heart Association, and the Jon HoldenDeHaan Foundation.
1. Distler JH, et al. (2003) Angiogenic and angiostatic factors in the molecular control ofangiogenesis. Q J Nucl Med 47:149–161.
2. Friedman E (2004) Update of the vascular model of AMD. Br J Ophthalmol 88:161–163.
3. Jager RD, Mieler WF, Miller JW (2008) Age-related macular degeneration. N Engl JMed 358:2606–2617.
4. Grisanti S, Tatar O (2008) The role of vascular endothelial growth factor and otherendogenous interplayers in age-related macular degeneration. Prog Retin Eye Res 27:372–390.
5. Brown DM, et al.; ANCHOR Study Group (2006) Ranibizumab versus verteporfin forneovascular age-related macular degeneration. N Engl J Med 355:1432–1444.
6. Rosenfeld PJ, et al.; MARINA Study Group (2006) Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 355:1419–1431.
7. Amrite A, Pugazhenthi V, Cheruvu N, Kompella U (2010) Delivery of celecoxib fortreating diseases of the eye: influence of pigment and diabetes. Expert Opin DrugDeliv 7:631–645.
8. Zachary I (2005) Neuroprotective role of vascular endothelial growth factor: signallingmechanisms, biological function, and therapeutic potential. Neurosignals 14:207–221.
9. Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology.Nature 469:336–342.
10. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell116:281–297.
11. Wang S, Olson EN (2009) AngiomiRs—key regulators of angiogenesis. Curr OpinGenet Dev 19:205–211.
12. Urbich C, Kuehbacher A, Dimmeler S (2008) Role of microRNAs in vascular diseases,inflammation, and angiogenesis. Cardiovasc Res 79:581–588.
13. Shen J, et al. (2008) MicroRNAs regulate ocular neovascularization. Mol Ther 16:1208–1216.
14. Poliseno L, et al. (2006) MicroRNAs modulate the angiogenic properties of HUVECs.Blood 108:3068–3071.
15. Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC (2007) Dicer dependentmicroRNAs regulate gene expression and functions in human endothelial cells. CircRes 100:1164–1173.
16. Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S (2007) Role of Dicer and Drosha forendothelial microRNA expression and angiogenesis. Circ Res 101:59–68.
17. Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ (2008) MicroRNA-126regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl AcadSci USA 105:1516–1521.
18. Chhabra R, Dubey R, Saini N (2010) Cooperative and individualistic functions of themicroRNAs in the miR-23a∼27a∼24-2 cluster and its implication in human diseases.Mol Cancer 9:232.
19. Friedman RC, Farh KK, Burge CB, Bartel DP (2009) Most mammalian mRNAs areconserved targets of microRNAs. Genome Res 19:92–105.
20. Maragkakis M, et al. (2009) DIANA-microT web server: Elucidating microRNA functionsthrough target prediction. Nucleic Acids Res 37(Web Server issue):W273–276.
21. Axton R, Wallis JA, Taylor H, Hanks M, Forrester LM (2008) Aminopeptidase Ocontains a functional nucleolar localization signal and is implicated in vascularbiology. J Cell Biochem 103:1171–1182.
22. Wang S, et al. (2008) The endothelial-specific microRNA miR-126 governs vascularintegrity and angiogenesis. Dev Cell 15:261–271.
23. Lee KS, et al. (2006) Troglitazone inhibits endothelial cell proliferation throughsuppression of casein kinase 2 activity. Biochem Biophys Res Commun 346:83–88.
24. Wang S, et al. (2008) Control of endothelial cell proliferation and migration by VEGFsignaling to histone deacetylase 7. Proc Natl Acad Sci USA 105:7738–7743.
25. Papadopoulos GL, Alexiou P, Maragkakis M, Reczko M, Hatzigeorgiou AG (2009)DIANA-mirPath: Integrating human andmouse microRNAs in pathways. Bioinformatics25:1991–1993.
26. Impagnatiello MA, et al. (2001) Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells.J Cell Biol 152:1087–1098.
27. Dhanabal M, et al. (2005) Recombinant semaphorin 6A-1 ectodomain inhibits in vivogrowth factor and tumor cell line-induced angiogenesis. Cancer Biol Ther 4:659–668.
28. Casci T, Vinós J, Freeman M (1999) Sprouty, an intracellular inhibitor of Ras signaling.Cell 96:655–665.
29. Toyofuku T, et al. (2004) Dual roles of Sema6D in cardiac morphogenesis throughregion-specific association of its receptor, Plexin-A1, with off-track and vascularendothelial growth factor receptor type 2. Genes Dev 18:435–447.
30. Stahl A, et al. (2010) The mouse retina as an angiogenesis model. Invest OphthalmolVis Sci 51:2813–2826.
31. Ryan SJ (1982) Subretinal neovascularization. Natural history of an experimentalmodel. Arch Ophthalmol 100:1804–1809.
32. Tobe T, et al. (1998) Targeted disruption of the FGF2 gene does not prevent choroidalneovascularization in a murine model. Am J Pathol 153:1641–1646.
33. Campa C, et al. (2008) Effects of an anti-VEGF-A monoclonal antibody on laser-induced choroidal neovascularization in mice: optimizing methods to quantifyvascular changes. Invest Ophthalmol Vis Sci 49:1178–1183.
34. Lal A, et al. (2009) miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and othercell-cycle genes via binding to “seedless” 3’UTR microRNA recognition elements. MolCell 35:610–625.
35. Walker JC, Harland RM (2009) microRNA-24a is required to repress apoptosis in thedeveloping neural retina. Genes Dev 23:1046–1051.
36. Yuan X, et al. (2009) Clustered microRNAs’ coordination in regulating protein-proteininteraction network. BMC Syst Biol 3:65.
37. Carmeliet P, Tessier-Lavigne M (2005) Common mechanisms of nerve and blood vesselwiring. Nature 436:193–200.
38. Ma Y, Yu S, Zhao W, Lu Z, Chen J (2010) miR-27a regulates the growth, colonyformation and migration of pancreatic cancer cells by targeting Sprouty2. Cancer Lett298:150–158.
39. BresslerSB(2009) Introduction:Understandingtheroleofangiogenesisandantiangiogenicagents in age-related macular degeneration.Ophthalmology 116(Suppl 10):S1–S7.
40. Augustin AJ, Kirchhof J (2009) Inflammation and the pathogenesis of age-relatedmacular degeneration. Expert Opin Ther Targets 13:641–651.
41. Jennewein C, von Knethen A, Schmid T, Brüne B (2010) MicroRNA-27b contributes tolipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma(PPARgamma) mRNA destabilization. J Biol Chem 285:11846–11853.
42. Zhou R, et al. (2009) NF-kappaB p65-dependent transactivation of miRNA genesfollowing Cryptosporidium parvum infection stimulates epithelial cell immuneresponses. PLoS Pathog 5:e1000681.
43. Matsuda T, Cepko CL (2004) Electroporation and RNA interference in the rodentretina in vivo and in vitro. Proc Natl Acad Sci USA 101:16–22.
8292 | www.pnas.org/cgi/doi/10.1073/pnas.1105254108 Zhou et al.

Supporting InformationZhou et al. 10.1073/pnas.1105254108Materials and MethodsLNA-Anti-miRs, Pre-miR Precursors, siRNAs, and miRNA ExpressingAdenoviruses. LNA-anti-miRs for miR-23a/b, miR-27a/b, orscramble controls for in vitro and in vivo studies were synthesizedfrom Exiqon. LNA-anti-miR-27a/b sequence for in vitro use is: 5′-GAACTTAGCCACTGTGAA-3′ (product no. 500150, batch no.604052). LNA-anti-miR-23a/b sequence for in vitro use is: 5′-AATCCCTGGCAATGTGAT-3′ (product no. 500150, batch no.604053). LNA-scramble control sequence for in vitro use is: 5′-GTGTAACACGTCTATACGCCCA-3′ (product no. 199004–00,batch no. 205157). LNA-anti-miR-27a/b sequence for in vivo useis: 5′-ACTTAGCCACTGTGA-3′ (product no. 500150, batch no.603226). LNA-anti-miR-23a/b sequence for in vivo use is: 5′-TC-CCTGGCAATGTGA-3′ (product no. 500150, batch no. 603227).LNA-scramble control sequence for in vivo use is: 5′-ACGTCTA-TACGCCCA-3′ (product no. 500150, batch no. 603228). LNA-anti-miRs were typically transfected at a final concentration of 50 nMusing Lipofectamine 2000 (Invitrogen) as transfection reagent.SiRNAs for SPROUTY2 were synthesized from Dharmacon.The target sequence for SPROUTY2 siRNA is 5′-GCAGGUA-CACGUCUUGUCU-3′. Ambion pre-miR miRNA precursor ofhsa-miR-23b, hsa-miR-27b, or negative control was synthesizedfrom Applied Biosystems. Adenovirus expressing miR-23b, miR-27b, or lacZ was generated from mouse genomic DNA encodingmiR-23b or -27 as described (1).
Cell Culture, Cell Proliferation, Scratch-Wound, in Vitro Matrigel, andAortic Ring Sprouting Assays.HUVEC (ATCC) cells were grown inEC growth medium (Lonza). SEMA6A recombinant protein wasobtained from R&D. For VEGF treatment, HUVECs werestarved with EC basal medium-2 with 0.1% FBS for 24 h and thentreated with VEGF (20 ng/mL) for the indicated periods of time.For LPS treatment, HUVECs were treated with LPS (100 ng/mL)for the indicated periods of time without starvation. Adenovirusinfection, siRNA, pre-miR miRNA precursor, or LNA-anti-miRtransfection in cell culture and aortic ring culture was performedas described (2). EC cell proliferation and scratch-wound assayswere performed using HUVEC cells as described (1, 3). For cel-lular proliferation assay, about 2 × 103 transfected HUVECs wereseeded in 24-well plates. After starvation with 0.1% serum forovernight, the cells were stimulated with 20 ng/mL VEGF-A for20 h and then subjected to BrDU labeling for 4 h. DNA synthesisas determined by BrDU incorporation was quantified usinga commercial ELISA kit from Roche according to the manu-facturer’s instructions. For scratch wound assay, scratch-woundwas made using a 200-μL pipette tip in LNA anti-miR–transfectedHUVEC monolayer before VEGF (20 ng/mL) stimulation. Then1 μM of 5-fluouracil (Sigma) was added to the cell right afterscratch wound to block cell proliferation. Post-scratch EC mi-gration was scored at 14 h after wound scratch. In vitro angio-genesis assays were performed as described (4). Briefly, 3 d afterLNA anti-miR transfection with Liptofectamine Plus reagent(Invitrogen), cells were harvested for either RNA analysis or invitro angiogenesis. Matrigel was purchased from Chemicon, andthe assays were performed according to manufacturer’s manual.Briefly, HUVECs were transfected with anti-miR or scramblecontrol using lipofectamine 2000 and cultured for 48 h, serumstarved overnight, then trypsinized and used for Matrigel assay.Quantification of branch points per field (20×) was done on sixfields and six sample repeats.
RNA, Western Blot Analysis, and Reporter Assay. Total RNA wasisolated from mouse tissues or cell lines using TRIzol reagent(Invitrogen). Northern blots to detect microRNAs were per-formed as described (2). Real-time RT-PCR using Sybergreenprobes was performed using 1 μg of RNA as a template withrandom hexamer primers to generate cDNA. miRNA real-timeRT-PCR was performed using miRCURY LNA Universal mi-croRNA RT-PCR system (Exiqon). For PCR and cloning ofmiR-23∼27∼24 pri-miRNAs, Race-Ready cDNA from mouseembryos (Ambion) was used. Sequences of PCR primers forSPROUTY2, SPROUTY2 (miR-23m), SPROUTY2 (miR-27m),SEMA6D 3′UTRs, and mouse miR-23a∼27a∼24–2 and miR-23b∼27b∼24–1 pri-miRNA are listed as follows:
i) hSPROUTY2-3′UTR (SacI): 5′-atcg gagctc AGCAA-CACAGACACTCCTAGGCA-3′
ii) hSPROUTY2-3′UTR (HindIII): 5′-atcg aagctt GCAT-CTGTAACCCCTCATTTGCAGC-3′
iii) hSPROUTY2 (miR-27mut) up: 5′-caataatatttgcacagactc-caaacaagttgtgc-3′
iv) hSPROUTY2 (miR-27mut) dn: 5′-gcacaacttgtttggagtctg-tgcaaatattattg-3′
v) hSPROUTY2 (miR-23mut1) up: 5′-gtacattcggaagccgaca-gatcaatcagtatg-3′
vi) hSPROUTY2 (miR-23mut1) dn: 5′-catactgattgatctgtcgg-cttccgaatgtac-3′
vii) hSPROUTY2 (miR-23mut2)dn: 5′-atcg aagctt gcatctgta-acccctcatttgcagcaactcgag tcgcctcataa aaggggc-3′
viii) hSEMA6D-3′UTR (SacI): 5′- atcggagctcCCCACTGGG-GCGAAGGTGGA-3′
ix) hSEMA6D-3′UTR (HindIII): 5′-atcgaagcttAGGGTT-GCGCATCATCAGCCGT-3′
x) mPri-miR-23a∼27a∼24–2 (up): 5′- CTGGTGCATTCG-GAAACCTTGTGT -3′
xi) mPri-miR-23a∼27a∼24–2 (dn): 5′- ATTGGAGCATT-CTTGCTTGCCTGC -3′
xii) mPri-miR-23b∼27b∼24–1 (up): 5′- ATGAAAGAGACG-CACTAGCCCACA -3′
xiii) mPri-miR-23b∼27b∼24–1 (dn): 5′- TTGGGTTCCTGG-CATGCTGATTTG -3′
ForWestern blot analysis, protein lysates were resolved by SDS/PAGE and blotted using standard procedures. Antibodies usedwere as follows: ERK1/2 (Cell Signaling), phospho-ERK1/2 (CellSignaling), phospho-AKT(Cell Signaling), SPROUTY2(Abcam),SEMA6A (R&D), SEMA6D (R&D), and GAPDH (Abcam) asloading control. Band intensity was quantified using NationalInstitutes of Health ImageJ software and normalized to corre-spondent GAPDH signal. For reporter assay, the 3′UTR ofSPROUTY2 or SEMA6D was inserted into the pMIR-REPORTvector (Ambion). SEMA6A 3′UTR construct was purchased fromGenecopeia. SPROUTY2 3′UTRs with mutations in the regioncomplementary to the miR-23 or miR-27 seed regions weregenerated by mutagenesis. Pre-miR miRNA precursors (ABI)were cotransfected with the UTR plasmids indicated into COS-7cells. Reporter assays were performed as described (5).
Neonatal Retinal Injection and Postnatal Retinal Angiogenesis. Invivo injection in the mouse retina was performed primarily asdescribed (6). Briefly, 1 μL of 5 mg/mL solution of LNA-anti-miR23 and miR-27 or LNA-scramble control was unilaterally in-jected intravitreously into the retina of P2 mice in the ICR back-ground. Mice were allowed to develop for RNA, protein isolation,
Zhou et al. www.pnas.org/cgi/content/short/1105254108 1 of 5

and histological analyses at P6. Visualization of the vasculaturewas performed by Alexa-594 conjugated isolectin B4 (MolecularProbes) or ICAM-2 staining of retinal flat mounts. Quantificationof vessel density was performed usingNational Institutes ofHealthImageJ software. The radial length of the vascular network wascalculated by measuring the distance from the optic disk to theperiphery of the vascular plexus. Student t tests were used to de-termine statistical significance between groups.
Laser-Induced CNV. Laser photocoagulation was induced in 6- to 8-wk-old male C57BL/6J mice as described (7). Briefly, the pupils ofanesthetized animals were dilated with 1% tropicamide (AlconLaboratories). Three 532-nm diode laser spots (140 mW, 100 ms,100 μm; OcuLight GL Photocoagulator, Iridex) were applied toeach fundus of adult mice using a coverslip as a contact lens. Sixlaser spots were applied when the samples were used for RNAisolation. Formation of a bubble at the time of laser applicationindicates rupture of Bruch’s membrane and successful laser injury.Animals were intravitreously injected with 1 μL of 5 mg/mL
solution of LNA-anti-miR23/27, LNA-scramble or PBS injectioncontrol after laser photocoagulation. A secondary injection wasadopted on the next day to ensure maximal knockdown of miR-23/27. The retina/choroid/sclera complexes from the treated eyeswere collected 7 d after laser injury for fluorescein angiography,RNA, and protein analyses. At 14 d after laser injury, the eyes werefixed in 4% paraformaldehyde for 30 min at room temperature.The retina/choroid/sclera complexes were then dissected and fixedfor frozen section or flatmount staining. For flatmount staining,the samples were postfixed for 1 h, incubated with blocking buffer(PBS with 0.5% Triton X-100 and 5% goat serum), and stainedwith ICAM-2 antibody at 4 °C overnight. After washing and sec-ondary antibody staining, the samples were flat mounted on glassslides. Images of CNV were captured using a Leica SP2 multi-photon Laser Scanning confocal microscope, and CNV volumewas quantified using National Institutes of Health ImageJ soft-ware. Student t tests were used to determine statistical significancebetween groups.
1. Wang S, et al. (2008) Control of endothelial cell proliferation and migration by VEGFsignaling to histone deacetylase 7. Proc Natl Acad Sci USA 105:7738–7743.
2. Wang S, et al. (2008) The endothelial-specific microRNA miR-126 governs vascularintegrity and angiogenesis. Dev Cell 15:261–271.
3. Lee KS, et al. (2006) Troglitazone inhibits endothelial cell proliferation throughsuppression of casein kinase 2 activity. Biochem Biophys Res Commun 346:83–88.
4. Chang S, et al. (2006) Histone deacetylase 7 maintains vascular integrity by repressingmatrix metalloproteinase 10. Cell 126:321–334.
5. Chang S, Bezprozvannaya S, Li S, Olson EN (2005) An expression screen revealsmodulators of class II histone deacetylase phosphorylation. Proc Natl Acad Sci USA 102:8120–8125.
6. Matsuda T, Cepko CL (2004) Electroporation and RNA interference in the rodent retinain vivo and in vitro. Proc Natl Acad Sci USA 101:16–22.
7. Tobe T, et al. (1998) Targeted disruption of the FGF2 gene does not prevent choroidalneovascularization in a murine model. Am J Pathol 153:1641–1646.
Zhou et al. www.pnas.org/cgi/content/short/1105254108 2 of 5
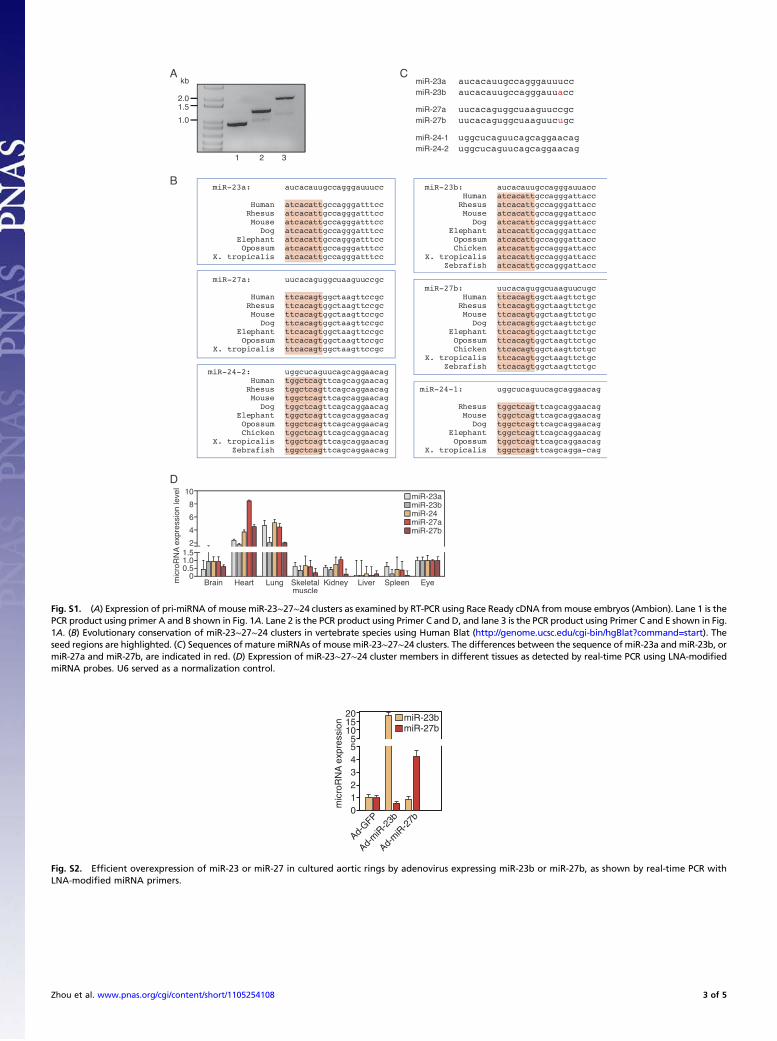
Fig. S1. (A) Expression of pri-miRNA of mouse miR-23∼27∼24 clusters as examined by RT-PCR using Race Ready cDNA frommouse embryos (Ambion). Lane 1 is thePCR product using primer A and B shown in Fig. 1A. Lane 2 is the PCR product using Primer C and D, and lane 3 is the PCR product using Primer C and E shown in Fig.1A. (B) Evolutionary conservation of miR-23∼27∼24 clusters in vertebrate species using Human Blat (http://genome.ucsc.edu/cgi-bin/hgBlat?command=start). Theseed regions are highlighted. (C) Sequences of mature miRNAs of mouse miR-23∼27∼24 clusters. The differences between the sequence of miR-23a and miR-23b, ormiR-27a and miR-27b, are indicated in red. (D) Expression of miR-23∼27∼24 cluster members in different tissues as detected by real-time PCR using LNA-modifiedmiRNA probes. U6 served as a normalization control.
20
0
Ad-GFP
Ad-m
iR-2
7b
Ad-m
iR-2
3b
1
55
mic
roR
NA
exp
ress
ion miR-23b
miR-27b1510
234
Fig. S2. Efficient overexpression of miR-23 or miR-27 in cultured aortic rings by adenovirus expressing miR-23b or miR-27b, as shown by real-time PCR withLNA-modified miRNA primers.
Zhou et al. www.pnas.org/cgi/content/short/1105254108 3 of 5
Pathways regulated by miR-23 and miR-27 predicted by DIANA Lab (DIANA-mirPath)
KEGG Pathway(Rank)
Gene Names Foundgenes
-ln(p-value)(p-value)
27 12.39(0.00018)
miR-23 ORmiR-27
Axon guidance (1) EFNB2, SRGAP3, SEMA6A, NCK2, SLIT1, CXCL12, ITGB1, LIMK1, LIMK2, EFNA3, EPHB2, MET, SEMA6D, SRGAP2, KRAS, PAK3, FYN, CFL2, EPHA7, SEMA4C, UNC5D, CDC42, PAK2, PAK6, ROBO2, SEMA3B, SEMA4F
11 14.54(4.2x10-5)
miR-23 ANDmiR-27
MAPK signalingpathway (1)
EVI1, CACNB2, MEF2C, DUSP5, SOS1, RPS6KA5, NLK, NF1, MAPK14, MAP3K12, SPROUTY2
5 3.85(0.07)
Axon guidance (5) MET, SEMA6A, SEMA6D, FYN, PAK6
37 4.78(0.013)
MAPK signalingpathway (8)
MAP4K3, MAP4K4, PRKY, PDGFRA, EVI1, TGFBR1, MAP3K4, MAP3K1, MAPKAPK3, KRAS, CACNB2, MEF2C, MKNK2, DUSP5, STK4, MAP2K4, SOS1, DUSP16, PRKX, MAP3K3, RPS6KA5, FGF1, CDC25B, CDC42, GRB2, PAK2, MAP2K7, RAP1B, NLK, NF1, TGFBR2, MAPK14, MAP3K12, RPS6KA3, MAP3K5, MAPK10, SPROUTY2
A
BGenename
SPROUTY2
SEMA6A
SEMA6D
miRNA
miR-23a/b
miR-27a/b
miR-23a/b
miR-27a/b
miR-23a/b
miR-27a/b
Position
436-464
678-706
360-388
1832-1860
546-574
1250-1278
1110-1128
2388-2416
1590-1618
2748-2776
2777-2805
3359-3387
3051-3079
2158-2186
1526-1554
Site
2 ; 8mer
1 ; 7mer
1 ; 8mer
1 ; 6mer
1 ; 8mer
1 ; 7mer
1 ; 7mer
1 ; 7mer
2 ; 8mer
2 ; 8mer
2 ; 8mer
2 ; 8mer
2 ; 8mer
1 ; 10mer
1 ; 7mer
Conservation(species#)
11
5
13
7
11
8
5
3
11
7
5
9
1
9
2
3’UTR (sequencecomplementary to miRNAseeds highlighted in red)
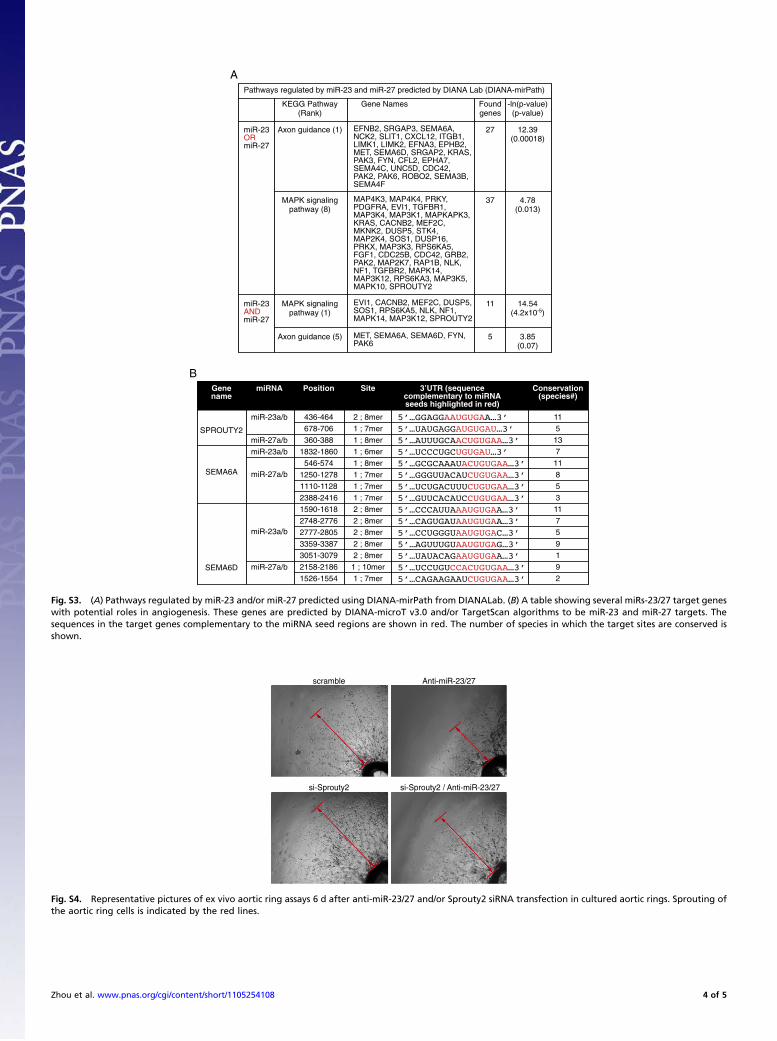
Fig. S3. (A) Pathways regulated by miR-23 and/or miR-27 predicted using DIANA-mirPath from DIANALab. (B) A table showing several miRs-23/27 target geneswith potential roles in angiogenesis. These genes are predicted by DIANA-microT v3.0 and/or TargetScan algorithms to be miR-23 and miR-27 targets. Thesequences in the target genes complementary to the miRNA seed regions are shown in red. The number of species in which the target sites are conserved isshown.
scramble Anti-miR-23/27
si-Sprouty2 si-Sprouty2 / Anti-miR-23/27y
Fig. S4. Representative pictures of ex vivo aortic ring assays 6 d after anti-miR-23/27 and/or Sprouty2 siRNA transfection in cultured aortic rings. Sprouting ofthe aortic ring cells is indicated by the red lines.
Zhou et al. www.pnas.org/cgi/content/short/1105254108 4 of 5

6000
0
2000
4000
CN
V a
rea
(µm
2 )
Scram
ble
No inj
ectio
n
Bns
A3
0
miR
-27a
miR
-24
miR
-16
miR
-27b
miR
-23b
miR
-23a
1
2
mic
roR
NA
exp
ress
ion
leve
l
controlLPS (4hr)p<0.03
*
p<0.0001*
nsns
p<0.002
*
p<0.0001
*
Fig. S5. (A) Expression of miR-23∼27∼24 cluster members in HUVECs induced by LPS, as detected using LNA-modified miRNA PCR primers. U6 served asa normalization control. (B) Quantification of CNV area from ICAM-2 staining of the choroid l flatmounts measured from control-injected retinas andno-injection control retinas 14 d after laser injury. ns, not significant.
Zhou et al. www.pnas.org/cgi/content/short/1105254108 5 of 5




















